User login
Treating comorbid posttraumatic stress disorder and cardiovascular disease
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1

Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.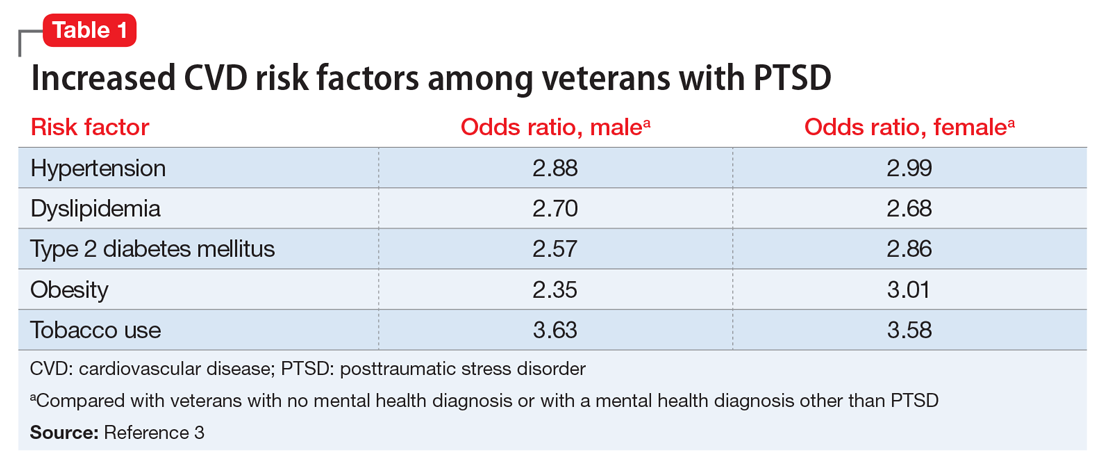
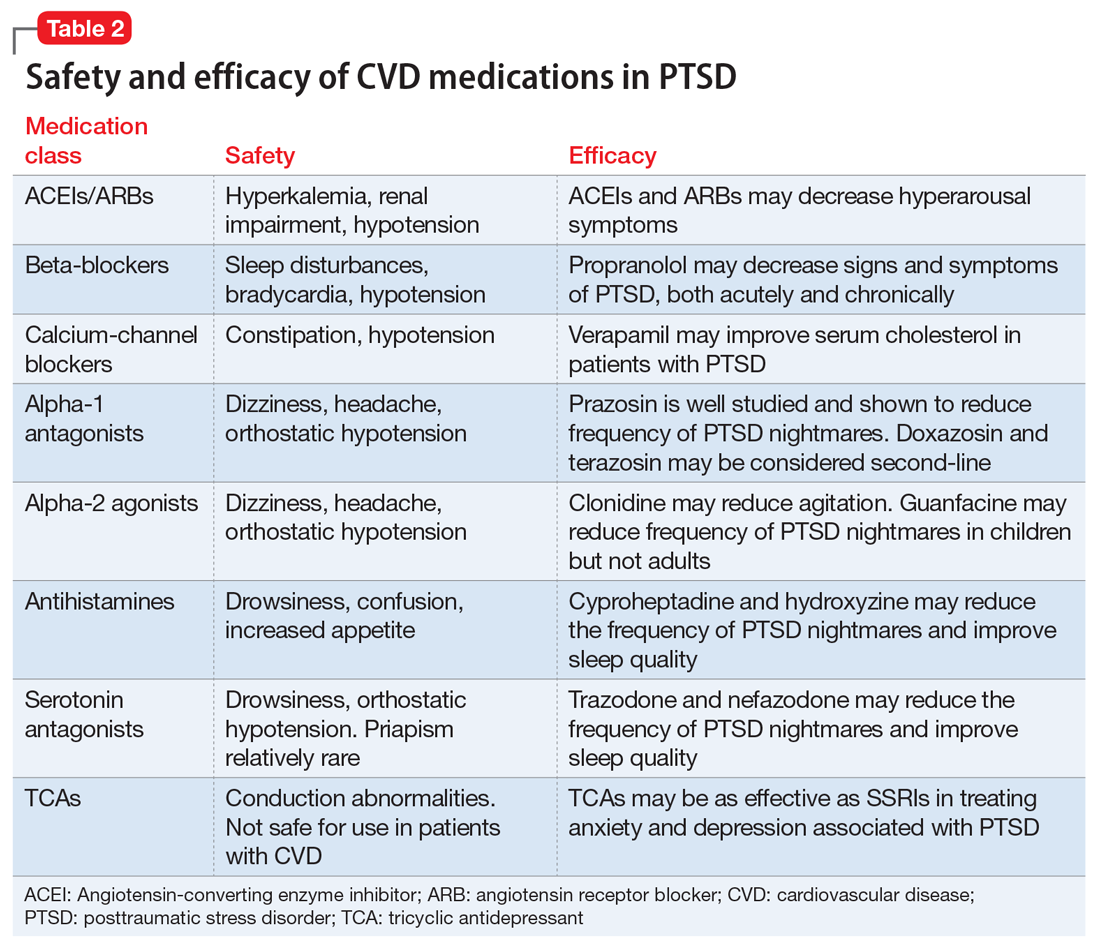
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15

Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1
Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15
Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1
Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15
Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
Paranoia and suicidality after starting treatment for lupus
CASE Unusual behavior, thoughts
Mr. L, age 28, an immigrant from Burma, is brought to his primary care physician’s clinic by his wife for follow-up on a rash. During the evaluation, his wife reports that Mr. L recently has had suicidal ideation, depression, and increased anger. She says Mr. L had made statements about wanting to kill himself with a gun. Mr. L had driven his car to a soccer field with a knife in hand and was contemplating suicide. She is concerned about her own safety and their children’s safety because of Mr. L’s anger. The physician refers Mr. L to the emergency department, and he is admitted to the medical floor for a rheumatological flare-up and suicidal ideation.
Mr. L starts displaying inappropriate behaviors, including masturbating in front of the patient safety attendant, telling the attendant “You are going to die today,” and assaulting a female attendant by trying to grab her breasts. He is given IM haloperidol, 2 mg, which effectively alleviates these behaviors. Between episodes of unusual behavior and outbursts, Mr. L is docile, quiet, and cooperative, and denies any memory of these episodes.
One month earlier, Mr. L had been hospitalized for progressive weakness and inability to ambulate. He was diagnosed with necrotizing myositis and a rash consistent with subacute cutaneous lupus. He was started on IV methylprednisolone, 1 g, and transitioned to oral prednisolone, 40 mg/d, which he continued taking after discharge. He also started taking azathioprine, which was increased from 50 to 100 mg/d. His condition improved shortly after beginning this regimen.
[polldaddy:9796586]
The authors’ observations
DSM-5 defines brief psychotic disorder as positive symptoms or disorganized or catatonic behavior appearing suddenly and lasting between 1 day to 1 month.1 Mr. L had a sudden onset of his symptoms and marked stressors as a result of his worsening health. However, the possibility of his general medical conditions or medications causing his symptoms needed to be investigated and ruled out before this diagnosis could be assigned.
Another consideration is the culture-bound syndrome amok. Although DSM-5 does not use the term “culture-bound syndrome,” which was used in DSM-IV, it does recognize cultural conceptualizations of distress. Amok is described as a dissociative episode in which an individual has a period of brooding followed by outbursts that include violent, aggressive, and suicidal and/or homicidal ideation. The individual may exhibit persecutory and paranoid thinking, amnesia of the outbursts, and a return to typical behavior when the episode concludes.2 However, it remained unclear whether Mr. L’s violent behavior was a manifestation of psychiatric or organic disease.
Identifying the possibility of amok is important not only for alleviating the patient’s distress but also for preventing violent outbursts that can result in injury or death.3 Amok should be considered only in the context of possible psychiatric or organic brain disease, such as corticosteroid-induced psychosis (CIP) or systemic lupus erythematosus-induced psychosis (SLEIP).4
EVALUATION Informants, labs
Mr. L immigrated to the United States when he was 5 years old. He does not speak English, and interviews are conducted with interpreting services at the hospital. Mr. L answers most questions with or 1 to 2 words. His medical and psychiatric histories are notable for hypothyroidism, hepatitis, non-ischemic cardiomyopathy, necrotizing myositis, subacute cutaneous lupus, and depression. Mr. L denies a personal or family history of mental illness; however, records show he has a history of unspecified depressive disorder.
Mr. L reports his current mood is “okay,” but he has felt different in the past few weeks. He denies auditory or visual hallucinations, or suicidal or homicidal ideation, but exhibits paranoid thoughts. Mr. L believes everyone “lied” to him, and he repeats this frequently. Collateral information from friends reveals that he had threatened to burn down their houses. A family friend states that Mr. L has been depressed and angry over the past 5 days.
During his prior and current hospitalizations, many labs were completed. Thyroid, urine drug screen, C-reactive protein, urine analysis, ethanol, complete blood count, and comprehensive metabolic panel were negative. Erythrocyte sedimentation rate was 30. Lumbar puncture cell count was notable for mildly elevated lymphocytes at 84%. Antinuclear antibody (ANA) was positive. Lupus anticoagulant panel revealed a mildly prolonged partial thromboplastin time at 38.9 seconds. DNA double-stranded antibody (anti-dsDNA) was positive. Anti-Smith antibody was negative. Anti-Ro/SSA and anti-La/SSB antibodies were elevated. Albumin was low. A MRI of the brain showed dystrophic-appearing right parieto-occipital calcification and mild cerebral volume loss.
Based on Mr. L’s presentation and imaging, the rheumatology team suspects CNS lupus and that his prescribed steroids could be playing a role in his behavior.
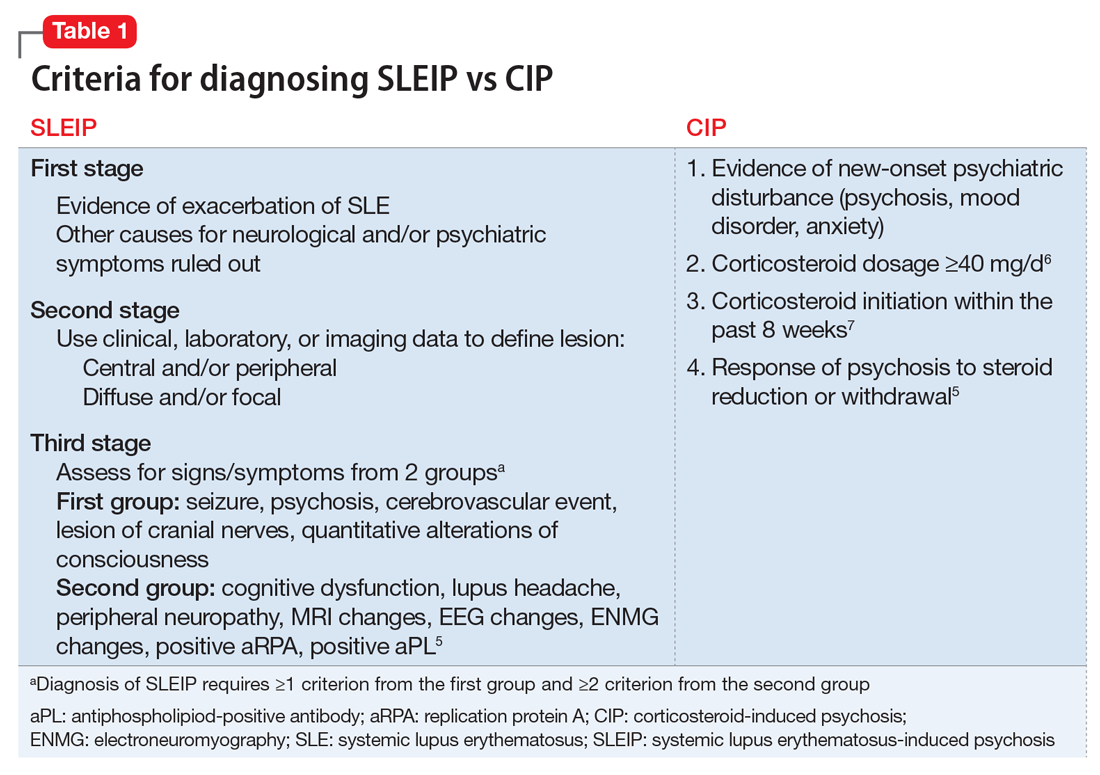
The authors’ observations
Differentiating CIP from SLEIP can be difficult. The clinical features and criteria for CIP and SLEIP are listed in Table 1.5-7 Several studies have highlighted the difficulties in separating the 2 diagnoses:
- Kampylafka et al8 found that CNS involvement, including stroke, myelopathy, seizures, optic neuritis, and meningitis, was present in 4.3% of their sample of patients with systematic lupus erythematosus (SLE), of whom 6.3% presented with SLEIP. Of patients with CNS involvement, 94% had positive ANA and 69% had positive anti-dsDNA antibodies. It remains difficult to definitively diagnose SLEIP rather than CIP, however, because 100% of patients in this study were taking corticosteroids, with 25% taking azathioprine, as was Mr. L.8
- Appenzeller et al9 found that acute psychosis was associated with SLE in 11.3% of their sample. Psychosis in patients with SLE was accompanied by other manifestations of CNS involvement. On follow-up these patients had mild increases in white blood cell count in their CSF, and MRI demonstrated hyperdense lesions and cerebral atrophy. Hypoalbuminemia, although often seen in SLEIP, also is observed in patients with CIP and cannot be used to differentiate these 2 conditions.9
- Monov and Monova5 recommended criteria for SLEIP that include 3 stages. The first stage is determining that there is evidence of an exacerbation of SLE, and ruling out other causes for neurologic and psychiatric symptoms. The second stage involves using clinical, laboratory, or imaging tests to define the lesion as central and/or peripheral and diffuse and/or focal. The third stage requires diagnosing SLEIP using criteria from 2 groups of signs and symptoms: the first group includes seizure, psychosis, cerebrovascular event, lesion of cranial nerves, and quantitative alterations of consciousness; the second group includes cognitive dysfunction, lupus headache, peripheral neuropathy, MRI changes, EEG changes, electroneuromyography changes, and a positive replication protein A or antiphospholipid-positive antibody. Diagnosing SLEIP requires ≥1 criterion from group 1 and ≥2 criteria from group 2.5
- Patten and Neutel6 found that patients taking prednisolone, Symbol Std<40 mg/d, had significantly higher rates of psychosis than those taking <40 mg/d.6
- Bhangle et Myriad Proal7 found that one of the major distinguishing factors between CIP and SLEIP is the timing of the onset of symptoms, with CIP occurring within 8 weeks of initiation of a corticosteroid, and SLEIP being more likely to occur when additional CNS symptoms are present.7
TREATMENT Decreased dosage
Mr. L starts quetiapine, 25 mg at bedtime, increased to 75 mg at bedtime. Prednisolone is decreased to 10 mg/d. Over the next few days Mr. L’s mood, psychosis, and aggression improve. He becomes calm and cooperative, and denies suicidal or homicidal ideation. Mr. L’s wife, who was initially scared to visit him, comes to see him and confirms that he has improved. After 3 consecutive days with no abnormal behaviors or psychiatric symptoms, Mr. L is discharged and continues taking quetiapine, 75 mg at bedtime, and prednisolone, 10 mg/d, with outpatient follow-up.
The authors’ observations
Table 210,11 describes approaches to treating CIP and SLEIP. Managing CIP typically consists of reducing the corticosteroid dosage. CIP treatment also includes adjunct therapy with psychotropics if the corticosteroid dose cannot be lowered enough to reduce psychiatric symptoms while suppressing symptoms of the disease for which the corticosteroid was prescribed.6

When treating SLEIP, the corticosteroid dosage often is increased. Corticosteroids often are used to treat SLEIP while suppressing symptoms of SLE.10 The main treatment of SLEIP is focused on the disease and using psychotropic medications to control symptoms that don’t respond after exacerbation of the disease has been controlled.10
The presence of Mr. L’s multiple SLE symptoms, as well as MRI findings, could indicate SLEIP. However, corticosteroids also were a possible cause of his psychotic symptoms. Mr. L’s psychosis began within 8 weeks of starting a corticosteroid (prednisolone, 40 mg/d), and his symptoms improved when the corticosteroid dosage was reduced. The difference between CIP and SLEIP may best be distinguished by reducing the corticosteroid dosage and seeing if psychotic symptoms improve. Because it is important to control SLE symptoms in those with CIP, prescribing psychotropics may be warranted, as well as alternative treatments for immunosuppression.
Because steroids are frequently prescribed for lupus, it is important for clinicians to be aware of their psychiatric effects as well as how to manage those effects. When distinguishing CIP from SLEIP, consider decreasing the corticosteroid dosage and see if psychotic symptoms improve. Use adjunct therapy as needed.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Saint Martin ML. Running amok: A modern perspective on a culture-bound syndrome. Prim Care Companion J Clin Psychiatry. 1999;1(3):66-70.
4. Flaskerud JH. Case studies in amok? Issues Ment Health Nurs. 2012;33(12):898-900.
5. Monov S, Monova D. Classification criteria for neuropsychiatric systemic lupus erythematosus: do they need a discussion? Hippokratia. 2008;12(2):103-107.
6. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf. 2000;22(2):111-122.
7. Bhangle SD, Kramer N, Rosenstein, ED. Corticosteroid-induced neuropsychiatric disorders: review and contrast with neuropsychiatric lupus. Rheumatol Int. 2013;33(8):1923-1932.
8. Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One. 2013;8(2):e55843. d
9. Appenzeller S, Cendes F, Costallat LT. Acute psychosisin systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
10. Sanna G, Bertolaccini ML, Khamashta MA. Neuropsychiatric involvement in systemic lupus erythematosus: current therapeutic approach. Curr Pharm Des. 2008;14(13):1261-1269.
11. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
CASE Unusual behavior, thoughts
Mr. L, age 28, an immigrant from Burma, is brought to his primary care physician’s clinic by his wife for follow-up on a rash. During the evaluation, his wife reports that Mr. L recently has had suicidal ideation, depression, and increased anger. She says Mr. L had made statements about wanting to kill himself with a gun. Mr. L had driven his car to a soccer field with a knife in hand and was contemplating suicide. She is concerned about her own safety and their children’s safety because of Mr. L’s anger. The physician refers Mr. L to the emergency department, and he is admitted to the medical floor for a rheumatological flare-up and suicidal ideation.
Mr. L starts displaying inappropriate behaviors, including masturbating in front of the patient safety attendant, telling the attendant “You are going to die today,” and assaulting a female attendant by trying to grab her breasts. He is given IM haloperidol, 2 mg, which effectively alleviates these behaviors. Between episodes of unusual behavior and outbursts, Mr. L is docile, quiet, and cooperative, and denies any memory of these episodes.
One month earlier, Mr. L had been hospitalized for progressive weakness and inability to ambulate. He was diagnosed with necrotizing myositis and a rash consistent with subacute cutaneous lupus. He was started on IV methylprednisolone, 1 g, and transitioned to oral prednisolone, 40 mg/d, which he continued taking after discharge. He also started taking azathioprine, which was increased from 50 to 100 mg/d. His condition improved shortly after beginning this regimen.
[polldaddy:9796586]
The authors’ observations
DSM-5 defines brief psychotic disorder as positive symptoms or disorganized or catatonic behavior appearing suddenly and lasting between 1 day to 1 month.1 Mr. L had a sudden onset of his symptoms and marked stressors as a result of his worsening health. However, the possibility of his general medical conditions or medications causing his symptoms needed to be investigated and ruled out before this diagnosis could be assigned.
Another consideration is the culture-bound syndrome amok. Although DSM-5 does not use the term “culture-bound syndrome,” which was used in DSM-IV, it does recognize cultural conceptualizations of distress. Amok is described as a dissociative episode in which an individual has a period of brooding followed by outbursts that include violent, aggressive, and suicidal and/or homicidal ideation. The individual may exhibit persecutory and paranoid thinking, amnesia of the outbursts, and a return to typical behavior when the episode concludes.2 However, it remained unclear whether Mr. L’s violent behavior was a manifestation of psychiatric or organic disease.
Identifying the possibility of amok is important not only for alleviating the patient’s distress but also for preventing violent outbursts that can result in injury or death.3 Amok should be considered only in the context of possible psychiatric or organic brain disease, such as corticosteroid-induced psychosis (CIP) or systemic lupus erythematosus-induced psychosis (SLEIP).4
EVALUATION Informants, labs
Mr. L immigrated to the United States when he was 5 years old. He does not speak English, and interviews are conducted with interpreting services at the hospital. Mr. L answers most questions with or 1 to 2 words. His medical and psychiatric histories are notable for hypothyroidism, hepatitis, non-ischemic cardiomyopathy, necrotizing myositis, subacute cutaneous lupus, and depression. Mr. L denies a personal or family history of mental illness; however, records show he has a history of unspecified depressive disorder.
Mr. L reports his current mood is “okay,” but he has felt different in the past few weeks. He denies auditory or visual hallucinations, or suicidal or homicidal ideation, but exhibits paranoid thoughts. Mr. L believes everyone “lied” to him, and he repeats this frequently. Collateral information from friends reveals that he had threatened to burn down their houses. A family friend states that Mr. L has been depressed and angry over the past 5 days.
During his prior and current hospitalizations, many labs were completed. Thyroid, urine drug screen, C-reactive protein, urine analysis, ethanol, complete blood count, and comprehensive metabolic panel were negative. Erythrocyte sedimentation rate was 30. Lumbar puncture cell count was notable for mildly elevated lymphocytes at 84%. Antinuclear antibody (ANA) was positive. Lupus anticoagulant panel revealed a mildly prolonged partial thromboplastin time at 38.9 seconds. DNA double-stranded antibody (anti-dsDNA) was positive. Anti-Smith antibody was negative. Anti-Ro/SSA and anti-La/SSB antibodies were elevated. Albumin was low. A MRI of the brain showed dystrophic-appearing right parieto-occipital calcification and mild cerebral volume loss.
Based on Mr. L’s presentation and imaging, the rheumatology team suspects CNS lupus and that his prescribed steroids could be playing a role in his behavior.
The authors’ observations
Differentiating CIP from SLEIP can be difficult. The clinical features and criteria for CIP and SLEIP are listed in Table 1.5-7 Several studies have highlighted the difficulties in separating the 2 diagnoses:
- Kampylafka et al8 found that CNS involvement, including stroke, myelopathy, seizures, optic neuritis, and meningitis, was present in 4.3% of their sample of patients with systematic lupus erythematosus (SLE), of whom 6.3% presented with SLEIP. Of patients with CNS involvement, 94% had positive ANA and 69% had positive anti-dsDNA antibodies. It remains difficult to definitively diagnose SLEIP rather than CIP, however, because 100% of patients in this study were taking corticosteroids, with 25% taking azathioprine, as was Mr. L.8
- Appenzeller et al9 found that acute psychosis was associated with SLE in 11.3% of their sample. Psychosis in patients with SLE was accompanied by other manifestations of CNS involvement. On follow-up these patients had mild increases in white blood cell count in their CSF, and MRI demonstrated hyperdense lesions and cerebral atrophy. Hypoalbuminemia, although often seen in SLEIP, also is observed in patients with CIP and cannot be used to differentiate these 2 conditions.9
- Monov and Monova5 recommended criteria for SLEIP that include 3 stages. The first stage is determining that there is evidence of an exacerbation of SLE, and ruling out other causes for neurologic and psychiatric symptoms. The second stage involves using clinical, laboratory, or imaging tests to define the lesion as central and/or peripheral and diffuse and/or focal. The third stage requires diagnosing SLEIP using criteria from 2 groups of signs and symptoms: the first group includes seizure, psychosis, cerebrovascular event, lesion of cranial nerves, and quantitative alterations of consciousness; the second group includes cognitive dysfunction, lupus headache, peripheral neuropathy, MRI changes, EEG changes, electroneuromyography changes, and a positive replication protein A or antiphospholipid-positive antibody. Diagnosing SLEIP requires ≥1 criterion from group 1 and ≥2 criteria from group 2.5
- Patten and Neutel6 found that patients taking prednisolone, Symbol Std<40 mg/d, had significantly higher rates of psychosis than those taking <40 mg/d.6
- Bhangle et Myriad Proal7 found that one of the major distinguishing factors between CIP and SLEIP is the timing of the onset of symptoms, with CIP occurring within 8 weeks of initiation of a corticosteroid, and SLEIP being more likely to occur when additional CNS symptoms are present.7
TREATMENT Decreased dosage
Mr. L starts quetiapine, 25 mg at bedtime, increased to 75 mg at bedtime. Prednisolone is decreased to 10 mg/d. Over the next few days Mr. L’s mood, psychosis, and aggression improve. He becomes calm and cooperative, and denies suicidal or homicidal ideation. Mr. L’s wife, who was initially scared to visit him, comes to see him and confirms that he has improved. After 3 consecutive days with no abnormal behaviors or psychiatric symptoms, Mr. L is discharged and continues taking quetiapine, 75 mg at bedtime, and prednisolone, 10 mg/d, with outpatient follow-up.
The authors’ observations
Table 210,11 describes approaches to treating CIP and SLEIP. Managing CIP typically consists of reducing the corticosteroid dosage. CIP treatment also includes adjunct therapy with psychotropics if the corticosteroid dose cannot be lowered enough to reduce psychiatric symptoms while suppressing symptoms of the disease for which the corticosteroid was prescribed.6
When treating SLEIP, the corticosteroid dosage often is increased. Corticosteroids often are used to treat SLEIP while suppressing symptoms of SLE.10 The main treatment of SLEIP is focused on the disease and using psychotropic medications to control symptoms that don’t respond after exacerbation of the disease has been controlled.10
The presence of Mr. L’s multiple SLE symptoms, as well as MRI findings, could indicate SLEIP. However, corticosteroids also were a possible cause of his psychotic symptoms. Mr. L’s psychosis began within 8 weeks of starting a corticosteroid (prednisolone, 40 mg/d), and his symptoms improved when the corticosteroid dosage was reduced. The difference between CIP and SLEIP may best be distinguished by reducing the corticosteroid dosage and seeing if psychotic symptoms improve. Because it is important to control SLE symptoms in those with CIP, prescribing psychotropics may be warranted, as well as alternative treatments for immunosuppression.
Because steroids are frequently prescribed for lupus, it is important for clinicians to be aware of their psychiatric effects as well as how to manage those effects. When distinguishing CIP from SLEIP, consider decreasing the corticosteroid dosage and see if psychotic symptoms improve. Use adjunct therapy as needed.
CASE Unusual behavior, thoughts
Mr. L, age 28, an immigrant from Burma, is brought to his primary care physician’s clinic by his wife for follow-up on a rash. During the evaluation, his wife reports that Mr. L recently has had suicidal ideation, depression, and increased anger. She says Mr. L had made statements about wanting to kill himself with a gun. Mr. L had driven his car to a soccer field with a knife in hand and was contemplating suicide. She is concerned about her own safety and their children’s safety because of Mr. L’s anger. The physician refers Mr. L to the emergency department, and he is admitted to the medical floor for a rheumatological flare-up and suicidal ideation.
Mr. L starts displaying inappropriate behaviors, including masturbating in front of the patient safety attendant, telling the attendant “You are going to die today,” and assaulting a female attendant by trying to grab her breasts. He is given IM haloperidol, 2 mg, which effectively alleviates these behaviors. Between episodes of unusual behavior and outbursts, Mr. L is docile, quiet, and cooperative, and denies any memory of these episodes.
One month earlier, Mr. L had been hospitalized for progressive weakness and inability to ambulate. He was diagnosed with necrotizing myositis and a rash consistent with subacute cutaneous lupus. He was started on IV methylprednisolone, 1 g, and transitioned to oral prednisolone, 40 mg/d, which he continued taking after discharge. He also started taking azathioprine, which was increased from 50 to 100 mg/d. His condition improved shortly after beginning this regimen.
[polldaddy:9796586]
The authors’ observations
DSM-5 defines brief psychotic disorder as positive symptoms or disorganized or catatonic behavior appearing suddenly and lasting between 1 day to 1 month.1 Mr. L had a sudden onset of his symptoms and marked stressors as a result of his worsening health. However, the possibility of his general medical conditions or medications causing his symptoms needed to be investigated and ruled out before this diagnosis could be assigned.
Another consideration is the culture-bound syndrome amok. Although DSM-5 does not use the term “culture-bound syndrome,” which was used in DSM-IV, it does recognize cultural conceptualizations of distress. Amok is described as a dissociative episode in which an individual has a period of brooding followed by outbursts that include violent, aggressive, and suicidal and/or homicidal ideation. The individual may exhibit persecutory and paranoid thinking, amnesia of the outbursts, and a return to typical behavior when the episode concludes.2 However, it remained unclear whether Mr. L’s violent behavior was a manifestation of psychiatric or organic disease.
Identifying the possibility of amok is important not only for alleviating the patient’s distress but also for preventing violent outbursts that can result in injury or death.3 Amok should be considered only in the context of possible psychiatric or organic brain disease, such as corticosteroid-induced psychosis (CIP) or systemic lupus erythematosus-induced psychosis (SLEIP).4
EVALUATION Informants, labs
Mr. L immigrated to the United States when he was 5 years old. He does not speak English, and interviews are conducted with interpreting services at the hospital. Mr. L answers most questions with or 1 to 2 words. His medical and psychiatric histories are notable for hypothyroidism, hepatitis, non-ischemic cardiomyopathy, necrotizing myositis, subacute cutaneous lupus, and depression. Mr. L denies a personal or family history of mental illness; however, records show he has a history of unspecified depressive disorder.
Mr. L reports his current mood is “okay,” but he has felt different in the past few weeks. He denies auditory or visual hallucinations, or suicidal or homicidal ideation, but exhibits paranoid thoughts. Mr. L believes everyone “lied” to him, and he repeats this frequently. Collateral information from friends reveals that he had threatened to burn down their houses. A family friend states that Mr. L has been depressed and angry over the past 5 days.
During his prior and current hospitalizations, many labs were completed. Thyroid, urine drug screen, C-reactive protein, urine analysis, ethanol, complete blood count, and comprehensive metabolic panel were negative. Erythrocyte sedimentation rate was 30. Lumbar puncture cell count was notable for mildly elevated lymphocytes at 84%. Antinuclear antibody (ANA) was positive. Lupus anticoagulant panel revealed a mildly prolonged partial thromboplastin time at 38.9 seconds. DNA double-stranded antibody (anti-dsDNA) was positive. Anti-Smith antibody was negative. Anti-Ro/SSA and anti-La/SSB antibodies were elevated. Albumin was low. A MRI of the brain showed dystrophic-appearing right parieto-occipital calcification and mild cerebral volume loss.
Based on Mr. L’s presentation and imaging, the rheumatology team suspects CNS lupus and that his prescribed steroids could be playing a role in his behavior.
The authors’ observations
Differentiating CIP from SLEIP can be difficult. The clinical features and criteria for CIP and SLEIP are listed in Table 1.5-7 Several studies have highlighted the difficulties in separating the 2 diagnoses:
- Kampylafka et al8 found that CNS involvement, including stroke, myelopathy, seizures, optic neuritis, and meningitis, was present in 4.3% of their sample of patients with systematic lupus erythematosus (SLE), of whom 6.3% presented with SLEIP. Of patients with CNS involvement, 94% had positive ANA and 69% had positive anti-dsDNA antibodies. It remains difficult to definitively diagnose SLEIP rather than CIP, however, because 100% of patients in this study were taking corticosteroids, with 25% taking azathioprine, as was Mr. L.8
- Appenzeller et al9 found that acute psychosis was associated with SLE in 11.3% of their sample. Psychosis in patients with SLE was accompanied by other manifestations of CNS involvement. On follow-up these patients had mild increases in white blood cell count in their CSF, and MRI demonstrated hyperdense lesions and cerebral atrophy. Hypoalbuminemia, although often seen in SLEIP, also is observed in patients with CIP and cannot be used to differentiate these 2 conditions.9
- Monov and Monova5 recommended criteria for SLEIP that include 3 stages. The first stage is determining that there is evidence of an exacerbation of SLE, and ruling out other causes for neurologic and psychiatric symptoms. The second stage involves using clinical, laboratory, or imaging tests to define the lesion as central and/or peripheral and diffuse and/or focal. The third stage requires diagnosing SLEIP using criteria from 2 groups of signs and symptoms: the first group includes seizure, psychosis, cerebrovascular event, lesion of cranial nerves, and quantitative alterations of consciousness; the second group includes cognitive dysfunction, lupus headache, peripheral neuropathy, MRI changes, EEG changes, electroneuromyography changes, and a positive replication protein A or antiphospholipid-positive antibody. Diagnosing SLEIP requires ≥1 criterion from group 1 and ≥2 criteria from group 2.5
- Patten and Neutel6 found that patients taking prednisolone, Symbol Std<40 mg/d, had significantly higher rates of psychosis than those taking <40 mg/d.6
- Bhangle et Myriad Proal7 found that one of the major distinguishing factors between CIP and SLEIP is the timing of the onset of symptoms, with CIP occurring within 8 weeks of initiation of a corticosteroid, and SLEIP being more likely to occur when additional CNS symptoms are present.7
TREATMENT Decreased dosage
Mr. L starts quetiapine, 25 mg at bedtime, increased to 75 mg at bedtime. Prednisolone is decreased to 10 mg/d. Over the next few days Mr. L’s mood, psychosis, and aggression improve. He becomes calm and cooperative, and denies suicidal or homicidal ideation. Mr. L’s wife, who was initially scared to visit him, comes to see him and confirms that he has improved. After 3 consecutive days with no abnormal behaviors or psychiatric symptoms, Mr. L is discharged and continues taking quetiapine, 75 mg at bedtime, and prednisolone, 10 mg/d, with outpatient follow-up.
The authors’ observations
Table 210,11 describes approaches to treating CIP and SLEIP. Managing CIP typically consists of reducing the corticosteroid dosage. CIP treatment also includes adjunct therapy with psychotropics if the corticosteroid dose cannot be lowered enough to reduce psychiatric symptoms while suppressing symptoms of the disease for which the corticosteroid was prescribed.6
When treating SLEIP, the corticosteroid dosage often is increased. Corticosteroids often are used to treat SLEIP while suppressing symptoms of SLE.10 The main treatment of SLEIP is focused on the disease and using psychotropic medications to control symptoms that don’t respond after exacerbation of the disease has been controlled.10
The presence of Mr. L’s multiple SLE symptoms, as well as MRI findings, could indicate SLEIP. However, corticosteroids also were a possible cause of his psychotic symptoms. Mr. L’s psychosis began within 8 weeks of starting a corticosteroid (prednisolone, 40 mg/d), and his symptoms improved when the corticosteroid dosage was reduced. The difference between CIP and SLEIP may best be distinguished by reducing the corticosteroid dosage and seeing if psychotic symptoms improve. Because it is important to control SLE symptoms in those with CIP, prescribing psychotropics may be warranted, as well as alternative treatments for immunosuppression.
Because steroids are frequently prescribed for lupus, it is important for clinicians to be aware of their psychiatric effects as well as how to manage those effects. When distinguishing CIP from SLEIP, consider decreasing the corticosteroid dosage and see if psychotic symptoms improve. Use adjunct therapy as needed.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Saint Martin ML. Running amok: A modern perspective on a culture-bound syndrome. Prim Care Companion J Clin Psychiatry. 1999;1(3):66-70.
4. Flaskerud JH. Case studies in amok? Issues Ment Health Nurs. 2012;33(12):898-900.
5. Monov S, Monova D. Classification criteria for neuropsychiatric systemic lupus erythematosus: do they need a discussion? Hippokratia. 2008;12(2):103-107.
6. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf. 2000;22(2):111-122.
7. Bhangle SD, Kramer N, Rosenstein, ED. Corticosteroid-induced neuropsychiatric disorders: review and contrast with neuropsychiatric lupus. Rheumatol Int. 2013;33(8):1923-1932.
8. Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One. 2013;8(2):e55843. d
9. Appenzeller S, Cendes F, Costallat LT. Acute psychosisin systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
10. Sanna G, Bertolaccini ML, Khamashta MA. Neuropsychiatric involvement in systemic lupus erythematosus: current therapeutic approach. Curr Pharm Des. 2008;14(13):1261-1269.
11. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
3. Saint Martin ML. Running amok: A modern perspective on a culture-bound syndrome. Prim Care Companion J Clin Psychiatry. 1999;1(3):66-70.
4. Flaskerud JH. Case studies in amok? Issues Ment Health Nurs. 2012;33(12):898-900.
5. Monov S, Monova D. Classification criteria for neuropsychiatric systemic lupus erythematosus: do they need a discussion? Hippokratia. 2008;12(2):103-107.
6. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf. 2000;22(2):111-122.
7. Bhangle SD, Kramer N, Rosenstein, ED. Corticosteroid-induced neuropsychiatric disorders: review and contrast with neuropsychiatric lupus. Rheumatol Int. 2013;33(8):1923-1932.
8. Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One. 2013;8(2):e55843. d
9. Appenzeller S, Cendes F, Costallat LT. Acute psychosisin systemic lupus erythematosus. Rheumatol Int. 2008;28(3):237-243.
10. Sanna G, Bertolaccini ML, Khamashta MA. Neuropsychiatric involvement in systemic lupus erythematosus: current therapeutic approach. Curr Pharm Des. 2008;14(13):1261-1269.
11. Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367.
Febrile Seizures: Evaluation and Treatment
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
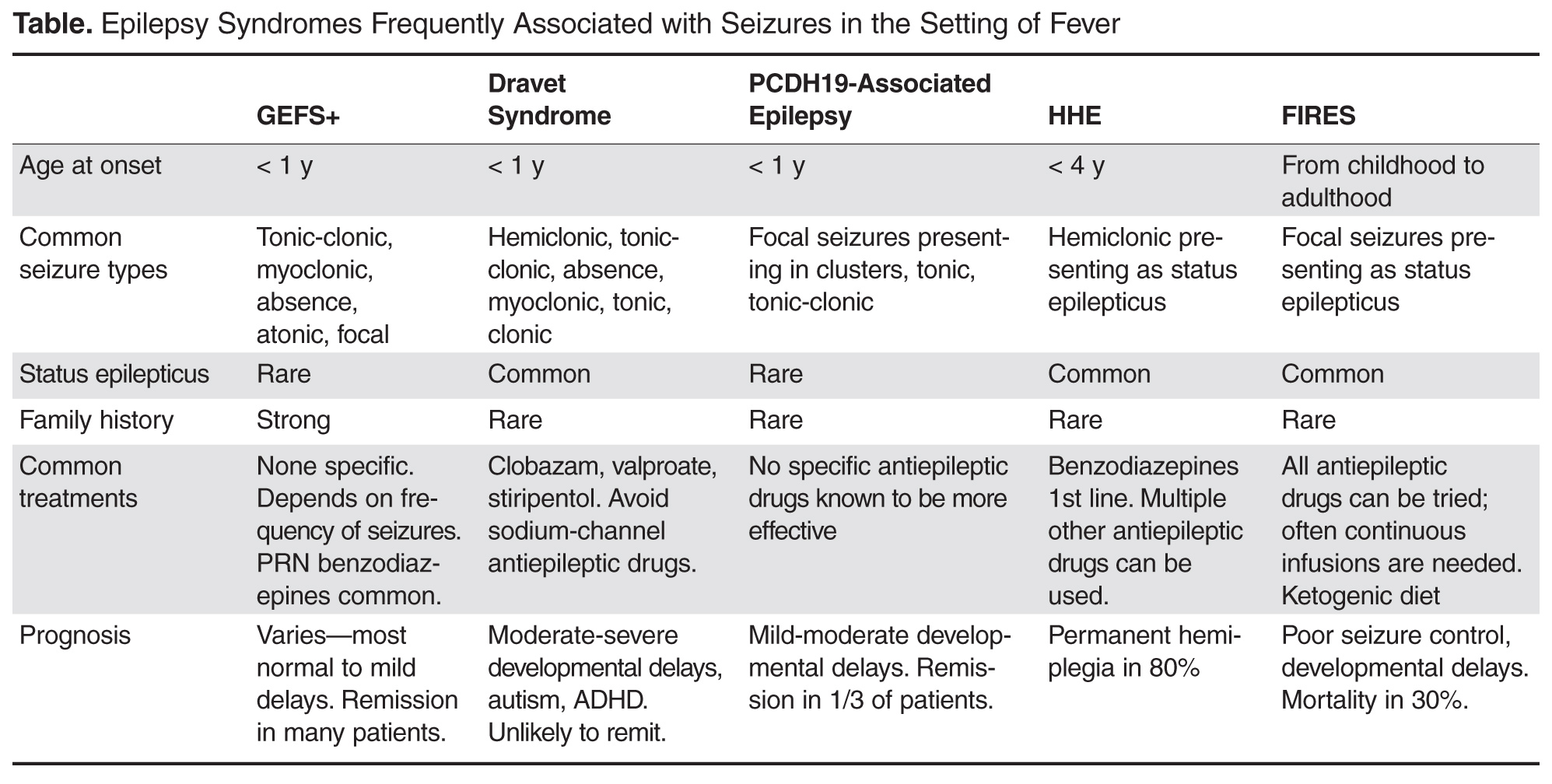
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, [email protected].
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, [email protected].
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, [email protected].
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
Suicidal and paranoid thoughts after starting hepatitis C virus treatment
CASE Suicidal and paranoid
Ms. B, age 53, has a 30-year history of bipolar disorder, a 1-year history of hepatitis C virus (HCV), and previous inpatient psychiatric hospitalizations secondary to acute mania. She presents to our hospital describing her symptoms as the “worst depression ever” and reports suicidal ideation and paranoid thoughts of people watching and following her. Ms. B describes significant neurovegetative symptoms of depression, including poor sleep, poor appetite, low energy and concentration, and chronic feelings of hopelessness with thoughts of “ending it all.” Ms. B reports that her symptoms started 3 weeks ago, a few days after she started taking sofosbuvir and ribavirin for refractory HCV.
Ms. B’s medication regimen consisted of quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, for bipolar disorder, when she started taking sofosbuvir and ribavirin. Ms. B admits she stopped taking her psychotropic and antiviral medications after she noticed progressively worsening depression with intrusive suicidal thoughts, including ruminative thoughts of overdosing on them.
At evaluation, Ms. B is casually dressed, pleasant, with fair hygiene and poor eye contact. Her speech is decreased in rate, volume, and tone; mood is “devastated and depressed”; affect is labile and tearful. Her thought process reveals occasional thought blocking and her thought content includes suicidal ideations and paranoid thoughts. Her cognition is intact; insight and judgment are poor. During evaluation, Ms. B reveals a history of alcohol and marijuana use, but reports that she has not used either for the past 15 years. She further states that she had agreed to a trial of medication first for her liver disease and had deferred any discussion of liver transplant at the time of her diagnosis with HCV.
Laboratory tests reveal a normal complete blood count, creatinine, and electrolytes. However, liver functions were elevated, including aspartate aminotransferase (AST) of 107 U/L (reference range, 8 to 48 U/L) and alanine aminotransferase of 117 U/L (reference range, 7 to 55 U/L). Although increased, the levels of AST and ALT were slightly less than her levels pre-sofosbuvir–ribavirin trial, indicating some response to the medication.
[polldaddy:9777325]
The authors’ observations
Approximately 170 million people worldwide suffer from chronic HCV infection, affecting 2.7 to 5.2 million people in the United States, with 350,000 deaths attributed to liver disease caused by HCV.1
The standard treatment of HCV genotype 1, which represents 70% of all cases of chronic HCV in the United States, is 12 to 32 weeks of an oral protease inhibitor combined with 24 to 48 weeks of peg-interferon (IFN)–alpha-2a plus ribavirin, with the duration of therapy guided by the on-treatment response and the stage of hepatic fibrosis.1
In 2013, the FDA approved sofosbuvir, a direct-acting antiviral drug for chronic HCV. It is a nucleotide analogue HCV NS5B polymerase inhibitor with similar in vitro activity against all HCV genotypes.1 This medication is efficient when used with an antiviral regimen in adults with HCV with liver disease, cirrhosis, HIV coinfection, and hepatocellular carcinoma awaiting liver transplant.2
TREATMENT Medication restarted
Ms. B is admitted to the psychiatric unit for management of severe depression and suicidal thoughts, and quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, are restarted. The hepatology team is consulted for further evaluation and management of her liver disease.
She receives supportive psychotherapy, art therapy, and group therapy to develop better coping skills for her depression and suicidal thoughts and psychoeducation about her medical and psychiatric illness to understand the importance of treatment adherence for symptom improvement. Over the course of her hospital stay, Ms. B has subjective and objective improvements of her depressive symptoms.
The authors’ observations
Psychiatric adverse effects associated with IFN-α therapy in chronic HCV patients are the main cause of antiviral treatment discontinuation, resulting in a decreased rate of sustained viral response.3 Chronic HCV is a major health burden; therefore there is a need for treatment options that are more efficient, safer, simpler, more convenient, and preferably IFN-free.
Sofosbuvir has met many of these criteria and has been found to be safe and well tolerated when administered alone or with ribavirin. Sofosbuvir represents a major breakthrough in HCV care to achieve cures and prevent IFN-associated morbidity and mortality.4,5
Our case report highlights, however, that significant depressive symptoms may be associated with sofosbuvir. Hepatologists should be cautious when prescribing sofosbuvir in patients with comorbid psychiatric illness to avoid exacerbating depressive symptoms and increasing the risk of suicidality.
[polldaddy:9777328]
OUTCOME Refuses treatment
Ms. B is seen by the hepatology team who discuss the best treatment options for HCV, including ledipasvir/sofosbuvir, daclatasvir and ribavirin, and ombitasvir/paritaprevir/ritonavir plus dasabuvir. However, she refuses treatment for HCV stating, “I would rather have no depression with hepatitis C than feel depressed and suicidal while getting treatment for hepatitis C.”
Ms. B is discharged with referral to the outpatient psychiatry clinic and hepatology clinic for monitoring her liver function and restarting sofosbuvir and ribavirin for HCV once her mood symptoms improved.
The authors’ observations
A robust psychiatric evaluation is required before initiating the previously mentioned antiviral therapy to identify high-risk patients to prevent emergence or exacerbation of new psychiatric symptoms, including depression and mania, when treating with IFN-free or IFN-containing regimens. Collaborative care involving a hepatologist and psychiatrist is necessary for comprehensive monitoring of a patient’s psychiatric symptoms and management with medication and psychotherapy. This will limit psychiatric morbidity in patients receiving antiviral treatment with sofosbuvir and ribavirin.
It’s imperative to improve medication adherence for patients by adopting strategies, such as:
- identifying factors leading to noncompliance
- establishing a strong rapport with the patients
- providing psychoeducation about the illness, discussing the benefits and risks of medications and the importance of maintenance treatment
- simplifying medication regimen.6
More research on medication management of HCV in patients with comorbid psychiatric illness should be encouraged and focused on initiating and monitoring non-IFN treatment regimens for patients with HCV and preexisting bipolar disorder or other mood disorders.
1. Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878-1887.
2. Centers for Disease Control and Prevention. Hepatitis C FAQ for health professionals. http://www.cdc.gov/hepatitis/HCV/HCVfaq.htm#section4. Updated January 27, 2017. Accessed June 2, 2017.
3. Lucaciu LA, Dumitrascu DL. Depression and suicide ideation in chronic hepatitis C patients untreated and treated with interferon: prevalence, prevention, and treatment. Ann Gastroenterol. 2015;28(4):440-447.
4. Lam B, Henry L, Younossi Z. Sofosbuvir (Sovaldi) for the treatment of hepatitis C. Expert Rev Clin Pharmacol. 2014;7(5):555-566.
5. Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomized, phase 2 trial. Lancet 2014;383(9916):515-523.
6. Balon R. Managing compliance. Psychiatric Times. www.psychiatrictimes.com/articles/managing-compliance. Published May 1, 2002. Accessed June 14, 2017.
CASE Suicidal and paranoid
Ms. B, age 53, has a 30-year history of bipolar disorder, a 1-year history of hepatitis C virus (HCV), and previous inpatient psychiatric hospitalizations secondary to acute mania. She presents to our hospital describing her symptoms as the “worst depression ever” and reports suicidal ideation and paranoid thoughts of people watching and following her. Ms. B describes significant neurovegetative symptoms of depression, including poor sleep, poor appetite, low energy and concentration, and chronic feelings of hopelessness with thoughts of “ending it all.” Ms. B reports that her symptoms started 3 weeks ago, a few days after she started taking sofosbuvir and ribavirin for refractory HCV.
Ms. B’s medication regimen consisted of quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, for bipolar disorder, when she started taking sofosbuvir and ribavirin. Ms. B admits she stopped taking her psychotropic and antiviral medications after she noticed progressively worsening depression with intrusive suicidal thoughts, including ruminative thoughts of overdosing on them.
At evaluation, Ms. B is casually dressed, pleasant, with fair hygiene and poor eye contact. Her speech is decreased in rate, volume, and tone; mood is “devastated and depressed”; affect is labile and tearful. Her thought process reveals occasional thought blocking and her thought content includes suicidal ideations and paranoid thoughts. Her cognition is intact; insight and judgment are poor. During evaluation, Ms. B reveals a history of alcohol and marijuana use, but reports that she has not used either for the past 15 years. She further states that she had agreed to a trial of medication first for her liver disease and had deferred any discussion of liver transplant at the time of her diagnosis with HCV.
Laboratory tests reveal a normal complete blood count, creatinine, and electrolytes. However, liver functions were elevated, including aspartate aminotransferase (AST) of 107 U/L (reference range, 8 to 48 U/L) and alanine aminotransferase of 117 U/L (reference range, 7 to 55 U/L). Although increased, the levels of AST and ALT were slightly less than her levels pre-sofosbuvir–ribavirin trial, indicating some response to the medication.
[polldaddy:9777325]
The authors’ observations
Approximately 170 million people worldwide suffer from chronic HCV infection, affecting 2.7 to 5.2 million people in the United States, with 350,000 deaths attributed to liver disease caused by HCV.1
The standard treatment of HCV genotype 1, which represents 70% of all cases of chronic HCV in the United States, is 12 to 32 weeks of an oral protease inhibitor combined with 24 to 48 weeks of peg-interferon (IFN)–alpha-2a plus ribavirin, with the duration of therapy guided by the on-treatment response and the stage of hepatic fibrosis.1
In 2013, the FDA approved sofosbuvir, a direct-acting antiviral drug for chronic HCV. It is a nucleotide analogue HCV NS5B polymerase inhibitor with similar in vitro activity against all HCV genotypes.1 This medication is efficient when used with an antiviral regimen in adults with HCV with liver disease, cirrhosis, HIV coinfection, and hepatocellular carcinoma awaiting liver transplant.2
TREATMENT Medication restarted
Ms. B is admitted to the psychiatric unit for management of severe depression and suicidal thoughts, and quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, are restarted. The hepatology team is consulted for further evaluation and management of her liver disease.
She receives supportive psychotherapy, art therapy, and group therapy to develop better coping skills for her depression and suicidal thoughts and psychoeducation about her medical and psychiatric illness to understand the importance of treatment adherence for symptom improvement. Over the course of her hospital stay, Ms. B has subjective and objective improvements of her depressive symptoms.
The authors’ observations
Psychiatric adverse effects associated with IFN-α therapy in chronic HCV patients are the main cause of antiviral treatment discontinuation, resulting in a decreased rate of sustained viral response.3 Chronic HCV is a major health burden; therefore there is a need for treatment options that are more efficient, safer, simpler, more convenient, and preferably IFN-free.
Sofosbuvir has met many of these criteria and has been found to be safe and well tolerated when administered alone or with ribavirin. Sofosbuvir represents a major breakthrough in HCV care to achieve cures and prevent IFN-associated morbidity and mortality.4,5
Our case report highlights, however, that significant depressive symptoms may be associated with sofosbuvir. Hepatologists should be cautious when prescribing sofosbuvir in patients with comorbid psychiatric illness to avoid exacerbating depressive symptoms and increasing the risk of suicidality.
[polldaddy:9777328]
OUTCOME Refuses treatment
Ms. B is seen by the hepatology team who discuss the best treatment options for HCV, including ledipasvir/sofosbuvir, daclatasvir and ribavirin, and ombitasvir/paritaprevir/ritonavir plus dasabuvir. However, she refuses treatment for HCV stating, “I would rather have no depression with hepatitis C than feel depressed and suicidal while getting treatment for hepatitis C.”
Ms. B is discharged with referral to the outpatient psychiatry clinic and hepatology clinic for monitoring her liver function and restarting sofosbuvir and ribavirin for HCV once her mood symptoms improved.
The authors’ observations
A robust psychiatric evaluation is required before initiating the previously mentioned antiviral therapy to identify high-risk patients to prevent emergence or exacerbation of new psychiatric symptoms, including depression and mania, when treating with IFN-free or IFN-containing regimens. Collaborative care involving a hepatologist and psychiatrist is necessary for comprehensive monitoring of a patient’s psychiatric symptoms and management with medication and psychotherapy. This will limit psychiatric morbidity in patients receiving antiviral treatment with sofosbuvir and ribavirin.
It’s imperative to improve medication adherence for patients by adopting strategies, such as:
- identifying factors leading to noncompliance
- establishing a strong rapport with the patients
- providing psychoeducation about the illness, discussing the benefits and risks of medications and the importance of maintenance treatment
- simplifying medication regimen.6
More research on medication management of HCV in patients with comorbid psychiatric illness should be encouraged and focused on initiating and monitoring non-IFN treatment regimens for patients with HCV and preexisting bipolar disorder or other mood disorders.
CASE Suicidal and paranoid
Ms. B, age 53, has a 30-year history of bipolar disorder, a 1-year history of hepatitis C virus (HCV), and previous inpatient psychiatric hospitalizations secondary to acute mania. She presents to our hospital describing her symptoms as the “worst depression ever” and reports suicidal ideation and paranoid thoughts of people watching and following her. Ms. B describes significant neurovegetative symptoms of depression, including poor sleep, poor appetite, low energy and concentration, and chronic feelings of hopelessness with thoughts of “ending it all.” Ms. B reports that her symptoms started 3 weeks ago, a few days after she started taking sofosbuvir and ribavirin for refractory HCV.
Ms. B’s medication regimen consisted of quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, for bipolar disorder, when she started taking sofosbuvir and ribavirin. Ms. B admits she stopped taking her psychotropic and antiviral medications after she noticed progressively worsening depression with intrusive suicidal thoughts, including ruminative thoughts of overdosing on them.
At evaluation, Ms. B is casually dressed, pleasant, with fair hygiene and poor eye contact. Her speech is decreased in rate, volume, and tone; mood is “devastated and depressed”; affect is labile and tearful. Her thought process reveals occasional thought blocking and her thought content includes suicidal ideations and paranoid thoughts. Her cognition is intact; insight and judgment are poor. During evaluation, Ms. B reveals a history of alcohol and marijuana use, but reports that she has not used either for the past 15 years. She further states that she had agreed to a trial of medication first for her liver disease and had deferred any discussion of liver transplant at the time of her diagnosis with HCV.
Laboratory tests reveal a normal complete blood count, creatinine, and electrolytes. However, liver functions were elevated, including aspartate aminotransferase (AST) of 107 U/L (reference range, 8 to 48 U/L) and alanine aminotransferase of 117 U/L (reference range, 7 to 55 U/L). Although increased, the levels of AST and ALT were slightly less than her levels pre-sofosbuvir–ribavirin trial, indicating some response to the medication.
[polldaddy:9777325]
The authors’ observations
Approximately 170 million people worldwide suffer from chronic HCV infection, affecting 2.7 to 5.2 million people in the United States, with 350,000 deaths attributed to liver disease caused by HCV.1
The standard treatment of HCV genotype 1, which represents 70% of all cases of chronic HCV in the United States, is 12 to 32 weeks of an oral protease inhibitor combined with 24 to 48 weeks of peg-interferon (IFN)–alpha-2a plus ribavirin, with the duration of therapy guided by the on-treatment response and the stage of hepatic fibrosis.1
In 2013, the FDA approved sofosbuvir, a direct-acting antiviral drug for chronic HCV. It is a nucleotide analogue HCV NS5B polymerase inhibitor with similar in vitro activity against all HCV genotypes.1 This medication is efficient when used with an antiviral regimen in adults with HCV with liver disease, cirrhosis, HIV coinfection, and hepatocellular carcinoma awaiting liver transplant.2
TREATMENT Medication restarted
Ms. B is admitted to the psychiatric unit for management of severe depression and suicidal thoughts, and quetiapine, 400 mg at bedtime, fluoxetine, 40 mg/d, and lamotrigine, 150 mg/d, are restarted. The hepatology team is consulted for further evaluation and management of her liver disease.
She receives supportive psychotherapy, art therapy, and group therapy to develop better coping skills for her depression and suicidal thoughts and psychoeducation about her medical and psychiatric illness to understand the importance of treatment adherence for symptom improvement. Over the course of her hospital stay, Ms. B has subjective and objective improvements of her depressive symptoms.
The authors’ observations
Psychiatric adverse effects associated with IFN-α therapy in chronic HCV patients are the main cause of antiviral treatment discontinuation, resulting in a decreased rate of sustained viral response.3 Chronic HCV is a major health burden; therefore there is a need for treatment options that are more efficient, safer, simpler, more convenient, and preferably IFN-free.
Sofosbuvir has met many of these criteria and has been found to be safe and well tolerated when administered alone or with ribavirin. Sofosbuvir represents a major breakthrough in HCV care to achieve cures and prevent IFN-associated morbidity and mortality.4,5
Our case report highlights, however, that significant depressive symptoms may be associated with sofosbuvir. Hepatologists should be cautious when prescribing sofosbuvir in patients with comorbid psychiatric illness to avoid exacerbating depressive symptoms and increasing the risk of suicidality.
[polldaddy:9777328]
OUTCOME Refuses treatment
Ms. B is seen by the hepatology team who discuss the best treatment options for HCV, including ledipasvir/sofosbuvir, daclatasvir and ribavirin, and ombitasvir/paritaprevir/ritonavir plus dasabuvir. However, she refuses treatment for HCV stating, “I would rather have no depression with hepatitis C than feel depressed and suicidal while getting treatment for hepatitis C.”
Ms. B is discharged with referral to the outpatient psychiatry clinic and hepatology clinic for monitoring her liver function and restarting sofosbuvir and ribavirin for HCV once her mood symptoms improved.
The authors’ observations
A robust psychiatric evaluation is required before initiating the previously mentioned antiviral therapy to identify high-risk patients to prevent emergence or exacerbation of new psychiatric symptoms, including depression and mania, when treating with IFN-free or IFN-containing regimens. Collaborative care involving a hepatologist and psychiatrist is necessary for comprehensive monitoring of a patient’s psychiatric symptoms and management with medication and psychotherapy. This will limit psychiatric morbidity in patients receiving antiviral treatment with sofosbuvir and ribavirin.
It’s imperative to improve medication adherence for patients by adopting strategies, such as:
- identifying factors leading to noncompliance
- establishing a strong rapport with the patients
- providing psychoeducation about the illness, discussing the benefits and risks of medications and the importance of maintenance treatment
- simplifying medication regimen.6
More research on medication management of HCV in patients with comorbid psychiatric illness should be encouraged and focused on initiating and monitoring non-IFN treatment regimens for patients with HCV and preexisting bipolar disorder or other mood disorders.
1. Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878-1887.
2. Centers for Disease Control and Prevention. Hepatitis C FAQ for health professionals. http://www.cdc.gov/hepatitis/HCV/HCVfaq.htm#section4. Updated January 27, 2017. Accessed June 2, 2017.
3. Lucaciu LA, Dumitrascu DL. Depression and suicide ideation in chronic hepatitis C patients untreated and treated with interferon: prevalence, prevention, and treatment. Ann Gastroenterol. 2015;28(4):440-447.
4. Lam B, Henry L, Younossi Z. Sofosbuvir (Sovaldi) for the treatment of hepatitis C. Expert Rev Clin Pharmacol. 2014;7(5):555-566.
5. Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomized, phase 2 trial. Lancet 2014;383(9916):515-523.
6. Balon R. Managing compliance. Psychiatric Times. www.psychiatrictimes.com/articles/managing-compliance. Published May 1, 2002. Accessed June 14, 2017.
1. Lawitz E, Mangia A, Wyles D, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878-1887.
2. Centers for Disease Control and Prevention. Hepatitis C FAQ for health professionals. http://www.cdc.gov/hepatitis/HCV/HCVfaq.htm#section4. Updated January 27, 2017. Accessed June 2, 2017.
3. Lucaciu LA, Dumitrascu DL. Depression and suicide ideation in chronic hepatitis C patients untreated and treated with interferon: prevalence, prevention, and treatment. Ann Gastroenterol. 2015;28(4):440-447.
4. Lam B, Henry L, Younossi Z. Sofosbuvir (Sovaldi) for the treatment of hepatitis C. Expert Rev Clin Pharmacol. 2014;7(5):555-566.
5. Lawitz E, Poordad FF, Pang PS, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomized, phase 2 trial. Lancet 2014;383(9916):515-523.
6. Balon R. Managing compliance. Psychiatric Times. www.psychiatrictimes.com/articles/managing-compliance. Published May 1, 2002. Accessed June 14, 2017.
Adjuvant Chemotherapy in the Treatment of Colon Cancer
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
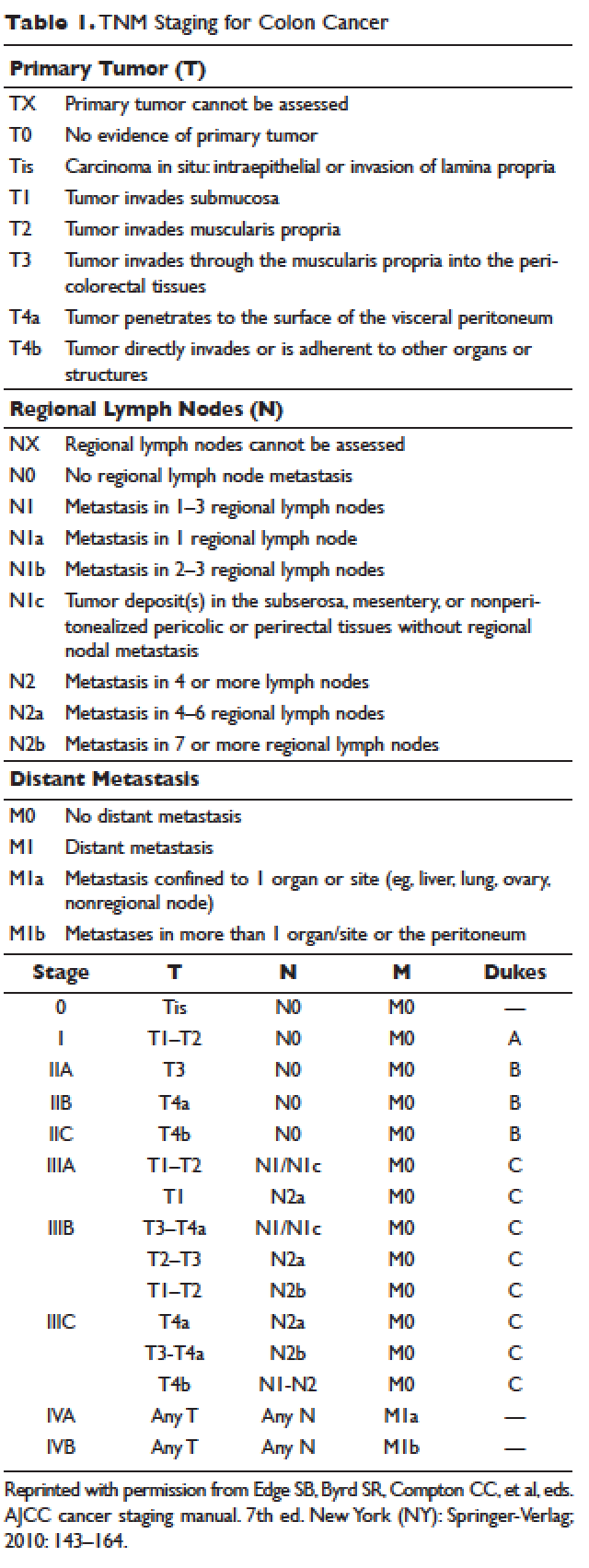
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.

SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
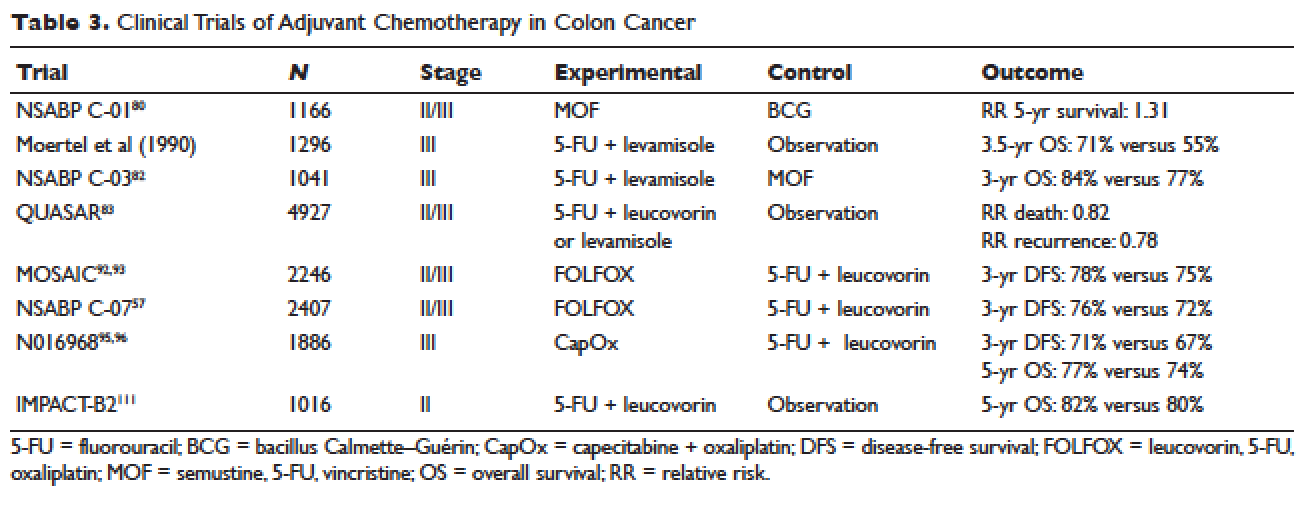
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
INTRODUCTION
Colorectal cancer (CRC) is one of the most prevalent malignancies and is the fourth most common cancer in the United States, with an estimated 133,490 new cases diagnosed in 2016. Of these, approximately 95,520 are located in the colon and 39,970 are in the rectum.1 CRC is the third leading cause of cancer death in women and the second leading cause of cancer death in men, with an estimated 49,190 total deaths in 2016.2 The incidence appears to be increasing,3 especially in patients younger than 55 years of age;4 the reason for this increase remains uncertain.
A number of risk factors for the development of CRC have been identified. Numerous hered-itary CRC syndromes have been described, including familial adenomatous polyposis,5 hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome,6 and MUTYH-associated polyposis.7,8 A family history of CRC doubles the risk of developing CRC,9 and current guidelines support lowering the age of screening in individuals with a family history of CRC to 10 years younger than the age of diagnosis of the family member or 40 years of age, whichever is lower.10 Patients with a personal history of adenomatous polyps are at increased risk for developing CRC, as are patients with a personal history of CRC, with a relative risk ranging from 3 to 6.11 Ulcerative colitis and Crohn’s disease are associated with the development of CRC and also influence screening, though evidence suggests good control of these diseases may mitigate risk.12 Finally, modifiable risk factors for the development of CRC include high red meat consumption,13 diets low in fiber,14 obesity,13 smoking, alcohol use,15 and physical inactivity16; lifestyle modification targeting these factors has been shown to decrease rates of CRC.17 The majority of colon cancers present with clinical symptoms, often with rectal bleeding, abdominal pain, change in bowel habits, or obstructive symptoms. More rarely, these tumors are detected during screening colonoscopy, in which case they tend to be at an early stage.
SURGICAL MANAGEMENT
A critical goal in the resection of early-stage colon cancer is attaining R0 resection. Patients who achieve R0 resection as compared to R1 (microscopic residual tumor) and R2 (macroscopic residual tumor)18 have significantly improved long-term overall survival.19 Traditionally, open resection of the involved colonic segment was employed, with end-end anastomosis of the uninvolved free margins. Laparoscopic resection for early-stage disease has been utilized in attempts to decrease morbidity of open procedures, with similar outcomes and node sampling.20 Laparoscopic resection appears to provide similar outcomes even in locally advanced disease.21 Right-sided lesions are treated with right colectomy and primary ileocolic anastomosis.22 For patients presenting with obstructing masses, the Hartmann procedure is the most commonly performed operation. This involves creation of an ostomy with subtotal colectomy and subsequent ostomy reversal in a 2- or 3-stage protocol.23 Patients with locally advanced disease and invasion into surrounding structures require multivisceral resection, which involves resection en bloc with secondarily involved organs.24 Intestinal perforation presents a unique challenge and is associated with surgical complications, infection, and lower overall survival (OS) and 5-year disease-free survival (DFS). Complete mesocolic excision is a newer technique that has been performed with reports of better oncologic outcome at some centers; however, this approach is not currently considered standard of care.25
STAGING
According to a report by the National Cancer Institute, the estimated 5-year relative survival rates for localized colon cancer (lymph node negative), regional (lymph node positive) disease, and distant (metastatic) disease are 89.9%, 71.3%, and 13.9%, respectively.1 However, efforts have been made to further classify patients into distinct categories to allow fine-tuning of prognostication. In the current system, staging of colon cancer utilizes the American Joint Committee on Cancer tumor/node/metastasis (TNM) system.20 Clinical and pathologic features include depth of invasion, local invasion of other organs, nodal involvement, and presence of distant metastasis (Table 1). Studies completed prior to the adoption of the TNM system used the Dukes criteria, which divided colon cancer into A, B, and C, corresponding to TNM stage I, stage IIA–IIC, and stage IIIA-IIIC. This classification is rarely used in more contemporary studies.
APPROACH TO ADJUVANT CHEMOTHERAPY
Adjuvant chemotherapy seeks to eliminate micrometastatic disease present following curative surgical resection. When stage 0 cancer is discovered incidentally during colonoscopy, endoscopic resection alone is the management of choice, as presence of micrometastatic disease is exceedingly unlikely.26 Stage I–III CRCs are treated with surgical resection withcurative intent. The 5-year survival rate for stage I and early-stage II CRC is estimated at 97% with surgery alone.27,28 The survival rate drops to about 60% for high-risk stage II tumors (T4aN0), and down to 50% or less for stage II-T4N0 or stage III cancers. Adjuvant chemotherapy is generally recommended to further decrease the rates of distant recurrence in certain cases of stage II and in all stage III tumors.
DETERMINATION OF BENEFIT FROM CHEMOTHERAPY: PROGNOSTIC MARKERS
Prior to administration of adjuvant chemotherapy, a clinical evaluation by the medical oncologist to determine appropriateness and safety of treatment is paramount. Poor performance status and comorbid conditions may indicate risk for excessive toxicity and minimal benefit from chemotherapy. CRC commonly presents in older individuals, with the median age at diagnosis of 69 years for men and 73 years for women.29 In this patient population, comorbidities such as cardiovascular disease, diabetes, and renal dysfunction are more prevalent.30 Decisions regarding adjuvant chemotherapy in this patient population have to take into consideration the fact that older patients may experience higher rates of toxicity with chemotherapy, including gastrointestinal toxicities and marrow suppression.31 Though some reports indicate patients older than 70 years derive similar benefit from adjuvant chemotherapy,32,33 a large pooled analysis of the ACCENT database, which included 7 adjuvant therapy trials and 14,528 patients, suggested limited benefit from the addition of oxaliplatin to fluorouracil in elderly patients.32 Other factors that weigh on the decision include stage, pathology, and presence of high-risk features. A common concern in the postoperative setting is delaying initiation of chemotherapy to allow adequate wound healing; however, evidence suggests that delays longer than 8 weeks leads to worse overall survival, with hazard ratios (HR) ranging from 1.4 to 1.7.34,35 Thus, the start of adjuvant therapy should ideally be within this time frame.
HIGH-RISK FEATURES
Multiple factors have been found to predict worse outcome and are classified as high-risk features (Table 2). Histologically, high-grade or poorly differentiated tumors are associated with higher recurrence rate and worse outcome.36 Certain histological subtypes, including mucinous and signet-ring, both appear to have more aggressive biology.37 Presence of microscopic invasion into surrounding blood vessels (vascular invasion) and nerves (perineural invasion) is associated with lower survival.38 Penetration of the cancer through the visceral peritoneum (T4a) or into surrounding structures (T4b) is associated with lower survival.36 During surgical resection, multiple lymph nodes are removed along with the primary tumor to evaluate for metastasis to the regional nodes. Multiple analyses have demonstrated that removal and pathologic assessment of fewer than 12 lymph nodes is associated with high risk of missing a positive node, and is thus equated with high risk.39–41 In addition, extension of tumor beyond the capsules of any single lymph node, termed extracapsular extension, is associated with an increased risk of all-cause mortality.42 Tumor deposits, or focal aggregates of adenocarcinoma in the pericolic fat that are not contiguous with the primary tumor and are not associated with lymph nodes, are currently classified as lymph nodes as N1c in the current TNM staging system. Presence of these deposits has been found to predict poor outcome stage for stage.43 Obstruction and/or perforation secondary to the tumor are also considered high-risk features that predict poor outcome.
SIDEDNESS
As reported at the 2016 American Society of Clinical Oncology annual meeting, tumor location predicts outcome in the metastatic setting. A report by Venook and colleagues based on a post-hoc analysis found that in the metastatic setting, location of the tumor primary in the left side is associated with longer OS (33.3 months) when compared to the right side of the colon (19.4 months).44 A retrospective analysis of multiple databases presented by Schrag and colleagues similarly reported inferior outcomes in patients with stage III and IV disease who had right-sided primary tumors.45 However, the prognostic implications for stage II disease remain uncertain.
BIOMARKERS
Given the controversy regarding adjuvant therapy of patients with stage II colon cancer, multiple biomarkers have been evaluated as possible predictive markers that can assist in this decision. The mismatch repair (MMR) system is a complex cellular enzymatic mechanism that identifies and corrects DNA errors during cell division and prevents mutagenesis.46 The familial cancer syndrome HNPCC is linked to alteration in a variety of MMR genes, leading to deficient mismatch repair (dMMR), also termed microsatellite instability-high (MSI-high).47,48 Epigenetic modification can also lead to silencing of the same implicated genes and accounts for 15% to 20% of sporadic colorectal cancer.49 These epigenetic modifications lead to hypermethylation of the promotor region of MLH1 in 70% of cases.50 The 4 MMR genes most commonly tested are MLH-1, MSH2, MSH6, and PMS2. Testing can be performed by immunohistochemistry or polymerase chain reaction.51 Across tumor histology and stage, MSI status is prognostic. Patients with MSI-high tumors have been shown to have improved prognosis and longer OS both in stage II and III disease52–54 and in the metastatic setting.55 However, despite this survival benefit, there is conflicting data as to whether patients with stage II, MSI-high colon cancer may benefit less from adjuvant chemotherapy. One early retrospective study compared outcomes of 70 patients with stage II and III disease and dMMR to those of 387 patients with stage II and III disease and proficient mismatch repair (pMMR). Adjuvant fluorouracil with leucovorin improved DFS for patients with pMMR (HR 0.67) but not for those with dMMR (HR 1.10). In addition, for patients with stage II disease and dMMR, the HR for OS was inferior at 2.95.56 Data collected from randomized clinical trials using fluorouracil-based adjuvant chemotherapy were analyzed in an attempt to predict benefit based on MSI status. Benefit was only seen in pMMR patients, with a HR of 0.72; this was not seen in the dMMR patients.57 Subsequent studies have had different findings and did not demonstrate a detrimental effect of fluorouracil in dMMR.58,59 For stage III patients, MSI status does not appear to affect benefit from chemotherapy, as analysis of data from the NSABP C-07 trial (Table 3) demonstrated benefit of FOLFOX (leucovorin, fluorouracil, oxaliplatin) in patients with dMMR status and stage III disease.59
Another genetic abnormality identified in colon cancers is chromosome 18q loss of heterozygosity (LOH). The presence of 18q LOH appears to be inversely associated with MSI-high status. Some reports have linked presence of 18q with worse outcome,60 but others question this, arguing the finding may simply be related to MSI status.61,62 This biomarker has not been established as a clear prognostic marker that can aid clinical decisions.
Most recently, expression of caudal-type homeobox transcription factor 2 (CDX2) has been reported as a novel prognostic and predictive tool. A 2015 report linked lack of expression of CDX2 to worse outcome; in this study, 5-year DFS was 41% in patients with CDX2-negative tumors versus 74% in the CDX2-positive tumors, with a HR of disease recurrence of 2.73 for CDX2-negative tumors.63 Similar numbers were observed in patients with stage II disease, with 5-year OS of 40% in patients with CDX2-negative tumors versus 70% in those with CDX2-positive tumors. Treatment of CDX2-negative patients with adjuvant chemotherapy improved outcomes: 5-year DFS in the stage II subgroup was 91% with chemotherapy versus 56% without, and in the stage III subgroup, 74% with chemotherapy versus 37% without. The authors concluded that patients with stage II and III colon cancer that is CDX2-negative may benefit from adjuvant chemotherapy. Importantly, CDX2-negativity is a rare event, occurring in only 6.9% of evaluable tumors.
RISK ASSESSMENT TOOLS
Several risk assessment tools have been developed in an attempt to aid clinical decision making regarding adjuvant chemotherapy for patients with stage II colon cancer. The Oncotype DX Colon Assay analyses a 12-gene signature in the pathologic sample and was developed with the goal to improve prognostication and aid in treatment decision making. The test utilizes reverse transcription-PCR on RNA extracted from the tumor.64 After evaluating 12 genes, a recurrence score is generated that predicts the risk of disease recurrence. This score was validated using data from 3 large clinical trials.65–67 Unlike the Oncotype Dx score used in breast cancer, the test in colon cancer has not been found to predict the benefit from chemotherapy and has not been incorporated widely into clinical practice.
Adjuvant! Online (available at www.adjuvantonline.com) is a web-based tool that combines clinical and histological features to estimate outcome. Calculations are based on US SEER tumor registry-reported outcomes.68 A second web-based tool, Numeracy (available at www.mayoclinic.com/calcs), was developed by the Mayo Clinic using pooled data from 7 randomized clinical trials including 3341 patients.68 Both tools seek to predict absolute benefit for patients treated with fluorouracil, though data suggests Adjuvant! Online may be more reliable in its predictive ability.69 Adjuvant! Online has also been validated in an Asian population70 and patients older than 70 years.71
MUTATIONAL ANALYSIS
Multiple mutations in proto-oncogenes have been found in colon cancer cells. One such proto-oncogene is BRAF, which encodes a serine-threonine kinase in the rapidly accelerated fibrosarcoma (RAF). Mutations in BRAF have been found in 5% to 10% of colon cancers and are associated with right-sided tumors.72 As a prognostic marker, some studies have associated BRAF mutations with worse prognosis, including shorter time to relapse and shorter OS.73,74 Two other proto-oncogenes are Kristen rat sarcoma viral oncogene homolog (KRAS) and neuroblastoma rat sarcoma viral oncogene homolog (NRAS), both of which encode proteins downstream of epidermal growth factor receptor (EGFR). KRAS and NRAS mutations have been shown to be predictive in the metastatic setting where they predict resistance to the EGFR inhibitors cetuximab and panitumumab.75,76 The effect of KRAS and NRAS mutations on outcome in stage II and III colon cancer is uncertain. Some studies suggest worse outcome in KRAS-mutated cancers,77 while others failed to demonstrate this finding.73
CASE PRESENTATION 1
A 53-year-old man with no past medical history presents to the emergency department with early satiety and generalized abdominal pain. Laboratory evaluation shows a microcytic anemia with normal white blood cell count, platelet count, renal function, and liver function tests. Computed tomography (CT) scan of the abdomen and pelvis show a 4-cm mass in the transverse colon without obstruction and without abnormality in the liver. CT scan of the chest does not demonstrate pathologic lymphadenopathy or other findings. He undergoes robotic laparoscopic transverse colon resection and appendectomy. Pathology confirms a 3.5-cm focus of adenocarcinoma of the colon with invasion through the muscularis propria and 5 of 27 regional lymph nodes positive for adenocarcinoma and uninvolved proximal, distal, and radial margins. He is given a stage of IIIB pT3 pN2a M0 and referred to medical oncology for further management, where 6 months of adjuvant FOLFOX chemotherapy is recommended.
ADJUVANT CHEMOTHERAPY IN STAGE III COLON CANCER
Postoperative adjuvant chemotherapy is the standard of care for patients with stage III disease. In the 1960s, infusional fluorouracil was first used to treat inoperable colon cancer.78,79 After encouraging results, the agent was used both intraluminally and intravenously as an adjuvant therapy for patients undergoing resection with curative intent; however, only modest benefits were described.80,81 The National Surgical Adjuvant Breast and Bowel Project (NSABP) C-01 trial (Table 3) was the first study to demonstrate a benefit from adjuvant chemotherapy in colon cancer. This study randomly assigned patients with stage II and III colon cancer to surgery alone, postoperative chemotherapy with fluorouracil, semustine, and vincristine (MOF), or postoperative bacillus Calmette-Guérin (BCG). DFS and OS were significantly improved with MOF chemotherapy.82 In 1990, a landmark study reported on outcomes after treatment of 1296 patients with stage III colon cancer with adjuvant fluorouracil and levamisole for 12 months. The combination was associated with a 41% reduction in risk of cancer recurrence and a 33% reduction in risk of death.83 The NSABP C-03 trial (Table 3) compared MOF to the combination of fluorouracil and leucovorin and demonstrated improved 3-year DFS (69% versus 73%) and 3-year OS (77% versus 84%) in patients with stage III disease.84 Building on these outcomes, the QUASAR study (Table 3) compared fluorouracil in combination with one of levamisole, low-dose leucovorin, or high-dose leucovorin. The study enrolled 4927 patients and found worse outcomes with fluorouracil plus levamisole and no difference in low-doseversus high-dose leucovorin.85 Levamisole fell out of use after associations with development of multifocal leukoencephalopathy,86 and was later shown to have inferior outcomes versus leucovorin when combined with fluorouracil.87,88 Intravenous fluorouracil has shown similar benefit when administered by bolus or infusion,89 although continuous infusion has been associated with lower incidence of severe toxicity.90 The efficacy of the oral fluoropyrimidine capecitabine has been shown to be equivalent to that of fluorouracil.91
Fluorouracil-based treatment remained the standard of care until the introduction of oxaliplatin in the mid-1990s. After encouraging results in the metastatic setting,92,93 the agent was moved to the adjuvant setting. The MOSAIC trial (Table 3) randomly assigned patients with stage II and III colon cancer to fluorouracil with leucovorin (FULV) versus FOLFOX given once every 2 weeks for 12 cycles. Analysis with respect to stage III patients showed a clear survival benefit, with a 10-year OS of 67.1% with FOLFOX chemotherapy versus 59% with fluorouracil and leucovorin.94,95 The NSABP C-07 (Table 3) trial used a similar trial design but employed bolus fluorouracil. More than 2400 patients with stage II and III colon cancer were randomly assigned to bolus FULV or bolus fluorouracil, leucovorin, and oxaliplatin (FLOX). The addition of oxaliplatin significantly improved outcomes, with 4-year DFS of 67% versus 71.8% for FULV and FLOX, respectively, and a HR of death of 0.80 with FLOX.59,96 The multicenter N016968 trial (Table 3) randomly assigned 1886 patients with stage III colon cancer to adjuvant capecitabine plus oxaliplatin (XELOX) or bolus fluorouracil plus leucovorin (FU/FA). The 3-year DFS was 70.9% versus 66.5% with XELOX and FU/FA, respectively, and 5-year OS was 77.6% versus 74.2%, respectively.97,98
In the metastatic setting, additional agents have shown efficacy, including irinotecan,99,100 bevacizumab,101,102 cetuximab,103,104 and regorafenib.105 This observation led to testing of these agents in earlier stage disease. The CALGB 89803 trial compared fluorouracil, leucovorin, and irinotecan to fluorouracil with leucovorin alone. No benefit in 5-year DFS or OS was seen.106 Similarly, infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) was not found to improve 5-year DFS as compared to fluorouracil with leucovorin alone in the PETACC-3 trial.107 The NSABP C-08 trial considered the addition of bevacizumab to FOLFOX. When compared to FOLFOX alone, the combination of bevacizumab to FOLFOX had similar 3-year DFS (77.9% versus 75.1%) and 5-year OS (82.5% versus 80.7%).108 This finding was confirmed in the Avant trial.109 The addition of cetuximab to FOLFOX was equally disappointing, as shown in the N0147 trial110 and PETACC-8 trial.111 Data on regorafenib in the adjuvant setting for stage III colon cancer is lacking; however, 2 ongoing clinical trials, NCT02425683 and NCT02664077, are each studying the use of regorafenib following completion of FOLFOX for patients with stage III disease.
Thus, after multiple trials comparing various regimens and despite attempts to improve outcomes by the addition of a third agent, the standard of care per National Comprehensive Cancer Network (NCCN) guidelines for management of stage III colon cancer remains 12 cycles of FOLFOX chemotherapy. Therapy should be initiated within 8 weeks of surgery. Data are emerging to support a short duration of therapy for patients with low-risk stage III tumors, as shown in an abstract presented at the 2017 American Society of Clinical Oncology annual meeting. The IDEA trial was a pooled analysis of 6 randomized clinical trials across multiple countries, all of which evaluated 3 versus 6 months of FOLFOX or capecitabine and oxaliplatin in the treatment of stage III colon cancer. The analysis was designed to test non-inferiority of 3 months of therapy as compared to 6 months. The analysis included 6088 patients across 244 centers in 6 countries. The overall analysis failed to establish noninferiority. The 3-year DFS rate was 74.6% for 3 months and 75.5% for 6 months, with a DFS HR of 1.07 and a confidence interval that did not meet the prespecified endpoint. Subgroup analysis suggested noninferiority for lower stage disease (T1–3 or N1) but not for higher stage disease (T4 or N2). Given the high rates of neuropathy with 6 months of oxaliplatin, these results suggest that 3 months of adjuvant therapy can be considered for patients with T1–3 or N1 disease in an attempt to limit toxicity.112
CASE PRESENTATION 2
A 57-year-old woman presents to the emergency department with fever and abdominal pain. CT of the abdomen and pelvis demonstrates a left-sided colonic mass with surrounding fat stranding and pelvic abscess. She is taken emergently for left hemicolectomy, cholecystectomy, and evacuation of pelvic abscess. Pathology reveals a 5-cm adenocarcinoma with invasion through the visceral peritoneum; 0/22 lymph nodes are involved. She is given a diagnosis of stage IIC and referred to medical oncology for further management. Due to her young age and presence of high-risk features, she is recommended adjuvant therapy with FOLFOX for 6 months.
ADJUVANT CHEMOTHERAPY IN STAGE II COLON CANCER
Because of excellent outcomes with surgical resection alone for stage II cancers, the use of adjuvant chemotherapy for patients with stage II disease is controversial. Limited prospective data is available to guide adjuvant treatment decisions for stage II patients. The QUASAR trial, which compared observation to adjuvant fluorouracil and leucovorin in patients with early-stage colon cancer, included 2963 patients with stage II disease and found a relative risk (RR) of death or recurrence of 0.82 and 0.78, respectively. Importantly, the absolute benefit of therapy was less than 5%.113 The IMPACT-B2 trial (Table 3) combined data from 5 separate trials and analyzed 1016 patients with stage II colon cancer who received fluorouracil with leucovorin or observation. Event-free survival was 0.86 versus 0.83 and 5-year OS was 82% versus 80%, suggesting no benefit.114 The benefit of addition of oxaliplatin to fluorouracil in stage II disease appears to be less than the benefit of adding this agent in the treatment of stage III CRC. As noted above, the MOSAIC trial randomly assigned patients with stage II and III colon cancer to receive adjuvant fluorouracil and leucovorin with or without oxaliplatin for 12 cycles. After a median follow-up of 9.5 years, 10-year OS rates for patients with stage II disease were 78.4% versus 79.5%. For patients with high-risk stage II disease (defined as T4, bowel perforation, or fewer than 10 lymph nodes examined), 10-year OS was 71.7% and 75.4% respectively, but these differences were not statistically significant.94
Because of conflicting data as to the benefit of adding oxaliplatin in stage II disease, oxaliplatin is not recommended for standard-risk stage II patients. The use of oxaliplatin in high-risk stage II tumors should be weighed carefully given the toxicity risk. Oxaliplatin is recognized to cause sensory neuropathy in many patients, which can become painful and debilitating.115 Two types of neuropathy are associated with oxaliplatin: acute and chronic. Acute neuropathy manifests most often as cold-induced paresthesias in the fingers and toes and is quite common, affecting up to 90% of patients. These symptoms are self-limited and resolve usually within 1 week of each treatment.116 Some patients, with reports ranging from 10% to 79%, develop chronic neuropathy that persists for 1 year or more and causes significant decrements in quality of life.117 Patients older than age 70 may be at greater risk for oxaliplatin-induced neuropathy, which would increase risk of falls in this population.118 In addition to neuropathy, oxaliplatin is associated with hypersensitivity reactions that can be severe and even fatal.119 In a single institution series, the incidence of severe reactions was 2%.120 Desensitization following hypersensitivity reactions is possible but requires a time-intensive protocol.121
Based on the inconclusive efficacy findings and due to concerns over toxicity, each decision must be individualized to fit patient characteristics and preferences. In general, for patients with stage II disease without high-risk features, an individualized discussion should be held as to the risks and benefits of single-agent fluorouracil, and this treatment should be offered in cases where the patient or provider would like to be aggressive. Patients with stage II cancer who have 1 or more high-risk features are often recommended adjuvant chemotherapy. Whether treatment with fluorouracil plus leucovorin or FOLFOX is preferred remains uncertain, and thus the risks and the potential gains of oxaliplatin must be discussed with the individual patient. MMR status can also influence the treatment recommendation for patients with stage II disease. In general, patients with standard-risk stage II tumors that are pMMR are offered MMR with leucovorin or oral capecitabine for 12 cycles. FOLFOX is considered for patients with MSI-high disease and those with multiple high-risk features.
MONITORING AFTER THERAPY
After completion of adjuvant chemotherapy, patients enter a period of survivorship. Patients are seen in clinic for symptom and laboratory monitoring of the complete blood count, liver function tests, and carcinoembryonic antigen (CEA). NCCN guidelines support history and physical examination with CEA testing every 3 to 6 months for the first 2 years, then every 6 months for the next 3 years, after which many patients continue to be seen annually. CT imaging of the chest, abdomen, and pelvis for monitoring of disease recurrence is recommended every 6 to 12 months for a total of 5 years. New elevations in CEA or liver function tests should prompt early imaging. Colonoscopy should be performed 1 year after completion of therapy; however, if no preoperative colonoscopy was performed, this should be done 3 to 6 months after completion. Colonoscopy is then repeated in 3 years and then every 5 years unless advanced adenomas are present.122
SUMMARY
The addition of chemotherapy to surgical management of colon cancer has lowered the rate of disease recurrence and improved long-term survival. Adjuvant FOLFOX for 12 cycles is the standard of care for patients with stage III colon cancer and for patients with stage II disease with certain high-risk features. Use of adjuvant chemotherapy in stage II disease without high-risk features is controversial, and treatment decisions should be individualized. Biologic markers such as MSI and CDX2 status as well as patient-related factors including age, overall health, and personal preferences can inform treatment decisions. If chemotherapy is recommended in this setting, it would be with single-agent fluorouracil in an infusional or oral formulation, unless the tumor has the MSI-high feature. Following completion of adjuvant therapy, patients should be followed with clinical evaluation, laboratory testing, and imaging for a total of 5 years as per recommended guidelines.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67(1):7–30.
- United States Cancer Statistics. 1999–2013 incidence and mortality web-based report. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute, 2016. www.cdc.gov/uscs. Accessed June 12, 2017.
- Ahnen DJ, Wade SW, Jones WF, et al. The increasing incidence of young-onset colorectal cancer: a call to action. Mayo Clin Proc 2014;89:216–24.
- Jemal A, Fedewa SA, Anderson WF, et al. Colorectal cancer incidence patterns in the United States, 1974–2013. J Natl Cancer Inst 2017;109(8).
- Boursi B, Sella T, Liberman E, et al. The APC p.I1307K polymorphism is a significant risk factor for CRC in average risk Ashkenazi Jews. Eur J Cancer 2013;49:3680–5.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60: 950–7.
- van Puijenbroek M, Nielsen M, Tops CM, et al. Identification of patients with (atypical) MUTYH-associated polyposis by KRAS2 c.34G > T prescreening followed by MUTYH hotspot analysis in formalin-fixed paraffin-embedded tissue. Clin Cancer Res 2008;14:139–42.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006;119:807–14.
- Tuohy TM, Rowe KG, Mineau GP, et al. Risk of colorectal cancer and adenomas in the families of patients with adenomas: a population-based study in Utah. Cancer 2014;120:35–42.
- Choi Y, Sateia HF, Peairs KS, Stewart RW. Screening for colorectal cancer. Semin Oncol 2017; 44:34–44.
- Atkin WS, Morson BC, Cuzick J. Long-term risk of colorectal cancer after excision of rectosigmoid adenomas. N Engl J Med 1992;326:658–62.
- Rutter MD. Surveillance programmes for neoplasia in colitis. J Gastroenterol 2011;46 Suppl 1:1–5.
- Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clin North Am 2002;31:925–43.
- Michels KB, Fuchs GS, Giovannucci E, et al. Fiber intake and incidence of colorectal cancer among 76,947 women and 47,279 men. Cancer Epidemiol Biomarkers Prev 2005;14:842–9.
- Omata F, Brown WR, Tokuda Y, et al. Modifiable risk factors for colorectal neoplasms and hyperplastic polyps. Intern Med 2009;48:123–8.
- Friedenreich CM, Neilson HK, Lynch BM. State of the epidemiological evidence on physical activity and cancer prevention. Eur J Cancer 2010;46:2593–604.
- Aleksandrova K, Pischon T, Jenab M, et al. Combined impact of healthy lifestyle factors on colorectal cancer: a large European cohort study. BMC Med 2014;12:168.
- Hermanek P, Wittekind C. The pathologist and the residual tumor (R) classification. Pathol Res Pract 1994;190:115–23.
- Lehnert T, Methner M, Pollok A, et al. Multivisceral resection for locally advanced primary colon and rectal cancer: an analysis of prognostic factors in 201 patients. Ann Surg 2002;235:217–25.
- Feinberg AE, et al. Oncologic outcomes following laparoscopic versus open resection of pT4 colon cancer: a systematic review and meta-analysis. Dis Colon Rectum 2017;60:116–125.
- Vignali A, et al. Laparoscopic treatment of advanced colonic cancer: a case-matched control with open surgery. Colorectal Dis 2013;15:944–8.
- Gainant A. Emergency management of acute colonic cancer obstruction. J Visc Surg 2012;149: e3–e10.
- Rosenman LD. Hartmann’s operation. Am J Surg 1994;168:283–4.
- Lee-Kong S, Lisle D. Surgical management of complicated colon cancer. Clin Colon Rectal Surg 2015;28:228–33.
- Bertelsen CA. Complete mesocolic excision an assessment of feasibility and outcome. Dan Med J 2017;64(2).
- Wolff WI SH. Definitive treatment of “malignant” polyps of the colon. Ann Surg 1975;182:516–25.
- Clinical Outcomes of Surgical Therapy Study Group, Nelson H, Sargent DJ, Wieand HS, et al. A comparison of laparoscopically assisted and open colectomy for colon cancer. N Engl J Med 2004;350:2050–9.
- Gunderson LL, Jessup JM, Sarjent DJ, et al. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J Clin Oncol 2010;28:256–63.
- Noone AM, Cronin KA, Altekruse SF, et al. Cancer incidence and survival trends by subtype using data from the Surveillance Epidemiology and End Results Program, 1992-2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41.
- Alves A, Panis Y, Mathieu P, et al. Postoperative mortality and morbidity in French patients undergoing colorectal surgery: results of a prospective multicenter study. Arch Surg 2005;140:278–83.
- Popescu RA, Norman A, Ross PJ, et al, Adjuvant or palliative chemotherapy for colorectal cancer in patients 70 years or older. J Clin Oncol 1999;17:2412–8.
- McCleary NJ, Meyerhardt JA, Green E, et al. Impact of age on the efficacy of newer adjuvant therapies in patients with stage II/III colon cancer: findings from the ACCENT database. J Clin Oncol 2013;31:2600–6.
- Tominaga T, Nonaka T, Sumida Y, et al. Effectiveness of adjuvant chemotherapy for elderly patients with lymph node-positive colorectal cancer. World J Surg Oncol 2016;14:197.
- Bos AC, van Erning FN, van Gestel YR, et al. Timing of adjuvant chemotherapy and its relation to survival among patients with stage III colon cancer. Eur J Cancer 2015;51:2553–61.
- Peixoto RD, Kumar A, Speers C, et al. Effect of delay in adjuvant oxaliplatin-based chemotherapy for stage III colon cancer. Clin Colorectal Cancer 2015;14:25–30.
- Compton CC, Fielding LP, Burgart LJ, et al. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. Arch Pathol Lab Med 2000;124:979–94.
- Lieu CH, Lambert LA, Wolff RA, et al. Systemic chemotherapy and surgical cytoreduction for poorly differentiated and signet ring cell adenocarcinomas of the appendix. Ann Oncol 2012;23:652–8.
- Krasna MJ, Flancbaum L, Cody RP, et al. Vascular and neural invasion in colorectal carcinoma. Incidence and prognostic significance. Cancer 1988;61:1018–23.
- Cianchi F, Palomba A, Boddi V, et al. Lymph node recovery from colorectal tumor specimens: recommendation for a minimum number of lymph nodes to be examined. World J Surg 2002;26:384–9.
- Yoshimatsu K, et al. How many lymph nodes should be examined in Dukes’ B colorectal cancer? Determination on the basis of cumulative survival rate. Hepatogastroenterology 2005;52:1703–6.
- Caplin S, Cerottini JP, Bosman FT, et al. For patients with Dukes’ B (TNM Stage II) colorectal carcinoma, examination of six or fewer lymph nodes is related to poor prognosis. Cancer 1998;83:666–72.
- Veronese N, Nottegar A, Pea A, et al. Prognostic impact and implications of extracapsular lymph node involvement in colorectal cancer: a systematic review with meta-analysis. Ann Oncol 2016;27:42–8.
- Li J, Yang S, Hu J, et al. Tumor deposits counted as positive lymph nodes in TNM staging for advanced colorectal cancer: a retrospective multicenter study. Oncotarget 2016;7:18269–79.
- Venook A, Niedzwiecki D, Innocenti Fet al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J Clin Oncol 2016;34 no. 15 suppl. Abstract 3504.
- Schrag D, Brooks G, Meyerhardt JA ,et al. The relationship between primary tumor sidedness and prognosis in colorectal cancer. J Clin Oncol 2016;34 no. 15 suppl. Abstract 3505.
- Larrea AA, Lujan SA, Kunkel TA. SnapShot: DNA mismatch repair. Cell 2010;141:730 e1.
- Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci 2000;910:62–73.
- Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer 1996;78:1149–67.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993;260:812–6.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol 2017;12:24.
- Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch-repair deficiency and others. J Gastrointest Oncol 2016;7:713–20.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609–18.
- Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77.
- Ogino S, Kuchiba A, Qian ZR, et al. Prognostic significance and molecular associations of 18q loss of heterozygosity: a cohort study of microsatellite stable colorectal cancers. J Clin Oncol 2009; 27:4591–8.
- Kim ST, Lee J, Park SH, et al. The effect of DNA mismatch repair (MMR) status on oxaliplatin-based first-line chemotherapy as in recurrent or metastatic colon cancer. Med Oncol 2010;27:1277–85.
- Sargent DJ, Monges G, Thibodeau SN, et al. Therapy in colon cancer. J Clin Oncol 2010;28:4664.
- Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med 2003;349:247–57.
- Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011;29:1261–270.
- Yothers G, O’Connell MJ, Allegra CJ, et al. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses J Clin Oncol 2011;29:3768–74.
- Chang SC, Lin JK, Lin TC, Liang WY. Loss of heterozygosity: an independent prognostic factor of colorectal cancer. World J Gastroenterol 2005;11:778–84.
- Bertagnolli MM, Niedzwiecki D, Compton CC, et al. Microsatellite instability predicts improved response to adjuvant therapy with irinotecan, fluorouracil, and leucovorin in stage III colon cancer: Cancer and Leukemia Group B Protocol 89803. J Clin Oncol 2009;27:1814–21.
- Bertagnolli MM, Redston M, Compton CC, et al. Microsatellite instability and loss of heterozygosity at chromosomal location 18q: prospective evaluation of biomarkers for stages II and III colon cancer--a study of CALGB 9581 and 89803. J Clin Oncol 2011;29:3153–62.
- Dalerba P, et al. CDX2 as a prognostic biomarker in stage II and stage III colon cancer. N Engl J Med 2016;374: 211–22.
- Clark-Langone KM, Wu JY, Sangli C, et al. Biomarker discovery for colon cancer using a 761 gene RT-PCR assay. BMC Genomics 2007;8:279.
- Gray RG, Quirke P, Handley K, et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol 2011;29:4611–9.
- Niedzwiecki D, Bertagnolli MM, Warren RS, et al. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: results from CALGB 9581. J Clin Oncol 2011;29:3146–52.
- Yothers G, O’Connell MJ, Lee M, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol 2013;31:4512–9.
- Gill S, Loprinzi CL, Sargent DJ, et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much? J Clin Oncol 2004;22:1797–806.
- Gill S, Loprinzi C, Kennecke H, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer 2011;117:4155–65.
- Jung M, Kim GW, Jung I, et al. Application of the Western-based adjuvant online model to Korean colon cancer patients; a single institution experience. BMC Cancer 2012;12:471.
- Papamichael D, Renfro LA, Matthaiou C, et al. Validity of Adjuvant! Online in older patients with stage III colon cancer based on 2967 patients from the ACCENT database. J Geriatr Oncol 2016;7:422–9.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 2011;117:4623–32.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–74.
- Lochhead P, Kuchiba A, Imamura Y, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 2013;105:1151–6.
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007;67:2643–8.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol 2014;53:852–64.
- Taieb J, Le Malicot K, Shi Q, et al. Prognostic value of BRAF and KRAS mutations in MSI and MSS stage III colon cancer. J Natl Cancer Inst 2017;109(5).
- Palumbo LT, Sharpe WS, Henry JS. Cancer of the colon and rectum; analysis of 300 cases. Am J Surg 1965;109:439–44.
- Sharp GS, Benefiel WW. 5-Fluorouracil in the treatment of inoperable carcinoma of the colon and rectum. Cancer Chemother Rep 1962;20:97–101.
- Lawrence W Jr, Terz JJ, Horsley JS 3rd, et al. Chemotherapy as an adjuvant to surgery for colorectal cancer. Ann Surg 1975;181:616–23.
- Grage TD, et al. Adjuvant chemotherapy with 5-fluorouracil after surgical resection of colorectal carcinoma (COG protocol 7041). A preliminary report. Am J Surg 1977;133:59–66.
- Wolmark N, Fisher B, Rockette H, et al. Postoperative adjuvant chemotherapy or BCG for colon cancer: results from NSABP protocol C-01. J Natl Cancer Inst 1988;80:30–6.
- Moertel CG, Fleming TR, Macdonald JS, et al. Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma. N Engl J Med 1990;322:352–8.
- Wolmark N, Rockette H, Fisher B, et al. The benefit of leucovorin-modulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol 1993;11:1879–87.
- Comparison of fluorouracil with additional levamisole, higher-dose folinic acid, or both, as adjuvant chemotherapy for colorectal cancer: a randomised trial. QUASAR Collaborative Group. Lancet 2000;355(9215):1588–96.
- Chen TC, Hinton DR, Leichman L, et al. Multifocal inflammatory leukoencephalopathy associated with levamisole and 5-fluorouracil: case report. Neurosurgery 1994;35:1138-42.
- Porschen R, Bermann A, Löffler T, et al. Fluorouracil plus leucovorin as effective adjuvant chemotherapy in curatively resected stage III colon cancer: results of the trial adjCCA-01. J Clin Oncol 2001;19:1787–94.
- Arkenau HT, Bermann A, Rettig K, et al. 5-Fluorouracil plus leucovorin is an effective adjuvant chemotherapy in curatively resected stage III colon cancer: long-term follow-up results of the adjCCA-01 trial. Ann Oncol 2003;14:395–9.
- Weinerman B, Shah A, Fields A, et al. Systemic infusion versus bolus chemotherapy with 5-fluorouracil in measurable metastatic colorectal cancer. Am J Clin Oncol 1992;15:518–23.
- Poplin EA, Benedetti JK, Estes NC, et al. Phase III Southwest Oncology Group 9415/Intergroup 0153 randomized trial of fluorouracil, leucovorin, and levamisole versus fluorouracil continuous infusion and levamisole for adjuvant treatment of stage III and high-risk stage II colon cancer. J Clin Oncol 2005;23:1819–25.
- Twelves C, Wong A, Nowacki MP, et al. Capecitabine as adjuvant treatment for stage III colon cancer. N Engl J Med 2005;352:2696–704.
- de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer 1997;33:214–9.
- Diaz-Rubio E, Sastre J, Zaniboni A, et al. Oxaliplatin as single agent in previously untreated colorectal carcinoma patients: a phase II multicentric study. Ann Oncol 1998;9:105–8.
- André T, de Gramont A, Vernerey D, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF mutation and mismatch repair status of the MOSAIC Study. J Clin Oncol 2015;33:4176–87.
- Andre T, Boni C, Mounedji-Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004;350:2343–51.
- Kuebler JP, Wieand HS, O’Connell MJ, et al. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. J Clin Oncol 2007;25:2198–204.
- Haller DG, Tabernero J, Maroun J, et al. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 2011;29:1465–71.
- Schmoll HJ, et al. Capecitabine plus oxaliplatin compared with fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results of the NO16968 randomized controlled phase III trial. J Clin Oncol 2015;33:3733–40.
- Colucci G, Gebbia V, Paoletti G, et al. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. J Clin Oncol 2005;23:4866–75.
- Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004;22:229–37.
- Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42.
- Saltz LB, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008;26:2013–9.
- Cremolini C, Loupakis F, Ruzzo A, et al. Predictors of benefit in colorectal cancer treated with cetuximab: are we getting “Lost in TranslationAL”? J Clin Oncol 2010;28:e173–4.
- Sorich MJ, Wiese MD, Rowland D, et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–21.
- Grothey A, van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013;381(9863):303–12.
- Saltz LB, Niedzwiecki D, Hollis D, et al. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: results of CALGB 89803. J Clin Oncol 2007;25:3456–61.
- Van Cutsem E, et al. Randomized phase III trial comparing biweekly infusional fluorouracil/leucovorin alone or with irinotecan in the adjuvant treatment of stage III colon cancer: PETACC-3. J Clin Oncol 2009;27:3117–25.
- Allegra CJ, et al. Bevacizumab in stage II-III colon cancer: 5-year update of the National Surgical Adjuvant Breast and Bowel Project C-08 trial. J Clin Oncol 2013;31:359–64.
- de Gramont A, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol 2012;13:1225–33.
- Alberts SR, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA 2012;307:1383–93.
- Taieb J, et al. Oxaliplatin, fluorouracil, and leucovorin with or without cetuximab in patients with resected stage III colon cancer (PETACC-8): an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:862–73.
- Shi Q, Sobrero AF, Shields AF, et al. Prospective pooled analysis of six phase III trials investigating duration of adjuvant (adjuvant) oxaliplatin-based therapy (3 vs 6 months) for patients (pts) with stage III colon cancer (CC): The IDEA (International Duration Evaluation of Adjuvant chemotherapy) collaboration. In: Proceedings from the American Society of Clinical Oncology; June 1–5, 2017; Chicago. Abstract LBA1.
- Quasar Collaborative Group; Gray R, Barnwell J, McConkey C, et al. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet 2007;370(9604):2020–9.
- Efficacy of adjuvant fluorouracil and folinic acid in B2 colon cancer. International Multicentre Pooled Analysis of B2 Colon Cancer Trials (IMPACT B2) Investigators. J Clin Oncol 1999;17:1356–63.
- Kidwell KM, et al. Long-term neurotoxicity effects of oxaliplatin added to fluorouracil and leucovorin as adjuvant therapy for colon cancer: results from National Surgical Adjuvant Breast and Bowel Project trials C-07 and LTS-01. Cancer 2012;118:5614–22.
- Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer 2014;22:1999–2007.
- Mols F, Beijers T, Lemmens V, et al. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol 2013;31:2699–707.
- Raphael MJ, Fischer HD, Fung K, et al. Neurotoxicity outcomes in a population-based cohort of elderly patients treated with adjuvant oxaliplatin for colorectal cancer. Clin Colorectal Cancer 2017 March 24.
- Toki MI, Saif MW, Syrigos KN. Hypersensitivity reactions associated with oxaliplatin and their clinical management. Expert Opin Drug Saf 2014;13:1545–54.
- Siu SW, Chan RT, Au GK. Hypersensitivity reactions to oxaliplatin: experience in a single institute. Ann Oncol 2006;17:259–61.
- Wong JT, Ling M, Patil S, et al. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract 2014;2:40–5.
- Benson AB 3rd, Venook AP, Cederquist L, et al. NCCN Guidelines Colon Cancer Version 2.2017. www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Accessed May 8, 2017.
- Wolmark N, Rockette H, Mamounas E, et al. Clinical trial to assess the relative efficacy of fluorouracil and leucovorin, fluorouracil and levamisole, and fluorouracil, leucovorin, and levamisole in patients with Dukes’ B and C carcinoma of the colon: results from National Surgical Adjuvant Breast and Bowel Project C-04. J Clin Oncol 1999;17:3553–9.
Management of Stable Chronic Obstructive Pulmonary Disease
From the Division of Pulmonary Critical Care Medicine, University of Florida, Gainesville, FL.
Abstract
- Objective:To review the management of stable chronic obstructive pulmonary disease (COPD).
- Methods: Review of the peer-reviewed literature.
- Results: Effective management of stable COPD requires the physician to apply a stepwise intensification of therapy depending on patient symptoms and functional reserve. Bronchodilators are the cornerstone of management. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
- Conclusion: Successful management of stable COPD requires a multidisciplinary approach that utilizes various medical therapies as well as nonpharmacologic interventions.
Key words: chronic obstructive pulmonary disease; exacerbation; bronchodilator; lung volume reduction; cough.
Chronic obstructive pulmonary disease (COPD) is a systemic inflammatory disease characterized by irreversible obstructive ventilatory defects [1–4]. It is a major cause of morbidity and mortality affecting 5% of the population in the United States and was the third leading cause of death in 2008 [5,6]. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. In this review, we will discuss the management of stable COPD in the context of 3 common clinical scenarios.
Case 1
A 65-year-old male with COPD underwent pulmonary function testing (PFT), which demonstrated an obstructive ventilatory defect (forced expiratory volume in 1 second/forced vital capacity ratio [FEV1/FVC], 0.45; FEV1, 2 L [65% of predicted]; and diffusing capacity of the lung for carbon monoxide [DLCO], 15 [65% of predicted]). He has dyspnea with strenuous exercise but is comfortable at rest and with minimal exercise. He has had 1 exacerbation in the last year that was treated on an outpatient basis with steroids and antibiotics. His medication regimen includes inhaled tiotropium once daily and inhaled albuterol as needed that he uses roughly twice a week.
What determines the appropriate therapy for a given COPD patient?
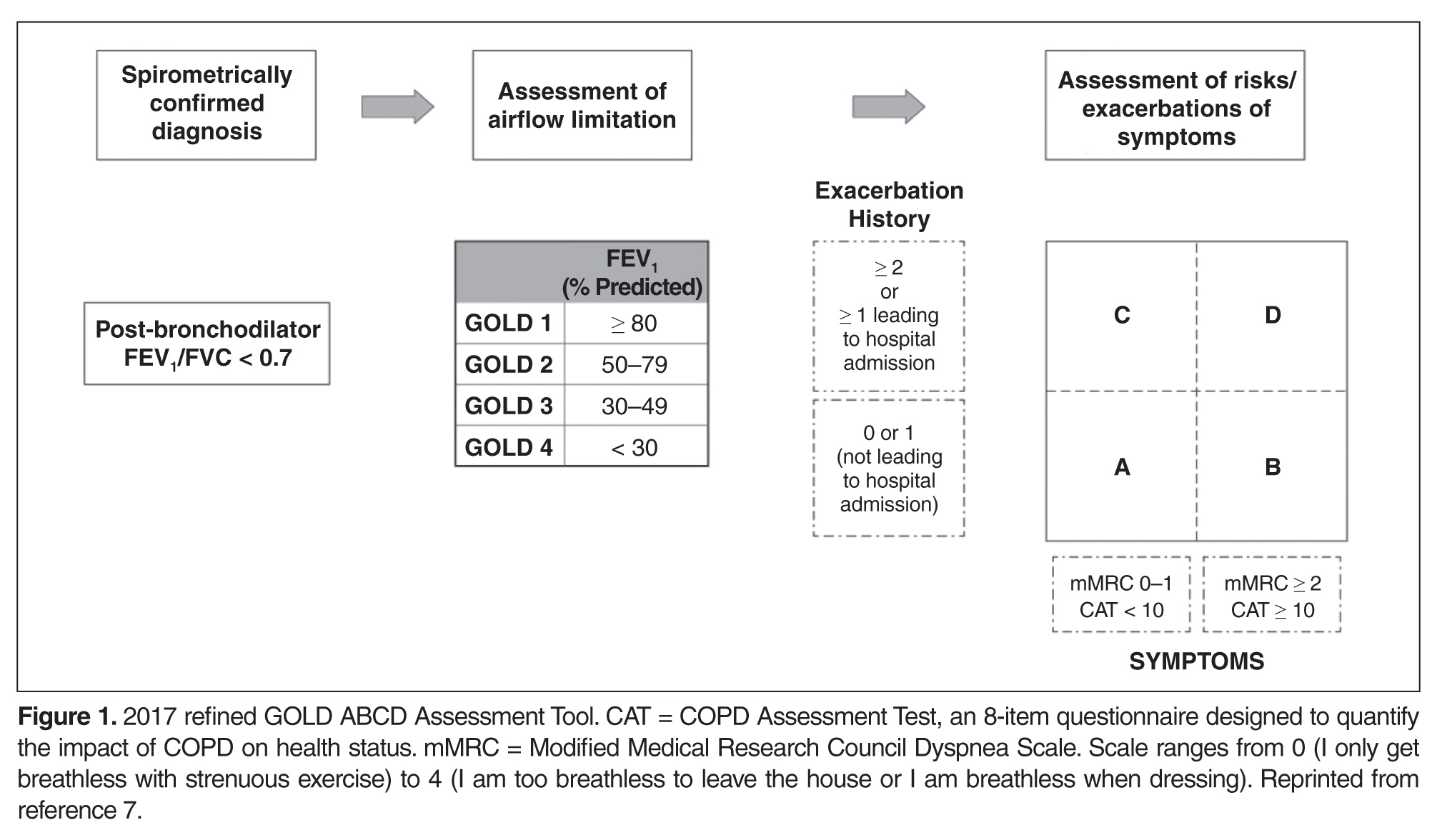
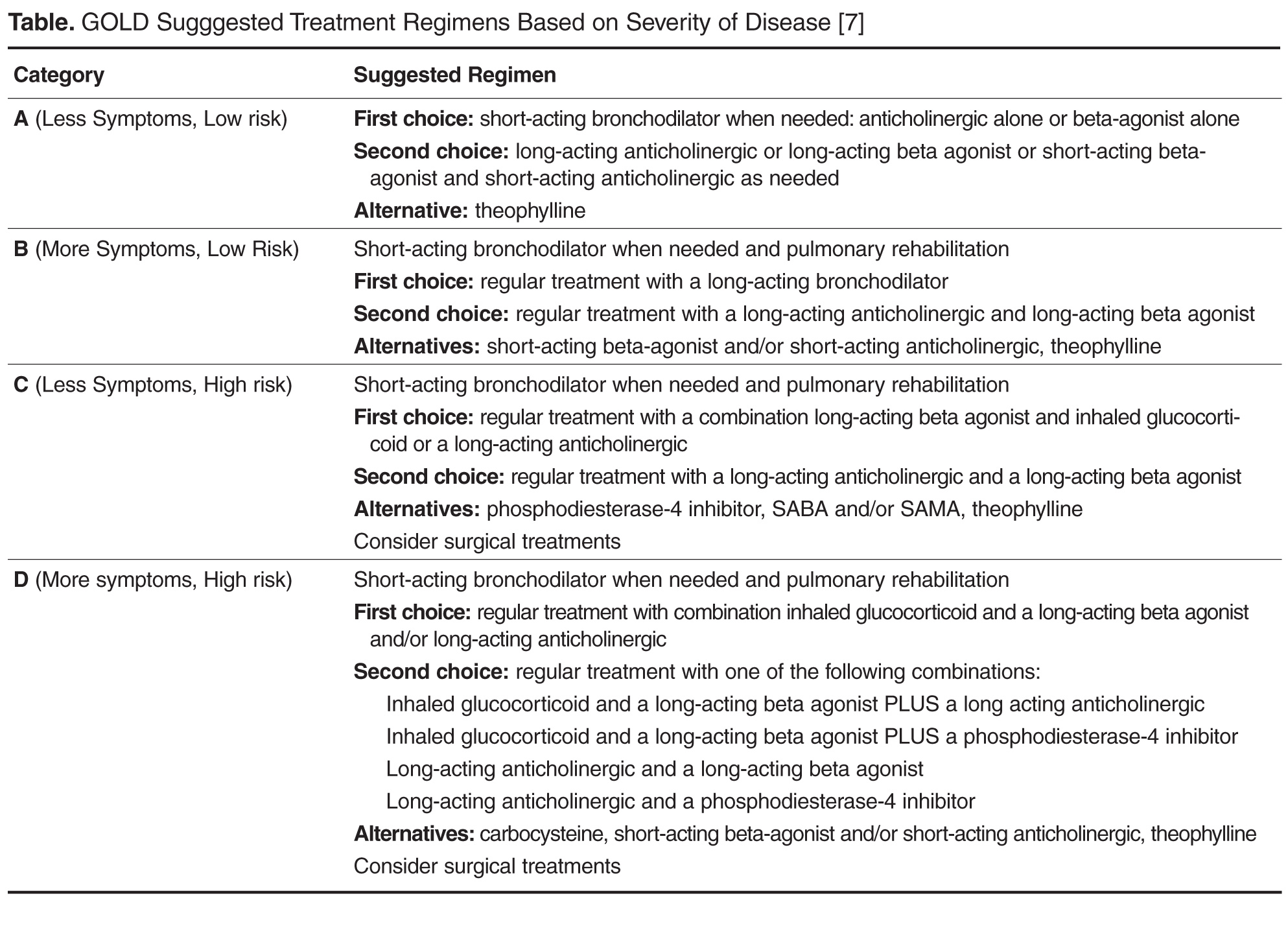
What is the approach to building a pharmacologic regimen for the patient with COPD?
The backbone of the pharmacologic regimen for COPD includes short- and long-acting bronchodilators. They are usually given in an inhaled form to maximize local effects on the lungs and minimize systemic side effects. There are 2 main classes of bronchodilators, beta agonists and muscarinic antagonists, and each targets specific receptors on the surface of airway smooth muscle cells. Beta agonists work by stimulating beta-2 receptors, resulting in bronchodilation, while muscarinic antagonists work by blocking the bronchoconstrictor action of M3 muscarinic receptors. Inhaled corticosteroids can be added to long-acting bronchodilator therapy but cannot be used as stand-alone therapy. Theophylline is an oral bronchodilator that is used infrequently due to its narrow therapeutic index, toxicity, and multiple drug interactions.
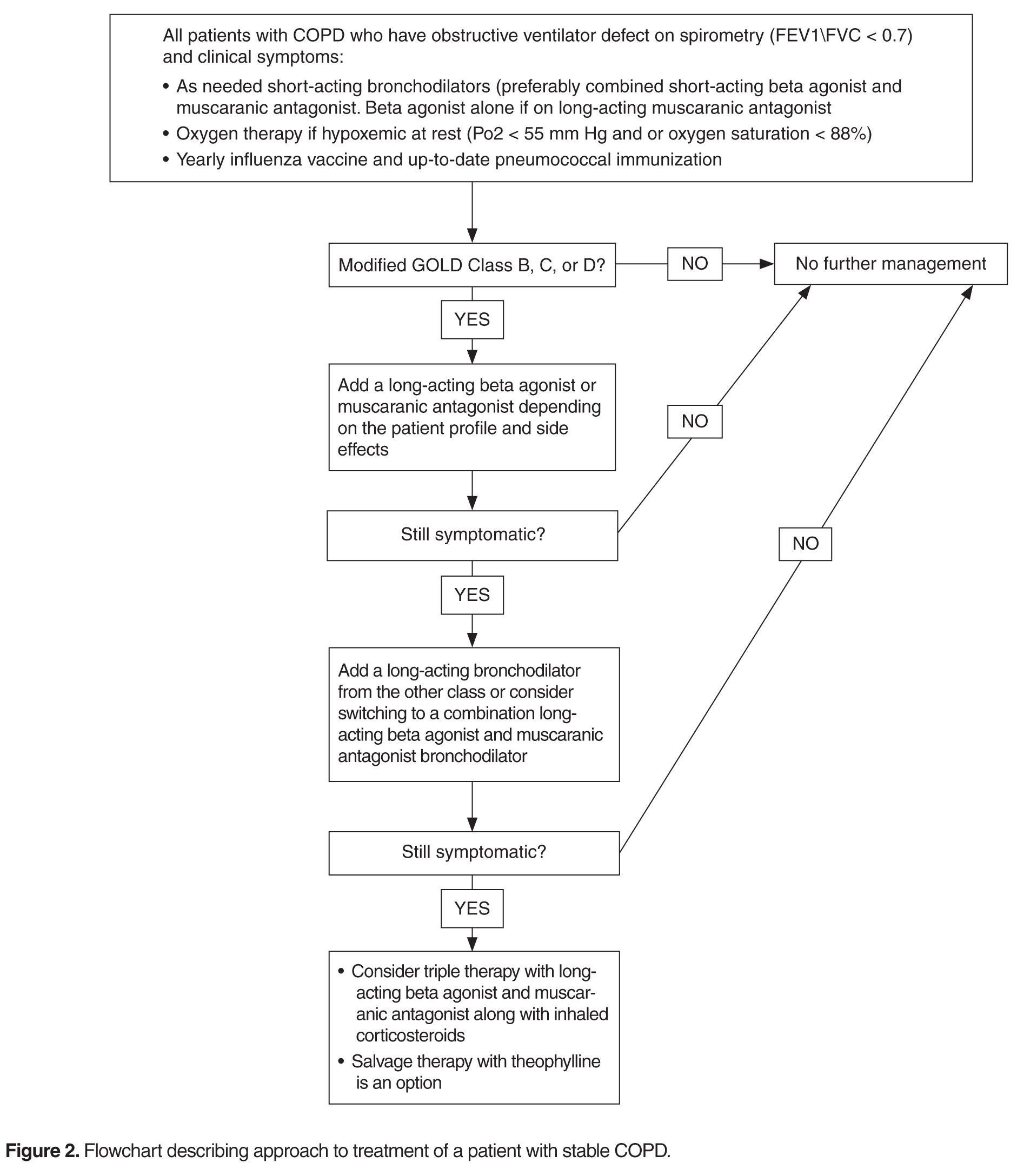
Who should be on short-acting bronchodilators? What is the best agent? Should it be scheduled or used as needed?
All patients with COPD should be an on inhaled short-acting bronchodilator as needed for relief of symptoms [7]. Both short-acting beta agonists (albuterol and levalbuterol) and short-acting muscarinic antagonists (ipratropium) have been shown in clinical trials and meta-analyses to improve symptoms and lung function in patients with stable COPD [9,10] and seem to have comparative efficacy when compared head-to-head in trials [11]. However, the airway bronchodilator effect achieved by both classes seems to be additive when used in combination and is also associated with less exacerbations compared to albuterol alone [12]. On the other hand, adding albuterol to ipratropium increased the bronchodilator response but did not reduce the exacerbation rate [11–13]. Inhaled short-acting beta agonists when used as needed rather than scheduled are associated with less medication use without any significant difference in symptoms or lung function [14].
The side effects related to using recommended doses of a short-acting bronchodilator are minimal. In retrospective studies, short-acting beta agonists increased the risk of severe cardiac arrhythmias [15]. Levalbuterol, the active enantiomer of albuterol (R-albuterol) developed for the theoretical benefits of reduced tachycardia, increased tolerability, and better or equal efficacy compared to racemic albuterol, failed to show a clinically significant difference in inducing tachycardia [16]. Beta agonist overuse is associated with tremor and in severe cases hypokalemia, which happens mainly when patients try to achieve maximal bronchodilation; the clinically used doses of beta agonists are associated with fewer side affects but achieve less than maximal bronchodilation [17]. Ipratropium can produce systemic anticholinergic side effects, urinary retention being the most clinically significant especially when combined with long-acting anticholinergic agents [18].
In light of the above discussion, a combination of short-acting beta agonist and muscarinic antagonist is recommended in all patients with COPD unless the patient is on a long-acting muscarinic antagonist [7,18]. In the latter case, a short-acting beta agonist used as a rescue inhaler is the best option. In our patient, albuterol was the choice for his short-acting bronchodilator as he was using the long-acting muscarinic antagonist tiotropium.
Are short-acting bronchodilators enough? What do we use for maintenance therapy?
All patients with COPD who are category B or higher according to the modified GOLD staging system should be on a long-acting bronchodilator [7,19]: either a long-acting beta agonist (LABA) or long-acting muscarinic antagonist (LAMA). Long-acting bronchodilators work on the same receptors as their short-acting counterparts but have structural differences. Salmeterol is the prototype for long-acting selective beta-2 agonist. It is structurally similar to albuterol but has an elongated side chain that allows it to bind firmly to the area of beta receptors and stimulate them repetitively, resulting in an extendedduration of action [20]. Tiotropium on the other hand is a quaternary ammonium of ipratropium that is a nonselective muscarinic antagonist [21]. Compared to ipratropium, tiotropium dissociates more quickly from M2 receptors, which is responsible for the undesired anticholinergic effects, while at the same time it binds M1 and M3 receptors for a prolonged time, resulting in extended duration of action [21].
The currently available long-acting beta agonists include salmeterol, formoterol, aformoterol, olodatetol, and indacaterol. The last two have the advantage of once-daily dosing rather than twice [22,23]. LABAs have been shown to improve lung function, exacerbation rate, and quality of life in multiple clinical trials [22–24]. Vilanterol is another LABA that has a long duration of action and can be used once daily [25], but is only available in a combination with umeclidinium, a LAMA. Several LAMAs are approved for use in COPD, including the prototype tiotropium in addition to aclidinium, umeclidinium, and glycopyrronium. These have been shown in clinical trials to improve lung function, symptoms, and exacerbation rate [26–29].
Patients can be started on either a LAMA or LABA depending on patient needs and side effects [7]. Both have comparable side effects and efficacy as detailed below. Concerning side effects, there is conflicting data concerning an association of cardiovascular events with both classes of long-acting bronchodilators. While clinical trials failed to show an increased risk [24,30,31], several retrospective studies showed an increased risk of emergency room visits and hospitalizations due to tachyarrhythmias, heart failure, myocardial infarction, and stroke upon initiation of long-acting bronchodilators [32,33]. There was no difference in risk for adverse cardiovascular events between LABA and LAMA in one Canadian study, and slightly more with LABA in a study using an American database [32,33]. Urinary retention is another possible complication of LAMA supported by evidence from meta-analyses and retrospective studies but not clinical trials and should be discussed with patients upon initiation [34,35]. There have been concerns about increased mortality with the soft mist formulation of tiotropium that were put to rest by the tiotropium safety and performance in Respimat (TIOSPIR) trial, which showed no increased mortality compared to Handihaler [36].
As far as efficacy and benefits, tiotropium and salmeterol were compared head-to-head in a clinical trial, and tiotropium increased the time before developing first exacerbation and decreased the overall rate of exacerbations [37]. No difference in hospitalization rate or mortality was noted in one meta-analysis, although tiotropium was more effective in reducing exacerbations [38]. The choice of agent should be made based on patient comorbidities and side effects. For example, an elderly patient with severe benign prostatic hyperplasia and urinary retention should try a LABA while for a patient with severe tachycardia induced by albuterol, LAMA would be a better first agent.
What is the role of inhaled corticosteroids in COPD?
Inhaled corticosteroids (ICS) are believed to work in COPD by reducing airway inflammation [39]. ICS should not be used alone for COPD management and are always combined with LABA [7]. Several inhaled corticosteroid formulations are approved for use in COPD, including budesonide and fluticasone. ICS has been shown to decrease symptoms and exacerbations with modest effect on lung function and no change in mortality [40]. Side effects include oral candidiasis, dysphonia, and skin bruising [41]. There is also an increased risk of pneumonia [42]. ICS are best reserved for patients with a component of asthma or asthma–COPD overlap syndrome (ACOS) [43]. ACOS is characterized by persistent airflow limitation with several features usually associated with asthma and several features usually associated with COPD [44].
What if the patient is still symptomatic on a LABA or LAMA?
For patients whose symptoms are not controlled on one class of long-acting bronchodilator, recommendations are to add a bronchodilator from the other class [7]. There are also multiple combined LAMA-LABA inhalers that are approved in the US and can possible improve adherence to therapy. These include tiotropium-oladeterol, umeclidinium-vilanterol, glycopyronnium-indacaterol, and glycopyrrolate-formoterol. In a large systematic review and meta-analysis comparing LABA-LAMA combination to either agent alone, there was a modest improvement in post bronchodilator FEV1 and quality of life with no change in hospital admissions, mortality, or side effects [45]. Interestingly, adding tiotropium to LABA reduced exacerbations although adding LABA to tiotropium did not [45].
Current guidelines recommend that patients in GOLD categories C and D that are not well controlled should receive a combination of LABA-ICS [7]. However, a new randomized trial showed better reduction of exacerbations and decreased occurrence of pneumonia in patients receiving LAMA-LABA compared to LABA-ICS [46]. In light of this new evidence, it is prudent to use a LAMA-LABA combination before adding ICS.
Triple therapy with LAMA, LABA, and ICS is a common approach for patients with severe uncontrolled disease and has been shown to decrease exacerbations and improve quality of life [7,47]. Adding tiotropium to LABA-ICS decreased exacerbations and improved quality of life and airflow in the landmark UPLIFT trial [26]. In another clinical trial, triple therapy with LAMA, LABA, and ICS compared to tiotropium alone decreased severe exacerbations, pre-bronchodilator FEV1, and morning symptoms [48].
Is there a role for theophylline? Other agents?
Theophylline
Theophylline is an oral adenosine diphosphate antagonist with indirect adrenergic activity, which is responsible for the bronchodilator therapeutic effect in patients with obstructive lung disease. It is also thought to work by an additional mechanism that decreases inflammation in the airways [49]. It has a serious side effect profile that includes ventricular arrhythmias, seizures, vomiting, and tremor [50]. It is metabolized in the liver and has multiple drug interactions and a narrow therapeutic index. It has been shown to improve lung function, gas exchange and symptoms in meta-analysis and clinical trials [51,52].
In light of the nature of the adverse effects and the wide array of safer and more effective pharmacologic agents available, theophylline should be avoided early on in patients with COPD. Its use can be justified as an add-on therapy in patients with refractory disease on triple therapy for symptomatic relief [50]. If used, the therapeutic range for COPD is 8–12 mcg/mL peak level measured 3 to 7 hours after morning dose and is usually achieved using a daily dose of 10 mg per kilogram of body weight for nonobese patients [53].
Systemic Steroids
Oral steroids are used in COPD exacerbations but should never be used chronically in COPD patients regardless of disease severity as they increase morbidity and mortality without improving symptoms or lung function [54,55]. The dose of systemic steroids should be tapered and finally discontinued.
Mucolytics
Classes of mucolytics include thiol derivatives, inhaled dornase alpha, hypertonic saline, and iodine preparations. Thiol derivatives such as N-acetylcysteine are the most widely studied [56].
There is no consistent evidence of beneficial role of mucolytics in COPD patient [7,56]. The PANTHEON trial showed decreased exacerbations with N-acetylcysteine (1.16 exacerbations per patient-year compared to 1.49 exacerbations per patient-year in the placebo group; risk ratio 0.78, 95% CI 0.67–0.90; P = 0.001) but had methodologic issues including high drop-out rate, exclusion of patients on oxygen, and a large of proportion of nonsmokers [57].
Chronic Antibiotics
There is no role for chronic antibiotics in the management of COPD [7]. Macrolides are an exception but are used for their anti-inflammatory effects rather than their antibiotic effects. They should be reserved for patient with frequent exacerbations on optimal therapy and will be discussed later in the review [58].
What nonpharmacologic treatments are recommended for COPD patients?
Smoking cessation, oxygen therapy for severe hypoxemia (resting O2 saturation ≤ 88 or PaO2 ≤ 55), vaccination for influenza and pneumococcus, and appropriate nutrition should be provided in all COPD patients. Pulmonary rehabilitation is indicated for patients in GOLD categories B, C, and D [7]. It improves symptoms, quality of life, exercise tolerance and health care utilization. Beneficial effects last for about 2 years [59,60].
What other diagnoses should be considered in patients who continue to be symptomatic on optimal therapy?
Other diseases that share the same risk factors as COPD and can contribute to dyspnea, including coronary heart disease, heart failure, thromboembolic disease, and pulmonary hypertension, should be considered. In addition, all patients with refractory disease should have a careful assessment of their inhaler technique, continued smoking, need for oxygen therapy, and associated deconditioning.
Case 2
A 70-year-old male with severe COPD on oxygen therapy and obstructive sleep apnea treated on nocturnal CPAP was seen in the pulmonary clinic for evaluation of his dyspnea. He was symptomatic with minimal activity and had chronic cough with some sputum production. He had been hospitalized 3 times over the past 12 months and had been to the emergency department (ED) the same number of times for dyspnea. Pertinent medications included as-needed albuterol inhaler, inhaled steroids, and tiotropium 18 mcg inhaled daily. He demonstrated good inhaler technique. On examination, his vital signs were pulse 99 bpm, SpO2 94% on 2L/min oxygen by nasal cannula, blood pressure 126/72 mm Hg, respiratory rate 15, and BMI 35 kg/m2. He appeared chronically ill but in no acute distress. No wheezing or rales were heard. He had no lower extremity edema. The remainder of the exam was within normal limits. His last pulmonary function test demonstrated moderate obstruction with significant bronchodilator response to 2 puffs of albuterol. The side effects of chronic steroid therapy were impressed upon the patient and 500 mg of roflumilast was started daily. Over the course of the next 3 months, he had no further exacerbations. Roflumilast was continued. He has not required any further hospitalizations, ED visits, or oral steroid use since the last clinic visit.
What is the significance of acute exacerbations of COPD?
Acute exacerbation of COPD (AECOPD) is a frequently observed complication for many patients with COPD [61,62]. AECOPD is associated with accelerated disease progression, augmented decline in health status and quality of life, and increased mortality [63]. Exacerbations account for most of the costs associated with COPD. Estimates suggest that the aggregate costs associated with the treatment of AECOPDs are between $3.2 and $3.8 billion, and that annual health care costs are 10-fold greater for patients with COPD associated with acute exacerbations than for patients with COPD but without exacerbations [64]. Hence, any intervention that could potentially minimize or prevent this complication will have far-reaching benefits to patients with COPD as well as provide significant cost saving.
How is acute exacerbation of COPD defined?
COPD exacerbation is defined as a baseline change of the patient’s dyspnea, cough, and/or sputum that is acute in onset, and may warrant a change in regular medication in a patient with underlying COPD [65]. Exacerbation in clinical trials has been defined on the basis of whether an increase in the level of care beyond regular care is required primarily in the hospital or ED [66]. Frequent exacerbations are defined as 3 symptom-defined exacerbations per year or 2 per year if defined by the need for therapy with corticosteroids, antibiotics, or both [67].
What is the underlying pathophysiology?
AECOPD is associated with enhanced upper and lower airway and systemic inflammation. The bronchial mucosa of stable COPD patients have increased numbers of CD8+ lymphocytes and macrophages. In mild AECOPD, eosinophils are increased in the bronchial mucosa and modest elevation of neutrophils, T lymphocytes (CD3), and TNF alpha positive cells has also been reported [62]. With more severe AECOPD, airway neutrophils are increased. Oxidative stress is a key factor in the development of airway inflammation in COPD [61]. Patients with severe exacerbations have augmented large airway interleukin-8 (IL-8) levels and increased oxidative stress as demonstrated by markers such as hydrogen peroxide and 8-isoprostane [66].
How do acute exacerbations affect the course of the disease?
In general, as the severity of the underlying COPD increases, exacerbations become both more severe and more frequent. The quality of life of patients with frequent exacerbations is worse than patients with a history of less frequent exacerbations [68]. Frequent exacerbations have also been linked to a decline in lung function, with studies suggesting that there might be a decline of 7 mL in FEV1 per lower respiratory tract infection per year [59,69] and approxi-mately 8 mL per year in patients with frequent exacerbations as compared to those with sporadic exacerbations [70].
What are the triggers for COPD exacerbation?
Respiratory infections are estimated to trigger approximately two-thirds of exacerbations [62]. Viral and bacterial infections cause most exacerbations. The effect of the infective triggers is to increase inflammation, cause bronchoconstriction, edema, and mucus production, with a resultant increase in dynamic hyperinflation [71]. Thus, any intervention that reduces inflammation in COPD reduces the number and severity of exacerbations, whereas bronchodilators have an impact on exacerbation by their effects on reducing dynamic hyperinflation. The triggers for the one-third of exacerbations not triggered by infection are postulated to be related to other medical conditions, including pulmonary embolism, aspiration, heart failure, and myocardial ischemia [66].
What are the pharmacologic options available for prevention of AECOPD?
In recognition of the importance of preventing COPD exacerbations, the American College of Chest Physicians and Canadian Thoracic Society [65] have published an evidence-informed clinical guideline specifically examining the prevention of AECOPD, with the goal of assisting clinicians in providing optimal management for COPD patients. The following pharmacologic agents have been recognized as being effective at reducing the frequency of acute exacerbations without any impact on the severity of COPD itself.
Roflumilast
Phosphodiesterase 4 (PDE4) inhibition appears to have inflammatory modulating properties in the airways, although the exact mechanism of action is unclear. Some have proposed that it reduces inflammation by inhibiting the breakdown of intracellular cyclic adenosine monophosphate [72]. In 2 large clinical trials [73,74], daily use of a PDE4 inhibitor (roflumilast) showed a significant (15%–18%) reduction in yearly AECOPD incidence (approximate number needed to treat: 4). This benefit was seen in patients with GOLD stage 3–4 disease (FEV1 < 50% predicted) with the chronic bronchitic phenotype and who had experienced at least 1 exacerbation in the previous year.
Importantly, these clinical trials specifically prohibited the use of ICS and LAMAs. Thus, it remains unclear if PDE4 inhibition should be used as an add-on to ICS/LAMA therapy in patients who continue to have frequent AECOPD or whether PDE4 inhibition could be used instead of these standard therapies in patients with well-controlled daily symptoms without ICS or LAMA therapy but who experience frequent exacerbations.
Of note, earlier trials with roflumilast included patients with ICS and LAMA use [73,75]. These trials were focused on FEV1 improvement and found no benefit. It was only in post ad hoc analyses that a reduction in AECOPD in patients with frequent exacerbations was found among those taking roflumilast, regardless of ICS or LAMA use [76]. While roflumilast has documented benefit in improving lung function and reducing the rate of exacerbations, it has not been reported to decrease hospitalizations [64]. This indicates that although the drug reduces the total number of exacerbations, it may not be as useful in preventing episodes of severe exacerbations of COPD.
Although PDE4 inhibitors are easy to administer (a once-daily pill), they are associated with significant GI side effects (diarrhea, nausea, reduced appetite), weight loss, headache, and sleep disturbance [77]. Adverse effects tend to occur early during treatment, are reversible, and lessen over time with treatment [66]. Studies reported an average unexplained weight loss of 2 kg, and monitoring weight during treatment is advised. In addition, it is important to avoid roflumilast in underweight patients. Roflumilast should also be used with caution in depressed patients [65].
N-acetylcysteine
N-acetylcysteine (NAC) reduces the viscosity of respiratory secretions as a result of the cleavage of the disulfide bonds and has been studied as a mucolytic agent to aid in the elimination of respiratory secretions [78]. Oral NAC is quickly absorbed and is rapidly present in an active form in lung tissue and respiratory secretions after ingestion. NAC is well tolerated except for occasional patients with GI adverse effects. The role of NAC in preventing AECOPD has been studied for more than 3 decades [79–81], although the largest clinical trial to date was reported in 2014 [57]. Taken together, the combined data demonstrate a significant reduction in the rate of COPD exacerbations associated with the use of NAC when compared with placebo (OR, 0.61; CI, 0.37–0.99). Clinical guidelines suggest that in patients with moderate to severe COPD (FEV1/FVC < 0.7, and FEV1 < 80% predicted) receiving maintenance bronchodilator therapy combined with ICS and history of 2 more exacerbations in the previous 2 years, treatment with oral NAC can be administered to prevent AECOPD.
Macrolides
Continuous prophylactic use of antibiotics in older studies had no effect on the frequency of AECOPD [82,83]. But it is known that macrolide antibiotics have several antimicrobial, anti-inflammatory and immunomodulating effects and have been used for many years in the management of other chronic airway disease, including diffuse pan-bronchiolitis and cystic fibrosis [65]. One recent study showed that the use of once-daily, generic azithromycin 5 days/week appeared to have an impact on AECOPD incidence [84]. In this study, AECOPD was reduced from 1.83 to 1.48 per patient-year (RR, 0.83; 95% CI, 0.72–0.95: P = 0.01). Azithromycin also prevented severe AECOPD. Greater benefit was obtained with milder forms of the disease and in the elderly. Azithromycin did not appear to provide any benefit in those who continued to smoke (hazard ratio, 0.99) [85]. Other studies have shown that azithromycin was associated with an increased incidence of bacterial resistance and impaired hearing [86]. Overall data from the available clinical trials are robust and demonstrate that regular macrolide therapy definitely reduces the risk of AECOPD. But due to potential side effects macrolide therapy is an option rather than a strong recommendation [65]. The prescribing clinician also needs to consider the potential of prolongation of the QT interval [84].
Immunostimulants
Immunostimulants have also been reported to reduce frequency of AECOPD [87,88]. Bacterial lysates, reconstituted mixtures of bacterial antigens present in the lower airways of COPD patients, act as immuno-stimulants through the induction of cellular maturation, stimulating lymphocyte chemotaxis, and increasing opsonization when administered to individuals with COPD [66]. Studies have demonstrated a reduction in the severe complications of exacerbations and hospital admissions in COPD patients with OM-85, a detoxified oral immunoactive bacterial extract [87,88]. However, most of these trials were conducted prior to the routine use of long-acting bronchodilators and ICS in COPD. A recent study by Braido et al evaluated the efficacy of ismigen, a bacterial lysate, in reducing AECOPD [89] and found no difference in the exacerbation rate between ismigen and placebo or the time to first exacerbation. Additional studies are needed to examine the long-term effects of this therapy in patients receiving currently recommended COPD maintenance therapy [66].
β Blockers
Observational studies of beta-blocker use in preventing AECOPD have yielded encouraging results, with one study showing a reduction in AECOPD risk (incidence risk ratio, 0.73; CI 0.60–0.90) in patients receiving beta blockers versus those not on beta blockers [90]. Based on these findings, a clinical trial investigating the impact of metoprolol on risk of AECOPD is ongoing [91].
Proton Pump Inhibitors
Gastroesophageal reflux disease is an independent risk factor for exacerbations [92]. Two small, single-center studies [93,94] have shown that use of lansoprazole decreases the risk and frequency of AECOPD. However, data from the Predicting Outcome using Systemic Markers in Severe Exacerbations of COPD (PROMISE-COPD) study [66], which was a multicenter prospective observational study, suggested that patients with stable COPD receiving a proton pump inhibitor were at high risk of frequent and severe exacerbations [95]. Thus, at this stage, their definitive role needs to be defined, possibly with a randomized, placebo-controlled study.
Case 3
A 65-year-old male with severe COPD (FEV1/FVC 27, FEV1 25% of predicted, residual volume 170% of predicted for his age and height) was seen in the pulmonary clinic. His medications include a LABA/LAMA combination that he uses twice daily as advised. He uses his rescue albuterol inhaler roughly once a week. The patient complains of severe disabling shortness of breath with exertion and severe limitation of his quality of life because of his inability to lead a normal active life. He is on 2 L/min of oxygen at all times. He has received pulmonary rehabilitation in hopes of improving his quality of life but can only climb a flight of stairs before he must stop to rest. He asks about options but does not want to consider lung transplantation today. His most recent chest CT scan demonstrates upper lobe predominant emphysematous changes with no masses or nodules.
What are the patient's options at this time?
Lung volume reduction surgery (LVRS) attempts to reduce space-occupying severely diseased, hyperexpanded lung, thus allowing the relatively normal adjoining lung parenchyma to expand into the vacated space and function effectively [96].Hence, such therapies are suitable for patients with emphysematous lungs and not those with bronchitic-predominant COPD. LVRS offers a greater chance of improvement in exercise capacity, lung function, quality of life, and dyspnea in the correctly chosen patient population as compared with pharmacologic management alone [97]. However, the procedure is associated with risks, including higher short-term morbidity and mortality [97]. Patients with predominantly upper-lobe emphysema and a low maximal workload after rehabilitation were noted to have lower mortality, a greater probability of improvement in exercise capacity, and a greater probability of improvement in symptoms if they underwent surgery compared to medical therapy alone [97]. On the contrary, patients with predominantly non–upper-lobe emphysema and a high maximal workload after rehabilitation had higher mortality if they underwent surgery compared to receiving medical therapy alone [97]. Thus, a subgroup of patients with homogeneous emphysema symmetrically affecting the upper and lower lobes are considered to be unlikely to benefit from this surgery [97,98].
Valves and other methods of lung volume reduction such as coils, sealants, intrapulmonary vents, and thermal vapor in the bronchi or subsegmental airways have emerged as new techniques for nonsurgical lung volume reduction [99–104]. Endobronchial-valve therapy is associated with improvement in lung function and with clinical benefits that are greatest in the presence of heterogeneous lung involvement. This works by the same principle as with LVRS, by reduction of the most severely diseased lung units, expansion of the more viable, less emphysematous lung results in substantial improvements in lung mechanics [105,106]. The most important complications of this procedure include pneumonia, pneumothorax, hemoptysis and increased frequency of COPD exacerbation in the following thirty days. The fact that high-heterogeneity subgroup had greater improvements in both the FEV1 and distance on the 6-minute walk test than did patients with lower heterogeneity supports the use of quantitative high-resolution computed tomography (HRCT) in selecting patients for endobronchial-valve therapy [107].The HRCT scans also help in identifying those with complete fissures; a marker of lack of collateral ventilation (CV+) between different lobes. Presence of CV+ state predicts failure of endobronchial valve and all forms of endoscopic lung volume reduction strategies [108]. Bronchoscopic thermal vapor ablation (BTVA) therapy can potentially work on a subsegmental level and be successful for treatment of emphysema with lack of intact fissures on CT scans. Other methods that have the potential to be effective in those with collateral ventilation would be endoscopic coil therapy and polymeric lung volume reduction [106,109].Unfortunately, there are no randomized controlled trial data demonstrating clinically meaningful improvement following coil therapy or polymeric lung volume reduction in this CV+ patient population. Vapor therapy is perhaps the only technique that has been found to be effective in upper lobe predominant emphysema even with CV+ status [108].
Our patient has evidence of air trapping and emphysema based on a high residual volume. A CT scan of the chest can determine the nature of the emphysema (heterogeneous versus homogenous) and based on these findings, further determination of the best strategy for lung volume reduction can be made.
Is there a role for long-term oxygen therapy?
Long-term oxygen therapy (LTOT) used for > 15 hours a day is thought to reduce mortality among patients with chronic obstructive pulmonary disease (COPD) and severe resting hypoxemia [110–113].More recent studies have failed to show similar beneficial effects of LTOT. A recent study examined the effects of LTOT in randomized fashion and determined that supplemental oxygen for patients with stable COPD and resting or exercise-induced moderate desaturation did not affect the time to death or first hospitalization, time to first COPD exacerbation, time to first hospitalization for a COPD exacerbation, the rate of all hospitalizations, the rate of all COPD exacerbations, or changes in measures of quality of life, depression, anxiety, or functional status [114].
Our patient is currently on long-term oxygen therapy and in spite of some uncertainty as to its benefit, it is prudent to order oxygen therapy until further clarification is available.
What is the role of pulmonary rehabilitation?
Pulmonary rehabilitation is an established treatment for patients with chronic lung disease [115]. Benefits include improvement in exercise tolerance, symptoms, and quality of life, with a reduction in the use of health care resources [116].A Spanish population-based cohort study that looked at the influence of regular physical activity on COPD showed that patients who reported low, moderate, or high physical activity had a lower risk of COPD admissions and all-cause mortality than patients with very low physical activity after adjusting for all confounders [117].
As previously mentioned, patients in GOLD categories B, C, and D should be offered pulmonary rehabilitation as part of their treatment [7]. The ideal patient is one who is not too sick to undergo rehabilitation and is motivated to his or her quality of life.
What is the current scope of lung transplantation in the management of severe COPD?
There is a indisputable role for lung transplantation in end-stage COPD. However, lung transplantation does not benefit all COPD patients. There is a subset of patients for whom the treatment provides a survival benefit. It has been reported that 79% of patients with an FEV1 < 16% predicted will survive at least 1 year additional after transplant, but only 11% of patients with an FEV1 > 25% will do so [118]. The pre-transplant BODE (body mass index, airflow obstruction/FEV1, dyspnea, and exercise capacity) index score is used to identify the patients who will benefit from lung transplantation [119,120]. International guidelines for the selection of lung transplant candidates identify the following patient characteristics [121]:
- The disease is progressive, despite maximal treatment including medication, pulmonary rehabilitation, and oxygen therapy
- The patient is not a candidate for endoscopic or surgical LVRS
- BODE index of 5 to 6
- The partial pressure of carbon dioxide is greater than 50 mm Hg or 6.6kPa and/or partial pressure of oxygen is less than 60 mm Hg or 8kPa
- FEV1 of 25% predicted
The perioperative mortality of lung transplantation surgery has been reduced to less than 10%. Risk of complications from surgery in the perioperative period, such as bronchial dehiscence, infectious complications, and acute rejection, have also been reduced but do occur. Chronic allograft dysfunction and the risk of lung cancer in cases of single lung transplant should be discussed with the patient before surgery [122].
How can we incorporate palliative care into the management plan for patients with COPD?
Among patients with end-stage COPD on home oxygen therapy who have required mechanical ventilation for an exacerbation, only 55% are alive at 1 year [123]. COPD patients at high risk of death within the next year of life as well as patients with refractory symptoms and unmet needs are candidates for early palliative care. Palliative care and palliative care specialists can aid in reducing symptom burden and improving quality of life among these patients and their family members and is recommended by multiple international societies for patients with advanced COPD [124,125]. In spite of these recommendations, the utilization of palliative care resources has been dismally low [126,127]. Improving physician-patient communication regarding palliative services and patients’ unmet care needs will help ensure that COPD patients receive adequate palliative care services at the appropriate time.
Conclusion
COPD is a leading cause of morbidity and mortality in the United States and represents a significant economic burden for both individuals and society. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. COPD management is guided by disease severity that is measured using the GOLD multimodal staging system and requires a multidisciplinary approach. Several classes of medication are available for treatment, and a step-wise approach should be applied in building an effective pharmacologic regimen. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
Corresponding author: Abhishek Biswas, MD, Division of Pulmonary and Critical Care Medicine, Rm. M452, University of Florida, 1600 SW Archer Rd, Gainesville, FL 32610, [email protected].
Financial disclosures: None.
1. Segreti A, Stirpe E, Rogliani P, Cazzola M. Defining phenotypes in COPD: an aid to personalized healthcare. Mol Diagn Ther 2014;18:381–8.
2. Han MK, Agusti A, Calverley PM, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010;182:598–604.
3. Aubier M, Marthan R, Berger P, et al. [COPD and inflammation: statement from a French expert group: inflammation and remodelling mechanisms]. Rev Mal Respir 2010;27:1254–66.
4. Wang ZL. Evolving role of systemic inflammation in comorbidities of chronic obstructive pulmonary disease. Chin Med J (Engl) 2010;123:3467–78.
5. Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet 2007;370:741–50.
6. Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011;59:1–126.
7. Global Initiative for Chronic Obstructive Lung Disease (GOLD): Global strategy for the diagnosis, management, and prevention of COPD 2017. Accessed at www.goldcopd.org.
8. Jones PW, Harding G, Berry P, et al. Development and first validation of the COPD Assessment Test. Eur Respir J 2009;34:648–54.
9. Wadbo M, Löfdahl CG, Larsson K, et al. Effects of formoterol and ipratropium bromide in COPD: a 3-month placebo-controlled study. Eur Respir J 2002;20:1138–46.
10. Ram FS, Sestini P. Regular inhaled short acting beta2 agonists for the management of stable chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. Thorax 2003;58:580–4.
11. Colice GL. Nebulized bronchodilators for outpatient management of stable chronic obstructive pulmonary disease. Am J Med 1996;100(1A):11S–8S.
12. In chronic obstructive pulmonary disease, a combination of ipratropium and albuterol is more effective than either agent alone. An 85-day multicenter trial. COMBIVENT Inhalation Aerosol Study Group. Chest 1994;105:1411–9.
13. Friedman M, Serby CW, Menjoge SS, et al. Pharmacoeconomic evaluation of a combination of ipratropium plus albuterol compared with ipratropium alone and albuterol alone in COPD. Chest 1999;115:635–41.
14. Cook D, Guyatt G, Wong E, et al. Regular versus as-needed short-acting inhaled beta-agonist therapy for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;163:85–90.
15. Wilchesky M, Ernst P, Brophy JM, et al. Bronchodilator use and the risk of arrhythmia in COPD: part 2: reassessment in the larger Quebec cohort. Chest 2012;142:305–11.
16. Scott VL, Frazee LA. Retrospective comparison of nebulized levalbuterol and albuterol for adverse events in patients with acute airflow obstruction. Am J Ther 2003;10:341–7.
17. Wong CS, Pavord ID, Williams J, et al. Bronchodilator, cardiovascular, and hypokalaemic effects of fenoterol, salbutamol, and terbutaline in asthma. Lancet 1990;336:1396–9.
18. Cole JM, Sheehan AH, Jordan JK. Concomitant use of ipratropium and tiotropium in chronic obstructive pulmonary disease. Ann Pharmacother 2012;46:1717–21.
19. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155 :179–91.
20. Pearlman DS, Chervinsky P, LaForce C, et al. A comparison of salmeterol with albuterol in the treatment of mild-to-moderate asthma. N Engl J Med 1992;327:1420–5.
21. Takahashi T, Belvisi MG, Patel H, et al. Effect of Ba 679 BR, a novel long-acting anticholinergic agent, on cholinergic neurotransmission in guinea pig and human airways. Am J Respir Crit Care Med 1994;150(6 Pt 1):1640–5.
22. Donohue JF, Fogarty C, Lötvall J, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010;182:155–62.
23. Koch A, Pizzichini E, Hamilton A, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat versus placebo and formoterol twice daily in patients with GOLD 2-4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis 2014;9:697–714.
24. Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007;356:775–89.
25. Hanania NA, Feldman G, Zachgo W, et al. The efficacy and safety of the novel long-acting β2 agonist vilanterol in patients with COPD: a randomized placebo-controlled trial. Chest 2012;142:119–27.
26. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54.
27. Decramer ML, Chapman KR, Dahl R, et al. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013;1:524–33.
28. Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012;40:830–6.
29. D’Urzo A, Ferguson GT, van Noord JA, et al. Efficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trial. Respir Res 2011;12:156.
30. Antoniu SA. UPLIFT Study: the effects of long-term therapy with inhaled tiotropium in chronic obstructive pulmonary disease. Evaluation of: Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54. Expert Opin Pharmacother 2009;10:719–22.
31. Nelson HS, Gross NJ, Levine B, et al. Cardiac safety profile of nebulized formoterol in adults with COPD: a 12-week, multicenter, randomized, double- blind, double-dummy, placebo- and active-controlled trial. Clin Ther 2007;29:2167–78.
32. Gershon A, Croxford R, Calzavara A, et al. Cardiovascular safety of inhaled long-acting bronchodilators in individuals with chronic obstructive pulmonary disease. JAMA Intern Med 2013;173:1175–85.
33. Aljaafareh A, Valle JR, Lin YL, et al. Risk of cardiovascular events after initiation of long-acting bronchodilators in patients with chronic obstructive lung disease: A population-based study. SAGE Open Med 2016;4:2050312116671337.
34. O’Connor AB. Tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2009;360:185–6.
35. Kesten S, Jara M, Wentworth C, Lanes S. Pooled clinical trial analysis of tiotropium safety. Chest 2006;130:1695–703.
36. Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat inhaler and the risk of death in COPD. N Engl J Med 2013;369:1491–501.
37. Vogelmeier C, Hederer B, Glaab T, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011;364:1093–103.
38. Chong J, Karner C, Poole P. Tiotropium versus long-acting beta-agonists for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(9):CD009157.
39. Gan WQ, Man SF, Sin DD. Effects of inhaled corticosteroids on sputum cell counts in stable chronic obstructive pulmonary disease: a systematic review and a meta-analysis. BMC Pulm Med 2005;5:3.
40. Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(7):CD002991.
41. Roland NJ, Bhalla RK, Earis J. The local side effects of inhaled corticosteroids: current understanding and review of the literature. Chest 2004;126:213–9.
42. Drummond MB, Dasenbrook EC, Pitz MW, et al. Inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta-analysis. JAMA 2008;300:2407–16.
43. Lee SY, Park HY, Kim EK, et al. Combination therapy of inhaled steroids and long-acting beta2-agonists in asthma-COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis 2016;11:2797–803.
44. Postma DS, Rabe KF. The asthma-COPD overlap syndrome. N Engl J Med 2015;373:1241–9.
45. Farne HA, Cates CJ. Long-acting beta2-agonist in addition to tiotropium versus either tiotropium or long-acting beta2-agonist alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015:CD008989.
46. Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol-glycopyrronium versus salmeterol-fluticasone for COPD. N Engl J Med 2016;374:2222–34.
47. Aaron SD, Vandemheen KL, Fergusson D, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007;146:545–55.
48. Welte T, Miravitlles M, Hernandez P, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009;180:741–50.
49. Gallelli L, Falcone D, Cannataro R, et al. Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: a therapeutic rationale approach. Drug Des Devel Ther 2017;11:265–72.
50. Paloucek FP, Rodvold KA. Evaluation of theophylline overdoses and toxicities. Ann Emerg Med 1988;17:135–44.
51. Ram FS, Jones PW, Castro AA, et al. Oral theophylline for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2002(4):CD003902.
52. Murciano D, Auclair MH, Pariente R, Aubier M. A randomized, controlled trial of theophylline in patients with severe chronic obstructive pulmonary disease. N Engl J Med 1989;320:1521–5.
53. Devereux G, Cotton S, Barnes P, et al. Use of low-dose oral theophylline as an adjunct to inhaled corticosteroids in preventing exacerbations of chronic obstructive pulmonary disease: study protocol for a randomised controlled trial. Trials 2015;16:267.
54. Walters JA, Walters EH, Wood-Baker R. Oral corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005(3):CD005374.
55. Horita N, Miyazawa N, Morita S, et al. Evidence suggesting that oral corticosteroids increase mortality in stable chronic obstructive pulmonary disease. Respir Res 2014;15:37.
56. Poole P, Chong J, Cates CJ. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(7):CD001287.
57. Zheng JP, Wen FQ, Bai CX, et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014;2:187–94.
58. Seemungal TA, Wilkinson TM, Hurst JR, et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med 2008;178:1139–47.
59. Ries AL, Kaplan RM, Limberg TM, Prewitt LM. Effects of pulmonary rehabilitation on physiologic and psychosocial outcomes in patients with chronic obstructive pulmonary disease. Ann Intern Med 1995;122:823– 32.
60. Güell R, Casan P, Belda J, et al. Long-term effects of outpatient rehabilitation of COPD: a randomized trial. Chest 2000;117:976–83.
61. Wedzicha JA, Singh R, Mackay AJ. Acute COPD exacerbations. Clin Chest Med 2014;35:157–63.
62. Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet 2007;370:786–96.
63. Spencer S, Calverley PMA, Burge PS, Jones PW. Impact of preventing exacerbations on deterioration of health status in COPD. Eur Respir J 2004;23:698–702.
64. Blanchette CM, Gross NJ, Altman P. Rising costs of COPD and the potential for maintenance therapy to slow the trend. Am Health Drug Benef 2014;7:98.
65. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest 2015;147:894–942.
66. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2017 Report. Respirology 2017;22:575–601.
67. Wedzicha JA, Brill SE, Allinson JP, Donaldson GC. Mechanisms and impact of the frequent exacerbator phenotype in chronic obstructive pulmonary disease. BMC Med 2013;11:181.
68. Seemungal TAR, Donaldson GC, Paul EA, et al. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:1418–22.
69. Kanner RE, Anthonisen NR, Connett JE. Lower respiratory illnesses promote FEV1 decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med 2001;164:358–64.
70. Donaldson GC, Seemungal TAR, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002;57:847–52.
71. Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006;173:1114–21.
72. Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163:53–67.
73. Calverley PMA, Rabe KF, Goehring U-M, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009;374:685–94.
74. Fabbri LM, Calverley PMA, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009;374:695–703.
75. Lee S, Hui DSC, Mahayiddin AA, et al. Roflumilast in Asian patients with COPD: a randomized placebo-controlled trial. Respirology 2011;16:1249–57.
76. Calverley PM, Martinez FJ, Fabbri LM, et al. Does roflumilast decrease exacerbations in severe COPD patients not controlled by inhaled combination therapy? The REACT study protocol. Int J Chron Obstruct Pulmon Dis 2012;7:375–82.
77. Chong J, Leung B, Poole P. Phosphodiesterase 4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2013(11):CD002309.
78. Sheffner AL, Medler EM, Jacobs LW, Sarett HP. The in vitro reduction in viscosity of human tracheobronchial secretions by acetylcysteine. Am Rev Respir Dis 1964;90:721–9.
79. Boman G, Bäcker U, Larsson S, et al. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis 1983;64:405–15.
80. Grassi C, Morandini GC. A controlled trial of intermittent oral acetylcysteine in the long-term treatment of chronic bronchitis. European journal of clinical pharmacology. 1976;9:393–6.
81. Hansen NCG, Skriver A, Brorsen-Riis L, et al. Orally administered N-acetylcysteine may improve general well-being in patients with mild chronic bronchitis. Respir Med 1994;88:531–5.
82. Francis RS, Spicer CC. Chemotherapy in chronic bronchitis: Influence of daily penicillin and tetracycline on exacerbations and their cost: A report to the research committee of the British Tuberculosis Association by Their Chronic Bronchitis Subcommittee. BMJ 1960;1:297–303.
83. Francis RS, May JR, Spicer CC. Chemotherapy of bronchitis. BMJ 1961;2:979.
84. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
85. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014;189:1503–8.
86. Uzun S, Djamin RS, Kluytmans JAJW, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014;2:361–8.
87. Collet JP, Shapiro S, Ernst P, et al. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997;156:1719–24.
88. Jing LI. Protective effect of a bacterial extract against acute exacerbation in patients with chronic bronchitis accompanied by chronic obstructive pulmonary. Age 2004;67:0–05.
89. Braido F, Tarantini F, Ghiglione V, et al. Bacterial lysate in the prevention of acute exacerbation of COPD and in respiratory recurrent infections. Int J Chron Obstruct Pulmon Dis 2007;2:335.
90. Bhatt SP, Wells JM, Kinney GL, et al. β-Blockers are associated with a reduction in COPD exacerbations. Thorax 2016;71:8–14.
91. Bhatt SP, Connett JE, Voelker H, et al. β-Blockers for the prevention of acute exacerbations of chronic obstructive pulmonary disease (βLOCK COPD): a randomised controlled study protocol. BMJ Open 2016;6:e012292.
92. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010;363:1128–8.
93. Sasaki T, Nakayama K, Yasuda H, et al. A randomized, single-blind study of lansoprazole for the prevention of exacerbations of chronic obstructive pulmonary disease in older patients. J Am Geriatr Soc 2009;57:1453–7.
94. Xiong W, Zhang Qs, Zhao W, et al. A 12-month follow-up study on the preventive effect of oral lansoprazole on acute exacerbation of chronic obstructive pulmonary disease. Int J Exper Pathol 2016;97:107–13.
95. Baumeler L, Papakonstantinou E, Milenkovic B, et al. Therapy with proton-pump inhibitors for gastroesophageal reflux disease does not reduce the risk for severe exacerbations in COPD. Respirology 2016;21:883–90.
96 Sabanathan A, Sabanathan S, Shah R, Richardson J. Lung volume reduction surgery for emphysema: a review. J Cardiovasc Surg 1998;39:237.
97. Group NETTR. Patients at high risk of death after lung-volume–reduction surgery. N Engl J Med 2001;345:1075–83.
98. Group NETTR. A randomized trial comparing lung-volume–reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003;348:2059–73.
99. Decker MR, Leverson GE, Jaoude WA, Maloney JD. Lung volume reduction surgery since the National Emphysema Treatment Trial: study of Society of Thoracic Surgeons database. J Thorac Cardiovasc Surg 2014;148:2651–8.
100. Deslée G, Mal H, Dutau H, et al. Lung volume reduction coil treatment vs usual care in patients with severe emphysema: the REVOLENS randomized clinical trial. JAMA 2016;315:175–84.
101. Hartman JE, Klooster K, Gortzak K, et al. Long-term follow-up after bronchoscopic lung volume reduction treatment with coils in patients with severe emphysema. Respirology 2015;20:319–26.
102. Snell GI, Hopkins P, Westall G, et al. A feasibility and safety study of bronchoscopic thermal vapor ablation: a novel emphysema therapy. Ann Thorac Surg 2009;88:1993–8.
103. Ingenito EP, Berger RL, Henderson AC, et al. Bronchoscopic lung volume reduction using tissue engineering principles. Am J Respir Crit Care Med 2003;167:771–8.
104. Ingenito EP, Loring SH, Moy ML, et al. Comparison of physiological and radiological screening for lung volume reduction surgery. Am J Respir Crit Care Med 2001;163:1068–73.
105. Shah P, Slebos D, Cardoso P, et al. Bronchoscopic lung-volume reduction with Exhale airway stents for emphysema (EASE trial): randomised, sham-controlled, multicentre trial. Lancet 2011;378:997–1005.
106. Sciurba FC, Ernst A, Herth FJ, et al. A randomized study of endobronchial valves for advanced emphysema. N Engl J Med 2010;363:1233–44.
107. Wan IY, Toma TP, Geddes DM, et al. Bronchoscopic lung volume reduction for end-stage emphysema: report on the first 98 patients. Chest 2006;129:518–26.
108. Gompelmann D, Eberhardt R, Schuhmann M, et al. Lung volume reduction with vapor ablation in the presence of incomplete fissures: 12-month results from the STEP-UP randomized controlled study. Respiration 2016;92:397–403.
109. Come CE, Kramer MR, Dransfield MT, et al. A randomised trial of lung sealant versus medical therapy for advanced emphysema. Eur Respir J 2015;46:651–62.
110. Group NOTT. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann Intern Med 1980;93:391–8.
111. Council M. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema: Report of the Medical Research Council Working Party. Lancet 1981;1:681–6.
112. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155:179–91.
113. Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013;187:347–65.
114. Group L-TOTTR. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016;375:1617–27.
115. McCarthy B, Casey D, Devane D, et al. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(2):CD003793.
116. Griffiths TL, Burr ML, Campbell IA, et al. Results at 1 year of outpatient multidisciplinary pulmonary rehabilitation: a randomised controlled trial. Lancet 2000;355:362–8.
117. Garcia-Aymerich J, Lange P, Benet M, et al. Regular physical activity reduces hospital admission and mortality in chronic obstructive pulmonary disease: a population based cohort study. Thorax 2006;61:772–8.
118. Thabut G, Ravaud P, Christie JD, et al. Determinants of the survival benefit of lung transplantation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008;177:1156–63.
119. Lahzami S, Bridevaux PO, Soccal PM, et al. Survival impact of lung transplantation for COPD. Eur Respir J 2010;36:74–80.
120. Cerón Navarro J, de Aguiar Quevedo K, Ansótegui Barrera E, et al. Functional outcomes after lung transplant in chronic obstructive pulmonary disease. Arch Bronconeumol 2015;51:109–14.
121. Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2015;34:1–15.
122. Minai OA, Shah S, Mazzone P, et al. Bronchogenic carcinoma after lung transplantation: characteristics and outcomes. J Thorac Oncol 2008;3:1404–9.
123. Hajizadeh N, Goldfeld K, Crothers K. What happens to patients with COPD with long-term oxygen treatment who receive mechanical ventilation for COPD exacerbation? A 1-year retrospective follow- up study. Thorax 2015;70:294–6.
124. Siouta N, van Beek K, Preston N, et al. Towards integration of palliative care in patients with chronic heart failure and chronic obstructive pulmonary disease: a systematic literature review of European guidelines and pathways. BMC Palliat Care 2016;15:18.
125. Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004;23:932–46.
126. Szekendi MK, Vaughn J, Lal A, et al. The prevalence of inpatients at thirty-three U.S. hospitals appropriate for and receiving referral to palliative care. J Palliat Med 2016;19:360–72.
127. Rush B, Hertz P, Bond A, et al. Use of palliative care in patients with end-stage COPD and receiving home oxygen: national trends and barriers to care in the United States. Chest 2017;151:41–6.
From the Division of Pulmonary Critical Care Medicine, University of Florida, Gainesville, FL.
Abstract
- Objective:To review the management of stable chronic obstructive pulmonary disease (COPD).
- Methods: Review of the peer-reviewed literature.
- Results: Effective management of stable COPD requires the physician to apply a stepwise intensification of therapy depending on patient symptoms and functional reserve. Bronchodilators are the cornerstone of management. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
- Conclusion: Successful management of stable COPD requires a multidisciplinary approach that utilizes various medical therapies as well as nonpharmacologic interventions.
Key words: chronic obstructive pulmonary disease; exacerbation; bronchodilator; lung volume reduction; cough.
Chronic obstructive pulmonary disease (COPD) is a systemic inflammatory disease characterized by irreversible obstructive ventilatory defects [1–4]. It is a major cause of morbidity and mortality affecting 5% of the population in the United States and was the third leading cause of death in 2008 [5,6]. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. In this review, we will discuss the management of stable COPD in the context of 3 common clinical scenarios.
Case 1
A 65-year-old male with COPD underwent pulmonary function testing (PFT), which demonstrated an obstructive ventilatory defect (forced expiratory volume in 1 second/forced vital capacity ratio [FEV1/FVC], 0.45; FEV1, 2 L [65% of predicted]; and diffusing capacity of the lung for carbon monoxide [DLCO], 15 [65% of predicted]). He has dyspnea with strenuous exercise but is comfortable at rest and with minimal exercise. He has had 1 exacerbation in the last year that was treated on an outpatient basis with steroids and antibiotics. His medication regimen includes inhaled tiotropium once daily and inhaled albuterol as needed that he uses roughly twice a week.
What determines the appropriate therapy for a given COPD patient?
What is the approach to building a pharmacologic regimen for the patient with COPD?
The backbone of the pharmacologic regimen for COPD includes short- and long-acting bronchodilators. They are usually given in an inhaled form to maximize local effects on the lungs and minimize systemic side effects. There are 2 main classes of bronchodilators, beta agonists and muscarinic antagonists, and each targets specific receptors on the surface of airway smooth muscle cells. Beta agonists work by stimulating beta-2 receptors, resulting in bronchodilation, while muscarinic antagonists work by blocking the bronchoconstrictor action of M3 muscarinic receptors. Inhaled corticosteroids can be added to long-acting bronchodilator therapy but cannot be used as stand-alone therapy. Theophylline is an oral bronchodilator that is used infrequently due to its narrow therapeutic index, toxicity, and multiple drug interactions.
Who should be on short-acting bronchodilators? What is the best agent? Should it be scheduled or used as needed?
All patients with COPD should be an on inhaled short-acting bronchodilator as needed for relief of symptoms [7]. Both short-acting beta agonists (albuterol and levalbuterol) and short-acting muscarinic antagonists (ipratropium) have been shown in clinical trials and meta-analyses to improve symptoms and lung function in patients with stable COPD [9,10] and seem to have comparative efficacy when compared head-to-head in trials [11]. However, the airway bronchodilator effect achieved by both classes seems to be additive when used in combination and is also associated with less exacerbations compared to albuterol alone [12]. On the other hand, adding albuterol to ipratropium increased the bronchodilator response but did not reduce the exacerbation rate [11–13]. Inhaled short-acting beta agonists when used as needed rather than scheduled are associated with less medication use without any significant difference in symptoms or lung function [14].
The side effects related to using recommended doses of a short-acting bronchodilator are minimal. In retrospective studies, short-acting beta agonists increased the risk of severe cardiac arrhythmias [15]. Levalbuterol, the active enantiomer of albuterol (R-albuterol) developed for the theoretical benefits of reduced tachycardia, increased tolerability, and better or equal efficacy compared to racemic albuterol, failed to show a clinically significant difference in inducing tachycardia [16]. Beta agonist overuse is associated with tremor and in severe cases hypokalemia, which happens mainly when patients try to achieve maximal bronchodilation; the clinically used doses of beta agonists are associated with fewer side affects but achieve less than maximal bronchodilation [17]. Ipratropium can produce systemic anticholinergic side effects, urinary retention being the most clinically significant especially when combined with long-acting anticholinergic agents [18].
In light of the above discussion, a combination of short-acting beta agonist and muscarinic antagonist is recommended in all patients with COPD unless the patient is on a long-acting muscarinic antagonist [7,18]. In the latter case, a short-acting beta agonist used as a rescue inhaler is the best option. In our patient, albuterol was the choice for his short-acting bronchodilator as he was using the long-acting muscarinic antagonist tiotropium.
Are short-acting bronchodilators enough? What do we use for maintenance therapy?
All patients with COPD who are category B or higher according to the modified GOLD staging system should be on a long-acting bronchodilator [7,19]: either a long-acting beta agonist (LABA) or long-acting muscarinic antagonist (LAMA). Long-acting bronchodilators work on the same receptors as their short-acting counterparts but have structural differences. Salmeterol is the prototype for long-acting selective beta-2 agonist. It is structurally similar to albuterol but has an elongated side chain that allows it to bind firmly to the area of beta receptors and stimulate them repetitively, resulting in an extendedduration of action [20]. Tiotropium on the other hand is a quaternary ammonium of ipratropium that is a nonselective muscarinic antagonist [21]. Compared to ipratropium, tiotropium dissociates more quickly from M2 receptors, which is responsible for the undesired anticholinergic effects, while at the same time it binds M1 and M3 receptors for a prolonged time, resulting in extended duration of action [21].
The currently available long-acting beta agonists include salmeterol, formoterol, aformoterol, olodatetol, and indacaterol. The last two have the advantage of once-daily dosing rather than twice [22,23]. LABAs have been shown to improve lung function, exacerbation rate, and quality of life in multiple clinical trials [22–24]. Vilanterol is another LABA that has a long duration of action and can be used once daily [25], but is only available in a combination with umeclidinium, a LAMA. Several LAMAs are approved for use in COPD, including the prototype tiotropium in addition to aclidinium, umeclidinium, and glycopyrronium. These have been shown in clinical trials to improve lung function, symptoms, and exacerbation rate [26–29].
Patients can be started on either a LAMA or LABA depending on patient needs and side effects [7]. Both have comparable side effects and efficacy as detailed below. Concerning side effects, there is conflicting data concerning an association of cardiovascular events with both classes of long-acting bronchodilators. While clinical trials failed to show an increased risk [24,30,31], several retrospective studies showed an increased risk of emergency room visits and hospitalizations due to tachyarrhythmias, heart failure, myocardial infarction, and stroke upon initiation of long-acting bronchodilators [32,33]. There was no difference in risk for adverse cardiovascular events between LABA and LAMA in one Canadian study, and slightly more with LABA in a study using an American database [32,33]. Urinary retention is another possible complication of LAMA supported by evidence from meta-analyses and retrospective studies but not clinical trials and should be discussed with patients upon initiation [34,35]. There have been concerns about increased mortality with the soft mist formulation of tiotropium that were put to rest by the tiotropium safety and performance in Respimat (TIOSPIR) trial, which showed no increased mortality compared to Handihaler [36].
As far as efficacy and benefits, tiotropium and salmeterol were compared head-to-head in a clinical trial, and tiotropium increased the time before developing first exacerbation and decreased the overall rate of exacerbations [37]. No difference in hospitalization rate or mortality was noted in one meta-analysis, although tiotropium was more effective in reducing exacerbations [38]. The choice of agent should be made based on patient comorbidities and side effects. For example, an elderly patient with severe benign prostatic hyperplasia and urinary retention should try a LABA while for a patient with severe tachycardia induced by albuterol, LAMA would be a better first agent.
What is the role of inhaled corticosteroids in COPD?
Inhaled corticosteroids (ICS) are believed to work in COPD by reducing airway inflammation [39]. ICS should not be used alone for COPD management and are always combined with LABA [7]. Several inhaled corticosteroid formulations are approved for use in COPD, including budesonide and fluticasone. ICS has been shown to decrease symptoms and exacerbations with modest effect on lung function and no change in mortality [40]. Side effects include oral candidiasis, dysphonia, and skin bruising [41]. There is also an increased risk of pneumonia [42]. ICS are best reserved for patients with a component of asthma or asthma–COPD overlap syndrome (ACOS) [43]. ACOS is characterized by persistent airflow limitation with several features usually associated with asthma and several features usually associated with COPD [44].
What if the patient is still symptomatic on a LABA or LAMA?
For patients whose symptoms are not controlled on one class of long-acting bronchodilator, recommendations are to add a bronchodilator from the other class [7]. There are also multiple combined LAMA-LABA inhalers that are approved in the US and can possible improve adherence to therapy. These include tiotropium-oladeterol, umeclidinium-vilanterol, glycopyronnium-indacaterol, and glycopyrrolate-formoterol. In a large systematic review and meta-analysis comparing LABA-LAMA combination to either agent alone, there was a modest improvement in post bronchodilator FEV1 and quality of life with no change in hospital admissions, mortality, or side effects [45]. Interestingly, adding tiotropium to LABA reduced exacerbations although adding LABA to tiotropium did not [45].
Current guidelines recommend that patients in GOLD categories C and D that are not well controlled should receive a combination of LABA-ICS [7]. However, a new randomized trial showed better reduction of exacerbations and decreased occurrence of pneumonia in patients receiving LAMA-LABA compared to LABA-ICS [46]. In light of this new evidence, it is prudent to use a LAMA-LABA combination before adding ICS.
Triple therapy with LAMA, LABA, and ICS is a common approach for patients with severe uncontrolled disease and has been shown to decrease exacerbations and improve quality of life [7,47]. Adding tiotropium to LABA-ICS decreased exacerbations and improved quality of life and airflow in the landmark UPLIFT trial [26]. In another clinical trial, triple therapy with LAMA, LABA, and ICS compared to tiotropium alone decreased severe exacerbations, pre-bronchodilator FEV1, and morning symptoms [48].
Is there a role for theophylline? Other agents?
Theophylline
Theophylline is an oral adenosine diphosphate antagonist with indirect adrenergic activity, which is responsible for the bronchodilator therapeutic effect in patients with obstructive lung disease. It is also thought to work by an additional mechanism that decreases inflammation in the airways [49]. It has a serious side effect profile that includes ventricular arrhythmias, seizures, vomiting, and tremor [50]. It is metabolized in the liver and has multiple drug interactions and a narrow therapeutic index. It has been shown to improve lung function, gas exchange and symptoms in meta-analysis and clinical trials [51,52].
In light of the nature of the adverse effects and the wide array of safer and more effective pharmacologic agents available, theophylline should be avoided early on in patients with COPD. Its use can be justified as an add-on therapy in patients with refractory disease on triple therapy for symptomatic relief [50]. If used, the therapeutic range for COPD is 8–12 mcg/mL peak level measured 3 to 7 hours after morning dose and is usually achieved using a daily dose of 10 mg per kilogram of body weight for nonobese patients [53].
Systemic Steroids
Oral steroids are used in COPD exacerbations but should never be used chronically in COPD patients regardless of disease severity as they increase morbidity and mortality without improving symptoms or lung function [54,55]. The dose of systemic steroids should be tapered and finally discontinued.
Mucolytics
Classes of mucolytics include thiol derivatives, inhaled dornase alpha, hypertonic saline, and iodine preparations. Thiol derivatives such as N-acetylcysteine are the most widely studied [56].
There is no consistent evidence of beneficial role of mucolytics in COPD patient [7,56]. The PANTHEON trial showed decreased exacerbations with N-acetylcysteine (1.16 exacerbations per patient-year compared to 1.49 exacerbations per patient-year in the placebo group; risk ratio 0.78, 95% CI 0.67–0.90; P = 0.001) but had methodologic issues including high drop-out rate, exclusion of patients on oxygen, and a large of proportion of nonsmokers [57].
Chronic Antibiotics
There is no role for chronic antibiotics in the management of COPD [7]. Macrolides are an exception but are used for their anti-inflammatory effects rather than their antibiotic effects. They should be reserved for patient with frequent exacerbations on optimal therapy and will be discussed later in the review [58].
What nonpharmacologic treatments are recommended for COPD patients?
Smoking cessation, oxygen therapy for severe hypoxemia (resting O2 saturation ≤ 88 or PaO2 ≤ 55), vaccination for influenza and pneumococcus, and appropriate nutrition should be provided in all COPD patients. Pulmonary rehabilitation is indicated for patients in GOLD categories B, C, and D [7]. It improves symptoms, quality of life, exercise tolerance and health care utilization. Beneficial effects last for about 2 years [59,60].
What other diagnoses should be considered in patients who continue to be symptomatic on optimal therapy?
Other diseases that share the same risk factors as COPD and can contribute to dyspnea, including coronary heart disease, heart failure, thromboembolic disease, and pulmonary hypertension, should be considered. In addition, all patients with refractory disease should have a careful assessment of their inhaler technique, continued smoking, need for oxygen therapy, and associated deconditioning.
Case 2
A 70-year-old male with severe COPD on oxygen therapy and obstructive sleep apnea treated on nocturnal CPAP was seen in the pulmonary clinic for evaluation of his dyspnea. He was symptomatic with minimal activity and had chronic cough with some sputum production. He had been hospitalized 3 times over the past 12 months and had been to the emergency department (ED) the same number of times for dyspnea. Pertinent medications included as-needed albuterol inhaler, inhaled steroids, and tiotropium 18 mcg inhaled daily. He demonstrated good inhaler technique. On examination, his vital signs were pulse 99 bpm, SpO2 94% on 2L/min oxygen by nasal cannula, blood pressure 126/72 mm Hg, respiratory rate 15, and BMI 35 kg/m2. He appeared chronically ill but in no acute distress. No wheezing or rales were heard. He had no lower extremity edema. The remainder of the exam was within normal limits. His last pulmonary function test demonstrated moderate obstruction with significant bronchodilator response to 2 puffs of albuterol. The side effects of chronic steroid therapy were impressed upon the patient and 500 mg of roflumilast was started daily. Over the course of the next 3 months, he had no further exacerbations. Roflumilast was continued. He has not required any further hospitalizations, ED visits, or oral steroid use since the last clinic visit.
What is the significance of acute exacerbations of COPD?
Acute exacerbation of COPD (AECOPD) is a frequently observed complication for many patients with COPD [61,62]. AECOPD is associated with accelerated disease progression, augmented decline in health status and quality of life, and increased mortality [63]. Exacerbations account for most of the costs associated with COPD. Estimates suggest that the aggregate costs associated with the treatment of AECOPDs are between $3.2 and $3.8 billion, and that annual health care costs are 10-fold greater for patients with COPD associated with acute exacerbations than for patients with COPD but without exacerbations [64]. Hence, any intervention that could potentially minimize or prevent this complication will have far-reaching benefits to patients with COPD as well as provide significant cost saving.
How is acute exacerbation of COPD defined?
COPD exacerbation is defined as a baseline change of the patient’s dyspnea, cough, and/or sputum that is acute in onset, and may warrant a change in regular medication in a patient with underlying COPD [65]. Exacerbation in clinical trials has been defined on the basis of whether an increase in the level of care beyond regular care is required primarily in the hospital or ED [66]. Frequent exacerbations are defined as 3 symptom-defined exacerbations per year or 2 per year if defined by the need for therapy with corticosteroids, antibiotics, or both [67].
What is the underlying pathophysiology?
AECOPD is associated with enhanced upper and lower airway and systemic inflammation. The bronchial mucosa of stable COPD patients have increased numbers of CD8+ lymphocytes and macrophages. In mild AECOPD, eosinophils are increased in the bronchial mucosa and modest elevation of neutrophils, T lymphocytes (CD3), and TNF alpha positive cells has also been reported [62]. With more severe AECOPD, airway neutrophils are increased. Oxidative stress is a key factor in the development of airway inflammation in COPD [61]. Patients with severe exacerbations have augmented large airway interleukin-8 (IL-8) levels and increased oxidative stress as demonstrated by markers such as hydrogen peroxide and 8-isoprostane [66].
How do acute exacerbations affect the course of the disease?
In general, as the severity of the underlying COPD increases, exacerbations become both more severe and more frequent. The quality of life of patients with frequent exacerbations is worse than patients with a history of less frequent exacerbations [68]. Frequent exacerbations have also been linked to a decline in lung function, with studies suggesting that there might be a decline of 7 mL in FEV1 per lower respiratory tract infection per year [59,69] and approxi-mately 8 mL per year in patients with frequent exacerbations as compared to those with sporadic exacerbations [70].
What are the triggers for COPD exacerbation?
Respiratory infections are estimated to trigger approximately two-thirds of exacerbations [62]. Viral and bacterial infections cause most exacerbations. The effect of the infective triggers is to increase inflammation, cause bronchoconstriction, edema, and mucus production, with a resultant increase in dynamic hyperinflation [71]. Thus, any intervention that reduces inflammation in COPD reduces the number and severity of exacerbations, whereas bronchodilators have an impact on exacerbation by their effects on reducing dynamic hyperinflation. The triggers for the one-third of exacerbations not triggered by infection are postulated to be related to other medical conditions, including pulmonary embolism, aspiration, heart failure, and myocardial ischemia [66].
What are the pharmacologic options available for prevention of AECOPD?
In recognition of the importance of preventing COPD exacerbations, the American College of Chest Physicians and Canadian Thoracic Society [65] have published an evidence-informed clinical guideline specifically examining the prevention of AECOPD, with the goal of assisting clinicians in providing optimal management for COPD patients. The following pharmacologic agents have been recognized as being effective at reducing the frequency of acute exacerbations without any impact on the severity of COPD itself.
Roflumilast
Phosphodiesterase 4 (PDE4) inhibition appears to have inflammatory modulating properties in the airways, although the exact mechanism of action is unclear. Some have proposed that it reduces inflammation by inhibiting the breakdown of intracellular cyclic adenosine monophosphate [72]. In 2 large clinical trials [73,74], daily use of a PDE4 inhibitor (roflumilast) showed a significant (15%–18%) reduction in yearly AECOPD incidence (approximate number needed to treat: 4). This benefit was seen in patients with GOLD stage 3–4 disease (FEV1 < 50% predicted) with the chronic bronchitic phenotype and who had experienced at least 1 exacerbation in the previous year.
Importantly, these clinical trials specifically prohibited the use of ICS and LAMAs. Thus, it remains unclear if PDE4 inhibition should be used as an add-on to ICS/LAMA therapy in patients who continue to have frequent AECOPD or whether PDE4 inhibition could be used instead of these standard therapies in patients with well-controlled daily symptoms without ICS or LAMA therapy but who experience frequent exacerbations.
Of note, earlier trials with roflumilast included patients with ICS and LAMA use [73,75]. These trials were focused on FEV1 improvement and found no benefit. It was only in post ad hoc analyses that a reduction in AECOPD in patients with frequent exacerbations was found among those taking roflumilast, regardless of ICS or LAMA use [76]. While roflumilast has documented benefit in improving lung function and reducing the rate of exacerbations, it has not been reported to decrease hospitalizations [64]. This indicates that although the drug reduces the total number of exacerbations, it may not be as useful in preventing episodes of severe exacerbations of COPD.
Although PDE4 inhibitors are easy to administer (a once-daily pill), they are associated with significant GI side effects (diarrhea, nausea, reduced appetite), weight loss, headache, and sleep disturbance [77]. Adverse effects tend to occur early during treatment, are reversible, and lessen over time with treatment [66]. Studies reported an average unexplained weight loss of 2 kg, and monitoring weight during treatment is advised. In addition, it is important to avoid roflumilast in underweight patients. Roflumilast should also be used with caution in depressed patients [65].
N-acetylcysteine
N-acetylcysteine (NAC) reduces the viscosity of respiratory secretions as a result of the cleavage of the disulfide bonds and has been studied as a mucolytic agent to aid in the elimination of respiratory secretions [78]. Oral NAC is quickly absorbed and is rapidly present in an active form in lung tissue and respiratory secretions after ingestion. NAC is well tolerated except for occasional patients with GI adverse effects. The role of NAC in preventing AECOPD has been studied for more than 3 decades [79–81], although the largest clinical trial to date was reported in 2014 [57]. Taken together, the combined data demonstrate a significant reduction in the rate of COPD exacerbations associated with the use of NAC when compared with placebo (OR, 0.61; CI, 0.37–0.99). Clinical guidelines suggest that in patients with moderate to severe COPD (FEV1/FVC < 0.7, and FEV1 < 80% predicted) receiving maintenance bronchodilator therapy combined with ICS and history of 2 more exacerbations in the previous 2 years, treatment with oral NAC can be administered to prevent AECOPD.
Macrolides
Continuous prophylactic use of antibiotics in older studies had no effect on the frequency of AECOPD [82,83]. But it is known that macrolide antibiotics have several antimicrobial, anti-inflammatory and immunomodulating effects and have been used for many years in the management of other chronic airway disease, including diffuse pan-bronchiolitis and cystic fibrosis [65]. One recent study showed that the use of once-daily, generic azithromycin 5 days/week appeared to have an impact on AECOPD incidence [84]. In this study, AECOPD was reduced from 1.83 to 1.48 per patient-year (RR, 0.83; 95% CI, 0.72–0.95: P = 0.01). Azithromycin also prevented severe AECOPD. Greater benefit was obtained with milder forms of the disease and in the elderly. Azithromycin did not appear to provide any benefit in those who continued to smoke (hazard ratio, 0.99) [85]. Other studies have shown that azithromycin was associated with an increased incidence of bacterial resistance and impaired hearing [86]. Overall data from the available clinical trials are robust and demonstrate that regular macrolide therapy definitely reduces the risk of AECOPD. But due to potential side effects macrolide therapy is an option rather than a strong recommendation [65]. The prescribing clinician also needs to consider the potential of prolongation of the QT interval [84].
Immunostimulants
Immunostimulants have also been reported to reduce frequency of AECOPD [87,88]. Bacterial lysates, reconstituted mixtures of bacterial antigens present in the lower airways of COPD patients, act as immuno-stimulants through the induction of cellular maturation, stimulating lymphocyte chemotaxis, and increasing opsonization when administered to individuals with COPD [66]. Studies have demonstrated a reduction in the severe complications of exacerbations and hospital admissions in COPD patients with OM-85, a detoxified oral immunoactive bacterial extract [87,88]. However, most of these trials were conducted prior to the routine use of long-acting bronchodilators and ICS in COPD. A recent study by Braido et al evaluated the efficacy of ismigen, a bacterial lysate, in reducing AECOPD [89] and found no difference in the exacerbation rate between ismigen and placebo or the time to first exacerbation. Additional studies are needed to examine the long-term effects of this therapy in patients receiving currently recommended COPD maintenance therapy [66].
β Blockers
Observational studies of beta-blocker use in preventing AECOPD have yielded encouraging results, with one study showing a reduction in AECOPD risk (incidence risk ratio, 0.73; CI 0.60–0.90) in patients receiving beta blockers versus those not on beta blockers [90]. Based on these findings, a clinical trial investigating the impact of metoprolol on risk of AECOPD is ongoing [91].
Proton Pump Inhibitors
Gastroesophageal reflux disease is an independent risk factor for exacerbations [92]. Two small, single-center studies [93,94] have shown that use of lansoprazole decreases the risk and frequency of AECOPD. However, data from the Predicting Outcome using Systemic Markers in Severe Exacerbations of COPD (PROMISE-COPD) study [66], which was a multicenter prospective observational study, suggested that patients with stable COPD receiving a proton pump inhibitor were at high risk of frequent and severe exacerbations [95]. Thus, at this stage, their definitive role needs to be defined, possibly with a randomized, placebo-controlled study.
Case 3
A 65-year-old male with severe COPD (FEV1/FVC 27, FEV1 25% of predicted, residual volume 170% of predicted for his age and height) was seen in the pulmonary clinic. His medications include a LABA/LAMA combination that he uses twice daily as advised. He uses his rescue albuterol inhaler roughly once a week. The patient complains of severe disabling shortness of breath with exertion and severe limitation of his quality of life because of his inability to lead a normal active life. He is on 2 L/min of oxygen at all times. He has received pulmonary rehabilitation in hopes of improving his quality of life but can only climb a flight of stairs before he must stop to rest. He asks about options but does not want to consider lung transplantation today. His most recent chest CT scan demonstrates upper lobe predominant emphysematous changes with no masses or nodules.
What are the patient's options at this time?
Lung volume reduction surgery (LVRS) attempts to reduce space-occupying severely diseased, hyperexpanded lung, thus allowing the relatively normal adjoining lung parenchyma to expand into the vacated space and function effectively [96].Hence, such therapies are suitable for patients with emphysematous lungs and not those with bronchitic-predominant COPD. LVRS offers a greater chance of improvement in exercise capacity, lung function, quality of life, and dyspnea in the correctly chosen patient population as compared with pharmacologic management alone [97]. However, the procedure is associated with risks, including higher short-term morbidity and mortality [97]. Patients with predominantly upper-lobe emphysema and a low maximal workload after rehabilitation were noted to have lower mortality, a greater probability of improvement in exercise capacity, and a greater probability of improvement in symptoms if they underwent surgery compared to medical therapy alone [97]. On the contrary, patients with predominantly non–upper-lobe emphysema and a high maximal workload after rehabilitation had higher mortality if they underwent surgery compared to receiving medical therapy alone [97]. Thus, a subgroup of patients with homogeneous emphysema symmetrically affecting the upper and lower lobes are considered to be unlikely to benefit from this surgery [97,98].
Valves and other methods of lung volume reduction such as coils, sealants, intrapulmonary vents, and thermal vapor in the bronchi or subsegmental airways have emerged as new techniques for nonsurgical lung volume reduction [99–104]. Endobronchial-valve therapy is associated with improvement in lung function and with clinical benefits that are greatest in the presence of heterogeneous lung involvement. This works by the same principle as with LVRS, by reduction of the most severely diseased lung units, expansion of the more viable, less emphysematous lung results in substantial improvements in lung mechanics [105,106]. The most important complications of this procedure include pneumonia, pneumothorax, hemoptysis and increased frequency of COPD exacerbation in the following thirty days. The fact that high-heterogeneity subgroup had greater improvements in both the FEV1 and distance on the 6-minute walk test than did patients with lower heterogeneity supports the use of quantitative high-resolution computed tomography (HRCT) in selecting patients for endobronchial-valve therapy [107].The HRCT scans also help in identifying those with complete fissures; a marker of lack of collateral ventilation (CV+) between different lobes. Presence of CV+ state predicts failure of endobronchial valve and all forms of endoscopic lung volume reduction strategies [108]. Bronchoscopic thermal vapor ablation (BTVA) therapy can potentially work on a subsegmental level and be successful for treatment of emphysema with lack of intact fissures on CT scans. Other methods that have the potential to be effective in those with collateral ventilation would be endoscopic coil therapy and polymeric lung volume reduction [106,109].Unfortunately, there are no randomized controlled trial data demonstrating clinically meaningful improvement following coil therapy or polymeric lung volume reduction in this CV+ patient population. Vapor therapy is perhaps the only technique that has been found to be effective in upper lobe predominant emphysema even with CV+ status [108].
Our patient has evidence of air trapping and emphysema based on a high residual volume. A CT scan of the chest can determine the nature of the emphysema (heterogeneous versus homogenous) and based on these findings, further determination of the best strategy for lung volume reduction can be made.
Is there a role for long-term oxygen therapy?
Long-term oxygen therapy (LTOT) used for > 15 hours a day is thought to reduce mortality among patients with chronic obstructive pulmonary disease (COPD) and severe resting hypoxemia [110–113].More recent studies have failed to show similar beneficial effects of LTOT. A recent study examined the effects of LTOT in randomized fashion and determined that supplemental oxygen for patients with stable COPD and resting or exercise-induced moderate desaturation did not affect the time to death or first hospitalization, time to first COPD exacerbation, time to first hospitalization for a COPD exacerbation, the rate of all hospitalizations, the rate of all COPD exacerbations, or changes in measures of quality of life, depression, anxiety, or functional status [114].
Our patient is currently on long-term oxygen therapy and in spite of some uncertainty as to its benefit, it is prudent to order oxygen therapy until further clarification is available.
What is the role of pulmonary rehabilitation?
Pulmonary rehabilitation is an established treatment for patients with chronic lung disease [115]. Benefits include improvement in exercise tolerance, symptoms, and quality of life, with a reduction in the use of health care resources [116].A Spanish population-based cohort study that looked at the influence of regular physical activity on COPD showed that patients who reported low, moderate, or high physical activity had a lower risk of COPD admissions and all-cause mortality than patients with very low physical activity after adjusting for all confounders [117].
As previously mentioned, patients in GOLD categories B, C, and D should be offered pulmonary rehabilitation as part of their treatment [7]. The ideal patient is one who is not too sick to undergo rehabilitation and is motivated to his or her quality of life.
What is the current scope of lung transplantation in the management of severe COPD?
There is a indisputable role for lung transplantation in end-stage COPD. However, lung transplantation does not benefit all COPD patients. There is a subset of patients for whom the treatment provides a survival benefit. It has been reported that 79% of patients with an FEV1 < 16% predicted will survive at least 1 year additional after transplant, but only 11% of patients with an FEV1 > 25% will do so [118]. The pre-transplant BODE (body mass index, airflow obstruction/FEV1, dyspnea, and exercise capacity) index score is used to identify the patients who will benefit from lung transplantation [119,120]. International guidelines for the selection of lung transplant candidates identify the following patient characteristics [121]:
- The disease is progressive, despite maximal treatment including medication, pulmonary rehabilitation, and oxygen therapy
- The patient is not a candidate for endoscopic or surgical LVRS
- BODE index of 5 to 6
- The partial pressure of carbon dioxide is greater than 50 mm Hg or 6.6kPa and/or partial pressure of oxygen is less than 60 mm Hg or 8kPa
- FEV1 of 25% predicted
The perioperative mortality of lung transplantation surgery has been reduced to less than 10%. Risk of complications from surgery in the perioperative period, such as bronchial dehiscence, infectious complications, and acute rejection, have also been reduced but do occur. Chronic allograft dysfunction and the risk of lung cancer in cases of single lung transplant should be discussed with the patient before surgery [122].
How can we incorporate palliative care into the management plan for patients with COPD?
Among patients with end-stage COPD on home oxygen therapy who have required mechanical ventilation for an exacerbation, only 55% are alive at 1 year [123]. COPD patients at high risk of death within the next year of life as well as patients with refractory symptoms and unmet needs are candidates for early palliative care. Palliative care and palliative care specialists can aid in reducing symptom burden and improving quality of life among these patients and their family members and is recommended by multiple international societies for patients with advanced COPD [124,125]. In spite of these recommendations, the utilization of palliative care resources has been dismally low [126,127]. Improving physician-patient communication regarding palliative services and patients’ unmet care needs will help ensure that COPD patients receive adequate palliative care services at the appropriate time.
Conclusion
COPD is a leading cause of morbidity and mortality in the United States and represents a significant economic burden for both individuals and society. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. COPD management is guided by disease severity that is measured using the GOLD multimodal staging system and requires a multidisciplinary approach. Several classes of medication are available for treatment, and a step-wise approach should be applied in building an effective pharmacologic regimen. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
Corresponding author: Abhishek Biswas, MD, Division of Pulmonary and Critical Care Medicine, Rm. M452, University of Florida, 1600 SW Archer Rd, Gainesville, FL 32610, [email protected].
Financial disclosures: None.
From the Division of Pulmonary Critical Care Medicine, University of Florida, Gainesville, FL.
Abstract
- Objective:To review the management of stable chronic obstructive pulmonary disease (COPD).
- Methods: Review of the peer-reviewed literature.
- Results: Effective management of stable COPD requires the physician to apply a stepwise intensification of therapy depending on patient symptoms and functional reserve. Bronchodilators are the cornerstone of management. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
- Conclusion: Successful management of stable COPD requires a multidisciplinary approach that utilizes various medical therapies as well as nonpharmacologic interventions.
Key words: chronic obstructive pulmonary disease; exacerbation; bronchodilator; lung volume reduction; cough.
Chronic obstructive pulmonary disease (COPD) is a systemic inflammatory disease characterized by irreversible obstructive ventilatory defects [1–4]. It is a major cause of morbidity and mortality affecting 5% of the population in the United States and was the third leading cause of death in 2008 [5,6]. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. In this review, we will discuss the management of stable COPD in the context of 3 common clinical scenarios.
Case 1
A 65-year-old male with COPD underwent pulmonary function testing (PFT), which demonstrated an obstructive ventilatory defect (forced expiratory volume in 1 second/forced vital capacity ratio [FEV1/FVC], 0.45; FEV1, 2 L [65% of predicted]; and diffusing capacity of the lung for carbon monoxide [DLCO], 15 [65% of predicted]). He has dyspnea with strenuous exercise but is comfortable at rest and with minimal exercise. He has had 1 exacerbation in the last year that was treated on an outpatient basis with steroids and antibiotics. His medication regimen includes inhaled tiotropium once daily and inhaled albuterol as needed that he uses roughly twice a week.
What determines the appropriate therapy for a given COPD patient?
What is the approach to building a pharmacologic regimen for the patient with COPD?
The backbone of the pharmacologic regimen for COPD includes short- and long-acting bronchodilators. They are usually given in an inhaled form to maximize local effects on the lungs and minimize systemic side effects. There are 2 main classes of bronchodilators, beta agonists and muscarinic antagonists, and each targets specific receptors on the surface of airway smooth muscle cells. Beta agonists work by stimulating beta-2 receptors, resulting in bronchodilation, while muscarinic antagonists work by blocking the bronchoconstrictor action of M3 muscarinic receptors. Inhaled corticosteroids can be added to long-acting bronchodilator therapy but cannot be used as stand-alone therapy. Theophylline is an oral bronchodilator that is used infrequently due to its narrow therapeutic index, toxicity, and multiple drug interactions.
Who should be on short-acting bronchodilators? What is the best agent? Should it be scheduled or used as needed?
All patients with COPD should be an on inhaled short-acting bronchodilator as needed for relief of symptoms [7]. Both short-acting beta agonists (albuterol and levalbuterol) and short-acting muscarinic antagonists (ipratropium) have been shown in clinical trials and meta-analyses to improve symptoms and lung function in patients with stable COPD [9,10] and seem to have comparative efficacy when compared head-to-head in trials [11]. However, the airway bronchodilator effect achieved by both classes seems to be additive when used in combination and is also associated with less exacerbations compared to albuterol alone [12]. On the other hand, adding albuterol to ipratropium increased the bronchodilator response but did not reduce the exacerbation rate [11–13]. Inhaled short-acting beta agonists when used as needed rather than scheduled are associated with less medication use without any significant difference in symptoms or lung function [14].
The side effects related to using recommended doses of a short-acting bronchodilator are minimal. In retrospective studies, short-acting beta agonists increased the risk of severe cardiac arrhythmias [15]. Levalbuterol, the active enantiomer of albuterol (R-albuterol) developed for the theoretical benefits of reduced tachycardia, increased tolerability, and better or equal efficacy compared to racemic albuterol, failed to show a clinically significant difference in inducing tachycardia [16]. Beta agonist overuse is associated with tremor and in severe cases hypokalemia, which happens mainly when patients try to achieve maximal bronchodilation; the clinically used doses of beta agonists are associated with fewer side affects but achieve less than maximal bronchodilation [17]. Ipratropium can produce systemic anticholinergic side effects, urinary retention being the most clinically significant especially when combined with long-acting anticholinergic agents [18].
In light of the above discussion, a combination of short-acting beta agonist and muscarinic antagonist is recommended in all patients with COPD unless the patient is on a long-acting muscarinic antagonist [7,18]. In the latter case, a short-acting beta agonist used as a rescue inhaler is the best option. In our patient, albuterol was the choice for his short-acting bronchodilator as he was using the long-acting muscarinic antagonist tiotropium.
Are short-acting bronchodilators enough? What do we use for maintenance therapy?
All patients with COPD who are category B or higher according to the modified GOLD staging system should be on a long-acting bronchodilator [7,19]: either a long-acting beta agonist (LABA) or long-acting muscarinic antagonist (LAMA). Long-acting bronchodilators work on the same receptors as their short-acting counterparts but have structural differences. Salmeterol is the prototype for long-acting selective beta-2 agonist. It is structurally similar to albuterol but has an elongated side chain that allows it to bind firmly to the area of beta receptors and stimulate them repetitively, resulting in an extendedduration of action [20]. Tiotropium on the other hand is a quaternary ammonium of ipratropium that is a nonselective muscarinic antagonist [21]. Compared to ipratropium, tiotropium dissociates more quickly from M2 receptors, which is responsible for the undesired anticholinergic effects, while at the same time it binds M1 and M3 receptors for a prolonged time, resulting in extended duration of action [21].
The currently available long-acting beta agonists include salmeterol, formoterol, aformoterol, olodatetol, and indacaterol. The last two have the advantage of once-daily dosing rather than twice [22,23]. LABAs have been shown to improve lung function, exacerbation rate, and quality of life in multiple clinical trials [22–24]. Vilanterol is another LABA that has a long duration of action and can be used once daily [25], but is only available in a combination with umeclidinium, a LAMA. Several LAMAs are approved for use in COPD, including the prototype tiotropium in addition to aclidinium, umeclidinium, and glycopyrronium. These have been shown in clinical trials to improve lung function, symptoms, and exacerbation rate [26–29].
Patients can be started on either a LAMA or LABA depending on patient needs and side effects [7]. Both have comparable side effects and efficacy as detailed below. Concerning side effects, there is conflicting data concerning an association of cardiovascular events with both classes of long-acting bronchodilators. While clinical trials failed to show an increased risk [24,30,31], several retrospective studies showed an increased risk of emergency room visits and hospitalizations due to tachyarrhythmias, heart failure, myocardial infarction, and stroke upon initiation of long-acting bronchodilators [32,33]. There was no difference in risk for adverse cardiovascular events between LABA and LAMA in one Canadian study, and slightly more with LABA in a study using an American database [32,33]. Urinary retention is another possible complication of LAMA supported by evidence from meta-analyses and retrospective studies but not clinical trials and should be discussed with patients upon initiation [34,35]. There have been concerns about increased mortality with the soft mist formulation of tiotropium that were put to rest by the tiotropium safety and performance in Respimat (TIOSPIR) trial, which showed no increased mortality compared to Handihaler [36].
As far as efficacy and benefits, tiotropium and salmeterol were compared head-to-head in a clinical trial, and tiotropium increased the time before developing first exacerbation and decreased the overall rate of exacerbations [37]. No difference in hospitalization rate or mortality was noted in one meta-analysis, although tiotropium was more effective in reducing exacerbations [38]. The choice of agent should be made based on patient comorbidities and side effects. For example, an elderly patient with severe benign prostatic hyperplasia and urinary retention should try a LABA while for a patient with severe tachycardia induced by albuterol, LAMA would be a better first agent.
What is the role of inhaled corticosteroids in COPD?
Inhaled corticosteroids (ICS) are believed to work in COPD by reducing airway inflammation [39]. ICS should not be used alone for COPD management and are always combined with LABA [7]. Several inhaled corticosteroid formulations are approved for use in COPD, including budesonide and fluticasone. ICS has been shown to decrease symptoms and exacerbations with modest effect on lung function and no change in mortality [40]. Side effects include oral candidiasis, dysphonia, and skin bruising [41]. There is also an increased risk of pneumonia [42]. ICS are best reserved for patients with a component of asthma or asthma–COPD overlap syndrome (ACOS) [43]. ACOS is characterized by persistent airflow limitation with several features usually associated with asthma and several features usually associated with COPD [44].
What if the patient is still symptomatic on a LABA or LAMA?
For patients whose symptoms are not controlled on one class of long-acting bronchodilator, recommendations are to add a bronchodilator from the other class [7]. There are also multiple combined LAMA-LABA inhalers that are approved in the US and can possible improve adherence to therapy. These include tiotropium-oladeterol, umeclidinium-vilanterol, glycopyronnium-indacaterol, and glycopyrrolate-formoterol. In a large systematic review and meta-analysis comparing LABA-LAMA combination to either agent alone, there was a modest improvement in post bronchodilator FEV1 and quality of life with no change in hospital admissions, mortality, or side effects [45]. Interestingly, adding tiotropium to LABA reduced exacerbations although adding LABA to tiotropium did not [45].
Current guidelines recommend that patients in GOLD categories C and D that are not well controlled should receive a combination of LABA-ICS [7]. However, a new randomized trial showed better reduction of exacerbations and decreased occurrence of pneumonia in patients receiving LAMA-LABA compared to LABA-ICS [46]. In light of this new evidence, it is prudent to use a LAMA-LABA combination before adding ICS.
Triple therapy with LAMA, LABA, and ICS is a common approach for patients with severe uncontrolled disease and has been shown to decrease exacerbations and improve quality of life [7,47]. Adding tiotropium to LABA-ICS decreased exacerbations and improved quality of life and airflow in the landmark UPLIFT trial [26]. In another clinical trial, triple therapy with LAMA, LABA, and ICS compared to tiotropium alone decreased severe exacerbations, pre-bronchodilator FEV1, and morning symptoms [48].
Is there a role for theophylline? Other agents?
Theophylline
Theophylline is an oral adenosine diphosphate antagonist with indirect adrenergic activity, which is responsible for the bronchodilator therapeutic effect in patients with obstructive lung disease. It is also thought to work by an additional mechanism that decreases inflammation in the airways [49]. It has a serious side effect profile that includes ventricular arrhythmias, seizures, vomiting, and tremor [50]. It is metabolized in the liver and has multiple drug interactions and a narrow therapeutic index. It has been shown to improve lung function, gas exchange and symptoms in meta-analysis and clinical trials [51,52].
In light of the nature of the adverse effects and the wide array of safer and more effective pharmacologic agents available, theophylline should be avoided early on in patients with COPD. Its use can be justified as an add-on therapy in patients with refractory disease on triple therapy for symptomatic relief [50]. If used, the therapeutic range for COPD is 8–12 mcg/mL peak level measured 3 to 7 hours after morning dose and is usually achieved using a daily dose of 10 mg per kilogram of body weight for nonobese patients [53].
Systemic Steroids
Oral steroids are used in COPD exacerbations but should never be used chronically in COPD patients regardless of disease severity as they increase morbidity and mortality without improving symptoms or lung function [54,55]. The dose of systemic steroids should be tapered and finally discontinued.
Mucolytics
Classes of mucolytics include thiol derivatives, inhaled dornase alpha, hypertonic saline, and iodine preparations. Thiol derivatives such as N-acetylcysteine are the most widely studied [56].
There is no consistent evidence of beneficial role of mucolytics in COPD patient [7,56]. The PANTHEON trial showed decreased exacerbations with N-acetylcysteine (1.16 exacerbations per patient-year compared to 1.49 exacerbations per patient-year in the placebo group; risk ratio 0.78, 95% CI 0.67–0.90; P = 0.001) but had methodologic issues including high drop-out rate, exclusion of patients on oxygen, and a large of proportion of nonsmokers [57].
Chronic Antibiotics
There is no role for chronic antibiotics in the management of COPD [7]. Macrolides are an exception but are used for their anti-inflammatory effects rather than their antibiotic effects. They should be reserved for patient with frequent exacerbations on optimal therapy and will be discussed later in the review [58].
What nonpharmacologic treatments are recommended for COPD patients?
Smoking cessation, oxygen therapy for severe hypoxemia (resting O2 saturation ≤ 88 or PaO2 ≤ 55), vaccination for influenza and pneumococcus, and appropriate nutrition should be provided in all COPD patients. Pulmonary rehabilitation is indicated for patients in GOLD categories B, C, and D [7]. It improves symptoms, quality of life, exercise tolerance and health care utilization. Beneficial effects last for about 2 years [59,60].
What other diagnoses should be considered in patients who continue to be symptomatic on optimal therapy?
Other diseases that share the same risk factors as COPD and can contribute to dyspnea, including coronary heart disease, heart failure, thromboembolic disease, and pulmonary hypertension, should be considered. In addition, all patients with refractory disease should have a careful assessment of their inhaler technique, continued smoking, need for oxygen therapy, and associated deconditioning.
Case 2
A 70-year-old male with severe COPD on oxygen therapy and obstructive sleep apnea treated on nocturnal CPAP was seen in the pulmonary clinic for evaluation of his dyspnea. He was symptomatic with minimal activity and had chronic cough with some sputum production. He had been hospitalized 3 times over the past 12 months and had been to the emergency department (ED) the same number of times for dyspnea. Pertinent medications included as-needed albuterol inhaler, inhaled steroids, and tiotropium 18 mcg inhaled daily. He demonstrated good inhaler technique. On examination, his vital signs were pulse 99 bpm, SpO2 94% on 2L/min oxygen by nasal cannula, blood pressure 126/72 mm Hg, respiratory rate 15, and BMI 35 kg/m2. He appeared chronically ill but in no acute distress. No wheezing or rales were heard. He had no lower extremity edema. The remainder of the exam was within normal limits. His last pulmonary function test demonstrated moderate obstruction with significant bronchodilator response to 2 puffs of albuterol. The side effects of chronic steroid therapy were impressed upon the patient and 500 mg of roflumilast was started daily. Over the course of the next 3 months, he had no further exacerbations. Roflumilast was continued. He has not required any further hospitalizations, ED visits, or oral steroid use since the last clinic visit.
What is the significance of acute exacerbations of COPD?
Acute exacerbation of COPD (AECOPD) is a frequently observed complication for many patients with COPD [61,62]. AECOPD is associated with accelerated disease progression, augmented decline in health status and quality of life, and increased mortality [63]. Exacerbations account for most of the costs associated with COPD. Estimates suggest that the aggregate costs associated with the treatment of AECOPDs are between $3.2 and $3.8 billion, and that annual health care costs are 10-fold greater for patients with COPD associated with acute exacerbations than for patients with COPD but without exacerbations [64]. Hence, any intervention that could potentially minimize or prevent this complication will have far-reaching benefits to patients with COPD as well as provide significant cost saving.
How is acute exacerbation of COPD defined?
COPD exacerbation is defined as a baseline change of the patient’s dyspnea, cough, and/or sputum that is acute in onset, and may warrant a change in regular medication in a patient with underlying COPD [65]. Exacerbation in clinical trials has been defined on the basis of whether an increase in the level of care beyond regular care is required primarily in the hospital or ED [66]. Frequent exacerbations are defined as 3 symptom-defined exacerbations per year or 2 per year if defined by the need for therapy with corticosteroids, antibiotics, or both [67].
What is the underlying pathophysiology?
AECOPD is associated with enhanced upper and lower airway and systemic inflammation. The bronchial mucosa of stable COPD patients have increased numbers of CD8+ lymphocytes and macrophages. In mild AECOPD, eosinophils are increased in the bronchial mucosa and modest elevation of neutrophils, T lymphocytes (CD3), and TNF alpha positive cells has also been reported [62]. With more severe AECOPD, airway neutrophils are increased. Oxidative stress is a key factor in the development of airway inflammation in COPD [61]. Patients with severe exacerbations have augmented large airway interleukin-8 (IL-8) levels and increased oxidative stress as demonstrated by markers such as hydrogen peroxide and 8-isoprostane [66].
How do acute exacerbations affect the course of the disease?
In general, as the severity of the underlying COPD increases, exacerbations become both more severe and more frequent. The quality of life of patients with frequent exacerbations is worse than patients with a history of less frequent exacerbations [68]. Frequent exacerbations have also been linked to a decline in lung function, with studies suggesting that there might be a decline of 7 mL in FEV1 per lower respiratory tract infection per year [59,69] and approxi-mately 8 mL per year in patients with frequent exacerbations as compared to those with sporadic exacerbations [70].
What are the triggers for COPD exacerbation?
Respiratory infections are estimated to trigger approximately two-thirds of exacerbations [62]. Viral and bacterial infections cause most exacerbations. The effect of the infective triggers is to increase inflammation, cause bronchoconstriction, edema, and mucus production, with a resultant increase in dynamic hyperinflation [71]. Thus, any intervention that reduces inflammation in COPD reduces the number and severity of exacerbations, whereas bronchodilators have an impact on exacerbation by their effects on reducing dynamic hyperinflation. The triggers for the one-third of exacerbations not triggered by infection are postulated to be related to other medical conditions, including pulmonary embolism, aspiration, heart failure, and myocardial ischemia [66].
What are the pharmacologic options available for prevention of AECOPD?
In recognition of the importance of preventing COPD exacerbations, the American College of Chest Physicians and Canadian Thoracic Society [65] have published an evidence-informed clinical guideline specifically examining the prevention of AECOPD, with the goal of assisting clinicians in providing optimal management for COPD patients. The following pharmacologic agents have been recognized as being effective at reducing the frequency of acute exacerbations without any impact on the severity of COPD itself.
Roflumilast
Phosphodiesterase 4 (PDE4) inhibition appears to have inflammatory modulating properties in the airways, although the exact mechanism of action is unclear. Some have proposed that it reduces inflammation by inhibiting the breakdown of intracellular cyclic adenosine monophosphate [72]. In 2 large clinical trials [73,74], daily use of a PDE4 inhibitor (roflumilast) showed a significant (15%–18%) reduction in yearly AECOPD incidence (approximate number needed to treat: 4). This benefit was seen in patients with GOLD stage 3–4 disease (FEV1 < 50% predicted) with the chronic bronchitic phenotype and who had experienced at least 1 exacerbation in the previous year.
Importantly, these clinical trials specifically prohibited the use of ICS and LAMAs. Thus, it remains unclear if PDE4 inhibition should be used as an add-on to ICS/LAMA therapy in patients who continue to have frequent AECOPD or whether PDE4 inhibition could be used instead of these standard therapies in patients with well-controlled daily symptoms without ICS or LAMA therapy but who experience frequent exacerbations.
Of note, earlier trials with roflumilast included patients with ICS and LAMA use [73,75]. These trials were focused on FEV1 improvement and found no benefit. It was only in post ad hoc analyses that a reduction in AECOPD in patients with frequent exacerbations was found among those taking roflumilast, regardless of ICS or LAMA use [76]. While roflumilast has documented benefit in improving lung function and reducing the rate of exacerbations, it has not been reported to decrease hospitalizations [64]. This indicates that although the drug reduces the total number of exacerbations, it may not be as useful in preventing episodes of severe exacerbations of COPD.
Although PDE4 inhibitors are easy to administer (a once-daily pill), they are associated with significant GI side effects (diarrhea, nausea, reduced appetite), weight loss, headache, and sleep disturbance [77]. Adverse effects tend to occur early during treatment, are reversible, and lessen over time with treatment [66]. Studies reported an average unexplained weight loss of 2 kg, and monitoring weight during treatment is advised. In addition, it is important to avoid roflumilast in underweight patients. Roflumilast should also be used with caution in depressed patients [65].
N-acetylcysteine
N-acetylcysteine (NAC) reduces the viscosity of respiratory secretions as a result of the cleavage of the disulfide bonds and has been studied as a mucolytic agent to aid in the elimination of respiratory secretions [78]. Oral NAC is quickly absorbed and is rapidly present in an active form in lung tissue and respiratory secretions after ingestion. NAC is well tolerated except for occasional patients with GI adverse effects. The role of NAC in preventing AECOPD has been studied for more than 3 decades [79–81], although the largest clinical trial to date was reported in 2014 [57]. Taken together, the combined data demonstrate a significant reduction in the rate of COPD exacerbations associated with the use of NAC when compared with placebo (OR, 0.61; CI, 0.37–0.99). Clinical guidelines suggest that in patients with moderate to severe COPD (FEV1/FVC < 0.7, and FEV1 < 80% predicted) receiving maintenance bronchodilator therapy combined with ICS and history of 2 more exacerbations in the previous 2 years, treatment with oral NAC can be administered to prevent AECOPD.
Macrolides
Continuous prophylactic use of antibiotics in older studies had no effect on the frequency of AECOPD [82,83]. But it is known that macrolide antibiotics have several antimicrobial, anti-inflammatory and immunomodulating effects and have been used for many years in the management of other chronic airway disease, including diffuse pan-bronchiolitis and cystic fibrosis [65]. One recent study showed that the use of once-daily, generic azithromycin 5 days/week appeared to have an impact on AECOPD incidence [84]. In this study, AECOPD was reduced from 1.83 to 1.48 per patient-year (RR, 0.83; 95% CI, 0.72–0.95: P = 0.01). Azithromycin also prevented severe AECOPD. Greater benefit was obtained with milder forms of the disease and in the elderly. Azithromycin did not appear to provide any benefit in those who continued to smoke (hazard ratio, 0.99) [85]. Other studies have shown that azithromycin was associated with an increased incidence of bacterial resistance and impaired hearing [86]. Overall data from the available clinical trials are robust and demonstrate that regular macrolide therapy definitely reduces the risk of AECOPD. But due to potential side effects macrolide therapy is an option rather than a strong recommendation [65]. The prescribing clinician also needs to consider the potential of prolongation of the QT interval [84].
Immunostimulants
Immunostimulants have also been reported to reduce frequency of AECOPD [87,88]. Bacterial lysates, reconstituted mixtures of bacterial antigens present in the lower airways of COPD patients, act as immuno-stimulants through the induction of cellular maturation, stimulating lymphocyte chemotaxis, and increasing opsonization when administered to individuals with COPD [66]. Studies have demonstrated a reduction in the severe complications of exacerbations and hospital admissions in COPD patients with OM-85, a detoxified oral immunoactive bacterial extract [87,88]. However, most of these trials were conducted prior to the routine use of long-acting bronchodilators and ICS in COPD. A recent study by Braido et al evaluated the efficacy of ismigen, a bacterial lysate, in reducing AECOPD [89] and found no difference in the exacerbation rate between ismigen and placebo or the time to first exacerbation. Additional studies are needed to examine the long-term effects of this therapy in patients receiving currently recommended COPD maintenance therapy [66].
β Blockers
Observational studies of beta-blocker use in preventing AECOPD have yielded encouraging results, with one study showing a reduction in AECOPD risk (incidence risk ratio, 0.73; CI 0.60–0.90) in patients receiving beta blockers versus those not on beta blockers [90]. Based on these findings, a clinical trial investigating the impact of metoprolol on risk of AECOPD is ongoing [91].
Proton Pump Inhibitors
Gastroesophageal reflux disease is an independent risk factor for exacerbations [92]. Two small, single-center studies [93,94] have shown that use of lansoprazole decreases the risk and frequency of AECOPD. However, data from the Predicting Outcome using Systemic Markers in Severe Exacerbations of COPD (PROMISE-COPD) study [66], which was a multicenter prospective observational study, suggested that patients with stable COPD receiving a proton pump inhibitor were at high risk of frequent and severe exacerbations [95]. Thus, at this stage, their definitive role needs to be defined, possibly with a randomized, placebo-controlled study.
Case 3
A 65-year-old male with severe COPD (FEV1/FVC 27, FEV1 25% of predicted, residual volume 170% of predicted for his age and height) was seen in the pulmonary clinic. His medications include a LABA/LAMA combination that he uses twice daily as advised. He uses his rescue albuterol inhaler roughly once a week. The patient complains of severe disabling shortness of breath with exertion and severe limitation of his quality of life because of his inability to lead a normal active life. He is on 2 L/min of oxygen at all times. He has received pulmonary rehabilitation in hopes of improving his quality of life but can only climb a flight of stairs before he must stop to rest. He asks about options but does not want to consider lung transplantation today. His most recent chest CT scan demonstrates upper lobe predominant emphysematous changes with no masses or nodules.
What are the patient's options at this time?
Lung volume reduction surgery (LVRS) attempts to reduce space-occupying severely diseased, hyperexpanded lung, thus allowing the relatively normal adjoining lung parenchyma to expand into the vacated space and function effectively [96].Hence, such therapies are suitable for patients with emphysematous lungs and not those with bronchitic-predominant COPD. LVRS offers a greater chance of improvement in exercise capacity, lung function, quality of life, and dyspnea in the correctly chosen patient population as compared with pharmacologic management alone [97]. However, the procedure is associated with risks, including higher short-term morbidity and mortality [97]. Patients with predominantly upper-lobe emphysema and a low maximal workload after rehabilitation were noted to have lower mortality, a greater probability of improvement in exercise capacity, and a greater probability of improvement in symptoms if they underwent surgery compared to medical therapy alone [97]. On the contrary, patients with predominantly non–upper-lobe emphysema and a high maximal workload after rehabilitation had higher mortality if they underwent surgery compared to receiving medical therapy alone [97]. Thus, a subgroup of patients with homogeneous emphysema symmetrically affecting the upper and lower lobes are considered to be unlikely to benefit from this surgery [97,98].
Valves and other methods of lung volume reduction such as coils, sealants, intrapulmonary vents, and thermal vapor in the bronchi or subsegmental airways have emerged as new techniques for nonsurgical lung volume reduction [99–104]. Endobronchial-valve therapy is associated with improvement in lung function and with clinical benefits that are greatest in the presence of heterogeneous lung involvement. This works by the same principle as with LVRS, by reduction of the most severely diseased lung units, expansion of the more viable, less emphysematous lung results in substantial improvements in lung mechanics [105,106]. The most important complications of this procedure include pneumonia, pneumothorax, hemoptysis and increased frequency of COPD exacerbation in the following thirty days. The fact that high-heterogeneity subgroup had greater improvements in both the FEV1 and distance on the 6-minute walk test than did patients with lower heterogeneity supports the use of quantitative high-resolution computed tomography (HRCT) in selecting patients for endobronchial-valve therapy [107].The HRCT scans also help in identifying those with complete fissures; a marker of lack of collateral ventilation (CV+) between different lobes. Presence of CV+ state predicts failure of endobronchial valve and all forms of endoscopic lung volume reduction strategies [108]. Bronchoscopic thermal vapor ablation (BTVA) therapy can potentially work on a subsegmental level and be successful for treatment of emphysema with lack of intact fissures on CT scans. Other methods that have the potential to be effective in those with collateral ventilation would be endoscopic coil therapy and polymeric lung volume reduction [106,109].Unfortunately, there are no randomized controlled trial data demonstrating clinically meaningful improvement following coil therapy or polymeric lung volume reduction in this CV+ patient population. Vapor therapy is perhaps the only technique that has been found to be effective in upper lobe predominant emphysema even with CV+ status [108].
Our patient has evidence of air trapping and emphysema based on a high residual volume. A CT scan of the chest can determine the nature of the emphysema (heterogeneous versus homogenous) and based on these findings, further determination of the best strategy for lung volume reduction can be made.
Is there a role for long-term oxygen therapy?
Long-term oxygen therapy (LTOT) used for > 15 hours a day is thought to reduce mortality among patients with chronic obstructive pulmonary disease (COPD) and severe resting hypoxemia [110–113].More recent studies have failed to show similar beneficial effects of LTOT. A recent study examined the effects of LTOT in randomized fashion and determined that supplemental oxygen for patients with stable COPD and resting or exercise-induced moderate desaturation did not affect the time to death or first hospitalization, time to first COPD exacerbation, time to first hospitalization for a COPD exacerbation, the rate of all hospitalizations, the rate of all COPD exacerbations, or changes in measures of quality of life, depression, anxiety, or functional status [114].
Our patient is currently on long-term oxygen therapy and in spite of some uncertainty as to its benefit, it is prudent to order oxygen therapy until further clarification is available.
What is the role of pulmonary rehabilitation?
Pulmonary rehabilitation is an established treatment for patients with chronic lung disease [115]. Benefits include improvement in exercise tolerance, symptoms, and quality of life, with a reduction in the use of health care resources [116].A Spanish population-based cohort study that looked at the influence of regular physical activity on COPD showed that patients who reported low, moderate, or high physical activity had a lower risk of COPD admissions and all-cause mortality than patients with very low physical activity after adjusting for all confounders [117].
As previously mentioned, patients in GOLD categories B, C, and D should be offered pulmonary rehabilitation as part of their treatment [7]. The ideal patient is one who is not too sick to undergo rehabilitation and is motivated to his or her quality of life.
What is the current scope of lung transplantation in the management of severe COPD?
There is a indisputable role for lung transplantation in end-stage COPD. However, lung transplantation does not benefit all COPD patients. There is a subset of patients for whom the treatment provides a survival benefit. It has been reported that 79% of patients with an FEV1 < 16% predicted will survive at least 1 year additional after transplant, but only 11% of patients with an FEV1 > 25% will do so [118]. The pre-transplant BODE (body mass index, airflow obstruction/FEV1, dyspnea, and exercise capacity) index score is used to identify the patients who will benefit from lung transplantation [119,120]. International guidelines for the selection of lung transplant candidates identify the following patient characteristics [121]:
- The disease is progressive, despite maximal treatment including medication, pulmonary rehabilitation, and oxygen therapy
- The patient is not a candidate for endoscopic or surgical LVRS
- BODE index of 5 to 6
- The partial pressure of carbon dioxide is greater than 50 mm Hg or 6.6kPa and/or partial pressure of oxygen is less than 60 mm Hg or 8kPa
- FEV1 of 25% predicted
The perioperative mortality of lung transplantation surgery has been reduced to less than 10%. Risk of complications from surgery in the perioperative period, such as bronchial dehiscence, infectious complications, and acute rejection, have also been reduced but do occur. Chronic allograft dysfunction and the risk of lung cancer in cases of single lung transplant should be discussed with the patient before surgery [122].
How can we incorporate palliative care into the management plan for patients with COPD?
Among patients with end-stage COPD on home oxygen therapy who have required mechanical ventilation for an exacerbation, only 55% are alive at 1 year [123]. COPD patients at high risk of death within the next year of life as well as patients with refractory symptoms and unmet needs are candidates for early palliative care. Palliative care and palliative care specialists can aid in reducing symptom burden and improving quality of life among these patients and their family members and is recommended by multiple international societies for patients with advanced COPD [124,125]. In spite of these recommendations, the utilization of palliative care resources has been dismally low [126,127]. Improving physician-patient communication regarding palliative services and patients’ unmet care needs will help ensure that COPD patients receive adequate palliative care services at the appropriate time.
Conclusion
COPD is a leading cause of morbidity and mortality in the United States and represents a significant economic burden for both individuals and society. The goals in COPD management are to provide symptom relief, improve the quality of life, preserve lung function, and reduce the frequency of exacerbations and mortality. COPD management is guided by disease severity that is measured using the GOLD multimodal staging system and requires a multidisciplinary approach. Several classes of medication are available for treatment, and a step-wise approach should be applied in building an effective pharmacologic regimen. In addition to pharmacologic therapies, nonpharmacologic therapies, including smoking cessation, vaccinations, proper nutrition, and maintaining physical activity, are an important part of long-term management. Those who continue to be symptomatic despite appropriate maximal therapy may be candidates for lung volume reduction. Palliative care services for COPD patients, which can aid in reducing symptom burden and improving quality of life, should not be overlooked.
Corresponding author: Abhishek Biswas, MD, Division of Pulmonary and Critical Care Medicine, Rm. M452, University of Florida, 1600 SW Archer Rd, Gainesville, FL 32610, [email protected].
Financial disclosures: None.
1. Segreti A, Stirpe E, Rogliani P, Cazzola M. Defining phenotypes in COPD: an aid to personalized healthcare. Mol Diagn Ther 2014;18:381–8.
2. Han MK, Agusti A, Calverley PM, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010;182:598–604.
3. Aubier M, Marthan R, Berger P, et al. [COPD and inflammation: statement from a French expert group: inflammation and remodelling mechanisms]. Rev Mal Respir 2010;27:1254–66.
4. Wang ZL. Evolving role of systemic inflammation in comorbidities of chronic obstructive pulmonary disease. Chin Med J (Engl) 2010;123:3467–78.
5. Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet 2007;370:741–50.
6. Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011;59:1–126.
7. Global Initiative for Chronic Obstructive Lung Disease (GOLD): Global strategy for the diagnosis, management, and prevention of COPD 2017. Accessed at www.goldcopd.org.
8. Jones PW, Harding G, Berry P, et al. Development and first validation of the COPD Assessment Test. Eur Respir J 2009;34:648–54.
9. Wadbo M, Löfdahl CG, Larsson K, et al. Effects of formoterol and ipratropium bromide in COPD: a 3-month placebo-controlled study. Eur Respir J 2002;20:1138–46.
10. Ram FS, Sestini P. Regular inhaled short acting beta2 agonists for the management of stable chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. Thorax 2003;58:580–4.
11. Colice GL. Nebulized bronchodilators for outpatient management of stable chronic obstructive pulmonary disease. Am J Med 1996;100(1A):11S–8S.
12. In chronic obstructive pulmonary disease, a combination of ipratropium and albuterol is more effective than either agent alone. An 85-day multicenter trial. COMBIVENT Inhalation Aerosol Study Group. Chest 1994;105:1411–9.
13. Friedman M, Serby CW, Menjoge SS, et al. Pharmacoeconomic evaluation of a combination of ipratropium plus albuterol compared with ipratropium alone and albuterol alone in COPD. Chest 1999;115:635–41.
14. Cook D, Guyatt G, Wong E, et al. Regular versus as-needed short-acting inhaled beta-agonist therapy for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;163:85–90.
15. Wilchesky M, Ernst P, Brophy JM, et al. Bronchodilator use and the risk of arrhythmia in COPD: part 2: reassessment in the larger Quebec cohort. Chest 2012;142:305–11.
16. Scott VL, Frazee LA. Retrospective comparison of nebulized levalbuterol and albuterol for adverse events in patients with acute airflow obstruction. Am J Ther 2003;10:341–7.
17. Wong CS, Pavord ID, Williams J, et al. Bronchodilator, cardiovascular, and hypokalaemic effects of fenoterol, salbutamol, and terbutaline in asthma. Lancet 1990;336:1396–9.
18. Cole JM, Sheehan AH, Jordan JK. Concomitant use of ipratropium and tiotropium in chronic obstructive pulmonary disease. Ann Pharmacother 2012;46:1717–21.
19. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155 :179–91.
20. Pearlman DS, Chervinsky P, LaForce C, et al. A comparison of salmeterol with albuterol in the treatment of mild-to-moderate asthma. N Engl J Med 1992;327:1420–5.
21. Takahashi T, Belvisi MG, Patel H, et al. Effect of Ba 679 BR, a novel long-acting anticholinergic agent, on cholinergic neurotransmission in guinea pig and human airways. Am J Respir Crit Care Med 1994;150(6 Pt 1):1640–5.
22. Donohue JF, Fogarty C, Lötvall J, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010;182:155–62.
23. Koch A, Pizzichini E, Hamilton A, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat versus placebo and formoterol twice daily in patients with GOLD 2-4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis 2014;9:697–714.
24. Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007;356:775–89.
25. Hanania NA, Feldman G, Zachgo W, et al. The efficacy and safety of the novel long-acting β2 agonist vilanterol in patients with COPD: a randomized placebo-controlled trial. Chest 2012;142:119–27.
26. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54.
27. Decramer ML, Chapman KR, Dahl R, et al. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013;1:524–33.
28. Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012;40:830–6.
29. D’Urzo A, Ferguson GT, van Noord JA, et al. Efficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trial. Respir Res 2011;12:156.
30. Antoniu SA. UPLIFT Study: the effects of long-term therapy with inhaled tiotropium in chronic obstructive pulmonary disease. Evaluation of: Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54. Expert Opin Pharmacother 2009;10:719–22.
31. Nelson HS, Gross NJ, Levine B, et al. Cardiac safety profile of nebulized formoterol in adults with COPD: a 12-week, multicenter, randomized, double- blind, double-dummy, placebo- and active-controlled trial. Clin Ther 2007;29:2167–78.
32. Gershon A, Croxford R, Calzavara A, et al. Cardiovascular safety of inhaled long-acting bronchodilators in individuals with chronic obstructive pulmonary disease. JAMA Intern Med 2013;173:1175–85.
33. Aljaafareh A, Valle JR, Lin YL, et al. Risk of cardiovascular events after initiation of long-acting bronchodilators in patients with chronic obstructive lung disease: A population-based study. SAGE Open Med 2016;4:2050312116671337.
34. O’Connor AB. Tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2009;360:185–6.
35. Kesten S, Jara M, Wentworth C, Lanes S. Pooled clinical trial analysis of tiotropium safety. Chest 2006;130:1695–703.
36. Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat inhaler and the risk of death in COPD. N Engl J Med 2013;369:1491–501.
37. Vogelmeier C, Hederer B, Glaab T, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011;364:1093–103.
38. Chong J, Karner C, Poole P. Tiotropium versus long-acting beta-agonists for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(9):CD009157.
39. Gan WQ, Man SF, Sin DD. Effects of inhaled corticosteroids on sputum cell counts in stable chronic obstructive pulmonary disease: a systematic review and a meta-analysis. BMC Pulm Med 2005;5:3.
40. Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(7):CD002991.
41. Roland NJ, Bhalla RK, Earis J. The local side effects of inhaled corticosteroids: current understanding and review of the literature. Chest 2004;126:213–9.
42. Drummond MB, Dasenbrook EC, Pitz MW, et al. Inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta-analysis. JAMA 2008;300:2407–16.
43. Lee SY, Park HY, Kim EK, et al. Combination therapy of inhaled steroids and long-acting beta2-agonists in asthma-COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis 2016;11:2797–803.
44. Postma DS, Rabe KF. The asthma-COPD overlap syndrome. N Engl J Med 2015;373:1241–9.
45. Farne HA, Cates CJ. Long-acting beta2-agonist in addition to tiotropium versus either tiotropium or long-acting beta2-agonist alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015:CD008989.
46. Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol-glycopyrronium versus salmeterol-fluticasone for COPD. N Engl J Med 2016;374:2222–34.
47. Aaron SD, Vandemheen KL, Fergusson D, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007;146:545–55.
48. Welte T, Miravitlles M, Hernandez P, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009;180:741–50.
49. Gallelli L, Falcone D, Cannataro R, et al. Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: a therapeutic rationale approach. Drug Des Devel Ther 2017;11:265–72.
50. Paloucek FP, Rodvold KA. Evaluation of theophylline overdoses and toxicities. Ann Emerg Med 1988;17:135–44.
51. Ram FS, Jones PW, Castro AA, et al. Oral theophylline for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2002(4):CD003902.
52. Murciano D, Auclair MH, Pariente R, Aubier M. A randomized, controlled trial of theophylline in patients with severe chronic obstructive pulmonary disease. N Engl J Med 1989;320:1521–5.
53. Devereux G, Cotton S, Barnes P, et al. Use of low-dose oral theophylline as an adjunct to inhaled corticosteroids in preventing exacerbations of chronic obstructive pulmonary disease: study protocol for a randomised controlled trial. Trials 2015;16:267.
54. Walters JA, Walters EH, Wood-Baker R. Oral corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005(3):CD005374.
55. Horita N, Miyazawa N, Morita S, et al. Evidence suggesting that oral corticosteroids increase mortality in stable chronic obstructive pulmonary disease. Respir Res 2014;15:37.
56. Poole P, Chong J, Cates CJ. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(7):CD001287.
57. Zheng JP, Wen FQ, Bai CX, et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014;2:187–94.
58. Seemungal TA, Wilkinson TM, Hurst JR, et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med 2008;178:1139–47.
59. Ries AL, Kaplan RM, Limberg TM, Prewitt LM. Effects of pulmonary rehabilitation on physiologic and psychosocial outcomes in patients with chronic obstructive pulmonary disease. Ann Intern Med 1995;122:823– 32.
60. Güell R, Casan P, Belda J, et al. Long-term effects of outpatient rehabilitation of COPD: a randomized trial. Chest 2000;117:976–83.
61. Wedzicha JA, Singh R, Mackay AJ. Acute COPD exacerbations. Clin Chest Med 2014;35:157–63.
62. Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet 2007;370:786–96.
63. Spencer S, Calverley PMA, Burge PS, Jones PW. Impact of preventing exacerbations on deterioration of health status in COPD. Eur Respir J 2004;23:698–702.
64. Blanchette CM, Gross NJ, Altman P. Rising costs of COPD and the potential for maintenance therapy to slow the trend. Am Health Drug Benef 2014;7:98.
65. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest 2015;147:894–942.
66. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2017 Report. Respirology 2017;22:575–601.
67. Wedzicha JA, Brill SE, Allinson JP, Donaldson GC. Mechanisms and impact of the frequent exacerbator phenotype in chronic obstructive pulmonary disease. BMC Med 2013;11:181.
68. Seemungal TAR, Donaldson GC, Paul EA, et al. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:1418–22.
69. Kanner RE, Anthonisen NR, Connett JE. Lower respiratory illnesses promote FEV1 decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med 2001;164:358–64.
70. Donaldson GC, Seemungal TAR, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002;57:847–52.
71. Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006;173:1114–21.
72. Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163:53–67.
73. Calverley PMA, Rabe KF, Goehring U-M, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009;374:685–94.
74. Fabbri LM, Calverley PMA, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009;374:695–703.
75. Lee S, Hui DSC, Mahayiddin AA, et al. Roflumilast in Asian patients with COPD: a randomized placebo-controlled trial. Respirology 2011;16:1249–57.
76. Calverley PM, Martinez FJ, Fabbri LM, et al. Does roflumilast decrease exacerbations in severe COPD patients not controlled by inhaled combination therapy? The REACT study protocol. Int J Chron Obstruct Pulmon Dis 2012;7:375–82.
77. Chong J, Leung B, Poole P. Phosphodiesterase 4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2013(11):CD002309.
78. Sheffner AL, Medler EM, Jacobs LW, Sarett HP. The in vitro reduction in viscosity of human tracheobronchial secretions by acetylcysteine. Am Rev Respir Dis 1964;90:721–9.
79. Boman G, Bäcker U, Larsson S, et al. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis 1983;64:405–15.
80. Grassi C, Morandini GC. A controlled trial of intermittent oral acetylcysteine in the long-term treatment of chronic bronchitis. European journal of clinical pharmacology. 1976;9:393–6.
81. Hansen NCG, Skriver A, Brorsen-Riis L, et al. Orally administered N-acetylcysteine may improve general well-being in patients with mild chronic bronchitis. Respir Med 1994;88:531–5.
82. Francis RS, Spicer CC. Chemotherapy in chronic bronchitis: Influence of daily penicillin and tetracycline on exacerbations and their cost: A report to the research committee of the British Tuberculosis Association by Their Chronic Bronchitis Subcommittee. BMJ 1960;1:297–303.
83. Francis RS, May JR, Spicer CC. Chemotherapy of bronchitis. BMJ 1961;2:979.
84. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
85. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014;189:1503–8.
86. Uzun S, Djamin RS, Kluytmans JAJW, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014;2:361–8.
87. Collet JP, Shapiro S, Ernst P, et al. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997;156:1719–24.
88. Jing LI. Protective effect of a bacterial extract against acute exacerbation in patients with chronic bronchitis accompanied by chronic obstructive pulmonary. Age 2004;67:0–05.
89. Braido F, Tarantini F, Ghiglione V, et al. Bacterial lysate in the prevention of acute exacerbation of COPD and in respiratory recurrent infections. Int J Chron Obstruct Pulmon Dis 2007;2:335.
90. Bhatt SP, Wells JM, Kinney GL, et al. β-Blockers are associated with a reduction in COPD exacerbations. Thorax 2016;71:8–14.
91. Bhatt SP, Connett JE, Voelker H, et al. β-Blockers for the prevention of acute exacerbations of chronic obstructive pulmonary disease (βLOCK COPD): a randomised controlled study protocol. BMJ Open 2016;6:e012292.
92. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010;363:1128–8.
93. Sasaki T, Nakayama K, Yasuda H, et al. A randomized, single-blind study of lansoprazole for the prevention of exacerbations of chronic obstructive pulmonary disease in older patients. J Am Geriatr Soc 2009;57:1453–7.
94. Xiong W, Zhang Qs, Zhao W, et al. A 12-month follow-up study on the preventive effect of oral lansoprazole on acute exacerbation of chronic obstructive pulmonary disease. Int J Exper Pathol 2016;97:107–13.
95. Baumeler L, Papakonstantinou E, Milenkovic B, et al. Therapy with proton-pump inhibitors for gastroesophageal reflux disease does not reduce the risk for severe exacerbations in COPD. Respirology 2016;21:883–90.
96 Sabanathan A, Sabanathan S, Shah R, Richardson J. Lung volume reduction surgery for emphysema: a review. J Cardiovasc Surg 1998;39:237.
97. Group NETTR. Patients at high risk of death after lung-volume–reduction surgery. N Engl J Med 2001;345:1075–83.
98. Group NETTR. A randomized trial comparing lung-volume–reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003;348:2059–73.
99. Decker MR, Leverson GE, Jaoude WA, Maloney JD. Lung volume reduction surgery since the National Emphysema Treatment Trial: study of Society of Thoracic Surgeons database. J Thorac Cardiovasc Surg 2014;148:2651–8.
100. Deslée G, Mal H, Dutau H, et al. Lung volume reduction coil treatment vs usual care in patients with severe emphysema: the REVOLENS randomized clinical trial. JAMA 2016;315:175–84.
101. Hartman JE, Klooster K, Gortzak K, et al. Long-term follow-up after bronchoscopic lung volume reduction treatment with coils in patients with severe emphysema. Respirology 2015;20:319–26.
102. Snell GI, Hopkins P, Westall G, et al. A feasibility and safety study of bronchoscopic thermal vapor ablation: a novel emphysema therapy. Ann Thorac Surg 2009;88:1993–8.
103. Ingenito EP, Berger RL, Henderson AC, et al. Bronchoscopic lung volume reduction using tissue engineering principles. Am J Respir Crit Care Med 2003;167:771–8.
104. Ingenito EP, Loring SH, Moy ML, et al. Comparison of physiological and radiological screening for lung volume reduction surgery. Am J Respir Crit Care Med 2001;163:1068–73.
105. Shah P, Slebos D, Cardoso P, et al. Bronchoscopic lung-volume reduction with Exhale airway stents for emphysema (EASE trial): randomised, sham-controlled, multicentre trial. Lancet 2011;378:997–1005.
106. Sciurba FC, Ernst A, Herth FJ, et al. A randomized study of endobronchial valves for advanced emphysema. N Engl J Med 2010;363:1233–44.
107. Wan IY, Toma TP, Geddes DM, et al. Bronchoscopic lung volume reduction for end-stage emphysema: report on the first 98 patients. Chest 2006;129:518–26.
108. Gompelmann D, Eberhardt R, Schuhmann M, et al. Lung volume reduction with vapor ablation in the presence of incomplete fissures: 12-month results from the STEP-UP randomized controlled study. Respiration 2016;92:397–403.
109. Come CE, Kramer MR, Dransfield MT, et al. A randomised trial of lung sealant versus medical therapy for advanced emphysema. Eur Respir J 2015;46:651–62.
110. Group NOTT. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann Intern Med 1980;93:391–8.
111. Council M. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema: Report of the Medical Research Council Working Party. Lancet 1981;1:681–6.
112. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155:179–91.
113. Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013;187:347–65.
114. Group L-TOTTR. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016;375:1617–27.
115. McCarthy B, Casey D, Devane D, et al. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(2):CD003793.
116. Griffiths TL, Burr ML, Campbell IA, et al. Results at 1 year of outpatient multidisciplinary pulmonary rehabilitation: a randomised controlled trial. Lancet 2000;355:362–8.
117. Garcia-Aymerich J, Lange P, Benet M, et al. Regular physical activity reduces hospital admission and mortality in chronic obstructive pulmonary disease: a population based cohort study. Thorax 2006;61:772–8.
118. Thabut G, Ravaud P, Christie JD, et al. Determinants of the survival benefit of lung transplantation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008;177:1156–63.
119. Lahzami S, Bridevaux PO, Soccal PM, et al. Survival impact of lung transplantation for COPD. Eur Respir J 2010;36:74–80.
120. Cerón Navarro J, de Aguiar Quevedo K, Ansótegui Barrera E, et al. Functional outcomes after lung transplant in chronic obstructive pulmonary disease. Arch Bronconeumol 2015;51:109–14.
121. Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2015;34:1–15.
122. Minai OA, Shah S, Mazzone P, et al. Bronchogenic carcinoma after lung transplantation: characteristics and outcomes. J Thorac Oncol 2008;3:1404–9.
123. Hajizadeh N, Goldfeld K, Crothers K. What happens to patients with COPD with long-term oxygen treatment who receive mechanical ventilation for COPD exacerbation? A 1-year retrospective follow- up study. Thorax 2015;70:294–6.
124. Siouta N, van Beek K, Preston N, et al. Towards integration of palliative care in patients with chronic heart failure and chronic obstructive pulmonary disease: a systematic literature review of European guidelines and pathways. BMC Palliat Care 2016;15:18.
125. Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004;23:932–46.
126. Szekendi MK, Vaughn J, Lal A, et al. The prevalence of inpatients at thirty-three U.S. hospitals appropriate for and receiving referral to palliative care. J Palliat Med 2016;19:360–72.
127. Rush B, Hertz P, Bond A, et al. Use of palliative care in patients with end-stage COPD and receiving home oxygen: national trends and barriers to care in the United States. Chest 2017;151:41–6.
1. Segreti A, Stirpe E, Rogliani P, Cazzola M. Defining phenotypes in COPD: an aid to personalized healthcare. Mol Diagn Ther 2014;18:381–8.
2. Han MK, Agusti A, Calverley PM, et al. Chronic obstructive pulmonary disease phenotypes: the future of COPD. Am J Respir Crit Care Med 2010;182:598–604.
3. Aubier M, Marthan R, Berger P, et al. [COPD and inflammation: statement from a French expert group: inflammation and remodelling mechanisms]. Rev Mal Respir 2010;27:1254–66.
4. Wang ZL. Evolving role of systemic inflammation in comorbidities of chronic obstructive pulmonary disease. Chin Med J (Engl) 2010;123:3467–78.
5. Buist AS, McBurnie MA, Vollmer WM, et al. International variation in the prevalence of COPD (the BOLD Study): a population-based prevalence study. Lancet 2007;370:741–50.
6. Miniño AM, Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2008. Natl Vital Stat Rep 2011;59:1–126.
7. Global Initiative for Chronic Obstructive Lung Disease (GOLD): Global strategy for the diagnosis, management, and prevention of COPD 2017. Accessed at www.goldcopd.org.
8. Jones PW, Harding G, Berry P, et al. Development and first validation of the COPD Assessment Test. Eur Respir J 2009;34:648–54.
9. Wadbo M, Löfdahl CG, Larsson K, et al. Effects of formoterol and ipratropium bromide in COPD: a 3-month placebo-controlled study. Eur Respir J 2002;20:1138–46.
10. Ram FS, Sestini P. Regular inhaled short acting beta2 agonists for the management of stable chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. Thorax 2003;58:580–4.
11. Colice GL. Nebulized bronchodilators for outpatient management of stable chronic obstructive pulmonary disease. Am J Med 1996;100(1A):11S–8S.
12. In chronic obstructive pulmonary disease, a combination of ipratropium and albuterol is more effective than either agent alone. An 85-day multicenter trial. COMBIVENT Inhalation Aerosol Study Group. Chest 1994;105:1411–9.
13. Friedman M, Serby CW, Menjoge SS, et al. Pharmacoeconomic evaluation of a combination of ipratropium plus albuterol compared with ipratropium alone and albuterol alone in COPD. Chest 1999;115:635–41.
14. Cook D, Guyatt G, Wong E, et al. Regular versus as-needed short-acting inhaled beta-agonist therapy for chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001;163:85–90.
15. Wilchesky M, Ernst P, Brophy JM, et al. Bronchodilator use and the risk of arrhythmia in COPD: part 2: reassessment in the larger Quebec cohort. Chest 2012;142:305–11.
16. Scott VL, Frazee LA. Retrospective comparison of nebulized levalbuterol and albuterol for adverse events in patients with acute airflow obstruction. Am J Ther 2003;10:341–7.
17. Wong CS, Pavord ID, Williams J, et al. Bronchodilator, cardiovascular, and hypokalaemic effects of fenoterol, salbutamol, and terbutaline in asthma. Lancet 1990;336:1396–9.
18. Cole JM, Sheehan AH, Jordan JK. Concomitant use of ipratropium and tiotropium in chronic obstructive pulmonary disease. Ann Pharmacother 2012;46:1717–21.
19. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155 :179–91.
20. Pearlman DS, Chervinsky P, LaForce C, et al. A comparison of salmeterol with albuterol in the treatment of mild-to-moderate asthma. N Engl J Med 1992;327:1420–5.
21. Takahashi T, Belvisi MG, Patel H, et al. Effect of Ba 679 BR, a novel long-acting anticholinergic agent, on cholinergic neurotransmission in guinea pig and human airways. Am J Respir Crit Care Med 1994;150(6 Pt 1):1640–5.
22. Donohue JF, Fogarty C, Lötvall J, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med 2010;182:155–62.
23. Koch A, Pizzichini E, Hamilton A, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat versus placebo and formoterol twice daily in patients with GOLD 2-4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis 2014;9:697–714.
24. Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007;356:775–89.
25. Hanania NA, Feldman G, Zachgo W, et al. The efficacy and safety of the novel long-acting β2 agonist vilanterol in patients with COPD: a randomized placebo-controlled trial. Chest 2012;142:119–27.
26. Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54.
27. Decramer ML, Chapman KR, Dahl R, et al. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med 2013;1:524–33.
28. Jones PW, Singh D, Bateman ED, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J 2012;40:830–6.
29. D’Urzo A, Ferguson GT, van Noord JA, et al. Efficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trial. Respir Res 2011;12:156.
30. Antoniu SA. UPLIFT Study: the effects of long-term therapy with inhaled tiotropium in chronic obstructive pulmonary disease. Evaluation of: Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008;359:1543–54. Expert Opin Pharmacother 2009;10:719–22.
31. Nelson HS, Gross NJ, Levine B, et al. Cardiac safety profile of nebulized formoterol in adults with COPD: a 12-week, multicenter, randomized, double- blind, double-dummy, placebo- and active-controlled trial. Clin Ther 2007;29:2167–78.
32. Gershon A, Croxford R, Calzavara A, et al. Cardiovascular safety of inhaled long-acting bronchodilators in individuals with chronic obstructive pulmonary disease. JAMA Intern Med 2013;173:1175–85.
33. Aljaafareh A, Valle JR, Lin YL, et al. Risk of cardiovascular events after initiation of long-acting bronchodilators in patients with chronic obstructive lung disease: A population-based study. SAGE Open Med 2016;4:2050312116671337.
34. O’Connor AB. Tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2009;360:185–6.
35. Kesten S, Jara M, Wentworth C, Lanes S. Pooled clinical trial analysis of tiotropium safety. Chest 2006;130:1695–703.
36. Wise RA, Anzueto A, Cotton D, et al. Tiotropium Respimat inhaler and the risk of death in COPD. N Engl J Med 2013;369:1491–501.
37. Vogelmeier C, Hederer B, Glaab T, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med 2011;364:1093–103.
38. Chong J, Karner C, Poole P. Tiotropium versus long-acting beta-agonists for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(9):CD009157.
39. Gan WQ, Man SF, Sin DD. Effects of inhaled corticosteroids on sputum cell counts in stable chronic obstructive pulmonary disease: a systematic review and a meta-analysis. BMC Pulm Med 2005;5:3.
40. Yang IA, Clarke MS, Sim EH, Fong KM. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2012(7):CD002991.
41. Roland NJ, Bhalla RK, Earis J. The local side effects of inhaled corticosteroids: current understanding and review of the literature. Chest 2004;126:213–9.
42. Drummond MB, Dasenbrook EC, Pitz MW, et al. Inhaled corticosteroids in patients with stable chronic obstructive pulmonary disease: a systematic review and meta-analysis. JAMA 2008;300:2407–16.
43. Lee SY, Park HY, Kim EK, et al. Combination therapy of inhaled steroids and long-acting beta2-agonists in asthma-COPD overlap syndrome. Int J Chron Obstruct Pulmon Dis 2016;11:2797–803.
44. Postma DS, Rabe KF. The asthma-COPD overlap syndrome. N Engl J Med 2015;373:1241–9.
45. Farne HA, Cates CJ. Long-acting beta2-agonist in addition to tiotropium versus either tiotropium or long-acting beta2-agonist alone for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015:CD008989.
46. Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol-glycopyrronium versus salmeterol-fluticasone for COPD. N Engl J Med 2016;374:2222–34.
47. Aaron SD, Vandemheen KL, Fergusson D, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone-salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2007;146:545–55.
48. Welte T, Miravitlles M, Hernandez P, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009;180:741–50.
49. Gallelli L, Falcone D, Cannataro R, et al. Theophylline action on primary human bronchial epithelial cells under proinflammatory stimuli and steroidal drugs: a therapeutic rationale approach. Drug Des Devel Ther 2017;11:265–72.
50. Paloucek FP, Rodvold KA. Evaluation of theophylline overdoses and toxicities. Ann Emerg Med 1988;17:135–44.
51. Ram FS, Jones PW, Castro AA, et al. Oral theophylline for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2002(4):CD003902.
52. Murciano D, Auclair MH, Pariente R, Aubier M. A randomized, controlled trial of theophylline in patients with severe chronic obstructive pulmonary disease. N Engl J Med 1989;320:1521–5.
53. Devereux G, Cotton S, Barnes P, et al. Use of low-dose oral theophylline as an adjunct to inhaled corticosteroids in preventing exacerbations of chronic obstructive pulmonary disease: study protocol for a randomised controlled trial. Trials 2015;16:267.
54. Walters JA, Walters EH, Wood-Baker R. Oral corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2005(3):CD005374.
55. Horita N, Miyazawa N, Morita S, et al. Evidence suggesting that oral corticosteroids increase mortality in stable chronic obstructive pulmonary disease. Respir Res 2014;15:37.
56. Poole P, Chong J, Cates CJ. Mucolytic agents versus placebo for chronic bronchitis or chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(7):CD001287.
57. Zheng JP, Wen FQ, Bai CX, et al. Twice daily N-acetylcysteine 600 mg for exacerbations of chronic obstructive pulmonary disease (PANTHEON): a randomised, double-blind placebo-controlled trial. Lancet Respir Med 2014;2:187–94.
58. Seemungal TA, Wilkinson TM, Hurst JR, et al. Long-term erythromycin therapy is associated with decreased chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med 2008;178:1139–47.
59. Ries AL, Kaplan RM, Limberg TM, Prewitt LM. Effects of pulmonary rehabilitation on physiologic and psychosocial outcomes in patients with chronic obstructive pulmonary disease. Ann Intern Med 1995;122:823– 32.
60. Güell R, Casan P, Belda J, et al. Long-term effects of outpatient rehabilitation of COPD: a randomized trial. Chest 2000;117:976–83.
61. Wedzicha JA, Singh R, Mackay AJ. Acute COPD exacerbations. Clin Chest Med 2014;35:157–63.
62. Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet 2007;370:786–96.
63. Spencer S, Calverley PMA, Burge PS, Jones PW. Impact of preventing exacerbations on deterioration of health status in COPD. Eur Respir J 2004;23:698–702.
64. Blanchette CM, Gross NJ, Altman P. Rising costs of COPD and the potential for maintenance therapy to slow the trend. Am Health Drug Benef 2014;7:98.
65. Criner GJ, Bourbeau J, Diekemper RL, et al. Prevention of acute exacerbations of COPD: American College of Chest Physicians and Canadian Thoracic Society Guideline. Chest 2015;147:894–942.
66. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Lung Disease 2017 Report. Respirology 2017;22:575–601.
67. Wedzicha JA, Brill SE, Allinson JP, Donaldson GC. Mechanisms and impact of the frequent exacerbator phenotype in chronic obstructive pulmonary disease. BMC Med 2013;11:181.
68. Seemungal TAR, Donaldson GC, Paul EA, et al. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:1418–22.
69. Kanner RE, Anthonisen NR, Connett JE. Lower respiratory illnesses promote FEV1 decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med 2001;164:358–64.
70. Donaldson GC, Seemungal TAR, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002;57:847–52.
71. Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006;173:1114–21.
72. Rabe KF. Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br J Pharmacol 2011;163:53–67.
73. Calverley PMA, Rabe KF, Goehring U-M, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 2009;374:685–94.
74. Fabbri LM, Calverley PMA, Izquierdo-Alonso JL, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 2009;374:695–703.
75. Lee S, Hui DSC, Mahayiddin AA, et al. Roflumilast in Asian patients with COPD: a randomized placebo-controlled trial. Respirology 2011;16:1249–57.
76. Calverley PM, Martinez FJ, Fabbri LM, et al. Does roflumilast decrease exacerbations in severe COPD patients not controlled by inhaled combination therapy? The REACT study protocol. Int J Chron Obstruct Pulmon Dis 2012;7:375–82.
77. Chong J, Leung B, Poole P. Phosphodiesterase 4 inhibitors for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2013(11):CD002309.
78. Sheffner AL, Medler EM, Jacobs LW, Sarett HP. The in vitro reduction in viscosity of human tracheobronchial secretions by acetylcysteine. Am Rev Respir Dis 1964;90:721–9.
79. Boman G, Bäcker U, Larsson S, et al. Oral acetylcysteine reduces exacerbation rate in chronic bronchitis: report of a trial organized by the Swedish Society for Pulmonary Diseases. Eur J Respir Dis 1983;64:405–15.
80. Grassi C, Morandini GC. A controlled trial of intermittent oral acetylcysteine in the long-term treatment of chronic bronchitis. European journal of clinical pharmacology. 1976;9:393–6.
81. Hansen NCG, Skriver A, Brorsen-Riis L, et al. Orally administered N-acetylcysteine may improve general well-being in patients with mild chronic bronchitis. Respir Med 1994;88:531–5.
82. Francis RS, Spicer CC. Chemotherapy in chronic bronchitis: Influence of daily penicillin and tetracycline on exacerbations and their cost: A report to the research committee of the British Tuberculosis Association by Their Chronic Bronchitis Subcommittee. BMJ 1960;1:297–303.
83. Francis RS, May JR, Spicer CC. Chemotherapy of bronchitis. BMJ 1961;2:979.
84. Albert RK, Connett J, Bailey WC, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011;365:689–98.
85. Han MK, Tayob N, Murray S, et al. Predictors of chronic obstructive pulmonary disease exacerbation reduction in response to daily azithromycin therapy. Am J Respir Crit Care Med 2014;189:1503–8.
86. Uzun S, Djamin RS, Kluytmans JAJW, et al. Azithromycin maintenance treatment in patients with frequent exacerbations of chronic obstructive pulmonary disease (COLUMBUS): a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2014;2:361–8.
87. Collet JP, Shapiro S, Ernst P, et al. Effects of an immunostimulating agent on acute exacerbations and hospitalizations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997;156:1719–24.
88. Jing LI. Protective effect of a bacterial extract against acute exacerbation in patients with chronic bronchitis accompanied by chronic obstructive pulmonary. Age 2004;67:0–05.
89. Braido F, Tarantini F, Ghiglione V, et al. Bacterial lysate in the prevention of acute exacerbation of COPD and in respiratory recurrent infections. Int J Chron Obstruct Pulmon Dis 2007;2:335.
90. Bhatt SP, Wells JM, Kinney GL, et al. β-Blockers are associated with a reduction in COPD exacerbations. Thorax 2016;71:8–14.
91. Bhatt SP, Connett JE, Voelker H, et al. β-Blockers for the prevention of acute exacerbations of chronic obstructive pulmonary disease (βLOCK COPD): a randomised controlled study protocol. BMJ Open 2016;6:e012292.
92. Hurst JR, Vestbo J, Anzueto A, et al. Susceptibility to exacerbation in chronic obstructive pulmonary disease. N Engl J Med 2010;363:1128–8.
93. Sasaki T, Nakayama K, Yasuda H, et al. A randomized, single-blind study of lansoprazole for the prevention of exacerbations of chronic obstructive pulmonary disease in older patients. J Am Geriatr Soc 2009;57:1453–7.
94. Xiong W, Zhang Qs, Zhao W, et al. A 12-month follow-up study on the preventive effect of oral lansoprazole on acute exacerbation of chronic obstructive pulmonary disease. Int J Exper Pathol 2016;97:107–13.
95. Baumeler L, Papakonstantinou E, Milenkovic B, et al. Therapy with proton-pump inhibitors for gastroesophageal reflux disease does not reduce the risk for severe exacerbations in COPD. Respirology 2016;21:883–90.
96 Sabanathan A, Sabanathan S, Shah R, Richardson J. Lung volume reduction surgery for emphysema: a review. J Cardiovasc Surg 1998;39:237.
97. Group NETTR. Patients at high risk of death after lung-volume–reduction surgery. N Engl J Med 2001;345:1075–83.
98. Group NETTR. A randomized trial comparing lung-volume–reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003;348:2059–73.
99. Decker MR, Leverson GE, Jaoude WA, Maloney JD. Lung volume reduction surgery since the National Emphysema Treatment Trial: study of Society of Thoracic Surgeons database. J Thorac Cardiovasc Surg 2014;148:2651–8.
100. Deslée G, Mal H, Dutau H, et al. Lung volume reduction coil treatment vs usual care in patients with severe emphysema: the REVOLENS randomized clinical trial. JAMA 2016;315:175–84.
101. Hartman JE, Klooster K, Gortzak K, et al. Long-term follow-up after bronchoscopic lung volume reduction treatment with coils in patients with severe emphysema. Respirology 2015;20:319–26.
102. Snell GI, Hopkins P, Westall G, et al. A feasibility and safety study of bronchoscopic thermal vapor ablation: a novel emphysema therapy. Ann Thorac Surg 2009;88:1993–8.
103. Ingenito EP, Berger RL, Henderson AC, et al. Bronchoscopic lung volume reduction using tissue engineering principles. Am J Respir Crit Care Med 2003;167:771–8.
104. Ingenito EP, Loring SH, Moy ML, et al. Comparison of physiological and radiological screening for lung volume reduction surgery. Am J Respir Crit Care Med 2001;163:1068–73.
105. Shah P, Slebos D, Cardoso P, et al. Bronchoscopic lung-volume reduction with Exhale airway stents for emphysema (EASE trial): randomised, sham-controlled, multicentre trial. Lancet 2011;378:997–1005.
106. Sciurba FC, Ernst A, Herth FJ, et al. A randomized study of endobronchial valves for advanced emphysema. N Engl J Med 2010;363:1233–44.
107. Wan IY, Toma TP, Geddes DM, et al. Bronchoscopic lung volume reduction for end-stage emphysema: report on the first 98 patients. Chest 2006;129:518–26.
108. Gompelmann D, Eberhardt R, Schuhmann M, et al. Lung volume reduction with vapor ablation in the presence of incomplete fissures: 12-month results from the STEP-UP randomized controlled study. Respiration 2016;92:397–403.
109. Come CE, Kramer MR, Dransfield MT, et al. A randomised trial of lung sealant versus medical therapy for advanced emphysema. Eur Respir J 2015;46:651–62.
110. Group NOTT. Continuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trial. Ann Intern Med 1980;93:391–8.
111. Council M. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema: Report of the Medical Research Council Working Party. Lancet 1981;1:681–6.
112. Qaseem A, Wilt TJ, Weinberger SE, et al. Diagnosis and management of stable chronic obstructive pulmonary disease: a clinical practice guideline update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann Intern Med 2011;155:179–91.
113. Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013;187:347–65.
114. Group L-TOTTR. A randomized trial of long-term oxygen for COPD with moderate desaturation. N Engl J Med 2016;375:1617–27.
115. McCarthy B, Casey D, Devane D, et al. Pulmonary rehabilitation for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2015(2):CD003793.
116. Griffiths TL, Burr ML, Campbell IA, et al. Results at 1 year of outpatient multidisciplinary pulmonary rehabilitation: a randomised controlled trial. Lancet 2000;355:362–8.
117. Garcia-Aymerich J, Lange P, Benet M, et al. Regular physical activity reduces hospital admission and mortality in chronic obstructive pulmonary disease: a population based cohort study. Thorax 2006;61:772–8.
118. Thabut G, Ravaud P, Christie JD, et al. Determinants of the survival benefit of lung transplantation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008;177:1156–63.
119. Lahzami S, Bridevaux PO, Soccal PM, et al. Survival impact of lung transplantation for COPD. Eur Respir J 2010;36:74–80.
120. Cerón Navarro J, de Aguiar Quevedo K, Ansótegui Barrera E, et al. Functional outcomes after lung transplant in chronic obstructive pulmonary disease. Arch Bronconeumol 2015;51:109–14.
121. Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2015;34:1–15.
122. Minai OA, Shah S, Mazzone P, et al. Bronchogenic carcinoma after lung transplantation: characteristics and outcomes. J Thorac Oncol 2008;3:1404–9.
123. Hajizadeh N, Goldfeld K, Crothers K. What happens to patients with COPD with long-term oxygen treatment who receive mechanical ventilation for COPD exacerbation? A 1-year retrospective follow- up study. Thorax 2015;70:294–6.
124. Siouta N, van Beek K, Preston N, et al. Towards integration of palliative care in patients with chronic heart failure and chronic obstructive pulmonary disease: a systematic literature review of European guidelines and pathways. BMC Palliat Care 2016;15:18.
125. Celli BR, MacNee W; ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004;23:932–46.
126. Szekendi MK, Vaughn J, Lal A, et al. The prevalence of inpatients at thirty-three U.S. hospitals appropriate for and receiving referral to palliative care. J Palliat Med 2016;19:360–72.
127. Rush B, Hertz P, Bond A, et al. Use of palliative care in patients with end-stage COPD and receiving home oxygen: national trends and barriers to care in the United States. Chest 2017;151:41–6.
Managing schizophrenia in a patient with cancer: A fine balance
CASE Stable with a new diagnosis
Ms. B, age 60, has a history of schizophrenia, which has been stable on clozapine, 500 mg/d, for more than 2 decades. After a series of hospitalizations in her 20s and 30s, clozapine was initiated and she has not required additional inpatient psychiatric care. She has been managed in the outpatient setting with standard biweekly absolute neutrophil count (ANC) monitoring. She lives independently and is an active member in her church.
After experiencing rectal bleeding, Ms. B is diagnosed with rectal carcinoma and is scheduled to undergo chemotherapy and radiation treatment.
[polldaddy:9754786]
The authors’ observations
Both clozapine and chemotherapy carry the risk of immunosuppression, presenting a clinical challenge when choosing an appropriate management strategy. However, the risks of stopping clozapine after a long period of symptom stability are substantial, with a relapse rate up to 50%.1 Among patients taking clozapine, the risk of agranulocytosis and neutropenia are approximately 0.8% and 3%, respectively, and >80% of agranulocyotis cases occur within the first 18 weeks of treatment.2,3 Although both clozapine and chemotherapy can lead to neutropenia and agranulocytosis, there currently is no evidence of a synergistic effect on bone marrow suppression with simultaneous use of these therapies2 nor is there evidence of the combination leading to sustained marrow suppression.4
Because of Ms. B’s positive response to clozapine, the risks associated with discontinuing the medication, and the relatively low risk of clozapine contributing to neutropenia after a long period of stabilization, her outpatient psychiatric providers decide to increase ANC monitoring to weekly while she undergoes cancer treatment.
TREATMENT Neutropenia, psychosis
Ms. B continues clozapine during radiation and chemotherapy, but develops leukopenia and neutropenia with a low of 1,220/μL white blood cells and an ANC of 610/μL. Clozapine is stopped, consistent with current recommendations to hold the drug if the neutrophil count is <1,000/μL in a patient without benign ethnic neutropenia, and her outpatient provider monitors her closely. The treatment team does not restart an antipsychotic immediately after discontinuing clozapine because of the risk that other antipsychotics can cause hematologic toxicity or prolong granulocytopenia associated with clozapine.5
Approximately 2 weeks later, Ms. B is admitted to a different hospital for altered mental status and is found to have hyponatremia and rectal bleeding. The workup suggests that her rectal carcinoma has not fully responded to initial therapies, and she likely will require further treatment. Her mental status improves after hyponatremia resolves, but she reports auditory hallucinations and paranoia. Risperidone, 4 mg/d, is initiated to target psychosis.
After discharge, Ms. B develops bilateral upper extremity tremor, which she finds intolerable and attributes to risperidone. She refuses to continue risperidone or try adjunctive medications to address the tremor, but is willing to consider a different antipsychotic. Olanzapine, 10 mg/d, is initiated and risperidone is slowly tapered. During this time, Ms. B experiences increased paranoia and believes that the Internal Revenue Service is calling her. She misses her next appointment.
Later, the fire department finds Ms. B wandering the streets and brings her to the psychiatric emergency room. During the examination, she is disheveled and withdrawn, and unable to reply to simple questions about diet and sleep. When asked why she was in the street, she says that she left her apartment because it was “too messy.” The treatment team learns that she had walked at least 10 miles from her apartment before sitting down by the side of the road and being picked up by the fire department. She reveals that she left her apartment and continued walking because “a man” told her to do so and threatened to harm her if she stopped.
When Ms. B is admitted to the psychiatric service, she is paranoid, disorganized, and guarded. She remains in her room for most of the day and either refuses to talk to providers or curses at them. She often is seen wearing soiled clothing with her hair mussed. She denies having rectal carcinoma, although she expressed understanding of her medical condition <2 months earlier.
[polldaddy:9754787]
The authors’ observations
Clozapine is considered the most efficacious agent for treatment-resistant schizophrenia.6 Although non-compliance is the most common reason for discontinuing clozapine, >20% of patients stop clozapine because of adverse effects.7 Clozapine often is a drug of last resort because of the need for frequent monitoring and significant side effects; therefore deciding on a next step when clozapine fails or cannot be continued because of other factors can pose a challenge.
Ms. B’s treatment team gave serious consideration to restarting clozapine. However, because it was likely that Ms. B would undergo another round of chemotherapy and possibly radiation, the risk of neutropenia recurring was considered too high. Lithium has been used successfully to manage neutropenia in patients taking clozapine and, for some, adding lithium could help boost white cell count and allow a successful rechallenge with clozapine.3,8 However, because of Ms. B’s medical comorbidities, including cancer and chronic kidney disease, adding lithium was not thought to be clinically prudent at that time and the treatment team considered other options.
Olanzapine. Although research is limited, studies suggest olanzapine is the most commonly prescribed medication when a patient has to discontinue clozapine,7 with comparable response rates in those with refractory schizophrenia.9 Therefore, Ms. B was initially maintained on olanzapine, and the dosage increased to 30 mg over the course of 16 days in the hospital. However, she did not respond to the medication, remaining disorganized and paranoid without any notable improvement in her symptoms therefore other treatment options were explored.
Loxapine. Previous limited case reports have shown loxapine to be effective in treating individuals with refractory schizophrenia, either alone or in combination with other antipsychotics.10,11 FDA-approved in 1975, loxapine was among the last of the typical antipsychotics brought to the U.S. market before the introduction of clozapine, the first atypical.12 Loxapine is a dibenzoxazepine that has a molecular structure similar to clozapine.13 Unlike clozapine, however, loxapine is not known to cause agranulocytosis.14 Research suggests that although clozapine is oxidized to metabolites that are cytotoxic, loxapine is not, potentially accounting for their different effects on neutrophils.15
The efficacy of loxapine has shown to be similar to other typical and atypical antipsychotics, with approximately 70% of patients showing improvement.14 However, loxapine may be overlooked as an option, possibly because it was not included in the CATIE trial and was the last typical antipsychotic to be approved before atypicals were introduced.12 First available in oral and IM formulations, there has been increased interest in loxapine recently because of the approval of an inhaled formulation in 2012.16
Although classified as a typical antipsychotic, studies have suggested that loxapine acts as an atypical at low dosages.17,18 Previous work suggests, however, that the side effect profile of loxapine is similar to typical antipsychotics.14 At dosages <50 mg, it results in fewer cases of extrapyramidal side effects than expected with a typical antipsychotic.18
Loxapine’s binding profile seems to exist along this spectrum of typical to atypical. Tissue-based binding studies have shown a higher 5-HT2 affinity relative to D2, consistent with atypical antipsychotics.19 Positron emission tomography studies in humans show 5-HT2 saturation of loxapine to be close to equal to D2 binding in loxapine, thus a slightly lower ratio of 5-HT2 to D2 relative to atypicals, but more than that seen with typical antipsychotics.20 These differences between in vitro and in vivo studies may be secondary to the binding of loxapine’s active metabolites, particularly 7- and 8-hydroxyloxapine, which have more dopaminergic activity. In addition to increased 5-HT2A binding compared with typical antipsychotics, loxapine also has a high affinity for the D4 receptors, as well as interacting with other serotonin receptors 5-HT3, 5-HT6, and 5-HT7. Of note this is a similar pattern of binding affinity as seen in clozapine.19

It should be noted, however, that loxapine may not be an appropriate treatment in all forms of cancer. Similar to other first-generation antipsychotics, it increases prolactin levels, and thus may have a negative clinical impact on patients with prolactin receptor positive breast cancers.21,22 Finally, although clozapine can result in significant weight gain, dyslipidemia, and hyperglycemia, unlike many antipsychotics, loxapine has been shown to be weight neutral or result in weight loss,14 making it an option to consider for patients with type 2 diabetes mellitus, metabolic syndrome, dyslipidemia, or cardiovascular disease.
OUTCOME Improvement, stability
Ms. B begins taking loxapine, 10 mg/d, gradually cross-tapered with olanzapine, increasing loxapine by 10 mg every 2 to 3 days (Figure). After 8 days, when the dosage has reached 40 mg/d, Ms. B’s treatment team begins to observe a consistent change in her behavior. Ms. B comes into the interview room, where previously the team had to see her in her own room because she refused to come out. She also tolerates an extensive interview, even sharing parts of her history without prompting, and is able to discuss her treatment. Ms. B continues to express some paranoia regarding the treatment team. On day 12, receiving loxapine, 50 mg/d, Ms. B says that she likes the new medication and feels she is doing well with it. She becomes less reclusive and begins socializing with other patients. By day 19, receiving loxapine, 80 mg/d, a nurse, who knows Ms. B from the outpatient facility, visits the unit and reports that Ms. B is at her baseline.
At discharge, Ms. B is noted to be “bright,” well organized, neatly dressed, and wearing makeup. Her paranoia and auditory hallucinations have almost completely resolved. She is social, engages appropriately with the treatment team, and is able to describe a plan for self-care after discharge including following up with her oncologist. Her white blood cell counts were carefully monitored throughout her admission and are within normal limits when she is discharged.
One year later, Ms. B remains taking loxapine, 70 mg/d. Although she continues to report mild paranoia, she is living independently in her apartment and attends church regularly.
2. Usta NG, Poyraz CA, Aktan M, et al. Clozapine treatment of refractory schizophrenia during essential chemotherapy: a case study and mini review of a clinical dilemma. Ther Adv Psychopharmacol. 2014;4(6):276-281.
3. Meyer N, Gee S, Whiskey E, et al. Optimizing outcomes in clozapine rechallenge following neutropenia: a cohort analysis. J Clin Psychiatry. 2015;76(11):e1410-e1416.
4. Cunningham NT, Dennis N, Dattilo W, et al. Continuation of clozapine during chemotherapy: a case report and review of literature. Psychosomatics. 2014;55(6):673-679.
5. Co¸sar B, Taner ME, Eser HY, et al. Does switching to another antipsychotic in patients with clozapine-associated granulocytopenia solve the problem? Case series of 18. J Clin Psychopharmacol. 2011;31(2):169-173.
6. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigation. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
7. Mustafa FA, Burke JG, Abukmeil SS, et al. “Schizophrenia past clozapine”: reasons for clozapine discontinuation, mortality, and alternative antipsychotic prescribing. Pharmacopsychiatry. 2015;48(1):11-14.
8. Aydin M, Ilhan BC, Calisir S, et al. Continuing clozapine treatment with lithium in schizophrenic patients with neutropenia or leukopenia: brief review of literature with case reports. Ther Adv Psychopharmacol. 2016;6(1):33-38.
9. Bitter I, Dossenbach MR, Brook S, et al; Olanzapine HGCK Study Group. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
10. Lehmann CR, Ereshefsky L, Saklad SR, et al. Very high dose loxapine in refractory schizophrenic patients. Am J Psychiatry. 1981;138(9):1212-1214.
11. Sokolski KN. Combination loxapine and aripiprazole for refractory hallucinations in schizophrenia. Ann Pharmacother. 2011;45(7-8):e36.
12. Shen WW. A history of antipsychotic drug development. Compr Psychiatry. 1999;40(6):407-414.
13. Mazzola CD, Miron S, Jenkins AJ. Loxapine intoxication: case report and literature review. J Anal Toxicol. 2000;24(7):638-641.
14. Chakrabarti A, Bagnall A, Chue P, et al. Loxapine for schizophrenia. Cochrane Database Syst Rev. 2007(4):CD001943.
15. Jegouzo A, Gressier B, Frimat B, et al. Comparative oxidation of loxapine and clozapine by human neutrophils. Fundam Clin Pharmacol. 1999;13(1):113-119.
16. Keating GM. Loxapine inhalation powder: a review of its use in the acute treatment of agitation in patients with bipolar disorder or schizophrenia. CNS Drugs. 2013;27(6):479-489.
1 7. Glazer WM. Does loxapine have “atypical” properties? Clinical evidence. J Clin Psychiatry. 1999;60(suppl 10):42-46.
18. Hellings JA, Jadhav M, Jain S, et al. Low dose loxapine: neuromotor side effects and tolerability in autism spectrum disorders. J Child Adolesc Psychopharmacol. 2015;25(8):618-624.
19. Singh AN, Barlas C, Singh S, et al. A neurochemical basis for the antipsychotic activity of loxapine: interactions with dopamine D1, D2, D4 and serotonin 5-HT2 receptor subtypes. J Psychiatry Neurosci. 1996;21(1):29-35.
20. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry. 1999;156(2):286-293.
21. Robertson AG, Berry R, Meltzer HY. Prolactin stimulating effects of amoxapine and loxapine in psychiatric patients. Psychopharmacology (Berl). 1982;78(3):287-292.
22. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
CASE Stable with a new diagnosis
Ms. B, age 60, has a history of schizophrenia, which has been stable on clozapine, 500 mg/d, for more than 2 decades. After a series of hospitalizations in her 20s and 30s, clozapine was initiated and she has not required additional inpatient psychiatric care. She has been managed in the outpatient setting with standard biweekly absolute neutrophil count (ANC) monitoring. She lives independently and is an active member in her church.
After experiencing rectal bleeding, Ms. B is diagnosed with rectal carcinoma and is scheduled to undergo chemotherapy and radiation treatment.
[polldaddy:9754786]
The authors’ observations
Both clozapine and chemotherapy carry the risk of immunosuppression, presenting a clinical challenge when choosing an appropriate management strategy. However, the risks of stopping clozapine after a long period of symptom stability are substantial, with a relapse rate up to 50%.1 Among patients taking clozapine, the risk of agranulocytosis and neutropenia are approximately 0.8% and 3%, respectively, and >80% of agranulocyotis cases occur within the first 18 weeks of treatment.2,3 Although both clozapine and chemotherapy can lead to neutropenia and agranulocytosis, there currently is no evidence of a synergistic effect on bone marrow suppression with simultaneous use of these therapies2 nor is there evidence of the combination leading to sustained marrow suppression.4
Because of Ms. B’s positive response to clozapine, the risks associated with discontinuing the medication, and the relatively low risk of clozapine contributing to neutropenia after a long period of stabilization, her outpatient psychiatric providers decide to increase ANC monitoring to weekly while she undergoes cancer treatment.
TREATMENT Neutropenia, psychosis
Ms. B continues clozapine during radiation and chemotherapy, but develops leukopenia and neutropenia with a low of 1,220/μL white blood cells and an ANC of 610/μL. Clozapine is stopped, consistent with current recommendations to hold the drug if the neutrophil count is <1,000/μL in a patient without benign ethnic neutropenia, and her outpatient provider monitors her closely. The treatment team does not restart an antipsychotic immediately after discontinuing clozapine because of the risk that other antipsychotics can cause hematologic toxicity or prolong granulocytopenia associated with clozapine.5
Approximately 2 weeks later, Ms. B is admitted to a different hospital for altered mental status and is found to have hyponatremia and rectal bleeding. The workup suggests that her rectal carcinoma has not fully responded to initial therapies, and she likely will require further treatment. Her mental status improves after hyponatremia resolves, but she reports auditory hallucinations and paranoia. Risperidone, 4 mg/d, is initiated to target psychosis.
After discharge, Ms. B develops bilateral upper extremity tremor, which she finds intolerable and attributes to risperidone. She refuses to continue risperidone or try adjunctive medications to address the tremor, but is willing to consider a different antipsychotic. Olanzapine, 10 mg/d, is initiated and risperidone is slowly tapered. During this time, Ms. B experiences increased paranoia and believes that the Internal Revenue Service is calling her. She misses her next appointment.
Later, the fire department finds Ms. B wandering the streets and brings her to the psychiatric emergency room. During the examination, she is disheveled and withdrawn, and unable to reply to simple questions about diet and sleep. When asked why she was in the street, she says that she left her apartment because it was “too messy.” The treatment team learns that she had walked at least 10 miles from her apartment before sitting down by the side of the road and being picked up by the fire department. She reveals that she left her apartment and continued walking because “a man” told her to do so and threatened to harm her if she stopped.
When Ms. B is admitted to the psychiatric service, she is paranoid, disorganized, and guarded. She remains in her room for most of the day and either refuses to talk to providers or curses at them. She often is seen wearing soiled clothing with her hair mussed. She denies having rectal carcinoma, although she expressed understanding of her medical condition <2 months earlier.
[polldaddy:9754787]
The authors’ observations
Clozapine is considered the most efficacious agent for treatment-resistant schizophrenia.6 Although non-compliance is the most common reason for discontinuing clozapine, >20% of patients stop clozapine because of adverse effects.7 Clozapine often is a drug of last resort because of the need for frequent monitoring and significant side effects; therefore deciding on a next step when clozapine fails or cannot be continued because of other factors can pose a challenge.
Ms. B’s treatment team gave serious consideration to restarting clozapine. However, because it was likely that Ms. B would undergo another round of chemotherapy and possibly radiation, the risk of neutropenia recurring was considered too high. Lithium has been used successfully to manage neutropenia in patients taking clozapine and, for some, adding lithium could help boost white cell count and allow a successful rechallenge with clozapine.3,8 However, because of Ms. B’s medical comorbidities, including cancer and chronic kidney disease, adding lithium was not thought to be clinically prudent at that time and the treatment team considered other options.
Olanzapine. Although research is limited, studies suggest olanzapine is the most commonly prescribed medication when a patient has to discontinue clozapine,7 with comparable response rates in those with refractory schizophrenia.9 Therefore, Ms. B was initially maintained on olanzapine, and the dosage increased to 30 mg over the course of 16 days in the hospital. However, she did not respond to the medication, remaining disorganized and paranoid without any notable improvement in her symptoms therefore other treatment options were explored.
Loxapine. Previous limited case reports have shown loxapine to be effective in treating individuals with refractory schizophrenia, either alone or in combination with other antipsychotics.10,11 FDA-approved in 1975, loxapine was among the last of the typical antipsychotics brought to the U.S. market before the introduction of clozapine, the first atypical.12 Loxapine is a dibenzoxazepine that has a molecular structure similar to clozapine.13 Unlike clozapine, however, loxapine is not known to cause agranulocytosis.14 Research suggests that although clozapine is oxidized to metabolites that are cytotoxic, loxapine is not, potentially accounting for their different effects on neutrophils.15
The efficacy of loxapine has shown to be similar to other typical and atypical antipsychotics, with approximately 70% of patients showing improvement.14 However, loxapine may be overlooked as an option, possibly because it was not included in the CATIE trial and was the last typical antipsychotic to be approved before atypicals were introduced.12 First available in oral and IM formulations, there has been increased interest in loxapine recently because of the approval of an inhaled formulation in 2012.16
Although classified as a typical antipsychotic, studies have suggested that loxapine acts as an atypical at low dosages.17,18 Previous work suggests, however, that the side effect profile of loxapine is similar to typical antipsychotics.14 At dosages <50 mg, it results in fewer cases of extrapyramidal side effects than expected with a typical antipsychotic.18
Loxapine’s binding profile seems to exist along this spectrum of typical to atypical. Tissue-based binding studies have shown a higher 5-HT2 affinity relative to D2, consistent with atypical antipsychotics.19 Positron emission tomography studies in humans show 5-HT2 saturation of loxapine to be close to equal to D2 binding in loxapine, thus a slightly lower ratio of 5-HT2 to D2 relative to atypicals, but more than that seen with typical antipsychotics.20 These differences between in vitro and in vivo studies may be secondary to the binding of loxapine’s active metabolites, particularly 7- and 8-hydroxyloxapine, which have more dopaminergic activity. In addition to increased 5-HT2A binding compared with typical antipsychotics, loxapine also has a high affinity for the D4 receptors, as well as interacting with other serotonin receptors 5-HT3, 5-HT6, and 5-HT7. Of note this is a similar pattern of binding affinity as seen in clozapine.19
It should be noted, however, that loxapine may not be an appropriate treatment in all forms of cancer. Similar to other first-generation antipsychotics, it increases prolactin levels, and thus may have a negative clinical impact on patients with prolactin receptor positive breast cancers.21,22 Finally, although clozapine can result in significant weight gain, dyslipidemia, and hyperglycemia, unlike many antipsychotics, loxapine has been shown to be weight neutral or result in weight loss,14 making it an option to consider for patients with type 2 diabetes mellitus, metabolic syndrome, dyslipidemia, or cardiovascular disease.
OUTCOME Improvement, stability
Ms. B begins taking loxapine, 10 mg/d, gradually cross-tapered with olanzapine, increasing loxapine by 10 mg every 2 to 3 days (Figure). After 8 days, when the dosage has reached 40 mg/d, Ms. B’s treatment team begins to observe a consistent change in her behavior. Ms. B comes into the interview room, where previously the team had to see her in her own room because she refused to come out. She also tolerates an extensive interview, even sharing parts of her history without prompting, and is able to discuss her treatment. Ms. B continues to express some paranoia regarding the treatment team. On day 12, receiving loxapine, 50 mg/d, Ms. B says that she likes the new medication and feels she is doing well with it. She becomes less reclusive and begins socializing with other patients. By day 19, receiving loxapine, 80 mg/d, a nurse, who knows Ms. B from the outpatient facility, visits the unit and reports that Ms. B is at her baseline.
At discharge, Ms. B is noted to be “bright,” well organized, neatly dressed, and wearing makeup. Her paranoia and auditory hallucinations have almost completely resolved. She is social, engages appropriately with the treatment team, and is able to describe a plan for self-care after discharge including following up with her oncologist. Her white blood cell counts were carefully monitored throughout her admission and are within normal limits when she is discharged.
One year later, Ms. B remains taking loxapine, 70 mg/d. Although she continues to report mild paranoia, she is living independently in her apartment and attends church regularly.
CASE Stable with a new diagnosis
Ms. B, age 60, has a history of schizophrenia, which has been stable on clozapine, 500 mg/d, for more than 2 decades. After a series of hospitalizations in her 20s and 30s, clozapine was initiated and she has not required additional inpatient psychiatric care. She has been managed in the outpatient setting with standard biweekly absolute neutrophil count (ANC) monitoring. She lives independently and is an active member in her church.
After experiencing rectal bleeding, Ms. B is diagnosed with rectal carcinoma and is scheduled to undergo chemotherapy and radiation treatment.
[polldaddy:9754786]
The authors’ observations
Both clozapine and chemotherapy carry the risk of immunosuppression, presenting a clinical challenge when choosing an appropriate management strategy. However, the risks of stopping clozapine after a long period of symptom stability are substantial, with a relapse rate up to 50%.1 Among patients taking clozapine, the risk of agranulocytosis and neutropenia are approximately 0.8% and 3%, respectively, and >80% of agranulocyotis cases occur within the first 18 weeks of treatment.2,3 Although both clozapine and chemotherapy can lead to neutropenia and agranulocytosis, there currently is no evidence of a synergistic effect on bone marrow suppression with simultaneous use of these therapies2 nor is there evidence of the combination leading to sustained marrow suppression.4
Because of Ms. B’s positive response to clozapine, the risks associated with discontinuing the medication, and the relatively low risk of clozapine contributing to neutropenia after a long period of stabilization, her outpatient psychiatric providers decide to increase ANC monitoring to weekly while she undergoes cancer treatment.
TREATMENT Neutropenia, psychosis
Ms. B continues clozapine during radiation and chemotherapy, but develops leukopenia and neutropenia with a low of 1,220/μL white blood cells and an ANC of 610/μL. Clozapine is stopped, consistent with current recommendations to hold the drug if the neutrophil count is <1,000/μL in a patient without benign ethnic neutropenia, and her outpatient provider monitors her closely. The treatment team does not restart an antipsychotic immediately after discontinuing clozapine because of the risk that other antipsychotics can cause hematologic toxicity or prolong granulocytopenia associated with clozapine.5
Approximately 2 weeks later, Ms. B is admitted to a different hospital for altered mental status and is found to have hyponatremia and rectal bleeding. The workup suggests that her rectal carcinoma has not fully responded to initial therapies, and she likely will require further treatment. Her mental status improves after hyponatremia resolves, but she reports auditory hallucinations and paranoia. Risperidone, 4 mg/d, is initiated to target psychosis.
After discharge, Ms. B develops bilateral upper extremity tremor, which she finds intolerable and attributes to risperidone. She refuses to continue risperidone or try adjunctive medications to address the tremor, but is willing to consider a different antipsychotic. Olanzapine, 10 mg/d, is initiated and risperidone is slowly tapered. During this time, Ms. B experiences increased paranoia and believes that the Internal Revenue Service is calling her. She misses her next appointment.
Later, the fire department finds Ms. B wandering the streets and brings her to the psychiatric emergency room. During the examination, she is disheveled and withdrawn, and unable to reply to simple questions about diet and sleep. When asked why she was in the street, she says that she left her apartment because it was “too messy.” The treatment team learns that she had walked at least 10 miles from her apartment before sitting down by the side of the road and being picked up by the fire department. She reveals that she left her apartment and continued walking because “a man” told her to do so and threatened to harm her if she stopped.
When Ms. B is admitted to the psychiatric service, she is paranoid, disorganized, and guarded. She remains in her room for most of the day and either refuses to talk to providers or curses at them. She often is seen wearing soiled clothing with her hair mussed. She denies having rectal carcinoma, although she expressed understanding of her medical condition <2 months earlier.
[polldaddy:9754787]
The authors’ observations
Clozapine is considered the most efficacious agent for treatment-resistant schizophrenia.6 Although non-compliance is the most common reason for discontinuing clozapine, >20% of patients stop clozapine because of adverse effects.7 Clozapine often is a drug of last resort because of the need for frequent monitoring and significant side effects; therefore deciding on a next step when clozapine fails or cannot be continued because of other factors can pose a challenge.
Ms. B’s treatment team gave serious consideration to restarting clozapine. However, because it was likely that Ms. B would undergo another round of chemotherapy and possibly radiation, the risk of neutropenia recurring was considered too high. Lithium has been used successfully to manage neutropenia in patients taking clozapine and, for some, adding lithium could help boost white cell count and allow a successful rechallenge with clozapine.3,8 However, because of Ms. B’s medical comorbidities, including cancer and chronic kidney disease, adding lithium was not thought to be clinically prudent at that time and the treatment team considered other options.
Olanzapine. Although research is limited, studies suggest olanzapine is the most commonly prescribed medication when a patient has to discontinue clozapine,7 with comparable response rates in those with refractory schizophrenia.9 Therefore, Ms. B was initially maintained on olanzapine, and the dosage increased to 30 mg over the course of 16 days in the hospital. However, she did not respond to the medication, remaining disorganized and paranoid without any notable improvement in her symptoms therefore other treatment options were explored.
Loxapine. Previous limited case reports have shown loxapine to be effective in treating individuals with refractory schizophrenia, either alone or in combination with other antipsychotics.10,11 FDA-approved in 1975, loxapine was among the last of the typical antipsychotics brought to the U.S. market before the introduction of clozapine, the first atypical.12 Loxapine is a dibenzoxazepine that has a molecular structure similar to clozapine.13 Unlike clozapine, however, loxapine is not known to cause agranulocytosis.14 Research suggests that although clozapine is oxidized to metabolites that are cytotoxic, loxapine is not, potentially accounting for their different effects on neutrophils.15
The efficacy of loxapine has shown to be similar to other typical and atypical antipsychotics, with approximately 70% of patients showing improvement.14 However, loxapine may be overlooked as an option, possibly because it was not included in the CATIE trial and was the last typical antipsychotic to be approved before atypicals were introduced.12 First available in oral and IM formulations, there has been increased interest in loxapine recently because of the approval of an inhaled formulation in 2012.16
Although classified as a typical antipsychotic, studies have suggested that loxapine acts as an atypical at low dosages.17,18 Previous work suggests, however, that the side effect profile of loxapine is similar to typical antipsychotics.14 At dosages <50 mg, it results in fewer cases of extrapyramidal side effects than expected with a typical antipsychotic.18
Loxapine’s binding profile seems to exist along this spectrum of typical to atypical. Tissue-based binding studies have shown a higher 5-HT2 affinity relative to D2, consistent with atypical antipsychotics.19 Positron emission tomography studies in humans show 5-HT2 saturation of loxapine to be close to equal to D2 binding in loxapine, thus a slightly lower ratio of 5-HT2 to D2 relative to atypicals, but more than that seen with typical antipsychotics.20 These differences between in vitro and in vivo studies may be secondary to the binding of loxapine’s active metabolites, particularly 7- and 8-hydroxyloxapine, which have more dopaminergic activity. In addition to increased 5-HT2A binding compared with typical antipsychotics, loxapine also has a high affinity for the D4 receptors, as well as interacting with other serotonin receptors 5-HT3, 5-HT6, and 5-HT7. Of note this is a similar pattern of binding affinity as seen in clozapine.19
It should be noted, however, that loxapine may not be an appropriate treatment in all forms of cancer. Similar to other first-generation antipsychotics, it increases prolactin levels, and thus may have a negative clinical impact on patients with prolactin receptor positive breast cancers.21,22 Finally, although clozapine can result in significant weight gain, dyslipidemia, and hyperglycemia, unlike many antipsychotics, loxapine has been shown to be weight neutral or result in weight loss,14 making it an option to consider for patients with type 2 diabetes mellitus, metabolic syndrome, dyslipidemia, or cardiovascular disease.
OUTCOME Improvement, stability
Ms. B begins taking loxapine, 10 mg/d, gradually cross-tapered with olanzapine, increasing loxapine by 10 mg every 2 to 3 days (Figure). After 8 days, when the dosage has reached 40 mg/d, Ms. B’s treatment team begins to observe a consistent change in her behavior. Ms. B comes into the interview room, where previously the team had to see her in her own room because she refused to come out. She also tolerates an extensive interview, even sharing parts of her history without prompting, and is able to discuss her treatment. Ms. B continues to express some paranoia regarding the treatment team. On day 12, receiving loxapine, 50 mg/d, Ms. B says that she likes the new medication and feels she is doing well with it. She becomes less reclusive and begins socializing with other patients. By day 19, receiving loxapine, 80 mg/d, a nurse, who knows Ms. B from the outpatient facility, visits the unit and reports that Ms. B is at her baseline.
At discharge, Ms. B is noted to be “bright,” well organized, neatly dressed, and wearing makeup. Her paranoia and auditory hallucinations have almost completely resolved. She is social, engages appropriately with the treatment team, and is able to describe a plan for self-care after discharge including following up with her oncologist. Her white blood cell counts were carefully monitored throughout her admission and are within normal limits when she is discharged.
One year later, Ms. B remains taking loxapine, 70 mg/d. Although she continues to report mild paranoia, she is living independently in her apartment and attends church regularly.
2. Usta NG, Poyraz CA, Aktan M, et al. Clozapine treatment of refractory schizophrenia during essential chemotherapy: a case study and mini review of a clinical dilemma. Ther Adv Psychopharmacol. 2014;4(6):276-281.
3. Meyer N, Gee S, Whiskey E, et al. Optimizing outcomes in clozapine rechallenge following neutropenia: a cohort analysis. J Clin Psychiatry. 2015;76(11):e1410-e1416.
4. Cunningham NT, Dennis N, Dattilo W, et al. Continuation of clozapine during chemotherapy: a case report and review of literature. Psychosomatics. 2014;55(6):673-679.
5. Co¸sar B, Taner ME, Eser HY, et al. Does switching to another antipsychotic in patients with clozapine-associated granulocytopenia solve the problem? Case series of 18. J Clin Psychopharmacol. 2011;31(2):169-173.
6. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigation. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
7. Mustafa FA, Burke JG, Abukmeil SS, et al. “Schizophrenia past clozapine”: reasons for clozapine discontinuation, mortality, and alternative antipsychotic prescribing. Pharmacopsychiatry. 2015;48(1):11-14.
8. Aydin M, Ilhan BC, Calisir S, et al. Continuing clozapine treatment with lithium in schizophrenic patients with neutropenia or leukopenia: brief review of literature with case reports. Ther Adv Psychopharmacol. 2016;6(1):33-38.
9. Bitter I, Dossenbach MR, Brook S, et al; Olanzapine HGCK Study Group. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
10. Lehmann CR, Ereshefsky L, Saklad SR, et al. Very high dose loxapine in refractory schizophrenic patients. Am J Psychiatry. 1981;138(9):1212-1214.
11. Sokolski KN. Combination loxapine and aripiprazole for refractory hallucinations in schizophrenia. Ann Pharmacother. 2011;45(7-8):e36.
12. Shen WW. A history of antipsychotic drug development. Compr Psychiatry. 1999;40(6):407-414.
13. Mazzola CD, Miron S, Jenkins AJ. Loxapine intoxication: case report and literature review. J Anal Toxicol. 2000;24(7):638-641.
14. Chakrabarti A, Bagnall A, Chue P, et al. Loxapine for schizophrenia. Cochrane Database Syst Rev. 2007(4):CD001943.
15. Jegouzo A, Gressier B, Frimat B, et al. Comparative oxidation of loxapine and clozapine by human neutrophils. Fundam Clin Pharmacol. 1999;13(1):113-119.
16. Keating GM. Loxapine inhalation powder: a review of its use in the acute treatment of agitation in patients with bipolar disorder or schizophrenia. CNS Drugs. 2013;27(6):479-489.
1 7. Glazer WM. Does loxapine have “atypical” properties? Clinical evidence. J Clin Psychiatry. 1999;60(suppl 10):42-46.
18. Hellings JA, Jadhav M, Jain S, et al. Low dose loxapine: neuromotor side effects and tolerability in autism spectrum disorders. J Child Adolesc Psychopharmacol. 2015;25(8):618-624.
19. Singh AN, Barlas C, Singh S, et al. A neurochemical basis for the antipsychotic activity of loxapine: interactions with dopamine D1, D2, D4 and serotonin 5-HT2 receptor subtypes. J Psychiatry Neurosci. 1996;21(1):29-35.
20. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry. 1999;156(2):286-293.
21. Robertson AG, Berry R, Meltzer HY. Prolactin stimulating effects of amoxapine and loxapine in psychiatric patients. Psychopharmacology (Berl). 1982;78(3):287-292.
22. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
2. Usta NG, Poyraz CA, Aktan M, et al. Clozapine treatment of refractory schizophrenia during essential chemotherapy: a case study and mini review of a clinical dilemma. Ther Adv Psychopharmacol. 2014;4(6):276-281.
3. Meyer N, Gee S, Whiskey E, et al. Optimizing outcomes in clozapine rechallenge following neutropenia: a cohort analysis. J Clin Psychiatry. 2015;76(11):e1410-e1416.
4. Cunningham NT, Dennis N, Dattilo W, et al. Continuation of clozapine during chemotherapy: a case report and review of literature. Psychosomatics. 2014;55(6):673-679.
5. Co¸sar B, Taner ME, Eser HY, et al. Does switching to another antipsychotic in patients with clozapine-associated granulocytopenia solve the problem? Case series of 18. J Clin Psychopharmacol. 2011;31(2):169-173.
6. McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigation. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-610.
7. Mustafa FA, Burke JG, Abukmeil SS, et al. “Schizophrenia past clozapine”: reasons for clozapine discontinuation, mortality, and alternative antipsychotic prescribing. Pharmacopsychiatry. 2015;48(1):11-14.
8. Aydin M, Ilhan BC, Calisir S, et al. Continuing clozapine treatment with lithium in schizophrenic patients with neutropenia or leukopenia: brief review of literature with case reports. Ther Adv Psychopharmacol. 2016;6(1):33-38.
9. Bitter I, Dossenbach MR, Brook S, et al; Olanzapine HGCK Study Group. Olanzapine versus clozapine in treatment-resistant or treatment-intolerant schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(1):173-180.
10. Lehmann CR, Ereshefsky L, Saklad SR, et al. Very high dose loxapine in refractory schizophrenic patients. Am J Psychiatry. 1981;138(9):1212-1214.
11. Sokolski KN. Combination loxapine and aripiprazole for refractory hallucinations in schizophrenia. Ann Pharmacother. 2011;45(7-8):e36.
12. Shen WW. A history of antipsychotic drug development. Compr Psychiatry. 1999;40(6):407-414.
13. Mazzola CD, Miron S, Jenkins AJ. Loxapine intoxication: case report and literature review. J Anal Toxicol. 2000;24(7):638-641.
14. Chakrabarti A, Bagnall A, Chue P, et al. Loxapine for schizophrenia. Cochrane Database Syst Rev. 2007(4):CD001943.
15. Jegouzo A, Gressier B, Frimat B, et al. Comparative oxidation of loxapine and clozapine by human neutrophils. Fundam Clin Pharmacol. 1999;13(1):113-119.
16. Keating GM. Loxapine inhalation powder: a review of its use in the acute treatment of agitation in patients with bipolar disorder or schizophrenia. CNS Drugs. 2013;27(6):479-489.
1 7. Glazer WM. Does loxapine have “atypical” properties? Clinical evidence. J Clin Psychiatry. 1999;60(suppl 10):42-46.
18. Hellings JA, Jadhav M, Jain S, et al. Low dose loxapine: neuromotor side effects and tolerability in autism spectrum disorders. J Child Adolesc Psychopharmacol. 2015;25(8):618-624.
19. Singh AN, Barlas C, Singh S, et al. A neurochemical basis for the antipsychotic activity of loxapine: interactions with dopamine D1, D2, D4 and serotonin 5-HT2 receptor subtypes. J Psychiatry Neurosci. 1996;21(1):29-35.
20. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry. 1999;156(2):286-293.
21. Robertson AG, Berry R, Meltzer HY. Prolactin stimulating effects of amoxapine and loxapine in psychiatric patients. Psychopharmacology (Berl). 1982;78(3):287-292.
22. Rahman T, Clevenger CV, Kaklamani V, et al. Antipsychotic treatment in breast cancer patients. Am J Psychiatry. 2014;171(6):616-621.
Herb–drug interactions: Caution patients when changing supplements
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels. 
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels.
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels.
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
Genomic Testing in the Management of Early-Stage Breast Cancer
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
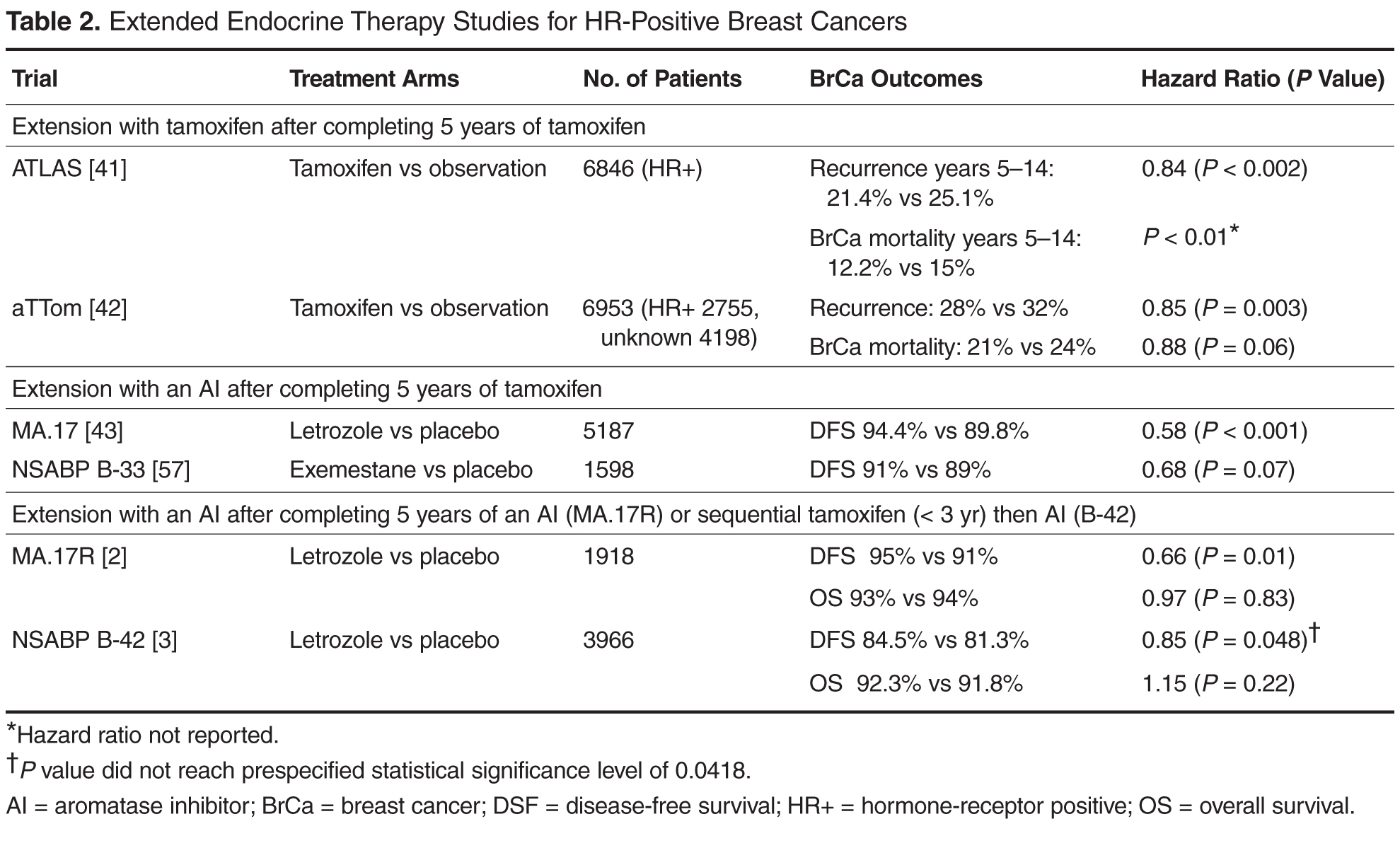
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
How you can simplify your patient’s medication regimen to enhance adherence
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2


Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.