User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
When the correct Dx is elusive
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
Editor-in-Chief
Editor-in-Chief
Editor-in-Chief
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
In this issue of JFP, Dr. Mendoza reminds us that “Parkinson’s disease can be a tough diagnosis to navigate.”1 Classically, Parkinson’s disease (PD) is associated with a resting tremor, but bradykinesia is actually the hallmark of the disease. PD can also present with subtle movement disorders, as well as depression and early dementia. It is, indeed, a difficult clinical diagnosis, and consultation with an expert to confirm or deny its presence can be quite helpful.
Other conundrums. PD, however, is not the only illness whose signs and symptoms can present a challenge. Chronic and intermittent shortness of breath, for example, can be very difficult to sort out. Is the shortness of breath due to congestive heart failure, chronic obstructive pulmonary disease, asthma, or a neurologic condition such as myasthenia gravis? Or is it the result of several causes?
When asthma isn’t asthma. Because it is a common illness, physicians often diagnose asthma in patients with shortness of breath or wheezing. But a recent study suggests that as many as 30% of primary care patients with a current diagnosis of asthma do not have asthma at all.2
In the study, Canadian researchers recruited 701 adults with physician-diagnosed asthma, all of whom were taking asthma medications regularly. The researchers did baseline pulmonary function testing (including methacholine challenge testing, if needed) and monitored symptoms frequently. Then they gradually withdrew asthma medications from those who did not appear to have a definitive diagnosis of asthma. They followed these patients for one year. One-third (203 of 613) of the patients with complete follow-up data were no longer taking asthma medications one year later and had no symptoms of asthma. Twelve patients had serious alternative diagnoses such as coronary artery disease and bronchiectasis.
Closer to home. In my practice, I found 2 patients with long-standing diagnoses of asthma who didn’t, in fact, have the condition at all. In both cases, my suspicion was raised by lung examination. In one case, fine bibasilar rales suggested pulmonary fibrosis, which was the correct diagnosis, and the patient is now on the lung transplant list. In the other case, a loud venous hum suggested an arteriovenous malformation. Surgery corrected the patient’s “asthma.”
I urge you to reevaluate your asthma patients to be sure they have the correct diagnosis and to keep PD in your differential for patients who present with atypical symptoms. Primary care clinicians must be expert diagnosticians, willing to question prior diagnoses.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
1. Young J, Mendoza M. Parkinson’s disease: a treatment guide. J Fam Pract. 2018;67:276-286.
2. Aaron SD, Vandemheen KL, FitzGerald JM, et al for the Canadian Respiratory Research Network. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017:317:269-279.
FDA: More COPD patients can use triple therapy
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.

“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.
“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).
The Food and Drug Administration has approved a new indication for the chronic obstructive pulmonary disease (COPD) therapy fluticasone furoate/umeclidinium/vilanterol (Trelegy Ellipta), which allows physicians to prescribe the drug to a broader class of COPD patients, according to a statement from two pharmaceutical companies.
“Following the initial approval of Trelegy Ellipta in September, we have analysed the data from the IMPACT study and identified additional benefits that this important medicine offers patients with [COPD],” said Hal Barron, MD, chief scientific officer and president of research and development at GlaxoSmithKline, in the statement. “We are pleased that the robust data from the IMPACT study has enabled the expanded indication announced today and the FDA action has been taken so swiftly.”
The results of the IMPACT trial, which was the first study to compare a single-inhaler triple therapy with two dual therapies, were published on April 18 (N Engl J Med 2018. doi: 10.1056/NEJMoa1713901).
This study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate, 62.5 mcg of umeclidinium, and 25 mcg of vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction, compared with vilanterol-umeclidinium (P less than .001 for both).
FDA approves epinephrine autoinjector for infants and toddlers
according to a press release from Kaléo, a privately-held pharmaceutical company.
“Anaphylactic reactions can be frightening and serious, and when experienced by the very young, some of whom can’t communicate about what’s happening, these episodes can be particularly alarming,” Vivian Hernandez-Trujillo, MD, a pediatric allergist and fellow of the American Academy of Allergy, Asthma and Immunology, said in a statement. “Now, caregivers can have the AUVI-Q 0.1 mg in hand to respond to an allergic emergency and safely administer epinephrine to infants and toddlers.”
The approval comes at a time when a higher percentage of children are being admitted to the hospital for food-related anaphylaxis: a 130% increase among children aged 0-4 years and a 196% increase in children aged 5-17 years.
The epinephrine autoinjector will be available for $0 out of pocket for commercially insured patients using the AUVI-Q AffordAbility Program and Direct Delivery Service starting May 1, 2018. More information concerning this EAI can be found here.
according to a press release from Kaléo, a privately-held pharmaceutical company.
“Anaphylactic reactions can be frightening and serious, and when experienced by the very young, some of whom can’t communicate about what’s happening, these episodes can be particularly alarming,” Vivian Hernandez-Trujillo, MD, a pediatric allergist and fellow of the American Academy of Allergy, Asthma and Immunology, said in a statement. “Now, caregivers can have the AUVI-Q 0.1 mg in hand to respond to an allergic emergency and safely administer epinephrine to infants and toddlers.”
The approval comes at a time when a higher percentage of children are being admitted to the hospital for food-related anaphylaxis: a 130% increase among children aged 0-4 years and a 196% increase in children aged 5-17 years.
The epinephrine autoinjector will be available for $0 out of pocket for commercially insured patients using the AUVI-Q AffordAbility Program and Direct Delivery Service starting May 1, 2018. More information concerning this EAI can be found here.
according to a press release from Kaléo, a privately-held pharmaceutical company.
“Anaphylactic reactions can be frightening and serious, and when experienced by the very young, some of whom can’t communicate about what’s happening, these episodes can be particularly alarming,” Vivian Hernandez-Trujillo, MD, a pediatric allergist and fellow of the American Academy of Allergy, Asthma and Immunology, said in a statement. “Now, caregivers can have the AUVI-Q 0.1 mg in hand to respond to an allergic emergency and safely administer epinephrine to infants and toddlers.”
The approval comes at a time when a higher percentage of children are being admitted to the hospital for food-related anaphylaxis: a 130% increase among children aged 0-4 years and a 196% increase in children aged 5-17 years.
The epinephrine autoinjector will be available for $0 out of pocket for commercially insured patients using the AUVI-Q AffordAbility Program and Direct Delivery Service starting May 1, 2018. More information concerning this EAI can be found here.
Stroke patients benefited from CPAP
the results of a randomized study suggest.
Obstructive sleep apnea is present in 50%-80% of patients with stroke, previous studies show, and its presence is associated with impaired function and cognition, delirium, and longer rehabilitation time, among other negative impacts, wrote Anupama Gupta, PhD, and her coauthors from the All India Institute of Medical Sciences, New Delhi, in the Journal of Clinical Sleep Medicine. Although multiple trials have shown a positive effect of CPAP on stroke recovery, relatively few investigations have looked specifically at whether the intervention prevents subsequent vascular events.
Patients’ clinical stroke outcomes were categorized in accordance with the Modified Rankin Scale (mRS), which is most widely used to assess disability and dependence outcomes among patients with stroke.
Significantly more patients who were treated with CPAP experienced an improvement in their mRS score by at least 1 point, when assessed at both 6 and 12 months following entrance into the study. Specifically, 53% (16) of patients in the CPAP group had an improvement of at least 1 point in their mRS score at 12 months, compared with 27% (11) of patients who did not use CPAP (P = .03).
“These differences are statistically significant, as well as clinically meaningful and relevant,” Dr. Gupta and her colleagues said in their report.
This finding was consistent with what researchers have seen in some earlier studies of stroke patients who used CPAP, the researchers wrote.
Additionally, CPAP-treated patients had fewer subsequent vascular events, compared with those who did not use CPAP, though the difference did not reach statistical significance. There was only one new vascular event (3.33%) in the CPAP group at 12-month follow-up, versus six events (15%) in the non-CPAP group (P = .23).
Nevertheless, the results provide more evidence for the potential benefit of CPAP in stroke patients with obstructive sleep apnea, the researchers noted.
“Our results indicate that new vascular events may be better prevented – and significantly more patients may make good stroke recovery – with CPAP treatment as compared to only best medical treatment,” Dr. Gupta and her colleagues wrote.
Before the study started, investigators determined that they would have needed 80 patients per arm for a power of 80%. A total of 679 patients were screened, but only 116 reported for polysomnography testing, and of those, 83 had at least moderate obstructive sleep apnea.
Due to a lack of CPAP devices, only 70 of those 83 patients made it all the way to randomization, investigators reported.
Dr. Gupta and her coauthors reported no conflicts of interest related to the study.
SOURCE: Gupta A et al. J Clin Sleep Med. 2018 Mar 30. pii:jc-17-00230.
the results of a randomized study suggest.
Obstructive sleep apnea is present in 50%-80% of patients with stroke, previous studies show, and its presence is associated with impaired function and cognition, delirium, and longer rehabilitation time, among other negative impacts, wrote Anupama Gupta, PhD, and her coauthors from the All India Institute of Medical Sciences, New Delhi, in the Journal of Clinical Sleep Medicine. Although multiple trials have shown a positive effect of CPAP on stroke recovery, relatively few investigations have looked specifically at whether the intervention prevents subsequent vascular events.
Patients’ clinical stroke outcomes were categorized in accordance with the Modified Rankin Scale (mRS), which is most widely used to assess disability and dependence outcomes among patients with stroke.
Significantly more patients who were treated with CPAP experienced an improvement in their mRS score by at least 1 point, when assessed at both 6 and 12 months following entrance into the study. Specifically, 53% (16) of patients in the CPAP group had an improvement of at least 1 point in their mRS score at 12 months, compared with 27% (11) of patients who did not use CPAP (P = .03).
“These differences are statistically significant, as well as clinically meaningful and relevant,” Dr. Gupta and her colleagues said in their report.
This finding was consistent with what researchers have seen in some earlier studies of stroke patients who used CPAP, the researchers wrote.
Additionally, CPAP-treated patients had fewer subsequent vascular events, compared with those who did not use CPAP, though the difference did not reach statistical significance. There was only one new vascular event (3.33%) in the CPAP group at 12-month follow-up, versus six events (15%) in the non-CPAP group (P = .23).
Nevertheless, the results provide more evidence for the potential benefit of CPAP in stroke patients with obstructive sleep apnea, the researchers noted.
“Our results indicate that new vascular events may be better prevented – and significantly more patients may make good stroke recovery – with CPAP treatment as compared to only best medical treatment,” Dr. Gupta and her colleagues wrote.
Before the study started, investigators determined that they would have needed 80 patients per arm for a power of 80%. A total of 679 patients were screened, but only 116 reported for polysomnography testing, and of those, 83 had at least moderate obstructive sleep apnea.
Due to a lack of CPAP devices, only 70 of those 83 patients made it all the way to randomization, investigators reported.
Dr. Gupta and her coauthors reported no conflicts of interest related to the study.
SOURCE: Gupta A et al. J Clin Sleep Med. 2018 Mar 30. pii:jc-17-00230.
the results of a randomized study suggest.
Obstructive sleep apnea is present in 50%-80% of patients with stroke, previous studies show, and its presence is associated with impaired function and cognition, delirium, and longer rehabilitation time, among other negative impacts, wrote Anupama Gupta, PhD, and her coauthors from the All India Institute of Medical Sciences, New Delhi, in the Journal of Clinical Sleep Medicine. Although multiple trials have shown a positive effect of CPAP on stroke recovery, relatively few investigations have looked specifically at whether the intervention prevents subsequent vascular events.
Patients’ clinical stroke outcomes were categorized in accordance with the Modified Rankin Scale (mRS), which is most widely used to assess disability and dependence outcomes among patients with stroke.
Significantly more patients who were treated with CPAP experienced an improvement in their mRS score by at least 1 point, when assessed at both 6 and 12 months following entrance into the study. Specifically, 53% (16) of patients in the CPAP group had an improvement of at least 1 point in their mRS score at 12 months, compared with 27% (11) of patients who did not use CPAP (P = .03).
“These differences are statistically significant, as well as clinically meaningful and relevant,” Dr. Gupta and her colleagues said in their report.
This finding was consistent with what researchers have seen in some earlier studies of stroke patients who used CPAP, the researchers wrote.
Additionally, CPAP-treated patients had fewer subsequent vascular events, compared with those who did not use CPAP, though the difference did not reach statistical significance. There was only one new vascular event (3.33%) in the CPAP group at 12-month follow-up, versus six events (15%) in the non-CPAP group (P = .23).
Nevertheless, the results provide more evidence for the potential benefit of CPAP in stroke patients with obstructive sleep apnea, the researchers noted.
“Our results indicate that new vascular events may be better prevented – and significantly more patients may make good stroke recovery – with CPAP treatment as compared to only best medical treatment,” Dr. Gupta and her colleagues wrote.
Before the study started, investigators determined that they would have needed 80 patients per arm for a power of 80%. A total of 679 patients were screened, but only 116 reported for polysomnography testing, and of those, 83 had at least moderate obstructive sleep apnea.
Due to a lack of CPAP devices, only 70 of those 83 patients made it all the way to randomization, investigators reported.
Dr. Gupta and her coauthors reported no conflicts of interest related to the study.
SOURCE: Gupta A et al. J Clin Sleep Med. 2018 Mar 30. pii:jc-17-00230.
FROM THE JOURNAL OF CLINICAL SLEEP MEDICINE
Key clinical point: Continuous positive airway pressure (CPAP) treatment may prevent vascular events in patients with stroke who have OSA.
Major finding: There was one vascular event (3.33%) at 12 months for CPAP-treated patients, versus six events (15%) in non-CPAP patients, though the difference was not significant (P = .23).
Study details: A single-blind, randomized, controlled trial including 70 patients with imaging-confirmed first arterial stroke and OSA.
Disclosures: The authors reported no conflicts of interest related to the study.
Source: Gupta A et al. J Clin Sleep Med. 2018 Mar 30. pii:jc-17-00230.
Adjunct treatments assist with persistent asthma
Asthma patients who struggle with poor control despite using inhaled corticosteroids can benefit from additional treatment with long-acting muscarinic antagonists (LAMAs) or single maintenance and reliever therapy, suggest data from a pair of systematic reviews and meta-analyses.
Asthma control remains a problem for many patients despite the daily use of inhaled corticosteroids. The current preferred adjunct therapy for patients aged 12 years and older is long-acting beta-agonists (LABAs), wrote Diana M. Sobieraj, PharmD, of the University of Connecticut School of Pharmacy, Storrs, and her colleagues in a study published in JAMA. The researchers examined the efficacy of other adjunct therapies and therapeutic regimes, including the use of a LAMA, in two studies of patients with persistent asthma.
Overall, patients who took a LAMA had a lower risk of asthma exacerbation requiring systemic corticosteroids and improved spirometry measures than did the patients who took a placebo or used another controller as an adjunct therapy.
In trials that compared LAMAs with placebo as an add-on to inhaled corticosteroids, LAMA patients experienced a significantly reduced risk of exacerbation requiring systemic corticosteroids (–1.8) and a significantly reduced risk of asthma worsening (–4.8). Another benefit seen in the patients who used a LAMA rather than those who used a placebo was improved spirometry measures, but the differences between these two patient groups’ numbers did not reach statistical significance.
The analysis also included studies that compared “triple therapy” – defined as use of a LAMA as add-on therapy to inhaled corticosteroids and LABAs – to LABA plus use of inhaled corticosteroids.
Triple therapy was significantly associated with a lower risk of asthma worsening, compared with inhaled corticosteroids and LABAs, but not with a reduced risk of exacerbation. In addition, no significant differences appeared in Asthma Control Questionnaire-7 scores or overall Asthma Quality of Life Questionnaire scores between the two patient groups.
“Triple therapy was not significantly associated with improvements in rescue medication use vs. combined inhaled corticosteroids and LABA therapy,” the researchers added.
The review of LAMAs used as add-on therapy was limited by several factors, including a primary focus on tiotropium, a lack of analysis of harms or the costs of the various therapies, the lack of data for children, and an inability to perform a subgroup analysis, the researchers said. Although LAMA use was associated with a lower risk of asthma exacerbation, compared with placebo use, the review could not adequately compare LAMA with controllers other than LABA, they added.
In the second analysis, which also was published in JAMA, the researchers evaluated the use of inhaled corticosteroids and LABAs as both a controller and quick-relief treatment, a strategy known as SMART, or Single Maintenance and Reliever Therapy. The SMART protocol, which is not approved in the United States, involved taking a combination of the corticosteroid budesonide and the LABA formoterol in a dry-powder inhaler in most of the studies reviewed.
Overall, in the analysis of 22,524 patients aged 12 years and older, an absolute risk difference of –2.8% for asthma exacerbations was seen in those who used the SMART protocol versus those who used a higher dose of inhaled corticosteroids and inhaled LABA as controller therapy.
In addition, data from 341 children aged 4-11 years showed a –12% absolute difference in risk of asthma exacerbation with the SMART therapy.
In trials that compared patients using the SMART protocol with those taking only the dose of inhaled corticosteroids called for by SMART, the protocol was associated with an improvement in forced expiratory volume in 1 second (FEV1) and a reduction in the need for rescue medication.
The SMART protocol also demonstrated advantages over taking the same dose of inhaled corticosteroid called for by SMART plus a LABA controller therapy or a higher dose of inhaled corticosteroids with a LABA controller therapy. Specifically, SMART patients experienced a –6.4% risk of asthma exacerbations, versus the first comparator group; and a –2.7% risk of asthma, compared with the group who took a higher dose of inhaled corticosteroids with LABA controller therapy.
No significant associations appeared in any of the studies between the SMART protocol and outcomes that included all-cause mortality or changes in FEV1, forced vital capacity, or the percentage of predicted FEV1, when compared with those for patients who used a LABA controller therapy plus inhaled corticosteroids at either dose.
The SMART protocol review was limited by factors that included a lack of data on adverse events, a lack of subgroup analysis, and the potential for bias, because of the open label nature of some of the studies, the researchers noted.
However, despite the limitations in both reviews, the results support the SMART strategy and LAMAs as alternatives for patients with persistent asthma, and highlight the need for further research, they noted.
The reviews were supported by the Agency for Healthcare Research and Quality. Dr. Sobieraj had no financial conflicts to disclose.
SOURCE: Sobieraj D et al. JAMA. 2018;319(14):1473-84. Sobieraj D et al. JAMA. 2018;319(14):1485-96.
Asthma remains a major public health problem in the United States, but 11 years have passed since the last update to treatment guidelines, and an update to the current guidelines for asthma treatment is needed, wrote Jerry A. Krishnan, MD, and David H. Au, MD, in an accompanying editorial (JAMA. 2018;319[14]:1441-3). “It is time to connect the efforts of the FDA, the evidence presented by Sobieraj et al., and the support from the National Education and Prevention Program to update the 2007 [Expert Panel Report 3] guidelines on asthma.”
Both reviews showed effectiveness for the treatments being assessed, compared with placebo, but each had limitations, the editorialists noted.
The study findings in the report on the efficacy of inhaled long-acting muscarinic antagonists (LAMAs) in adolescents and adults with uncontrolled asthma were limited by several factors including a focus primarily on tiotropium, absence of data on potential harms and relative costs of treatment, and a lack of data on children younger than 12 years, they noted. The findings in the analysis of the strategy known as Single Maintenance and Reliever Therapy (SMART) containing formoterol, a long-acting beta2-agonist, were similarly limited by a lack of assessment of potential harm and a data on children within the same age group, they said.
However, the effectiveness of the treatments seen in both reviews suggest that the forthcoming revision of the Expert Panel Report 3 guidelines on asthma from the National Asthma Education and Prevention Program should include the option for inhaled tiotropium, a LAMA, and for the formoterol-based SMART protocol, the editorialists wrote.
“For patients and clinicians, the results from these meta-analyses suggest that dual therapy with scheduled doses of inhaled corticosteroids and LABA or inhaled corticosteroids and LAMA should help reduce the risk of future asthma exacerbations in patients with inadequate asthma control while using inhaled corticosteroids alone,” they said. The new guidelines should include evidence for the SMART therapy as well, but “studies assessing the efficacy of SMART using combination formoterol and budesonide via a metered-dose inhaler are needed,” they concluded.
Dr. Krishnan is affiliated with the division of pulmonary, critical care, sleep, and allergy at the University of Illinois, Chicago, and disclosed having received compensation from Sanofi for participation on an independent data-monitoring committee. Dr. Au is affiliated with the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle, and disclosed having received compensation from Novartis for participation on a data-monitoring committee and for serving as a consultant to Gilead Sciences.
Asthma remains a major public health problem in the United States, but 11 years have passed since the last update to treatment guidelines, and an update to the current guidelines for asthma treatment is needed, wrote Jerry A. Krishnan, MD, and David H. Au, MD, in an accompanying editorial (JAMA. 2018;319[14]:1441-3). “It is time to connect the efforts of the FDA, the evidence presented by Sobieraj et al., and the support from the National Education and Prevention Program to update the 2007 [Expert Panel Report 3] guidelines on asthma.”
Both reviews showed effectiveness for the treatments being assessed, compared with placebo, but each had limitations, the editorialists noted.
The study findings in the report on the efficacy of inhaled long-acting muscarinic antagonists (LAMAs) in adolescents and adults with uncontrolled asthma were limited by several factors including a focus primarily on tiotropium, absence of data on potential harms and relative costs of treatment, and a lack of data on children younger than 12 years, they noted. The findings in the analysis of the strategy known as Single Maintenance and Reliever Therapy (SMART) containing formoterol, a long-acting beta2-agonist, were similarly limited by a lack of assessment of potential harm and a data on children within the same age group, they said.
However, the effectiveness of the treatments seen in both reviews suggest that the forthcoming revision of the Expert Panel Report 3 guidelines on asthma from the National Asthma Education and Prevention Program should include the option for inhaled tiotropium, a LAMA, and for the formoterol-based SMART protocol, the editorialists wrote.
“For patients and clinicians, the results from these meta-analyses suggest that dual therapy with scheduled doses of inhaled corticosteroids and LABA or inhaled corticosteroids and LAMA should help reduce the risk of future asthma exacerbations in patients with inadequate asthma control while using inhaled corticosteroids alone,” they said. The new guidelines should include evidence for the SMART therapy as well, but “studies assessing the efficacy of SMART using combination formoterol and budesonide via a metered-dose inhaler are needed,” they concluded.
Dr. Krishnan is affiliated with the division of pulmonary, critical care, sleep, and allergy at the University of Illinois, Chicago, and disclosed having received compensation from Sanofi for participation on an independent data-monitoring committee. Dr. Au is affiliated with the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle, and disclosed having received compensation from Novartis for participation on a data-monitoring committee and for serving as a consultant to Gilead Sciences.
Asthma remains a major public health problem in the United States, but 11 years have passed since the last update to treatment guidelines, and an update to the current guidelines for asthma treatment is needed, wrote Jerry A. Krishnan, MD, and David H. Au, MD, in an accompanying editorial (JAMA. 2018;319[14]:1441-3). “It is time to connect the efforts of the FDA, the evidence presented by Sobieraj et al., and the support from the National Education and Prevention Program to update the 2007 [Expert Panel Report 3] guidelines on asthma.”
Both reviews showed effectiveness for the treatments being assessed, compared with placebo, but each had limitations, the editorialists noted.
The study findings in the report on the efficacy of inhaled long-acting muscarinic antagonists (LAMAs) in adolescents and adults with uncontrolled asthma were limited by several factors including a focus primarily on tiotropium, absence of data on potential harms and relative costs of treatment, and a lack of data on children younger than 12 years, they noted. The findings in the analysis of the strategy known as Single Maintenance and Reliever Therapy (SMART) containing formoterol, a long-acting beta2-agonist, were similarly limited by a lack of assessment of potential harm and a data on children within the same age group, they said.
However, the effectiveness of the treatments seen in both reviews suggest that the forthcoming revision of the Expert Panel Report 3 guidelines on asthma from the National Asthma Education and Prevention Program should include the option for inhaled tiotropium, a LAMA, and for the formoterol-based SMART protocol, the editorialists wrote.
“For patients and clinicians, the results from these meta-analyses suggest that dual therapy with scheduled doses of inhaled corticosteroids and LABA or inhaled corticosteroids and LAMA should help reduce the risk of future asthma exacerbations in patients with inadequate asthma control while using inhaled corticosteroids alone,” they said. The new guidelines should include evidence for the SMART therapy as well, but “studies assessing the efficacy of SMART using combination formoterol and budesonide via a metered-dose inhaler are needed,” they concluded.
Dr. Krishnan is affiliated with the division of pulmonary, critical care, sleep, and allergy at the University of Illinois, Chicago, and disclosed having received compensation from Sanofi for participation on an independent data-monitoring committee. Dr. Au is affiliated with the division of pulmonary, critical care, and sleep medicine at the University of Washington, Seattle, and disclosed having received compensation from Novartis for participation on a data-monitoring committee and for serving as a consultant to Gilead Sciences.
Asthma patients who struggle with poor control despite using inhaled corticosteroids can benefit from additional treatment with long-acting muscarinic antagonists (LAMAs) or single maintenance and reliever therapy, suggest data from a pair of systematic reviews and meta-analyses.
Asthma control remains a problem for many patients despite the daily use of inhaled corticosteroids. The current preferred adjunct therapy for patients aged 12 years and older is long-acting beta-agonists (LABAs), wrote Diana M. Sobieraj, PharmD, of the University of Connecticut School of Pharmacy, Storrs, and her colleagues in a study published in JAMA. The researchers examined the efficacy of other adjunct therapies and therapeutic regimes, including the use of a LAMA, in two studies of patients with persistent asthma.
Overall, patients who took a LAMA had a lower risk of asthma exacerbation requiring systemic corticosteroids and improved spirometry measures than did the patients who took a placebo or used another controller as an adjunct therapy.
In trials that compared LAMAs with placebo as an add-on to inhaled corticosteroids, LAMA patients experienced a significantly reduced risk of exacerbation requiring systemic corticosteroids (–1.8) and a significantly reduced risk of asthma worsening (–4.8). Another benefit seen in the patients who used a LAMA rather than those who used a placebo was improved spirometry measures, but the differences between these two patient groups’ numbers did not reach statistical significance.
The analysis also included studies that compared “triple therapy” – defined as use of a LAMA as add-on therapy to inhaled corticosteroids and LABAs – to LABA plus use of inhaled corticosteroids.
Triple therapy was significantly associated with a lower risk of asthma worsening, compared with inhaled corticosteroids and LABAs, but not with a reduced risk of exacerbation. In addition, no significant differences appeared in Asthma Control Questionnaire-7 scores or overall Asthma Quality of Life Questionnaire scores between the two patient groups.
“Triple therapy was not significantly associated with improvements in rescue medication use vs. combined inhaled corticosteroids and LABA therapy,” the researchers added.
The review of LAMAs used as add-on therapy was limited by several factors, including a primary focus on tiotropium, a lack of analysis of harms or the costs of the various therapies, the lack of data for children, and an inability to perform a subgroup analysis, the researchers said. Although LAMA use was associated with a lower risk of asthma exacerbation, compared with placebo use, the review could not adequately compare LAMA with controllers other than LABA, they added.
In the second analysis, which also was published in JAMA, the researchers evaluated the use of inhaled corticosteroids and LABAs as both a controller and quick-relief treatment, a strategy known as SMART, or Single Maintenance and Reliever Therapy. The SMART protocol, which is not approved in the United States, involved taking a combination of the corticosteroid budesonide and the LABA formoterol in a dry-powder inhaler in most of the studies reviewed.
Overall, in the analysis of 22,524 patients aged 12 years and older, an absolute risk difference of –2.8% for asthma exacerbations was seen in those who used the SMART protocol versus those who used a higher dose of inhaled corticosteroids and inhaled LABA as controller therapy.
In addition, data from 341 children aged 4-11 years showed a –12% absolute difference in risk of asthma exacerbation with the SMART therapy.
In trials that compared patients using the SMART protocol with those taking only the dose of inhaled corticosteroids called for by SMART, the protocol was associated with an improvement in forced expiratory volume in 1 second (FEV1) and a reduction in the need for rescue medication.
The SMART protocol also demonstrated advantages over taking the same dose of inhaled corticosteroid called for by SMART plus a LABA controller therapy or a higher dose of inhaled corticosteroids with a LABA controller therapy. Specifically, SMART patients experienced a –6.4% risk of asthma exacerbations, versus the first comparator group; and a –2.7% risk of asthma, compared with the group who took a higher dose of inhaled corticosteroids with LABA controller therapy.
No significant associations appeared in any of the studies between the SMART protocol and outcomes that included all-cause mortality or changes in FEV1, forced vital capacity, or the percentage of predicted FEV1, when compared with those for patients who used a LABA controller therapy plus inhaled corticosteroids at either dose.
The SMART protocol review was limited by factors that included a lack of data on adverse events, a lack of subgroup analysis, and the potential for bias, because of the open label nature of some of the studies, the researchers noted.
However, despite the limitations in both reviews, the results support the SMART strategy and LAMAs as alternatives for patients with persistent asthma, and highlight the need for further research, they noted.
The reviews were supported by the Agency for Healthcare Research and Quality. Dr. Sobieraj had no financial conflicts to disclose.
SOURCE: Sobieraj D et al. JAMA. 2018;319(14):1473-84. Sobieraj D et al. JAMA. 2018;319(14):1485-96.
Asthma patients who struggle with poor control despite using inhaled corticosteroids can benefit from additional treatment with long-acting muscarinic antagonists (LAMAs) or single maintenance and reliever therapy, suggest data from a pair of systematic reviews and meta-analyses.
Asthma control remains a problem for many patients despite the daily use of inhaled corticosteroids. The current preferred adjunct therapy for patients aged 12 years and older is long-acting beta-agonists (LABAs), wrote Diana M. Sobieraj, PharmD, of the University of Connecticut School of Pharmacy, Storrs, and her colleagues in a study published in JAMA. The researchers examined the efficacy of other adjunct therapies and therapeutic regimes, including the use of a LAMA, in two studies of patients with persistent asthma.
Overall, patients who took a LAMA had a lower risk of asthma exacerbation requiring systemic corticosteroids and improved spirometry measures than did the patients who took a placebo or used another controller as an adjunct therapy.
In trials that compared LAMAs with placebo as an add-on to inhaled corticosteroids, LAMA patients experienced a significantly reduced risk of exacerbation requiring systemic corticosteroids (–1.8) and a significantly reduced risk of asthma worsening (–4.8). Another benefit seen in the patients who used a LAMA rather than those who used a placebo was improved spirometry measures, but the differences between these two patient groups’ numbers did not reach statistical significance.
The analysis also included studies that compared “triple therapy” – defined as use of a LAMA as add-on therapy to inhaled corticosteroids and LABAs – to LABA plus use of inhaled corticosteroids.
Triple therapy was significantly associated with a lower risk of asthma worsening, compared with inhaled corticosteroids and LABAs, but not with a reduced risk of exacerbation. In addition, no significant differences appeared in Asthma Control Questionnaire-7 scores or overall Asthma Quality of Life Questionnaire scores between the two patient groups.
“Triple therapy was not significantly associated with improvements in rescue medication use vs. combined inhaled corticosteroids and LABA therapy,” the researchers added.
The review of LAMAs used as add-on therapy was limited by several factors, including a primary focus on tiotropium, a lack of analysis of harms or the costs of the various therapies, the lack of data for children, and an inability to perform a subgroup analysis, the researchers said. Although LAMA use was associated with a lower risk of asthma exacerbation, compared with placebo use, the review could not adequately compare LAMA with controllers other than LABA, they added.
In the second analysis, which also was published in JAMA, the researchers evaluated the use of inhaled corticosteroids and LABAs as both a controller and quick-relief treatment, a strategy known as SMART, or Single Maintenance and Reliever Therapy. The SMART protocol, which is not approved in the United States, involved taking a combination of the corticosteroid budesonide and the LABA formoterol in a dry-powder inhaler in most of the studies reviewed.
Overall, in the analysis of 22,524 patients aged 12 years and older, an absolute risk difference of –2.8% for asthma exacerbations was seen in those who used the SMART protocol versus those who used a higher dose of inhaled corticosteroids and inhaled LABA as controller therapy.
In addition, data from 341 children aged 4-11 years showed a –12% absolute difference in risk of asthma exacerbation with the SMART therapy.
In trials that compared patients using the SMART protocol with those taking only the dose of inhaled corticosteroids called for by SMART, the protocol was associated with an improvement in forced expiratory volume in 1 second (FEV1) and a reduction in the need for rescue medication.
The SMART protocol also demonstrated advantages over taking the same dose of inhaled corticosteroid called for by SMART plus a LABA controller therapy or a higher dose of inhaled corticosteroids with a LABA controller therapy. Specifically, SMART patients experienced a –6.4% risk of asthma exacerbations, versus the first comparator group; and a –2.7% risk of asthma, compared with the group who took a higher dose of inhaled corticosteroids with LABA controller therapy.
No significant associations appeared in any of the studies between the SMART protocol and outcomes that included all-cause mortality or changes in FEV1, forced vital capacity, or the percentage of predicted FEV1, when compared with those for patients who used a LABA controller therapy plus inhaled corticosteroids at either dose.
The SMART protocol review was limited by factors that included a lack of data on adverse events, a lack of subgroup analysis, and the potential for bias, because of the open label nature of some of the studies, the researchers noted.
However, despite the limitations in both reviews, the results support the SMART strategy and LAMAs as alternatives for patients with persistent asthma, and highlight the need for further research, they noted.
The reviews were supported by the Agency for Healthcare Research and Quality. Dr. Sobieraj had no financial conflicts to disclose.
SOURCE: Sobieraj D et al. JAMA. 2018;319(14):1473-84. Sobieraj D et al. JAMA. 2018;319(14):1485-96.
FROM JAMA
Key clinical point:
Major finding: LAMA patients had a risk difference of –1.8 for asthma exacerbations, compared with placebo users, while patients using the SMART protocol had an absolute risk difference of –2.8%, compared with those taking a higher dose of inhaled corticosteroids plus LABAs as a controller.
Study details: The data came from two reviews of randomized clinical trials; the first included 7,122 patients and the second included 22,524 patients.
Disclosures: The reviews were supported in part by the Agency for Healthcare Research and Quality. Dr. Sobieraj had no financial conflicts to disclose.
Source: Sobieraj D et al. JAMA. 2018;319(14):1473-84. Sobieraj D et al. JAMA. 2018;319(14):1485-96.
VTE risk after bariatric surgery should be assessed
SEATTLE – Preop thromboelastometry can identify patients who need extra according to a prospective investigation of 40 patients at Conemaugh Memorial Medical Center in Johnstown, Pa.
Enoxaparin 40 mg twice daily just wasn’t enough for people who were hypercoagulable before surgery. The goal of the study was to find the best way to prevent venous thromboembolism (VTE) after weight loss surgery. At present, there’s no consensus on prophylaxis dosing, timing, duration, or even what agent to use for these patients. Conemaugh uses postop enoxaparin, a low-molecular-weight heparin. Among many other options, some hospitals opt for preop dosing with traditional heparin, which is less expensive.
The Conemaugh team turned to thromboelastometry (TEM) to look at the question of VTE risk in bariatric surgery patients. The test gauges coagulation status by measuring elasticity as a small blood sample clots over a few minutes. The investigators found that patients who were hypercoagulable before surgery were likely to be hypercoagulable afterwards. The finding argues for baseline TEM testing to guide postop anticoagulation.
The problem is that bariatric services don’t often have access to TEM equipment, and insurance doesn’t cover the $60 test. In this instance, the Lake Erie College of Osteopathic Medicine in Erie, Pa., had the equipment and covered the testing for the study.
The patients had TEM at baseline and then received 40 mg of enoxaparin about 4 hours after surgery – mostly laparoscopic gastric bypasses – and a second dose about 12 hours after the first. TEM was repeated about 2 hours after the second dose.
At baseline, 2 (5%) of the patients were hypocoagulable, 15 (37.5%) were normal, and 23 (57.5%) were hypercoagulable. On postop TEM, 17 patients (42.5%) were normal and 23 (57.5%) were hypercoagulable: “These 23 were inadequately anticoagulated,” said lead investigator Daniel Urias, MD, a general surgery resident at the medical center.
“There was an association between being normal at baseline and being normal postop, and being hypercoagulable at baseline and hypercoagulable postop. We didn’t anticipate finding such similarity between the numbers. Our suspicion that baseline status plays a major role is holding true,” Dr. Urias said at the World Congress of Endoscopic Surgery hosted by SAGES & CAGS.
When patients test hypercoagulable at baseline, “we are [now] leaning towards [enoxaparin] 60 mg twice daily,” he said.
Ultimately, anticoagulation TEM could be used to titrate patients into the normal range. For best outcomes, it’s likely that “obese patients require goal-directed therapy instead of weight-based or fixed dosing,” he said, but nothing is going to happen until insurance steps up.
The patients did not have underlying coagulopathies, and 33 (82.5%) were women; the average age was 44 years and average body mass index was 43.6 kg/m2. The mean preop Caprini score was 4, indicating moderate VTE risk. Surgery lasted about 200 minutes. Patients were out of bed and walking on postop day 0.
The investigators had no relevant disclosures.
SOURCE: Urias D et al. World Congress of Endoscopic Surgery hosted by SAGES & CAGS abstract S023.
SEATTLE – Preop thromboelastometry can identify patients who need extra according to a prospective investigation of 40 patients at Conemaugh Memorial Medical Center in Johnstown, Pa.
Enoxaparin 40 mg twice daily just wasn’t enough for people who were hypercoagulable before surgery. The goal of the study was to find the best way to prevent venous thromboembolism (VTE) after weight loss surgery. At present, there’s no consensus on prophylaxis dosing, timing, duration, or even what agent to use for these patients. Conemaugh uses postop enoxaparin, a low-molecular-weight heparin. Among many other options, some hospitals opt for preop dosing with traditional heparin, which is less expensive.
The Conemaugh team turned to thromboelastometry (TEM) to look at the question of VTE risk in bariatric surgery patients. The test gauges coagulation status by measuring elasticity as a small blood sample clots over a few minutes. The investigators found that patients who were hypercoagulable before surgery were likely to be hypercoagulable afterwards. The finding argues for baseline TEM testing to guide postop anticoagulation.
The problem is that bariatric services don’t often have access to TEM equipment, and insurance doesn’t cover the $60 test. In this instance, the Lake Erie College of Osteopathic Medicine in Erie, Pa., had the equipment and covered the testing for the study.
The patients had TEM at baseline and then received 40 mg of enoxaparin about 4 hours after surgery – mostly laparoscopic gastric bypasses – and a second dose about 12 hours after the first. TEM was repeated about 2 hours after the second dose.
At baseline, 2 (5%) of the patients were hypocoagulable, 15 (37.5%) were normal, and 23 (57.5%) were hypercoagulable. On postop TEM, 17 patients (42.5%) were normal and 23 (57.5%) were hypercoagulable: “These 23 were inadequately anticoagulated,” said lead investigator Daniel Urias, MD, a general surgery resident at the medical center.
“There was an association between being normal at baseline and being normal postop, and being hypercoagulable at baseline and hypercoagulable postop. We didn’t anticipate finding such similarity between the numbers. Our suspicion that baseline status plays a major role is holding true,” Dr. Urias said at the World Congress of Endoscopic Surgery hosted by SAGES & CAGS.
When patients test hypercoagulable at baseline, “we are [now] leaning towards [enoxaparin] 60 mg twice daily,” he said.
Ultimately, anticoagulation TEM could be used to titrate patients into the normal range. For best outcomes, it’s likely that “obese patients require goal-directed therapy instead of weight-based or fixed dosing,” he said, but nothing is going to happen until insurance steps up.
The patients did not have underlying coagulopathies, and 33 (82.5%) were women; the average age was 44 years and average body mass index was 43.6 kg/m2. The mean preop Caprini score was 4, indicating moderate VTE risk. Surgery lasted about 200 minutes. Patients were out of bed and walking on postop day 0.
The investigators had no relevant disclosures.
SOURCE: Urias D et al. World Congress of Endoscopic Surgery hosted by SAGES & CAGS abstract S023.
SEATTLE – Preop thromboelastometry can identify patients who need extra according to a prospective investigation of 40 patients at Conemaugh Memorial Medical Center in Johnstown, Pa.
Enoxaparin 40 mg twice daily just wasn’t enough for people who were hypercoagulable before surgery. The goal of the study was to find the best way to prevent venous thromboembolism (VTE) after weight loss surgery. At present, there’s no consensus on prophylaxis dosing, timing, duration, or even what agent to use for these patients. Conemaugh uses postop enoxaparin, a low-molecular-weight heparin. Among many other options, some hospitals opt for preop dosing with traditional heparin, which is less expensive.
The Conemaugh team turned to thromboelastometry (TEM) to look at the question of VTE risk in bariatric surgery patients. The test gauges coagulation status by measuring elasticity as a small blood sample clots over a few minutes. The investigators found that patients who were hypercoagulable before surgery were likely to be hypercoagulable afterwards. The finding argues for baseline TEM testing to guide postop anticoagulation.
The problem is that bariatric services don’t often have access to TEM equipment, and insurance doesn’t cover the $60 test. In this instance, the Lake Erie College of Osteopathic Medicine in Erie, Pa., had the equipment and covered the testing for the study.
The patients had TEM at baseline and then received 40 mg of enoxaparin about 4 hours after surgery – mostly laparoscopic gastric bypasses – and a second dose about 12 hours after the first. TEM was repeated about 2 hours after the second dose.
At baseline, 2 (5%) of the patients were hypocoagulable, 15 (37.5%) were normal, and 23 (57.5%) were hypercoagulable. On postop TEM, 17 patients (42.5%) were normal and 23 (57.5%) were hypercoagulable: “These 23 were inadequately anticoagulated,” said lead investigator Daniel Urias, MD, a general surgery resident at the medical center.
“There was an association between being normal at baseline and being normal postop, and being hypercoagulable at baseline and hypercoagulable postop. We didn’t anticipate finding such similarity between the numbers. Our suspicion that baseline status plays a major role is holding true,” Dr. Urias said at the World Congress of Endoscopic Surgery hosted by SAGES & CAGS.
When patients test hypercoagulable at baseline, “we are [now] leaning towards [enoxaparin] 60 mg twice daily,” he said.
Ultimately, anticoagulation TEM could be used to titrate patients into the normal range. For best outcomes, it’s likely that “obese patients require goal-directed therapy instead of weight-based or fixed dosing,” he said, but nothing is going to happen until insurance steps up.
The patients did not have underlying coagulopathies, and 33 (82.5%) were women; the average age was 44 years and average body mass index was 43.6 kg/m2. The mean preop Caprini score was 4, indicating moderate VTE risk. Surgery lasted about 200 minutes. Patients were out of bed and walking on postop day 0.
The investigators had no relevant disclosures.
SOURCE: Urias D et al. World Congress of Endoscopic Surgery hosted by SAGES & CAGS abstract S023.
REPORTING FROM SAGES 2018
Key clinical point: Preoperative thromboelastometry identifies patients who need extra anticoagulation against venous thromboembolism following bariatric surgery.
Major finding: Baseline and postop coagulation were similar: 37.5% vs. 42.5% were normal and 57.5% vs 57.5% were hypercoagulable.
Study details: Prospective study of 40 bariatric surgery patients.
Disclosures: The investigators did not have any relevant disclosures. The Lake Erie College of Osteopathic Medicine paid for the testing.
Source: Urias D et al. World Congress of Endoscopic Surgery hosted by SAGES & CAGS abstract S023.
Triple-therapy cuts COPD exacerbations
Triple therapy for chronic obstructive pulmonary disease (COPD) achieved reductions in moderate to severe exacerbations when compared with two kinds of dual therapy, in a study published online in the New England Journal of Medicine.
The trial compared the outcomes of COPD patients using an inhaled therapy comprising a corticosteroid, a long-acting muscarinic antagonist (LAMA), and a long-acting beta2-agonist (LABA) with the outcomes of similar patients taking one of two other therapy combinations – a corticosteroid and a LABA, or a LABA and a LAMA. This trial – Informing the Pathway of COPD Treatment (IMPACT) – included 10,355 patients with symptomatic COPD in 37 countries, according to David A. Lipson, MD, and his colleagues (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901).
The study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate (a corticosteroid), 62.5 mcg of the LAMA umeclidinium and 25 mcg of the LABA vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction compared with vilanterol-umeclidinium (P less than .001 for both).
When the analysis was limited to severe exacerbations alone, the difference was significant only between the triple therapy, which GSK is marketing as Trelegy Ellipta, and the vilanterol-umeclidinium dual therapy.
Dr. Lipson, of GSK and the University of Pennsylvania, and his coauthors noted that their finding of a greater benefit with the glucocorticoid-containing dual-therapy compared with the LABA-LAMA vilanterol-umeclidinium combination contradicted the findings of the earlier FLAME trial. This was likely due to differences in patient populations and design, as all patients in the FLAME trial had a 1-month run-in treatment with the bronchodilator tiotropium, the researchers explained.
“Therefore any patients who would require an inhaled glucocorticoid may have had an increase in exacerbations and a decrease in lung function during the run-in period and would have been forced to leave the trial,” they wrote.
Patients with higher eosinophil levels seemed to do even better with triple therapy. In those with eosinophil levels of 150 cells per microliter or above, the annual rate of moderate to severe exacerbations was 0.95 with triple therapy, 1.08 with fluticasone furoate–vilanterol, and 1.39 with vilanterol-umeclidinium.
Triple therapy also was associated with a significantly longer time to first event and greater improvements in quality of life, compared with the dual therapies.
Overall, the adverse event profile of triple therapy was similar to that of dual therapy. Contrasting that finding were differences in the incidences of physician-diagnosed pneumonia between the treatment groups. Physician-diagnosed pneumonia was 53% higher among patients who received fluticasone furoate – either in dual or triple therapy combinations. Eight percent of patients in the triple therapy group experienced pneumonia, compared with 7% of patients in the fluticasone furoate–vilanterol group and 5% in the vilanterol-umeclidinium group.
All-cause mortality was significantly lower in patients who received the inhaled glucocorticoid, although the authors said this finding was “fragile” and needed further investigation.
The rate of discontinuation or withdrawal from the trial was 6% for the triple therapy group, 8% for the fluticasone furoate–vilanterol group, and 9% for the vilanterol-umeclidinium group. The rates of serious adverse events in each group were 22%, 21%, and 23%, respectively.
At trial entry, 38% of patients were already receiving triple therapy and 29% were taking an inhaled glucocorticoid. The authors noted that any patients taking an inhaled glucocorticoid who were randomized to the vilanterol-umeclidinium group would have had to abruptly stop taking their inhaled glucocorticoids.
“It is unknown whether the abrupt discontinuation of inhaled glucocorticoids would have contributed to our finding of a lower rate of exacerbations in the inhaled glucocorticoid groups than in the LAMA-LABA group,” they wrote.
Fernando Martinez, MD, chief of the division of pulmonary and critical care medicine at New York–Presbyterian Hospital/Weill Cornell Medical Center, said the study advanced the understanding of COPD management by addressing some key evidence gaps, in a statement issued by GSK.
“By comparing various combinations of effective medications in the same device the study clarifies which type of patient gains greatest benefit from each class of medicine,” Dr Martinez said in the statement. “As many patients experience frequent exacerbations or ‘flare ups,’ which can often result in hospitalization, these data will be highly relevant to patients and clinicians as they consider the optimal treatment.”
The study was funded by GSK, which manufactures Trelegy Ellipta triple therapy for COPD. Eight authors were employees of GSK and two were on advisory boards for the company. Seven authors declared funding from a range of pharmaceutical companies including GSK. One author had no conflicts of interest to declare.
SOURCE: Lipson D et al. N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901.
The data from the IMPACT study fills a gap in the evidence supporting a step-up from dual to triple inhaled therapy for COPD, which so far has been recommended only for patients with severe loss of lung function and those with frequent exacerbations despite maximum bronchodilator treatment. The study has the strengths of comparing the step-up to triple therapy with the GOLD guideline–recommended dual therapies and using the same dosages in the triple therapy as in the dual therapy
However, it is important to note that nearly 40% of patients enrolled in the trial were already being treated with triple therapy, 70% were receiving a glucocorticoid, and patients with a history of asthma were not excluded. This means patients assigned to the dual therapy without glucocorticoids would have had an abrupt cessation of their glucocorticoid therapy, which may explain a rapid surge in exacerbations in the first month and the lower rate of exacerbations in the dual-therapy group that did include glucocorticoids. The choice of patients for the study could potentially have artificially inflated the observed effectiveness of triple therapy over dual bronchodilator treatment.
As such, we suggest clinicians stick with the GOLD 2017 recommendations that escalation to triple therapy only occur after maximization of bronchodilator treatment.
Dr. Samy Suissa (PhD) is with the Center for Clinical Epidemiology at Lady Davis Institute–Jewish General Hospital, and the departments of epidemiology and biostatistics and medicine at McGill University, Montreal. Dr. Jeffrey M. Drazen is editor-in-chief of the New England Journal of Medicine. These comments are taken from an editorial (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMe1716802 ). Dr. Suissa declared personal fees and grants from the pharmaceutical industry outside the submitted work.
The data from the IMPACT study fills a gap in the evidence supporting a step-up from dual to triple inhaled therapy for COPD, which so far has been recommended only for patients with severe loss of lung function and those with frequent exacerbations despite maximum bronchodilator treatment. The study has the strengths of comparing the step-up to triple therapy with the GOLD guideline–recommended dual therapies and using the same dosages in the triple therapy as in the dual therapy
However, it is important to note that nearly 40% of patients enrolled in the trial were already being treated with triple therapy, 70% were receiving a glucocorticoid, and patients with a history of asthma were not excluded. This means patients assigned to the dual therapy without glucocorticoids would have had an abrupt cessation of their glucocorticoid therapy, which may explain a rapid surge in exacerbations in the first month and the lower rate of exacerbations in the dual-therapy group that did include glucocorticoids. The choice of patients for the study could potentially have artificially inflated the observed effectiveness of triple therapy over dual bronchodilator treatment.
As such, we suggest clinicians stick with the GOLD 2017 recommendations that escalation to triple therapy only occur after maximization of bronchodilator treatment.
Dr. Samy Suissa (PhD) is with the Center for Clinical Epidemiology at Lady Davis Institute–Jewish General Hospital, and the departments of epidemiology and biostatistics and medicine at McGill University, Montreal. Dr. Jeffrey M. Drazen is editor-in-chief of the New England Journal of Medicine. These comments are taken from an editorial (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMe1716802 ). Dr. Suissa declared personal fees and grants from the pharmaceutical industry outside the submitted work.
The data from the IMPACT study fills a gap in the evidence supporting a step-up from dual to triple inhaled therapy for COPD, which so far has been recommended only for patients with severe loss of lung function and those with frequent exacerbations despite maximum bronchodilator treatment. The study has the strengths of comparing the step-up to triple therapy with the GOLD guideline–recommended dual therapies and using the same dosages in the triple therapy as in the dual therapy
However, it is important to note that nearly 40% of patients enrolled in the trial were already being treated with triple therapy, 70% were receiving a glucocorticoid, and patients with a history of asthma were not excluded. This means patients assigned to the dual therapy without glucocorticoids would have had an abrupt cessation of their glucocorticoid therapy, which may explain a rapid surge in exacerbations in the first month and the lower rate of exacerbations in the dual-therapy group that did include glucocorticoids. The choice of patients for the study could potentially have artificially inflated the observed effectiveness of triple therapy over dual bronchodilator treatment.
As such, we suggest clinicians stick with the GOLD 2017 recommendations that escalation to triple therapy only occur after maximization of bronchodilator treatment.
Dr. Samy Suissa (PhD) is with the Center for Clinical Epidemiology at Lady Davis Institute–Jewish General Hospital, and the departments of epidemiology and biostatistics and medicine at McGill University, Montreal. Dr. Jeffrey M. Drazen is editor-in-chief of the New England Journal of Medicine. These comments are taken from an editorial (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMe1716802 ). Dr. Suissa declared personal fees and grants from the pharmaceutical industry outside the submitted work.
Triple therapy for chronic obstructive pulmonary disease (COPD) achieved reductions in moderate to severe exacerbations when compared with two kinds of dual therapy, in a study published online in the New England Journal of Medicine.
The trial compared the outcomes of COPD patients using an inhaled therapy comprising a corticosteroid, a long-acting muscarinic antagonist (LAMA), and a long-acting beta2-agonist (LABA) with the outcomes of similar patients taking one of two other therapy combinations – a corticosteroid and a LABA, or a LABA and a LAMA. This trial – Informing the Pathway of COPD Treatment (IMPACT) – included 10,355 patients with symptomatic COPD in 37 countries, according to David A. Lipson, MD, and his colleagues (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901).
The study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate (a corticosteroid), 62.5 mcg of the LAMA umeclidinium and 25 mcg of the LABA vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction compared with vilanterol-umeclidinium (P less than .001 for both).
When the analysis was limited to severe exacerbations alone, the difference was significant only between the triple therapy, which GSK is marketing as Trelegy Ellipta, and the vilanterol-umeclidinium dual therapy.
Dr. Lipson, of GSK and the University of Pennsylvania, and his coauthors noted that their finding of a greater benefit with the glucocorticoid-containing dual-therapy compared with the LABA-LAMA vilanterol-umeclidinium combination contradicted the findings of the earlier FLAME trial. This was likely due to differences in patient populations and design, as all patients in the FLAME trial had a 1-month run-in treatment with the bronchodilator tiotropium, the researchers explained.
“Therefore any patients who would require an inhaled glucocorticoid may have had an increase in exacerbations and a decrease in lung function during the run-in period and would have been forced to leave the trial,” they wrote.
Patients with higher eosinophil levels seemed to do even better with triple therapy. In those with eosinophil levels of 150 cells per microliter or above, the annual rate of moderate to severe exacerbations was 0.95 with triple therapy, 1.08 with fluticasone furoate–vilanterol, and 1.39 with vilanterol-umeclidinium.
Triple therapy also was associated with a significantly longer time to first event and greater improvements in quality of life, compared with the dual therapies.
Overall, the adverse event profile of triple therapy was similar to that of dual therapy. Contrasting that finding were differences in the incidences of physician-diagnosed pneumonia between the treatment groups. Physician-diagnosed pneumonia was 53% higher among patients who received fluticasone furoate – either in dual or triple therapy combinations. Eight percent of patients in the triple therapy group experienced pneumonia, compared with 7% of patients in the fluticasone furoate–vilanterol group and 5% in the vilanterol-umeclidinium group.
All-cause mortality was significantly lower in patients who received the inhaled glucocorticoid, although the authors said this finding was “fragile” and needed further investigation.
The rate of discontinuation or withdrawal from the trial was 6% for the triple therapy group, 8% for the fluticasone furoate–vilanterol group, and 9% for the vilanterol-umeclidinium group. The rates of serious adverse events in each group were 22%, 21%, and 23%, respectively.
At trial entry, 38% of patients were already receiving triple therapy and 29% were taking an inhaled glucocorticoid. The authors noted that any patients taking an inhaled glucocorticoid who were randomized to the vilanterol-umeclidinium group would have had to abruptly stop taking their inhaled glucocorticoids.
“It is unknown whether the abrupt discontinuation of inhaled glucocorticoids would have contributed to our finding of a lower rate of exacerbations in the inhaled glucocorticoid groups than in the LAMA-LABA group,” they wrote.
Fernando Martinez, MD, chief of the division of pulmonary and critical care medicine at New York–Presbyterian Hospital/Weill Cornell Medical Center, said the study advanced the understanding of COPD management by addressing some key evidence gaps, in a statement issued by GSK.
“By comparing various combinations of effective medications in the same device the study clarifies which type of patient gains greatest benefit from each class of medicine,” Dr Martinez said in the statement. “As many patients experience frequent exacerbations or ‘flare ups,’ which can often result in hospitalization, these data will be highly relevant to patients and clinicians as they consider the optimal treatment.”
The study was funded by GSK, which manufactures Trelegy Ellipta triple therapy for COPD. Eight authors were employees of GSK and two were on advisory boards for the company. Seven authors declared funding from a range of pharmaceutical companies including GSK. One author had no conflicts of interest to declare.
SOURCE: Lipson D et al. N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901.
Triple therapy for chronic obstructive pulmonary disease (COPD) achieved reductions in moderate to severe exacerbations when compared with two kinds of dual therapy, in a study published online in the New England Journal of Medicine.
The trial compared the outcomes of COPD patients using an inhaled therapy comprising a corticosteroid, a long-acting muscarinic antagonist (LAMA), and a long-acting beta2-agonist (LABA) with the outcomes of similar patients taking one of two other therapy combinations – a corticosteroid and a LABA, or a LABA and a LAMA. This trial – Informing the Pathway of COPD Treatment (IMPACT) – included 10,355 patients with symptomatic COPD in 37 countries, according to David A. Lipson, MD, and his colleagues (N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901).
The study randomized patients to 52 weeks of either triple inhaled therapy involving a once-daily combination of 100 mcg fluticasone furoate (a corticosteroid), 62.5 mcg of the LAMA umeclidinium and 25 mcg of the LABA vilanterol; or dual inhaled therapy involving either 100 mcg fluticasone furoate plus 25 mcg of vilanterol, or 62.5 mcg of umeclidinium plus 25 mcg of vilanterol.
After 1 year, the rate of moderate to severe COPD exacerbations in the triple-therapy group was 0.91 per year, compared with 1.07 in the fluticasone furoate–vilanterol group and 1.21 in the vilanterol-umeclidinium group. This translated to a 15% reduction with triple therapy compared with fluticasone furoate–vilanterol and a 25% reduction compared with vilanterol-umeclidinium (P less than .001 for both).
When the analysis was limited to severe exacerbations alone, the difference was significant only between the triple therapy, which GSK is marketing as Trelegy Ellipta, and the vilanterol-umeclidinium dual therapy.
Dr. Lipson, of GSK and the University of Pennsylvania, and his coauthors noted that their finding of a greater benefit with the glucocorticoid-containing dual-therapy compared with the LABA-LAMA vilanterol-umeclidinium combination contradicted the findings of the earlier FLAME trial. This was likely due to differences in patient populations and design, as all patients in the FLAME trial had a 1-month run-in treatment with the bronchodilator tiotropium, the researchers explained.
“Therefore any patients who would require an inhaled glucocorticoid may have had an increase in exacerbations and a decrease in lung function during the run-in period and would have been forced to leave the trial,” they wrote.
Patients with higher eosinophil levels seemed to do even better with triple therapy. In those with eosinophil levels of 150 cells per microliter or above, the annual rate of moderate to severe exacerbations was 0.95 with triple therapy, 1.08 with fluticasone furoate–vilanterol, and 1.39 with vilanterol-umeclidinium.
Triple therapy also was associated with a significantly longer time to first event and greater improvements in quality of life, compared with the dual therapies.
Overall, the adverse event profile of triple therapy was similar to that of dual therapy. Contrasting that finding were differences in the incidences of physician-diagnosed pneumonia between the treatment groups. Physician-diagnosed pneumonia was 53% higher among patients who received fluticasone furoate – either in dual or triple therapy combinations. Eight percent of patients in the triple therapy group experienced pneumonia, compared with 7% of patients in the fluticasone furoate–vilanterol group and 5% in the vilanterol-umeclidinium group.
All-cause mortality was significantly lower in patients who received the inhaled glucocorticoid, although the authors said this finding was “fragile” and needed further investigation.
The rate of discontinuation or withdrawal from the trial was 6% for the triple therapy group, 8% for the fluticasone furoate–vilanterol group, and 9% for the vilanterol-umeclidinium group. The rates of serious adverse events in each group were 22%, 21%, and 23%, respectively.
At trial entry, 38% of patients were already receiving triple therapy and 29% were taking an inhaled glucocorticoid. The authors noted that any patients taking an inhaled glucocorticoid who were randomized to the vilanterol-umeclidinium group would have had to abruptly stop taking their inhaled glucocorticoids.
“It is unknown whether the abrupt discontinuation of inhaled glucocorticoids would have contributed to our finding of a lower rate of exacerbations in the inhaled glucocorticoid groups than in the LAMA-LABA group,” they wrote.
Fernando Martinez, MD, chief of the division of pulmonary and critical care medicine at New York–Presbyterian Hospital/Weill Cornell Medical Center, said the study advanced the understanding of COPD management by addressing some key evidence gaps, in a statement issued by GSK.
“By comparing various combinations of effective medications in the same device the study clarifies which type of patient gains greatest benefit from each class of medicine,” Dr Martinez said in the statement. “As many patients experience frequent exacerbations or ‘flare ups,’ which can often result in hospitalization, these data will be highly relevant to patients and clinicians as they consider the optimal treatment.”
The study was funded by GSK, which manufactures Trelegy Ellipta triple therapy for COPD. Eight authors were employees of GSK and two were on advisory boards for the company. Seven authors declared funding from a range of pharmaceutical companies including GSK. One author had no conflicts of interest to declare.
SOURCE: Lipson D et al. N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Triple COPD therapy shows fewer exacerbations than does dual therapy.
Major finding: Triple COPD therapy achieves a 15%-25% greater reduction in exacerbations compared with dual therapy.
Study details: Randomized controlled trial of 10,355 patients with symptomatic COPD.
Disclosures: The study was funded by GlaxoSmithKline, which manufactures Trelegy Ellipta triple therapy for COPD. Eight authors were employees of GlaxoSmithKline and two were on advisory boards for the company. Seven authors declared funding from a range of pharmaceutical companies including GlaxoSmithKline. One author had no conflicts of interest to declare.
Source: Lipson D et al. N Engl J Med. 2018 Apr 18. doi: 10.1056/NEJMoa1713901.
Don’t use cannabis to treat OSA, AASM recommends
, according to a position statement published in the Journal of Clinical Sleep Medicine’s April issue.

In the statement, the professional society recommends that state legislators, regulators, and health departments exclude obstructive sleep apnea (OSA) as an indication for medical cannabis programs.
The “unreliable delivery methods and insufficient evidence of treatment effectiveness, tolerability, and safety” of medical cannabis and its synthetic extracts are among the reasons the AASM gave for making its recommendations. “Further research is needed to better understand the mechanistic actions of medical cannabis and its synthetic extracts, the long-term role of these synthetic extracts on OSA treatment, and harms and benefits,” the AASM concluded in its statement, authored by Kannan Ramar, MD, and other members of a panel of experts on sleep medicine.
Dronabinol is the only cannabis product that has been tested on patients with OSA for the treatment of this disorder. While some synthetic cannabis products are approved by the Food and Drug Administration for other medical indications, the synthetic-based cannabis product dronabinol has not received FDA approval for the treatment of OSA.
Researchers have examined dronabinol’s use for treating OSA in small pilot and proof-of-concept studies and most patients in these studies reported experiencing treatment-related side effects, such as somnolence, wrote Dr. Ramar, of the division of pulmonary and critical care medicine at the Center for Sleep Medicine, Mayo Clinic, Rochester Minn., and his colleagues.
These trials involved patients having taken dronabinol pills in strengths ranging from 2.5 mg to 10 mg. One such study (Front Psychiatry. 2013 Jan 22. doi: 10.3389/fpsyt.2013.00001), authored by Bharati Prasad of the University of Illinois, Chicago, and colleagues, showed a significant improvement in apnea-hypopnea index (AHI) of 32%, after 17 patients used dronabinol for 3 weeks, when compared with baseline AHIs (–14.1; P = .007).
A placebo-controlled randomized study of 73 adults with moderate or severe OSA similarly found a 33% decline in AHI in patients following 6 weeks of treatment with 10-mg doses of dronabinol (Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184).
In the placebo-controlled study, 73 patients were randomized to receive 2.5 mg of dronabinol or 10 mg of dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the AHI among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo. The difference between the placebo and treatment arms was significant for both dosages, and the AHI decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline AHI, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater. Dronabinol treatment also was associated with significant decreases, compared with placebo, in non-REM AHI and REM AHI.
Overall, nearly 90% of patients in this trial reported at least one adverse event, with the rates having not differed significantly between the treatment and placebo arms. The most frequently reported adverse events were “sleepiness/drowsiness” (n = 25; 8% of total adverse events reported), headache (n = 24; 8%), “nausea/vomiting” (n = 23; 8%), and “dizziness/lightheadedness” (n = 12; 4%). In addition, one patient experienced diarrhea and vomiting that required admission to a hospital, which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events, including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting.
“Synthetic medical cannabis may have differential side effects, with variable efficacy and side effects in the treatment of OSA. Therefore, it is the position of the American Academy of Sleep Medicine that medical cannabis and/or its synthetic extracts should not be used for the treatment of OSA,” Dr. Ramar and his associates wrote.
, according to a position statement published in the Journal of Clinical Sleep Medicine’s April issue.
In the statement, the professional society recommends that state legislators, regulators, and health departments exclude obstructive sleep apnea (OSA) as an indication for medical cannabis programs.
The “unreliable delivery methods and insufficient evidence of treatment effectiveness, tolerability, and safety” of medical cannabis and its synthetic extracts are among the reasons the AASM gave for making its recommendations. “Further research is needed to better understand the mechanistic actions of medical cannabis and its synthetic extracts, the long-term role of these synthetic extracts on OSA treatment, and harms and benefits,” the AASM concluded in its statement, authored by Kannan Ramar, MD, and other members of a panel of experts on sleep medicine.
Dronabinol is the only cannabis product that has been tested on patients with OSA for the treatment of this disorder. While some synthetic cannabis products are approved by the Food and Drug Administration for other medical indications, the synthetic-based cannabis product dronabinol has not received FDA approval for the treatment of OSA.
Researchers have examined dronabinol’s use for treating OSA in small pilot and proof-of-concept studies and most patients in these studies reported experiencing treatment-related side effects, such as somnolence, wrote Dr. Ramar, of the division of pulmonary and critical care medicine at the Center for Sleep Medicine, Mayo Clinic, Rochester Minn., and his colleagues.
These trials involved patients having taken dronabinol pills in strengths ranging from 2.5 mg to 10 mg. One such study (Front Psychiatry. 2013 Jan 22. doi: 10.3389/fpsyt.2013.00001), authored by Bharati Prasad of the University of Illinois, Chicago, and colleagues, showed a significant improvement in apnea-hypopnea index (AHI) of 32%, after 17 patients used dronabinol for 3 weeks, when compared with baseline AHIs (–14.1; P = .007).
A placebo-controlled randomized study of 73 adults with moderate or severe OSA similarly found a 33% decline in AHI in patients following 6 weeks of treatment with 10-mg doses of dronabinol (Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184).
In the placebo-controlled study, 73 patients were randomized to receive 2.5 mg of dronabinol or 10 mg of dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the AHI among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo. The difference between the placebo and treatment arms was significant for both dosages, and the AHI decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline AHI, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater. Dronabinol treatment also was associated with significant decreases, compared with placebo, in non-REM AHI and REM AHI.
Overall, nearly 90% of patients in this trial reported at least one adverse event, with the rates having not differed significantly between the treatment and placebo arms. The most frequently reported adverse events were “sleepiness/drowsiness” (n = 25; 8% of total adverse events reported), headache (n = 24; 8%), “nausea/vomiting” (n = 23; 8%), and “dizziness/lightheadedness” (n = 12; 4%). In addition, one patient experienced diarrhea and vomiting that required admission to a hospital, which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events, including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting.
“Synthetic medical cannabis may have differential side effects, with variable efficacy and side effects in the treatment of OSA. Therefore, it is the position of the American Academy of Sleep Medicine that medical cannabis and/or its synthetic extracts should not be used for the treatment of OSA,” Dr. Ramar and his associates wrote.
, according to a position statement published in the Journal of Clinical Sleep Medicine’s April issue.
In the statement, the professional society recommends that state legislators, regulators, and health departments exclude obstructive sleep apnea (OSA) as an indication for medical cannabis programs.
The “unreliable delivery methods and insufficient evidence of treatment effectiveness, tolerability, and safety” of medical cannabis and its synthetic extracts are among the reasons the AASM gave for making its recommendations. “Further research is needed to better understand the mechanistic actions of medical cannabis and its synthetic extracts, the long-term role of these synthetic extracts on OSA treatment, and harms and benefits,” the AASM concluded in its statement, authored by Kannan Ramar, MD, and other members of a panel of experts on sleep medicine.
Dronabinol is the only cannabis product that has been tested on patients with OSA for the treatment of this disorder. While some synthetic cannabis products are approved by the Food and Drug Administration for other medical indications, the synthetic-based cannabis product dronabinol has not received FDA approval for the treatment of OSA.
Researchers have examined dronabinol’s use for treating OSA in small pilot and proof-of-concept studies and most patients in these studies reported experiencing treatment-related side effects, such as somnolence, wrote Dr. Ramar, of the division of pulmonary and critical care medicine at the Center for Sleep Medicine, Mayo Clinic, Rochester Minn., and his colleagues.
These trials involved patients having taken dronabinol pills in strengths ranging from 2.5 mg to 10 mg. One such study (Front Psychiatry. 2013 Jan 22. doi: 10.3389/fpsyt.2013.00001), authored by Bharati Prasad of the University of Illinois, Chicago, and colleagues, showed a significant improvement in apnea-hypopnea index (AHI) of 32%, after 17 patients used dronabinol for 3 weeks, when compared with baseline AHIs (–14.1; P = .007).
A placebo-controlled randomized study of 73 adults with moderate or severe OSA similarly found a 33% decline in AHI in patients following 6 weeks of treatment with 10-mg doses of dronabinol (Sleep. 2018 Jan 1. doi: 10.1093/sleep/zsx184).
In the placebo-controlled study, 73 patients were randomized to receive 2.5 mg of dronabinol or 10 mg of dronabinol daily for up to 6 weeks, or placebo. At the end of treatment, researchers saw significant increases in the AHI among the patients on placebo, while those who received dronabinol showed decreases in the number of apnea and hypopnea events per hour. Patients given the 2.5-mg dose of dronabinol had a mean decrease of 10.7 events per hour, and those on the 10-mg dose had a mean decrease of 12.9 events per hour compared with placebo. The difference between the placebo and treatment arms was significant for both dosages, and the AHI decreases were similar between the two dosages of dronabinol.
These effects were largely due to reductions in apnea events; the largest reduction was seen in the REM apnea index in patients treated with the 10-mg dose of dronabinol. However, there were few effects on the expression of hypopneas, except in the higher-dose group.
After adjustment for age, race, ethnicity, and baseline AHI, the increases seen in the placebo group were no longer significant, but the decreases from baseline seen in the treatment arms were greater. Dronabinol treatment also was associated with significant decreases, compared with placebo, in non-REM AHI and REM AHI.
Overall, nearly 90% of patients in this trial reported at least one adverse event, with the rates having not differed significantly between the treatment and placebo arms. The most frequently reported adverse events were “sleepiness/drowsiness” (n = 25; 8% of total adverse events reported), headache (n = 24; 8%), “nausea/vomiting” (n = 23; 8%), and “dizziness/lightheadedness” (n = 12; 4%). In addition, one patient experienced diarrhea and vomiting that required admission to a hospital, which was judged as possibly related to the study medication. There were six other withdrawals due to adverse events, including dizziness and vision changes, vertigo, ECG arrhythmias, and headache with dizziness and vomiting.
“Synthetic medical cannabis may have differential side effects, with variable efficacy and side effects in the treatment of OSA. Therefore, it is the position of the American Academy of Sleep Medicine that medical cannabis and/or its synthetic extracts should not be used for the treatment of OSA,” Dr. Ramar and his associates wrote.
FROM THE JOURNAL OF CLINICAL SLEEP MEDICINE
Nivolumab shows promise in early-stage resectable NSCLC
(NSCLC), according to the results of a 21-patient pilot trial.
Eighty percent of patients were alive and recurrence-free a year after surgery, said Patrick M. Forde, MBBCh, and his colleagues from Johns Hopkins University, Baltimore. The only grade 3 or higher adverse event was treatment-related pneumonia, which did not prevent surgery. The findings were reported at the annual meeting of the American Association for Cancer Research and simultaneously in the New England Journal of Medicine.
For the study (NCT02259621), 21 patients with treatment-naive, stage I, II, or III NSCLC received two preoperative doses of nivolumab (3 mg/kg) 2 weeks apart, with surgery timed for 4 weeks after the first dose. In all, 62% of patients had adenocarcinoma, 81% had stage II or IIIa disease, and 86% were current or former smokers. Patients were followed for a median of 12 months after surgery (range, 0.8-19.7 months), and the researchers assessed safety, tumor response, programmed death ligand 1 mutational burden, and T-cell response.
Among 20 patients with evaluable resected primary tumors, nine (45%) showed a major pathologic response, defined as having 10% or fewer residual viable tumor cells. Twelve-month, recurrence-free survival was 83% (95% confidence interval, 66%-100%). The three progressors included one patient with 75% residual tumor at resection who subsequently developed a brain lesion, a patient with 5% residual tumor at resection who developed mediastinal lymph node recurrence, and a patient with 80% residual tumor at resection. The first two patients had durable responses to stereotactic radiotherapy or chemoradiotherapy, while the third patient developed fatal distal metastatic disease.
Sequencing of 11 completely resected tumors linked major pathologic response with higher tumor mutational burden (P = .01). Mutational burden did not correlate with tumor programmed death ligand 1 expression. Deep sequencing of T-cell receptor–beta chain CDR3 regions also correlated major pathologic response with increased clonality of tumor-infiltrating T-cell clones that also expanded into peripheral blood. “Many of these clones were not detected in peripheral blood before treatment,” the investigators wrote.
In all, five (23%) patients developed treatment-related adverse events, and many developed more than one side effect. Grade 1-2 anorexia, taste distortion, vomiting, and diarrhea were most common, with isolated cases of grade 1-2 fever, infusion reaction, abdominal pain, abnormal liver function, dry skin, and delirium. The case of grade 3 pneumonia developed after the first dose of nivolumab. The patient stopped treatment and underwent uncomplicated surgical resection.
Funders included Cancer Research Institute–Stand Up 2 Cancer; Johns Hopkins Bloomberg–Kimmel Institute for Cancer Immunotherapy; Bristol-Myers Squibb; International Immuno-Oncology Network, LUNGevity Foundation; International Association for the Study of Lung Cancer; Lung Cancer Foundation of America; and numerous other foundations and universities. Bristol-Myers Squibb makes nivolumab and supplied the study drug. Dr. Forde disclosed study grant support from Bristol-Myers Squibb. He reported ties to Bristol-Myers Squibb, AbbVie, and other pharmaceutical companies outside the submitted work.
SOURCE: Forde PM. AACR Annual Meeting 2018. Forde PM et al. N Engl J Med. 2018 Apr 16. doi: 10.1056/NEJMoa1716078.
(NSCLC), according to the results of a 21-patient pilot trial.
Eighty percent of patients were alive and recurrence-free a year after surgery, said Patrick M. Forde, MBBCh, and his colleagues from Johns Hopkins University, Baltimore. The only grade 3 or higher adverse event was treatment-related pneumonia, which did not prevent surgery. The findings were reported at the annual meeting of the American Association for Cancer Research and simultaneously in the New England Journal of Medicine.
For the study (NCT02259621), 21 patients with treatment-naive, stage I, II, or III NSCLC received two preoperative doses of nivolumab (3 mg/kg) 2 weeks apart, with surgery timed for 4 weeks after the first dose. In all, 62% of patients had adenocarcinoma, 81% had stage II or IIIa disease, and 86% were current or former smokers. Patients were followed for a median of 12 months after surgery (range, 0.8-19.7 months), and the researchers assessed safety, tumor response, programmed death ligand 1 mutational burden, and T-cell response.
Among 20 patients with evaluable resected primary tumors, nine (45%) showed a major pathologic response, defined as having 10% or fewer residual viable tumor cells. Twelve-month, recurrence-free survival was 83% (95% confidence interval, 66%-100%). The three progressors included one patient with 75% residual tumor at resection who subsequently developed a brain lesion, a patient with 5% residual tumor at resection who developed mediastinal lymph node recurrence, and a patient with 80% residual tumor at resection. The first two patients had durable responses to stereotactic radiotherapy or chemoradiotherapy, while the third patient developed fatal distal metastatic disease.
Sequencing of 11 completely resected tumors linked major pathologic response with higher tumor mutational burden (P = .01). Mutational burden did not correlate with tumor programmed death ligand 1 expression. Deep sequencing of T-cell receptor–beta chain CDR3 regions also correlated major pathologic response with increased clonality of tumor-infiltrating T-cell clones that also expanded into peripheral blood. “Many of these clones were not detected in peripheral blood before treatment,” the investigators wrote.
In all, five (23%) patients developed treatment-related adverse events, and many developed more than one side effect. Grade 1-2 anorexia, taste distortion, vomiting, and diarrhea were most common, with isolated cases of grade 1-2 fever, infusion reaction, abdominal pain, abnormal liver function, dry skin, and delirium. The case of grade 3 pneumonia developed after the first dose of nivolumab. The patient stopped treatment and underwent uncomplicated surgical resection.
Funders included Cancer Research Institute–Stand Up 2 Cancer; Johns Hopkins Bloomberg–Kimmel Institute for Cancer Immunotherapy; Bristol-Myers Squibb; International Immuno-Oncology Network, LUNGevity Foundation; International Association for the Study of Lung Cancer; Lung Cancer Foundation of America; and numerous other foundations and universities. Bristol-Myers Squibb makes nivolumab and supplied the study drug. Dr. Forde disclosed study grant support from Bristol-Myers Squibb. He reported ties to Bristol-Myers Squibb, AbbVie, and other pharmaceutical companies outside the submitted work.
SOURCE: Forde PM. AACR Annual Meeting 2018. Forde PM et al. N Engl J Med. 2018 Apr 16. doi: 10.1056/NEJMoa1716078.
(NSCLC), according to the results of a 21-patient pilot trial.
Eighty percent of patients were alive and recurrence-free a year after surgery, said Patrick M. Forde, MBBCh, and his colleagues from Johns Hopkins University, Baltimore. The only grade 3 or higher adverse event was treatment-related pneumonia, which did not prevent surgery. The findings were reported at the annual meeting of the American Association for Cancer Research and simultaneously in the New England Journal of Medicine.
For the study (NCT02259621), 21 patients with treatment-naive, stage I, II, or III NSCLC received two preoperative doses of nivolumab (3 mg/kg) 2 weeks apart, with surgery timed for 4 weeks after the first dose. In all, 62% of patients had adenocarcinoma, 81% had stage II or IIIa disease, and 86% were current or former smokers. Patients were followed for a median of 12 months after surgery (range, 0.8-19.7 months), and the researchers assessed safety, tumor response, programmed death ligand 1 mutational burden, and T-cell response.
Among 20 patients with evaluable resected primary tumors, nine (45%) showed a major pathologic response, defined as having 10% or fewer residual viable tumor cells. Twelve-month, recurrence-free survival was 83% (95% confidence interval, 66%-100%). The three progressors included one patient with 75% residual tumor at resection who subsequently developed a brain lesion, a patient with 5% residual tumor at resection who developed mediastinal lymph node recurrence, and a patient with 80% residual tumor at resection. The first two patients had durable responses to stereotactic radiotherapy or chemoradiotherapy, while the third patient developed fatal distal metastatic disease.
Sequencing of 11 completely resected tumors linked major pathologic response with higher tumor mutational burden (P = .01). Mutational burden did not correlate with tumor programmed death ligand 1 expression. Deep sequencing of T-cell receptor–beta chain CDR3 regions also correlated major pathologic response with increased clonality of tumor-infiltrating T-cell clones that also expanded into peripheral blood. “Many of these clones were not detected in peripheral blood before treatment,” the investigators wrote.
In all, five (23%) patients developed treatment-related adverse events, and many developed more than one side effect. Grade 1-2 anorexia, taste distortion, vomiting, and diarrhea were most common, with isolated cases of grade 1-2 fever, infusion reaction, abdominal pain, abnormal liver function, dry skin, and delirium. The case of grade 3 pneumonia developed after the first dose of nivolumab. The patient stopped treatment and underwent uncomplicated surgical resection.
Funders included Cancer Research Institute–Stand Up 2 Cancer; Johns Hopkins Bloomberg–Kimmel Institute for Cancer Immunotherapy; Bristol-Myers Squibb; International Immuno-Oncology Network, LUNGevity Foundation; International Association for the Study of Lung Cancer; Lung Cancer Foundation of America; and numerous other foundations and universities. Bristol-Myers Squibb makes nivolumab and supplied the study drug. Dr. Forde disclosed study grant support from Bristol-Myers Squibb. He reported ties to Bristol-Myers Squibb, AbbVie, and other pharmaceutical companies outside the submitted work.
SOURCE: Forde PM. AACR Annual Meeting 2018. Forde PM et al. N Engl J Med. 2018 Apr 16. doi: 10.1056/NEJMoa1716078.
REPORTING FROM AACR ANNUAL MEETING
Key clinical point: Neoadjuvant nivolumab was tolerable and induced robust responses in resectable non–small cell lung cancers.
Major finding: In all, 45% of evaluable tumors showed a major pathological response. Eighty percent of resected patients were alive and recurrence-free a median of 12 months after surgery. One patient (5%) developed a grade 3 treatment-related adverse event.
Study details: Pilot study of 21 adults with early-stage resectable non–small cell lung cancer (NCT02259621).
Disclosures: Funders included Cancer Research Institute–Stand Up 2 Cancer; Johns Hopkins Bloomberg–Kimmel Institute for Cancer Immunotherapy; Bristol-Myers Squibb; International Immuno-Oncology Network, LUNGevity Foundation; International Association for the Study of Lung Cancer; Lung Cancer Foundation of America; and numerous other foundations and universities. Bristol-Myers Squibb makes nivolumab and supplied the study drug. Dr. Forde disclosed study grant support from Bristol-Myers Squibb. He reported ties to Bristol-Myers Squibb, AbbVie, and other pharmaceutical companies outside the submitted work.
Source: Forde PM. AACR Annual Meeting 2018. Forde PM et al. N Engl J Med. 2018 Apr 16. doi: 10.1056/NEJMoa1716078.
Pharmacologic Treatments for Idiopathic Pulmonary Fibrosis
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
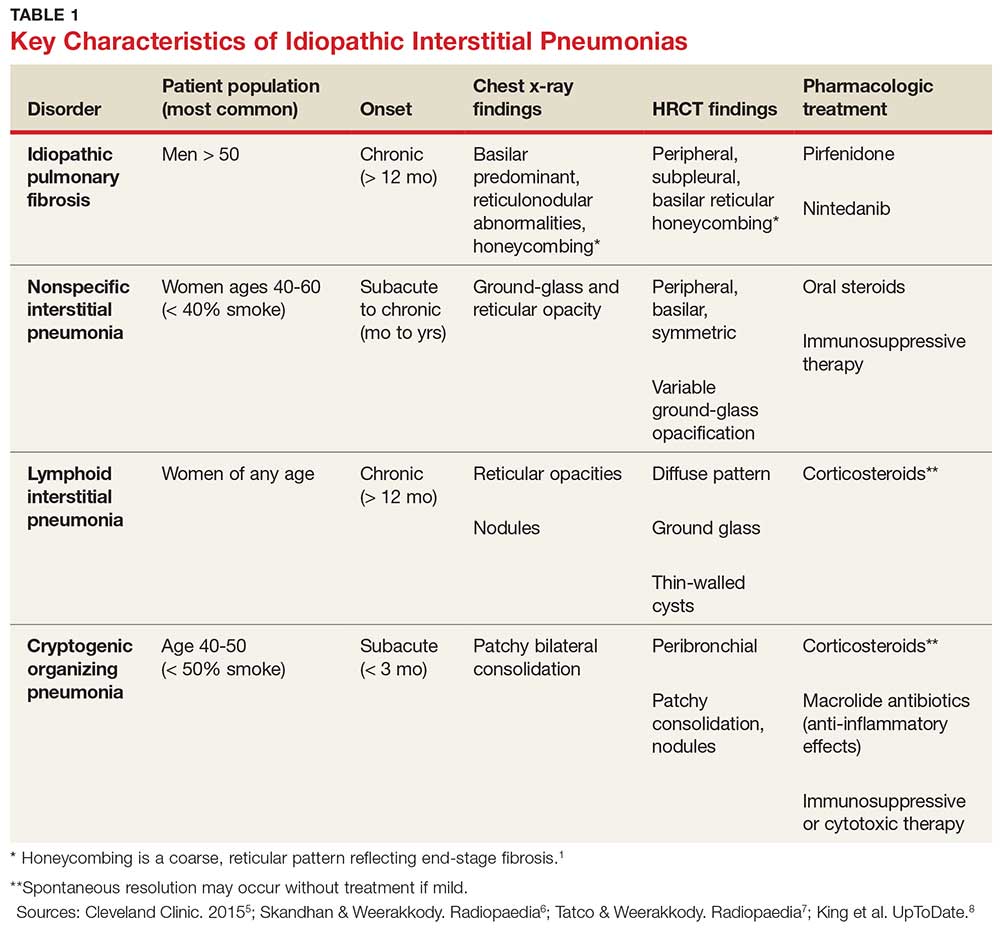
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
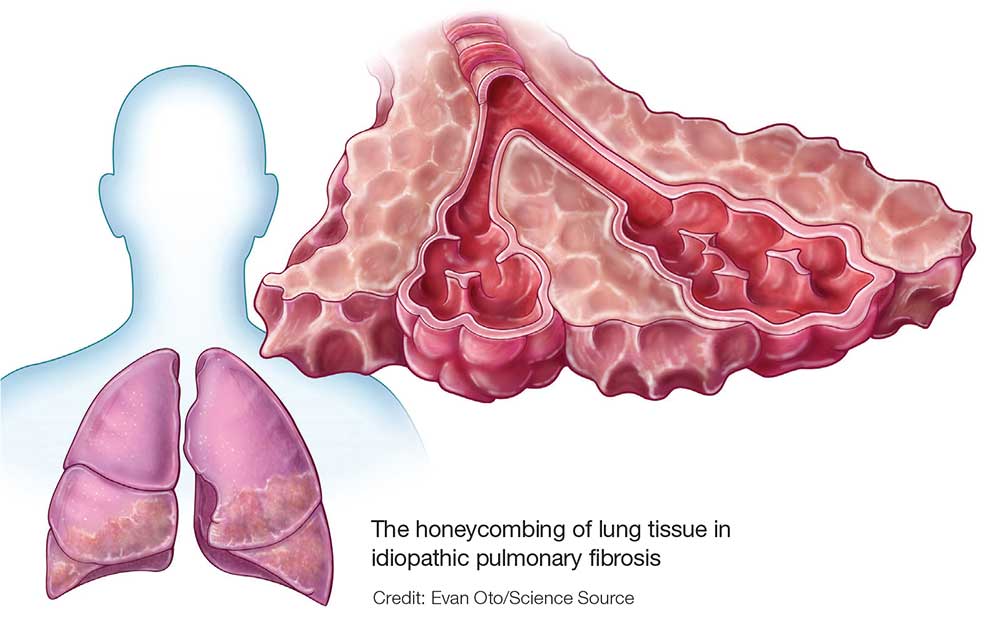
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
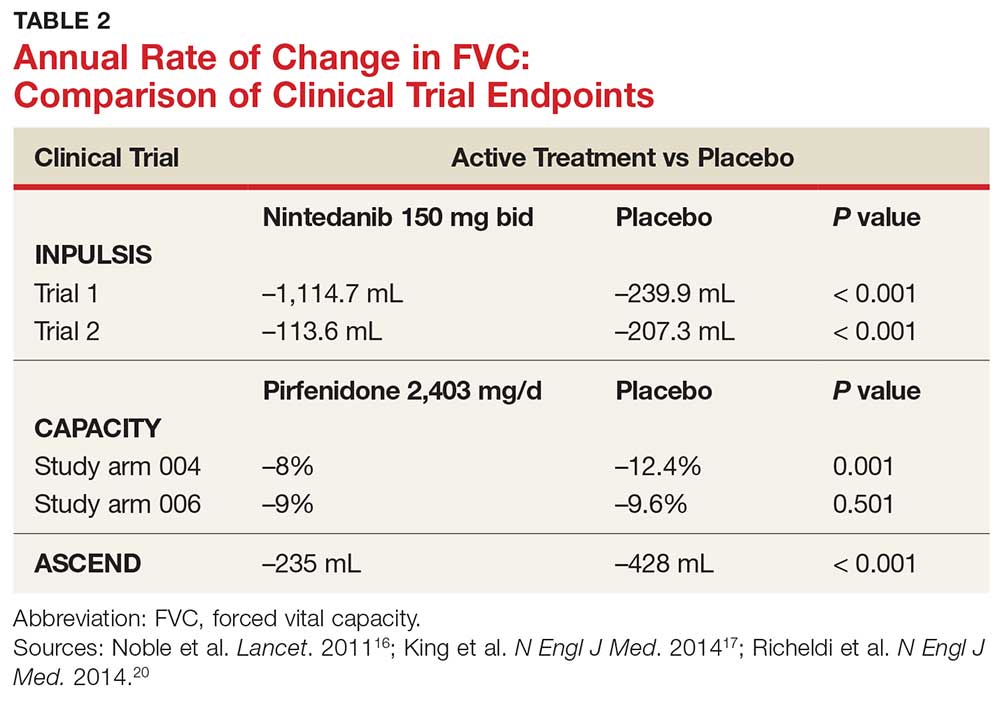
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.

The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.
The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
IN THIS ARTICLE
- Confirming the diagnosis
- Pirfenidone treatment
- Nintedanib treatment
A 64-year-old man has a one-year history of dyspnea on exertion and a nonproductive cough. His symptoms are gradually worsening and increasingly bothersome to him.
His medical history includes mild seasonal allergies and GERD, which is well-controlled by oral antihistamines and proton pump inhibitors. He has spent the past 30 years working a desk job as an accountant. He denies a history of smoking, exposure to secondhand smoke, and initiation of new medication.
He admits to increased fatigue, but denies fever, chills, lymphadenopathy, weight change, chest pain, wheezing, abdominal pain, diarrhea, vomiting, claudication, and swelling in the extremities. The rest of the review of systems is negative.
Lab results—complete blood count, comprehensive metabolic panel, TSH, antinuclear antibodies, erythrocyte sedimentation rate, and C-reactive protein—are within normal limits. Spirometry shows very mild restriction. A chest x-ray is abnormal but nonspecific, showing peripheral opacities. An ECG shows normal sinus rhythm.
The patient is given a trial of an inhaled steroid, which yields no improvement. Six months later, the patient is seen by a pulmonologist. Idiopathic pulmonary fibrosis (IPF) is diagnosed based on high-resolution CT (HRCT) and lung biopsy results.
IPF is a chronic, progressive, fibrosing interstitial disease that is limited to lung tissue. It most commonly manifests in older adults with vague symptoms of dyspnea on exertion and nonproductive cough, but symptoms can also include fatigue, muscle and joint aches, clubbing of the fingernails, and weight loss.1 The average life expectancy following diagnosis of IPF is two to five years, and the mortality rate is estimated at 64.3 per million men and 58.4 per million women per year.2,3
Continue to: DIAGNOSIS
DIAGNOSIS
IPF belongs in the general class of idiopathic interstitial pneumonias (IIPs), which are characterized by varying degrees of inflammation and fibrosis of lung interstitium.4 All subtypes of IIPs cause dyspnea and diffuse abnormalities on HRCT, and all vary from each other histologically. Table 1 outlines the key features of each.5-8
Because of its vague symptomology and the extensive workup needed to rule out other diseases, patients with IPF often have symptoms for one to two years before a diagnosis is made.1 Physical exam may reveal fine inspiratory rales in both lung bases and digital clubbing; eventual signs of pulmonary hypertension and right-sided heart failure may be appreciated.1,9
There are no specific diagnostic laboratory tests to confirm IPF; however, baseline labwork (as outlined in the case presentation) is typically ordered to rule out infection, thyroid disease, or connective tissue disease.10 Many patients are referred to a cardiologist before being seen by a pulmonologist; cardiac stress testing may be done, and an echocardiogram may be performed to rule out heart failure.
Diagnostic testing may include pulmonary function testing, HRCT of the chest, and lung biopsy.10 Tissue samples from patients with IPF reveal different stages of disease, including dense fibrosis with honeycombing, subpleural or paraseptal distribution, fibroblast foci, and normal tissue.11 Pulmonary function test results will show a restrictive pattern. Both forced expiratory volume in one second (FEV1) and forced vital capacity (FVC) will be reduced, and the FEV1/FVC ratio preserved. Due to decreased functional lung volume, diffusing capacity of the lung for carbon monoxide (DLCO) will also be reduced.4,12
The differential is broad and includes allergic asthma, bronchitis, COPD, lung cancer, hypersensitivity pneumonitis, asbestosis, or pulmonary embolism.
Continue to: TREATMENT HISTORY
TREATMENT HISTORY
IPF has a long history of tried and failed treatment options. The American Thoracic Society (ATS), in concert with other professional organizations, has published comprehensive guidelines and recommendations pertaining to the use of pharmacologic medications to control disease progression. Warfarin and other anticoagulants have been studied, based on the observation that a procoagulant state promotes fibrotic changes in the lung tissue.13 However, anticoagulant use is not recommended in patients with IPF due to lack of efficacy and high potential for harm.13
Immunosuppressants have also been in the spotlight as possible treatment for IPF, but a clinical study investigating the efficacy of a three-drug regimen including prednisone, azathioprine, and N-acetylcysteine was stopped early due to increased risk for harm. Endothelin antagonists and potent tyrosine kinase inhibitors are also not recommended in the most recent edition of IPF guidelines, as they lack benefit.13
In fact, prior to the 2015 edition of the guidelines, no single medication was routinely recommended for patients with IPF. But this is now changing, following the 2014 FDA approval of two new drugs, nintedanib and pirfenidone, designed specifically to treat IPF.14 These drugs have shown promise in clinical trials (results of which are summarized in Table 2).
Continue to: NEW PHARMACOLOGIC OPTIONS
NEW PHARMACOLOGIC OPTIONS
Pirfenidone
In 2008, a study was conducted in Japan to determine the mechanism of action of pirfenidone.15 Through in vitro studies of healthy adult lung fibroblasts with added pro-fibrotic factor and transforming growth factor (TGF-ß 1), the researchers found that pirfenidone was effective at decreasing the production of a collagen-binding protein called HSP47. This protein is ubiquitous in fibrotic tissue. The study also showed that pirfenidone decreased the production of collagen type 1, which, when uninhibited, increases fibrosis.15
CAPACITY trials. In the CAPACITY trials, two phase 3 multinational studies conducted from 2006 to 2008, patients were given either pirfenidone or placebo.16 In the first study arm, patients were assigned to pirfenidone 2,403 mg/d (n = 174), pirfenidone 1,197 mg/d (n = 87), or placebo (n = 174). In the second study arm, 171 patients received pirfenidone 2,403 mg/d and 173 patients received placebo. Endpoints were measured at baseline and up to week 72.
The first study arm found that the mean rate of decline of FVC—the primary endpoint—was 4.4% less in the treatment group than in the placebo group (p = 0.001), and there was a 36% decrease in risk for death or disease progression in the treatment group (HR, 0.64; p, 0.023). (Endpoints were defined as: time to confirmed > 10% decline in percentage predicted FVC, > 15% decline in percentage predicted DLCO, or death.) The researchers found no clinically significant change in the six-minute walk test—a secondary endpoint of the study.16
The second study arm, however, found no statistically significant change in FVC between the treatment and placebo groups (with a 0.6% smaller decrease in FVC in the pirfenidone group), nor did they see a difference in progression-free survival. However, there was a significant change in the six-minute walk test between the treatment and placebo groups (p = 0.0009). Throughout the study, the most common adverse effects included nausea (36%), rash (32%), and dyspepsia (19%).16
ASCEND trial. The 2014 Assessment of Pirfenidone to Confirm Efficacy and Safety in Idiopathic Pulmonary Fibrosis (ASCEND) trial was a phase 3, multinational, randomized, double-blind, placebo-controlled study of the use of pirfenidone 2,403 mg/d.17 The study was conducted from 2012 to 2013. Of the total number of patients (N = 522), half received pirfenidone and half received placebo. After 52 weeks of treatment (the end of the study), the researchers found a smaller decline in FVC—the primary endpoint—in the treatment group compared to placebo (mean decline, 235 mL vs 428 mL, respectively [p < 0.001]). Regarding the six-minute walk test, the investigators found that 25.9% of the treatment group exhibited a decrease of ≥ 50 meters, compared to 35.7% of the placebo group (p = 0.04). (Progression-free survival was defined as a confirmed ≥ 10% decrease in predicted FVC, a confirmed decrease of 50 meters in the six-minute walk test, or death.)
The pirfenidone group in the ASCEND trial showed a 43% reduced risk for death or disease progression (HR, 0.57; p, < 0.001).16,17 All-cause mortality was lower in the pirfenidone group (4%) than in the placebo group (7.2%), but this was not statistically significant. Deaths from IPF in the pirfenidone group totaled three patients (1.1%) versus seven patients (2.5%) in the placebo group; this was also not statistically significant. The most common adverse effects seen during the study were nausea (36%), rash (28.1%), and headache (25.9%).17
Recommendations for use. Liver function testing should be performed at baseline, monthly for six months, and every three months afterward, as elevations in liver enzymes have been observed.18 Pirfenidone is a CYP1A2 substrate; moderate-to-strong CYP1A2 inhibitors should therefore be discontinued prior to initiation, as they are likely to decrease exposure and efficacy of pirfenidone. There are currently no black box warnings.18
Continue to: Nintedanib
Nintedanib
Hostettler et al studied lung samples from patients with IPF to determine the mechanism of action of nintedanib.19 Evaluation of fibroblasts derived from IPF samples revealed that they contained higher levels of platelet-derived growth factor (PDGF) than did nonfibrotic control cells. They also found that nintedanib, a tyrosine kinase inhibitor, significantly inhibited the phosphorylation of fibrotic-inducing growth factors—PDGF as well as vascular endothelial growth factor (VEGF).
INPULSIS trials. A phase 3 replicate of randomized, double-blind, multinational studies, the INPULSIS trials were performed between 2011 and 2012.20 Two study arms were used to evaluate a total of 638 patients who received nintedanib 150 mg bid for 52 weeks. The primary endpoint was annual rate of decline of FVC.
The researchers also evaluated efficacy through two other endpoints: patient-reported quality of life and symptoms via the St. George’s Respiratory Questionnaire (SGRQ) and evaluation of time to acute exacerbation. The latter was defined as worsening or new dyspnea, new diffuse pulmonary infiltrates visualized on chest radiography and/or HRCT, or the development of parenchymal abnormalities with no pneumothorax or pleural effusion since the preceding visit; and exclusion of any known causes of acute worsening, including infection, heart failure, pulmonary embolism, and any identifiable cause of acute lung injury.20
INPULSIS 1 (first arm) included 309 patients in the treatment group. Results showed an adjusted annual rate of decline in FVC of 114.7 mL/year, versus 239.9 mL/year in the placebo group (p < 0.001). In the treatment group, 52.8% exhibited ≤ 5% decline in FVC, compared to 38.2% in the placebo group (p = 0.001). No significant between-group differences were found in SGRQ score or time to acute exacerbation.20
INPULSIS 2 had 329 patients receiving nintedanib. An annual rate of decline in FVC of 113.6 mL/year from baseline was observed in the treatment group, compared to 207.3 mL/year in the placebo group (p < 0.001). In the treatment group, 53.2% showed ≤ 5% decline in FVC, versus 39.3% in the placebo group (p = 0.001). There was also a significantly smaller increase in total SGRQ score (meaning, less deterioration in quality of life) in the nintedanib group versus the placebo group (p = 0.02). A statistically significant increase in time to first acute exacerbation was observed in the nintedanib group (p = 0.005).20
There was no significant difference between groups in death from any cause, death from respiratory causation, or death that occurred between randomization and 28 days post treatment. The most common adverse effects seen throughout the two trials included diarrhea (trial 1, 61.5%; trial 2, 63.2%), nausea (trial 1, 22.7%; trial 2, 26.1%), and nasopharyngitis (trial 1, 12.6%; trial 2, 14.6%).20
Recommendations for use. Liver function testing should be performed at baseline, at regular intervals during the first three months, then periodically thereafter; patients in the treatment group of both INPULSIS trials had elevated liver enzymes, and cases of drug-induced liver injury have been observed with use of nintedanib.21 This medication may increase risk for bleeding due to its mechanism of action (VEGFR inhibition). Coadministration with CYP3A4 inhibitors may increase concentration of nintedanib; therefore, close monitoring is recommended. Avoid coadministration with CYP3A4 inducers, as this may decrease concentration of nintedanib by 50%. There are currently no black box warnings.21
Continue to: Patient monitoring
Patient monitoring
The ATS recommends measuring FVC and DLCO every three to six months, or sooner if clinically indicated.13 Pulse oximetry should be measured at rest and on exertion in all patients, regardless of symptoms, to assure proper saturation and identify the need for supplemental oxygen; this should also be done every three to six months.
The ATS recommends prompt detection and treatment of comorbidities such as pulmonary hypertension, emphysema, airflow obstruction, GERD, sleep apnea, and coronary artery disease.13 These recommendations are based on the organization’s 2015 guidelines.
OUTCOME FOR THE CASE PATIENT
The patient was started on pirfenidone (2,403 mg/d). He is continuing treatment and showing improvements in quality of life and slowed deterioration of lung function.
CONCLUSION
IPF causes progressive fibrosis of lung interstitium. The etiology is unknown, the symptoms and signs are vague, and mean life expectancy following diagnosis is two to five years. The most recent IPF guidelines recommend avoiding use of anticoagulants and immunosuppressants (eg, steroids, azathioprine, and N-acetylcysteine), due to their proven ineffectiveness and harm to patients with IPF.
Since the FDA’s approval of pirfenidone and nintedanib, the ATS has made recommendations for their use in patients with IPF. Despite mixed results in clinical trials, both drugs have demonstrated the ability to slow the decline in FVC over time, with relatively benign adverse effects. It is difficult to compare pirfenidone and nintedanib, or to recommend use of one drug over the other. However, it is promising that patients with this routinely fatal disease now have treatment options that can potentially modulate their disease progression.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.
1. Kim DS, Collard HR, King TE Jr. Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):285-292.
2. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009;15(5):463-469.
3. Olson AL, Swigris JJ, Lezotte DC, et al. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176(3):277-284.
4. Chapman JT. Interstitial lung disease. Cleveland Clinic. August 2010. www.clevelandclinicmeded.com/medical pubs/diseasemanagement/pulmonary/interstitial-lung-disease. Accessed March 12, 2018.
5. Cleveland Clinic. Nonspecific interstitial pneumonia. January 16, 2015. https://my.clevelandclinic.org/health/articles/nonspecific-interstitial-pneumonia. Accessed March 12, 2018.
6. Skandhan AKP, Weerakkody Y. Non-specific interstitial pneumonia. Radiopaedia. https://radiopaedia.org/articles/non-specific-interstitial-pneumonia-1. Accessed March 12, 2018.
7. Tatco V, Weerakkody Y. Lymphocytic interstitial pneumonitis. Radiopaedia. https://radiopaedia.org/articles/lymphocytic-interstitial-pneumonitis-1. Accessed March 12, 2018.
8. King TE Jr, Flaherty KR, Hollingsworth H. Cryptogenic organizing pneumonia. UpToDate. www.uptodate.com/contents/cryptogenic-organizing-pneumonia#H12. Accessed March 12, 2018.
9. Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007; 132(3):998-1006.
10. Lee J. Overview of idiopathic interstitial pneumonias. April 2016. www.merckmanuals.com/professional/pulmonary-disorders/interstitial-lung-diseases/overview-of-idiopathic-interstitial-pneumonias. Accessed March 12, 2018.
11. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138-153.
12. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006; 3(4):315-321.
13. Raghu G, Rochwerg B, Zhang Y, et al; American Thoracic Society; European Respiratory Society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015; 192(2):e3-e19.
14. Chowdhury BA; FDA. Two FDA drug approvals for idiopathic pulmonary fibrosis (IPF). October 15, 2014. https://blogs.fda.gov/fdavoice/index.php/2014/10/two-fda-drug-approvals-for-idiopathic-pulmonary-fibrosis-ipf/. Accessed March 12, 2018.
15. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008; 82(3-4):210-217.
16. Noble PW, Albera C, Bradford WZ, et al; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomized trials. Lancet. 2011;377: 1760-1769.
17. King TE Jr, Bradford WZ, Castro-Bernardini S, et al; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22): 2083-2092.
18. Esbriet [package insert]. South San Francisco, CA: Genentech, Inc; 2016.
19. Hostettler KE, Zhong J, Papakonstantinou E, et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15(1):157.
20. Richeldi L, du Bois RM, Raghu G, et al; INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071-2082.
21. OFEV [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018.