User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
Dupilumab reduces exacerbations, cuts glucocorticoid use in moderate to severe asthma
Among patients with moderate to severe asthma, dupilumab reduced exacerbations by almost 50%, while also allowing glucocorticoid-treated patients to cut their use of that medication by 70%, with no increased risk of exacerbation.
The pair of placebo-controlled studies – Liberty Asthma Quest and Liberty Asthma Venture – also showed treatment-associated stability in forced expiratory volume (FEV1) evidence of lung remodeling among those who took the antibody, Mario Castro, MD, of Washington University, St. Louis, and his colleagues reported in the New England Journal of Medicine.
By week 12, FEV1 it had already increased by 0.32 L, they said.
“An analysis of the postbronchodilator FEV1 slope showed a loss of lung function in patients who received placebo and no loss in those who received dupilumab, findings that suggest a potential effect of dupilumab on airway remodeling,” wrote Dr. Castro and his colleagues. “The slope analysis showed that patients who received placebo lost, on average, approximately 40 mL annually, which is consistent with data from other cohorts of patients with asthma.”
Dupilumab is an anti–interleukin-4 alpha antibody that blocks both IL-4 and IL-13. The Quest trial examined efficacy and safety of two doses (200 mg and 300 mg every 2 weeks), compared with placebo in patients with uncontrolled asthma. Venture examined efficacy and safety of 300 mg or placebo as add-on therapy for patients with severe asthma who were taking glucocorticoids.
Liberty Asthma Quest
This 52-week study randomized 1,902 patients with severe, uncontrolled asthma to placebo or dupilumab 200 mg or 300 mg every other week. The primary endpoints were annual rate of severe asthma exacerbations and the change in FEV1 by week 12. The study also looked at these endpoints in patients whose baseline eosinophil count was greater than 300 per cubic millimeter.
Patients were a mean of 48 years old with a mean baseline FEV1 of about 1.75 L (about 58% of the predicted normal value). They had a mean of two exacerbations per year and an average eosinophil count of about 350 per cubic millimeter.
Both doses outperformed placebo in all endpoints.
Among those taking 200 mg, the annual relapse rate was 0.46 versus 0.87 among those taking placebo – a significant 47.7% risk reduction. Among those taking 300 mg, the exacerbation rate was 0.52 versus 0.97; this translated to a significant 46% risk reduction.
The response rate was even greater among those with an eosinophil count greater than 300 per cubic millimeter: 0.37 for 200 mg and 0.40 for 300 mg versus the placebo rates of 1.08 and 1.24. This translated to risk reductions of 65.8% and 67.4%, respectively.
By week 12, FEV1 had significantly increased by 0.32 L in the 200-mg group and by 0.34 L in the 300-mg group, compared with nonsignificant increases among those taking placebo.
Again, patients with the high eosinophil counts experienced the greatest benefits, with FEV1 increasing by a mean of 0.43 L at 12 weeks in the 200-mg group and by 0.47 L in the 300-mg group, significantly better than either placebo comparator.
The benefit was already noticeable by the 2-week evaluation, the investigators noted.
Dupilumab appeared safe; injection-site reactions were the most common adverse event, occurring in 15% of the low-dose group and 18% of the high-dose group. However, 52 patients taking the drug experienced eosinophilia, compared with four of those taking placebo (4.1% vs. 0.6%).
Four of those taking the study drug experienced clinical symptoms associated with eosinophilia, including worsening eosinophilia and chronic eosinophilic pneumonia.
Liberty Asthma Venture
In this study, the effect of dupilumab on glucocorticoid use among 210 patients with severe asthma was examined. Patients were randomized to add-on dupilumab 300 mg every 2 weeks for 24 weeks. Glucocorticoids were tapered downward from weeks 4 to 20. The primary endpoints were percent reduction in glucocorticoid dose at week 24, and the percentage of patients who experienced a reduction of at least 50% in glucocorticoid dose.
These patients were a mean of 51 years old, with a mean of two severe asthma exacerbations in the past year. Their mean daily oral glucocorticoid dose at randomization was about 11 mg per day. Their mean prebronchodilator FEV1 was about 1.6 liters – about 52% of predicted value.
Oral glucocorticoid use decreased by a mean of 70.1% in the active group, compared with 41.9% in the placebo group, a statistically significant difference, Klaus F. Rabe, MD, of Christian Albrechts University, Kiel, Germany, and his coauthors wrote in the New England Journal of Medicine. The median change was even better: A 100% reduction in the active group and 50% reduction in the placebo group.
By week 24, 80% of those taking dupilumab had decreased their glucocorticoid intake by at least 50%, compared with 50% of the placebo group reaching this goal. The glucocorticoid dose was less than 5 mg/day in 69% of the dupilumab group, compared with 33% of the placebo group.
Like Quest, Venture showed a treatment advantage among patients with high baseline eosinophil count. “The magnitude of the effect was largest in patients with a higher eosinophil count at baseline,” the investigators wrote. “… The odds ratios [a 50% glucocorticoid reduction] for dupilumab versus placebo were 6.59 among patients with 300 or more cells per cubic millimeter at baseline and 2.91 among those with less than 300 cells per cubic millimeter at baseline.”
In a fully adjusted model at week 24, 48% of the patients in the dupilumab group were able to stop oral glucocorticoids entirely, compared with 25% of the placebo group.
Dupilumab was also associated with a significant 59% reduction in severe annual asthma exacerbations. FEV1 among the active group was 0.22 L better than that in the placebo group at week 24.
Again, patients with a higher baseline blood eosinophil count experienced greater treatment benefit; among these, the rate of severe asthma exacerbations was 71% lower than the rate in the placebo group, and FEV1 was 0.32 L higher.
The most frequent adverse events were viral infections (9% of the patients in the dupilumab group vs. 18% of those in the placebo group), bronchitis (7% vs. 6%), sinusitis (7% vs. 4%), influenza (3% vs. 6%), and eosinophilia (14% vs. 1%). Injection-site reactions occurred in 9% of those taking dupilumab and 4% of those taking placebo.
Antidrug antibodies developed in five patients in each group, without clinical effect.
Both trials were funded by Sanofi and Regeneron. Dr. Castro has received grant support from Sanofi. Dr. Rabe has received consulting and lecture fees from AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, and Teva Pharmaceutical Industries.
SOURCES: Castro M et al. N Engl J Med. 2018;378:2486-96; KF Rabe et al. N Engl J Med. 2018;378:2475-85.
Among patients with moderate to severe asthma, dupilumab reduced exacerbations by almost 50%, while also allowing glucocorticoid-treated patients to cut their use of that medication by 70%, with no increased risk of exacerbation.
The pair of placebo-controlled studies – Liberty Asthma Quest and Liberty Asthma Venture – also showed treatment-associated stability in forced expiratory volume (FEV1) evidence of lung remodeling among those who took the antibody, Mario Castro, MD, of Washington University, St. Louis, and his colleagues reported in the New England Journal of Medicine.
By week 12, FEV1 it had already increased by 0.32 L, they said.
“An analysis of the postbronchodilator FEV1 slope showed a loss of lung function in patients who received placebo and no loss in those who received dupilumab, findings that suggest a potential effect of dupilumab on airway remodeling,” wrote Dr. Castro and his colleagues. “The slope analysis showed that patients who received placebo lost, on average, approximately 40 mL annually, which is consistent with data from other cohorts of patients with asthma.”
Dupilumab is an anti–interleukin-4 alpha antibody that blocks both IL-4 and IL-13. The Quest trial examined efficacy and safety of two doses (200 mg and 300 mg every 2 weeks), compared with placebo in patients with uncontrolled asthma. Venture examined efficacy and safety of 300 mg or placebo as add-on therapy for patients with severe asthma who were taking glucocorticoids.
Liberty Asthma Quest
This 52-week study randomized 1,902 patients with severe, uncontrolled asthma to placebo or dupilumab 200 mg or 300 mg every other week. The primary endpoints were annual rate of severe asthma exacerbations and the change in FEV1 by week 12. The study also looked at these endpoints in patients whose baseline eosinophil count was greater than 300 per cubic millimeter.
Patients were a mean of 48 years old with a mean baseline FEV1 of about 1.75 L (about 58% of the predicted normal value). They had a mean of two exacerbations per year and an average eosinophil count of about 350 per cubic millimeter.
Both doses outperformed placebo in all endpoints.
Among those taking 200 mg, the annual relapse rate was 0.46 versus 0.87 among those taking placebo – a significant 47.7% risk reduction. Among those taking 300 mg, the exacerbation rate was 0.52 versus 0.97; this translated to a significant 46% risk reduction.
The response rate was even greater among those with an eosinophil count greater than 300 per cubic millimeter: 0.37 for 200 mg and 0.40 for 300 mg versus the placebo rates of 1.08 and 1.24. This translated to risk reductions of 65.8% and 67.4%, respectively.
By week 12, FEV1 had significantly increased by 0.32 L in the 200-mg group and by 0.34 L in the 300-mg group, compared with nonsignificant increases among those taking placebo.
Again, patients with the high eosinophil counts experienced the greatest benefits, with FEV1 increasing by a mean of 0.43 L at 12 weeks in the 200-mg group and by 0.47 L in the 300-mg group, significantly better than either placebo comparator.
The benefit was already noticeable by the 2-week evaluation, the investigators noted.
Dupilumab appeared safe; injection-site reactions were the most common adverse event, occurring in 15% of the low-dose group and 18% of the high-dose group. However, 52 patients taking the drug experienced eosinophilia, compared with four of those taking placebo (4.1% vs. 0.6%).
Four of those taking the study drug experienced clinical symptoms associated with eosinophilia, including worsening eosinophilia and chronic eosinophilic pneumonia.
Liberty Asthma Venture
In this study, the effect of dupilumab on glucocorticoid use among 210 patients with severe asthma was examined. Patients were randomized to add-on dupilumab 300 mg every 2 weeks for 24 weeks. Glucocorticoids were tapered downward from weeks 4 to 20. The primary endpoints were percent reduction in glucocorticoid dose at week 24, and the percentage of patients who experienced a reduction of at least 50% in glucocorticoid dose.
These patients were a mean of 51 years old, with a mean of two severe asthma exacerbations in the past year. Their mean daily oral glucocorticoid dose at randomization was about 11 mg per day. Their mean prebronchodilator FEV1 was about 1.6 liters – about 52% of predicted value.
Oral glucocorticoid use decreased by a mean of 70.1% in the active group, compared with 41.9% in the placebo group, a statistically significant difference, Klaus F. Rabe, MD, of Christian Albrechts University, Kiel, Germany, and his coauthors wrote in the New England Journal of Medicine. The median change was even better: A 100% reduction in the active group and 50% reduction in the placebo group.
By week 24, 80% of those taking dupilumab had decreased their glucocorticoid intake by at least 50%, compared with 50% of the placebo group reaching this goal. The glucocorticoid dose was less than 5 mg/day in 69% of the dupilumab group, compared with 33% of the placebo group.
Like Quest, Venture showed a treatment advantage among patients with high baseline eosinophil count. “The magnitude of the effect was largest in patients with a higher eosinophil count at baseline,” the investigators wrote. “… The odds ratios [a 50% glucocorticoid reduction] for dupilumab versus placebo were 6.59 among patients with 300 or more cells per cubic millimeter at baseline and 2.91 among those with less than 300 cells per cubic millimeter at baseline.”
In a fully adjusted model at week 24, 48% of the patients in the dupilumab group were able to stop oral glucocorticoids entirely, compared with 25% of the placebo group.
Dupilumab was also associated with a significant 59% reduction in severe annual asthma exacerbations. FEV1 among the active group was 0.22 L better than that in the placebo group at week 24.
Again, patients with a higher baseline blood eosinophil count experienced greater treatment benefit; among these, the rate of severe asthma exacerbations was 71% lower than the rate in the placebo group, and FEV1 was 0.32 L higher.
The most frequent adverse events were viral infections (9% of the patients in the dupilumab group vs. 18% of those in the placebo group), bronchitis (7% vs. 6%), sinusitis (7% vs. 4%), influenza (3% vs. 6%), and eosinophilia (14% vs. 1%). Injection-site reactions occurred in 9% of those taking dupilumab and 4% of those taking placebo.
Antidrug antibodies developed in five patients in each group, without clinical effect.
Both trials were funded by Sanofi and Regeneron. Dr. Castro has received grant support from Sanofi. Dr. Rabe has received consulting and lecture fees from AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, and Teva Pharmaceutical Industries.
SOURCES: Castro M et al. N Engl J Med. 2018;378:2486-96; KF Rabe et al. N Engl J Med. 2018;378:2475-85.
Among patients with moderate to severe asthma, dupilumab reduced exacerbations by almost 50%, while also allowing glucocorticoid-treated patients to cut their use of that medication by 70%, with no increased risk of exacerbation.
The pair of placebo-controlled studies – Liberty Asthma Quest and Liberty Asthma Venture – also showed treatment-associated stability in forced expiratory volume (FEV1) evidence of lung remodeling among those who took the antibody, Mario Castro, MD, of Washington University, St. Louis, and his colleagues reported in the New England Journal of Medicine.
By week 12, FEV1 it had already increased by 0.32 L, they said.
“An analysis of the postbronchodilator FEV1 slope showed a loss of lung function in patients who received placebo and no loss in those who received dupilumab, findings that suggest a potential effect of dupilumab on airway remodeling,” wrote Dr. Castro and his colleagues. “The slope analysis showed that patients who received placebo lost, on average, approximately 40 mL annually, which is consistent with data from other cohorts of patients with asthma.”
Dupilumab is an anti–interleukin-4 alpha antibody that blocks both IL-4 and IL-13. The Quest trial examined efficacy and safety of two doses (200 mg and 300 mg every 2 weeks), compared with placebo in patients with uncontrolled asthma. Venture examined efficacy and safety of 300 mg or placebo as add-on therapy for patients with severe asthma who were taking glucocorticoids.
Liberty Asthma Quest
This 52-week study randomized 1,902 patients with severe, uncontrolled asthma to placebo or dupilumab 200 mg or 300 mg every other week. The primary endpoints were annual rate of severe asthma exacerbations and the change in FEV1 by week 12. The study also looked at these endpoints in patients whose baseline eosinophil count was greater than 300 per cubic millimeter.
Patients were a mean of 48 years old with a mean baseline FEV1 of about 1.75 L (about 58% of the predicted normal value). They had a mean of two exacerbations per year and an average eosinophil count of about 350 per cubic millimeter.
Both doses outperformed placebo in all endpoints.
Among those taking 200 mg, the annual relapse rate was 0.46 versus 0.87 among those taking placebo – a significant 47.7% risk reduction. Among those taking 300 mg, the exacerbation rate was 0.52 versus 0.97; this translated to a significant 46% risk reduction.
The response rate was even greater among those with an eosinophil count greater than 300 per cubic millimeter: 0.37 for 200 mg and 0.40 for 300 mg versus the placebo rates of 1.08 and 1.24. This translated to risk reductions of 65.8% and 67.4%, respectively.
By week 12, FEV1 had significantly increased by 0.32 L in the 200-mg group and by 0.34 L in the 300-mg group, compared with nonsignificant increases among those taking placebo.
Again, patients with the high eosinophil counts experienced the greatest benefits, with FEV1 increasing by a mean of 0.43 L at 12 weeks in the 200-mg group and by 0.47 L in the 300-mg group, significantly better than either placebo comparator.
The benefit was already noticeable by the 2-week evaluation, the investigators noted.
Dupilumab appeared safe; injection-site reactions were the most common adverse event, occurring in 15% of the low-dose group and 18% of the high-dose group. However, 52 patients taking the drug experienced eosinophilia, compared with four of those taking placebo (4.1% vs. 0.6%).
Four of those taking the study drug experienced clinical symptoms associated with eosinophilia, including worsening eosinophilia and chronic eosinophilic pneumonia.
Liberty Asthma Venture
In this study, the effect of dupilumab on glucocorticoid use among 210 patients with severe asthma was examined. Patients were randomized to add-on dupilumab 300 mg every 2 weeks for 24 weeks. Glucocorticoids were tapered downward from weeks 4 to 20. The primary endpoints were percent reduction in glucocorticoid dose at week 24, and the percentage of patients who experienced a reduction of at least 50% in glucocorticoid dose.
These patients were a mean of 51 years old, with a mean of two severe asthma exacerbations in the past year. Their mean daily oral glucocorticoid dose at randomization was about 11 mg per day. Their mean prebronchodilator FEV1 was about 1.6 liters – about 52% of predicted value.
Oral glucocorticoid use decreased by a mean of 70.1% in the active group, compared with 41.9% in the placebo group, a statistically significant difference, Klaus F. Rabe, MD, of Christian Albrechts University, Kiel, Germany, and his coauthors wrote in the New England Journal of Medicine. The median change was even better: A 100% reduction in the active group and 50% reduction in the placebo group.
By week 24, 80% of those taking dupilumab had decreased their glucocorticoid intake by at least 50%, compared with 50% of the placebo group reaching this goal. The glucocorticoid dose was less than 5 mg/day in 69% of the dupilumab group, compared with 33% of the placebo group.
Like Quest, Venture showed a treatment advantage among patients with high baseline eosinophil count. “The magnitude of the effect was largest in patients with a higher eosinophil count at baseline,” the investigators wrote. “… The odds ratios [a 50% glucocorticoid reduction] for dupilumab versus placebo were 6.59 among patients with 300 or more cells per cubic millimeter at baseline and 2.91 among those with less than 300 cells per cubic millimeter at baseline.”
In a fully adjusted model at week 24, 48% of the patients in the dupilumab group were able to stop oral glucocorticoids entirely, compared with 25% of the placebo group.
Dupilumab was also associated with a significant 59% reduction in severe annual asthma exacerbations. FEV1 among the active group was 0.22 L better than that in the placebo group at week 24.
Again, patients with a higher baseline blood eosinophil count experienced greater treatment benefit; among these, the rate of severe asthma exacerbations was 71% lower than the rate in the placebo group, and FEV1 was 0.32 L higher.
The most frequent adverse events were viral infections (9% of the patients in the dupilumab group vs. 18% of those in the placebo group), bronchitis (7% vs. 6%), sinusitis (7% vs. 4%), influenza (3% vs. 6%), and eosinophilia (14% vs. 1%). Injection-site reactions occurred in 9% of those taking dupilumab and 4% of those taking placebo.
Antidrug antibodies developed in five patients in each group, without clinical effect.
Both trials were funded by Sanofi and Regeneron. Dr. Castro has received grant support from Sanofi. Dr. Rabe has received consulting and lecture fees from AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, and Teva Pharmaceutical Industries.
SOURCES: Castro M et al. N Engl J Med. 2018;378:2486-96; KF Rabe et al. N Engl J Med. 2018;378:2475-85.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Dupilumab allowed asthma patients to decrease glucocorticoids with no risk of asthma exacerbation.
Major finding: Dupilumab reduced exacerbations by almost 50%, while also allowing glucocorticoid-treated patients to cut their use of that medication by 70%.
Study details: Liberty Asthma Quest comprised 1,902 patients and Liberty Asthma Venture comprised 210. Both were randomized, placebo-controlled trials.
Disclosures: Both trials were funded by Sanofi and Regeneron. Dr. Castro has received grant support from Sanofi. Dr. Rabe has received consulting and lecture fees from AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, and Teva Pharmaceutical Industries.
Sources: Castro M et al. N Engl J Med. 2018;378:2486-96; Rabe KF et al. N Engl J Med. 2018;378:2475-85.
ACIP votes to recommend new strains for the 2018-2019 flu vaccine
Thirteen members of the Advisory Committee on Immunization Practices (ACIP) voted to approve the influenza vaccine recommendations for 2018-2019, while one member abstained from voting at the summer ACIP meeting.
The 2018-2019 recommendation maintains the core recommendation that influenza vaccines should be administered to all persons 6 months or older who have no contraindications.
FluMist Quadrivalent (LAIV4) also is being updated for the 2018-2019 season. At the February meeting of ACIP, the committee approved language that providers may provide any licensed, age-appropriate influenza vaccine, and LAIV4 is considered in this set of vaccine options.
Prior to this approval, there was a discussion of the safety of the 2017-2018 vaccine. For many of the available vaccines, there were no new safety concerns raised from reports during the flu season. Monitoring during the 2018-2019 will yield more safety monitoring data concerning pregnancy and influenza vaccinations and anaphylaxis in persons with an egg allergy.
The committee’s recommendations must be approved by the Centers for Disease Control and Prevention’s director before they are considered official recommendations.
Thirteen members of the Advisory Committee on Immunization Practices (ACIP) voted to approve the influenza vaccine recommendations for 2018-2019, while one member abstained from voting at the summer ACIP meeting.
The 2018-2019 recommendation maintains the core recommendation that influenza vaccines should be administered to all persons 6 months or older who have no contraindications.
FluMist Quadrivalent (LAIV4) also is being updated for the 2018-2019 season. At the February meeting of ACIP, the committee approved language that providers may provide any licensed, age-appropriate influenza vaccine, and LAIV4 is considered in this set of vaccine options.
Prior to this approval, there was a discussion of the safety of the 2017-2018 vaccine. For many of the available vaccines, there were no new safety concerns raised from reports during the flu season. Monitoring during the 2018-2019 will yield more safety monitoring data concerning pregnancy and influenza vaccinations and anaphylaxis in persons with an egg allergy.
The committee’s recommendations must be approved by the Centers for Disease Control and Prevention’s director before they are considered official recommendations.
Thirteen members of the Advisory Committee on Immunization Practices (ACIP) voted to approve the influenza vaccine recommendations for 2018-2019, while one member abstained from voting at the summer ACIP meeting.
The 2018-2019 recommendation maintains the core recommendation that influenza vaccines should be administered to all persons 6 months or older who have no contraindications.
FluMist Quadrivalent (LAIV4) also is being updated for the 2018-2019 season. At the February meeting of ACIP, the committee approved language that providers may provide any licensed, age-appropriate influenza vaccine, and LAIV4 is considered in this set of vaccine options.
Prior to this approval, there was a discussion of the safety of the 2017-2018 vaccine. For many of the available vaccines, there were no new safety concerns raised from reports during the flu season. Monitoring during the 2018-2019 will yield more safety monitoring data concerning pregnancy and influenza vaccinations and anaphylaxis in persons with an egg allergy.
The committee’s recommendations must be approved by the Centers for Disease Control and Prevention’s director before they are considered official recommendations.
REPORTING FROM AN ACIP MEETING
Additional training may be warranted for clinicians administering DTaP
Additional training may be needed for providers who administer DTaP vaccine to prevent errors in vaccination, but reported Pedro Moro, MD, MPH, of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases and his associates in Pediatrics.
After Dr. Moro and his associates performed an automated analysis of all reports included in the Vaccine Adverse Event Reporting System (VAERS), which is coadministered by the CDC and the Food and Drug Administration, as well as a clinical review of reported deaths and a random sampling of serious reports in the database, they concluded that safety findings concerning DTaP were consistent with those from prelicensure trials and postlicensure studies.
DTaP vaccines, which included Infanrix, Daptacel, Pediarix, Kinrix, and Pentacel, were coadministered with one or more other vaccines in 43,984 (88%) of cases reported; of the reports included in the data mining, 5,627 (11%) were classified as serious, including 844 (2%) deaths. Of all reports received in the prelicensure clinical trials, injection site reactions and systemic reactions, such as fever and vomiting, were the most common reactions to DTaP vaccine.
In a 5% random sample of the 4,783 serious nondeath reports included in the study, 25% were neurologic, 23% gastrointestinal, and 20% were caused by general disorders and vaccine site conditions. Fully 80% of those flagged as neurologic were seizure related. In another 79%, for which intussusception was the most common gastrointestinal condition, all but two cases had rotavirus vaccine coadministered with DTaP. Altogether, there were 182 cases of anaphylaxis reported.
Serious events were characterized as death, life-threatening illness, hospitalization, lengthening of existing hospital stay, or permanent disability. In cases of death, reports that followed DTaP vaccine were manually reviewed by a physician, who evaluated autopsy report, death certificate, or medical records. The authors also included in their evaluation of records any reports of postvaccine anaphylaxis.
Of the 844 deaths, death certificates, autopsy reports, or medical records were obtained for 86%. Among these, sudden infant death syndrome (SIDS) was found to be the most frequent cause of death in 48%; of these, 62% were male infants, and 91% were infants under 6 months of age.
“It would not be uncommon to observe a coincidental close temporal relationship between vaccination and SIDS because this condition peaks at a time when children receive a relatively large number of recommended vaccinations,” said Dr. Moro and his associates. “There is a large body of evidence in which it is shown that vaccination is not causally associated with SIDS.”
The authors identified disproportional reporting for injection site reactions, as well as other events and conditions, to which they attribute, at least in part, administration of the wrong vaccine or formulation and administration at the wrong site. Such mistakes can be lessened or even prevented with provider education and training on appropriate recommendations and package insert specifications put forth by the CDC’s Advisory Committee on Immunization Practices, they advised.
While the authors praised VAERS for the wealth of timely data it has offered in detecting potential safety issues that may require further investigation, Dr. Moro cautioned that it is a passive surveillance system with limitations that warrant “careful interpretation of its findings.” Its purpose is to improve immunization programs.
Because it does not “meet the definition of research,” the work performed in this study was not subject to institutional review board evaluation and informed consent requirements, the authors added. VAERS generally is not able to assess whether vaccines are the direct cause of adverse events, primarily because of underreporting or overreporting, biased reporting, and inconsistency in quality and completeness of information reported. Because it does not tally number of vaccines administered, it is also unable to provide data needed to calculate incidence rates.
The authors had no relevant financial disclosures. The study was funded by the CDC and the FDA.
SOURCE: Moro P et al. Pediatrics. 2018. doi: 10.1542/peds.2017-4171.
The Vaccine Adverse Event Reporting System offers confirmation that DTaP vaccines are safe and have a reasonably low frequency of adverse events. Despite this, the U.S.-based resurgence of pertussis shortly after acellular vaccines were introduced legitimately raised concerns over the efficacy of DTaP, which is now known to have a shorter duration of protection than its predecessor, the diphtheria, tetanus toxoids, whole-cell pertussis vaccine. Consequently, older children, adolescents, and adults are left unprotected without periodic booster doses, Flor M. Muñoz, MD, wrote in an editorial accompanying the study by Moro et al.
The World Health Organization’s recommendation to countries that never made the switch to DTaP is to continue using the whole-cell vaccines “because of their consistent higher efficacy” points to “an imperative need to develop more immunogenic pertussis vaccines that are also safe,” she observed.
“Active research is ongoing for the development of novel vaccines, including live attenuated vaccines, whole-cell vaccines with reduced endotoxin content to be less reactogenic, outer membrane vesicles–based vaccines, and acellular vaccine formulations prepared with new adjuvants or additional and novel antigens.
“As we go back to the drawing board in the fight against Bordetella pertussis, much work is needed to learn more about this fascinating pathogen and its interactions with humans to improve our understanding of how immunity and long-lasting protection can be achieved, to engineer and produce novel vaccines, and to design and perform the clinical studies that will eventually lead to the control of pertussis disease and its global impact with safe and effective vaccines for all,” Dr. Muñoz added.
Dr. Muñoz is affiliated with the section of infectious diseases in the department of pediatrics at Baylor College of Medicine and Texas Children’s Hospital, both in Houston. Her comments here were summarized from her editorial accompanying the article by Moro et al (Pediatrics. 2018. doi: 10.1542/peds.2018-1036). Dr. Munoz said she had no relevant financial disclosures and received no external funding.
The Vaccine Adverse Event Reporting System offers confirmation that DTaP vaccines are safe and have a reasonably low frequency of adverse events. Despite this, the U.S.-based resurgence of pertussis shortly after acellular vaccines were introduced legitimately raised concerns over the efficacy of DTaP, which is now known to have a shorter duration of protection than its predecessor, the diphtheria, tetanus toxoids, whole-cell pertussis vaccine. Consequently, older children, adolescents, and adults are left unprotected without periodic booster doses, Flor M. Muñoz, MD, wrote in an editorial accompanying the study by Moro et al.
The World Health Organization’s recommendation to countries that never made the switch to DTaP is to continue using the whole-cell vaccines “because of their consistent higher efficacy” points to “an imperative need to develop more immunogenic pertussis vaccines that are also safe,” she observed.
“Active research is ongoing for the development of novel vaccines, including live attenuated vaccines, whole-cell vaccines with reduced endotoxin content to be less reactogenic, outer membrane vesicles–based vaccines, and acellular vaccine formulations prepared with new adjuvants or additional and novel antigens.
“As we go back to the drawing board in the fight against Bordetella pertussis, much work is needed to learn more about this fascinating pathogen and its interactions with humans to improve our understanding of how immunity and long-lasting protection can be achieved, to engineer and produce novel vaccines, and to design and perform the clinical studies that will eventually lead to the control of pertussis disease and its global impact with safe and effective vaccines for all,” Dr. Muñoz added.
Dr. Muñoz is affiliated with the section of infectious diseases in the department of pediatrics at Baylor College of Medicine and Texas Children’s Hospital, both in Houston. Her comments here were summarized from her editorial accompanying the article by Moro et al (Pediatrics. 2018. doi: 10.1542/peds.2018-1036). Dr. Munoz said she had no relevant financial disclosures and received no external funding.
The Vaccine Adverse Event Reporting System offers confirmation that DTaP vaccines are safe and have a reasonably low frequency of adverse events. Despite this, the U.S.-based resurgence of pertussis shortly after acellular vaccines were introduced legitimately raised concerns over the efficacy of DTaP, which is now known to have a shorter duration of protection than its predecessor, the diphtheria, tetanus toxoids, whole-cell pertussis vaccine. Consequently, older children, adolescents, and adults are left unprotected without periodic booster doses, Flor M. Muñoz, MD, wrote in an editorial accompanying the study by Moro et al.
The World Health Organization’s recommendation to countries that never made the switch to DTaP is to continue using the whole-cell vaccines “because of their consistent higher efficacy” points to “an imperative need to develop more immunogenic pertussis vaccines that are also safe,” she observed.
“Active research is ongoing for the development of novel vaccines, including live attenuated vaccines, whole-cell vaccines with reduced endotoxin content to be less reactogenic, outer membrane vesicles–based vaccines, and acellular vaccine formulations prepared with new adjuvants or additional and novel antigens.
“As we go back to the drawing board in the fight against Bordetella pertussis, much work is needed to learn more about this fascinating pathogen and its interactions with humans to improve our understanding of how immunity and long-lasting protection can be achieved, to engineer and produce novel vaccines, and to design and perform the clinical studies that will eventually lead to the control of pertussis disease and its global impact with safe and effective vaccines for all,” Dr. Muñoz added.
Dr. Muñoz is affiliated with the section of infectious diseases in the department of pediatrics at Baylor College of Medicine and Texas Children’s Hospital, both in Houston. Her comments here were summarized from her editorial accompanying the article by Moro et al (Pediatrics. 2018. doi: 10.1542/peds.2018-1036). Dr. Munoz said she had no relevant financial disclosures and received no external funding.
Additional training may be needed for providers who administer DTaP vaccine to prevent errors in vaccination, but reported Pedro Moro, MD, MPH, of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases and his associates in Pediatrics.
After Dr. Moro and his associates performed an automated analysis of all reports included in the Vaccine Adverse Event Reporting System (VAERS), which is coadministered by the CDC and the Food and Drug Administration, as well as a clinical review of reported deaths and a random sampling of serious reports in the database, they concluded that safety findings concerning DTaP were consistent with those from prelicensure trials and postlicensure studies.
DTaP vaccines, which included Infanrix, Daptacel, Pediarix, Kinrix, and Pentacel, were coadministered with one or more other vaccines in 43,984 (88%) of cases reported; of the reports included in the data mining, 5,627 (11%) were classified as serious, including 844 (2%) deaths. Of all reports received in the prelicensure clinical trials, injection site reactions and systemic reactions, such as fever and vomiting, were the most common reactions to DTaP vaccine.
In a 5% random sample of the 4,783 serious nondeath reports included in the study, 25% were neurologic, 23% gastrointestinal, and 20% were caused by general disorders and vaccine site conditions. Fully 80% of those flagged as neurologic were seizure related. In another 79%, for which intussusception was the most common gastrointestinal condition, all but two cases had rotavirus vaccine coadministered with DTaP. Altogether, there were 182 cases of anaphylaxis reported.
Serious events were characterized as death, life-threatening illness, hospitalization, lengthening of existing hospital stay, or permanent disability. In cases of death, reports that followed DTaP vaccine were manually reviewed by a physician, who evaluated autopsy report, death certificate, or medical records. The authors also included in their evaluation of records any reports of postvaccine anaphylaxis.
Of the 844 deaths, death certificates, autopsy reports, or medical records were obtained for 86%. Among these, sudden infant death syndrome (SIDS) was found to be the most frequent cause of death in 48%; of these, 62% were male infants, and 91% were infants under 6 months of age.
“It would not be uncommon to observe a coincidental close temporal relationship between vaccination and SIDS because this condition peaks at a time when children receive a relatively large number of recommended vaccinations,” said Dr. Moro and his associates. “There is a large body of evidence in which it is shown that vaccination is not causally associated with SIDS.”
The authors identified disproportional reporting for injection site reactions, as well as other events and conditions, to which they attribute, at least in part, administration of the wrong vaccine or formulation and administration at the wrong site. Such mistakes can be lessened or even prevented with provider education and training on appropriate recommendations and package insert specifications put forth by the CDC’s Advisory Committee on Immunization Practices, they advised.
While the authors praised VAERS for the wealth of timely data it has offered in detecting potential safety issues that may require further investigation, Dr. Moro cautioned that it is a passive surveillance system with limitations that warrant “careful interpretation of its findings.” Its purpose is to improve immunization programs.
Because it does not “meet the definition of research,” the work performed in this study was not subject to institutional review board evaluation and informed consent requirements, the authors added. VAERS generally is not able to assess whether vaccines are the direct cause of adverse events, primarily because of underreporting or overreporting, biased reporting, and inconsistency in quality and completeness of information reported. Because it does not tally number of vaccines administered, it is also unable to provide data needed to calculate incidence rates.
The authors had no relevant financial disclosures. The study was funded by the CDC and the FDA.
SOURCE: Moro P et al. Pediatrics. 2018. doi: 10.1542/peds.2017-4171.
Additional training may be needed for providers who administer DTaP vaccine to prevent errors in vaccination, but reported Pedro Moro, MD, MPH, of the Centers for Disease Control and Prevention’s National Center for Immunization and Respiratory Diseases and his associates in Pediatrics.
After Dr. Moro and his associates performed an automated analysis of all reports included in the Vaccine Adverse Event Reporting System (VAERS), which is coadministered by the CDC and the Food and Drug Administration, as well as a clinical review of reported deaths and a random sampling of serious reports in the database, they concluded that safety findings concerning DTaP were consistent with those from prelicensure trials and postlicensure studies.
DTaP vaccines, which included Infanrix, Daptacel, Pediarix, Kinrix, and Pentacel, were coadministered with one or more other vaccines in 43,984 (88%) of cases reported; of the reports included in the data mining, 5,627 (11%) were classified as serious, including 844 (2%) deaths. Of all reports received in the prelicensure clinical trials, injection site reactions and systemic reactions, such as fever and vomiting, were the most common reactions to DTaP vaccine.
In a 5% random sample of the 4,783 serious nondeath reports included in the study, 25% were neurologic, 23% gastrointestinal, and 20% were caused by general disorders and vaccine site conditions. Fully 80% of those flagged as neurologic were seizure related. In another 79%, for which intussusception was the most common gastrointestinal condition, all but two cases had rotavirus vaccine coadministered with DTaP. Altogether, there were 182 cases of anaphylaxis reported.
Serious events were characterized as death, life-threatening illness, hospitalization, lengthening of existing hospital stay, or permanent disability. In cases of death, reports that followed DTaP vaccine were manually reviewed by a physician, who evaluated autopsy report, death certificate, or medical records. The authors also included in their evaluation of records any reports of postvaccine anaphylaxis.
Of the 844 deaths, death certificates, autopsy reports, or medical records were obtained for 86%. Among these, sudden infant death syndrome (SIDS) was found to be the most frequent cause of death in 48%; of these, 62% were male infants, and 91% were infants under 6 months of age.
“It would not be uncommon to observe a coincidental close temporal relationship between vaccination and SIDS because this condition peaks at a time when children receive a relatively large number of recommended vaccinations,” said Dr. Moro and his associates. “There is a large body of evidence in which it is shown that vaccination is not causally associated with SIDS.”
The authors identified disproportional reporting for injection site reactions, as well as other events and conditions, to which they attribute, at least in part, administration of the wrong vaccine or formulation and administration at the wrong site. Such mistakes can be lessened or even prevented with provider education and training on appropriate recommendations and package insert specifications put forth by the CDC’s Advisory Committee on Immunization Practices, they advised.
While the authors praised VAERS for the wealth of timely data it has offered in detecting potential safety issues that may require further investigation, Dr. Moro cautioned that it is a passive surveillance system with limitations that warrant “careful interpretation of its findings.” Its purpose is to improve immunization programs.
Because it does not “meet the definition of research,” the work performed in this study was not subject to institutional review board evaluation and informed consent requirements, the authors added. VAERS generally is not able to assess whether vaccines are the direct cause of adverse events, primarily because of underreporting or overreporting, biased reporting, and inconsistency in quality and completeness of information reported. Because it does not tally number of vaccines administered, it is also unable to provide data needed to calculate incidence rates.
The authors had no relevant financial disclosures. The study was funded by the CDC and the FDA.
SOURCE: Moro P et al. Pediatrics. 2018. doi: 10.1542/peds.2017-4171.
FROM PEDIATRICS
Key clinical point: No new or unexpected safety issues were found with DTaP.
Major finding: Nearly 90% of adverse events reported were not considered serious.
Study details: Large-scale data mining and records review from the Vaccine Adverse Event Reporting System.
Disclosures: The authors had no relevant financial disclosures. The study was funded by the Centers for Disease Control and Prevention and the Food and Drug Administration.
Source: Moro P et al. Pediatrics. 2018. doi: 10.1542/peds.2017-4171.
New NIH consortium aims to coordinate pediatric research programs
across its institutes and centers.
Almost all of the 27 institutes and centers of the NIH fund at least some kind of child health research, totaling more than $4 billion in the 2017 fiscal year, according to an NIH statement. “The new consortium aims to harmonize these activities, explore gaps and opportunities in the overall pediatric research portfolio, and set priorities.”
Research funded by NIH “has resulted in tremendous advances against diseases and conditions that affect child health and well-being, including asthma, cancer, autism, obesity, and intellectual and developmental disabilities,” explained Diana W. Bianchi, MD, in the statement. Dr. Bianchi is director of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the lead NIH institute for the consortium.
The new consortium, which will be led by the NICHD director, will meet several times a year.
across its institutes and centers.
Almost all of the 27 institutes and centers of the NIH fund at least some kind of child health research, totaling more than $4 billion in the 2017 fiscal year, according to an NIH statement. “The new consortium aims to harmonize these activities, explore gaps and opportunities in the overall pediatric research portfolio, and set priorities.”
Research funded by NIH “has resulted in tremendous advances against diseases and conditions that affect child health and well-being, including asthma, cancer, autism, obesity, and intellectual and developmental disabilities,” explained Diana W. Bianchi, MD, in the statement. Dr. Bianchi is director of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the lead NIH institute for the consortium.
The new consortium, which will be led by the NICHD director, will meet several times a year.
across its institutes and centers.
Almost all of the 27 institutes and centers of the NIH fund at least some kind of child health research, totaling more than $4 billion in the 2017 fiscal year, according to an NIH statement. “The new consortium aims to harmonize these activities, explore gaps and opportunities in the overall pediatric research portfolio, and set priorities.”
Research funded by NIH “has resulted in tremendous advances against diseases and conditions that affect child health and well-being, including asthma, cancer, autism, obesity, and intellectual and developmental disabilities,” explained Diana W. Bianchi, MD, in the statement. Dr. Bianchi is director of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, the lead NIH institute for the consortium.
The new consortium, which will be led by the NICHD director, will meet several times a year.
Benefits of nicotine preloading undercut by reduced varenicline usage
Nicotine preloading with patches 4 weeks before making a quit attempt was not significantly associated with according to Paul Aveyard, PhD, and his associates at Nuffield Department of Primary Care Health Sciences, University of Oxford (England).
The primary study outcome, biochemically validated abstinence at 6 months, was achieved by 17.5% of the 899 people who preloaded with a 21-mg/24-hr nicotine patch for 4 weeks and by 14.4% of the 893 in the control group. After 1 year, 14.0% of people in the preloading group maintained long-term abstinence, compared with 11.3% in the control group. In addition, 35.5% of the preloading group and 32.3% of the control group achieved abstinence 4 weeks from baseline.
The unadjusted odds ratio for the effect of preloading at 6 months was 1.25 (95% confidence interval, 0.97-1.62; P = .08) and not statistically significant. However, when reduced varenicline usage in the preloading group was taken into account, the effect of preloading did reach statistical significance (OR, 1.34; 95% CI, 1.03-1.73; P = .03). Similar results were found at 1 year and at 4 weeks, where the preloading effect did not reach significance until adjusted for varenicline usage.
“Nicotine preloading with a 21-mg/24-hr nicotine patch for 4 weeks seems to be efficacious, safe, and well tolerated, but probably deters the use of varenicline, the most effective smoking cessation drug. If it were possible to overcome this unintended consequence, preloading could lead to a worthwhile increase in long-term smoking abstinence,” the investigators concluded.
SOURCE: Aveyard P et al. BMJ. 2018 Jun 13. doi: 10.1136/bmj.k2164.
Nicotine preloading with patches 4 weeks before making a quit attempt was not significantly associated with according to Paul Aveyard, PhD, and his associates at Nuffield Department of Primary Care Health Sciences, University of Oxford (England).
The primary study outcome, biochemically validated abstinence at 6 months, was achieved by 17.5% of the 899 people who preloaded with a 21-mg/24-hr nicotine patch for 4 weeks and by 14.4% of the 893 in the control group. After 1 year, 14.0% of people in the preloading group maintained long-term abstinence, compared with 11.3% in the control group. In addition, 35.5% of the preloading group and 32.3% of the control group achieved abstinence 4 weeks from baseline.
The unadjusted odds ratio for the effect of preloading at 6 months was 1.25 (95% confidence interval, 0.97-1.62; P = .08) and not statistically significant. However, when reduced varenicline usage in the preloading group was taken into account, the effect of preloading did reach statistical significance (OR, 1.34; 95% CI, 1.03-1.73; P = .03). Similar results were found at 1 year and at 4 weeks, where the preloading effect did not reach significance until adjusted for varenicline usage.
“Nicotine preloading with a 21-mg/24-hr nicotine patch for 4 weeks seems to be efficacious, safe, and well tolerated, but probably deters the use of varenicline, the most effective smoking cessation drug. If it were possible to overcome this unintended consequence, preloading could lead to a worthwhile increase in long-term smoking abstinence,” the investigators concluded.
SOURCE: Aveyard P et al. BMJ. 2018 Jun 13. doi: 10.1136/bmj.k2164.
Nicotine preloading with patches 4 weeks before making a quit attempt was not significantly associated with according to Paul Aveyard, PhD, and his associates at Nuffield Department of Primary Care Health Sciences, University of Oxford (England).
The primary study outcome, biochemically validated abstinence at 6 months, was achieved by 17.5% of the 899 people who preloaded with a 21-mg/24-hr nicotine patch for 4 weeks and by 14.4% of the 893 in the control group. After 1 year, 14.0% of people in the preloading group maintained long-term abstinence, compared with 11.3% in the control group. In addition, 35.5% of the preloading group and 32.3% of the control group achieved abstinence 4 weeks from baseline.
The unadjusted odds ratio for the effect of preloading at 6 months was 1.25 (95% confidence interval, 0.97-1.62; P = .08) and not statistically significant. However, when reduced varenicline usage in the preloading group was taken into account, the effect of preloading did reach statistical significance (OR, 1.34; 95% CI, 1.03-1.73; P = .03). Similar results were found at 1 year and at 4 weeks, where the preloading effect did not reach significance until adjusted for varenicline usage.
“Nicotine preloading with a 21-mg/24-hr nicotine patch for 4 weeks seems to be efficacious, safe, and well tolerated, but probably deters the use of varenicline, the most effective smoking cessation drug. If it were possible to overcome this unintended consequence, preloading could lead to a worthwhile increase in long-term smoking abstinence,” the investigators concluded.
SOURCE: Aveyard P et al. BMJ. 2018 Jun 13. doi: 10.1136/bmj.k2164.
FROM THE BMJ
E-cigarette flavorings foster cardiovascular dysfunction
Flavored tobacco products are popular among current smokers, including youth, and the flavorings have been deemed ingestible, but their impact on heart health has not been studied, wrote Jennifer Fetterman, PhD, of Boston University, and her colleagues. The report was published in Arteriosclerosis, Thrombosis, and Vascular Biology.
The researchers studied nine types of flavorings used in alternative tobacco products to assess their impact on cardiovascular health.
The first part of the study comprised a population of nine nonsmokers, six nonmenthol cigarette smokers, and six menthol cigarette smokers without cardiovascular disease. The researchers isolated venous endothelial cells from each participant.
Overall, cells from both nonmenthol and menthol cigarette smokers had significantly lower nitric oxide production compared with nonsmokers (P = .003 and P = .012, respectively). In addition, the flavoring compounds menthol and eugenol impaired nitric oxide production in the cells of healthy individuals.
“Increased inflammation and a loss of nitric oxide are some of the first changes to occur leading up to cardiovascular disease and events like heart attacks and stroke, so they are considered early predictors of heart disease,” Dr. Fetterman said in a statement, adding that the “findings suggest that these flavoring additives may have serious health consequences.”
All nine flavorings induced cell death at the highest concentration tested, ranging from 10 to 100 mmol/L).
The study findings were limited by several factors, primarily a lack of data on how heating the flavorings in the in vitro part of the study might have affected toxicity in the body, the researchers noted.
“Future studies will focus on how the toxicity of the flavorings is altered with heating and characterization of the levels obtained in the circulation after use of an e-cigarette,” they said.
However, data support the need for regulation and limits on the level of flavorings used in e-cigarettes and other tobacco products, they emphasized.
“These findings suggest that flavoring compounds induce endothelial cell dysfunction in human cells similarly to the abnormal function in active cigarette smokers,” the researchers noted.
The study was funded by the National Heart, Lung, and Blood Institute; Food and Drug Administration Center for Tobacco Products; and the American Heart Association. The researchers had no financial conflicts to disclose.
SOURCE: Fetterman J et al. Arterioscler Thromb Vasc Biol. 2018. doi: 10.1161/ATVBAHA.118.311156.
Flavored tobacco products are popular among current smokers, including youth, and the flavorings have been deemed ingestible, but their impact on heart health has not been studied, wrote Jennifer Fetterman, PhD, of Boston University, and her colleagues. The report was published in Arteriosclerosis, Thrombosis, and Vascular Biology.
The researchers studied nine types of flavorings used in alternative tobacco products to assess their impact on cardiovascular health.
The first part of the study comprised a population of nine nonsmokers, six nonmenthol cigarette smokers, and six menthol cigarette smokers without cardiovascular disease. The researchers isolated venous endothelial cells from each participant.
Overall, cells from both nonmenthol and menthol cigarette smokers had significantly lower nitric oxide production compared with nonsmokers (P = .003 and P = .012, respectively). In addition, the flavoring compounds menthol and eugenol impaired nitric oxide production in the cells of healthy individuals.
“Increased inflammation and a loss of nitric oxide are some of the first changes to occur leading up to cardiovascular disease and events like heart attacks and stroke, so they are considered early predictors of heart disease,” Dr. Fetterman said in a statement, adding that the “findings suggest that these flavoring additives may have serious health consequences.”
All nine flavorings induced cell death at the highest concentration tested, ranging from 10 to 100 mmol/L).
The study findings were limited by several factors, primarily a lack of data on how heating the flavorings in the in vitro part of the study might have affected toxicity in the body, the researchers noted.
“Future studies will focus on how the toxicity of the flavorings is altered with heating and characterization of the levels obtained in the circulation after use of an e-cigarette,” they said.
However, data support the need for regulation and limits on the level of flavorings used in e-cigarettes and other tobacco products, they emphasized.
“These findings suggest that flavoring compounds induce endothelial cell dysfunction in human cells similarly to the abnormal function in active cigarette smokers,” the researchers noted.
The study was funded by the National Heart, Lung, and Blood Institute; Food and Drug Administration Center for Tobacco Products; and the American Heart Association. The researchers had no financial conflicts to disclose.
SOURCE: Fetterman J et al. Arterioscler Thromb Vasc Biol. 2018. doi: 10.1161/ATVBAHA.118.311156.
Flavored tobacco products are popular among current smokers, including youth, and the flavorings have been deemed ingestible, but their impact on heart health has not been studied, wrote Jennifer Fetterman, PhD, of Boston University, and her colleagues. The report was published in Arteriosclerosis, Thrombosis, and Vascular Biology.
The researchers studied nine types of flavorings used in alternative tobacco products to assess their impact on cardiovascular health.
The first part of the study comprised a population of nine nonsmokers, six nonmenthol cigarette smokers, and six menthol cigarette smokers without cardiovascular disease. The researchers isolated venous endothelial cells from each participant.
Overall, cells from both nonmenthol and menthol cigarette smokers had significantly lower nitric oxide production compared with nonsmokers (P = .003 and P = .012, respectively). In addition, the flavoring compounds menthol and eugenol impaired nitric oxide production in the cells of healthy individuals.
“Increased inflammation and a loss of nitric oxide are some of the first changes to occur leading up to cardiovascular disease and events like heart attacks and stroke, so they are considered early predictors of heart disease,” Dr. Fetterman said in a statement, adding that the “findings suggest that these flavoring additives may have serious health consequences.”
All nine flavorings induced cell death at the highest concentration tested, ranging from 10 to 100 mmol/L).
The study findings were limited by several factors, primarily a lack of data on how heating the flavorings in the in vitro part of the study might have affected toxicity in the body, the researchers noted.
“Future studies will focus on how the toxicity of the flavorings is altered with heating and characterization of the levels obtained in the circulation after use of an e-cigarette,” they said.
However, data support the need for regulation and limits on the level of flavorings used in e-cigarettes and other tobacco products, they emphasized.
“These findings suggest that flavoring compounds induce endothelial cell dysfunction in human cells similarly to the abnormal function in active cigarette smokers,” the researchers noted.
The study was funded by the National Heart, Lung, and Blood Institute; Food and Drug Administration Center for Tobacco Products; and the American Heart Association. The researchers had no financial conflicts to disclose.
SOURCE: Fetterman J et al. Arterioscler Thromb Vasc Biol. 2018. doi: 10.1161/ATVBAHA.118.311156.
FROM ARTERIOSCLEROSIS, THROMBOSIS, AND VASCULAR BIOLOGY
Key clinical point: Nitric oxide production was impaired in cells exposed to compounds used in alternative tobacco products.
Major finding: Nitric oxide products were significantly lower in nonmenthol and menthol cigarette smokers compared with nonsmokers (P = .003 and P = .012, respectively).
Study details: The data come from nine nonsmokers, six menthol cigarette smokers, and six nonmenthol cigarette smokers, plus in vitro cells.
Disclosures: The study was funded by the National Heart, Lung, and Blood Institute; Food and Drug Administration Center for Tobacco Products; and the American Heart Association. The researchers had no financial conflicts to disclose.
Source: Fetterman J et al. Arterioscler Thromb Vasc Biol. 2018. doi: 10.1161/ATVBAHA.118.311156.
Vaccine nonmedical exemptions creating metro ‘hotspots’
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.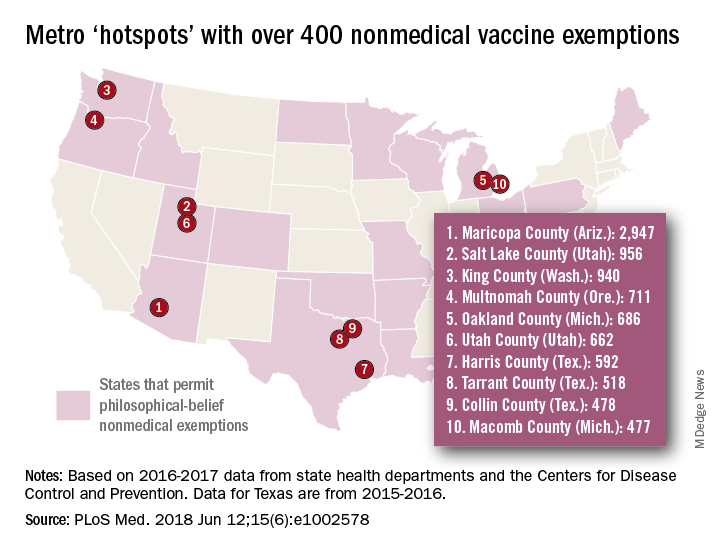
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
Recent increases in nonmedical exemptions (NMEs) to vaccination have created metropolitan “hotspots” with large numbers of unvaccinated children, according to a report published June 12 in PLoS Medicine.
although rates seem to have plateaued in some states since 2014. As a result of those increases, there were, during the 2016-2017 school year, 15 metro areas with kindergarten NME populations over 400, reported Jacqueline K. Olive, and her associates at Baylor College of Medicine. Their report was based on data from state health departments and the Centers for Disease Control and Prevention.
Leading the way was Maricopa County, Ariz., home of Phoenix and 2,947 unvaccinated kindergartners, which was more than triple the number in county/city No. 2, Salt Lake County/Salt Lake City (NME total: 956). Close behind in third was King County, Wash. (Seattle) at 940, followed by Multnomah County, Ore. (Portland) at 711 and Oakland County, Mich. (Troy) at 686, the investigators said.
[There was only room for 10 in the map, so here are hotspots 11-15: Wayne County, Mich. (Detroit); Allegheny County, Pa. (Pittsburgh); Travis County, Tex. (Austin); Jackson County, Mo. (Kansas City); and Spokane County, Wash. (Spokane).]
In addition to the large-population hotspots, there are also a number of mainly rural counties with smaller populations but high NME rates. Eight of the 10 highest such rates can be found in Idaho, and at the top of that list is Camas County, which had an NME rate of 27% in 2016-2017, the researchers reported.
Analysis of the relationship between NMEs and MMR vaccination showed that “states with more NME students exhibited lower MMR vaccination rates. In contrast, states that have banned NMEs – Mississippi, California, and West Virginia – exhibit the highest MMR vaccine uptake and lowest incidence of vaccine preventable diseases,” the investigators wrote.
Ms. Olive and her associates said that there was no specific funding for the study and that no conflicts of interest existed.
SOURCE: Olive JK et al. PLoS Med. 2018 Jun 12;15(6): e1002578. doi: 10.1371/journal.pmed.1002578.
FROM PLOS MEDICINE
Aclidinium bromide for COPD: No impact on MACE
SAN DIEGO – The use of aclidinium bromide 400 mcg b.i.d. did not increase the risk of major adverse cardiac events or mortality in patients with moderate to very severe , compared with placebo.

Those are two key findings from the ASCENT COPD trial presented by Robert A. Wise, MD, at an international conference of the American Thoracic Society. “Cardiovascular risk factors and comorbidities are prevalent in patients with COPD, and about 30% of COPD patients die of cardiovascular disease,” said Dr. Wise, who serves as director of research for the division of pulmonary and critical care medicine at the Johns Hopkins University School of Medicine, Baltimore. “However, patients who have cardiovascular disease are often excluded from, or not enrolled in, COPD clinical trials. Moreover, there has been controversy as to whether or not treatment with a long-acting muscarinic antagonist is associated with an increased risk of cardiovascular events. That’s been seen in randomized trials, meta-analyses, as well as in observational studies.”
Aclidinium bromide 400 mcg b.i.d., administered by the Pressair inhaler, is approved as a maintenance treatment for patients with COPD. However, during the registration studies, there were not an adequate number of cardiovascular events in order to ascertain clearly whether or not the drug was associated with increased risk, Dr. Wise said. Therefore, he and his associates in the ASCENT COPD study set out to assess the long-term cardiovascular safety profile of aclidinium 400 mcg b.i.d. in patients with moderate to very severe COPD at risk of major adverse cardiovascular events (MACE) for up to 3 years (Chronic Obstr Pulm Dis. 2018;5[1]:5-15). For the randomized, placebo-controlled, parallel-group study, patients received treatment with aclidinium bromide or a placebo inhaler of similar appearance. The study was designed to be terminated when at least 122 patients experienced an adjudicated MACE. The primary safety endpoint was time to first MACE during follow-up of up to 3 years, while the primary efficacy endpoint was the rate of moderate to severe exacerbations per patient per year during the first year of treatment.
To be included in the study, patients had to be at least 40 years of age with moderate to very severe stable COPD, have a smoking history of at least 10 pack-years, and have at least one of the following significant risk factors: cerebrovascular disease; coronary artery disease; peripheral vascular disease, or history of claudication; or at least two atherothrombotic risk factors (male at least 65 years of age, female at least 70 years of age; waist circumference of at least 40 inches among males or at least 38 inches among females; an estimated glomerular filtration rate of less than 60 mL/min and microalbuminuria; dyslipidemia; or hypertension).
The researchers randomized 1,791 patients to the aclidinium group and 1,798 to the placebo group. Their mean age was 67 years, and about 60% of patients had an exacerbation in the preceding year. Nearly two-thirds of patients (63%) were receiving concomitant long-acting beta 2-agonists (LABA) or LABA/inhaled corticosteroid therapy. In addition, 44% of patients entered the study with a history of a prior cardiovascular event plus at least two atherothrombotic risk factors, 52% reported at least two atherothrombotic risk factors without any prior cardiovascular events, and 4% had a history of a prior cardiovascular event only.
Dr. Wise reported that aclidinium did not increase the risk of MACE in patients with moderate to very severe COPD with significant cardiovascular risk factors, compared with placebo (hazard ratio 0.89; P = .469); non-inferiority was concluded as the upper bound of the 95% confidence interval was less than 1.8). In terms of all-cause mortality, aclidinium did not increase the risk of death, compared with placebo (HR 0.99; P = .929).
During the first year of treatment, Dr. Wise and his associates also observed a 22% reduction in COPD exacerbation rate for aclidinium vs. placebo groups (HR 0.44 vs. 0.57, respectively; P less than .001), and a 35% reduction in the rate of COPD exacerbations leading to hospitalizations (HR 0.07 vs. 0.10; P = .006). “The reduction in exacerbation risk was similar, whether or not patients had an exacerbation in the past year,” Dr. Wise said. He reported being a consultant to, and receiving research support from, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and ContraFect.
[email protected]
SOURCE: Wise, R., et al., Abstract 7711, ATS 2018.
SAN DIEGO – The use of aclidinium bromide 400 mcg b.i.d. did not increase the risk of major adverse cardiac events or mortality in patients with moderate to very severe , compared with placebo.
Those are two key findings from the ASCENT COPD trial presented by Robert A. Wise, MD, at an international conference of the American Thoracic Society. “Cardiovascular risk factors and comorbidities are prevalent in patients with COPD, and about 30% of COPD patients die of cardiovascular disease,” said Dr. Wise, who serves as director of research for the division of pulmonary and critical care medicine at the Johns Hopkins University School of Medicine, Baltimore. “However, patients who have cardiovascular disease are often excluded from, or not enrolled in, COPD clinical trials. Moreover, there has been controversy as to whether or not treatment with a long-acting muscarinic antagonist is associated with an increased risk of cardiovascular events. That’s been seen in randomized trials, meta-analyses, as well as in observational studies.”
Aclidinium bromide 400 mcg b.i.d., administered by the Pressair inhaler, is approved as a maintenance treatment for patients with COPD. However, during the registration studies, there were not an adequate number of cardiovascular events in order to ascertain clearly whether or not the drug was associated with increased risk, Dr. Wise said. Therefore, he and his associates in the ASCENT COPD study set out to assess the long-term cardiovascular safety profile of aclidinium 400 mcg b.i.d. in patients with moderate to very severe COPD at risk of major adverse cardiovascular events (MACE) for up to 3 years (Chronic Obstr Pulm Dis. 2018;5[1]:5-15). For the randomized, placebo-controlled, parallel-group study, patients received treatment with aclidinium bromide or a placebo inhaler of similar appearance. The study was designed to be terminated when at least 122 patients experienced an adjudicated MACE. The primary safety endpoint was time to first MACE during follow-up of up to 3 years, while the primary efficacy endpoint was the rate of moderate to severe exacerbations per patient per year during the first year of treatment.
To be included in the study, patients had to be at least 40 years of age with moderate to very severe stable COPD, have a smoking history of at least 10 pack-years, and have at least one of the following significant risk factors: cerebrovascular disease; coronary artery disease; peripheral vascular disease, or history of claudication; or at least two atherothrombotic risk factors (male at least 65 years of age, female at least 70 years of age; waist circumference of at least 40 inches among males or at least 38 inches among females; an estimated glomerular filtration rate of less than 60 mL/min and microalbuminuria; dyslipidemia; or hypertension).
The researchers randomized 1,791 patients to the aclidinium group and 1,798 to the placebo group. Their mean age was 67 years, and about 60% of patients had an exacerbation in the preceding year. Nearly two-thirds of patients (63%) were receiving concomitant long-acting beta 2-agonists (LABA) or LABA/inhaled corticosteroid therapy. In addition, 44% of patients entered the study with a history of a prior cardiovascular event plus at least two atherothrombotic risk factors, 52% reported at least two atherothrombotic risk factors without any prior cardiovascular events, and 4% had a history of a prior cardiovascular event only.
Dr. Wise reported that aclidinium did not increase the risk of MACE in patients with moderate to very severe COPD with significant cardiovascular risk factors, compared with placebo (hazard ratio 0.89; P = .469); non-inferiority was concluded as the upper bound of the 95% confidence interval was less than 1.8). In terms of all-cause mortality, aclidinium did not increase the risk of death, compared with placebo (HR 0.99; P = .929).
During the first year of treatment, Dr. Wise and his associates also observed a 22% reduction in COPD exacerbation rate for aclidinium vs. placebo groups (HR 0.44 vs. 0.57, respectively; P less than .001), and a 35% reduction in the rate of COPD exacerbations leading to hospitalizations (HR 0.07 vs. 0.10; P = .006). “The reduction in exacerbation risk was similar, whether or not patients had an exacerbation in the past year,” Dr. Wise said. He reported being a consultant to, and receiving research support from, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and ContraFect.
[email protected]
SOURCE: Wise, R., et al., Abstract 7711, ATS 2018.
SAN DIEGO – The use of aclidinium bromide 400 mcg b.i.d. did not increase the risk of major adverse cardiac events or mortality in patients with moderate to very severe , compared with placebo.
Those are two key findings from the ASCENT COPD trial presented by Robert A. Wise, MD, at an international conference of the American Thoracic Society. “Cardiovascular risk factors and comorbidities are prevalent in patients with COPD, and about 30% of COPD patients die of cardiovascular disease,” said Dr. Wise, who serves as director of research for the division of pulmonary and critical care medicine at the Johns Hopkins University School of Medicine, Baltimore. “However, patients who have cardiovascular disease are often excluded from, or not enrolled in, COPD clinical trials. Moreover, there has been controversy as to whether or not treatment with a long-acting muscarinic antagonist is associated with an increased risk of cardiovascular events. That’s been seen in randomized trials, meta-analyses, as well as in observational studies.”
Aclidinium bromide 400 mcg b.i.d., administered by the Pressair inhaler, is approved as a maintenance treatment for patients with COPD. However, during the registration studies, there were not an adequate number of cardiovascular events in order to ascertain clearly whether or not the drug was associated with increased risk, Dr. Wise said. Therefore, he and his associates in the ASCENT COPD study set out to assess the long-term cardiovascular safety profile of aclidinium 400 mcg b.i.d. in patients with moderate to very severe COPD at risk of major adverse cardiovascular events (MACE) for up to 3 years (Chronic Obstr Pulm Dis. 2018;5[1]:5-15). For the randomized, placebo-controlled, parallel-group study, patients received treatment with aclidinium bromide or a placebo inhaler of similar appearance. The study was designed to be terminated when at least 122 patients experienced an adjudicated MACE. The primary safety endpoint was time to first MACE during follow-up of up to 3 years, while the primary efficacy endpoint was the rate of moderate to severe exacerbations per patient per year during the first year of treatment.
To be included in the study, patients had to be at least 40 years of age with moderate to very severe stable COPD, have a smoking history of at least 10 pack-years, and have at least one of the following significant risk factors: cerebrovascular disease; coronary artery disease; peripheral vascular disease, or history of claudication; or at least two atherothrombotic risk factors (male at least 65 years of age, female at least 70 years of age; waist circumference of at least 40 inches among males or at least 38 inches among females; an estimated glomerular filtration rate of less than 60 mL/min and microalbuminuria; dyslipidemia; or hypertension).
The researchers randomized 1,791 patients to the aclidinium group and 1,798 to the placebo group. Their mean age was 67 years, and about 60% of patients had an exacerbation in the preceding year. Nearly two-thirds of patients (63%) were receiving concomitant long-acting beta 2-agonists (LABA) or LABA/inhaled corticosteroid therapy. In addition, 44% of patients entered the study with a history of a prior cardiovascular event plus at least two atherothrombotic risk factors, 52% reported at least two atherothrombotic risk factors without any prior cardiovascular events, and 4% had a history of a prior cardiovascular event only.
Dr. Wise reported that aclidinium did not increase the risk of MACE in patients with moderate to very severe COPD with significant cardiovascular risk factors, compared with placebo (hazard ratio 0.89; P = .469); non-inferiority was concluded as the upper bound of the 95% confidence interval was less than 1.8). In terms of all-cause mortality, aclidinium did not increase the risk of death, compared with placebo (HR 0.99; P = .929).
During the first year of treatment, Dr. Wise and his associates also observed a 22% reduction in COPD exacerbation rate for aclidinium vs. placebo groups (HR 0.44 vs. 0.57, respectively; P less than .001), and a 35% reduction in the rate of COPD exacerbations leading to hospitalizations (HR 0.07 vs. 0.10; P = .006). “The reduction in exacerbation risk was similar, whether or not patients had an exacerbation in the past year,” Dr. Wise said. He reported being a consultant to, and receiving research support from, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and ContraFect.
[email protected]
SOURCE: Wise, R., et al., Abstract 7711, ATS 2018.
AT ATS 2018
Key clinical point: Researchers found no increased risk of MACE in at-risk patients with COPD receiving aclidinium.
Major finding: MACE risk and mortality in COPD patients with significant cardiovascular risk given aclidinium bromide had a hazard ratio 0.89 (P = .469), compared to placebo.
Study details: A randomized, placebo-controlled, parallel-group study of 3,589 patients with moderate to very severe COPD at risk of major adverse cardiovascular events.
Disclosures: Dr. Wise reported being a consultant to, and receiving research support from, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, and ContraFect.
Source: Wise, R. et al, ATS 2018, Abstract 7711.
Education sessions upped COPD patients’ knowledge of their disease
A brief patient-directed education program delivered at the time of hospitalization for an acute exacerbation of chronic obstructive pulmonary disease (AECOPD) improved disease-specific knowledge, according to results of a pilot randomized trial.
Patients who participated in education sessions had a significant improvement in their scores on the Bristol COPD Knowledge Questionnaire (BCKQ), compared with control patients who received no education, study investigators reported in the journal Chest.
“Early education may be a bridge to more active approaches and could provide an important contribution to self-management interventions post-AECOPD,” wrote first author Tania Janaudis-Ferreira, PhD, of the School of Physical and Occupational Therapy, McGill University, Montreal, and her co-authors.
In the study, patients admitted to a community hospital with an AECOPD were randomized to standard care plus brief education or standard care alone. The education consisted of two 30-minute sessions delivered by a physiotherapist, either in the hospital or at home up to 2 weeks after the admission.
Before and after the intervention period, participant knowledge was measured using both the BCKQ and the Lung Information Needs Questionnaire (LINQ).
A total of 31 patients participated, including 15 in the intervention group and 16 in the control group, although 3 patients in the control group did not complete the follow-up testing, investigators said in their report.
The mean change in BCKQ was 8 points for the educational intervention group, and 3.4 for the control group (P = 0.02). That result was in keeping with findings of a previous randomized study noting an 8.3-point change in BCKQ scores for COPD patients who received education in the primary care setting, Dr. Janaudis-Ferreira and co-authors said.
“The change itself is relatively modest, suggesting more frequent sessions might result in greater improvements,” they wrote. For example, they said, an 8-week educational intervention delivered in the context of pulmonary rehabilitation program in one study yielded a mean change of 18.3 points on the BCKQ in the intervention group.
By contrast, the investigators found no significant difference in LINQ score changes between the intervention and control groups (P = .8).
That may indicate that two 30-minute sessions were not sufficient to attend to patients’ learning needs, authors said, though it could also have been an issue with the instrument itself in the setting of this study.
“The majority of the questions in the LINQ ask whether or not a doctor or nurse has explained a specific question to the patient,” authors explained. “Since a physiotherapist delivered the program, had the wording been altered to include physiotherapists or a more general term for healthcare professionals, we may have seen a change in these results.”
Dr. Janaudis-Ferreira and co-authors had no conflicts of interest to disclose. The study was funded by a grant from the Canadian Respiratory Health Professionals, which did not have input in research or manuscript development.
SOURCE: Janaudis-Ferreira T, et al. Chest
A brief patient-directed education program delivered at the time of hospitalization for an acute exacerbation of chronic obstructive pulmonary disease (AECOPD) improved disease-specific knowledge, according to results of a pilot randomized trial.
Patients who participated in education sessions had a significant improvement in their scores on the Bristol COPD Knowledge Questionnaire (BCKQ), compared with control patients who received no education, study investigators reported in the journal Chest.
“Early education may be a bridge to more active approaches and could provide an important contribution to self-management interventions post-AECOPD,” wrote first author Tania Janaudis-Ferreira, PhD, of the School of Physical and Occupational Therapy, McGill University, Montreal, and her co-authors.
In the study, patients admitted to a community hospital with an AECOPD were randomized to standard care plus brief education or standard care alone. The education consisted of two 30-minute sessions delivered by a physiotherapist, either in the hospital or at home up to 2 weeks after the admission.
Before and after the intervention period, participant knowledge was measured using both the BCKQ and the Lung Information Needs Questionnaire (LINQ).
A total of 31 patients participated, including 15 in the intervention group and 16 in the control group, although 3 patients in the control group did not complete the follow-up testing, investigators said in their report.
The mean change in BCKQ was 8 points for the educational intervention group, and 3.4 for the control group (P = 0.02). That result was in keeping with findings of a previous randomized study noting an 8.3-point change in BCKQ scores for COPD patients who received education in the primary care setting, Dr. Janaudis-Ferreira and co-authors said.
“The change itself is relatively modest, suggesting more frequent sessions might result in greater improvements,” they wrote. For example, they said, an 8-week educational intervention delivered in the context of pulmonary rehabilitation program in one study yielded a mean change of 18.3 points on the BCKQ in the intervention group.
By contrast, the investigators found no significant difference in LINQ score changes between the intervention and control groups (P = .8).
That may indicate that two 30-minute sessions were not sufficient to attend to patients’ learning needs, authors said, though it could also have been an issue with the instrument itself in the setting of this study.
“The majority of the questions in the LINQ ask whether or not a doctor or nurse has explained a specific question to the patient,” authors explained. “Since a physiotherapist delivered the program, had the wording been altered to include physiotherapists or a more general term for healthcare professionals, we may have seen a change in these results.”
Dr. Janaudis-Ferreira and co-authors had no conflicts of interest to disclose. The study was funded by a grant from the Canadian Respiratory Health Professionals, which did not have input in research or manuscript development.
SOURCE: Janaudis-Ferreira T, et al. Chest
A brief patient-directed education program delivered at the time of hospitalization for an acute exacerbation of chronic obstructive pulmonary disease (AECOPD) improved disease-specific knowledge, according to results of a pilot randomized trial.
Patients who participated in education sessions had a significant improvement in their scores on the Bristol COPD Knowledge Questionnaire (BCKQ), compared with control patients who received no education, study investigators reported in the journal Chest.
“Early education may be a bridge to more active approaches and could provide an important contribution to self-management interventions post-AECOPD,” wrote first author Tania Janaudis-Ferreira, PhD, of the School of Physical and Occupational Therapy, McGill University, Montreal, and her co-authors.
In the study, patients admitted to a community hospital with an AECOPD were randomized to standard care plus brief education or standard care alone. The education consisted of two 30-minute sessions delivered by a physiotherapist, either in the hospital or at home up to 2 weeks after the admission.
Before and after the intervention period, participant knowledge was measured using both the BCKQ and the Lung Information Needs Questionnaire (LINQ).
A total of 31 patients participated, including 15 in the intervention group and 16 in the control group, although 3 patients in the control group did not complete the follow-up testing, investigators said in their report.
The mean change in BCKQ was 8 points for the educational intervention group, and 3.4 for the control group (P = 0.02). That result was in keeping with findings of a previous randomized study noting an 8.3-point change in BCKQ scores for COPD patients who received education in the primary care setting, Dr. Janaudis-Ferreira and co-authors said.
“The change itself is relatively modest, suggesting more frequent sessions might result in greater improvements,” they wrote. For example, they said, an 8-week educational intervention delivered in the context of pulmonary rehabilitation program in one study yielded a mean change of 18.3 points on the BCKQ in the intervention group.
By contrast, the investigators found no significant difference in LINQ score changes between the intervention and control groups (P = .8).
That may indicate that two 30-minute sessions were not sufficient to attend to patients’ learning needs, authors said, though it could also have been an issue with the instrument itself in the setting of this study.
“The majority of the questions in the LINQ ask whether or not a doctor or nurse has explained a specific question to the patient,” authors explained. “Since a physiotherapist delivered the program, had the wording been altered to include physiotherapists or a more general term for healthcare professionals, we may have seen a change in these results.”
Dr. Janaudis-Ferreira and co-authors had no conflicts of interest to disclose. The study was funded by a grant from the Canadian Respiratory Health Professionals, which did not have input in research or manuscript development.
SOURCE: Janaudis-Ferreira T, et al. Chest
FROM CHEST
Key clinical point: Two 30-minute education sessions improved patients’ disease-specific knowledge for an acute exacerbation of COPD.
Major finding: Mean change on the Bristol COPD Knowledge Questionnaire (BCKQ) was 8 points for the educational intervention, and 3.4 for controls.
Study details: A pilot randomized controlled trial of 31 patients admitted to a community hospital.
Disclosures: Authors had no conflicts of interest to disclose. The study was funded by a grant from the Canadian Respiratory Health Professionals, which did not have input in research or manuscript development.
Source: Janaudis-Ferreira T, et al. Chest 2018 Jun 4.
Sleep apnea treatment may not prevent sleepiness
Physicians who treat obstructive sleep apnea reported that patients with OSA can experience excessive sleepiness despite being on optimized airway treatment, findings from an online surgery of clinicians show.
Jazz Pharmaceuticals and the social media network Sermo conducted an online questionnaire on topics in excessive sleepiness and obstructive sleep apnea. The study was conducted March 30-31, 2018.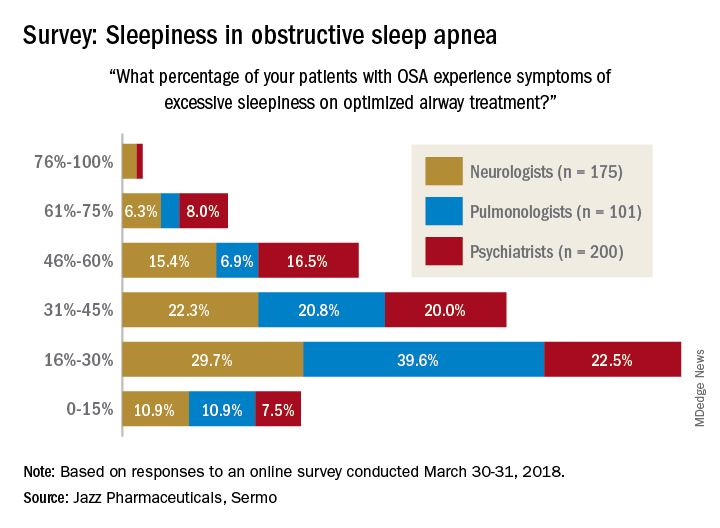
When asked how often they assess their OSA patients’ sleepiness, 46% of respondents said every 3 months, 28% said every 6 months, 19% said once a month, 6% said once a year, and 0.4% (one neurologist and one pulmonologist) said never. The method of evaluation varied by specialty: 82% of pulmonologists most often use the Epworth Sleepiness Scale and 76% of psychiatrists primarily use an informal set of questions, with neurologists in between but leaning toward informal questions, Jazz reported.
“As more scientific evidence emerges around the neuronal injury occurring due to OSA and the potential neurocognitive effects of excessive sleepiness, it’s imperative that pulmonologists, neurologists and psychiatrists understand the impact ES [excessive sleepiness] can have on patients’ lives,” Richard K. Bogan, MD, of the University of South Carolina, Columbia, a paid consultant to Jazz, said in a written statement.
Physicians who treat obstructive sleep apnea reported that patients with OSA can experience excessive sleepiness despite being on optimized airway treatment, findings from an online surgery of clinicians show.
Jazz Pharmaceuticals and the social media network Sermo conducted an online questionnaire on topics in excessive sleepiness and obstructive sleep apnea. The study was conducted March 30-31, 2018.
When asked how often they assess their OSA patients’ sleepiness, 46% of respondents said every 3 months, 28% said every 6 months, 19% said once a month, 6% said once a year, and 0.4% (one neurologist and one pulmonologist) said never. The method of evaluation varied by specialty: 82% of pulmonologists most often use the Epworth Sleepiness Scale and 76% of psychiatrists primarily use an informal set of questions, with neurologists in between but leaning toward informal questions, Jazz reported.
“As more scientific evidence emerges around the neuronal injury occurring due to OSA and the potential neurocognitive effects of excessive sleepiness, it’s imperative that pulmonologists, neurologists and psychiatrists understand the impact ES [excessive sleepiness] can have on patients’ lives,” Richard K. Bogan, MD, of the University of South Carolina, Columbia, a paid consultant to Jazz, said in a written statement.
Physicians who treat obstructive sleep apnea reported that patients with OSA can experience excessive sleepiness despite being on optimized airway treatment, findings from an online surgery of clinicians show.
Jazz Pharmaceuticals and the social media network Sermo conducted an online questionnaire on topics in excessive sleepiness and obstructive sleep apnea. The study was conducted March 30-31, 2018.
When asked how often they assess their OSA patients’ sleepiness, 46% of respondents said every 3 months, 28% said every 6 months, 19% said once a month, 6% said once a year, and 0.4% (one neurologist and one pulmonologist) said never. The method of evaluation varied by specialty: 82% of pulmonologists most often use the Epworth Sleepiness Scale and 76% of psychiatrists primarily use an informal set of questions, with neurologists in between but leaning toward informal questions, Jazz reported.
“As more scientific evidence emerges around the neuronal injury occurring due to OSA and the potential neurocognitive effects of excessive sleepiness, it’s imperative that pulmonologists, neurologists and psychiatrists understand the impact ES [excessive sleepiness] can have on patients’ lives,” Richard K. Bogan, MD, of the University of South Carolina, Columbia, a paid consultant to Jazz, said in a written statement.