User login
Symptom-related emergency department visits and hospital admissions during ambulatory cancer treatment
Background People with cancer experience symptoms related to the disease and treatments. Symptom distress has a negative impact on quality of life (QoL). Attending to symptoms and side effects of treatment promotes safe and effective delivery of therapies and may prevent or reduce emergency department visits (EDVs) and unplanned hospital admissions (HAs). There is limited evidence examining symptom-related EDVs or HAs (sx-EDV/HAs) and interventions in ambulatory oncology patients.
Objective To examine factors associated with sx-EDV/HAs in ambulatory oncology patients receiving chemotherapy and/or radiation.
Methods This secondary analysis used data from a randomized controlled trial of ambulatory oncology patients (n = 663) who received the web-based Electronic Self-Report Assessment – Cancer intervention (symptom self-monitoring, tailored education, and communication coaching) or usual care with symptom self-monitoring alone. Group differences were described by summary statistics and compared by t test. Factors associated with the odds of at least 1 sx-EDV/HA were modeled using logistic regression.
Results 98 patients had a total of 171 sx-EDV/HAs with no difference between groups. Higher odds of at least 1 sx-EDV/HA were associated with socioeconomic and clinical factors. The multivariable model indicated that work status, education level, treatment modality, and on-treatment Symptom Distress Scale-15 scores were significantly associated with having at least 1 sx-EDV/HA.
Limitations This is a secondary analysis not sized to determine cause and effect. The results have limited generalizability.
Conclusion Most patients did not experience a sx-EDV/HA. Demographic and clinical factors predicted a sx-EDV/HA.
Funding National Institute of Nursing Research, National Institutes of Health, R01 NR008726; 2008-2011
Click on the PDF icon at the top of this introduction to read the full article.
Background People with cancer experience symptoms related to the disease and treatments. Symptom distress has a negative impact on quality of life (QoL). Attending to symptoms and side effects of treatment promotes safe and effective delivery of therapies and may prevent or reduce emergency department visits (EDVs) and unplanned hospital admissions (HAs). There is limited evidence examining symptom-related EDVs or HAs (sx-EDV/HAs) and interventions in ambulatory oncology patients.
Objective To examine factors associated with sx-EDV/HAs in ambulatory oncology patients receiving chemotherapy and/or radiation.
Methods This secondary analysis used data from a randomized controlled trial of ambulatory oncology patients (n = 663) who received the web-based Electronic Self-Report Assessment – Cancer intervention (symptom self-monitoring, tailored education, and communication coaching) or usual care with symptom self-monitoring alone. Group differences were described by summary statistics and compared by t test. Factors associated with the odds of at least 1 sx-EDV/HA were modeled using logistic regression.
Results 98 patients had a total of 171 sx-EDV/HAs with no difference between groups. Higher odds of at least 1 sx-EDV/HA were associated with socioeconomic and clinical factors. The multivariable model indicated that work status, education level, treatment modality, and on-treatment Symptom Distress Scale-15 scores were significantly associated with having at least 1 sx-EDV/HA.
Limitations This is a secondary analysis not sized to determine cause and effect. The results have limited generalizability.
Conclusion Most patients did not experience a sx-EDV/HA. Demographic and clinical factors predicted a sx-EDV/HA.
Funding National Institute of Nursing Research, National Institutes of Health, R01 NR008726; 2008-2011
Click on the PDF icon at the top of this introduction to read the full article.
Background People with cancer experience symptoms related to the disease and treatments. Symptom distress has a negative impact on quality of life (QoL). Attending to symptoms and side effects of treatment promotes safe and effective delivery of therapies and may prevent or reduce emergency department visits (EDVs) and unplanned hospital admissions (HAs). There is limited evidence examining symptom-related EDVs or HAs (sx-EDV/HAs) and interventions in ambulatory oncology patients.
Objective To examine factors associated with sx-EDV/HAs in ambulatory oncology patients receiving chemotherapy and/or radiation.
Methods This secondary analysis used data from a randomized controlled trial of ambulatory oncology patients (n = 663) who received the web-based Electronic Self-Report Assessment – Cancer intervention (symptom self-monitoring, tailored education, and communication coaching) or usual care with symptom self-monitoring alone. Group differences were described by summary statistics and compared by t test. Factors associated with the odds of at least 1 sx-EDV/HA were modeled using logistic regression.
Results 98 patients had a total of 171 sx-EDV/HAs with no difference between groups. Higher odds of at least 1 sx-EDV/HA were associated with socioeconomic and clinical factors. The multivariable model indicated that work status, education level, treatment modality, and on-treatment Symptom Distress Scale-15 scores were significantly associated with having at least 1 sx-EDV/HA.
Limitations This is a secondary analysis not sized to determine cause and effect. The results have limited generalizability.
Conclusion Most patients did not experience a sx-EDV/HA. Demographic and clinical factors predicted a sx-EDV/HA.
Funding National Institute of Nursing Research, National Institutes of Health, R01 NR008726; 2008-2011
Click on the PDF icon at the top of this introduction to read the full article.
Asymptomatic carotid stenosis and central sleep apnea linked
More than two-thirds of patients with asymptomatic carotid stenosis are likely have sleep apnea, according to an observational study.
The polysomnography results of 96 patients with asymptomatic extracranial carotid stenosis revealed that 69% had sleep apnea. Obstructive sleep apnea was present in 42% of patients and central sleep apnea in 27%.
Stenosis severity was significantly associated with central sleep apnea, but not with obstructive sleep apnea. Researchers found that central sleep apnea, but not obstructive sleep apnea, was associated with arterial hypertension and diabetes mellitus in those patients with asymptomatic carotid stenosis (CHEST 2015;147:1029-1036 [doi:10.1378/chest.14-1655]).
The patients ranged in age from 39 to 86 years (mean age, 70 years); 64 were men. Of the 96 patients, 21 had mild/moderate stenosis and 75 had severe carotid stenosis. Patients with severe stenosis were older, average age 67 years, than were those with mild/moderate stenosis, average age 61 years. The frequency of arterial hypertension and diabetes mellitus was higher in the severe stenosis group than in the mild/moderate stenosis group.
The prevalence of sleep apnea was 76% in patients with severe stenosis compared with 29% in those with mild/moderate carotid stenosis. Total apnea-hypopnea index was higher in the severe stenosis group compared with the mild/moderate stenosis group (P less than or equal to .009). Increase in sleep apnea severity was based on an increase in central apnea-hypopnea index (P less than or equal to .001) but not in obstructive apnea-hypopnea index, reflecting an augmentation of central sleep apnea and not of obstructive sleep apnea in patients with severe compared with mild/moderate carotid stenosis.
“This vascular risk constellation seems to be more strongly connected with CSA [central sleep apnea] than with OSA [obstructive sleep apnea], possibly attributable to carotid chemoreceptor dysfunction,” wrote Dr. Jens Ehrhardt and colleagues at Jena University Hospital, Germany.
No conflicts of interest were declared.
More than two-thirds of patients with asymptomatic carotid stenosis are likely have sleep apnea, according to an observational study.
The polysomnography results of 96 patients with asymptomatic extracranial carotid stenosis revealed that 69% had sleep apnea. Obstructive sleep apnea was present in 42% of patients and central sleep apnea in 27%.
Stenosis severity was significantly associated with central sleep apnea, but not with obstructive sleep apnea. Researchers found that central sleep apnea, but not obstructive sleep apnea, was associated with arterial hypertension and diabetes mellitus in those patients with asymptomatic carotid stenosis (CHEST 2015;147:1029-1036 [doi:10.1378/chest.14-1655]).
The patients ranged in age from 39 to 86 years (mean age, 70 years); 64 were men. Of the 96 patients, 21 had mild/moderate stenosis and 75 had severe carotid stenosis. Patients with severe stenosis were older, average age 67 years, than were those with mild/moderate stenosis, average age 61 years. The frequency of arterial hypertension and diabetes mellitus was higher in the severe stenosis group than in the mild/moderate stenosis group.
The prevalence of sleep apnea was 76% in patients with severe stenosis compared with 29% in those with mild/moderate carotid stenosis. Total apnea-hypopnea index was higher in the severe stenosis group compared with the mild/moderate stenosis group (P less than or equal to .009). Increase in sleep apnea severity was based on an increase in central apnea-hypopnea index (P less than or equal to .001) but not in obstructive apnea-hypopnea index, reflecting an augmentation of central sleep apnea and not of obstructive sleep apnea in patients with severe compared with mild/moderate carotid stenosis.
“This vascular risk constellation seems to be more strongly connected with CSA [central sleep apnea] than with OSA [obstructive sleep apnea], possibly attributable to carotid chemoreceptor dysfunction,” wrote Dr. Jens Ehrhardt and colleagues at Jena University Hospital, Germany.
No conflicts of interest were declared.
More than two-thirds of patients with asymptomatic carotid stenosis are likely have sleep apnea, according to an observational study.
The polysomnography results of 96 patients with asymptomatic extracranial carotid stenosis revealed that 69% had sleep apnea. Obstructive sleep apnea was present in 42% of patients and central sleep apnea in 27%.
Stenosis severity was significantly associated with central sleep apnea, but not with obstructive sleep apnea. Researchers found that central sleep apnea, but not obstructive sleep apnea, was associated with arterial hypertension and diabetes mellitus in those patients with asymptomatic carotid stenosis (CHEST 2015;147:1029-1036 [doi:10.1378/chest.14-1655]).
The patients ranged in age from 39 to 86 years (mean age, 70 years); 64 were men. Of the 96 patients, 21 had mild/moderate stenosis and 75 had severe carotid stenosis. Patients with severe stenosis were older, average age 67 years, than were those with mild/moderate stenosis, average age 61 years. The frequency of arterial hypertension and diabetes mellitus was higher in the severe stenosis group than in the mild/moderate stenosis group.
The prevalence of sleep apnea was 76% in patients with severe stenosis compared with 29% in those with mild/moderate carotid stenosis. Total apnea-hypopnea index was higher in the severe stenosis group compared with the mild/moderate stenosis group (P less than or equal to .009). Increase in sleep apnea severity was based on an increase in central apnea-hypopnea index (P less than or equal to .001) but not in obstructive apnea-hypopnea index, reflecting an augmentation of central sleep apnea and not of obstructive sleep apnea in patients with severe compared with mild/moderate carotid stenosis.
“This vascular risk constellation seems to be more strongly connected with CSA [central sleep apnea] than with OSA [obstructive sleep apnea], possibly attributable to carotid chemoreceptor dysfunction,” wrote Dr. Jens Ehrhardt and colleagues at Jena University Hospital, Germany.
No conflicts of interest were declared.
FROM CHEST
Key clinical point: More than two-thirds of patients with asymptomatic carotid stenosis are likely to have sleep apnea.
Major finding: The prevalence of sleep apnea was 76% in patients with severe stenosis compared with 29% in those with mild/moderate carotid stenosis.
Data source: Study of 96 patients with asymptomatic extracranial carotid stenosis.
Disclosures: No conflicts of interest were declared.
Spacing out
The number of parents asking their pediatricians to stray from the recommended immunization schedule by spreading out the vaccines is increasing, and so is the number of pediatricians who are agreeing to follow these spaced-out schedules.
One of the two reasons most often given by pediatricians for agreeing to the less than optimal immunization schedules is that by showing a willingness to compromise, that physician may be helping to build a trusting relationship with these families. The other reason is a concern – let’s be honest and call it a fear – that a dissatisfied family will move its care to another physician/provider.
When we scratch the surface of these two rationales, neither seems to make much sense. The conflict over immunization spacing comes to a head at the 2-month well-child visit recommended call for six injections. If the infant has had an unremarkable neonatal course, there may not have been any situation in which the physician was forced to demonstrate her trustworthiness. As long as she has dressed professionally, showed up on time for appointments, washed her hands, and appeared genuinely interested in the child’s well-being, that’s about all she has had to do.
The physician may give the impression that she can be trusted, but real trust is usually something that must accumulate over time, in monthly – or more likely yearly – increments. Occasionally a crisis allows the physician to behave so heroically that her route to a trusting relationship is compressed to just a few hours, but fortunately these crises are rare.
Does agreeing to an unnecessary and unsubstantiated diversion from the recommended immunization schedule play a role in trust building? It may signal that the physician is willing to compromise, which in some situations may not be a bad attribute. For example, the mother who has struggled and failed at breastfeeding her 6 weeks despite everyone’s best efforts will appreciate her pediatrician’s willingness to compromise. But should compromise of scientifically validated practices really be one of the cornerstones of a physician-patient relationship?
I have never had a family request that the immunization schedule be spread out for their second child because they have seen for themselves that the process is not what they have feared. I gave all the immunizations myself, and my administration style was quick and matter-of-fact. The problem, of course, is getting hesitant parents up to and over that hurdle of the 2-month visit. Unfortunately, the evidence seems to be that education and extra time and reassurance are of little value in getting them to that point of trust.
The more difficult issue is a physician’s fear that by failing to agree to a spaced-out schedule, she will open a spigot and families will flow out of her practice to other more compromising providers. Is this just an ego thing? No one likes to feel rejected. Will the feared patient exodus seriously depress the physician’s income or will it be merely a trickle that can be ignored? Obviously, the answer varies from community to community. Do families have so many options that they will easily be able to find a provider who is eager to grow his or her practice, and is less concerned about the immunization level of the community? Or, is the pediatrician so busy that a firm adherence to the standard schedule might provide a welcome opportunity to have a more manageable panel size, and at the same time shift the patient mix toward families that don’t require the extra time in fruitless “educational” discussions?
These are questions that don’t seem to be getting asked. What are the numbers? Is the loss of patients just an irrational fear for physicians created by an irrational fear of a small segment of the population? If the physician practices in a group, could her fear of patient loss be eased if the entire group committed itself to following the standard immunization schedule? Are group members discussing this issue among themselves and with their practice managers? Or, is everyone just spacing out?
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” E-mail him at [email protected]. Scan this QR code to read similar articles or go to pediatricnews.com.
The number of parents asking their pediatricians to stray from the recommended immunization schedule by spreading out the vaccines is increasing, and so is the number of pediatricians who are agreeing to follow these spaced-out schedules.
One of the two reasons most often given by pediatricians for agreeing to the less than optimal immunization schedules is that by showing a willingness to compromise, that physician may be helping to build a trusting relationship with these families. The other reason is a concern – let’s be honest and call it a fear – that a dissatisfied family will move its care to another physician/provider.
When we scratch the surface of these two rationales, neither seems to make much sense. The conflict over immunization spacing comes to a head at the 2-month well-child visit recommended call for six injections. If the infant has had an unremarkable neonatal course, there may not have been any situation in which the physician was forced to demonstrate her trustworthiness. As long as she has dressed professionally, showed up on time for appointments, washed her hands, and appeared genuinely interested in the child’s well-being, that’s about all she has had to do.
The physician may give the impression that she can be trusted, but real trust is usually something that must accumulate over time, in monthly – or more likely yearly – increments. Occasionally a crisis allows the physician to behave so heroically that her route to a trusting relationship is compressed to just a few hours, but fortunately these crises are rare.
Does agreeing to an unnecessary and unsubstantiated diversion from the recommended immunization schedule play a role in trust building? It may signal that the physician is willing to compromise, which in some situations may not be a bad attribute. For example, the mother who has struggled and failed at breastfeeding her 6 weeks despite everyone’s best efforts will appreciate her pediatrician’s willingness to compromise. But should compromise of scientifically validated practices really be one of the cornerstones of a physician-patient relationship?
I have never had a family request that the immunization schedule be spread out for their second child because they have seen for themselves that the process is not what they have feared. I gave all the immunizations myself, and my administration style was quick and matter-of-fact. The problem, of course, is getting hesitant parents up to and over that hurdle of the 2-month visit. Unfortunately, the evidence seems to be that education and extra time and reassurance are of little value in getting them to that point of trust.
The more difficult issue is a physician’s fear that by failing to agree to a spaced-out schedule, she will open a spigot and families will flow out of her practice to other more compromising providers. Is this just an ego thing? No one likes to feel rejected. Will the feared patient exodus seriously depress the physician’s income or will it be merely a trickle that can be ignored? Obviously, the answer varies from community to community. Do families have so many options that they will easily be able to find a provider who is eager to grow his or her practice, and is less concerned about the immunization level of the community? Or, is the pediatrician so busy that a firm adherence to the standard schedule might provide a welcome opportunity to have a more manageable panel size, and at the same time shift the patient mix toward families that don’t require the extra time in fruitless “educational” discussions?
These are questions that don’t seem to be getting asked. What are the numbers? Is the loss of patients just an irrational fear for physicians created by an irrational fear of a small segment of the population? If the physician practices in a group, could her fear of patient loss be eased if the entire group committed itself to following the standard immunization schedule? Are group members discussing this issue among themselves and with their practice managers? Or, is everyone just spacing out?
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” E-mail him at [email protected]. Scan this QR code to read similar articles or go to pediatricnews.com.
The number of parents asking their pediatricians to stray from the recommended immunization schedule by spreading out the vaccines is increasing, and so is the number of pediatricians who are agreeing to follow these spaced-out schedules.
One of the two reasons most often given by pediatricians for agreeing to the less than optimal immunization schedules is that by showing a willingness to compromise, that physician may be helping to build a trusting relationship with these families. The other reason is a concern – let’s be honest and call it a fear – that a dissatisfied family will move its care to another physician/provider.
When we scratch the surface of these two rationales, neither seems to make much sense. The conflict over immunization spacing comes to a head at the 2-month well-child visit recommended call for six injections. If the infant has had an unremarkable neonatal course, there may not have been any situation in which the physician was forced to demonstrate her trustworthiness. As long as she has dressed professionally, showed up on time for appointments, washed her hands, and appeared genuinely interested in the child’s well-being, that’s about all she has had to do.
The physician may give the impression that she can be trusted, but real trust is usually something that must accumulate over time, in monthly – or more likely yearly – increments. Occasionally a crisis allows the physician to behave so heroically that her route to a trusting relationship is compressed to just a few hours, but fortunately these crises are rare.
Does agreeing to an unnecessary and unsubstantiated diversion from the recommended immunization schedule play a role in trust building? It may signal that the physician is willing to compromise, which in some situations may not be a bad attribute. For example, the mother who has struggled and failed at breastfeeding her 6 weeks despite everyone’s best efforts will appreciate her pediatrician’s willingness to compromise. But should compromise of scientifically validated practices really be one of the cornerstones of a physician-patient relationship?
I have never had a family request that the immunization schedule be spread out for their second child because they have seen for themselves that the process is not what they have feared. I gave all the immunizations myself, and my administration style was quick and matter-of-fact. The problem, of course, is getting hesitant parents up to and over that hurdle of the 2-month visit. Unfortunately, the evidence seems to be that education and extra time and reassurance are of little value in getting them to that point of trust.
The more difficult issue is a physician’s fear that by failing to agree to a spaced-out schedule, she will open a spigot and families will flow out of her practice to other more compromising providers. Is this just an ego thing? No one likes to feel rejected. Will the feared patient exodus seriously depress the physician’s income or will it be merely a trickle that can be ignored? Obviously, the answer varies from community to community. Do families have so many options that they will easily be able to find a provider who is eager to grow his or her practice, and is less concerned about the immunization level of the community? Or, is the pediatrician so busy that a firm adherence to the standard schedule might provide a welcome opportunity to have a more manageable panel size, and at the same time shift the patient mix toward families that don’t require the extra time in fruitless “educational” discussions?
These are questions that don’t seem to be getting asked. What are the numbers? Is the loss of patients just an irrational fear for physicians created by an irrational fear of a small segment of the population? If the physician practices in a group, could her fear of patient loss be eased if the entire group committed itself to following the standard immunization schedule? Are group members discussing this issue among themselves and with their practice managers? Or, is everyone just spacing out?
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “Coping with a Picky Eater.” E-mail him at [email protected]. Scan this QR code to read similar articles or go to pediatricnews.com.
Acne Scarring: A Review of Cosmetic Therapies
Acne vulgaris is one of the most common inflammatory dermatoses affecting nearly all adolescents and a large proportion of adults.1 Incidence rates trend downward with age, but prevalence has been reported to be as high as 51% in individuals aged 20 to 29 years.2 Notably, recent evidence suggests there is an increasing incidence rate of acne among postadolescent women, with the severity associated with the menstrual cycle.3,4 Scarring is a common result of acne and may even occur in the setting of appropriate medical therapy. In particular, some form of facial scarring has been reported to occur in up to 95% of acne patients, with severe scarring in 30% of these patients.5 The detrimental effects of acne scarring are not only limited to impaired cosmetic appearance, as it also has been associated with depression symptoms, suicidal ideation, mental health problems, and general social impairment.6 Given the negative impact of acne scarring on overall health and well-being as well as its permanent nature, early and effective treatment is essential to maximize cosmetic outcomes and minimize long-term deleterious effects.
Acne scarring can be broadly divided into 2 major categories: atrophic and hypertrophic. Atrophic scarring is more common and is characterized by an overall localized reduction in collagen content. Clinically, atrophic scars present as depressions in the skin secondary to inflammatory fibrous contractions induced by acne. This type of scarring can be further divided into various subtypes based on morphologic criteria (eg, size, depth), such as boxcar, ice pick, and rolling scars.7 Conversely, hypertrophic scarring is characterized by an overall increase in collagen content and presents as firm raised lesions. Hypertrophic scars should be distinguished from keloid scars, as the former will not outgrow the margins of the original wound while the latter will.8 Treatment of acne scarring is based on scar type and can be accomplished through a variety of medical and surgical modalities (Table). In this article, we review some of the most commonly utilized therapies for both atrophic and hypertrophic acne scarring with a focus on cosmetic outcomes. It is important to keep in mind, however, that the best treatment is to prevent the occurrence of acne scarring through early and proactive treatment of acne.9
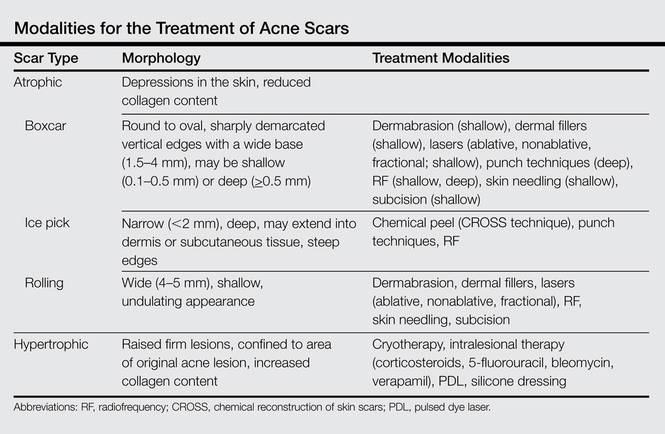
Dermabrasion
Dermabrasion is a decades-old technique that employs the use of a motorized device equipped with an abrasive material to physically remove the superficial layers of the skin, thus inducing the wound-healing process with subsequent formation of new collagen.10 In the same vein, microdermabrasion utilizes aluminum oxide crystals ejected from a nozzle to induce superficial microlacerations.11 This technique is most successful when used to soften scar edges in superficial atrophic scars of the rolling or boxcar subtypes.12 Dermabrasion has been shown to be equally as effective as laser therapy in the treatment of facial scars but is reported to have a much greater risk for adverse effects (AEs)(eg, erythema, edema) that may last for several weeks posttherapy.13,14 Dermabrasion is a particularly operator-dependent technique for which outcomes may vary depending on operator experience. As such, it is not generally recommended as a first-line therapy given its risks and relatively modest results; however, dermabrasion can be a useful adjunct when performed in the right setting. This technique, in addition to laser resurfacing, should be used with caution in patients who have recently taken or currently are taking isotretinoin, as several case series have reported postprocedural development of hypertrophic or keloid scars,15-17 but these findings subsequently were questioned in the literature.18
Laser Therapy
Laser technology has advanced tremendously over the last few decades and there are now a multitude of available lasers that are capable of variable depth penetration and energy delivery patterns. Common to all, however, is the ability to induce localized thermal damage with eventual collagen remodeling. Lasers can be divided into 2 major categories: ablative and nonablative. Ablative lasers cause epidermal destruction, while nonablative lasers are able to selectively target dermal layers without disrupting the overlying epithelium. Generally speaking, ablative lasers are more effective than nonablative lasers in the treatment of atrophic scars, with reported mean improvements of up to 81%.19 This increased efficacy comes with an increased risk for AEs such as postinflammatory hyperpigmentation, prolonged posttreatment erythema, and formation of additional scarring.20 Both ablative and nonablative lasers can be applied in the more recently developed technology of fractional photothermolysis. With this method, noncontiguous microscopic columns of thermal injury surrounded by zones of viable tissue are created, which is in contrast to the traditional manner of inducing broad thermal injury. Fractional ablative lasers can achieve efficacy rates similar to traditional ablative lasers with a reduced risk for permanent scarring or dispigmentation.21 Notably, recent studies have shown promising results for the use of fractional ablative lasers as a mechanism to enhance drug delivery of topically applied medications such as poly-L-lactic acid and triamcinolone acetonide in the treatment of atrophic and hypertrophic scars, respectively.22,23
Lasers also play a role in the treatment of hypertrophic acne scars with the use of nonablative pulsed dye lasers. These lasers cause selective thermolysis of dermal vasculature, and average clinical improvements in hypertrophic scars of 67.5% after a single treatment have been reported.24 Temporary postoperative purpura and long-term hyperpigmentation are reported outcomes of this therapy.20
Radiofrequency
Nonablative radiofrequency (RF) is a relatively novel technique that creates an electric current in the dermis at preset depths to induce thermal damage and eventual collagen synthesis. There are a variety of modalities for which RF can be applied, but microneedle bipolar RF and fractional bipolar RF treatments offer the best results for atrophic acne scars. Improvements in scar appearance of 25% to 75% have been reported after several treatment sessions.25 Better results have been reported in the treatment of ice pick scars as compared to more superficial scars,26 but additional studies will be necessary to validate this claim. Adverse effects are largely limited to temporary erythema and posttreatment scabbing.27
Subcision
Subcision is a more physically intensive technique useful for treatment of superficial atrophic acne scars. This method involves the use of a small needle that is inserted into the periphery of a scar before being moved in a back-and-forth manner underneath the base of the scar to loosen the fibrotic adhesions that result in the depressed appearance of the scar. Additionally, loosening of the tissue and resultant bleeding creates a potential space for future collagen deposition during the subsequent wound-healing phase. Subcision has a reported success rate of 50% to 60% in the treatment of rolling scars, and prospective, randomized, split-face trials have indicated that the short-term outcomes of subcision are superior to dermal fillers while being equally effective long-term.28,29 Of note, a small percentage of patients may develop a localized nodule at the site of treatment, which can be resolved with intralesional steroids.11
Skin Needling
Skin needling, also referred to as collagen induction therapy, utilizes vertical needle punctures rather than the horizontally directed punctures that are used in subcision and can be used to treat rolling and boxcar scars. Traditionally, a small roller equipped with rows of small needles typically ranging in size from 0.5 to 3.0 mm in length is passed over the skin using gentle pressure, puncturing the superficial layers of the skin to loosen fibrotic adhesions and induce collagen synthesis. This procedure may be repeated several times within a single session or over multiple sessions depending on the depth and quality of the scars. This technique has been reported to reduce scar depth up to 25% after 2 sessions.30
Punch Techniques
Punch techniques are useful for treatment of deeper atrophic acne scarring, for which most other treatment modalities are not particularly effective. A punch excision approximately equal to the scar size is first performed, which may then be followed by either removal of the scar tissue with subsequent suturing, graft replacement of the removed tissue, or elevation of the already established scar tissue to the level of surrounding skin where it is then held in place by sutures or adhesive skin closure material. Success rates with this method are largely limited to case series, but punch techniques are reported to be efficacious, especially for treatment of ice pick scars. Risks for this method include graft failure, graft depression, and formation of sinus tracts.31
Chemical Peels
Chemicals peels traditionally employ the use of acidic compounds to strip away the outer layers of skin to variable depths depending on the concentration of the agent being applied. Chemical peels are not generally recommended for application in a nonspecific manner in the treatment of acne scars given the relatively mild cosmetic improvements seen and the high rate of AEs such as pigmentary alterations and additional scar formation.12 Rather, clinicians should employ the CROSS (chemical reconstruction of skin scars) technique, in which peel agents such as trichloroacetic acid are applied in high concentrations only to areas of atrophic scarring. Use of this method can minimize AEs while simultaneously achieving high success rates, with excellent results in 100% (32/32) of patients after 5 to 6 treatment sessions.32 This method has been successful for hard-to-treat ice pick scars.33
Soft-Tissue Augmentation
Soft-tissue augmentation is another effective treatment of superficial atrophic acne scarring that utilizes injections of collagen fillers such as hyaluronic acid, calcium hydroxylapatite, poly-L-lactic acid, silicone, and even autologous fat to replace lost tissue volume while simultaneously inducing collagen production via stretching of dermal fibroblasts.34 These treatments may require multiple sessions for cosmetic improvement but have shown considerable efficacy in the treatment of atrophic acne scars. Hyaluronic acid has been reported to be particularly effective for rolling scars.12 However, these compounds only provide temporary results, thus requiring repeated treatments to maintain cosmetic outcomes. Permanent options include the recently US Food and Drug Administration–approved polymethylmethacrylate microspheres suspended in bovine collagen as well as the novel technique of autologous fibroblast transfer. These options are relatively new, but initial double-blind, randomized, controlled trials have shown minimal AEs with substantial improvements in 64% to 100% of atrophic scars treated.35,36
Intralesional Therapy
Intralesional corticosteroid injections are a mainstay treatment of hypertrophic acne scarring and are believed to exert their effects by decreasing fibroblast proliferation and promoting collagen degradation.37 Treatment with steroids generally is effective, with reported improvement in 75% (6/8) of patients and complete flattening in 50% (4/8) of lesions according to one study.38 Development of hypopigmentation, dermal atrophy, and telangiectasia are potential sequelae of this treatment.37
5-Fluorouracil, bleomycin, and verapamil also have been used with good results as intralesional treatments of hypertrophic scars, but these agents typically are reserved for cases of corticosteroid failure. Such compounds are thought to mediate their effects through inhibition of dermal fibroblast proliferation.39 Results with these therapies are varied, but greater than 75% improvement is seen in most cases. Adverse effects include injection-site ulceration and hyperpigmentation.39
Cryotherapy
Contact cryotherapy has been studied as treatment of hypertrophic acne scars. The exact mechanism through which scars are reduced is unclear, but it is hypothesized that the physical damage caused by freezing and thrombosis lead to collagen restructuring. According to one study, cryotherapy was reported to achieve good or excellent results in 76% (29/38) of cases.40 Permanent pigmentary alterations are a possible AE.
Silicone Dressings
Silicone dressings are a reasonable treatment option for hypertrophic acne scarring given their proven efficacy and minimal risk for AEs. Thin sheets of silicone gels or membranes are applied daily in a topical manner to acne scars and are believed to be therapeutic through a combination of pressure and hydration, which subsequently inhibits fibroblast production of collagen. Notable reductions in scar appearance and size are seen in 60% to 80% of individuals using this method.41 Adverse effects are limited to pruritus and local skin maceration. Patient noncompliance may be an issue, as the silicone dressings may be applied on highly visible areas such as the face. Patients may apply the dressings at night, but efficacy may be reduced.
Conclusion
When determining which treatment options to use in a patient with acne scarring, it is important to first determine the patient’s treatment goals while simultaneously establishing realistic expectations. Important factors to consider are the patient’s preferences regarding treatment risk, duration, and permanence, as well as budget and social or work requirements. As such, treatment plans for each patient should be determined on a case-by-case basis. It also is important to note that a combination of different treatment modalities often is necessary and superior to monotherapy in achieving satisfactory cosmetic outcomes.
1. Ghodsi SZ, Orawa H, Zouboulis CC. Prevalence, severity, and severity risk factors of acne in high school pupils: a community-based study. J Invest Dermatol. 2009;129:2136-2141.
2. Collier CN, Harper JC, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
3. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
4. Geller L, Rosen J, Frankel A, et al. Perimenstrual flare of adult acne. J Clin Aesthet Dermatol. 2014;7:30-34.
5. Layton AM, Henderson CA, Cunliffe WJ. A clinical evaluation of acne scarring and its incidence. Clin Exp Dermatol. 1994;19:303-308.
6. Halvorsen JA, Stern RS, Dalgard F, et al. Suicidal ideation, mental health problems, and social impairment are increased in adolescents with acne: a population-based study. J Invest Dermatol. 2011;131:363-370.
7. Jacob CI, Dover JS, Kaminer MS. Acne scarring: a classification system and review of treatment options. J Am Acad Dermatol. 2001;45:109-117.
8. Rivera AE. Acne scarring: a review and current treatment modalities. J Am Acad Dermatol. 2008;59:659-676.
9. Goodman GJ. Acne and acne scarring: why should we treat? Med J Aust. 1999;171:62-63.
10. Frank W. Therapeutic dermabrasion. back to the future. Arch Dermatol. 1994;130:1187-1189.
11. Goodman GJ. Postacne scarring: a review of its pathophysiology and treatment. Dermatol Surg. 2000;26:857-871.
12. Hession MT, Graber EM. Atrophic acne scarring: a review of treatment options. J Clin Aesthet Dermatol. 2015;8:50-58.
13. Levy LL, Zeichner JA. Management of acne scarring, part II: a comparative review of non-laser-based, minimally invasive approaches. Am J Clin Dermatol. 2012;13:331-340.
14. Christophel JJ, Elm C, Endrizzi BT, et al. A randomized controlled trial of fractional laser therapy and dermabrasion for scar resurfacing. Dermatol Surg. 2012;38:595-602.
15. Katz BE, McFarlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853.
16. Bernestein LJ, Geronemus RG. Keloid formation with the 585-nm pulsed dye laser during isotretinoin treatment. Arch Dermatol. 1997;133:111-112.
17. Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706.
18. Wootton CI, Cartwright RP, Manning P, et al. Should isotretinoin be stopped prior to surgery? a critically appraised topic. Br J Dermatol. 2014;170:239-244.
19. Alster TS, West TB. Resurfacing of atrophic facial acne scars with a high-energy, pulsed carbon dioxide laser. Dermatol Surg. 1996;22:151-155.
20. Sobanko JF, Alster TS. Management of acne scarring, part I: a comparative review of laser surgical approaches. Am J Clin Dermatol. 2012;13:319-330.
21. Cho SB, Lee SJ, Oh SH, et al. Non-ablative 1550nm erbium-glass and ablative 10,600nm carbon dioxide fractional lasers for acne scar: a randomized split-face study with blinded response evaluation. J Eur Acad Dermatol Venereol. 2010;24:921-925.
22. Rkein A, Ozog D, Waibel JS. Treatment of atrophic scars with fractionated CO2 laser facilitating delivery of topically applied poly-L-lactic acid. Dermatol Surg. 2014;40:624-631.
23. Waibel JS, Wulkan AJ, Shumaker PR. Treatment of hypertrophic scars using laser and laser assisted corticosteroid delivery. Lasers Surg Med. 2013;45:135-140.
24. Alster TS, McMeekin TO. Improvement of facial acne scars by the 585-nm flashlamp-pumped pulsed dye laser. J Am Acad Dermatol. 1996;35:79-81.
25. Simmons BJ, Griffith RD, Falto-Aizpurua LA, et al. Use of radiofrequency in cosmetic dermatology: focus on nonablative treatment of acne scars. Clin Cosmet Investig Dermatol. 2014;7:335-339.
26. Ramesh M, Gopal M, Kumar S, et al. Novel technology in the treatment of acne scars: the matrix-tunable radiofrequency technology. J Cutan Aesthet Surg. 2010;3:97-101.
27. Johnson WC. Treatment of pitted scars; punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
28. Alam M, Omura N, Kaminer MS. Subcision for acne scarring: technique and outcomes in 40 patients. Dermatol Surg. 2005;31:310-317.
29. Sage R, Lopiccolo M, Liu A, et al. Subcuticular incision versus naturally sourced porcine collagen filler for acne scars: a randomized split-face comparison. Dermatol Surg. 2011;37:426-431.
30. Fabbrocini G, Annunziata MC, D’arco V, et al. Acne scars: pathogenesis, classification and treatment [published online ahead of print October 14, 2010]. Dermatol Res Pract. 2010;2010:893080.
31. Johnson WC. Treatment of pitted scars: punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
32. Lee JB, Chung WG, Kwahck H, et al. Focal treatment of acne scars with trichloroacetic acid: chemical reconstruction of skin scars method. Dermatol Surg. 2002;28:1017-1021.
33. Bhardwaj D, Khunger N. An assessment of the efficacy and safety of CROSS technique with 100% TCA in the management of ice pick acne scars. J Cutan Aesthet Surg. 2010;3:93-96.
34. Wang F, Garza LA, Kang S, et al. In vivo stimulation of de novo collagen production caused by cross-linked hyaluronic acid dermal filler injections in photodamaged human skin. Arch Dermatol. 2007;143:155-163.
35. Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
36. Munavalli GS, Smith S, Maslowski JM, et al. Successful treatment of depressed, distensible acne scars using autologous fibroblasts: a multi-site, prospective, double blind, placebo-controlled clinical trial. Dermatol Surg. 2013;39:1226-1236.
37. Leventhal D, Furr M, Reiter D. Treatment of keloids and hypertrophic scars: a meta-analysis and review of the literature. Arch Facial Plast Surg. 2006;8:362-368.
38. Darzi MA, Chowdri NA, Kaul SK, et al. Evaluation of various methods of treating keloids and hypertrophic scars: a 10-year follow-up study. Br J Plast Surg. 1992;45:374-379.
39. Ledon JA, Savas J, Franca K, et al. Intralesional treatment for keloids and hypertrophic scars: a review. Dermatol Surg. 2013;39:1745-1757.
40. Zouboulis CC, Blume U, Büttner P, et al. Outcomes of cryosurgery in keloids and hypertrophic scars. a prospective consecutive trial of case series. Arch Dermatol. 1993;129:1146-1151.
41. Puri N, Talwar A. The efficacy of silicone gel for the treatment of hypertrophic scars and keloids. J Cutan Aesthet Surg. 2009;2:104-106.
Acne vulgaris is one of the most common inflammatory dermatoses affecting nearly all adolescents and a large proportion of adults.1 Incidence rates trend downward with age, but prevalence has been reported to be as high as 51% in individuals aged 20 to 29 years.2 Notably, recent evidence suggests there is an increasing incidence rate of acne among postadolescent women, with the severity associated with the menstrual cycle.3,4 Scarring is a common result of acne and may even occur in the setting of appropriate medical therapy. In particular, some form of facial scarring has been reported to occur in up to 95% of acne patients, with severe scarring in 30% of these patients.5 The detrimental effects of acne scarring are not only limited to impaired cosmetic appearance, as it also has been associated with depression symptoms, suicidal ideation, mental health problems, and general social impairment.6 Given the negative impact of acne scarring on overall health and well-being as well as its permanent nature, early and effective treatment is essential to maximize cosmetic outcomes and minimize long-term deleterious effects.
Acne scarring can be broadly divided into 2 major categories: atrophic and hypertrophic. Atrophic scarring is more common and is characterized by an overall localized reduction in collagen content. Clinically, atrophic scars present as depressions in the skin secondary to inflammatory fibrous contractions induced by acne. This type of scarring can be further divided into various subtypes based on morphologic criteria (eg, size, depth), such as boxcar, ice pick, and rolling scars.7 Conversely, hypertrophic scarring is characterized by an overall increase in collagen content and presents as firm raised lesions. Hypertrophic scars should be distinguished from keloid scars, as the former will not outgrow the margins of the original wound while the latter will.8 Treatment of acne scarring is based on scar type and can be accomplished through a variety of medical and surgical modalities (Table). In this article, we review some of the most commonly utilized therapies for both atrophic and hypertrophic acne scarring with a focus on cosmetic outcomes. It is important to keep in mind, however, that the best treatment is to prevent the occurrence of acne scarring through early and proactive treatment of acne.9
Dermabrasion
Dermabrasion is a decades-old technique that employs the use of a motorized device equipped with an abrasive material to physically remove the superficial layers of the skin, thus inducing the wound-healing process with subsequent formation of new collagen.10 In the same vein, microdermabrasion utilizes aluminum oxide crystals ejected from a nozzle to induce superficial microlacerations.11 This technique is most successful when used to soften scar edges in superficial atrophic scars of the rolling or boxcar subtypes.12 Dermabrasion has been shown to be equally as effective as laser therapy in the treatment of facial scars but is reported to have a much greater risk for adverse effects (AEs)(eg, erythema, edema) that may last for several weeks posttherapy.13,14 Dermabrasion is a particularly operator-dependent technique for which outcomes may vary depending on operator experience. As such, it is not generally recommended as a first-line therapy given its risks and relatively modest results; however, dermabrasion can be a useful adjunct when performed in the right setting. This technique, in addition to laser resurfacing, should be used with caution in patients who have recently taken or currently are taking isotretinoin, as several case series have reported postprocedural development of hypertrophic or keloid scars,15-17 but these findings subsequently were questioned in the literature.18
Laser Therapy
Laser technology has advanced tremendously over the last few decades and there are now a multitude of available lasers that are capable of variable depth penetration and energy delivery patterns. Common to all, however, is the ability to induce localized thermal damage with eventual collagen remodeling. Lasers can be divided into 2 major categories: ablative and nonablative. Ablative lasers cause epidermal destruction, while nonablative lasers are able to selectively target dermal layers without disrupting the overlying epithelium. Generally speaking, ablative lasers are more effective than nonablative lasers in the treatment of atrophic scars, with reported mean improvements of up to 81%.19 This increased efficacy comes with an increased risk for AEs such as postinflammatory hyperpigmentation, prolonged posttreatment erythema, and formation of additional scarring.20 Both ablative and nonablative lasers can be applied in the more recently developed technology of fractional photothermolysis. With this method, noncontiguous microscopic columns of thermal injury surrounded by zones of viable tissue are created, which is in contrast to the traditional manner of inducing broad thermal injury. Fractional ablative lasers can achieve efficacy rates similar to traditional ablative lasers with a reduced risk for permanent scarring or dispigmentation.21 Notably, recent studies have shown promising results for the use of fractional ablative lasers as a mechanism to enhance drug delivery of topically applied medications such as poly-L-lactic acid and triamcinolone acetonide in the treatment of atrophic and hypertrophic scars, respectively.22,23
Lasers also play a role in the treatment of hypertrophic acne scars with the use of nonablative pulsed dye lasers. These lasers cause selective thermolysis of dermal vasculature, and average clinical improvements in hypertrophic scars of 67.5% after a single treatment have been reported.24 Temporary postoperative purpura and long-term hyperpigmentation are reported outcomes of this therapy.20
Radiofrequency
Nonablative radiofrequency (RF) is a relatively novel technique that creates an electric current in the dermis at preset depths to induce thermal damage and eventual collagen synthesis. There are a variety of modalities for which RF can be applied, but microneedle bipolar RF and fractional bipolar RF treatments offer the best results for atrophic acne scars. Improvements in scar appearance of 25% to 75% have been reported after several treatment sessions.25 Better results have been reported in the treatment of ice pick scars as compared to more superficial scars,26 but additional studies will be necessary to validate this claim. Adverse effects are largely limited to temporary erythema and posttreatment scabbing.27
Subcision
Subcision is a more physically intensive technique useful for treatment of superficial atrophic acne scars. This method involves the use of a small needle that is inserted into the periphery of a scar before being moved in a back-and-forth manner underneath the base of the scar to loosen the fibrotic adhesions that result in the depressed appearance of the scar. Additionally, loosening of the tissue and resultant bleeding creates a potential space for future collagen deposition during the subsequent wound-healing phase. Subcision has a reported success rate of 50% to 60% in the treatment of rolling scars, and prospective, randomized, split-face trials have indicated that the short-term outcomes of subcision are superior to dermal fillers while being equally effective long-term.28,29 Of note, a small percentage of patients may develop a localized nodule at the site of treatment, which can be resolved with intralesional steroids.11
Skin Needling
Skin needling, also referred to as collagen induction therapy, utilizes vertical needle punctures rather than the horizontally directed punctures that are used in subcision and can be used to treat rolling and boxcar scars. Traditionally, a small roller equipped with rows of small needles typically ranging in size from 0.5 to 3.0 mm in length is passed over the skin using gentle pressure, puncturing the superficial layers of the skin to loosen fibrotic adhesions and induce collagen synthesis. This procedure may be repeated several times within a single session or over multiple sessions depending on the depth and quality of the scars. This technique has been reported to reduce scar depth up to 25% after 2 sessions.30
Punch Techniques
Punch techniques are useful for treatment of deeper atrophic acne scarring, for which most other treatment modalities are not particularly effective. A punch excision approximately equal to the scar size is first performed, which may then be followed by either removal of the scar tissue with subsequent suturing, graft replacement of the removed tissue, or elevation of the already established scar tissue to the level of surrounding skin where it is then held in place by sutures or adhesive skin closure material. Success rates with this method are largely limited to case series, but punch techniques are reported to be efficacious, especially for treatment of ice pick scars. Risks for this method include graft failure, graft depression, and formation of sinus tracts.31
Chemical Peels
Chemicals peels traditionally employ the use of acidic compounds to strip away the outer layers of skin to variable depths depending on the concentration of the agent being applied. Chemical peels are not generally recommended for application in a nonspecific manner in the treatment of acne scars given the relatively mild cosmetic improvements seen and the high rate of AEs such as pigmentary alterations and additional scar formation.12 Rather, clinicians should employ the CROSS (chemical reconstruction of skin scars) technique, in which peel agents such as trichloroacetic acid are applied in high concentrations only to areas of atrophic scarring. Use of this method can minimize AEs while simultaneously achieving high success rates, with excellent results in 100% (32/32) of patients after 5 to 6 treatment sessions.32 This method has been successful for hard-to-treat ice pick scars.33
Soft-Tissue Augmentation
Soft-tissue augmentation is another effective treatment of superficial atrophic acne scarring that utilizes injections of collagen fillers such as hyaluronic acid, calcium hydroxylapatite, poly-L-lactic acid, silicone, and even autologous fat to replace lost tissue volume while simultaneously inducing collagen production via stretching of dermal fibroblasts.34 These treatments may require multiple sessions for cosmetic improvement but have shown considerable efficacy in the treatment of atrophic acne scars. Hyaluronic acid has been reported to be particularly effective for rolling scars.12 However, these compounds only provide temporary results, thus requiring repeated treatments to maintain cosmetic outcomes. Permanent options include the recently US Food and Drug Administration–approved polymethylmethacrylate microspheres suspended in bovine collagen as well as the novel technique of autologous fibroblast transfer. These options are relatively new, but initial double-blind, randomized, controlled trials have shown minimal AEs with substantial improvements in 64% to 100% of atrophic scars treated.35,36
Intralesional Therapy
Intralesional corticosteroid injections are a mainstay treatment of hypertrophic acne scarring and are believed to exert their effects by decreasing fibroblast proliferation and promoting collagen degradation.37 Treatment with steroids generally is effective, with reported improvement in 75% (6/8) of patients and complete flattening in 50% (4/8) of lesions according to one study.38 Development of hypopigmentation, dermal atrophy, and telangiectasia are potential sequelae of this treatment.37
5-Fluorouracil, bleomycin, and verapamil also have been used with good results as intralesional treatments of hypertrophic scars, but these agents typically are reserved for cases of corticosteroid failure. Such compounds are thought to mediate their effects through inhibition of dermal fibroblast proliferation.39 Results with these therapies are varied, but greater than 75% improvement is seen in most cases. Adverse effects include injection-site ulceration and hyperpigmentation.39
Cryotherapy
Contact cryotherapy has been studied as treatment of hypertrophic acne scars. The exact mechanism through which scars are reduced is unclear, but it is hypothesized that the physical damage caused by freezing and thrombosis lead to collagen restructuring. According to one study, cryotherapy was reported to achieve good or excellent results in 76% (29/38) of cases.40 Permanent pigmentary alterations are a possible AE.
Silicone Dressings
Silicone dressings are a reasonable treatment option for hypertrophic acne scarring given their proven efficacy and minimal risk for AEs. Thin sheets of silicone gels or membranes are applied daily in a topical manner to acne scars and are believed to be therapeutic through a combination of pressure and hydration, which subsequently inhibits fibroblast production of collagen. Notable reductions in scar appearance and size are seen in 60% to 80% of individuals using this method.41 Adverse effects are limited to pruritus and local skin maceration. Patient noncompliance may be an issue, as the silicone dressings may be applied on highly visible areas such as the face. Patients may apply the dressings at night, but efficacy may be reduced.
Conclusion
When determining which treatment options to use in a patient with acne scarring, it is important to first determine the patient’s treatment goals while simultaneously establishing realistic expectations. Important factors to consider are the patient’s preferences regarding treatment risk, duration, and permanence, as well as budget and social or work requirements. As such, treatment plans for each patient should be determined on a case-by-case basis. It also is important to note that a combination of different treatment modalities often is necessary and superior to monotherapy in achieving satisfactory cosmetic outcomes.
Acne vulgaris is one of the most common inflammatory dermatoses affecting nearly all adolescents and a large proportion of adults.1 Incidence rates trend downward with age, but prevalence has been reported to be as high as 51% in individuals aged 20 to 29 years.2 Notably, recent evidence suggests there is an increasing incidence rate of acne among postadolescent women, with the severity associated with the menstrual cycle.3,4 Scarring is a common result of acne and may even occur in the setting of appropriate medical therapy. In particular, some form of facial scarring has been reported to occur in up to 95% of acne patients, with severe scarring in 30% of these patients.5 The detrimental effects of acne scarring are not only limited to impaired cosmetic appearance, as it also has been associated with depression symptoms, suicidal ideation, mental health problems, and general social impairment.6 Given the negative impact of acne scarring on overall health and well-being as well as its permanent nature, early and effective treatment is essential to maximize cosmetic outcomes and minimize long-term deleterious effects.
Acne scarring can be broadly divided into 2 major categories: atrophic and hypertrophic. Atrophic scarring is more common and is characterized by an overall localized reduction in collagen content. Clinically, atrophic scars present as depressions in the skin secondary to inflammatory fibrous contractions induced by acne. This type of scarring can be further divided into various subtypes based on morphologic criteria (eg, size, depth), such as boxcar, ice pick, and rolling scars.7 Conversely, hypertrophic scarring is characterized by an overall increase in collagen content and presents as firm raised lesions. Hypertrophic scars should be distinguished from keloid scars, as the former will not outgrow the margins of the original wound while the latter will.8 Treatment of acne scarring is based on scar type and can be accomplished through a variety of medical and surgical modalities (Table). In this article, we review some of the most commonly utilized therapies for both atrophic and hypertrophic acne scarring with a focus on cosmetic outcomes. It is important to keep in mind, however, that the best treatment is to prevent the occurrence of acne scarring through early and proactive treatment of acne.9
Dermabrasion
Dermabrasion is a decades-old technique that employs the use of a motorized device equipped with an abrasive material to physically remove the superficial layers of the skin, thus inducing the wound-healing process with subsequent formation of new collagen.10 In the same vein, microdermabrasion utilizes aluminum oxide crystals ejected from a nozzle to induce superficial microlacerations.11 This technique is most successful when used to soften scar edges in superficial atrophic scars of the rolling or boxcar subtypes.12 Dermabrasion has been shown to be equally as effective as laser therapy in the treatment of facial scars but is reported to have a much greater risk for adverse effects (AEs)(eg, erythema, edema) that may last for several weeks posttherapy.13,14 Dermabrasion is a particularly operator-dependent technique for which outcomes may vary depending on operator experience. As such, it is not generally recommended as a first-line therapy given its risks and relatively modest results; however, dermabrasion can be a useful adjunct when performed in the right setting. This technique, in addition to laser resurfacing, should be used with caution in patients who have recently taken or currently are taking isotretinoin, as several case series have reported postprocedural development of hypertrophic or keloid scars,15-17 but these findings subsequently were questioned in the literature.18
Laser Therapy
Laser technology has advanced tremendously over the last few decades and there are now a multitude of available lasers that are capable of variable depth penetration and energy delivery patterns. Common to all, however, is the ability to induce localized thermal damage with eventual collagen remodeling. Lasers can be divided into 2 major categories: ablative and nonablative. Ablative lasers cause epidermal destruction, while nonablative lasers are able to selectively target dermal layers without disrupting the overlying epithelium. Generally speaking, ablative lasers are more effective than nonablative lasers in the treatment of atrophic scars, with reported mean improvements of up to 81%.19 This increased efficacy comes with an increased risk for AEs such as postinflammatory hyperpigmentation, prolonged posttreatment erythema, and formation of additional scarring.20 Both ablative and nonablative lasers can be applied in the more recently developed technology of fractional photothermolysis. With this method, noncontiguous microscopic columns of thermal injury surrounded by zones of viable tissue are created, which is in contrast to the traditional manner of inducing broad thermal injury. Fractional ablative lasers can achieve efficacy rates similar to traditional ablative lasers with a reduced risk for permanent scarring or dispigmentation.21 Notably, recent studies have shown promising results for the use of fractional ablative lasers as a mechanism to enhance drug delivery of topically applied medications such as poly-L-lactic acid and triamcinolone acetonide in the treatment of atrophic and hypertrophic scars, respectively.22,23
Lasers also play a role in the treatment of hypertrophic acne scars with the use of nonablative pulsed dye lasers. These lasers cause selective thermolysis of dermal vasculature, and average clinical improvements in hypertrophic scars of 67.5% after a single treatment have been reported.24 Temporary postoperative purpura and long-term hyperpigmentation are reported outcomes of this therapy.20
Radiofrequency
Nonablative radiofrequency (RF) is a relatively novel technique that creates an electric current in the dermis at preset depths to induce thermal damage and eventual collagen synthesis. There are a variety of modalities for which RF can be applied, but microneedle bipolar RF and fractional bipolar RF treatments offer the best results for atrophic acne scars. Improvements in scar appearance of 25% to 75% have been reported after several treatment sessions.25 Better results have been reported in the treatment of ice pick scars as compared to more superficial scars,26 but additional studies will be necessary to validate this claim. Adverse effects are largely limited to temporary erythema and posttreatment scabbing.27
Subcision
Subcision is a more physically intensive technique useful for treatment of superficial atrophic acne scars. This method involves the use of a small needle that is inserted into the periphery of a scar before being moved in a back-and-forth manner underneath the base of the scar to loosen the fibrotic adhesions that result in the depressed appearance of the scar. Additionally, loosening of the tissue and resultant bleeding creates a potential space for future collagen deposition during the subsequent wound-healing phase. Subcision has a reported success rate of 50% to 60% in the treatment of rolling scars, and prospective, randomized, split-face trials have indicated that the short-term outcomes of subcision are superior to dermal fillers while being equally effective long-term.28,29 Of note, a small percentage of patients may develop a localized nodule at the site of treatment, which can be resolved with intralesional steroids.11
Skin Needling
Skin needling, also referred to as collagen induction therapy, utilizes vertical needle punctures rather than the horizontally directed punctures that are used in subcision and can be used to treat rolling and boxcar scars. Traditionally, a small roller equipped with rows of small needles typically ranging in size from 0.5 to 3.0 mm in length is passed over the skin using gentle pressure, puncturing the superficial layers of the skin to loosen fibrotic adhesions and induce collagen synthesis. This procedure may be repeated several times within a single session or over multiple sessions depending on the depth and quality of the scars. This technique has been reported to reduce scar depth up to 25% after 2 sessions.30
Punch Techniques
Punch techniques are useful for treatment of deeper atrophic acne scarring, for which most other treatment modalities are not particularly effective. A punch excision approximately equal to the scar size is first performed, which may then be followed by either removal of the scar tissue with subsequent suturing, graft replacement of the removed tissue, or elevation of the already established scar tissue to the level of surrounding skin where it is then held in place by sutures or adhesive skin closure material. Success rates with this method are largely limited to case series, but punch techniques are reported to be efficacious, especially for treatment of ice pick scars. Risks for this method include graft failure, graft depression, and formation of sinus tracts.31
Chemical Peels
Chemicals peels traditionally employ the use of acidic compounds to strip away the outer layers of skin to variable depths depending on the concentration of the agent being applied. Chemical peels are not generally recommended for application in a nonspecific manner in the treatment of acne scars given the relatively mild cosmetic improvements seen and the high rate of AEs such as pigmentary alterations and additional scar formation.12 Rather, clinicians should employ the CROSS (chemical reconstruction of skin scars) technique, in which peel agents such as trichloroacetic acid are applied in high concentrations only to areas of atrophic scarring. Use of this method can minimize AEs while simultaneously achieving high success rates, with excellent results in 100% (32/32) of patients after 5 to 6 treatment sessions.32 This method has been successful for hard-to-treat ice pick scars.33
Soft-Tissue Augmentation
Soft-tissue augmentation is another effective treatment of superficial atrophic acne scarring that utilizes injections of collagen fillers such as hyaluronic acid, calcium hydroxylapatite, poly-L-lactic acid, silicone, and even autologous fat to replace lost tissue volume while simultaneously inducing collagen production via stretching of dermal fibroblasts.34 These treatments may require multiple sessions for cosmetic improvement but have shown considerable efficacy in the treatment of atrophic acne scars. Hyaluronic acid has been reported to be particularly effective for rolling scars.12 However, these compounds only provide temporary results, thus requiring repeated treatments to maintain cosmetic outcomes. Permanent options include the recently US Food and Drug Administration–approved polymethylmethacrylate microspheres suspended in bovine collagen as well as the novel technique of autologous fibroblast transfer. These options are relatively new, but initial double-blind, randomized, controlled trials have shown minimal AEs with substantial improvements in 64% to 100% of atrophic scars treated.35,36
Intralesional Therapy
Intralesional corticosteroid injections are a mainstay treatment of hypertrophic acne scarring and are believed to exert their effects by decreasing fibroblast proliferation and promoting collagen degradation.37 Treatment with steroids generally is effective, with reported improvement in 75% (6/8) of patients and complete flattening in 50% (4/8) of lesions according to one study.38 Development of hypopigmentation, dermal atrophy, and telangiectasia are potential sequelae of this treatment.37
5-Fluorouracil, bleomycin, and verapamil also have been used with good results as intralesional treatments of hypertrophic scars, but these agents typically are reserved for cases of corticosteroid failure. Such compounds are thought to mediate their effects through inhibition of dermal fibroblast proliferation.39 Results with these therapies are varied, but greater than 75% improvement is seen in most cases. Adverse effects include injection-site ulceration and hyperpigmentation.39
Cryotherapy
Contact cryotherapy has been studied as treatment of hypertrophic acne scars. The exact mechanism through which scars are reduced is unclear, but it is hypothesized that the physical damage caused by freezing and thrombosis lead to collagen restructuring. According to one study, cryotherapy was reported to achieve good or excellent results in 76% (29/38) of cases.40 Permanent pigmentary alterations are a possible AE.
Silicone Dressings
Silicone dressings are a reasonable treatment option for hypertrophic acne scarring given their proven efficacy and minimal risk for AEs. Thin sheets of silicone gels or membranes are applied daily in a topical manner to acne scars and are believed to be therapeutic through a combination of pressure and hydration, which subsequently inhibits fibroblast production of collagen. Notable reductions in scar appearance and size are seen in 60% to 80% of individuals using this method.41 Adverse effects are limited to pruritus and local skin maceration. Patient noncompliance may be an issue, as the silicone dressings may be applied on highly visible areas such as the face. Patients may apply the dressings at night, but efficacy may be reduced.
Conclusion
When determining which treatment options to use in a patient with acne scarring, it is important to first determine the patient’s treatment goals while simultaneously establishing realistic expectations. Important factors to consider are the patient’s preferences regarding treatment risk, duration, and permanence, as well as budget and social or work requirements. As such, treatment plans for each patient should be determined on a case-by-case basis. It also is important to note that a combination of different treatment modalities often is necessary and superior to monotherapy in achieving satisfactory cosmetic outcomes.
1. Ghodsi SZ, Orawa H, Zouboulis CC. Prevalence, severity, and severity risk factors of acne in high school pupils: a community-based study. J Invest Dermatol. 2009;129:2136-2141.
2. Collier CN, Harper JC, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
3. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
4. Geller L, Rosen J, Frankel A, et al. Perimenstrual flare of adult acne. J Clin Aesthet Dermatol. 2014;7:30-34.
5. Layton AM, Henderson CA, Cunliffe WJ. A clinical evaluation of acne scarring and its incidence. Clin Exp Dermatol. 1994;19:303-308.
6. Halvorsen JA, Stern RS, Dalgard F, et al. Suicidal ideation, mental health problems, and social impairment are increased in adolescents with acne: a population-based study. J Invest Dermatol. 2011;131:363-370.
7. Jacob CI, Dover JS, Kaminer MS. Acne scarring: a classification system and review of treatment options. J Am Acad Dermatol. 2001;45:109-117.
8. Rivera AE. Acne scarring: a review and current treatment modalities. J Am Acad Dermatol. 2008;59:659-676.
9. Goodman GJ. Acne and acne scarring: why should we treat? Med J Aust. 1999;171:62-63.
10. Frank W. Therapeutic dermabrasion. back to the future. Arch Dermatol. 1994;130:1187-1189.
11. Goodman GJ. Postacne scarring: a review of its pathophysiology and treatment. Dermatol Surg. 2000;26:857-871.
12. Hession MT, Graber EM. Atrophic acne scarring: a review of treatment options. J Clin Aesthet Dermatol. 2015;8:50-58.
13. Levy LL, Zeichner JA. Management of acne scarring, part II: a comparative review of non-laser-based, minimally invasive approaches. Am J Clin Dermatol. 2012;13:331-340.
14. Christophel JJ, Elm C, Endrizzi BT, et al. A randomized controlled trial of fractional laser therapy and dermabrasion for scar resurfacing. Dermatol Surg. 2012;38:595-602.
15. Katz BE, McFarlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853.
16. Bernestein LJ, Geronemus RG. Keloid formation with the 585-nm pulsed dye laser during isotretinoin treatment. Arch Dermatol. 1997;133:111-112.
17. Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706.
18. Wootton CI, Cartwright RP, Manning P, et al. Should isotretinoin be stopped prior to surgery? a critically appraised topic. Br J Dermatol. 2014;170:239-244.
19. Alster TS, West TB. Resurfacing of atrophic facial acne scars with a high-energy, pulsed carbon dioxide laser. Dermatol Surg. 1996;22:151-155.
20. Sobanko JF, Alster TS. Management of acne scarring, part I: a comparative review of laser surgical approaches. Am J Clin Dermatol. 2012;13:319-330.
21. Cho SB, Lee SJ, Oh SH, et al. Non-ablative 1550nm erbium-glass and ablative 10,600nm carbon dioxide fractional lasers for acne scar: a randomized split-face study with blinded response evaluation. J Eur Acad Dermatol Venereol. 2010;24:921-925.
22. Rkein A, Ozog D, Waibel JS. Treatment of atrophic scars with fractionated CO2 laser facilitating delivery of topically applied poly-L-lactic acid. Dermatol Surg. 2014;40:624-631.
23. Waibel JS, Wulkan AJ, Shumaker PR. Treatment of hypertrophic scars using laser and laser assisted corticosteroid delivery. Lasers Surg Med. 2013;45:135-140.
24. Alster TS, McMeekin TO. Improvement of facial acne scars by the 585-nm flashlamp-pumped pulsed dye laser. J Am Acad Dermatol. 1996;35:79-81.
25. Simmons BJ, Griffith RD, Falto-Aizpurua LA, et al. Use of radiofrequency in cosmetic dermatology: focus on nonablative treatment of acne scars. Clin Cosmet Investig Dermatol. 2014;7:335-339.
26. Ramesh M, Gopal M, Kumar S, et al. Novel technology in the treatment of acne scars: the matrix-tunable radiofrequency technology. J Cutan Aesthet Surg. 2010;3:97-101.
27. Johnson WC. Treatment of pitted scars; punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
28. Alam M, Omura N, Kaminer MS. Subcision for acne scarring: technique and outcomes in 40 patients. Dermatol Surg. 2005;31:310-317.
29. Sage R, Lopiccolo M, Liu A, et al. Subcuticular incision versus naturally sourced porcine collagen filler for acne scars: a randomized split-face comparison. Dermatol Surg. 2011;37:426-431.
30. Fabbrocini G, Annunziata MC, D’arco V, et al. Acne scars: pathogenesis, classification and treatment [published online ahead of print October 14, 2010]. Dermatol Res Pract. 2010;2010:893080.
31. Johnson WC. Treatment of pitted scars: punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
32. Lee JB, Chung WG, Kwahck H, et al. Focal treatment of acne scars with trichloroacetic acid: chemical reconstruction of skin scars method. Dermatol Surg. 2002;28:1017-1021.
33. Bhardwaj D, Khunger N. An assessment of the efficacy and safety of CROSS technique with 100% TCA in the management of ice pick acne scars. J Cutan Aesthet Surg. 2010;3:93-96.
34. Wang F, Garza LA, Kang S, et al. In vivo stimulation of de novo collagen production caused by cross-linked hyaluronic acid dermal filler injections in photodamaged human skin. Arch Dermatol. 2007;143:155-163.
35. Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
36. Munavalli GS, Smith S, Maslowski JM, et al. Successful treatment of depressed, distensible acne scars using autologous fibroblasts: a multi-site, prospective, double blind, placebo-controlled clinical trial. Dermatol Surg. 2013;39:1226-1236.
37. Leventhal D, Furr M, Reiter D. Treatment of keloids and hypertrophic scars: a meta-analysis and review of the literature. Arch Facial Plast Surg. 2006;8:362-368.
38. Darzi MA, Chowdri NA, Kaul SK, et al. Evaluation of various methods of treating keloids and hypertrophic scars: a 10-year follow-up study. Br J Plast Surg. 1992;45:374-379.
39. Ledon JA, Savas J, Franca K, et al. Intralesional treatment for keloids and hypertrophic scars: a review. Dermatol Surg. 2013;39:1745-1757.
40. Zouboulis CC, Blume U, Büttner P, et al. Outcomes of cryosurgery in keloids and hypertrophic scars. a prospective consecutive trial of case series. Arch Dermatol. 1993;129:1146-1151.
41. Puri N, Talwar A. The efficacy of silicone gel for the treatment of hypertrophic scars and keloids. J Cutan Aesthet Surg. 2009;2:104-106.
1. Ghodsi SZ, Orawa H, Zouboulis CC. Prevalence, severity, and severity risk factors of acne in high school pupils: a community-based study. J Invest Dermatol. 2009;129:2136-2141.
2. Collier CN, Harper JC, Cafardi JA, et al. The prevalence of acne in adults 20 years and older. J Am Acad Dermatol. 2008;58:56-59.
3. Kim GK, Michaels BB. Post-adolescent acne in women: more common and more clinical considerations. J Drugs Dermatol. 2012;11:708-713.
4. Geller L, Rosen J, Frankel A, et al. Perimenstrual flare of adult acne. J Clin Aesthet Dermatol. 2014;7:30-34.
5. Layton AM, Henderson CA, Cunliffe WJ. A clinical evaluation of acne scarring and its incidence. Clin Exp Dermatol. 1994;19:303-308.
6. Halvorsen JA, Stern RS, Dalgard F, et al. Suicidal ideation, mental health problems, and social impairment are increased in adolescents with acne: a population-based study. J Invest Dermatol. 2011;131:363-370.
7. Jacob CI, Dover JS, Kaminer MS. Acne scarring: a classification system and review of treatment options. J Am Acad Dermatol. 2001;45:109-117.
8. Rivera AE. Acne scarring: a review and current treatment modalities. J Am Acad Dermatol. 2008;59:659-676.
9. Goodman GJ. Acne and acne scarring: why should we treat? Med J Aust. 1999;171:62-63.
10. Frank W. Therapeutic dermabrasion. back to the future. Arch Dermatol. 1994;130:1187-1189.
11. Goodman GJ. Postacne scarring: a review of its pathophysiology and treatment. Dermatol Surg. 2000;26:857-871.
12. Hession MT, Graber EM. Atrophic acne scarring: a review of treatment options. J Clin Aesthet Dermatol. 2015;8:50-58.
13. Levy LL, Zeichner JA. Management of acne scarring, part II: a comparative review of non-laser-based, minimally invasive approaches. Am J Clin Dermatol. 2012;13:331-340.
14. Christophel JJ, Elm C, Endrizzi BT, et al. A randomized controlled trial of fractional laser therapy and dermabrasion for scar resurfacing. Dermatol Surg. 2012;38:595-602.
15. Katz BE, McFarlane DF. Atypical facial scarring after isotretinoin therapy in a patient with previous dermabrasion. J Am Acad Dermatol. 1994;30:852-853.
16. Bernestein LJ, Geronemus RG. Keloid formation with the 585-nm pulsed dye laser during isotretinoin treatment. Arch Dermatol. 1997;133:111-112.
17. Zachariae H. Delayed wound healing and keloid formation following argon laser treatment or dermabrasion during isotretinoin treatment. Br J Dermatol. 1988;118:703-706.
18. Wootton CI, Cartwright RP, Manning P, et al. Should isotretinoin be stopped prior to surgery? a critically appraised topic. Br J Dermatol. 2014;170:239-244.
19. Alster TS, West TB. Resurfacing of atrophic facial acne scars with a high-energy, pulsed carbon dioxide laser. Dermatol Surg. 1996;22:151-155.
20. Sobanko JF, Alster TS. Management of acne scarring, part I: a comparative review of laser surgical approaches. Am J Clin Dermatol. 2012;13:319-330.
21. Cho SB, Lee SJ, Oh SH, et al. Non-ablative 1550nm erbium-glass and ablative 10,600nm carbon dioxide fractional lasers for acne scar: a randomized split-face study with blinded response evaluation. J Eur Acad Dermatol Venereol. 2010;24:921-925.
22. Rkein A, Ozog D, Waibel JS. Treatment of atrophic scars with fractionated CO2 laser facilitating delivery of topically applied poly-L-lactic acid. Dermatol Surg. 2014;40:624-631.
23. Waibel JS, Wulkan AJ, Shumaker PR. Treatment of hypertrophic scars using laser and laser assisted corticosteroid delivery. Lasers Surg Med. 2013;45:135-140.
24. Alster TS, McMeekin TO. Improvement of facial acne scars by the 585-nm flashlamp-pumped pulsed dye laser. J Am Acad Dermatol. 1996;35:79-81.
25. Simmons BJ, Griffith RD, Falto-Aizpurua LA, et al. Use of radiofrequency in cosmetic dermatology: focus on nonablative treatment of acne scars. Clin Cosmet Investig Dermatol. 2014;7:335-339.
26. Ramesh M, Gopal M, Kumar S, et al. Novel technology in the treatment of acne scars: the matrix-tunable radiofrequency technology. J Cutan Aesthet Surg. 2010;3:97-101.
27. Johnson WC. Treatment of pitted scars; punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
28. Alam M, Omura N, Kaminer MS. Subcision for acne scarring: technique and outcomes in 40 patients. Dermatol Surg. 2005;31:310-317.
29. Sage R, Lopiccolo M, Liu A, et al. Subcuticular incision versus naturally sourced porcine collagen filler for acne scars: a randomized split-face comparison. Dermatol Surg. 2011;37:426-431.
30. Fabbrocini G, Annunziata MC, D’arco V, et al. Acne scars: pathogenesis, classification and treatment [published online ahead of print October 14, 2010]. Dermatol Res Pract. 2010;2010:893080.
31. Johnson WC. Treatment of pitted scars: punch transplant technique. J Dermatol Surg Oncol. 1986;12:260-265.
32. Lee JB, Chung WG, Kwahck H, et al. Focal treatment of acne scars with trichloroacetic acid: chemical reconstruction of skin scars method. Dermatol Surg. 2002;28:1017-1021.
33. Bhardwaj D, Khunger N. An assessment of the efficacy and safety of CROSS technique with 100% TCA in the management of ice pick acne scars. J Cutan Aesthet Surg. 2010;3:93-96.
34. Wang F, Garza LA, Kang S, et al. In vivo stimulation of de novo collagen production caused by cross-linked hyaluronic acid dermal filler injections in photodamaged human skin. Arch Dermatol. 2007;143:155-163.
35. Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
36. Munavalli GS, Smith S, Maslowski JM, et al. Successful treatment of depressed, distensible acne scars using autologous fibroblasts: a multi-site, prospective, double blind, placebo-controlled clinical trial. Dermatol Surg. 2013;39:1226-1236.
37. Leventhal D, Furr M, Reiter D. Treatment of keloids and hypertrophic scars: a meta-analysis and review of the literature. Arch Facial Plast Surg. 2006;8:362-368.
38. Darzi MA, Chowdri NA, Kaul SK, et al. Evaluation of various methods of treating keloids and hypertrophic scars: a 10-year follow-up study. Br J Plast Surg. 1992;45:374-379.
39. Ledon JA, Savas J, Franca K, et al. Intralesional treatment for keloids and hypertrophic scars: a review. Dermatol Surg. 2013;39:1745-1757.
40. Zouboulis CC, Blume U, Büttner P, et al. Outcomes of cryosurgery in keloids and hypertrophic scars. a prospective consecutive trial of case series. Arch Dermatol. 1993;129:1146-1151.
41. Puri N, Talwar A. The efficacy of silicone gel for the treatment of hypertrophic scars and keloids. J Cutan Aesthet Surg. 2009;2:104-106.
Practice Points
- Scarring is a common and undesirable outcome of acne vulgaris that can occur even in the setting of appropriate medical management.
- Acne scars can be classified into several different types based on scar quality and appearance. The choice of treatment with medical or surgical measures should be made with respect to the type of scar present.
- A combination of therapeutic modalities often is necessary to achieve optimal cosmetic outcomes in the treatment of both atrophic and hypertrophic acne scars.
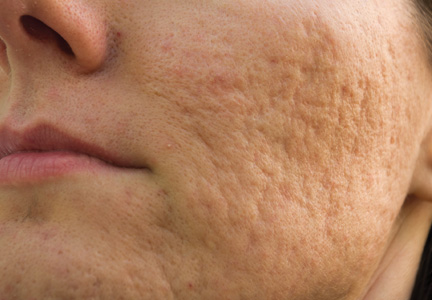
Portulaca oleracea (purslane)
Portulaca oleracea, also known as purslane, has long been used in various traditional medicine systems to relieve pain and edema.1Portulaca oleracea is a warm-climate annual plant originally found in the Middle East, North Africa, and the Indian subcontinent and now cultivated in the Arabian peninsula; Japan, where it is an abundant garden plant from spring to fall;2,3 and throughout the world.
The use of P. oleracea, a member of the Portulacaceae family, as a vegetable as well as herbal medicine dates back several centuries.4 In modern times, purslane has been found to be rich in antioxidants, particularly omega-3 fatty acids, vitamins C and E, beta-carotene, melatonin, and glutathione, as well as several minerals.5,6,7 Currently, it is considered one of the top ten most common plants in the world, and one of the most-used medical plants according to the World Health Organization.6 It is considered a weed in the United States, but is eaten in many parts of the world.

Antioxidant activity
Using two different assays, Uddin et al. determined in 2012 that P. oleracea cultivars exhibited significant antioxidant activity through various growth stages. In addition, the researchers suggested that purslane could provide multiple minerals as well as antioxidants in the context of nutraceutical products and functional food.7 Early this year, Silva et al. studied the antioxidant activity of P. oleracea leaves, flowers, and stems from two different locations in Portugal, with assays revealing significantly greater antioxidant activity in the stems of both samples compared to the leaves and flowers. However, the phenolic extracts of all three plant sections from both samples were found to protect DNA against hydroxyl radicals. The investigators concluded that their findings, particularly related to high antioxidant activity, support the potential benefits of purslane consumption to human health.8
A 2014 analysis of 13 collected purslane accessions revealed significant mineral content (particularly potassium, followed by nitrogen, sodium, calcium, magnesium, phosphorus, iron, zinc, and manganese) and showed that antioxidant activity was more strongly associated with ornamental as opposed to common purslane, the latter of which was richer in mineral content.6
Anti-inflammatory activity
In 2000, Chan et al. found that a 10% ethanolic extract of the dried leaves and stem of a P. oleracea cultivar displayed significant anti-inflammatory and analgesic properties after topical and intraperitoneal, but not oral, administration in comparison to diclofenac sodium, a synthetic drug used as active control. They added that these activities corresponded to the reputed effects of the traditional uses of the wild species.9
Wound healing activity
Rashed et al. reported in 2003 that a crude extract of P. oleracea accelerates wound healing. They used Mus musculus JVI-1 to show that fresh homogenized crude aerial parts of the plant topically applied on excision wound surfaces reduced wound surface areas and increased tensile strength. The best documented contraction was associated with a single dose of 50 mg, followed by two doses of 25 mg each.10
Oral lichen planus treatment
In 2010, Agha-Hosseini et al. conducted a randomized double-blind placebo-controlled 3-month study to assess the effectiveness of purslane in the treatment of oral lichen planus. Thirty-seven symptomatic patients (confirmed by biopsy) were divided into a purslane treatment group (n = 20) and a placebo group (n = 17). The investigators reported that partial to complete clinical improvement was observed in 83% of the treatment group, with no response in the remaining 17%, whereas partial improvement was seen in 17% of the placebo group, 73% had no response, and the condition was aggravated in 10% of the placebo group. No adverse side effects were reported in either group, and the researchers concluded that purslane was clinically effective in treating oral lichen planus and warrants consideration as a treatment option for the disorder.5
Other activities
In 2001, Radhakrishnan et al. identified several neuropharmacological actions, particularly anti-nociceptive and muscle-relaxing activity, with a range of effects on the central and peripheral nervous system observed in animal studies.1 The betacyanins found in P. oleracea have subsequently been found to confer a protective effect against neurotoxicity, specifically, ameliorating the D-galactose-induced cognitive deficits in senescent mice.11P. oleracea also has been shown to efficiently eliminate the endocrine-disrupting chemical bisphenol A from a hydroponic solution.12
In 2012, Yan et al. showed that three newly isolated homoisoflavonoids, known as portulacanones, and the compound 2,2’-dihydroxy-4’,6’-dimethoxychalcone selectively exhibited in vitro cytotoxic activities against four human cancer cell lines.2
Conclusions
This antioxidant-rich plant is found throughout the world and has long been associated with traditional health care. Modern research into its potential dermatologic uses is ongoing, but the evidence is relatively scarce. There are indications that the antioxidant, anti-inflammatory, and wound healing activity reportedly exhibited by purslane may be harnessed for various cutaneous applications. However, much more research is necessary to determine how extensive a role purslane may play in skin care.
References
1.J. Ethnopharmacol. 2001;76:171-6
2.Phytochemistry 2012;80:37-41
3.J. Biosci. Bioeng. 2007;103:420-6
4.J. Ethnopharmacol. 2000;73:445-51
5.Phytother. Res. 2010;24:240-4
6.Biomed. Res. Int. 2014;2014:296063
7.Int. J. Mol. Sci. 2012;13:10257-67
8.Nat Prod. Commun. 2014;9:45-50
9. J. Ethnopharmacol. 2000;73:445-51
10. J. Ethnopharmacol. 2003;88:131-6
11. Phytomedicine. 2010 Jun;17:527-32
12. Biosci. Biotechnol. Biochem. 2012;76:1015-7
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.
Portulaca oleracea, also known as purslane, has long been used in various traditional medicine systems to relieve pain and edema.1Portulaca oleracea is a warm-climate annual plant originally found in the Middle East, North Africa, and the Indian subcontinent and now cultivated in the Arabian peninsula; Japan, where it is an abundant garden plant from spring to fall;2,3 and throughout the world.
The use of P. oleracea, a member of the Portulacaceae family, as a vegetable as well as herbal medicine dates back several centuries.4 In modern times, purslane has been found to be rich in antioxidants, particularly omega-3 fatty acids, vitamins C and E, beta-carotene, melatonin, and glutathione, as well as several minerals.5,6,7 Currently, it is considered one of the top ten most common plants in the world, and one of the most-used medical plants according to the World Health Organization.6 It is considered a weed in the United States, but is eaten in many parts of the world.
Antioxidant activity
Using two different assays, Uddin et al. determined in 2012 that P. oleracea cultivars exhibited significant antioxidant activity through various growth stages. In addition, the researchers suggested that purslane could provide multiple minerals as well as antioxidants in the context of nutraceutical products and functional food.7 Early this year, Silva et al. studied the antioxidant activity of P. oleracea leaves, flowers, and stems from two different locations in Portugal, with assays revealing significantly greater antioxidant activity in the stems of both samples compared to the leaves and flowers. However, the phenolic extracts of all three plant sections from both samples were found to protect DNA against hydroxyl radicals. The investigators concluded that their findings, particularly related to high antioxidant activity, support the potential benefits of purslane consumption to human health.8
A 2014 analysis of 13 collected purslane accessions revealed significant mineral content (particularly potassium, followed by nitrogen, sodium, calcium, magnesium, phosphorus, iron, zinc, and manganese) and showed that antioxidant activity was more strongly associated with ornamental as opposed to common purslane, the latter of which was richer in mineral content.6
Anti-inflammatory activity
In 2000, Chan et al. found that a 10% ethanolic extract of the dried leaves and stem of a P. oleracea cultivar displayed significant anti-inflammatory and analgesic properties after topical and intraperitoneal, but not oral, administration in comparison to diclofenac sodium, a synthetic drug used as active control. They added that these activities corresponded to the reputed effects of the traditional uses of the wild species.9
Wound healing activity
Rashed et al. reported in 2003 that a crude extract of P. oleracea accelerates wound healing. They used Mus musculus JVI-1 to show that fresh homogenized crude aerial parts of the plant topically applied on excision wound surfaces reduced wound surface areas and increased tensile strength. The best documented contraction was associated with a single dose of 50 mg, followed by two doses of 25 mg each.10
Oral lichen planus treatment
In 2010, Agha-Hosseini et al. conducted a randomized double-blind placebo-controlled 3-month study to assess the effectiveness of purslane in the treatment of oral lichen planus. Thirty-seven symptomatic patients (confirmed by biopsy) were divided into a purslane treatment group (n = 20) and a placebo group (n = 17). The investigators reported that partial to complete clinical improvement was observed in 83% of the treatment group, with no response in the remaining 17%, whereas partial improvement was seen in 17% of the placebo group, 73% had no response, and the condition was aggravated in 10% of the placebo group. No adverse side effects were reported in either group, and the researchers concluded that purslane was clinically effective in treating oral lichen planus and warrants consideration as a treatment option for the disorder.5
Other activities
In 2001, Radhakrishnan et al. identified several neuropharmacological actions, particularly anti-nociceptive and muscle-relaxing activity, with a range of effects on the central and peripheral nervous system observed in animal studies.1 The betacyanins found in P. oleracea have subsequently been found to confer a protective effect against neurotoxicity, specifically, ameliorating the D-galactose-induced cognitive deficits in senescent mice.11P. oleracea also has been shown to efficiently eliminate the endocrine-disrupting chemical bisphenol A from a hydroponic solution.12
In 2012, Yan et al. showed that three newly isolated homoisoflavonoids, known as portulacanones, and the compound 2,2’-dihydroxy-4’,6’-dimethoxychalcone selectively exhibited in vitro cytotoxic activities against four human cancer cell lines.2
Conclusions
This antioxidant-rich plant is found throughout the world and has long been associated with traditional health care. Modern research into its potential dermatologic uses is ongoing, but the evidence is relatively scarce. There are indications that the antioxidant, anti-inflammatory, and wound healing activity reportedly exhibited by purslane may be harnessed for various cutaneous applications. However, much more research is necessary to determine how extensive a role purslane may play in skin care.
References
1.J. Ethnopharmacol. 2001;76:171-6
2.Phytochemistry 2012;80:37-41
3.J. Biosci. Bioeng. 2007;103:420-6
4.J. Ethnopharmacol. 2000;73:445-51
5.Phytother. Res. 2010;24:240-4
6.Biomed. Res. Int. 2014;2014:296063
7.Int. J. Mol. Sci. 2012;13:10257-67
8.Nat Prod. Commun. 2014;9:45-50
9. J. Ethnopharmacol. 2000;73:445-51
10. J. Ethnopharmacol. 2003;88:131-6
11. Phytomedicine. 2010 Jun;17:527-32
12. Biosci. Biotechnol. Biochem. 2012;76:1015-7
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.
Portulaca oleracea, also known as purslane, has long been used in various traditional medicine systems to relieve pain and edema.1Portulaca oleracea is a warm-climate annual plant originally found in the Middle East, North Africa, and the Indian subcontinent and now cultivated in the Arabian peninsula; Japan, where it is an abundant garden plant from spring to fall;2,3 and throughout the world.
The use of P. oleracea, a member of the Portulacaceae family, as a vegetable as well as herbal medicine dates back several centuries.4 In modern times, purslane has been found to be rich in antioxidants, particularly omega-3 fatty acids, vitamins C and E, beta-carotene, melatonin, and glutathione, as well as several minerals.5,6,7 Currently, it is considered one of the top ten most common plants in the world, and one of the most-used medical plants according to the World Health Organization.6 It is considered a weed in the United States, but is eaten in many parts of the world.
Antioxidant activity
Using two different assays, Uddin et al. determined in 2012 that P. oleracea cultivars exhibited significant antioxidant activity through various growth stages. In addition, the researchers suggested that purslane could provide multiple minerals as well as antioxidants in the context of nutraceutical products and functional food.7 Early this year, Silva et al. studied the antioxidant activity of P. oleracea leaves, flowers, and stems from two different locations in Portugal, with assays revealing significantly greater antioxidant activity in the stems of both samples compared to the leaves and flowers. However, the phenolic extracts of all three plant sections from both samples were found to protect DNA against hydroxyl radicals. The investigators concluded that their findings, particularly related to high antioxidant activity, support the potential benefits of purslane consumption to human health.8
A 2014 analysis of 13 collected purslane accessions revealed significant mineral content (particularly potassium, followed by nitrogen, sodium, calcium, magnesium, phosphorus, iron, zinc, and manganese) and showed that antioxidant activity was more strongly associated with ornamental as opposed to common purslane, the latter of which was richer in mineral content.6
Anti-inflammatory activity
In 2000, Chan et al. found that a 10% ethanolic extract of the dried leaves and stem of a P. oleracea cultivar displayed significant anti-inflammatory and analgesic properties after topical and intraperitoneal, but not oral, administration in comparison to diclofenac sodium, a synthetic drug used as active control. They added that these activities corresponded to the reputed effects of the traditional uses of the wild species.9
Wound healing activity
Rashed et al. reported in 2003 that a crude extract of P. oleracea accelerates wound healing. They used Mus musculus JVI-1 to show that fresh homogenized crude aerial parts of the plant topically applied on excision wound surfaces reduced wound surface areas and increased tensile strength. The best documented contraction was associated with a single dose of 50 mg, followed by two doses of 25 mg each.10
Oral lichen planus treatment
In 2010, Agha-Hosseini et al. conducted a randomized double-blind placebo-controlled 3-month study to assess the effectiveness of purslane in the treatment of oral lichen planus. Thirty-seven symptomatic patients (confirmed by biopsy) were divided into a purslane treatment group (n = 20) and a placebo group (n = 17). The investigators reported that partial to complete clinical improvement was observed in 83% of the treatment group, with no response in the remaining 17%, whereas partial improvement was seen in 17% of the placebo group, 73% had no response, and the condition was aggravated in 10% of the placebo group. No adverse side effects were reported in either group, and the researchers concluded that purslane was clinically effective in treating oral lichen planus and warrants consideration as a treatment option for the disorder.5
Other activities
In 2001, Radhakrishnan et al. identified several neuropharmacological actions, particularly anti-nociceptive and muscle-relaxing activity, with a range of effects on the central and peripheral nervous system observed in animal studies.1 The betacyanins found in P. oleracea have subsequently been found to confer a protective effect against neurotoxicity, specifically, ameliorating the D-galactose-induced cognitive deficits in senescent mice.11P. oleracea also has been shown to efficiently eliminate the endocrine-disrupting chemical bisphenol A from a hydroponic solution.12
In 2012, Yan et al. showed that three newly isolated homoisoflavonoids, known as portulacanones, and the compound 2,2’-dihydroxy-4’,6’-dimethoxychalcone selectively exhibited in vitro cytotoxic activities against four human cancer cell lines.2
Conclusions
This antioxidant-rich plant is found throughout the world and has long been associated with traditional health care. Modern research into its potential dermatologic uses is ongoing, but the evidence is relatively scarce. There are indications that the antioxidant, anti-inflammatory, and wound healing activity reportedly exhibited by purslane may be harnessed for various cutaneous applications. However, much more research is necessary to determine how extensive a role purslane may play in skin care.
References
1.J. Ethnopharmacol. 2001;76:171-6
2.Phytochemistry 2012;80:37-41
3.J. Biosci. Bioeng. 2007;103:420-6
4.J. Ethnopharmacol. 2000;73:445-51
5.Phytother. Res. 2010;24:240-4
6.Biomed. Res. Int. 2014;2014:296063
7.Int. J. Mol. Sci. 2012;13:10257-67
8.Nat Prod. Commun. 2014;9:45-50
9. J. Ethnopharmacol. 2000;73:445-51
10. J. Ethnopharmacol. 2003;88:131-6
11. Phytomedicine. 2010 Jun;17:527-32
12. Biosci. Biotechnol. Biochem. 2012;76:1015-7
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera Biopharmaceuticals, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.

NASPAG: Obesity raises unique contraceptive concerns in teens
ORLANDO – The data with respect to the effects of obesity on the efficacy of contraceptives in adolescents are limited, but the general consensus is that if efficacy is reduced, it isn’t by enough to make a real difference, according to Dr. Alene Toulany.
The effect of obesity is likely to be very small, and studies that have looked at pharmacokinetics in obesity have estimated that body weight accounts for only about 10%-20% of the variability of hormone levels, Dr. Toulany said at the North American Society for Pediatric and Adolescent Gynecology annual meeting.
“We know that this is within the normal range for individuals who are not obese, she said.

The concerns regarding efficacy in obese patients are understandable, as obesity increases the metabolic rate, increases clearance of hepatically metabolized drugs, increases circulating blood volume, and affects the absorption of contraceptive steroids through the adipose tissue, she said, adding that “it makes sense that the serum drug levels may be insufficient to maintain contraceptive effects, but the data are very limited and inconsistent.”
That’s not to say obesity isn’t a concern, added Dr. Toulany, an adolescent medicine specialist at the Hospital for Sick Children, Toronto, and the University of Toronto.
“Without fail, all of us will be seeing patients with obesity,” she said. The rate of adolescent obesity has quadrupled in the last 3 decades, increasing from 5% among those aged 12-19 years in 1980 to more than 20% now. A third are currently overweight or obese.
Further, sexually active obese women, regardless of age, are significantly less likely to use contraception, and obese teens are more likely to engage in risky sexual behaviors than are nonobese teens.
For these reasons, it is important to find the most effective contraceptive method, taking into account other risk factors and the likelihood of compliance, she said, noting that obesity is an independent risk factor for venous thromboembolism (VTE) and that studies suggest the risk is additive in users of estrogen-containing contraceptives.
However, she said, the benefits outweigh the risks of pregnancy in obesity – especially of unintended pregnancy.
The absolute risk of VTE in healthy women of reproductive age is small, and in adolescents it’s even smaller, she explained.
“The presence of risk factors for VTE should be taken into account when we see these youth in our clinics, but we can and should offer estrogen-containing contraceptives as long as there are no other risk factors,” she said.
Contraceptive options in young obese patients include:
• Intrauterine devices. There is no evidence that either copper IUDs or progestin-releasing IUDs have reduced efficacy in obese adolescents.
• Implants. These are highly effective in obese women, and even though the concentrations may be 30%-60% lower in obese women, they do remain above the contraceptive threshold for at least the first 3 years, Dr. Toulany said.
• Depot medroxyprogesterone acetate. There is some concern about weight gain with this injectable progestin-only contraceptive, particularly in those who are already obese, but it remains an option, as the levels do remain above what is needed to prevent ovulation. Interestingly, the persistence of ovulation suppression following discontinuation is different in obese women, and may be prolonged, compared with nonobese women; it is important to counsel patients about this, she said.
“So although randomized, controlled trials report no significant weight gain, we do agree with these observational studies that show that overweight and obese teens gain more weight with Depo-Provera than with oral contraceptives or with no contraceptives,” she said.
• Oral contraceptive pills. Although these may be less effective in obese adolescents, they remain an option and may be the best option in a given patient. Combined oral contraceptives are believed to be generally effective for pregnancy prevention, but “may be less forgiving of imperfect use,” and thus may not be the best choice in those who may have problems with adherence, for example.
The contraceptive patch is probably not a good option, because efficacy may be diminished as a result of absorption through the adipose tissues in those weighing more than 90 kg, Dr .Toulany said. Evidence is insufficient regarding the use of contraceptive rings in obese patients.
Bariatric surgery is increasingly being performed in adolescents, and this raises unique concerns with respect to contraception, Dr. Toulany said.
“We recommend discontinuing estrogen-containing contraceptives 1 month before surgery to reduce the risk of VTE postoperatively,” she said.
After bariatric surgery, those who undergo a restrictive procedure such as gastric banding or a gastric sleeve procedure that reduces the volume of the stomach can use oral contraceptives, but postsurgery vomiting and diarrhea could increase the risk of complications. In those who undergo surgery using a technique that involves a significant malabsorption component, such as Roux-en-Y gastric bypass, nonoral contraceptives are the best option.
“Most patients going for bariatric surgery have an IUD inserted at the time of surgery, and that’s what we would recommend,” Dr. Toulany said.
She reported having no relevant financial disclosures.
ORLANDO – The data with respect to the effects of obesity on the efficacy of contraceptives in adolescents are limited, but the general consensus is that if efficacy is reduced, it isn’t by enough to make a real difference, according to Dr. Alene Toulany.
The effect of obesity is likely to be very small, and studies that have looked at pharmacokinetics in obesity have estimated that body weight accounts for only about 10%-20% of the variability of hormone levels, Dr. Toulany said at the North American Society for Pediatric and Adolescent Gynecology annual meeting.
“We know that this is within the normal range for individuals who are not obese, she said.
The concerns regarding efficacy in obese patients are understandable, as obesity increases the metabolic rate, increases clearance of hepatically metabolized drugs, increases circulating blood volume, and affects the absorption of contraceptive steroids through the adipose tissue, she said, adding that “it makes sense that the serum drug levels may be insufficient to maintain contraceptive effects, but the data are very limited and inconsistent.”
That’s not to say obesity isn’t a concern, added Dr. Toulany, an adolescent medicine specialist at the Hospital for Sick Children, Toronto, and the University of Toronto.
“Without fail, all of us will be seeing patients with obesity,” she said. The rate of adolescent obesity has quadrupled in the last 3 decades, increasing from 5% among those aged 12-19 years in 1980 to more than 20% now. A third are currently overweight or obese.
Further, sexually active obese women, regardless of age, are significantly less likely to use contraception, and obese teens are more likely to engage in risky sexual behaviors than are nonobese teens.
For these reasons, it is important to find the most effective contraceptive method, taking into account other risk factors and the likelihood of compliance, she said, noting that obesity is an independent risk factor for venous thromboembolism (VTE) and that studies suggest the risk is additive in users of estrogen-containing contraceptives.
However, she said, the benefits outweigh the risks of pregnancy in obesity – especially of unintended pregnancy.
The absolute risk of VTE in healthy women of reproductive age is small, and in adolescents it’s even smaller, she explained.
“The presence of risk factors for VTE should be taken into account when we see these youth in our clinics, but we can and should offer estrogen-containing contraceptives as long as there are no other risk factors,” she said.
Contraceptive options in young obese patients include:
• Intrauterine devices. There is no evidence that either copper IUDs or progestin-releasing IUDs have reduced efficacy in obese adolescents.
• Implants. These are highly effective in obese women, and even though the concentrations may be 30%-60% lower in obese women, they do remain above the contraceptive threshold for at least the first 3 years, Dr. Toulany said.
• Depot medroxyprogesterone acetate. There is some concern about weight gain with this injectable progestin-only contraceptive, particularly in those who are already obese, but it remains an option, as the levels do remain above what is needed to prevent ovulation. Interestingly, the persistence of ovulation suppression following discontinuation is different in obese women, and may be prolonged, compared with nonobese women; it is important to counsel patients about this, she said.
“So although randomized, controlled trials report no significant weight gain, we do agree with these observational studies that show that overweight and obese teens gain more weight with Depo-Provera than with oral contraceptives or with no contraceptives,” she said.
• Oral contraceptive pills. Although these may be less effective in obese adolescents, they remain an option and may be the best option in a given patient. Combined oral contraceptives are believed to be generally effective for pregnancy prevention, but “may be less forgiving of imperfect use,” and thus may not be the best choice in those who may have problems with adherence, for example.
The contraceptive patch is probably not a good option, because efficacy may be diminished as a result of absorption through the adipose tissues in those weighing more than 90 kg, Dr .Toulany said. Evidence is insufficient regarding the use of contraceptive rings in obese patients.
Bariatric surgery is increasingly being performed in adolescents, and this raises unique concerns with respect to contraception, Dr. Toulany said.
“We recommend discontinuing estrogen-containing contraceptives 1 month before surgery to reduce the risk of VTE postoperatively,” she said.
After bariatric surgery, those who undergo a restrictive procedure such as gastric banding or a gastric sleeve procedure that reduces the volume of the stomach can use oral contraceptives, but postsurgery vomiting and diarrhea could increase the risk of complications. In those who undergo surgery using a technique that involves a significant malabsorption component, such as Roux-en-Y gastric bypass, nonoral contraceptives are the best option.
“Most patients going for bariatric surgery have an IUD inserted at the time of surgery, and that’s what we would recommend,” Dr. Toulany said.
She reported having no relevant financial disclosures.
ORLANDO – The data with respect to the effects of obesity on the efficacy of contraceptives in adolescents are limited, but the general consensus is that if efficacy is reduced, it isn’t by enough to make a real difference, according to Dr. Alene Toulany.
The effect of obesity is likely to be very small, and studies that have looked at pharmacokinetics in obesity have estimated that body weight accounts for only about 10%-20% of the variability of hormone levels, Dr. Toulany said at the North American Society for Pediatric and Adolescent Gynecology annual meeting.
“We know that this is within the normal range for individuals who are not obese, she said.
The concerns regarding efficacy in obese patients are understandable, as obesity increases the metabolic rate, increases clearance of hepatically metabolized drugs, increases circulating blood volume, and affects the absorption of contraceptive steroids through the adipose tissue, she said, adding that “it makes sense that the serum drug levels may be insufficient to maintain contraceptive effects, but the data are very limited and inconsistent.”
That’s not to say obesity isn’t a concern, added Dr. Toulany, an adolescent medicine specialist at the Hospital for Sick Children, Toronto, and the University of Toronto.
“Without fail, all of us will be seeing patients with obesity,” she said. The rate of adolescent obesity has quadrupled in the last 3 decades, increasing from 5% among those aged 12-19 years in 1980 to more than 20% now. A third are currently overweight or obese.
Further, sexually active obese women, regardless of age, are significantly less likely to use contraception, and obese teens are more likely to engage in risky sexual behaviors than are nonobese teens.
For these reasons, it is important to find the most effective contraceptive method, taking into account other risk factors and the likelihood of compliance, she said, noting that obesity is an independent risk factor for venous thromboembolism (VTE) and that studies suggest the risk is additive in users of estrogen-containing contraceptives.
However, she said, the benefits outweigh the risks of pregnancy in obesity – especially of unintended pregnancy.
The absolute risk of VTE in healthy women of reproductive age is small, and in adolescents it’s even smaller, she explained.
“The presence of risk factors for VTE should be taken into account when we see these youth in our clinics, but we can and should offer estrogen-containing contraceptives as long as there are no other risk factors,” she said.
Contraceptive options in young obese patients include:
• Intrauterine devices. There is no evidence that either copper IUDs or progestin-releasing IUDs have reduced efficacy in obese adolescents.
• Implants. These are highly effective in obese women, and even though the concentrations may be 30%-60% lower in obese women, they do remain above the contraceptive threshold for at least the first 3 years, Dr. Toulany said.
• Depot medroxyprogesterone acetate. There is some concern about weight gain with this injectable progestin-only contraceptive, particularly in those who are already obese, but it remains an option, as the levels do remain above what is needed to prevent ovulation. Interestingly, the persistence of ovulation suppression following discontinuation is different in obese women, and may be prolonged, compared with nonobese women; it is important to counsel patients about this, she said.
“So although randomized, controlled trials report no significant weight gain, we do agree with these observational studies that show that overweight and obese teens gain more weight with Depo-Provera than with oral contraceptives or with no contraceptives,” she said.
• Oral contraceptive pills. Although these may be less effective in obese adolescents, they remain an option and may be the best option in a given patient. Combined oral contraceptives are believed to be generally effective for pregnancy prevention, but “may be less forgiving of imperfect use,” and thus may not be the best choice in those who may have problems with adherence, for example.
The contraceptive patch is probably not a good option, because efficacy may be diminished as a result of absorption through the adipose tissues in those weighing more than 90 kg, Dr .Toulany said. Evidence is insufficient regarding the use of contraceptive rings in obese patients.
Bariatric surgery is increasingly being performed in adolescents, and this raises unique concerns with respect to contraception, Dr. Toulany said.
“We recommend discontinuing estrogen-containing contraceptives 1 month before surgery to reduce the risk of VTE postoperatively,” she said.
After bariatric surgery, those who undergo a restrictive procedure such as gastric banding or a gastric sleeve procedure that reduces the volume of the stomach can use oral contraceptives, but postsurgery vomiting and diarrhea could increase the risk of complications. In those who undergo surgery using a technique that involves a significant malabsorption component, such as Roux-en-Y gastric bypass, nonoral contraceptives are the best option.
“Most patients going for bariatric surgery have an IUD inserted at the time of surgery, and that’s what we would recommend,” Dr. Toulany said.
She reported having no relevant financial disclosures.
EXPERT ANALYSIS FROM THE NASPAG ANNUAL MEETING

Vitiligo Disease Triggers: Psychological Stressors Preceding the Onset of Disease
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.

Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
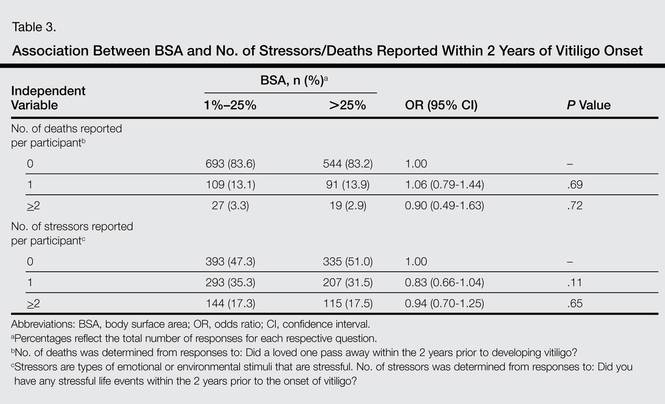
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
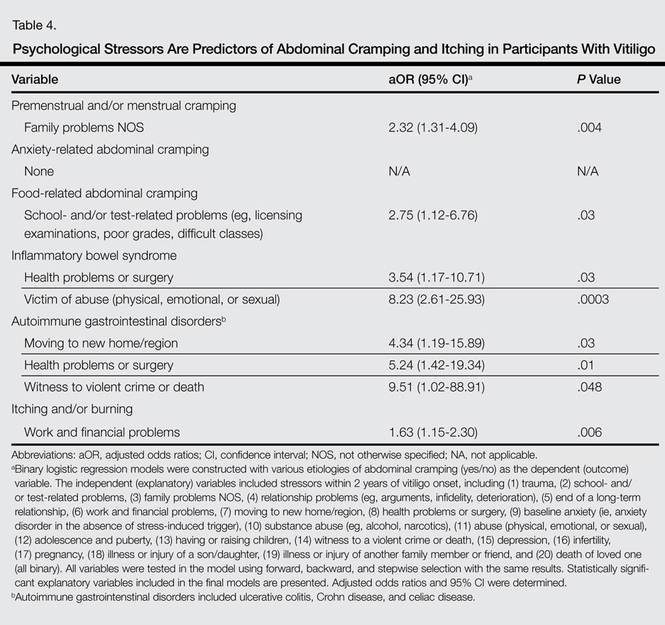
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.
Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.
Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
Practice Points
- Psychological stressors (eg, loss of a loved one) that occurred within 2 years prior to vitiligo onset should be considered as potential disease triggers.
- Psychological stressors have been associated with symptoms of abdominal cramping and itching/burning in vitiligo patients but not disease extent or distribution.
Aesthetic Dermatology: Sun protection after aesthetic procedures
As summertime approaches, opportunities for outdoor activities increase. For many of our patients, summer inspires a desire to have aesthetic procedures in preparation for outdoor events, such as weddings and vacations. We must, however, be mindful that increased sun exposure after some aesthetic procedures can mean an increased risk of complications.
The main complication we worry about with sun exposure is, of course, hyperpigmentation. The risk is low with injectable procedures such as botulinum toxin and fillers, but sun protection is still encouraged, especially in skin types III-VI. The risk increases greatly with chemical peels and laser and light-based procedures, such as intense pulsed light, vascular lasers, pigment lasers, laser hair removal, and especially nonablative and ablative resurfacing (including nonlaser resurfacing such as dermabrasion).
Sun protection should be encouraged, even with seemingly less invasive procedures, such as electrodessication. I once had a patient with type-IV skin tell me at her first visit that, years before, she had electrodessication on her face for DPN (dermatosis papulosa nigra), a procedure she had done on several occasions without complications and great results. However, she went to a party on a boat the weekend after the procedure and developed hyperpigmentation at the procedure areas, and she still had a few dark macules several years later.
At a follow-up visit, she said the doctor told her she should not have gone out on the boat and should have worn sunscreen. Of course, she was highly upset that she wasn’t advised about sun protection at the time of the procedure. This is one of several stories I’ve heard or seen of complications and postinflammatory hyperpigmentation after an aesthetic procedure, when the patients felt that the treating physician or practitioner did not counsel them about sun exposure during the consultation or treatment visit. It seems intuitive, but I’ve made it a habit to make sun protection part of my counseling routine.
In my practice, we often give patients sunscreen to apply immediately after a procedure. Specifically encouraging the use of zinc- and/or titanium-based, broad-spectrum, noncomedogenic physical blockers that are SPF 30 or higher may help reduce the risk of potential irritation or allergy and subsequent postinflammatory pigmentary alteration from chemical blocking ingredients. We provide a postprocedure handout, and the medical assistant also will counsel about sun protection when applying it to the patient or reviewing postprocedure instructions. So the patient is counseled at least three times: By me during consultation or pre-procedure, by the medical assistant post procedure, and by written instructions.
Vigorous sun protection is encouraged for at least 1 week after any aesthetic procedure (and longer if the downtime is longer or if multiple treatments are required). Some practices also use antioxidant serums to reduce free radicals, encourage healing, and reduce the risk of hyperpigmentation after procedures. Wide-brimmed hats also are encouraged, particularly after resurfacing or photodynamic therapy (PDT). We give patients sun-protective hats when they leave our office after PDT. We counsel them to practice vigorous sun protection for at least 1 week and to avoid sitting by a window for 48 hours after the procedure so as to not reactivate the levulan.
Delaying more high-risk procedures, such as laser treatments, until after the summer months may be appropriate if sun cannot be avoided to mitigate the risk of complications. If a patient comes to the office for a laser procedure and is visibly more tan than at the time of the last treatment, I will counsel about risks, adjust the settings appropriately, or even delay the treatment altogether to a time when the tan has faded. This is particularly important for lasers and light treatments for which melanin is the target chromosphere, such as intense pulsed light and laser hair removal. Although UV exposure is more intense in the summer, in our practice in Southern California we follow these principles year-round for the safety of our patients.
Dr. Wesley and Dr. Talakoub are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.
As summertime approaches, opportunities for outdoor activities increase. For many of our patients, summer inspires a desire to have aesthetic procedures in preparation for outdoor events, such as weddings and vacations. We must, however, be mindful that increased sun exposure after some aesthetic procedures can mean an increased risk of complications.
The main complication we worry about with sun exposure is, of course, hyperpigmentation. The risk is low with injectable procedures such as botulinum toxin and fillers, but sun protection is still encouraged, especially in skin types III-VI. The risk increases greatly with chemical peels and laser and light-based procedures, such as intense pulsed light, vascular lasers, pigment lasers, laser hair removal, and especially nonablative and ablative resurfacing (including nonlaser resurfacing such as dermabrasion).
Sun protection should be encouraged, even with seemingly less invasive procedures, such as electrodessication. I once had a patient with type-IV skin tell me at her first visit that, years before, she had electrodessication on her face for DPN (dermatosis papulosa nigra), a procedure she had done on several occasions without complications and great results. However, she went to a party on a boat the weekend after the procedure and developed hyperpigmentation at the procedure areas, and she still had a few dark macules several years later.
At a follow-up visit, she said the doctor told her she should not have gone out on the boat and should have worn sunscreen. Of course, she was highly upset that she wasn’t advised about sun protection at the time of the procedure. This is one of several stories I’ve heard or seen of complications and postinflammatory hyperpigmentation after an aesthetic procedure, when the patients felt that the treating physician or practitioner did not counsel them about sun exposure during the consultation or treatment visit. It seems intuitive, but I’ve made it a habit to make sun protection part of my counseling routine.
In my practice, we often give patients sunscreen to apply immediately after a procedure. Specifically encouraging the use of zinc- and/or titanium-based, broad-spectrum, noncomedogenic physical blockers that are SPF 30 or higher may help reduce the risk of potential irritation or allergy and subsequent postinflammatory pigmentary alteration from chemical blocking ingredients. We provide a postprocedure handout, and the medical assistant also will counsel about sun protection when applying it to the patient or reviewing postprocedure instructions. So the patient is counseled at least three times: By me during consultation or pre-procedure, by the medical assistant post procedure, and by written instructions.
Vigorous sun protection is encouraged for at least 1 week after any aesthetic procedure (and longer if the downtime is longer or if multiple treatments are required). Some practices also use antioxidant serums to reduce free radicals, encourage healing, and reduce the risk of hyperpigmentation after procedures. Wide-brimmed hats also are encouraged, particularly after resurfacing or photodynamic therapy (PDT). We give patients sun-protective hats when they leave our office after PDT. We counsel them to practice vigorous sun protection for at least 1 week and to avoid sitting by a window for 48 hours after the procedure so as to not reactivate the levulan.
Delaying more high-risk procedures, such as laser treatments, until after the summer months may be appropriate if sun cannot be avoided to mitigate the risk of complications. If a patient comes to the office for a laser procedure and is visibly more tan than at the time of the last treatment, I will counsel about risks, adjust the settings appropriately, or even delay the treatment altogether to a time when the tan has faded. This is particularly important for lasers and light treatments for which melanin is the target chromosphere, such as intense pulsed light and laser hair removal. Although UV exposure is more intense in the summer, in our practice in Southern California we follow these principles year-round for the safety of our patients.
Dr. Wesley and Dr. Talakoub are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.
As summertime approaches, opportunities for outdoor activities increase. For many of our patients, summer inspires a desire to have aesthetic procedures in preparation for outdoor events, such as weddings and vacations. We must, however, be mindful that increased sun exposure after some aesthetic procedures can mean an increased risk of complications.
The main complication we worry about with sun exposure is, of course, hyperpigmentation. The risk is low with injectable procedures such as botulinum toxin and fillers, but sun protection is still encouraged, especially in skin types III-VI. The risk increases greatly with chemical peels and laser and light-based procedures, such as intense pulsed light, vascular lasers, pigment lasers, laser hair removal, and especially nonablative and ablative resurfacing (including nonlaser resurfacing such as dermabrasion).
Sun protection should be encouraged, even with seemingly less invasive procedures, such as electrodessication. I once had a patient with type-IV skin tell me at her first visit that, years before, she had electrodessication on her face for DPN (dermatosis papulosa nigra), a procedure she had done on several occasions without complications and great results. However, she went to a party on a boat the weekend after the procedure and developed hyperpigmentation at the procedure areas, and she still had a few dark macules several years later.
At a follow-up visit, she said the doctor told her she should not have gone out on the boat and should have worn sunscreen. Of course, she was highly upset that she wasn’t advised about sun protection at the time of the procedure. This is one of several stories I’ve heard or seen of complications and postinflammatory hyperpigmentation after an aesthetic procedure, when the patients felt that the treating physician or practitioner did not counsel them about sun exposure during the consultation or treatment visit. It seems intuitive, but I’ve made it a habit to make sun protection part of my counseling routine.
In my practice, we often give patients sunscreen to apply immediately after a procedure. Specifically encouraging the use of zinc- and/or titanium-based, broad-spectrum, noncomedogenic physical blockers that are SPF 30 or higher may help reduce the risk of potential irritation or allergy and subsequent postinflammatory pigmentary alteration from chemical blocking ingredients. We provide a postprocedure handout, and the medical assistant also will counsel about sun protection when applying it to the patient or reviewing postprocedure instructions. So the patient is counseled at least three times: By me during consultation or pre-procedure, by the medical assistant post procedure, and by written instructions.
Vigorous sun protection is encouraged for at least 1 week after any aesthetic procedure (and longer if the downtime is longer or if multiple treatments are required). Some practices also use antioxidant serums to reduce free radicals, encourage healing, and reduce the risk of hyperpigmentation after procedures. Wide-brimmed hats also are encouraged, particularly after resurfacing or photodynamic therapy (PDT). We give patients sun-protective hats when they leave our office after PDT. We counsel them to practice vigorous sun protection for at least 1 week and to avoid sitting by a window for 48 hours after the procedure so as to not reactivate the levulan.
Delaying more high-risk procedures, such as laser treatments, until after the summer months may be appropriate if sun cannot be avoided to mitigate the risk of complications. If a patient comes to the office for a laser procedure and is visibly more tan than at the time of the last treatment, I will counsel about risks, adjust the settings appropriately, or even delay the treatment altogether to a time when the tan has faded. This is particularly important for lasers and light treatments for which melanin is the target chromosphere, such as intense pulsed light and laser hair removal. Although UV exposure is more intense in the summer, in our practice in Southern California we follow these principles year-round for the safety of our patients.
Dr. Wesley and Dr. Talakoub are co-contributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Wesley.

The dangers of desonide
In a previous column, I warned about the high cost of generic desonide. This month, I alert you to the many potential dangers of this drug. By the time I’m done, you may not want to go near the stuff.
To approve e-scribe refills, we all need to acknowledge warnings and dangers and click “Benefit outweighs risk” or “Previously tolerated” or some other option. But some of these warnings make me wonder who on earth writes them.
Desonide comes with more warnings than almost any other medicine I prescribe electronically. I counted 21 such warnings. Here are some examples:
1. Desonide External Cream 0.05% should be used cautiously in Bacterial Infection, especially in Systemic Bacterial Infection. Since Folliculitis is a specific form of Bacterial Infection, the same precaution may apply.
I confess that I never thought of prescribing desonide for Bacterial Infection, Systemic or otherwise. Have you? (By the way, what’s with the excess use of capital letters?)
The second warning is even more dramatic.
2. Desonide External Cream 0.05% should be used cautiously in Viral Infection, especially in Systemic Viral Infection.
What makes this even more curious is the Viral Infections the warnings go on to enumerate.
2a. Since Actinic Keratosis is a specific form of Viral Infection, the same precaution may apply.
Actinic Keratosis is a Viral Infection? I didn’t know that.
3. Since Actinic Keratosis of the Hands and Arms is a specific form of Viral Infection, the same precaution may apply.
Now we learn of different subgroups of Actinic Keratoses that are Viral Infections. Did they teach you these in Dermatology School? (Please see Warnings 6-10, below.)
4. This warning refers to a specific Bacterial Infection called Folliculitis Nares Perforans. I don’t know what that is, but it sounds bad. Glad they warned me.
5. Since Pseudofolliculitis Barbae is a specific form of Bacterial Infection, the same precaution may apply.
I never used much desonide for pseudofolliculitis, cautiously or otherwise.
Warnings 6-10 describe more specific forms of Viral Infection: (6) Non-Hyperkeratotic Actinic Keratosis, (7) Actinic Keratosis of Face and Anterior Scalp, (8) Non-Hyperkeratotic Non-Pigmented Actinic Keratosis, (9) Non-Hyperkeratotic Face and Scalp Actinic Keratosis, (10) Pigmented Actinic Keratosis.
This is most disturbing. What Systemic Viral Infections did they leave me to use desonide on? Hyperkeratotic Non-Pigmented Actinic Keratoses of the Posterior Scalp?
Warning 11 is another specific Bacterial Infection: Local Folliculitis. What is the opposite of Local Folliculitis? Express Folliculitis?
Warning 12 is Perioral Dermatitis. Steroids on rosacea? Really? Maybe a cheaper one.
I will now skip to warning 16: Hirsutism has been associated with Desonide External Cream 0.05%. Since Hair Disease is a more general form of Hirsutism, it may also be considered a drug-related medical condition.
Did you know that desonide causes unwanted hair growth? Or realize that Hair Disease is a more general form of Hirsutism? I myself have male-pattern baldness. (Sorry, Male-Pattern BALDNESS.) Since Baldness is a Hair Disease, is it also a more general form of Hirsutism? Instead of having too little hair, do I now have too much?
The same is true for warning 17, which is identical to 16, except that it substitutes “Hypertrichosis” for “Hirsutism.”
Okay, colleagues, it’s time for a personal reckoning. You trained, practiced, took CME, but you didn’t know about any of these risks, did you? You’ve just been just heedlessly, incautiously, throwing around desonide, producing hairy patients with Systemic Bacterial and Viral Infections. And on “Non-Hyperkeratotic Non-Pigmented Actinic Keratosis,” no less. Aren’t you disappointed in yourselves?
When I first read warnings like these, I wrote my EMR provider to ask who puts together this stuff, and which consultants vet it. They never answered. It is very hard to believe that a dermatologist was involved at any stage of developing these warnings, with their irrelevant caveats and absurd classification schemes.
Who would develop electronic prescribing guidelines without at least consulting the physicians who do the prescribing? Why would they want to?
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.
In a previous column, I warned about the high cost of generic desonide. This month, I alert you to the many potential dangers of this drug. By the time I’m done, you may not want to go near the stuff.
To approve e-scribe refills, we all need to acknowledge warnings and dangers and click “Benefit outweighs risk” or “Previously tolerated” or some other option. But some of these warnings make me wonder who on earth writes them.
Desonide comes with more warnings than almost any other medicine I prescribe electronically. I counted 21 such warnings. Here are some examples:
1. Desonide External Cream 0.05% should be used cautiously in Bacterial Infection, especially in Systemic Bacterial Infection. Since Folliculitis is a specific form of Bacterial Infection, the same precaution may apply.
I confess that I never thought of prescribing desonide for Bacterial Infection, Systemic or otherwise. Have you? (By the way, what’s with the excess use of capital letters?)
The second warning is even more dramatic.
2. Desonide External Cream 0.05% should be used cautiously in Viral Infection, especially in Systemic Viral Infection.
What makes this even more curious is the Viral Infections the warnings go on to enumerate.
2a. Since Actinic Keratosis is a specific form of Viral Infection, the same precaution may apply.
Actinic Keratosis is a Viral Infection? I didn’t know that.
3. Since Actinic Keratosis of the Hands and Arms is a specific form of Viral Infection, the same precaution may apply.
Now we learn of different subgroups of Actinic Keratoses that are Viral Infections. Did they teach you these in Dermatology School? (Please see Warnings 6-10, below.)
4. This warning refers to a specific Bacterial Infection called Folliculitis Nares Perforans. I don’t know what that is, but it sounds bad. Glad they warned me.
5. Since Pseudofolliculitis Barbae is a specific form of Bacterial Infection, the same precaution may apply.
I never used much desonide for pseudofolliculitis, cautiously or otherwise.
Warnings 6-10 describe more specific forms of Viral Infection: (6) Non-Hyperkeratotic Actinic Keratosis, (7) Actinic Keratosis of Face and Anterior Scalp, (8) Non-Hyperkeratotic Non-Pigmented Actinic Keratosis, (9) Non-Hyperkeratotic Face and Scalp Actinic Keratosis, (10) Pigmented Actinic Keratosis.
This is most disturbing. What Systemic Viral Infections did they leave me to use desonide on? Hyperkeratotic Non-Pigmented Actinic Keratoses of the Posterior Scalp?
Warning 11 is another specific Bacterial Infection: Local Folliculitis. What is the opposite of Local Folliculitis? Express Folliculitis?
Warning 12 is Perioral Dermatitis. Steroids on rosacea? Really? Maybe a cheaper one.
I will now skip to warning 16: Hirsutism has been associated with Desonide External Cream 0.05%. Since Hair Disease is a more general form of Hirsutism, it may also be considered a drug-related medical condition.
Did you know that desonide causes unwanted hair growth? Or realize that Hair Disease is a more general form of Hirsutism? I myself have male-pattern baldness. (Sorry, Male-Pattern BALDNESS.) Since Baldness is a Hair Disease, is it also a more general form of Hirsutism? Instead of having too little hair, do I now have too much?
The same is true for warning 17, which is identical to 16, except that it substitutes “Hypertrichosis” for “Hirsutism.”
Okay, colleagues, it’s time for a personal reckoning. You trained, practiced, took CME, but you didn’t know about any of these risks, did you? You’ve just been just heedlessly, incautiously, throwing around desonide, producing hairy patients with Systemic Bacterial and Viral Infections. And on “Non-Hyperkeratotic Non-Pigmented Actinic Keratosis,” no less. Aren’t you disappointed in yourselves?
When I first read warnings like these, I wrote my EMR provider to ask who puts together this stuff, and which consultants vet it. They never answered. It is very hard to believe that a dermatologist was involved at any stage of developing these warnings, with their irrelevant caveats and absurd classification schemes.
Who would develop electronic prescribing guidelines without at least consulting the physicians who do the prescribing? Why would they want to?
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.
In a previous column, I warned about the high cost of generic desonide. This month, I alert you to the many potential dangers of this drug. By the time I’m done, you may not want to go near the stuff.
To approve e-scribe refills, we all need to acknowledge warnings and dangers and click “Benefit outweighs risk” or “Previously tolerated” or some other option. But some of these warnings make me wonder who on earth writes them.
Desonide comes with more warnings than almost any other medicine I prescribe electronically. I counted 21 such warnings. Here are some examples:
1. Desonide External Cream 0.05% should be used cautiously in Bacterial Infection, especially in Systemic Bacterial Infection. Since Folliculitis is a specific form of Bacterial Infection, the same precaution may apply.
I confess that I never thought of prescribing desonide for Bacterial Infection, Systemic or otherwise. Have you? (By the way, what’s with the excess use of capital letters?)
The second warning is even more dramatic.
2. Desonide External Cream 0.05% should be used cautiously in Viral Infection, especially in Systemic Viral Infection.
What makes this even more curious is the Viral Infections the warnings go on to enumerate.
2a. Since Actinic Keratosis is a specific form of Viral Infection, the same precaution may apply.
Actinic Keratosis is a Viral Infection? I didn’t know that.
3. Since Actinic Keratosis of the Hands and Arms is a specific form of Viral Infection, the same precaution may apply.
Now we learn of different subgroups of Actinic Keratoses that are Viral Infections. Did they teach you these in Dermatology School? (Please see Warnings 6-10, below.)
4. This warning refers to a specific Bacterial Infection called Folliculitis Nares Perforans. I don’t know what that is, but it sounds bad. Glad they warned me.
5. Since Pseudofolliculitis Barbae is a specific form of Bacterial Infection, the same precaution may apply.
I never used much desonide for pseudofolliculitis, cautiously or otherwise.
Warnings 6-10 describe more specific forms of Viral Infection: (6) Non-Hyperkeratotic Actinic Keratosis, (7) Actinic Keratosis of Face and Anterior Scalp, (8) Non-Hyperkeratotic Non-Pigmented Actinic Keratosis, (9) Non-Hyperkeratotic Face and Scalp Actinic Keratosis, (10) Pigmented Actinic Keratosis.
This is most disturbing. What Systemic Viral Infections did they leave me to use desonide on? Hyperkeratotic Non-Pigmented Actinic Keratoses of the Posterior Scalp?
Warning 11 is another specific Bacterial Infection: Local Folliculitis. What is the opposite of Local Folliculitis? Express Folliculitis?
Warning 12 is Perioral Dermatitis. Steroids on rosacea? Really? Maybe a cheaper one.
I will now skip to warning 16: Hirsutism has been associated with Desonide External Cream 0.05%. Since Hair Disease is a more general form of Hirsutism, it may also be considered a drug-related medical condition.
Did you know that desonide causes unwanted hair growth? Or realize that Hair Disease is a more general form of Hirsutism? I myself have male-pattern baldness. (Sorry, Male-Pattern BALDNESS.) Since Baldness is a Hair Disease, is it also a more general form of Hirsutism? Instead of having too little hair, do I now have too much?
The same is true for warning 17, which is identical to 16, except that it substitutes “Hypertrichosis” for “Hirsutism.”
Okay, colleagues, it’s time for a personal reckoning. You trained, practiced, took CME, but you didn’t know about any of these risks, did you? You’ve just been just heedlessly, incautiously, throwing around desonide, producing hairy patients with Systemic Bacterial and Viral Infections. And on “Non-Hyperkeratotic Non-Pigmented Actinic Keratosis,” no less. Aren’t you disappointed in yourselves?
When I first read warnings like these, I wrote my EMR provider to ask who puts together this stuff, and which consultants vet it. They never answered. It is very hard to believe that a dermatologist was involved at any stage of developing these warnings, with their irrelevant caveats and absurd classification schemes.
Who would develop electronic prescribing guidelines without at least consulting the physicians who do the prescribing? Why would they want to?
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years.

Resident debt is ruining medicine
Most physicians accumulate near $300,000 in debt by the time they finish residency and go to work. This debt is crushing, and it is having negative effects.
College and medical school tuition has soared in the last 20 years, keeping pace with federal loan availability. There is no clear relationship with increased quality of the educational experience or with increased knowledge of the students. Colleges are part of the problem, but, in my opinion, the medical schools are even more egregious. After all, how much does it cost to rent a lecture hall for 2 years and provide some lecturers?

I will never forget when I complained about the increasing expense of medical school to a dean once. He explained that all of the faculty’s salary is considered an expense to the medical school, and that students are only paying a fraction of the true cost.
I must object! If a professor makes a guest appearance for an afternoon or two, the medical students are expected to pay him for a year of work?
I will never forget the goofy physiology professor who gave us two afternoons of demonstrations and lectures. He hooked live frogs to electrodes and made waves on a monitor. It was interesting, but his salary is $200,000 a year. Was it worth $2,000 a student (a class of 100 medical students) to watch him make frogs twitch two afternoons? I think not.
Caribbean medical schools charge about the same as those in North America and make a large profit. Medical students have become a “profit center.” This is occurring in an age when medical students write and share much of their own educational content in an interactive environment. This cannot be sustained.
For the last 2 years of medical school, the students are turned loose on the hospital wards and become slaves. This is called running scut, and it includes running specimens to the lab, wheeling patients to x-ray, drawing blood, fetching lab results, doing much chart work (completing the chart is the most important thing), and learning a lot in spite of the grunt work. The older physicians do teach on rounds, and there is always a resident physician around.
Again, these practicing physicians (they bill for their services) would be there anyway for the residents, and if there were not residents, would have to be there for their patients. The medical students cannot possibly add much additional cost, but these attending physicians and residents salaries are included in the educational cost justification.
The debt introduces a toxic calculus to specialty selection. Residencies are chosen for their fiscal attractiveness, which is not a correct or sustaining reason in a long hard career. Also, it may become economically impossible to practice in a lower-paying specialty.
Let me tell you about Razor Rick, a college student I mentored for several years. I hire these kids in their college summers and breaks to do not much but make a little money, and in exchange they let me pontificate and smile at me. Well, Rick got through med school and then dropped out of his primary care residency, owing several hundred thousand dollars. I was stunned when his parents called me, and I insisted he come in to talk to me.
He patiently explained that his board scores were passing but not good enough to get into to a well-paying specialty. He logically explained that he would have a better life, and have a better return on his investment, if he simply went into pharma with his expensive degree.
I helped him find some interviews.
Some criticize this generation of doctors for not being engaged, for not joining organized medicine, and for avoiding the glorious heart of difficult medicine. I don’t blame them for being distracted. They have, on average, 300,000 good reasons to be more self-concerned.
Dermatology News is proud to introduce the inaugural column of Cold Iron Truth by Dr. Brett Coldiron. Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of over 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics.
Most physicians accumulate near $300,000 in debt by the time they finish residency and go to work. This debt is crushing, and it is having negative effects.
College and medical school tuition has soared in the last 20 years, keeping pace with federal loan availability. There is no clear relationship with increased quality of the educational experience or with increased knowledge of the students. Colleges are part of the problem, but, in my opinion, the medical schools are even more egregious. After all, how much does it cost to rent a lecture hall for 2 years and provide some lecturers?
I will never forget when I complained about the increasing expense of medical school to a dean once. He explained that all of the faculty’s salary is considered an expense to the medical school, and that students are only paying a fraction of the true cost.
I must object! If a professor makes a guest appearance for an afternoon or two, the medical students are expected to pay him for a year of work?
I will never forget the goofy physiology professor who gave us two afternoons of demonstrations and lectures. He hooked live frogs to electrodes and made waves on a monitor. It was interesting, but his salary is $200,000 a year. Was it worth $2,000 a student (a class of 100 medical students) to watch him make frogs twitch two afternoons? I think not.
Caribbean medical schools charge about the same as those in North America and make a large profit. Medical students have become a “profit center.” This is occurring in an age when medical students write and share much of their own educational content in an interactive environment. This cannot be sustained.
For the last 2 years of medical school, the students are turned loose on the hospital wards and become slaves. This is called running scut, and it includes running specimens to the lab, wheeling patients to x-ray, drawing blood, fetching lab results, doing much chart work (completing the chart is the most important thing), and learning a lot in spite of the grunt work. The older physicians do teach on rounds, and there is always a resident physician around.
Again, these practicing physicians (they bill for their services) would be there anyway for the residents, and if there were not residents, would have to be there for their patients. The medical students cannot possibly add much additional cost, but these attending physicians and residents salaries are included in the educational cost justification.
The debt introduces a toxic calculus to specialty selection. Residencies are chosen for their fiscal attractiveness, which is not a correct or sustaining reason in a long hard career. Also, it may become economically impossible to practice in a lower-paying specialty.
Let me tell you about Razor Rick, a college student I mentored for several years. I hire these kids in their college summers and breaks to do not much but make a little money, and in exchange they let me pontificate and smile at me. Well, Rick got through med school and then dropped out of his primary care residency, owing several hundred thousand dollars. I was stunned when his parents called me, and I insisted he come in to talk to me.
He patiently explained that his board scores were passing but not good enough to get into to a well-paying specialty. He logically explained that he would have a better life, and have a better return on his investment, if he simply went into pharma with his expensive degree.
I helped him find some interviews.
Some criticize this generation of doctors for not being engaged, for not joining organized medicine, and for avoiding the glorious heart of difficult medicine. I don’t blame them for being distracted. They have, on average, 300,000 good reasons to be more self-concerned.
Dermatology News is proud to introduce the inaugural column of Cold Iron Truth by Dr. Brett Coldiron. Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of over 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics.
Most physicians accumulate near $300,000 in debt by the time they finish residency and go to work. This debt is crushing, and it is having negative effects.
College and medical school tuition has soared in the last 20 years, keeping pace with federal loan availability. There is no clear relationship with increased quality of the educational experience or with increased knowledge of the students. Colleges are part of the problem, but, in my opinion, the medical schools are even more egregious. After all, how much does it cost to rent a lecture hall for 2 years and provide some lecturers?
I will never forget when I complained about the increasing expense of medical school to a dean once. He explained that all of the faculty’s salary is considered an expense to the medical school, and that students are only paying a fraction of the true cost.
I must object! If a professor makes a guest appearance for an afternoon or two, the medical students are expected to pay him for a year of work?
I will never forget the goofy physiology professor who gave us two afternoons of demonstrations and lectures. He hooked live frogs to electrodes and made waves on a monitor. It was interesting, but his salary is $200,000 a year. Was it worth $2,000 a student (a class of 100 medical students) to watch him make frogs twitch two afternoons? I think not.
Caribbean medical schools charge about the same as those in North America and make a large profit. Medical students have become a “profit center.” This is occurring in an age when medical students write and share much of their own educational content in an interactive environment. This cannot be sustained.
For the last 2 years of medical school, the students are turned loose on the hospital wards and become slaves. This is called running scut, and it includes running specimens to the lab, wheeling patients to x-ray, drawing blood, fetching lab results, doing much chart work (completing the chart is the most important thing), and learning a lot in spite of the grunt work. The older physicians do teach on rounds, and there is always a resident physician around.
Again, these practicing physicians (they bill for their services) would be there anyway for the residents, and if there were not residents, would have to be there for their patients. The medical students cannot possibly add much additional cost, but these attending physicians and residents salaries are included in the educational cost justification.
The debt introduces a toxic calculus to specialty selection. Residencies are chosen for their fiscal attractiveness, which is not a correct or sustaining reason in a long hard career. Also, it may become economically impossible to practice in a lower-paying specialty.
Let me tell you about Razor Rick, a college student I mentored for several years. I hire these kids in their college summers and breaks to do not much but make a little money, and in exchange they let me pontificate and smile at me. Well, Rick got through med school and then dropped out of his primary care residency, owing several hundred thousand dollars. I was stunned when his parents called me, and I insisted he come in to talk to me.
He patiently explained that his board scores were passing but not good enough to get into to a well-paying specialty. He logically explained that he would have a better life, and have a better return on his investment, if he simply went into pharma with his expensive degree.
I helped him find some interviews.
Some criticize this generation of doctors for not being engaged, for not joining organized medicine, and for avoiding the glorious heart of difficult medicine. I don’t blame them for being distracted. They have, on average, 300,000 good reasons to be more self-concerned.
Dermatology News is proud to introduce the inaugural column of Cold Iron Truth by Dr. Brett Coldiron. Dr. Coldiron is a past president of the American Academy of Dermatology. He is currently in private practice, but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of over 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics.
