User login
Novel, noninvasive skin cancer detection device shows promise
DALLAS – An investigational device that couples laser spectroscopy with a machine-learning algorithm demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions in real time, results from a single-center study showed.
“More than 5.4 million cases of nonmelanoma skin cancer were treated in 2012, but the accuracy of skin cancer screening prior to biopsy is pretty low, about 70%, and is individual dependent,” lead study author Sung Hyun Pyun, PhD, said at the annual conference of the American Society for Laser Medicine and Surgery. “There have been several in vivo skin cancer screening devices based on noninvasive techniques such as multispectral imaging, Raman spectroscopy, and electrical impedance spectroscopy, but their diagnostic accuracies were not sufficient for clinical use and could not be applied in real time.”

For the single-site study, carried out in Australia, the researchers collected 502 emission spectra from skin cancers confirmed with biopsy results. They also collected 1,429 emission spectra from benign lesions. They achieved a sensitivity of 92% and a specificity of 90% out of 1,931 spectral data sets. No adverse events occurred and no microscopic damage of the irradiated skin was observed.
“Pathologic diagnosis-based cancer detection is considered to be time- and labor-consuming, and can sometimes be individual dependent,” Dr. Pyun said. “Our real-time, noninvasive, in vivo skin cancer detection device demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions.” He added that the device could be helpful in office-based cancer screening and real-time, on-site cancer detection during skin cancer surgeries.
Larger, multicenter studies of the device are being planned. Dr. Pyun holds ownership interests with Speclipse, and is an employee of the company.
DALLAS – An investigational device that couples laser spectroscopy with a machine-learning algorithm demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions in real time, results from a single-center study showed.
“More than 5.4 million cases of nonmelanoma skin cancer were treated in 2012, but the accuracy of skin cancer screening prior to biopsy is pretty low, about 70%, and is individual dependent,” lead study author Sung Hyun Pyun, PhD, said at the annual conference of the American Society for Laser Medicine and Surgery. “There have been several in vivo skin cancer screening devices based on noninvasive techniques such as multispectral imaging, Raman spectroscopy, and electrical impedance spectroscopy, but their diagnostic accuracies were not sufficient for clinical use and could not be applied in real time.”
For the single-site study, carried out in Australia, the researchers collected 502 emission spectra from skin cancers confirmed with biopsy results. They also collected 1,429 emission spectra from benign lesions. They achieved a sensitivity of 92% and a specificity of 90% out of 1,931 spectral data sets. No adverse events occurred and no microscopic damage of the irradiated skin was observed.
“Pathologic diagnosis-based cancer detection is considered to be time- and labor-consuming, and can sometimes be individual dependent,” Dr. Pyun said. “Our real-time, noninvasive, in vivo skin cancer detection device demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions.” He added that the device could be helpful in office-based cancer screening and real-time, on-site cancer detection during skin cancer surgeries.
Larger, multicenter studies of the device are being planned. Dr. Pyun holds ownership interests with Speclipse, and is an employee of the company.
DALLAS – An investigational device that couples laser spectroscopy with a machine-learning algorithm demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions in real time, results from a single-center study showed.
“More than 5.4 million cases of nonmelanoma skin cancer were treated in 2012, but the accuracy of skin cancer screening prior to biopsy is pretty low, about 70%, and is individual dependent,” lead study author Sung Hyun Pyun, PhD, said at the annual conference of the American Society for Laser Medicine and Surgery. “There have been several in vivo skin cancer screening devices based on noninvasive techniques such as multispectral imaging, Raman spectroscopy, and electrical impedance spectroscopy, but their diagnostic accuracies were not sufficient for clinical use and could not be applied in real time.”
For the single-site study, carried out in Australia, the researchers collected 502 emission spectra from skin cancers confirmed with biopsy results. They also collected 1,429 emission spectra from benign lesions. They achieved a sensitivity of 92% and a specificity of 90% out of 1,931 spectral data sets. No adverse events occurred and no microscopic damage of the irradiated skin was observed.
“Pathologic diagnosis-based cancer detection is considered to be time- and labor-consuming, and can sometimes be individual dependent,” Dr. Pyun said. “Our real-time, noninvasive, in vivo skin cancer detection device demonstrated a high sensitivity and specificity for discriminating skin cancers from benign lesions.” He added that the device could be helpful in office-based cancer screening and real-time, on-site cancer detection during skin cancer surgeries.
Larger, multicenter studies of the device are being planned. Dr. Pyun holds ownership interests with Speclipse, and is an employee of the company.
REPORTING FROM ASLMS 2018
Key clinical point: A novel device that uses spectroscopy and machine-learning algorithms was found to be a promising tool for the detection of skin cancer.
Major finding: Out of 1,931 spectral data sets, the device achieved a sensitivity of 92% and a specificity of 90%.
Study details: A single-center analysis of 502 emission spectra from skin cancers confirmed with biopsy results.
Disclosures: Dr. Pyun holds ownership interests with Speclipse and is an employee of the company.
Physicians voice concern over ibrutinib flat pricing
A group of oncologists is urging the Food and Drug Administration to review a new, flat-pricing strategy for all doses of ibrutinib (Imbruvica), calling the shift potentially dangerous for patients.
Pharmacyclics, an AbbVie company based in Sunnyvale, Calif., recently introduced a new, single-tablet formulation of ibrutinib in varying strengths (140 mg, 280 mg, 420 mg, and 560 mg) and set a flat price across all doses. At the same time, they removed the drug’s original 140-mg capsules – which cost about a third of the new, flat-rate price – from the market. The drug company says the new, single-tablet formulations give patients a convenient, once-a-day dosing regimen that could improve therapy adherence.
Ibrutinib is approved to treat a number of hematologic cancers, including mantle cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, Waldenstrom macroglobulinemia, and marginal zone lymphoma.
But in an April letter published in the Cancer Letter, nine physicians from cancer centers across the country expressed concern that the new dosage and pricing scheme could compromise patient safety and limit doctors’ dosage decisions.
“While the prescribing information is complex, prescribers and patients have had the flexibility of taking anywhere from one to four 140-mg capsules, readily permitting dose reduction to all but the lowest labeled dose [70 mg],” the authors wrote. “However, the new formulation and its associated marketing scheme [YOU&i] greatly impact the ability of the prescriber and patient to follow the prescribing information, resulting in the potential for a greatly enhanced risk of toxicities relative to that observed in the clinical trials which utilized multiples of the 140-mg capsules.”
The problems stem from a cancer drug that often requires dosage adjustments, the authors noted. The specific dosage for an ibrutinib patient depends on the indication, prior toxicities, concomitant medications, occurrence of ibrutinib intolerance, and current hepatic function. Because of such diverse criteria, dosage modifications are made frequently.
The new pricing system makes it harder for physicians to adjust the dose without running afoul of insurers and potentially affecting access for patients.
“In order to ensure that all patients receive a single tablet rather than multiple 140-mg tablets, the manufacturer has priced all tablet strengths at the same price, so that a physician who wished to prescribe 420 mg as three 140-mg tablets would be unlikely to get payer approval to do so, since the cost would be 300% of the single 420-mg tablet,” the letter states. “Furthermore, patients who have been on a daily dose of 140 mg now find that the cost of their 140-mg tablet is more than threefold higher than the cost of their prior 140-mg capsule.”
Pharmacyclics – which jointly markets the drug with Janssen – defended the new pricing regimen, asserting that it was designed to help patients take their medication as prescribed.
“The price is based on the most widely prescribed and lower of the two FDA-approved dosages, which is 420 mg per day. While a patient’s out-of-pocket cost for Imbruvica is ultimately determined by their insurance plan, the vast majority of patients [i.e., patients taking 420-mg and 560-mg doses of Imbruvica] will likely see no increase in out-of-pocket costs when transitioning to the single-tablet formulation,” Pharmacyclics said in a statement. “In fact, current patients on the 560-mg dose will likely see a decrease in their out-of-pocket costs. Out-of-pocket expenses may increase for patients who are taking a lower dose of Imbruvica [140 mg or 280 mg].”
Pharmacyclics stressed that there is “extremely limited data” on the use of lower doses of the drug and said they do not know if lower doses will yield the same clinical outcomes.
The authors of the letter on the other hand, want the FDA to review the safety of the You&i program in the context of the approved drug label.
The FDA should take another look at ibrutinib and make some necessary changes, said letter coauthor Mark J. Ratain, MD, of the University of Chicago.
“The FDA should reconsider whether the drug can be prescribed as labeled,” Dr. Ratain said in an interview. “I would like to see the pricing changed to linear [constant $/mg] pricing, so that prescribers and patients can select the most appropriate strength.”

Brian T. Hill, MD, PhD, director of the Lymphoid Malignancies Program at the Cleveland Clinic’s Taussig Cancer Institute, prescribes ibrutinib fairly regularly. He was not involved in the drafting of the letter but said he supports the intent.
“This is a significant inconvenience and a potential problem for patients taking this medication because patients frequently require dosage modifications, typically dose reductions,” Dr. Hill said in an interview. “Having this new program in which the medication has to be exchanged for a single-tablet dose makes it much more difficult and onerous to make those changes.”
A group of oncologists is urging the Food and Drug Administration to review a new, flat-pricing strategy for all doses of ibrutinib (Imbruvica), calling the shift potentially dangerous for patients.
Pharmacyclics, an AbbVie company based in Sunnyvale, Calif., recently introduced a new, single-tablet formulation of ibrutinib in varying strengths (140 mg, 280 mg, 420 mg, and 560 mg) and set a flat price across all doses. At the same time, they removed the drug’s original 140-mg capsules – which cost about a third of the new, flat-rate price – from the market. The drug company says the new, single-tablet formulations give patients a convenient, once-a-day dosing regimen that could improve therapy adherence.
Ibrutinib is approved to treat a number of hematologic cancers, including mantle cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, Waldenstrom macroglobulinemia, and marginal zone lymphoma.
But in an April letter published in the Cancer Letter, nine physicians from cancer centers across the country expressed concern that the new dosage and pricing scheme could compromise patient safety and limit doctors’ dosage decisions.
“While the prescribing information is complex, prescribers and patients have had the flexibility of taking anywhere from one to four 140-mg capsules, readily permitting dose reduction to all but the lowest labeled dose [70 mg],” the authors wrote. “However, the new formulation and its associated marketing scheme [YOU&i] greatly impact the ability of the prescriber and patient to follow the prescribing information, resulting in the potential for a greatly enhanced risk of toxicities relative to that observed in the clinical trials which utilized multiples of the 140-mg capsules.”
The problems stem from a cancer drug that often requires dosage adjustments, the authors noted. The specific dosage for an ibrutinib patient depends on the indication, prior toxicities, concomitant medications, occurrence of ibrutinib intolerance, and current hepatic function. Because of such diverse criteria, dosage modifications are made frequently.
The new pricing system makes it harder for physicians to adjust the dose without running afoul of insurers and potentially affecting access for patients.
“In order to ensure that all patients receive a single tablet rather than multiple 140-mg tablets, the manufacturer has priced all tablet strengths at the same price, so that a physician who wished to prescribe 420 mg as three 140-mg tablets would be unlikely to get payer approval to do so, since the cost would be 300% of the single 420-mg tablet,” the letter states. “Furthermore, patients who have been on a daily dose of 140 mg now find that the cost of their 140-mg tablet is more than threefold higher than the cost of their prior 140-mg capsule.”
Pharmacyclics – which jointly markets the drug with Janssen – defended the new pricing regimen, asserting that it was designed to help patients take their medication as prescribed.
“The price is based on the most widely prescribed and lower of the two FDA-approved dosages, which is 420 mg per day. While a patient’s out-of-pocket cost for Imbruvica is ultimately determined by their insurance plan, the vast majority of patients [i.e., patients taking 420-mg and 560-mg doses of Imbruvica] will likely see no increase in out-of-pocket costs when transitioning to the single-tablet formulation,” Pharmacyclics said in a statement. “In fact, current patients on the 560-mg dose will likely see a decrease in their out-of-pocket costs. Out-of-pocket expenses may increase for patients who are taking a lower dose of Imbruvica [140 mg or 280 mg].”
Pharmacyclics stressed that there is “extremely limited data” on the use of lower doses of the drug and said they do not know if lower doses will yield the same clinical outcomes.
The authors of the letter on the other hand, want the FDA to review the safety of the You&i program in the context of the approved drug label.
The FDA should take another look at ibrutinib and make some necessary changes, said letter coauthor Mark J. Ratain, MD, of the University of Chicago.
“The FDA should reconsider whether the drug can be prescribed as labeled,” Dr. Ratain said in an interview. “I would like to see the pricing changed to linear [constant $/mg] pricing, so that prescribers and patients can select the most appropriate strength.”
Brian T. Hill, MD, PhD, director of the Lymphoid Malignancies Program at the Cleveland Clinic’s Taussig Cancer Institute, prescribes ibrutinib fairly regularly. He was not involved in the drafting of the letter but said he supports the intent.
“This is a significant inconvenience and a potential problem for patients taking this medication because patients frequently require dosage modifications, typically dose reductions,” Dr. Hill said in an interview. “Having this new program in which the medication has to be exchanged for a single-tablet dose makes it much more difficult and onerous to make those changes.”
A group of oncologists is urging the Food and Drug Administration to review a new, flat-pricing strategy for all doses of ibrutinib (Imbruvica), calling the shift potentially dangerous for patients.
Pharmacyclics, an AbbVie company based in Sunnyvale, Calif., recently introduced a new, single-tablet formulation of ibrutinib in varying strengths (140 mg, 280 mg, 420 mg, and 560 mg) and set a flat price across all doses. At the same time, they removed the drug’s original 140-mg capsules – which cost about a third of the new, flat-rate price – from the market. The drug company says the new, single-tablet formulations give patients a convenient, once-a-day dosing regimen that could improve therapy adherence.
Ibrutinib is approved to treat a number of hematologic cancers, including mantle cell lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, Waldenstrom macroglobulinemia, and marginal zone lymphoma.
But in an April letter published in the Cancer Letter, nine physicians from cancer centers across the country expressed concern that the new dosage and pricing scheme could compromise patient safety and limit doctors’ dosage decisions.
“While the prescribing information is complex, prescribers and patients have had the flexibility of taking anywhere from one to four 140-mg capsules, readily permitting dose reduction to all but the lowest labeled dose [70 mg],” the authors wrote. “However, the new formulation and its associated marketing scheme [YOU&i] greatly impact the ability of the prescriber and patient to follow the prescribing information, resulting in the potential for a greatly enhanced risk of toxicities relative to that observed in the clinical trials which utilized multiples of the 140-mg capsules.”
The problems stem from a cancer drug that often requires dosage adjustments, the authors noted. The specific dosage for an ibrutinib patient depends on the indication, prior toxicities, concomitant medications, occurrence of ibrutinib intolerance, and current hepatic function. Because of such diverse criteria, dosage modifications are made frequently.
The new pricing system makes it harder for physicians to adjust the dose without running afoul of insurers and potentially affecting access for patients.
“In order to ensure that all patients receive a single tablet rather than multiple 140-mg tablets, the manufacturer has priced all tablet strengths at the same price, so that a physician who wished to prescribe 420 mg as three 140-mg tablets would be unlikely to get payer approval to do so, since the cost would be 300% of the single 420-mg tablet,” the letter states. “Furthermore, patients who have been on a daily dose of 140 mg now find that the cost of their 140-mg tablet is more than threefold higher than the cost of their prior 140-mg capsule.”
Pharmacyclics – which jointly markets the drug with Janssen – defended the new pricing regimen, asserting that it was designed to help patients take their medication as prescribed.
“The price is based on the most widely prescribed and lower of the two FDA-approved dosages, which is 420 mg per day. While a patient’s out-of-pocket cost for Imbruvica is ultimately determined by their insurance plan, the vast majority of patients [i.e., patients taking 420-mg and 560-mg doses of Imbruvica] will likely see no increase in out-of-pocket costs when transitioning to the single-tablet formulation,” Pharmacyclics said in a statement. “In fact, current patients on the 560-mg dose will likely see a decrease in their out-of-pocket costs. Out-of-pocket expenses may increase for patients who are taking a lower dose of Imbruvica [140 mg or 280 mg].”
Pharmacyclics stressed that there is “extremely limited data” on the use of lower doses of the drug and said they do not know if lower doses will yield the same clinical outcomes.
The authors of the letter on the other hand, want the FDA to review the safety of the You&i program in the context of the approved drug label.
The FDA should take another look at ibrutinib and make some necessary changes, said letter coauthor Mark J. Ratain, MD, of the University of Chicago.
“The FDA should reconsider whether the drug can be prescribed as labeled,” Dr. Ratain said in an interview. “I would like to see the pricing changed to linear [constant $/mg] pricing, so that prescribers and patients can select the most appropriate strength.”
Brian T. Hill, MD, PhD, director of the Lymphoid Malignancies Program at the Cleveland Clinic’s Taussig Cancer Institute, prescribes ibrutinib fairly regularly. He was not involved in the drafting of the letter but said he supports the intent.
“This is a significant inconvenience and a potential problem for patients taking this medication because patients frequently require dosage modifications, typically dose reductions,” Dr. Hill said in an interview. “Having this new program in which the medication has to be exchanged for a single-tablet dose makes it much more difficult and onerous to make those changes.”
U.S. youth suicide prevention saved nearly 900 lives
WASHINGTON – A U.S. grant program that was started in 2004 to fund youth suicide prevention efforts throughout the country was linked with nearly 900 youth suicide deaths prevented by 2015, according to a matched, case-control analysis.

U.S. counties with a youth suicide prevention effort funded by a grant from the federal Garrett Lee Smith (GLS) Memorial Suicide Prevention program had an average 1.09 fewer youth suicide deaths per 100,000 youths (10-24 years old) 2 years after ongoing funding began, when compared with matched U.S. counties without a GLS-funded program, said Mr. Garraza, an analyst with ICF International in New York. Counties that had GLS-funded programs operating for 3 years showed an average reduction in youth suicide death of roughly 2 fewer deaths per 100,000 population, compared with control counties, and counties with programs in operation for 4 years showed a reduction of roughly 3 fewer deaths per 100,000 population.
“These extremely important data show convincingly that if we want to save lives, suicide prevention can’t be a one-and-done. said Richard McKeon, PhD, chief of the Suicide Prevention Branch of the Substance Abuse and Mental Health Services Administration, the U.S. agency that administers the GLS grant program and commissioned ICF International to analyze the grant program’s effects.

As of October 2017, the GLS grant initiative had made nearly 200 awards to youth suicide prevention programs that operated in all 50 states, the District of Columbia, one territory, and among 49 Native American tribes, Mr. Garraza said.
To assess the effects of the GLS grants on suicide death rates in people aged 10-24 years, Mr. Garraza and his associates used U.S. suicide data collected in 2,095 counties throughout the country that each had more than 3,000 resident youths, including 1,126 counties that had, as of 2015, been exposed to at least 1 year of a program sponsored by a GLS grant that began before 2010 and 969 counties without any GLS-grant exposure. After the counties in each subgroup were propensity-score matched using several criteria, including numbers of youth by specific age, race, household income, employment rates, health insurance status, and urbanization, the researchers analyzed suicide death rates in 481 counties with 1-4 years of exposure of a GLS-funded prevention program and in 851 counties with no exposure.
Their analysis estimated that 882 fewer suicide deaths occurred among youths in counties with GLS-funded programs, compared with the expected suicide mortality based on the unexposed counties.
In addition to showing statistically significant mortality reductions among youths in the counties with GLS-funded programs, compared with the counties with no such programs, the analysis showed, as expected, no difference between the intervention and control counties in rates of suicide deaths among adults and no difference in youth-mortality from causes other than suicide, which indicated that the observed differences linked with GLS funding were specific for youth suicides.
The current analysis looking at the effects of GLS funding on youth suicide deaths rates follows a prior report from the same researchers with similar findings using data collected through 2010 (Am J Public Health. 2015 May;105[5]:986-93). They also published two prior reports that used a similar analysis to assess the effects of GLS-funded suicide prevention programs on suicide attempt rates. One of those reports showed that GLS-funded programs linked with a cut in suicide attempts of nearly 5 per 1,000 population (JAMA Psychiatry. 2015 Nov;72[11]:1143-9), and the second showed that this effect on suicide attempts was cost effective when the cost of the grants was compared with the money saved from avoided emergency department visits and hospitalizations (Suicide Life Threat Behav. 2018 Feb;48[1]:3-11).
WASHINGTON – A U.S. grant program that was started in 2004 to fund youth suicide prevention efforts throughout the country was linked with nearly 900 youth suicide deaths prevented by 2015, according to a matched, case-control analysis.
U.S. counties with a youth suicide prevention effort funded by a grant from the federal Garrett Lee Smith (GLS) Memorial Suicide Prevention program had an average 1.09 fewer youth suicide deaths per 100,000 youths (10-24 years old) 2 years after ongoing funding began, when compared with matched U.S. counties without a GLS-funded program, said Mr. Garraza, an analyst with ICF International in New York. Counties that had GLS-funded programs operating for 3 years showed an average reduction in youth suicide death of roughly 2 fewer deaths per 100,000 population, compared with control counties, and counties with programs in operation for 4 years showed a reduction of roughly 3 fewer deaths per 100,000 population.
“These extremely important data show convincingly that if we want to save lives, suicide prevention can’t be a one-and-done. said Richard McKeon, PhD, chief of the Suicide Prevention Branch of the Substance Abuse and Mental Health Services Administration, the U.S. agency that administers the GLS grant program and commissioned ICF International to analyze the grant program’s effects.
As of October 2017, the GLS grant initiative had made nearly 200 awards to youth suicide prevention programs that operated in all 50 states, the District of Columbia, one territory, and among 49 Native American tribes, Mr. Garraza said.
To assess the effects of the GLS grants on suicide death rates in people aged 10-24 years, Mr. Garraza and his associates used U.S. suicide data collected in 2,095 counties throughout the country that each had more than 3,000 resident youths, including 1,126 counties that had, as of 2015, been exposed to at least 1 year of a program sponsored by a GLS grant that began before 2010 and 969 counties without any GLS-grant exposure. After the counties in each subgroup were propensity-score matched using several criteria, including numbers of youth by specific age, race, household income, employment rates, health insurance status, and urbanization, the researchers analyzed suicide death rates in 481 counties with 1-4 years of exposure of a GLS-funded prevention program and in 851 counties with no exposure.
Their analysis estimated that 882 fewer suicide deaths occurred among youths in counties with GLS-funded programs, compared with the expected suicide mortality based on the unexposed counties.
In addition to showing statistically significant mortality reductions among youths in the counties with GLS-funded programs, compared with the counties with no such programs, the analysis showed, as expected, no difference between the intervention and control counties in rates of suicide deaths among adults and no difference in youth-mortality from causes other than suicide, which indicated that the observed differences linked with GLS funding were specific for youth suicides.
The current analysis looking at the effects of GLS funding on youth suicide deaths rates follows a prior report from the same researchers with similar findings using data collected through 2010 (Am J Public Health. 2015 May;105[5]:986-93). They also published two prior reports that used a similar analysis to assess the effects of GLS-funded suicide prevention programs on suicide attempt rates. One of those reports showed that GLS-funded programs linked with a cut in suicide attempts of nearly 5 per 1,000 population (JAMA Psychiatry. 2015 Nov;72[11]:1143-9), and the second showed that this effect on suicide attempts was cost effective when the cost of the grants was compared with the money saved from avoided emergency department visits and hospitalizations (Suicide Life Threat Behav. 2018 Feb;48[1]:3-11).
WASHINGTON – A U.S. grant program that was started in 2004 to fund youth suicide prevention efforts throughout the country was linked with nearly 900 youth suicide deaths prevented by 2015, according to a matched, case-control analysis.
U.S. counties with a youth suicide prevention effort funded by a grant from the federal Garrett Lee Smith (GLS) Memorial Suicide Prevention program had an average 1.09 fewer youth suicide deaths per 100,000 youths (10-24 years old) 2 years after ongoing funding began, when compared with matched U.S. counties without a GLS-funded program, said Mr. Garraza, an analyst with ICF International in New York. Counties that had GLS-funded programs operating for 3 years showed an average reduction in youth suicide death of roughly 2 fewer deaths per 100,000 population, compared with control counties, and counties with programs in operation for 4 years showed a reduction of roughly 3 fewer deaths per 100,000 population.
“These extremely important data show convincingly that if we want to save lives, suicide prevention can’t be a one-and-done. said Richard McKeon, PhD, chief of the Suicide Prevention Branch of the Substance Abuse and Mental Health Services Administration, the U.S. agency that administers the GLS grant program and commissioned ICF International to analyze the grant program’s effects.
As of October 2017, the GLS grant initiative had made nearly 200 awards to youth suicide prevention programs that operated in all 50 states, the District of Columbia, one territory, and among 49 Native American tribes, Mr. Garraza said.
To assess the effects of the GLS grants on suicide death rates in people aged 10-24 years, Mr. Garraza and his associates used U.S. suicide data collected in 2,095 counties throughout the country that each had more than 3,000 resident youths, including 1,126 counties that had, as of 2015, been exposed to at least 1 year of a program sponsored by a GLS grant that began before 2010 and 969 counties without any GLS-grant exposure. After the counties in each subgroup were propensity-score matched using several criteria, including numbers of youth by specific age, race, household income, employment rates, health insurance status, and urbanization, the researchers analyzed suicide death rates in 481 counties with 1-4 years of exposure of a GLS-funded prevention program and in 851 counties with no exposure.
Their analysis estimated that 882 fewer suicide deaths occurred among youths in counties with GLS-funded programs, compared with the expected suicide mortality based on the unexposed counties.
In addition to showing statistically significant mortality reductions among youths in the counties with GLS-funded programs, compared with the counties with no such programs, the analysis showed, as expected, no difference between the intervention and control counties in rates of suicide deaths among adults and no difference in youth-mortality from causes other than suicide, which indicated that the observed differences linked with GLS funding were specific for youth suicides.
The current analysis looking at the effects of GLS funding on youth suicide deaths rates follows a prior report from the same researchers with similar findings using data collected through 2010 (Am J Public Health. 2015 May;105[5]:986-93). They also published two prior reports that used a similar analysis to assess the effects of GLS-funded suicide prevention programs on suicide attempt rates. One of those reports showed that GLS-funded programs linked with a cut in suicide attempts of nearly 5 per 1,000 population (JAMA Psychiatry. 2015 Nov;72[11]:1143-9), and the second showed that this effect on suicide attempts was cost effective when the cost of the grants was compared with the money saved from avoided emergency department visits and hospitalizations (Suicide Life Threat Behav. 2018 Feb;48[1]:3-11).
REPORTING FROM THE AAS ANNUAL CONFERENCE
Key clinical point: A U.S. youth suicide prevention grant program seems to be making a difference.
Major finding: During 2007-2015, U.S. counties with youth suicide prevention funding had 882 fewer deaths than expected.
Study details: A case-control analysis of U.S. counties that received federal funding for youth-suicide prevention.
Disclosures: Mr. Garraza and Dr. McKeon had no disclosures.
Novartis CAR T-cell therapy adds a lymphoma indication
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
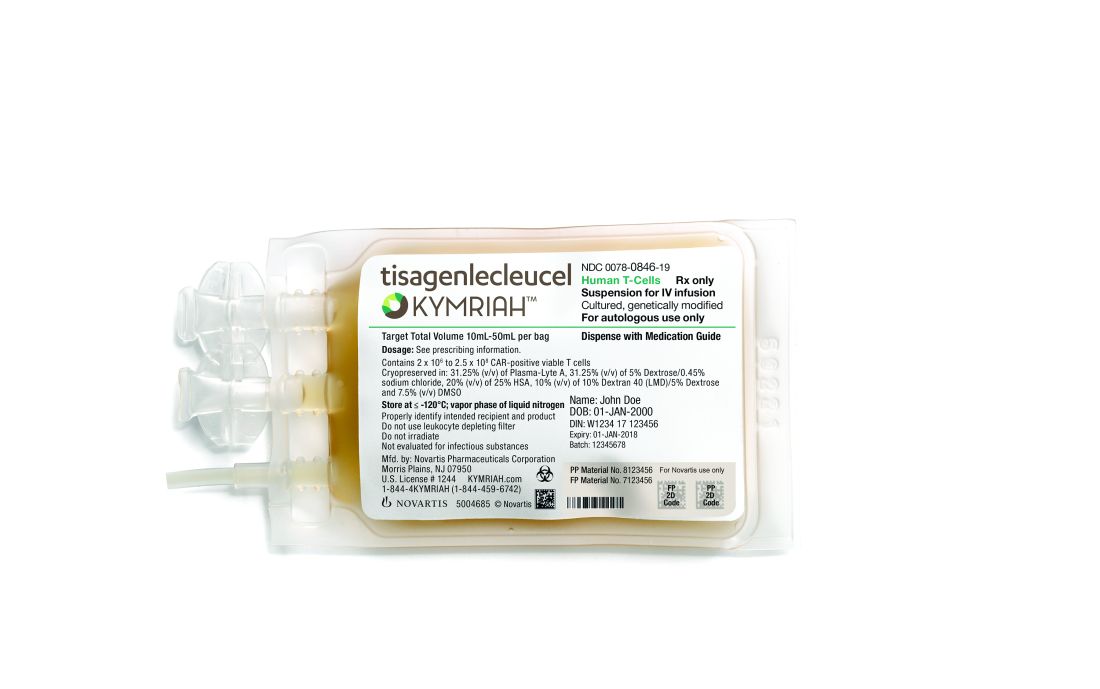
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Novartis’s after failure of two or more lines of systemic therapy.
The Food and Drug Administration approved the expanded indication on May 1. The chimeric antigen receptor (CAR) T-cell therapy was initially approved in Aug. 2017 for refractory or relapsed B-cell precursor acute lymphoblastic leukemia (ALL) in patients up to 25 years old. The new approval brings tisagenlecleucel into direct competition with Gilead Science’s CAR T-cell therapy axicabtagene ciloleucel (Yescarta), which was approved in Oct. 2017 for B-cell lymphoma.
Besides matching the competition, she said the lower price is because tisagenlecleucel takes longer to work for lymphoma, and the response isn’t as potent as for childhood ALL. Novartis is looking into chronic lymphocytic leukemia, multiple myeloma, and solid tumor indications for tisagenlecleucel and other CAR T-cell agents, she added.
The Centers for Medicare & Medicaid Services recently committed to covering outpatient administration of both agents for their initial indications; Novartis is working with CMS for coverage of the new lymphoma indication.
With both products, T cells are collected then shipped off to a company facility where a CAR gene is spliced into their DNA, essentially programming the T cells to attack the targeted cancer. The cells are then infused back into the patient.
In the phase 2 JULIET trial, tisagenlecleucel showed an overall response rate of 50% among 68 B-cell lymphoma patients, with 32% achieving complete response (CR) and 18% achieving partial response (PR). The median duration of response was not reached.
Axicabtagene ciloleucel’s label reports an objective response rate of 72% among 101 patients, with CR in 51% and PR in 21%. Median duration of response was 9.2 months but was also not reached among complete responders.
“Different trials. Different CARTs. Different levels of disease. Our drug is cryopreserved and theirs is not. No way to compare them,” the Novartis spokeswoman said when asked about the response differences.
T-cell reprogramming isn’t clean at this point in medical history; both agents carry black box warnings of potentially fatal cytokine release syndrome and neurologic toxicity, and both are subject to Risk Evaluation and Mitigation Strategy programs.
The B-cell lymphoma indication for both therapies includes diffuse large B-cell lymphoma (DLBCL), high grade B-cell lymphoma, and DLBCL arising from follicular lymphoma. The Gilead product carries an additional indication for primary mediastinal large B-cell lymphoma. Neither agent is indicated for primary central nervous system lymphoma. Both labels say that patients should not donate blood, organs, or tissues after treatment. Tisagenlecleucel labeling also notes that some commercial HIV nucleic acid tests may yield false positives after treatment.
Novartis said in a press release that T cells are treated at the company’s Morris Plains, N.J., facility with a turnaround time of about 22 days. Cryopreservation of the harvested cells gives providers some flexibility in treatment timing.
Deep Gray Matter Volume Loss May Drive Disability Worsening in MS
Deep gray matter volume loss drives disability accumulation in multiple sclerosis (MS), according to a multicenter, longitudinal study published in the February issue of Annals of Neurology.
The results “suggest that the development of deep gray matter atrophy may drive disability accumulation irrespective of clinical phenotypes, thereby becoming a useful outcome measure in neuroprotective clinical trials,” said Arman Eshaghi, MD, of the Queen Square MS Centre at the University College London Institute of Neurology, and colleagues.

Gray matter atrophy occurs in all MS phenotypes. To investigate whether a spatiotemporal pattern of gray matter atrophy is associated with faster disability accumulation in MS, the investigators analyzed 3,604 high-resolution T1-weighted MRI brain scans from 1,417 participants. The investigators retrospectively collected the scans from seven European centers in the MRI in MS (MAGNIMS) network.
The study included 253 patients with clinically isolated syndrome (CIS), 708 patients with relapsing-remitting MS, 128 patients with secondary progressive MS and 125 patients with primary progressive MS with an average follow-up of 2.41 years, as well as 203 healthy controls with an average follow-up of 1.83 years. The researchers assessed disability using the Expanded Disability Status Scale (EDSS). They obtained volumes of deep gray matter; temporal, frontal, parietal, occipital, and cerebellar gray matter; brainstem; and cerebral white matter. Hierarchical mixed models assessed annual percentage rate of regional tissue loss and identified regional volumes associated with time to EDSS progression.
Of all baseline regional volumes, only deep gray matter predicted time to EDSS progression. For every standard deviation decrease in baseline deep gray matter volume, the risk of having a shorter time to EDSS worsening during follow-up increased by 27%. Of all longitudinal measures, deep gray matter had the fastest annual rate of atrophy. This rate
—Jake Remaly
Suggested Reading
Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210-222.
Deep gray matter volume loss drives disability accumulation in multiple sclerosis (MS), according to a multicenter, longitudinal study published in the February issue of Annals of Neurology.
The results “suggest that the development of deep gray matter atrophy may drive disability accumulation irrespective of clinical phenotypes, thereby becoming a useful outcome measure in neuroprotective clinical trials,” said Arman Eshaghi, MD, of the Queen Square MS Centre at the University College London Institute of Neurology, and colleagues.
Gray matter atrophy occurs in all MS phenotypes. To investigate whether a spatiotemporal pattern of gray matter atrophy is associated with faster disability accumulation in MS, the investigators analyzed 3,604 high-resolution T1-weighted MRI brain scans from 1,417 participants. The investigators retrospectively collected the scans from seven European centers in the MRI in MS (MAGNIMS) network.
The study included 253 patients with clinically isolated syndrome (CIS), 708 patients with relapsing-remitting MS, 128 patients with secondary progressive MS and 125 patients with primary progressive MS with an average follow-up of 2.41 years, as well as 203 healthy controls with an average follow-up of 1.83 years. The researchers assessed disability using the Expanded Disability Status Scale (EDSS). They obtained volumes of deep gray matter; temporal, frontal, parietal, occipital, and cerebellar gray matter; brainstem; and cerebral white matter. Hierarchical mixed models assessed annual percentage rate of regional tissue loss and identified regional volumes associated with time to EDSS progression.
Of all baseline regional volumes, only deep gray matter predicted time to EDSS progression. For every standard deviation decrease in baseline deep gray matter volume, the risk of having a shorter time to EDSS worsening during follow-up increased by 27%. Of all longitudinal measures, deep gray matter had the fastest annual rate of atrophy. This rate
—Jake Remaly
Suggested Reading
Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210-222.
Deep gray matter volume loss drives disability accumulation in multiple sclerosis (MS), according to a multicenter, longitudinal study published in the February issue of Annals of Neurology.
The results “suggest that the development of deep gray matter atrophy may drive disability accumulation irrespective of clinical phenotypes, thereby becoming a useful outcome measure in neuroprotective clinical trials,” said Arman Eshaghi, MD, of the Queen Square MS Centre at the University College London Institute of Neurology, and colleagues.
Gray matter atrophy occurs in all MS phenotypes. To investigate whether a spatiotemporal pattern of gray matter atrophy is associated with faster disability accumulation in MS, the investigators analyzed 3,604 high-resolution T1-weighted MRI brain scans from 1,417 participants. The investigators retrospectively collected the scans from seven European centers in the MRI in MS (MAGNIMS) network.
The study included 253 patients with clinically isolated syndrome (CIS), 708 patients with relapsing-remitting MS, 128 patients with secondary progressive MS and 125 patients with primary progressive MS with an average follow-up of 2.41 years, as well as 203 healthy controls with an average follow-up of 1.83 years. The researchers assessed disability using the Expanded Disability Status Scale (EDSS). They obtained volumes of deep gray matter; temporal, frontal, parietal, occipital, and cerebellar gray matter; brainstem; and cerebral white matter. Hierarchical mixed models assessed annual percentage rate of regional tissue loss and identified regional volumes associated with time to EDSS progression.
Of all baseline regional volumes, only deep gray matter predicted time to EDSS progression. For every standard deviation decrease in baseline deep gray matter volume, the risk of having a shorter time to EDSS worsening during follow-up increased by 27%. Of all longitudinal measures, deep gray matter had the fastest annual rate of atrophy. This rate
—Jake Remaly
Suggested Reading
Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210-222.
Heart disease in GPA exacts high toll in year 2 and beyond
LIVERPOOL, ENGLAND – Cardiovascular disease was the predominant cause of death of patients with granulomatosis with polyangiitis 1-5 years after a diagnosis in a study by U.K. researchers, suggesting that this could be a target for future intervention.
While active disease was the No. 1 cause of death within the first year of diagnosis in 40% of patients with granulomatosis with polyangiitis (GPA), it was overtaken by cardiovascular disease (CVD) as the main cause of death in 37.5% of patients in the next 4 years from diagnosis.

“The idea for this study came from patients with vasculitis who were polled by Vasculitis UK,” Fiona A. Pearce, MBBS, explained at the British Society for Rheumatology annual conference.
“Further research into mortality was one of their top priorities as patients want to know the honest truth about what is going to happen to them,” added Dr. Pearce, of the division of epidemiology and public health at the University of Nottingham (England).
GPA is a rare type of vasculitis that is estimated to occur in around 1,350 people every year in the United Kingdom. Mortality is known to be high, with around 11%-14% of patients dying in the first year of diagnosis, but there are few data on what happens over a longer term.
The aim of the study was therefore to examine patient survival in the long term – what were the mortality rates several years post diagnosis? Did the risk of death remain high throughout this time and did the causes of death change?
“Of course, the important clinical question was, “Can we then improve things?’ ” Dr. Pearce said.
Two U.K. databases – Clinical Practice Research Datalink and Hospital Episode Statistics – were used to identify patients with GPA diagnosed between 1998 and 2014 and match each case to 10 controls based on age, gender, and family practice. Data on the cause and date of any deaths were then obtained from the Office of National Statistics.
Overall, 465 cases of GPA were matched to 4,610 controls. The median age of participants was 61 years and 57% were male. There were 50 cases with more than 10 years of follow-up data available and data on 139 deaths could be analyzed.
A survival analysis showed that there was a significant reduction in cases versus controls, “but it’s not a constant,” Dr. Pearce noted.
“In the time immediately after diagnosis, the risk of death in people with GPA is very high, and over the first 6 months it tails off.” Then the mortalities are very similar, albeit much lower, to those of controls, but with another dip around 8 years.
The number at risk of death in the second year was 310 for GPA vs. 3,543 for controls and 230 vs. 2,622 at 4 years, 138 vs. 1,704 at 6 years, 93 vs. 1,136 at 8 years, and 49 vs. 658 at 10 years.
Looking at the data another way, the hazard ratios for death comparing GPA cases to controls was 24.5 in the first month, 14.6 in months 1-2, 7.5 by 2-3 months, 4.3 at 3-6 months, 1.6 at 6 months to 8 years, and 3.2 at 8-10 years.
Mortality seems to have improved with time. Splitting the cohort into two time periods based on their diagnosis, those diagnosed between 2008 and 2014 had a lower risk of death than did those diagnosed between 1998 and 2007, although it was still more than four times higher than the background population.
The leading cause of death in patients with GPA 5-10 years after diagnosis was cancer (30.3% of cases), but when this was compared against the general population, the risk was no greater (hazard ratio, 1.0).
GPA cases were also 2.9 times more likely than controls to die as a result of an infection, suggesting that this together with CVD could be a target for mortality reduction strategies.
“We can’t get away from the fact that although this is a large study, there are still small numbers of patients because this is rare disease,” Dr. Pearce observed. “We also don’t have detailed clinical information on each patient, so we can’t look for associations or clinical phenotypes at diagnosis, and there are no biomarkers.”
From a clinical perspective, she noted, it is important to remember that deaths in the first year are mainly from active disease and to continue to try to diagnose and treat the condition as early as possible.
Arthritis Research UK funded the trial. Dr. Pearce had nothing to disclose.
SOURCE: Pearce F et al. Rheumatology. 2018;57(Suppl. 3):key075.204.
LIVERPOOL, ENGLAND – Cardiovascular disease was the predominant cause of death of patients with granulomatosis with polyangiitis 1-5 years after a diagnosis in a study by U.K. researchers, suggesting that this could be a target for future intervention.
While active disease was the No. 1 cause of death within the first year of diagnosis in 40% of patients with granulomatosis with polyangiitis (GPA), it was overtaken by cardiovascular disease (CVD) as the main cause of death in 37.5% of patients in the next 4 years from diagnosis.
“The idea for this study came from patients with vasculitis who were polled by Vasculitis UK,” Fiona A. Pearce, MBBS, explained at the British Society for Rheumatology annual conference.
“Further research into mortality was one of their top priorities as patients want to know the honest truth about what is going to happen to them,” added Dr. Pearce, of the division of epidemiology and public health at the University of Nottingham (England).
GPA is a rare type of vasculitis that is estimated to occur in around 1,350 people every year in the United Kingdom. Mortality is known to be high, with around 11%-14% of patients dying in the first year of diagnosis, but there are few data on what happens over a longer term.
The aim of the study was therefore to examine patient survival in the long term – what were the mortality rates several years post diagnosis? Did the risk of death remain high throughout this time and did the causes of death change?
“Of course, the important clinical question was, “Can we then improve things?’ ” Dr. Pearce said.
Two U.K. databases – Clinical Practice Research Datalink and Hospital Episode Statistics – were used to identify patients with GPA diagnosed between 1998 and 2014 and match each case to 10 controls based on age, gender, and family practice. Data on the cause and date of any deaths were then obtained from the Office of National Statistics.
Overall, 465 cases of GPA were matched to 4,610 controls. The median age of participants was 61 years and 57% were male. There were 50 cases with more than 10 years of follow-up data available and data on 139 deaths could be analyzed.
A survival analysis showed that there was a significant reduction in cases versus controls, “but it’s not a constant,” Dr. Pearce noted.
“In the time immediately after diagnosis, the risk of death in people with GPA is very high, and over the first 6 months it tails off.” Then the mortalities are very similar, albeit much lower, to those of controls, but with another dip around 8 years.
The number at risk of death in the second year was 310 for GPA vs. 3,543 for controls and 230 vs. 2,622 at 4 years, 138 vs. 1,704 at 6 years, 93 vs. 1,136 at 8 years, and 49 vs. 658 at 10 years.
Looking at the data another way, the hazard ratios for death comparing GPA cases to controls was 24.5 in the first month, 14.6 in months 1-2, 7.5 by 2-3 months, 4.3 at 3-6 months, 1.6 at 6 months to 8 years, and 3.2 at 8-10 years.
Mortality seems to have improved with time. Splitting the cohort into two time periods based on their diagnosis, those diagnosed between 2008 and 2014 had a lower risk of death than did those diagnosed between 1998 and 2007, although it was still more than four times higher than the background population.
The leading cause of death in patients with GPA 5-10 years after diagnosis was cancer (30.3% of cases), but when this was compared against the general population, the risk was no greater (hazard ratio, 1.0).
GPA cases were also 2.9 times more likely than controls to die as a result of an infection, suggesting that this together with CVD could be a target for mortality reduction strategies.
“We can’t get away from the fact that although this is a large study, there are still small numbers of patients because this is rare disease,” Dr. Pearce observed. “We also don’t have detailed clinical information on each patient, so we can’t look for associations or clinical phenotypes at diagnosis, and there are no biomarkers.”
From a clinical perspective, she noted, it is important to remember that deaths in the first year are mainly from active disease and to continue to try to diagnose and treat the condition as early as possible.
Arthritis Research UK funded the trial. Dr. Pearce had nothing to disclose.
SOURCE: Pearce F et al. Rheumatology. 2018;57(Suppl. 3):key075.204.
LIVERPOOL, ENGLAND – Cardiovascular disease was the predominant cause of death of patients with granulomatosis with polyangiitis 1-5 years after a diagnosis in a study by U.K. researchers, suggesting that this could be a target for future intervention.
While active disease was the No. 1 cause of death within the first year of diagnosis in 40% of patients with granulomatosis with polyangiitis (GPA), it was overtaken by cardiovascular disease (CVD) as the main cause of death in 37.5% of patients in the next 4 years from diagnosis.
“The idea for this study came from patients with vasculitis who were polled by Vasculitis UK,” Fiona A. Pearce, MBBS, explained at the British Society for Rheumatology annual conference.
“Further research into mortality was one of their top priorities as patients want to know the honest truth about what is going to happen to them,” added Dr. Pearce, of the division of epidemiology and public health at the University of Nottingham (England).
GPA is a rare type of vasculitis that is estimated to occur in around 1,350 people every year in the United Kingdom. Mortality is known to be high, with around 11%-14% of patients dying in the first year of diagnosis, but there are few data on what happens over a longer term.
The aim of the study was therefore to examine patient survival in the long term – what were the mortality rates several years post diagnosis? Did the risk of death remain high throughout this time and did the causes of death change?
“Of course, the important clinical question was, “Can we then improve things?’ ” Dr. Pearce said.
Two U.K. databases – Clinical Practice Research Datalink and Hospital Episode Statistics – were used to identify patients with GPA diagnosed between 1998 and 2014 and match each case to 10 controls based on age, gender, and family practice. Data on the cause and date of any deaths were then obtained from the Office of National Statistics.
Overall, 465 cases of GPA were matched to 4,610 controls. The median age of participants was 61 years and 57% were male. There were 50 cases with more than 10 years of follow-up data available and data on 139 deaths could be analyzed.
A survival analysis showed that there was a significant reduction in cases versus controls, “but it’s not a constant,” Dr. Pearce noted.
“In the time immediately after diagnosis, the risk of death in people with GPA is very high, and over the first 6 months it tails off.” Then the mortalities are very similar, albeit much lower, to those of controls, but with another dip around 8 years.
The number at risk of death in the second year was 310 for GPA vs. 3,543 for controls and 230 vs. 2,622 at 4 years, 138 vs. 1,704 at 6 years, 93 vs. 1,136 at 8 years, and 49 vs. 658 at 10 years.
Looking at the data another way, the hazard ratios for death comparing GPA cases to controls was 24.5 in the first month, 14.6 in months 1-2, 7.5 by 2-3 months, 4.3 at 3-6 months, 1.6 at 6 months to 8 years, and 3.2 at 8-10 years.
Mortality seems to have improved with time. Splitting the cohort into two time periods based on their diagnosis, those diagnosed between 2008 and 2014 had a lower risk of death than did those diagnosed between 1998 and 2007, although it was still more than four times higher than the background population.
The leading cause of death in patients with GPA 5-10 years after diagnosis was cancer (30.3% of cases), but when this was compared against the general population, the risk was no greater (hazard ratio, 1.0).
GPA cases were also 2.9 times more likely than controls to die as a result of an infection, suggesting that this together with CVD could be a target for mortality reduction strategies.
“We can’t get away from the fact that although this is a large study, there are still small numbers of patients because this is rare disease,” Dr. Pearce observed. “We also don’t have detailed clinical information on each patient, so we can’t look for associations or clinical phenotypes at diagnosis, and there are no biomarkers.”
From a clinical perspective, she noted, it is important to remember that deaths in the first year are mainly from active disease and to continue to try to diagnose and treat the condition as early as possible.
Arthritis Research UK funded the trial. Dr. Pearce had nothing to disclose.
SOURCE: Pearce F et al. Rheumatology. 2018;57(Suppl. 3):key075.204.
REPORTING FROM RHEUMATOLOGY 2018
Key clinical point:
Major finding: The main cause of death changed from active disease (40% of all cases) within the first year to cardiovascular disease at years 1-5 (37.5%).
Study details: Retrospective cohort study of 465 individuals newly diagnosed with granulomatosis with polyangiitis matched to 4,610 controls from the general U.K. primary care population.
Disclosures: Arthritis Research UK funded the trial. Dr. Pearce had nothing to disclose.
Source: Pearce F et al. Rheumatology. 2018;57(Suppl. 3):key075.204.
Hospitalist movers and shakers – May 2018

Dr. Lenchus serves as the Society of Hospital Medicine’s Public Policy Committee chair. He is noted for his work as a clinician, hospital administrator, educator, and researcher.
J. Kevin Shushtari, MD, FHM, recently was named chief medical officer at the New Britain (Conn.) Hospital for Special Care, where he will focus on managing the medical staff, admissions, credentials, infection prevention, and clinical affiliations.

Tianzhong Yang, MD, has been selected as the new long-term care medical director for Van Dyk Healthcare in Montclair, N.J. Dr. Yang began his career 3 decades ago in China and has been a hospitalist at Hackensack University Medical Center Mountainside, also in Montclair, since 2013.
Dr. Yang has been an instructor at Brigham and Women’s Hospital in Boston, specializing in anesthesiology. At Van Dyk, he will work with the nursing staff to help patients recover their independence outside of the hospital setting.
Brent W. Burkey, MD, SFHM, a longtime hospitalist at the Cleveland Clinic, has been named the president of Fisher-Titus Medical Center in Norwalk, Ohio. Dr. Burkey has been the chief medical officer at the Cleveland Clinic’s Avon, Ohio, location the past 2 years. He helped the clinic open the Avon hospital in 2016 when he served as vice president of medical affairs.
Dr. Burkey has been a clinical hospitalist since 2004, and he has a master’s degree in business administration from Cleveland State University. He will run all hospital and medical center operations at Fisher-Titus, including quality and safety.

Dr. Maiona has extensive experience as a practicing hospital physician and as an executive. Most recently, he served as national medical director for TeamHealth of Knoxville, Tenn. He also has been an instructor at Tufts University and Harvard Medical School, both in Boston.
Tom Cummins, MD, has been appointed chief medical officer at Bon Secours St. Francis Health System in Greenville, S.C. Dr. Cummins comes to Bon Secours from Catholic Health Initiatives St. Vincent, Arkansas, where he was senior vice president and CMO; he also first served as a hospitalist within that system.
At Bon Secours, Dr. Cummins will oversee the regional health system’s 11 facilities, including St. Francis Downtown in Judson, S.C., and St. Francis Eastside in Greenville.
BUSINESS MOVES
The South Korean government recently announced that it has given permission for all general hospitals that use integrated nursing care to take part in a hospitalist system.
The Korean hospitalist program is a pilot in which those physicians provide all medical care for inpatients. It was adopted in September 2016, and 15 hospitals take part in the program. Prior to the recent ruling, those facilities with integrated nursing services were not eligible for the hospitalist program.
Surgical Affiliates (Sacramento, Calif.), a provider of surgical hospitalist services, has announced a partnership with Regional Medical Center in San Jose, Calif. The surgical hospitalists will assist and support local providers, providing 24/7 access to RMC of San Jose, a Level II trauma center.
Dr. Lenchus serves as the Society of Hospital Medicine’s Public Policy Committee chair. He is noted for his work as a clinician, hospital administrator, educator, and researcher.
J. Kevin Shushtari, MD, FHM, recently was named chief medical officer at the New Britain (Conn.) Hospital for Special Care, where he will focus on managing the medical staff, admissions, credentials, infection prevention, and clinical affiliations.
Tianzhong Yang, MD, has been selected as the new long-term care medical director for Van Dyk Healthcare in Montclair, N.J. Dr. Yang began his career 3 decades ago in China and has been a hospitalist at Hackensack University Medical Center Mountainside, also in Montclair, since 2013.
Dr. Yang has been an instructor at Brigham and Women’s Hospital in Boston, specializing in anesthesiology. At Van Dyk, he will work with the nursing staff to help patients recover their independence outside of the hospital setting.
Brent W. Burkey, MD, SFHM, a longtime hospitalist at the Cleveland Clinic, has been named the president of Fisher-Titus Medical Center in Norwalk, Ohio. Dr. Burkey has been the chief medical officer at the Cleveland Clinic’s Avon, Ohio, location the past 2 years. He helped the clinic open the Avon hospital in 2016 when he served as vice president of medical affairs.
Dr. Burkey has been a clinical hospitalist since 2004, and he has a master’s degree in business administration from Cleveland State University. He will run all hospital and medical center operations at Fisher-Titus, including quality and safety.
Dr. Maiona has extensive experience as a practicing hospital physician and as an executive. Most recently, he served as national medical director for TeamHealth of Knoxville, Tenn. He also has been an instructor at Tufts University and Harvard Medical School, both in Boston.
Tom Cummins, MD, has been appointed chief medical officer at Bon Secours St. Francis Health System in Greenville, S.C. Dr. Cummins comes to Bon Secours from Catholic Health Initiatives St. Vincent, Arkansas, where he was senior vice president and CMO; he also first served as a hospitalist within that system.
At Bon Secours, Dr. Cummins will oversee the regional health system’s 11 facilities, including St. Francis Downtown in Judson, S.C., and St. Francis Eastside in Greenville.
BUSINESS MOVES
The South Korean government recently announced that it has given permission for all general hospitals that use integrated nursing care to take part in a hospitalist system.
The Korean hospitalist program is a pilot in which those physicians provide all medical care for inpatients. It was adopted in September 2016, and 15 hospitals take part in the program. Prior to the recent ruling, those facilities with integrated nursing services were not eligible for the hospitalist program.
Surgical Affiliates (Sacramento, Calif.), a provider of surgical hospitalist services, has announced a partnership with Regional Medical Center in San Jose, Calif. The surgical hospitalists will assist and support local providers, providing 24/7 access to RMC of San Jose, a Level II trauma center.
Dr. Lenchus serves as the Society of Hospital Medicine’s Public Policy Committee chair. He is noted for his work as a clinician, hospital administrator, educator, and researcher.
J. Kevin Shushtari, MD, FHM, recently was named chief medical officer at the New Britain (Conn.) Hospital for Special Care, where he will focus on managing the medical staff, admissions, credentials, infection prevention, and clinical affiliations.
Tianzhong Yang, MD, has been selected as the new long-term care medical director for Van Dyk Healthcare in Montclair, N.J. Dr. Yang began his career 3 decades ago in China and has been a hospitalist at Hackensack University Medical Center Mountainside, also in Montclair, since 2013.
Dr. Yang has been an instructor at Brigham and Women’s Hospital in Boston, specializing in anesthesiology. At Van Dyk, he will work with the nursing staff to help patients recover their independence outside of the hospital setting.
Brent W. Burkey, MD, SFHM, a longtime hospitalist at the Cleveland Clinic, has been named the president of Fisher-Titus Medical Center in Norwalk, Ohio. Dr. Burkey has been the chief medical officer at the Cleveland Clinic’s Avon, Ohio, location the past 2 years. He helped the clinic open the Avon hospital in 2016 when he served as vice president of medical affairs.
Dr. Burkey has been a clinical hospitalist since 2004, and he has a master’s degree in business administration from Cleveland State University. He will run all hospital and medical center operations at Fisher-Titus, including quality and safety.
Dr. Maiona has extensive experience as a practicing hospital physician and as an executive. Most recently, he served as national medical director for TeamHealth of Knoxville, Tenn. He also has been an instructor at Tufts University and Harvard Medical School, both in Boston.
Tom Cummins, MD, has been appointed chief medical officer at Bon Secours St. Francis Health System in Greenville, S.C. Dr. Cummins comes to Bon Secours from Catholic Health Initiatives St. Vincent, Arkansas, where he was senior vice president and CMO; he also first served as a hospitalist within that system.
At Bon Secours, Dr. Cummins will oversee the regional health system’s 11 facilities, including St. Francis Downtown in Judson, S.C., and St. Francis Eastside in Greenville.
BUSINESS MOVES
The South Korean government recently announced that it has given permission for all general hospitals that use integrated nursing care to take part in a hospitalist system.
The Korean hospitalist program is a pilot in which those physicians provide all medical care for inpatients. It was adopted in September 2016, and 15 hospitals take part in the program. Prior to the recent ruling, those facilities with integrated nursing services were not eligible for the hospitalist program.
Surgical Affiliates (Sacramento, Calif.), a provider of surgical hospitalist services, has announced a partnership with Regional Medical Center in San Jose, Calif. The surgical hospitalists will assist and support local providers, providing 24/7 access to RMC of San Jose, a Level II trauma center.
Basic technique of vaginal hysterectomy

Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
This video is brought to you by![]()
Targetable genetic alterations found in 41% of soft tissue sarcomas
, reported Carlo Lucchesi, PhD, of Institut Bergonié in Bordeaux, France, and his associates.
In a cross-sectional study of next-generation sequencing results from 584 patients with soft tissue sarcomas in the American Association for Cancer Research’s GENIE Database, 57% of patients had complex genomics sarcomas (sarcomas with multiple, complex karyotypic abnormalities with no specific pattern), 25% had translocation-related sarcomas (sarcomas with specific reciprocal translocations resulting in oncogenic fusion transcripts), and 18% had simple amplicon sarcomas or sarcomas with inactivating mutations.
A total of 2,697 alterations (1,154 substitutions, 765 gene amplifications, 364 short indels and splicing variants, 346 gene homozygous deletions, and 68 gene rearrangements) were identified in 451 genes. A median of four alterations per case were detected, the researchers wrote in a study published online May 3 in JAMA Oncology.
The researchers identified the 20 genes that were most often altered. The top 5 were TP53, MDM2, CDK4, RB1, and ATRX.
Among these 584 samples, 85% had at least one alteration. The proportions of affected patients in each sarcoma group varied significantly among groups, with the other sarcomas group being the most altered (90.8%) and translocation-related sarcomas being the least mutated (77.8%).
At least one relevant gene alteration that could potentially be used to guide targeted therapy was found in 239 cases (41%) with a statistically significant higher number in other sarcomas (89 cases) and complex genomics sarcomas (131 cases) than in translocation-related sarcomas (19 cases).
This finding of an “unexpectedly high frequency” of clinically relevant genetic alterations supports the premise of the soon-to-be-launched MULTISARC trial, which posits that next-generation sequencing results can be used to guide and improve the treatment outcomes of patients with advanced soft tissue sarcomas. For MULTISARC, such patients will be randomized either to an experimental group that will undergo exome and RNA sequencing – and their results will be discussed in a molecular tumor board to tailor the treatment – or to a control group that will not undergo molecular profiling and will receive conventional therapy. The program will include 16 targeted therapies.
The researchers reported having no relevant financial conflicts of interest.
SOURCE: Lucchesi C et al. JAMA Oncol. doi: 10.1001/jamaoncol.2018.0723.
, reported Carlo Lucchesi, PhD, of Institut Bergonié in Bordeaux, France, and his associates.
In a cross-sectional study of next-generation sequencing results from 584 patients with soft tissue sarcomas in the American Association for Cancer Research’s GENIE Database, 57% of patients had complex genomics sarcomas (sarcomas with multiple, complex karyotypic abnormalities with no specific pattern), 25% had translocation-related sarcomas (sarcomas with specific reciprocal translocations resulting in oncogenic fusion transcripts), and 18% had simple amplicon sarcomas or sarcomas with inactivating mutations.
A total of 2,697 alterations (1,154 substitutions, 765 gene amplifications, 364 short indels and splicing variants, 346 gene homozygous deletions, and 68 gene rearrangements) were identified in 451 genes. A median of four alterations per case were detected, the researchers wrote in a study published online May 3 in JAMA Oncology.
The researchers identified the 20 genes that were most often altered. The top 5 were TP53, MDM2, CDK4, RB1, and ATRX.
Among these 584 samples, 85% had at least one alteration. The proportions of affected patients in each sarcoma group varied significantly among groups, with the other sarcomas group being the most altered (90.8%) and translocation-related sarcomas being the least mutated (77.8%).
At least one relevant gene alteration that could potentially be used to guide targeted therapy was found in 239 cases (41%) with a statistically significant higher number in other sarcomas (89 cases) and complex genomics sarcomas (131 cases) than in translocation-related sarcomas (19 cases).
This finding of an “unexpectedly high frequency” of clinically relevant genetic alterations supports the premise of the soon-to-be-launched MULTISARC trial, which posits that next-generation sequencing results can be used to guide and improve the treatment outcomes of patients with advanced soft tissue sarcomas. For MULTISARC, such patients will be randomized either to an experimental group that will undergo exome and RNA sequencing – and their results will be discussed in a molecular tumor board to tailor the treatment – or to a control group that will not undergo molecular profiling and will receive conventional therapy. The program will include 16 targeted therapies.
The researchers reported having no relevant financial conflicts of interest.
SOURCE: Lucchesi C et al. JAMA Oncol. doi: 10.1001/jamaoncol.2018.0723.
, reported Carlo Lucchesi, PhD, of Institut Bergonié in Bordeaux, France, and his associates.
In a cross-sectional study of next-generation sequencing results from 584 patients with soft tissue sarcomas in the American Association for Cancer Research’s GENIE Database, 57% of patients had complex genomics sarcomas (sarcomas with multiple, complex karyotypic abnormalities with no specific pattern), 25% had translocation-related sarcomas (sarcomas with specific reciprocal translocations resulting in oncogenic fusion transcripts), and 18% had simple amplicon sarcomas or sarcomas with inactivating mutations.
A total of 2,697 alterations (1,154 substitutions, 765 gene amplifications, 364 short indels and splicing variants, 346 gene homozygous deletions, and 68 gene rearrangements) were identified in 451 genes. A median of four alterations per case were detected, the researchers wrote in a study published online May 3 in JAMA Oncology.
The researchers identified the 20 genes that were most often altered. The top 5 were TP53, MDM2, CDK4, RB1, and ATRX.
Among these 584 samples, 85% had at least one alteration. The proportions of affected patients in each sarcoma group varied significantly among groups, with the other sarcomas group being the most altered (90.8%) and translocation-related sarcomas being the least mutated (77.8%).
At least one relevant gene alteration that could potentially be used to guide targeted therapy was found in 239 cases (41%) with a statistically significant higher number in other sarcomas (89 cases) and complex genomics sarcomas (131 cases) than in translocation-related sarcomas (19 cases).
This finding of an “unexpectedly high frequency” of clinically relevant genetic alterations supports the premise of the soon-to-be-launched MULTISARC trial, which posits that next-generation sequencing results can be used to guide and improve the treatment outcomes of patients with advanced soft tissue sarcomas. For MULTISARC, such patients will be randomized either to an experimental group that will undergo exome and RNA sequencing – and their results will be discussed in a molecular tumor board to tailor the treatment – or to a control group that will not undergo molecular profiling and will receive conventional therapy. The program will include 16 targeted therapies.
The researchers reported having no relevant financial conflicts of interest.
SOURCE: Lucchesi C et al. JAMA Oncol. doi: 10.1001/jamaoncol.2018.0723.
FROM JAMA ONCOLOGY
Key clinical point: Next-generation sequencing results might prove useful for guiding targeted therapy that could improve the treatment outcomes of patients with advanced soft tissue sarcomas.
Major finding: At least one targetable genetic alteration was found in 41% of 584 soft tissue sarcomas, and the probability of an alteration was higher in sarcomas with complex genomics than in translocation-related sarcomas.
Study details: A cross-sectional study of next-generation sequencing results from 584 patients with soft tissue sarcomas in the AACR GENIE Database.
Disclosures: The researchers reported having no relevant financial conflicts of interest.
Source: Lucchesi C et al. JAMA Oncol. doi: 10.1001/jamaoncol.2018.0723.
Did unsafe oxytocin dose cause uterine rupture? $3.5M settlement
Did unsafe oxytocin dose cause uterine rupture? $3.5M settlement

When a mother presented to the hospital in labor, the on-call ObGyn ordered oxytocin with the dosage to be increased by 2 mU/min until she was receiving 30 mU/min or until an adequate contraction pattern was achieved and maintained. Over the next few hours, the labor and delivery nurse increased the dosage of the infusion several times.
As the patient began to push, a trickle of bright red blood was seen coming from her vagina and the baby's heart tones were temporarily lost. When the fetal heart tones were restored, his heart rate was approximately 50 bpm. After vaginal delivery was attempted using vacuum extraction and forceps, an emergency cesarean delivery was performed, leading to the finding that the mother's uterus had ruptured.
The baby suffered a permanent brain injury due to hypoxic-ischemic encephalopathy.
PATIENT'S CLAIM: The mother sued the hospital and on-call ObGyn. She alleged that the health care providers breached the standard of care by negligently increasing and maintaining the oxytocin at unsafe levels, which caused the mother's uterus to be overworked and eventually rupture. The rupture led to the child's hypoxia. An expert ObGyn noted that the patient's contractions were adequate by the time the oxytocin dose reached 14 mU/min, but the dosage continued to be increased.
DEFENDANTS' DEFENSE: The case was settled during the trial.
VERDICT: A $3.5 million Kansas settlement was reached.
When did the bowel injury occur?
One day after undergoing a hysterectomy, a woman went to the emergency department (ED) because she was feeling ill. She received a diagnosis of a pulmonary embolism for which she was given anticoagulant medications. The patient's symptoms persisted. Computed tomography (CT) imaging showed a bowel injury, and, 17 days after the initial surgery, an emergency laparotomy was performed.
PATIENT'S CLAIM: The patient sued the surgeon and the hospital. The hospital settled before the trial and the case continued against the surgeon.
The patient's early symptoms after surgery were evidence of a bowel injury, but imaging was not undertaken for several days. If the imaging had been undertaken earlier, the bowel injury would have been detected before it caused a rectovaginal fistula. An expert pathologist testified that the microscopic findings he detected postlaparotomy could only exist if a bowel perforation had been there for a significant period of time before the fistula developed. The patient's experts argued that the injury was not a "free perforation," but had been contained by her body, preventing the spread of the infection.
DEFENDANTS' DEFENSE: The surgeon maintained that the injury did not occur during the hysterectomy but developed in the days just before it was discovered. Over time, a collection of infected fluid at the vaginal cuff eroded into the bowel above it, creating an entryway for stool to pass through. Continuous leakage from the bowel for 17 days (the length of time between development of symptoms and discovery of the bowel injury) would have likely resulted in the patient's death.
VERDICT: A Missouri defense verdict was returned.
Did improper delivery techniques caused brachial plexus injury? $950,000 settlement
In April, a woman began receiving prenatal care for her 7th pregnancy. Her history of maternal obesity and diabetes mellitus, physical examinations, and tests suggested that she was at increased risk for having a macrosomic baby and encountering shoulder dystocia during vaginal delivery.
At 37 weeks' gestation, the mother was admitted to the hospital for induction of labor. Shoulder dystocia was encountered during delivery. At birth, the baby weighed 9 lb 10 oz and her right arm was limp. She was found to have a right brachial plexus injury involving C5‒C8 nerve roots and muscles. Two nerve root avulsions were evident on MRI and visualized by the surgeon during an extensive nerve graft operation. Arm function and range of motion improved after surgery, but the child has not recovered normal use of her arm.
PARENTS' CLAIM: Under the standard of care, the ObGyn was required to obtain informed consent, including a discussion of the risks of vaginal delivery (shoulder dystocia and brachial plexus injury), and the option of cesarean delivery. The patient claimed that the ObGyn neither obtained informed consent nor discussed these risks with her.
The labor and delivery nurse reported that the ObGyn told her, before delivery, that he was expecting a large baby and, perhaps, shoulder dystocia.
The ObGyn deviated from the standard of care by applying more than gentle traction to the fetal head while the shoulder was still impacted. The injury to the baby's right brachial plexus resulted from excessive lateral traction used by the ObGyn. The injury would not have occurred if a cesarean delivery had been performed. The mechanism of maternal forces injuring a brachial plexus nerve has never been visualized by any physician and is an unproven hypothesis.
PHYSICIAN'S DEFENSE: The ObGyn reported using standard maneuvers to deliver the baby. He applied traction on the fetal head 3 times: once after McRoberts maneuver, once after suprapubic pressure, and once after delivery of the posterior arm. He dictated into his notes that "We had to be careful to avoid excessive tractive forces." He claimed that shoulder dystocia is an unpredictable and unpreventable obstetric emergency, and that the injury was caused by the maternal forces of labor.
VERDICT: A $950,000 Virginia settlement was reached.
Wrongful death claim
On March 13, a 76-year-old woman went to her primary care physician's office because of a vaginal discharge. A nurse practitioner (NP) diagnosed a urinary tract infection (UTI) and prescribed cefixime (Suprax). Four days later, the patient began to experience severe diarrhea and blamed the medication. At a follow-up visit on March 20, the NP switched the patient to sulfamethoxazole-trimethoprim (Bactrim).
The following day, the patient was found unresponsive on her bathroom floor and was taken to the ED. It was determined that she had Clostridium difficile colitis (C difficile) and was admitted to the hospital. She developed acute renal failure, metabolic acidosis, hypovolemia, hypotension, and tachycardia. When she went into cardiac arrest, attempts to resuscitate her failed. She died on March 22.
ESTATE'S CLAIM: The estate sued the NP, claiming that the patient's symptoms did not meet the criteria for a UTI. If appropriate tests had been performed, the correct diagnosis would have been made and she could have received potentially life-saving treatment.
DEFENDANTS' DEFENSE: The NP claimed there was no negligence. Her diagnosis and treatment of the patient's condition were appropriate in all respects. The development of C difficile is a risk of any antibiotic.
VERDICT: An Indiana defense verdict was returned.
Nuchal cord: Undisclosed settlement
During delivery, the labor and delivery nurses lost the fetal heart-rate (FHR) monitor tracing, resulting in their being unaware of increasing signs of fetal intolerance to labor. The nurses continued to administer oxytocin to induce labor.
At birth, a nuchal cord was identified. The baby was born without signs of life but was successfully resuscitated by hospital staff.
The baby was found to have sustained severe brain damage as a result of profound fetal hypoxia. The child will require 24-hour nursing and supportive care for as long as she lives.
PARENTS' CLAIM: The nurses and ObGyn breached the standard of care resulting in her child's severe brain damage. The hospital nurses failed to continuously monitor the FHR. Profound brain damage was preventable in this case.
DEFENDANTS' DEFENSE: The nurses continuously monitored by listening to sounds coming out of the bedside monitor even though no taping of FHR was occurring on the central monitors or FHR monitor strip. A nuchal cord is an unforeseeable medical emergency; nothing different could have been done to change the outcome.
VERDICT: An undisclosed Texas settlement was reached.
Surgical needle left near bladder
The patient underwent a hysterectomy on July 9. Because of an injury sustained during the operation, bladder repair surgery was performed on July 19. After that surgery, she reported bleeding and urinary incontinence. Results of a computed tomography (CT) scan showed that the bladder repair was not successful, a vesicovaginal fistula had developed, and a 13-mm C-shaped needle was found near her bladder. A third operation to remove the needle from her abdomen took place on August 16.
PATIENT'S CLAIM: The needle left behind after the second surgery caused a fistula to develop. The patient suffered mental and emotional distress from knowing the needle was in her abdomen. Foreign objects left in a patient during surgery are strong evidence of negligence.
DEFENDANTS' DEFENSE: The primary defense claim was the absence of causation--any negligence did not cause the injury. Defense experts testified that the needle was on top of the bladder and did move anywhere to cause damage. The patient developed a vesicovaginal fistula due to complications from the bladder repair operation and not from the needle. In addition, there was testimony that the third surgery was unnecessary because the needle would eventually flush out of the abdomen without causing damage.
VERDICT: A Michigan defense verdict was returned.
Claims cancer diagnosis was delayed
On October 1, 2008, a woman saw her family physician (FP) for routine care. Blood work results showed an elevated white blood cell (WBC) count. The patient claimed that the FP did not inform her of these test results.
One year later, the patient went to an urgent care facility where blood work was performed; the results showed a high WBC count. After a work-up, the patient was given a presumptive diagnosis of mantel cell lymphoma. By December 9, she had undergone the first round of chemotherapy. Subsequent tests revealed that the presumptive diagnosis was incorrect; she actually had low-grade lymphoproliferative disorder.
In lieu of this new diagnosis, the medical team offered the patient the option of discontinuing chemotherapy. She decided, however, to continue the treatment. Chemotherapy was followed by 2 years of rituximab/hyaluronidase human maintenance therapy.
PATIENT'S CLAIM: The patient presented her case to a medical review board. The course of treatment (chemotherapy plus maintenance therapy) left her with permanent heart damage and an elevated risk of developing secondary cancer.
The patient claimed that none of this would have happened if her FP had informed her of the October 2008 test results and recommended appropriate follow-up studies. The results of those studies would have given her the correct diagnosis and allowed her to receive prompt, proper treatment. The medical review board responded unanimously that the FP's conduct constituted a breach of the standard of care, but concluded that the breach was not a factor in the patient's damages.
The patient filed suit against the FP. An expert in internal medicine commented that, based on the 2008 WBC count, the tests should have been repeated and the patient should have been referred to a hematologist/oncologist. Failure to do so increased the patient's risk of developing cancer in the future.
DEFENDANTS' DEFENSE: The FP denied any breach of standard of care. According to her notes, she had shared test results with the patient on November 26 and recommended following up with repeat blood work. The FP blamed the patient for failing to follow-up as recommended.
VERDICT: An Indiana defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Did unsafe oxytocin dose cause uterine rupture? $3.5M settlement
When a mother presented to the hospital in labor, the on-call ObGyn ordered oxytocin with the dosage to be increased by 2 mU/min until she was receiving 30 mU/min or until an adequate contraction pattern was achieved and maintained. Over the next few hours, the labor and delivery nurse increased the dosage of the infusion several times.
As the patient began to push, a trickle of bright red blood was seen coming from her vagina and the baby's heart tones were temporarily lost. When the fetal heart tones were restored, his heart rate was approximately 50 bpm. After vaginal delivery was attempted using vacuum extraction and forceps, an emergency cesarean delivery was performed, leading to the finding that the mother's uterus had ruptured.
The baby suffered a permanent brain injury due to hypoxic-ischemic encephalopathy.
PATIENT'S CLAIM: The mother sued the hospital and on-call ObGyn. She alleged that the health care providers breached the standard of care by negligently increasing and maintaining the oxytocin at unsafe levels, which caused the mother's uterus to be overworked and eventually rupture. The rupture led to the child's hypoxia. An expert ObGyn noted that the patient's contractions were adequate by the time the oxytocin dose reached 14 mU/min, but the dosage continued to be increased.
DEFENDANTS' DEFENSE: The case was settled during the trial.
VERDICT: A $3.5 million Kansas settlement was reached.
When did the bowel injury occur?
One day after undergoing a hysterectomy, a woman went to the emergency department (ED) because she was feeling ill. She received a diagnosis of a pulmonary embolism for which she was given anticoagulant medications. The patient's symptoms persisted. Computed tomography (CT) imaging showed a bowel injury, and, 17 days after the initial surgery, an emergency laparotomy was performed.
PATIENT'S CLAIM: The patient sued the surgeon and the hospital. The hospital settled before the trial and the case continued against the surgeon.
The patient's early symptoms after surgery were evidence of a bowel injury, but imaging was not undertaken for several days. If the imaging had been undertaken earlier, the bowel injury would have been detected before it caused a rectovaginal fistula. An expert pathologist testified that the microscopic findings he detected postlaparotomy could only exist if a bowel perforation had been there for a significant period of time before the fistula developed. The patient's experts argued that the injury was not a "free perforation," but had been contained by her body, preventing the spread of the infection.
DEFENDANTS' DEFENSE: The surgeon maintained that the injury did not occur during the hysterectomy but developed in the days just before it was discovered. Over time, a collection of infected fluid at the vaginal cuff eroded into the bowel above it, creating an entryway for stool to pass through. Continuous leakage from the bowel for 17 days (the length of time between development of symptoms and discovery of the bowel injury) would have likely resulted in the patient's death.
VERDICT: A Missouri defense verdict was returned.
Did improper delivery techniques caused brachial plexus injury? $950,000 settlement
In April, a woman began receiving prenatal care for her 7th pregnancy. Her history of maternal obesity and diabetes mellitus, physical examinations, and tests suggested that she was at increased risk for having a macrosomic baby and encountering shoulder dystocia during vaginal delivery.
At 37 weeks' gestation, the mother was admitted to the hospital for induction of labor. Shoulder dystocia was encountered during delivery. At birth, the baby weighed 9 lb 10 oz and her right arm was limp. She was found to have a right brachial plexus injury involving C5‒C8 nerve roots and muscles. Two nerve root avulsions were evident on MRI and visualized by the surgeon during an extensive nerve graft operation. Arm function and range of motion improved after surgery, but the child has not recovered normal use of her arm.
PARENTS' CLAIM: Under the standard of care, the ObGyn was required to obtain informed consent, including a discussion of the risks of vaginal delivery (shoulder dystocia and brachial plexus injury), and the option of cesarean delivery. The patient claimed that the ObGyn neither obtained informed consent nor discussed these risks with her.
The labor and delivery nurse reported that the ObGyn told her, before delivery, that he was expecting a large baby and, perhaps, shoulder dystocia.
The ObGyn deviated from the standard of care by applying more than gentle traction to the fetal head while the shoulder was still impacted. The injury to the baby's right brachial plexus resulted from excessive lateral traction used by the ObGyn. The injury would not have occurred if a cesarean delivery had been performed. The mechanism of maternal forces injuring a brachial plexus nerve has never been visualized by any physician and is an unproven hypothesis.
PHYSICIAN'S DEFENSE: The ObGyn reported using standard maneuvers to deliver the baby. He applied traction on the fetal head 3 times: once after McRoberts maneuver, once after suprapubic pressure, and once after delivery of the posterior arm. He dictated into his notes that "We had to be careful to avoid excessive tractive forces." He claimed that shoulder dystocia is an unpredictable and unpreventable obstetric emergency, and that the injury was caused by the maternal forces of labor.
VERDICT: A $950,000 Virginia settlement was reached.
Wrongful death claim
On March 13, a 76-year-old woman went to her primary care physician's office because of a vaginal discharge. A nurse practitioner (NP) diagnosed a urinary tract infection (UTI) and prescribed cefixime (Suprax). Four days later, the patient began to experience severe diarrhea and blamed the medication. At a follow-up visit on March 20, the NP switched the patient to sulfamethoxazole-trimethoprim (Bactrim).
The following day, the patient was found unresponsive on her bathroom floor and was taken to the ED. It was determined that she had Clostridium difficile colitis (C difficile) and was admitted to the hospital. She developed acute renal failure, metabolic acidosis, hypovolemia, hypotension, and tachycardia. When she went into cardiac arrest, attempts to resuscitate her failed. She died on March 22.
ESTATE'S CLAIM: The estate sued the NP, claiming that the patient's symptoms did not meet the criteria for a UTI. If appropriate tests had been performed, the correct diagnosis would have been made and she could have received potentially life-saving treatment.
DEFENDANTS' DEFENSE: The NP claimed there was no negligence. Her diagnosis and treatment of the patient's condition were appropriate in all respects. The development of C difficile is a risk of any antibiotic.
VERDICT: An Indiana defense verdict was returned.
Nuchal cord: Undisclosed settlement
During delivery, the labor and delivery nurses lost the fetal heart-rate (FHR) monitor tracing, resulting in their being unaware of increasing signs of fetal intolerance to labor. The nurses continued to administer oxytocin to induce labor.
At birth, a nuchal cord was identified. The baby was born without signs of life but was successfully resuscitated by hospital staff.
The baby was found to have sustained severe brain damage as a result of profound fetal hypoxia. The child will require 24-hour nursing and supportive care for as long as she lives.
PARENTS' CLAIM: The nurses and ObGyn breached the standard of care resulting in her child's severe brain damage. The hospital nurses failed to continuously monitor the FHR. Profound brain damage was preventable in this case.
DEFENDANTS' DEFENSE: The nurses continuously monitored by listening to sounds coming out of the bedside monitor even though no taping of FHR was occurring on the central monitors or FHR monitor strip. A nuchal cord is an unforeseeable medical emergency; nothing different could have been done to change the outcome.
VERDICT: An undisclosed Texas settlement was reached.
Surgical needle left near bladder
The patient underwent a hysterectomy on July 9. Because of an injury sustained during the operation, bladder repair surgery was performed on July 19. After that surgery, she reported bleeding and urinary incontinence. Results of a computed tomography (CT) scan showed that the bladder repair was not successful, a vesicovaginal fistula had developed, and a 13-mm C-shaped needle was found near her bladder. A third operation to remove the needle from her abdomen took place on August 16.
PATIENT'S CLAIM: The needle left behind after the second surgery caused a fistula to develop. The patient suffered mental and emotional distress from knowing the needle was in her abdomen. Foreign objects left in a patient during surgery are strong evidence of negligence.
DEFENDANTS' DEFENSE: The primary defense claim was the absence of causation--any negligence did not cause the injury. Defense experts testified that the needle was on top of the bladder and did move anywhere to cause damage. The patient developed a vesicovaginal fistula due to complications from the bladder repair operation and not from the needle. In addition, there was testimony that the third surgery was unnecessary because the needle would eventually flush out of the abdomen without causing damage.
VERDICT: A Michigan defense verdict was returned.
Claims cancer diagnosis was delayed
On October 1, 2008, a woman saw her family physician (FP) for routine care. Blood work results showed an elevated white blood cell (WBC) count. The patient claimed that the FP did not inform her of these test results.
One year later, the patient went to an urgent care facility where blood work was performed; the results showed a high WBC count. After a work-up, the patient was given a presumptive diagnosis of mantel cell lymphoma. By December 9, she had undergone the first round of chemotherapy. Subsequent tests revealed that the presumptive diagnosis was incorrect; she actually had low-grade lymphoproliferative disorder.
In lieu of this new diagnosis, the medical team offered the patient the option of discontinuing chemotherapy. She decided, however, to continue the treatment. Chemotherapy was followed by 2 years of rituximab/hyaluronidase human maintenance therapy.
PATIENT'S CLAIM: The patient presented her case to a medical review board. The course of treatment (chemotherapy plus maintenance therapy) left her with permanent heart damage and an elevated risk of developing secondary cancer.
The patient claimed that none of this would have happened if her FP had informed her of the October 2008 test results and recommended appropriate follow-up studies. The results of those studies would have given her the correct diagnosis and allowed her to receive prompt, proper treatment. The medical review board responded unanimously that the FP's conduct constituted a breach of the standard of care, but concluded that the breach was not a factor in the patient's damages.
The patient filed suit against the FP. An expert in internal medicine commented that, based on the 2008 WBC count, the tests should have been repeated and the patient should have been referred to a hematologist/oncologist. Failure to do so increased the patient's risk of developing cancer in the future.
DEFENDANTS' DEFENSE: The FP denied any breach of standard of care. According to her notes, she had shared test results with the patient on November 26 and recommended following up with repeat blood work. The FP blamed the patient for failing to follow-up as recommended.
VERDICT: An Indiana defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Did unsafe oxytocin dose cause uterine rupture? $3.5M settlement
When a mother presented to the hospital in labor, the on-call ObGyn ordered oxytocin with the dosage to be increased by 2 mU/min until she was receiving 30 mU/min or until an adequate contraction pattern was achieved and maintained. Over the next few hours, the labor and delivery nurse increased the dosage of the infusion several times.
As the patient began to push, a trickle of bright red blood was seen coming from her vagina and the baby's heart tones were temporarily lost. When the fetal heart tones were restored, his heart rate was approximately 50 bpm. After vaginal delivery was attempted using vacuum extraction and forceps, an emergency cesarean delivery was performed, leading to the finding that the mother's uterus had ruptured.
The baby suffered a permanent brain injury due to hypoxic-ischemic encephalopathy.
PATIENT'S CLAIM: The mother sued the hospital and on-call ObGyn. She alleged that the health care providers breached the standard of care by negligently increasing and maintaining the oxytocin at unsafe levels, which caused the mother's uterus to be overworked and eventually rupture. The rupture led to the child's hypoxia. An expert ObGyn noted that the patient's contractions were adequate by the time the oxytocin dose reached 14 mU/min, but the dosage continued to be increased.
DEFENDANTS' DEFENSE: The case was settled during the trial.
VERDICT: A $3.5 million Kansas settlement was reached.
When did the bowel injury occur?
One day after undergoing a hysterectomy, a woman went to the emergency department (ED) because she was feeling ill. She received a diagnosis of a pulmonary embolism for which she was given anticoagulant medications. The patient's symptoms persisted. Computed tomography (CT) imaging showed a bowel injury, and, 17 days after the initial surgery, an emergency laparotomy was performed.
PATIENT'S CLAIM: The patient sued the surgeon and the hospital. The hospital settled before the trial and the case continued against the surgeon.
The patient's early symptoms after surgery were evidence of a bowel injury, but imaging was not undertaken for several days. If the imaging had been undertaken earlier, the bowel injury would have been detected before it caused a rectovaginal fistula. An expert pathologist testified that the microscopic findings he detected postlaparotomy could only exist if a bowel perforation had been there for a significant period of time before the fistula developed. The patient's experts argued that the injury was not a "free perforation," but had been contained by her body, preventing the spread of the infection.
DEFENDANTS' DEFENSE: The surgeon maintained that the injury did not occur during the hysterectomy but developed in the days just before it was discovered. Over time, a collection of infected fluid at the vaginal cuff eroded into the bowel above it, creating an entryway for stool to pass through. Continuous leakage from the bowel for 17 days (the length of time between development of symptoms and discovery of the bowel injury) would have likely resulted in the patient's death.
VERDICT: A Missouri defense verdict was returned.
Did improper delivery techniques caused brachial plexus injury? $950,000 settlement
In April, a woman began receiving prenatal care for her 7th pregnancy. Her history of maternal obesity and diabetes mellitus, physical examinations, and tests suggested that she was at increased risk for having a macrosomic baby and encountering shoulder dystocia during vaginal delivery.
At 37 weeks' gestation, the mother was admitted to the hospital for induction of labor. Shoulder dystocia was encountered during delivery. At birth, the baby weighed 9 lb 10 oz and her right arm was limp. She was found to have a right brachial plexus injury involving C5‒C8 nerve roots and muscles. Two nerve root avulsions were evident on MRI and visualized by the surgeon during an extensive nerve graft operation. Arm function and range of motion improved after surgery, but the child has not recovered normal use of her arm.
PARENTS' CLAIM: Under the standard of care, the ObGyn was required to obtain informed consent, including a discussion of the risks of vaginal delivery (shoulder dystocia and brachial plexus injury), and the option of cesarean delivery. The patient claimed that the ObGyn neither obtained informed consent nor discussed these risks with her.
The labor and delivery nurse reported that the ObGyn told her, before delivery, that he was expecting a large baby and, perhaps, shoulder dystocia.
The ObGyn deviated from the standard of care by applying more than gentle traction to the fetal head while the shoulder was still impacted. The injury to the baby's right brachial plexus resulted from excessive lateral traction used by the ObGyn. The injury would not have occurred if a cesarean delivery had been performed. The mechanism of maternal forces injuring a brachial plexus nerve has never been visualized by any physician and is an unproven hypothesis.
PHYSICIAN'S DEFENSE: The ObGyn reported using standard maneuvers to deliver the baby. He applied traction on the fetal head 3 times: once after McRoberts maneuver, once after suprapubic pressure, and once after delivery of the posterior arm. He dictated into his notes that "We had to be careful to avoid excessive tractive forces." He claimed that shoulder dystocia is an unpredictable and unpreventable obstetric emergency, and that the injury was caused by the maternal forces of labor.
VERDICT: A $950,000 Virginia settlement was reached.
Wrongful death claim
On March 13, a 76-year-old woman went to her primary care physician's office because of a vaginal discharge. A nurse practitioner (NP) diagnosed a urinary tract infection (UTI) and prescribed cefixime (Suprax). Four days later, the patient began to experience severe diarrhea and blamed the medication. At a follow-up visit on March 20, the NP switched the patient to sulfamethoxazole-trimethoprim (Bactrim).
The following day, the patient was found unresponsive on her bathroom floor and was taken to the ED. It was determined that she had Clostridium difficile colitis (C difficile) and was admitted to the hospital. She developed acute renal failure, metabolic acidosis, hypovolemia, hypotension, and tachycardia. When she went into cardiac arrest, attempts to resuscitate her failed. She died on March 22.
ESTATE'S CLAIM: The estate sued the NP, claiming that the patient's symptoms did not meet the criteria for a UTI. If appropriate tests had been performed, the correct diagnosis would have been made and she could have received potentially life-saving treatment.
DEFENDANTS' DEFENSE: The NP claimed there was no negligence. Her diagnosis and treatment of the patient's condition were appropriate in all respects. The development of C difficile is a risk of any antibiotic.
VERDICT: An Indiana defense verdict was returned.
Nuchal cord: Undisclosed settlement
During delivery, the labor and delivery nurses lost the fetal heart-rate (FHR) monitor tracing, resulting in their being unaware of increasing signs of fetal intolerance to labor. The nurses continued to administer oxytocin to induce labor.
At birth, a nuchal cord was identified. The baby was born without signs of life but was successfully resuscitated by hospital staff.
The baby was found to have sustained severe brain damage as a result of profound fetal hypoxia. The child will require 24-hour nursing and supportive care for as long as she lives.
PARENTS' CLAIM: The nurses and ObGyn breached the standard of care resulting in her child's severe brain damage. The hospital nurses failed to continuously monitor the FHR. Profound brain damage was preventable in this case.
DEFENDANTS' DEFENSE: The nurses continuously monitored by listening to sounds coming out of the bedside monitor even though no taping of FHR was occurring on the central monitors or FHR monitor strip. A nuchal cord is an unforeseeable medical emergency; nothing different could have been done to change the outcome.
VERDICT: An undisclosed Texas settlement was reached.
Surgical needle left near bladder
The patient underwent a hysterectomy on July 9. Because of an injury sustained during the operation, bladder repair surgery was performed on July 19. After that surgery, she reported bleeding and urinary incontinence. Results of a computed tomography (CT) scan showed that the bladder repair was not successful, a vesicovaginal fistula had developed, and a 13-mm C-shaped needle was found near her bladder. A third operation to remove the needle from her abdomen took place on August 16.
PATIENT'S CLAIM: The needle left behind after the second surgery caused a fistula to develop. The patient suffered mental and emotional distress from knowing the needle was in her abdomen. Foreign objects left in a patient during surgery are strong evidence of negligence.
DEFENDANTS' DEFENSE: The primary defense claim was the absence of causation--any negligence did not cause the injury. Defense experts testified that the needle was on top of the bladder and did move anywhere to cause damage. The patient developed a vesicovaginal fistula due to complications from the bladder repair operation and not from the needle. In addition, there was testimony that the third surgery was unnecessary because the needle would eventually flush out of the abdomen without causing damage.
VERDICT: A Michigan defense verdict was returned.
Claims cancer diagnosis was delayed
On October 1, 2008, a woman saw her family physician (FP) for routine care. Blood work results showed an elevated white blood cell (WBC) count. The patient claimed that the FP did not inform her of these test results.
One year later, the patient went to an urgent care facility where blood work was performed; the results showed a high WBC count. After a work-up, the patient was given a presumptive diagnosis of mantel cell lymphoma. By December 9, she had undergone the first round of chemotherapy. Subsequent tests revealed that the presumptive diagnosis was incorrect; she actually had low-grade lymphoproliferative disorder.
In lieu of this new diagnosis, the medical team offered the patient the option of discontinuing chemotherapy. She decided, however, to continue the treatment. Chemotherapy was followed by 2 years of rituximab/hyaluronidase human maintenance therapy.
PATIENT'S CLAIM: The patient presented her case to a medical review board. The course of treatment (chemotherapy plus maintenance therapy) left her with permanent heart damage and an elevated risk of developing secondary cancer.
The patient claimed that none of this would have happened if her FP had informed her of the October 2008 test results and recommended appropriate follow-up studies. The results of those studies would have given her the correct diagnosis and allowed her to receive prompt, proper treatment. The medical review board responded unanimously that the FP's conduct constituted a breach of the standard of care, but concluded that the breach was not a factor in the patient's damages.
The patient filed suit against the FP. An expert in internal medicine commented that, based on the 2008 WBC count, the tests should have been repeated and the patient should have been referred to a hematologist/oncologist. Failure to do so increased the patient's risk of developing cancer in the future.
DEFENDANTS' DEFENSE: The FP denied any breach of standard of care. According to her notes, she had shared test results with the patient on November 26 and recommended following up with repeat blood work. The FP blamed the patient for failing to follow-up as recommended.
VERDICT: An Indiana defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.