User login
CHEST Foundation support for young career clinicians
As the CHEST Foundation continues to grow, so does our ability to impact the careers of early career clinicians. What began as a small travel grants program for the 2015 winners of the NetWorks Challenge to help offset their trainee members’ travel to CHEST 2015 in Montreal, was quickly identified as opportunity for the CHEST Foundation to deepen their engagement with early career clinicians. The CHEST Foundation travel grants program has grown immensely since then, but the core tenants of the program remain unchanged – to provide excellent trainees, medical students, and all other members of the care team with the fiscal support they need to become successful clinicians and faithfully treat their patients and community. Some of the ways our travel grants are put to good use is to attend the CHEST Annual Meeting and to further engage them as active members of CHEST. In addition to travel grant support to offset the costs of attending the annual meeting, recipients of these competitive grants receive free registration to the meeting; individualized mentorship from a CHEST member who is currently or has been part of CHEST leadership (ie, served on one of the boards, as faculty, on committees, as well as chairs and vice-chairs of the NetWorks); learn best practices for applying for research and community service grants from previous grant winners; invitations to exclusive receptions to network with peers and potential employers; and access to several sessions at the annual meeting intended to strengthen their clinical skill set. All of these programmatic pieces come together to help propel these young leaders’ careers and invest in the future of our discipline as CHEST clinicians.
Due to your overwhelming philanthropic support, CHEST Foundation’s travel grant programs continue to flourish. In 2017, the CHEST Foundation supported
a total of 43 early career clinicians’ travel to attend the CHEST Annual Meeting in Toronto. Through continued donor support, a successful NetWorks Challenge
fundraiser, and an overwhelming number of qualified early career applicants for the travel grants, that number swelled to 72 clinicians for the 2018 CHEST Annual Meeting in San Antonio. In total, the CHEST Foundation dispensed over $70,000 in travel grants for CHEST 2018. We can’t thank you enough for the impact you have made in these early career clinicians’ professional lives, and we urge you to increase your gifts, so we can advance these important professional development opportunities for clinicians by CHEST 2019!
“I’m so thankful to be a recipient of the CHEST travel grant! It enabled me to connect with such a wide array of health-care professionals and learn from my peers. It was wonderful to discover that there are many ways for me as a respiratory therapist to become involved in CHEST! Thank you to all the donors who made these awards a reality!”
- Maya Jenkins, RRT
“As an international medical graduate fellow, I experience challenges spanning from economic (inability to moonlight), professional (scarce funding and sponsorship opportunities, mentorship) to immigration-related difficulties. The CHEST Foundation grant is a superbly structured and implemented opportunity that allowed me a chance to address most of these challenges as I advance in my academic career. The grant itinerary permitted me to network with mentors and, subsequently, resulted in critical leads: A collaborative research project, offers to write letters in support of my visa situation, interest from a journal for one my manuscripts, plans to submit proposals for #CHEST2019, and, most importantly, support from leaders in our field who offered guidance and sponsorship (huge shout out to Dr. Chris Carroll)! I would like to thank the Foundation for awarding this grant as it isn’t just the grant but the slew of opportunities that came along with it that can, and, in my case, catapult fledgling careers in the field of pulmonary and critical care medicine.”
-Viren Kaul, MD
“CHEST education is the cornerstone of pulmonary medicine and delivering world-class health care. CHEST and the CHEST Foundation care about me and the importance of being the best practitioner I can be for my patients. Having impactful conversations with other clinicians, seeing new innovations, and learning through a diverse number of ways while at CHEST 2018 gave me meaningful lessons to apply in my daily practice. The travel grant made this possible!”
- Sarah Brundidge, MSc, RRT
As the CHEST Foundation continues to grow, so does our ability to impact the careers of early career clinicians. What began as a small travel grants program for the 2015 winners of the NetWorks Challenge to help offset their trainee members’ travel to CHEST 2015 in Montreal, was quickly identified as opportunity for the CHEST Foundation to deepen their engagement with early career clinicians. The CHEST Foundation travel grants program has grown immensely since then, but the core tenants of the program remain unchanged – to provide excellent trainees, medical students, and all other members of the care team with the fiscal support they need to become successful clinicians and faithfully treat their patients and community. Some of the ways our travel grants are put to good use is to attend the CHEST Annual Meeting and to further engage them as active members of CHEST. In addition to travel grant support to offset the costs of attending the annual meeting, recipients of these competitive grants receive free registration to the meeting; individualized mentorship from a CHEST member who is currently or has been part of CHEST leadership (ie, served on one of the boards, as faculty, on committees, as well as chairs and vice-chairs of the NetWorks); learn best practices for applying for research and community service grants from previous grant winners; invitations to exclusive receptions to network with peers and potential employers; and access to several sessions at the annual meeting intended to strengthen their clinical skill set. All of these programmatic pieces come together to help propel these young leaders’ careers and invest in the future of our discipline as CHEST clinicians.
Due to your overwhelming philanthropic support, CHEST Foundation’s travel grant programs continue to flourish. In 2017, the CHEST Foundation supported
a total of 43 early career clinicians’ travel to attend the CHEST Annual Meeting in Toronto. Through continued donor support, a successful NetWorks Challenge
fundraiser, and an overwhelming number of qualified early career applicants for the travel grants, that number swelled to 72 clinicians for the 2018 CHEST Annual Meeting in San Antonio. In total, the CHEST Foundation dispensed over $70,000 in travel grants for CHEST 2018. We can’t thank you enough for the impact you have made in these early career clinicians’ professional lives, and we urge you to increase your gifts, so we can advance these important professional development opportunities for clinicians by CHEST 2019!
“I’m so thankful to be a recipient of the CHEST travel grant! It enabled me to connect with such a wide array of health-care professionals and learn from my peers. It was wonderful to discover that there are many ways for me as a respiratory therapist to become involved in CHEST! Thank you to all the donors who made these awards a reality!”
- Maya Jenkins, RRT
“As an international medical graduate fellow, I experience challenges spanning from economic (inability to moonlight), professional (scarce funding and sponsorship opportunities, mentorship) to immigration-related difficulties. The CHEST Foundation grant is a superbly structured and implemented opportunity that allowed me a chance to address most of these challenges as I advance in my academic career. The grant itinerary permitted me to network with mentors and, subsequently, resulted in critical leads: A collaborative research project, offers to write letters in support of my visa situation, interest from a journal for one my manuscripts, plans to submit proposals for #CHEST2019, and, most importantly, support from leaders in our field who offered guidance and sponsorship (huge shout out to Dr. Chris Carroll)! I would like to thank the Foundation for awarding this grant as it isn’t just the grant but the slew of opportunities that came along with it that can, and, in my case, catapult fledgling careers in the field of pulmonary and critical care medicine.”
-Viren Kaul, MD
“CHEST education is the cornerstone of pulmonary medicine and delivering world-class health care. CHEST and the CHEST Foundation care about me and the importance of being the best practitioner I can be for my patients. Having impactful conversations with other clinicians, seeing new innovations, and learning through a diverse number of ways while at CHEST 2018 gave me meaningful lessons to apply in my daily practice. The travel grant made this possible!”
- Sarah Brundidge, MSc, RRT
As the CHEST Foundation continues to grow, so does our ability to impact the careers of early career clinicians. What began as a small travel grants program for the 2015 winners of the NetWorks Challenge to help offset their trainee members’ travel to CHEST 2015 in Montreal, was quickly identified as opportunity for the CHEST Foundation to deepen their engagement with early career clinicians. The CHEST Foundation travel grants program has grown immensely since then, but the core tenants of the program remain unchanged – to provide excellent trainees, medical students, and all other members of the care team with the fiscal support they need to become successful clinicians and faithfully treat their patients and community. Some of the ways our travel grants are put to good use is to attend the CHEST Annual Meeting and to further engage them as active members of CHEST. In addition to travel grant support to offset the costs of attending the annual meeting, recipients of these competitive grants receive free registration to the meeting; individualized mentorship from a CHEST member who is currently or has been part of CHEST leadership (ie, served on one of the boards, as faculty, on committees, as well as chairs and vice-chairs of the NetWorks); learn best practices for applying for research and community service grants from previous grant winners; invitations to exclusive receptions to network with peers and potential employers; and access to several sessions at the annual meeting intended to strengthen their clinical skill set. All of these programmatic pieces come together to help propel these young leaders’ careers and invest in the future of our discipline as CHEST clinicians.
Due to your overwhelming philanthropic support, CHEST Foundation’s travel grant programs continue to flourish. In 2017, the CHEST Foundation supported
a total of 43 early career clinicians’ travel to attend the CHEST Annual Meeting in Toronto. Through continued donor support, a successful NetWorks Challenge
fundraiser, and an overwhelming number of qualified early career applicants for the travel grants, that number swelled to 72 clinicians for the 2018 CHEST Annual Meeting in San Antonio. In total, the CHEST Foundation dispensed over $70,000 in travel grants for CHEST 2018. We can’t thank you enough for the impact you have made in these early career clinicians’ professional lives, and we urge you to increase your gifts, so we can advance these important professional development opportunities for clinicians by CHEST 2019!
“I’m so thankful to be a recipient of the CHEST travel grant! It enabled me to connect with such a wide array of health-care professionals and learn from my peers. It was wonderful to discover that there are many ways for me as a respiratory therapist to become involved in CHEST! Thank you to all the donors who made these awards a reality!”
- Maya Jenkins, RRT
“As an international medical graduate fellow, I experience challenges spanning from economic (inability to moonlight), professional (scarce funding and sponsorship opportunities, mentorship) to immigration-related difficulties. The CHEST Foundation grant is a superbly structured and implemented opportunity that allowed me a chance to address most of these challenges as I advance in my academic career. The grant itinerary permitted me to network with mentors and, subsequently, resulted in critical leads: A collaborative research project, offers to write letters in support of my visa situation, interest from a journal for one my manuscripts, plans to submit proposals for #CHEST2019, and, most importantly, support from leaders in our field who offered guidance and sponsorship (huge shout out to Dr. Chris Carroll)! I would like to thank the Foundation for awarding this grant as it isn’t just the grant but the slew of opportunities that came along with it that can, and, in my case, catapult fledgling careers in the field of pulmonary and critical care medicine.”
-Viren Kaul, MD
“CHEST education is the cornerstone of pulmonary medicine and delivering world-class health care. CHEST and the CHEST Foundation care about me and the importance of being the best practitioner I can be for my patients. Having impactful conversations with other clinicians, seeing new innovations, and learning through a diverse number of ways while at CHEST 2018 gave me meaningful lessons to apply in my daily practice. The travel grant made this possible!”
- Sarah Brundidge, MSc, RRT
Winners all
Everyone who attended CHEST Annual Meeting 2018 is a winner, but we would like to call out the winners participating in CHEST’s special categories of awards and events. Congratulations to all!
ANNUAL CHEST AWARDS
Master FCCP
David Gutterman, MD, Master FCCP
Distinguished Service Award
David Gutterman, MD, Master FCCP
College Medalist Award
Ghada Bourjeily, MD, FCCP
Master Clinician Educator
Lisa Moores, MD, FCCP
Early Career Clinician Educator
Amy Morris, MD, FCCP
Alfred Soffer Award for Editorial Excellence
Jean Rice
Presidential Citation
Darcy Marciniuk, MD, FCCP
Presidential Citation
D. Robert McCaffree, MD, Master FCCP
HONOR LECTURES AND MEMORIAL AWARDS
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture Accelerated Aging in COPD and Its Comorbidities: Novel Therapeutic Targets
Peter Barnes, MD, Master FCCP
The lecture is generously funded by the CHEST Foundation.
Distinguished Scientist Honor Lecture in Cardiopulmonary Physiology
Understanding Diaphragm Performance: The Role of Ultrasound
F. Dennis McCool, MD, FCCP
The lecture is generously funded by the CHEST Foundation.
Presidential Honor Lecture
Asthma: Past, Present, and Future
Jay Peters, MD, FCCP
Thomas L. Petty, MD, Master FCCP Memorial Lecture
Recent Developments in Pulmonary Rehabilitation and Long-Term Oxygen Therapy: Would Tom Petty be Pleased?
Richard Casaburi, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation
Saving Lives…One Ventilator at a Time - HMV in 2018 and Beyond
Douglas McKim, MD, FCCP
The Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation is generously supported by International Ventilator Users Network of Post-Polio Health International and the CHEST Foundation.
Pasquale Ciaglia Memorial Lecture in Interventional Medicine
Evolution of Endobronchial Ultrasound: From Diagnostics to Therapeutics
Kazuhiro Yasufuku, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Roger C. Bone Memorial Lecture in Critical Care
Methylprednisolone in ARDS: A Highly Effective Treatment. How it Works, How to Use it
G. Umberto Meduri, MD
The lecture is generously funded by the CHEST Foundation.
CHEST FOUNDATION GRANT WINNERS
Distinguished Scholar
Robert C. Hyzy, MD, FCCP
Eli Lilly and Company Distinguished Scholar in Critical Care MedicineGrant Title: The Use of Electrical Impedance Tomography to Assess Mechanical Ventilation in Acute Respiratory Distress Syndrome
This grant is made possible due to the philanthropic support from Eli Lilly and Company.
Community Service Grantees
Deborah Haisch, MD
Columbia University Medical Center – New York, NY
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: East African Training Initiative in Pulmonary and Critical Care Medicine
Pamela Garrett, CCRN, MN
Gwinnett Medical Center – Lawrenceville, GA
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Breathe Better Gwinnett
Phillip Sheridan
Mobile Care Chicago – Chicago, IL
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Home Environment Education for Children with Asthma
These grants are supported in full by the CHEST Foundation.
Research Grant Winners
Ayodeji Adegunsoye, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Impact of Telomere Length on Pulmonary Fibrosis Clusters Across Diverse Racial Cohorts
Justin Oldham, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Plasma Biomarkers to Predict Outcomes and Treatment Response in Patients with Pulmonary Fibrosis
These grants above are supported by Boehringer Ingelheim Pharmaceuticals, Inc and Genentech.
Jacob Brenner, MD, PhD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Ambulatory Cuirass Ventilation for Relief of Exertional Dyspnea in Severe COPD Patients
William Zhang, MD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Pulmonary Iron Overload as a Novel COPD Endotype
These grants above are supported by AstraZeneca LP and Sunovion Pharmaceuticals Inc.
Margaret Bublitz, PhD
CHEST Foundation Research Grant in Women’s Lung Health
Grant Title: Sex as a Predictor of Sleep-Disordered Breathing and Its Consequences in Pregnancy
This grant is supported in full by the CHEST Foundation.
Tim Morris, MD, FCCP
CHEST Foundation Research Grant in Venous Thromboembolism
Grant Title: Long-term Follow-up of Acute Pulmonary Embolism
This grant is supported in full by the CHEST Foundation.
Monica Mukherjee, MD, MPH
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Grant Title: Exercise Provocation in the Noninvasive Detection of Occult Right Ventricular Dysfunction and Emerging Pulmonary Hypertension in Systemic Sclerosis
This grant is supported in full by the CHEST Foundation.
Don Sanders, MD, MS
CHEST Foundation Research Grant in Cystic Fibrosis
Grant Title: Whole-genome Shotgun Sequencing of Oropharyngeal Swabs in Infants With CF
This grant is supported by Vertex Pharmaceuticals.
Imran Sulaiman, MD, PhD
CHEST Foundation Research Grant in Nontuberculosis Mycobacteria Diseases
Grant Title: Lower Airway Microbiota Signatures Associated W ith Impaired Immune Response in Non-Tuberculous Mycobacterium
This grant is supported by Insmed.
Samira Shojaee, MD, MPH, FCCP
CHEST Foundation Research Grant in Lung Cancer
Grant Title: Extracellular Vesicle miRNA as a Biomarker in Malignant Pleural Effusion
This grant is supported in full by the CHEST Foundation.
Anna Volerman, MD
CHEST Foundation Research Grant in Severe Asthma
Grant Title: A Randomized Clinical Trial Evaluating the Effectiveness of Virtual Teach-to-Goal(TM) Education versus Brief Intervention for Children with Severe Asthma
This grant is supported by AstraZeneca LP.
ABSTRACT AND CASE REPORT WINNERS
Alfred Soffer Research Award Winners
Clauden Louis, MD: Left ventricular assist devices in Intermacs 1 acute cardiogenic shock patients
Babith J. Mankidy, MBBS, FCCP: Reduction in in-hospital cardiac arrest with early interventions in the emergency department and non-ICU units by a novel approach of rapid response teams and mobile ICU management
Young Investigator Award Winners
Fayez Kheir, MD, MSc: Intrapleural tissue plasminogen activator and deoxyribonuclease therapy vs early medical thoracoscopy for treatment of pleural infection: a randomized clinical trial
Michael Rosman, MD: The utility of end tidal CO2 (ETCO2) monitoring during in-hospital cardiac arrest to predict return of spontaneous circulation
Top 5 Abstract Poster Winners
Neha Agarwal, MD: The 3 wishes project: a feasible intervention to improve end of life care in the ICU at UCLA
Hiroaki Harada, MD: Usefulness of comprehensive preoperative pulmonary rehabilitation program including intensive nutritional support concomitant with physical exercise through an interdisciplinary team approach
Joseph M. Carrington, DO, MHA: Targeting the trans-IL-6 signaling pathway to reduce agriculture organic dust exposure-induced airway inflammation in mice
Yu Kuang Lai, MBBCh: The utility of parametric response mapping in pulmonary graft vs host disease following hematopoietic stem cell transplant
Top Abstract Poster Finalists
Ligia M. Puiu, MD, PhD, FCCP: Association between echocardiographic and lipid parameters to workers in the metalliferous mines
Kush R. Dholakia, MD: Colloids vs crystalloids for postoperative resuscitation in patients undergoing off-pump coronary artery bypass surgery
Kulothungan Gunasekaran, MD, MBBS: Risk of VTE in idiopathic pulmonary fibrosis: a systematic review
Laura B. Sutton, PharmD: Ease and correct use of Ellipta by age in patients with asthma and COPD
Ankur Mogla, MD: To assess the utilization of pulmonary function testing for perioperative respiratory complications in bariatric surgery patients
Ali Ammar: Tracheostomy and admission diagnosis as predictors for an extended length of stay (ELOS)
Charlene Kalani, PharmD: Efficacy and safety of direct oral anticoagulants (DOACS) in morbidly obese patients
Jonghoo Lee, MD: Performances of modified CRB-65 score compared to SIRS and QSOFA as a rapid screening tool for sepsis among infected patients in initial emergency department: a propensity score matching study
Frank J. Trudo, MD, FCCP: Clinical burden of eosinophilic COPD
Elise L. Stephenson, MD: Vitamin C and point of care glucose measurements: a retrospective, observational study
Faisal Siddiqi, MD: Implementation of an early mobility program in the medical ICU
Eileen Harder, MD: Connective tissue disease-associated pulmonary arterial hypertension hospitalizations from 2001-2014
Sophie Korzan, MD: Exhaled nitric oxide and asthma-COPD overlap in patients hospitalized with exacerbations of airway disease: preliminary observations
Andreas Grove, MD: MicroRNA (MIRNA) and biological markers discriminate between normotensive and prehypertensive young men in hypobaric hypoxic environments
Snigdha Nutalapati, MBBS: Large cell neuroendocrine cancer of the lung: SEER 2004-2014 analysis
Anubhav Jain, MBBS: Survival benefit of beta-blockers in patients hospitalized for acute exacerbation of COPD
Case Report Slide Winners
Ze Ying Tan: All that wheezes is not asthma
Jason Lam: Pulmonary mucor mycetoma
Adam Young: Nonresolving pneumonia and cyclic fevers in an immunocompetent patient
Ritu Modi: Histopathological misdiagnosis of pulmonary coccidiodes
Argun Can: A rare inborn error of fatty acid oxidation presenting with severe hyperammonemia in the ICU
Morgan Gilani: A colorful cause of cardiovascular collapse
Katie Jeans: A sweet surprise
Anthony Mattox: Unusual case of interstitial lung disease
Andrew Berglund: Pulmonary light chain deposition disease in a 29-year-old army soldier
Cristia Maysol Morales: A case report of a primary malignant melanoma of anterior mediastinum
Anthony McClafferty: Fibrosing mediastinitis and rheumatoid arthritis: an autoimmune inflammatory connection
Ahmed Munir: HIV with disseminated tularemia: a rare presentation Benjamin Garren: Mycobacterium avium complex mediastinal lymphadenitis in an immunocompetent adolescent with erosion into the airway
Robert Hilton: Obtunded with a chest mass: a case of a rare neurologic paraneoplastic syndrome,
Audra Schwalk: Mucoepidermoid carcinoma: a rare malignancy treated endobronchially
Jessica Riggs: Successful transplantation defies genetics: a case of rapidly-progressive pulmonary fibrosis due to Hermansky-Pudlak syndrome
Meghan Cirulis: Acute vasodilator testing: an opportunity to refine study design and provide precision care in pulmonary hypertension
Patrick Chan: VATS lobectomy for bronchial atresia in an adult
Andrew Mehlman: Multivessel coronary artery aneurysms presenting as myocardial ischemia
Scott Maughan: Diagnosing milliary Mycobacterium bovis from the prostate of an immunocompetent host
Adam Austin: Survived ECMO, death by BLASTO: the first reported fatal case of disseminated blastomycosis in pregnancy
Tie: Donnie Carter: Subclinical polycythemia vera presenting as extensive thrombosis due to massive transfusion, and
Lindsay Hammons: Rare case of Serratia pneumonia causing transient aplastic anemia
Paola Baskin: Novel observations during point-of-care ultrasound (POCUS) in cardiopulmonary resuscitation: a case of ultrasound-guided probe pressure to reduce esophageal insufflation during bag-valve-mask ventilator
David Dennis: Pulmonary alveolar proteinosis presenting as intracerebral nocardiosis
Rakin Choudhury: Severe asthma caused by therapy-resistant asthmatic granulomatosis
Andrew Lytle: Lung adenocarcinoma in a patient with Turcot syndrome
Chelsea Leipold: Case of a granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency disorder
Galyna Ivashchuk: Double trouble: ANCA vasculitis with concomitant IGA nephropathy presenting as massive diffuse alveolar hemorrhage and fulminant renal failure
Case Report Poster Winners
Christine Zhou: Role of transbronchial lung cryobiopsy in the diagnosis of adenocarcinoma in situ
Parin Shah: A rare case of Erdheim-Chester disease masquerading as metastatic lung cancer
Avanthika Wynn : A rare asthma mimic
Muhammad S. Ali: Severe pancolitis: a rare adverse effect of nintedanib
Brian Foster: Don’t forget to breathe: a case of hypoxemia after carotid body resection
Kelly Pennington: Intra-cardiac embolization of an inferior vena cava filter resulting in cardiac arrest
George Elkomos-Botros: Acute generalized exanthematous pustulosis presenting as distributive shock with multi-organ failure
Ashley M. Scott: Avian occupational hypersensitivity pneumonitis in a restaurant employee
Andrew Polito: Pulmonary amyloidosis: an unusual presentation of a rare disease
CHEST B-I-N-G-O WINNERS
Stella Ogake, MD
Erin E. Peterson, APRN, CNP
Megan J. Castillo, PA-C
Gretchen R. Winter, MD
Jeanette P. Brown, MD, PhD
Yu Hong Chan, MBBS
Anita Naik, DO
Gary A. Aaronson, DO, FCCP
Allison S. Cowl, MD
Kyle Halligan, MD
Palaniappan Muthappan, MD
Faizullah S. Lokhandwala, MBBS, FCCP
Jamie R. Chua, MD
Francis L. Ervin, MD, FCCP
Robyn Luper
CHEST CHALLENGE WINNER (AND RUNNER’S-UP)
Emory University (First Place)
Mirza Haider Ali, MD
Mohleen Kang, MD
Matthew Schimmel, MD
University of Michigan (Second Place)
Patrick Bradley, MD
Matthew Hensley, MD
Bonnie Wang, MD
Cleveland Clinic (Third Place)
Jorge Mirales-Estrella, MD
Apostolos Perelas, MD
Gretchen Winter, MD
2018 DISTINGUISHED CHEST EDUCATORS
Michael H Ackerman, DNSc
Sandra G Adams, MD, MS, FCCP
Doreen J Addrizzo-Harris, MD, FCCP
Cara Lyn Agerstrand, MD, BS
Jason A Akulian, MD, FCCP
Raed H Alalawi, MD, FCCP
A. Christine Argento, MD, FCCP
Robert Arntfield, MD, FCCP
Alex A Balekian, MD
Meyer S Balter, MD, FCCP
Gisela I Banauch, MD, MS, FCCP
Robert P Baughman, MD, FCCP
David G Bell, MD, FCCP
Michel A Boivin, MD, FCCP
Gabriel T Bosslet, MD, FCCP
Jean Bourbeau, MD, MS, FCCP
Ghada R Bourjeily, MD, FCCP
David L Bowton, MD, FCCM
Jack D Buckley, MD, MPH, FCCP
Marie M Budev, DO, MPH, FCCP
Kristin M Burkart, MD, MS, FCCP
Brian Carlin, MD, FCCP
Christopher L Carroll, MD, FCCP
Roberto F Casal, MD
Kevin M Chan, MD, FCCP
Subani Chandra, MD, FCCP
Ching-Fei Chang, MD
Alexander C Chen, MD
Nancy A Collop, MD, FCCP
Clayton T Cowl, MD, MS, FCCP
Angel O Coz Yataco, MD, FCCP
Gerard J Criner, MD, FCCP
Carolyn M D’Ambrosio, MD, FCCP
Mauricio Danckers, MD, FCCP
Aneesa M Das, MD, FCCP
John Davies, RRT, MA, FCCP
Zachary S DePew, MD, FCCP
Frank C Detterbeck, MD, FCCP
Naresh A. Dewan, MBBS, FCCP
Kevin C Doerschug, MD, MS, FCCP
Meagan Dubosky, RRT-ACCS
Kevin M Dushay, MD, FCCP
Eric S Edell, MD, FCCP
Jean M Elwing, MD, FCCP
William Enfinger
Michael E Ezzie, MD, FCCP
Kevin J Felner, MD, FCCP
Mark E Fenton, MD, MSc, FCCP
Jason Filopei, MD
Neil S Freedman, MD, FCCP
Laura Kathleen Frye, MD
Thomas M Fuhrman, MD, MS, FCCP
John P Gaillard, MD, FCCP
Colin T Gillespie, MD
Yonatan Y Greenstein, MD
Maritza L Groth, MD, FCCP
Keith P Guevarra, DO, FCCP
Jesse B Hall, MD, FCCP
Nicola A Hanania, MD, MBBS, FCCP
D Kyle Hogarth, MD, FCCP
Steven M Hollenberg, MD, FCCP
David W Hsia, MD, FCCP
Candace A Huebert, MD, FCCP
Robert C Hyzy, MD, FCCP
Octavian C Ioachimescu, MD, PhD, FCCP
Richard S Irwin, MD, Master FCCP
Kirk D Jones, MD
Nader Kamangar, MD, MS, FCCP
Carl A Kaplan, MD, FCCP
Brian S Kaufman, MD, FCCP
William F Kelly, MD, FCCP
Marcus P Kennedy, MD, FCCP
Sandhya Khurana, MD, FCCP
James R Klinger, MD, FCCP
Seth J Koenig, MD, FCCP
Lindsey Kreisher, RRT
Karol Kremens, MD, FCCP
Patricia A Kritek, MD, FCCP
Sunita Kumar, MD, MBBS, FCCP
Rudy P Lackner, MD, FCCP
Viera Lakticova, MD
Carla R Lamb, MD, FCCP
Hans J Lee, MD, FCCP
Peter H Lenz, MD, MEd, FCCP
Stephanie M Levine, MD, FCCP
Deborah Jo Levine, MD, MS, FCCP
Andrea Loiselle, MD
Kenneth E Lyn-Kew, MD
Michael S Machuzak, MD, FCCP
Neil R MacIntyre, MD, FCCP
Donald A Mahler, MD, FCCP
Fabien Maldonado, MD, FCCP
Atul Malhotra, MD, FCCP
Darcy D Marciniuk, MD, FCCP
Diego J Maselli Caceres, MD, FCCP
Paul H Mayo, MD, FCCP
Peter J Mazzone, MD, MPH, FCCP
John K McIlwaine, DO, MBA, FCCP
Matthew C Miles, MD, FCCP
Scott Millington, MD
Taro Minami, MD, FCCP
Lisa K Moores, MD, FCCP
Amy E Morris, MD, FCCP
John J Mullon, MD, FCCP
Septimiu D Murgu, MD, FCCP
Mangala Narasimhan, DO, FCCP
Michael S Niederman, MD, FCCP
Alexander S Niven, MD, FCCP
Anne E O’Donnell, MD, FCCP
Erik C Osborn, MD
David E Ost, MD, MPH, FCCP
Ronald J Oudiz, MD, FCCP
Daniel R Ouellette, MD, MS, FCCP
Amit D Parulekar, MD, MS, FCCP
Nicholas J Pastis, MD, FCCP
Nina M Patel, MD, FCCP
Paru S Patrawalla, MD, FCCP
Jay I Peters, MD, FCCP
Barbara A Phillips, MD, MSPH, FCCP
Margaret A Pisani, MD, MS, FCCP
Janos Porszasz, MD, PhD
Whitney S Prince, MD, FCCP
Suhail Raoof, MBBS, Master FCCP
Ruben D Restrepo, RRT, FCCP
Marcos I Restrepo, MD, PhD, FCCP
Otis B Rickman, DO, FCCP
Roy D Ridgeway
Mary Ried, RN, CCRN
Linda Rogers, MD, FCCP
Mark J Rosen, MD, Master FCCP
Bernard J Roth, MD, FCCP
Ashutosh Sachdeva, MBBS, FCCP
Anthony G Saleh, MD, FCCP
Juan F Sanchez, MD, FCCP
Pralay K Sarkar, MBBS, FCCP
Lewis G Satterwhite, MD, BA, FCCP
Gregory A Schmidt, MD, FCCP
Mary Beth Scholand, MD, FCCP
David A Schulman, MD, MPH, FCCP
Brady Scott, RRT, MS, FCCP
Bernardo Selim, MD, FCCP
Curtis N Sessler, MD, FCCP
Rakesh D Shah, MD, FCCP
Ray Wes Shepherd, MD, FCCP
John H Sherner, MD, FCCP
Ariel L Shiloh, MD
Samira Shojaee, MD, FCCP
Marcos Silva Restrepo
Gerard A Silvestri, MD, MS, FCCP
Steven Q Simpson, MD, FCCP
James K Stoller, MD, MS, FCCP
Charlie Strange, MD, FCCP
Mary E Strek, MD, FCCP
William W Stringer, MD, FCCP
Eleanor M Summerhill, MD, FCCP
Maximiliano A Tamae Kakazu, MD, FCCP
Nichole T Tanner, MD, MS, FCCP
Lynn T Tanoue, MD, FCCP
Victor J Test, MD, FCCP
Arthur J Tokarczyk, MD, FCCP
Alain Tremblay, MD, FCCP
Adey Tsegaye, MD, FCCP
Anil Vachani, MD, FCCP
Momen M Wahidi, MD, MBA, FCCP
Keith M Wille, MD, FCCP
Lisa F Wolfe, MD
Richard G Wunderink, MD, FCCP
Lonny B Yarmus, DO, FCCP
Kazuhiro Yasufuku, MD, PhD, FCCP
Gulrukh Zaidi, MD, FCCP
David Zielinski, MD, FCCP
Everyone who attended CHEST Annual Meeting 2018 is a winner, but we would like to call out the winners participating in CHEST’s special categories of awards and events. Congratulations to all!
ANNUAL CHEST AWARDS
Master FCCP
David Gutterman, MD, Master FCCP
Distinguished Service Award
David Gutterman, MD, Master FCCP
College Medalist Award
Ghada Bourjeily, MD, FCCP
Master Clinician Educator
Lisa Moores, MD, FCCP
Early Career Clinician Educator
Amy Morris, MD, FCCP
Alfred Soffer Award for Editorial Excellence
Jean Rice
Presidential Citation
Darcy Marciniuk, MD, FCCP
Presidential Citation
D. Robert McCaffree, MD, Master FCCP
HONOR LECTURES AND MEMORIAL AWARDS
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture Accelerated Aging in COPD and Its Comorbidities: Novel Therapeutic Targets
Peter Barnes, MD, Master FCCP
The lecture is generously funded by the CHEST Foundation.
Distinguished Scientist Honor Lecture in Cardiopulmonary Physiology
Understanding Diaphragm Performance: The Role of Ultrasound
F. Dennis McCool, MD, FCCP
The lecture is generously funded by the CHEST Foundation.
Presidential Honor Lecture
Asthma: Past, Present, and Future
Jay Peters, MD, FCCP
Thomas L. Petty, MD, Master FCCP Memorial Lecture
Recent Developments in Pulmonary Rehabilitation and Long-Term Oxygen Therapy: Would Tom Petty be Pleased?
Richard Casaburi, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation
Saving Lives…One Ventilator at a Time - HMV in 2018 and Beyond
Douglas McKim, MD, FCCP
The Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation is generously supported by International Ventilator Users Network of Post-Polio Health International and the CHEST Foundation.
Pasquale Ciaglia Memorial Lecture in Interventional Medicine
Evolution of Endobronchial Ultrasound: From Diagnostics to Therapeutics
Kazuhiro Yasufuku, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Roger C. Bone Memorial Lecture in Critical Care
Methylprednisolone in ARDS: A Highly Effective Treatment. How it Works, How to Use it
G. Umberto Meduri, MD
The lecture is generously funded by the CHEST Foundation.
CHEST FOUNDATION GRANT WINNERS
Distinguished Scholar
Robert C. Hyzy, MD, FCCP
Eli Lilly and Company Distinguished Scholar in Critical Care MedicineGrant Title: The Use of Electrical Impedance Tomography to Assess Mechanical Ventilation in Acute Respiratory Distress Syndrome
This grant is made possible due to the philanthropic support from Eli Lilly and Company.
Community Service Grantees
Deborah Haisch, MD
Columbia University Medical Center – New York, NY
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: East African Training Initiative in Pulmonary and Critical Care Medicine
Pamela Garrett, CCRN, MN
Gwinnett Medical Center – Lawrenceville, GA
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Breathe Better Gwinnett
Phillip Sheridan
Mobile Care Chicago – Chicago, IL
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Home Environment Education for Children with Asthma
These grants are supported in full by the CHEST Foundation.
Research Grant Winners
Ayodeji Adegunsoye, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Impact of Telomere Length on Pulmonary Fibrosis Clusters Across Diverse Racial Cohorts
Justin Oldham, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Plasma Biomarkers to Predict Outcomes and Treatment Response in Patients with Pulmonary Fibrosis
These grants above are supported by Boehringer Ingelheim Pharmaceuticals, Inc and Genentech.
Jacob Brenner, MD, PhD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Ambulatory Cuirass Ventilation for Relief of Exertional Dyspnea in Severe COPD Patients
William Zhang, MD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Pulmonary Iron Overload as a Novel COPD Endotype
These grants above are supported by AstraZeneca LP and Sunovion Pharmaceuticals Inc.
Margaret Bublitz, PhD
CHEST Foundation Research Grant in Women’s Lung Health
Grant Title: Sex as a Predictor of Sleep-Disordered Breathing and Its Consequences in Pregnancy
This grant is supported in full by the CHEST Foundation.
Tim Morris, MD, FCCP
CHEST Foundation Research Grant in Venous Thromboembolism
Grant Title: Long-term Follow-up of Acute Pulmonary Embolism
This grant is supported in full by the CHEST Foundation.
Monica Mukherjee, MD, MPH
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Grant Title: Exercise Provocation in the Noninvasive Detection of Occult Right Ventricular Dysfunction and Emerging Pulmonary Hypertension in Systemic Sclerosis
This grant is supported in full by the CHEST Foundation.
Don Sanders, MD, MS
CHEST Foundation Research Grant in Cystic Fibrosis
Grant Title: Whole-genome Shotgun Sequencing of Oropharyngeal Swabs in Infants With CF
This grant is supported by Vertex Pharmaceuticals.
Imran Sulaiman, MD, PhD
CHEST Foundation Research Grant in Nontuberculosis Mycobacteria Diseases
Grant Title: Lower Airway Microbiota Signatures Associated W ith Impaired Immune Response in Non-Tuberculous Mycobacterium
This grant is supported by Insmed.
Samira Shojaee, MD, MPH, FCCP
CHEST Foundation Research Grant in Lung Cancer
Grant Title: Extracellular Vesicle miRNA as a Biomarker in Malignant Pleural Effusion
This grant is supported in full by the CHEST Foundation.
Anna Volerman, MD
CHEST Foundation Research Grant in Severe Asthma
Grant Title: A Randomized Clinical Trial Evaluating the Effectiveness of Virtual Teach-to-Goal(TM) Education versus Brief Intervention for Children with Severe Asthma
This grant is supported by AstraZeneca LP.
ABSTRACT AND CASE REPORT WINNERS
Alfred Soffer Research Award Winners
Clauden Louis, MD: Left ventricular assist devices in Intermacs 1 acute cardiogenic shock patients
Babith J. Mankidy, MBBS, FCCP: Reduction in in-hospital cardiac arrest with early interventions in the emergency department and non-ICU units by a novel approach of rapid response teams and mobile ICU management
Young Investigator Award Winners
Fayez Kheir, MD, MSc: Intrapleural tissue plasminogen activator and deoxyribonuclease therapy vs early medical thoracoscopy for treatment of pleural infection: a randomized clinical trial
Michael Rosman, MD: The utility of end tidal CO2 (ETCO2) monitoring during in-hospital cardiac arrest to predict return of spontaneous circulation
Top 5 Abstract Poster Winners
Neha Agarwal, MD: The 3 wishes project: a feasible intervention to improve end of life care in the ICU at UCLA
Hiroaki Harada, MD: Usefulness of comprehensive preoperative pulmonary rehabilitation program including intensive nutritional support concomitant with physical exercise through an interdisciplinary team approach
Joseph M. Carrington, DO, MHA: Targeting the trans-IL-6 signaling pathway to reduce agriculture organic dust exposure-induced airway inflammation in mice
Yu Kuang Lai, MBBCh: The utility of parametric response mapping in pulmonary graft vs host disease following hematopoietic stem cell transplant
Top Abstract Poster Finalists
Ligia M. Puiu, MD, PhD, FCCP: Association between echocardiographic and lipid parameters to workers in the metalliferous mines
Kush R. Dholakia, MD: Colloids vs crystalloids for postoperative resuscitation in patients undergoing off-pump coronary artery bypass surgery
Kulothungan Gunasekaran, MD, MBBS: Risk of VTE in idiopathic pulmonary fibrosis: a systematic review
Laura B. Sutton, PharmD: Ease and correct use of Ellipta by age in patients with asthma and COPD
Ankur Mogla, MD: To assess the utilization of pulmonary function testing for perioperative respiratory complications in bariatric surgery patients
Ali Ammar: Tracheostomy and admission diagnosis as predictors for an extended length of stay (ELOS)
Charlene Kalani, PharmD: Efficacy and safety of direct oral anticoagulants (DOACS) in morbidly obese patients
Jonghoo Lee, MD: Performances of modified CRB-65 score compared to SIRS and QSOFA as a rapid screening tool for sepsis among infected patients in initial emergency department: a propensity score matching study
Frank J. Trudo, MD, FCCP: Clinical burden of eosinophilic COPD
Elise L. Stephenson, MD: Vitamin C and point of care glucose measurements: a retrospective, observational study
Faisal Siddiqi, MD: Implementation of an early mobility program in the medical ICU
Eileen Harder, MD: Connective tissue disease-associated pulmonary arterial hypertension hospitalizations from 2001-2014
Sophie Korzan, MD: Exhaled nitric oxide and asthma-COPD overlap in patients hospitalized with exacerbations of airway disease: preliminary observations
Andreas Grove, MD: MicroRNA (MIRNA) and biological markers discriminate between normotensive and prehypertensive young men in hypobaric hypoxic environments
Snigdha Nutalapati, MBBS: Large cell neuroendocrine cancer of the lung: SEER 2004-2014 analysis
Anubhav Jain, MBBS: Survival benefit of beta-blockers in patients hospitalized for acute exacerbation of COPD
Case Report Slide Winners
Ze Ying Tan: All that wheezes is not asthma
Jason Lam: Pulmonary mucor mycetoma
Adam Young: Nonresolving pneumonia and cyclic fevers in an immunocompetent patient
Ritu Modi: Histopathological misdiagnosis of pulmonary coccidiodes
Argun Can: A rare inborn error of fatty acid oxidation presenting with severe hyperammonemia in the ICU
Morgan Gilani: A colorful cause of cardiovascular collapse
Katie Jeans: A sweet surprise
Anthony Mattox: Unusual case of interstitial lung disease
Andrew Berglund: Pulmonary light chain deposition disease in a 29-year-old army soldier
Cristia Maysol Morales: A case report of a primary malignant melanoma of anterior mediastinum
Anthony McClafferty: Fibrosing mediastinitis and rheumatoid arthritis: an autoimmune inflammatory connection
Ahmed Munir: HIV with disseminated tularemia: a rare presentation Benjamin Garren: Mycobacterium avium complex mediastinal lymphadenitis in an immunocompetent adolescent with erosion into the airway
Robert Hilton: Obtunded with a chest mass: a case of a rare neurologic paraneoplastic syndrome,
Audra Schwalk: Mucoepidermoid carcinoma: a rare malignancy treated endobronchially
Jessica Riggs: Successful transplantation defies genetics: a case of rapidly-progressive pulmonary fibrosis due to Hermansky-Pudlak syndrome
Meghan Cirulis: Acute vasodilator testing: an opportunity to refine study design and provide precision care in pulmonary hypertension
Patrick Chan: VATS lobectomy for bronchial atresia in an adult
Andrew Mehlman: Multivessel coronary artery aneurysms presenting as myocardial ischemia
Scott Maughan: Diagnosing milliary Mycobacterium bovis from the prostate of an immunocompetent host
Adam Austin: Survived ECMO, death by BLASTO: the first reported fatal case of disseminated blastomycosis in pregnancy
Tie: Donnie Carter: Subclinical polycythemia vera presenting as extensive thrombosis due to massive transfusion, and
Lindsay Hammons: Rare case of Serratia pneumonia causing transient aplastic anemia
Paola Baskin: Novel observations during point-of-care ultrasound (POCUS) in cardiopulmonary resuscitation: a case of ultrasound-guided probe pressure to reduce esophageal insufflation during bag-valve-mask ventilator
David Dennis: Pulmonary alveolar proteinosis presenting as intracerebral nocardiosis
Rakin Choudhury: Severe asthma caused by therapy-resistant asthmatic granulomatosis
Andrew Lytle: Lung adenocarcinoma in a patient with Turcot syndrome
Chelsea Leipold: Case of a granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency disorder
Galyna Ivashchuk: Double trouble: ANCA vasculitis with concomitant IGA nephropathy presenting as massive diffuse alveolar hemorrhage and fulminant renal failure
Case Report Poster Winners
Christine Zhou: Role of transbronchial lung cryobiopsy in the diagnosis of adenocarcinoma in situ
Parin Shah: A rare case of Erdheim-Chester disease masquerading as metastatic lung cancer
Avanthika Wynn : A rare asthma mimic
Muhammad S. Ali: Severe pancolitis: a rare adverse effect of nintedanib
Brian Foster: Don’t forget to breathe: a case of hypoxemia after carotid body resection
Kelly Pennington: Intra-cardiac embolization of an inferior vena cava filter resulting in cardiac arrest
George Elkomos-Botros: Acute generalized exanthematous pustulosis presenting as distributive shock with multi-organ failure
Ashley M. Scott: Avian occupational hypersensitivity pneumonitis in a restaurant employee
Andrew Polito: Pulmonary amyloidosis: an unusual presentation of a rare disease
CHEST B-I-N-G-O WINNERS
Stella Ogake, MD
Erin E. Peterson, APRN, CNP
Megan J. Castillo, PA-C
Gretchen R. Winter, MD
Jeanette P. Brown, MD, PhD
Yu Hong Chan, MBBS
Anita Naik, DO
Gary A. Aaronson, DO, FCCP
Allison S. Cowl, MD
Kyle Halligan, MD
Palaniappan Muthappan, MD
Faizullah S. Lokhandwala, MBBS, FCCP
Jamie R. Chua, MD
Francis L. Ervin, MD, FCCP
Robyn Luper
CHEST CHALLENGE WINNER (AND RUNNER’S-UP)
Emory University (First Place)
Mirza Haider Ali, MD
Mohleen Kang, MD
Matthew Schimmel, MD
University of Michigan (Second Place)
Patrick Bradley, MD
Matthew Hensley, MD
Bonnie Wang, MD
Cleveland Clinic (Third Place)
Jorge Mirales-Estrella, MD
Apostolos Perelas, MD
Gretchen Winter, MD
2018 DISTINGUISHED CHEST EDUCATORS
Michael H Ackerman, DNSc
Sandra G Adams, MD, MS, FCCP
Doreen J Addrizzo-Harris, MD, FCCP
Cara Lyn Agerstrand, MD, BS
Jason A Akulian, MD, FCCP
Raed H Alalawi, MD, FCCP
A. Christine Argento, MD, FCCP
Robert Arntfield, MD, FCCP
Alex A Balekian, MD
Meyer S Balter, MD, FCCP
Gisela I Banauch, MD, MS, FCCP
Robert P Baughman, MD, FCCP
David G Bell, MD, FCCP
Michel A Boivin, MD, FCCP
Gabriel T Bosslet, MD, FCCP
Jean Bourbeau, MD, MS, FCCP
Ghada R Bourjeily, MD, FCCP
David L Bowton, MD, FCCM
Jack D Buckley, MD, MPH, FCCP
Marie M Budev, DO, MPH, FCCP
Kristin M Burkart, MD, MS, FCCP
Brian Carlin, MD, FCCP
Christopher L Carroll, MD, FCCP
Roberto F Casal, MD
Kevin M Chan, MD, FCCP
Subani Chandra, MD, FCCP
Ching-Fei Chang, MD
Alexander C Chen, MD
Nancy A Collop, MD, FCCP
Clayton T Cowl, MD, MS, FCCP
Angel O Coz Yataco, MD, FCCP
Gerard J Criner, MD, FCCP
Carolyn M D’Ambrosio, MD, FCCP
Mauricio Danckers, MD, FCCP
Aneesa M Das, MD, FCCP
John Davies, RRT, MA, FCCP
Zachary S DePew, MD, FCCP
Frank C Detterbeck, MD, FCCP
Naresh A. Dewan, MBBS, FCCP
Kevin C Doerschug, MD, MS, FCCP
Meagan Dubosky, RRT-ACCS
Kevin M Dushay, MD, FCCP
Eric S Edell, MD, FCCP
Jean M Elwing, MD, FCCP
William Enfinger
Michael E Ezzie, MD, FCCP
Kevin J Felner, MD, FCCP
Mark E Fenton, MD, MSc, FCCP
Jason Filopei, MD
Neil S Freedman, MD, FCCP
Laura Kathleen Frye, MD
Thomas M Fuhrman, MD, MS, FCCP
John P Gaillard, MD, FCCP
Colin T Gillespie, MD
Yonatan Y Greenstein, MD
Maritza L Groth, MD, FCCP
Keith P Guevarra, DO, FCCP
Jesse B Hall, MD, FCCP
Nicola A Hanania, MD, MBBS, FCCP
D Kyle Hogarth, MD, FCCP
Steven M Hollenberg, MD, FCCP
David W Hsia, MD, FCCP
Candace A Huebert, MD, FCCP
Robert C Hyzy, MD, FCCP
Octavian C Ioachimescu, MD, PhD, FCCP
Richard S Irwin, MD, Master FCCP
Kirk D Jones, MD
Nader Kamangar, MD, MS, FCCP
Carl A Kaplan, MD, FCCP
Brian S Kaufman, MD, FCCP
William F Kelly, MD, FCCP
Marcus P Kennedy, MD, FCCP
Sandhya Khurana, MD, FCCP
James R Klinger, MD, FCCP
Seth J Koenig, MD, FCCP
Lindsey Kreisher, RRT
Karol Kremens, MD, FCCP
Patricia A Kritek, MD, FCCP
Sunita Kumar, MD, MBBS, FCCP
Rudy P Lackner, MD, FCCP
Viera Lakticova, MD
Carla R Lamb, MD, FCCP
Hans J Lee, MD, FCCP
Peter H Lenz, MD, MEd, FCCP
Stephanie M Levine, MD, FCCP
Deborah Jo Levine, MD, MS, FCCP
Andrea Loiselle, MD
Kenneth E Lyn-Kew, MD
Michael S Machuzak, MD, FCCP
Neil R MacIntyre, MD, FCCP
Donald A Mahler, MD, FCCP
Fabien Maldonado, MD, FCCP
Atul Malhotra, MD, FCCP
Darcy D Marciniuk, MD, FCCP
Diego J Maselli Caceres, MD, FCCP
Paul H Mayo, MD, FCCP
Peter J Mazzone, MD, MPH, FCCP
John K McIlwaine, DO, MBA, FCCP
Matthew C Miles, MD, FCCP
Scott Millington, MD
Taro Minami, MD, FCCP
Lisa K Moores, MD, FCCP
Amy E Morris, MD, FCCP
John J Mullon, MD, FCCP
Septimiu D Murgu, MD, FCCP
Mangala Narasimhan, DO, FCCP
Michael S Niederman, MD, FCCP
Alexander S Niven, MD, FCCP
Anne E O’Donnell, MD, FCCP
Erik C Osborn, MD
David E Ost, MD, MPH, FCCP
Ronald J Oudiz, MD, FCCP
Daniel R Ouellette, MD, MS, FCCP
Amit D Parulekar, MD, MS, FCCP
Nicholas J Pastis, MD, FCCP
Nina M Patel, MD, FCCP
Paru S Patrawalla, MD, FCCP
Jay I Peters, MD, FCCP
Barbara A Phillips, MD, MSPH, FCCP
Margaret A Pisani, MD, MS, FCCP
Janos Porszasz, MD, PhD
Whitney S Prince, MD, FCCP
Suhail Raoof, MBBS, Master FCCP
Ruben D Restrepo, RRT, FCCP
Marcos I Restrepo, MD, PhD, FCCP
Otis B Rickman, DO, FCCP
Roy D Ridgeway
Mary Ried, RN, CCRN
Linda Rogers, MD, FCCP
Mark J Rosen, MD, Master FCCP
Bernard J Roth, MD, FCCP
Ashutosh Sachdeva, MBBS, FCCP
Anthony G Saleh, MD, FCCP
Juan F Sanchez, MD, FCCP
Pralay K Sarkar, MBBS, FCCP
Lewis G Satterwhite, MD, BA, FCCP
Gregory A Schmidt, MD, FCCP
Mary Beth Scholand, MD, FCCP
David A Schulman, MD, MPH, FCCP
Brady Scott, RRT, MS, FCCP
Bernardo Selim, MD, FCCP
Curtis N Sessler, MD, FCCP
Rakesh D Shah, MD, FCCP
Ray Wes Shepherd, MD, FCCP
John H Sherner, MD, FCCP
Ariel L Shiloh, MD
Samira Shojaee, MD, FCCP
Marcos Silva Restrepo
Gerard A Silvestri, MD, MS, FCCP
Steven Q Simpson, MD, FCCP
James K Stoller, MD, MS, FCCP
Charlie Strange, MD, FCCP
Mary E Strek, MD, FCCP
William W Stringer, MD, FCCP
Eleanor M Summerhill, MD, FCCP
Maximiliano A Tamae Kakazu, MD, FCCP
Nichole T Tanner, MD, MS, FCCP
Lynn T Tanoue, MD, FCCP
Victor J Test, MD, FCCP
Arthur J Tokarczyk, MD, FCCP
Alain Tremblay, MD, FCCP
Adey Tsegaye, MD, FCCP
Anil Vachani, MD, FCCP
Momen M Wahidi, MD, MBA, FCCP
Keith M Wille, MD, FCCP
Lisa F Wolfe, MD
Richard G Wunderink, MD, FCCP
Lonny B Yarmus, DO, FCCP
Kazuhiro Yasufuku, MD, PhD, FCCP
Gulrukh Zaidi, MD, FCCP
David Zielinski, MD, FCCP
Everyone who attended CHEST Annual Meeting 2018 is a winner, but we would like to call out the winners participating in CHEST’s special categories of awards and events. Congratulations to all!
ANNUAL CHEST AWARDS
Master FCCP
David Gutterman, MD, Master FCCP
Distinguished Service Award
David Gutterman, MD, Master FCCP
College Medalist Award
Ghada Bourjeily, MD, FCCP
Master Clinician Educator
Lisa Moores, MD, FCCP
Early Career Clinician Educator
Amy Morris, MD, FCCP
Alfred Soffer Award for Editorial Excellence
Jean Rice
Presidential Citation
Darcy Marciniuk, MD, FCCP
Presidential Citation
D. Robert McCaffree, MD, Master FCCP
HONOR LECTURES AND MEMORIAL AWARDS
Edward C. Rosenow III, MD, Master FCCP/Master Teacher Honor Lecture Accelerated Aging in COPD and Its Comorbidities: Novel Therapeutic Targets
Peter Barnes, MD, Master FCCP
The lecture is generously funded by the CHEST Foundation.
Distinguished Scientist Honor Lecture in Cardiopulmonary Physiology
Understanding Diaphragm Performance: The Role of Ultrasound
F. Dennis McCool, MD, FCCP
The lecture is generously funded by the CHEST Foundation.
Presidential Honor Lecture
Asthma: Past, Present, and Future
Jay Peters, MD, FCCP
Thomas L. Petty, MD, Master FCCP Memorial Lecture
Recent Developments in Pulmonary Rehabilitation and Long-Term Oxygen Therapy: Would Tom Petty be Pleased?
Richard Casaburi, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation
Saving Lives…One Ventilator at a Time - HMV in 2018 and Beyond
Douglas McKim, MD, FCCP
The Margaret Pfrommer Memorial Lecture in Long-term Mechanical Ventilation is generously supported by International Ventilator Users Network of Post-Polio Health International and the CHEST Foundation.
Pasquale Ciaglia Memorial Lecture in Interventional Medicine
Evolution of Endobronchial Ultrasound: From Diagnostics to Therapeutics
Kazuhiro Yasufuku, MD, PhD, FCCP
The lecture is generously funded by the CHEST Foundation.
Roger C. Bone Memorial Lecture in Critical Care
Methylprednisolone in ARDS: A Highly Effective Treatment. How it Works, How to Use it
G. Umberto Meduri, MD
The lecture is generously funded by the CHEST Foundation.
CHEST FOUNDATION GRANT WINNERS
Distinguished Scholar
Robert C. Hyzy, MD, FCCP
Eli Lilly and Company Distinguished Scholar in Critical Care MedicineGrant Title: The Use of Electrical Impedance Tomography to Assess Mechanical Ventilation in Acute Respiratory Distress Syndrome
This grant is made possible due to the philanthropic support from Eli Lilly and Company.
Community Service Grantees
Deborah Haisch, MD
Columbia University Medical Center – New York, NY
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: East African Training Initiative in Pulmonary and Critical Care Medicine
Pamela Garrett, CCRN, MN
Gwinnett Medical Center – Lawrenceville, GA
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Breathe Better Gwinnett
Phillip Sheridan
Mobile Care Chicago – Chicago, IL
CHEST Foundation Community Service Grant Honoring D. Robert McCaffree, MD, Master FCCP
Grant Title: Home Environment Education for Children with Asthma
These grants are supported in full by the CHEST Foundation.
Research Grant Winners
Ayodeji Adegunsoye, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Impact of Telomere Length on Pulmonary Fibrosis Clusters Across Diverse Racial Cohorts
Justin Oldham, MD, MS
Research Grant in Pulmonary Fibrosis
Grant Title: Plasma Biomarkers to Predict Outcomes and Treatment Response in Patients with Pulmonary Fibrosis
These grants above are supported by Boehringer Ingelheim Pharmaceuticals, Inc and Genentech.
Jacob Brenner, MD, PhD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Ambulatory Cuirass Ventilation for Relief of Exertional Dyspnea in Severe COPD Patients
William Zhang, MD
Research Grant in Chronic Obstructive Pulmonary Disease
Grant Title: Pulmonary Iron Overload as a Novel COPD Endotype
These grants above are supported by AstraZeneca LP and Sunovion Pharmaceuticals Inc.
Margaret Bublitz, PhD
CHEST Foundation Research Grant in Women’s Lung Health
Grant Title: Sex as a Predictor of Sleep-Disordered Breathing and Its Consequences in Pregnancy
This grant is supported in full by the CHEST Foundation.
Tim Morris, MD, FCCP
CHEST Foundation Research Grant in Venous Thromboembolism
Grant Title: Long-term Follow-up of Acute Pulmonary Embolism
This grant is supported in full by the CHEST Foundation.
Monica Mukherjee, MD, MPH
CHEST Foundation Research Grant in Pulmonary Arterial Hypertension
Grant Title: Exercise Provocation in the Noninvasive Detection of Occult Right Ventricular Dysfunction and Emerging Pulmonary Hypertension in Systemic Sclerosis
This grant is supported in full by the CHEST Foundation.
Don Sanders, MD, MS
CHEST Foundation Research Grant in Cystic Fibrosis
Grant Title: Whole-genome Shotgun Sequencing of Oropharyngeal Swabs in Infants With CF
This grant is supported by Vertex Pharmaceuticals.
Imran Sulaiman, MD, PhD
CHEST Foundation Research Grant in Nontuberculosis Mycobacteria Diseases
Grant Title: Lower Airway Microbiota Signatures Associated W ith Impaired Immune Response in Non-Tuberculous Mycobacterium
This grant is supported by Insmed.
Samira Shojaee, MD, MPH, FCCP
CHEST Foundation Research Grant in Lung Cancer
Grant Title: Extracellular Vesicle miRNA as a Biomarker in Malignant Pleural Effusion
This grant is supported in full by the CHEST Foundation.
Anna Volerman, MD
CHEST Foundation Research Grant in Severe Asthma
Grant Title: A Randomized Clinical Trial Evaluating the Effectiveness of Virtual Teach-to-Goal(TM) Education versus Brief Intervention for Children with Severe Asthma
This grant is supported by AstraZeneca LP.
ABSTRACT AND CASE REPORT WINNERS
Alfred Soffer Research Award Winners
Clauden Louis, MD: Left ventricular assist devices in Intermacs 1 acute cardiogenic shock patients
Babith J. Mankidy, MBBS, FCCP: Reduction in in-hospital cardiac arrest with early interventions in the emergency department and non-ICU units by a novel approach of rapid response teams and mobile ICU management
Young Investigator Award Winners
Fayez Kheir, MD, MSc: Intrapleural tissue plasminogen activator and deoxyribonuclease therapy vs early medical thoracoscopy for treatment of pleural infection: a randomized clinical trial
Michael Rosman, MD: The utility of end tidal CO2 (ETCO2) monitoring during in-hospital cardiac arrest to predict return of spontaneous circulation
Top 5 Abstract Poster Winners
Neha Agarwal, MD: The 3 wishes project: a feasible intervention to improve end of life care in the ICU at UCLA
Hiroaki Harada, MD: Usefulness of comprehensive preoperative pulmonary rehabilitation program including intensive nutritional support concomitant with physical exercise through an interdisciplinary team approach
Joseph M. Carrington, DO, MHA: Targeting the trans-IL-6 signaling pathway to reduce agriculture organic dust exposure-induced airway inflammation in mice
Yu Kuang Lai, MBBCh: The utility of parametric response mapping in pulmonary graft vs host disease following hematopoietic stem cell transplant
Top Abstract Poster Finalists
Ligia M. Puiu, MD, PhD, FCCP: Association between echocardiographic and lipid parameters to workers in the metalliferous mines
Kush R. Dholakia, MD: Colloids vs crystalloids for postoperative resuscitation in patients undergoing off-pump coronary artery bypass surgery
Kulothungan Gunasekaran, MD, MBBS: Risk of VTE in idiopathic pulmonary fibrosis: a systematic review
Laura B. Sutton, PharmD: Ease and correct use of Ellipta by age in patients with asthma and COPD
Ankur Mogla, MD: To assess the utilization of pulmonary function testing for perioperative respiratory complications in bariatric surgery patients
Ali Ammar: Tracheostomy and admission diagnosis as predictors for an extended length of stay (ELOS)
Charlene Kalani, PharmD: Efficacy and safety of direct oral anticoagulants (DOACS) in morbidly obese patients
Jonghoo Lee, MD: Performances of modified CRB-65 score compared to SIRS and QSOFA as a rapid screening tool for sepsis among infected patients in initial emergency department: a propensity score matching study
Frank J. Trudo, MD, FCCP: Clinical burden of eosinophilic COPD
Elise L. Stephenson, MD: Vitamin C and point of care glucose measurements: a retrospective, observational study
Faisal Siddiqi, MD: Implementation of an early mobility program in the medical ICU
Eileen Harder, MD: Connective tissue disease-associated pulmonary arterial hypertension hospitalizations from 2001-2014
Sophie Korzan, MD: Exhaled nitric oxide and asthma-COPD overlap in patients hospitalized with exacerbations of airway disease: preliminary observations
Andreas Grove, MD: MicroRNA (MIRNA) and biological markers discriminate between normotensive and prehypertensive young men in hypobaric hypoxic environments
Snigdha Nutalapati, MBBS: Large cell neuroendocrine cancer of the lung: SEER 2004-2014 analysis
Anubhav Jain, MBBS: Survival benefit of beta-blockers in patients hospitalized for acute exacerbation of COPD
Case Report Slide Winners
Ze Ying Tan: All that wheezes is not asthma
Jason Lam: Pulmonary mucor mycetoma
Adam Young: Nonresolving pneumonia and cyclic fevers in an immunocompetent patient
Ritu Modi: Histopathological misdiagnosis of pulmonary coccidiodes
Argun Can: A rare inborn error of fatty acid oxidation presenting with severe hyperammonemia in the ICU
Morgan Gilani: A colorful cause of cardiovascular collapse
Katie Jeans: A sweet surprise
Anthony Mattox: Unusual case of interstitial lung disease
Andrew Berglund: Pulmonary light chain deposition disease in a 29-year-old army soldier
Cristia Maysol Morales: A case report of a primary malignant melanoma of anterior mediastinum
Anthony McClafferty: Fibrosing mediastinitis and rheumatoid arthritis: an autoimmune inflammatory connection
Ahmed Munir: HIV with disseminated tularemia: a rare presentation Benjamin Garren: Mycobacterium avium complex mediastinal lymphadenitis in an immunocompetent adolescent with erosion into the airway
Robert Hilton: Obtunded with a chest mass: a case of a rare neurologic paraneoplastic syndrome,
Audra Schwalk: Mucoepidermoid carcinoma: a rare malignancy treated endobronchially
Jessica Riggs: Successful transplantation defies genetics: a case of rapidly-progressive pulmonary fibrosis due to Hermansky-Pudlak syndrome
Meghan Cirulis: Acute vasodilator testing: an opportunity to refine study design and provide precision care in pulmonary hypertension
Patrick Chan: VATS lobectomy for bronchial atresia in an adult
Andrew Mehlman: Multivessel coronary artery aneurysms presenting as myocardial ischemia
Scott Maughan: Diagnosing milliary Mycobacterium bovis from the prostate of an immunocompetent host
Adam Austin: Survived ECMO, death by BLASTO: the first reported fatal case of disseminated blastomycosis in pregnancy
Tie: Donnie Carter: Subclinical polycythemia vera presenting as extensive thrombosis due to massive transfusion, and
Lindsay Hammons: Rare case of Serratia pneumonia causing transient aplastic anemia
Paola Baskin: Novel observations during point-of-care ultrasound (POCUS) in cardiopulmonary resuscitation: a case of ultrasound-guided probe pressure to reduce esophageal insufflation during bag-valve-mask ventilator
David Dennis: Pulmonary alveolar proteinosis presenting as intracerebral nocardiosis
Rakin Choudhury: Severe asthma caused by therapy-resistant asthmatic granulomatosis
Andrew Lytle: Lung adenocarcinoma in a patient with Turcot syndrome
Chelsea Leipold: Case of a granulomatous-lymphocytic interstitial lung disease in a patient with common variable immunodeficiency disorder
Galyna Ivashchuk: Double trouble: ANCA vasculitis with concomitant IGA nephropathy presenting as massive diffuse alveolar hemorrhage and fulminant renal failure
Case Report Poster Winners
Christine Zhou: Role of transbronchial lung cryobiopsy in the diagnosis of adenocarcinoma in situ
Parin Shah: A rare case of Erdheim-Chester disease masquerading as metastatic lung cancer
Avanthika Wynn : A rare asthma mimic
Muhammad S. Ali: Severe pancolitis: a rare adverse effect of nintedanib
Brian Foster: Don’t forget to breathe: a case of hypoxemia after carotid body resection
Kelly Pennington: Intra-cardiac embolization of an inferior vena cava filter resulting in cardiac arrest
George Elkomos-Botros: Acute generalized exanthematous pustulosis presenting as distributive shock with multi-organ failure
Ashley M. Scott: Avian occupational hypersensitivity pneumonitis in a restaurant employee
Andrew Polito: Pulmonary amyloidosis: an unusual presentation of a rare disease
CHEST B-I-N-G-O WINNERS
Stella Ogake, MD
Erin E. Peterson, APRN, CNP
Megan J. Castillo, PA-C
Gretchen R. Winter, MD
Jeanette P. Brown, MD, PhD
Yu Hong Chan, MBBS
Anita Naik, DO
Gary A. Aaronson, DO, FCCP
Allison S. Cowl, MD
Kyle Halligan, MD
Palaniappan Muthappan, MD
Faizullah S. Lokhandwala, MBBS, FCCP
Jamie R. Chua, MD
Francis L. Ervin, MD, FCCP
Robyn Luper
CHEST CHALLENGE WINNER (AND RUNNER’S-UP)
Emory University (First Place)
Mirza Haider Ali, MD
Mohleen Kang, MD
Matthew Schimmel, MD
University of Michigan (Second Place)
Patrick Bradley, MD
Matthew Hensley, MD
Bonnie Wang, MD
Cleveland Clinic (Third Place)
Jorge Mirales-Estrella, MD
Apostolos Perelas, MD
Gretchen Winter, MD
2018 DISTINGUISHED CHEST EDUCATORS
Michael H Ackerman, DNSc
Sandra G Adams, MD, MS, FCCP
Doreen J Addrizzo-Harris, MD, FCCP
Cara Lyn Agerstrand, MD, BS
Jason A Akulian, MD, FCCP
Raed H Alalawi, MD, FCCP
A. Christine Argento, MD, FCCP
Robert Arntfield, MD, FCCP
Alex A Balekian, MD
Meyer S Balter, MD, FCCP
Gisela I Banauch, MD, MS, FCCP
Robert P Baughman, MD, FCCP
David G Bell, MD, FCCP
Michel A Boivin, MD, FCCP
Gabriel T Bosslet, MD, FCCP
Jean Bourbeau, MD, MS, FCCP
Ghada R Bourjeily, MD, FCCP
David L Bowton, MD, FCCM
Jack D Buckley, MD, MPH, FCCP
Marie M Budev, DO, MPH, FCCP
Kristin M Burkart, MD, MS, FCCP
Brian Carlin, MD, FCCP
Christopher L Carroll, MD, FCCP
Roberto F Casal, MD
Kevin M Chan, MD, FCCP
Subani Chandra, MD, FCCP
Ching-Fei Chang, MD
Alexander C Chen, MD
Nancy A Collop, MD, FCCP
Clayton T Cowl, MD, MS, FCCP
Angel O Coz Yataco, MD, FCCP
Gerard J Criner, MD, FCCP
Carolyn M D’Ambrosio, MD, FCCP
Mauricio Danckers, MD, FCCP
Aneesa M Das, MD, FCCP
John Davies, RRT, MA, FCCP
Zachary S DePew, MD, FCCP
Frank C Detterbeck, MD, FCCP
Naresh A. Dewan, MBBS, FCCP
Kevin C Doerschug, MD, MS, FCCP
Meagan Dubosky, RRT-ACCS
Kevin M Dushay, MD, FCCP
Eric S Edell, MD, FCCP
Jean M Elwing, MD, FCCP
William Enfinger
Michael E Ezzie, MD, FCCP
Kevin J Felner, MD, FCCP
Mark E Fenton, MD, MSc, FCCP
Jason Filopei, MD
Neil S Freedman, MD, FCCP
Laura Kathleen Frye, MD
Thomas M Fuhrman, MD, MS, FCCP
John P Gaillard, MD, FCCP
Colin T Gillespie, MD
Yonatan Y Greenstein, MD
Maritza L Groth, MD, FCCP
Keith P Guevarra, DO, FCCP
Jesse B Hall, MD, FCCP
Nicola A Hanania, MD, MBBS, FCCP
D Kyle Hogarth, MD, FCCP
Steven M Hollenberg, MD, FCCP
David W Hsia, MD, FCCP
Candace A Huebert, MD, FCCP
Robert C Hyzy, MD, FCCP
Octavian C Ioachimescu, MD, PhD, FCCP
Richard S Irwin, MD, Master FCCP
Kirk D Jones, MD
Nader Kamangar, MD, MS, FCCP
Carl A Kaplan, MD, FCCP
Brian S Kaufman, MD, FCCP
William F Kelly, MD, FCCP
Marcus P Kennedy, MD, FCCP
Sandhya Khurana, MD, FCCP
James R Klinger, MD, FCCP
Seth J Koenig, MD, FCCP
Lindsey Kreisher, RRT
Karol Kremens, MD, FCCP
Patricia A Kritek, MD, FCCP
Sunita Kumar, MD, MBBS, FCCP
Rudy P Lackner, MD, FCCP
Viera Lakticova, MD
Carla R Lamb, MD, FCCP
Hans J Lee, MD, FCCP
Peter H Lenz, MD, MEd, FCCP
Stephanie M Levine, MD, FCCP
Deborah Jo Levine, MD, MS, FCCP
Andrea Loiselle, MD
Kenneth E Lyn-Kew, MD
Michael S Machuzak, MD, FCCP
Neil R MacIntyre, MD, FCCP
Donald A Mahler, MD, FCCP
Fabien Maldonado, MD, FCCP
Atul Malhotra, MD, FCCP
Darcy D Marciniuk, MD, FCCP
Diego J Maselli Caceres, MD, FCCP
Paul H Mayo, MD, FCCP
Peter J Mazzone, MD, MPH, FCCP
John K McIlwaine, DO, MBA, FCCP
Matthew C Miles, MD, FCCP
Scott Millington, MD
Taro Minami, MD, FCCP
Lisa K Moores, MD, FCCP
Amy E Morris, MD, FCCP
John J Mullon, MD, FCCP
Septimiu D Murgu, MD, FCCP
Mangala Narasimhan, DO, FCCP
Michael S Niederman, MD, FCCP
Alexander S Niven, MD, FCCP
Anne E O’Donnell, MD, FCCP
Erik C Osborn, MD
David E Ost, MD, MPH, FCCP
Ronald J Oudiz, MD, FCCP
Daniel R Ouellette, MD, MS, FCCP
Amit D Parulekar, MD, MS, FCCP
Nicholas J Pastis, MD, FCCP
Nina M Patel, MD, FCCP
Paru S Patrawalla, MD, FCCP
Jay I Peters, MD, FCCP
Barbara A Phillips, MD, MSPH, FCCP
Margaret A Pisani, MD, MS, FCCP
Janos Porszasz, MD, PhD
Whitney S Prince, MD, FCCP
Suhail Raoof, MBBS, Master FCCP
Ruben D Restrepo, RRT, FCCP
Marcos I Restrepo, MD, PhD, FCCP
Otis B Rickman, DO, FCCP
Roy D Ridgeway
Mary Ried, RN, CCRN
Linda Rogers, MD, FCCP
Mark J Rosen, MD, Master FCCP
Bernard J Roth, MD, FCCP
Ashutosh Sachdeva, MBBS, FCCP
Anthony G Saleh, MD, FCCP
Juan F Sanchez, MD, FCCP
Pralay K Sarkar, MBBS, FCCP
Lewis G Satterwhite, MD, BA, FCCP
Gregory A Schmidt, MD, FCCP
Mary Beth Scholand, MD, FCCP
David A Schulman, MD, MPH, FCCP
Brady Scott, RRT, MS, FCCP
Bernardo Selim, MD, FCCP
Curtis N Sessler, MD, FCCP
Rakesh D Shah, MD, FCCP
Ray Wes Shepherd, MD, FCCP
John H Sherner, MD, FCCP
Ariel L Shiloh, MD
Samira Shojaee, MD, FCCP
Marcos Silva Restrepo
Gerard A Silvestri, MD, MS, FCCP
Steven Q Simpson, MD, FCCP
James K Stoller, MD, MS, FCCP
Charlie Strange, MD, FCCP
Mary E Strek, MD, FCCP
William W Stringer, MD, FCCP
Eleanor M Summerhill, MD, FCCP
Maximiliano A Tamae Kakazu, MD, FCCP
Nichole T Tanner, MD, MS, FCCP
Lynn T Tanoue, MD, FCCP
Victor J Test, MD, FCCP
Arthur J Tokarczyk, MD, FCCP
Alain Tremblay, MD, FCCP
Adey Tsegaye, MD, FCCP
Anil Vachani, MD, FCCP
Momen M Wahidi, MD, MBA, FCCP
Keith M Wille, MD, FCCP
Lisa F Wolfe, MD
Richard G Wunderink, MD, FCCP
Lonny B Yarmus, DO, FCCP
Kazuhiro Yasufuku, MD, PhD, FCCP
Gulrukh Zaidi, MD, FCCP
David Zielinski, MD, FCCP
Use of ECMO in the management of influenza-associated ARDS
Now that we are in the midst of flu season, many discussions regarding the management of patients with influenza virus infections are ensuing. While prevention is always preferable, and we encourage everyone to get vaccinated, influenza remains a rapidly widespread infection. In the United States during last year’s flu season (2017-18), there was an estimated 49 million cases of influenza, 960,000 hospitalizations, and 79,000 deaths. Approximately 86% of all deaths were estimated to occur in those aged 65 and older (Centers for Disease Control and Prevention webpage on Burden of Influenza).

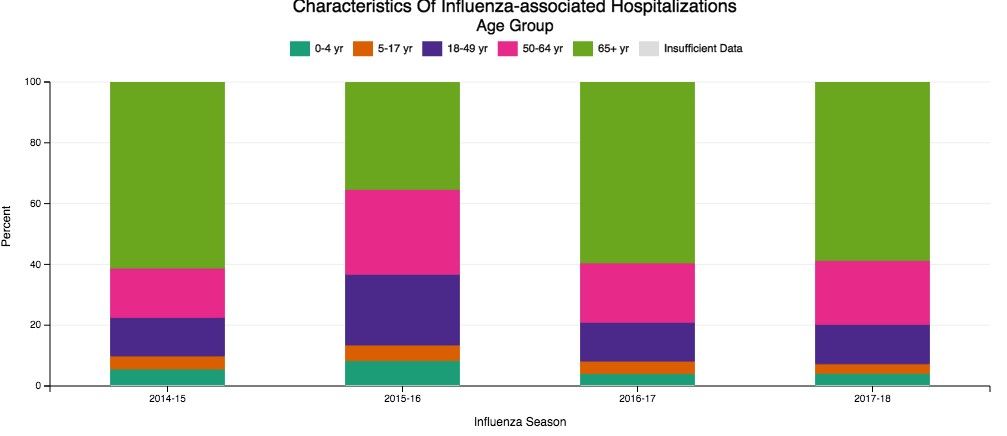
Despite our best efforts, there are inevitable times when some patients become ill enough to require hospitalization. Patients aged 65 and older make up the overwhelming majority of patients with influenza who eventually require hospitalization (Fig 1) (The Centers for Disease Control and Prevention FluView Database). Comorbidities also confer higher risk for more severe illness and potential hospitalization irrespective of age (Fig 2). In children with known medical conditions, asthma confers highest risk of hospitalization, as 27% of those with asthma were hospitalized after developing the flu. In adults, 52% of those with cardiovascular disease and 30% of adult patients with chronic lung disease who were confirmed to have influenza required hospitalization for treatment (Fig 2, The Centers for Disease Control and Prevention FluView Database).
The most severe cases of influenza can require ICU care and advanced management of respiratory failure as a result of the acute respiratory distress syndrome (ARDS). The lungs suffer significant injury due to the viral infection, and they lose their ability to effectively oxygenate the blood. Secondary bacterial infections can also occur as a complication, which compounds the injury. Given the fact that so many patients have significant comorbidities and are of advanced age, it is reasonable to expect that a fair proportion of those with influenza would develop respiratory failure as a consequence. For some of these patients, the hypoxemia that develops as a result of the lung injury can be exceptionally challenging to manage. In extreme cases, conventional ventilator management is insufficient, and the need for additional, advanced therapies arise.
Studies of VV ECMO in severe influenza
ECMO (extracorporeal membrane oxygenation) is a treatment that has been employed to help support patients with severe hypoxemic respiratory failure while their lungs recover from acute injury. Venovenous (VV) ECMO requires peripheral insertion of large cannulae into the venous system to take deoxygenated blood, deliver it through the membrane oxygenator and return the oxygenated blood back to the venous system. In simplest terms, the membrane of ECMO circuit serves as a substitute for the gas exchange function of the lungs and provides the oxygenation that the injured alveoli of the lung are unable to provide. The overall intent is to have the external ECMO circuit do all of the gas exchange work while the lungs heal.
Much research has been done on VV ECMO as an adjunct or salvage therapy in patients with refractory hypoxemic respiratory failure due to ARDS. Historical and recent studies have shown that approximately 60% of patients with ARDS have viral (approximately 20%) or bacterial (approximately 40%) pneumonia as the underlying cause (Zapol, et al. JAMA. 1979; 242[20]:2193; Combes A, et al. N Engl J Med. 2018;378:1965). Naturally, given the frequency of infection as a cause for ARDS, and the severity of illness that can develop with influenza infection in particular, an interest has arisen in the applicability of ECMO in cases of severe influenza-related ARDS.
In 2009, during the H1N1 influenza pandemic, the ANZ ECMO investigators in Australia and New Zealand described a 78% survival rate for their patients with severe H1N1 associated ARDS treated with VV ECMO between June and August of that year (Davies A, et al. JAMA. 2009;302[17]:1888). The eagerly awaited results of the randomized, controlled CESAR trial (Peek G, et al. Lancet. 2009;374:1351) that studied patients aged 18 to 64 with severe, refractory respiratory failure transferred to a specialized center for ECMO care had additional impact in catalyzing interest in ECMO use. This trial showed improved survival with ECMO (63% in ECMO vs 47% control, RR 0.69; 95% CI 0.05-0.97 P=.03) with a gain of 0.03 QALY (quality-adjusted life years) with additional cost of 40,000 pounds sterling. However, a major critique is that 24% of patients transferred to the specialized center never were treated with ECMO. Significantly, there was incomplete follow-up data on nearly half of the patients, as well. Many conclude that the survival benefit seen in this study may be more reflective of the expertise in respiratory failure management (especially as it relates to lung protective ventilation) at this center than therapy with ECMO itself.
Additional cohort studies in the United Kingdom (Noah MA, et al. JAMA. 2011;306[15]:1659) and Italy (Pappalardo F, et al. Intensive Care Med. 2013;39[2]:275) showed approximately 70% in-hospital survival rates for patients with H1N1 influenza transferred to a specialized ECMO center and treated with ECMO.
Nonetheless, the information gained from the observational data from ANZ ECMO, along with data published in European cohort studies and the randomized controlled CESAR trial after the 2009 H1N1 influenza pandemic, greatly contributed to the rise in use of ECMO for refractory ARDS due to influenza. Subsequently, there has been a rapid establishment and expansion of ECMO centers over the past decade, primarily to meet the anticipated demands of treating severe influenza-related ARDS.

The recently published EOLIA trial (Combes A, et al. N Engl J Med. 2018;378:1965) was designed to study the benefit of VV ECMO vs conventional mechanical ventilation in ARDS and demonstrated an 11% absolute reduction in 60-day mortality, which did not reach statistical significance. Like the CESAR trial, there are critiques of the outcome, especially as it relates to stopping the trial early due to the inability to show a significant benefit of VV ECMO over mechanical ventilation.
All of the aforementioned studies evaluated adults under age 65. Interestingly, there are no specific age contraindications for the use of ECMO (ELSO Guidelines for Cardiopulmonary Extracorporeal Life Support, Extracorporeal Life Support Organization, Version 1.4 August 2017), but many consider older age as a risk for poor outcome. Approximately 2,300 adult patients in the United States have been treated with ECMO for respiratory failure each year, and only 10% of those are over age 65 (CMS Changes in ECMO Reimbursements – ELSO Report). The outcome benefit of ECMO for a relatively healthy patient over age 65 is not known, as those patients have not been evaluated in studies thus far. When comparison to data from decades ago is made, one must keep in mind that populations worldwide are living longer, and a continued increase in number of adults over the age 65 is expected.
While the overall interpretation of the outcomes of studies of ECMO may be fraught with controversy, there is little debate that providing care for patients with refractory respiratory failure in centers that provide high-level skill and expertise in management of respiratory failure has a clear benefit, irrespective of whether the patient eventually receives therapy with ECMO. What is also clear is that ECMO is costly, with per-patient costs demonstrated to be at least double that of those receiving mechanical ventilation alone (Peek G, et al. Lancet. 2009;374:1351). This substantial cost associated with ECMO cannot be ignored in today’s era of value-based care.
Fortuitously, CMS recently released new DRG reimbursement scales for the use of ECMO effective Oct 1, 2018. VV ECMO could have as much as a 70% reduction in reimbursement, and many insurance companies are expected to follow suit (CMS Changes in ECMO Reimbursements –ELSO Report). Only time will tell what impact this, along with the current evidence, will have on long-term provision of ECMO care for our sickest of patients with influenza and associated respiratory illnesses.
Dr. Tatem is with the Division of Pulmonary and Critical Care Medicine, Department of Medicine, Henry Ford Hospital, Detroit, Michigan.
Now that we are in the midst of flu season, many discussions regarding the management of patients with influenza virus infections are ensuing. While prevention is always preferable, and we encourage everyone to get vaccinated, influenza remains a rapidly widespread infection. In the United States during last year’s flu season (2017-18), there was an estimated 49 million cases of influenza, 960,000 hospitalizations, and 79,000 deaths. Approximately 86% of all deaths were estimated to occur in those aged 65 and older (Centers for Disease Control and Prevention webpage on Burden of Influenza).
Despite our best efforts, there are inevitable times when some patients become ill enough to require hospitalization. Patients aged 65 and older make up the overwhelming majority of patients with influenza who eventually require hospitalization (Fig 1) (The Centers for Disease Control and Prevention FluView Database). Comorbidities also confer higher risk for more severe illness and potential hospitalization irrespective of age (Fig 2). In children with known medical conditions, asthma confers highest risk of hospitalization, as 27% of those with asthma were hospitalized after developing the flu. In adults, 52% of those with cardiovascular disease and 30% of adult patients with chronic lung disease who were confirmed to have influenza required hospitalization for treatment (Fig 2, The Centers for Disease Control and Prevention FluView Database).
The most severe cases of influenza can require ICU care and advanced management of respiratory failure as a result of the acute respiratory distress syndrome (ARDS). The lungs suffer significant injury due to the viral infection, and they lose their ability to effectively oxygenate the blood. Secondary bacterial infections can also occur as a complication, which compounds the injury. Given the fact that so many patients have significant comorbidities and are of advanced age, it is reasonable to expect that a fair proportion of those with influenza would develop respiratory failure as a consequence. For some of these patients, the hypoxemia that develops as a result of the lung injury can be exceptionally challenging to manage. In extreme cases, conventional ventilator management is insufficient, and the need for additional, advanced therapies arise.
Studies of VV ECMO in severe influenza
ECMO (extracorporeal membrane oxygenation) is a treatment that has been employed to help support patients with severe hypoxemic respiratory failure while their lungs recover from acute injury. Venovenous (VV) ECMO requires peripheral insertion of large cannulae into the venous system to take deoxygenated blood, deliver it through the membrane oxygenator and return the oxygenated blood back to the venous system. In simplest terms, the membrane of ECMO circuit serves as a substitute for the gas exchange function of the lungs and provides the oxygenation that the injured alveoli of the lung are unable to provide. The overall intent is to have the external ECMO circuit do all of the gas exchange work while the lungs heal.
Much research has been done on VV ECMO as an adjunct or salvage therapy in patients with refractory hypoxemic respiratory failure due to ARDS. Historical and recent studies have shown that approximately 60% of patients with ARDS have viral (approximately 20%) or bacterial (approximately 40%) pneumonia as the underlying cause (Zapol, et al. JAMA. 1979; 242[20]:2193; Combes A, et al. N Engl J Med. 2018;378:1965). Naturally, given the frequency of infection as a cause for ARDS, and the severity of illness that can develop with influenza infection in particular, an interest has arisen in the applicability of ECMO in cases of severe influenza-related ARDS.
In 2009, during the H1N1 influenza pandemic, the ANZ ECMO investigators in Australia and New Zealand described a 78% survival rate for their patients with severe H1N1 associated ARDS treated with VV ECMO between June and August of that year (Davies A, et al. JAMA. 2009;302[17]:1888). The eagerly awaited results of the randomized, controlled CESAR trial (Peek G, et al. Lancet. 2009;374:1351) that studied patients aged 18 to 64 with severe, refractory respiratory failure transferred to a specialized center for ECMO care had additional impact in catalyzing interest in ECMO use. This trial showed improved survival with ECMO (63% in ECMO vs 47% control, RR 0.69; 95% CI 0.05-0.97 P=.03) with a gain of 0.03 QALY (quality-adjusted life years) with additional cost of 40,000 pounds sterling. However, a major critique is that 24% of patients transferred to the specialized center never were treated with ECMO. Significantly, there was incomplete follow-up data on nearly half of the patients, as well. Many conclude that the survival benefit seen in this study may be more reflective of the expertise in respiratory failure management (especially as it relates to lung protective ventilation) at this center than therapy with ECMO itself.
Additional cohort studies in the United Kingdom (Noah MA, et al. JAMA. 2011;306[15]:1659) and Italy (Pappalardo F, et al. Intensive Care Med. 2013;39[2]:275) showed approximately 70% in-hospital survival rates for patients with H1N1 influenza transferred to a specialized ECMO center and treated with ECMO.
Nonetheless, the information gained from the observational data from ANZ ECMO, along with data published in European cohort studies and the randomized controlled CESAR trial after the 2009 H1N1 influenza pandemic, greatly contributed to the rise in use of ECMO for refractory ARDS due to influenza. Subsequently, there has been a rapid establishment and expansion of ECMO centers over the past decade, primarily to meet the anticipated demands of treating severe influenza-related ARDS.
The recently published EOLIA trial (Combes A, et al. N Engl J Med. 2018;378:1965) was designed to study the benefit of VV ECMO vs conventional mechanical ventilation in ARDS and demonstrated an 11% absolute reduction in 60-day mortality, which did not reach statistical significance. Like the CESAR trial, there are critiques of the outcome, especially as it relates to stopping the trial early due to the inability to show a significant benefit of VV ECMO over mechanical ventilation.
All of the aforementioned studies evaluated adults under age 65. Interestingly, there are no specific age contraindications for the use of ECMO (ELSO Guidelines for Cardiopulmonary Extracorporeal Life Support, Extracorporeal Life Support Organization, Version 1.4 August 2017), but many consider older age as a risk for poor outcome. Approximately 2,300 adult patients in the United States have been treated with ECMO for respiratory failure each year, and only 10% of those are over age 65 (CMS Changes in ECMO Reimbursements – ELSO Report). The outcome benefit of ECMO for a relatively healthy patient over age 65 is not known, as those patients have not been evaluated in studies thus far. When comparison to data from decades ago is made, one must keep in mind that populations worldwide are living longer, and a continued increase in number of adults over the age 65 is expected.
While the overall interpretation of the outcomes of studies of ECMO may be fraught with controversy, there is little debate that providing care for patients with refractory respiratory failure in centers that provide high-level skill and expertise in management of respiratory failure has a clear benefit, irrespective of whether the patient eventually receives therapy with ECMO. What is also clear is that ECMO is costly, with per-patient costs demonstrated to be at least double that of those receiving mechanical ventilation alone (Peek G, et al. Lancet. 2009;374:1351). This substantial cost associated with ECMO cannot be ignored in today’s era of value-based care.
Fortuitously, CMS recently released new DRG reimbursement scales for the use of ECMO effective Oct 1, 2018. VV ECMO could have as much as a 70% reduction in reimbursement, and many insurance companies are expected to follow suit (CMS Changes in ECMO Reimbursements –ELSO Report). Only time will tell what impact this, along with the current evidence, will have on long-term provision of ECMO care for our sickest of patients with influenza and associated respiratory illnesses.
Dr. Tatem is with the Division of Pulmonary and Critical Care Medicine, Department of Medicine, Henry Ford Hospital, Detroit, Michigan.
Now that we are in the midst of flu season, many discussions regarding the management of patients with influenza virus infections are ensuing. While prevention is always preferable, and we encourage everyone to get vaccinated, influenza remains a rapidly widespread infection. In the United States during last year’s flu season (2017-18), there was an estimated 49 million cases of influenza, 960,000 hospitalizations, and 79,000 deaths. Approximately 86% of all deaths were estimated to occur in those aged 65 and older (Centers for Disease Control and Prevention webpage on Burden of Influenza).
Despite our best efforts, there are inevitable times when some patients become ill enough to require hospitalization. Patients aged 65 and older make up the overwhelming majority of patients with influenza who eventually require hospitalization (Fig 1) (The Centers for Disease Control and Prevention FluView Database). Comorbidities also confer higher risk for more severe illness and potential hospitalization irrespective of age (Fig 2). In children with known medical conditions, asthma confers highest risk of hospitalization, as 27% of those with asthma were hospitalized after developing the flu. In adults, 52% of those with cardiovascular disease and 30% of adult patients with chronic lung disease who were confirmed to have influenza required hospitalization for treatment (Fig 2, The Centers for Disease Control and Prevention FluView Database).
The most severe cases of influenza can require ICU care and advanced management of respiratory failure as a result of the acute respiratory distress syndrome (ARDS). The lungs suffer significant injury due to the viral infection, and they lose their ability to effectively oxygenate the blood. Secondary bacterial infections can also occur as a complication, which compounds the injury. Given the fact that so many patients have significant comorbidities and are of advanced age, it is reasonable to expect that a fair proportion of those with influenza would develop respiratory failure as a consequence. For some of these patients, the hypoxemia that develops as a result of the lung injury can be exceptionally challenging to manage. In extreme cases, conventional ventilator management is insufficient, and the need for additional, advanced therapies arise.
Studies of VV ECMO in severe influenza
ECMO (extracorporeal membrane oxygenation) is a treatment that has been employed to help support patients with severe hypoxemic respiratory failure while their lungs recover from acute injury. Venovenous (VV) ECMO requires peripheral insertion of large cannulae into the venous system to take deoxygenated blood, deliver it through the membrane oxygenator and return the oxygenated blood back to the venous system. In simplest terms, the membrane of ECMO circuit serves as a substitute for the gas exchange function of the lungs and provides the oxygenation that the injured alveoli of the lung are unable to provide. The overall intent is to have the external ECMO circuit do all of the gas exchange work while the lungs heal.
Much research has been done on VV ECMO as an adjunct or salvage therapy in patients with refractory hypoxemic respiratory failure due to ARDS. Historical and recent studies have shown that approximately 60% of patients with ARDS have viral (approximately 20%) or bacterial (approximately 40%) pneumonia as the underlying cause (Zapol, et al. JAMA. 1979; 242[20]:2193; Combes A, et al. N Engl J Med. 2018;378:1965). Naturally, given the frequency of infection as a cause for ARDS, and the severity of illness that can develop with influenza infection in particular, an interest has arisen in the applicability of ECMO in cases of severe influenza-related ARDS.
In 2009, during the H1N1 influenza pandemic, the ANZ ECMO investigators in Australia and New Zealand described a 78% survival rate for their patients with severe H1N1 associated ARDS treated with VV ECMO between June and August of that year (Davies A, et al. JAMA. 2009;302[17]:1888). The eagerly awaited results of the randomized, controlled CESAR trial (Peek G, et al. Lancet. 2009;374:1351) that studied patients aged 18 to 64 with severe, refractory respiratory failure transferred to a specialized center for ECMO care had additional impact in catalyzing interest in ECMO use. This trial showed improved survival with ECMO (63% in ECMO vs 47% control, RR 0.69; 95% CI 0.05-0.97 P=.03) with a gain of 0.03 QALY (quality-adjusted life years) with additional cost of 40,000 pounds sterling. However, a major critique is that 24% of patients transferred to the specialized center never were treated with ECMO. Significantly, there was incomplete follow-up data on nearly half of the patients, as well. Many conclude that the survival benefit seen in this study may be more reflective of the expertise in respiratory failure management (especially as it relates to lung protective ventilation) at this center than therapy with ECMO itself.
Additional cohort studies in the United Kingdom (Noah MA, et al. JAMA. 2011;306[15]:1659) and Italy (Pappalardo F, et al. Intensive Care Med. 2013;39[2]:275) showed approximately 70% in-hospital survival rates for patients with H1N1 influenza transferred to a specialized ECMO center and treated with ECMO.
Nonetheless, the information gained from the observational data from ANZ ECMO, along with data published in European cohort studies and the randomized controlled CESAR trial after the 2009 H1N1 influenza pandemic, greatly contributed to the rise in use of ECMO for refractory ARDS due to influenza. Subsequently, there has been a rapid establishment and expansion of ECMO centers over the past decade, primarily to meet the anticipated demands of treating severe influenza-related ARDS.
The recently published EOLIA trial (Combes A, et al. N Engl J Med. 2018;378:1965) was designed to study the benefit of VV ECMO vs conventional mechanical ventilation in ARDS and demonstrated an 11% absolute reduction in 60-day mortality, which did not reach statistical significance. Like the CESAR trial, there are critiques of the outcome, especially as it relates to stopping the trial early due to the inability to show a significant benefit of VV ECMO over mechanical ventilation.
All of the aforementioned studies evaluated adults under age 65. Interestingly, there are no specific age contraindications for the use of ECMO (ELSO Guidelines for Cardiopulmonary Extracorporeal Life Support, Extracorporeal Life Support Organization, Version 1.4 August 2017), but many consider older age as a risk for poor outcome. Approximately 2,300 adult patients in the United States have been treated with ECMO for respiratory failure each year, and only 10% of those are over age 65 (CMS Changes in ECMO Reimbursements – ELSO Report). The outcome benefit of ECMO for a relatively healthy patient over age 65 is not known, as those patients have not been evaluated in studies thus far. When comparison to data from decades ago is made, one must keep in mind that populations worldwide are living longer, and a continued increase in number of adults over the age 65 is expected.
While the overall interpretation of the outcomes of studies of ECMO may be fraught with controversy, there is little debate that providing care for patients with refractory respiratory failure in centers that provide high-level skill and expertise in management of respiratory failure has a clear benefit, irrespective of whether the patient eventually receives therapy with ECMO. What is also clear is that ECMO is costly, with per-patient costs demonstrated to be at least double that of those receiving mechanical ventilation alone (Peek G, et al. Lancet. 2009;374:1351). This substantial cost associated with ECMO cannot be ignored in today’s era of value-based care.
Fortuitously, CMS recently released new DRG reimbursement scales for the use of ECMO effective Oct 1, 2018. VV ECMO could have as much as a 70% reduction in reimbursement, and many insurance companies are expected to follow suit (CMS Changes in ECMO Reimbursements –ELSO Report). Only time will tell what impact this, along with the current evidence, will have on long-term provision of ECMO care for our sickest of patients with influenza and associated respiratory illnesses.
Dr. Tatem is with the Division of Pulmonary and Critical Care Medicine, Department of Medicine, Henry Ford Hospital, Detroit, Michigan.
The 1-hour sepsis bundle is serious—serious like a heart attack
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.

In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.
In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
In 2002, the European Society of Intensive Care Medicine, the Society of Critical Care Medicine, and the International Sepsis Forum formed the Surviving Sepsis Campaign (SSC) aiming to reduce sepsis-related mortality by 25% within 5 years, mimicking the progress made in the management of STEMI (http://www.survivingsepsis.org/About-SSC/Pages/History.aspx).
SSC bundles: a historic perspective
The first guidelines were published in 2004. Recognizing that guidelines may not influence bedside practice for many years, the SSC partnered with the Institute for Healthcare Improvement to apply performance improvement methodology to sepsis management, developing the “sepsis change bundles.” In addition to hospital resources for education, screening, and data collection, the 6-hour resuscitation and 24-hour management bundles were created. Subsequent data, collected as part of the initiative, demonstrated an association between bundle compliance and survival.
In 2008, the SSC guidelines were revised, and the National Quality Forum (NQF) adopted sepsis bundle compliance as a quality measure. NQF endorsement is often the first step toward the creation of mandates by the Centers for Medicare and Medicaid Services (CMS), but that did not occur at the time.
In 2012, the SSC guidelines were updated and published with new 3- and 6-hour bundles. That year, Rory Staunton, an otherwise healthy 12-year-old boy, died of septic shock in New York. The public discussion of this case, among other factors, prompted New York state to develop a sepsis care mandate that became state law in 2014. An annual public report details each hospital’s compliance with process measures and risk-adjusted mortality. The correlation between measure compliance and survival also holds true in this data set.
In 2015, CMS developed the SEP-1 measure. While the symbolic importance of a sepsis federal mandate and its potential to improve patient outcomes is recognized, concerns remain about the measure itself. The detailed and specific way data must be collected may disconnect clinical care provided from measured compliance. The time pressure and the “all-or-nothing” approach might incentivize interventions potentially harmful in some patients. No patient-centered outcomes are reported. This measure might be tied to reimbursement in the future.
The original version of SEP-1 was based on the 2012 SSC bundles, which reflected the best evidence available at the time (the 2001 Early Goal-Directed Therapy trial). By 2015, elements of that strategy had been challenged, and the PROCESS, PROMISE, and ARISE trials contested the notion that protcolized resuscitation decreased mortality. Moreover, new definitions of sepsis syndromes (Sepsis-3) were published in 2016 (Singer M, et al. JAMA. 2016;315[8]:801).
The 2016 SSC guidelines adopted the new definitions and recommended that patients with sepsis-induced hypoperfusion immediately receive a 30 mL/kg crystalloid bolus, followed by frequent reassessment. CMS did not adopt the Sepsis-3 definitions, but updates were made to allow the clinicians flexibility to demonstrate reassessment of the patient.
Comparing the 1-hour bundle to STEMI care
This year, the SSC published a 1-hour bundle to replace the 3- and 6-hour bundles (Levy MM et al. Crit Care Med. 2018;46[6]:997). Whereas previous bundles set time frames for completion of the elements, the 1-hour bundle focuses on the initiation of these components. The authors revisited the parallel between early management of sepsis and STEMI. The 1-hour bundle includes serum lactate, blood cultures prior to antibiotics, broad-spectrum antibiotics, a 30 mL/kg crystalloid bolus for patients with hypotension or lactate greater than or equal to 4 mmol/L, and vasopressors for persistent hypotension.
Elements of controversy after the publication of this bundle include:
1. One hour seems insufficient for complex clinical decision making and interventions for a syndrome with no specific diagnostic test: sepsis often mimics, or is mimicked by, other conditions.
2. Some bundle elements are not supported by high-quality evidence. No controlled studies exist regarding the appropriate volume of initial fluids or the impact of timing of antibiotics on outcomes.
3. The 1-hour time frame will encourage empiric delivery of fluids and antibiotics to patients who are not septic, potentially leading to harm.
4. While the 1-hour bundle is a quality improvement tool and not for public reporting, former bundles have been adopted as federally regulated measures.
Has the SSC gone too far? Are these concerns enough to abandon the 1-hour bundle? Or are the concerns regarding the 1-hour bundle an example of “perfect is the enemy of better”? To understand the potential for imperfect guidelines to drive tremendous patient-level improvements, one must consider the evolution of STEMI management.
Since the 1970s, the in-hospital mortality for STEMI has decreased from 25% to around 5%. The most significant factor in this achievement was the recognition that early reperfusion improves outcomes and that doing it consistently requires complex coordination. In 2004, a Door-to-Balloon (D2B) time of less than 90 minutes was included as a guideline recommendation (Antman EM, et al. Circulation. 2004;110[5]:588). CMS started collecting performance data on this metric, made that data public, and later tied the performance to hospital reimbursement.
Initially, the 90-minute goal was achieved in only 44% of cases. In 2006, the D2B initiative was launched, providing recommendations for public education, coordination of care, and emergent management of STEMI. Compliance with these recommendations required significant education and changes to STEMI care at multiple levels. Data were collected and submitted to inform the process. Six years later, compliance with the D2B goal had increased from 44% to 91%. The median D2B dropped from 96 to 64 minutes. Based on high compliance, CMS discontinued the use of this metric for reimbursement as the variation between high and low performers was minimal. Put simply, the entire country had gotten better at treating STEMI. The “time-zero” for STEMI was pushed back further, and D2B has been replaced with first-medical-contact (FMC) to device time. The recommendation is to achieve this as quickly as possible, and in less than 90 minutes (O’Gara P, et al. JACC. 2013;61[4]:485).
Consider the complexity of getting a patient from their home to a catheterization lab within 90 minutes, even in ideal circumstances. This short time frame encourages, by design, a low threshold to activate the system. We accept that some patients will receive an unnecessary catheterization or systemic fibrinolysis although the recommendation is based on level B evidence.
Compliance with the STEMI guidelines is more labor-intensive and complex than compliance with the 1-hour sepsis bundle. So, is STEMI a fair comparison to sepsis? Both syndromes are common, potentially deadly, and time-sensitive. Both require early recognition, but neither has a definitive diagnostic test. Instead, diagnosis requires an integration of multiple complex clinical factors. Both are backed by imperfect science that continues to evolve. Over-diagnosis of either will expose the patient to potentially harmful therapies.
The early management of STEMI is a valid comparison to the early management of sepsis. We must consider this comparison as we ponder the 1-hour sepsis bundle.
Is triage time the appropriate time-zero? In either condition, triage time is too early in some cases and too late in others. Unfortunately, there is no better alternative, and STEMI guidelines have evolved to start the clock before triage. Using a point such as “recognition of sepsis” would fail to capture delayed recognition.
Is it possible to diagnose and initiate treatment for sepsis in such a short time frame? Consider the treatment received by the usual care group of the PROCESS trial (The ProCESS Investigators. N Engl J Med. 2014;370:1683). Prior to meeting entry criteria, which occurred in less than 1 hour, patients in this group received an initial fluid bolus and had a lactate assessment. Prior to randomization, which occurred at around 90 minutes, this group completed 28 mL/kg of crystalloid fluid, and 76% received antibiotics. Thus, the usual-care group in this study nearly achieved the 1-hour bundle currently being contested.
Is it appropriate for a guideline to strongly recommend interventions not backed by level A evidence? The recommendation for FMC to catheterization within 90 minutes has not been studied in a controlled way. The precise dosing and timing of fibrinolysis is also not based on controlled data. Reperfusion devices and antiplatelet agents continue to be rigorously studied, sometimes with conflicting results.
Finally, should the 1-hour bundle be abandoned out of concern that it will be used as a national performance metric? First, there is currently no indication that the 1-hour bundle will be adopted as a performance metric. For the sake of argument, let’s assume the 1-hour bundle will be regulated and used to compare hospitals. Is there reason to think this bundle favors some hospitals over others and will lead to an unfair comparison? Is there significant inequity in the ability to draw blood cultures, send a lactate, start IV fluids, and initiate antibiotics?
Certainly, national compliance with such a metric would be very low at first. Therein lies the actual problem: a person who suffers a STEMI anywhere in the country is very likely to receive high-quality care. Currently, the same cannot be said about a patient with sepsis. Perhaps that should be the focus of our concern.
Dr. Uppal is Assistant Professor, NYU School of Medicine, Bellevue Hospital Center, New York, New York.
Introducing CHEST’s new CEO/EVP
Greetings! My name is Robert Musacchio; I am proud to introduce myself as the new Chief Executive Officer and Executive Vice President of CHEST. I am honored to join this team of distinguished clinicians as we spearhead progress in the fight against lung disease.

I have had the pleasure of working at CHEST for the last 4 years, first joining CHEST Enterprises as Senior Vice President of Business Development. In that role, I focused on revenue growth and product diversification before becoming COO of CHEST. As COO, I dedicated myself to strengthening our team by mentoring staff and collaborating with senior leadership, a challenge that I have deeply enjoyed.
Before joining CHEST, I worked at the American Medical Association for 35 years in roles encompassing research, advocacy, membership, and publishing. I also worked with boards and membership groups as a member of the AMA’s CPT® Editorial Panel and as a publisher for JAMA.
As CEO, I hope to leverage those experiences to support CHEST in its mission, which is to improve lung health not just for 1 year but for the next 25 years. For that reason, our leadership team has outlined an organizational culture that fosters short-term success and long-term innovation, focusing on four key areas:
People: How do we attract and retain the right people?
Strategy: How do we create a truly differentiated strategy?
Execution: How do we improve our process to drive flawless execution?
Resources: How do we ensure that we have sufficient resources to invest in our mission?
Those questions in mind, we have established several standards that guide the way we work. We are focusing on leading with integrity, cultivating passion and innovation, honoring our team, and having fun while we deliver cutting-edge education and create community for our members. With these norms, we can continue to foster an environment that generates results. This, in turn, will enable CHEST to fulfill its core purpose of crushing lung disease.
We can crush lung disease by arming our members with industry-leading education offerings—including simulation experiences and live lab courses—and expanding them worldwide to Thailand and Greece in 2019. We can crush lung disease by using cutting-edge technologies—including interactive gaming platforms—to glean further insights. We can crush lung disease by connecting our membership of nearly 20,000 pulmonary, critical care, and sleep medicine professionals to innovative education tools, along with a network of prestigious colleagues to deliver the highest quality patient care.
Most importantly, we can crush lung disease by empowering our members in their work—if you care about lung disease, we care about you!
And, if you care about lung disease, I am excited to partner with you in this cause. I am thankful for this opportunity to lead CHEST and the CHEST Foundation into the future and look forward to working with you.
Greetings! My name is Robert Musacchio; I am proud to introduce myself as the new Chief Executive Officer and Executive Vice President of CHEST. I am honored to join this team of distinguished clinicians as we spearhead progress in the fight against lung disease.
I have had the pleasure of working at CHEST for the last 4 years, first joining CHEST Enterprises as Senior Vice President of Business Development. In that role, I focused on revenue growth and product diversification before becoming COO of CHEST. As COO, I dedicated myself to strengthening our team by mentoring staff and collaborating with senior leadership, a challenge that I have deeply enjoyed.
Before joining CHEST, I worked at the American Medical Association for 35 years in roles encompassing research, advocacy, membership, and publishing. I also worked with boards and membership groups as a member of the AMA’s CPT® Editorial Panel and as a publisher for JAMA.
As CEO, I hope to leverage those experiences to support CHEST in its mission, which is to improve lung health not just for 1 year but for the next 25 years. For that reason, our leadership team has outlined an organizational culture that fosters short-term success and long-term innovation, focusing on four key areas:
People: How do we attract and retain the right people?
Strategy: How do we create a truly differentiated strategy?
Execution: How do we improve our process to drive flawless execution?
Resources: How do we ensure that we have sufficient resources to invest in our mission?
Those questions in mind, we have established several standards that guide the way we work. We are focusing on leading with integrity, cultivating passion and innovation, honoring our team, and having fun while we deliver cutting-edge education and create community for our members. With these norms, we can continue to foster an environment that generates results. This, in turn, will enable CHEST to fulfill its core purpose of crushing lung disease.
We can crush lung disease by arming our members with industry-leading education offerings—including simulation experiences and live lab courses—and expanding them worldwide to Thailand and Greece in 2019. We can crush lung disease by using cutting-edge technologies—including interactive gaming platforms—to glean further insights. We can crush lung disease by connecting our membership of nearly 20,000 pulmonary, critical care, and sleep medicine professionals to innovative education tools, along with a network of prestigious colleagues to deliver the highest quality patient care.
Most importantly, we can crush lung disease by empowering our members in their work—if you care about lung disease, we care about you!
And, if you care about lung disease, I am excited to partner with you in this cause. I am thankful for this opportunity to lead CHEST and the CHEST Foundation into the future and look forward to working with you.
Greetings! My name is Robert Musacchio; I am proud to introduce myself as the new Chief Executive Officer and Executive Vice President of CHEST. I am honored to join this team of distinguished clinicians as we spearhead progress in the fight against lung disease.
I have had the pleasure of working at CHEST for the last 4 years, first joining CHEST Enterprises as Senior Vice President of Business Development. In that role, I focused on revenue growth and product diversification before becoming COO of CHEST. As COO, I dedicated myself to strengthening our team by mentoring staff and collaborating with senior leadership, a challenge that I have deeply enjoyed.
Before joining CHEST, I worked at the American Medical Association for 35 years in roles encompassing research, advocacy, membership, and publishing. I also worked with boards and membership groups as a member of the AMA’s CPT® Editorial Panel and as a publisher for JAMA.
As CEO, I hope to leverage those experiences to support CHEST in its mission, which is to improve lung health not just for 1 year but for the next 25 years. For that reason, our leadership team has outlined an organizational culture that fosters short-term success and long-term innovation, focusing on four key areas:
People: How do we attract and retain the right people?
Strategy: How do we create a truly differentiated strategy?
Execution: How do we improve our process to drive flawless execution?
Resources: How do we ensure that we have sufficient resources to invest in our mission?
Those questions in mind, we have established several standards that guide the way we work. We are focusing on leading with integrity, cultivating passion and innovation, honoring our team, and having fun while we deliver cutting-edge education and create community for our members. With these norms, we can continue to foster an environment that generates results. This, in turn, will enable CHEST to fulfill its core purpose of crushing lung disease.
We can crush lung disease by arming our members with industry-leading education offerings—including simulation experiences and live lab courses—and expanding them worldwide to Thailand and Greece in 2019. We can crush lung disease by using cutting-edge technologies—including interactive gaming platforms—to glean further insights. We can crush lung disease by connecting our membership of nearly 20,000 pulmonary, critical care, and sleep medicine professionals to innovative education tools, along with a network of prestigious colleagues to deliver the highest quality patient care.
Most importantly, we can crush lung disease by empowering our members in their work—if you care about lung disease, we care about you!
And, if you care about lung disease, I am excited to partner with you in this cause. I am thankful for this opportunity to lead CHEST and the CHEST Foundation into the future and look forward to working with you.
TOURMALINE-MM3: Ixazomib improves PFS after myeloma transplant
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.

Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.
Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
SAN DIEGO – Ixazomib improved progression-free survival following autologous stem cell transplantation (ASCT) in patients with newly diagnosed multiple myeloma, according to results of a double-blind, randomized, placebo-controlled, phase 3 trial.
Treatment with the oral proteasome inhibitor for 24 months was well tolerated, had a low discontinuation rate, and improved progression-free survival by 39% versus placebo, according to Meletios A. Dimopoulos, MD, of the National and Kapodistrian University of Athens.
These findings suggest ixazomib (Ninlaro) represents a “new treatment option for maintenance after transplantation,” Dr. Dimopoulos said at the annual meeting of the American Society of Hematology.
The trial, known as TOURMALINE-MM3, is the first-ever randomized, double-blind, placebo-controlled study of a proteasome inhibitor used as maintenance after ASCT, according to Dr. Dimopoulos. Lenalidomide is approved in that setting, but 29% of patients who start the treatment discontinue because of treatment-related adverse events.
“Proteasome inhibitors have a different mechanism of action and may provide an alternative to lenalidomide,” Dr. Dimopoulos said at an oral abstract session. Ixazomib has a manageable toxicity profile and “convenient” weekly oral dosing, making it “well suited” for maintenance.
When asked by an attendee whether ixazomib would become “the standard of care” for younger patients with myeloma in this setting, Dr. Dimopoulos replied the results show that ixazomib “is an option for patients, especially for those where a physician may believe that a proteasome inhibitor may be indicated.”
However, when pressed by an attendee to comment on how ixazomib compares with lenalidomide for maintenance, Dr. Dimopoulos remarked that current maintenance studies are moving in the direction of combining therapies.
“I think that instead of saying, ‘is ixazomib better than lenalidomide?’ or vice versa, it is better to see how one may combine those drugs in subsets of patients, or even combine these drugs with other agents,” he said.
The TOURMALINE-MM3 study included 656 patients randomized posttransplantation to receive weekly ixazomib or placebo for up to 2 years.
The median progression-free survival was 26.5 months for ixazomib versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890). Median overall survival had not been reached in either ixazomib or placebo arms as of this report, with a median follow-up of 31 months.
The discontinuation rate was 7% for ixazomib versus 5% for placebo, according to the investigator. Moreover, ixazomib was associated with “low toxicity” and no difference in the rates of new primary malignancies, at 3% in both arms.
A manuscript describing results of the TOURMALINE-MM3 study is in press in the Lancet, with an expected online publication date of Dec. 10, Dr. Dimopoulos told attendees. Other studies are ongoing to evaluate ixazomib combinations and treatment to progression in this setting.
TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
SOURCE: Dimopoulos MA et al. ASH 2018, Abstract 301.
REPORTING FROM ASH 2018
Key clinical point: The proteasome inhibitor ixazomib significantly improved progression-free survival following autologous stem cell transplantation in patients with newly diagnosed multiple myeloma.
Major finding: The median progression-free survival was 26.5 months for ixazomib, versus 21.3 months for placebo (P = .002; hazard ratio, 0.720; 95% confidence interval, 0.582-0.890).
Study details: TOURMALINE-MM3, a randomized, placebo-controlled trial, includes 656 patients with newly diagnosed myeloma who had undergone autologous stem cell transplantation.
Disclosures: TOURMALINE-MM3 is sponsored by Takeda (Millennium), the maker of ixazomib. Dr. Dimopoulos reported honoraria and consultancy with Janssen, Takeda Pharmaceutical, Amgen, Bristol-Myers Squibb, and Celgene.
Source: Dimopoulos MA et al. ASH 2018, Abstract 301.
JULIET: CAR T cells go the distance in r/r DLBCL
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
SAN DIEGO – Two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma who had early responses to chimeric antigen receptor T-cell (CAR T) therapy with tisagenlecleucel (Kymriah) remain in remission with no evidence of minimal residual disease, according to an updated analysis of the JULIET trial.
In the single-arm, open-label trial, the overall response rate after 19 months of follow-up was 54%, including 40% complete remissions and 14% partial remissions. The median duration of response had not been reached at the time of data cutoff, and the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events were similar to those previously reported and were manageable, according to investigator Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland.
In this video interview at the annual meeting of the American Society of Hematology, Dr. Maziarz discusses the promising results using CAR T cells in this difficult to treat population.
REPORTING FROM ASH 2018

Beat AML trial delivers genomic results in 7 days
SAN DIEGO – Investigators demonstrated the feasibility of delivering genomic results in 7 days in a population of older, newly diagnosed patients with acute myeloid leukemia (AML).
The Beat AML Master Trial is an ongoing umbrella study that harnesses cytogenetic information and next generation sequencing to match patients with targeted therapies across a number of substudies or outside of the trial’s multicenter network.
The researchers chose AML for this precision-medicine study because of its rapid onset and lethal nature, its heterogeneity, and the availability of more-targeted therapies, said Amy Burd, PhD, of the Leukemia & Lymphoma Society, which is sponsoring the study.
Initial data from the trial showed that more than 95% of patients were assigned to treatment in 7 days or less, based on their personalized genomic information.
Overall, 285 patients had usable genomic screening data and were assigned to treatment. Of those patients, 273 were assigned to a treatment within 7 days, Dr. Burd reported at the annual meeting of the American Society of Hematology.
The speed of delivering these results is critical, said Joseph Mikhael, MD, chief medical officer for the International Myeloma Foundation in Phoenix, who moderated a media briefing on personalized medicine.
“One of the greatest challenges we faced in the concept of personalized medicine is by the time you’ve determined what is best for that patient ... the horse is already out of the barn,” Dr. Mikhael said. “You have to have started the patient on treatment already or else their disease could have progressed quite rapidly.”
In the past, genomic results might come back a month after the patient started therapy. “It was really almost academic,” he said.
In the Beat AML study, more than half (146 patients) were treated based on their AML subtype. The remaining patients (139) were not treated: 2.5% of patients died within 7 days, 7% of patients chose an alternative treatment prior to assignment, 20% chose standard of care, 9.1% chose an alternative trial after assignment, 8.1% chose palliative care, and the remainder had a reason that was not specified.
“The treatment decisions are made for what’s best for the patient even if that means a study outside of Beat AML,” Dr. Burd said.
Currently, there are 11 substudies offering treatment to trial participants across 13 clinical sites. There has been promising efficacy in many of the treatment arms, Dr. Burd said.
In the future, the researchers are looking to expand the substudies to look into novel drug combinations for certain AML subtypes, specifically isocitrate dehydrogenase 2–mutated groups.
Dr. Burd is an employee of the Leukemia & Lymphoma Society. Other coinvestigators reported financial relationships with the pharmaceutical industry. Dr. Mikhael reported research funding from AbbVie, Celgene, Onyx Pharmaceuticals, and Sanofi.
SOURCE: Burd A et al. ASH 2018, Abstract 559.
SAN DIEGO – Investigators demonstrated the feasibility of delivering genomic results in 7 days in a population of older, newly diagnosed patients with acute myeloid leukemia (AML).
The Beat AML Master Trial is an ongoing umbrella study that harnesses cytogenetic information and next generation sequencing to match patients with targeted therapies across a number of substudies or outside of the trial’s multicenter network.
The researchers chose AML for this precision-medicine study because of its rapid onset and lethal nature, its heterogeneity, and the availability of more-targeted therapies, said Amy Burd, PhD, of the Leukemia & Lymphoma Society, which is sponsoring the study.
Initial data from the trial showed that more than 95% of patients were assigned to treatment in 7 days or less, based on their personalized genomic information.
Overall, 285 patients had usable genomic screening data and were assigned to treatment. Of those patients, 273 were assigned to a treatment within 7 days, Dr. Burd reported at the annual meeting of the American Society of Hematology.
The speed of delivering these results is critical, said Joseph Mikhael, MD, chief medical officer for the International Myeloma Foundation in Phoenix, who moderated a media briefing on personalized medicine.
“One of the greatest challenges we faced in the concept of personalized medicine is by the time you’ve determined what is best for that patient ... the horse is already out of the barn,” Dr. Mikhael said. “You have to have started the patient on treatment already or else their disease could have progressed quite rapidly.”
In the past, genomic results might come back a month after the patient started therapy. “It was really almost academic,” he said.
In the Beat AML study, more than half (146 patients) were treated based on their AML subtype. The remaining patients (139) were not treated: 2.5% of patients died within 7 days, 7% of patients chose an alternative treatment prior to assignment, 20% chose standard of care, 9.1% chose an alternative trial after assignment, 8.1% chose palliative care, and the remainder had a reason that was not specified.
“The treatment decisions are made for what’s best for the patient even if that means a study outside of Beat AML,” Dr. Burd said.
Currently, there are 11 substudies offering treatment to trial participants across 13 clinical sites. There has been promising efficacy in many of the treatment arms, Dr. Burd said.
In the future, the researchers are looking to expand the substudies to look into novel drug combinations for certain AML subtypes, specifically isocitrate dehydrogenase 2–mutated groups.
Dr. Burd is an employee of the Leukemia & Lymphoma Society. Other coinvestigators reported financial relationships with the pharmaceutical industry. Dr. Mikhael reported research funding from AbbVie, Celgene, Onyx Pharmaceuticals, and Sanofi.
SOURCE: Burd A et al. ASH 2018, Abstract 559.
SAN DIEGO – Investigators demonstrated the feasibility of delivering genomic results in 7 days in a population of older, newly diagnosed patients with acute myeloid leukemia (AML).
The Beat AML Master Trial is an ongoing umbrella study that harnesses cytogenetic information and next generation sequencing to match patients with targeted therapies across a number of substudies or outside of the trial’s multicenter network.
The researchers chose AML for this precision-medicine study because of its rapid onset and lethal nature, its heterogeneity, and the availability of more-targeted therapies, said Amy Burd, PhD, of the Leukemia & Lymphoma Society, which is sponsoring the study.
Initial data from the trial showed that more than 95% of patients were assigned to treatment in 7 days or less, based on their personalized genomic information.
Overall, 285 patients had usable genomic screening data and were assigned to treatment. Of those patients, 273 were assigned to a treatment within 7 days, Dr. Burd reported at the annual meeting of the American Society of Hematology.
The speed of delivering these results is critical, said Joseph Mikhael, MD, chief medical officer for the International Myeloma Foundation in Phoenix, who moderated a media briefing on personalized medicine.
“One of the greatest challenges we faced in the concept of personalized medicine is by the time you’ve determined what is best for that patient ... the horse is already out of the barn,” Dr. Mikhael said. “You have to have started the patient on treatment already or else their disease could have progressed quite rapidly.”
In the past, genomic results might come back a month after the patient started therapy. “It was really almost academic,” he said.
In the Beat AML study, more than half (146 patients) were treated based on their AML subtype. The remaining patients (139) were not treated: 2.5% of patients died within 7 days, 7% of patients chose an alternative treatment prior to assignment, 20% chose standard of care, 9.1% chose an alternative trial after assignment, 8.1% chose palliative care, and the remainder had a reason that was not specified.
“The treatment decisions are made for what’s best for the patient even if that means a study outside of Beat AML,” Dr. Burd said.
Currently, there are 11 substudies offering treatment to trial participants across 13 clinical sites. There has been promising efficacy in many of the treatment arms, Dr. Burd said.
In the future, the researchers are looking to expand the substudies to look into novel drug combinations for certain AML subtypes, specifically isocitrate dehydrogenase 2–mutated groups.
Dr. Burd is an employee of the Leukemia & Lymphoma Society. Other coinvestigators reported financial relationships with the pharmaceutical industry. Dr. Mikhael reported research funding from AbbVie, Celgene, Onyx Pharmaceuticals, and Sanofi.
SOURCE: Burd A et al. ASH 2018, Abstract 559.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: More than 95% of patients in the trial were assigned to treatment within 7 days based on results of their genomic screening.
Study details: An umbrella study of 285 patients aged 60 years and older with newly diagnosed acute myeloid leukemia.
Disclosures: The study is sponsored by the Leukemia & Lymphoma Society. Dr. Burd is an employee of the Society and other investigators reported funding from multiple pharmaceutical companies.
Source: Burd A et al. ASH 2018, Abstract 559.

Gut bacteria influence HCT outcomes
A multinational study of intestinal microbiota in the United States, Europe, and Japan showed that in all four geographic regions patients scheduled for HCT had about a 100% lower median diversity of intestinal bacteria, compared with healthy volunteers, and that enterococcal species predominated in the transplant candidates, reported Jonathan U. Peled, MD, PhD, from the bone marrow transplantation service at Memorial Sloan Kettering Cancer Center in New York.
The investigators also found that intestinal microbial diversity was significantly associated with overall survival following an HCT.
In a video interview at the annual meeting of the American Society of Hematology, Dr. Peled elaborated on the study findings and described potential pre- and posttransplant interventions that could improve results and increase survival following HCT.
Dr. Peled reported current or prior relationships with Seres Therapeutics, the Parker Institute for Cancer Immunotherapy, and Merck/Society for Immunotherapy of Cancer.
A multinational study of intestinal microbiota in the United States, Europe, and Japan showed that in all four geographic regions patients scheduled for HCT had about a 100% lower median diversity of intestinal bacteria, compared with healthy volunteers, and that enterococcal species predominated in the transplant candidates, reported Jonathan U. Peled, MD, PhD, from the bone marrow transplantation service at Memorial Sloan Kettering Cancer Center in New York.
The investigators also found that intestinal microbial diversity was significantly associated with overall survival following an HCT.
In a video interview at the annual meeting of the American Society of Hematology, Dr. Peled elaborated on the study findings and described potential pre- and posttransplant interventions that could improve results and increase survival following HCT.
Dr. Peled reported current or prior relationships with Seres Therapeutics, the Parker Institute for Cancer Immunotherapy, and Merck/Society for Immunotherapy of Cancer.
A multinational study of intestinal microbiota in the United States, Europe, and Japan showed that in all four geographic regions patients scheduled for HCT had about a 100% lower median diversity of intestinal bacteria, compared with healthy volunteers, and that enterococcal species predominated in the transplant candidates, reported Jonathan U. Peled, MD, PhD, from the bone marrow transplantation service at Memorial Sloan Kettering Cancer Center in New York.
The investigators also found that intestinal microbial diversity was significantly associated with overall survival following an HCT.
In a video interview at the annual meeting of the American Society of Hematology, Dr. Peled elaborated on the study findings and described potential pre- and posttransplant interventions that could improve results and increase survival following HCT.
Dr. Peled reported current or prior relationships with Seres Therapeutics, the Parker Institute for Cancer Immunotherapy, and Merck/Society for Immunotherapy of Cancer.
REPORTING FROM ASH 2018

JULIET: CAR T cells keep trucking against DLBCL
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.

After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.
After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
SAN DIEGO – Chimeric antigen receptor T-cell therapy with tisagenlecleucel (Kymriah) is associated with a high rate of durable responses in adults with relapsed or refractory diffuse large B-cell lymphoma, an updated analysis of the JULIET trial showed.
After a median follow-up of 19 months, two-thirds of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who had early responses to chimeric antigen receptor (CAR) T-cell therapy with tisagenlecleucel remained in remission with no evidence of minimal residual disease, reported Richard Thomas Maziarz, MD, from the Oregon Health & Science Knight Cancer Institute in Portland, at the annual meeting of the American Society of Hematology.
“Since the previous report, no new deaths have been reported due to any cause other than patient disease progression. No treatment-related mortality was seen throughout the study, and there were three early deaths, all related to lymphoma that progressed,” he said in a briefing prior to presentation of the data in a scientific poster.
The updated study results were published simultaneously online in the New England Journal of Medicine.
JULIET then
In the phase 2, single-arm trial, investigators enrolled adults with DLBCL that had relapsed or was refractory after two or more prior lines of therapy and who were either ineligible for hematopoietic stem cell transplant or who experienced disease progression after transplant.
Interim results of the study were previously reported at the European Hematology Association Congress in 2017.
At that meeting, Gilles Salles, MD, PhD, from the University of Lyon (France), presented results of an analysis of available efficacy data on 51 patients with at least 3 months of follow-up. In this population, the best overall response rate was 59%. The 3-month overall response rate was 45%, consisting of 37% complete responses and 8% partial responses. Relapse-free survival at 6 months was 79% and all patients who had responses at 3 months continued to have responses at the time of data cutoff.
JULIET now
In the most recent analysis, completed after a median time from infusion to data cutoff of 14 months, the investigators reported on efficacy in 93 patients who received CAR T-cell infusions.
The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses. The response rates were consistent across all prognostic subgroups, including age, sex, previous response status, International Prognostic Index score at enrollment, prior therapy, molecular subtype, and other factors.
Estimated relapse-free survival 12 months after documentation of an initial response was 65%, and was 79% among patients who had complete responses.
The median duration of response had not been reached at the time of data cutoff; the median overall survival had not been reached for patients with a complete remission. Overall survival in this heavily pretreated population as a whole (all patients who received CAR T-cell infusions) was 11.1 months.
Adverse events of special interest included grade 3 or 4 cytokine release syndrome (CRS) in 23% of patients, prolonged cytopenia in 34%, infections in 19%, neurologic events in 11%, febrile neutropenia in 15%, and tumor lysis syndrome in 2%.
There were no deaths attributable to CRS or to cerebral edema, a complication of CAR T-cell therapy that appears to be related to the costimulatory molecule used in various constructs.
“Patients with relapsed or refractory DLBCL who are not eligible for high-dose therapy and hematopoietic cell transplantation or for whom such therapy was not successful have very few treatment options. For these patients, tisagenlecleucel shows promise that will need to be confirmed through larger studies with longer follow-up,” the investigators wrote in the New England Journal of Medicine.
The JULIET Trial is supported by Novartis. Dr. Maziar reported personal fees from Incyte, Kite Therapeutics, and Athersys.
SOURCE: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.
REPORTING FROM ASH 2018
Key clinical point: Chimeric antigen receptor T-cell therapy produced durable responses in patients with heavily pretreated diffuse large B-cell lymphoma.
Major finding: The best overall response rate, the primary endpoint, was 52%, comprising 40% complete responses and 12% partial responses.
Study details: A single-arm, open-label study of tisagenlecleucel in adults with relapsed or refractory diffuse large B-cell lymphoma.
Disclosures: The JULIET trial is supported by Novartis. Dr. Maziarz reported personal fees from Incyte, Kite Therapeutics, and Athersys.
Source: Maziarz RT et al. N Engl J Med. 2018 Dec 1. doi: 10.1056/NEJMoa1804980.