User login
Top AGA Community patient cases
Physicians with difficult patient scenarios regularly bring their questions to the AGA Community (https://community.gastro.org) to seek advice from colleagues about therapy and disease management options, best practices, and diagnoses. In case you missed it, here are the most popular clinical discussions shared in the forum recently:
1. Severe lower GI bleed (http://ow.ly/iTrS30pOKaP) – A 15-year-old male patient was sent to the ER with severe lower GI bleed after a general physical exam revealed he was experiencing hypotension and tachycardia. The GI community discusses diagnostic possibilities for severe lower GI bleed at such young age and management options.
2. Refractory nausea and vomiting in a transgender patient (http://ow.ly/Di9C30pOKbq) – In this case of a 45-year-old transgender male-to-female patient, the community deliberates on several clinical issues, including a non-binary gender option on patient identification forms, treatment options for the patient and if hormonal therapy is contributing to GI symptoms.
3. Multidisciplinary guidelines (http://ow.ly/BtUK30pOKC8) – Are multidisciplinary guidelines with related specialty societies “the need of the hour” or too rare and short-lived for the effort?
Also in the forum: The AGA Clinical Practice Updates Committee is soliciting topics for future clinical expert review and commentaries commissioned by AGA. Share your ideas with the GI community (http://ow.ly/siV930pOJS1).
Access these clinical cases and more discussions at https://community.gastro.org/discussions.
Physicians with difficult patient scenarios regularly bring their questions to the AGA Community (https://community.gastro.org) to seek advice from colleagues about therapy and disease management options, best practices, and diagnoses. In case you missed it, here are the most popular clinical discussions shared in the forum recently:
1. Severe lower GI bleed (http://ow.ly/iTrS30pOKaP) – A 15-year-old male patient was sent to the ER with severe lower GI bleed after a general physical exam revealed he was experiencing hypotension and tachycardia. The GI community discusses diagnostic possibilities for severe lower GI bleed at such young age and management options.
2. Refractory nausea and vomiting in a transgender patient (http://ow.ly/Di9C30pOKbq) – In this case of a 45-year-old transgender male-to-female patient, the community deliberates on several clinical issues, including a non-binary gender option on patient identification forms, treatment options for the patient and if hormonal therapy is contributing to GI symptoms.
3. Multidisciplinary guidelines (http://ow.ly/BtUK30pOKC8) – Are multidisciplinary guidelines with related specialty societies “the need of the hour” or too rare and short-lived for the effort?
Also in the forum: The AGA Clinical Practice Updates Committee is soliciting topics for future clinical expert review and commentaries commissioned by AGA. Share your ideas with the GI community (http://ow.ly/siV930pOJS1).
Access these clinical cases and more discussions at https://community.gastro.org/discussions.
Physicians with difficult patient scenarios regularly bring their questions to the AGA Community (https://community.gastro.org) to seek advice from colleagues about therapy and disease management options, best practices, and diagnoses. In case you missed it, here are the most popular clinical discussions shared in the forum recently:
1. Severe lower GI bleed (http://ow.ly/iTrS30pOKaP) – A 15-year-old male patient was sent to the ER with severe lower GI bleed after a general physical exam revealed he was experiencing hypotension and tachycardia. The GI community discusses diagnostic possibilities for severe lower GI bleed at such young age and management options.
2. Refractory nausea and vomiting in a transgender patient (http://ow.ly/Di9C30pOKbq) – In this case of a 45-year-old transgender male-to-female patient, the community deliberates on several clinical issues, including a non-binary gender option on patient identification forms, treatment options for the patient and if hormonal therapy is contributing to GI symptoms.
3. Multidisciplinary guidelines (http://ow.ly/BtUK30pOKC8) – Are multidisciplinary guidelines with related specialty societies “the need of the hour” or too rare and short-lived for the effort?
Also in the forum: The AGA Clinical Practice Updates Committee is soliciting topics for future clinical expert review and commentaries commissioned by AGA. Share your ideas with the GI community (http://ow.ly/siV930pOJS1).
Access these clinical cases and more discussions at https://community.gastro.org/discussions.
Meet Donald Payne, Jr. – A staunch advocate for increasing access to colorectal cancer screening
Rep. Donald Payne Jr., D-N.J., has been representing the 10th congressional district of New Jersey which includes the Newark area and the thriving life-sciences community in the region since 2012. Rep. Payne Jr. ran to serve in the seat that his father, the late Rep.

Donald M. Payne, D-N.J., held for eleven terms until his untimely death in March 2012. The elder Payne succumbed to colon cancer 1 month after his initial diagnosis, and Rep. Payne Jr. has made it his personal mission since assuming his father’s seat to increase awareness of colorectal cancer screenings. In fact, shortly after entering office, Rep. Payne Jr. wrote an op-ed with AGA member Carla Ginsburg, MD, MPH, AGAF, on the importance of screening, relaying in deeply personal terms the cost of not getting screened.
Rep. Payne Jr. also made it a top priority to push national awareness of colon cancer screening beyond the halls of Congress. To that end, Rep. Payne Jr. successfully lobbied the Obama administration in 2014 to designate March as National Colorectal Cancer Awareness Month – the first colorectal cancer presidential proclamation in over than a decade. The presidential proclamation was subsequently reissued in both 2015 and 2016 by the Obama administration and in 2018 and 2019 by the Trump administration. Additionally, Rep. Payne Jr. introduces a resolution in the House of Representatives every year to designate March as National Colorectal Cancer Awareness Month in an effort to further promote awareness and educational activities of colorectal cancer screening in the chamber.
Most importantly, Rep. Payne Jr. is the lead champion of legislative efforts in the House to increase access to colorectal cancer screenings. Specifically, Rep. Payne Jr. is the lead sponsor of H.R. 1570, the Removing Barriers to Colorectal Cancer Screening Act, legislation that has been one of AGA’s top policy priorities. The legislation, which enjoys broad bipartisan support with over 300 cosponsors in the House, would correct the Medicare beneficiary coinsurance issue when a screening colonoscopy becomes therapeutic. He also is a strong supporter of biomedical research funding, noting in an op-ed with former Rep. Charlie Dent, R-Pa., that “scientists need stable, consistent, and robust funding to ensure that we can continue ... breakthroughs for the colorectal cancer community and beyond.”
AGA looks forward to continuing to work with Rep. Payne Jr. and his office in the 116th Congress on these critical issues and on policies affecting our patients and the field of gastroenterology.
Rep. Donald Payne Jr., D-N.J., has been representing the 10th congressional district of New Jersey which includes the Newark area and the thriving life-sciences community in the region since 2012. Rep. Payne Jr. ran to serve in the seat that his father, the late Rep.
Donald M. Payne, D-N.J., held for eleven terms until his untimely death in March 2012. The elder Payne succumbed to colon cancer 1 month after his initial diagnosis, and Rep. Payne Jr. has made it his personal mission since assuming his father’s seat to increase awareness of colorectal cancer screenings. In fact, shortly after entering office, Rep. Payne Jr. wrote an op-ed with AGA member Carla Ginsburg, MD, MPH, AGAF, on the importance of screening, relaying in deeply personal terms the cost of not getting screened.
Rep. Payne Jr. also made it a top priority to push national awareness of colon cancer screening beyond the halls of Congress. To that end, Rep. Payne Jr. successfully lobbied the Obama administration in 2014 to designate March as National Colorectal Cancer Awareness Month – the first colorectal cancer presidential proclamation in over than a decade. The presidential proclamation was subsequently reissued in both 2015 and 2016 by the Obama administration and in 2018 and 2019 by the Trump administration. Additionally, Rep. Payne Jr. introduces a resolution in the House of Representatives every year to designate March as National Colorectal Cancer Awareness Month in an effort to further promote awareness and educational activities of colorectal cancer screening in the chamber.
Most importantly, Rep. Payne Jr. is the lead champion of legislative efforts in the House to increase access to colorectal cancer screenings. Specifically, Rep. Payne Jr. is the lead sponsor of H.R. 1570, the Removing Barriers to Colorectal Cancer Screening Act, legislation that has been one of AGA’s top policy priorities. The legislation, which enjoys broad bipartisan support with over 300 cosponsors in the House, would correct the Medicare beneficiary coinsurance issue when a screening colonoscopy becomes therapeutic. He also is a strong supporter of biomedical research funding, noting in an op-ed with former Rep. Charlie Dent, R-Pa., that “scientists need stable, consistent, and robust funding to ensure that we can continue ... breakthroughs for the colorectal cancer community and beyond.”
AGA looks forward to continuing to work with Rep. Payne Jr. and his office in the 116th Congress on these critical issues and on policies affecting our patients and the field of gastroenterology.
Rep. Donald Payne Jr., D-N.J., has been representing the 10th congressional district of New Jersey which includes the Newark area and the thriving life-sciences community in the region since 2012. Rep. Payne Jr. ran to serve in the seat that his father, the late Rep.
Donald M. Payne, D-N.J., held for eleven terms until his untimely death in March 2012. The elder Payne succumbed to colon cancer 1 month after his initial diagnosis, and Rep. Payne Jr. has made it his personal mission since assuming his father’s seat to increase awareness of colorectal cancer screenings. In fact, shortly after entering office, Rep. Payne Jr. wrote an op-ed with AGA member Carla Ginsburg, MD, MPH, AGAF, on the importance of screening, relaying in deeply personal terms the cost of not getting screened.
Rep. Payne Jr. also made it a top priority to push national awareness of colon cancer screening beyond the halls of Congress. To that end, Rep. Payne Jr. successfully lobbied the Obama administration in 2014 to designate March as National Colorectal Cancer Awareness Month – the first colorectal cancer presidential proclamation in over than a decade. The presidential proclamation was subsequently reissued in both 2015 and 2016 by the Obama administration and in 2018 and 2019 by the Trump administration. Additionally, Rep. Payne Jr. introduces a resolution in the House of Representatives every year to designate March as National Colorectal Cancer Awareness Month in an effort to further promote awareness and educational activities of colorectal cancer screening in the chamber.
Most importantly, Rep. Payne Jr. is the lead champion of legislative efforts in the House to increase access to colorectal cancer screenings. Specifically, Rep. Payne Jr. is the lead sponsor of H.R. 1570, the Removing Barriers to Colorectal Cancer Screening Act, legislation that has been one of AGA’s top policy priorities. The legislation, which enjoys broad bipartisan support with over 300 cosponsors in the House, would correct the Medicare beneficiary coinsurance issue when a screening colonoscopy becomes therapeutic. He also is a strong supporter of biomedical research funding, noting in an op-ed with former Rep. Charlie Dent, R-Pa., that “scientists need stable, consistent, and robust funding to ensure that we can continue ... breakthroughs for the colorectal cancer community and beyond.”
AGA looks forward to continuing to work with Rep. Payne Jr. and his office in the 116th Congress on these critical issues and on policies affecting our patients and the field of gastroenterology.
HVPG predicts clinical benefit after sustained virologic response
BOSTON – For patients with hepatitis C virus infection who achieve sustained virologic response to interferon-free therapy, changes in hepatic venous pressure gradient (HVPG) predict clinical benefit, according to investigators.

This finding will allow investigators to use HVPG as a surrogate endpoint for etiologic therapies, which could accelerate future research, reported lead author Mattias Mandorfer, MD, PhD, of the Medical University of Vienna and colleagues.
“Sustained virologic response to interferon-free therapies ameliorates portal hypertension,” Dr. Mandorfer said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. “[Previous research has shown that] nearly two-thirds of patients with pretreatment clinically significant portal hypertension had an HVPG decrease above or equal to 10%, which denotes a clinically meaningful change according to current recommendations. However, evidence is limited to studies evaluating the impact of HVPG response to nonselective beta-blockers, and nonselective beta-blockers have a completely different mode of action than etiological therapies. Accordingly, it is unclear whether a decrease in HVPG after the cure of hepatitis C translates into the same clinical benefit.”
To find out, the investigators enrolled 90 patients with hepatitis C virus who had an elevated HVPG of 6 mm Hg or higher prior to sustained virologic response. Before and after interferon-free therapy, patients underwent paired HVPG measurement. In addition, to evaluate noninvasive methods of HVPG assessment, transient elastography and von Willebrand factor to platelet count ratio testing were performed.
Analysis showed that HVPG measurements after, but not before, interferon-free therapy predicted liver decompensation. Specifically, HVPG was associated with an 18% increased risk of hepatic decompensation per mm Hg. After 3 years, 40.1% of patients with posttherapy HVPG measurements of 16 mm Hg or more developed hepatic decompensation, an event that occurred in none of the patients with a posttherapy HVPG of 9 mm Hg or less. Among patients who had a baseline HVPG of 10 mm Hg or more, which is considered a clinically significant level of portal hypertension, a decrease in HVPG of least 10% after therapy was associated with a similar level of protection against decompensation, compared with those who had no such decrease (2.5% vs. 31.8%).
While the two noninvasive methods (transient elastography and von Willebrand factor to platelet count ratio) were able to detect clinically significant portal hypertension (at least 10 mm Hg), they were not accurate enough to detect the protective 10% drop in HVPG.
“These results support the concept of applying HVPG as a surrogate endpoint for interventions that primarily aim at decreasing intrahepatic resistance (e.g., etiological therapies),” the investigators concluded in their abstract.
Jaime Bosch, MD, PhD, of the University of Barcelona provided some expert insight into the findings.
“The significance of the work is very important,” Dr. Bosch said in a public comment. “This provides, for the first time, firm evidence that HVPG can be taken as a surrogate endpoint ... for studies involving portal hypertension and cirrhosis in general.”
In an interview, Dr. Bosch elaborated on this statement. “The problem is, it takes a long time to get rid of cirrhosis [after sustained virologic response], and meanwhile, as long as portal hypertension remains, there is a risk for decompensation, so the patients cannot be said to be cured. They are cured of the infection, of the consequences of the infection, but it may take 10 years or more [to resolve cirrhosis], so the patient needs clinical surveillance and treatment after curing the cause of the disease.
“An academic consequence of these findings is that they’ve proved that decreasing HVPG by means of achieving sustained virologic response is followed by an improvement in prognosis. ... And when you can influence prognosis, and the influence in prognosis is reflected by a measurement independent from the way that we achieve this effect on the measurement, it means that this measurement is robust and now has to be used as a surrogate marker of resolution of cirrhosis.”
The study was funded by the Medical Scientific Fund of the city of Vienna. The investigators disclosed relationships with AbbVie, Bristol-Myers Squibb, Gilead, and others.
SOURCE: Mandorfer M et al. The Liver Meeting 2019, Abstract 146.
BOSTON – For patients with hepatitis C virus infection who achieve sustained virologic response to interferon-free therapy, changes in hepatic venous pressure gradient (HVPG) predict clinical benefit, according to investigators.
This finding will allow investigators to use HVPG as a surrogate endpoint for etiologic therapies, which could accelerate future research, reported lead author Mattias Mandorfer, MD, PhD, of the Medical University of Vienna and colleagues.
“Sustained virologic response to interferon-free therapies ameliorates portal hypertension,” Dr. Mandorfer said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. “[Previous research has shown that] nearly two-thirds of patients with pretreatment clinically significant portal hypertension had an HVPG decrease above or equal to 10%, which denotes a clinically meaningful change according to current recommendations. However, evidence is limited to studies evaluating the impact of HVPG response to nonselective beta-blockers, and nonselective beta-blockers have a completely different mode of action than etiological therapies. Accordingly, it is unclear whether a decrease in HVPG after the cure of hepatitis C translates into the same clinical benefit.”
To find out, the investigators enrolled 90 patients with hepatitis C virus who had an elevated HVPG of 6 mm Hg or higher prior to sustained virologic response. Before and after interferon-free therapy, patients underwent paired HVPG measurement. In addition, to evaluate noninvasive methods of HVPG assessment, transient elastography and von Willebrand factor to platelet count ratio testing were performed.
Analysis showed that HVPG measurements after, but not before, interferon-free therapy predicted liver decompensation. Specifically, HVPG was associated with an 18% increased risk of hepatic decompensation per mm Hg. After 3 years, 40.1% of patients with posttherapy HVPG measurements of 16 mm Hg or more developed hepatic decompensation, an event that occurred in none of the patients with a posttherapy HVPG of 9 mm Hg or less. Among patients who had a baseline HVPG of 10 mm Hg or more, which is considered a clinically significant level of portal hypertension, a decrease in HVPG of least 10% after therapy was associated with a similar level of protection against decompensation, compared with those who had no such decrease (2.5% vs. 31.8%).
While the two noninvasive methods (transient elastography and von Willebrand factor to platelet count ratio) were able to detect clinically significant portal hypertension (at least 10 mm Hg), they were not accurate enough to detect the protective 10% drop in HVPG.
“These results support the concept of applying HVPG as a surrogate endpoint for interventions that primarily aim at decreasing intrahepatic resistance (e.g., etiological therapies),” the investigators concluded in their abstract.
Jaime Bosch, MD, PhD, of the University of Barcelona provided some expert insight into the findings.
“The significance of the work is very important,” Dr. Bosch said in a public comment. “This provides, for the first time, firm evidence that HVPG can be taken as a surrogate endpoint ... for studies involving portal hypertension and cirrhosis in general.”
In an interview, Dr. Bosch elaborated on this statement. “The problem is, it takes a long time to get rid of cirrhosis [after sustained virologic response], and meanwhile, as long as portal hypertension remains, there is a risk for decompensation, so the patients cannot be said to be cured. They are cured of the infection, of the consequences of the infection, but it may take 10 years or more [to resolve cirrhosis], so the patient needs clinical surveillance and treatment after curing the cause of the disease.
“An academic consequence of these findings is that they’ve proved that decreasing HVPG by means of achieving sustained virologic response is followed by an improvement in prognosis. ... And when you can influence prognosis, and the influence in prognosis is reflected by a measurement independent from the way that we achieve this effect on the measurement, it means that this measurement is robust and now has to be used as a surrogate marker of resolution of cirrhosis.”
The study was funded by the Medical Scientific Fund of the city of Vienna. The investigators disclosed relationships with AbbVie, Bristol-Myers Squibb, Gilead, and others.
SOURCE: Mandorfer M et al. The Liver Meeting 2019, Abstract 146.
BOSTON – For patients with hepatitis C virus infection who achieve sustained virologic response to interferon-free therapy, changes in hepatic venous pressure gradient (HVPG) predict clinical benefit, according to investigators.
This finding will allow investigators to use HVPG as a surrogate endpoint for etiologic therapies, which could accelerate future research, reported lead author Mattias Mandorfer, MD, PhD, of the Medical University of Vienna and colleagues.
“Sustained virologic response to interferon-free therapies ameliorates portal hypertension,” Dr. Mandorfer said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. “[Previous research has shown that] nearly two-thirds of patients with pretreatment clinically significant portal hypertension had an HVPG decrease above or equal to 10%, which denotes a clinically meaningful change according to current recommendations. However, evidence is limited to studies evaluating the impact of HVPG response to nonselective beta-blockers, and nonselective beta-blockers have a completely different mode of action than etiological therapies. Accordingly, it is unclear whether a decrease in HVPG after the cure of hepatitis C translates into the same clinical benefit.”
To find out, the investigators enrolled 90 patients with hepatitis C virus who had an elevated HVPG of 6 mm Hg or higher prior to sustained virologic response. Before and after interferon-free therapy, patients underwent paired HVPG measurement. In addition, to evaluate noninvasive methods of HVPG assessment, transient elastography and von Willebrand factor to platelet count ratio testing were performed.
Analysis showed that HVPG measurements after, but not before, interferon-free therapy predicted liver decompensation. Specifically, HVPG was associated with an 18% increased risk of hepatic decompensation per mm Hg. After 3 years, 40.1% of patients with posttherapy HVPG measurements of 16 mm Hg or more developed hepatic decompensation, an event that occurred in none of the patients with a posttherapy HVPG of 9 mm Hg or less. Among patients who had a baseline HVPG of 10 mm Hg or more, which is considered a clinically significant level of portal hypertension, a decrease in HVPG of least 10% after therapy was associated with a similar level of protection against decompensation, compared with those who had no such decrease (2.5% vs. 31.8%).
While the two noninvasive methods (transient elastography and von Willebrand factor to platelet count ratio) were able to detect clinically significant portal hypertension (at least 10 mm Hg), they were not accurate enough to detect the protective 10% drop in HVPG.
“These results support the concept of applying HVPG as a surrogate endpoint for interventions that primarily aim at decreasing intrahepatic resistance (e.g., etiological therapies),” the investigators concluded in their abstract.
Jaime Bosch, MD, PhD, of the University of Barcelona provided some expert insight into the findings.
“The significance of the work is very important,” Dr. Bosch said in a public comment. “This provides, for the first time, firm evidence that HVPG can be taken as a surrogate endpoint ... for studies involving portal hypertension and cirrhosis in general.”
In an interview, Dr. Bosch elaborated on this statement. “The problem is, it takes a long time to get rid of cirrhosis [after sustained virologic response], and meanwhile, as long as portal hypertension remains, there is a risk for decompensation, so the patients cannot be said to be cured. They are cured of the infection, of the consequences of the infection, but it may take 10 years or more [to resolve cirrhosis], so the patient needs clinical surveillance and treatment after curing the cause of the disease.
“An academic consequence of these findings is that they’ve proved that decreasing HVPG by means of achieving sustained virologic response is followed by an improvement in prognosis. ... And when you can influence prognosis, and the influence in prognosis is reflected by a measurement independent from the way that we achieve this effect on the measurement, it means that this measurement is robust and now has to be used as a surrogate marker of resolution of cirrhosis.”
The study was funded by the Medical Scientific Fund of the city of Vienna. The investigators disclosed relationships with AbbVie, Bristol-Myers Squibb, Gilead, and others.
SOURCE: Mandorfer M et al. The Liver Meeting 2019, Abstract 146.
REPORTING FROM THE LIVER MEETING 2019
Health policy Q&A: Oncology Care Model
The Oncology Care Model is a value-based payment approach aimed at encouraging coordinated cancer care through targeted bonus payments to practices. The payment experiment was launched by the Centers for Medicare & Medicaid Services in 2016 and now includes 175 practices and 10 payers. It is set to end in 2021. As agency officials consider whether to continue the program, Stephen S. Grubbs, MD, vice president for clinical affairs at the American Society of Clinical Oncology, weighs in on the model’s track record and its future.

Question: How would you rate the Oncology Care Model in helping to drive practice transformation?
Dr. Grubbs: Participants in the Oncology Care Model (OCM) have demonstrated improved care coordination, psychosocial support, use of risk assessment tools, and other strategies to lower costs and adverse events. Over the past 2 years, ASCO has accepted numerous posters, articles, and abstracts from OCM participants on their outstanding work to advance cancer care delivery.
Question: Should the model be extended beyond 2021?
Dr. Grubbs: Changes to the model are necessary prior to a significant extension or expansion. Some have suggested that CMS extend OCM for an additional year with current participants. This would give CMS time to consider input from all stakeholders on its eventual replacement.
Question: What additional resources or payments do oncology practices need to be more successful in meeting the goals of the Oncology Care Model?
Dr. Grubbs: OCM has shown that by providing oncologists with payment for care management – OCM participants receive $160 per patient, per month – the results are better care coordination and reduced hospital and emergency department visits. If CMS chooses to expand payments to all oncology providers, we could expect to see improved care for cancer patients.
Question: ASCO has advanced its own Patient-Centered Oncology Payment model. What are the main elements of this strategy and how does it differ from the Oncology Care Model?
Dr. Grubbs: The Patient-Centered Oncology Payment (PCOP) model is the result of input from a wide group of stakeholders, including providers, employers, and managed care organizations. In the coming months, ASCO will publish an updated copy of the PCOP model.

Our review of OCM is that the included prediction model and two-sided risk options place small, rural, and certain other practices at considerable peril because of imprecise and inconsistent cost predictions. PCOP takes a different approach. Rather than requiring that practices take on actuarial risk for total cost of care, PCOP includes a three-part performance methodology. Practices are measured on adherence to clinical treatment pathways; electronically capturable quality measures; and select, targeted cost-of-care measures. Practices that perform well in PCOP’s performance methodology receive increased incentive payments to fund further advancements in care.
Question: The PCOP model includes payments to oncology practices for participation in clinical trials. How might that drive a change in behavior in a typical practice?
Dr. Grubbs: Practices that enroll patients in clinical trials have the same or greater storage and handling requirements as those treated with standard treatments, yet forgo revenue associated with the Medicare Part B average sales price methodology. PCOP ensures that such practices are not disadvantaged for supporting clinical research.
Question: Are there other areas – such as tumor biomarker tests – in which a tailored payment approach would improve the quality of care?
Dr. Grubbs: Recent studies have shown that not all patients receive the appropriate genomic profiling and other tests necessary to ensure that they benefit from personalized therapies. Clinical treatment pathways have the ability to inform and measure diagnostic completeness to improve the quality of care.
Question: What are the barriers that are keeping oncology practices from participating in alternative payment models designed to improve care?
Dr. Grubbs: Some alternative payment models, such as OCM, place a high administrative burden on their participants. Manual reporting of measures and clinical data, complicated billing requirements, and lack of support from electronic health record vendors create barriers for expanded participation. Practices are also concerned about the financial risks placed upon participants; it is impractical to expect that physicians hire actuaries in order to participate in a Medicare program.
ASCO has offered support for OCM practices through its PracticeNET benchmarking program, but we have also proposed PCOP as an appropriate alternative, applicable to practices of all types and sizes.
Dr. Grubbs joined ASCO in 2015 as the vice president of the newly launched clinical affairs department. Before joining ASCO, Dr. Grubbs worked as a community oncologist and managing partner at Medical Oncology Hematology Consultants in Newark, Del. Dr. Grubbs is a volunteer and the principal investigator of the Delaware Christiana Care National Cancer Institute Community Oncology Research Program. Dr. Grubbs reported having no financial disclosures.
The Oncology Care Model is a value-based payment approach aimed at encouraging coordinated cancer care through targeted bonus payments to practices. The payment experiment was launched by the Centers for Medicare & Medicaid Services in 2016 and now includes 175 practices and 10 payers. It is set to end in 2021. As agency officials consider whether to continue the program, Stephen S. Grubbs, MD, vice president for clinical affairs at the American Society of Clinical Oncology, weighs in on the model’s track record and its future.
Question: How would you rate the Oncology Care Model in helping to drive practice transformation?
Dr. Grubbs: Participants in the Oncology Care Model (OCM) have demonstrated improved care coordination, psychosocial support, use of risk assessment tools, and other strategies to lower costs and adverse events. Over the past 2 years, ASCO has accepted numerous posters, articles, and abstracts from OCM participants on their outstanding work to advance cancer care delivery.
Question: Should the model be extended beyond 2021?
Dr. Grubbs: Changes to the model are necessary prior to a significant extension or expansion. Some have suggested that CMS extend OCM for an additional year with current participants. This would give CMS time to consider input from all stakeholders on its eventual replacement.
Question: What additional resources or payments do oncology practices need to be more successful in meeting the goals of the Oncology Care Model?
Dr. Grubbs: OCM has shown that by providing oncologists with payment for care management – OCM participants receive $160 per patient, per month – the results are better care coordination and reduced hospital and emergency department visits. If CMS chooses to expand payments to all oncology providers, we could expect to see improved care for cancer patients.
Question: ASCO has advanced its own Patient-Centered Oncology Payment model. What are the main elements of this strategy and how does it differ from the Oncology Care Model?
Dr. Grubbs: The Patient-Centered Oncology Payment (PCOP) model is the result of input from a wide group of stakeholders, including providers, employers, and managed care organizations. In the coming months, ASCO will publish an updated copy of the PCOP model.
Our review of OCM is that the included prediction model and two-sided risk options place small, rural, and certain other practices at considerable peril because of imprecise and inconsistent cost predictions. PCOP takes a different approach. Rather than requiring that practices take on actuarial risk for total cost of care, PCOP includes a three-part performance methodology. Practices are measured on adherence to clinical treatment pathways; electronically capturable quality measures; and select, targeted cost-of-care measures. Practices that perform well in PCOP’s performance methodology receive increased incentive payments to fund further advancements in care.
Question: The PCOP model includes payments to oncology practices for participation in clinical trials. How might that drive a change in behavior in a typical practice?
Dr. Grubbs: Practices that enroll patients in clinical trials have the same or greater storage and handling requirements as those treated with standard treatments, yet forgo revenue associated with the Medicare Part B average sales price methodology. PCOP ensures that such practices are not disadvantaged for supporting clinical research.
Question: Are there other areas – such as tumor biomarker tests – in which a tailored payment approach would improve the quality of care?
Dr. Grubbs: Recent studies have shown that not all patients receive the appropriate genomic profiling and other tests necessary to ensure that they benefit from personalized therapies. Clinical treatment pathways have the ability to inform and measure diagnostic completeness to improve the quality of care.
Question: What are the barriers that are keeping oncology practices from participating in alternative payment models designed to improve care?
Dr. Grubbs: Some alternative payment models, such as OCM, place a high administrative burden on their participants. Manual reporting of measures and clinical data, complicated billing requirements, and lack of support from electronic health record vendors create barriers for expanded participation. Practices are also concerned about the financial risks placed upon participants; it is impractical to expect that physicians hire actuaries in order to participate in a Medicare program.
ASCO has offered support for OCM practices through its PracticeNET benchmarking program, but we have also proposed PCOP as an appropriate alternative, applicable to practices of all types and sizes.
Dr. Grubbs joined ASCO in 2015 as the vice president of the newly launched clinical affairs department. Before joining ASCO, Dr. Grubbs worked as a community oncologist and managing partner at Medical Oncology Hematology Consultants in Newark, Del. Dr. Grubbs is a volunteer and the principal investigator of the Delaware Christiana Care National Cancer Institute Community Oncology Research Program. Dr. Grubbs reported having no financial disclosures.
The Oncology Care Model is a value-based payment approach aimed at encouraging coordinated cancer care through targeted bonus payments to practices. The payment experiment was launched by the Centers for Medicare & Medicaid Services in 2016 and now includes 175 practices and 10 payers. It is set to end in 2021. As agency officials consider whether to continue the program, Stephen S. Grubbs, MD, vice president for clinical affairs at the American Society of Clinical Oncology, weighs in on the model’s track record and its future.
Question: How would you rate the Oncology Care Model in helping to drive practice transformation?
Dr. Grubbs: Participants in the Oncology Care Model (OCM) have demonstrated improved care coordination, psychosocial support, use of risk assessment tools, and other strategies to lower costs and adverse events. Over the past 2 years, ASCO has accepted numerous posters, articles, and abstracts from OCM participants on their outstanding work to advance cancer care delivery.
Question: Should the model be extended beyond 2021?
Dr. Grubbs: Changes to the model are necessary prior to a significant extension or expansion. Some have suggested that CMS extend OCM for an additional year with current participants. This would give CMS time to consider input from all stakeholders on its eventual replacement.
Question: What additional resources or payments do oncology practices need to be more successful in meeting the goals of the Oncology Care Model?
Dr. Grubbs: OCM has shown that by providing oncologists with payment for care management – OCM participants receive $160 per patient, per month – the results are better care coordination and reduced hospital and emergency department visits. If CMS chooses to expand payments to all oncology providers, we could expect to see improved care for cancer patients.
Question: ASCO has advanced its own Patient-Centered Oncology Payment model. What are the main elements of this strategy and how does it differ from the Oncology Care Model?
Dr. Grubbs: The Patient-Centered Oncology Payment (PCOP) model is the result of input from a wide group of stakeholders, including providers, employers, and managed care organizations. In the coming months, ASCO will publish an updated copy of the PCOP model.
Our review of OCM is that the included prediction model and two-sided risk options place small, rural, and certain other practices at considerable peril because of imprecise and inconsistent cost predictions. PCOP takes a different approach. Rather than requiring that practices take on actuarial risk for total cost of care, PCOP includes a three-part performance methodology. Practices are measured on adherence to clinical treatment pathways; electronically capturable quality measures; and select, targeted cost-of-care measures. Practices that perform well in PCOP’s performance methodology receive increased incentive payments to fund further advancements in care.
Question: The PCOP model includes payments to oncology practices for participation in clinical trials. How might that drive a change in behavior in a typical practice?
Dr. Grubbs: Practices that enroll patients in clinical trials have the same or greater storage and handling requirements as those treated with standard treatments, yet forgo revenue associated with the Medicare Part B average sales price methodology. PCOP ensures that such practices are not disadvantaged for supporting clinical research.
Question: Are there other areas – such as tumor biomarker tests – in which a tailored payment approach would improve the quality of care?
Dr. Grubbs: Recent studies have shown that not all patients receive the appropriate genomic profiling and other tests necessary to ensure that they benefit from personalized therapies. Clinical treatment pathways have the ability to inform and measure diagnostic completeness to improve the quality of care.
Question: What are the barriers that are keeping oncology practices from participating in alternative payment models designed to improve care?
Dr. Grubbs: Some alternative payment models, such as OCM, place a high administrative burden on their participants. Manual reporting of measures and clinical data, complicated billing requirements, and lack of support from electronic health record vendors create barriers for expanded participation. Practices are also concerned about the financial risks placed upon participants; it is impractical to expect that physicians hire actuaries in order to participate in a Medicare program.
ASCO has offered support for OCM practices through its PracticeNET benchmarking program, but we have also proposed PCOP as an appropriate alternative, applicable to practices of all types and sizes.
Dr. Grubbs joined ASCO in 2015 as the vice president of the newly launched clinical affairs department. Before joining ASCO, Dr. Grubbs worked as a community oncologist and managing partner at Medical Oncology Hematology Consultants in Newark, Del. Dr. Grubbs is a volunteer and the principal investigator of the Delaware Christiana Care National Cancer Institute Community Oncology Research Program. Dr. Grubbs reported having no financial disclosures.
Pediatric study characterizes recurrent PSC
BOSTON – Children who have recurrence of primary sclerosing cholangitis after liver transplant tend to be younger and have more rapidly progressive disease, based on an international retrospective analysis.

Within 5 years of transplant, the probability of primary sclerosing cholangitis (PSC) recurrence in pediatric patients is 26%, reported lead author Mercedes Martinez, MD, of Columbia University, New York, and colleagues.
“The aim of our study was to identify risk factors for primary sclerosing cholangitis recurrence following transplant,” Dr. Martinez said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. This may be the largest pediatric study evaluating recurrent PSC to date, she added.
The investigators drew data from 35 centers around the world via the Pediatric PSC Consortium database. Recurrence was defined by cholestatic biochemistry with nonanastomotic biliary strictures and beading of bile ducts on cholangiography. Recurrences caused by hepatic artery thrombosis or chronic rejection were excluded, as were any cases that recurred within 6 months of transplant.
The final analysis included 149 patients with a median age at diagnosis and liver transplant of 12 years and 15.4 years, respectively. Of these, 31 patients had recurrence after a median of 3.3 years. A closer look at the data showed that recurrence was linked with younger median age at time of transplant (13.2 vs. 16.2 years). In cases of recurrence, PSC was generally more aggressive prior to transplant, with a shorter interval between diagnosis and transplant (1.6 vs. 4.1 years), higher total bilirubin (7.8 vs. 3.8 mg/dL), and higher ALT (118 vs. 62 U/L). Furthermore, almost half of the patients (45%) who had recurrence also had pretransplant autoimmune hepatitis overlap, compared with approximately one-quarter of the patients (27%) who did not have recurrence, although this trend was not statistically significant (P = .06).
Recurrent PSC was also associated with poorer outcomes; almost half of those with recurrence (48%) were relisted for liver transplant, developed portal hypertension, or died within 2 years of diagnosis. Mean rejection rates were higher in recurrent versus nonrecurrent cases (3 vs. 1); recurrent cases also had shorter time until rejection (3 vs. 6 months) and greater prevalence of rejection that was refractory to steroids (23% vs. 12%). Moreover, a significantly greater proportion of patients with recurrence had Epstein-Barr viremia (41% vs. 21%).
Dr. Martinez noted that ongoing therapy involving mammalian target of rapamycin inhibition was associated with lower rates of recurrence and suggested that this deserves further investigation; however, owing to small population size, she urged a cautious interpretation of this finding.
“We have to do prospective research,” Dr. Martinez said, emphasizing that tissue immunophenotyping was needed, as a better understanding of underlying immune processes and disease subtypes may open doors to more effective therapies.
The investigators disclosed relationships with Gilead, Merck, Novartis, and others.
SOURCE: Martinez M et al. The Liver Meeting 2019, Abstract 44.
BOSTON – Children who have recurrence of primary sclerosing cholangitis after liver transplant tend to be younger and have more rapidly progressive disease, based on an international retrospective analysis.
Within 5 years of transplant, the probability of primary sclerosing cholangitis (PSC) recurrence in pediatric patients is 26%, reported lead author Mercedes Martinez, MD, of Columbia University, New York, and colleagues.
“The aim of our study was to identify risk factors for primary sclerosing cholangitis recurrence following transplant,” Dr. Martinez said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. This may be the largest pediatric study evaluating recurrent PSC to date, she added.
The investigators drew data from 35 centers around the world via the Pediatric PSC Consortium database. Recurrence was defined by cholestatic biochemistry with nonanastomotic biliary strictures and beading of bile ducts on cholangiography. Recurrences caused by hepatic artery thrombosis or chronic rejection were excluded, as were any cases that recurred within 6 months of transplant.
The final analysis included 149 patients with a median age at diagnosis and liver transplant of 12 years and 15.4 years, respectively. Of these, 31 patients had recurrence after a median of 3.3 years. A closer look at the data showed that recurrence was linked with younger median age at time of transplant (13.2 vs. 16.2 years). In cases of recurrence, PSC was generally more aggressive prior to transplant, with a shorter interval between diagnosis and transplant (1.6 vs. 4.1 years), higher total bilirubin (7.8 vs. 3.8 mg/dL), and higher ALT (118 vs. 62 U/L). Furthermore, almost half of the patients (45%) who had recurrence also had pretransplant autoimmune hepatitis overlap, compared with approximately one-quarter of the patients (27%) who did not have recurrence, although this trend was not statistically significant (P = .06).
Recurrent PSC was also associated with poorer outcomes; almost half of those with recurrence (48%) were relisted for liver transplant, developed portal hypertension, or died within 2 years of diagnosis. Mean rejection rates were higher in recurrent versus nonrecurrent cases (3 vs. 1); recurrent cases also had shorter time until rejection (3 vs. 6 months) and greater prevalence of rejection that was refractory to steroids (23% vs. 12%). Moreover, a significantly greater proportion of patients with recurrence had Epstein-Barr viremia (41% vs. 21%).
Dr. Martinez noted that ongoing therapy involving mammalian target of rapamycin inhibition was associated with lower rates of recurrence and suggested that this deserves further investigation; however, owing to small population size, she urged a cautious interpretation of this finding.
“We have to do prospective research,” Dr. Martinez said, emphasizing that tissue immunophenotyping was needed, as a better understanding of underlying immune processes and disease subtypes may open doors to more effective therapies.
The investigators disclosed relationships with Gilead, Merck, Novartis, and others.
SOURCE: Martinez M et al. The Liver Meeting 2019, Abstract 44.
BOSTON – Children who have recurrence of primary sclerosing cholangitis after liver transplant tend to be younger and have more rapidly progressive disease, based on an international retrospective analysis.
Within 5 years of transplant, the probability of primary sclerosing cholangitis (PSC) recurrence in pediatric patients is 26%, reported lead author Mercedes Martinez, MD, of Columbia University, New York, and colleagues.
“The aim of our study was to identify risk factors for primary sclerosing cholangitis recurrence following transplant,” Dr. Martinez said during a presentation at the annual meeting of the American Association for the Study of Liver Diseases. This may be the largest pediatric study evaluating recurrent PSC to date, she added.
The investigators drew data from 35 centers around the world via the Pediatric PSC Consortium database. Recurrence was defined by cholestatic biochemistry with nonanastomotic biliary strictures and beading of bile ducts on cholangiography. Recurrences caused by hepatic artery thrombosis or chronic rejection were excluded, as were any cases that recurred within 6 months of transplant.
The final analysis included 149 patients with a median age at diagnosis and liver transplant of 12 years and 15.4 years, respectively. Of these, 31 patients had recurrence after a median of 3.3 years. A closer look at the data showed that recurrence was linked with younger median age at time of transplant (13.2 vs. 16.2 years). In cases of recurrence, PSC was generally more aggressive prior to transplant, with a shorter interval between diagnosis and transplant (1.6 vs. 4.1 years), higher total bilirubin (7.8 vs. 3.8 mg/dL), and higher ALT (118 vs. 62 U/L). Furthermore, almost half of the patients (45%) who had recurrence also had pretransplant autoimmune hepatitis overlap, compared with approximately one-quarter of the patients (27%) who did not have recurrence, although this trend was not statistically significant (P = .06).
Recurrent PSC was also associated with poorer outcomes; almost half of those with recurrence (48%) were relisted for liver transplant, developed portal hypertension, or died within 2 years of diagnosis. Mean rejection rates were higher in recurrent versus nonrecurrent cases (3 vs. 1); recurrent cases also had shorter time until rejection (3 vs. 6 months) and greater prevalence of rejection that was refractory to steroids (23% vs. 12%). Moreover, a significantly greater proportion of patients with recurrence had Epstein-Barr viremia (41% vs. 21%).
Dr. Martinez noted that ongoing therapy involving mammalian target of rapamycin inhibition was associated with lower rates of recurrence and suggested that this deserves further investigation; however, owing to small population size, she urged a cautious interpretation of this finding.
“We have to do prospective research,” Dr. Martinez said, emphasizing that tissue immunophenotyping was needed, as a better understanding of underlying immune processes and disease subtypes may open doors to more effective therapies.
The investigators disclosed relationships with Gilead, Merck, Novartis, and others.
SOURCE: Martinez M et al. The Liver Meeting 2019, Abstract 44.
REPORTING FROM THE LIVER MEETING 2019
Could the biosimilar market stall before it ever really started?
NATIONAL HARBOR, MD. – If the United States does not step up and create a thriving biosimilars market soon, it risks destroying the market not only domestically but internationally as well.

This was the warning Gillian Woollett, senior vice president at Avalere, provided to attendees at the annual meeting of the Academy of Managed Care Pharmacy.
She prefaced her warning by quoting Alex Azar, secretary of Health & Human Services, who said that those “trying to hold back biosimilars are simply on the wrong side of history,” though Ms. Woollett said they “may be on the right side of the current economic model in the United States.”
And despite the probusiness, procompetition philosophy of current HHS leadership, there has been very little movement on creating a competitive market for biosimilars in the United States, evidenced by the very expensive regulatory requirements that biosimilar manufacturers need to meet in order to get products to market.
“It’s not that we won’t have competition in the U.S.,” she said. “I think we will. We do have that innovation. ... It’s just that biosimilars may not ultimately be part of that competition. And for that, we will pay a price, and I actually think the whole world will pay a price because if we are not providing the [return on investment], I am not sure the other markets can sustain it.”
One issue biosimilars have is the lack of recognition of the value that they bring.
“That biosimilars offer the same clinical outcomes at a lower price is yet to be a recognized value,” she said. “To me that’s a really surprising situation in the United States.”
Ms. Woollett disclosed no relevant conflicts of interest.
To prepare for the entry of biosimilars to the market, AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
NATIONAL HARBOR, MD. – If the United States does not step up and create a thriving biosimilars market soon, it risks destroying the market not only domestically but internationally as well.
This was the warning Gillian Woollett, senior vice president at Avalere, provided to attendees at the annual meeting of the Academy of Managed Care Pharmacy.
She prefaced her warning by quoting Alex Azar, secretary of Health & Human Services, who said that those “trying to hold back biosimilars are simply on the wrong side of history,” though Ms. Woollett said they “may be on the right side of the current economic model in the United States.”
And despite the probusiness, procompetition philosophy of current HHS leadership, there has been very little movement on creating a competitive market for biosimilars in the United States, evidenced by the very expensive regulatory requirements that biosimilar manufacturers need to meet in order to get products to market.
“It’s not that we won’t have competition in the U.S.,” she said. “I think we will. We do have that innovation. ... It’s just that biosimilars may not ultimately be part of that competition. And for that, we will pay a price, and I actually think the whole world will pay a price because if we are not providing the [return on investment], I am not sure the other markets can sustain it.”
One issue biosimilars have is the lack of recognition of the value that they bring.
“That biosimilars offer the same clinical outcomes at a lower price is yet to be a recognized value,” she said. “To me that’s a really surprising situation in the United States.”
Ms. Woollett disclosed no relevant conflicts of interest.
To prepare for the entry of biosimilars to the market, AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
NATIONAL HARBOR, MD. – If the United States does not step up and create a thriving biosimilars market soon, it risks destroying the market not only domestically but internationally as well.
This was the warning Gillian Woollett, senior vice president at Avalere, provided to attendees at the annual meeting of the Academy of Managed Care Pharmacy.
She prefaced her warning by quoting Alex Azar, secretary of Health & Human Services, who said that those “trying to hold back biosimilars are simply on the wrong side of history,” though Ms. Woollett said they “may be on the right side of the current economic model in the United States.”
And despite the probusiness, procompetition philosophy of current HHS leadership, there has been very little movement on creating a competitive market for biosimilars in the United States, evidenced by the very expensive regulatory requirements that biosimilar manufacturers need to meet in order to get products to market.
“It’s not that we won’t have competition in the U.S.,” she said. “I think we will. We do have that innovation. ... It’s just that biosimilars may not ultimately be part of that competition. And for that, we will pay a price, and I actually think the whole world will pay a price because if we are not providing the [return on investment], I am not sure the other markets can sustain it.”
One issue biosimilars have is the lack of recognition of the value that they bring.
“That biosimilars offer the same clinical outcomes at a lower price is yet to be a recognized value,” she said. “To me that’s a really surprising situation in the United States.”
Ms. Woollett disclosed no relevant conflicts of interest.
To prepare for the entry of biosimilars to the market, AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
Apremilast for Behçet’s oral ulcers: Benefits maintained at 64 weeks
MADRID – of the long-term extension phase of the pivotal RELIEF trial, Alfred Mahr, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.

“We now have strong evidence that apremilast is an effective and safe therapy to treat oral ulcers in patients with Behçet’s syndrome. I think this is a major advance in the field,” declared Dr. Mahr, a rheumatologist at St. Gallen (Switzerland) Cantonal Hospital.
Based largely upon the results of the 12-week, double-blind portion of the phase 3 RELIEF trial, the Food and Drug Administration approved apremilast (Otezla) for the treatment of oral ulcers in patients with Behçet’s disease in the summer of 2019.
The safety profile of the oral phosphodiesterase-4 inhibitor was as seen in other studies, including in patients with psoriatic arthritis, an FDA-approved indication for the drug since 2014. The main side effects in the long-term extension of RELIEF were diarrhea and nausea, typically mild or moderate in nature and roughly twice as frequent as in placebo-treated controls in the double-blind study phase.
“At the end of the day, at week 64, only 12% of patients treated with apremilast during the entire 64 weeks discontinued the drug due to a treatment-emergent adverse event, which I believe is a good indicator of the safety of this medication,” the rheumatologist said. “The overall feeling is that the benefit-to-risk ratio is very good and it’s a safe drug to prescribe.”
At the close of the initial 12-week, double-blind phase of RELIEF, 178 of the original 207 participants elected to enter the long-term extension, either staying on apremilast at 30 mg twice a day for an additional 52 weeks or switching to that regimen from placebo.
The focus of the long-term extension was on disease activity and quality of life outcomes. The results in patients who had switched from placebo to apremilast after 12 weeks proved to be reassuringly similar to outcomes in patients on the drug for the full duration. For example, the mean improvement on the patient-reported Behçet’s Syndrome Activity Scale was 18.6 points after 12 weeks of double-blind apremilast, 16.9 points after 64 weeks of continuous apremilast, and 16.8 points with 12 weeks of placebo followed by 52 weeks of active therapy.
After 12 weeks of double-blind apremilast, patients averaged a 3.4-point improvement on the Behçet’s Disease Quality of Life measure. After 64 weeks on the drug, the improvement over baseline was 3.6 points, while in the switch group it was 3.4 points. Similarly, on all three components of the SF-36 quality of life metric, the continuous apremilast group showed maintenance of effect from week 12 to week 64, while the placebo-to-apremilast group caught up. The same was true with regards to the Behçet’s Disease Current Activity Index, which encompasses measures of both the patient’s and clinician’s perception of disease activity.
At the outset of the RELIEF trial, participants averaged four oral ulcers. At week 64, the continuous apremilast group averaged 1.4 and the switch group 0.8, a nonsignificant difference.
Asked if apremilast had a favorable impact upon other manifestations of Behçet’s disease besides the oral ulcers, Dr. Mahr replied, “This is a very good question. People often wonder about it. We do, too. But this trial was not designed to capture less common manifestations of Behçet’s syndrome, such as genital ulcers. There have been some analyses done, but the number of patients who had genital ulcers at 12 weeks were very few. The same was true for eye manifestations. There was sort of a signal that it works, but we can’t prove it in a placebo-controlled trial.”
Dr. Mahr reported receiving research funding from and serving as a consultant to Celgene, the study sponsor.
MADRID – of the long-term extension phase of the pivotal RELIEF trial, Alfred Mahr, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We now have strong evidence that apremilast is an effective and safe therapy to treat oral ulcers in patients with Behçet’s syndrome. I think this is a major advance in the field,” declared Dr. Mahr, a rheumatologist at St. Gallen (Switzerland) Cantonal Hospital.
Based largely upon the results of the 12-week, double-blind portion of the phase 3 RELIEF trial, the Food and Drug Administration approved apremilast (Otezla) for the treatment of oral ulcers in patients with Behçet’s disease in the summer of 2019.
The safety profile of the oral phosphodiesterase-4 inhibitor was as seen in other studies, including in patients with psoriatic arthritis, an FDA-approved indication for the drug since 2014. The main side effects in the long-term extension of RELIEF were diarrhea and nausea, typically mild or moderate in nature and roughly twice as frequent as in placebo-treated controls in the double-blind study phase.
“At the end of the day, at week 64, only 12% of patients treated with apremilast during the entire 64 weeks discontinued the drug due to a treatment-emergent adverse event, which I believe is a good indicator of the safety of this medication,” the rheumatologist said. “The overall feeling is that the benefit-to-risk ratio is very good and it’s a safe drug to prescribe.”
At the close of the initial 12-week, double-blind phase of RELIEF, 178 of the original 207 participants elected to enter the long-term extension, either staying on apremilast at 30 mg twice a day for an additional 52 weeks or switching to that regimen from placebo.
The focus of the long-term extension was on disease activity and quality of life outcomes. The results in patients who had switched from placebo to apremilast after 12 weeks proved to be reassuringly similar to outcomes in patients on the drug for the full duration. For example, the mean improvement on the patient-reported Behçet’s Syndrome Activity Scale was 18.6 points after 12 weeks of double-blind apremilast, 16.9 points after 64 weeks of continuous apremilast, and 16.8 points with 12 weeks of placebo followed by 52 weeks of active therapy.
After 12 weeks of double-blind apremilast, patients averaged a 3.4-point improvement on the Behçet’s Disease Quality of Life measure. After 64 weeks on the drug, the improvement over baseline was 3.6 points, while in the switch group it was 3.4 points. Similarly, on all three components of the SF-36 quality of life metric, the continuous apremilast group showed maintenance of effect from week 12 to week 64, while the placebo-to-apremilast group caught up. The same was true with regards to the Behçet’s Disease Current Activity Index, which encompasses measures of both the patient’s and clinician’s perception of disease activity.
At the outset of the RELIEF trial, participants averaged four oral ulcers. At week 64, the continuous apremilast group averaged 1.4 and the switch group 0.8, a nonsignificant difference.
Asked if apremilast had a favorable impact upon other manifestations of Behçet’s disease besides the oral ulcers, Dr. Mahr replied, “This is a very good question. People often wonder about it. We do, too. But this trial was not designed to capture less common manifestations of Behçet’s syndrome, such as genital ulcers. There have been some analyses done, but the number of patients who had genital ulcers at 12 weeks were very few. The same was true for eye manifestations. There was sort of a signal that it works, but we can’t prove it in a placebo-controlled trial.”
Dr. Mahr reported receiving research funding from and serving as a consultant to Celgene, the study sponsor.
MADRID – of the long-term extension phase of the pivotal RELIEF trial, Alfred Mahr, MD, PhD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“We now have strong evidence that apremilast is an effective and safe therapy to treat oral ulcers in patients with Behçet’s syndrome. I think this is a major advance in the field,” declared Dr. Mahr, a rheumatologist at St. Gallen (Switzerland) Cantonal Hospital.
Based largely upon the results of the 12-week, double-blind portion of the phase 3 RELIEF trial, the Food and Drug Administration approved apremilast (Otezla) for the treatment of oral ulcers in patients with Behçet’s disease in the summer of 2019.
The safety profile of the oral phosphodiesterase-4 inhibitor was as seen in other studies, including in patients with psoriatic arthritis, an FDA-approved indication for the drug since 2014. The main side effects in the long-term extension of RELIEF were diarrhea and nausea, typically mild or moderate in nature and roughly twice as frequent as in placebo-treated controls in the double-blind study phase.
“At the end of the day, at week 64, only 12% of patients treated with apremilast during the entire 64 weeks discontinued the drug due to a treatment-emergent adverse event, which I believe is a good indicator of the safety of this medication,” the rheumatologist said. “The overall feeling is that the benefit-to-risk ratio is very good and it’s a safe drug to prescribe.”
At the close of the initial 12-week, double-blind phase of RELIEF, 178 of the original 207 participants elected to enter the long-term extension, either staying on apremilast at 30 mg twice a day for an additional 52 weeks or switching to that regimen from placebo.
The focus of the long-term extension was on disease activity and quality of life outcomes. The results in patients who had switched from placebo to apremilast after 12 weeks proved to be reassuringly similar to outcomes in patients on the drug for the full duration. For example, the mean improvement on the patient-reported Behçet’s Syndrome Activity Scale was 18.6 points after 12 weeks of double-blind apremilast, 16.9 points after 64 weeks of continuous apremilast, and 16.8 points with 12 weeks of placebo followed by 52 weeks of active therapy.
After 12 weeks of double-blind apremilast, patients averaged a 3.4-point improvement on the Behçet’s Disease Quality of Life measure. After 64 weeks on the drug, the improvement over baseline was 3.6 points, while in the switch group it was 3.4 points. Similarly, on all three components of the SF-36 quality of life metric, the continuous apremilast group showed maintenance of effect from week 12 to week 64, while the placebo-to-apremilast group caught up. The same was true with regards to the Behçet’s Disease Current Activity Index, which encompasses measures of both the patient’s and clinician’s perception of disease activity.
At the outset of the RELIEF trial, participants averaged four oral ulcers. At week 64, the continuous apremilast group averaged 1.4 and the switch group 0.8, a nonsignificant difference.
Asked if apremilast had a favorable impact upon other manifestations of Behçet’s disease besides the oral ulcers, Dr. Mahr replied, “This is a very good question. People often wonder about it. We do, too. But this trial was not designed to capture less common manifestations of Behçet’s syndrome, such as genital ulcers. There have been some analyses done, but the number of patients who had genital ulcers at 12 weeks were very few. The same was true for eye manifestations. There was sort of a signal that it works, but we can’t prove it in a placebo-controlled trial.”
Dr. Mahr reported receiving research funding from and serving as a consultant to Celgene, the study sponsor.
REPORTING FROM EADV 2019
Is It More Than a Cold?

ANSWER
The radiograph does not demonstrate any evidence of infiltrate or pleural effusion. However, of note is a rather large lytic lesion involving the posterior aspect of the right fourth rib. This finding is very concerning for either a primary bone neoplasm or (more likely) a metastatic one.
The patient denied any history of cancer. She was promptly referred to Hematology/Oncology for further evaluation and workup. At last update, she had undergone a bone marrow biopsy, with preliminary pathology results suggestive of a plasma cell neoplasm.
ANSWER
The radiograph does not demonstrate any evidence of infiltrate or pleural effusion. However, of note is a rather large lytic lesion involving the posterior aspect of the right fourth rib. This finding is very concerning for either a primary bone neoplasm or (more likely) a metastatic one.
The patient denied any history of cancer. She was promptly referred to Hematology/Oncology for further evaluation and workup. At last update, she had undergone a bone marrow biopsy, with preliminary pathology results suggestive of a plasma cell neoplasm.
ANSWER
The radiograph does not demonstrate any evidence of infiltrate or pleural effusion. However, of note is a rather large lytic lesion involving the posterior aspect of the right fourth rib. This finding is very concerning for either a primary bone neoplasm or (more likely) a metastatic one.
The patient denied any history of cancer. She was promptly referred to Hematology/Oncology for further evaluation and workup. At last update, she had undergone a bone marrow biopsy, with preliminary pathology results suggestive of a plasma cell neoplasm.

A 70-year-old woman presents to the urgent care clinic with a week-long history of cold and cough that she feels is getting worse. She reports subjective fever and chills, as well as an occasional pain in the right side of her chest when she breathes. She has been taking OTC products with limited relief.
Her medical history is significant for mild hypertension. She denies smoking. On physical exam, you note an elderly female in no obvious distress. She is afebrile, with normal vital signs. Pulse oximetry reveals an O2 saturation of 98% on room air. Auscultation of her lungs demonstrates a little bit of mid bronchial congestion and perhaps some bibasilar crackles.
You order a complete blood count as well as a chest radiograph (shown). What is your impression?

Alzheimer’s disease subtypes follow neuropathologic patterns seen in the nucleus basalis of Meynert
Cholinergic neurons in the nucleus basalis of Meynert appear more susceptible to neurofibrillary tangles and neuronal destruction in women, patients carrying the apolipoprotein E–epsilon 4 (APOE4) allele, and people with hippocampal-sparing Alzheimer’s disease, a subtype characterized by early onset and rapid cognitive decline.

Those findings and others from a postmortem study published in JAMA Neurology also suggests that the nucleus basalis of Meynert (nbM) could be the first place that neuronal damage appears in Alzheimer’s disease (AD), according to first author Fadi S. Hanna Al-Shaikh and colleagues.
The study also confirmed the authors’ previous categorization of three AD subtypes: early-onset, rapidly declining hippocampal-sparing AD (HpSp), typical sporadic AD, and limbic predominant AD, a later-onset form with a slower rate of decline.
“We observed a wave of vulnerability in which the exacerbation of nbM neurofibrillary tangles [NFTs] in HpSp AD may leave the cortex more vulnerable to [tangle] accumulation, perhaps via a biologically accelerated process or through a mechanism of disinhibition,” wrote Mr. Al-Shaikh, of the Mayo Clinic, Jacksonville, Fla., and colleagues. “By contrast, the limbic predominant AD cases had an exacerbation of areas vulnerable early in the Braak-like pattern of NFT accumulation, perhaps via a biologically restrictive process that relatively confines pathology to limbic areas.”
The nbM is of interest to researchers because 90% of its neurons are cholinergic with cortical penetration. “Postmortem studies of AD and more recent neuroimaging studies provide evidence that involvement of the nucleus basalis of Meynert may be critical and early in the molecular cascade of events,” the authors said. “The accumulation of NFTs in the nbM may precede entorhinal cortex and locus coeruleus involvement, making the nbM potentially one of the earliest sites where NFT accumulation occurs.”
Previously, this team had identified three AD subtypes based on patterns of corticolimbic neurofibrillary tangling. In HpSp, the hippocampus is relatively spared, while the cortex has a greater number of tangles. In limbic predominant AD, the cortex is relatively spared, and the hippocampus is severely involved. Typical AD shows the expected patterns of hippocampal and cortical tangling.
Cases in this study came from the Florida Autopsied Multi-Ethnic (FLAME) cohort, comprising 1,361 brain tissue samples from confirmed AD cases and 103 nondemented controls. The investigators sought to understand the patterns of neuronal demise in the nbM, and any associations with clinical signs, demographics, and the recently described three subtypes.
In the cohort, AD subtypes included 175 with HpSp, 1,014 with typical AD, and 172 with limbic predominant AD. Patients with HpSp were the youngest, with a median disease onset age of 65 years, compared with 71 years in typical AD and 78 in limbic predominant. There were fewer women in the HpSp group (35%), compared with the typical AD group (54%) and the limbic group (70%). More patients with HpSp had atypical presentation (38%) in comparison with typical (11%) and limbic predominant AD (2%). But patients with HpSp were less likely to be APOE4 positive (46%), whereas those with limbic predominant AD were most likely to be APOE4 positive (72%).
Cognitively, HpSp patients declined more rapidly, losing a median of 4 points per year on the Mini Mental State Exam (MMSE), compared with 2 and 1 points in those with typical and limbic predominant AD. At death, the HpSp patients had a median MMSE score of 7, versus 13 in the typical AD group and 18 in the limbic group.
Patients with HpSp had the highest concentration of tangles and the lowest neuronal density in the nbM. Limbic predominant cases had the lowest tangle burden and the highest neuronal density. Typical AD cases lay between these extremes on both measures.
A multivariate regression analysis determined the overlap of neuronal findings and AD subtypes. A younger age at symptom onset was significantly associated with higher tangle counts in the nbM regions among patients with HpSp. In women with typical AD, there were 2.5 times more tangles than in men. APOE4 carriers had 1.3 times more tangles than did noncarriers.
There were also associations with cognition. “For every 10-point decrease in final MMSE of typical AD cases, the number of nbM NFTs was expected to increase by 1.8,” the authors wrote.
Although limbic predominant AD wasn’t associated with any clinical or demographic variables in this analysis, it was associated with neuronal changes in the nbM. “For every 10 years’ younger age at onset, the number of neurons was expected to be lower by 4.6 [per mm2]. … In addition, limbic predominant cases were observed to have 4.3 [per mm2] fewer neurons for every 10-point decrease in MMSE,” the authors said.
This study was supported by the National Institute on Aging, the Florida Department of Health, the Ed and Ethel Moore Alzheimer’s Disease Research Program, a Gerstner Family Career Development Award, and the Alzheimer’s Association. Two authors reported financial relationships with industry outside the submitted work.
SOURCE: Al Shaikh FSH et al. JAMA Neurol. 2019 Oct 28. doi: 10.1001/jamaneurol.2019.3606.
Cholinergic neurons in the nucleus basalis of Meynert appear more susceptible to neurofibrillary tangles and neuronal destruction in women, patients carrying the apolipoprotein E–epsilon 4 (APOE4) allele, and people with hippocampal-sparing Alzheimer’s disease, a subtype characterized by early onset and rapid cognitive decline.
Those findings and others from a postmortem study published in JAMA Neurology also suggests that the nucleus basalis of Meynert (nbM) could be the first place that neuronal damage appears in Alzheimer’s disease (AD), according to first author Fadi S. Hanna Al-Shaikh and colleagues.
The study also confirmed the authors’ previous categorization of three AD subtypes: early-onset, rapidly declining hippocampal-sparing AD (HpSp), typical sporadic AD, and limbic predominant AD, a later-onset form with a slower rate of decline.
“We observed a wave of vulnerability in which the exacerbation of nbM neurofibrillary tangles [NFTs] in HpSp AD may leave the cortex more vulnerable to [tangle] accumulation, perhaps via a biologically accelerated process or through a mechanism of disinhibition,” wrote Mr. Al-Shaikh, of the Mayo Clinic, Jacksonville, Fla., and colleagues. “By contrast, the limbic predominant AD cases had an exacerbation of areas vulnerable early in the Braak-like pattern of NFT accumulation, perhaps via a biologically restrictive process that relatively confines pathology to limbic areas.”
The nbM is of interest to researchers because 90% of its neurons are cholinergic with cortical penetration. “Postmortem studies of AD and more recent neuroimaging studies provide evidence that involvement of the nucleus basalis of Meynert may be critical and early in the molecular cascade of events,” the authors said. “The accumulation of NFTs in the nbM may precede entorhinal cortex and locus coeruleus involvement, making the nbM potentially one of the earliest sites where NFT accumulation occurs.”
Previously, this team had identified three AD subtypes based on patterns of corticolimbic neurofibrillary tangling. In HpSp, the hippocampus is relatively spared, while the cortex has a greater number of tangles. In limbic predominant AD, the cortex is relatively spared, and the hippocampus is severely involved. Typical AD shows the expected patterns of hippocampal and cortical tangling.
Cases in this study came from the Florida Autopsied Multi-Ethnic (FLAME) cohort, comprising 1,361 brain tissue samples from confirmed AD cases and 103 nondemented controls. The investigators sought to understand the patterns of neuronal demise in the nbM, and any associations with clinical signs, demographics, and the recently described three subtypes.
In the cohort, AD subtypes included 175 with HpSp, 1,014 with typical AD, and 172 with limbic predominant AD. Patients with HpSp were the youngest, with a median disease onset age of 65 years, compared with 71 years in typical AD and 78 in limbic predominant. There were fewer women in the HpSp group (35%), compared with the typical AD group (54%) and the limbic group (70%). More patients with HpSp had atypical presentation (38%) in comparison with typical (11%) and limbic predominant AD (2%). But patients with HpSp were less likely to be APOE4 positive (46%), whereas those with limbic predominant AD were most likely to be APOE4 positive (72%).
Cognitively, HpSp patients declined more rapidly, losing a median of 4 points per year on the Mini Mental State Exam (MMSE), compared with 2 and 1 points in those with typical and limbic predominant AD. At death, the HpSp patients had a median MMSE score of 7, versus 13 in the typical AD group and 18 in the limbic group.
Patients with HpSp had the highest concentration of tangles and the lowest neuronal density in the nbM. Limbic predominant cases had the lowest tangle burden and the highest neuronal density. Typical AD cases lay between these extremes on both measures.
A multivariate regression analysis determined the overlap of neuronal findings and AD subtypes. A younger age at symptom onset was significantly associated with higher tangle counts in the nbM regions among patients with HpSp. In women with typical AD, there were 2.5 times more tangles than in men. APOE4 carriers had 1.3 times more tangles than did noncarriers.
There were also associations with cognition. “For every 10-point decrease in final MMSE of typical AD cases, the number of nbM NFTs was expected to increase by 1.8,” the authors wrote.
Although limbic predominant AD wasn’t associated with any clinical or demographic variables in this analysis, it was associated with neuronal changes in the nbM. “For every 10 years’ younger age at onset, the number of neurons was expected to be lower by 4.6 [per mm2]. … In addition, limbic predominant cases were observed to have 4.3 [per mm2] fewer neurons for every 10-point decrease in MMSE,” the authors said.
This study was supported by the National Institute on Aging, the Florida Department of Health, the Ed and Ethel Moore Alzheimer’s Disease Research Program, a Gerstner Family Career Development Award, and the Alzheimer’s Association. Two authors reported financial relationships with industry outside the submitted work.
SOURCE: Al Shaikh FSH et al. JAMA Neurol. 2019 Oct 28. doi: 10.1001/jamaneurol.2019.3606.
Cholinergic neurons in the nucleus basalis of Meynert appear more susceptible to neurofibrillary tangles and neuronal destruction in women, patients carrying the apolipoprotein E–epsilon 4 (APOE4) allele, and people with hippocampal-sparing Alzheimer’s disease, a subtype characterized by early onset and rapid cognitive decline.
Those findings and others from a postmortem study published in JAMA Neurology also suggests that the nucleus basalis of Meynert (nbM) could be the first place that neuronal damage appears in Alzheimer’s disease (AD), according to first author Fadi S. Hanna Al-Shaikh and colleagues.
The study also confirmed the authors’ previous categorization of three AD subtypes: early-onset, rapidly declining hippocampal-sparing AD (HpSp), typical sporadic AD, and limbic predominant AD, a later-onset form with a slower rate of decline.
“We observed a wave of vulnerability in which the exacerbation of nbM neurofibrillary tangles [NFTs] in HpSp AD may leave the cortex more vulnerable to [tangle] accumulation, perhaps via a biologically accelerated process or through a mechanism of disinhibition,” wrote Mr. Al-Shaikh, of the Mayo Clinic, Jacksonville, Fla., and colleagues. “By contrast, the limbic predominant AD cases had an exacerbation of areas vulnerable early in the Braak-like pattern of NFT accumulation, perhaps via a biologically restrictive process that relatively confines pathology to limbic areas.”
The nbM is of interest to researchers because 90% of its neurons are cholinergic with cortical penetration. “Postmortem studies of AD and more recent neuroimaging studies provide evidence that involvement of the nucleus basalis of Meynert may be critical and early in the molecular cascade of events,” the authors said. “The accumulation of NFTs in the nbM may precede entorhinal cortex and locus coeruleus involvement, making the nbM potentially one of the earliest sites where NFT accumulation occurs.”
Previously, this team had identified three AD subtypes based on patterns of corticolimbic neurofibrillary tangling. In HpSp, the hippocampus is relatively spared, while the cortex has a greater number of tangles. In limbic predominant AD, the cortex is relatively spared, and the hippocampus is severely involved. Typical AD shows the expected patterns of hippocampal and cortical tangling.
Cases in this study came from the Florida Autopsied Multi-Ethnic (FLAME) cohort, comprising 1,361 brain tissue samples from confirmed AD cases and 103 nondemented controls. The investigators sought to understand the patterns of neuronal demise in the nbM, and any associations with clinical signs, demographics, and the recently described three subtypes.
In the cohort, AD subtypes included 175 with HpSp, 1,014 with typical AD, and 172 with limbic predominant AD. Patients with HpSp were the youngest, with a median disease onset age of 65 years, compared with 71 years in typical AD and 78 in limbic predominant. There were fewer women in the HpSp group (35%), compared with the typical AD group (54%) and the limbic group (70%). More patients with HpSp had atypical presentation (38%) in comparison with typical (11%) and limbic predominant AD (2%). But patients with HpSp were less likely to be APOE4 positive (46%), whereas those with limbic predominant AD were most likely to be APOE4 positive (72%).
Cognitively, HpSp patients declined more rapidly, losing a median of 4 points per year on the Mini Mental State Exam (MMSE), compared with 2 and 1 points in those with typical and limbic predominant AD. At death, the HpSp patients had a median MMSE score of 7, versus 13 in the typical AD group and 18 in the limbic group.
Patients with HpSp had the highest concentration of tangles and the lowest neuronal density in the nbM. Limbic predominant cases had the lowest tangle burden and the highest neuronal density. Typical AD cases lay between these extremes on both measures.
A multivariate regression analysis determined the overlap of neuronal findings and AD subtypes. A younger age at symptom onset was significantly associated with higher tangle counts in the nbM regions among patients with HpSp. In women with typical AD, there were 2.5 times more tangles than in men. APOE4 carriers had 1.3 times more tangles than did noncarriers.
There were also associations with cognition. “For every 10-point decrease in final MMSE of typical AD cases, the number of nbM NFTs was expected to increase by 1.8,” the authors wrote.
Although limbic predominant AD wasn’t associated with any clinical or demographic variables in this analysis, it was associated with neuronal changes in the nbM. “For every 10 years’ younger age at onset, the number of neurons was expected to be lower by 4.6 [per mm2]. … In addition, limbic predominant cases were observed to have 4.3 [per mm2] fewer neurons for every 10-point decrease in MMSE,” the authors said.
This study was supported by the National Institute on Aging, the Florida Department of Health, the Ed and Ethel Moore Alzheimer’s Disease Research Program, a Gerstner Family Career Development Award, and the Alzheimer’s Association. Two authors reported financial relationships with industry outside the submitted work.
SOURCE: Al Shaikh FSH et al. JAMA Neurol. 2019 Oct 28. doi: 10.1001/jamaneurol.2019.3606.
FROM JAMA NEUROLOGY
Inpatient care declining among family physicians
and by 2017, only one of four FPs was practicing hospital medicine, according to the American Academy of Family Physicians.
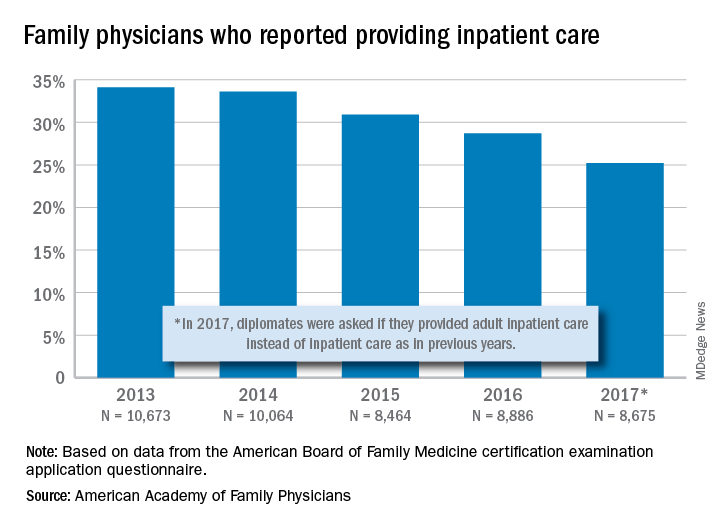
The share of family physicians who provided hospital care went from 34.1% in 2013 to 25.2% in 2017, for a relative decrease of 26% that left only a quarter of FPs seeing inpatients, based on data from the annual American Board of Family Medicine certification exam application questionnaire. For the 5-year period, 46,762 individuals were included in the study sample of FPs in direct patient care.
“As observed in other domains (prenatal care, home visits, nursing home care, and obstetric care), this study adds to the evidence demonstrating contracting scope of practice among FPs,” Anuradha Jetty, MPH, of the AAFP’s Robert Graham Center in Washington, D.C., and associates said in a recent Policy Brief published in the Journal of the American Board of Family Medicine.
Much of that contraction is occurring among new family physicians who can’t “find positions that allow them to use all their expertise,” the investigators said in a separate statement. The AAFP had previously reported that about 40% of family physicians had full hospital privileges in 2018, compared with 56% in 2012.
Many new FPs now work in large multispecialty practices or hospital systems, and “[some] of these employers dictate scope of practice, limiting family physicians to coordinating outpatient care and relying on subspecialists or hospitalists to provide inpatient care,” they noted.
and by 2017, only one of four FPs was practicing hospital medicine, according to the American Academy of Family Physicians.
The share of family physicians who provided hospital care went from 34.1% in 2013 to 25.2% in 2017, for a relative decrease of 26% that left only a quarter of FPs seeing inpatients, based on data from the annual American Board of Family Medicine certification exam application questionnaire. For the 5-year period, 46,762 individuals were included in the study sample of FPs in direct patient care.
“As observed in other domains (prenatal care, home visits, nursing home care, and obstetric care), this study adds to the evidence demonstrating contracting scope of practice among FPs,” Anuradha Jetty, MPH, of the AAFP’s Robert Graham Center in Washington, D.C., and associates said in a recent Policy Brief published in the Journal of the American Board of Family Medicine.
Much of that contraction is occurring among new family physicians who can’t “find positions that allow them to use all their expertise,” the investigators said in a separate statement. The AAFP had previously reported that about 40% of family physicians had full hospital privileges in 2018, compared with 56% in 2012.
Many new FPs now work in large multispecialty practices or hospital systems, and “[some] of these employers dictate scope of practice, limiting family physicians to coordinating outpatient care and relying on subspecialists or hospitalists to provide inpatient care,” they noted.
and by 2017, only one of four FPs was practicing hospital medicine, according to the American Academy of Family Physicians.
The share of family physicians who provided hospital care went from 34.1% in 2013 to 25.2% in 2017, for a relative decrease of 26% that left only a quarter of FPs seeing inpatients, based on data from the annual American Board of Family Medicine certification exam application questionnaire. For the 5-year period, 46,762 individuals were included in the study sample of FPs in direct patient care.
“As observed in other domains (prenatal care, home visits, nursing home care, and obstetric care), this study adds to the evidence demonstrating contracting scope of practice among FPs,” Anuradha Jetty, MPH, of the AAFP’s Robert Graham Center in Washington, D.C., and associates said in a recent Policy Brief published in the Journal of the American Board of Family Medicine.
Much of that contraction is occurring among new family physicians who can’t “find positions that allow them to use all their expertise,” the investigators said in a separate statement. The AAFP had previously reported that about 40% of family physicians had full hospital privileges in 2018, compared with 56% in 2012.
Many new FPs now work in large multispecialty practices or hospital systems, and “[some] of these employers dictate scope of practice, limiting family physicians to coordinating outpatient care and relying on subspecialists or hospitalists to provide inpatient care,” they noted.