User login
Development of an Integrative Medicine Rotation for Family Medicine and Preventive Medicine Residency
Development of an Integrative Medicine Rotation for Family Medicine and Preventive Medicine Residency
Integrative medicine or complementary alternative medicine (IM/CAM) is increasingly being recognized as an integral part of optimal health and healing. IM/CAM “reaffirms the importance of the relationship between practitioner and patient, focuses on the whole person, is informed by evidence, and makes use of all appropriate therapeutic approaches, healthcare professionals and disciplines.”1 IM/CAM encompasses a wide range of therapies, conceptual frameworks, and health care-related professions, such as acupuncture, massage, dietary supplements, mindfulness, yoga, meditation and guided imagery.1 Research has found that 30% to 98% of patients with chronic conditions seek IM/CAM therapies.1-3
Despite the high prevalence of patients utilizing IM/CAM therapies and the National Institutes of Health grants for IM/CAM education, implementation of IM/CAM instruction in graduate medical education programs remains inconsistent.1 Barriers cited by programs include a lack of IM/CAM experts in the program, faculty training, competing financial resources, and an already full resident education schedule.4 As a result, many physicians have limited or no training in IM/CAM.1,5
The US Department of Veterans Affairs (VA) offers IM/CAM health programs to veterans and caregivers as part of its whole health care initiative.6 Several VA health care systems have adopted whole health and IM/CAM through programs for mental health integration into primary care; women’s health; integrative pain care; geriatrics, through adoption of Age-Friendly Health Systems standards; and nutrition and physical activity.7-13 The VA provides training to more medical students than any other health system: > 95% of US medical schools are affiliated with a VA medical center (VAMC).14 As part of the training mission, VA seeks to encourage students of diverse professions to consider careers in the VA.14
Residency is a time for newly licensed physicians to acquire additional experience and training to translate knowledge and skills acquired during medical school directly to patient care.15 However, residency curricula have limited time to incorporate IM/CAM training. Residency training is also physically and psychosocially demanding, often resulting in inadequate self-care, poor work-life balance, and disrupted sleep.16-18 Resident wellness is at a historic low, resulting in high rates of burnout during training.4,15
Residency programs are required to provide wellness education; however, most programs include minimal content.19 Despite high rates of burnout, formal curricula on the topic have not been established. 20 IM/CAM education also can provide a path for residents to learn about and engage in mindfulness-based training or cognitive stress reduction for self-care.
INTEGRATIVE WHOLE HEALTH ROTATION
In 2017, the Baltimore Geriatric Research Education and Clinical Center (GRECC) established an IM/whole health residency rotation and created a structured curriculum incorporating self-assessment, active reflection, and self-care to complement training in specific IM/CAM modalities for residents in family medicine. The curriculum evaluated how this training improved residents’ perceptions of IM/CAM and how it personally and professionally impacted the practice of self-care as a strategy to decrease burnout. We hypothesized that this structured experience would increase IM/CAM knowledge among clinicians while promoting the importance and practice of self-care to reduce burnout.
The 2-week IM/CAM curriculum was developed by University of Maryland School of Medicine faculty in partnership with the Baltimore GRECC and staff at the VA Maryland Health Care System. The curriculum was designed to expose residents to the 8 components of the whole health Circle of Health (moving the body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind) in addition to IM/ CAM modalities the VA is mandated to offer to veterans (acupuncture, chiropractic, meditation, massage therapy, biofeedback, clinical hypnosis, guided imagery, yoga, and tai chi).21 Twelve residents (1 preventive medicine and 11 third-year family medicine residents) rotated individually throughout the year as part of their behavioral health block rotation. All residents completed the 2-week curriculum as their schedules allowed. The curriculum consisted of didactics sessions and activities at the Baltimore, Loch Raven, and Perry Point VAMCs. Residents completed evaluations before and after the rotation. The experience described in this article by the residents and the survey data were collected from the 2018/2019 training year. A rotation syllabus, competencies adapted from Locke and colleagues and skills residents obtain during this rotation that support these competencies, as well as a resident sample schedule were developed (eAppendix is available at doi:10.12788/fp.0544).1

Rotation Overview
for each resident were built around instructional opportunities, which included 1-on-1 didactics, direct observation of treatment modalities, and personal reflection of the residents’ self-care practices. While each resident’s rotation schedule varied slightly due to their schedules, the foundational instruction elements were the same. Didactic session themes included an overview of IM/CAM, nutrition, narrative medicine, pain psychology, music therapy, chaplain services, motor-cognitive training, and exercise guidelines. Assigned readings, including peer-reviewed literature on IM/CAM therapies, complemented all sessions. Residents created an evidence-supported integrative treatment plan for a patient with a condition of interest to them.
Residents observed clinician-led veteran group sessions on IM/CAM treatment modalities, including guided meditation, mindfulness and relaxation, self-awareness, living well with chronic pain, tai chi, drumming for health and balance, anger management, recovery group, acceptance and commitment therapy, and Gerofit exercise. The group classes allowed residents to actively participate in the activity or discussion. Residents also shadowed VA clinicians in sleep, pain, nutrition, acupuncture, and mental health clinics.
Residents were encouraged to practice self-care during the 2-week rotation. The rotation schedule built in free time, including a 1-hour daily lunch period, for residents to consider their own health habits, complete a personal health inventory, and try self-care activities outlined on the syllabus with links to resources. These resources also served as educational materials that residents could share with patients. All materials, including didactic lectures, journal articles and self-care resources, were provided to each resident through a free online course to ensure residents had access throughout and following completion of the rotation. This content, including the rotation evaluation metrics, is available upon request from the corresponding author.
Evaluations
Residents completed a survey before and after the rotation to measure IM/CAM knowledge and application and self-care/ burnout perceptions. Residents were asked to evaluate rotation sessions and comment on whether this rotation benefited them personally and professionally (Table 1). Descriptive statistics were analyzed using Microsoft Excel. Given the small sample size and lack of statistical power, only mean survey results are reported in this article. Because this opportunity is specific to the University of Maryland School of Medicine and the proposed project was part of ordinary educational practice, the study was deemed not human subject research by the University of Maryland Institutional Review Board (HP-00089256).

Perceptions and attitudes toward IM/CAM were assessed using a survey designed by the University of Minnesota Academic Health Center. It included 18 items scored on a 5-point semantic rating scale (1, strongly disagree; 5, strongly agree).22 Residents rated their level of agreement with statements reflecting both positive (eg, clinical care should integrate the best of conventional and CAM practices) and negative (eg, CAM is a threat to public health) views. Three questions adapted from the NHIS Adult Complementary Health Questionnaire and UC Irvine Survey of Health Care Use and Practice assessed the use of IM/CAM resources.23,24
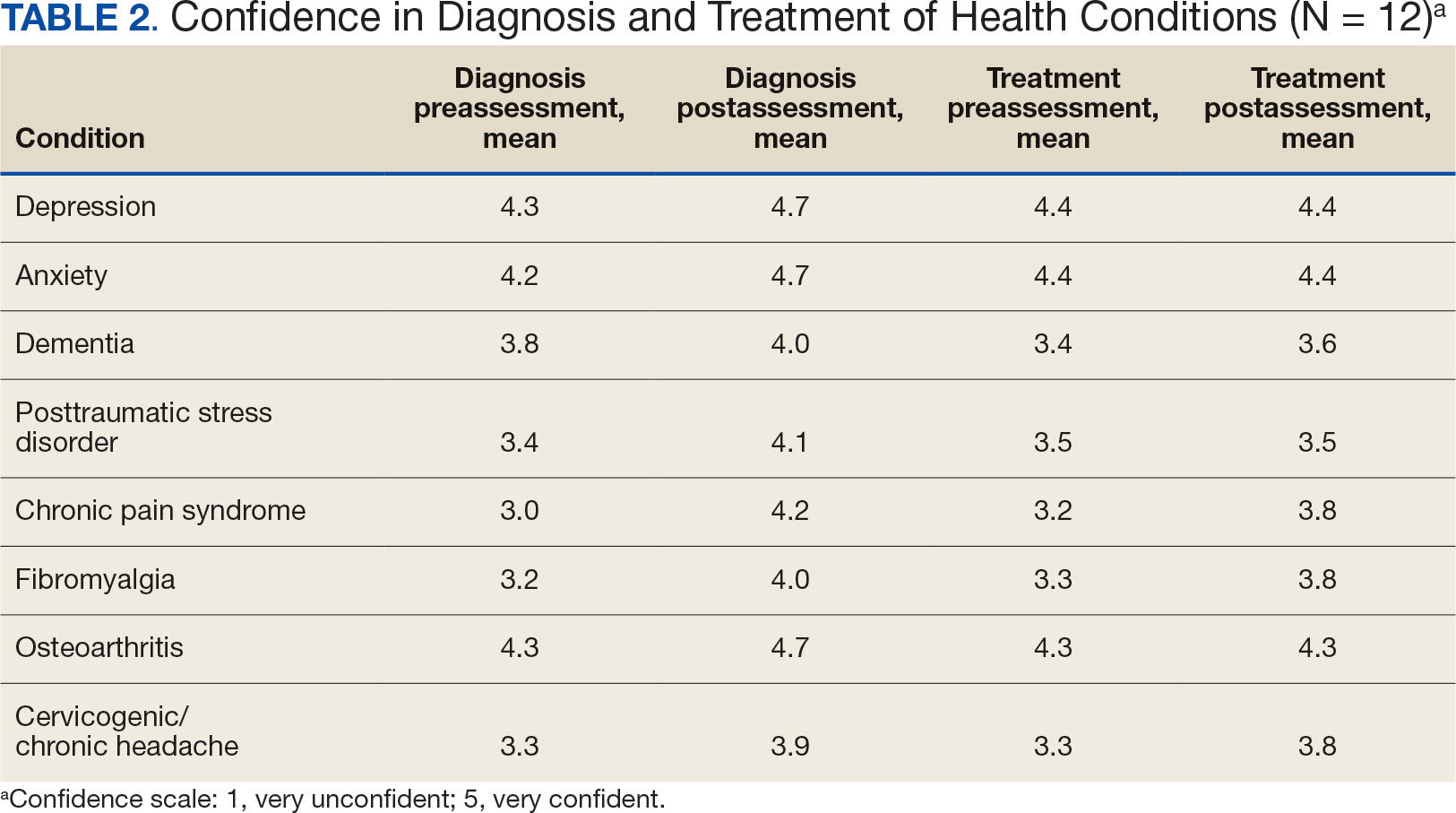
Resident knowledge and application of IM/CAM were measured using a case study designed by the course faculty. The case listed a chief complaint of nerve pain, with a history of chronic pain, neuropathic pain, anxiety, chronic fatigue, depression, insomnia, posttraumatic stress disorder, history of present illness, past surgical history, medication list, review of symptoms, laboratory values, and physical examination. The residents completed an assessment before and after the rotation. Residents rated their confidence in the diagnosis and treatment of 8 medical conditions using a 5-point semantic rating scale (Table 2). Self-care importance and selfcare frequency were measured by a variety of means, including 3 survey questions, the Five Facet Mindfulness Questionnaire, 2 prompts on a 7-point semantic scale, and a slightly modified version of the validated Perceived Stress Scale.25-28
Survey Results
Residents gave the rotation positive feedback with a mean score of 8.5 out of 10. They reported the beneficial impact of seeing the nontraditional and nonpharmacological practices in treating patients, chronic pain management team approaches, and enjoyed being able to participate in group classes with patients. Many residents expressed a desire for a longer rotation to have more time to experience the behavioral health-focused sessions. Residents also requested additional information on nutritional supplements/natural medicines, battlefield acupuncture training and osteopathic manipulative therapy practices. All residents reported the rotation personally and/or professionally benefited them (Appendix).
Given the sample of 12 residents, values are presented as prerotation to postrotation comparisons without statistical analysis. There was a trend towards an increase in the reported use and recommendation of 26 modalities of nonconventional therapies following the rotation. There was also a slight increase in resource knowledge and use of these resources, and residents reported accessing more types of resources. Mean scores of the case study to gauge knowledge and application of IM increased from 7.5 at baseline to 11.0 after the rotation. Resident confidence in diagnosis increased for all 8 conditions, but confidence in treatment only increased for 4 conditions.
Results of self-care importance, self-care frequency and mindfulness were consistent baseline to postrotation. The mean time residents spent regularly practicing self-care during a work week increased slightly while feelings of burnout decreased. The perceived stress scale average score decreased from 13.4 at baseline to 10.5 after rotation.
DISCUSSION
The implementation of an IM residency rotation that incorporates whole health and interprofessional practices demonstrated improved perception and increased use of IM/CAM resources and knowledge among a small sample of third-year residents. Residents reported they had a positive experience participating in the rotation and gained knowledge, resources, and skills they felt confident discussing with their patients.
Many studies reported favorable attitudes and perceptions of IM/CAM use among physicians, but few have assessed these measures while implementing a training curriculum.3,4,22 Gardiner and colleagues reported on the perception and use of IM resources among family medicine residents.4 The study found that while 58% of all residents reported IM/CAM as an important part of their training, only 60% reported they received it or had specific learning objectives in their curriculum. 4 The program outlined in this study and previous research illustrate that physicians recognize the importance of IM/CAM education in training programs, but most were unaware of the resources available or did not feel comfortable counseling patients about most IM/CAM applications.
Residents in this program slightly increased their use of IM/CAM to diagnose and treat medical conditions after the rotation. A study by Wahner-Roedler and colleagues assessed physician knowledge regarding common IM/CAM therapies.3 On average, physicians only felt knowledgeable and comfortable counseling patients for 3 of 13 listed treatments/techniques and few natural herbal treatments. The study also found that most physicians had difficulty accessing IM/CAM information at their institution despite having free access to electronic databases. However, this study only assessed physician attitudes of IM/CAM and did not include an educational component to increase their knowledge of the modalities.3 This evaluation supports the need for interventions like the program described in this article that provide physicians with access to evidence-based resources combined with the applied experiences to increase their comfort within this growing field.
Though the sample size in this study was small, its results support existing research indicating that clinicians view selfcare as important. Many residents were already using a self-care plan at baseline, but there was slight increase in the practice of self-care during the rotation and a slight decrease in burnout. Previous research reflects high rates of burnout and relatively poor quality of life among primary care physicians.15 Burnout is associated with lower quality of care, lower patient satisfaction and contributes to medical errors. Studies suggest as many as 60% of primary care physicians report symptoms of burnout, which negatively affected the quality of patient care they provide.15
Despite the profound effects burnout has on physicians and patient care, a standardized wellness education or self-care tool kit is not currently available. The University of Massachusetts recently introduced a pilot program to promote resident wellness that demonstrated favorable results.15 A meta-analysis of physicians and medical trainees found decreases in anxiety and symptoms of anxiety as well as a decrease in burnout among participants in cognitive, behavioral and mindfulness interventions.29 However, unlike our program, these programs focused solely on the well-being of medical trainees, residents, and physicians and didn’t focus on the patient-clinician interactions. Given the impact on patient care, there is a need to develop and implement additional programs like our residency rotation that promote health and wellness among physicians while also evaluating how physicians may translate these skills to patient education.
While this program st i l l exists for third-year residents at Baltimore GRECC, it has significantly changed since the COVID-19 pandemic. For about the first 6 months of the pandemic, when physical distancing requirements were in place, family medicine trainees were not able to rotate. Upon return to the facility, many group classes were cancelled and some clinicians no longer offered the sessions. The rotation has evolved to a hybrid format, where many group classes for veteran patients are offered virtually, and residents observe a mix of virtual and in-person shadowing opportunities. Our formal evaluation included administering the survey and occurred from July 2018 to July 2019 but wasn’t implemented upon return to post-COVID activities due to the inconsistent experiences offered to residents over the past few years. Future research should evaluate the impact of this hybrid program on the clinicians and explore dissemination to other VAMCs and their academic affiliates.
Limitations
Project recruitment was limited to 11 family medicine and 1 preventive medicine resident. Perceptions, use of IM/CAM, and knowledge about IM/CAM could be considerably different in different departments with varying schedules, hours worked, and patient volumes. Secondly, the survey was conducted 2 weeks apart. Indications of self-care and burnout may not reflect long-term effects, adoption, or maintenance. Future research should include longer follow up to examine how this type of educational activity may impact burnout rates of physicians following the completion of residency, as well as changes in perspectives of IM/CAM while practicing as a physician. Trainees were exposed to a wide range of health care professions, but additional research is needed regarding medical resident perceptions of the roles of specific professions in a collaborative health care team.30,31
CONCLUSIONS
The residency rotation program illustrates the benefits of establishing a standardized IM/CAM rotation that includes self-care resources in family medicine programs to adequately train clinicians to practice wellness and promote it to their patients. The results of this project suggest this type of training will help residents assess the literature to better counsel patients on IM/CAM options while also providing strategies for maintaining optimal health and well-being for health care professionals. Broadening and shifting the scope of medicine from treatment to prevention, personal wellness, and optimal healing should be a top priority.
- Locke AB, Gordon A, Guerrera MP, Gardiner P, Lebensohn P. Recommended integrative medicine competencies for family medicine residents. Explore (NY). 2013;9(5):308-313. doi:10.1016/j.explore.2013.06.005
- Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA. 1998;280(18):1569-1575. doi:10.1001/jama.280.18.1569
- Wahner-Roedler DL, Vincent A, Elkin PL, Loehrer LL, Cha SS, Bauer BA. Physicians’ attitudes toward complementary and alternative medicine and their knowledge of specific therapies: a survey at an academic medical center. Evid Based Complement Alternat Med. 2006;3(4):495-501. doi:10.1093/ecam/nel036
- Gardiner P, Filippelli AC, Lebensohn P, Bonakdar R. Family medicine residency program directors attitudes and knowledge of family medicine CAM competencies. Explore (NY). 2013;9(5):299-307. doi:10.1016/j.explore.2013.06.002
- Sierpina V, Levine R, Astin J, Tan A. Use of mind-body therapies in psychiatry and family medicine faculty and residents: attitudes, barriers, and gender differences. Explore (NY). 2007;3(2):129-135. doi:10.1016/j.explore.2006.12.001
- Krist AH, South-Paul J, Meisnere M, eds. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023.
- Bokhour BG, DeFaccio R, Gaj L, et al. Changes in patientreported outcomes associated with receiving whole health in the Veteran Health Administration (VHA)’s National Demonstration Project. J Gen Intern Med. 2024;39(1):84-94. doi:10.1007/s11606-023-08376-0
- Courtney RE, Schadegg MJ, Bolton R, Smith S, Harden SM. Using a whole health approach to build biopsychosocial- spiritual personal health plans for veterans with chronic pain. Pain Manag Nurs. 2024;25(1):69-74. doi:10.1016/j.pmn.2023.09.010
- Gabrielian S, Jones AL, Hoge AE, et al. Enhancing primary care experiences for homeless patients with serious mental illness: results from a national survey. J Prim Care Community Health. 2021;12:2150132721993654. doi:10.1177/2150132721993654
- Matthieu MM, Church KA, Taylor LD, et al. Integrating the age-friendly health systems movement in Veterans Health Administration: national advance care planning via group visits and the 4Ms framework. Health Soc Work. 2023;48(4):277-280. doi:10.1093/hsw/hlad022
- Meisler AW, Gianoli MO, Na PJ, Pietrzak RH. Functional disability in US military veterans: the importance of integrated whole health initiatives. Prim Care Companion CNS Disord. 2023;25(4):22m03461. doi:10.4088/PCC.22m03461
- Ortmeyer HK, Giffuni J, Etchberger D, Katzel L. The role of companion dogs in the VA Maryland Health Care System Whole Health(y) GeroFit Program. Animals (Basel). 2023;13(19):3047. doi:10.3390/ani13193047
- Sullivan MB, Hill K, Ballengee LA, et al. Remotely delivered psychologically informed mindful movement physical therapy for pain care: a framework for operationalization. Glob Adv Integr Med Health. 2023;12:27536130231209751. doi:10.1177/27536130231209751
- (OAA) OoAA. 75th Anniversary: Passion to learn. Power to heal. Washington DC.: US Department of Veterans Affairs; 2021. https://content.yudu.com/web/448fx/0A448g9/75thAnniversary2021/html/index.html?page=24&origin=reader
- Runyan C, Savageau JA, Potts S, Weinreb L. Impact of a family medicine resident wellness curriculum: a feasibility study. Med Educ Online. 2016;21:30648. doi:10.3402/meo.v21.30648
- Lafreniere JP, Rios R, Packer H, Ghazarian S, Wright SM, Levine RB. Burned out at the bedside: patient perceptions of physician burnout in an internal medicine resident continuity clinic. J Gen Intern Med. 2016;31(2):203-208. doi:10.1007/s11606-015-3503-3
- Freedy JR, Staley C, Mims LD, et al. Social, individual, and environmental characteristics of family medicine resident burnout: a CERA study. Fam Med. 2022;54(4):270-276. doi:10.22454/FamMed.2022.526799
- Alrishan MA, Alshammari SA. Prevalence of sleep deprivation and its effect on the performance of family medicine residents in Riyadh, Saudi Arabia. J Family Community Med. 2020;27(2):125-130. doi:10.4103/jfcm.JFCM_9_20
- ACGME. ACGME Program Requirements for Graduate Medical Education in Family Medicine. https://www.acgme.org/globalassets/pfassets/programrequirements/120_familymedicine_2024.pdf
- Nene Y, Tadi P. Resident Burnout. In: StatPearls; 2023.
- Bokhour BG, Haun JN, Hyde J, Charns M, Kligler B. Transforming the veterans affairs to a whole health system of care: time for action and research. Med Care. 2020;58(4):295-300. doi:10.1097/MLF.0000000000001316
- Kreitzer MJ, Mitten D, Harris I, Shandeling J. Attitudes toward CAM among medical, nursing, and pharmacy faculty and students: a comparative analysis. Altern Ther Health Med. 2002;8(6):44-53.
- Clarke TC, Black LI, Stussman BJ, Barnes PM, Nahin RL. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Report. 2015(79):1-16.
- Nguyen J, Liu MA, Patel RJ, Tahara K, Nguyen AL. Use and interest in complementary and alternative medicine among college students seeking healthcare at a university campus student health center. Complement Ther Clin Pract. 2016;24:103-108. doi:10.1016/j.ctcp.2016.06.001
- Baer RA, Smith GT, Hopkins J, Krietemeyer J, Toney L. Using self-report assessment methods to explore facets of mindfulness. Assessment. 2006;13(1):27-45. doi:10.1177/1073191105283504
- Baer RA, Smith GT, Lykins E, et al. Construct validity of the five facet mindfulness questionnaire in meditating and nonmeditating samples. Assessment. 2008;15(3):329-342. doi:10.1177/1073191107313003
- West CP, Dyrbye LN, Sloan JA, Shanafelt TD. Single item measures of emotional exhaustion and depersonalization are useful for assessing burnout in medical professionals. J Gen Intern Med. 2009;24(12):1318- 1321. doi:10.1007/s11606-009-1129-z
- Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24(4):385-396.
- Regehr C, Glancy D, Pitts A, LeBlanc VR. Interventions to reduce the consequences of stress in physicians: a review and meta-analysis. J Nerv Ment Dis. 2014;202(5):353-359. doi:10.1097/NMD.0000000000000130
- Visser CLF, Ket JCF, Croiset G, Kusurkar RA. Perceptions of residents, medical and nursing students about interprofessional education: a systematic review of the quantitative and qualitative literature. BMC Med Educ. 2017;17(1):77. doi:10.1186/s12909-017-0909-0
- Lingard L, Espin S, Evans C, Hawryluck L. The rules of the game: interprofessional collaboration on the intensive care unit team. Crit Care. 2004;8(6):R403-408. doi:10.1186/cc2958
Integrative medicine or complementary alternative medicine (IM/CAM) is increasingly being recognized as an integral part of optimal health and healing. IM/CAM “reaffirms the importance of the relationship between practitioner and patient, focuses on the whole person, is informed by evidence, and makes use of all appropriate therapeutic approaches, healthcare professionals and disciplines.”1 IM/CAM encompasses a wide range of therapies, conceptual frameworks, and health care-related professions, such as acupuncture, massage, dietary supplements, mindfulness, yoga, meditation and guided imagery.1 Research has found that 30% to 98% of patients with chronic conditions seek IM/CAM therapies.1-3
Despite the high prevalence of patients utilizing IM/CAM therapies and the National Institutes of Health grants for IM/CAM education, implementation of IM/CAM instruction in graduate medical education programs remains inconsistent.1 Barriers cited by programs include a lack of IM/CAM experts in the program, faculty training, competing financial resources, and an already full resident education schedule.4 As a result, many physicians have limited or no training in IM/CAM.1,5
The US Department of Veterans Affairs (VA) offers IM/CAM health programs to veterans and caregivers as part of its whole health care initiative.6 Several VA health care systems have adopted whole health and IM/CAM through programs for mental health integration into primary care; women’s health; integrative pain care; geriatrics, through adoption of Age-Friendly Health Systems standards; and nutrition and physical activity.7-13 The VA provides training to more medical students than any other health system: > 95% of US medical schools are affiliated with a VA medical center (VAMC).14 As part of the training mission, VA seeks to encourage students of diverse professions to consider careers in the VA.14
Residency is a time for newly licensed physicians to acquire additional experience and training to translate knowledge and skills acquired during medical school directly to patient care.15 However, residency curricula have limited time to incorporate IM/CAM training. Residency training is also physically and psychosocially demanding, often resulting in inadequate self-care, poor work-life balance, and disrupted sleep.16-18 Resident wellness is at a historic low, resulting in high rates of burnout during training.4,15
Residency programs are required to provide wellness education; however, most programs include minimal content.19 Despite high rates of burnout, formal curricula on the topic have not been established. 20 IM/CAM education also can provide a path for residents to learn about and engage in mindfulness-based training or cognitive stress reduction for self-care.
INTEGRATIVE WHOLE HEALTH ROTATION
In 2017, the Baltimore Geriatric Research Education and Clinical Center (GRECC) established an IM/whole health residency rotation and created a structured curriculum incorporating self-assessment, active reflection, and self-care to complement training in specific IM/CAM modalities for residents in family medicine. The curriculum evaluated how this training improved residents’ perceptions of IM/CAM and how it personally and professionally impacted the practice of self-care as a strategy to decrease burnout. We hypothesized that this structured experience would increase IM/CAM knowledge among clinicians while promoting the importance and practice of self-care to reduce burnout.
The 2-week IM/CAM curriculum was developed by University of Maryland School of Medicine faculty in partnership with the Baltimore GRECC and staff at the VA Maryland Health Care System. The curriculum was designed to expose residents to the 8 components of the whole health Circle of Health (moving the body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind) in addition to IM/ CAM modalities the VA is mandated to offer to veterans (acupuncture, chiropractic, meditation, massage therapy, biofeedback, clinical hypnosis, guided imagery, yoga, and tai chi).21 Twelve residents (1 preventive medicine and 11 third-year family medicine residents) rotated individually throughout the year as part of their behavioral health block rotation. All residents completed the 2-week curriculum as their schedules allowed. The curriculum consisted of didactics sessions and activities at the Baltimore, Loch Raven, and Perry Point VAMCs. Residents completed evaluations before and after the rotation. The experience described in this article by the residents and the survey data were collected from the 2018/2019 training year. A rotation syllabus, competencies adapted from Locke and colleagues and skills residents obtain during this rotation that support these competencies, as well as a resident sample schedule were developed (eAppendix is available at doi:10.12788/fp.0544).1
Rotation Overview
for each resident were built around instructional opportunities, which included 1-on-1 didactics, direct observation of treatment modalities, and personal reflection of the residents’ self-care practices. While each resident’s rotation schedule varied slightly due to their schedules, the foundational instruction elements were the same. Didactic session themes included an overview of IM/CAM, nutrition, narrative medicine, pain psychology, music therapy, chaplain services, motor-cognitive training, and exercise guidelines. Assigned readings, including peer-reviewed literature on IM/CAM therapies, complemented all sessions. Residents created an evidence-supported integrative treatment plan for a patient with a condition of interest to them.
Residents observed clinician-led veteran group sessions on IM/CAM treatment modalities, including guided meditation, mindfulness and relaxation, self-awareness, living well with chronic pain, tai chi, drumming for health and balance, anger management, recovery group, acceptance and commitment therapy, and Gerofit exercise. The group classes allowed residents to actively participate in the activity or discussion. Residents also shadowed VA clinicians in sleep, pain, nutrition, acupuncture, and mental health clinics.
Residents were encouraged to practice self-care during the 2-week rotation. The rotation schedule built in free time, including a 1-hour daily lunch period, for residents to consider their own health habits, complete a personal health inventory, and try self-care activities outlined on the syllabus with links to resources. These resources also served as educational materials that residents could share with patients. All materials, including didactic lectures, journal articles and self-care resources, were provided to each resident through a free online course to ensure residents had access throughout and following completion of the rotation. This content, including the rotation evaluation metrics, is available upon request from the corresponding author.
Evaluations
Residents completed a survey before and after the rotation to measure IM/CAM knowledge and application and self-care/ burnout perceptions. Residents were asked to evaluate rotation sessions and comment on whether this rotation benefited them personally and professionally (Table 1). Descriptive statistics were analyzed using Microsoft Excel. Given the small sample size and lack of statistical power, only mean survey results are reported in this article. Because this opportunity is specific to the University of Maryland School of Medicine and the proposed project was part of ordinary educational practice, the study was deemed not human subject research by the University of Maryland Institutional Review Board (HP-00089256).
Perceptions and attitudes toward IM/CAM were assessed using a survey designed by the University of Minnesota Academic Health Center. It included 18 items scored on a 5-point semantic rating scale (1, strongly disagree; 5, strongly agree).22 Residents rated their level of agreement with statements reflecting both positive (eg, clinical care should integrate the best of conventional and CAM practices) and negative (eg, CAM is a threat to public health) views. Three questions adapted from the NHIS Adult Complementary Health Questionnaire and UC Irvine Survey of Health Care Use and Practice assessed the use of IM/CAM resources.23,24
Resident knowledge and application of IM/CAM were measured using a case study designed by the course faculty. The case listed a chief complaint of nerve pain, with a history of chronic pain, neuropathic pain, anxiety, chronic fatigue, depression, insomnia, posttraumatic stress disorder, history of present illness, past surgical history, medication list, review of symptoms, laboratory values, and physical examination. The residents completed an assessment before and after the rotation. Residents rated their confidence in the diagnosis and treatment of 8 medical conditions using a 5-point semantic rating scale (Table 2). Self-care importance and selfcare frequency were measured by a variety of means, including 3 survey questions, the Five Facet Mindfulness Questionnaire, 2 prompts on a 7-point semantic scale, and a slightly modified version of the validated Perceived Stress Scale.25-28
Survey Results
Residents gave the rotation positive feedback with a mean score of 8.5 out of 10. They reported the beneficial impact of seeing the nontraditional and nonpharmacological practices in treating patients, chronic pain management team approaches, and enjoyed being able to participate in group classes with patients. Many residents expressed a desire for a longer rotation to have more time to experience the behavioral health-focused sessions. Residents also requested additional information on nutritional supplements/natural medicines, battlefield acupuncture training and osteopathic manipulative therapy practices. All residents reported the rotation personally and/or professionally benefited them (Appendix).
Given the sample of 12 residents, values are presented as prerotation to postrotation comparisons without statistical analysis. There was a trend towards an increase in the reported use and recommendation of 26 modalities of nonconventional therapies following the rotation. There was also a slight increase in resource knowledge and use of these resources, and residents reported accessing more types of resources. Mean scores of the case study to gauge knowledge and application of IM increased from 7.5 at baseline to 11.0 after the rotation. Resident confidence in diagnosis increased for all 8 conditions, but confidence in treatment only increased for 4 conditions.
Results of self-care importance, self-care frequency and mindfulness were consistent baseline to postrotation. The mean time residents spent regularly practicing self-care during a work week increased slightly while feelings of burnout decreased. The perceived stress scale average score decreased from 13.4 at baseline to 10.5 after rotation.
DISCUSSION
The implementation of an IM residency rotation that incorporates whole health and interprofessional practices demonstrated improved perception and increased use of IM/CAM resources and knowledge among a small sample of third-year residents. Residents reported they had a positive experience participating in the rotation and gained knowledge, resources, and skills they felt confident discussing with their patients.
Many studies reported favorable attitudes and perceptions of IM/CAM use among physicians, but few have assessed these measures while implementing a training curriculum.3,4,22 Gardiner and colleagues reported on the perception and use of IM resources among family medicine residents.4 The study found that while 58% of all residents reported IM/CAM as an important part of their training, only 60% reported they received it or had specific learning objectives in their curriculum. 4 The program outlined in this study and previous research illustrate that physicians recognize the importance of IM/CAM education in training programs, but most were unaware of the resources available or did not feel comfortable counseling patients about most IM/CAM applications.
Residents in this program slightly increased their use of IM/CAM to diagnose and treat medical conditions after the rotation. A study by Wahner-Roedler and colleagues assessed physician knowledge regarding common IM/CAM therapies.3 On average, physicians only felt knowledgeable and comfortable counseling patients for 3 of 13 listed treatments/techniques and few natural herbal treatments. The study also found that most physicians had difficulty accessing IM/CAM information at their institution despite having free access to electronic databases. However, this study only assessed physician attitudes of IM/CAM and did not include an educational component to increase their knowledge of the modalities.3 This evaluation supports the need for interventions like the program described in this article that provide physicians with access to evidence-based resources combined with the applied experiences to increase their comfort within this growing field.
Though the sample size in this study was small, its results support existing research indicating that clinicians view selfcare as important. Many residents were already using a self-care plan at baseline, but there was slight increase in the practice of self-care during the rotation and a slight decrease in burnout. Previous research reflects high rates of burnout and relatively poor quality of life among primary care physicians.15 Burnout is associated with lower quality of care, lower patient satisfaction and contributes to medical errors. Studies suggest as many as 60% of primary care physicians report symptoms of burnout, which negatively affected the quality of patient care they provide.15
Despite the profound effects burnout has on physicians and patient care, a standardized wellness education or self-care tool kit is not currently available. The University of Massachusetts recently introduced a pilot program to promote resident wellness that demonstrated favorable results.15 A meta-analysis of physicians and medical trainees found decreases in anxiety and symptoms of anxiety as well as a decrease in burnout among participants in cognitive, behavioral and mindfulness interventions.29 However, unlike our program, these programs focused solely on the well-being of medical trainees, residents, and physicians and didn’t focus on the patient-clinician interactions. Given the impact on patient care, there is a need to develop and implement additional programs like our residency rotation that promote health and wellness among physicians while also evaluating how physicians may translate these skills to patient education.
While this program st i l l exists for third-year residents at Baltimore GRECC, it has significantly changed since the COVID-19 pandemic. For about the first 6 months of the pandemic, when physical distancing requirements were in place, family medicine trainees were not able to rotate. Upon return to the facility, many group classes were cancelled and some clinicians no longer offered the sessions. The rotation has evolved to a hybrid format, where many group classes for veteran patients are offered virtually, and residents observe a mix of virtual and in-person shadowing opportunities. Our formal evaluation included administering the survey and occurred from July 2018 to July 2019 but wasn’t implemented upon return to post-COVID activities due to the inconsistent experiences offered to residents over the past few years. Future research should evaluate the impact of this hybrid program on the clinicians and explore dissemination to other VAMCs and their academic affiliates.
Limitations
Project recruitment was limited to 11 family medicine and 1 preventive medicine resident. Perceptions, use of IM/CAM, and knowledge about IM/CAM could be considerably different in different departments with varying schedules, hours worked, and patient volumes. Secondly, the survey was conducted 2 weeks apart. Indications of self-care and burnout may not reflect long-term effects, adoption, or maintenance. Future research should include longer follow up to examine how this type of educational activity may impact burnout rates of physicians following the completion of residency, as well as changes in perspectives of IM/CAM while practicing as a physician. Trainees were exposed to a wide range of health care professions, but additional research is needed regarding medical resident perceptions of the roles of specific professions in a collaborative health care team.30,31
CONCLUSIONS
The residency rotation program illustrates the benefits of establishing a standardized IM/CAM rotation that includes self-care resources in family medicine programs to adequately train clinicians to practice wellness and promote it to their patients. The results of this project suggest this type of training will help residents assess the literature to better counsel patients on IM/CAM options while also providing strategies for maintaining optimal health and well-being for health care professionals. Broadening and shifting the scope of medicine from treatment to prevention, personal wellness, and optimal healing should be a top priority.
Integrative medicine or complementary alternative medicine (IM/CAM) is increasingly being recognized as an integral part of optimal health and healing. IM/CAM “reaffirms the importance of the relationship between practitioner and patient, focuses on the whole person, is informed by evidence, and makes use of all appropriate therapeutic approaches, healthcare professionals and disciplines.”1 IM/CAM encompasses a wide range of therapies, conceptual frameworks, and health care-related professions, such as acupuncture, massage, dietary supplements, mindfulness, yoga, meditation and guided imagery.1 Research has found that 30% to 98% of patients with chronic conditions seek IM/CAM therapies.1-3
Despite the high prevalence of patients utilizing IM/CAM therapies and the National Institutes of Health grants for IM/CAM education, implementation of IM/CAM instruction in graduate medical education programs remains inconsistent.1 Barriers cited by programs include a lack of IM/CAM experts in the program, faculty training, competing financial resources, and an already full resident education schedule.4 As a result, many physicians have limited or no training in IM/CAM.1,5
The US Department of Veterans Affairs (VA) offers IM/CAM health programs to veterans and caregivers as part of its whole health care initiative.6 Several VA health care systems have adopted whole health and IM/CAM through programs for mental health integration into primary care; women’s health; integrative pain care; geriatrics, through adoption of Age-Friendly Health Systems standards; and nutrition and physical activity.7-13 The VA provides training to more medical students than any other health system: > 95% of US medical schools are affiliated with a VA medical center (VAMC).14 As part of the training mission, VA seeks to encourage students of diverse professions to consider careers in the VA.14
Residency is a time for newly licensed physicians to acquire additional experience and training to translate knowledge and skills acquired during medical school directly to patient care.15 However, residency curricula have limited time to incorporate IM/CAM training. Residency training is also physically and psychosocially demanding, often resulting in inadequate self-care, poor work-life balance, and disrupted sleep.16-18 Resident wellness is at a historic low, resulting in high rates of burnout during training.4,15
Residency programs are required to provide wellness education; however, most programs include minimal content.19 Despite high rates of burnout, formal curricula on the topic have not been established. 20 IM/CAM education also can provide a path for residents to learn about and engage in mindfulness-based training or cognitive stress reduction for self-care.
INTEGRATIVE WHOLE HEALTH ROTATION
In 2017, the Baltimore Geriatric Research Education and Clinical Center (GRECC) established an IM/whole health residency rotation and created a structured curriculum incorporating self-assessment, active reflection, and self-care to complement training in specific IM/CAM modalities for residents in family medicine. The curriculum evaluated how this training improved residents’ perceptions of IM/CAM and how it personally and professionally impacted the practice of self-care as a strategy to decrease burnout. We hypothesized that this structured experience would increase IM/CAM knowledge among clinicians while promoting the importance and practice of self-care to reduce burnout.
The 2-week IM/CAM curriculum was developed by University of Maryland School of Medicine faculty in partnership with the Baltimore GRECC and staff at the VA Maryland Health Care System. The curriculum was designed to expose residents to the 8 components of the whole health Circle of Health (moving the body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind) in addition to IM/ CAM modalities the VA is mandated to offer to veterans (acupuncture, chiropractic, meditation, massage therapy, biofeedback, clinical hypnosis, guided imagery, yoga, and tai chi).21 Twelve residents (1 preventive medicine and 11 third-year family medicine residents) rotated individually throughout the year as part of their behavioral health block rotation. All residents completed the 2-week curriculum as their schedules allowed. The curriculum consisted of didactics sessions and activities at the Baltimore, Loch Raven, and Perry Point VAMCs. Residents completed evaluations before and after the rotation. The experience described in this article by the residents and the survey data were collected from the 2018/2019 training year. A rotation syllabus, competencies adapted from Locke and colleagues and skills residents obtain during this rotation that support these competencies, as well as a resident sample schedule were developed (eAppendix is available at doi:10.12788/fp.0544).1
Rotation Overview
for each resident were built around instructional opportunities, which included 1-on-1 didactics, direct observation of treatment modalities, and personal reflection of the residents’ self-care practices. While each resident’s rotation schedule varied slightly due to their schedules, the foundational instruction elements were the same. Didactic session themes included an overview of IM/CAM, nutrition, narrative medicine, pain psychology, music therapy, chaplain services, motor-cognitive training, and exercise guidelines. Assigned readings, including peer-reviewed literature on IM/CAM therapies, complemented all sessions. Residents created an evidence-supported integrative treatment plan for a patient with a condition of interest to them.
Residents observed clinician-led veteran group sessions on IM/CAM treatment modalities, including guided meditation, mindfulness and relaxation, self-awareness, living well with chronic pain, tai chi, drumming for health and balance, anger management, recovery group, acceptance and commitment therapy, and Gerofit exercise. The group classes allowed residents to actively participate in the activity or discussion. Residents also shadowed VA clinicians in sleep, pain, nutrition, acupuncture, and mental health clinics.
Residents were encouraged to practice self-care during the 2-week rotation. The rotation schedule built in free time, including a 1-hour daily lunch period, for residents to consider their own health habits, complete a personal health inventory, and try self-care activities outlined on the syllabus with links to resources. These resources also served as educational materials that residents could share with patients. All materials, including didactic lectures, journal articles and self-care resources, were provided to each resident through a free online course to ensure residents had access throughout and following completion of the rotation. This content, including the rotation evaluation metrics, is available upon request from the corresponding author.
Evaluations
Residents completed a survey before and after the rotation to measure IM/CAM knowledge and application and self-care/ burnout perceptions. Residents were asked to evaluate rotation sessions and comment on whether this rotation benefited them personally and professionally (Table 1). Descriptive statistics were analyzed using Microsoft Excel. Given the small sample size and lack of statistical power, only mean survey results are reported in this article. Because this opportunity is specific to the University of Maryland School of Medicine and the proposed project was part of ordinary educational practice, the study was deemed not human subject research by the University of Maryland Institutional Review Board (HP-00089256).
Perceptions and attitudes toward IM/CAM were assessed using a survey designed by the University of Minnesota Academic Health Center. It included 18 items scored on a 5-point semantic rating scale (1, strongly disagree; 5, strongly agree).22 Residents rated their level of agreement with statements reflecting both positive (eg, clinical care should integrate the best of conventional and CAM practices) and negative (eg, CAM is a threat to public health) views. Three questions adapted from the NHIS Adult Complementary Health Questionnaire and UC Irvine Survey of Health Care Use and Practice assessed the use of IM/CAM resources.23,24
Resident knowledge and application of IM/CAM were measured using a case study designed by the course faculty. The case listed a chief complaint of nerve pain, with a history of chronic pain, neuropathic pain, anxiety, chronic fatigue, depression, insomnia, posttraumatic stress disorder, history of present illness, past surgical history, medication list, review of symptoms, laboratory values, and physical examination. The residents completed an assessment before and after the rotation. Residents rated their confidence in the diagnosis and treatment of 8 medical conditions using a 5-point semantic rating scale (Table 2). Self-care importance and selfcare frequency were measured by a variety of means, including 3 survey questions, the Five Facet Mindfulness Questionnaire, 2 prompts on a 7-point semantic scale, and a slightly modified version of the validated Perceived Stress Scale.25-28
Survey Results
Residents gave the rotation positive feedback with a mean score of 8.5 out of 10. They reported the beneficial impact of seeing the nontraditional and nonpharmacological practices in treating patients, chronic pain management team approaches, and enjoyed being able to participate in group classes with patients. Many residents expressed a desire for a longer rotation to have more time to experience the behavioral health-focused sessions. Residents also requested additional information on nutritional supplements/natural medicines, battlefield acupuncture training and osteopathic manipulative therapy practices. All residents reported the rotation personally and/or professionally benefited them (Appendix).
Given the sample of 12 residents, values are presented as prerotation to postrotation comparisons without statistical analysis. There was a trend towards an increase in the reported use and recommendation of 26 modalities of nonconventional therapies following the rotation. There was also a slight increase in resource knowledge and use of these resources, and residents reported accessing more types of resources. Mean scores of the case study to gauge knowledge and application of IM increased from 7.5 at baseline to 11.0 after the rotation. Resident confidence in diagnosis increased for all 8 conditions, but confidence in treatment only increased for 4 conditions.
Results of self-care importance, self-care frequency and mindfulness were consistent baseline to postrotation. The mean time residents spent regularly practicing self-care during a work week increased slightly while feelings of burnout decreased. The perceived stress scale average score decreased from 13.4 at baseline to 10.5 after rotation.
DISCUSSION
The implementation of an IM residency rotation that incorporates whole health and interprofessional practices demonstrated improved perception and increased use of IM/CAM resources and knowledge among a small sample of third-year residents. Residents reported they had a positive experience participating in the rotation and gained knowledge, resources, and skills they felt confident discussing with their patients.
Many studies reported favorable attitudes and perceptions of IM/CAM use among physicians, but few have assessed these measures while implementing a training curriculum.3,4,22 Gardiner and colleagues reported on the perception and use of IM resources among family medicine residents.4 The study found that while 58% of all residents reported IM/CAM as an important part of their training, only 60% reported they received it or had specific learning objectives in their curriculum. 4 The program outlined in this study and previous research illustrate that physicians recognize the importance of IM/CAM education in training programs, but most were unaware of the resources available or did not feel comfortable counseling patients about most IM/CAM applications.
Residents in this program slightly increased their use of IM/CAM to diagnose and treat medical conditions after the rotation. A study by Wahner-Roedler and colleagues assessed physician knowledge regarding common IM/CAM therapies.3 On average, physicians only felt knowledgeable and comfortable counseling patients for 3 of 13 listed treatments/techniques and few natural herbal treatments. The study also found that most physicians had difficulty accessing IM/CAM information at their institution despite having free access to electronic databases. However, this study only assessed physician attitudes of IM/CAM and did not include an educational component to increase their knowledge of the modalities.3 This evaluation supports the need for interventions like the program described in this article that provide physicians with access to evidence-based resources combined with the applied experiences to increase their comfort within this growing field.
Though the sample size in this study was small, its results support existing research indicating that clinicians view selfcare as important. Many residents were already using a self-care plan at baseline, but there was slight increase in the practice of self-care during the rotation and a slight decrease in burnout. Previous research reflects high rates of burnout and relatively poor quality of life among primary care physicians.15 Burnout is associated with lower quality of care, lower patient satisfaction and contributes to medical errors. Studies suggest as many as 60% of primary care physicians report symptoms of burnout, which negatively affected the quality of patient care they provide.15
Despite the profound effects burnout has on physicians and patient care, a standardized wellness education or self-care tool kit is not currently available. The University of Massachusetts recently introduced a pilot program to promote resident wellness that demonstrated favorable results.15 A meta-analysis of physicians and medical trainees found decreases in anxiety and symptoms of anxiety as well as a decrease in burnout among participants in cognitive, behavioral and mindfulness interventions.29 However, unlike our program, these programs focused solely on the well-being of medical trainees, residents, and physicians and didn’t focus on the patient-clinician interactions. Given the impact on patient care, there is a need to develop and implement additional programs like our residency rotation that promote health and wellness among physicians while also evaluating how physicians may translate these skills to patient education.
While this program st i l l exists for third-year residents at Baltimore GRECC, it has significantly changed since the COVID-19 pandemic. For about the first 6 months of the pandemic, when physical distancing requirements were in place, family medicine trainees were not able to rotate. Upon return to the facility, many group classes were cancelled and some clinicians no longer offered the sessions. The rotation has evolved to a hybrid format, where many group classes for veteran patients are offered virtually, and residents observe a mix of virtual and in-person shadowing opportunities. Our formal evaluation included administering the survey and occurred from July 2018 to July 2019 but wasn’t implemented upon return to post-COVID activities due to the inconsistent experiences offered to residents over the past few years. Future research should evaluate the impact of this hybrid program on the clinicians and explore dissemination to other VAMCs and their academic affiliates.
Limitations
Project recruitment was limited to 11 family medicine and 1 preventive medicine resident. Perceptions, use of IM/CAM, and knowledge about IM/CAM could be considerably different in different departments with varying schedules, hours worked, and patient volumes. Secondly, the survey was conducted 2 weeks apart. Indications of self-care and burnout may not reflect long-term effects, adoption, or maintenance. Future research should include longer follow up to examine how this type of educational activity may impact burnout rates of physicians following the completion of residency, as well as changes in perspectives of IM/CAM while practicing as a physician. Trainees were exposed to a wide range of health care professions, but additional research is needed regarding medical resident perceptions of the roles of specific professions in a collaborative health care team.30,31
CONCLUSIONS
The residency rotation program illustrates the benefits of establishing a standardized IM/CAM rotation that includes self-care resources in family medicine programs to adequately train clinicians to practice wellness and promote it to their patients. The results of this project suggest this type of training will help residents assess the literature to better counsel patients on IM/CAM options while also providing strategies for maintaining optimal health and well-being for health care professionals. Broadening and shifting the scope of medicine from treatment to prevention, personal wellness, and optimal healing should be a top priority.
- Locke AB, Gordon A, Guerrera MP, Gardiner P, Lebensohn P. Recommended integrative medicine competencies for family medicine residents. Explore (NY). 2013;9(5):308-313. doi:10.1016/j.explore.2013.06.005
- Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA. 1998;280(18):1569-1575. doi:10.1001/jama.280.18.1569
- Wahner-Roedler DL, Vincent A, Elkin PL, Loehrer LL, Cha SS, Bauer BA. Physicians’ attitudes toward complementary and alternative medicine and their knowledge of specific therapies: a survey at an academic medical center. Evid Based Complement Alternat Med. 2006;3(4):495-501. doi:10.1093/ecam/nel036
- Gardiner P, Filippelli AC, Lebensohn P, Bonakdar R. Family medicine residency program directors attitudes and knowledge of family medicine CAM competencies. Explore (NY). 2013;9(5):299-307. doi:10.1016/j.explore.2013.06.002
- Sierpina V, Levine R, Astin J, Tan A. Use of mind-body therapies in psychiatry and family medicine faculty and residents: attitudes, barriers, and gender differences. Explore (NY). 2007;3(2):129-135. doi:10.1016/j.explore.2006.12.001
- Krist AH, South-Paul J, Meisnere M, eds. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023.
- Bokhour BG, DeFaccio R, Gaj L, et al. Changes in patientreported outcomes associated with receiving whole health in the Veteran Health Administration (VHA)’s National Demonstration Project. J Gen Intern Med. 2024;39(1):84-94. doi:10.1007/s11606-023-08376-0
- Courtney RE, Schadegg MJ, Bolton R, Smith S, Harden SM. Using a whole health approach to build biopsychosocial- spiritual personal health plans for veterans with chronic pain. Pain Manag Nurs. 2024;25(1):69-74. doi:10.1016/j.pmn.2023.09.010
- Gabrielian S, Jones AL, Hoge AE, et al. Enhancing primary care experiences for homeless patients with serious mental illness: results from a national survey. J Prim Care Community Health. 2021;12:2150132721993654. doi:10.1177/2150132721993654
- Matthieu MM, Church KA, Taylor LD, et al. Integrating the age-friendly health systems movement in Veterans Health Administration: national advance care planning via group visits and the 4Ms framework. Health Soc Work. 2023;48(4):277-280. doi:10.1093/hsw/hlad022
- Meisler AW, Gianoli MO, Na PJ, Pietrzak RH. Functional disability in US military veterans: the importance of integrated whole health initiatives. Prim Care Companion CNS Disord. 2023;25(4):22m03461. doi:10.4088/PCC.22m03461
- Ortmeyer HK, Giffuni J, Etchberger D, Katzel L. The role of companion dogs in the VA Maryland Health Care System Whole Health(y) GeroFit Program. Animals (Basel). 2023;13(19):3047. doi:10.3390/ani13193047
- Sullivan MB, Hill K, Ballengee LA, et al. Remotely delivered psychologically informed mindful movement physical therapy for pain care: a framework for operationalization. Glob Adv Integr Med Health. 2023;12:27536130231209751. doi:10.1177/27536130231209751
- (OAA) OoAA. 75th Anniversary: Passion to learn. Power to heal. Washington DC.: US Department of Veterans Affairs; 2021. https://content.yudu.com/web/448fx/0A448g9/75thAnniversary2021/html/index.html?page=24&origin=reader
- Runyan C, Savageau JA, Potts S, Weinreb L. Impact of a family medicine resident wellness curriculum: a feasibility study. Med Educ Online. 2016;21:30648. doi:10.3402/meo.v21.30648
- Lafreniere JP, Rios R, Packer H, Ghazarian S, Wright SM, Levine RB. Burned out at the bedside: patient perceptions of physician burnout in an internal medicine resident continuity clinic. J Gen Intern Med. 2016;31(2):203-208. doi:10.1007/s11606-015-3503-3
- Freedy JR, Staley C, Mims LD, et al. Social, individual, and environmental characteristics of family medicine resident burnout: a CERA study. Fam Med. 2022;54(4):270-276. doi:10.22454/FamMed.2022.526799
- Alrishan MA, Alshammari SA. Prevalence of sleep deprivation and its effect on the performance of family medicine residents in Riyadh, Saudi Arabia. J Family Community Med. 2020;27(2):125-130. doi:10.4103/jfcm.JFCM_9_20
- ACGME. ACGME Program Requirements for Graduate Medical Education in Family Medicine. https://www.acgme.org/globalassets/pfassets/programrequirements/120_familymedicine_2024.pdf
- Nene Y, Tadi P. Resident Burnout. In: StatPearls; 2023.
- Bokhour BG, Haun JN, Hyde J, Charns M, Kligler B. Transforming the veterans affairs to a whole health system of care: time for action and research. Med Care. 2020;58(4):295-300. doi:10.1097/MLF.0000000000001316
- Kreitzer MJ, Mitten D, Harris I, Shandeling J. Attitudes toward CAM among medical, nursing, and pharmacy faculty and students: a comparative analysis. Altern Ther Health Med. 2002;8(6):44-53.
- Clarke TC, Black LI, Stussman BJ, Barnes PM, Nahin RL. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Report. 2015(79):1-16.
- Nguyen J, Liu MA, Patel RJ, Tahara K, Nguyen AL. Use and interest in complementary and alternative medicine among college students seeking healthcare at a university campus student health center. Complement Ther Clin Pract. 2016;24:103-108. doi:10.1016/j.ctcp.2016.06.001
- Baer RA, Smith GT, Hopkins J, Krietemeyer J, Toney L. Using self-report assessment methods to explore facets of mindfulness. Assessment. 2006;13(1):27-45. doi:10.1177/1073191105283504
- Baer RA, Smith GT, Lykins E, et al. Construct validity of the five facet mindfulness questionnaire in meditating and nonmeditating samples. Assessment. 2008;15(3):329-342. doi:10.1177/1073191107313003
- West CP, Dyrbye LN, Sloan JA, Shanafelt TD. Single item measures of emotional exhaustion and depersonalization are useful for assessing burnout in medical professionals. J Gen Intern Med. 2009;24(12):1318- 1321. doi:10.1007/s11606-009-1129-z
- Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24(4):385-396.
- Regehr C, Glancy D, Pitts A, LeBlanc VR. Interventions to reduce the consequences of stress in physicians: a review and meta-analysis. J Nerv Ment Dis. 2014;202(5):353-359. doi:10.1097/NMD.0000000000000130
- Visser CLF, Ket JCF, Croiset G, Kusurkar RA. Perceptions of residents, medical and nursing students about interprofessional education: a systematic review of the quantitative and qualitative literature. BMC Med Educ. 2017;17(1):77. doi:10.1186/s12909-017-0909-0
- Lingard L, Espin S, Evans C, Hawryluck L. The rules of the game: interprofessional collaboration on the intensive care unit team. Crit Care. 2004;8(6):R403-408. doi:10.1186/cc2958
- Locke AB, Gordon A, Guerrera MP, Gardiner P, Lebensohn P. Recommended integrative medicine competencies for family medicine residents. Explore (NY). 2013;9(5):308-313. doi:10.1016/j.explore.2013.06.005
- Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA. 1998;280(18):1569-1575. doi:10.1001/jama.280.18.1569
- Wahner-Roedler DL, Vincent A, Elkin PL, Loehrer LL, Cha SS, Bauer BA. Physicians’ attitudes toward complementary and alternative medicine and their knowledge of specific therapies: a survey at an academic medical center. Evid Based Complement Alternat Med. 2006;3(4):495-501. doi:10.1093/ecam/nel036
- Gardiner P, Filippelli AC, Lebensohn P, Bonakdar R. Family medicine residency program directors attitudes and knowledge of family medicine CAM competencies. Explore (NY). 2013;9(5):299-307. doi:10.1016/j.explore.2013.06.002
- Sierpina V, Levine R, Astin J, Tan A. Use of mind-body therapies in psychiatry and family medicine faculty and residents: attitudes, barriers, and gender differences. Explore (NY). 2007;3(2):129-135. doi:10.1016/j.explore.2006.12.001
- Krist AH, South-Paul J, Meisnere M, eds. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023.
- Bokhour BG, DeFaccio R, Gaj L, et al. Changes in patientreported outcomes associated with receiving whole health in the Veteran Health Administration (VHA)’s National Demonstration Project. J Gen Intern Med. 2024;39(1):84-94. doi:10.1007/s11606-023-08376-0
- Courtney RE, Schadegg MJ, Bolton R, Smith S, Harden SM. Using a whole health approach to build biopsychosocial- spiritual personal health plans for veterans with chronic pain. Pain Manag Nurs. 2024;25(1):69-74. doi:10.1016/j.pmn.2023.09.010
- Gabrielian S, Jones AL, Hoge AE, et al. Enhancing primary care experiences for homeless patients with serious mental illness: results from a national survey. J Prim Care Community Health. 2021;12:2150132721993654. doi:10.1177/2150132721993654
- Matthieu MM, Church KA, Taylor LD, et al. Integrating the age-friendly health systems movement in Veterans Health Administration: national advance care planning via group visits and the 4Ms framework. Health Soc Work. 2023;48(4):277-280. doi:10.1093/hsw/hlad022
- Meisler AW, Gianoli MO, Na PJ, Pietrzak RH. Functional disability in US military veterans: the importance of integrated whole health initiatives. Prim Care Companion CNS Disord. 2023;25(4):22m03461. doi:10.4088/PCC.22m03461
- Ortmeyer HK, Giffuni J, Etchberger D, Katzel L. The role of companion dogs in the VA Maryland Health Care System Whole Health(y) GeroFit Program. Animals (Basel). 2023;13(19):3047. doi:10.3390/ani13193047
- Sullivan MB, Hill K, Ballengee LA, et al. Remotely delivered psychologically informed mindful movement physical therapy for pain care: a framework for operationalization. Glob Adv Integr Med Health. 2023;12:27536130231209751. doi:10.1177/27536130231209751
- (OAA) OoAA. 75th Anniversary: Passion to learn. Power to heal. Washington DC.: US Department of Veterans Affairs; 2021. https://content.yudu.com/web/448fx/0A448g9/75thAnniversary2021/html/index.html?page=24&origin=reader
- Runyan C, Savageau JA, Potts S, Weinreb L. Impact of a family medicine resident wellness curriculum: a feasibility study. Med Educ Online. 2016;21:30648. doi:10.3402/meo.v21.30648
- Lafreniere JP, Rios R, Packer H, Ghazarian S, Wright SM, Levine RB. Burned out at the bedside: patient perceptions of physician burnout in an internal medicine resident continuity clinic. J Gen Intern Med. 2016;31(2):203-208. doi:10.1007/s11606-015-3503-3
- Freedy JR, Staley C, Mims LD, et al. Social, individual, and environmental characteristics of family medicine resident burnout: a CERA study. Fam Med. 2022;54(4):270-276. doi:10.22454/FamMed.2022.526799
- Alrishan MA, Alshammari SA. Prevalence of sleep deprivation and its effect on the performance of family medicine residents in Riyadh, Saudi Arabia. J Family Community Med. 2020;27(2):125-130. doi:10.4103/jfcm.JFCM_9_20
- ACGME. ACGME Program Requirements for Graduate Medical Education in Family Medicine. https://www.acgme.org/globalassets/pfassets/programrequirements/120_familymedicine_2024.pdf
- Nene Y, Tadi P. Resident Burnout. In: StatPearls; 2023.
- Bokhour BG, Haun JN, Hyde J, Charns M, Kligler B. Transforming the veterans affairs to a whole health system of care: time for action and research. Med Care. 2020;58(4):295-300. doi:10.1097/MLF.0000000000001316
- Kreitzer MJ, Mitten D, Harris I, Shandeling J. Attitudes toward CAM among medical, nursing, and pharmacy faculty and students: a comparative analysis. Altern Ther Health Med. 2002;8(6):44-53.
- Clarke TC, Black LI, Stussman BJ, Barnes PM, Nahin RL. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Report. 2015(79):1-16.
- Nguyen J, Liu MA, Patel RJ, Tahara K, Nguyen AL. Use and interest in complementary and alternative medicine among college students seeking healthcare at a university campus student health center. Complement Ther Clin Pract. 2016;24:103-108. doi:10.1016/j.ctcp.2016.06.001
- Baer RA, Smith GT, Hopkins J, Krietemeyer J, Toney L. Using self-report assessment methods to explore facets of mindfulness. Assessment. 2006;13(1):27-45. doi:10.1177/1073191105283504
- Baer RA, Smith GT, Lykins E, et al. Construct validity of the five facet mindfulness questionnaire in meditating and nonmeditating samples. Assessment. 2008;15(3):329-342. doi:10.1177/1073191107313003
- West CP, Dyrbye LN, Sloan JA, Shanafelt TD. Single item measures of emotional exhaustion and depersonalization are useful for assessing burnout in medical professionals. J Gen Intern Med. 2009;24(12):1318- 1321. doi:10.1007/s11606-009-1129-z
- Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. J Health Soc Behav. 1983;24(4):385-396.
- Regehr C, Glancy D, Pitts A, LeBlanc VR. Interventions to reduce the consequences of stress in physicians: a review and meta-analysis. J Nerv Ment Dis. 2014;202(5):353-359. doi:10.1097/NMD.0000000000000130
- Visser CLF, Ket JCF, Croiset G, Kusurkar RA. Perceptions of residents, medical and nursing students about interprofessional education: a systematic review of the quantitative and qualitative literature. BMC Med Educ. 2017;17(1):77. doi:10.1186/s12909-017-0909-0
- Lingard L, Espin S, Evans C, Hawryluck L. The rules of the game: interprofessional collaboration on the intensive care unit team. Crit Care. 2004;8(6):R403-408. doi:10.1186/cc2958
Development of an Integrative Medicine Rotation for Family Medicine and Preventive Medicine Residency
Development of an Integrative Medicine Rotation for Family Medicine and Preventive Medicine Residency

Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
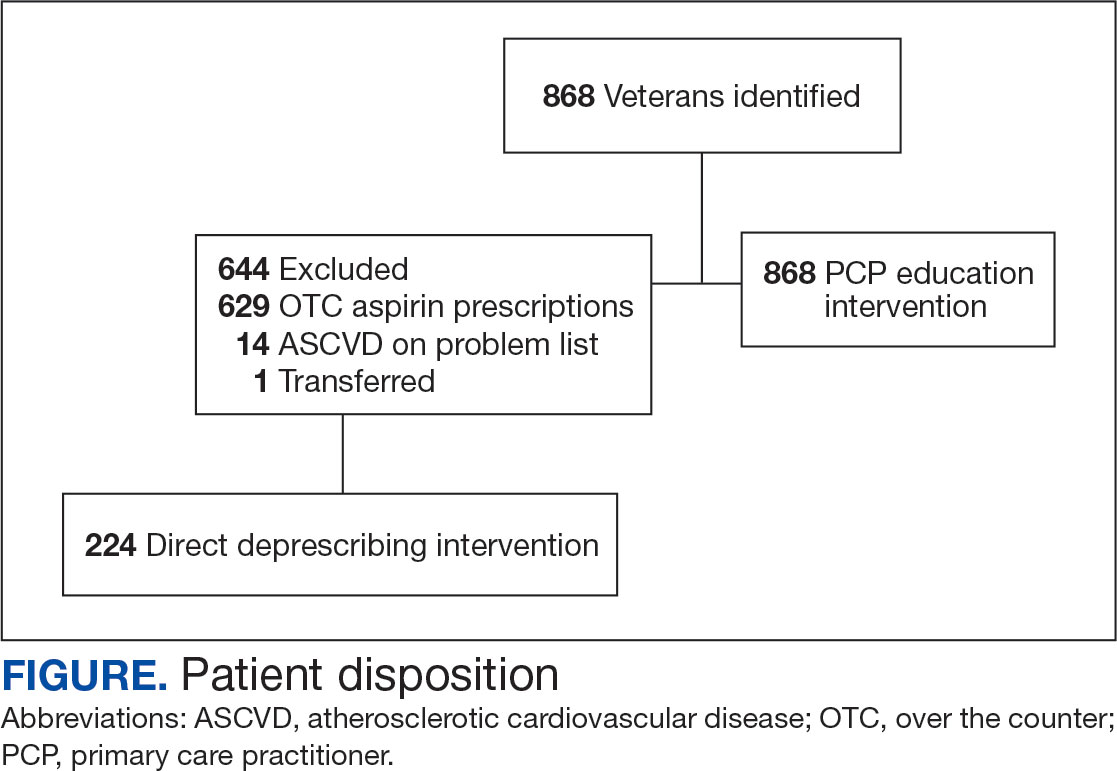
Results
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
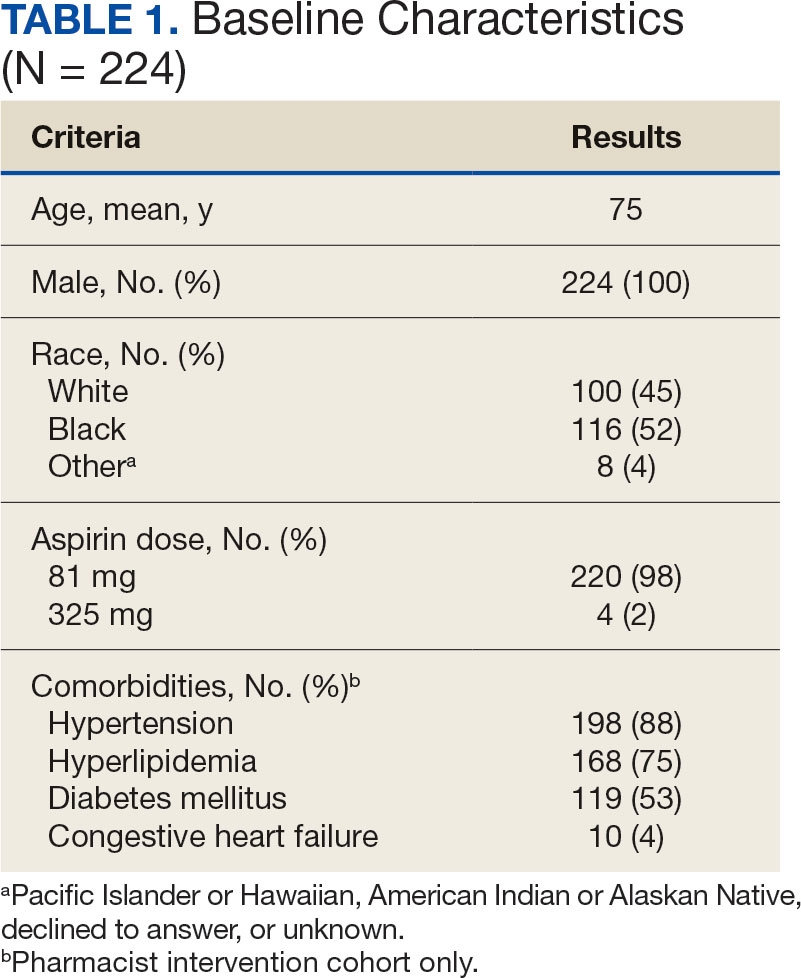
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).

The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.

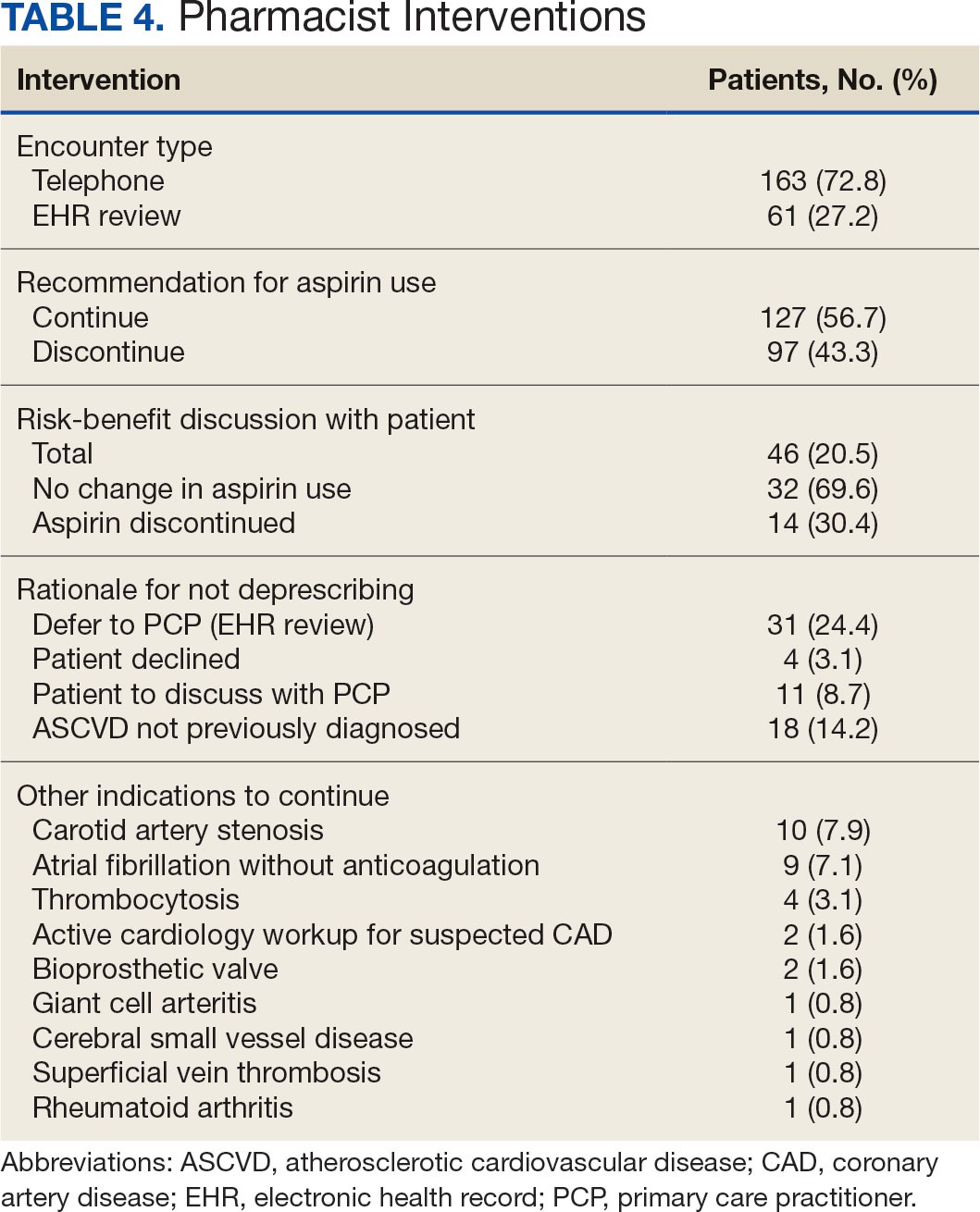
Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
Results
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).
The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.
Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
Results
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).
The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.
Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Treating GERD: Lifestyle Modifications vs Medication
Dear colleagues,
Gastroesophageal reflux disease (GERD) is a common reason for referral to gastroenterology. It affects a broad cross-section of our population and is often managed through a combination of lifestyle modifications and proton pump inhibitors (PPIs). However,
While PPIs are highly effective, concerns about their potential side effects frequently make headlines. Moreover, the financial burden of lifelong PPI use is a growing consideration. In this issue of Perspectives, Dr. Brijesh B. Patel and Dr. Juan D. Gomez Cifuentes explore these questions. Dr. Gomez Cifuentes highlights the benefits of lifestyle changes and identifies which strategies have proved most effective in his practice. Dr. Patel examines the ubiquitous use of PPIs and the challenges of sustaining adherence to lifestyle modifications. We hope these discussions will spark new ideas for managing GERD in your own practice.

We also welcome your thoughts on this topic — join the conversation on X at @AGA_GIHN.
Gyanprakash A. Ketwaroo, MD, MSc, is associate professor of medicine, Yale University, New Haven, and chief of endoscopy at West Haven VA Medical Center, both in Connecticut. He is an associate editor for GI & Hepatology News.
Do Lifestyle Changes Still Apply in the Treatment of GERD?
BY JUAN D. GOMEZ CIFUENTES, MD
Lifestyle changes are an essential part of managing gastroesophageal reflux disease (GERD). Increasingly, patients are asking about non-medication approaches to control their symptoms. These lifestyle modifications can be categorized into four main areas: 1) Weight loss, the cornerstone intervention, with significant symptom improvement observed after losing as little as 1.7 BMI points. 2) Dietary modifications, which includes both the traditional avoidance of trigger foods and the newer focus on a diet low in simple carbohydrates. 3) Bedtime adjustments, strategies that include elevating the head of the bed, sleeping on the left side, using anti-reflux pillows, and avoiding late-night meals. 4) Tobacco cessation, a key measure for reducing GERD symptoms and promoting overall health. I routinely discuss these changes with my patients, as they not only help manage GERD but also foster healthy habits and have a positive impact beyond the gastrointestinal tract.

Weight loss is the most impactful lifestyle intervention for GERD. Research shows a clear linear improvement in symptoms with weight reduction. Traditionally, losing 10% of body weight is a widely accepted goal, extrapolated from other obesity-associated conditions. A reduction in 3.5 points of BMI led to significant symptom improvement in landmark studies but also a modest reduction of 1.7 BMI points has been shown to provide symptom relief.1 Abdominal circumference is another key metric used to track progress, as central obesity rather than BMI alone is strongly linked with GERD. Goals are typically set at less than 40 inches for men and 35 inches for women. Patients using GLP-1 agonists should be informed that these medications may temporarily worsen GERD symptoms due to delayed gastric emptying, however in the long-term these symptoms are expected to improve once significant weight loss is achieved.
Food triggers vary among individuals, with common culprits including fatty meals, spicy foods, chocolate, tomato sauce, citrus fruits, and carbonated beverages. Patients tend to overemphasize diet elimination based on triggers and engage in strict diets. Patients are frequently afraid of these foods causing direct damage to the esophageal mucosa but the hypothesis is that these triggers worsen GERD by increasing transient relaxations of the lower esophageal sphincter. The evidence behind this and diet elimination based on triggers has always been weak. In my practice, I encourage patients to follow a diet low in simple carbohydrates. Simple carbohydrates are present in highly processed food, the average western diet contains ~140 g/day. In a trial, a diet low in simple sugars (monosaccharides and disaccharides < 62 g/day) without reducing total daily calories, objectively improved total acid exposure time in pH study.2
Thanks to gravity, nocturnal GERD symptoms are the culprit of many restless nights in these patients. I recommend avoiding food 3 hours before lying down. Since the stomach empties approximately 90% of its contents after 4 hours, waiting longer is not recommended and may result in hunger, making it harder to fall asleep. Sleeping on the left side, which takes advantage of the gastric anatomy, has proved to objectively decrease nocturnal acid exposure time, though some patients may find it challenging to maintain this position all night.3
Elevating the head of the bed is another effective intervention, but it must involve raising the upper body from the waist. Patients should avoid stacking ordinary pillows as this will only elevate the neck and place the body in an unnatural position for sleeping. The most effective strategies are putting blocks/bricks under the feet of the bed, using a bed wedge between the mattress and the box spring or using an adjustable bed frame. There are two types of pillows that have been shown to improve nocturnal GERD symptoms. The classic wedge pillows and the more expensive Medcline reflux relief system®. The Medcline pillow has a dual mechanism that elevates the upper body but also keeps the body on the left side position.4
Tobacco cessation is strongly recommended. Tobacco worsens GERD symptoms by reducing the lower esophageal sphincter pressure and decreasing saliva production which is one of the key components of the normal esophageal acid barrier. Moreover, it is a known risk factor for esophageal cancer. Alcohol has a variety of negative health impacts and decreasing alcohol intake is advised; however, the link between alcohol and GERD symptoms is less robust, especially in patients with low occasional consumption.
In summary, lifestyle modifications play a pivotal role in managing GERD symptoms, offering patients effective, non-pharmacologic strategies to complement medical treatments. Weight loss remains the cornerstone, with even modest reductions in BMI showing significant symptom relief. Dietary adjustments, particularly adopting a low-simple-carbohydrate diet, provide an evidence-based approach. Various bedtime interventions are available to improve nocturnal GERD symptoms. Finally, tobacco cessation is essential, not only for GERD symptom relief but also for overall health. By integrating these lifestyle changes into their routine, patients can improve GERD symptoms while building healthy habits.
Dr. Gomez Cifuentes is vice-chair in the section of gastroenterology at Presbyterian Healthcare Services, Albuquerque, New Mexico. He declares no conflicts of interest.
References
1. Ness-Jensen E et al. Lifestyle Intervention in Gastroesophageal Reflux Disease. Clin Gastroenterol Hepatol. 2016 Feb;14(2):175-82.e1-3. doi: 10.1016/j.cgh.2015.04.176.
2. Gu C et al. The Effects of Modifying Amount and Type of Dietary Carbohydrate on Esophageal Acid Exposure Time and Esophageal Reflux Symptoms: A Randomized Controlled Trial. Am J Gastroenterol. 2022 Oct 1;117(10):1655-1667. doi: 10.14309/ajg.0000000000001889.
3. Schuitenmaker JM et al. Associations Between Sleep Position and Nocturnal Gastroesophageal Reflux: A Study Using Concurrent Monitoring of Sleep Position and Esophageal pH and Impedance. Am J Gastroenterol. 2022 Feb 1;117(2):346-351. doi: 10.14309/ajg.0000000000001588.
4. Person E et al. A Novel Sleep Positioning Device Reduces Gastroesophageal Reflux: A Randomized Controlled Trial. J Clin Gastroenterol. 2015 Sep;49(8):655-9. doi: 10.1097/MCG.0000000000000359.
Medical Therapy Is the Cornerstone of Effective GERD Treatment
BY BRIJESH B. PATEL, MD
Today, I saw Mr. S in the office for gastroesophageal reflux disease (GERD). He has been on a trial of proton pump inhibitors (PPIs) and has implemented several lifestyle modifications to manage his reflux. He shared his frustrations, saying, “Doctor, I’ve tried changing my diet, sleeping in a recliner, and adjusting the timing of my meals. I’m practically not enjoying food anymore, and these lifestyle changes have affected my quality of life. Despite all this, I still wake up in the middle of the night with a ‘horrible taste’ in my mouth, and it’s ruining my sleep.”

Later that day, during a discussion with my trainees, one posed an important question: “What about lifestyle measures in the treatment of GERD?” This is a common query in both clinical and academic settings. GERD, with a prevalence estimated at ~20%, is often underreported as many patients begin self-medicating with over-the-counter acid suppressive therapies before seeking medical care. For gastroenterologists, PPIs, histamine-2 receptor antagonists (H2RAs), and now potassium-competitive acid blockers (PCABs) form the cornerstone of GERD management.
When I lecture medical students, residents, and fellows about GERD, I emphasize a standard approach: initiating an 8- to 12-week trial of PPIs followed by reassessment. I also stress the importance of combining medical therapy with lifestyle measures. However, the question remains: How adherent are our patients to these lifestyle changes? Similarly, how effectively are trainees integrating the value of lifestyle modifications into their practice? As an academic gastroenterologist, I can teach the theory, but is it being translated into real-world patient care?
The advent of PPIs has been a game changer for managing GERD symptoms and preventing disease progression. PPIs are the backbone of treatment in both gastroenterology and primary care, and they have profoundly improved patients’ quality of life. Most of my patients who present with GERD — whether due to uncontrolled reflux or acid exposure — have already been on a trial of PPIs before seeing me. My role often involves optimizing their timing of PPI administration, addressing incorrect usage, and reinforcing the importance of adherence. In some cases, I incorporate H2RAs as adjunctive therapy for patients who fail to respond adequately to PPIs, particularly when objective disease activity is confirmed through pH studies. These studies also highlight how challenging it is for many patients to maintain a refluxogenic-free lifestyle.
Lifestyle modifications should supplement and support GERD management. Regardless of medical specialty, lifestyle measures should be the first line of treatment. However, adherence and effectiveness vary widely. In reality, achieving sustained weight loss, meal timing adjustments, and dietary modifications (e.g., eliminating trigger foods like red wine, chocolate, coffee, and tomato-based sauces) is a significant challenge for patients. While these measures can reduce the need for PPIs in some cases, they are rarely sufficient as standalone treatments. Until lifestyle modifications are consistently and sustainably incorporated into daily routines, acid-suppressive therapy will remain the mainstay of GERD management.
Turning to newer therapies, PCABs are now FDA-approved for treating GERD. Early efficacy data suggest that PCABs are non-inferior to PPIs, with promising results in managing LA Class C and D esophagitis and maintaining symptom-free days. However, like PPIs, PCABs are associated with potential adverse effects, including C. difficile colitis, impacts on bone health, renal impairment, and mineral deficiencies. While these risks must be carefully discussed with patients, the benefits of medical therapy far outweigh the risks, especially for those with erosive esophagitis, Barrett’s esophagus, or a high-risk profile for esophageal cancer. In such cases, medical therapies provide superior disease control compared to lifestyle measures, supported by both subjective and objective data.
Managing GERD requires a multipronged approach. Relying solely on lifestyle measures rarely provides complete benefit, as restrictive dietary regimens are difficult to sustain long term. Like many, I can maintain a restrictive diet temporarily but find it unsustainable over time. Conversely, adherence to daily or twice-daily medications tends to be much higher than compliance with multi-level lifestyle changes (e.g., restrictive diets, weight loss, and trigger-food avoidance).
Our therapeutic arsenal for GERD continues to expand, enabling more effective management of patients with uncontrolled acid reflux. While I will continue to counsel patients and educate trainees on the value of lifestyle modifications, I emphasize the importance of adherence to timely medical therapy — whether with PPIs, H2RAs, or PCABs — as the cornerstone of effective GERD treatment.
Dr. Patel is associate program director in the division of digestive diseases & nutrition, at USF Health, Tampa, Fla. He declares no conflicts of interest.
Dear colleagues,
Gastroesophageal reflux disease (GERD) is a common reason for referral to gastroenterology. It affects a broad cross-section of our population and is often managed through a combination of lifestyle modifications and proton pump inhibitors (PPIs). However,
While PPIs are highly effective, concerns about their potential side effects frequently make headlines. Moreover, the financial burden of lifelong PPI use is a growing consideration. In this issue of Perspectives, Dr. Brijesh B. Patel and Dr. Juan D. Gomez Cifuentes explore these questions. Dr. Gomez Cifuentes highlights the benefits of lifestyle changes and identifies which strategies have proved most effective in his practice. Dr. Patel examines the ubiquitous use of PPIs and the challenges of sustaining adherence to lifestyle modifications. We hope these discussions will spark new ideas for managing GERD in your own practice.
We also welcome your thoughts on this topic — join the conversation on X at @AGA_GIHN.
Gyanprakash A. Ketwaroo, MD, MSc, is associate professor of medicine, Yale University, New Haven, and chief of endoscopy at West Haven VA Medical Center, both in Connecticut. He is an associate editor for GI & Hepatology News.
Do Lifestyle Changes Still Apply in the Treatment of GERD?
BY JUAN D. GOMEZ CIFUENTES, MD
Lifestyle changes are an essential part of managing gastroesophageal reflux disease (GERD). Increasingly, patients are asking about non-medication approaches to control their symptoms. These lifestyle modifications can be categorized into four main areas: 1) Weight loss, the cornerstone intervention, with significant symptom improvement observed after losing as little as 1.7 BMI points. 2) Dietary modifications, which includes both the traditional avoidance of trigger foods and the newer focus on a diet low in simple carbohydrates. 3) Bedtime adjustments, strategies that include elevating the head of the bed, sleeping on the left side, using anti-reflux pillows, and avoiding late-night meals. 4) Tobacco cessation, a key measure for reducing GERD symptoms and promoting overall health. I routinely discuss these changes with my patients, as they not only help manage GERD but also foster healthy habits and have a positive impact beyond the gastrointestinal tract.
Weight loss is the most impactful lifestyle intervention for GERD. Research shows a clear linear improvement in symptoms with weight reduction. Traditionally, losing 10% of body weight is a widely accepted goal, extrapolated from other obesity-associated conditions. A reduction in 3.5 points of BMI led to significant symptom improvement in landmark studies but also a modest reduction of 1.7 BMI points has been shown to provide symptom relief.1 Abdominal circumference is another key metric used to track progress, as central obesity rather than BMI alone is strongly linked with GERD. Goals are typically set at less than 40 inches for men and 35 inches for women. Patients using GLP-1 agonists should be informed that these medications may temporarily worsen GERD symptoms due to delayed gastric emptying, however in the long-term these symptoms are expected to improve once significant weight loss is achieved.
Food triggers vary among individuals, with common culprits including fatty meals, spicy foods, chocolate, tomato sauce, citrus fruits, and carbonated beverages. Patients tend to overemphasize diet elimination based on triggers and engage in strict diets. Patients are frequently afraid of these foods causing direct damage to the esophageal mucosa but the hypothesis is that these triggers worsen GERD by increasing transient relaxations of the lower esophageal sphincter. The evidence behind this and diet elimination based on triggers has always been weak. In my practice, I encourage patients to follow a diet low in simple carbohydrates. Simple carbohydrates are present in highly processed food, the average western diet contains ~140 g/day. In a trial, a diet low in simple sugars (monosaccharides and disaccharides < 62 g/day) without reducing total daily calories, objectively improved total acid exposure time in pH study.2
Thanks to gravity, nocturnal GERD symptoms are the culprit of many restless nights in these patients. I recommend avoiding food 3 hours before lying down. Since the stomach empties approximately 90% of its contents after 4 hours, waiting longer is not recommended and may result in hunger, making it harder to fall asleep. Sleeping on the left side, which takes advantage of the gastric anatomy, has proved to objectively decrease nocturnal acid exposure time, though some patients may find it challenging to maintain this position all night.3
Elevating the head of the bed is another effective intervention, but it must involve raising the upper body from the waist. Patients should avoid stacking ordinary pillows as this will only elevate the neck and place the body in an unnatural position for sleeping. The most effective strategies are putting blocks/bricks under the feet of the bed, using a bed wedge between the mattress and the box spring or using an adjustable bed frame. There are two types of pillows that have been shown to improve nocturnal GERD symptoms. The classic wedge pillows and the more expensive Medcline reflux relief system®. The Medcline pillow has a dual mechanism that elevates the upper body but also keeps the body on the left side position.4
Tobacco cessation is strongly recommended. Tobacco worsens GERD symptoms by reducing the lower esophageal sphincter pressure and decreasing saliva production which is one of the key components of the normal esophageal acid barrier. Moreover, it is a known risk factor for esophageal cancer. Alcohol has a variety of negative health impacts and decreasing alcohol intake is advised; however, the link between alcohol and GERD symptoms is less robust, especially in patients with low occasional consumption.
In summary, lifestyle modifications play a pivotal role in managing GERD symptoms, offering patients effective, non-pharmacologic strategies to complement medical treatments. Weight loss remains the cornerstone, with even modest reductions in BMI showing significant symptom relief. Dietary adjustments, particularly adopting a low-simple-carbohydrate diet, provide an evidence-based approach. Various bedtime interventions are available to improve nocturnal GERD symptoms. Finally, tobacco cessation is essential, not only for GERD symptom relief but also for overall health. By integrating these lifestyle changes into their routine, patients can improve GERD symptoms while building healthy habits.
Dr. Gomez Cifuentes is vice-chair in the section of gastroenterology at Presbyterian Healthcare Services, Albuquerque, New Mexico. He declares no conflicts of interest.
References
1. Ness-Jensen E et al. Lifestyle Intervention in Gastroesophageal Reflux Disease. Clin Gastroenterol Hepatol. 2016 Feb;14(2):175-82.e1-3. doi: 10.1016/j.cgh.2015.04.176.
2. Gu C et al. The Effects of Modifying Amount and Type of Dietary Carbohydrate on Esophageal Acid Exposure Time and Esophageal Reflux Symptoms: A Randomized Controlled Trial. Am J Gastroenterol. 2022 Oct 1;117(10):1655-1667. doi: 10.14309/ajg.0000000000001889.
3. Schuitenmaker JM et al. Associations Between Sleep Position and Nocturnal Gastroesophageal Reflux: A Study Using Concurrent Monitoring of Sleep Position and Esophageal pH and Impedance. Am J Gastroenterol. 2022 Feb 1;117(2):346-351. doi: 10.14309/ajg.0000000000001588.
4. Person E et al. A Novel Sleep Positioning Device Reduces Gastroesophageal Reflux: A Randomized Controlled Trial. J Clin Gastroenterol. 2015 Sep;49(8):655-9. doi: 10.1097/MCG.0000000000000359.
Medical Therapy Is the Cornerstone of Effective GERD Treatment
BY BRIJESH B. PATEL, MD
Today, I saw Mr. S in the office for gastroesophageal reflux disease (GERD). He has been on a trial of proton pump inhibitors (PPIs) and has implemented several lifestyle modifications to manage his reflux. He shared his frustrations, saying, “Doctor, I’ve tried changing my diet, sleeping in a recliner, and adjusting the timing of my meals. I’m practically not enjoying food anymore, and these lifestyle changes have affected my quality of life. Despite all this, I still wake up in the middle of the night with a ‘horrible taste’ in my mouth, and it’s ruining my sleep.”
Later that day, during a discussion with my trainees, one posed an important question: “What about lifestyle measures in the treatment of GERD?” This is a common query in both clinical and academic settings. GERD, with a prevalence estimated at ~20%, is often underreported as many patients begin self-medicating with over-the-counter acid suppressive therapies before seeking medical care. For gastroenterologists, PPIs, histamine-2 receptor antagonists (H2RAs), and now potassium-competitive acid blockers (PCABs) form the cornerstone of GERD management.
When I lecture medical students, residents, and fellows about GERD, I emphasize a standard approach: initiating an 8- to 12-week trial of PPIs followed by reassessment. I also stress the importance of combining medical therapy with lifestyle measures. However, the question remains: How adherent are our patients to these lifestyle changes? Similarly, how effectively are trainees integrating the value of lifestyle modifications into their practice? As an academic gastroenterologist, I can teach the theory, but is it being translated into real-world patient care?
The advent of PPIs has been a game changer for managing GERD symptoms and preventing disease progression. PPIs are the backbone of treatment in both gastroenterology and primary care, and they have profoundly improved patients’ quality of life. Most of my patients who present with GERD — whether due to uncontrolled reflux or acid exposure — have already been on a trial of PPIs before seeing me. My role often involves optimizing their timing of PPI administration, addressing incorrect usage, and reinforcing the importance of adherence. In some cases, I incorporate H2RAs as adjunctive therapy for patients who fail to respond adequately to PPIs, particularly when objective disease activity is confirmed through pH studies. These studies also highlight how challenging it is for many patients to maintain a refluxogenic-free lifestyle.
Lifestyle modifications should supplement and support GERD management. Regardless of medical specialty, lifestyle measures should be the first line of treatment. However, adherence and effectiveness vary widely. In reality, achieving sustained weight loss, meal timing adjustments, and dietary modifications (e.g., eliminating trigger foods like red wine, chocolate, coffee, and tomato-based sauces) is a significant challenge for patients. While these measures can reduce the need for PPIs in some cases, they are rarely sufficient as standalone treatments. Until lifestyle modifications are consistently and sustainably incorporated into daily routines, acid-suppressive therapy will remain the mainstay of GERD management.
Turning to newer therapies, PCABs are now FDA-approved for treating GERD. Early efficacy data suggest that PCABs are non-inferior to PPIs, with promising results in managing LA Class C and D esophagitis and maintaining symptom-free days. However, like PPIs, PCABs are associated with potential adverse effects, including C. difficile colitis, impacts on bone health, renal impairment, and mineral deficiencies. While these risks must be carefully discussed with patients, the benefits of medical therapy far outweigh the risks, especially for those with erosive esophagitis, Barrett’s esophagus, or a high-risk profile for esophageal cancer. In such cases, medical therapies provide superior disease control compared to lifestyle measures, supported by both subjective and objective data.
Managing GERD requires a multipronged approach. Relying solely on lifestyle measures rarely provides complete benefit, as restrictive dietary regimens are difficult to sustain long term. Like many, I can maintain a restrictive diet temporarily but find it unsustainable over time. Conversely, adherence to daily or twice-daily medications tends to be much higher than compliance with multi-level lifestyle changes (e.g., restrictive diets, weight loss, and trigger-food avoidance).
Our therapeutic arsenal for GERD continues to expand, enabling more effective management of patients with uncontrolled acid reflux. While I will continue to counsel patients and educate trainees on the value of lifestyle modifications, I emphasize the importance of adherence to timely medical therapy — whether with PPIs, H2RAs, or PCABs — as the cornerstone of effective GERD treatment.
Dr. Patel is associate program director in the division of digestive diseases & nutrition, at USF Health, Tampa, Fla. He declares no conflicts of interest.
Dear colleagues,
Gastroesophageal reflux disease (GERD) is a common reason for referral to gastroenterology. It affects a broad cross-section of our population and is often managed through a combination of lifestyle modifications and proton pump inhibitors (PPIs). However,
While PPIs are highly effective, concerns about their potential side effects frequently make headlines. Moreover, the financial burden of lifelong PPI use is a growing consideration. In this issue of Perspectives, Dr. Brijesh B. Patel and Dr. Juan D. Gomez Cifuentes explore these questions. Dr. Gomez Cifuentes highlights the benefits of lifestyle changes and identifies which strategies have proved most effective in his practice. Dr. Patel examines the ubiquitous use of PPIs and the challenges of sustaining adherence to lifestyle modifications. We hope these discussions will spark new ideas for managing GERD in your own practice.
We also welcome your thoughts on this topic — join the conversation on X at @AGA_GIHN.
Gyanprakash A. Ketwaroo, MD, MSc, is associate professor of medicine, Yale University, New Haven, and chief of endoscopy at West Haven VA Medical Center, both in Connecticut. He is an associate editor for GI & Hepatology News.
Do Lifestyle Changes Still Apply in the Treatment of GERD?
BY JUAN D. GOMEZ CIFUENTES, MD
Lifestyle changes are an essential part of managing gastroesophageal reflux disease (GERD). Increasingly, patients are asking about non-medication approaches to control their symptoms. These lifestyle modifications can be categorized into four main areas: 1) Weight loss, the cornerstone intervention, with significant symptom improvement observed after losing as little as 1.7 BMI points. 2) Dietary modifications, which includes both the traditional avoidance of trigger foods and the newer focus on a diet low in simple carbohydrates. 3) Bedtime adjustments, strategies that include elevating the head of the bed, sleeping on the left side, using anti-reflux pillows, and avoiding late-night meals. 4) Tobacco cessation, a key measure for reducing GERD symptoms and promoting overall health. I routinely discuss these changes with my patients, as they not only help manage GERD but also foster healthy habits and have a positive impact beyond the gastrointestinal tract.
Weight loss is the most impactful lifestyle intervention for GERD. Research shows a clear linear improvement in symptoms with weight reduction. Traditionally, losing 10% of body weight is a widely accepted goal, extrapolated from other obesity-associated conditions. A reduction in 3.5 points of BMI led to significant symptom improvement in landmark studies but also a modest reduction of 1.7 BMI points has been shown to provide symptom relief.1 Abdominal circumference is another key metric used to track progress, as central obesity rather than BMI alone is strongly linked with GERD. Goals are typically set at less than 40 inches for men and 35 inches for women. Patients using GLP-1 agonists should be informed that these medications may temporarily worsen GERD symptoms due to delayed gastric emptying, however in the long-term these symptoms are expected to improve once significant weight loss is achieved.
Food triggers vary among individuals, with common culprits including fatty meals, spicy foods, chocolate, tomato sauce, citrus fruits, and carbonated beverages. Patients tend to overemphasize diet elimination based on triggers and engage in strict diets. Patients are frequently afraid of these foods causing direct damage to the esophageal mucosa but the hypothesis is that these triggers worsen GERD by increasing transient relaxations of the lower esophageal sphincter. The evidence behind this and diet elimination based on triggers has always been weak. In my practice, I encourage patients to follow a diet low in simple carbohydrates. Simple carbohydrates are present in highly processed food, the average western diet contains ~140 g/day. In a trial, a diet low in simple sugars (monosaccharides and disaccharides < 62 g/day) without reducing total daily calories, objectively improved total acid exposure time in pH study.2
Thanks to gravity, nocturnal GERD symptoms are the culprit of many restless nights in these patients. I recommend avoiding food 3 hours before lying down. Since the stomach empties approximately 90% of its contents after 4 hours, waiting longer is not recommended and may result in hunger, making it harder to fall asleep. Sleeping on the left side, which takes advantage of the gastric anatomy, has proved to objectively decrease nocturnal acid exposure time, though some patients may find it challenging to maintain this position all night.3
Elevating the head of the bed is another effective intervention, but it must involve raising the upper body from the waist. Patients should avoid stacking ordinary pillows as this will only elevate the neck and place the body in an unnatural position for sleeping. The most effective strategies are putting blocks/bricks under the feet of the bed, using a bed wedge between the mattress and the box spring or using an adjustable bed frame. There are two types of pillows that have been shown to improve nocturnal GERD symptoms. The classic wedge pillows and the more expensive Medcline reflux relief system®. The Medcline pillow has a dual mechanism that elevates the upper body but also keeps the body on the left side position.4
Tobacco cessation is strongly recommended. Tobacco worsens GERD symptoms by reducing the lower esophageal sphincter pressure and decreasing saliva production which is one of the key components of the normal esophageal acid barrier. Moreover, it is a known risk factor for esophageal cancer. Alcohol has a variety of negative health impacts and decreasing alcohol intake is advised; however, the link between alcohol and GERD symptoms is less robust, especially in patients with low occasional consumption.
In summary, lifestyle modifications play a pivotal role in managing GERD symptoms, offering patients effective, non-pharmacologic strategies to complement medical treatments. Weight loss remains the cornerstone, with even modest reductions in BMI showing significant symptom relief. Dietary adjustments, particularly adopting a low-simple-carbohydrate diet, provide an evidence-based approach. Various bedtime interventions are available to improve nocturnal GERD symptoms. Finally, tobacco cessation is essential, not only for GERD symptom relief but also for overall health. By integrating these lifestyle changes into their routine, patients can improve GERD symptoms while building healthy habits.
Dr. Gomez Cifuentes is vice-chair in the section of gastroenterology at Presbyterian Healthcare Services, Albuquerque, New Mexico. He declares no conflicts of interest.
References
1. Ness-Jensen E et al. Lifestyle Intervention in Gastroesophageal Reflux Disease. Clin Gastroenterol Hepatol. 2016 Feb;14(2):175-82.e1-3. doi: 10.1016/j.cgh.2015.04.176.
2. Gu C et al. The Effects of Modifying Amount and Type of Dietary Carbohydrate on Esophageal Acid Exposure Time and Esophageal Reflux Symptoms: A Randomized Controlled Trial. Am J Gastroenterol. 2022 Oct 1;117(10):1655-1667. doi: 10.14309/ajg.0000000000001889.
3. Schuitenmaker JM et al. Associations Between Sleep Position and Nocturnal Gastroesophageal Reflux: A Study Using Concurrent Monitoring of Sleep Position and Esophageal pH and Impedance. Am J Gastroenterol. 2022 Feb 1;117(2):346-351. doi: 10.14309/ajg.0000000000001588.
4. Person E et al. A Novel Sleep Positioning Device Reduces Gastroesophageal Reflux: A Randomized Controlled Trial. J Clin Gastroenterol. 2015 Sep;49(8):655-9. doi: 10.1097/MCG.0000000000000359.
Medical Therapy Is the Cornerstone of Effective GERD Treatment
BY BRIJESH B. PATEL, MD
Today, I saw Mr. S in the office for gastroesophageal reflux disease (GERD). He has been on a trial of proton pump inhibitors (PPIs) and has implemented several lifestyle modifications to manage his reflux. He shared his frustrations, saying, “Doctor, I’ve tried changing my diet, sleeping in a recliner, and adjusting the timing of my meals. I’m practically not enjoying food anymore, and these lifestyle changes have affected my quality of life. Despite all this, I still wake up in the middle of the night with a ‘horrible taste’ in my mouth, and it’s ruining my sleep.”
Later that day, during a discussion with my trainees, one posed an important question: “What about lifestyle measures in the treatment of GERD?” This is a common query in both clinical and academic settings. GERD, with a prevalence estimated at ~20%, is often underreported as many patients begin self-medicating with over-the-counter acid suppressive therapies before seeking medical care. For gastroenterologists, PPIs, histamine-2 receptor antagonists (H2RAs), and now potassium-competitive acid blockers (PCABs) form the cornerstone of GERD management.
When I lecture medical students, residents, and fellows about GERD, I emphasize a standard approach: initiating an 8- to 12-week trial of PPIs followed by reassessment. I also stress the importance of combining medical therapy with lifestyle measures. However, the question remains: How adherent are our patients to these lifestyle changes? Similarly, how effectively are trainees integrating the value of lifestyle modifications into their practice? As an academic gastroenterologist, I can teach the theory, but is it being translated into real-world patient care?
The advent of PPIs has been a game changer for managing GERD symptoms and preventing disease progression. PPIs are the backbone of treatment in both gastroenterology and primary care, and they have profoundly improved patients’ quality of life. Most of my patients who present with GERD — whether due to uncontrolled reflux or acid exposure — have already been on a trial of PPIs before seeing me. My role often involves optimizing their timing of PPI administration, addressing incorrect usage, and reinforcing the importance of adherence. In some cases, I incorporate H2RAs as adjunctive therapy for patients who fail to respond adequately to PPIs, particularly when objective disease activity is confirmed through pH studies. These studies also highlight how challenging it is for many patients to maintain a refluxogenic-free lifestyle.
Lifestyle modifications should supplement and support GERD management. Regardless of medical specialty, lifestyle measures should be the first line of treatment. However, adherence and effectiveness vary widely. In reality, achieving sustained weight loss, meal timing adjustments, and dietary modifications (e.g., eliminating trigger foods like red wine, chocolate, coffee, and tomato-based sauces) is a significant challenge for patients. While these measures can reduce the need for PPIs in some cases, they are rarely sufficient as standalone treatments. Until lifestyle modifications are consistently and sustainably incorporated into daily routines, acid-suppressive therapy will remain the mainstay of GERD management.
Turning to newer therapies, PCABs are now FDA-approved for treating GERD. Early efficacy data suggest that PCABs are non-inferior to PPIs, with promising results in managing LA Class C and D esophagitis and maintaining symptom-free days. However, like PPIs, PCABs are associated with potential adverse effects, including C. difficile colitis, impacts on bone health, renal impairment, and mineral deficiencies. While these risks must be carefully discussed with patients, the benefits of medical therapy far outweigh the risks, especially for those with erosive esophagitis, Barrett’s esophagus, or a high-risk profile for esophageal cancer. In such cases, medical therapies provide superior disease control compared to lifestyle measures, supported by both subjective and objective data.
Managing GERD requires a multipronged approach. Relying solely on lifestyle measures rarely provides complete benefit, as restrictive dietary regimens are difficult to sustain long term. Like many, I can maintain a restrictive diet temporarily but find it unsustainable over time. Conversely, adherence to daily or twice-daily medications tends to be much higher than compliance with multi-level lifestyle changes (e.g., restrictive diets, weight loss, and trigger-food avoidance).
Our therapeutic arsenal for GERD continues to expand, enabling more effective management of patients with uncontrolled acid reflux. While I will continue to counsel patients and educate trainees on the value of lifestyle modifications, I emphasize the importance of adherence to timely medical therapy — whether with PPIs, H2RAs, or PCABs — as the cornerstone of effective GERD treatment.
Dr. Patel is associate program director in the division of digestive diseases & nutrition, at USF Health, Tampa, Fla. He declares no conflicts of interest.
An Exciting Time to Be a Gastroenterologist
Happy New Year, everyone! As we enter 2025, I’ve been reflecting on just how much has changed in the field of gastroenterology since I completed my fellowship a decade ago.
After developing and disseminating highly effective treatments for hepatitis C, the field of hepatology has shifted rapidly toward identifying and managing other significant causes of liver disease, particularly alcohol-associated liver disease and metabolic dysfunction–associated steatotic liver disease (MASLD). New disease nomenclatures have been developed that have changed the way we describe common diseases – most notably, NALFD is now MASLD and FGID are now DGBI.

There have been marked advances in obesity management, including not only innovations in endobariatric therapies such as intragastric balloons and endoscopic sleeve gastroplasty, but also the introduction of glucagon-like peptide 1 (GLP-1) agonists, which offer new hope in effectively tackling the obesity epidemic. Our growing understanding of the microbiome’s role in health has opened new avenues for treating GI diseases and introduced the potential for more personalized treatment approaches based on individual microbiome profiles. New inflammatory bowel disease (IBD) pharmacotherapeutics have been developed at a dizzying pace – our IBD patients have so many more treatment options today than they did just a decade ago, making treatment decisions much more complex.
Finally, we are just beginning to unleash the potential of artificial intelligence, which is likely to transform the field of medicine and GI clinical practice over the next decade. To be sure, it is an exciting time to be a gastroenterologist, and I can’t wait to see to what the next decade of innovation and discovery will bring.
From the recent AASLD meeting, we bring you exciting new data demonstrating the effectiveness of GLP-1 agonists (specifically, semaglutide) in treating MASH. In January’s Member Spotlight column, we introduce you to Drs. Mindy, Amy, and Kristen Engevik, who share their fascinating career journeys as GI researchers (and sisters!). In our quarterly Perspectives column, Dr. Brijesh Patel and Dr. Gomez Cifuentes share their experiences counseling patients regarding lifestyle modifications for gastroesophageal reflux disease and what strategies have proven to be the most effective adjuncts to pharmacotherapy. We hope you enjoy this and all the exciting content in our January issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Happy New Year, everyone! As we enter 2025, I’ve been reflecting on just how much has changed in the field of gastroenterology since I completed my fellowship a decade ago.
After developing and disseminating highly effective treatments for hepatitis C, the field of hepatology has shifted rapidly toward identifying and managing other significant causes of liver disease, particularly alcohol-associated liver disease and metabolic dysfunction–associated steatotic liver disease (MASLD). New disease nomenclatures have been developed that have changed the way we describe common diseases – most notably, NALFD is now MASLD and FGID are now DGBI.
There have been marked advances in obesity management, including not only innovations in endobariatric therapies such as intragastric balloons and endoscopic sleeve gastroplasty, but also the introduction of glucagon-like peptide 1 (GLP-1) agonists, which offer new hope in effectively tackling the obesity epidemic. Our growing understanding of the microbiome’s role in health has opened new avenues for treating GI diseases and introduced the potential for more personalized treatment approaches based on individual microbiome profiles. New inflammatory bowel disease (IBD) pharmacotherapeutics have been developed at a dizzying pace – our IBD patients have so many more treatment options today than they did just a decade ago, making treatment decisions much more complex.
Finally, we are just beginning to unleash the potential of artificial intelligence, which is likely to transform the field of medicine and GI clinical practice over the next decade. To be sure, it is an exciting time to be a gastroenterologist, and I can’t wait to see to what the next decade of innovation and discovery will bring.
From the recent AASLD meeting, we bring you exciting new data demonstrating the effectiveness of GLP-1 agonists (specifically, semaglutide) in treating MASH. In January’s Member Spotlight column, we introduce you to Drs. Mindy, Amy, and Kristen Engevik, who share their fascinating career journeys as GI researchers (and sisters!). In our quarterly Perspectives column, Dr. Brijesh Patel and Dr. Gomez Cifuentes share their experiences counseling patients regarding lifestyle modifications for gastroesophageal reflux disease and what strategies have proven to be the most effective adjuncts to pharmacotherapy. We hope you enjoy this and all the exciting content in our January issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Happy New Year, everyone! As we enter 2025, I’ve been reflecting on just how much has changed in the field of gastroenterology since I completed my fellowship a decade ago.
After developing and disseminating highly effective treatments for hepatitis C, the field of hepatology has shifted rapidly toward identifying and managing other significant causes of liver disease, particularly alcohol-associated liver disease and metabolic dysfunction–associated steatotic liver disease (MASLD). New disease nomenclatures have been developed that have changed the way we describe common diseases – most notably, NALFD is now MASLD and FGID are now DGBI.
There have been marked advances in obesity management, including not only innovations in endobariatric therapies such as intragastric balloons and endoscopic sleeve gastroplasty, but also the introduction of glucagon-like peptide 1 (GLP-1) agonists, which offer new hope in effectively tackling the obesity epidemic. Our growing understanding of the microbiome’s role in health has opened new avenues for treating GI diseases and introduced the potential for more personalized treatment approaches based on individual microbiome profiles. New inflammatory bowel disease (IBD) pharmacotherapeutics have been developed at a dizzying pace – our IBD patients have so many more treatment options today than they did just a decade ago, making treatment decisions much more complex.
Finally, we are just beginning to unleash the potential of artificial intelligence, which is likely to transform the field of medicine and GI clinical practice over the next decade. To be sure, it is an exciting time to be a gastroenterologist, and I can’t wait to see to what the next decade of innovation and discovery will bring.
From the recent AASLD meeting, we bring you exciting new data demonstrating the effectiveness of GLP-1 agonists (specifically, semaglutide) in treating MASH. In January’s Member Spotlight column, we introduce you to Drs. Mindy, Amy, and Kristen Engevik, who share their fascinating career journeys as GI researchers (and sisters!). In our quarterly Perspectives column, Dr. Brijesh Patel and Dr. Gomez Cifuentes share their experiences counseling patients regarding lifestyle modifications for gastroesophageal reflux disease and what strategies have proven to be the most effective adjuncts to pharmacotherapy. We hope you enjoy this and all the exciting content in our January issue.
Megan A. Adams, MD, JD, MSc
Editor in Chief
Noninvasive Microbiome Test May Specifically Identify Crohn’s and Ulcerative Colitis
International researchers have uncovered potentially diagnostic gut microbiome signatures and metabolic pathways associated specifically with ulcerative colitis (UC) and Crohn’s disease (CD).
Targeted droplet digital polymerase chain reaction (ddPCR)‒based quantification of bacterial species led to convenient inflammatory bowel disease (IBD) diagnostic assays that “are sufficiently robust, sensitive and cost-effective for clinical application,” the investigators wrote in a recent study published in Nature Medicine.
“Although traditional modalities used for diagnosis of IBD, including colonoscopy and cross-sectional imaging, are well established, the inconvenience of bowel preparation and radiation represents relevant concerns,” senior author Siew C. Ng, MBBS, PhD, a professor in the Department of Medicine and Therapeutics at the Chinese University of Hong Kong, said in an interview. “Furthermore, existing serological and fecal markers indicate inflammation but lack specificity for IBD.”

Identifying reproducible bacterial biomarkers specific to CD and IBD should enable precise and personalized approaches to detection and management.
As a starting point, the researchers hypothesized that changes in the gut microbiome of IBD patients may reflect underlying functional associations, if not causes, of the disease, said Ng, who is also director of Hong Kong’s Microbiota I-Center (MagIC). “Unlike inflammation, which is a manifestation of the disease, the gut microbiome may serve as a more reliable biomarker less affected by the disease’s fluctuating cycle.”
The study findings showed that bacterial markers remain consistent even during the inactive disease phase. , she added. “With a better performance than the commonly used noninvasive test, fecal calprotectin, we believe the test will be a valuable addition to clinician’s toolbox and a strong option for first-line diagnostics.”
The Study
The group used metagenomic data from 5979 fecal samples from persons with and without IBD from different regions (including the United States) and of different ethnicities. Identifying several microbiota alterations in IBD, they selected bacterial species to construct diagnostic models for UC (n = 10) and CD (n = 9). Some species were deleted and some were enriched in IBD.
Metagenomic findings confirmed, for example, enrichments of Escherichia coli and Bacteroides fragilis in the guts of CD patients, with adherent invasive E coli present in more than half of these. This pathogen has been linked to mucosal dysbiosis and functional alteration, and has been associated with disease activity and endoscopic recurrence following surgery. B fragilis may induce intestinal inflammation through toxin production.
The researchers also identified a new oral bacterium, Actinomyces species oral taxon 181, which was significantly enriched in stool samples with both CD and UC.
The diagnostic models achieved areas under the curve of >.90 for distinguishing IBD patients from controls in the discovery cohort and maintained satisfactory performance in transethnic validation cohorts from eight populations.
Ng’s group further developed a multiplex droplet digital PCR test targeting selected IBD-associated bacterial species. Models based on this test showed numerically higher performance than fecal calprotectin in discriminating UC and CD samples from controls. These universally IBD-associated bacteria suggest the potential applicability of a biomarker panel for noninvasive diagnosis.
Commenting on the paper but not involved in it, Ashwin N. Ananthakrishnan, MBBS, MPH, AGAF, director of the Crohn’s and Colitis Center at Massachusetts General Hospital in Boston and associate professor of medicine at Harvard Medical School, called it “a very important study that highlights the potential role of a microbiome-based diagnostic for screening. It could have application in a wide variety of settings and is very promising.”

More work, however, is necessary to clarify such testing’s role. “The study’s validation in independent cohorts is an important strength, but the sizes of those cohorts are still quite small,” he said in an interview. “It’s important to understand its accuracy across a spectrum of IBD phenotypes and severity.”
Furthermore, endoscopic evaluation at diagnosis is important to establish severity and extent of disease. “It’s not clear this diagnostic biomarker can help supplant that role. But I see potential value to it for patients for whom we may not be considering endoscopy yet but who would like to risk-stratify.”
The Test’s Future
“We expect to see a real shift in clinical practice,” Ng said. “As a cost-effective test, it will help millions of people dealing with nonspecific gastrointestinal symptoms get the diagnoses they need.” Because the bacterial test can identify IBD at an inactive stage, it has the potential for early diagnosis. “This capability allows clinicians to initiate treatment sooner, helping to prevent progression from subclinical to clinical stages of the disease.”
The next research steps involve prospective studies with a larger and more diverse group of patients with various gastrointestinal symptoms. “This will enable a comprehensive evaluation of bacterial biomarkers in real-world populations,” she said. In vivo and in vitro experiments are expected to provide mechanistic insights into the causal role of these bacteria and metabolic dysregulations in the pathogenesis of IBD, as well as their future clinical utility in disease monitoring and predicting treatment response.
Her group plans to work with the biotech industry and regulatory agencies to transform these biomarkers into an approved test kit. “The rollout is likely to be gradual, but we’re optimistic that supportive international and national guidelines will be developed and will pave the way for widespread implementation.”
This study was supported by various academic, charitable, and governmental research-funding bodies, including the governments of Hong Kong and the People’s Republic of China. Ng has served as an advisory board member or speaker for Pfizer, Ferring, Janssen, AbbVie, Tillotts, Menarini, and Takeda. She has received research grants through her institutions from Olympus, Ferring, and AbbVie and is a founding member and shareholder of GenieBiome. She receives patent royalties through her institutions, including MagIC, which holds patents on the therapeutic and diagnostic use of the microbiome in IBD. Several co-authors reported various relationships, including patent holding, with private-sector companies. Ananthakrishnan had no relevant competing interests.
A version of this article first appeared on Medscape.com.
International researchers have uncovered potentially diagnostic gut microbiome signatures and metabolic pathways associated specifically with ulcerative colitis (UC) and Crohn’s disease (CD).
Targeted droplet digital polymerase chain reaction (ddPCR)‒based quantification of bacterial species led to convenient inflammatory bowel disease (IBD) diagnostic assays that “are sufficiently robust, sensitive and cost-effective for clinical application,” the investigators wrote in a recent study published in Nature Medicine.
“Although traditional modalities used for diagnosis of IBD, including colonoscopy and cross-sectional imaging, are well established, the inconvenience of bowel preparation and radiation represents relevant concerns,” senior author Siew C. Ng, MBBS, PhD, a professor in the Department of Medicine and Therapeutics at the Chinese University of Hong Kong, said in an interview. “Furthermore, existing serological and fecal markers indicate inflammation but lack specificity for IBD.”
Identifying reproducible bacterial biomarkers specific to CD and IBD should enable precise and personalized approaches to detection and management.
As a starting point, the researchers hypothesized that changes in the gut microbiome of IBD patients may reflect underlying functional associations, if not causes, of the disease, said Ng, who is also director of Hong Kong’s Microbiota I-Center (MagIC). “Unlike inflammation, which is a manifestation of the disease, the gut microbiome may serve as a more reliable biomarker less affected by the disease’s fluctuating cycle.”
The study findings showed that bacterial markers remain consistent even during the inactive disease phase. , she added. “With a better performance than the commonly used noninvasive test, fecal calprotectin, we believe the test will be a valuable addition to clinician’s toolbox and a strong option for first-line diagnostics.”
The Study
The group used metagenomic data from 5979 fecal samples from persons with and without IBD from different regions (including the United States) and of different ethnicities. Identifying several microbiota alterations in IBD, they selected bacterial species to construct diagnostic models for UC (n = 10) and CD (n = 9). Some species were deleted and some were enriched in IBD.
Metagenomic findings confirmed, for example, enrichments of Escherichia coli and Bacteroides fragilis in the guts of CD patients, with adherent invasive E coli present in more than half of these. This pathogen has been linked to mucosal dysbiosis and functional alteration, and has been associated with disease activity and endoscopic recurrence following surgery. B fragilis may induce intestinal inflammation through toxin production.
The researchers also identified a new oral bacterium, Actinomyces species oral taxon 181, which was significantly enriched in stool samples with both CD and UC.
The diagnostic models achieved areas under the curve of >.90 for distinguishing IBD patients from controls in the discovery cohort and maintained satisfactory performance in transethnic validation cohorts from eight populations.
Ng’s group further developed a multiplex droplet digital PCR test targeting selected IBD-associated bacterial species. Models based on this test showed numerically higher performance than fecal calprotectin in discriminating UC and CD samples from controls. These universally IBD-associated bacteria suggest the potential applicability of a biomarker panel for noninvasive diagnosis.
Commenting on the paper but not involved in it, Ashwin N. Ananthakrishnan, MBBS, MPH, AGAF, director of the Crohn’s and Colitis Center at Massachusetts General Hospital in Boston and associate professor of medicine at Harvard Medical School, called it “a very important study that highlights the potential role of a microbiome-based diagnostic for screening. It could have application in a wide variety of settings and is very promising.”
More work, however, is necessary to clarify such testing’s role. “The study’s validation in independent cohorts is an important strength, but the sizes of those cohorts are still quite small,” he said in an interview. “It’s important to understand its accuracy across a spectrum of IBD phenotypes and severity.”
Furthermore, endoscopic evaluation at diagnosis is important to establish severity and extent of disease. “It’s not clear this diagnostic biomarker can help supplant that role. But I see potential value to it for patients for whom we may not be considering endoscopy yet but who would like to risk-stratify.”
The Test’s Future
“We expect to see a real shift in clinical practice,” Ng said. “As a cost-effective test, it will help millions of people dealing with nonspecific gastrointestinal symptoms get the diagnoses they need.” Because the bacterial test can identify IBD at an inactive stage, it has the potential for early diagnosis. “This capability allows clinicians to initiate treatment sooner, helping to prevent progression from subclinical to clinical stages of the disease.”
The next research steps involve prospective studies with a larger and more diverse group of patients with various gastrointestinal symptoms. “This will enable a comprehensive evaluation of bacterial biomarkers in real-world populations,” she said. In vivo and in vitro experiments are expected to provide mechanistic insights into the causal role of these bacteria and metabolic dysregulations in the pathogenesis of IBD, as well as their future clinical utility in disease monitoring and predicting treatment response.
Her group plans to work with the biotech industry and regulatory agencies to transform these biomarkers into an approved test kit. “The rollout is likely to be gradual, but we’re optimistic that supportive international and national guidelines will be developed and will pave the way for widespread implementation.”
This study was supported by various academic, charitable, and governmental research-funding bodies, including the governments of Hong Kong and the People’s Republic of China. Ng has served as an advisory board member or speaker for Pfizer, Ferring, Janssen, AbbVie, Tillotts, Menarini, and Takeda. She has received research grants through her institutions from Olympus, Ferring, and AbbVie and is a founding member and shareholder of GenieBiome. She receives patent royalties through her institutions, including MagIC, which holds patents on the therapeutic and diagnostic use of the microbiome in IBD. Several co-authors reported various relationships, including patent holding, with private-sector companies. Ananthakrishnan had no relevant competing interests.
A version of this article first appeared on Medscape.com.
International researchers have uncovered potentially diagnostic gut microbiome signatures and metabolic pathways associated specifically with ulcerative colitis (UC) and Crohn’s disease (CD).
Targeted droplet digital polymerase chain reaction (ddPCR)‒based quantification of bacterial species led to convenient inflammatory bowel disease (IBD) diagnostic assays that “are sufficiently robust, sensitive and cost-effective for clinical application,” the investigators wrote in a recent study published in Nature Medicine.
“Although traditional modalities used for diagnosis of IBD, including colonoscopy and cross-sectional imaging, are well established, the inconvenience of bowel preparation and radiation represents relevant concerns,” senior author Siew C. Ng, MBBS, PhD, a professor in the Department of Medicine and Therapeutics at the Chinese University of Hong Kong, said in an interview. “Furthermore, existing serological and fecal markers indicate inflammation but lack specificity for IBD.”
Identifying reproducible bacterial biomarkers specific to CD and IBD should enable precise and personalized approaches to detection and management.
As a starting point, the researchers hypothesized that changes in the gut microbiome of IBD patients may reflect underlying functional associations, if not causes, of the disease, said Ng, who is also director of Hong Kong’s Microbiota I-Center (MagIC). “Unlike inflammation, which is a manifestation of the disease, the gut microbiome may serve as a more reliable biomarker less affected by the disease’s fluctuating cycle.”
The study findings showed that bacterial markers remain consistent even during the inactive disease phase. , she added. “With a better performance than the commonly used noninvasive test, fecal calprotectin, we believe the test will be a valuable addition to clinician’s toolbox and a strong option for first-line diagnostics.”
The Study
The group used metagenomic data from 5979 fecal samples from persons with and without IBD from different regions (including the United States) and of different ethnicities. Identifying several microbiota alterations in IBD, they selected bacterial species to construct diagnostic models for UC (n = 10) and CD (n = 9). Some species were deleted and some were enriched in IBD.
Metagenomic findings confirmed, for example, enrichments of Escherichia coli and Bacteroides fragilis in the guts of CD patients, with adherent invasive E coli present in more than half of these. This pathogen has been linked to mucosal dysbiosis and functional alteration, and has been associated with disease activity and endoscopic recurrence following surgery. B fragilis may induce intestinal inflammation through toxin production.
The researchers also identified a new oral bacterium, Actinomyces species oral taxon 181, which was significantly enriched in stool samples with both CD and UC.
The diagnostic models achieved areas under the curve of >.90 for distinguishing IBD patients from controls in the discovery cohort and maintained satisfactory performance in transethnic validation cohorts from eight populations.
Ng’s group further developed a multiplex droplet digital PCR test targeting selected IBD-associated bacterial species. Models based on this test showed numerically higher performance than fecal calprotectin in discriminating UC and CD samples from controls. These universally IBD-associated bacteria suggest the potential applicability of a biomarker panel for noninvasive diagnosis.
Commenting on the paper but not involved in it, Ashwin N. Ananthakrishnan, MBBS, MPH, AGAF, director of the Crohn’s and Colitis Center at Massachusetts General Hospital in Boston and associate professor of medicine at Harvard Medical School, called it “a very important study that highlights the potential role of a microbiome-based diagnostic for screening. It could have application in a wide variety of settings and is very promising.”
More work, however, is necessary to clarify such testing’s role. “The study’s validation in independent cohorts is an important strength, but the sizes of those cohorts are still quite small,” he said in an interview. “It’s important to understand its accuracy across a spectrum of IBD phenotypes and severity.”
Furthermore, endoscopic evaluation at diagnosis is important to establish severity and extent of disease. “It’s not clear this diagnostic biomarker can help supplant that role. But I see potential value to it for patients for whom we may not be considering endoscopy yet but who would like to risk-stratify.”
The Test’s Future
“We expect to see a real shift in clinical practice,” Ng said. “As a cost-effective test, it will help millions of people dealing with nonspecific gastrointestinal symptoms get the diagnoses they need.” Because the bacterial test can identify IBD at an inactive stage, it has the potential for early diagnosis. “This capability allows clinicians to initiate treatment sooner, helping to prevent progression from subclinical to clinical stages of the disease.”
The next research steps involve prospective studies with a larger and more diverse group of patients with various gastrointestinal symptoms. “This will enable a comprehensive evaluation of bacterial biomarkers in real-world populations,” she said. In vivo and in vitro experiments are expected to provide mechanistic insights into the causal role of these bacteria and metabolic dysregulations in the pathogenesis of IBD, as well as their future clinical utility in disease monitoring and predicting treatment response.
Her group plans to work with the biotech industry and regulatory agencies to transform these biomarkers into an approved test kit. “The rollout is likely to be gradual, but we’re optimistic that supportive international and national guidelines will be developed and will pave the way for widespread implementation.”
This study was supported by various academic, charitable, and governmental research-funding bodies, including the governments of Hong Kong and the People’s Republic of China. Ng has served as an advisory board member or speaker for Pfizer, Ferring, Janssen, AbbVie, Tillotts, Menarini, and Takeda. She has received research grants through her institutions from Olympus, Ferring, and AbbVie and is a founding member and shareholder of GenieBiome. She receives patent royalties through her institutions, including MagIC, which holds patents on the therapeutic and diagnostic use of the microbiome in IBD. Several co-authors reported various relationships, including patent holding, with private-sector companies. Ananthakrishnan had no relevant competing interests.
A version of this article first appeared on Medscape.com.
FROM NATURE MEDICINE
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
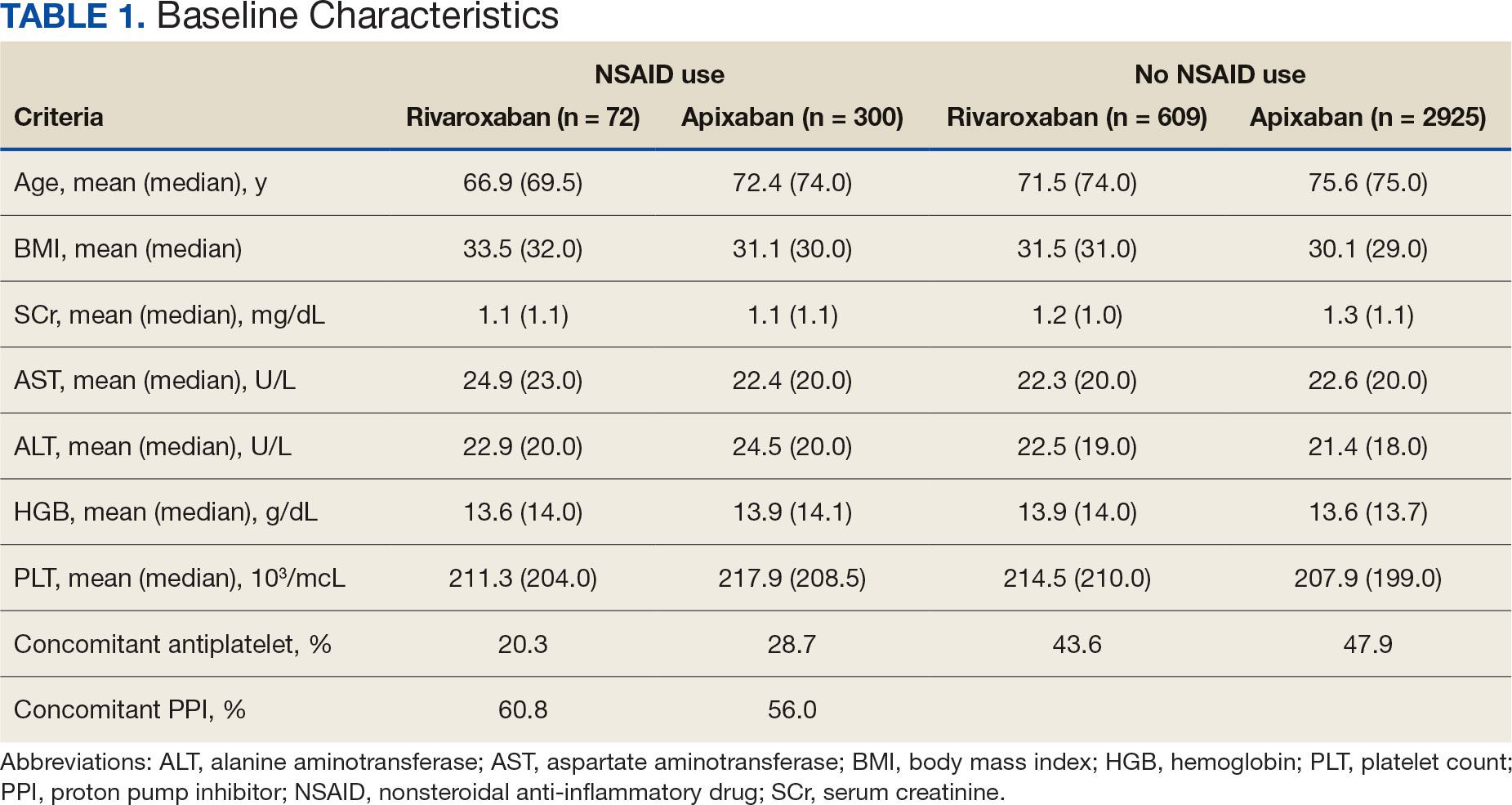
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
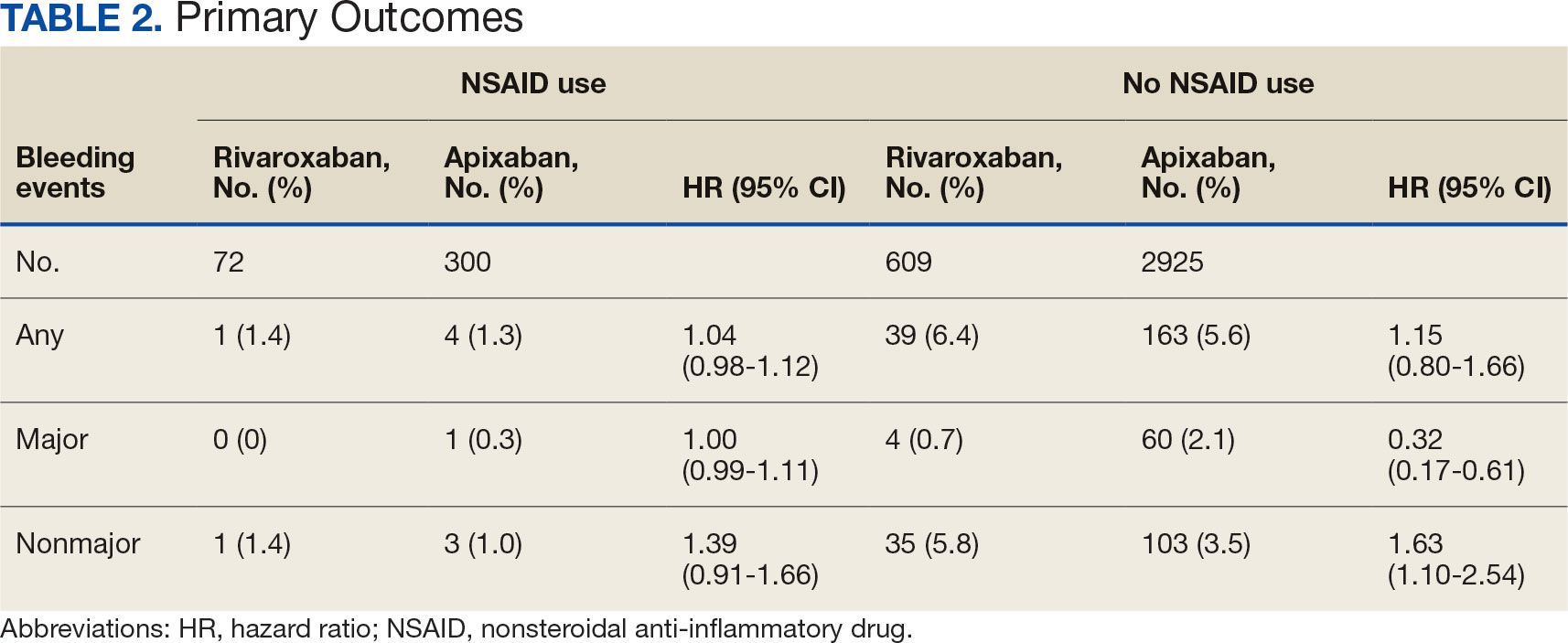
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
PPI-Responsive Disease a Subtype of EoE Rather Than GERD
, according to comparative proteomic analyses.
Notably, after PPI therapy, the protein profiles of responsive patients reverted and appeared similar to non-EoE patients, whereas the profiles of nonresponsive patients remained largely unchanged.
“Identifying protein biomarkers associated with PPI response may help distinguish EoE phenotypes and guide therapy selections,” said senior author Walter Chan, MD, AGAF, associate professor of medicine in the Division of Gastroenterology, Hepatology, and Endoscopy at Harvard Medical School and director of the center for gastrointestinal motility at Brigham and Women’s Hospital, Boston.
“These findings may provide the framework for developing protein biomarkers to assess response to therapy and monitor disease activity,” he added.
The study was published online in Gastroenterology.
Comparative Proteomic Analyses
Chan and colleagues conducted a prospective exploratory pilot study to identify the differences in esophageal protein profiles among PPI-responsive-EoE (PPI-R-EoE), PPI-nonresponsive-EoE (PPI-NR-EoE), and non-EoE controls using SOMAscan, a proteomics platform that allows simultaneous detection of 1305 human proteins.
The research team prospectively enrolled patients undergoing endoscopy for esophageal symptoms or for EoE follow-up, obtaining clinically indicated biopsies as well as extra samples from the midesophagus.
Patients who were diagnosed with EoE (at 15 or greater eosinophils per high-power field, or eos/hpf) were treated with 20 mg of omeprazole twice daily for 8 weeks, followed by repeat biopsies to assess treatment response.
Patients with histologic remission (fewer than 15 eos/hpf) were classified as PPI-R-EoE, whereas those with persistently active disease were classified as PPI-NR-EoE. Patients without EoE served as controls and were categorized as having erosive esophagitis (EE) or no esophagitis.
Overall, the study enrolled 32 patients, including 15 with PPI-R-EoE, eight with PPI-NR-EoE, three with EE, and six with no esophagitis. The demographics, symptoms, and endoscopic findings were similar between the PPI-R-EoE and PPI-NR-EoE patients.
At the index endoscopy, the PPI-R-EoE and PPI-NR-EoE patients had similar esophageal protein profiles, with only 20 proteins differentially expressed at a relaxed cutoff of P < .1. An analysis of the 20 proteins predicted lower expression of six proteins that may be associated with gastrointestinal inflammation in nonresponsive patients, including STAT1, STAT3, CFB, interleukin (IL)-17RA, TNFRSF1A, and SERPINA3.
In addition, 136 proteins — including 15 with corrected P < .05 — clearly discriminated PPI-R-EoE patients from non-EoE controls, and 255 proteins — including 249 with P < .05 — discriminated PPI-NR-EoE patients from controls. Both types of EoE patients had proteins associated with enhanced inflammation and vasculogenesis, as well as down-regulation of CRISP3 and DSG1 and upregulation of TNFAIP6.
The comparative analyses also showed that the follow-up biopsies of PPI-R-EoE patients had protein profiles that resembled non-EoE controls after PPI therapy.
“This further supports the hypothesis that despite the PPI response, PPI-R-EoE represents a subtype of EoE rather than gastroesophageal reflux disease (GERD),” Chan said.
Future EoE Considerations
Although most expressed proteins appeared similar between PPI-responsive and nonresponsive patients before treatment, a few proteins differed related to gastrointestinal inflammation, the study authors wrote, including some previously implicated in IL4 and IL13 inflammatory pathways.
“Further study of these proteins may provide insights into the EoE pathogenic pathway, explore their potential to predict PPI response at diagnosis, and identify possible therapeutic targets,” they wrote.
The authors pointed to the small study size as the primary limitation, noting that the pilot study was intended to explore the feasibility of using SomaScan to assess esophageal protein profiles in different EoE phenotypes. In the future, larger studies with more expansive candidate proteins could help characterize the differences and better identify specific proteins and pathways in EoE, they wrote.
“The takeaway is that PPI responsiveness does not distinguish EoE from GERD but rather PPI is a primary therapy for EoE independent of GERD,” said Marc Rothenberg, MD, director of allergy and immunology and director of the Cincinnati Center for Eosinophilic Disorders at Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio.
Rothenberg, who wasn’t involved with this study, has conducted transcriptome analyses of PPI-R-EoE, which showed PPI-reversible allergic inflammation.
“PPI-R-EoE and PPI-NR-EoE look the same at the molecular level,” he said. “After therapy, PPI-R-EoE normalizes, as per its definition.”
This study was supported by the Campaign Urging Research for Eosinophilic Disease Foundation Grant, the Kenneth and Louise Goldberg Junior Faculty Award, and a National Institutes of Health award. Chan declared advisory board positions with several pharmaceutical companies and Rothenberg reported no relevant disclosures.
A version of this article appeared on Medscape.com.
, according to comparative proteomic analyses.
Notably, after PPI therapy, the protein profiles of responsive patients reverted and appeared similar to non-EoE patients, whereas the profiles of nonresponsive patients remained largely unchanged.
“Identifying protein biomarkers associated with PPI response may help distinguish EoE phenotypes and guide therapy selections,” said senior author Walter Chan, MD, AGAF, associate professor of medicine in the Division of Gastroenterology, Hepatology, and Endoscopy at Harvard Medical School and director of the center for gastrointestinal motility at Brigham and Women’s Hospital, Boston.
“These findings may provide the framework for developing protein biomarkers to assess response to therapy and monitor disease activity,” he added.
The study was published online in Gastroenterology.
Comparative Proteomic Analyses
Chan and colleagues conducted a prospective exploratory pilot study to identify the differences in esophageal protein profiles among PPI-responsive-EoE (PPI-R-EoE), PPI-nonresponsive-EoE (PPI-NR-EoE), and non-EoE controls using SOMAscan, a proteomics platform that allows simultaneous detection of 1305 human proteins.
The research team prospectively enrolled patients undergoing endoscopy for esophageal symptoms or for EoE follow-up, obtaining clinically indicated biopsies as well as extra samples from the midesophagus.
Patients who were diagnosed with EoE (at 15 or greater eosinophils per high-power field, or eos/hpf) were treated with 20 mg of omeprazole twice daily for 8 weeks, followed by repeat biopsies to assess treatment response.
Patients with histologic remission (fewer than 15 eos/hpf) were classified as PPI-R-EoE, whereas those with persistently active disease were classified as PPI-NR-EoE. Patients without EoE served as controls and were categorized as having erosive esophagitis (EE) or no esophagitis.
Overall, the study enrolled 32 patients, including 15 with PPI-R-EoE, eight with PPI-NR-EoE, three with EE, and six with no esophagitis. The demographics, symptoms, and endoscopic findings were similar between the PPI-R-EoE and PPI-NR-EoE patients.
At the index endoscopy, the PPI-R-EoE and PPI-NR-EoE patients had similar esophageal protein profiles, with only 20 proteins differentially expressed at a relaxed cutoff of P < .1. An analysis of the 20 proteins predicted lower expression of six proteins that may be associated with gastrointestinal inflammation in nonresponsive patients, including STAT1, STAT3, CFB, interleukin (IL)-17RA, TNFRSF1A, and SERPINA3.
In addition, 136 proteins — including 15 with corrected P < .05 — clearly discriminated PPI-R-EoE patients from non-EoE controls, and 255 proteins — including 249 with P < .05 — discriminated PPI-NR-EoE patients from controls. Both types of EoE patients had proteins associated with enhanced inflammation and vasculogenesis, as well as down-regulation of CRISP3 and DSG1 and upregulation of TNFAIP6.
The comparative analyses also showed that the follow-up biopsies of PPI-R-EoE patients had protein profiles that resembled non-EoE controls after PPI therapy.
“This further supports the hypothesis that despite the PPI response, PPI-R-EoE represents a subtype of EoE rather than gastroesophageal reflux disease (GERD),” Chan said.
Future EoE Considerations
Although most expressed proteins appeared similar between PPI-responsive and nonresponsive patients before treatment, a few proteins differed related to gastrointestinal inflammation, the study authors wrote, including some previously implicated in IL4 and IL13 inflammatory pathways.
“Further study of these proteins may provide insights into the EoE pathogenic pathway, explore their potential to predict PPI response at diagnosis, and identify possible therapeutic targets,” they wrote.
The authors pointed to the small study size as the primary limitation, noting that the pilot study was intended to explore the feasibility of using SomaScan to assess esophageal protein profiles in different EoE phenotypes. In the future, larger studies with more expansive candidate proteins could help characterize the differences and better identify specific proteins and pathways in EoE, they wrote.
“The takeaway is that PPI responsiveness does not distinguish EoE from GERD but rather PPI is a primary therapy for EoE independent of GERD,” said Marc Rothenberg, MD, director of allergy and immunology and director of the Cincinnati Center for Eosinophilic Disorders at Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio.
Rothenberg, who wasn’t involved with this study, has conducted transcriptome analyses of PPI-R-EoE, which showed PPI-reversible allergic inflammation.
“PPI-R-EoE and PPI-NR-EoE look the same at the molecular level,” he said. “After therapy, PPI-R-EoE normalizes, as per its definition.”
This study was supported by the Campaign Urging Research for Eosinophilic Disease Foundation Grant, the Kenneth and Louise Goldberg Junior Faculty Award, and a National Institutes of Health award. Chan declared advisory board positions with several pharmaceutical companies and Rothenberg reported no relevant disclosures.
A version of this article appeared on Medscape.com.
, according to comparative proteomic analyses.
Notably, after PPI therapy, the protein profiles of responsive patients reverted and appeared similar to non-EoE patients, whereas the profiles of nonresponsive patients remained largely unchanged.
“Identifying protein biomarkers associated with PPI response may help distinguish EoE phenotypes and guide therapy selections,” said senior author Walter Chan, MD, AGAF, associate professor of medicine in the Division of Gastroenterology, Hepatology, and Endoscopy at Harvard Medical School and director of the center for gastrointestinal motility at Brigham and Women’s Hospital, Boston.
“These findings may provide the framework for developing protein biomarkers to assess response to therapy and monitor disease activity,” he added.
The study was published online in Gastroenterology.
Comparative Proteomic Analyses
Chan and colleagues conducted a prospective exploratory pilot study to identify the differences in esophageal protein profiles among PPI-responsive-EoE (PPI-R-EoE), PPI-nonresponsive-EoE (PPI-NR-EoE), and non-EoE controls using SOMAscan, a proteomics platform that allows simultaneous detection of 1305 human proteins.
The research team prospectively enrolled patients undergoing endoscopy for esophageal symptoms or for EoE follow-up, obtaining clinically indicated biopsies as well as extra samples from the midesophagus.
Patients who were diagnosed with EoE (at 15 or greater eosinophils per high-power field, or eos/hpf) were treated with 20 mg of omeprazole twice daily for 8 weeks, followed by repeat biopsies to assess treatment response.
Patients with histologic remission (fewer than 15 eos/hpf) were classified as PPI-R-EoE, whereas those with persistently active disease were classified as PPI-NR-EoE. Patients without EoE served as controls and were categorized as having erosive esophagitis (EE) or no esophagitis.
Overall, the study enrolled 32 patients, including 15 with PPI-R-EoE, eight with PPI-NR-EoE, three with EE, and six with no esophagitis. The demographics, symptoms, and endoscopic findings were similar between the PPI-R-EoE and PPI-NR-EoE patients.
At the index endoscopy, the PPI-R-EoE and PPI-NR-EoE patients had similar esophageal protein profiles, with only 20 proteins differentially expressed at a relaxed cutoff of P < .1. An analysis of the 20 proteins predicted lower expression of six proteins that may be associated with gastrointestinal inflammation in nonresponsive patients, including STAT1, STAT3, CFB, interleukin (IL)-17RA, TNFRSF1A, and SERPINA3.
In addition, 136 proteins — including 15 with corrected P < .05 — clearly discriminated PPI-R-EoE patients from non-EoE controls, and 255 proteins — including 249 with P < .05 — discriminated PPI-NR-EoE patients from controls. Both types of EoE patients had proteins associated with enhanced inflammation and vasculogenesis, as well as down-regulation of CRISP3 and DSG1 and upregulation of TNFAIP6.
The comparative analyses also showed that the follow-up biopsies of PPI-R-EoE patients had protein profiles that resembled non-EoE controls after PPI therapy.
“This further supports the hypothesis that despite the PPI response, PPI-R-EoE represents a subtype of EoE rather than gastroesophageal reflux disease (GERD),” Chan said.
Future EoE Considerations
Although most expressed proteins appeared similar between PPI-responsive and nonresponsive patients before treatment, a few proteins differed related to gastrointestinal inflammation, the study authors wrote, including some previously implicated in IL4 and IL13 inflammatory pathways.
“Further study of these proteins may provide insights into the EoE pathogenic pathway, explore their potential to predict PPI response at diagnosis, and identify possible therapeutic targets,” they wrote.
The authors pointed to the small study size as the primary limitation, noting that the pilot study was intended to explore the feasibility of using SomaScan to assess esophageal protein profiles in different EoE phenotypes. In the future, larger studies with more expansive candidate proteins could help characterize the differences and better identify specific proteins and pathways in EoE, they wrote.
“The takeaway is that PPI responsiveness does not distinguish EoE from GERD but rather PPI is a primary therapy for EoE independent of GERD,” said Marc Rothenberg, MD, director of allergy and immunology and director of the Cincinnati Center for Eosinophilic Disorders at Cincinnati Children’s Hospital Medical Center, Cincinnati, Ohio.
Rothenberg, who wasn’t involved with this study, has conducted transcriptome analyses of PPI-R-EoE, which showed PPI-reversible allergic inflammation.
“PPI-R-EoE and PPI-NR-EoE look the same at the molecular level,” he said. “After therapy, PPI-R-EoE normalizes, as per its definition.”
This study was supported by the Campaign Urging Research for Eosinophilic Disease Foundation Grant, the Kenneth and Louise Goldberg Junior Faculty Award, and a National Institutes of Health award. Chan declared advisory board positions with several pharmaceutical companies and Rothenberg reported no relevant disclosures.
A version of this article appeared on Medscape.com.
Tapering Corticosteroids in Severe Alcohol-Associated Hepatitis Appears Safe
SAN DIEGO — , according to new research.
“Although several drugs have been evaluated for severe alcohol-associated hepatitis, none have succeeded in practice. Corticosteroids remain the mainstay of treatment; however, infections remain a major concern in 25%-40% of cases,” said Anand Kulkarni, MD, senior consultant and director of critical care hepatology at the Asian Institute of Gastroenterology in Hyderabad, India.
“There are no standard society guidelines for steroid dosing, and our current practices stem from studies in the 1970s, so there’s a major knowledge gap around optimal dosing and if stepwise tapering helps,” said Kulkarni, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Assessing Tapered Doses
In a multicenter, open-label randomized controlled trial, 254 patients with SAH from four Indian centers and one Canadian center were randomized to receive either a fixed or tapering dose of 40 mg prednisolone daily for 4 weeks. The patients in the tapering group received a starting dose of 40 mg, which was reduced by 10 mg weekly over 4 weeks.
While taking corticosteroids, 66% of those in the fixed dose group and 55% of those in the tapering group also received prophylactic antibiotics.
The mean age of participants was 41.1 years, the median Model For End-Stage Liver Disease score was 25.6, and 98.4% were men.
The primary objective was to compare the incidence of drug-related adverse events, infections, hospitalization, and mortality through day 90.
The duration of corticosteroid therapy was 22 days in the fixed dose group and 23 days in the tapering dose group.
Overall, the proportion of steroid responders was similar in both groups, at 80.3% in the fixed dose group and 82.5% in the tapering dose group.
However, the incidence of drug-related adverse events was significantly higher in the fixed dose group (52%) than in the tapering dose group (36.2%). The most common adverse events in both groups were infection, hyperglycemia, and hematochezia.
At 90 days, the incidence of infection was significantly lower in the tapering group (19.7%) than in the fixed dose group (33.1%). In both groups, the most common infection sites were the lungs (28.3%) and urinary tract (22.4%).
In terms of liver-related outcomes, some patients developed hepatic encephalopathy (11.8% in fixed dose vs 6.3% in tapering dose) and acute variceal bleed (3.1% in each group), as well as acute kidney injury (26.8% in fixed dose vs 18.9% in tapering dose).
Hospitalization within 90 days was required in 44.1% of the fixed dose group and 33.1% of the tapering dose group.
Survival at day 90 was 83.5% in the fixed dose group and 86.6% in the tapering dose group. Four patients in the fixed dose group and three patients in the tapering dose group underwent living donor liver transplantation by day 90.
Relapse of alcohol use by day 90 occurred in 13.4% of the fixed dose group and 12.6% of the tapering dose group.
“Rapid tapering in severe alcohol-associated hepatitis reduces infections and hospitalizations but doesn’t have a significant impact on survival,” Kulkarni concluded.
Considering Alternative Therapies
Given the high risk for infection in patients with SAH and limited certainty around benefits, the data may also call into question whether to give steroids to these patients at all, said session co-moderator Aleksander Krag, MD, professor of clinical medicine at the University of Southern Denmark, Odense, Denmark, and secretary general of the European Association for the Study of Liver 2023-2025.
“Since there are no other treatments available as of now, we’ll still continue to give steroids,” Kulkarni noted. But “tapering the dose should be beneficial.”
Although steroid therapy has been considered the “mainstay treatment” for SAH for 50 years, it doesn’t always lead to long-term improvement in liver values or survival, said Prasun Jalal, MD, the Stan and Sue Partee Endowed Chair in Hepatology at Baylor College of Medicine, Houston, who wasn’t involved with the study.
Researchers are looking to other connections, such as the gut microbiome, to find treatments for advanced alcoholic liver disease, Jalal said in an interview. In a small pilot study, he and colleagues found that intestinal microbiota transplantation (IMT) appears to be safe and effective for these patients.
“Early analyses suggest that IMT has a favorable outcome on the prognosis of patients with severe alcohol-associated hepatitis and is safe,” Jalal said. “A longer follow-up study with a larger sample size is in progress.”
Kulkarni and Krag reported no relevant disclosures. Jalal has speaking and teaching relationships with AbbVie and Madrigal.
A version of this article appeared on Medscape.com.
SAN DIEGO — , according to new research.
“Although several drugs have been evaluated for severe alcohol-associated hepatitis, none have succeeded in practice. Corticosteroids remain the mainstay of treatment; however, infections remain a major concern in 25%-40% of cases,” said Anand Kulkarni, MD, senior consultant and director of critical care hepatology at the Asian Institute of Gastroenterology in Hyderabad, India.
“There are no standard society guidelines for steroid dosing, and our current practices stem from studies in the 1970s, so there’s a major knowledge gap around optimal dosing and if stepwise tapering helps,” said Kulkarni, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Assessing Tapered Doses
In a multicenter, open-label randomized controlled trial, 254 patients with SAH from four Indian centers and one Canadian center were randomized to receive either a fixed or tapering dose of 40 mg prednisolone daily for 4 weeks. The patients in the tapering group received a starting dose of 40 mg, which was reduced by 10 mg weekly over 4 weeks.
While taking corticosteroids, 66% of those in the fixed dose group and 55% of those in the tapering group also received prophylactic antibiotics.
The mean age of participants was 41.1 years, the median Model For End-Stage Liver Disease score was 25.6, and 98.4% were men.
The primary objective was to compare the incidence of drug-related adverse events, infections, hospitalization, and mortality through day 90.
The duration of corticosteroid therapy was 22 days in the fixed dose group and 23 days in the tapering dose group.
Overall, the proportion of steroid responders was similar in both groups, at 80.3% in the fixed dose group and 82.5% in the tapering dose group.
However, the incidence of drug-related adverse events was significantly higher in the fixed dose group (52%) than in the tapering dose group (36.2%). The most common adverse events in both groups were infection, hyperglycemia, and hematochezia.
At 90 days, the incidence of infection was significantly lower in the tapering group (19.7%) than in the fixed dose group (33.1%). In both groups, the most common infection sites were the lungs (28.3%) and urinary tract (22.4%).
In terms of liver-related outcomes, some patients developed hepatic encephalopathy (11.8% in fixed dose vs 6.3% in tapering dose) and acute variceal bleed (3.1% in each group), as well as acute kidney injury (26.8% in fixed dose vs 18.9% in tapering dose).
Hospitalization within 90 days was required in 44.1% of the fixed dose group and 33.1% of the tapering dose group.
Survival at day 90 was 83.5% in the fixed dose group and 86.6% in the tapering dose group. Four patients in the fixed dose group and three patients in the tapering dose group underwent living donor liver transplantation by day 90.
Relapse of alcohol use by day 90 occurred in 13.4% of the fixed dose group and 12.6% of the tapering dose group.
“Rapid tapering in severe alcohol-associated hepatitis reduces infections and hospitalizations but doesn’t have a significant impact on survival,” Kulkarni concluded.
Considering Alternative Therapies
Given the high risk for infection in patients with SAH and limited certainty around benefits, the data may also call into question whether to give steroids to these patients at all, said session co-moderator Aleksander Krag, MD, professor of clinical medicine at the University of Southern Denmark, Odense, Denmark, and secretary general of the European Association for the Study of Liver 2023-2025.
“Since there are no other treatments available as of now, we’ll still continue to give steroids,” Kulkarni noted. But “tapering the dose should be beneficial.”
Although steroid therapy has been considered the “mainstay treatment” for SAH for 50 years, it doesn’t always lead to long-term improvement in liver values or survival, said Prasun Jalal, MD, the Stan and Sue Partee Endowed Chair in Hepatology at Baylor College of Medicine, Houston, who wasn’t involved with the study.
Researchers are looking to other connections, such as the gut microbiome, to find treatments for advanced alcoholic liver disease, Jalal said in an interview. In a small pilot study, he and colleagues found that intestinal microbiota transplantation (IMT) appears to be safe and effective for these patients.
“Early analyses suggest that IMT has a favorable outcome on the prognosis of patients with severe alcohol-associated hepatitis and is safe,” Jalal said. “A longer follow-up study with a larger sample size is in progress.”
Kulkarni and Krag reported no relevant disclosures. Jalal has speaking and teaching relationships with AbbVie and Madrigal.
A version of this article appeared on Medscape.com.
SAN DIEGO — , according to new research.
“Although several drugs have been evaluated for severe alcohol-associated hepatitis, none have succeeded in practice. Corticosteroids remain the mainstay of treatment; however, infections remain a major concern in 25%-40% of cases,” said Anand Kulkarni, MD, senior consultant and director of critical care hepatology at the Asian Institute of Gastroenterology in Hyderabad, India.
“There are no standard society guidelines for steroid dosing, and our current practices stem from studies in the 1970s, so there’s a major knowledge gap around optimal dosing and if stepwise tapering helps,” said Kulkarni, who presented the findings at The Liver Meeting 2024: American Association for the Study of Liver Diseases (AASLD).
Assessing Tapered Doses
In a multicenter, open-label randomized controlled trial, 254 patients with SAH from four Indian centers and one Canadian center were randomized to receive either a fixed or tapering dose of 40 mg prednisolone daily for 4 weeks. The patients in the tapering group received a starting dose of 40 mg, which was reduced by 10 mg weekly over 4 weeks.
While taking corticosteroids, 66% of those in the fixed dose group and 55% of those in the tapering group also received prophylactic antibiotics.
The mean age of participants was 41.1 years, the median Model For End-Stage Liver Disease score was 25.6, and 98.4% were men.
The primary objective was to compare the incidence of drug-related adverse events, infections, hospitalization, and mortality through day 90.
The duration of corticosteroid therapy was 22 days in the fixed dose group and 23 days in the tapering dose group.
Overall, the proportion of steroid responders was similar in both groups, at 80.3% in the fixed dose group and 82.5% in the tapering dose group.
However, the incidence of drug-related adverse events was significantly higher in the fixed dose group (52%) than in the tapering dose group (36.2%). The most common adverse events in both groups were infection, hyperglycemia, and hematochezia.
At 90 days, the incidence of infection was significantly lower in the tapering group (19.7%) than in the fixed dose group (33.1%). In both groups, the most common infection sites were the lungs (28.3%) and urinary tract (22.4%).
In terms of liver-related outcomes, some patients developed hepatic encephalopathy (11.8% in fixed dose vs 6.3% in tapering dose) and acute variceal bleed (3.1% in each group), as well as acute kidney injury (26.8% in fixed dose vs 18.9% in tapering dose).
Hospitalization within 90 days was required in 44.1% of the fixed dose group and 33.1% of the tapering dose group.
Survival at day 90 was 83.5% in the fixed dose group and 86.6% in the tapering dose group. Four patients in the fixed dose group and three patients in the tapering dose group underwent living donor liver transplantation by day 90.
Relapse of alcohol use by day 90 occurred in 13.4% of the fixed dose group and 12.6% of the tapering dose group.
“Rapid tapering in severe alcohol-associated hepatitis reduces infections and hospitalizations but doesn’t have a significant impact on survival,” Kulkarni concluded.
Considering Alternative Therapies
Given the high risk for infection in patients with SAH and limited certainty around benefits, the data may also call into question whether to give steroids to these patients at all, said session co-moderator Aleksander Krag, MD, professor of clinical medicine at the University of Southern Denmark, Odense, Denmark, and secretary general of the European Association for the Study of Liver 2023-2025.
“Since there are no other treatments available as of now, we’ll still continue to give steroids,” Kulkarni noted. But “tapering the dose should be beneficial.”
Although steroid therapy has been considered the “mainstay treatment” for SAH for 50 years, it doesn’t always lead to long-term improvement in liver values or survival, said Prasun Jalal, MD, the Stan and Sue Partee Endowed Chair in Hepatology at Baylor College of Medicine, Houston, who wasn’t involved with the study.
Researchers are looking to other connections, such as the gut microbiome, to find treatments for advanced alcoholic liver disease, Jalal said in an interview. In a small pilot study, he and colleagues found that intestinal microbiota transplantation (IMT) appears to be safe and effective for these patients.
“Early analyses suggest that IMT has a favorable outcome on the prognosis of patients with severe alcohol-associated hepatitis and is safe,” Jalal said. “A longer follow-up study with a larger sample size is in progress.”
Kulkarni and Krag reported no relevant disclosures. Jalal has speaking and teaching relationships with AbbVie and Madrigal.
A version of this article appeared on Medscape.com.
FROM AASLD 2024