User login
The Journal of Clinical Outcomes Management® is an independent, peer-reviewed journal offering evidence-based, practical information for improving the quality, safety, and value of health care.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Is the WHO’s HPV vaccination target within reach?
The WHO’s goal is to have HPV vaccines delivered to 90% of all adolescent girls by 2030, part of the organization’s larger goal to “eliminate” cervical cancer, or reduce the annual incidence of cervical cancer to below 4 cases per 100,000 people globally.
Laia Bruni, MD, PhD, of Catalan Institute of Oncology in Barcelona, and colleagues outlined the progress made thus far toward reaching the WHO’s goals in an article published in Preventive Medicine.
The authors noted that cervical cancer caused by HPV is a “major public health problem, especially in low- and middle-income countries (LMIC).”
However, vaccines against HPV have been available since 2006 and have been recommended by the WHO since 2009.
HPV vaccines have been introduced into many national immunization schedules. Among the 194 WHO member states, 107 (55%) had introduced HPV vaccination as of June 2020, according to estimates from the WHO and the United Nations International Children’s Emergency Fund (UNICEF).
Still, vaccine introduction and coverages are suboptimal, according to several studies and international agencies.
In their article, Dr. Bruni and colleagues describe the mid-2020 status of HPV vaccine introduction, based on WHO/UNICEF estimates of national HPV immunization coverage from 2010 to 2019.
HPV vaccination by region
The Americas and Europe are by far the WHO regions with the highest rates of HPV vaccination, with 85% and 77% of their countries, respectively, having already introduced HPV vaccination, either partially or nationwide.
In 2019, a record number of introductions, 16, were reported, mostly in LMICs where access has been limited. In prior years, the average had been a relatively steady 7-8 introductions per year.
The percentage of high-income countries (HICs) that have introduced HPV vaccination exceeds 80%. LMICs started introducing HPV vaccination later and at a slower pace, compared with HICs. By the end of 2019, only 41% of LMICs had introduced vaccination. However, of the new introductions in 2019, 87% were in LMICs.
In 2019, the average performance coverage for HPV vaccination programs in 99 countries (both HICs and LMICs) was around 67% for the first vaccine dose and 53% for the final dose.
Median performance coverage was higher in LMICs than in HICs for the first dose (80% and 72%, respectively), but mean dropout rates were higher in LMICs than in HICs (18% and 11%, respectively).
Coverage of more than 90% was achieved for the last dose in only five countries (6%). Twenty-two countries (21%) achieved coverages of 75% or higher, while 35 countries (40%) had final dose coverages of 50% or less.
Global coverage of the final HPV vaccine dose (weighted by population size) was estimated at 15%. According to the authors, that low percentage can be explained by the fact that many of the most populous countries have either not yet introduced HPV vaccination or have low performance.
The countries with highest cervical cancer burden have had limited secondary prevention and have been less likely to provide access to vaccination, the authors noted. However, this trend appears to be reversing, with 14 new LMICs providing HPV vaccination in 2019.
HPV vaccination by sex
By 2019, almost a third of the 107 HPV vaccination programs (n = 33) were “gender neutral,” with girls and boys receiving HPV vaccines. Generally, LMICs targeted younger girls (9-10 years) compared with HICs (11-13 years).
Dr. Bruni and colleagues estimated that 15% of girls and 4% of boys were vaccinated globally with the full course of vaccine. At least one dose was received by 20% of girls and 5% of boys.
From 2010 to 2019, HPV vaccination rates in HICs rose from 42% in girls and 0% in boys to 88% and 44%, respectively. In LMICs, over the same period, rates rose from 4% in girls and 0% in boys to 40% and 5%, respectively.
Obstacles and the path forward
The COVID-19 pandemic has halted HPV vaccine delivery in the majority of countries, Dr. Bruni and colleagues noted. About 70 countries had reported program interruptions by August 2020, and delays to HPV vaccine introductions were anticipated for other countries.
An economic downturn could have further far-reaching effects on plans to introduce HPV vaccines, Dr. Bruni and colleagues observed.
While meeting the 2030 target will be challenging, the authors noted that, in every geographic area, some programs are meeting the 90% target.
“HPV national programs should aim to get 90+% of girls vaccinated before the age of 15,” Dr. Bruni said in an interview. “This is a feasible goal, and some countries have succeeded, such as Norway and Rwanda. Average performance, however, is around 55%, and that shows that it is not an easy task.”
Dr. Bruni underscored the four main actions that should be taken to achieve 90% coverage of HPV vaccination, as outlined in the WHO cervical cancer elimination strategy:
- Secure sufficient and affordable HPV vaccines.
- Increase the quality and coverage of vaccination.
- Improve communication and social mobilization.
- Innovate to improve efficiency of vaccine delivery.
“Addressing vaccine hesitancy adequately is one of the biggest challenges we face, especially for the HPV vaccine,” Dr. Bruni said. “As the WHO document states, understanding social, cultural, societal, and other barriers affecting acceptance and uptake of the vaccine will be critical for overcoming vaccine hesitancy and countering misinformation.”
This research was funded by a grant from Instituto de Salud Carlos III and various other grants. Dr. Bruni and coauthors said they have no relevant disclosures.
The WHO’s goal is to have HPV vaccines delivered to 90% of all adolescent girls by 2030, part of the organization’s larger goal to “eliminate” cervical cancer, or reduce the annual incidence of cervical cancer to below 4 cases per 100,000 people globally.
Laia Bruni, MD, PhD, of Catalan Institute of Oncology in Barcelona, and colleagues outlined the progress made thus far toward reaching the WHO’s goals in an article published in Preventive Medicine.
The authors noted that cervical cancer caused by HPV is a “major public health problem, especially in low- and middle-income countries (LMIC).”
However, vaccines against HPV have been available since 2006 and have been recommended by the WHO since 2009.
HPV vaccines have been introduced into many national immunization schedules. Among the 194 WHO member states, 107 (55%) had introduced HPV vaccination as of June 2020, according to estimates from the WHO and the United Nations International Children’s Emergency Fund (UNICEF).
Still, vaccine introduction and coverages are suboptimal, according to several studies and international agencies.
In their article, Dr. Bruni and colleagues describe the mid-2020 status of HPV vaccine introduction, based on WHO/UNICEF estimates of national HPV immunization coverage from 2010 to 2019.
HPV vaccination by region
The Americas and Europe are by far the WHO regions with the highest rates of HPV vaccination, with 85% and 77% of their countries, respectively, having already introduced HPV vaccination, either partially or nationwide.
In 2019, a record number of introductions, 16, were reported, mostly in LMICs where access has been limited. In prior years, the average had been a relatively steady 7-8 introductions per year.
The percentage of high-income countries (HICs) that have introduced HPV vaccination exceeds 80%. LMICs started introducing HPV vaccination later and at a slower pace, compared with HICs. By the end of 2019, only 41% of LMICs had introduced vaccination. However, of the new introductions in 2019, 87% were in LMICs.
In 2019, the average performance coverage for HPV vaccination programs in 99 countries (both HICs and LMICs) was around 67% for the first vaccine dose and 53% for the final dose.
Median performance coverage was higher in LMICs than in HICs for the first dose (80% and 72%, respectively), but mean dropout rates were higher in LMICs than in HICs (18% and 11%, respectively).
Coverage of more than 90% was achieved for the last dose in only five countries (6%). Twenty-two countries (21%) achieved coverages of 75% or higher, while 35 countries (40%) had final dose coverages of 50% or less.
Global coverage of the final HPV vaccine dose (weighted by population size) was estimated at 15%. According to the authors, that low percentage can be explained by the fact that many of the most populous countries have either not yet introduced HPV vaccination or have low performance.
The countries with highest cervical cancer burden have had limited secondary prevention and have been less likely to provide access to vaccination, the authors noted. However, this trend appears to be reversing, with 14 new LMICs providing HPV vaccination in 2019.
HPV vaccination by sex
By 2019, almost a third of the 107 HPV vaccination programs (n = 33) were “gender neutral,” with girls and boys receiving HPV vaccines. Generally, LMICs targeted younger girls (9-10 years) compared with HICs (11-13 years).
Dr. Bruni and colleagues estimated that 15% of girls and 4% of boys were vaccinated globally with the full course of vaccine. At least one dose was received by 20% of girls and 5% of boys.
From 2010 to 2019, HPV vaccination rates in HICs rose from 42% in girls and 0% in boys to 88% and 44%, respectively. In LMICs, over the same period, rates rose from 4% in girls and 0% in boys to 40% and 5%, respectively.
Obstacles and the path forward
The COVID-19 pandemic has halted HPV vaccine delivery in the majority of countries, Dr. Bruni and colleagues noted. About 70 countries had reported program interruptions by August 2020, and delays to HPV vaccine introductions were anticipated for other countries.
An economic downturn could have further far-reaching effects on plans to introduce HPV vaccines, Dr. Bruni and colleagues observed.
While meeting the 2030 target will be challenging, the authors noted that, in every geographic area, some programs are meeting the 90% target.
“HPV national programs should aim to get 90+% of girls vaccinated before the age of 15,” Dr. Bruni said in an interview. “This is a feasible goal, and some countries have succeeded, such as Norway and Rwanda. Average performance, however, is around 55%, and that shows that it is not an easy task.”
Dr. Bruni underscored the four main actions that should be taken to achieve 90% coverage of HPV vaccination, as outlined in the WHO cervical cancer elimination strategy:
- Secure sufficient and affordable HPV vaccines.
- Increase the quality and coverage of vaccination.
- Improve communication and social mobilization.
- Innovate to improve efficiency of vaccine delivery.
“Addressing vaccine hesitancy adequately is one of the biggest challenges we face, especially for the HPV vaccine,” Dr. Bruni said. “As the WHO document states, understanding social, cultural, societal, and other barriers affecting acceptance and uptake of the vaccine will be critical for overcoming vaccine hesitancy and countering misinformation.”
This research was funded by a grant from Instituto de Salud Carlos III and various other grants. Dr. Bruni and coauthors said they have no relevant disclosures.
The WHO’s goal is to have HPV vaccines delivered to 90% of all adolescent girls by 2030, part of the organization’s larger goal to “eliminate” cervical cancer, or reduce the annual incidence of cervical cancer to below 4 cases per 100,000 people globally.
Laia Bruni, MD, PhD, of Catalan Institute of Oncology in Barcelona, and colleagues outlined the progress made thus far toward reaching the WHO’s goals in an article published in Preventive Medicine.
The authors noted that cervical cancer caused by HPV is a “major public health problem, especially in low- and middle-income countries (LMIC).”
However, vaccines against HPV have been available since 2006 and have been recommended by the WHO since 2009.
HPV vaccines have been introduced into many national immunization schedules. Among the 194 WHO member states, 107 (55%) had introduced HPV vaccination as of June 2020, according to estimates from the WHO and the United Nations International Children’s Emergency Fund (UNICEF).
Still, vaccine introduction and coverages are suboptimal, according to several studies and international agencies.
In their article, Dr. Bruni and colleagues describe the mid-2020 status of HPV vaccine introduction, based on WHO/UNICEF estimates of national HPV immunization coverage from 2010 to 2019.
HPV vaccination by region
The Americas and Europe are by far the WHO regions with the highest rates of HPV vaccination, with 85% and 77% of their countries, respectively, having already introduced HPV vaccination, either partially or nationwide.
In 2019, a record number of introductions, 16, were reported, mostly in LMICs where access has been limited. In prior years, the average had been a relatively steady 7-8 introductions per year.
The percentage of high-income countries (HICs) that have introduced HPV vaccination exceeds 80%. LMICs started introducing HPV vaccination later and at a slower pace, compared with HICs. By the end of 2019, only 41% of LMICs had introduced vaccination. However, of the new introductions in 2019, 87% were in LMICs.
In 2019, the average performance coverage for HPV vaccination programs in 99 countries (both HICs and LMICs) was around 67% for the first vaccine dose and 53% for the final dose.
Median performance coverage was higher in LMICs than in HICs for the first dose (80% and 72%, respectively), but mean dropout rates were higher in LMICs than in HICs (18% and 11%, respectively).
Coverage of more than 90% was achieved for the last dose in only five countries (6%). Twenty-two countries (21%) achieved coverages of 75% or higher, while 35 countries (40%) had final dose coverages of 50% or less.
Global coverage of the final HPV vaccine dose (weighted by population size) was estimated at 15%. According to the authors, that low percentage can be explained by the fact that many of the most populous countries have either not yet introduced HPV vaccination or have low performance.
The countries with highest cervical cancer burden have had limited secondary prevention and have been less likely to provide access to vaccination, the authors noted. However, this trend appears to be reversing, with 14 new LMICs providing HPV vaccination in 2019.
HPV vaccination by sex
By 2019, almost a third of the 107 HPV vaccination programs (n = 33) were “gender neutral,” with girls and boys receiving HPV vaccines. Generally, LMICs targeted younger girls (9-10 years) compared with HICs (11-13 years).
Dr. Bruni and colleagues estimated that 15% of girls and 4% of boys were vaccinated globally with the full course of vaccine. At least one dose was received by 20% of girls and 5% of boys.
From 2010 to 2019, HPV vaccination rates in HICs rose from 42% in girls and 0% in boys to 88% and 44%, respectively. In LMICs, over the same period, rates rose from 4% in girls and 0% in boys to 40% and 5%, respectively.
Obstacles and the path forward
The COVID-19 pandemic has halted HPV vaccine delivery in the majority of countries, Dr. Bruni and colleagues noted. About 70 countries had reported program interruptions by August 2020, and delays to HPV vaccine introductions were anticipated for other countries.
An economic downturn could have further far-reaching effects on plans to introduce HPV vaccines, Dr. Bruni and colleagues observed.
While meeting the 2030 target will be challenging, the authors noted that, in every geographic area, some programs are meeting the 90% target.
“HPV national programs should aim to get 90+% of girls vaccinated before the age of 15,” Dr. Bruni said in an interview. “This is a feasible goal, and some countries have succeeded, such as Norway and Rwanda. Average performance, however, is around 55%, and that shows that it is not an easy task.”
Dr. Bruni underscored the four main actions that should be taken to achieve 90% coverage of HPV vaccination, as outlined in the WHO cervical cancer elimination strategy:
- Secure sufficient and affordable HPV vaccines.
- Increase the quality and coverage of vaccination.
- Improve communication and social mobilization.
- Innovate to improve efficiency of vaccine delivery.
“Addressing vaccine hesitancy adequately is one of the biggest challenges we face, especially for the HPV vaccine,” Dr. Bruni said. “As the WHO document states, understanding social, cultural, societal, and other barriers affecting acceptance and uptake of the vaccine will be critical for overcoming vaccine hesitancy and countering misinformation.”
This research was funded by a grant from Instituto de Salud Carlos III and various other grants. Dr. Bruni and coauthors said they have no relevant disclosures.
FROM PREVENTIVE MEDICINE
FDA scrutinizes cancer therapies granted accelerated approval
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
U.S. regulators are stepping up scrutiny of therapies that were granted an accelerated approval to treat cancers on the basis of surrogate endpoints but have failed to show clinical or survival benefits upon more extensive testing.
At issue are a number of cancer indications for immunotherapies. Four have already been withdrawn (voluntarily by the manufacturer), and six more will be reviewed at an upcoming meeting.
In recent years, the US Food and Drug Administration has granted accelerated approvals to oncology medicines on the basis of evidence that suggests a benefit for patients. Examples of such evidence relate to response rates and estimates of tumor shrinkage. But these approvals are granted on the condition that the manufacturer conducts larger clinical trials that show clinical benefit, including benefit in overall survival.
Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, has argued that the point of these conditional approvals is to find acceptable surrogate markers to allow people with “desperate illnesses” to have access to potentially helpful drugs while work continues to determine the drug’s actual benefit to patients.
Oncologists are now questioning whether the FDA has become too lenient in its approach, Daniel A. Goldstein, MD, a senior physician in medical oncology and internal medicine at the Rabin Medical Center, Petah Tikva, Israel, told this news organization.
“The main two things you want from a cancer drug is to live longer and live a higher quality of life,” said Goldstein. “But these endpoints that they’ve been using over the past few years are not really giving us confidence that these drugs are actually going to help to live longer or better.”
Dr. Pazdur said the FDA will consider withdrawing its accelerated approvals when results of further studies do not confirm expected benefit for patients.
“This is like the pendulum has swung as far as it was going to swing and now is on the backswing,” said Dr. Goldstein, also of the department of health policy and management at the University of North Carolina at Chapel Hill. “You could call this a watershed moment.”
Although there’s near universal interest in allowing people with advanced cancer access to promising medicines, there’s also rising concern about exposing patients needlessly to costly drugs with potentially tough side effects. That may prompt a shift in the standards U.S. regulators apply to cancer medicines, Dr. Goldstein said.
Indications withdrawn and under review
In a meeting scheduled for April 27-29, the FDA’s Oncologic Drugs Advisory Committee will review indications granted through the accelerated approval process for three immunotherapies: pembrolizumab (Keytruda), atezolizumab (Tecentriq), and nivolumab (Opdivo).
It is part of an industry-wide evaluation of accelerated approvals for cancer indications in which confirmatory trials did not confirm clinical benefit, the FDA noted.
The process has already led to voluntary withdrawals of four cancer indications by the manufacturers, including one indication each for pembrolizumab, atezolizumab, and nivolumab, and one for durvalumab (Imfinzi).
All of these immunotherapies are approved for numerous cancer indications, and they all remain on the market. It is only the U.S. approvals for particular cancer indications that have been withdrawn.
In the past, olaratumab (Lartruvo) was withdrawn from the market altogether. The FDA granted accelerated approval of the drug for soft tissue sarcoma, but clinical benefit was not confirmed in a phase 3 trial.
Issue highlighted by Dr. Prasad and Dr. Gyawali
In recent years, much of the attention on accelerated approvals was spurred by the work of a few researchers, particularly Vinay Prasad, MD, MPH, associate professor in the department of epidemiology and biostatistics, University of California, San Francisco, and Bishal Gyawali, MD, PhD, from Queen’s University Cancer Research Institute, Kingston, Ont. (Both are regular contributors to the oncology section of this news organization.)
Dr. Goldstein made this point in a tweet about the FDA’s announcement of the April ODAC meetings:
“Well done to @oncology_bg and @VPrasadMDMPH among others for highlighting in their papers that the FDA wasn’t properly evaluating accelerated approval drugs.
FDA have listened.
And I thought that the impact of academia was limited!”
Dr. Prasad has made the case for closer scrutiny of accelerated approvals in a number of journal articles and in his 2020 book, “Malignant: How Bad Policy and Bad Evidence Harm People with Cancer,” published by Johns Hopkins University Press.
The book includes highlights of a 2016 article published in Mayo Clinic Proceedings that focused on surrogate endpoints used for FDA approvals. In the article, Dr. Prasad and his coauthor report that they did not find formal analyses of the strength of the surrogate-survival correlation in 14 of 25 cases of accelerated approvals (56%) and in 11 of 30 traditional approvals (37%).
“Our results were concerning. They imply that many surrogates are based on little more than a gut feeling. You might rationalize that and argue a gut feeling is the same as ‘reasonably likely to predict,’ but no reasonable person could think a gut feeling means established,” Dr. Prasad writes in his book. “Our result suggests the FDA is using surrogate endpoints far beyond what may be fair or reasonable.”
Dr. Gyawali has argued that the process by which the FDA assesses cancer drugs for approvals has undergone a profound shift. He has most recently remarked on this in an October 2020 commentary on Medscape.
“Until the recent floodgate of approvals based on response rates from single-arm trials, the majority of cancer therapy decisions were supported by evidence generated from randomized controlled trials (RCTs),” Dr. Gyawali wrote. “The evidence base to support clinical decisions in managing therapeutic side effects has been comparatively sparse.”
Accelerated approval to improve access
The FDA has struggled for about 2 decades with questions of where to set the bar on evidence for promising cancer drugs.
The agency’s accelerated approval program for drugs began in 1992. During the first decade, the focus was largely on medicines related to HIV.
In the early 2000s, oncology drugs began to dominate the program.
Dr. Pazdur has presided over the FDA’s marked changes regarding the use of surrogate markers when weighing whether to allow sales of cancer medicines. Formerly a professor at the University of Texas MD Anderson Cancer Center, Houston, Dr. Pazdur joined the FDA as director of the Division of Oncology Drug Products in 1999.
Soon after his appointment, he had to field inquiries from pharmaceutical companies about how much evidence they needed to receive accelerated approvals.
Early on, he publicly expressed impatience about the drugmakers’ approach. “The purpose of accelerated approval was not accelerated drug company profits,” Dr. Padzur said at a 2004 ODAC meeting.
Rather, the point is to allow access to potentially helpful drugs while work continues to determine their actual benefit to patients, he explained.
“It wasn’t a license to do less, less, less, and less to a point now that we may be getting companies that are coming in” intent on determining the minimum evidence the FDA will take, Dr. Pazdur said. “It shouldn’t be what is the lowest. It is what is a sufficient amount to give patients and physicians a real understanding of what their drug will do.”
In a 2016 interview with The New York Times, Dr. Pazdur said that his views on cancer drug approvals have evolved with time. He described himself as being “on a jihad to streamline the review process and get things out the door faster.”
“I have evolved from regulator to regulator-advocate,” Dr. Pazdur told the newspaper.
His attitude reflected his personal experience in losing his wife to ovarian cancer in 2015, as well as shifts in science and law. In 2012, Congress passed a law that gave the FDA new resources to speed medicines for life-threatening diseases to market. In addition, advances in genetics appeared to be making some medications more effective and easier to test, Dr. Pazdur said in The New York Times interview.
Withdrawals seen as sign of success
Since the program’s inception, only 6% of accelerated approvals for oncology indications have been withdrawn, the FDA said.
It would be a sign that the program is working if the April meetings lead to further withdrawals of indications that have been granted accelerated approval, Julie R. Gralow, MD, chief medical officer of the American Society of Clinical Oncology, said in an interview with this news organization.
“It shouldn’t be seen as a failure,” Dr. Gralow said.
In her own practice at the Fred Hutchinson Cancer Research Center, Seattle, she has seen the value of emerging therapies for patients fighting advanced cancers. During her 25 years of clinical practice in an academic setting, she has gained access to drugs through single-patient investigative new drug applications.
However, this path is not an option for many patients who undergo treatment in facilities other than academic centers, she commented. She noted that the accelerated approval process is a way to expand access to emerging medicines, but she sees a need for caution in the use of drugs that have been given only this conditional approval. She emphasizes that such drugs may be suitable only for certain patients.
“I would say that, for metastatic patients, patients with incurable disease, we are willing to take some risk,” Dr. Gralow said. “We don’t have other options. They can’t wait the years that it would take to get a drug approved.”
One such patient is David Mitchell, who serves as the consumer representative on ODAC. He told this news organization that he is taking three drugs for multiple myeloma that received accelerated approvals: pomalidomide, bortezomib, and daratumumab.
“I want the FDA to have the option to approve drugs in an accelerated pathway, because as a patient taking three drugs granted accelerated approval, I’m benefiting – I’ve lived the benefit,” Mr. Mitchell said, “and I want other patients to have the opportunity to have that benefit.”
He believes that the FDA’s approach regarding accelerated approvals serves to get potentially beneficial medicines to patients who have few options and also fulfills the FDA’s mandate to protect the public from treatments that have little benefit but can cause harm.
Accelerated approval also offers needed flexibility to drugmakers as they develop more specifically targeted drugs for diseases that affect relatively few people, such as multiple myeloma, he said. “As the targeting of your therapies gets tighter and for smaller groups of patients, you have a harder time following the traditional model,” such as conducting large, double-blind, placebo-controlled trials that may indicate increased overall survival, he said.
“To me, this is the way the FDA intended it to work,” he added. “It’s going to offer the accelerated approval based on a surrogate endpoint for a safe drug, but it’s going to require the confirmatory trial, and if the confirmatory trial fails, it will pull the drug off the market.”
Some medicines that have received accelerated approvals may ultimately be found not to benefit patients, Mr. Mitchell acknowledged. But people in his situation, whose disease has progressed despite treatments, may want to take that risk, he added.
Four cancer indications recently withdrawn voluntarily by the manufacturer
- December 2020: Nivolumab for the treatment of patients with metastatic small cell lung cancer with progression after platinum-based chemotherapy and at least one other line of therapy (Bristol Myers Squibb).
- February 2021: Durvalumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease has progressed during or following platinum-based chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy (AstraZeneca).
- March 2021: Pembrolizumab for the treatment of patients with metastatic small cell lung cancer with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy (Merck).
- March 2021: Atezolizumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who experience disease progression during or following platinum-containing atezolizumab chemotherapy or disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy (Genentech).
Six cancer indications under review at the April 2021 ODAC meeting
- Atezolizumab indicated in combination with protein-bound for the treatment of adults with unresectable locally advanced or metastatic triple-negative whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells of any intensity covering ≥1% of the tumor area), as determined by an FDA-approved test.
- Atezolizumab indicated for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
- Pembrolizumab indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1 (Combined Positive Score ≥1), as determined by an FDA-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
- Pembrolizumab indicated for the treatment of patients with who have been previously treated with .
- Nivolumab indicated as a single agent for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib.
A version of this article first appeared on Medscape.com.
Updated recommendations released on COVID-19 and pediatric ALL
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
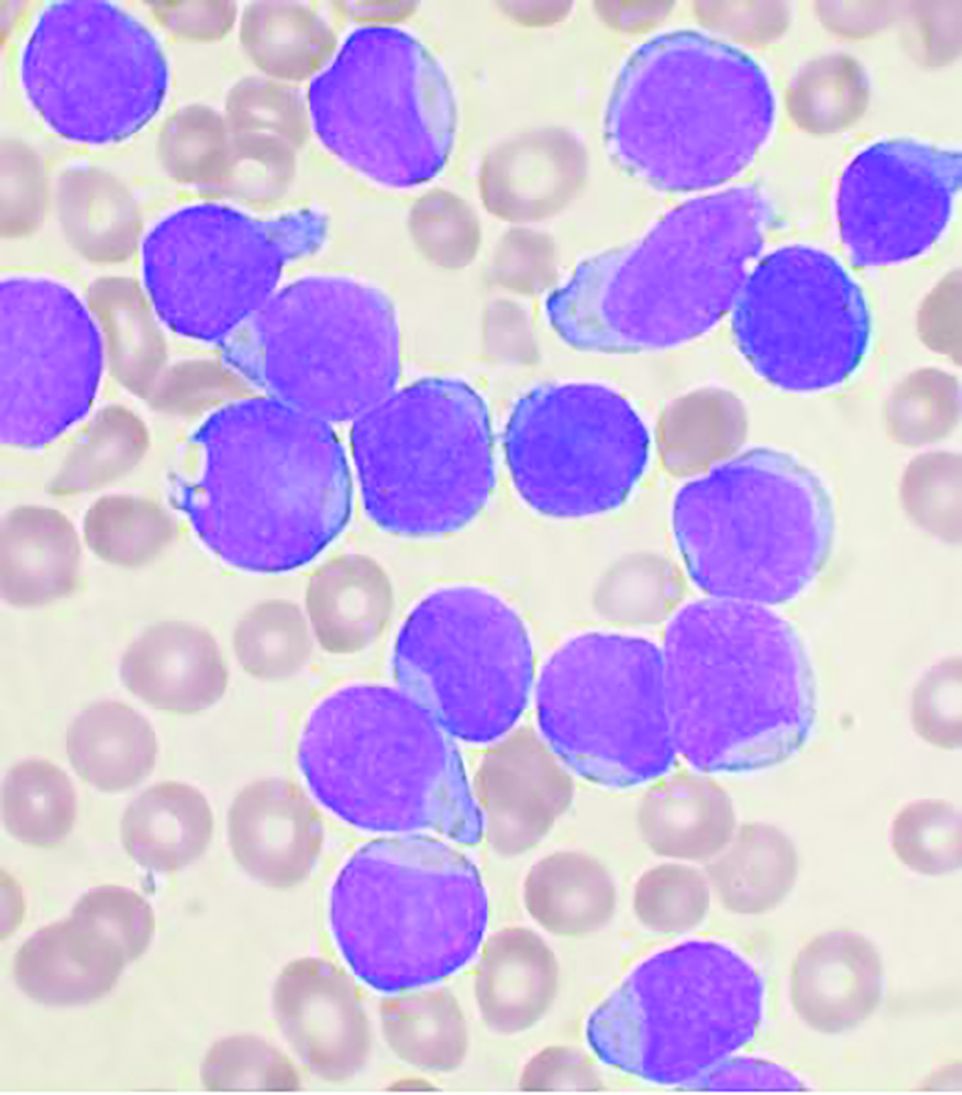
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
FROM BULLETIN DU CANCER
Type 2 diabetes linked to increased risk for Parkinson’s
New analyses of both observational and genetic data have provided “convincing evidence” that type 2 diabetes is associated with an increased risk for Parkinson’s disease.

“The fact that we see the same effects in both types of analysis separately makes it more likely that these results are real – that type 2 diabetes really is a driver of Parkinson’s disease risk,” Alastair Noyce, PhD, senior author of the new studies, said in an interview.
The two analyses are reported in one paper published online March 8 in the journal Movement Disorders.
Dr. Noyce, clinical senior lecturer in the preventive neurology unit at the Wolfson Institute of Preventive Medicine, Queen Mary University of London, explained that his group is interested in risk factors for Parkinson’s disease, particularly those relevant at the population level and which might be modifiable.
“Several studies have looked at diabetes as a risk factor for Parkinson’s but very few have focused on type 2 diabetes, and, as this is such a growing health issue, we wanted to look at that in more detail,” he said.
The researchers performed two different analyses: a meta-analysis of observational studies investigating an association between type 2 diabetes and Parkinson’s; and a separate Mendelian randomization analysis of genetic data on the two conditions.
They found similar results in both studies, with the observational data suggesting type 2 diabetes was associated with a 21% increased risk for Parkinson’s disease and the genetic data suggesting an 8% increased risk. There were also hints that type 2 diabetes might also be associated with faster progression of Parkinson’s symptoms.
“I don’t think type 2 diabetes is a major cause of Parkinson’s, but it probably makes some contribution and may increase the risk of a more aggressive form of the condition,” Dr. Noyce said.
“I would say the increased risk of Parkinson’s disease attributable to type 2 diabetes may be similar to that of head injury or pesticide exposure, but it is important, as type 2 diabetes is very prevalent and is increasing,” he added. “As we see the growth in type 2 diabetes, this could lead to a later increase in Parkinson’s, which is already one of the fastest-growing diseases worldwide.”
For the meta-analysis of observational data, the researchers included nine studies that investigated preceding type 2 diabetes specifically and its effect on the risk for Parkinson’s disease and progression.
The pooled effect estimates showed that type 2 diabetes was associated with an increased risk for Parkinson’s disease (odds ratio, 1.21; 95% confidence interval, 1.07-1.36), and there was some evidence that type 2 diabetes was associated with faster progression of motor symptoms (standardized mean difference [SMD], 0.55) and cognitive decline (SMD, −0.92).
The observational meta-analysis included seven cohort studies and two case-control studies, and these different types of studies showed different results in regard to the association between diabetes and Parkinson’s. While the cohort studies showed a detrimental effect of diabetes on Parkinson’s risk (OR, 1.29), the case-control studies suggested protective effect (OR, 0.51).
Addressing this, Dr. Noyce noted that the case-control studies may be less reliable as they suffered more from survivor bias. “Diabetes may cause deaths in mid-life before people go on to develop Parkinson’s, and this would cause a protective effect to be seen, but we believe this to be a spurious result. Cohort studies are generally more reliable and are less susceptible to survivor bias,” he said.
For the genetic analysis, the researchers combined results from two large publicly available genome-wide association studies – one for type 2 diabetes and one for Parkinson’s disease to assess whether individuals with a genetic tendency to type 2 diabetes had a higher risk of developing Parkinson’s.
Results showed an increased risk for Parkinson’s in those individuals with genetic variants associated with type 2 diabetes, with an odds ratio of 1.08 (P = .010). There was also some evidence of an effect on motor progression (OR, 1.10; P = .032) but not on cognitive progression.
On the possible mechanism behind this observation, Dr. Noyce noted type 2 diabetes and Parkinson’s have some similarities in biology, including abnormal protein aggregation.
In the study, the authors also suggest that circulating insulin may have a neuroprotective role, whereas systemic and local insulin resistance can influence pathways known to be important in Parkinson’s pathogenesis, including those that relate to mitochondrial dysfunction, neuroinflammation, synaptic plasticity, and mitochondrial dysfunction.
Dr. Noyce further pointed out that several drugs used for the treatment of type 2 diabetes have been repurposed as possible treatments for Parkinson’s disease and are now being tested for this new indication. “Our results support that approach and raise the idea that some of these drugs may even prevent Parkinson’s in people at risk,” he said.
Most people who have type 2 diabetes won’t get Parkinson’s disease, he added. Other outcomes such as heart disease, kidney disease, and microvascular complications are far more likely, and the main aim of preventing and treating type 2 diabetes is to prevent these far more common outcomes. “But our data suggests that this could also have a possible benefit in reducing future Parkinson’s risk,” he said.
Not on the horizon at present is the possibility of screening patients with type 2 diabetes for signs of early Parkinson’s, Dr. Noyce said.
“There isn’t a test for identifying presymptomatic neurodegenerative diseases such as Parkinson’s yet, but perhaps in the future there will be, and type 2 diabetes may be one risk factor to take into account when considering such screening,” he added.
This work was financially supported by grants from The Michael J. Fox Foundation; the Canadian Consortium on Neurodegeneration in Aging (CCNA); the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) initiative; and Parkinson Canada, and the Intramural Research Program of the NIH, National Institute on Aging.
Dr. Noyce reports grants from the Barts Charity, Parkinson’s UK, Aligning Science Across Parkinson’s and Michael J. Fox Foundation, and the Virginia Keiley Benefaction; and personal fees/honoraria from Britannia, BIAL, AbbVie, Global Kinetics Corporation, Profile, Biogen, Roche, and UCB outside of the submitted work.
A version of this article first appeared on Medscape.com.
New analyses of both observational and genetic data have provided “convincing evidence” that type 2 diabetes is associated with an increased risk for Parkinson’s disease.
“The fact that we see the same effects in both types of analysis separately makes it more likely that these results are real – that type 2 diabetes really is a driver of Parkinson’s disease risk,” Alastair Noyce, PhD, senior author of the new studies, said in an interview.
The two analyses are reported in one paper published online March 8 in the journal Movement Disorders.
Dr. Noyce, clinical senior lecturer in the preventive neurology unit at the Wolfson Institute of Preventive Medicine, Queen Mary University of London, explained that his group is interested in risk factors for Parkinson’s disease, particularly those relevant at the population level and which might be modifiable.
“Several studies have looked at diabetes as a risk factor for Parkinson’s but very few have focused on type 2 diabetes, and, as this is such a growing health issue, we wanted to look at that in more detail,” he said.
The researchers performed two different analyses: a meta-analysis of observational studies investigating an association between type 2 diabetes and Parkinson’s; and a separate Mendelian randomization analysis of genetic data on the two conditions.
They found similar results in both studies, with the observational data suggesting type 2 diabetes was associated with a 21% increased risk for Parkinson’s disease and the genetic data suggesting an 8% increased risk. There were also hints that type 2 diabetes might also be associated with faster progression of Parkinson’s symptoms.
“I don’t think type 2 diabetes is a major cause of Parkinson’s, but it probably makes some contribution and may increase the risk of a more aggressive form of the condition,” Dr. Noyce said.
“I would say the increased risk of Parkinson’s disease attributable to type 2 diabetes may be similar to that of head injury or pesticide exposure, but it is important, as type 2 diabetes is very prevalent and is increasing,” he added. “As we see the growth in type 2 diabetes, this could lead to a later increase in Parkinson’s, which is already one of the fastest-growing diseases worldwide.”
For the meta-analysis of observational data, the researchers included nine studies that investigated preceding type 2 diabetes specifically and its effect on the risk for Parkinson’s disease and progression.
The pooled effect estimates showed that type 2 diabetes was associated with an increased risk for Parkinson’s disease (odds ratio, 1.21; 95% confidence interval, 1.07-1.36), and there was some evidence that type 2 diabetes was associated with faster progression of motor symptoms (standardized mean difference [SMD], 0.55) and cognitive decline (SMD, −0.92).
The observational meta-analysis included seven cohort studies and two case-control studies, and these different types of studies showed different results in regard to the association between diabetes and Parkinson’s. While the cohort studies showed a detrimental effect of diabetes on Parkinson’s risk (OR, 1.29), the case-control studies suggested protective effect (OR, 0.51).
Addressing this, Dr. Noyce noted that the case-control studies may be less reliable as they suffered more from survivor bias. “Diabetes may cause deaths in mid-life before people go on to develop Parkinson’s, and this would cause a protective effect to be seen, but we believe this to be a spurious result. Cohort studies are generally more reliable and are less susceptible to survivor bias,” he said.
For the genetic analysis, the researchers combined results from two large publicly available genome-wide association studies – one for type 2 diabetes and one for Parkinson’s disease to assess whether individuals with a genetic tendency to type 2 diabetes had a higher risk of developing Parkinson’s.
Results showed an increased risk for Parkinson’s in those individuals with genetic variants associated with type 2 diabetes, with an odds ratio of 1.08 (P = .010). There was also some evidence of an effect on motor progression (OR, 1.10; P = .032) but not on cognitive progression.
On the possible mechanism behind this observation, Dr. Noyce noted type 2 diabetes and Parkinson’s have some similarities in biology, including abnormal protein aggregation.
In the study, the authors also suggest that circulating insulin may have a neuroprotective role, whereas systemic and local insulin resistance can influence pathways known to be important in Parkinson’s pathogenesis, including those that relate to mitochondrial dysfunction, neuroinflammation, synaptic plasticity, and mitochondrial dysfunction.
Dr. Noyce further pointed out that several drugs used for the treatment of type 2 diabetes have been repurposed as possible treatments for Parkinson’s disease and are now being tested for this new indication. “Our results support that approach and raise the idea that some of these drugs may even prevent Parkinson’s in people at risk,” he said.
Most people who have type 2 diabetes won’t get Parkinson’s disease, he added. Other outcomes such as heart disease, kidney disease, and microvascular complications are far more likely, and the main aim of preventing and treating type 2 diabetes is to prevent these far more common outcomes. “But our data suggests that this could also have a possible benefit in reducing future Parkinson’s risk,” he said.
Not on the horizon at present is the possibility of screening patients with type 2 diabetes for signs of early Parkinson’s, Dr. Noyce said.
“There isn’t a test for identifying presymptomatic neurodegenerative diseases such as Parkinson’s yet, but perhaps in the future there will be, and type 2 diabetes may be one risk factor to take into account when considering such screening,” he added.
This work was financially supported by grants from The Michael J. Fox Foundation; the Canadian Consortium on Neurodegeneration in Aging (CCNA); the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) initiative; and Parkinson Canada, and the Intramural Research Program of the NIH, National Institute on Aging.
Dr. Noyce reports grants from the Barts Charity, Parkinson’s UK, Aligning Science Across Parkinson’s and Michael J. Fox Foundation, and the Virginia Keiley Benefaction; and personal fees/honoraria from Britannia, BIAL, AbbVie, Global Kinetics Corporation, Profile, Biogen, Roche, and UCB outside of the submitted work.
A version of this article first appeared on Medscape.com.
New analyses of both observational and genetic data have provided “convincing evidence” that type 2 diabetes is associated with an increased risk for Parkinson’s disease.
“The fact that we see the same effects in both types of analysis separately makes it more likely that these results are real – that type 2 diabetes really is a driver of Parkinson’s disease risk,” Alastair Noyce, PhD, senior author of the new studies, said in an interview.
The two analyses are reported in one paper published online March 8 in the journal Movement Disorders.
Dr. Noyce, clinical senior lecturer in the preventive neurology unit at the Wolfson Institute of Preventive Medicine, Queen Mary University of London, explained that his group is interested in risk factors for Parkinson’s disease, particularly those relevant at the population level and which might be modifiable.
“Several studies have looked at diabetes as a risk factor for Parkinson’s but very few have focused on type 2 diabetes, and, as this is such a growing health issue, we wanted to look at that in more detail,” he said.
The researchers performed two different analyses: a meta-analysis of observational studies investigating an association between type 2 diabetes and Parkinson’s; and a separate Mendelian randomization analysis of genetic data on the two conditions.
They found similar results in both studies, with the observational data suggesting type 2 diabetes was associated with a 21% increased risk for Parkinson’s disease and the genetic data suggesting an 8% increased risk. There were also hints that type 2 diabetes might also be associated with faster progression of Parkinson’s symptoms.
“I don’t think type 2 diabetes is a major cause of Parkinson’s, but it probably makes some contribution and may increase the risk of a more aggressive form of the condition,” Dr. Noyce said.
“I would say the increased risk of Parkinson’s disease attributable to type 2 diabetes may be similar to that of head injury or pesticide exposure, but it is important, as type 2 diabetes is very prevalent and is increasing,” he added. “As we see the growth in type 2 diabetes, this could lead to a later increase in Parkinson’s, which is already one of the fastest-growing diseases worldwide.”
For the meta-analysis of observational data, the researchers included nine studies that investigated preceding type 2 diabetes specifically and its effect on the risk for Parkinson’s disease and progression.
The pooled effect estimates showed that type 2 diabetes was associated with an increased risk for Parkinson’s disease (odds ratio, 1.21; 95% confidence interval, 1.07-1.36), and there was some evidence that type 2 diabetes was associated with faster progression of motor symptoms (standardized mean difference [SMD], 0.55) and cognitive decline (SMD, −0.92).
The observational meta-analysis included seven cohort studies and two case-control studies, and these different types of studies showed different results in regard to the association between diabetes and Parkinson’s. While the cohort studies showed a detrimental effect of diabetes on Parkinson’s risk (OR, 1.29), the case-control studies suggested protective effect (OR, 0.51).
Addressing this, Dr. Noyce noted that the case-control studies may be less reliable as they suffered more from survivor bias. “Diabetes may cause deaths in mid-life before people go on to develop Parkinson’s, and this would cause a protective effect to be seen, but we believe this to be a spurious result. Cohort studies are generally more reliable and are less susceptible to survivor bias,” he said.
For the genetic analysis, the researchers combined results from two large publicly available genome-wide association studies – one for type 2 diabetes and one for Parkinson’s disease to assess whether individuals with a genetic tendency to type 2 diabetes had a higher risk of developing Parkinson’s.
Results showed an increased risk for Parkinson’s in those individuals with genetic variants associated with type 2 diabetes, with an odds ratio of 1.08 (P = .010). There was also some evidence of an effect on motor progression (OR, 1.10; P = .032) but not on cognitive progression.
On the possible mechanism behind this observation, Dr. Noyce noted type 2 diabetes and Parkinson’s have some similarities in biology, including abnormal protein aggregation.
In the study, the authors also suggest that circulating insulin may have a neuroprotective role, whereas systemic and local insulin resistance can influence pathways known to be important in Parkinson’s pathogenesis, including those that relate to mitochondrial dysfunction, neuroinflammation, synaptic plasticity, and mitochondrial dysfunction.
Dr. Noyce further pointed out that several drugs used for the treatment of type 2 diabetes have been repurposed as possible treatments for Parkinson’s disease and are now being tested for this new indication. “Our results support that approach and raise the idea that some of these drugs may even prevent Parkinson’s in people at risk,” he said.
Most people who have type 2 diabetes won’t get Parkinson’s disease, he added. Other outcomes such as heart disease, kidney disease, and microvascular complications are far more likely, and the main aim of preventing and treating type 2 diabetes is to prevent these far more common outcomes. “But our data suggests that this could also have a possible benefit in reducing future Parkinson’s risk,” he said.
Not on the horizon at present is the possibility of screening patients with type 2 diabetes for signs of early Parkinson’s, Dr. Noyce said.
“There isn’t a test for identifying presymptomatic neurodegenerative diseases such as Parkinson’s yet, but perhaps in the future there will be, and type 2 diabetes may be one risk factor to take into account when considering such screening,” he added.
This work was financially supported by grants from The Michael J. Fox Foundation; the Canadian Consortium on Neurodegeneration in Aging (CCNA); the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) initiative; and Parkinson Canada, and the Intramural Research Program of the NIH, National Institute on Aging.
Dr. Noyce reports grants from the Barts Charity, Parkinson’s UK, Aligning Science Across Parkinson’s and Michael J. Fox Foundation, and the Virginia Keiley Benefaction; and personal fees/honoraria from Britannia, BIAL, AbbVie, Global Kinetics Corporation, Profile, Biogen, Roche, and UCB outside of the submitted work.
A version of this article first appeared on Medscape.com.
Some with long COVID see relief after vaccination
Several weeks after getting his second dose of an mRNA vaccine, Aaron Goyang thinks his long bout with COVID-19 has finally come to an end.
Mr. Goyang, who is 33 and is a radiology technician in Austin, Tex., thinks he got COVID-19 from some of the coughing, gasping patients he treated last spring.
At the time, testing was scarce, and by the time he was tested – several weeks into his illness – it came back negative. He fought off the initial symptoms but experienced relapse a week later.
Mr. Goyang says that, for the next 8 or 9 months, he was on a roller coaster with extreme shortness of breath and chest tightness that could be so severe it would send him to the emergency department. He had to use an inhaler to get through his workdays.
“Even if I was just sitting around, it would come and take me,” he says. “It almost felt like someone was bear-hugging me constantly, and I just couldn’t get in a good enough breath.”
On his best days, he would walk around his neighborhood, being careful not to overdo it. He tried running once, and it nearly sent him to the hospital.
“Very honestly, I didn’t know if I would ever be able to do it again,” he says.
But Mr. Goyang says that, several weeks after getting the Pfizer vaccine, he was able to run a mile again with no problems. “I was very thankful for that,” he says.
Mr. Goyang is not alone. Some social media groups are dedicated to patients who are living with a condition that’s been known as long COVID and that was recently termed postacute sequelae of SARS-CoV-2 infection (PASC). These patients are sometimes referred to as long haulers.
On social media, patients with PASC are eagerly and anxiously quizzing each other about the vaccines and their effects.
Survivor Corps, which has a public Facebook group with 159,000 members, recently took a poll to see whether there was any substance to rumors that those with long COVID were feeling better after being vaccinated.
“Out of 400 people, 36% showed an improvement in symptoms, anywhere between a mild improvement to complete resolution of symptoms,” said Diana Berrent, a long-COVID patient who founded the group. Survivor Corps has become active in patient advocacy and is a resource for researchers studying the new condition.
Ms. Berrent has become such a trusted voice during the pandemic. She interviewed Anthony Fauci, MD, head of the National Institutes of Allergy and Infectious Diseases, last October.
“The implications are huge,” she says.
“Some of this damage is permanent damage. It’s not going to cure the scarring of your heart tissue, it’s not going to cure the irreparable damage to your lungs, but if it’s making people feel better, then that’s an indication there’s viral persistence going on,” says Ms. Berrent.
“I’ve been saying for months and months, we shouldn’t be calling this postacute anything,” she adds.
Patients report improvement
Daniel Griffin, MD, PhD, is equally excited. He’s an infectious disease specialist at Columbia University, New York. He says about one in five patients he treated for COVID-19 last year never got better. Many of them, such as Mr. Goyang, were health care workers.
“I don’t know if people actually catch this, but a lot of our coworkers are either permanently disabled or died,” Dr. Griffin says.
Health care workers were also among the first to be vaccinated. Dr. Griffin says many of his patients began reaching out to him about a week or two after being vaccinated “and saying, ‘You know, I actually feel better.’ And some of them were saying, ‘I feel all better,’ after being sick – a lot of them – for a year.”
Then he was getting calls and texts from other doctors, asking, “Hey, are you seeing this?”
The benefits of vaccination for some long-haulers came as a surprise. Dr. Griffin says that, before the vaccines came out, many of his patients were worried that getting vaccinated might overstimulate their immune systems and cause symptoms to get worse.
Indeed, a small percentage of people – about 3%-5%, based on informal polls on social media – report that they do experience worsening of symptoms after getting the shot. It’s not clear why.
Dr. Griffin estimates that between 30% and 50% of patients’ symptoms improve after they receive the mRNA vaccines. “I’m seeing this chunk of people – they tell me their brain fog has improved, their fatigue is gone, the fevers that wouldn’t resolve have now gone,” he says. “I’m seeing that personally, and I’m hearing it from my colleagues.”
Dr. Griffin says the observation has launched several studies and that there are several theories about how the vaccines might be affecting long COVID.
An immune system boost?
One possibility is that the virus continues to stimulate the immune system, which continues to fight the virus for months. If that is the case, Dr. Griffin says, the vaccine may be giving the immune system the boost it needs to finally clear the virus away.
Donna Farber, PhD, a professor of microbiology and immunology at Columbia University, has heard the stories, too.
“It is possible that the persisting virus in long COVID-19 may be at a low level – not enough to stimulate a potent immune response to clear the virus, but enough to cause symptoms. Activating the immune response therefore is therapeutic in directing viral clearance,” she says.
Dr. Farber explains that long COVID may be a bit like Lyme disease. Some patients with Lyme disease must take antibiotics for months before their symptoms disappear.
Dr. Griffin says there’s another possibility. Several studies have now shown that people with lingering COVID-19 symptoms develop autoantibodies. There’s a theory that SARS-CoV-2 may create an autoimmune condition that leads to long-term symptoms.
If that is the case, Dr. Griffin says, the vaccine may be helping the body to reset its tolerance to itself, “so maybe now you’re getting a healthy immune response.”
More studies are needed to know for sure.
Either way, the vaccines are a much-needed bit of hope for the long-COVID community, and Dr. Griffin tells his patients who are still worried that, at the very least, they’ll be protected from another SARS-CoV-2 infection.
A version of this article first appeared on Medscape.com.
Several weeks after getting his second dose of an mRNA vaccine, Aaron Goyang thinks his long bout with COVID-19 has finally come to an end.
Mr. Goyang, who is 33 and is a radiology technician in Austin, Tex., thinks he got COVID-19 from some of the coughing, gasping patients he treated last spring.
At the time, testing was scarce, and by the time he was tested – several weeks into his illness – it came back negative. He fought off the initial symptoms but experienced relapse a week later.
Mr. Goyang says that, for the next 8 or 9 months, he was on a roller coaster with extreme shortness of breath and chest tightness that could be so severe it would send him to the emergency department. He had to use an inhaler to get through his workdays.
“Even if I was just sitting around, it would come and take me,” he says. “It almost felt like someone was bear-hugging me constantly, and I just couldn’t get in a good enough breath.”
On his best days, he would walk around his neighborhood, being careful not to overdo it. He tried running once, and it nearly sent him to the hospital.
“Very honestly, I didn’t know if I would ever be able to do it again,” he says.
But Mr. Goyang says that, several weeks after getting the Pfizer vaccine, he was able to run a mile again with no problems. “I was very thankful for that,” he says.
Mr. Goyang is not alone. Some social media groups are dedicated to patients who are living with a condition that’s been known as long COVID and that was recently termed postacute sequelae of SARS-CoV-2 infection (PASC). These patients are sometimes referred to as long haulers.
On social media, patients with PASC are eagerly and anxiously quizzing each other about the vaccines and their effects.
Survivor Corps, which has a public Facebook group with 159,000 members, recently took a poll to see whether there was any substance to rumors that those with long COVID were feeling better after being vaccinated.
“Out of 400 people, 36% showed an improvement in symptoms, anywhere between a mild improvement to complete resolution of symptoms,” said Diana Berrent, a long-COVID patient who founded the group. Survivor Corps has become active in patient advocacy and is a resource for researchers studying the new condition.
Ms. Berrent has become such a trusted voice during the pandemic. She interviewed Anthony Fauci, MD, head of the National Institutes of Allergy and Infectious Diseases, last October.
“The implications are huge,” she says.
“Some of this damage is permanent damage. It’s not going to cure the scarring of your heart tissue, it’s not going to cure the irreparable damage to your lungs, but if it’s making people feel better, then that’s an indication there’s viral persistence going on,” says Ms. Berrent.
“I’ve been saying for months and months, we shouldn’t be calling this postacute anything,” she adds.
Patients report improvement
Daniel Griffin, MD, PhD, is equally excited. He’s an infectious disease specialist at Columbia University, New York. He says about one in five patients he treated for COVID-19 last year never got better. Many of them, such as Mr. Goyang, were health care workers.
“I don’t know if people actually catch this, but a lot of our coworkers are either permanently disabled or died,” Dr. Griffin says.
Health care workers were also among the first to be vaccinated. Dr. Griffin says many of his patients began reaching out to him about a week or two after being vaccinated “and saying, ‘You know, I actually feel better.’ And some of them were saying, ‘I feel all better,’ after being sick – a lot of them – for a year.”
Then he was getting calls and texts from other doctors, asking, “Hey, are you seeing this?”
The benefits of vaccination for some long-haulers came as a surprise. Dr. Griffin says that, before the vaccines came out, many of his patients were worried that getting vaccinated might overstimulate their immune systems and cause symptoms to get worse.
Indeed, a small percentage of people – about 3%-5%, based on informal polls on social media – report that they do experience worsening of symptoms after getting the shot. It’s not clear why.
Dr. Griffin estimates that between 30% and 50% of patients’ symptoms improve after they receive the mRNA vaccines. “I’m seeing this chunk of people – they tell me their brain fog has improved, their fatigue is gone, the fevers that wouldn’t resolve have now gone,” he says. “I’m seeing that personally, and I’m hearing it from my colleagues.”
Dr. Griffin says the observation has launched several studies and that there are several theories about how the vaccines might be affecting long COVID.
An immune system boost?
One possibility is that the virus continues to stimulate the immune system, which continues to fight the virus for months. If that is the case, Dr. Griffin says, the vaccine may be giving the immune system the boost it needs to finally clear the virus away.
Donna Farber, PhD, a professor of microbiology and immunology at Columbia University, has heard the stories, too.
“It is possible that the persisting virus in long COVID-19 may be at a low level – not enough to stimulate a potent immune response to clear the virus, but enough to cause symptoms. Activating the immune response therefore is therapeutic in directing viral clearance,” she says.
Dr. Farber explains that long COVID may be a bit like Lyme disease. Some patients with Lyme disease must take antibiotics for months before their symptoms disappear.
Dr. Griffin says there’s another possibility. Several studies have now shown that people with lingering COVID-19 symptoms develop autoantibodies. There’s a theory that SARS-CoV-2 may create an autoimmune condition that leads to long-term symptoms.
If that is the case, Dr. Griffin says, the vaccine may be helping the body to reset its tolerance to itself, “so maybe now you’re getting a healthy immune response.”
More studies are needed to know for sure.
Either way, the vaccines are a much-needed bit of hope for the long-COVID community, and Dr. Griffin tells his patients who are still worried that, at the very least, they’ll be protected from another SARS-CoV-2 infection.
A version of this article first appeared on Medscape.com.
Several weeks after getting his second dose of an mRNA vaccine, Aaron Goyang thinks his long bout with COVID-19 has finally come to an end.
Mr. Goyang, who is 33 and is a radiology technician in Austin, Tex., thinks he got COVID-19 from some of the coughing, gasping patients he treated last spring.
At the time, testing was scarce, and by the time he was tested – several weeks into his illness – it came back negative. He fought off the initial symptoms but experienced relapse a week later.
Mr. Goyang says that, for the next 8 or 9 months, he was on a roller coaster with extreme shortness of breath and chest tightness that could be so severe it would send him to the emergency department. He had to use an inhaler to get through his workdays.
“Even if I was just sitting around, it would come and take me,” he says. “It almost felt like someone was bear-hugging me constantly, and I just couldn’t get in a good enough breath.”
On his best days, he would walk around his neighborhood, being careful not to overdo it. He tried running once, and it nearly sent him to the hospital.
“Very honestly, I didn’t know if I would ever be able to do it again,” he says.
But Mr. Goyang says that, several weeks after getting the Pfizer vaccine, he was able to run a mile again with no problems. “I was very thankful for that,” he says.
Mr. Goyang is not alone. Some social media groups are dedicated to patients who are living with a condition that’s been known as long COVID and that was recently termed postacute sequelae of SARS-CoV-2 infection (PASC). These patients are sometimes referred to as long haulers.
On social media, patients with PASC are eagerly and anxiously quizzing each other about the vaccines and their effects.
Survivor Corps, which has a public Facebook group with 159,000 members, recently took a poll to see whether there was any substance to rumors that those with long COVID were feeling better after being vaccinated.
“Out of 400 people, 36% showed an improvement in symptoms, anywhere between a mild improvement to complete resolution of symptoms,” said Diana Berrent, a long-COVID patient who founded the group. Survivor Corps has become active in patient advocacy and is a resource for researchers studying the new condition.
Ms. Berrent has become such a trusted voice during the pandemic. She interviewed Anthony Fauci, MD, head of the National Institutes of Allergy and Infectious Diseases, last October.
“The implications are huge,” she says.
“Some of this damage is permanent damage. It’s not going to cure the scarring of your heart tissue, it’s not going to cure the irreparable damage to your lungs, but if it’s making people feel better, then that’s an indication there’s viral persistence going on,” says Ms. Berrent.
“I’ve been saying for months and months, we shouldn’t be calling this postacute anything,” she adds.
Patients report improvement
Daniel Griffin, MD, PhD, is equally excited. He’s an infectious disease specialist at Columbia University, New York. He says about one in five patients he treated for COVID-19 last year never got better. Many of them, such as Mr. Goyang, were health care workers.
“I don’t know if people actually catch this, but a lot of our coworkers are either permanently disabled or died,” Dr. Griffin says.
Health care workers were also among the first to be vaccinated. Dr. Griffin says many of his patients began reaching out to him about a week or two after being vaccinated “and saying, ‘You know, I actually feel better.’ And some of them were saying, ‘I feel all better,’ after being sick – a lot of them – for a year.”
Then he was getting calls and texts from other doctors, asking, “Hey, are you seeing this?”
The benefits of vaccination for some long-haulers came as a surprise. Dr. Griffin says that, before the vaccines came out, many of his patients were worried that getting vaccinated might overstimulate their immune systems and cause symptoms to get worse.
Indeed, a small percentage of people – about 3%-5%, based on informal polls on social media – report that they do experience worsening of symptoms after getting the shot. It’s not clear why.
Dr. Griffin estimates that between 30% and 50% of patients’ symptoms improve after they receive the mRNA vaccines. “I’m seeing this chunk of people – they tell me their brain fog has improved, their fatigue is gone, the fevers that wouldn’t resolve have now gone,” he says. “I’m seeing that personally, and I’m hearing it from my colleagues.”
Dr. Griffin says the observation has launched several studies and that there are several theories about how the vaccines might be affecting long COVID.
An immune system boost?
One possibility is that the virus continues to stimulate the immune system, which continues to fight the virus for months. If that is the case, Dr. Griffin says, the vaccine may be giving the immune system the boost it needs to finally clear the virus away.
Donna Farber, PhD, a professor of microbiology and immunology at Columbia University, has heard the stories, too.
“It is possible that the persisting virus in long COVID-19 may be at a low level – not enough to stimulate a potent immune response to clear the virus, but enough to cause symptoms. Activating the immune response therefore is therapeutic in directing viral clearance,” she says.
Dr. Farber explains that long COVID may be a bit like Lyme disease. Some patients with Lyme disease must take antibiotics for months before their symptoms disappear.
Dr. Griffin says there’s another possibility. Several studies have now shown that people with lingering COVID-19 symptoms develop autoantibodies. There’s a theory that SARS-CoV-2 may create an autoimmune condition that leads to long-term symptoms.
If that is the case, Dr. Griffin says, the vaccine may be helping the body to reset its tolerance to itself, “so maybe now you’re getting a healthy immune response.”
More studies are needed to know for sure.
Either way, the vaccines are a much-needed bit of hope for the long-COVID community, and Dr. Griffin tells his patients who are still worried that, at the very least, they’ll be protected from another SARS-CoV-2 infection.
A version of this article first appeared on Medscape.com.
Colchicine before PCI for acute MI fails to improve major outcomes
In a placebo-controlled randomized trial, a preprocedural dose of colchicine administered immediately before percutaneous coronary intervention (PCI) for an acute ST-segment elevated myocardial infarction (STEMI) did not reduce the no-reflow phenomenon or improve outcomes.
No-reflow, in which insufficient myocardial perfusion is present even though the coronary artery appears patent, was the primary outcome, and the proportion of patients experiencing this event was exactly the same (14.4%) in the colchicine and placebo groups, reported Yaser Jenab, MD, at CRT 2021 sponsored by MedStar Heart & Vascular Institute.
The hypothesis that colchicine would offer benefit in this setting was largely based on the Colchicine Cardiovascular Outcomes Trial (COLCOT). In that study, colchicine was associated with a 23% reduction in risk for major adverse cardiovascular events (MACE) relative to placebo when administered within 30 days after a myocardial infarction (hazard ratio, 0.77; P = .02).
The benefit in that trial was attributed to an anti-inflammatory effect, according to Dr. Jenab, associate professor of cardiology at Tehran (Iran) Heart Center. In particular as it relates to vascular disease, he cited experimental studies associating colchicine with a reduction in neutrophil activation and adherence to vascular endothelium.
The rationale for a preprocedural approach to colchicine was supplied by a subsequent time-to-treatment COLCOT analysis. In this study, MACE risk reduction for colchicine climbed to 48% (HR 0.52) for those treated within 3 days of the MI but largely disappeared (HR 0.96) if treatment was started at least 8 days post MI.
PodCAST-PCI trial
In the preprocedural study, called the PodCAST-PCI trial, 321 acute STEMI patients were randomized. Patients received a 1-mg dose of oral colchicine or placebo at the time PCI was scheduled. Another dose of colchicine (0.5 mg) or placebo was administered 1 hour after the procedure.
Of secondary outcomes, which included MACE at 1 month and 1 year, ST-segment resolution at 1 month, and change in inflammatory markers at 1 month, none were significant. Few even trended for significance.
For MACE, which included cardiac death, stroke, nonfatal MI, new hospitalization due to heart failure, or target vessel revascularization, the rates were lower in the colchicine group at 1 month (4.3% vs. 7.5%) and 1 year (9.3% vs. 11.2%), but neither approached significance.
For ST-segment resolution, the proportions were generally comparable among the colchicine and placebo groups, respectively, for the proportion below 50% (18.6% vs. 23.1%), between 50% and 70% (16.8% vs. 15.6%), and above 70% (64.6% vs. 61.3%).
The average troponin levels were nonsignificantly lower at 6 hours (1,847 vs. 2,883 ng/mL) in the colchicine group but higher at 48 hours (1,197 vs. 1,147 ng/mL). The average C-reactive protein (CRP) levels at 48 hours were nonsignificantly lower on colchicine (176.5 vs. 244.5 mg/L).
There were no significant differences in postprocedural perfusion, as measured with TIMI blood flow, or in the rate of stent thrombosis, which occurred in roughly 3% of each group of patients.
The small sample size was one limitation of this study, Dr. Jenab acknowledged. For this and other reasons, he cautioned that these data are not definitive and do not preclude a benefit on clinical outcomes in a study with a larger size, a different design, or different dosing.
Timing might be the issue
However, even if colchicine has a potential benefit in this setting, timing might be a major obstacle, according to Binata Shah, MD, associate director of research for the Cardiac Catheterization Laboratory at New York University.
“We have learned from our rheumatology colleagues that peak plasma levels of colchicine are not achieved for at least 1 hour after the full loading dose,” Dr. Shah said. “With us moving so quickly in a primary PCI setting, it is hard to imagine that colchicine would have had time to really kick in and exert its anti-inflammatory effect.”
Indeed, the problem might be worse than reaching the peak plasma level.
“Even though peak plasma levels occur as early as 1 hour after a full loading dose, we see that it takes about 24 hours to really see the effects translate downstream into more systemic inflammatory markers such as CRP and interleukin-6,” she added. If lowering these signals of inflammation is predictive of benefit, than this might be the biggest obstacle to benefit from colchicine in an urgent treatment setting.
Dr. Jenab and Dr. Shah reported no potential conflicts of interest.
In a placebo-controlled randomized trial, a preprocedural dose of colchicine administered immediately before percutaneous coronary intervention (PCI) for an acute ST-segment elevated myocardial infarction (STEMI) did not reduce the no-reflow phenomenon or improve outcomes.
No-reflow, in which insufficient myocardial perfusion is present even though the coronary artery appears patent, was the primary outcome, and the proportion of patients experiencing this event was exactly the same (14.4%) in the colchicine and placebo groups, reported Yaser Jenab, MD, at CRT 2021 sponsored by MedStar Heart & Vascular Institute.
The hypothesis that colchicine would offer benefit in this setting was largely based on the Colchicine Cardiovascular Outcomes Trial (COLCOT). In that study, colchicine was associated with a 23% reduction in risk for major adverse cardiovascular events (MACE) relative to placebo when administered within 30 days after a myocardial infarction (hazard ratio, 0.77; P = .02).
The benefit in that trial was attributed to an anti-inflammatory effect, according to Dr. Jenab, associate professor of cardiology at Tehran (Iran) Heart Center. In particular as it relates to vascular disease, he cited experimental studies associating colchicine with a reduction in neutrophil activation and adherence to vascular endothelium.
The rationale for a preprocedural approach to colchicine was supplied by a subsequent time-to-treatment COLCOT analysis. In this study, MACE risk reduction for colchicine climbed to 48% (HR 0.52) for those treated within 3 days of the MI but largely disappeared (HR 0.96) if treatment was started at least 8 days post MI.
PodCAST-PCI trial
In the preprocedural study, called the PodCAST-PCI trial, 321 acute STEMI patients were randomized. Patients received a 1-mg dose of oral colchicine or placebo at the time PCI was scheduled. Another dose of colchicine (0.5 mg) or placebo was administered 1 hour after the procedure.
Of secondary outcomes, which included MACE at 1 month and 1 year, ST-segment resolution at 1 month, and change in inflammatory markers at 1 month, none were significant. Few even trended for significance.
For MACE, which included cardiac death, stroke, nonfatal MI, new hospitalization due to heart failure, or target vessel revascularization, the rates were lower in the colchicine group at 1 month (4.3% vs. 7.5%) and 1 year (9.3% vs. 11.2%), but neither approached significance.
For ST-segment resolution, the proportions were generally comparable among the colchicine and placebo groups, respectively, for the proportion below 50% (18.6% vs. 23.1%), between 50% and 70% (16.8% vs. 15.6%), and above 70% (64.6% vs. 61.3%).
The average troponin levels were nonsignificantly lower at 6 hours (1,847 vs. 2,883 ng/mL) in the colchicine group but higher at 48 hours (1,197 vs. 1,147 ng/mL). The average C-reactive protein (CRP) levels at 48 hours were nonsignificantly lower on colchicine (176.5 vs. 244.5 mg/L).
There were no significant differences in postprocedural perfusion, as measured with TIMI blood flow, or in the rate of stent thrombosis, which occurred in roughly 3% of each group of patients.
The small sample size was one limitation of this study, Dr. Jenab acknowledged. For this and other reasons, he cautioned that these data are not definitive and do not preclude a benefit on clinical outcomes in a study with a larger size, a different design, or different dosing.
Timing might be the issue
However, even if colchicine has a potential benefit in this setting, timing might be a major obstacle, according to Binata Shah, MD, associate director of research for the Cardiac Catheterization Laboratory at New York University.
“We have learned from our rheumatology colleagues that peak plasma levels of colchicine are not achieved for at least 1 hour after the full loading dose,” Dr. Shah said. “With us moving so quickly in a primary PCI setting, it is hard to imagine that colchicine would have had time to really kick in and exert its anti-inflammatory effect.”
Indeed, the problem might be worse than reaching the peak plasma level.
“Even though peak plasma levels occur as early as 1 hour after a full loading dose, we see that it takes about 24 hours to really see the effects translate downstream into more systemic inflammatory markers such as CRP and interleukin-6,” she added. If lowering these signals of inflammation is predictive of benefit, than this might be the biggest obstacle to benefit from colchicine in an urgent treatment setting.
Dr. Jenab and Dr. Shah reported no potential conflicts of interest.
In a placebo-controlled randomized trial, a preprocedural dose of colchicine administered immediately before percutaneous coronary intervention (PCI) for an acute ST-segment elevated myocardial infarction (STEMI) did not reduce the no-reflow phenomenon or improve outcomes.
No-reflow, in which insufficient myocardial perfusion is present even though the coronary artery appears patent, was the primary outcome, and the proportion of patients experiencing this event was exactly the same (14.4%) in the colchicine and placebo groups, reported Yaser Jenab, MD, at CRT 2021 sponsored by MedStar Heart & Vascular Institute.
The hypothesis that colchicine would offer benefit in this setting was largely based on the Colchicine Cardiovascular Outcomes Trial (COLCOT). In that study, colchicine was associated with a 23% reduction in risk for major adverse cardiovascular events (MACE) relative to placebo when administered within 30 days after a myocardial infarction (hazard ratio, 0.77; P = .02).
The benefit in that trial was attributed to an anti-inflammatory effect, according to Dr. Jenab, associate professor of cardiology at Tehran (Iran) Heart Center. In particular as it relates to vascular disease, he cited experimental studies associating colchicine with a reduction in neutrophil activation and adherence to vascular endothelium.
The rationale for a preprocedural approach to colchicine was supplied by a subsequent time-to-treatment COLCOT analysis. In this study, MACE risk reduction for colchicine climbed to 48% (HR 0.52) for those treated within 3 days of the MI but largely disappeared (HR 0.96) if treatment was started at least 8 days post MI.
PodCAST-PCI trial
In the preprocedural study, called the PodCAST-PCI trial, 321 acute STEMI patients were randomized. Patients received a 1-mg dose of oral colchicine or placebo at the time PCI was scheduled. Another dose of colchicine (0.5 mg) or placebo was administered 1 hour after the procedure.
Of secondary outcomes, which included MACE at 1 month and 1 year, ST-segment resolution at 1 month, and change in inflammatory markers at 1 month, none were significant. Few even trended for significance.
For MACE, which included cardiac death, stroke, nonfatal MI, new hospitalization due to heart failure, or target vessel revascularization, the rates were lower in the colchicine group at 1 month (4.3% vs. 7.5%) and 1 year (9.3% vs. 11.2%), but neither approached significance.
For ST-segment resolution, the proportions were generally comparable among the colchicine and placebo groups, respectively, for the proportion below 50% (18.6% vs. 23.1%), between 50% and 70% (16.8% vs. 15.6%), and above 70% (64.6% vs. 61.3%).
The average troponin levels were nonsignificantly lower at 6 hours (1,847 vs. 2,883 ng/mL) in the colchicine group but higher at 48 hours (1,197 vs. 1,147 ng/mL). The average C-reactive protein (CRP) levels at 48 hours were nonsignificantly lower on colchicine (176.5 vs. 244.5 mg/L).
There were no significant differences in postprocedural perfusion, as measured with TIMI blood flow, or in the rate of stent thrombosis, which occurred in roughly 3% of each group of patients.
The small sample size was one limitation of this study, Dr. Jenab acknowledged. For this and other reasons, he cautioned that these data are not definitive and do not preclude a benefit on clinical outcomes in a study with a larger size, a different design, or different dosing.
Timing might be the issue
However, even if colchicine has a potential benefit in this setting, timing might be a major obstacle, according to Binata Shah, MD, associate director of research for the Cardiac Catheterization Laboratory at New York University.
“We have learned from our rheumatology colleagues that peak plasma levels of colchicine are not achieved for at least 1 hour after the full loading dose,” Dr. Shah said. “With us moving so quickly in a primary PCI setting, it is hard to imagine that colchicine would have had time to really kick in and exert its anti-inflammatory effect.”
Indeed, the problem might be worse than reaching the peak plasma level.
“Even though peak plasma levels occur as early as 1 hour after a full loading dose, we see that it takes about 24 hours to really see the effects translate downstream into more systemic inflammatory markers such as CRP and interleukin-6,” she added. If lowering these signals of inflammation is predictive of benefit, than this might be the biggest obstacle to benefit from colchicine in an urgent treatment setting.
Dr. Jenab and Dr. Shah reported no potential conflicts of interest.
FROM CRT 2021
Neurologic drug prices jump 50% in five years
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).

“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).
“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
, new research shows. Results of the retrospective study also showed that most of the increased costs for these agents were due to rising costs for neuroimmunology drugs, mainly for those used to treat multiple sclerosis (MS).
“The same brand name medication in 2017 cost approximately 50% more than in 2013,” said Adam de Havenon, MD, assistant professor of neurology, University of Utah, Salt Lake City.
“An analogy would be if you bought an iPhone 5 in 2013 for $500, and then in 2017, you were asked to pay $750 for the exact same iPhone 5,” Dr. de Havenon added.
The study findings were published online March 10 in the journal Neurology.
$26 billion in payments
Both neurologists and patients are concerned about the high cost of prescription drugs for neurologic diseases, and Medicare Part D data indicate that these drugs are the most expensive component of neurologic care, the researchers noted. In addition, out-of-pocket costs have increased significantly for patients with neurologic disease such as Parkinson’s disease, epilepsy, and MS.
To understand trends in payments for neurologic drugs, Dr. de Havenon and colleagues analyzed Medicare Part D claims filed from 2013 to 2017. The payments include costs paid by Medicare, the patient, government subsidies, and other third-party payers.
In addition to examining more current Medicare Part D data than previous studies, the current analysis examined all medications prescribed by neurologists that consistently remained branded or generic during the 5-year study period, said Dr. de Havenon. This approach resulted in a large number of claims and a large total cost.
To calculate the percentage change in annual payment claims, the researchers used 2013 prices as a reference point. They identified drugs named in 2013 claims and classified them as generic, brand-name only, or brand-name with generic equivalent. Researchers also divided the drugs by neurologic subspecialty.
The analysis included 520 drugs, all of which were available in each year of the study period. Of these drugs, 322 were generic, 61 were brand-name only, and 137 were brand-name with a generic equivalent. There were 90.7 million total claims.
Results showed total payments amounted to $26.65 billion. Yearly total payments increased from $4.05 billion in 2013 to $6.09 billion in 2017, representing a 50.4% increase, even after adjusting for inflation. Total claims increased by 7.6% – from 17.1 million in 2013 to 18.4 million in 2017.
From 2013 to 2017, claim payments increased by 0.6% for generic drugs, 42.4% for brand-name only drugs, and 45% for brand-name drugs with generic equivalents. The proportion of claims increased from 81.9% to 88% for generic drugs and from 4.9% to 6.2% for brand-name only drugs.
However, the proportion of claims for brand-name drugs with generic equivalents decreased from 13.3% to 5.8%.
Treatment barrier
Neuroimmunologic drugs, most of which were prescribed for MS, had exceptional cost, the researchers noted. These drugs accounted for more than 50% of payments but only 4.3% of claims. Claim payment for these drugs increased by 46.9% during the study period, from $3,337 to $4,902.
When neuroimmunologic drugs were removed from the analysis there was still significant increase in claim payments for brand-name only drugs (50.4%) and brand-name drugs with generic equivalents (45.6%).
Although neuroimmunologic medicines, including monoclonal antibodies, are more expensive to produce, this factor alone does not explain their exceptional cost, said Dr. de Havenon. “The high cost of brand-name drugs in this speciality is likely because the market bears it,” he added. “In other words, MS is a disabling disease and the medications work, so historically the Centers for Medicare & Medicaid Services have been willing to tolerate the high cost of these primarily brand-name medications.”
Several countries have controlled drug costs by negotiating with pharmaceutical companies and through legislation, Dr. de Havenon noted.
“My intent with this article was to raise awareness on the topic, which I struggle with frequently as a clinician. I know I want my patients to have a medication, but the cost prevents it,” he said.
‘Unfettered’ price-setting
Commenting on the findings, Robert J. Fox, MD, vice chair for research at the Neurological Institute of the Cleveland Clinic, said the study “brings into clear light” what neurologists, particularly those who treat MS, have long suspected but did not really know. These neurologists “are typically distanced from the payment aspects of the medications they prescribe,” said Dr. Fox, who was not involved with the research.
Although a particular strength of the study was its comprehensiveness, the researchers excluded infusion claims – which account for a large portion of total patient care costs for many disorders, he noted.
Drugs for MS historically have been expensive, ostensibly because of their high cost of development. In addition, the large and continued price increase that occurs long after these drugs have been approved remains unexplained, said Dr. Fox.
He noted that the study findings might not directly affect clinical practice because neurologists will continue prescribing medications they think are best for their patients. “Instead, I think this is a lesson to lawmakers about the massive error in the Medicare Modernization Act of 2003, where the federal government was prohibited from negotiating drug prices. If the seller is unfettered in setting a price, then no one should be surprised when the price rises,” Dr. Fox said.
Because many new drugs and new generic formulations for treating MS have become available during the past year, “repeating these types of economic studies for the period 2020-2025 will help us understand if generic competition – as well as new laws if they are passed – alter price,” he concluded.
The study was funded by the American Academy of Neurology, which publishes Neurology. Dr. de Havenon has received clinical research funding from AMAG Pharmaceuticals and Regeneron Pharmaceuticals. Dr. Fox receives consulting fees from many pharmaceutical companies involved in the development of therapies for MS.
A version of this article first appeared on Medscape.com.
FROM NEUROLOGY
Despite risks and warnings, CNS polypharmacy is prevalent among patients with dementia
, new research suggests.
Investigators found that 14% of these individuals were receiving CNS-active polypharmacy, defined as combinations of multiple psychotropic and opioid medications taken for more than 30 days.
“For most patients, the risks of these medications, particularly in combination, are almost certainly greater than the potential benefits,” said Donovan Maust, MD, associate director of the geriatric psychiatry program, University of Michigan, Ann Arbor.
The study was published online March 9 in JAMA.
Serious risks
Memory impairment is the cardinal feature of dementia, but behavioral and psychological symptoms, which can include apathy, delusions, and agitation, are common during all stages of illness and cause significant caregiver distress, the researchers noted.
They noted that there is a dearth of high-quality evidence to support prescribing these medications in this patient population, yet “clinicians regularly prescribe psychotropic medications to community-dwelling persons with dementia in rates that far exceed use in the general older adult population.”
The Beers Criteria, from the American Geriatrics Society, advise against the practice of CNS polypharmacy because of the significant increase in risk for falls as well as impaired cognition, cardiac conduction abnormalities, respiratory suppression, and death when polypharmacy involves opioids.
They note that previous studies from Europe of polypharmacy for patients with dementia have not included antiepileptic medications or opioids, so the true extent of CNS-active polypharmacy may be “significantly” underestimated.
To determine the prevalence of polypharmacy with CNS-active medications among community-dwelling older adults with dementia, the researchers analyzed data on prescription fills for nearly 1.2 million community-dwelling Medicare patients with dementia.
The primary outcome was the prevalence of CNS-active polypharmacy in 2018. They defined CNS-active polypharmacy as exposure to three or more medications for more than 30 consecutive days from the following drug classes: antidepressants, antipsychotics, antiepileptics, benzodiazepines, nonbenzodiazepines, benzodiazepine receptor agonist hypnotics, and opioids.
They found that roughly one in seven (13.9%) patients met criteria for CNS-active polypharmacy. Of those receiving a CNS-active polypharmacy regimen, 57.8% had been doing so for longer than 180 days, and 6.8% had been doing so for a year. Nearly 30% of patients were exposed to five or more medications, and 5.2% were exposed to five or more medication classes.
Conservative approach warranted
Nearly all (92%) patients taking three or more CNS-active medications were taking an antidepressant, “consistent with their place as the psychotropic class most commonly prescribed both to older adults overall and those with dementia,” the investigators noted.
There is minimal high-quality evidence to support the efficacy of antidepressants for the treatment of depression for patients with dementia, they pointed out.
Nearly half (47%) of patients who were taking three or more CNS-active medications received at least one antipsychotic, most often quetiapine. Antipsychotics are not approved for people with dementia but are often prescribed off label for agitation, anxiety, and sleep problems, the researchers noted.
Nearly two thirds (62%) of patients with dementia who were taking three or more CNS drugs were taking an antiepileptic (most commonly, gabapentin); 41%, benzodiazepines; 32%, opioids; and 6%, Z-drugs.
The most common polypharmacy class combination included at least one antidepressant, one antiepileptic, and one antipsychotic. These accounted for 12.9% of polypharmacy days.
Despite limited high-quality evidence of efficacy, the prescribing of psychotropic medications and opioids is “pervasive” for adults with dementia in the United States, the investigators noted.
“Especially given that older adults with dementia might not be able to convey side effects they are experiencing, I think clinicians should be more conservative in how they are prescribing these medications and skeptical about the potential for benefit,” said Dr. Maust.
Regarding study limitations, the researchers noted that prescription medication claims may have led to an overestimation of the exposure to polypharmacy, insofar as the prescriptions may have been filled but not taken or were taken only on an as-needed basis.
In addition, the investigators were unable to determine the appropriateness of the particular combinations used or to examine the specific harms associated with CNS-active polypharmacy.
A major clinical challenge
Weighing in on the results, Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation, said the study is important because polypharmacy is one of the “geriatric giants, and the question is, what do you do about it?”
Dr. Fillit said it is important to conduct a careful medication review for all older patients, “making sure that the use of each drug is appropriate. The most important thing is to define what is the appropriate utilization of these kinds of drugs. That goes for both overutilization or misuse of these drugs and underutilization, where people are undertreated for symptoms that can’t be managed by behavioral management, for example,” Dr. Fillit said.
Dr. Fillit also said the finding that about 14% of dementia patients were receiving three or more of these drugs “may not be an outrageous number, because these patients, especially as they get into moderate and severe stages of disease, can be incredibly difficult to manage.
“Very often, dementia patients have depression, and up to 90% will have agitation and even psychosis during the course of dementia. And many of these patients need these types of drugs,” said Dr. Fillit.
Echoing the authors, Dr. Fillit said a key limitation of the study is not knowing whether the prescribing was appropriate or not.
The study was supported by a grant from the National Institute on Aging. Dr. Maust and Dr. Fillit have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Investigators found that 14% of these individuals were receiving CNS-active polypharmacy, defined as combinations of multiple psychotropic and opioid medications taken for more than 30 days.
“For most patients, the risks of these medications, particularly in combination, are almost certainly greater than the potential benefits,” said Donovan Maust, MD, associate director of the geriatric psychiatry program, University of Michigan, Ann Arbor.
The study was published online March 9 in JAMA.
Serious risks
Memory impairment is the cardinal feature of dementia, but behavioral and psychological symptoms, which can include apathy, delusions, and agitation, are common during all stages of illness and cause significant caregiver distress, the researchers noted.
They noted that there is a dearth of high-quality evidence to support prescribing these medications in this patient population, yet “clinicians regularly prescribe psychotropic medications to community-dwelling persons with dementia in rates that far exceed use in the general older adult population.”
The Beers Criteria, from the American Geriatrics Society, advise against the practice of CNS polypharmacy because of the significant increase in risk for falls as well as impaired cognition, cardiac conduction abnormalities, respiratory suppression, and death when polypharmacy involves opioids.
They note that previous studies from Europe of polypharmacy for patients with dementia have not included antiepileptic medications or opioids, so the true extent of CNS-active polypharmacy may be “significantly” underestimated.
To determine the prevalence of polypharmacy with CNS-active medications among community-dwelling older adults with dementia, the researchers analyzed data on prescription fills for nearly 1.2 million community-dwelling Medicare patients with dementia.
The primary outcome was the prevalence of CNS-active polypharmacy in 2018. They defined CNS-active polypharmacy as exposure to three or more medications for more than 30 consecutive days from the following drug classes: antidepressants, antipsychotics, antiepileptics, benzodiazepines, nonbenzodiazepines, benzodiazepine receptor agonist hypnotics, and opioids.
They found that roughly one in seven (13.9%) patients met criteria for CNS-active polypharmacy. Of those receiving a CNS-active polypharmacy regimen, 57.8% had been doing so for longer than 180 days, and 6.8% had been doing so for a year. Nearly 30% of patients were exposed to five or more medications, and 5.2% were exposed to five or more medication classes.
Conservative approach warranted
Nearly all (92%) patients taking three or more CNS-active medications were taking an antidepressant, “consistent with their place as the psychotropic class most commonly prescribed both to older adults overall and those with dementia,” the investigators noted.
There is minimal high-quality evidence to support the efficacy of antidepressants for the treatment of depression for patients with dementia, they pointed out.
Nearly half (47%) of patients who were taking three or more CNS-active medications received at least one antipsychotic, most often quetiapine. Antipsychotics are not approved for people with dementia but are often prescribed off label for agitation, anxiety, and sleep problems, the researchers noted.
Nearly two thirds (62%) of patients with dementia who were taking three or more CNS drugs were taking an antiepileptic (most commonly, gabapentin); 41%, benzodiazepines; 32%, opioids; and 6%, Z-drugs.
The most common polypharmacy class combination included at least one antidepressant, one antiepileptic, and one antipsychotic. These accounted for 12.9% of polypharmacy days.
Despite limited high-quality evidence of efficacy, the prescribing of psychotropic medications and opioids is “pervasive” for adults with dementia in the United States, the investigators noted.
“Especially given that older adults with dementia might not be able to convey side effects they are experiencing, I think clinicians should be more conservative in how they are prescribing these medications and skeptical about the potential for benefit,” said Dr. Maust.
Regarding study limitations, the researchers noted that prescription medication claims may have led to an overestimation of the exposure to polypharmacy, insofar as the prescriptions may have been filled but not taken or were taken only on an as-needed basis.
In addition, the investigators were unable to determine the appropriateness of the particular combinations used or to examine the specific harms associated with CNS-active polypharmacy.
A major clinical challenge
Weighing in on the results, Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation, said the study is important because polypharmacy is one of the “geriatric giants, and the question is, what do you do about it?”
Dr. Fillit said it is important to conduct a careful medication review for all older patients, “making sure that the use of each drug is appropriate. The most important thing is to define what is the appropriate utilization of these kinds of drugs. That goes for both overutilization or misuse of these drugs and underutilization, where people are undertreated for symptoms that can’t be managed by behavioral management, for example,” Dr. Fillit said.
Dr. Fillit also said the finding that about 14% of dementia patients were receiving three or more of these drugs “may not be an outrageous number, because these patients, especially as they get into moderate and severe stages of disease, can be incredibly difficult to manage.
“Very often, dementia patients have depression, and up to 90% will have agitation and even psychosis during the course of dementia. And many of these patients need these types of drugs,” said Dr. Fillit.
Echoing the authors, Dr. Fillit said a key limitation of the study is not knowing whether the prescribing was appropriate or not.
The study was supported by a grant from the National Institute on Aging. Dr. Maust and Dr. Fillit have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Investigators found that 14% of these individuals were receiving CNS-active polypharmacy, defined as combinations of multiple psychotropic and opioid medications taken for more than 30 days.
“For most patients, the risks of these medications, particularly in combination, are almost certainly greater than the potential benefits,” said Donovan Maust, MD, associate director of the geriatric psychiatry program, University of Michigan, Ann Arbor.
The study was published online March 9 in JAMA.
Serious risks
Memory impairment is the cardinal feature of dementia, but behavioral and psychological symptoms, which can include apathy, delusions, and agitation, are common during all stages of illness and cause significant caregiver distress, the researchers noted.
They noted that there is a dearth of high-quality evidence to support prescribing these medications in this patient population, yet “clinicians regularly prescribe psychotropic medications to community-dwelling persons with dementia in rates that far exceed use in the general older adult population.”
The Beers Criteria, from the American Geriatrics Society, advise against the practice of CNS polypharmacy because of the significant increase in risk for falls as well as impaired cognition, cardiac conduction abnormalities, respiratory suppression, and death when polypharmacy involves opioids.
They note that previous studies from Europe of polypharmacy for patients with dementia have not included antiepileptic medications or opioids, so the true extent of CNS-active polypharmacy may be “significantly” underestimated.
To determine the prevalence of polypharmacy with CNS-active medications among community-dwelling older adults with dementia, the researchers analyzed data on prescription fills for nearly 1.2 million community-dwelling Medicare patients with dementia.
The primary outcome was the prevalence of CNS-active polypharmacy in 2018. They defined CNS-active polypharmacy as exposure to three or more medications for more than 30 consecutive days from the following drug classes: antidepressants, antipsychotics, antiepileptics, benzodiazepines, nonbenzodiazepines, benzodiazepine receptor agonist hypnotics, and opioids.
They found that roughly one in seven (13.9%) patients met criteria for CNS-active polypharmacy. Of those receiving a CNS-active polypharmacy regimen, 57.8% had been doing so for longer than 180 days, and 6.8% had been doing so for a year. Nearly 30% of patients were exposed to five or more medications, and 5.2% were exposed to five or more medication classes.
Conservative approach warranted
Nearly all (92%) patients taking three or more CNS-active medications were taking an antidepressant, “consistent with their place as the psychotropic class most commonly prescribed both to older adults overall and those with dementia,” the investigators noted.
There is minimal high-quality evidence to support the efficacy of antidepressants for the treatment of depression for patients with dementia, they pointed out.
Nearly half (47%) of patients who were taking three or more CNS-active medications received at least one antipsychotic, most often quetiapine. Antipsychotics are not approved for people with dementia but are often prescribed off label for agitation, anxiety, and sleep problems, the researchers noted.
Nearly two thirds (62%) of patients with dementia who were taking three or more CNS drugs were taking an antiepileptic (most commonly, gabapentin); 41%, benzodiazepines; 32%, opioids; and 6%, Z-drugs.
The most common polypharmacy class combination included at least one antidepressant, one antiepileptic, and one antipsychotic. These accounted for 12.9% of polypharmacy days.
Despite limited high-quality evidence of efficacy, the prescribing of psychotropic medications and opioids is “pervasive” for adults with dementia in the United States, the investigators noted.
“Especially given that older adults with dementia might not be able to convey side effects they are experiencing, I think clinicians should be more conservative in how they are prescribing these medications and skeptical about the potential for benefit,” said Dr. Maust.
Regarding study limitations, the researchers noted that prescription medication claims may have led to an overestimation of the exposure to polypharmacy, insofar as the prescriptions may have been filled but not taken or were taken only on an as-needed basis.
In addition, the investigators were unable to determine the appropriateness of the particular combinations used or to examine the specific harms associated with CNS-active polypharmacy.
A major clinical challenge
Weighing in on the results, Howard Fillit, MD, founding executive director and chief science officer of the Alzheimer’s Drug Discovery Foundation, said the study is important because polypharmacy is one of the “geriatric giants, and the question is, what do you do about it?”
Dr. Fillit said it is important to conduct a careful medication review for all older patients, “making sure that the use of each drug is appropriate. The most important thing is to define what is the appropriate utilization of these kinds of drugs. That goes for both overutilization or misuse of these drugs and underutilization, where people are undertreated for symptoms that can’t be managed by behavioral management, for example,” Dr. Fillit said.
Dr. Fillit also said the finding that about 14% of dementia patients were receiving three or more of these drugs “may not be an outrageous number, because these patients, especially as they get into moderate and severe stages of disease, can be incredibly difficult to manage.
“Very often, dementia patients have depression, and up to 90% will have agitation and even psychosis during the course of dementia. And many of these patients need these types of drugs,” said Dr. Fillit.
Echoing the authors, Dr. Fillit said a key limitation of the study is not knowing whether the prescribing was appropriate or not.
The study was supported by a grant from the National Institute on Aging. Dr. Maust and Dr. Fillit have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA
Telerheumatology will thrive post pandemic
Telemedicine has had a profound effect upon the practice of rheumatology during the COVID-19 pandemic and will continue to do so afterward, speakers predicted at the 2021 Rheumatology Winter Clinical Symposium.

“Telemedicine will change the way we do business. It already has,” observed Eric M. Ruderman, MD, professor of medicine (rheumatology) at Northwestern University in Chicago.
“All of a sudden in March of last year we all turned on a dime and went 100% remote, and we made it work. And it has worked well. It’s not the same as seeing people in person, but I’m pretty sure that going forward probably somewhere in the range of 30% of our visits are going to be telemedicine. It’s an incredible way to deal with people who are stable and are driving in from an hour-and-a-half away to get their prescription refilled,” he said.
Conditions well suited for video patient visits are those where the physical exam isn’t informative or necessary, such as polymyalgia rheumatica, axial spondyloarthritis with axial disease only, childhood periodic fever syndromes, and even many cases of rheumatoid arthritis, in Dr. Ruderman’s view.
“People who are stable – maybe not in remission, but we’ve decided they’re at that their target – a lot of those visits can be done remotely. It’s way more efficient. Everybody loves it: We like it, the patients like it. But we have to get to where we can do it better. The technology is clumsy right now,” he said.
“We do need better and smarter platforms,” agreed Alvin F. Wells, MD, PhD, a telerheumatology pioneer who has been involved in digital/video communication with his patients for nearly 6 years. “But the biggest issue is connectivity. Not all of our patients can get on the Internet.”
The telerheumatology paradigm he has used during the pandemic and will continue to use afterward is to see every new patient in the office, then do the follow-up visits virtually.
“They don’t need to come back into the office in 4 weeks. I’ve done my physical exam, ordered the x-rays and lab work. At the virtual 4-week follow-up we go over everything and I tell them if they need to come in for training in giving their injections,” explained Dr. Wells, a rheumatologist in Franklin, Wisc.
“The telemedicine visit doesn’t take the place of an in-person visit, but it allows you to stratify, to say who needs to be seen sooner rather than later,” he added.
While he anticipates that physician-patient virtual visits will continue to be an important part of clinical practice post pandemic, he predicted the major growth areas for telerheumatology once COVID-19 is squashed will be in clinician-to-clinician interactions and remote patient monitoring using smart devices.
Dr. Wells hasn’t gone into the hospital once since the pandemic began. Initially, that was because he didn’t want to deal with the personal protective equipment shortage or expose himself to the virus. Now, it’s because it’s just a more efficient use of his time to conduct virtual – and billable – 15-minute e-consults with clinicians in the hospital.
“I’ve had a lot of appropriate consults with the hospitalists,” he said. He can walk a hospitalist through a real-time physical exam at a gout patient’s bedside and order the right laboratory tests.
“I don’t need to go into the hospital. The interventional radiologist can tap an ankle or toe as well as I can,” the rheumatologist said.
Dermatologist George Martin, MD, rose from the audience to say that while he recognizes that pandemic telemedicine has been a good fit for rheumatologists, it’s been a very different story in dermatology.
“I realize telemedicine works really well when you don’t have to lay your hands on a patient, or when you’re just doing a stable follow-up and talking about test results. But we in dermatology have found as a group that telemedicine is pretty worthless. When patients are trying to send you a video stream of what their melanoma looks like, or maybe it’s a benign seborrheic keratosis, you’re going to hang their life on that? Dermatology is a very hands-on, visual thing, and unless the camera work becomes better telemedicine is worthless, with the exception of a laboratory follow-up or a stable visit where a physical exam is not required,” declared Dr. Martin, who is in private practice in Maui.
Dr. Wells reported serving as a consultant to MiCare Path, a remote health and monitoring company.
Telemedicine has had a profound effect upon the practice of rheumatology during the COVID-19 pandemic and will continue to do so afterward, speakers predicted at the 2021 Rheumatology Winter Clinical Symposium.
“Telemedicine will change the way we do business. It already has,” observed Eric M. Ruderman, MD, professor of medicine (rheumatology) at Northwestern University in Chicago.
“All of a sudden in March of last year we all turned on a dime and went 100% remote, and we made it work. And it has worked well. It’s not the same as seeing people in person, but I’m pretty sure that going forward probably somewhere in the range of 30% of our visits are going to be telemedicine. It’s an incredible way to deal with people who are stable and are driving in from an hour-and-a-half away to get their prescription refilled,” he said.
Conditions well suited for video patient visits are those where the physical exam isn’t informative or necessary, such as polymyalgia rheumatica, axial spondyloarthritis with axial disease only, childhood periodic fever syndromes, and even many cases of rheumatoid arthritis, in Dr. Ruderman’s view.
“People who are stable – maybe not in remission, but we’ve decided they’re at that their target – a lot of those visits can be done remotely. It’s way more efficient. Everybody loves it: We like it, the patients like it. But we have to get to where we can do it better. The technology is clumsy right now,” he said.
“We do need better and smarter platforms,” agreed Alvin F. Wells, MD, PhD, a telerheumatology pioneer who has been involved in digital/video communication with his patients for nearly 6 years. “But the biggest issue is connectivity. Not all of our patients can get on the Internet.”
The telerheumatology paradigm he has used during the pandemic and will continue to use afterward is to see every new patient in the office, then do the follow-up visits virtually.
“They don’t need to come back into the office in 4 weeks. I’ve done my physical exam, ordered the x-rays and lab work. At the virtual 4-week follow-up we go over everything and I tell them if they need to come in for training in giving their injections,” explained Dr. Wells, a rheumatologist in Franklin, Wisc.
“The telemedicine visit doesn’t take the place of an in-person visit, but it allows you to stratify, to say who needs to be seen sooner rather than later,” he added.
While he anticipates that physician-patient virtual visits will continue to be an important part of clinical practice post pandemic, he predicted the major growth areas for telerheumatology once COVID-19 is squashed will be in clinician-to-clinician interactions and remote patient monitoring using smart devices.
Dr. Wells hasn’t gone into the hospital once since the pandemic began. Initially, that was because he didn’t want to deal with the personal protective equipment shortage or expose himself to the virus. Now, it’s because it’s just a more efficient use of his time to conduct virtual – and billable – 15-minute e-consults with clinicians in the hospital.
“I’ve had a lot of appropriate consults with the hospitalists,” he said. He can walk a hospitalist through a real-time physical exam at a gout patient’s bedside and order the right laboratory tests.
“I don’t need to go into the hospital. The interventional radiologist can tap an ankle or toe as well as I can,” the rheumatologist said.
Dermatologist George Martin, MD, rose from the audience to say that while he recognizes that pandemic telemedicine has been a good fit for rheumatologists, it’s been a very different story in dermatology.
“I realize telemedicine works really well when you don’t have to lay your hands on a patient, or when you’re just doing a stable follow-up and talking about test results. But we in dermatology have found as a group that telemedicine is pretty worthless. When patients are trying to send you a video stream of what their melanoma looks like, or maybe it’s a benign seborrheic keratosis, you’re going to hang their life on that? Dermatology is a very hands-on, visual thing, and unless the camera work becomes better telemedicine is worthless, with the exception of a laboratory follow-up or a stable visit where a physical exam is not required,” declared Dr. Martin, who is in private practice in Maui.
Dr. Wells reported serving as a consultant to MiCare Path, a remote health and monitoring company.
Telemedicine has had a profound effect upon the practice of rheumatology during the COVID-19 pandemic and will continue to do so afterward, speakers predicted at the 2021 Rheumatology Winter Clinical Symposium.
“Telemedicine will change the way we do business. It already has,” observed Eric M. Ruderman, MD, professor of medicine (rheumatology) at Northwestern University in Chicago.
“All of a sudden in March of last year we all turned on a dime and went 100% remote, and we made it work. And it has worked well. It’s not the same as seeing people in person, but I’m pretty sure that going forward probably somewhere in the range of 30% of our visits are going to be telemedicine. It’s an incredible way to deal with people who are stable and are driving in from an hour-and-a-half away to get their prescription refilled,” he said.
Conditions well suited for video patient visits are those where the physical exam isn’t informative or necessary, such as polymyalgia rheumatica, axial spondyloarthritis with axial disease only, childhood periodic fever syndromes, and even many cases of rheumatoid arthritis, in Dr. Ruderman’s view.
“People who are stable – maybe not in remission, but we’ve decided they’re at that their target – a lot of those visits can be done remotely. It’s way more efficient. Everybody loves it: We like it, the patients like it. But we have to get to where we can do it better. The technology is clumsy right now,” he said.
“We do need better and smarter platforms,” agreed Alvin F. Wells, MD, PhD, a telerheumatology pioneer who has been involved in digital/video communication with his patients for nearly 6 years. “But the biggest issue is connectivity. Not all of our patients can get on the Internet.”
The telerheumatology paradigm he has used during the pandemic and will continue to use afterward is to see every new patient in the office, then do the follow-up visits virtually.
“They don’t need to come back into the office in 4 weeks. I’ve done my physical exam, ordered the x-rays and lab work. At the virtual 4-week follow-up we go over everything and I tell them if they need to come in for training in giving their injections,” explained Dr. Wells, a rheumatologist in Franklin, Wisc.
“The telemedicine visit doesn’t take the place of an in-person visit, but it allows you to stratify, to say who needs to be seen sooner rather than later,” he added.
While he anticipates that physician-patient virtual visits will continue to be an important part of clinical practice post pandemic, he predicted the major growth areas for telerheumatology once COVID-19 is squashed will be in clinician-to-clinician interactions and remote patient monitoring using smart devices.
Dr. Wells hasn’t gone into the hospital once since the pandemic began. Initially, that was because he didn’t want to deal with the personal protective equipment shortage or expose himself to the virus. Now, it’s because it’s just a more efficient use of his time to conduct virtual – and billable – 15-minute e-consults with clinicians in the hospital.
“I’ve had a lot of appropriate consults with the hospitalists,” he said. He can walk a hospitalist through a real-time physical exam at a gout patient’s bedside and order the right laboratory tests.
“I don’t need to go into the hospital. The interventional radiologist can tap an ankle or toe as well as I can,” the rheumatologist said.
Dermatologist George Martin, MD, rose from the audience to say that while he recognizes that pandemic telemedicine has been a good fit for rheumatologists, it’s been a very different story in dermatology.
“I realize telemedicine works really well when you don’t have to lay your hands on a patient, or when you’re just doing a stable follow-up and talking about test results. But we in dermatology have found as a group that telemedicine is pretty worthless. When patients are trying to send you a video stream of what their melanoma looks like, or maybe it’s a benign seborrheic keratosis, you’re going to hang their life on that? Dermatology is a very hands-on, visual thing, and unless the camera work becomes better telemedicine is worthless, with the exception of a laboratory follow-up or a stable visit where a physical exam is not required,” declared Dr. Martin, who is in private practice in Maui.
Dr. Wells reported serving as a consultant to MiCare Path, a remote health and monitoring company.
FROM RWCS 2021
Decline in child COVID-19 cases picks up after 2-week slowdown
, according to data gathered by the American Academy of Pediatrics and the Children’s Hospital Association.
From Feb. 19 to March 4, the drop in new cases averaged just 5% each week, compared with 13.3% per week over the 5-week period from Jan. 15 to Feb. 18. For the week of March 5-11, a total of 52,695 COVID-19 cases were reported in children, down from 63,562 the previous week and the lowest number since late October, based on data from 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam.
In those jurisdictions, 3.28 million children have been infected with SARS-CoV-2, representing 13.2% of all cases since the beginning of the pandemic. The cumulative rate of COVID-19 has now risen to 4,364 cases per 100,000 children nationally, with state rates ranging from 1,062 per 100,000 in Hawaii to 8,692 per 100,000 in North Dakota, the AAP and CHA said in their weekly COVID-19 report.
Hospitalization data are more limited – 24 states and New York City – but continue to show that serious illness is much less common in younger individuals: Children represent just 1.9% of all hospitalizations, and only 0.8% of the children who have been infected were hospitalized. Neither rate has changed since early February, the AAP and CHA said.
The number of deaths in children, however, rose from 253 to 266, the largest 1-week increase since early February in the 43 states (along with New York City, Puerto Rico, and Guam) that are tracking mortality data by age, the AAP and CHA reported.
Among those 46 jurisdictions, there are 10 (9 states and the District of Columbia) that have not yet reported a COVID-19–related child death, while Texas has almost twice as many deaths, 47, as the next state, Arizona, which has 24. Meanwhile, California’s total of 452,000 cases is almost 2½ times higher than the 183,000 recorded by Illinois, according to the report.
, according to data gathered by the American Academy of Pediatrics and the Children’s Hospital Association.
From Feb. 19 to March 4, the drop in new cases averaged just 5% each week, compared with 13.3% per week over the 5-week period from Jan. 15 to Feb. 18. For the week of March 5-11, a total of 52,695 COVID-19 cases were reported in children, down from 63,562 the previous week and the lowest number since late October, based on data from 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam.
In those jurisdictions, 3.28 million children have been infected with SARS-CoV-2, representing 13.2% of all cases since the beginning of the pandemic. The cumulative rate of COVID-19 has now risen to 4,364 cases per 100,000 children nationally, with state rates ranging from 1,062 per 100,000 in Hawaii to 8,692 per 100,000 in North Dakota, the AAP and CHA said in their weekly COVID-19 report.
Hospitalization data are more limited – 24 states and New York City – but continue to show that serious illness is much less common in younger individuals: Children represent just 1.9% of all hospitalizations, and only 0.8% of the children who have been infected were hospitalized. Neither rate has changed since early February, the AAP and CHA said.
The number of deaths in children, however, rose from 253 to 266, the largest 1-week increase since early February in the 43 states (along with New York City, Puerto Rico, and Guam) that are tracking mortality data by age, the AAP and CHA reported.
Among those 46 jurisdictions, there are 10 (9 states and the District of Columbia) that have not yet reported a COVID-19–related child death, while Texas has almost twice as many deaths, 47, as the next state, Arizona, which has 24. Meanwhile, California’s total of 452,000 cases is almost 2½ times higher than the 183,000 recorded by Illinois, according to the report.
, according to data gathered by the American Academy of Pediatrics and the Children’s Hospital Association.
From Feb. 19 to March 4, the drop in new cases averaged just 5% each week, compared with 13.3% per week over the 5-week period from Jan. 15 to Feb. 18. For the week of March 5-11, a total of 52,695 COVID-19 cases were reported in children, down from 63,562 the previous week and the lowest number since late October, based on data from 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam.
In those jurisdictions, 3.28 million children have been infected with SARS-CoV-2, representing 13.2% of all cases since the beginning of the pandemic. The cumulative rate of COVID-19 has now risen to 4,364 cases per 100,000 children nationally, with state rates ranging from 1,062 per 100,000 in Hawaii to 8,692 per 100,000 in North Dakota, the AAP and CHA said in their weekly COVID-19 report.
Hospitalization data are more limited – 24 states and New York City – but continue to show that serious illness is much less common in younger individuals: Children represent just 1.9% of all hospitalizations, and only 0.8% of the children who have been infected were hospitalized. Neither rate has changed since early February, the AAP and CHA said.
The number of deaths in children, however, rose from 253 to 266, the largest 1-week increase since early February in the 43 states (along with New York City, Puerto Rico, and Guam) that are tracking mortality data by age, the AAP and CHA reported.
Among those 46 jurisdictions, there are 10 (9 states and the District of Columbia) that have not yet reported a COVID-19–related child death, while Texas has almost twice as many deaths, 47, as the next state, Arizona, which has 24. Meanwhile, California’s total of 452,000 cases is almost 2½ times higher than the 183,000 recorded by Illinois, according to the report.