User login
Transfusion Rates Vary Widely at Academic Hospitals
Wide variations in perioperative blood transfusion rates among patients undergoing major noncardiac procedures across U.S. hospitals highlight the need to further investigate evidence-based "transfusion triggers" in this population of surgical patients, according to a study published ahead of print in Annals of Surgery.
"In light of the increased risk of mortality and major complications associated with blood transfusion, the extensive variability in hospital transfusion practice in noncardiac surgery may represent an important opportunity to improve surgical outcomes," wrote Feng Qian, Ph.D., of the University of Rochester (N.Y.), and associates.
The researchers used the University HealthSystem Consortium hospital database to compare transfusion rates of allogeneic red blood cells, fresh frozen plasma, and platelets in patients undergoing elective primary total hip replacement (54,405 patients), colectomy (21,334), or pancreaticoduodenectomy (7,929) at 77 hospitals between June 2006 and September 2010. Most of the hospitals were teaching hospitals with at least 500 beds.
Transfusion rates varied widely before and after adjustment for comorbidities and other patient risk factors. Patients who were treated in hospitals with high rates of transfusions were about twice as likely to receive a blood transfusion as were patients at hospitals with average transfusion rates (Ann. Surg. 2012 July 13[doi:10.1097/SLA.0b013e31825ffc37]).
In hospitals where the transfusion rate for one procedure was high, transfusion rates also tended to be high for the other two procedures. There was some evidence indicating that a higher volume of surgical cases was associated with lower transfusion rates.
After adjusting for patient risk factors, the authors determined that transfusion rates for the different blood components among those undergoing a total hip replacement ranged from 1.3% to almost 75% (red blood cells), from 0.1% to 7.7% (fresh frozen plasma), and from 0.1% to 2% (platelets). Among colectomy patients, transfusion rates ranged from 1.9% to 47.8% (RBCs), from 1.4% to 17.7% (fresh frozen plasma), and from 1.3% to 6.2% (platelets). Among those undergoing a pancreaticoduodenectomy, the rates ranged from 3% to 78.6% (RBCs), from 1% to 47% (fresh frozen plasma), and from 1.4% to 12.6% (platelets).
The variability, the authors said, "reflects, in part, the complexity of the medical decision-making process underlying transfusion therapy." Because the data included patients from 90% of academic medical centers in the United States, the results provide "a broad and contemporary picture of transfusion practices in academic surgical centers" and "reflect transfusion practices that are being taught to the next generation of academic and private-practice clinicians during residency training," they noted.
To the best of their knowledge, the authors said, there are no large randomized studies that have compared liberal and restrictive transfusion strategies in noncardiac surgery patients, and they believe that such trials are "urgently needed to better define evidence-based transfusion triggers for patients undergoing noncardiac surgery."
The study was supported by a grant from the Agency for Healthcare and Quality Research and funding from the department of anesthesiology at the University of Rochester. No disclosures were reported by the authors.
Wide variations in perioperative blood transfusion rates among patients undergoing major noncardiac procedures across U.S. hospitals highlight the need to further investigate evidence-based "transfusion triggers" in this population of surgical patients, according to a study published ahead of print in Annals of Surgery.
"In light of the increased risk of mortality and major complications associated with blood transfusion, the extensive variability in hospital transfusion practice in noncardiac surgery may represent an important opportunity to improve surgical outcomes," wrote Feng Qian, Ph.D., of the University of Rochester (N.Y.), and associates.
The researchers used the University HealthSystem Consortium hospital database to compare transfusion rates of allogeneic red blood cells, fresh frozen plasma, and platelets in patients undergoing elective primary total hip replacement (54,405 patients), colectomy (21,334), or pancreaticoduodenectomy (7,929) at 77 hospitals between June 2006 and September 2010. Most of the hospitals were teaching hospitals with at least 500 beds.
Transfusion rates varied widely before and after adjustment for comorbidities and other patient risk factors. Patients who were treated in hospitals with high rates of transfusions were about twice as likely to receive a blood transfusion as were patients at hospitals with average transfusion rates (Ann. Surg. 2012 July 13[doi:10.1097/SLA.0b013e31825ffc37]).
In hospitals where the transfusion rate for one procedure was high, transfusion rates also tended to be high for the other two procedures. There was some evidence indicating that a higher volume of surgical cases was associated with lower transfusion rates.
After adjusting for patient risk factors, the authors determined that transfusion rates for the different blood components among those undergoing a total hip replacement ranged from 1.3% to almost 75% (red blood cells), from 0.1% to 7.7% (fresh frozen plasma), and from 0.1% to 2% (platelets). Among colectomy patients, transfusion rates ranged from 1.9% to 47.8% (RBCs), from 1.4% to 17.7% (fresh frozen plasma), and from 1.3% to 6.2% (platelets). Among those undergoing a pancreaticoduodenectomy, the rates ranged from 3% to 78.6% (RBCs), from 1% to 47% (fresh frozen plasma), and from 1.4% to 12.6% (platelets).
The variability, the authors said, "reflects, in part, the complexity of the medical decision-making process underlying transfusion therapy." Because the data included patients from 90% of academic medical centers in the United States, the results provide "a broad and contemporary picture of transfusion practices in academic surgical centers" and "reflect transfusion practices that are being taught to the next generation of academic and private-practice clinicians during residency training," they noted.
To the best of their knowledge, the authors said, there are no large randomized studies that have compared liberal and restrictive transfusion strategies in noncardiac surgery patients, and they believe that such trials are "urgently needed to better define evidence-based transfusion triggers for patients undergoing noncardiac surgery."
The study was supported by a grant from the Agency for Healthcare and Quality Research and funding from the department of anesthesiology at the University of Rochester. No disclosures were reported by the authors.
Wide variations in perioperative blood transfusion rates among patients undergoing major noncardiac procedures across U.S. hospitals highlight the need to further investigate evidence-based "transfusion triggers" in this population of surgical patients, according to a study published ahead of print in Annals of Surgery.
"In light of the increased risk of mortality and major complications associated with blood transfusion, the extensive variability in hospital transfusion practice in noncardiac surgery may represent an important opportunity to improve surgical outcomes," wrote Feng Qian, Ph.D., of the University of Rochester (N.Y.), and associates.
The researchers used the University HealthSystem Consortium hospital database to compare transfusion rates of allogeneic red blood cells, fresh frozen plasma, and platelets in patients undergoing elective primary total hip replacement (54,405 patients), colectomy (21,334), or pancreaticoduodenectomy (7,929) at 77 hospitals between June 2006 and September 2010. Most of the hospitals were teaching hospitals with at least 500 beds.
Transfusion rates varied widely before and after adjustment for comorbidities and other patient risk factors. Patients who were treated in hospitals with high rates of transfusions were about twice as likely to receive a blood transfusion as were patients at hospitals with average transfusion rates (Ann. Surg. 2012 July 13[doi:10.1097/SLA.0b013e31825ffc37]).
In hospitals where the transfusion rate for one procedure was high, transfusion rates also tended to be high for the other two procedures. There was some evidence indicating that a higher volume of surgical cases was associated with lower transfusion rates.
After adjusting for patient risk factors, the authors determined that transfusion rates for the different blood components among those undergoing a total hip replacement ranged from 1.3% to almost 75% (red blood cells), from 0.1% to 7.7% (fresh frozen plasma), and from 0.1% to 2% (platelets). Among colectomy patients, transfusion rates ranged from 1.9% to 47.8% (RBCs), from 1.4% to 17.7% (fresh frozen plasma), and from 1.3% to 6.2% (platelets). Among those undergoing a pancreaticoduodenectomy, the rates ranged from 3% to 78.6% (RBCs), from 1% to 47% (fresh frozen plasma), and from 1.4% to 12.6% (platelets).
The variability, the authors said, "reflects, in part, the complexity of the medical decision-making process underlying transfusion therapy." Because the data included patients from 90% of academic medical centers in the United States, the results provide "a broad and contemporary picture of transfusion practices in academic surgical centers" and "reflect transfusion practices that are being taught to the next generation of academic and private-practice clinicians during residency training," they noted.
To the best of their knowledge, the authors said, there are no large randomized studies that have compared liberal and restrictive transfusion strategies in noncardiac surgery patients, and they believe that such trials are "urgently needed to better define evidence-based transfusion triggers for patients undergoing noncardiac surgery."
The study was supported by a grant from the Agency for Healthcare and Quality Research and funding from the department of anesthesiology at the University of Rochester. No disclosures were reported by the authors.
FROM THE ANNALS OF SURGERY
Major Finding: Transfusion rates of red blood cells, fresh frozen plasma, and platelets among patients undergoing noncardiac procedures varied widely across different U.S. academic-affiliated hospitals.
Data Source: Data from a national database of academic medical centers were used to compare transfusions in patients undergoing one of three elective noncardiac surgical procedures at 77 academic hospitals between June 2006 and September 2010.
Disclosures: The study was supported by a grant from the Agency for Healthcare and Quality Research and funding from the department of anesthesiology at the University of Rochester (N.Y.). The authors reported no disclosures.
Understanding PTSD

Getting to Goal: How Thiazide-Type Diuretics, Following the Guidelines, and Improving Patient Adherence Can Help
An estimated 1 of every 3 Americans has hypertension, putting them at an increased risk for cardiovascular disease, heart failure, stroke, and kidney disease. Despite the availability of effective medications to control high blood pressure, only half of the patients with hypertension under treatment are meeting their blood pressure goals. To address these gaps in the quality of care patients receive, this supplement will focus on the following topics in hypertension management: key clinical trials and their influence on sequencing algorithms; the differences between thiazide-type diuretics; the use of thiazide-type diuretics in African American patients; and strategies to improve patient adherence to hypertensive therapy.
Webcast—October 2012
An estimated 1 of every 3 Americans has hypertension, putting them at an increased risk for cardiovascular disease, heart failure, stroke, and kidney disease. Despite the availability of effective medications to control high blood pressure, only half of the patients with hypertension under treatment are meeting their blood pressure goals. To address these gaps in the quality of care patients receive, this supplement will focus on the following topics in hypertension management: key clinical trials and their influence on sequencing algorithms; the differences between thiazide-type diuretics; the use of thiazide-type diuretics in African American patients; and strategies to improve patient adherence to hypertensive therapy.
Webcast—October 2012
An estimated 1 of every 3 Americans has hypertension, putting them at an increased risk for cardiovascular disease, heart failure, stroke, and kidney disease. Despite the availability of effective medications to control high blood pressure, only half of the patients with hypertension under treatment are meeting their blood pressure goals. To address these gaps in the quality of care patients receive, this supplement will focus on the following topics in hypertension management: key clinical trials and their influence on sequencing algorithms; the differences between thiazide-type diuretics; the use of thiazide-type diuretics in African American patients; and strategies to improve patient adherence to hypertensive therapy.
Webcast—October 2012

Synthetic legal intoxicating drugs
To the Editor: I greatly appreciate the well-presented article by Drs. Jerry, Collins, and Streem in your April 2012 issue.1
As a specialist in integrative addiction medicine, I have had first-hand experience with many of the medical concerns described by the authors, and I expect to learn more about optimal management strategies as we learn more as a profession.
The lone case report cited in the article suggests a relatively short time to onset of seizure of 30 minutes following intentional ingestion of synthetic cannabinoids (JWH-018).2
In the residential treatment (“rehab”) setting where I work, I am seeing a latency to seizure onset of 24 to 72 hours with patients reporting use of synthetic cannabinoids.
Given this experience to date, I have two questions for the authors regarding new-onset seizures.
Are the authors aware of this trend in patients who present to non-emergency-department treatment settings such as residential treatment facilities? And in these cases, what if any recommendations would the authors make regarding seizure prophylaxis in patients with no history of seizure?
- Jerry J, Collins G, Streem D. Synthetic legal intoxicating drugs: the emerging ‘incense’ and ‘bath salt’ phenomenon. Cleve Clin J Med 2012; 79:258–264.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
To the Editor: I greatly appreciate the well-presented article by Drs. Jerry, Collins, and Streem in your April 2012 issue.1
As a specialist in integrative addiction medicine, I have had first-hand experience with many of the medical concerns described by the authors, and I expect to learn more about optimal management strategies as we learn more as a profession.
The lone case report cited in the article suggests a relatively short time to onset of seizure of 30 minutes following intentional ingestion of synthetic cannabinoids (JWH-018).2
In the residential treatment (“rehab”) setting where I work, I am seeing a latency to seizure onset of 24 to 72 hours with patients reporting use of synthetic cannabinoids.
Given this experience to date, I have two questions for the authors regarding new-onset seizures.
Are the authors aware of this trend in patients who present to non-emergency-department treatment settings such as residential treatment facilities? And in these cases, what if any recommendations would the authors make regarding seizure prophylaxis in patients with no history of seizure?
To the Editor: I greatly appreciate the well-presented article by Drs. Jerry, Collins, and Streem in your April 2012 issue.1
As a specialist in integrative addiction medicine, I have had first-hand experience with many of the medical concerns described by the authors, and I expect to learn more about optimal management strategies as we learn more as a profession.
The lone case report cited in the article suggests a relatively short time to onset of seizure of 30 minutes following intentional ingestion of synthetic cannabinoids (JWH-018).2
In the residential treatment (“rehab”) setting where I work, I am seeing a latency to seizure onset of 24 to 72 hours with patients reporting use of synthetic cannabinoids.
Given this experience to date, I have two questions for the authors regarding new-onset seizures.
Are the authors aware of this trend in patients who present to non-emergency-department treatment settings such as residential treatment facilities? And in these cases, what if any recommendations would the authors make regarding seizure prophylaxis in patients with no history of seizure?
- Jerry J, Collins G, Streem D. Synthetic legal intoxicating drugs: the emerging ‘incense’ and ‘bath salt’ phenomenon. Cleve Clin J Med 2012; 79:258–264.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
- Jerry J, Collins G, Streem D. Synthetic legal intoxicating drugs: the emerging ‘incense’ and ‘bath salt’ phenomenon. Cleve Clin J Med 2012; 79:258–264.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
In reply: Synthetic legal intoxicating drugs
In Reply: We thank Dr. Chandiramani for his thoughtful comments.
Only four cases of seizure-like activity associated with synthetic cannabinoids have been reported in the literature. In addition to the case reported in our paper,1 there was another in which a 19-year-old had two seizures soon after smoking a spice product, and the second seizure was witnessed by paramedics on the way to the hospital.2 Though this patient’s urine was not analyzed for synthetic cannabinoids, the spice product that was reportedly smoked by the patient was later sent to a laboratory for analysis and was found to contain four synthetic cannabinoids: JWH-018, JWH-081, JWH-250, and AM-2201.
In another case,3 seizure occurred after use of an incense product called “Spicy XXX,” but neither the incense sample nor the patient’s urine was tested for synthetic cannabinoids.
The final case reported in the literature involved a 25-year-old man who was brought to an emergency department by coworkers who had witnessed seizure-like activity.4 He was reported to have smoked an incense product about “45 minutes prior to presentation,”4 indicating that the seizure-like activity happened within that time frame. Two synthetic cannabinoids (JWH-018 and JWH-073) were detected in the patient’s urine.
In the case by Lapoint et al1 that we referred to in our paper,1 seizure activity recurred in the hospital and was successfully treated with lorazepam. The case reported by Schneir and Baumbacher2 described treatment of the second seizure with intranasal midazolam, with no recurrence of seizure activity.
In summary, the literature on seizure activity related to synthetic cannabinoids is sparse. When the time course has been documented in these few cases, seizures seem to occur “soon” after using these products,2 or from 45 minutes to 1 hour after use.1,4 Although benzodiazepines have been used to treat seizure activity, there have been no published reports of using medications to prevent seizures in individuals who have been using spice products. Furthermore, the routine employment of seizure prophylaxis of any kind would probably be premature at this point given the uncertainty of the actual seizure risk among all synthetic cannabinoid users. We would consider giving a benzodiazepine to prevent possible seizures after drug ingestion in cases in which prior seizures have occurred, in cases of extreme excitement or agitation, or in those with marked alterations of mental state.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
- Simmons JR, Skinner CG, Williams J, Kang CS, Schwartz MD, Wills BK. Intoxication from smoking “spice” (letter). Ann Emerg Med 2011; 57:187–188.
- Simmons J, Cookman L, Kang C, Skinner C. Three cases of ‘spice’ exposure. Clin Toxicol (Phila) 2011; 49:431–433.
In Reply: We thank Dr. Chandiramani for his thoughtful comments.
Only four cases of seizure-like activity associated with synthetic cannabinoids have been reported in the literature. In addition to the case reported in our paper,1 there was another in which a 19-year-old had two seizures soon after smoking a spice product, and the second seizure was witnessed by paramedics on the way to the hospital.2 Though this patient’s urine was not analyzed for synthetic cannabinoids, the spice product that was reportedly smoked by the patient was later sent to a laboratory for analysis and was found to contain four synthetic cannabinoids: JWH-018, JWH-081, JWH-250, and AM-2201.
In another case,3 seizure occurred after use of an incense product called “Spicy XXX,” but neither the incense sample nor the patient’s urine was tested for synthetic cannabinoids.
The final case reported in the literature involved a 25-year-old man who was brought to an emergency department by coworkers who had witnessed seizure-like activity.4 He was reported to have smoked an incense product about “45 minutes prior to presentation,”4 indicating that the seizure-like activity happened within that time frame. Two synthetic cannabinoids (JWH-018 and JWH-073) were detected in the patient’s urine.
In the case by Lapoint et al1 that we referred to in our paper,1 seizure activity recurred in the hospital and was successfully treated with lorazepam. The case reported by Schneir and Baumbacher2 described treatment of the second seizure with intranasal midazolam, with no recurrence of seizure activity.
In summary, the literature on seizure activity related to synthetic cannabinoids is sparse. When the time course has been documented in these few cases, seizures seem to occur “soon” after using these products,2 or from 45 minutes to 1 hour after use.1,4 Although benzodiazepines have been used to treat seizure activity, there have been no published reports of using medications to prevent seizures in individuals who have been using spice products. Furthermore, the routine employment of seizure prophylaxis of any kind would probably be premature at this point given the uncertainty of the actual seizure risk among all synthetic cannabinoid users. We would consider giving a benzodiazepine to prevent possible seizures after drug ingestion in cases in which prior seizures have occurred, in cases of extreme excitement or agitation, or in those with marked alterations of mental state.
In Reply: We thank Dr. Chandiramani for his thoughtful comments.
Only four cases of seizure-like activity associated with synthetic cannabinoids have been reported in the literature. In addition to the case reported in our paper,1 there was another in which a 19-year-old had two seizures soon after smoking a spice product, and the second seizure was witnessed by paramedics on the way to the hospital.2 Though this patient’s urine was not analyzed for synthetic cannabinoids, the spice product that was reportedly smoked by the patient was later sent to a laboratory for analysis and was found to contain four synthetic cannabinoids: JWH-018, JWH-081, JWH-250, and AM-2201.
In another case,3 seizure occurred after use of an incense product called “Spicy XXX,” but neither the incense sample nor the patient’s urine was tested for synthetic cannabinoids.
The final case reported in the literature involved a 25-year-old man who was brought to an emergency department by coworkers who had witnessed seizure-like activity.4 He was reported to have smoked an incense product about “45 minutes prior to presentation,”4 indicating that the seizure-like activity happened within that time frame. Two synthetic cannabinoids (JWH-018 and JWH-073) were detected in the patient’s urine.
In the case by Lapoint et al1 that we referred to in our paper,1 seizure activity recurred in the hospital and was successfully treated with lorazepam. The case reported by Schneir and Baumbacher2 described treatment of the second seizure with intranasal midazolam, with no recurrence of seizure activity.
In summary, the literature on seizure activity related to synthetic cannabinoids is sparse. When the time course has been documented in these few cases, seizures seem to occur “soon” after using these products,2 or from 45 minutes to 1 hour after use.1,4 Although benzodiazepines have been used to treat seizure activity, there have been no published reports of using medications to prevent seizures in individuals who have been using spice products. Furthermore, the routine employment of seizure prophylaxis of any kind would probably be premature at this point given the uncertainty of the actual seizure risk among all synthetic cannabinoid users. We would consider giving a benzodiazepine to prevent possible seizures after drug ingestion in cases in which prior seizures have occurred, in cases of extreme excitement or agitation, or in those with marked alterations of mental state.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
- Simmons JR, Skinner CG, Williams J, Kang CS, Schwartz MD, Wills BK. Intoxication from smoking “spice” (letter). Ann Emerg Med 2011; 57:187–188.
- Simmons J, Cookman L, Kang C, Skinner C. Three cases of ‘spice’ exposure. Clin Toxicol (Phila) 2011; 49:431–433.
- Lapoint J, James LP, Moran CL, Nelson LS, Hoffman RS, Moran JH. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol (Phila) 2011; 49:760–764.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
- Simmons JR, Skinner CG, Williams J, Kang CS, Schwartz MD, Wills BK. Intoxication from smoking “spice” (letter). Ann Emerg Med 2011; 57:187–188.
- Simmons J, Cookman L, Kang C, Skinner C. Three cases of ‘spice’ exposure. Clin Toxicol (Phila) 2011; 49:431–433.
Geriatric patient-centered medical home
To the Editor: The discussion by Gennari and colleagues1 on how to obtain certification from the National Committee for Quality Assurance (NCQA) for a geriatric patient-centered medical home was very timely and instructive. The great effort that must be put into getting one’s practice certified was thoroughly documented.
Some community-based physicians will not require financial incentives to undertake this laborious process, finding sufficient reward in continuous quality improvement. However, economic reality dictates that time spent on certification must be taken away from other, productive (ie, income-generating) activities. Therefore, it is reasonable to ask what kind of financial incentives will be provided to physicians who obtain NCQA certification, and which organization or entity will pay for these incentives.
- Gennari A, Fedor K, Bakow E, Resnick NM. A geriatric patient-centered medical home: how to obtain NCQA certification. Cleve Clin J Med 2012; 79:359–366.
To the Editor: The discussion by Gennari and colleagues1 on how to obtain certification from the National Committee for Quality Assurance (NCQA) for a geriatric patient-centered medical home was very timely and instructive. The great effort that must be put into getting one’s practice certified was thoroughly documented.
Some community-based physicians will not require financial incentives to undertake this laborious process, finding sufficient reward in continuous quality improvement. However, economic reality dictates that time spent on certification must be taken away from other, productive (ie, income-generating) activities. Therefore, it is reasonable to ask what kind of financial incentives will be provided to physicians who obtain NCQA certification, and which organization or entity will pay for these incentives.
To the Editor: The discussion by Gennari and colleagues1 on how to obtain certification from the National Committee for Quality Assurance (NCQA) for a geriatric patient-centered medical home was very timely and instructive. The great effort that must be put into getting one’s practice certified was thoroughly documented.
Some community-based physicians will not require financial incentives to undertake this laborious process, finding sufficient reward in continuous quality improvement. However, economic reality dictates that time spent on certification must be taken away from other, productive (ie, income-generating) activities. Therefore, it is reasonable to ask what kind of financial incentives will be provided to physicians who obtain NCQA certification, and which organization or entity will pay for these incentives.
- Gennari A, Fedor K, Bakow E, Resnick NM. A geriatric patient-centered medical home: how to obtain NCQA certification. Cleve Clin J Med 2012; 79:359–366.
- Gennari A, Fedor K, Bakow E, Resnick NM. A geriatric patient-centered medical home: how to obtain NCQA certification. Cleve Clin J Med 2012; 79:359–366.
In reply: Geriatric patient-centered medical home
In Reply: At this time, the financial incentives for acquiring NCQA medical home certification depend on your geographic location. According to a June 5th publication in Health Care Payer News,1 26 states have adopted policies to make payments to healthcare providers that have met medical home standards. These payments and their specific requirements vary from state to state.
Your question underscores the importance of our recommendation to partner with your local health insurance provider. By reaching out to them, you can learn about what incentive programs are in place in your area or are under development. The model that many insurance companies have used is to give higher reimbursements for practices that are medical homes or that meet certain quality insurance markers. If you align your medical home quality insurance markers with your local insurance company’s incentive plan, then your medical home work can translate into real dollars for your practice. This concept of an incentive plan for quality care is becoming more and more prevalent. Furthermore, the public (ie, patients) are also becoming more savvy about the concepts of the medical home and quality. Becoming a medical home has great marketing potential that can turn into financial benefits for a practice, as well.
- Mosquera M. States make progress with medical homes. Healthcare Payer News. June 5, 2012. Available at www.healthcarepayernews.com/content/states-make-progressmedical-homes. Accessed July 5, 2012.
In Reply: At this time, the financial incentives for acquiring NCQA medical home certification depend on your geographic location. According to a June 5th publication in Health Care Payer News,1 26 states have adopted policies to make payments to healthcare providers that have met medical home standards. These payments and their specific requirements vary from state to state.
Your question underscores the importance of our recommendation to partner with your local health insurance provider. By reaching out to them, you can learn about what incentive programs are in place in your area or are under development. The model that many insurance companies have used is to give higher reimbursements for practices that are medical homes or that meet certain quality insurance markers. If you align your medical home quality insurance markers with your local insurance company’s incentive plan, then your medical home work can translate into real dollars for your practice. This concept of an incentive plan for quality care is becoming more and more prevalent. Furthermore, the public (ie, patients) are also becoming more savvy about the concepts of the medical home and quality. Becoming a medical home has great marketing potential that can turn into financial benefits for a practice, as well.
In Reply: At this time, the financial incentives for acquiring NCQA medical home certification depend on your geographic location. According to a June 5th publication in Health Care Payer News,1 26 states have adopted policies to make payments to healthcare providers that have met medical home standards. These payments and their specific requirements vary from state to state.
Your question underscores the importance of our recommendation to partner with your local health insurance provider. By reaching out to them, you can learn about what incentive programs are in place in your area or are under development. The model that many insurance companies have used is to give higher reimbursements for practices that are medical homes or that meet certain quality insurance markers. If you align your medical home quality insurance markers with your local insurance company’s incentive plan, then your medical home work can translate into real dollars for your practice. This concept of an incentive plan for quality care is becoming more and more prevalent. Furthermore, the public (ie, patients) are also becoming more savvy about the concepts of the medical home and quality. Becoming a medical home has great marketing potential that can turn into financial benefits for a practice, as well.
- Mosquera M. States make progress with medical homes. Healthcare Payer News. June 5, 2012. Available at www.healthcarepayernews.com/content/states-make-progressmedical-homes. Accessed July 5, 2012.
- Mosquera M. States make progress with medical homes. Healthcare Payer News. June 5, 2012. Available at www.healthcarepayernews.com/content/states-make-progressmedical-homes. Accessed July 5, 2012.
Autoinflammatory syndromes: Fever is not always a sign of infection
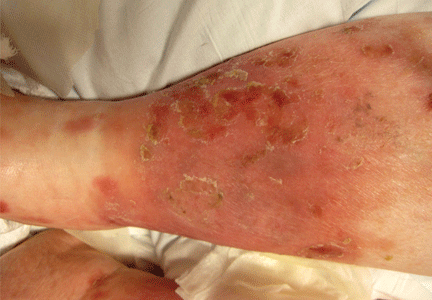
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
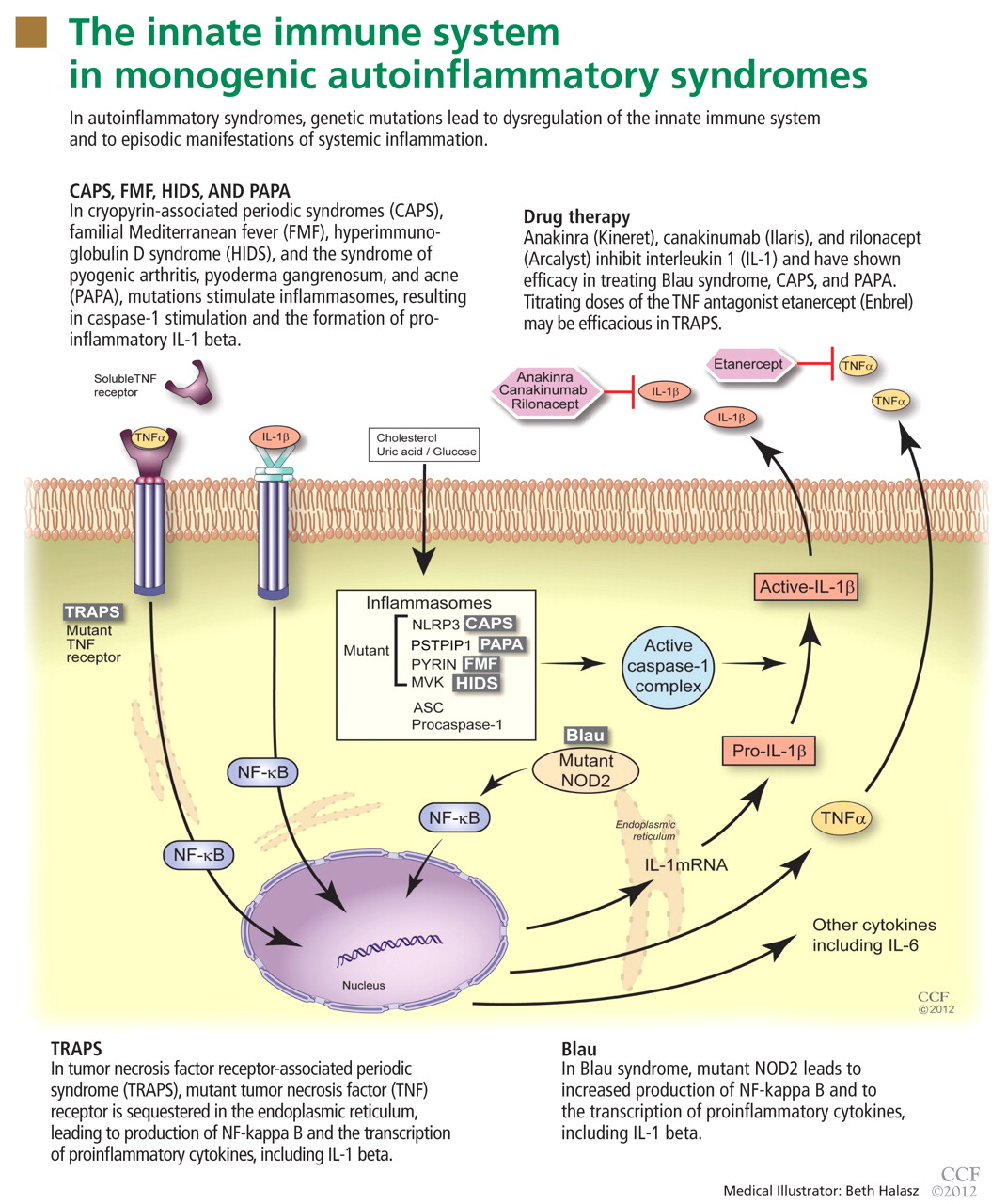
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
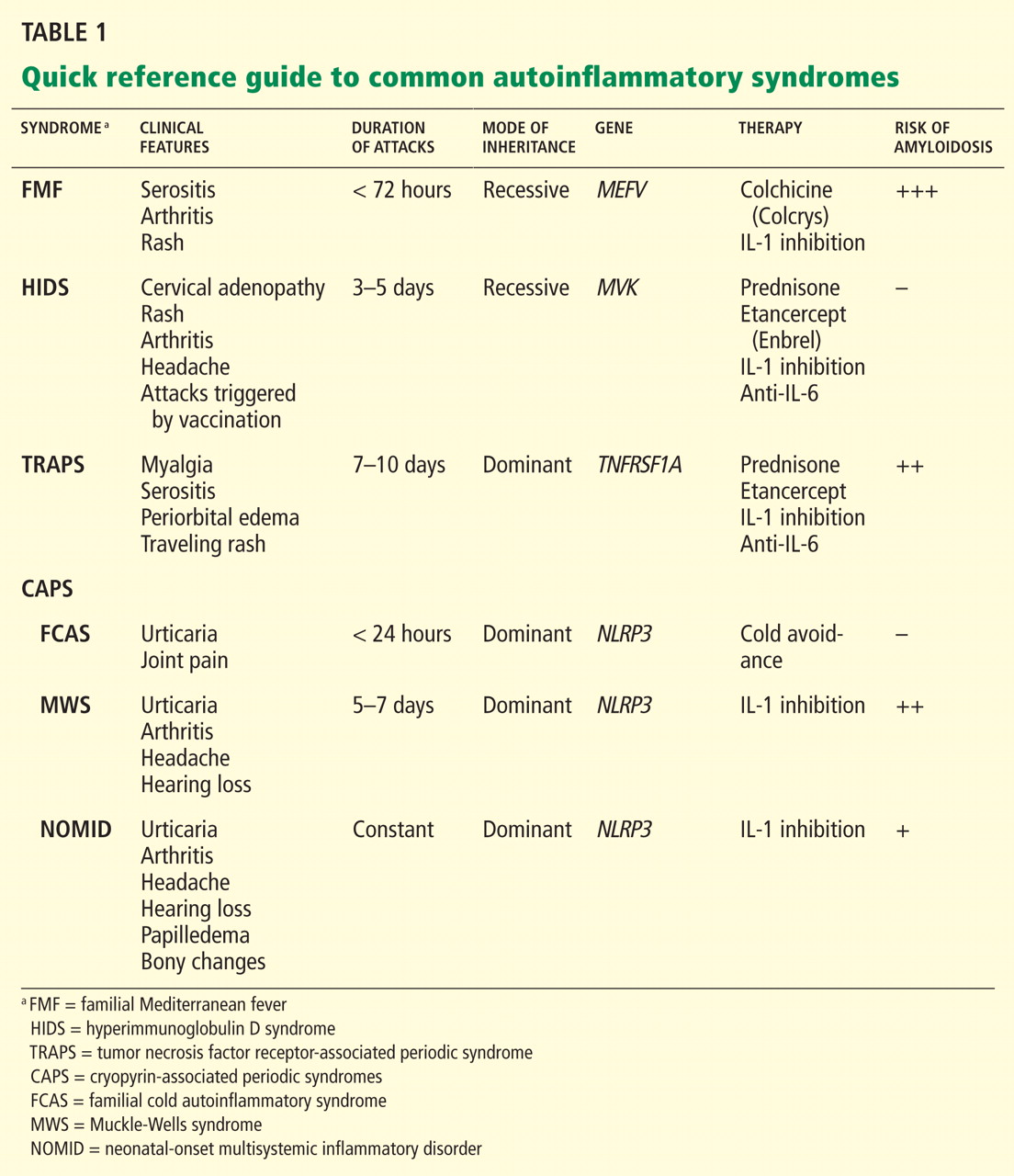
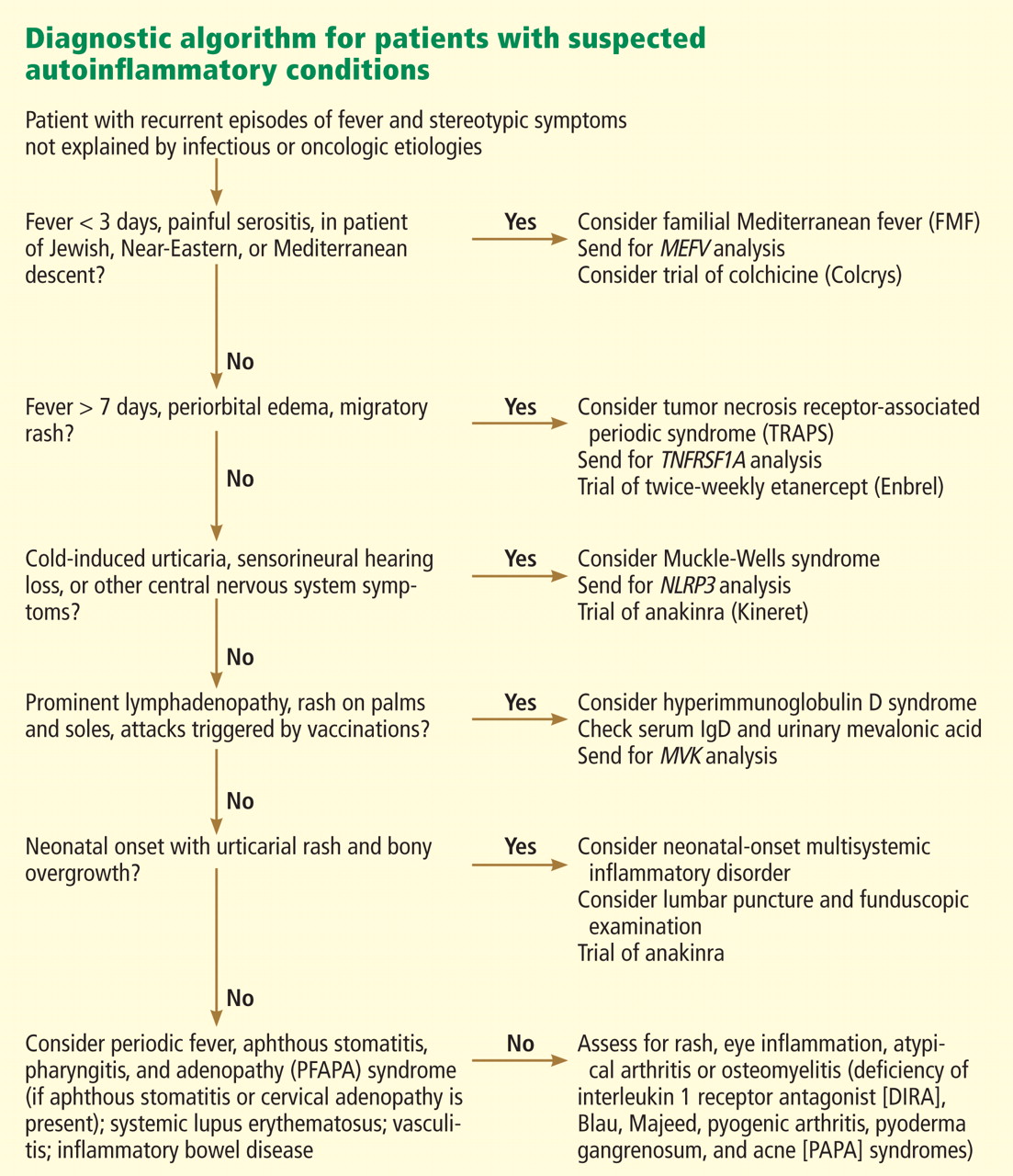
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74

Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74
Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74
Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
KEY POINTS
- In many of the autoinflammatory syndromes, genetic abnormalities and consequent disordered regulation of the innate immune system lead to overactivity of proinflammatory cytokines and subsequent inflammatory symptoms.
- Early recognition and treatment with immunoregulatory agents may improve quality of life and reduce the risk of disease sequelae.
- Abnormal regulation of the innate inflammatory pathway has also been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.
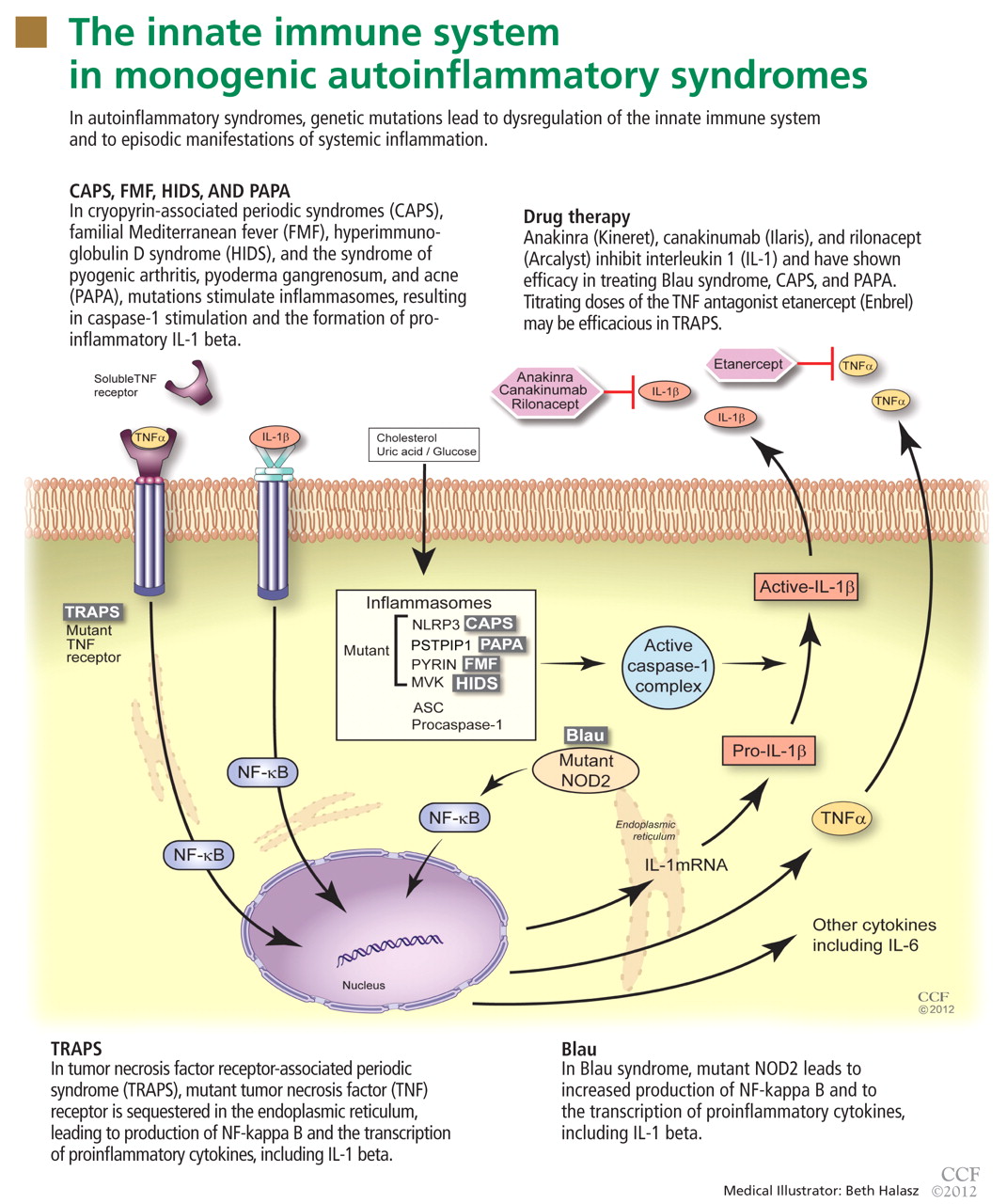
Genetic counselors: Your partners in clinical practice
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?

The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34

At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?
The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34
At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?
The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34
At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
KEY POINTS
- The sequencing of the human genome has provided valuable information about the genetic causes of many conditions, but it has also uncovered tremendous complexities.
- Genetic counselors are master’s-trained allied health care professionals with specific expertise in identifying and educating patients at risk for inherited conditions.
- Genetic testing should not be ordered without informed consent and without appropriate counseling before and after the test.
- Huntington disease, which is inherited in an autosomal dominant manner, illustrates the need for genetic counseling before predictive testing.
- The National Society of Genetic Counselors (www.nsgc.org) and the American Board of Genetic Counseling (www.abgc.net) provide searchable databases of genetic counselors.
Distinguishing cellulitis from its mimics
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
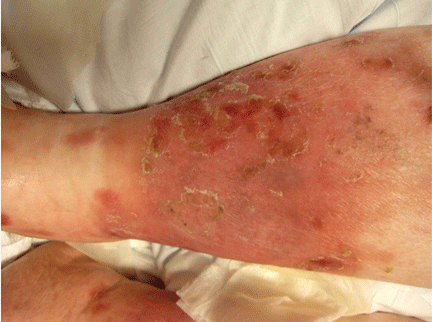
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
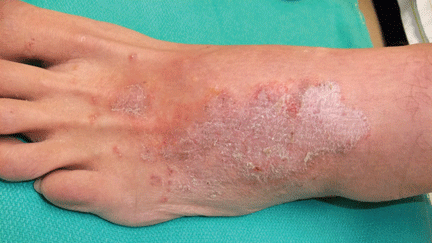
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
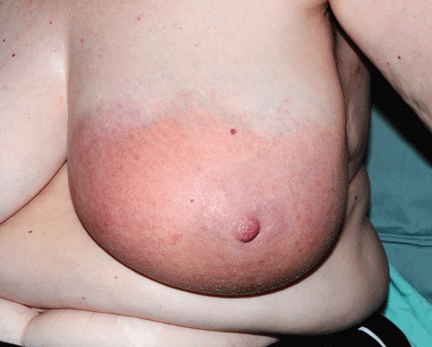
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
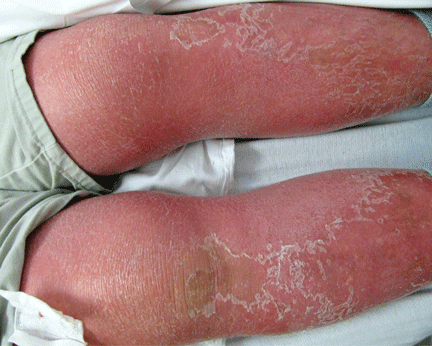
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
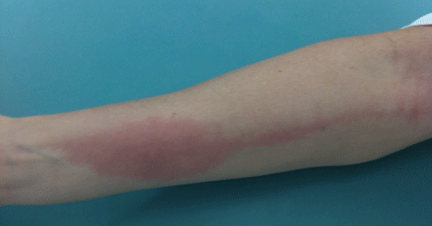
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
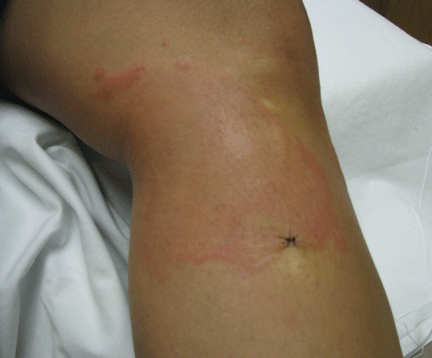
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
KEY POINTS
- Cellulitis is rarely bilateral.
- Patients with cellulitis often have systemic symptoms, such as fever and leukocytosis.
- A chronic course points to a diagnosis other than cellulitis.
- Plaques with a “bound-down” appearance or dark pigmentation point to a chronic disease rather than cellulitis.
- Stasis dermatitis is the most common mimic of cellulitis.