User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Strategic Initiatives for Veterans with Lung Cancer (FULL)
The Veterans Health Administration (VHA) facilitates care for > 7,700 veterans with newly diagnosed lung cancer each year.1 This includes comprehensive clinical evaluations and management that are facilitated through interdisciplinary networks of pulmonologists, radiologists, thoracic surgeons, radiation oncologists, and medical oncologists. Veterans with lung cancer have access to advanced medical technologies at US Department of Veterans Affairs (VA) medical centers (VAMCs), including the latest US Food and Drug Administration (FDA)-approved targeted radiation delivery systems and novel immunotherapies, as well as precision oncology-driven clinical trials.2
Despite access to high-quality care, lung cancer remains the leading cause of cancer-related mortality among VHA enrollees as well as the US population.3 About 15 veterans die of lung cancer each day; most are diagnosed with advanced stage III or stage IV disease. To address this issue, VHA launched 3 new initiatives between 2016 and 2017 to improve outcomes for veterans impacted by lung cancer. The VA Partnership to increase Access to Lung Screening (VA-PALS) is a clinical implementation project to increase access to early detection lung screening scans at 10 VAMCs. The Veterans Affairs Lung cancer surgery Or stereotactic Radiotherapy (VALOR) is a phase 3 randomized trial that investigates the role of stereotactic body radiation therapy (SBRT) as a potential alternative to surgery for veterans with operable stage I non-small cell lung cancer (NSCLC). The VA Radiation Oncology Quality Surveillance program (VA-ROQS) established national expert-derived benchmarks for the quality assurance of lung cancer therapy.
VA-PALS
The central mission of VA-PALS is to reduce lung cancer mortality among veterans at risk by increasing access to low-dose computed tomography (LDCT) lung screening scans.4,5 The program was developed as a public-private partnership to introduce structured lung cancer screening programs at 10 VAMCs to safely manage large cohorts of veterans undergoing annual screening scans. The VA-PALS project brings together pulmonologists, radiologists, thoracic surgeons, radiation oncologists, medical oncologists, and computer scientists who have experience developing open-source electronic health record systems for VHA networks. The project was launched in 2017 after an earlier clinical demonstration project identified substantial variability and challenges with efforts to implement new lung cancer screening programs in the VA.6
Each of the 10 VA-PALS-designated lung cancer screening programs (Atlanta, Georgia; Phoenix, Arizona; Indianapolis, Indiana; Chicago, Illinois; Nashville, Tennessee; Philadelphia, Pennsylvania; St. Louis, Missouri; Denver, Colorado; Milwaukee, Wisconsin; and Cleveland, Ohio) assumes a major responsibility for ordering and evaluating the results of LDCT scans to ensure appropriate follow-up care of veterans with abnormal radiographic findings. Lung cancer screening programs are supported with a full-time navigator (nurse practitioner or physician assistant) who has received training from the VA-PALS project team with direct supervision by a local site director who is a pulmonologist, thoracic surgeon, or medical oncologist. Lung cancer screening programs establish a centralized approach that aims to reduce the burden on primary care providers for remembering to order annual baseline and repeat LDCT scans. The lung screening programs also manage radiographic findings that usually are benign to facilitate appropriate decisions to minimize the risk of unnecessary tests and procedures. Program implementation across VA-PALS sites includes a strong connection among participants through meetings, newsletters, and attendance at conferences to create a collaborative learning network, which has been shown to improve dissemination of best practices across the VHA.7,8
The International Early Lung Cancer Action Program (I-ELCAP), which pioneered the use of LDCT to reduce lung cancer mortality, is a leading partner for VA-PALS.9 This group has > 25 years of experience overcoming many of the obstacles and challenges that new lung cancer screening programs face.10 The I-ELCAP has successfully implemented new lung cancer screening programs at > 70 health care institutions worldwide. Their implementation processes provide continuous oversight for each center. As a result, the I-ELCAP team has developed a large and detailed lung cancer screening registry with > 75,000 patients enrolled globally, comprising a vast database of clinical data that has produced > 270 scientific publications focusing on improving the quality and safety of lung cancer screening.11,12
These reports have helped guide evidence-based recommendations for lung cancer screening in several countries and include standardized processes for patient counseling and smoking cessation, data acquisition and interpretation of LDCT images, and clinical management of abnormal findings to facilitate timely transition from diagnosis to treatment.13-15 The I-ELCAP management system detects 10% abnormal findings in the baseline screening study, which declines to 6% in subsequent years.12 The scientific findings from this approach have provided additional insights into technical CT scanning errors that can affect tumor nodule measurements.16 The vast amount of clinical data and expertise have helped explore genetic markers.17 The I-ELCAP has facilitated cost-effectiveness investigations to determine the value of screening, and their research portfolio includes investigations into the longer-term outcomes after primary treatment for patients with screen-detected lung cancers.18,19
I-ELCAP gifted its comprehensive clinical software management system that has been in use for the above contributions for use in the VHA through an open source agreement without licensing fees. The I-ELCAP software management system was rewritten in MUMPS, the software programming language that is used by the VA Computerized Patient Record System (CPRS). The newly adapted VA-PALS/I-ELCAP system underwent modifications with VHA clinicians’ input, and was successfully installed at the Phoenix VA Health Care System in Arizona, which has assumed a leading role for the VA-PALS project.
The VA-PALS/I-ELCAP clinical management system currently is under review by the VA Office of Information and Technology for broad distribution across the VHA through the VA Enterprise Cloud. Once in use across the VHA, the VA-PALS/I-ELCAP clinical management system will offer a longitudinal central database that can support numerous quality improvement and quality assurance initiatives, as well as innovative research projects. Research opportunities include: (1) large-scale examination of LDCT images with artificial intelligence and machine learning techniques; (2) epidemiologic investigations of environmental and genetic risk factors to better understand the high percentage of veterans diagnosed with lung cancer who were never smokers or had quit many years ago; and (3) multisite clinical trials that explore early detection blood screening tests that are under development.
The VA-PALS project is sponsored by the VHA Office of Rural Health as an enterprise-wide initiative that focuses on reaching rural veterans at risk. The project received additional support through the VA Secretary’s Center for Strategic Partnerships with a $5.8 million grant from the Bristol-Myers Squibb Foundation. The VistA (Veterans Health Information Systems and Technology Architecture) Expertise Network is an additional key partner that helped adapt the VAPALS-ELCAP system for use on VHA networks.
VALOR Trial
The VA Cooperative Studies Program (CSP) #2005 VALOR study is a randomized phase 3 clinical trial that evaluates optimal treatment for participants with operable early-stage NSCLC.20 The trial is sponsored by the CSP, which is responsible for and provides resources for the planning and conduct of large multicenter surgical and clinical trials in VHA.21 The CSP #2005 VALOR study plans to enroll veterans with stage I NSCLC who will be treated with a surgical lobectomy or SBRT according to random assignment. An alternative surgical approach with a segmentectomy is acceptable, although patients in poor health who are only qualify for a wedge resection will not be enrolled. The CSP will follow each participant for at least 5 years to evaluate which treatment, if either, results in a higher overall survival rate. Secondary outcome measures are quality of life, pulmonary function, health state utilities, patterns of failure, and causes of death.
Although the study design of the VALOR trial is relatively straightforward, recruitment of participants to similar randomized trials of surgery vs SBRT for operable stage I NSCLC outside the VA has historically been very difficult. Three earlier phase 3 trials in the Netherlands and US closed prematurely after collectively enrolling only 4% of planned participants. Although a pooled analysis of 2 of these trials demonstrated a statistically significant difference of 95% vs 79% survival in favor of SBRT at a median follow-up of 40 months, the analysis was underpowered because only 58 of the planned 1,380 participants were enrolled.22,23
The CSP #2005 VALOR study team was keenly aware of these past challenges and addressed many of the obstacles to enrollment by optimizing eligibility criteria and follow-up requirements. Enrollment sites were carefully selected after confirming equipoise between the 2 treatments, and study coordinators at each enrollment site were empowered to provide a leading role with recruitment. Multiple communication channels were established for constant contact to disseminate new best practices for recruitment as they were identified. Furthermore, a veteran-centric educational recruitment video, approved by the VA Central Institutional Review Board, was designed to help study participants better understand the purpose of participating in a clinical trial (www.vacsp.research.va.gov/CSP_2005/CSP_2005.asp).
After the first year of recruitment, researchers identified individual clinician and patient preferences as the predominant difficulty with recruitment, which was not easy to address. The CSP #2005 VALOR study team opted to partner directly with the Qualitative Research Integrated within Trials (QuinteT) team in the United Kingdom to adopt its methods to successfully support randomized clinical trials with serious recruitment challenges.24,25 By working directly with the QuinteT director, the CSP #2005 VALOR team made a major revision to the informed consent forms by shifting focus away from disclosing potential harms of research to an informative document that emphasized the purpose of the study. The work with QuinteT also led to the creation of balanced narratives for study teams to use and for potential participants to read. These provide a more consistent message that describes why the study is important and why clinicians are no longer certain that surgery is the optimal treatment for all patients with operable stage I NSCLC.
The VALOR clinical trial, opened in 2017, remains open at only 9 VAMCs. As of early 2020, it has enrolled more participants than all previous phase 3 trials combined. Once completed, the results from CSP #2005 VALOR study will help clinicians and veterans with operable stage I NSCLC better understand the tradeoffs of surgery vs SBRT as an initial treatment option. Plans are under way to expand the scope of the trial and include investigations of pretreatment radiomic signatures and genetic markers from biopsy tissue and blood samples, to better predict when surgery or SBRT might be the best treatment option for an individual patient.
VA-ROQS
The VA-ROQS was created in 2016 to compare treatment of veterans with lung cancer in the VHA with quality standards recommended by nationally recognized experts in lung cancer care. Partnering with Washington University in St. Louis, Missouri and the American Society for Radiation Oncology, the VHA established a blue-ribbon panel of experts to review clinical trial data and medical literature to provide evidence-based quality metrics for lung cancer therapy. As a result, 26 metrics applicable to each patient’s case were developed, published, and used to assess lung cancer care in each VHA radiation oncology practice.26
By 2019, the resulting data led to a report on 773 lung cancer cases accumulated from all VHA radiation oncology practices. Performance data for each quality metric were compared for each practice within the VHA, which found that VHA practices met > 80% of all 1,278 metrics scored. Quality metrics included those documented within each patient health record and the specific radiation delivery parameters that reflected each health care provider’s treatment. After team investigators visited each center and recorded treatment data, VA-ROQS is now maturing to permit continuous, electronic monitoring of all lung cancer treatment delivered within VHA. As each veteran’s case is planned, the quality of the therapy is monitored, assessed, and reported to the treating physician. Each VHA radiation oncologist will receive up-to-date evaluation of each case compared with these evidence-based quality standards. The quality standards are reviewed by the blue-ribbon panel to keep the process current and valid.
Future of VHA Lung Cancer Care
As VHA continues to prioritize resources to improve and assure optimal outcomes for veterans with lung cancer, it is now looking to create a national network of Lung Cancer Centers of Excellence (LCCE) as described in the VA Budget Submission for fiscal year 2021. If Congress approves funding, LCCEs will soon be developed within the VA regional Veteran Integrated Service Network system to ensure that treatment decisions for veterans with lung cancer are based on all available molecular information, including data on pharmacogenomic profiles. Such a network would create more opportunities to leverage public–private partnerships similar to the VA-PALS project. Creation of LCCEs would help the VA leverage an even stronger learning network to support more research so that all veterans who are impacted by lung cancer have access to personalized care that optimizes safety, quality of life, and overall survival. The lessons learned, networks developed, and partnerships established through VA-PALS, VALOR, and VA-ROQS are instrumental toward achieving these goals.
1. Zullig LL, Jackson GL, Dorn RA, et al. Cancer incidence among patients of the U.S. Veterans Affairs Health Care System. Mil Med. 2012;177(6):693-701. doi:10.7205/milmed-d-11-00434
2. Dawson GA, Cheuk AV, Lutz S, et al. The availability of advanced radiation oncology technology within the Veterans Health Administration radiation oncology centers. Fed Pract. 2016;33(suppl 4):18S-22S.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
4. National Lung Screening Trial Research Team. Lung cancer incidence and mortality with extended follow-up in the National Lung Screening Trial. J Thorac Oncol. 2019;14(10):1732-1742. doi:10.1016/j.jtho.2019.05.044
5. de Koning HJ, van der Aalst CM, de Jong PA, et al. Reduced lung-cancer mortality with volume CT Screening in a randomized trial. N Engl J Med. 2020;382(6):503-513. doi:10.1056/NEJMoa1911793
6. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406. doi:10.1001/jamainternmed.2016.9022
7. Clancy C. Creating World-class care and service for our nation’s finest: how Veterans Health Administration Diffusion of Excellence Initiative Is innovating and transforming Veterans Affairs health care. Perm J. 2019;23:18.301. doi:10.7812/TPP/18.301
8. Elnahal SM, Clancy CM, Shulkin DJ. A framework for disseminating clinical best practices in the VA health system. JAMA. 2017;317(3):255-256. doi:10.1001/jama.2016.18764
9. Henschke CI, McCauley DI, Yankelevitz DF, et al. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet. 1999;354(9173):99-105. doi:10.1016/S0140-6736(99)06093-6
10. Mulshine JL, Henschke CI. Lung cancer screening: achieving more by intervening less. Lancet Oncol. 2014;15(12):1284-1285. doi:10.1016/S1470-2045(14)70418-8
11. Henschke CI, Li K, Yip R, Salvatore M, Yankelevitz DF. The importance of the regimen of screening in maximizing the benefit and minimizing the harms. Ann Transl Med. 2016;4(8):153. doi:10.21037/atm.2016.04.06
12. Henschke CI, Yip R, Yankelevitz DF, Smith JP; International Early Lung Cancer Action Program Investigators*. Definition of a positive test result in computed tomography screening for lung cancer: a cohort study. Ann Intern Med. 2013;158(4):246-252. doi:10.7326/0003-4819-158-4-201302190-00004
13. Zeliadt SB, Heffner JL, Sayre G, et al. Attitudes and perceptions about smoking cessation in the context of lung cancer screening. JAMA Intern Med. 2015;175(9):1530-1537. doi:10.1001/jamainternmed.2015.3558
14. Henschke CI, Yankelevitz DF, Yip R, et al. Tumor volume measurement error using computed tomography imaging in a phase II clinical trial in lung cancer. J Med Imaging (Bellingham). 2016;3(3):035505. doi:10.1117/1.JMI.3.3.035505
15. Yip R, Henschke CI, Yankelevitz DF, Boffetta P, Smith JP; International Early Lung Cancer Investigators. The impact of the regimen of screening on lung cancer cure: a comparison of I-ELCAP and NLST. Eur J Cancer Prev. 2015;24(3):201-208. doi:10.1097/CEJ.0000000000000065
16. Armato SG 3rd, McLennan G, Bidaut L, et al. The Lung Image Database Consortium (LIDC) and Image Database Resource Initiative (IDRI): a completed reference database of lung nodules on CT scans. Med Phys. 2011;38(2):915-931. doi:10.1118/1.3528204
17. Gill RK, Vazquez MF, Kramer A, et al. The use of genetic markers to identify lung cancer in fine needle aspiration samples. Clin Cancer Res. 2008;14(22):7481-7487. doi:10.1158/1078-0432.CCR-07-5242
18. Pyenson BS, Henschke CI, Yankelevitz DF, Yip R, Dec E. Offering lung cancer screening to high-risk medicare beneficiaries saves lives and is cost-effective: an actuarial analysis. Am Health Drug Benefits. 2014;7(5):272-282.
19. Schwartz RM, Yip R, Olkin I, et al. Impact of surgery for stage IA non-small-cell lung cancer on patient quality of life. J Community Support Oncol. 2016;14(1):37-44. doi:10.12788/jcso.0205
20. Moghanaki D, Chang JY. Is surgery still the optimal treatment for stage I non-small cell lung cancer? Transl Lung Cancer Res. 2016;5(2):183-189. doi:10.21037/tlcr.2016.04.05
21. Bakaeen FG, Reda DJ, Gelijns AC, et al. Department of Veterans Affairs Cooperative Studies Program network of dedicated enrollment sites: implications for surgical trials [published correction appears in JAMA Surg. 2014 Sep;149(9):961]. JAMA Surg. 2014;149(6):507-513. doi:10.1001/jamasurg.2013.4150
22. Chang JY, Senan S, Paul MA, et al. Stereotactic ablative radiotherapy versus lobectomy for operable stage I non-small-cell lung cancer: a pooled analysis of two randomised trials [published correction appears in Lancet Oncol. 2015 Sep;16(9):e427]. Lancet Oncol. 2015;16(6):630-637. doi:10.1016/S1470-2045(15)70168-3
23. Samson P, Keogan K, Crabtree T, et al. Interpreting survival data from clinical trials of surgery versus stereotactic body radiation therapy in operable Stage I non-small cell lung cancer patients. Lung Cancer. 2017;103:6-10. doi:10.1016/j.lungcan.2016.11.005
24. Donovan JL, Rooshenas L, Jepson M, et al. Optimising recruitment and informed consent in randomised controlled trials: the development and implementation of the Quintet Recruitment Intervention (QRI). Trials. 2016;17(1):283. Published 2016 Jun 8. doi:10.1186/s13063-016-1391-4
25. Rooshenas L, Scott LJ, Blazeby JM, et al. The QuinteT Recruitment Intervention supported five randomized trials to recruit to target: a mixed-methods evaluation. J Clin Epidemiol. 2019;106:108-120. doi:10.1016/j.jclinepi.2018.10.004
26. Hagan M, Kapoor R, Michalski J, et al. VA-Radiation Oncology Quality Surveillance Program. Int J Radiat Oncol Biol Phys. 2020;106(3):639-647. doi:10.1016/j.ijrobp.2019.08.064
The Veterans Health Administration (VHA) facilitates care for > 7,700 veterans with newly diagnosed lung cancer each year.1 This includes comprehensive clinical evaluations and management that are facilitated through interdisciplinary networks of pulmonologists, radiologists, thoracic surgeons, radiation oncologists, and medical oncologists. Veterans with lung cancer have access to advanced medical technologies at US Department of Veterans Affairs (VA) medical centers (VAMCs), including the latest US Food and Drug Administration (FDA)-approved targeted radiation delivery systems and novel immunotherapies, as well as precision oncology-driven clinical trials.2
Despite access to high-quality care, lung cancer remains the leading cause of cancer-related mortality among VHA enrollees as well as the US population.3 About 15 veterans die of lung cancer each day; most are diagnosed with advanced stage III or stage IV disease. To address this issue, VHA launched 3 new initiatives between 2016 and 2017 to improve outcomes for veterans impacted by lung cancer. The VA Partnership to increase Access to Lung Screening (VA-PALS) is a clinical implementation project to increase access to early detection lung screening scans at 10 VAMCs. The Veterans Affairs Lung cancer surgery Or stereotactic Radiotherapy (VALOR) is a phase 3 randomized trial that investigates the role of stereotactic body radiation therapy (SBRT) as a potential alternative to surgery for veterans with operable stage I non-small cell lung cancer (NSCLC). The VA Radiation Oncology Quality Surveillance program (VA-ROQS) established national expert-derived benchmarks for the quality assurance of lung cancer therapy.
VA-PALS
The central mission of VA-PALS is to reduce lung cancer mortality among veterans at risk by increasing access to low-dose computed tomography (LDCT) lung screening scans.4,5 The program was developed as a public-private partnership to introduce structured lung cancer screening programs at 10 VAMCs to safely manage large cohorts of veterans undergoing annual screening scans. The VA-PALS project brings together pulmonologists, radiologists, thoracic surgeons, radiation oncologists, medical oncologists, and computer scientists who have experience developing open-source electronic health record systems for VHA networks. The project was launched in 2017 after an earlier clinical demonstration project identified substantial variability and challenges with efforts to implement new lung cancer screening programs in the VA.6
Each of the 10 VA-PALS-designated lung cancer screening programs (Atlanta, Georgia; Phoenix, Arizona; Indianapolis, Indiana; Chicago, Illinois; Nashville, Tennessee; Philadelphia, Pennsylvania; St. Louis, Missouri; Denver, Colorado; Milwaukee, Wisconsin; and Cleveland, Ohio) assumes a major responsibility for ordering and evaluating the results of LDCT scans to ensure appropriate follow-up care of veterans with abnormal radiographic findings. Lung cancer screening programs are supported with a full-time navigator (nurse practitioner or physician assistant) who has received training from the VA-PALS project team with direct supervision by a local site director who is a pulmonologist, thoracic surgeon, or medical oncologist. Lung cancer screening programs establish a centralized approach that aims to reduce the burden on primary care providers for remembering to order annual baseline and repeat LDCT scans. The lung screening programs also manage radiographic findings that usually are benign to facilitate appropriate decisions to minimize the risk of unnecessary tests and procedures. Program implementation across VA-PALS sites includes a strong connection among participants through meetings, newsletters, and attendance at conferences to create a collaborative learning network, which has been shown to improve dissemination of best practices across the VHA.7,8
The International Early Lung Cancer Action Program (I-ELCAP), which pioneered the use of LDCT to reduce lung cancer mortality, is a leading partner for VA-PALS.9 This group has > 25 years of experience overcoming many of the obstacles and challenges that new lung cancer screening programs face.10 The I-ELCAP has successfully implemented new lung cancer screening programs at > 70 health care institutions worldwide. Their implementation processes provide continuous oversight for each center. As a result, the I-ELCAP team has developed a large and detailed lung cancer screening registry with > 75,000 patients enrolled globally, comprising a vast database of clinical data that has produced > 270 scientific publications focusing on improving the quality and safety of lung cancer screening.11,12
These reports have helped guide evidence-based recommendations for lung cancer screening in several countries and include standardized processes for patient counseling and smoking cessation, data acquisition and interpretation of LDCT images, and clinical management of abnormal findings to facilitate timely transition from diagnosis to treatment.13-15 The I-ELCAP management system detects 10% abnormal findings in the baseline screening study, which declines to 6% in subsequent years.12 The scientific findings from this approach have provided additional insights into technical CT scanning errors that can affect tumor nodule measurements.16 The vast amount of clinical data and expertise have helped explore genetic markers.17 The I-ELCAP has facilitated cost-effectiveness investigations to determine the value of screening, and their research portfolio includes investigations into the longer-term outcomes after primary treatment for patients with screen-detected lung cancers.18,19
I-ELCAP gifted its comprehensive clinical software management system that has been in use for the above contributions for use in the VHA through an open source agreement without licensing fees. The I-ELCAP software management system was rewritten in MUMPS, the software programming language that is used by the VA Computerized Patient Record System (CPRS). The newly adapted VA-PALS/I-ELCAP system underwent modifications with VHA clinicians’ input, and was successfully installed at the Phoenix VA Health Care System in Arizona, which has assumed a leading role for the VA-PALS project.
The VA-PALS/I-ELCAP clinical management system currently is under review by the VA Office of Information and Technology for broad distribution across the VHA through the VA Enterprise Cloud. Once in use across the VHA, the VA-PALS/I-ELCAP clinical management system will offer a longitudinal central database that can support numerous quality improvement and quality assurance initiatives, as well as innovative research projects. Research opportunities include: (1) large-scale examination of LDCT images with artificial intelligence and machine learning techniques; (2) epidemiologic investigations of environmental and genetic risk factors to better understand the high percentage of veterans diagnosed with lung cancer who were never smokers or had quit many years ago; and (3) multisite clinical trials that explore early detection blood screening tests that are under development.
The VA-PALS project is sponsored by the VHA Office of Rural Health as an enterprise-wide initiative that focuses on reaching rural veterans at risk. The project received additional support through the VA Secretary’s Center for Strategic Partnerships with a $5.8 million grant from the Bristol-Myers Squibb Foundation. The VistA (Veterans Health Information Systems and Technology Architecture) Expertise Network is an additional key partner that helped adapt the VAPALS-ELCAP system for use on VHA networks.
VALOR Trial
The VA Cooperative Studies Program (CSP) #2005 VALOR study is a randomized phase 3 clinical trial that evaluates optimal treatment for participants with operable early-stage NSCLC.20 The trial is sponsored by the CSP, which is responsible for and provides resources for the planning and conduct of large multicenter surgical and clinical trials in VHA.21 The CSP #2005 VALOR study plans to enroll veterans with stage I NSCLC who will be treated with a surgical lobectomy or SBRT according to random assignment. An alternative surgical approach with a segmentectomy is acceptable, although patients in poor health who are only qualify for a wedge resection will not be enrolled. The CSP will follow each participant for at least 5 years to evaluate which treatment, if either, results in a higher overall survival rate. Secondary outcome measures are quality of life, pulmonary function, health state utilities, patterns of failure, and causes of death.
Although the study design of the VALOR trial is relatively straightforward, recruitment of participants to similar randomized trials of surgery vs SBRT for operable stage I NSCLC outside the VA has historically been very difficult. Three earlier phase 3 trials in the Netherlands and US closed prematurely after collectively enrolling only 4% of planned participants. Although a pooled analysis of 2 of these trials demonstrated a statistically significant difference of 95% vs 79% survival in favor of SBRT at a median follow-up of 40 months, the analysis was underpowered because only 58 of the planned 1,380 participants were enrolled.22,23
The CSP #2005 VALOR study team was keenly aware of these past challenges and addressed many of the obstacles to enrollment by optimizing eligibility criteria and follow-up requirements. Enrollment sites were carefully selected after confirming equipoise between the 2 treatments, and study coordinators at each enrollment site were empowered to provide a leading role with recruitment. Multiple communication channels were established for constant contact to disseminate new best practices for recruitment as they were identified. Furthermore, a veteran-centric educational recruitment video, approved by the VA Central Institutional Review Board, was designed to help study participants better understand the purpose of participating in a clinical trial (www.vacsp.research.va.gov/CSP_2005/CSP_2005.asp).
After the first year of recruitment, researchers identified individual clinician and patient preferences as the predominant difficulty with recruitment, which was not easy to address. The CSP #2005 VALOR study team opted to partner directly with the Qualitative Research Integrated within Trials (QuinteT) team in the United Kingdom to adopt its methods to successfully support randomized clinical trials with serious recruitment challenges.24,25 By working directly with the QuinteT director, the CSP #2005 VALOR team made a major revision to the informed consent forms by shifting focus away from disclosing potential harms of research to an informative document that emphasized the purpose of the study. The work with QuinteT also led to the creation of balanced narratives for study teams to use and for potential participants to read. These provide a more consistent message that describes why the study is important and why clinicians are no longer certain that surgery is the optimal treatment for all patients with operable stage I NSCLC.
The VALOR clinical trial, opened in 2017, remains open at only 9 VAMCs. As of early 2020, it has enrolled more participants than all previous phase 3 trials combined. Once completed, the results from CSP #2005 VALOR study will help clinicians and veterans with operable stage I NSCLC better understand the tradeoffs of surgery vs SBRT as an initial treatment option. Plans are under way to expand the scope of the trial and include investigations of pretreatment radiomic signatures and genetic markers from biopsy tissue and blood samples, to better predict when surgery or SBRT might be the best treatment option for an individual patient.
VA-ROQS
The VA-ROQS was created in 2016 to compare treatment of veterans with lung cancer in the VHA with quality standards recommended by nationally recognized experts in lung cancer care. Partnering with Washington University in St. Louis, Missouri and the American Society for Radiation Oncology, the VHA established a blue-ribbon panel of experts to review clinical trial data and medical literature to provide evidence-based quality metrics for lung cancer therapy. As a result, 26 metrics applicable to each patient’s case were developed, published, and used to assess lung cancer care in each VHA radiation oncology practice.26
By 2019, the resulting data led to a report on 773 lung cancer cases accumulated from all VHA radiation oncology practices. Performance data for each quality metric were compared for each practice within the VHA, which found that VHA practices met > 80% of all 1,278 metrics scored. Quality metrics included those documented within each patient health record and the specific radiation delivery parameters that reflected each health care provider’s treatment. After team investigators visited each center and recorded treatment data, VA-ROQS is now maturing to permit continuous, electronic monitoring of all lung cancer treatment delivered within VHA. As each veteran’s case is planned, the quality of the therapy is monitored, assessed, and reported to the treating physician. Each VHA radiation oncologist will receive up-to-date evaluation of each case compared with these evidence-based quality standards. The quality standards are reviewed by the blue-ribbon panel to keep the process current and valid.
Future of VHA Lung Cancer Care
As VHA continues to prioritize resources to improve and assure optimal outcomes for veterans with lung cancer, it is now looking to create a national network of Lung Cancer Centers of Excellence (LCCE) as described in the VA Budget Submission for fiscal year 2021. If Congress approves funding, LCCEs will soon be developed within the VA regional Veteran Integrated Service Network system to ensure that treatment decisions for veterans with lung cancer are based on all available molecular information, including data on pharmacogenomic profiles. Such a network would create more opportunities to leverage public–private partnerships similar to the VA-PALS project. Creation of LCCEs would help the VA leverage an even stronger learning network to support more research so that all veterans who are impacted by lung cancer have access to personalized care that optimizes safety, quality of life, and overall survival. The lessons learned, networks developed, and partnerships established through VA-PALS, VALOR, and VA-ROQS are instrumental toward achieving these goals.
The Veterans Health Administration (VHA) facilitates care for > 7,700 veterans with newly diagnosed lung cancer each year.1 This includes comprehensive clinical evaluations and management that are facilitated through interdisciplinary networks of pulmonologists, radiologists, thoracic surgeons, radiation oncologists, and medical oncologists. Veterans with lung cancer have access to advanced medical technologies at US Department of Veterans Affairs (VA) medical centers (VAMCs), including the latest US Food and Drug Administration (FDA)-approved targeted radiation delivery systems and novel immunotherapies, as well as precision oncology-driven clinical trials.2
Despite access to high-quality care, lung cancer remains the leading cause of cancer-related mortality among VHA enrollees as well as the US population.3 About 15 veterans die of lung cancer each day; most are diagnosed with advanced stage III or stage IV disease. To address this issue, VHA launched 3 new initiatives between 2016 and 2017 to improve outcomes for veterans impacted by lung cancer. The VA Partnership to increase Access to Lung Screening (VA-PALS) is a clinical implementation project to increase access to early detection lung screening scans at 10 VAMCs. The Veterans Affairs Lung cancer surgery Or stereotactic Radiotherapy (VALOR) is a phase 3 randomized trial that investigates the role of stereotactic body radiation therapy (SBRT) as a potential alternative to surgery for veterans with operable stage I non-small cell lung cancer (NSCLC). The VA Radiation Oncology Quality Surveillance program (VA-ROQS) established national expert-derived benchmarks for the quality assurance of lung cancer therapy.
VA-PALS
The central mission of VA-PALS is to reduce lung cancer mortality among veterans at risk by increasing access to low-dose computed tomography (LDCT) lung screening scans.4,5 The program was developed as a public-private partnership to introduce structured lung cancer screening programs at 10 VAMCs to safely manage large cohorts of veterans undergoing annual screening scans. The VA-PALS project brings together pulmonologists, radiologists, thoracic surgeons, radiation oncologists, medical oncologists, and computer scientists who have experience developing open-source electronic health record systems for VHA networks. The project was launched in 2017 after an earlier clinical demonstration project identified substantial variability and challenges with efforts to implement new lung cancer screening programs in the VA.6
Each of the 10 VA-PALS-designated lung cancer screening programs (Atlanta, Georgia; Phoenix, Arizona; Indianapolis, Indiana; Chicago, Illinois; Nashville, Tennessee; Philadelphia, Pennsylvania; St. Louis, Missouri; Denver, Colorado; Milwaukee, Wisconsin; and Cleveland, Ohio) assumes a major responsibility for ordering and evaluating the results of LDCT scans to ensure appropriate follow-up care of veterans with abnormal radiographic findings. Lung cancer screening programs are supported with a full-time navigator (nurse practitioner or physician assistant) who has received training from the VA-PALS project team with direct supervision by a local site director who is a pulmonologist, thoracic surgeon, or medical oncologist. Lung cancer screening programs establish a centralized approach that aims to reduce the burden on primary care providers for remembering to order annual baseline and repeat LDCT scans. The lung screening programs also manage radiographic findings that usually are benign to facilitate appropriate decisions to minimize the risk of unnecessary tests and procedures. Program implementation across VA-PALS sites includes a strong connection among participants through meetings, newsletters, and attendance at conferences to create a collaborative learning network, which has been shown to improve dissemination of best practices across the VHA.7,8
The International Early Lung Cancer Action Program (I-ELCAP), which pioneered the use of LDCT to reduce lung cancer mortality, is a leading partner for VA-PALS.9 This group has > 25 years of experience overcoming many of the obstacles and challenges that new lung cancer screening programs face.10 The I-ELCAP has successfully implemented new lung cancer screening programs at > 70 health care institutions worldwide. Their implementation processes provide continuous oversight for each center. As a result, the I-ELCAP team has developed a large and detailed lung cancer screening registry with > 75,000 patients enrolled globally, comprising a vast database of clinical data that has produced > 270 scientific publications focusing on improving the quality and safety of lung cancer screening.11,12
These reports have helped guide evidence-based recommendations for lung cancer screening in several countries and include standardized processes for patient counseling and smoking cessation, data acquisition and interpretation of LDCT images, and clinical management of abnormal findings to facilitate timely transition from diagnosis to treatment.13-15 The I-ELCAP management system detects 10% abnormal findings in the baseline screening study, which declines to 6% in subsequent years.12 The scientific findings from this approach have provided additional insights into technical CT scanning errors that can affect tumor nodule measurements.16 The vast amount of clinical data and expertise have helped explore genetic markers.17 The I-ELCAP has facilitated cost-effectiveness investigations to determine the value of screening, and their research portfolio includes investigations into the longer-term outcomes after primary treatment for patients with screen-detected lung cancers.18,19
I-ELCAP gifted its comprehensive clinical software management system that has been in use for the above contributions for use in the VHA through an open source agreement without licensing fees. The I-ELCAP software management system was rewritten in MUMPS, the software programming language that is used by the VA Computerized Patient Record System (CPRS). The newly adapted VA-PALS/I-ELCAP system underwent modifications with VHA clinicians’ input, and was successfully installed at the Phoenix VA Health Care System in Arizona, which has assumed a leading role for the VA-PALS project.
The VA-PALS/I-ELCAP clinical management system currently is under review by the VA Office of Information and Technology for broad distribution across the VHA through the VA Enterprise Cloud. Once in use across the VHA, the VA-PALS/I-ELCAP clinical management system will offer a longitudinal central database that can support numerous quality improvement and quality assurance initiatives, as well as innovative research projects. Research opportunities include: (1) large-scale examination of LDCT images with artificial intelligence and machine learning techniques; (2) epidemiologic investigations of environmental and genetic risk factors to better understand the high percentage of veterans diagnosed with lung cancer who were never smokers or had quit many years ago; and (3) multisite clinical trials that explore early detection blood screening tests that are under development.
The VA-PALS project is sponsored by the VHA Office of Rural Health as an enterprise-wide initiative that focuses on reaching rural veterans at risk. The project received additional support through the VA Secretary’s Center for Strategic Partnerships with a $5.8 million grant from the Bristol-Myers Squibb Foundation. The VistA (Veterans Health Information Systems and Technology Architecture) Expertise Network is an additional key partner that helped adapt the VAPALS-ELCAP system for use on VHA networks.
VALOR Trial
The VA Cooperative Studies Program (CSP) #2005 VALOR study is a randomized phase 3 clinical trial that evaluates optimal treatment for participants with operable early-stage NSCLC.20 The trial is sponsored by the CSP, which is responsible for and provides resources for the planning and conduct of large multicenter surgical and clinical trials in VHA.21 The CSP #2005 VALOR study plans to enroll veterans with stage I NSCLC who will be treated with a surgical lobectomy or SBRT according to random assignment. An alternative surgical approach with a segmentectomy is acceptable, although patients in poor health who are only qualify for a wedge resection will not be enrolled. The CSP will follow each participant for at least 5 years to evaluate which treatment, if either, results in a higher overall survival rate. Secondary outcome measures are quality of life, pulmonary function, health state utilities, patterns of failure, and causes of death.
Although the study design of the VALOR trial is relatively straightforward, recruitment of participants to similar randomized trials of surgery vs SBRT for operable stage I NSCLC outside the VA has historically been very difficult. Three earlier phase 3 trials in the Netherlands and US closed prematurely after collectively enrolling only 4% of planned participants. Although a pooled analysis of 2 of these trials demonstrated a statistically significant difference of 95% vs 79% survival in favor of SBRT at a median follow-up of 40 months, the analysis was underpowered because only 58 of the planned 1,380 participants were enrolled.22,23
The CSP #2005 VALOR study team was keenly aware of these past challenges and addressed many of the obstacles to enrollment by optimizing eligibility criteria and follow-up requirements. Enrollment sites were carefully selected after confirming equipoise between the 2 treatments, and study coordinators at each enrollment site were empowered to provide a leading role with recruitment. Multiple communication channels were established for constant contact to disseminate new best practices for recruitment as they were identified. Furthermore, a veteran-centric educational recruitment video, approved by the VA Central Institutional Review Board, was designed to help study participants better understand the purpose of participating in a clinical trial (www.vacsp.research.va.gov/CSP_2005/CSP_2005.asp).
After the first year of recruitment, researchers identified individual clinician and patient preferences as the predominant difficulty with recruitment, which was not easy to address. The CSP #2005 VALOR study team opted to partner directly with the Qualitative Research Integrated within Trials (QuinteT) team in the United Kingdom to adopt its methods to successfully support randomized clinical trials with serious recruitment challenges.24,25 By working directly with the QuinteT director, the CSP #2005 VALOR team made a major revision to the informed consent forms by shifting focus away from disclosing potential harms of research to an informative document that emphasized the purpose of the study. The work with QuinteT also led to the creation of balanced narratives for study teams to use and for potential participants to read. These provide a more consistent message that describes why the study is important and why clinicians are no longer certain that surgery is the optimal treatment for all patients with operable stage I NSCLC.
The VALOR clinical trial, opened in 2017, remains open at only 9 VAMCs. As of early 2020, it has enrolled more participants than all previous phase 3 trials combined. Once completed, the results from CSP #2005 VALOR study will help clinicians and veterans with operable stage I NSCLC better understand the tradeoffs of surgery vs SBRT as an initial treatment option. Plans are under way to expand the scope of the trial and include investigations of pretreatment radiomic signatures and genetic markers from biopsy tissue and blood samples, to better predict when surgery or SBRT might be the best treatment option for an individual patient.
VA-ROQS
The VA-ROQS was created in 2016 to compare treatment of veterans with lung cancer in the VHA with quality standards recommended by nationally recognized experts in lung cancer care. Partnering with Washington University in St. Louis, Missouri and the American Society for Radiation Oncology, the VHA established a blue-ribbon panel of experts to review clinical trial data and medical literature to provide evidence-based quality metrics for lung cancer therapy. As a result, 26 metrics applicable to each patient’s case were developed, published, and used to assess lung cancer care in each VHA radiation oncology practice.26
By 2019, the resulting data led to a report on 773 lung cancer cases accumulated from all VHA radiation oncology practices. Performance data for each quality metric were compared for each practice within the VHA, which found that VHA practices met > 80% of all 1,278 metrics scored. Quality metrics included those documented within each patient health record and the specific radiation delivery parameters that reflected each health care provider’s treatment. After team investigators visited each center and recorded treatment data, VA-ROQS is now maturing to permit continuous, electronic monitoring of all lung cancer treatment delivered within VHA. As each veteran’s case is planned, the quality of the therapy is monitored, assessed, and reported to the treating physician. Each VHA radiation oncologist will receive up-to-date evaluation of each case compared with these evidence-based quality standards. The quality standards are reviewed by the blue-ribbon panel to keep the process current and valid.
Future of VHA Lung Cancer Care
As VHA continues to prioritize resources to improve and assure optimal outcomes for veterans with lung cancer, it is now looking to create a national network of Lung Cancer Centers of Excellence (LCCE) as described in the VA Budget Submission for fiscal year 2021. If Congress approves funding, LCCEs will soon be developed within the VA regional Veteran Integrated Service Network system to ensure that treatment decisions for veterans with lung cancer are based on all available molecular information, including data on pharmacogenomic profiles. Such a network would create more opportunities to leverage public–private partnerships similar to the VA-PALS project. Creation of LCCEs would help the VA leverage an even stronger learning network to support more research so that all veterans who are impacted by lung cancer have access to personalized care that optimizes safety, quality of life, and overall survival. The lessons learned, networks developed, and partnerships established through VA-PALS, VALOR, and VA-ROQS are instrumental toward achieving these goals.
1. Zullig LL, Jackson GL, Dorn RA, et al. Cancer incidence among patients of the U.S. Veterans Affairs Health Care System. Mil Med. 2012;177(6):693-701. doi:10.7205/milmed-d-11-00434
2. Dawson GA, Cheuk AV, Lutz S, et al. The availability of advanced radiation oncology technology within the Veterans Health Administration radiation oncology centers. Fed Pract. 2016;33(suppl 4):18S-22S.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
4. National Lung Screening Trial Research Team. Lung cancer incidence and mortality with extended follow-up in the National Lung Screening Trial. J Thorac Oncol. 2019;14(10):1732-1742. doi:10.1016/j.jtho.2019.05.044
5. de Koning HJ, van der Aalst CM, de Jong PA, et al. Reduced lung-cancer mortality with volume CT Screening in a randomized trial. N Engl J Med. 2020;382(6):503-513. doi:10.1056/NEJMoa1911793
6. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406. doi:10.1001/jamainternmed.2016.9022
7. Clancy C. Creating World-class care and service for our nation’s finest: how Veterans Health Administration Diffusion of Excellence Initiative Is innovating and transforming Veterans Affairs health care. Perm J. 2019;23:18.301. doi:10.7812/TPP/18.301
8. Elnahal SM, Clancy CM, Shulkin DJ. A framework for disseminating clinical best practices in the VA health system. JAMA. 2017;317(3):255-256. doi:10.1001/jama.2016.18764
9. Henschke CI, McCauley DI, Yankelevitz DF, et al. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet. 1999;354(9173):99-105. doi:10.1016/S0140-6736(99)06093-6
10. Mulshine JL, Henschke CI. Lung cancer screening: achieving more by intervening less. Lancet Oncol. 2014;15(12):1284-1285. doi:10.1016/S1470-2045(14)70418-8
11. Henschke CI, Li K, Yip R, Salvatore M, Yankelevitz DF. The importance of the regimen of screening in maximizing the benefit and minimizing the harms. Ann Transl Med. 2016;4(8):153. doi:10.21037/atm.2016.04.06
12. Henschke CI, Yip R, Yankelevitz DF, Smith JP; International Early Lung Cancer Action Program Investigators*. Definition of a positive test result in computed tomography screening for lung cancer: a cohort study. Ann Intern Med. 2013;158(4):246-252. doi:10.7326/0003-4819-158-4-201302190-00004
13. Zeliadt SB, Heffner JL, Sayre G, et al. Attitudes and perceptions about smoking cessation in the context of lung cancer screening. JAMA Intern Med. 2015;175(9):1530-1537. doi:10.1001/jamainternmed.2015.3558
14. Henschke CI, Yankelevitz DF, Yip R, et al. Tumor volume measurement error using computed tomography imaging in a phase II clinical trial in lung cancer. J Med Imaging (Bellingham). 2016;3(3):035505. doi:10.1117/1.JMI.3.3.035505
15. Yip R, Henschke CI, Yankelevitz DF, Boffetta P, Smith JP; International Early Lung Cancer Investigators. The impact of the regimen of screening on lung cancer cure: a comparison of I-ELCAP and NLST. Eur J Cancer Prev. 2015;24(3):201-208. doi:10.1097/CEJ.0000000000000065
16. Armato SG 3rd, McLennan G, Bidaut L, et al. The Lung Image Database Consortium (LIDC) and Image Database Resource Initiative (IDRI): a completed reference database of lung nodules on CT scans. Med Phys. 2011;38(2):915-931. doi:10.1118/1.3528204
17. Gill RK, Vazquez MF, Kramer A, et al. The use of genetic markers to identify lung cancer in fine needle aspiration samples. Clin Cancer Res. 2008;14(22):7481-7487. doi:10.1158/1078-0432.CCR-07-5242
18. Pyenson BS, Henschke CI, Yankelevitz DF, Yip R, Dec E. Offering lung cancer screening to high-risk medicare beneficiaries saves lives and is cost-effective: an actuarial analysis. Am Health Drug Benefits. 2014;7(5):272-282.
19. Schwartz RM, Yip R, Olkin I, et al. Impact of surgery for stage IA non-small-cell lung cancer on patient quality of life. J Community Support Oncol. 2016;14(1):37-44. doi:10.12788/jcso.0205
20. Moghanaki D, Chang JY. Is surgery still the optimal treatment for stage I non-small cell lung cancer? Transl Lung Cancer Res. 2016;5(2):183-189. doi:10.21037/tlcr.2016.04.05
21. Bakaeen FG, Reda DJ, Gelijns AC, et al. Department of Veterans Affairs Cooperative Studies Program network of dedicated enrollment sites: implications for surgical trials [published correction appears in JAMA Surg. 2014 Sep;149(9):961]. JAMA Surg. 2014;149(6):507-513. doi:10.1001/jamasurg.2013.4150
22. Chang JY, Senan S, Paul MA, et al. Stereotactic ablative radiotherapy versus lobectomy for operable stage I non-small-cell lung cancer: a pooled analysis of two randomised trials [published correction appears in Lancet Oncol. 2015 Sep;16(9):e427]. Lancet Oncol. 2015;16(6):630-637. doi:10.1016/S1470-2045(15)70168-3
23. Samson P, Keogan K, Crabtree T, et al. Interpreting survival data from clinical trials of surgery versus stereotactic body radiation therapy in operable Stage I non-small cell lung cancer patients. Lung Cancer. 2017;103:6-10. doi:10.1016/j.lungcan.2016.11.005
24. Donovan JL, Rooshenas L, Jepson M, et al. Optimising recruitment and informed consent in randomised controlled trials: the development and implementation of the Quintet Recruitment Intervention (QRI). Trials. 2016;17(1):283. Published 2016 Jun 8. doi:10.1186/s13063-016-1391-4
25. Rooshenas L, Scott LJ, Blazeby JM, et al. The QuinteT Recruitment Intervention supported five randomized trials to recruit to target: a mixed-methods evaluation. J Clin Epidemiol. 2019;106:108-120. doi:10.1016/j.jclinepi.2018.10.004
26. Hagan M, Kapoor R, Michalski J, et al. VA-Radiation Oncology Quality Surveillance Program. Int J Radiat Oncol Biol Phys. 2020;106(3):639-647. doi:10.1016/j.ijrobp.2019.08.064
1. Zullig LL, Jackson GL, Dorn RA, et al. Cancer incidence among patients of the U.S. Veterans Affairs Health Care System. Mil Med. 2012;177(6):693-701. doi:10.7205/milmed-d-11-00434
2. Dawson GA, Cheuk AV, Lutz S, et al. The availability of advanced radiation oncology technology within the Veterans Health Administration radiation oncology centers. Fed Pract. 2016;33(suppl 4):18S-22S.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7-34. doi:10.3322/caac.21551
4. National Lung Screening Trial Research Team. Lung cancer incidence and mortality with extended follow-up in the National Lung Screening Trial. J Thorac Oncol. 2019;14(10):1732-1742. doi:10.1016/j.jtho.2019.05.044
5. de Koning HJ, van der Aalst CM, de Jong PA, et al. Reduced lung-cancer mortality with volume CT Screening in a randomized trial. N Engl J Med. 2020;382(6):503-513. doi:10.1056/NEJMoa1911793
6. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406. doi:10.1001/jamainternmed.2016.9022
7. Clancy C. Creating World-class care and service for our nation’s finest: how Veterans Health Administration Diffusion of Excellence Initiative Is innovating and transforming Veterans Affairs health care. Perm J. 2019;23:18.301. doi:10.7812/TPP/18.301
8. Elnahal SM, Clancy CM, Shulkin DJ. A framework for disseminating clinical best practices in the VA health system. JAMA. 2017;317(3):255-256. doi:10.1001/jama.2016.18764
9. Henschke CI, McCauley DI, Yankelevitz DF, et al. Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet. 1999;354(9173):99-105. doi:10.1016/S0140-6736(99)06093-6
10. Mulshine JL, Henschke CI. Lung cancer screening: achieving more by intervening less. Lancet Oncol. 2014;15(12):1284-1285. doi:10.1016/S1470-2045(14)70418-8
11. Henschke CI, Li K, Yip R, Salvatore M, Yankelevitz DF. The importance of the regimen of screening in maximizing the benefit and minimizing the harms. Ann Transl Med. 2016;4(8):153. doi:10.21037/atm.2016.04.06
12. Henschke CI, Yip R, Yankelevitz DF, Smith JP; International Early Lung Cancer Action Program Investigators*. Definition of a positive test result in computed tomography screening for lung cancer: a cohort study. Ann Intern Med. 2013;158(4):246-252. doi:10.7326/0003-4819-158-4-201302190-00004
13. Zeliadt SB, Heffner JL, Sayre G, et al. Attitudes and perceptions about smoking cessation in the context of lung cancer screening. JAMA Intern Med. 2015;175(9):1530-1537. doi:10.1001/jamainternmed.2015.3558
14. Henschke CI, Yankelevitz DF, Yip R, et al. Tumor volume measurement error using computed tomography imaging in a phase II clinical trial in lung cancer. J Med Imaging (Bellingham). 2016;3(3):035505. doi:10.1117/1.JMI.3.3.035505
15. Yip R, Henschke CI, Yankelevitz DF, Boffetta P, Smith JP; International Early Lung Cancer Investigators. The impact of the regimen of screening on lung cancer cure: a comparison of I-ELCAP and NLST. Eur J Cancer Prev. 2015;24(3):201-208. doi:10.1097/CEJ.0000000000000065
16. Armato SG 3rd, McLennan G, Bidaut L, et al. The Lung Image Database Consortium (LIDC) and Image Database Resource Initiative (IDRI): a completed reference database of lung nodules on CT scans. Med Phys. 2011;38(2):915-931. doi:10.1118/1.3528204
17. Gill RK, Vazquez MF, Kramer A, et al. The use of genetic markers to identify lung cancer in fine needle aspiration samples. Clin Cancer Res. 2008;14(22):7481-7487. doi:10.1158/1078-0432.CCR-07-5242
18. Pyenson BS, Henschke CI, Yankelevitz DF, Yip R, Dec E. Offering lung cancer screening to high-risk medicare beneficiaries saves lives and is cost-effective: an actuarial analysis. Am Health Drug Benefits. 2014;7(5):272-282.
19. Schwartz RM, Yip R, Olkin I, et al. Impact of surgery for stage IA non-small-cell lung cancer on patient quality of life. J Community Support Oncol. 2016;14(1):37-44. doi:10.12788/jcso.0205
20. Moghanaki D, Chang JY. Is surgery still the optimal treatment for stage I non-small cell lung cancer? Transl Lung Cancer Res. 2016;5(2):183-189. doi:10.21037/tlcr.2016.04.05
21. Bakaeen FG, Reda DJ, Gelijns AC, et al. Department of Veterans Affairs Cooperative Studies Program network of dedicated enrollment sites: implications for surgical trials [published correction appears in JAMA Surg. 2014 Sep;149(9):961]. JAMA Surg. 2014;149(6):507-513. doi:10.1001/jamasurg.2013.4150
22. Chang JY, Senan S, Paul MA, et al. Stereotactic ablative radiotherapy versus lobectomy for operable stage I non-small-cell lung cancer: a pooled analysis of two randomised trials [published correction appears in Lancet Oncol. 2015 Sep;16(9):e427]. Lancet Oncol. 2015;16(6):630-637. doi:10.1016/S1470-2045(15)70168-3
23. Samson P, Keogan K, Crabtree T, et al. Interpreting survival data from clinical trials of surgery versus stereotactic body radiation therapy in operable Stage I non-small cell lung cancer patients. Lung Cancer. 2017;103:6-10. doi:10.1016/j.lungcan.2016.11.005
24. Donovan JL, Rooshenas L, Jepson M, et al. Optimising recruitment and informed consent in randomised controlled trials: the development and implementation of the Quintet Recruitment Intervention (QRI). Trials. 2016;17(1):283. Published 2016 Jun 8. doi:10.1186/s13063-016-1391-4
25. Rooshenas L, Scott LJ, Blazeby JM, et al. The QuinteT Recruitment Intervention supported five randomized trials to recruit to target: a mixed-methods evaluation. J Clin Epidemiol. 2019;106:108-120. doi:10.1016/j.jclinepi.2018.10.004
26. Hagan M, Kapoor R, Michalski J, et al. VA-Radiation Oncology Quality Surveillance Program. Int J Radiat Oncol Biol Phys. 2020;106(3):639-647. doi:10.1016/j.ijrobp.2019.08.064
Leveraging Veterans Health Administration Clinical and Research Resources to Accelerate Discovery and Testing in Precision Oncology(FULL)
In May 2020, the US Food and Drug Administration (FDA) approved the first 2 targeted treatments for prostate cancer, specifically, the poly-(adenosine diphosphate-ribose) polymerase (PARP) inhibitors rucaparib and olaparib.1,2 For these medications to work, the tumor must have a homologous recombination deficiency (HRD), which is a form of DNA repair deficiency. The PARP pathway is important for DNA repair, and PARP inhibition leads to “synthetic lethality” in cancer cells that already are deficient in DNA repair mechanisms.3 Now, there is evidence that patients with prostate cancer who have HRD tumors and receive PARP inhibitors live longer when compared with those who receive standard of care options.4 These findings offer hope for patients with prostate cancer. They also demonstrate the process and potential benefits of precision oncology efforts; namely, targeted treatments for specific tumor types in cancer patients.
This article discusses the challenges and opportunities of precision oncology for US Department of Veterans Affairs (VA) Veterans Health Administration (VHA). First, the article will discuss working with relatively rare mutations. Second, the article will examine how the trials of olaparib and rucaparib illuminate the VHA contribution to research on new therapies for patients with cancer. Finally, the article will explore the ways in which VHA is becoming a major national contributor in drug discovery and approval of precision medications.
Precision Oncology
Despite advances in screening and treatment, an estimated 600,000 people in the US will die of cancer in 2020.5 Meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa. Successful translational research requires many components. These include talented scientists to form hypotheses and perform the work; money for supplies and equipment; platforms for timely dissemination of knowledge; well-trained clinicians to treat patients and lead research teams; and patients to participate in clinical trials. In precision oncology, the ability to find patients for the trials can be daunting, particularly in cases where the frequency of the mutation of interest is low.
During the 20th century, with few exceptions, physicians caring for patients with cancer had blunt instruments at their disposal. Surgery and radiation could lead to survival if the cancer was caught early enough. Systemic therapies, such as chemotherapy, rarely cured but could prolong life in some patients. However, chemotherapy is imprecise and targets any cell growing rapidly, including blood, hair, and gastrointestinal tract cells, which often leads to adverse effects. Sometimes complications from chemotherapy may shorten a person’s life, and certainly the quality of life during and after these treatments could be diminished. The improvements in cancer care occurred more rapidly once scientists had the tools to learn about individual tumors.
In the summer of 2000, researchers announced that the human genome had been sequenced.6 The genome (ie, DNA) consists of introns and exons that form a map for human development. Exons can be converted to proteins that carry out specific actions, such as helping in cell growth, cell death, or DNA repair. Solving the human genome itself did not lead directly to cures, but it did represent a huge advance in medical research. As time passed, sequencing genomes became more affordable, and sequencing just the exome alone was even cheaper.7 Treatments for cancer began to expand with the help of these tools, but questions as to the true benefit of targeted therapy also grew.8
Physicians and scientists have amassed more information about cancer cells and have applied this knowledge to active drug development. In 2001, the FDA approved the first targeted therapy, imatinib, for the treatment of chronic myelogenous leukemia (CML). This rapidly improved patient survival through targeting the mutated protein that leads to CML, rather than just aiming for rapidly dividing cells.9 Those mutations for which there is a drug to target, such as the BCR-ABL translocation in CML, are called actionable mutations.
Precision Oncology Program for Prostate Cancer
In 2016, the VA and the Prostate Cancer Foundation (PCF) established the Precision Oncology Program for Prostate Cancer (POPCaP) Centers of Excellence (COE). This partnership was formed to accelerate treatment and cure for veterans with prostate cancer. The VA Greater Los Angeles Healthcare System in California and VA Puget Sound Health Care System in Washington led this effort, and their principal investigators continue to co-lead POPCaP. Since its inception, 9 additional funded POPCaP COEs have joined, each with a mandate to sequence the tumors of men with metastatic prostate cancer.
The more that is learned about a tumor, the more likely it is that researchers can find mutations that are that tumor’s Achilles heel and defeat it. In fact, many drugs that can target mutations are already available. For example, BRCA2 is an actionable mutation that can be exploited by knocking out another key DNA repair mechanism in the cell, PARP. Today, the effort of sequencing has led to a rich database of mutations present in men with metastatic prostate cancer.
Although there are many targeted therapies, most have not been studied formally in prostate cancer. Occasionally, clinicians treating patients will use these drugs in an unapproved way, hoping that there will be anticancer activity. It is difficult to estimate the likelihood of success with a drug in this situation, and the safety profile may not be well described in that setting. Treatment decisions for incurable cancers must be made knowing the risks and benefits. This helps in shared decision making between the clinician and patient and informs choices concerning which laboratory tests to order and how often to see the patient. However, treatment decisions are sometimes made with the hope of activity when a cancer is known to be incurable. Very little data, which are critical to determine whether this helps or hurts patients, support this approach.
Some data suggest that sequencing and giving a drug for an actionable mutation may lead to better outcomes for patients. Sequencing of pancreatic tumors by Pishvaian and colleagues revealed that 282 of 1,082 (26%) samples harbored actionable mutations.10 Those patients who received a drug that targeted their actionable mutation (n = 46; 24%) lived longer when compared with those who had an actionable mutation but did not receive a drug that targeted it (hazard ratio [HR] 0.42 [95% CI, 0.26-0.68; P = .0004]). Additionally, those who received therapy for an actionable mutation lived longer when compared with those who did not have an actionable mutation (HR 0.34 [95% CI, 0.22-0.53; P < .001]). While this finding is intriguing, it does not mean that treating actionable mutations outside of a clinical trial should be done. To this end, VA established Prostate cancer Analysis for Therapy CHoice (PATCH) as a clinical trials network within POPCaP.
Prostate Cancer Analysis
The overall PATCH vision is designed for clinical care and research work to together toward improved care for those with prostate cancer (Figure 1). The resources necessary for successful translational research are substantial, and PATCH aims to streamline those resources. PATCH will support innovative, precision-based clinical research at the POPCaP COEs through its 5 arms.
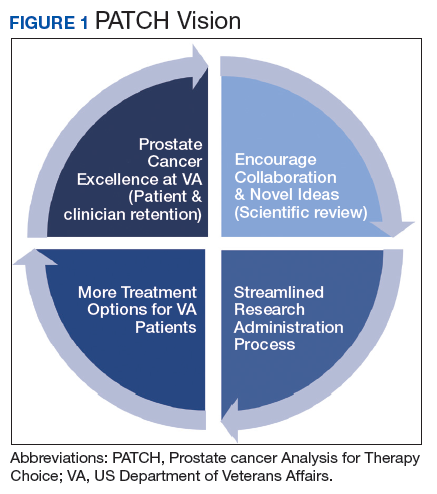
Arm 1. Dedicated personnel ensure veteran access to trials in PATCH by giving patients and providers accurate information about available trial options; aiding veterans in traveling from home VA to a POPCaP COE for participation on a study; and maintaining the Committee for Veteran Participation in PATCH, where veterans will be represented and asked to provide input into the PATCH process.
Arm 2. Coordinators at the coordinating COE in Portland, Orgeon, train investigators and study staff at the local POPCaP COEs to ensure research can be performed in a safe and responsible way.
Arm 3. Personnel experienced in conducting clinical trials liaise with investigators at VA Central Institutional Review Board, monitor trials, build databases for appropriate and efficient data collection, and manage high-risk studies conducted under an Investigational New Drug application. This group works closely with biostatisticians to choose appropriate trial designs, estimate numbers of patients needed, and interpret data once they are collected.
Arm 4. Protocol development and data dissemination is coordinated by a group to assist investigators in drafting protocols and reviewing abstracts and manuscripts.
Arm 5. A core group manages contracts and budgets, as well as relationships between VA and industry, where funding and drugs may be obtained.
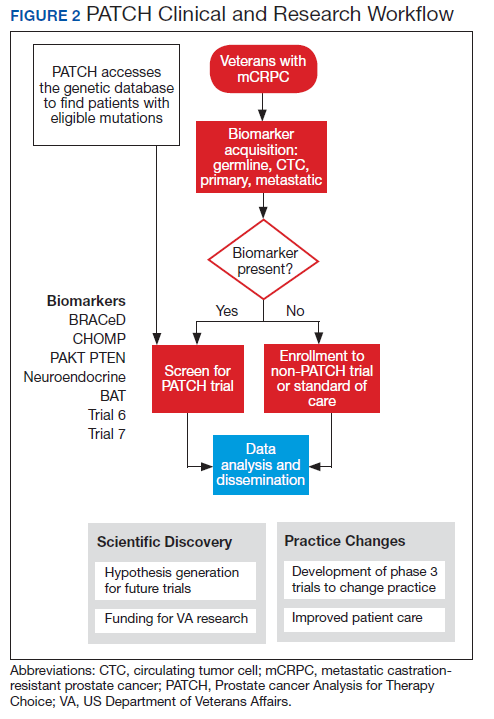
Perhaps most importantly, PATCH leverages the genetic data collected by POPCaP COEs to find patients for clinical trials. For example, the trials that examined olaparib and rucaparib assumed that the prevalence of HRD was about 25% in men with advanced prostate cancer.11 As these trials began enrollment, however, researchers discovered that the prevalence was < 20%. In fact, the study of olaparib screened 4,425 patients at 206 sites in 20 countries to identify 778 (18% of screened) patients with HRD.4 With widespread sequencing within VA, it could be possible to identify a substantial number of patients who are already known to have the mutation of interest (Figure 2).
Clinical Trials
There are currently 2 clinical trials in PATCH; 4 additional trials await funding decisions, and more trials are in the concept stage. BRACeD (NCT04038502) is a phase 2 trial examining platinum and taxane chemotherapy in tumors with HRD (specifically, BRCA1, BRCA2, and PALB2). About 15% to 20% of men with advanced prostate cancer will have a DNA repair defect in the tumor that could make them eligible for this study. The primary endpoint is progression-free survival.
A second study, CHOMP (NCT04104893), is a phase 2 trial examining the efficacy of immunotherapy (PD-1 inhibition) in tumors having mismatch repair deficiency or CDK12-/-. Each of those is found in about 7% of men with metastatic prostate cancer, and full accrual of a trial with rare mutations could take 5 to 10 years without a systematic approach of sequencing and identifying potential participants. The primary endpoint is a composite of radiographic response by iRECIST (immune response evaluation criteria in solid tumors), progression-free survival at 6 months and prostate specific antigen reduction by ≥ 50% in ≤ 12 weeks. With 11 POPCaP COEs sequencing the tumors of every man with metastatic prostate cancer, identifying men with the appropriate mutation is possible. PATCH will aid the sites in recruitment through outreach and coordination of travel.
Industry Partnerships
PATCH depends upon pharmaceutical industry partners, as clinical trials of even 40 patients can require significant funding and trial resources to operate. Furthermore, many drugs of interest are not available outside of a clinical trial, and partnerships enable VA researchers to access these medications. PATCH also benefits greatly from foundation partners, such as the PCF, which has made POPCaP possible and will continue to connect talented researchers with VA resources. Finally, access to other publicly available research funds, such as those from VA Office of Research and Development, National Institutes of Health, and US Department of Defense (DoD) Congressionally Directed Research Program are needed for trials.
Funding for these trials remains limited despite public health and broader interests in addressing important questions. Accelerated accrual through PATCH may be an attractive partnership opportunity for companies, foundations and government funding agencies to support the PATCH efforts.
Both POPCaP and PATCH highlight the potential promise of precision oncology within the nation’s largest integrated health care system. The VHA patient population enables prostate cancer researchers to serve an important early target. It also provides a foundational platform for a broader set of activities. These include a tailored approach to identifying tumor profiles and other patient characteristics that may help to elevate standard of care for other common cancers including ones affecting the lungs and/or head and neck.
To this end, VA has been working with the National Cancer Institute (NCI) and DoD to establish a national infrastructure for precision oncology across multiple cancer types.12 In addition to clinical capabilities and the ability to run clinical trials that can accrue sufficient patients to answer key questions, we have developed capabilities for data collection and sharing, and analytical tools to support a learning health care system approach as a core element to precision oncology.
Besides having a research-specific context, such informatics and information technology systems enable clinicians to obtain and apply decision-making data rapidly for a specific patient and cancer type. These systems take particular advantage of the extensive electronic health record that underlies the VHA system, integrating real-world evidence into rigorous trials for precision oncology and other diseases. This is important for facilitating prerequisite activities for quality assessments for incorporation into databases (with appropriate permissions) to enhance treatment options. These activities are a key focus of the APOLLO initiative.13 While a more in-depth discussion of the importance of informatics is beyond the scope of this article, the field represents an important investment that is needed to achieve the goals of precision oncology.
In addition to informatics and data handling capabilities, VA has a longstanding tradition of designing and coordinating multisite clinical trials. This dates to the time of World War II when returning veterans had a high prevalence of tuberculosis. Since then, VA has contributed extensively to landmark findings in cardiovascular disease and surgery, mental health, infectious disease, and cancer. It was a VA study that helped establish colonoscopy as a standard for colorectal cancer screening by detecting colonic neoplasms in asymptomatic patients.14
From such investigations, the VA Cooperative Studies Program (CSP) has developed many strategies to conduct multisite clinical trials. But, CSP also has organized its sites methodically for operational efficiency and the ability to maintain institutional knowledge that crosses different types of studies and diseases. Using its Network of Dedicated Enrollment Sites (NODES) model, VA partnered with NCI to more effectively address administrative and regulatory requirements for initiating trials and recruiting veterans into cancer clinical trials.15 This partnership—the NCI And VA Interagency Group to Accelerate Trials Enrollment (NAVIGATE)—supports 12 sites with a central CSP Coordinating Center (CSPCC).
CSPCC provides support, shares best practices and provides organizational commitment at the senior levels of both agencies to overcome potential barriers. The goals and strategies are described by Schiller and colleagues.16 While still in its early stage as a cancer research network, NAVIGATE may be integrated with POPCaP and other parts of VA clinical research enterprise. This would allow us to specialize in advancing oncology care and to leverage capabilities more specifically to precision oncology. With an emphasis on recruitment, NAVIGATE has established capabilities with VA Informatics and Computing Infrastructure to quickly identify patients who may be eligible for particular clinical trials. We envision further refining these capabilities for precision oncology trials that incorporate genetic and other information for individual patients. VA also hopes to inform trial sponsors about design considerations. This is important since networked investigators will have direct insights into patient-level factors, which may help with more effectively identifying and enrolling them into trials for their particular cancers.
Conclusions
VA may have an opportunity to reach out to veterans who may not have immediate access to facilities running clinical trials. As it develops capabilities to bring the trial to the veteran, VA could have more virtual and/or centralized recruitment strategies. This would broaden opportunities for considering novel approaches that may not rely on a more traditional facility-based recruitment approach.
Ultimately, VA can be a critical part of a national effort to fight and, perhaps even, defeat cancers. With its extensive resources and capabilities, VA has the ability to advance a precision oncology agenda that provides veterans with the highest standard of care. It has built upon many key elements in clinical, technological and scientific fields of study that would challenge most health care systems given the extensive costs involved. In addition, creating strong partnerships with organizations such as PCF, NCI, and DoD that are complementary in resources and expertise will help VA to build a national network for cancer care. Putting this all together will support and facilitate a vision for more precise care for any veteran with cancer by more rapidly enabling the testing and approval of medications developed for this purpose.
Acknowledgments
The authors would like to thank Daphne Swancutt for comments and edits on drafts of this article.
1. Lynparza (Olaparib) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP Inc, 2019.
2. Rubraca (rucaparib) [package insert]: Clovis Oncology, Inc., Boulder, CO: 2018.
3. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725-1735. doi:10.1056/NEJMra1407390
4. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7-30. doi:10.3322/caac.21590
6. Bentley DR. Decoding the human genome sequence. Hum Mol Genet. 2000;9(16):2353-2358. doi:10.1093/hmg/9.16.2353
7. National Human Genome Research institute. The cost of sequencing a human genome. https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost. Updated October 30, 2019. Accessed July 31, 2020. 8. Paggio JCD, Sullivan R, Booth CM. Targeting the value of targeted therapy. Oncotarget. 2017;8(53):90612-90613. Published 2017 Oct 7. doi:10.18632/oncotarget.21596
9. Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408-2417. doi:10.1056/NEJMoa062867
10. Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial [published correction appears in Lancet Oncol. 2020 Apr;21(4):e182]. Lancet Oncol. 2020;21(4):508-518. doi:10.1016/S1470-2045(20)30074-7
11. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
12. Fiore LD, Brophy MT, Ferguson RE, et al. Data sharing, clinical trials, and biomarkers in precision oncology: challenges, opportunities, and programs at the Department of Veterans Affairs. Clin Pharmacol Ther. 2017;101(5):586-589. doi:10.1002/cpt.660
13. Lee JSH, Darcy KM, Hu H, et al. From discovery to practice and survivorship: building a national real-world data learning healthcare framework for military and veteran cancer patients. Clin Pharmacol Ther. 2019;106(1):52-57. doi:10.1002/cpt.1425
14. Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380 [published correction appears in N Engl J Med 2000 Oct 19;343(16):1204]. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
15. Condon DL, Beck D, Kenworthy-Heinige T, et al. A cross-cutting approach to enhancing clinical trial site success: The Department of Veterans Affairs’ Network of Dedicated Enrollment Sites (NODES) model. Contemp Clin Trials Commun. 2017;6:78-84. Published 2017 Mar 29. doi:10.1016/j.conctc.2017.03.006
16. Schiller SJ, Shannon C, Brophy MT, et al. The National Cancer Institute and Department of Veterans Affairs Interagency Group to Accelerate Trials Enrollment (NAVIGATE): A federal collaboration to improve cancer care. Semin Oncol. 2019;46(4-5):308-313. doi:10.1053/j.seminoncol.2019.09.005
In May 2020, the US Food and Drug Administration (FDA) approved the first 2 targeted treatments for prostate cancer, specifically, the poly-(adenosine diphosphate-ribose) polymerase (PARP) inhibitors rucaparib and olaparib.1,2 For these medications to work, the tumor must have a homologous recombination deficiency (HRD), which is a form of DNA repair deficiency. The PARP pathway is important for DNA repair, and PARP inhibition leads to “synthetic lethality” in cancer cells that already are deficient in DNA repair mechanisms.3 Now, there is evidence that patients with prostate cancer who have HRD tumors and receive PARP inhibitors live longer when compared with those who receive standard of care options.4 These findings offer hope for patients with prostate cancer. They also demonstrate the process and potential benefits of precision oncology efforts; namely, targeted treatments for specific tumor types in cancer patients.
This article discusses the challenges and opportunities of precision oncology for US Department of Veterans Affairs (VA) Veterans Health Administration (VHA). First, the article will discuss working with relatively rare mutations. Second, the article will examine how the trials of olaparib and rucaparib illuminate the VHA contribution to research on new therapies for patients with cancer. Finally, the article will explore the ways in which VHA is becoming a major national contributor in drug discovery and approval of precision medications.
Precision Oncology
Despite advances in screening and treatment, an estimated 600,000 people in the US will die of cancer in 2020.5 Meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa. Successful translational research requires many components. These include talented scientists to form hypotheses and perform the work; money for supplies and equipment; platforms for timely dissemination of knowledge; well-trained clinicians to treat patients and lead research teams; and patients to participate in clinical trials. In precision oncology, the ability to find patients for the trials can be daunting, particularly in cases where the frequency of the mutation of interest is low.
During the 20th century, with few exceptions, physicians caring for patients with cancer had blunt instruments at their disposal. Surgery and radiation could lead to survival if the cancer was caught early enough. Systemic therapies, such as chemotherapy, rarely cured but could prolong life in some patients. However, chemotherapy is imprecise and targets any cell growing rapidly, including blood, hair, and gastrointestinal tract cells, which often leads to adverse effects. Sometimes complications from chemotherapy may shorten a person’s life, and certainly the quality of life during and after these treatments could be diminished. The improvements in cancer care occurred more rapidly once scientists had the tools to learn about individual tumors.
In the summer of 2000, researchers announced that the human genome had been sequenced.6 The genome (ie, DNA) consists of introns and exons that form a map for human development. Exons can be converted to proteins that carry out specific actions, such as helping in cell growth, cell death, or DNA repair. Solving the human genome itself did not lead directly to cures, but it did represent a huge advance in medical research. As time passed, sequencing genomes became more affordable, and sequencing just the exome alone was even cheaper.7 Treatments for cancer began to expand with the help of these tools, but questions as to the true benefit of targeted therapy also grew.8
Physicians and scientists have amassed more information about cancer cells and have applied this knowledge to active drug development. In 2001, the FDA approved the first targeted therapy, imatinib, for the treatment of chronic myelogenous leukemia (CML). This rapidly improved patient survival through targeting the mutated protein that leads to CML, rather than just aiming for rapidly dividing cells.9 Those mutations for which there is a drug to target, such as the BCR-ABL translocation in CML, are called actionable mutations.
Precision Oncology Program for Prostate Cancer
In 2016, the VA and the Prostate Cancer Foundation (PCF) established the Precision Oncology Program for Prostate Cancer (POPCaP) Centers of Excellence (COE). This partnership was formed to accelerate treatment and cure for veterans with prostate cancer. The VA Greater Los Angeles Healthcare System in California and VA Puget Sound Health Care System in Washington led this effort, and their principal investigators continue to co-lead POPCaP. Since its inception, 9 additional funded POPCaP COEs have joined, each with a mandate to sequence the tumors of men with metastatic prostate cancer.
The more that is learned about a tumor, the more likely it is that researchers can find mutations that are that tumor’s Achilles heel and defeat it. In fact, many drugs that can target mutations are already available. For example, BRCA2 is an actionable mutation that can be exploited by knocking out another key DNA repair mechanism in the cell, PARP. Today, the effort of sequencing has led to a rich database of mutations present in men with metastatic prostate cancer.
Although there are many targeted therapies, most have not been studied formally in prostate cancer. Occasionally, clinicians treating patients will use these drugs in an unapproved way, hoping that there will be anticancer activity. It is difficult to estimate the likelihood of success with a drug in this situation, and the safety profile may not be well described in that setting. Treatment decisions for incurable cancers must be made knowing the risks and benefits. This helps in shared decision making between the clinician and patient and informs choices concerning which laboratory tests to order and how often to see the patient. However, treatment decisions are sometimes made with the hope of activity when a cancer is known to be incurable. Very little data, which are critical to determine whether this helps or hurts patients, support this approach.
Some data suggest that sequencing and giving a drug for an actionable mutation may lead to better outcomes for patients. Sequencing of pancreatic tumors by Pishvaian and colleagues revealed that 282 of 1,082 (26%) samples harbored actionable mutations.10 Those patients who received a drug that targeted their actionable mutation (n = 46; 24%) lived longer when compared with those who had an actionable mutation but did not receive a drug that targeted it (hazard ratio [HR] 0.42 [95% CI, 0.26-0.68; P = .0004]). Additionally, those who received therapy for an actionable mutation lived longer when compared with those who did not have an actionable mutation (HR 0.34 [95% CI, 0.22-0.53; P < .001]). While this finding is intriguing, it does not mean that treating actionable mutations outside of a clinical trial should be done. To this end, VA established Prostate cancer Analysis for Therapy CHoice (PATCH) as a clinical trials network within POPCaP.
Prostate Cancer Analysis
The overall PATCH vision is designed for clinical care and research work to together toward improved care for those with prostate cancer (Figure 1). The resources necessary for successful translational research are substantial, and PATCH aims to streamline those resources. PATCH will support innovative, precision-based clinical research at the POPCaP COEs through its 5 arms.
Arm 1. Dedicated personnel ensure veteran access to trials in PATCH by giving patients and providers accurate information about available trial options; aiding veterans in traveling from home VA to a POPCaP COE for participation on a study; and maintaining the Committee for Veteran Participation in PATCH, where veterans will be represented and asked to provide input into the PATCH process.
Arm 2. Coordinators at the coordinating COE in Portland, Orgeon, train investigators and study staff at the local POPCaP COEs to ensure research can be performed in a safe and responsible way.
Arm 3. Personnel experienced in conducting clinical trials liaise with investigators at VA Central Institutional Review Board, monitor trials, build databases for appropriate and efficient data collection, and manage high-risk studies conducted under an Investigational New Drug application. This group works closely with biostatisticians to choose appropriate trial designs, estimate numbers of patients needed, and interpret data once they are collected.
Arm 4. Protocol development and data dissemination is coordinated by a group to assist investigators in drafting protocols and reviewing abstracts and manuscripts.
Arm 5. A core group manages contracts and budgets, as well as relationships between VA and industry, where funding and drugs may be obtained.
Perhaps most importantly, PATCH leverages the genetic data collected by POPCaP COEs to find patients for clinical trials. For example, the trials that examined olaparib and rucaparib assumed that the prevalence of HRD was about 25% in men with advanced prostate cancer.11 As these trials began enrollment, however, researchers discovered that the prevalence was < 20%. In fact, the study of olaparib screened 4,425 patients at 206 sites in 20 countries to identify 778 (18% of screened) patients with HRD.4 With widespread sequencing within VA, it could be possible to identify a substantial number of patients who are already known to have the mutation of interest (Figure 2).
Clinical Trials
There are currently 2 clinical trials in PATCH; 4 additional trials await funding decisions, and more trials are in the concept stage. BRACeD (NCT04038502) is a phase 2 trial examining platinum and taxane chemotherapy in tumors with HRD (specifically, BRCA1, BRCA2, and PALB2). About 15% to 20% of men with advanced prostate cancer will have a DNA repair defect in the tumor that could make them eligible for this study. The primary endpoint is progression-free survival.
A second study, CHOMP (NCT04104893), is a phase 2 trial examining the efficacy of immunotherapy (PD-1 inhibition) in tumors having mismatch repair deficiency or CDK12-/-. Each of those is found in about 7% of men with metastatic prostate cancer, and full accrual of a trial with rare mutations could take 5 to 10 years without a systematic approach of sequencing and identifying potential participants. The primary endpoint is a composite of radiographic response by iRECIST (immune response evaluation criteria in solid tumors), progression-free survival at 6 months and prostate specific antigen reduction by ≥ 50% in ≤ 12 weeks. With 11 POPCaP COEs sequencing the tumors of every man with metastatic prostate cancer, identifying men with the appropriate mutation is possible. PATCH will aid the sites in recruitment through outreach and coordination of travel.
Industry Partnerships
PATCH depends upon pharmaceutical industry partners, as clinical trials of even 40 patients can require significant funding and trial resources to operate. Furthermore, many drugs of interest are not available outside of a clinical trial, and partnerships enable VA researchers to access these medications. PATCH also benefits greatly from foundation partners, such as the PCF, which has made POPCaP possible and will continue to connect talented researchers with VA resources. Finally, access to other publicly available research funds, such as those from VA Office of Research and Development, National Institutes of Health, and US Department of Defense (DoD) Congressionally Directed Research Program are needed for trials.
Funding for these trials remains limited despite public health and broader interests in addressing important questions. Accelerated accrual through PATCH may be an attractive partnership opportunity for companies, foundations and government funding agencies to support the PATCH efforts.
Both POPCaP and PATCH highlight the potential promise of precision oncology within the nation’s largest integrated health care system. The VHA patient population enables prostate cancer researchers to serve an important early target. It also provides a foundational platform for a broader set of activities. These include a tailored approach to identifying tumor profiles and other patient characteristics that may help to elevate standard of care for other common cancers including ones affecting the lungs and/or head and neck.
To this end, VA has been working with the National Cancer Institute (NCI) and DoD to establish a national infrastructure for precision oncology across multiple cancer types.12 In addition to clinical capabilities and the ability to run clinical trials that can accrue sufficient patients to answer key questions, we have developed capabilities for data collection and sharing, and analytical tools to support a learning health care system approach as a core element to precision oncology.
Besides having a research-specific context, such informatics and information technology systems enable clinicians to obtain and apply decision-making data rapidly for a specific patient and cancer type. These systems take particular advantage of the extensive electronic health record that underlies the VHA system, integrating real-world evidence into rigorous trials for precision oncology and other diseases. This is important for facilitating prerequisite activities for quality assessments for incorporation into databases (with appropriate permissions) to enhance treatment options. These activities are a key focus of the APOLLO initiative.13 While a more in-depth discussion of the importance of informatics is beyond the scope of this article, the field represents an important investment that is needed to achieve the goals of precision oncology.
In addition to informatics and data handling capabilities, VA has a longstanding tradition of designing and coordinating multisite clinical trials. This dates to the time of World War II when returning veterans had a high prevalence of tuberculosis. Since then, VA has contributed extensively to landmark findings in cardiovascular disease and surgery, mental health, infectious disease, and cancer. It was a VA study that helped establish colonoscopy as a standard for colorectal cancer screening by detecting colonic neoplasms in asymptomatic patients.14
From such investigations, the VA Cooperative Studies Program (CSP) has developed many strategies to conduct multisite clinical trials. But, CSP also has organized its sites methodically for operational efficiency and the ability to maintain institutional knowledge that crosses different types of studies and diseases. Using its Network of Dedicated Enrollment Sites (NODES) model, VA partnered with NCI to more effectively address administrative and regulatory requirements for initiating trials and recruiting veterans into cancer clinical trials.15 This partnership—the NCI And VA Interagency Group to Accelerate Trials Enrollment (NAVIGATE)—supports 12 sites with a central CSP Coordinating Center (CSPCC).
CSPCC provides support, shares best practices and provides organizational commitment at the senior levels of both agencies to overcome potential barriers. The goals and strategies are described by Schiller and colleagues.16 While still in its early stage as a cancer research network, NAVIGATE may be integrated with POPCaP and other parts of VA clinical research enterprise. This would allow us to specialize in advancing oncology care and to leverage capabilities more specifically to precision oncology. With an emphasis on recruitment, NAVIGATE has established capabilities with VA Informatics and Computing Infrastructure to quickly identify patients who may be eligible for particular clinical trials. We envision further refining these capabilities for precision oncology trials that incorporate genetic and other information for individual patients. VA also hopes to inform trial sponsors about design considerations. This is important since networked investigators will have direct insights into patient-level factors, which may help with more effectively identifying and enrolling them into trials for their particular cancers.
Conclusions
VA may have an opportunity to reach out to veterans who may not have immediate access to facilities running clinical trials. As it develops capabilities to bring the trial to the veteran, VA could have more virtual and/or centralized recruitment strategies. This would broaden opportunities for considering novel approaches that may not rely on a more traditional facility-based recruitment approach.
Ultimately, VA can be a critical part of a national effort to fight and, perhaps even, defeat cancers. With its extensive resources and capabilities, VA has the ability to advance a precision oncology agenda that provides veterans with the highest standard of care. It has built upon many key elements in clinical, technological and scientific fields of study that would challenge most health care systems given the extensive costs involved. In addition, creating strong partnerships with organizations such as PCF, NCI, and DoD that are complementary in resources and expertise will help VA to build a national network for cancer care. Putting this all together will support and facilitate a vision for more precise care for any veteran with cancer by more rapidly enabling the testing and approval of medications developed for this purpose.
Acknowledgments
The authors would like to thank Daphne Swancutt for comments and edits on drafts of this article.
In May 2020, the US Food and Drug Administration (FDA) approved the first 2 targeted treatments for prostate cancer, specifically, the poly-(adenosine diphosphate-ribose) polymerase (PARP) inhibitors rucaparib and olaparib.1,2 For these medications to work, the tumor must have a homologous recombination deficiency (HRD), which is a form of DNA repair deficiency. The PARP pathway is important for DNA repair, and PARP inhibition leads to “synthetic lethality” in cancer cells that already are deficient in DNA repair mechanisms.3 Now, there is evidence that patients with prostate cancer who have HRD tumors and receive PARP inhibitors live longer when compared with those who receive standard of care options.4 These findings offer hope for patients with prostate cancer. They also demonstrate the process and potential benefits of precision oncology efforts; namely, targeted treatments for specific tumor types in cancer patients.
This article discusses the challenges and opportunities of precision oncology for US Department of Veterans Affairs (VA) Veterans Health Administration (VHA). First, the article will discuss working with relatively rare mutations. Second, the article will examine how the trials of olaparib and rucaparib illuminate the VHA contribution to research on new therapies for patients with cancer. Finally, the article will explore the ways in which VHA is becoming a major national contributor in drug discovery and approval of precision medications.
Precision Oncology
Despite advances in screening and treatment, an estimated 600,000 people in the US will die of cancer in 2020.5 Meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa. Successful translational research requires many components. These include talented scientists to form hypotheses and perform the work; money for supplies and equipment; platforms for timely dissemination of knowledge; well-trained clinicians to treat patients and lead research teams; and patients to participate in clinical trials. In precision oncology, the ability to find patients for the trials can be daunting, particularly in cases where the frequency of the mutation of interest is low.
During the 20th century, with few exceptions, physicians caring for patients with cancer had blunt instruments at their disposal. Surgery and radiation could lead to survival if the cancer was caught early enough. Systemic therapies, such as chemotherapy, rarely cured but could prolong life in some patients. However, chemotherapy is imprecise and targets any cell growing rapidly, including blood, hair, and gastrointestinal tract cells, which often leads to adverse effects. Sometimes complications from chemotherapy may shorten a person’s life, and certainly the quality of life during and after these treatments could be diminished. The improvements in cancer care occurred more rapidly once scientists had the tools to learn about individual tumors.
In the summer of 2000, researchers announced that the human genome had been sequenced.6 The genome (ie, DNA) consists of introns and exons that form a map for human development. Exons can be converted to proteins that carry out specific actions, such as helping in cell growth, cell death, or DNA repair. Solving the human genome itself did not lead directly to cures, but it did represent a huge advance in medical research. As time passed, sequencing genomes became more affordable, and sequencing just the exome alone was even cheaper.7 Treatments for cancer began to expand with the help of these tools, but questions as to the true benefit of targeted therapy also grew.8
Physicians and scientists have amassed more information about cancer cells and have applied this knowledge to active drug development. In 2001, the FDA approved the first targeted therapy, imatinib, for the treatment of chronic myelogenous leukemia (CML). This rapidly improved patient survival through targeting the mutated protein that leads to CML, rather than just aiming for rapidly dividing cells.9 Those mutations for which there is a drug to target, such as the BCR-ABL translocation in CML, are called actionable mutations.
Precision Oncology Program for Prostate Cancer
In 2016, the VA and the Prostate Cancer Foundation (PCF) established the Precision Oncology Program for Prostate Cancer (POPCaP) Centers of Excellence (COE). This partnership was formed to accelerate treatment and cure for veterans with prostate cancer. The VA Greater Los Angeles Healthcare System in California and VA Puget Sound Health Care System in Washington led this effort, and their principal investigators continue to co-lead POPCaP. Since its inception, 9 additional funded POPCaP COEs have joined, each with a mandate to sequence the tumors of men with metastatic prostate cancer.
The more that is learned about a tumor, the more likely it is that researchers can find mutations that are that tumor’s Achilles heel and defeat it. In fact, many drugs that can target mutations are already available. For example, BRCA2 is an actionable mutation that can be exploited by knocking out another key DNA repair mechanism in the cell, PARP. Today, the effort of sequencing has led to a rich database of mutations present in men with metastatic prostate cancer.
Although there are many targeted therapies, most have not been studied formally in prostate cancer. Occasionally, clinicians treating patients will use these drugs in an unapproved way, hoping that there will be anticancer activity. It is difficult to estimate the likelihood of success with a drug in this situation, and the safety profile may not be well described in that setting. Treatment decisions for incurable cancers must be made knowing the risks and benefits. This helps in shared decision making between the clinician and patient and informs choices concerning which laboratory tests to order and how often to see the patient. However, treatment decisions are sometimes made with the hope of activity when a cancer is known to be incurable. Very little data, which are critical to determine whether this helps or hurts patients, support this approach.
Some data suggest that sequencing and giving a drug for an actionable mutation may lead to better outcomes for patients. Sequencing of pancreatic tumors by Pishvaian and colleagues revealed that 282 of 1,082 (26%) samples harbored actionable mutations.10 Those patients who received a drug that targeted their actionable mutation (n = 46; 24%) lived longer when compared with those who had an actionable mutation but did not receive a drug that targeted it (hazard ratio [HR] 0.42 [95% CI, 0.26-0.68; P = .0004]). Additionally, those who received therapy for an actionable mutation lived longer when compared with those who did not have an actionable mutation (HR 0.34 [95% CI, 0.22-0.53; P < .001]). While this finding is intriguing, it does not mean that treating actionable mutations outside of a clinical trial should be done. To this end, VA established Prostate cancer Analysis for Therapy CHoice (PATCH) as a clinical trials network within POPCaP.
Prostate Cancer Analysis
The overall PATCH vision is designed for clinical care and research work to together toward improved care for those with prostate cancer (Figure 1). The resources necessary for successful translational research are substantial, and PATCH aims to streamline those resources. PATCH will support innovative, precision-based clinical research at the POPCaP COEs through its 5 arms.
Arm 1. Dedicated personnel ensure veteran access to trials in PATCH by giving patients and providers accurate information about available trial options; aiding veterans in traveling from home VA to a POPCaP COE for participation on a study; and maintaining the Committee for Veteran Participation in PATCH, where veterans will be represented and asked to provide input into the PATCH process.
Arm 2. Coordinators at the coordinating COE in Portland, Orgeon, train investigators and study staff at the local POPCaP COEs to ensure research can be performed in a safe and responsible way.
Arm 3. Personnel experienced in conducting clinical trials liaise with investigators at VA Central Institutional Review Board, monitor trials, build databases for appropriate and efficient data collection, and manage high-risk studies conducted under an Investigational New Drug application. This group works closely with biostatisticians to choose appropriate trial designs, estimate numbers of patients needed, and interpret data once they are collected.
Arm 4. Protocol development and data dissemination is coordinated by a group to assist investigators in drafting protocols and reviewing abstracts and manuscripts.
Arm 5. A core group manages contracts and budgets, as well as relationships between VA and industry, where funding and drugs may be obtained.
Perhaps most importantly, PATCH leverages the genetic data collected by POPCaP COEs to find patients for clinical trials. For example, the trials that examined olaparib and rucaparib assumed that the prevalence of HRD was about 25% in men with advanced prostate cancer.11 As these trials began enrollment, however, researchers discovered that the prevalence was < 20%. In fact, the study of olaparib screened 4,425 patients at 206 sites in 20 countries to identify 778 (18% of screened) patients with HRD.4 With widespread sequencing within VA, it could be possible to identify a substantial number of patients who are already known to have the mutation of interest (Figure 2).
Clinical Trials
There are currently 2 clinical trials in PATCH; 4 additional trials await funding decisions, and more trials are in the concept stage. BRACeD (NCT04038502) is a phase 2 trial examining platinum and taxane chemotherapy in tumors with HRD (specifically, BRCA1, BRCA2, and PALB2). About 15% to 20% of men with advanced prostate cancer will have a DNA repair defect in the tumor that could make them eligible for this study. The primary endpoint is progression-free survival.
A second study, CHOMP (NCT04104893), is a phase 2 trial examining the efficacy of immunotherapy (PD-1 inhibition) in tumors having mismatch repair deficiency or CDK12-/-. Each of those is found in about 7% of men with metastatic prostate cancer, and full accrual of a trial with rare mutations could take 5 to 10 years without a systematic approach of sequencing and identifying potential participants. The primary endpoint is a composite of radiographic response by iRECIST (immune response evaluation criteria in solid tumors), progression-free survival at 6 months and prostate specific antigen reduction by ≥ 50% in ≤ 12 weeks. With 11 POPCaP COEs sequencing the tumors of every man with metastatic prostate cancer, identifying men with the appropriate mutation is possible. PATCH will aid the sites in recruitment through outreach and coordination of travel.
Industry Partnerships
PATCH depends upon pharmaceutical industry partners, as clinical trials of even 40 patients can require significant funding and trial resources to operate. Furthermore, many drugs of interest are not available outside of a clinical trial, and partnerships enable VA researchers to access these medications. PATCH also benefits greatly from foundation partners, such as the PCF, which has made POPCaP possible and will continue to connect talented researchers with VA resources. Finally, access to other publicly available research funds, such as those from VA Office of Research and Development, National Institutes of Health, and US Department of Defense (DoD) Congressionally Directed Research Program are needed for trials.
Funding for these trials remains limited despite public health and broader interests in addressing important questions. Accelerated accrual through PATCH may be an attractive partnership opportunity for companies, foundations and government funding agencies to support the PATCH efforts.
Both POPCaP and PATCH highlight the potential promise of precision oncology within the nation’s largest integrated health care system. The VHA patient population enables prostate cancer researchers to serve an important early target. It also provides a foundational platform for a broader set of activities. These include a tailored approach to identifying tumor profiles and other patient characteristics that may help to elevate standard of care for other common cancers including ones affecting the lungs and/or head and neck.
To this end, VA has been working with the National Cancer Institute (NCI) and DoD to establish a national infrastructure for precision oncology across multiple cancer types.12 In addition to clinical capabilities and the ability to run clinical trials that can accrue sufficient patients to answer key questions, we have developed capabilities for data collection and sharing, and analytical tools to support a learning health care system approach as a core element to precision oncology.
Besides having a research-specific context, such informatics and information technology systems enable clinicians to obtain and apply decision-making data rapidly for a specific patient and cancer type. These systems take particular advantage of the extensive electronic health record that underlies the VHA system, integrating real-world evidence into rigorous trials for precision oncology and other diseases. This is important for facilitating prerequisite activities for quality assessments for incorporation into databases (with appropriate permissions) to enhance treatment options. These activities are a key focus of the APOLLO initiative.13 While a more in-depth discussion of the importance of informatics is beyond the scope of this article, the field represents an important investment that is needed to achieve the goals of precision oncology.
In addition to informatics and data handling capabilities, VA has a longstanding tradition of designing and coordinating multisite clinical trials. This dates to the time of World War II when returning veterans had a high prevalence of tuberculosis. Since then, VA has contributed extensively to landmark findings in cardiovascular disease and surgery, mental health, infectious disease, and cancer. It was a VA study that helped establish colonoscopy as a standard for colorectal cancer screening by detecting colonic neoplasms in asymptomatic patients.14
From such investigations, the VA Cooperative Studies Program (CSP) has developed many strategies to conduct multisite clinical trials. But, CSP also has organized its sites methodically for operational efficiency and the ability to maintain institutional knowledge that crosses different types of studies and diseases. Using its Network of Dedicated Enrollment Sites (NODES) model, VA partnered with NCI to more effectively address administrative and regulatory requirements for initiating trials and recruiting veterans into cancer clinical trials.15 This partnership—the NCI And VA Interagency Group to Accelerate Trials Enrollment (NAVIGATE)—supports 12 sites with a central CSP Coordinating Center (CSPCC).
CSPCC provides support, shares best practices and provides organizational commitment at the senior levels of both agencies to overcome potential barriers. The goals and strategies are described by Schiller and colleagues.16 While still in its early stage as a cancer research network, NAVIGATE may be integrated with POPCaP and other parts of VA clinical research enterprise. This would allow us to specialize in advancing oncology care and to leverage capabilities more specifically to precision oncology. With an emphasis on recruitment, NAVIGATE has established capabilities with VA Informatics and Computing Infrastructure to quickly identify patients who may be eligible for particular clinical trials. We envision further refining these capabilities for precision oncology trials that incorporate genetic and other information for individual patients. VA also hopes to inform trial sponsors about design considerations. This is important since networked investigators will have direct insights into patient-level factors, which may help with more effectively identifying and enrolling them into trials for their particular cancers.
Conclusions
VA may have an opportunity to reach out to veterans who may not have immediate access to facilities running clinical trials. As it develops capabilities to bring the trial to the veteran, VA could have more virtual and/or centralized recruitment strategies. This would broaden opportunities for considering novel approaches that may not rely on a more traditional facility-based recruitment approach.
Ultimately, VA can be a critical part of a national effort to fight and, perhaps even, defeat cancers. With its extensive resources and capabilities, VA has the ability to advance a precision oncology agenda that provides veterans with the highest standard of care. It has built upon many key elements in clinical, technological and scientific fields of study that would challenge most health care systems given the extensive costs involved. In addition, creating strong partnerships with organizations such as PCF, NCI, and DoD that are complementary in resources and expertise will help VA to build a national network for cancer care. Putting this all together will support and facilitate a vision for more precise care for any veteran with cancer by more rapidly enabling the testing and approval of medications developed for this purpose.
Acknowledgments
The authors would like to thank Daphne Swancutt for comments and edits on drafts of this article.
1. Lynparza (Olaparib) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP Inc, 2019.
2. Rubraca (rucaparib) [package insert]: Clovis Oncology, Inc., Boulder, CO: 2018.
3. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725-1735. doi:10.1056/NEJMra1407390
4. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7-30. doi:10.3322/caac.21590
6. Bentley DR. Decoding the human genome sequence. Hum Mol Genet. 2000;9(16):2353-2358. doi:10.1093/hmg/9.16.2353
7. National Human Genome Research institute. The cost of sequencing a human genome. https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost. Updated October 30, 2019. Accessed July 31, 2020. 8. Paggio JCD, Sullivan R, Booth CM. Targeting the value of targeted therapy. Oncotarget. 2017;8(53):90612-90613. Published 2017 Oct 7. doi:10.18632/oncotarget.21596
9. Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408-2417. doi:10.1056/NEJMoa062867
10. Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial [published correction appears in Lancet Oncol. 2020 Apr;21(4):e182]. Lancet Oncol. 2020;21(4):508-518. doi:10.1016/S1470-2045(20)30074-7
11. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
12. Fiore LD, Brophy MT, Ferguson RE, et al. Data sharing, clinical trials, and biomarkers in precision oncology: challenges, opportunities, and programs at the Department of Veterans Affairs. Clin Pharmacol Ther. 2017;101(5):586-589. doi:10.1002/cpt.660
13. Lee JSH, Darcy KM, Hu H, et al. From discovery to practice and survivorship: building a national real-world data learning healthcare framework for military and veteran cancer patients. Clin Pharmacol Ther. 2019;106(1):52-57. doi:10.1002/cpt.1425
14. Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380 [published correction appears in N Engl J Med 2000 Oct 19;343(16):1204]. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
15. Condon DL, Beck D, Kenworthy-Heinige T, et al. A cross-cutting approach to enhancing clinical trial site success: The Department of Veterans Affairs’ Network of Dedicated Enrollment Sites (NODES) model. Contemp Clin Trials Commun. 2017;6:78-84. Published 2017 Mar 29. doi:10.1016/j.conctc.2017.03.006
16. Schiller SJ, Shannon C, Brophy MT, et al. The National Cancer Institute and Department of Veterans Affairs Interagency Group to Accelerate Trials Enrollment (NAVIGATE): A federal collaboration to improve cancer care. Semin Oncol. 2019;46(4-5):308-313. doi:10.1053/j.seminoncol.2019.09.005
1. Lynparza (Olaparib) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP Inc, 2019.
2. Rubraca (rucaparib) [package insert]: Clovis Oncology, Inc., Boulder, CO: 2018.
3. McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725-1735. doi:10.1056/NEJMra1407390
4. de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091-2102. doi:10.1056/NEJMoa1911440
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7-30. doi:10.3322/caac.21590
6. Bentley DR. Decoding the human genome sequence. Hum Mol Genet. 2000;9(16):2353-2358. doi:10.1093/hmg/9.16.2353
7. National Human Genome Research institute. The cost of sequencing a human genome. https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost. Updated October 30, 2019. Accessed July 31, 2020. 8. Paggio JCD, Sullivan R, Booth CM. Targeting the value of targeted therapy. Oncotarget. 2017;8(53):90612-90613. Published 2017 Oct 7. doi:10.18632/oncotarget.21596
9. Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408-2417. doi:10.1056/NEJMoa062867
10. Pishvaian MJ, Blais EM, Brody JR, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial [published correction appears in Lancet Oncol. 2020 Apr;21(4):e182]. Lancet Oncol. 2020;21(4):508-518. doi:10.1016/S1470-2045(20)30074-7
11. Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer [published correction appears in Cell. 2015 Jul 16;162(2):454]. Cell. 2015;161(5):1215-1228. doi:10.1016/j.cell.2015.05.001
12. Fiore LD, Brophy MT, Ferguson RE, et al. Data sharing, clinical trials, and biomarkers in precision oncology: challenges, opportunities, and programs at the Department of Veterans Affairs. Clin Pharmacol Ther. 2017;101(5):586-589. doi:10.1002/cpt.660
13. Lee JSH, Darcy KM, Hu H, et al. From discovery to practice and survivorship: building a national real-world data learning healthcare framework for military and veteran cancer patients. Clin Pharmacol Ther. 2019;106(1):52-57. doi:10.1002/cpt.1425
14. Lieberman DA, Weiss DG, Bond JH, Ahnen DJ, Garewal H, Chejfec G. Use of colonoscopy to screen asymptomatic adults for colorectal cancer. Veterans Affairs Cooperative Study Group 380 [published correction appears in N Engl J Med 2000 Oct 19;343(16):1204]. N Engl J Med. 2000;343(3):162-168. doi:10.1056/NEJM200007203430301
15. Condon DL, Beck D, Kenworthy-Heinige T, et al. A cross-cutting approach to enhancing clinical trial site success: The Department of Veterans Affairs’ Network of Dedicated Enrollment Sites (NODES) model. Contemp Clin Trials Commun. 2017;6:78-84. Published 2017 Mar 29. doi:10.1016/j.conctc.2017.03.006
16. Schiller SJ, Shannon C, Brophy MT, et al. The National Cancer Institute and Department of Veterans Affairs Interagency Group to Accelerate Trials Enrollment (NAVIGATE): A federal collaboration to improve cancer care. Semin Oncol. 2019;46(4-5):308-313. doi:10.1053/j.seminoncol.2019.09.005
Advances in Precision Oncology: Foreword (FULL)
For > 90 years, the US Department of Veterans Affairs (VA) has been in the vanguard of cancer research and treatment—improving the lives of veterans and all Americans. In 1932, recognizing the intrinsic link between research and clinical care, the Edward Hines, Jr. VA Hospital in Chicago, Illinois, established a tumor research laboratory to complement the work of its cancer treatment center. As the first VA laboratory to receive funding specifically for research, the new facility symbolized a paradigm shift in thinking about cancer treatment.
Today, through its National Precision Oncology Program (NPOP), the Veterans Health Administration (VHA) has embarked upon another paradigm shift—one that also puts research front and center by leveraging VHA’s unique assets as a learning health care system. As noted by Montgomery and colleagues, “given its size, integration and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.”1 The articles in this special issue, which focus on the 2 cancers that affects the most veterans—prostate and lung—show the transformative work underway to develop a new model of collaboration in cancer care.
At VHA, research and practice are not just proximal; they are truly integrated in the service of enhancing veterans’ outcomes. For example, > 60% of VA researchers are clinicians who also provide direct patient care. As observed by Levine and colleagues, “meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa.”2
For example, it was physician-scientist Donald Gleason, MD, PhD, who in the 1960s pioneered the standardized system that helps doctors better assess and treat prostate cancer (the Gleason score). More recently, physician-scientists Matthew Rettig, MD, and Bruce Montgomery, MD, both leading experts in prostate cancer research, were instrumental to VA’s partnership with the Prostate Cancer Foundation (PCF) to establish a national network for oncology trials serving veterans.
Having an embedded research program within the nation’s largest integrated health care system also provides the VA with the ability to conduct large-scale, multisite clinical trials. Since the 1940s, the VA Cooperative Studies Program (CSP) has generated key research findings across a range of diseases, including cancer, and provided definitive evidence and learning. In 1994, CSP launched its Prostate Cancer Intervention vs Observation Trial (PIVOT) study to determine whether observation is as effective as surgery for early-stage prostate cancer. Today, through the CSP, VA researchers are conducting a randomized, phase 3 clinical trial called VA Lung cancer surgery Or stereotactic Radiotherapy trial (VALOR) that will assess which of the 2 modalities is better when treating veterans with operable early-stage non-small cell lung cancer.
Additionally, VA is privileged to serve a patient population so dedicated to their country that many volunteer to serve again as participants in VA research clinical trials. In fact, Levine and colleagues credit the patients willing to enter clinical trials for the collective call to action and “critical philanthropic investment” that led to the Precision Oncology Program for Cancer of the Prostate (POPCaP).2
As a learning health care system, we also have been mindful of lessons drawn from the ongoing COVID-19 public health crisis. Almost overnight, VHA shifted from in-person to virtual visits to minimize the risk for veterans and their families. At the same time, we limited in-person clinical research visits to those that were required for the Veterans’ health or well-being and conducted large numbers of virtual research visits. (Notably, the current crisis motivated accelerated study regarding virtual research trials, clarifying which touchpoints must be face-to-face and which have been face-to-face due mainly to convention.) In parallel, we also launched numerous clinical studies focused on the fight against COVID-19. Our capacity to transition both clinical care and research is due in no small part to our preexisting and strong foundation in telehealth.
With one-third of our patient population living in rural areas, these achievements are vital to our commitment of “no veteran left behind.” These efforts were recently boosted by VHA’s newest partnership with the Bristol Myers Squibb Foundation to establish a national teleoncology center that will enable all veterans to benefit from new research advances no matter where they live.
Precision oncology represents a new model of collaboration in cancer care among clinicians, operations leaders, researchers and veterans. By leveraging the many assets that have contributed to VA’s success as a learning health care system, we can fulfill the promise of providing leading edge cancer care to all veterans.
1. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37(suppl 4):S48-S53. doi:10.12788/fp.0021
2. Levine RD, Ekanayake RN, Martin AC, et al. Prostate Cancer Foundation-Department of Veterans Affairs Partnership: a model of public-private collaboration to advance treatment and care of invasive cancers. Fed Pract. 2020;37(suppl 4):S32-S37. doi:10.12788/fp.0035
For > 90 years, the US Department of Veterans Affairs (VA) has been in the vanguard of cancer research and treatment—improving the lives of veterans and all Americans. In 1932, recognizing the intrinsic link between research and clinical care, the Edward Hines, Jr. VA Hospital in Chicago, Illinois, established a tumor research laboratory to complement the work of its cancer treatment center. As the first VA laboratory to receive funding specifically for research, the new facility symbolized a paradigm shift in thinking about cancer treatment.
Today, through its National Precision Oncology Program (NPOP), the Veterans Health Administration (VHA) has embarked upon another paradigm shift—one that also puts research front and center by leveraging VHA’s unique assets as a learning health care system. As noted by Montgomery and colleagues, “given its size, integration and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.”1 The articles in this special issue, which focus on the 2 cancers that affects the most veterans—prostate and lung—show the transformative work underway to develop a new model of collaboration in cancer care.
At VHA, research and practice are not just proximal; they are truly integrated in the service of enhancing veterans’ outcomes. For example, > 60% of VA researchers are clinicians who also provide direct patient care. As observed by Levine and colleagues, “meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa.”2
For example, it was physician-scientist Donald Gleason, MD, PhD, who in the 1960s pioneered the standardized system that helps doctors better assess and treat prostate cancer (the Gleason score). More recently, physician-scientists Matthew Rettig, MD, and Bruce Montgomery, MD, both leading experts in prostate cancer research, were instrumental to VA’s partnership with the Prostate Cancer Foundation (PCF) to establish a national network for oncology trials serving veterans.
Having an embedded research program within the nation’s largest integrated health care system also provides the VA with the ability to conduct large-scale, multisite clinical trials. Since the 1940s, the VA Cooperative Studies Program (CSP) has generated key research findings across a range of diseases, including cancer, and provided definitive evidence and learning. In 1994, CSP launched its Prostate Cancer Intervention vs Observation Trial (PIVOT) study to determine whether observation is as effective as surgery for early-stage prostate cancer. Today, through the CSP, VA researchers are conducting a randomized, phase 3 clinical trial called VA Lung cancer surgery Or stereotactic Radiotherapy trial (VALOR) that will assess which of the 2 modalities is better when treating veterans with operable early-stage non-small cell lung cancer.
Additionally, VA is privileged to serve a patient population so dedicated to their country that many volunteer to serve again as participants in VA research clinical trials. In fact, Levine and colleagues credit the patients willing to enter clinical trials for the collective call to action and “critical philanthropic investment” that led to the Precision Oncology Program for Cancer of the Prostate (POPCaP).2
As a learning health care system, we also have been mindful of lessons drawn from the ongoing COVID-19 public health crisis. Almost overnight, VHA shifted from in-person to virtual visits to minimize the risk for veterans and their families. At the same time, we limited in-person clinical research visits to those that were required for the Veterans’ health or well-being and conducted large numbers of virtual research visits. (Notably, the current crisis motivated accelerated study regarding virtual research trials, clarifying which touchpoints must be face-to-face and which have been face-to-face due mainly to convention.) In parallel, we also launched numerous clinical studies focused on the fight against COVID-19. Our capacity to transition both clinical care and research is due in no small part to our preexisting and strong foundation in telehealth.
With one-third of our patient population living in rural areas, these achievements are vital to our commitment of “no veteran left behind.” These efforts were recently boosted by VHA’s newest partnership with the Bristol Myers Squibb Foundation to establish a national teleoncology center that will enable all veterans to benefit from new research advances no matter where they live.
Precision oncology represents a new model of collaboration in cancer care among clinicians, operations leaders, researchers and veterans. By leveraging the many assets that have contributed to VA’s success as a learning health care system, we can fulfill the promise of providing leading edge cancer care to all veterans.
For > 90 years, the US Department of Veterans Affairs (VA) has been in the vanguard of cancer research and treatment—improving the lives of veterans and all Americans. In 1932, recognizing the intrinsic link between research and clinical care, the Edward Hines, Jr. VA Hospital in Chicago, Illinois, established a tumor research laboratory to complement the work of its cancer treatment center. As the first VA laboratory to receive funding specifically for research, the new facility symbolized a paradigm shift in thinking about cancer treatment.
Today, through its National Precision Oncology Program (NPOP), the Veterans Health Administration (VHA) has embarked upon another paradigm shift—one that also puts research front and center by leveraging VHA’s unique assets as a learning health care system. As noted by Montgomery and colleagues, “given its size, integration and capabilities, the VA is an ideal setting for rapid learning cycles of testing and implementing best practices at scale.”1 The articles in this special issue, which focus on the 2 cancers that affects the most veterans—prostate and lung—show the transformative work underway to develop a new model of collaboration in cancer care.
At VHA, research and practice are not just proximal; they are truly integrated in the service of enhancing veterans’ outcomes. For example, > 60% of VA researchers are clinicians who also provide direct patient care. As observed by Levine and colleagues, “meaningful advances in cancer care depend on both laboratory and clinical research. This combination, known as translational research, takes discoveries in the laboratory and applies them to patients and vice versa.”2
For example, it was physician-scientist Donald Gleason, MD, PhD, who in the 1960s pioneered the standardized system that helps doctors better assess and treat prostate cancer (the Gleason score). More recently, physician-scientists Matthew Rettig, MD, and Bruce Montgomery, MD, both leading experts in prostate cancer research, were instrumental to VA’s partnership with the Prostate Cancer Foundation (PCF) to establish a national network for oncology trials serving veterans.
Having an embedded research program within the nation’s largest integrated health care system also provides the VA with the ability to conduct large-scale, multisite clinical trials. Since the 1940s, the VA Cooperative Studies Program (CSP) has generated key research findings across a range of diseases, including cancer, and provided definitive evidence and learning. In 1994, CSP launched its Prostate Cancer Intervention vs Observation Trial (PIVOT) study to determine whether observation is as effective as surgery for early-stage prostate cancer. Today, through the CSP, VA researchers are conducting a randomized, phase 3 clinical trial called VA Lung cancer surgery Or stereotactic Radiotherapy trial (VALOR) that will assess which of the 2 modalities is better when treating veterans with operable early-stage non-small cell lung cancer.
Additionally, VA is privileged to serve a patient population so dedicated to their country that many volunteer to serve again as participants in VA research clinical trials. In fact, Levine and colleagues credit the patients willing to enter clinical trials for the collective call to action and “critical philanthropic investment” that led to the Precision Oncology Program for Cancer of the Prostate (POPCaP).2
As a learning health care system, we also have been mindful of lessons drawn from the ongoing COVID-19 public health crisis. Almost overnight, VHA shifted from in-person to virtual visits to minimize the risk for veterans and their families. At the same time, we limited in-person clinical research visits to those that were required for the Veterans’ health or well-being and conducted large numbers of virtual research visits. (Notably, the current crisis motivated accelerated study regarding virtual research trials, clarifying which touchpoints must be face-to-face and which have been face-to-face due mainly to convention.) In parallel, we also launched numerous clinical studies focused on the fight against COVID-19. Our capacity to transition both clinical care and research is due in no small part to our preexisting and strong foundation in telehealth.
With one-third of our patient population living in rural areas, these achievements are vital to our commitment of “no veteran left behind.” These efforts were recently boosted by VHA’s newest partnership with the Bristol Myers Squibb Foundation to establish a national teleoncology center that will enable all veterans to benefit from new research advances no matter where they live.
Precision oncology represents a new model of collaboration in cancer care among clinicians, operations leaders, researchers and veterans. By leveraging the many assets that have contributed to VA’s success as a learning health care system, we can fulfill the promise of providing leading edge cancer care to all veterans.
1. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37(suppl 4):S48-S53. doi:10.12788/fp.0021
2. Levine RD, Ekanayake RN, Martin AC, et al. Prostate Cancer Foundation-Department of Veterans Affairs Partnership: a model of public-private collaboration to advance treatment and care of invasive cancers. Fed Pract. 2020;37(suppl 4):S32-S37. doi:10.12788/fp.0035
1. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37(suppl 4):S48-S53. doi:10.12788/fp.0021
2. Levine RD, Ekanayake RN, Martin AC, et al. Prostate Cancer Foundation-Department of Veterans Affairs Partnership: a model of public-private collaboration to advance treatment and care of invasive cancers. Fed Pract. 2020;37(suppl 4):S32-S37. doi:10.12788/fp.0035
Prostate Cancer Foundation-Department of Veterans Affairs Partnership: A Model of Public-Private Collaboration to Advance Treatment and Care of Invasive Cancers(FULL)
In late 2016, the US Department of Veterans Affairs (VA) and the Prostate Cancer Foundation (PCF) announced a multiyear public-private partnership to deliver precision oncology and best-in-class care to all veterans battling prostate cancer.1 The creation of this partnership was due to several favorable factors. At that time, VA Secretary Robert A. McDonald had created the Secretary’s Center for Strategic Partnerships. This Center provided a mechanism for nonprofit and industry partners to collaborate with the VA, thereby advancing partnerships that served the VA mission of “serving and honoring…America’s veterans.”1,2 Concurrently, Vice President Joseph Biden’s Cancer Moonshot (later renamed the Beau Biden Cancer Moonshot) charged PCF and other cancer-focused organizations with the ambitious goal of making a decade’s worth of advancements in cancer prevention, diagnosis, and treatment in 5 years.3 As such, both organizations were positioned to recognize and address the unique prostate cancer challenges faced by male veterans, which ultimately led to the PCF-VA partnership.
A number of factors have allowed the PCF-VA partnership to scale the Centers of Excellence (COE) program. This article seeks to highlight the strategic organizing and mobilization techniques employed by the PCF-VA partnership, which can inform future public-private hybrid initiatives focused on precision medicine.
Executive Leadership as Patient Advocates
From its creation, the PCF-VA partnership placed as much importance on veteran patient care as it has on making oncologic advances. The fact that this focus came primarily from executive leadership was critical to the partnership’s success. PCF board members emphasized the significance of prioritizing veterans and military families in cancer research efforts.
A notable example is S. Ward “Trip” Casscells, MD, a veteran who was deployed to Iraq in 2006 and subsequently served as US Department of Defense Assistant Secretary of Defense for Health Affairs. He focused much of his leadership on ensuring that veterans and military families, having performed a critical service for the country, were served with this same degree of excellence when it came to health.4 Fellow PCF Board member Lawrence Stupski, spoke publicly about his drug-resistant form of prostate cancer, bringing awareness to the complexity of ending death and suffering from the disease.5 Like Casscells, Stupski has a military service background, and served in Vietnam in 1968 as an officer in the US Navy. Both participated in multiple prostate cancer clinical trials themselves, serving as models of veteran trial participants. This visibility and leadership created a culture where veterans were not just instrumental in advancing cancer research, but also representative of a responsibility to ensure high-quality care for an underserved and at-risk community (Figure 1).

Executive advocacy and visionary philanthropy on behalf of veterans were vital to catalyzing the PCF-VA partnership framework, allowing both organizations to act on shared goals through a joint venture. Stupski’s legacy also jump-started the partnership itself, as the Stupski Foundation provided the crucial initial funding to launch a pilot version of the partnership.
Ultimately, this suggests that entrepreneurial philanthropy, top-level patient-led advocacy, and executive leadership can bolster the success of future health partnerships by advocating for specific missions, thus allowing convergence of goals between public and private entities. Visibility of leaders also encourages participation in the initiative itself, specifically in regard to patients being willing to enroll in clinical trials.
During the Launch Pad: Pathways to Cancer InnoVAtion PCF-VA summit in November 2016, PCF and the VA signed a memorandum of understanding (MOU) that solidified joint goals and accountability practices to create a scalable model for veteran-centered, genomics-based precision oncology care. Special focus was placed upon developing clinical trials for vulnerable veteran populations (Figure 2). PCF dedicated $50 million of funding to this partnership, facilitated largely in part by several philanthropists who stepped up after the MOU was signed, and early, life-extending successes from the pilot were demonstrated. This “snowballing” of funding indicates that the establishment of a public-private health partnership—with clear and compelling goals and early proof-of-concept—galvanizes efforts to further advance the partnership by garnering critical philanthropic investment.
VHA Economy of Scale
Utilizing the vast capacity of the Veterans Health Administration (VHA) for care was integral to the success of the partnership. The VHA serves 9 million veterans each year in 1,255 health care facilities, which include 170 medical centers and 1,075 outpatient clinics.6 As the nation’s largest integrated health care system, the VHA approaches cancer care with a single electronic health record system across all of its facilities, featuring comprehensive clinical outcome documentation.7 The VHA’s systemwide DNA sequence platform, through the National Precision Oncology Program (NPOP), also provided an optimal area for research and standardization of precision oncology practices on a national scale.8
Centers of Excellence: An Adaptable Model
The primary thrust of the partnership centers on the PCF-VA COEs, which form the Precision Oncology Program for Cancer of the Prostate (POPCaP) network. Over the last 4 years, PCF-deployed philanthropy has established 12 PCF-VA COEs, located in the Bronx and Manhattan, New York; Tampa Bay, Florida; Los Angeles, California; Seattle, Washington; Chicago, Illinois; Philadelphia, Pennsylvania; Ann Arbor, Michigan; Durham, North Carolina; Washington, DC; Boston, Massachusetts; and Portland, Oregon. Sites were initially chosen based on strong connections to academic medical centers, National Cancer Institute-designated comprehensive care centers, and physician-scientists who were professionally invested in precision prostate cancer oncology. Drawing on PCF’s existing networks helped to identify these areas, which were already rich in human and technological capital, before expanding to areas that were less resource rich. Future health partnerships may therefore consider capitalizing on existing relationships to spark initial growth, which can provide pathways for scaling.
In collaboration with NPOP, COEs work to sequence genomic and somatic tissue from veterans with metastatic prostate cancer, connect patients to appropriate clinical trials and treatment pathways, and advance guidelines for precision cancer care. Certain aspects of COE operations remain constant across all facilities. Annual progress reports, comprising of a written report, slide deck of accomplishments, and bulleted delineation of challenges and future plans are required of all COE-funded investigators. All COEs also are tasked with hiring a center coordinator, instituting a standardized sequencing and mutation reporting protocol, participating in consortium-wide phase 3 studies, and engaging in monthly conference calls to assess progress. A complete list of requirements is found in the Table.

However, the methods through which these goals must be completed is at the discretion of the COE investigators. Each COE, due to institutional and patient variance, experiences distinctive challenges and must mold its practice to fit existing capacities. For example, certain sites optimized workflow by training coordinators to analyze specimens, thereby improving care speed for veteran patients. Other COEs maximized nearby resources by hiring offsite specialists such as genetic counselors and interventional radiologists. By providing the freedom to design site-specific methodology, the PCF-VA partnership allows each COE to meet the award goals through any appropriate path using the funds provided, increasing efficiency and optimizing progress. This diversity of protocol also helped to expand the capabilities of the POPCaP Network, allowing sites to specialize in areas of interest in precision oncology. This eventually helped to inform future initiatives.
Accelerating Clinical Trials
A critical feature of the POPCaP network is the Prostate Cancer Analysis for Therapy Choice (PATCH) plexus.9 Through this investigative umbrella, veterans who are sequenced at any COE are given access to clinical trials at sites across POPCaP. Funding is available to support veteran travel to these sites, decreasing the chance that a veteran’s location is a barrier to treatment. In this way, the PCF-VA partnership continues to broaden treatment scopes for tens of thousands of veterans while simultaneously advancing clinical knowledge of precision oncology.
Fostering a Scientific Community
The PCF-VA partnership’s COE initiative capitalizes on resources from both nonprofit and public sectors to cultivate dynamic scientific discourse and investigative support. Through monthly meetings of the NPOP Molecular Oncology Tumor Board, COE investigators receive guidance and education to better assist veterans sequenced through their programs. Another example of enriched scientific collaboration are the Dream Team investigators, who were collaboratively funded by PCF, Stand Up 2 Cancer, and the American Association for Cancer Research.10 These teams made significant strides in genomic profiling of advanced prostate cancer and outpatient computed tomography-guided metastatic bone biopsy techniques. Through the PCF-VA partnership, COE researchers benefited from these investigators’ insight and expertise during regular check-in calls with investigators. PCF’s Prescription Pad, also connects all investigators to current therapies and trials, better informing them of future directions for their own work (Figure 3).11,12
The PCF-VA partnership also facilitates peer-to-peer communication through regular inperson and virtual meetings of investigators, coordinators, and other stakeholders. These meetings allow the creation of focused working groups composed of COE leaders across the nation. The working groups seek to improve all aspects of functionality, including operational roadblocks, sequencing and phenotyping protocols, and addressing health service disparities. The VA Puget Sound Health Care System in Seattle, Washington, and the West Los Angeles VA Medical Center in California both are mentorship sites that play instrumental roles in guiding newer sites through challenges, such as obtaining rapid pathology results and navigating the VA system. This interinvestigator communication also helps to recruit new junior and senior investigators to POPCaP, thereby broadening the network’s reach.
Future Pathways
In line with the mission outlined in the MOU of developing treatments for veteran populations, the PCF-VA partnership has actively pursued addressing veteran health inequities. In 2018, a $2.5 million gift from Robert F. Smith, Founder, Chairman, and Chief Executive Officer of Vista Equity Partners, set up the Chicago COE with the express purpose of serving African American veterans, who represent men at highest risk of prostate cancer incidence and mortality.13 A regularly convened health disparities working group explores future efforts. This group, composed of VA investigators, epidemiologists, geneticists, and other field leaders, seeks to advance the most compelling approaches to eliminate inequities in prostate cancer care.
A novel nursing initiative that focuses on the role of nurses in providing genetic services for prostate cancer is being developed. The need for new genetic care models and significant barriers to genetic service delivery have been well-documented for prostate cancer.14 The initiative provides nurses with opportunities to train with POPCaP and VA geneticists, enroll in a City of Hope genetics course, and to join a collaborative of geneticists, medical oncologists, and nurse practitioners.15 By furthering nursing education and leadership, the initiative empowers nurses to fill the gaps in veteran health care, particularly in genomics-based precision oncology.
The COE platform also has provided the foundation for the building of COEs for other cancers relevant to veterans, such as lung cancer. This expansion of COE function helps to further the VA goal of not only creating COEs, but a system of excellence. More recently, COE infrastructure has been leveraged in the fight against COVID-19 through HITCH, a clinical trial investigating the use of temporary androgen suppression in improving clinical outcomes of veterans with COVID-19.16 This expansion of function also provides a mechanism for COEs to continue to be funded in the future: attracting federal capital, private philanthropy, and industrial support is dependent on realized and expanded goals, as well as demonstrable progress in veteran care.
Conclusions
The PCF-VA partnership serves as an example of a public-private health partnership pursuing strategic pathways and bold goals to ensure that every eligible veteran has access to precision oncology. These pathways include advocacy on the part of executive leadership, recognizing existing economies of scale, building compelling narratives to maximize funding, creating flexible requirements, and facilitating a robust, resource-rich scientific network. This partnership already has opened doors to future initiatives and continues to adapt to a rapidly changing health landscape. The discussed strategies have the potential to inform future health initiatives and showcase how a systemic approach to eradicating health inequities can greatly benefit underserved communities.
The success of the PCF-VA partnership represents more than just an efficient partnership model. The partnership’s emphasis on veterans, who exemplify service, highlights the extent to which cancer patients sacrifice to contribute to medical research. This service necessitates a service in kind: all health stakeholders share the responsibility to rapidly advance therapies and care, both to honor the patients who have come before, and to meet the needs of patients with treatment resistant forms of the disease urgently awaiting precision breakthroughs and cures.
1. US Department of Veterans Affairs. Secretary’s Center for Strategic Partnerships (SCSP): about us. https://www.va.gov/scsp/about/. Updated January 22, 2020. Accessed July 27, 2020.
2. US Department of Veterans Affairs. About VA. https://www.va.gov/about_va/mission.asp. Updated August 20, 2015. Accessed July 27, 2020.
3. American Association for Cancer Research. National Cancer Moonshot Initiative. https://www.aacr.org/professionals/policy-and-advocacy/science-policy-government-affairs/national-cancer-moonshot-initiative. Accessed July 30, 2020.
4. Zogby J, Fighting cancer is a Defense Department obligation. https://www.huffpost.com/entry/fighting-cancer-is-our-co_b_837535. Updated May 25, 2011. Accessed July 30, 2020.
5. Colliver V. Lawrence Stupski, former Schwab exec, dies. San Francisco Chronicle June 12, 2013. https://www.sfchronicle.com/bayarea/article/Lawrence-Stupski-former-Schwab-exec-dies-4597329.php. Accessed July 30, 2020.
6. US Department of Veterans Affairs, Veterans Health Administration. About VHA. https://www.va.gov/health/aboutvha.asp. Updated July 14, 2019. Accessed July 27, 2020.
7. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) Network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37 (suppl 4):S48-S53. doi:10.12788/fp.0021
8. US Department of Veterans Affairs, National Oncology Program Office: about us. https://www.cancer.va.gov/CANCER/about.asp. Accessed July 28, 2020.
9. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(8):S62-S67. doi:10.12788/fp.0028
10. Prostate Cancer Foundation. Prostate Cancer Foundation and Stand Up To Cancer announce new dream team [press release]. https://www.pcf.org/news/prostate-cancer-foundation-and-stand-up-to-cancer-announce-new-dream-team/. Published April 1, 2020. Accessed July 30, 2020.
11. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
12. Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer [published correction appears in Nat Genet. 2019 Jul;51(7):1194]. Nat Genet. 2018;50(5):645-651. doi:10.1038/s41588-018-0078-z
13. Prostate Cancer Foundation. $2.5 million gift from Robert Frederick Smith to the Prostate Cancer Foundation is the largest donation ever dedicated to advancing prostate cancer research in African-American men [press release]. https://www.pcf.org/news/robert-frederick-smith-gift/. Published January 14, 2018. Accessed July 27, 2020.
14. Carlo MI, Giri VN, Paller CJ, et al. Evolving intersection between inherited cancer genetics and therapeutic clinical trials in prostate cancer: a white paper from the Germline Genetics Working Group of the Prostate Cancer Clinical Trials Consortium. JCO Precis Oncol. 2018;2018:10.1200/PO.18.00060. doi:10.1200/PO.18.00060
15. City of Hope. Intensive course in genomic cancer risk assessment. https://www.cityofhope.org/education/health-professional-education/cancer-genomics-education-program/intensive-course-in-cancer-risk-assessment-overview. Accessed July 28, 2020.
16. US National Library of Medicine, Clinicaltrial.gov. Hormonal Intervention for the Treatment in Veterans with COVID-19 Requiring Hospitalization (HITCH): NCT04397718. https://clinicaltrials.gov/ct2/show/NCT04397718. Updated July 23, 2020. Accessed July 30, 2020.
In late 2016, the US Department of Veterans Affairs (VA) and the Prostate Cancer Foundation (PCF) announced a multiyear public-private partnership to deliver precision oncology and best-in-class care to all veterans battling prostate cancer.1 The creation of this partnership was due to several favorable factors. At that time, VA Secretary Robert A. McDonald had created the Secretary’s Center for Strategic Partnerships. This Center provided a mechanism for nonprofit and industry partners to collaborate with the VA, thereby advancing partnerships that served the VA mission of “serving and honoring…America’s veterans.”1,2 Concurrently, Vice President Joseph Biden’s Cancer Moonshot (later renamed the Beau Biden Cancer Moonshot) charged PCF and other cancer-focused organizations with the ambitious goal of making a decade’s worth of advancements in cancer prevention, diagnosis, and treatment in 5 years.3 As such, both organizations were positioned to recognize and address the unique prostate cancer challenges faced by male veterans, which ultimately led to the PCF-VA partnership.
A number of factors have allowed the PCF-VA partnership to scale the Centers of Excellence (COE) program. This article seeks to highlight the strategic organizing and mobilization techniques employed by the PCF-VA partnership, which can inform future public-private hybrid initiatives focused on precision medicine.
Executive Leadership as Patient Advocates
From its creation, the PCF-VA partnership placed as much importance on veteran patient care as it has on making oncologic advances. The fact that this focus came primarily from executive leadership was critical to the partnership’s success. PCF board members emphasized the significance of prioritizing veterans and military families in cancer research efforts.
A notable example is S. Ward “Trip” Casscells, MD, a veteran who was deployed to Iraq in 2006 and subsequently served as US Department of Defense Assistant Secretary of Defense for Health Affairs. He focused much of his leadership on ensuring that veterans and military families, having performed a critical service for the country, were served with this same degree of excellence when it came to health.4 Fellow PCF Board member Lawrence Stupski, spoke publicly about his drug-resistant form of prostate cancer, bringing awareness to the complexity of ending death and suffering from the disease.5 Like Casscells, Stupski has a military service background, and served in Vietnam in 1968 as an officer in the US Navy. Both participated in multiple prostate cancer clinical trials themselves, serving as models of veteran trial participants. This visibility and leadership created a culture where veterans were not just instrumental in advancing cancer research, but also representative of a responsibility to ensure high-quality care for an underserved and at-risk community (Figure 1).
Executive advocacy and visionary philanthropy on behalf of veterans were vital to catalyzing the PCF-VA partnership framework, allowing both organizations to act on shared goals through a joint venture. Stupski’s legacy also jump-started the partnership itself, as the Stupski Foundation provided the crucial initial funding to launch a pilot version of the partnership.
Ultimately, this suggests that entrepreneurial philanthropy, top-level patient-led advocacy, and executive leadership can bolster the success of future health partnerships by advocating for specific missions, thus allowing convergence of goals between public and private entities. Visibility of leaders also encourages participation in the initiative itself, specifically in regard to patients being willing to enroll in clinical trials.
During the Launch Pad: Pathways to Cancer InnoVAtion PCF-VA summit in November 2016, PCF and the VA signed a memorandum of understanding (MOU) that solidified joint goals and accountability practices to create a scalable model for veteran-centered, genomics-based precision oncology care. Special focus was placed upon developing clinical trials for vulnerable veteran populations (Figure 2). PCF dedicated $50 million of funding to this partnership, facilitated largely in part by several philanthropists who stepped up after the MOU was signed, and early, life-extending successes from the pilot were demonstrated. This “snowballing” of funding indicates that the establishment of a public-private health partnership—with clear and compelling goals and early proof-of-concept—galvanizes efforts to further advance the partnership by garnering critical philanthropic investment.
VHA Economy of Scale
Utilizing the vast capacity of the Veterans Health Administration (VHA) for care was integral to the success of the partnership. The VHA serves 9 million veterans each year in 1,255 health care facilities, which include 170 medical centers and 1,075 outpatient clinics.6 As the nation’s largest integrated health care system, the VHA approaches cancer care with a single electronic health record system across all of its facilities, featuring comprehensive clinical outcome documentation.7 The VHA’s systemwide DNA sequence platform, through the National Precision Oncology Program (NPOP), also provided an optimal area for research and standardization of precision oncology practices on a national scale.8
Centers of Excellence: An Adaptable Model
The primary thrust of the partnership centers on the PCF-VA COEs, which form the Precision Oncology Program for Cancer of the Prostate (POPCaP) network. Over the last 4 years, PCF-deployed philanthropy has established 12 PCF-VA COEs, located in the Bronx and Manhattan, New York; Tampa Bay, Florida; Los Angeles, California; Seattle, Washington; Chicago, Illinois; Philadelphia, Pennsylvania; Ann Arbor, Michigan; Durham, North Carolina; Washington, DC; Boston, Massachusetts; and Portland, Oregon. Sites were initially chosen based on strong connections to academic medical centers, National Cancer Institute-designated comprehensive care centers, and physician-scientists who were professionally invested in precision prostate cancer oncology. Drawing on PCF’s existing networks helped to identify these areas, which were already rich in human and technological capital, before expanding to areas that were less resource rich. Future health partnerships may therefore consider capitalizing on existing relationships to spark initial growth, which can provide pathways for scaling.
In collaboration with NPOP, COEs work to sequence genomic and somatic tissue from veterans with metastatic prostate cancer, connect patients to appropriate clinical trials and treatment pathways, and advance guidelines for precision cancer care. Certain aspects of COE operations remain constant across all facilities. Annual progress reports, comprising of a written report, slide deck of accomplishments, and bulleted delineation of challenges and future plans are required of all COE-funded investigators. All COEs also are tasked with hiring a center coordinator, instituting a standardized sequencing and mutation reporting protocol, participating in consortium-wide phase 3 studies, and engaging in monthly conference calls to assess progress. A complete list of requirements is found in the Table.
However, the methods through which these goals must be completed is at the discretion of the COE investigators. Each COE, due to institutional and patient variance, experiences distinctive challenges and must mold its practice to fit existing capacities. For example, certain sites optimized workflow by training coordinators to analyze specimens, thereby improving care speed for veteran patients. Other COEs maximized nearby resources by hiring offsite specialists such as genetic counselors and interventional radiologists. By providing the freedom to design site-specific methodology, the PCF-VA partnership allows each COE to meet the award goals through any appropriate path using the funds provided, increasing efficiency and optimizing progress. This diversity of protocol also helped to expand the capabilities of the POPCaP Network, allowing sites to specialize in areas of interest in precision oncology. This eventually helped to inform future initiatives.
Accelerating Clinical Trials
A critical feature of the POPCaP network is the Prostate Cancer Analysis for Therapy Choice (PATCH) plexus.9 Through this investigative umbrella, veterans who are sequenced at any COE are given access to clinical trials at sites across POPCaP. Funding is available to support veteran travel to these sites, decreasing the chance that a veteran’s location is a barrier to treatment. In this way, the PCF-VA partnership continues to broaden treatment scopes for tens of thousands of veterans while simultaneously advancing clinical knowledge of precision oncology.
Fostering a Scientific Community
The PCF-VA partnership’s COE initiative capitalizes on resources from both nonprofit and public sectors to cultivate dynamic scientific discourse and investigative support. Through monthly meetings of the NPOP Molecular Oncology Tumor Board, COE investigators receive guidance and education to better assist veterans sequenced through their programs. Another example of enriched scientific collaboration are the Dream Team investigators, who were collaboratively funded by PCF, Stand Up 2 Cancer, and the American Association for Cancer Research.10 These teams made significant strides in genomic profiling of advanced prostate cancer and outpatient computed tomography-guided metastatic bone biopsy techniques. Through the PCF-VA partnership, COE researchers benefited from these investigators’ insight and expertise during regular check-in calls with investigators. PCF’s Prescription Pad, also connects all investigators to current therapies and trials, better informing them of future directions for their own work (Figure 3).11,12
The PCF-VA partnership also facilitates peer-to-peer communication through regular inperson and virtual meetings of investigators, coordinators, and other stakeholders. These meetings allow the creation of focused working groups composed of COE leaders across the nation. The working groups seek to improve all aspects of functionality, including operational roadblocks, sequencing and phenotyping protocols, and addressing health service disparities. The VA Puget Sound Health Care System in Seattle, Washington, and the West Los Angeles VA Medical Center in California both are mentorship sites that play instrumental roles in guiding newer sites through challenges, such as obtaining rapid pathology results and navigating the VA system. This interinvestigator communication also helps to recruit new junior and senior investigators to POPCaP, thereby broadening the network’s reach.
Future Pathways
In line with the mission outlined in the MOU of developing treatments for veteran populations, the PCF-VA partnership has actively pursued addressing veteran health inequities. In 2018, a $2.5 million gift from Robert F. Smith, Founder, Chairman, and Chief Executive Officer of Vista Equity Partners, set up the Chicago COE with the express purpose of serving African American veterans, who represent men at highest risk of prostate cancer incidence and mortality.13 A regularly convened health disparities working group explores future efforts. This group, composed of VA investigators, epidemiologists, geneticists, and other field leaders, seeks to advance the most compelling approaches to eliminate inequities in prostate cancer care.
A novel nursing initiative that focuses on the role of nurses in providing genetic services for prostate cancer is being developed. The need for new genetic care models and significant barriers to genetic service delivery have been well-documented for prostate cancer.14 The initiative provides nurses with opportunities to train with POPCaP and VA geneticists, enroll in a City of Hope genetics course, and to join a collaborative of geneticists, medical oncologists, and nurse practitioners.15 By furthering nursing education and leadership, the initiative empowers nurses to fill the gaps in veteran health care, particularly in genomics-based precision oncology.
The COE platform also has provided the foundation for the building of COEs for other cancers relevant to veterans, such as lung cancer. This expansion of COE function helps to further the VA goal of not only creating COEs, but a system of excellence. More recently, COE infrastructure has been leveraged in the fight against COVID-19 through HITCH, a clinical trial investigating the use of temporary androgen suppression in improving clinical outcomes of veterans with COVID-19.16 This expansion of function also provides a mechanism for COEs to continue to be funded in the future: attracting federal capital, private philanthropy, and industrial support is dependent on realized and expanded goals, as well as demonstrable progress in veteran care.
Conclusions
The PCF-VA partnership serves as an example of a public-private health partnership pursuing strategic pathways and bold goals to ensure that every eligible veteran has access to precision oncology. These pathways include advocacy on the part of executive leadership, recognizing existing economies of scale, building compelling narratives to maximize funding, creating flexible requirements, and facilitating a robust, resource-rich scientific network. This partnership already has opened doors to future initiatives and continues to adapt to a rapidly changing health landscape. The discussed strategies have the potential to inform future health initiatives and showcase how a systemic approach to eradicating health inequities can greatly benefit underserved communities.
The success of the PCF-VA partnership represents more than just an efficient partnership model. The partnership’s emphasis on veterans, who exemplify service, highlights the extent to which cancer patients sacrifice to contribute to medical research. This service necessitates a service in kind: all health stakeholders share the responsibility to rapidly advance therapies and care, both to honor the patients who have come before, and to meet the needs of patients with treatment resistant forms of the disease urgently awaiting precision breakthroughs and cures.
In late 2016, the US Department of Veterans Affairs (VA) and the Prostate Cancer Foundation (PCF) announced a multiyear public-private partnership to deliver precision oncology and best-in-class care to all veterans battling prostate cancer.1 The creation of this partnership was due to several favorable factors. At that time, VA Secretary Robert A. McDonald had created the Secretary’s Center for Strategic Partnerships. This Center provided a mechanism for nonprofit and industry partners to collaborate with the VA, thereby advancing partnerships that served the VA mission of “serving and honoring…America’s veterans.”1,2 Concurrently, Vice President Joseph Biden’s Cancer Moonshot (later renamed the Beau Biden Cancer Moonshot) charged PCF and other cancer-focused organizations with the ambitious goal of making a decade’s worth of advancements in cancer prevention, diagnosis, and treatment in 5 years.3 As such, both organizations were positioned to recognize and address the unique prostate cancer challenges faced by male veterans, which ultimately led to the PCF-VA partnership.
A number of factors have allowed the PCF-VA partnership to scale the Centers of Excellence (COE) program. This article seeks to highlight the strategic organizing and mobilization techniques employed by the PCF-VA partnership, which can inform future public-private hybrid initiatives focused on precision medicine.
Executive Leadership as Patient Advocates
From its creation, the PCF-VA partnership placed as much importance on veteran patient care as it has on making oncologic advances. The fact that this focus came primarily from executive leadership was critical to the partnership’s success. PCF board members emphasized the significance of prioritizing veterans and military families in cancer research efforts.
A notable example is S. Ward “Trip” Casscells, MD, a veteran who was deployed to Iraq in 2006 and subsequently served as US Department of Defense Assistant Secretary of Defense for Health Affairs. He focused much of his leadership on ensuring that veterans and military families, having performed a critical service for the country, were served with this same degree of excellence when it came to health.4 Fellow PCF Board member Lawrence Stupski, spoke publicly about his drug-resistant form of prostate cancer, bringing awareness to the complexity of ending death and suffering from the disease.5 Like Casscells, Stupski has a military service background, and served in Vietnam in 1968 as an officer in the US Navy. Both participated in multiple prostate cancer clinical trials themselves, serving as models of veteran trial participants. This visibility and leadership created a culture where veterans were not just instrumental in advancing cancer research, but also representative of a responsibility to ensure high-quality care for an underserved and at-risk community (Figure 1).
Executive advocacy and visionary philanthropy on behalf of veterans were vital to catalyzing the PCF-VA partnership framework, allowing both organizations to act on shared goals through a joint venture. Stupski’s legacy also jump-started the partnership itself, as the Stupski Foundation provided the crucial initial funding to launch a pilot version of the partnership.
Ultimately, this suggests that entrepreneurial philanthropy, top-level patient-led advocacy, and executive leadership can bolster the success of future health partnerships by advocating for specific missions, thus allowing convergence of goals between public and private entities. Visibility of leaders also encourages participation in the initiative itself, specifically in regard to patients being willing to enroll in clinical trials.
During the Launch Pad: Pathways to Cancer InnoVAtion PCF-VA summit in November 2016, PCF and the VA signed a memorandum of understanding (MOU) that solidified joint goals and accountability practices to create a scalable model for veteran-centered, genomics-based precision oncology care. Special focus was placed upon developing clinical trials for vulnerable veteran populations (Figure 2). PCF dedicated $50 million of funding to this partnership, facilitated largely in part by several philanthropists who stepped up after the MOU was signed, and early, life-extending successes from the pilot were demonstrated. This “snowballing” of funding indicates that the establishment of a public-private health partnership—with clear and compelling goals and early proof-of-concept—galvanizes efforts to further advance the partnership by garnering critical philanthropic investment.
VHA Economy of Scale
Utilizing the vast capacity of the Veterans Health Administration (VHA) for care was integral to the success of the partnership. The VHA serves 9 million veterans each year in 1,255 health care facilities, which include 170 medical centers and 1,075 outpatient clinics.6 As the nation’s largest integrated health care system, the VHA approaches cancer care with a single electronic health record system across all of its facilities, featuring comprehensive clinical outcome documentation.7 The VHA’s systemwide DNA sequence platform, through the National Precision Oncology Program (NPOP), also provided an optimal area for research and standardization of precision oncology practices on a national scale.8
Centers of Excellence: An Adaptable Model
The primary thrust of the partnership centers on the PCF-VA COEs, which form the Precision Oncology Program for Cancer of the Prostate (POPCaP) network. Over the last 4 years, PCF-deployed philanthropy has established 12 PCF-VA COEs, located in the Bronx and Manhattan, New York; Tampa Bay, Florida; Los Angeles, California; Seattle, Washington; Chicago, Illinois; Philadelphia, Pennsylvania; Ann Arbor, Michigan; Durham, North Carolina; Washington, DC; Boston, Massachusetts; and Portland, Oregon. Sites were initially chosen based on strong connections to academic medical centers, National Cancer Institute-designated comprehensive care centers, and physician-scientists who were professionally invested in precision prostate cancer oncology. Drawing on PCF’s existing networks helped to identify these areas, which were already rich in human and technological capital, before expanding to areas that were less resource rich. Future health partnerships may therefore consider capitalizing on existing relationships to spark initial growth, which can provide pathways for scaling.
In collaboration with NPOP, COEs work to sequence genomic and somatic tissue from veterans with metastatic prostate cancer, connect patients to appropriate clinical trials and treatment pathways, and advance guidelines for precision cancer care. Certain aspects of COE operations remain constant across all facilities. Annual progress reports, comprising of a written report, slide deck of accomplishments, and bulleted delineation of challenges and future plans are required of all COE-funded investigators. All COEs also are tasked with hiring a center coordinator, instituting a standardized sequencing and mutation reporting protocol, participating in consortium-wide phase 3 studies, and engaging in monthly conference calls to assess progress. A complete list of requirements is found in the Table.
However, the methods through which these goals must be completed is at the discretion of the COE investigators. Each COE, due to institutional and patient variance, experiences distinctive challenges and must mold its practice to fit existing capacities. For example, certain sites optimized workflow by training coordinators to analyze specimens, thereby improving care speed for veteran patients. Other COEs maximized nearby resources by hiring offsite specialists such as genetic counselors and interventional radiologists. By providing the freedom to design site-specific methodology, the PCF-VA partnership allows each COE to meet the award goals through any appropriate path using the funds provided, increasing efficiency and optimizing progress. This diversity of protocol also helped to expand the capabilities of the POPCaP Network, allowing sites to specialize in areas of interest in precision oncology. This eventually helped to inform future initiatives.
Accelerating Clinical Trials
A critical feature of the POPCaP network is the Prostate Cancer Analysis for Therapy Choice (PATCH) plexus.9 Through this investigative umbrella, veterans who are sequenced at any COE are given access to clinical trials at sites across POPCaP. Funding is available to support veteran travel to these sites, decreasing the chance that a veteran’s location is a barrier to treatment. In this way, the PCF-VA partnership continues to broaden treatment scopes for tens of thousands of veterans while simultaneously advancing clinical knowledge of precision oncology.
Fostering a Scientific Community
The PCF-VA partnership’s COE initiative capitalizes on resources from both nonprofit and public sectors to cultivate dynamic scientific discourse and investigative support. Through monthly meetings of the NPOP Molecular Oncology Tumor Board, COE investigators receive guidance and education to better assist veterans sequenced through their programs. Another example of enriched scientific collaboration are the Dream Team investigators, who were collaboratively funded by PCF, Stand Up 2 Cancer, and the American Association for Cancer Research.10 These teams made significant strides in genomic profiling of advanced prostate cancer and outpatient computed tomography-guided metastatic bone biopsy techniques. Through the PCF-VA partnership, COE researchers benefited from these investigators’ insight and expertise during regular check-in calls with investigators. PCF’s Prescription Pad, also connects all investigators to current therapies and trials, better informing them of future directions for their own work (Figure 3).11,12
The PCF-VA partnership also facilitates peer-to-peer communication through regular inperson and virtual meetings of investigators, coordinators, and other stakeholders. These meetings allow the creation of focused working groups composed of COE leaders across the nation. The working groups seek to improve all aspects of functionality, including operational roadblocks, sequencing and phenotyping protocols, and addressing health service disparities. The VA Puget Sound Health Care System in Seattle, Washington, and the West Los Angeles VA Medical Center in California both are mentorship sites that play instrumental roles in guiding newer sites through challenges, such as obtaining rapid pathology results and navigating the VA system. This interinvestigator communication also helps to recruit new junior and senior investigators to POPCaP, thereby broadening the network’s reach.
Future Pathways
In line with the mission outlined in the MOU of developing treatments for veteran populations, the PCF-VA partnership has actively pursued addressing veteran health inequities. In 2018, a $2.5 million gift from Robert F. Smith, Founder, Chairman, and Chief Executive Officer of Vista Equity Partners, set up the Chicago COE with the express purpose of serving African American veterans, who represent men at highest risk of prostate cancer incidence and mortality.13 A regularly convened health disparities working group explores future efforts. This group, composed of VA investigators, epidemiologists, geneticists, and other field leaders, seeks to advance the most compelling approaches to eliminate inequities in prostate cancer care.
A novel nursing initiative that focuses on the role of nurses in providing genetic services for prostate cancer is being developed. The need for new genetic care models and significant barriers to genetic service delivery have been well-documented for prostate cancer.14 The initiative provides nurses with opportunities to train with POPCaP and VA geneticists, enroll in a City of Hope genetics course, and to join a collaborative of geneticists, medical oncologists, and nurse practitioners.15 By furthering nursing education and leadership, the initiative empowers nurses to fill the gaps in veteran health care, particularly in genomics-based precision oncology.
The COE platform also has provided the foundation for the building of COEs for other cancers relevant to veterans, such as lung cancer. This expansion of COE function helps to further the VA goal of not only creating COEs, but a system of excellence. More recently, COE infrastructure has been leveraged in the fight against COVID-19 through HITCH, a clinical trial investigating the use of temporary androgen suppression in improving clinical outcomes of veterans with COVID-19.16 This expansion of function also provides a mechanism for COEs to continue to be funded in the future: attracting federal capital, private philanthropy, and industrial support is dependent on realized and expanded goals, as well as demonstrable progress in veteran care.
Conclusions
The PCF-VA partnership serves as an example of a public-private health partnership pursuing strategic pathways and bold goals to ensure that every eligible veteran has access to precision oncology. These pathways include advocacy on the part of executive leadership, recognizing existing economies of scale, building compelling narratives to maximize funding, creating flexible requirements, and facilitating a robust, resource-rich scientific network. This partnership already has opened doors to future initiatives and continues to adapt to a rapidly changing health landscape. The discussed strategies have the potential to inform future health initiatives and showcase how a systemic approach to eradicating health inequities can greatly benefit underserved communities.
The success of the PCF-VA partnership represents more than just an efficient partnership model. The partnership’s emphasis on veterans, who exemplify service, highlights the extent to which cancer patients sacrifice to contribute to medical research. This service necessitates a service in kind: all health stakeholders share the responsibility to rapidly advance therapies and care, both to honor the patients who have come before, and to meet the needs of patients with treatment resistant forms of the disease urgently awaiting precision breakthroughs and cures.
1. US Department of Veterans Affairs. Secretary’s Center for Strategic Partnerships (SCSP): about us. https://www.va.gov/scsp/about/. Updated January 22, 2020. Accessed July 27, 2020.
2. US Department of Veterans Affairs. About VA. https://www.va.gov/about_va/mission.asp. Updated August 20, 2015. Accessed July 27, 2020.
3. American Association for Cancer Research. National Cancer Moonshot Initiative. https://www.aacr.org/professionals/policy-and-advocacy/science-policy-government-affairs/national-cancer-moonshot-initiative. Accessed July 30, 2020.
4. Zogby J, Fighting cancer is a Defense Department obligation. https://www.huffpost.com/entry/fighting-cancer-is-our-co_b_837535. Updated May 25, 2011. Accessed July 30, 2020.
5. Colliver V. Lawrence Stupski, former Schwab exec, dies. San Francisco Chronicle June 12, 2013. https://www.sfchronicle.com/bayarea/article/Lawrence-Stupski-former-Schwab-exec-dies-4597329.php. Accessed July 30, 2020.
6. US Department of Veterans Affairs, Veterans Health Administration. About VHA. https://www.va.gov/health/aboutvha.asp. Updated July 14, 2019. Accessed July 27, 2020.
7. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) Network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37 (suppl 4):S48-S53. doi:10.12788/fp.0021
8. US Department of Veterans Affairs, National Oncology Program Office: about us. https://www.cancer.va.gov/CANCER/about.asp. Accessed July 28, 2020.
9. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(8):S62-S67. doi:10.12788/fp.0028
10. Prostate Cancer Foundation. Prostate Cancer Foundation and Stand Up To Cancer announce new dream team [press release]. https://www.pcf.org/news/prostate-cancer-foundation-and-stand-up-to-cancer-announce-new-dream-team/. Published April 1, 2020. Accessed July 30, 2020.
11. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
12. Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer [published correction appears in Nat Genet. 2019 Jul;51(7):1194]. Nat Genet. 2018;50(5):645-651. doi:10.1038/s41588-018-0078-z
13. Prostate Cancer Foundation. $2.5 million gift from Robert Frederick Smith to the Prostate Cancer Foundation is the largest donation ever dedicated to advancing prostate cancer research in African-American men [press release]. https://www.pcf.org/news/robert-frederick-smith-gift/. Published January 14, 2018. Accessed July 27, 2020.
14. Carlo MI, Giri VN, Paller CJ, et al. Evolving intersection between inherited cancer genetics and therapeutic clinical trials in prostate cancer: a white paper from the Germline Genetics Working Group of the Prostate Cancer Clinical Trials Consortium. JCO Precis Oncol. 2018;2018:10.1200/PO.18.00060. doi:10.1200/PO.18.00060
15. City of Hope. Intensive course in genomic cancer risk assessment. https://www.cityofhope.org/education/health-professional-education/cancer-genomics-education-program/intensive-course-in-cancer-risk-assessment-overview. Accessed July 28, 2020.
16. US National Library of Medicine, Clinicaltrial.gov. Hormonal Intervention for the Treatment in Veterans with COVID-19 Requiring Hospitalization (HITCH): NCT04397718. https://clinicaltrials.gov/ct2/show/NCT04397718. Updated July 23, 2020. Accessed July 30, 2020.
1. US Department of Veterans Affairs. Secretary’s Center for Strategic Partnerships (SCSP): about us. https://www.va.gov/scsp/about/. Updated January 22, 2020. Accessed July 27, 2020.
2. US Department of Veterans Affairs. About VA. https://www.va.gov/about_va/mission.asp. Updated August 20, 2015. Accessed July 27, 2020.
3. American Association for Cancer Research. National Cancer Moonshot Initiative. https://www.aacr.org/professionals/policy-and-advocacy/science-policy-government-affairs/national-cancer-moonshot-initiative. Accessed July 30, 2020.
4. Zogby J, Fighting cancer is a Defense Department obligation. https://www.huffpost.com/entry/fighting-cancer-is-our-co_b_837535. Updated May 25, 2011. Accessed July 30, 2020.
5. Colliver V. Lawrence Stupski, former Schwab exec, dies. San Francisco Chronicle June 12, 2013. https://www.sfchronicle.com/bayarea/article/Lawrence-Stupski-former-Schwab-exec-dies-4597329.php. Accessed July 30, 2020.
6. US Department of Veterans Affairs, Veterans Health Administration. About VHA. https://www.va.gov/health/aboutvha.asp. Updated July 14, 2019. Accessed July 27, 2020.
7. Montgomery B, Rettig M, Kasten J, Muralidhar S, Myrie K, Ramoni R. The Precision Oncology Program for Cancer of the Prostate (POPCaP) Network: a Veterans Affairs/Prostate Cancer Foundation collaboration. Fed Pract. 2020;37 (suppl 4):S48-S53. doi:10.12788/fp.0021
8. US Department of Veterans Affairs, National Oncology Program Office: about us. https://www.cancer.va.gov/CANCER/about.asp. Accessed July 28, 2020.
9. Graff JN, Huang GD. Leveraging Veterans Health Administration clinical and research resources to accelerate discovery and testing in precision oncology. Fed Pract. 2020;37(8):S62-S67. doi:10.12788/fp.0028
10. Prostate Cancer Foundation. Prostate Cancer Foundation and Stand Up To Cancer announce new dream team [press release]. https://www.pcf.org/news/prostate-cancer-foundation-and-stand-up-to-cancer-announce-new-dream-team/. Published April 1, 2020. Accessed July 30, 2020.
11. Quigley DA, Dang HX, Zhao SG, et al. Genomic hallmarks and structural variation in metastatic prostate cancer [published correction appears in Cell. 2018 Oct 18;175(3):889]. Cell. 2018;174(3):758-769.e9. doi:10.1016/j.cell.2018.06.039
12. Armenia J, Wankowicz SAM, Liu D, et al. The long tail of oncogenic drivers in prostate cancer [published correction appears in Nat Genet. 2019 Jul;51(7):1194]. Nat Genet. 2018;50(5):645-651. doi:10.1038/s41588-018-0078-z
13. Prostate Cancer Foundation. $2.5 million gift from Robert Frederick Smith to the Prostate Cancer Foundation is the largest donation ever dedicated to advancing prostate cancer research in African-American men [press release]. https://www.pcf.org/news/robert-frederick-smith-gift/. Published January 14, 2018. Accessed July 27, 2020.
14. Carlo MI, Giri VN, Paller CJ, et al. Evolving intersection between inherited cancer genetics and therapeutic clinical trials in prostate cancer: a white paper from the Germline Genetics Working Group of the Prostate Cancer Clinical Trials Consortium. JCO Precis Oncol. 2018;2018:10.1200/PO.18.00060. doi:10.1200/PO.18.00060
15. City of Hope. Intensive course in genomic cancer risk assessment. https://www.cityofhope.org/education/health-professional-education/cancer-genomics-education-program/intensive-course-in-cancer-risk-assessment-overview. Accessed July 28, 2020.
16. US National Library of Medicine, Clinicaltrial.gov. Hormonal Intervention for the Treatment in Veterans with COVID-19 Requiring Hospitalization (HITCH): NCT04397718. https://clinicaltrials.gov/ct2/show/NCT04397718. Updated July 23, 2020. Accessed July 30, 2020.
Remote 24-hour monitoring improves life for patients on chemo
A remote monitoring system was highly effective in managing symptoms and improving quality of life among patients with cancer who were receiving chemotherapy, say researchers reporting the first clinical trial of the new approach.
The study tested the Advanced Symptom Management System (ASyMS) for patients with various cancer types who were undergoing treatment at cancer centers in several European countries. The study primarily focused on patients who were being treated with curative intent.
The 24-hour monitoring system optimized symptom management in a manner safe, secure, and in “real time,” the team reports. This is particularly relevant during the COVID-19 pandemic, they note.
“Our findings suggest that an evidence based remote monitoring intervention, such as ASyMS, has potential for implementation into routine care to make a meaningful difference to people with cancer,” the authors conclude.
The findings were published online in BMJ.
The results show that “ASyMS can be implemented across multiple countries within diverse health care systems,” commented lead author Roma Maguire, PhD, a professor of digital health and care at the University of Strathclyde, in Glasgow, and director of the Health and Care Futures initiative.
So far, the system has only been used in clinical research studies, but “our findings do suggest that it is feasible to implement our system on a wider scale,” she added.
The study cohort included 829 patients with various cancers, including nonmetastatic breast cancer, colorectal cancer, Hodgkin disease, and non-Hodgkin lymphoma. The patients were receiving first-line adjuvant chemotherapy or chemotherapy for the first time in 5 years. They were recruited from 12 cancer centers in Austria, Greece, Norway, the Republic of Ireland, and the United Kingdom.
Patients were randomly assigned to receive ASyMS (n = 415) or standard care (n = 414) during six cycles of chemotherapy.
The primary outcome was symptom burden, as determined using the Memorial Symptom Assessment Scale. Secondary outcomes included health-related quality of life, as determined by results on the Functional Assessment of Cancer Therapy–General, the Supportive Care Needs Survey–Short Form, the State-Trait Anxiety Inventory–Revised, the Communication and Attitudinal Self-Efficacy scale for cancer, and the Work Limitations Questionnaire.
Patients in the intervention group completed a daily symptom questionnaire on a handheld ASyMS device, which generated alerts to health care professionals if any intervention was needed. The patients were also provided with advice and information on how to manage their symptoms themselves.
Among patients using ASyMS, symptom burden remained at prechemotherapy levels over all six chemotherapy cycles. Conversely, the control group reported an increase in symptom burden from cycle 1; symptom burden slowly decreased during the remaining chemotherapy cycles.
Overall, the investigators found that, among the patients who used ASyMS, psychological and physical symptoms were significantly reduced, along with the level of distress associated with each symptom.
In addition, for the patients who used ASyMS, health-related quality-of-life scores were higher across all cycles. The authors note that the improvements in health-related quality of life are consistent with findings from recent trials of the use of remote monitoring systems in chemotherapy care. The intervention group also experienced significant improvements regarding the need for supportive care.
Improvements in symptom burden differed among countries. The greatest improvements were seen among patients with breast cancer, Hodgkin disease, or non-Hodgkin lymphoma in Austria, Ireland, and the United Kingdom. The reasons for these differences are unclear, the authors note. ASyMS was developed in the United Kingdom, and it’s possible that ASyMS is more effective in countries that have health care systems similar to the system in the United Kingdom, they suggest.
The incidence of adverse events was similar for the two groups, although the rate of neutropenia was higher among patients using ASyMS (n = 125; 64%) in comparison with the standard-care group ( n = 71; 36%). Three deaths occurred in each study arm. The number of planned hospital admissions was similar between the two groups (34 vs. 38), as was the number of unplanned hospital admissions (120 vs. 109). No ASyMS device-related incidents were reported.
The trial was funded by the European Commission and was sponsored by the University of Strathclyde. Dr. Maguire has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A remote monitoring system was highly effective in managing symptoms and improving quality of life among patients with cancer who were receiving chemotherapy, say researchers reporting the first clinical trial of the new approach.
The study tested the Advanced Symptom Management System (ASyMS) for patients with various cancer types who were undergoing treatment at cancer centers in several European countries. The study primarily focused on patients who were being treated with curative intent.
The 24-hour monitoring system optimized symptom management in a manner safe, secure, and in “real time,” the team reports. This is particularly relevant during the COVID-19 pandemic, they note.
“Our findings suggest that an evidence based remote monitoring intervention, such as ASyMS, has potential for implementation into routine care to make a meaningful difference to people with cancer,” the authors conclude.
The findings were published online in BMJ.
The results show that “ASyMS can be implemented across multiple countries within diverse health care systems,” commented lead author Roma Maguire, PhD, a professor of digital health and care at the University of Strathclyde, in Glasgow, and director of the Health and Care Futures initiative.
So far, the system has only been used in clinical research studies, but “our findings do suggest that it is feasible to implement our system on a wider scale,” she added.
The study cohort included 829 patients with various cancers, including nonmetastatic breast cancer, colorectal cancer, Hodgkin disease, and non-Hodgkin lymphoma. The patients were receiving first-line adjuvant chemotherapy or chemotherapy for the first time in 5 years. They were recruited from 12 cancer centers in Austria, Greece, Norway, the Republic of Ireland, and the United Kingdom.
Patients were randomly assigned to receive ASyMS (n = 415) or standard care (n = 414) during six cycles of chemotherapy.
The primary outcome was symptom burden, as determined using the Memorial Symptom Assessment Scale. Secondary outcomes included health-related quality of life, as determined by results on the Functional Assessment of Cancer Therapy–General, the Supportive Care Needs Survey–Short Form, the State-Trait Anxiety Inventory–Revised, the Communication and Attitudinal Self-Efficacy scale for cancer, and the Work Limitations Questionnaire.
Patients in the intervention group completed a daily symptom questionnaire on a handheld ASyMS device, which generated alerts to health care professionals if any intervention was needed. The patients were also provided with advice and information on how to manage their symptoms themselves.
Among patients using ASyMS, symptom burden remained at prechemotherapy levels over all six chemotherapy cycles. Conversely, the control group reported an increase in symptom burden from cycle 1; symptom burden slowly decreased during the remaining chemotherapy cycles.
Overall, the investigators found that, among the patients who used ASyMS, psychological and physical symptoms were significantly reduced, along with the level of distress associated with each symptom.
In addition, for the patients who used ASyMS, health-related quality-of-life scores were higher across all cycles. The authors note that the improvements in health-related quality of life are consistent with findings from recent trials of the use of remote monitoring systems in chemotherapy care. The intervention group also experienced significant improvements regarding the need for supportive care.
Improvements in symptom burden differed among countries. The greatest improvements were seen among patients with breast cancer, Hodgkin disease, or non-Hodgkin lymphoma in Austria, Ireland, and the United Kingdom. The reasons for these differences are unclear, the authors note. ASyMS was developed in the United Kingdom, and it’s possible that ASyMS is more effective in countries that have health care systems similar to the system in the United Kingdom, they suggest.
The incidence of adverse events was similar for the two groups, although the rate of neutropenia was higher among patients using ASyMS (n = 125; 64%) in comparison with the standard-care group ( n = 71; 36%). Three deaths occurred in each study arm. The number of planned hospital admissions was similar between the two groups (34 vs. 38), as was the number of unplanned hospital admissions (120 vs. 109). No ASyMS device-related incidents were reported.
The trial was funded by the European Commission and was sponsored by the University of Strathclyde. Dr. Maguire has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A remote monitoring system was highly effective in managing symptoms and improving quality of life among patients with cancer who were receiving chemotherapy, say researchers reporting the first clinical trial of the new approach.
The study tested the Advanced Symptom Management System (ASyMS) for patients with various cancer types who were undergoing treatment at cancer centers in several European countries. The study primarily focused on patients who were being treated with curative intent.
The 24-hour monitoring system optimized symptom management in a manner safe, secure, and in “real time,” the team reports. This is particularly relevant during the COVID-19 pandemic, they note.
“Our findings suggest that an evidence based remote monitoring intervention, such as ASyMS, has potential for implementation into routine care to make a meaningful difference to people with cancer,” the authors conclude.
The findings were published online in BMJ.
The results show that “ASyMS can be implemented across multiple countries within diverse health care systems,” commented lead author Roma Maguire, PhD, a professor of digital health and care at the University of Strathclyde, in Glasgow, and director of the Health and Care Futures initiative.
So far, the system has only been used in clinical research studies, but “our findings do suggest that it is feasible to implement our system on a wider scale,” she added.
The study cohort included 829 patients with various cancers, including nonmetastatic breast cancer, colorectal cancer, Hodgkin disease, and non-Hodgkin lymphoma. The patients were receiving first-line adjuvant chemotherapy or chemotherapy for the first time in 5 years. They were recruited from 12 cancer centers in Austria, Greece, Norway, the Republic of Ireland, and the United Kingdom.
Patients were randomly assigned to receive ASyMS (n = 415) or standard care (n = 414) during six cycles of chemotherapy.
The primary outcome was symptom burden, as determined using the Memorial Symptom Assessment Scale. Secondary outcomes included health-related quality of life, as determined by results on the Functional Assessment of Cancer Therapy–General, the Supportive Care Needs Survey–Short Form, the State-Trait Anxiety Inventory–Revised, the Communication and Attitudinal Self-Efficacy scale for cancer, and the Work Limitations Questionnaire.
Patients in the intervention group completed a daily symptom questionnaire on a handheld ASyMS device, which generated alerts to health care professionals if any intervention was needed. The patients were also provided with advice and information on how to manage their symptoms themselves.
Among patients using ASyMS, symptom burden remained at prechemotherapy levels over all six chemotherapy cycles. Conversely, the control group reported an increase in symptom burden from cycle 1; symptom burden slowly decreased during the remaining chemotherapy cycles.
Overall, the investigators found that, among the patients who used ASyMS, psychological and physical symptoms were significantly reduced, along with the level of distress associated with each symptom.
In addition, for the patients who used ASyMS, health-related quality-of-life scores were higher across all cycles. The authors note that the improvements in health-related quality of life are consistent with findings from recent trials of the use of remote monitoring systems in chemotherapy care. The intervention group also experienced significant improvements regarding the need for supportive care.
Improvements in symptom burden differed among countries. The greatest improvements were seen among patients with breast cancer, Hodgkin disease, or non-Hodgkin lymphoma in Austria, Ireland, and the United Kingdom. The reasons for these differences are unclear, the authors note. ASyMS was developed in the United Kingdom, and it’s possible that ASyMS is more effective in countries that have health care systems similar to the system in the United Kingdom, they suggest.
The incidence of adverse events was similar for the two groups, although the rate of neutropenia was higher among patients using ASyMS (n = 125; 64%) in comparison with the standard-care group ( n = 71; 36%). Three deaths occurred in each study arm. The number of planned hospital admissions was similar between the two groups (34 vs. 38), as was the number of unplanned hospital admissions (120 vs. 109). No ASyMS device-related incidents were reported.
The trial was funded by the European Commission and was sponsored by the University of Strathclyde. Dr. Maguire has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Surgeon marks ‘right’ instead of ‘left’ testicle, then operates
Wrong-site surgery
Florida regulators have imposed a fine and other measures on a Tampa doctor who made a crucial error prior to his patient’s testicular surgery, as a story in the Miami Herald, among other news sites, reports.
On Sept. 10, 2019, a patient referred to in state documents as “C.F.” showed up for a procedure – a varicocelectomy – that would remove the enlarged veins in his left testicle. His doctor that day was Raul Fernandez-Crespo, MD, a urologist who had been licensed to practice in Florida since April of the same year. Dr. Fernandez-Crespo completed his urology residency at the University of Puerto Rico in 2019.
Following a conversation with C.F., Dr. Fernandez-Crespo designated what he believed was the proper surgical site – his patient’s right testicle.
He then proceeded to operate, but at some point during the procedure – news accounts don’t make clear when or how he became aware of his error – he realized C.F. had actually consented to a left-testicle varicocelectomy. With his patient still sedated, Dr. Fernandez-Crespo also completed the second procedure.
His mistake came to the attention of the Department of Health, which filed an administrative complaint against the surgeon. On June 17, 2021, the department’s medical licensing body, the Florida Board of Medicine, handed down its final order about the case.
In addition to imposing a $2,500 fine on Dr. Fernandez-Crespo and issuing “a letter of concern” – a public document that can be used as evidence in any relevant future disciplinary action against him – regulators said the surgeon must reimburse $2,045.56 to the department for its case-related administrative costs; take a 5-hour CME course in risk management or attend 8 hours of board disciplinary hearings; and, finally, give a 1-hour lecture on wrong-site surgeries at a board-approved medical facility.
Before this, Dr. Fernandez-Crespo had no previous disciplinary history with the Florida Board of Medicine.
Huge judgment after fertility procedure goes wrong
A Connecticut couple whose fertility and prenatal care at a state university health center proved disastrous will receive millions of dollars in damages, according to a report in the Hartford Courant.
In 2014, Jean-Marie Monroe-Lynch and her husband, Aaron Lynch, went to UConn Health, in Farmington, for treatment of Jean-Marie’s infertility. Her care was overseen by the Center for Advanced Reproductive Services (CARS), a private company then under contract with UConn Health. (The contract, which ended in 2014, obligated UConn to provide CARS providers with medical malpractice coverage.)
There, Jean-Marie was inseminated with sperm from a donor who turned out to be a carrier for cytomegalovirus (CMV), the herpes virus that can cause severe birth defects, or fetal death, when contracted by a pregnant woman. The insemination resulted in a twin pregnancy, a boy and a girl. The girl, Shay, died in utero after several of her organs became infected with CMV; the boy, Joshua, was born with severe mental and physical disabilities.
In their suit, Ms. Monroe-Lynch and her husband alleged that they were never cautioned about the risks associated with using a sperm donor whose blood had tested positive for CMV antibodies. Their suit further alleged that, at the 20-week ultrasound, UConn’s prenatal team failed to detect evidence of congenital CMV infection and again failed, at the 22-week ultrasound, to properly recognize and respond to abnormal findings.
“They totally dropped the ball,” said the couple’s attorney. “If you’re a pregnant woman and contract the virus for the first time, the results can be devastating.”
CARS disputes this conclusion, arguing that the plaintiffs failed to prove as a “matter of scientific fact” that Ms. Monroe-Lynch was infected with CMV as the result of her intrauterine insemination.
But Superior Court Judge Mark H. Taylor disagreed. In his 107-page ruling, he said that the court “agrees with the vast majority of superior courts, concluding that a physician providing obstetric care owes a direct duty to a mother to prevent harm to her child during gestation and delivery.”
Jean-Marie Monroe-Lynch and Aaron Lynch received a $37.6 million award, consisting of $24.1 million in economic damages and $13.5 million in noneconomic damages.
Their surviving child, Joshua, will reportedly require a lifetime of medical and other care. In the meantime, UConn Health vows to appeal the Superior Court’s decision.
COVID patient’s relative demands justice for fatal outcome
An Indiana man whose grandfather recently died after suffering a stroke is calling on state lawmakers to rethink legislation passed earlier this year to protect health care providers during the COVID-19 pandemic, according to a story reported by CBS4Indy.
Late last year, Daniel Enlow’s 83-year-old grandfather, Edward Rigney, was checked into Eskenazi Hospital, in Indianapolis. Mr. Rigney suffered from COPD and had also been diagnosed with COVID-19.
At some point during his hospitalization, medical staff attempted to place what seems to have been an arterial line in order to monitor his condition. During the procedure, or at some point shortly thereafter, an “iatrogenic air embolus” was released into his veins and caused a stroke, according to medical records and Mr. Rigney’s death certificate.
“I started asking for medical records because I wanted to know what was happening leading up to it in black and white in front of me,” said Mr. Enlow, who wished to present his evidence to a medical review panel, as required by Indiana law. The first step in this process would have been to consult with a medical malpractice attorney, but several declined to take his case.
Why? Because a pair of bills passed by Indiana legislators in early 2021 make COVID-19–related suits – even tangentially related ones – potentially difficult to take to court.
The bills raised the bar to file a medical malpractice claim in COVID-19 cases and to allow only those that involve “gross negligence or willful or wanton misconduct.”
“In the vast majority of cases, it’s impossible to prove that,” said Fred Schultz, immediate past president of the Indiana Trial Lawyers Association, who lobbied against the legislation.
The bills were never designed to offer “blanket freedom,” said GOP State Senator Aaron Freeman, sponsor of one of the bills. “If something is being used in a way that it is a complete bar to certain claims, then maybe we need to go back and look at it and open that up a little bit and make it less restrictive. I’m certainly open to having those conversations.”
Meanwhile, Mr. Enlow has vowed to keep pushing in the name of his late grandfather. The hospital’s parent company, Eskenazi Health, has declined to comment.
A version of this article first appeared on Medscape.com.
Wrong-site surgery
Florida regulators have imposed a fine and other measures on a Tampa doctor who made a crucial error prior to his patient’s testicular surgery, as a story in the Miami Herald, among other news sites, reports.
On Sept. 10, 2019, a patient referred to in state documents as “C.F.” showed up for a procedure – a varicocelectomy – that would remove the enlarged veins in his left testicle. His doctor that day was Raul Fernandez-Crespo, MD, a urologist who had been licensed to practice in Florida since April of the same year. Dr. Fernandez-Crespo completed his urology residency at the University of Puerto Rico in 2019.
Following a conversation with C.F., Dr. Fernandez-Crespo designated what he believed was the proper surgical site – his patient’s right testicle.
He then proceeded to operate, but at some point during the procedure – news accounts don’t make clear when or how he became aware of his error – he realized C.F. had actually consented to a left-testicle varicocelectomy. With his patient still sedated, Dr. Fernandez-Crespo also completed the second procedure.
His mistake came to the attention of the Department of Health, which filed an administrative complaint against the surgeon. On June 17, 2021, the department’s medical licensing body, the Florida Board of Medicine, handed down its final order about the case.
In addition to imposing a $2,500 fine on Dr. Fernandez-Crespo and issuing “a letter of concern” – a public document that can be used as evidence in any relevant future disciplinary action against him – regulators said the surgeon must reimburse $2,045.56 to the department for its case-related administrative costs; take a 5-hour CME course in risk management or attend 8 hours of board disciplinary hearings; and, finally, give a 1-hour lecture on wrong-site surgeries at a board-approved medical facility.
Before this, Dr. Fernandez-Crespo had no previous disciplinary history with the Florida Board of Medicine.
Huge judgment after fertility procedure goes wrong
A Connecticut couple whose fertility and prenatal care at a state university health center proved disastrous will receive millions of dollars in damages, according to a report in the Hartford Courant.
In 2014, Jean-Marie Monroe-Lynch and her husband, Aaron Lynch, went to UConn Health, in Farmington, for treatment of Jean-Marie’s infertility. Her care was overseen by the Center for Advanced Reproductive Services (CARS), a private company then under contract with UConn Health. (The contract, which ended in 2014, obligated UConn to provide CARS providers with medical malpractice coverage.)
There, Jean-Marie was inseminated with sperm from a donor who turned out to be a carrier for cytomegalovirus (CMV), the herpes virus that can cause severe birth defects, or fetal death, when contracted by a pregnant woman. The insemination resulted in a twin pregnancy, a boy and a girl. The girl, Shay, died in utero after several of her organs became infected with CMV; the boy, Joshua, was born with severe mental and physical disabilities.
In their suit, Ms. Monroe-Lynch and her husband alleged that they were never cautioned about the risks associated with using a sperm donor whose blood had tested positive for CMV antibodies. Their suit further alleged that, at the 20-week ultrasound, UConn’s prenatal team failed to detect evidence of congenital CMV infection and again failed, at the 22-week ultrasound, to properly recognize and respond to abnormal findings.
“They totally dropped the ball,” said the couple’s attorney. “If you’re a pregnant woman and contract the virus for the first time, the results can be devastating.”
CARS disputes this conclusion, arguing that the plaintiffs failed to prove as a “matter of scientific fact” that Ms. Monroe-Lynch was infected with CMV as the result of her intrauterine insemination.
But Superior Court Judge Mark H. Taylor disagreed. In his 107-page ruling, he said that the court “agrees with the vast majority of superior courts, concluding that a physician providing obstetric care owes a direct duty to a mother to prevent harm to her child during gestation and delivery.”
Jean-Marie Monroe-Lynch and Aaron Lynch received a $37.6 million award, consisting of $24.1 million in economic damages and $13.5 million in noneconomic damages.
Their surviving child, Joshua, will reportedly require a lifetime of medical and other care. In the meantime, UConn Health vows to appeal the Superior Court’s decision.
COVID patient’s relative demands justice for fatal outcome
An Indiana man whose grandfather recently died after suffering a stroke is calling on state lawmakers to rethink legislation passed earlier this year to protect health care providers during the COVID-19 pandemic, according to a story reported by CBS4Indy.
Late last year, Daniel Enlow’s 83-year-old grandfather, Edward Rigney, was checked into Eskenazi Hospital, in Indianapolis. Mr. Rigney suffered from COPD and had also been diagnosed with COVID-19.
At some point during his hospitalization, medical staff attempted to place what seems to have been an arterial line in order to monitor his condition. During the procedure, or at some point shortly thereafter, an “iatrogenic air embolus” was released into his veins and caused a stroke, according to medical records and Mr. Rigney’s death certificate.
“I started asking for medical records because I wanted to know what was happening leading up to it in black and white in front of me,” said Mr. Enlow, who wished to present his evidence to a medical review panel, as required by Indiana law. The first step in this process would have been to consult with a medical malpractice attorney, but several declined to take his case.
Why? Because a pair of bills passed by Indiana legislators in early 2021 make COVID-19–related suits – even tangentially related ones – potentially difficult to take to court.
The bills raised the bar to file a medical malpractice claim in COVID-19 cases and to allow only those that involve “gross negligence or willful or wanton misconduct.”
“In the vast majority of cases, it’s impossible to prove that,” said Fred Schultz, immediate past president of the Indiana Trial Lawyers Association, who lobbied against the legislation.
The bills were never designed to offer “blanket freedom,” said GOP State Senator Aaron Freeman, sponsor of one of the bills. “If something is being used in a way that it is a complete bar to certain claims, then maybe we need to go back and look at it and open that up a little bit and make it less restrictive. I’m certainly open to having those conversations.”
Meanwhile, Mr. Enlow has vowed to keep pushing in the name of his late grandfather. The hospital’s parent company, Eskenazi Health, has declined to comment.
A version of this article first appeared on Medscape.com.
Wrong-site surgery
Florida regulators have imposed a fine and other measures on a Tampa doctor who made a crucial error prior to his patient’s testicular surgery, as a story in the Miami Herald, among other news sites, reports.
On Sept. 10, 2019, a patient referred to in state documents as “C.F.” showed up for a procedure – a varicocelectomy – that would remove the enlarged veins in his left testicle. His doctor that day was Raul Fernandez-Crespo, MD, a urologist who had been licensed to practice in Florida since April of the same year. Dr. Fernandez-Crespo completed his urology residency at the University of Puerto Rico in 2019.
Following a conversation with C.F., Dr. Fernandez-Crespo designated what he believed was the proper surgical site – his patient’s right testicle.
He then proceeded to operate, but at some point during the procedure – news accounts don’t make clear when or how he became aware of his error – he realized C.F. had actually consented to a left-testicle varicocelectomy. With his patient still sedated, Dr. Fernandez-Crespo also completed the second procedure.
His mistake came to the attention of the Department of Health, which filed an administrative complaint against the surgeon. On June 17, 2021, the department’s medical licensing body, the Florida Board of Medicine, handed down its final order about the case.
In addition to imposing a $2,500 fine on Dr. Fernandez-Crespo and issuing “a letter of concern” – a public document that can be used as evidence in any relevant future disciplinary action against him – regulators said the surgeon must reimburse $2,045.56 to the department for its case-related administrative costs; take a 5-hour CME course in risk management or attend 8 hours of board disciplinary hearings; and, finally, give a 1-hour lecture on wrong-site surgeries at a board-approved medical facility.
Before this, Dr. Fernandez-Crespo had no previous disciplinary history with the Florida Board of Medicine.
Huge judgment after fertility procedure goes wrong
A Connecticut couple whose fertility and prenatal care at a state university health center proved disastrous will receive millions of dollars in damages, according to a report in the Hartford Courant.
In 2014, Jean-Marie Monroe-Lynch and her husband, Aaron Lynch, went to UConn Health, in Farmington, for treatment of Jean-Marie’s infertility. Her care was overseen by the Center for Advanced Reproductive Services (CARS), a private company then under contract with UConn Health. (The contract, which ended in 2014, obligated UConn to provide CARS providers with medical malpractice coverage.)
There, Jean-Marie was inseminated with sperm from a donor who turned out to be a carrier for cytomegalovirus (CMV), the herpes virus that can cause severe birth defects, or fetal death, when contracted by a pregnant woman. The insemination resulted in a twin pregnancy, a boy and a girl. The girl, Shay, died in utero after several of her organs became infected with CMV; the boy, Joshua, was born with severe mental and physical disabilities.
In their suit, Ms. Monroe-Lynch and her husband alleged that they were never cautioned about the risks associated with using a sperm donor whose blood had tested positive for CMV antibodies. Their suit further alleged that, at the 20-week ultrasound, UConn’s prenatal team failed to detect evidence of congenital CMV infection and again failed, at the 22-week ultrasound, to properly recognize and respond to abnormal findings.
“They totally dropped the ball,” said the couple’s attorney. “If you’re a pregnant woman and contract the virus for the first time, the results can be devastating.”
CARS disputes this conclusion, arguing that the plaintiffs failed to prove as a “matter of scientific fact” that Ms. Monroe-Lynch was infected with CMV as the result of her intrauterine insemination.
But Superior Court Judge Mark H. Taylor disagreed. In his 107-page ruling, he said that the court “agrees with the vast majority of superior courts, concluding that a physician providing obstetric care owes a direct duty to a mother to prevent harm to her child during gestation and delivery.”
Jean-Marie Monroe-Lynch and Aaron Lynch received a $37.6 million award, consisting of $24.1 million in economic damages and $13.5 million in noneconomic damages.
Their surviving child, Joshua, will reportedly require a lifetime of medical and other care. In the meantime, UConn Health vows to appeal the Superior Court’s decision.
COVID patient’s relative demands justice for fatal outcome
An Indiana man whose grandfather recently died after suffering a stroke is calling on state lawmakers to rethink legislation passed earlier this year to protect health care providers during the COVID-19 pandemic, according to a story reported by CBS4Indy.
Late last year, Daniel Enlow’s 83-year-old grandfather, Edward Rigney, was checked into Eskenazi Hospital, in Indianapolis. Mr. Rigney suffered from COPD and had also been diagnosed with COVID-19.
At some point during his hospitalization, medical staff attempted to place what seems to have been an arterial line in order to monitor his condition. During the procedure, or at some point shortly thereafter, an “iatrogenic air embolus” was released into his veins and caused a stroke, according to medical records and Mr. Rigney’s death certificate.
“I started asking for medical records because I wanted to know what was happening leading up to it in black and white in front of me,” said Mr. Enlow, who wished to present his evidence to a medical review panel, as required by Indiana law. The first step in this process would have been to consult with a medical malpractice attorney, but several declined to take his case.
Why? Because a pair of bills passed by Indiana legislators in early 2021 make COVID-19–related suits – even tangentially related ones – potentially difficult to take to court.
The bills raised the bar to file a medical malpractice claim in COVID-19 cases and to allow only those that involve “gross negligence or willful or wanton misconduct.”
“In the vast majority of cases, it’s impossible to prove that,” said Fred Schultz, immediate past president of the Indiana Trial Lawyers Association, who lobbied against the legislation.
The bills were never designed to offer “blanket freedom,” said GOP State Senator Aaron Freeman, sponsor of one of the bills. “If something is being used in a way that it is a complete bar to certain claims, then maybe we need to go back and look at it and open that up a little bit and make it less restrictive. I’m certainly open to having those conversations.”
Meanwhile, Mr. Enlow has vowed to keep pushing in the name of his late grandfather. The hospital’s parent company, Eskenazi Health, has declined to comment.
A version of this article first appeared on Medscape.com.
New investigational helmet device shrinks glioblastoma
This is the first time that the wearable Oncomagnetic device was tried with a patient.
The patient had end-stage recurrent glioblastoma and had undergone all standard therapy options. He wore the device for 5 weeks but died from an unrelated injury, so the treatment period was cut short.
A brain scan showed a 31% reduction of contrast-enhanced tumor volume, and an autopsy of his brain confirmed the rapid response to the treatment.
The case study was published online on July 22, 2021, in Frontiers in Oncology.
“I believe that there is a great potential with this device,” said study author David S. Baskin, MD, director of the Kenneth R. Peak Center for Brain and Pituitary Tumor Treatment in the department of neurosurgery at Houston Methodist Hospital. “This is a very exciting time.”
The team is now treating several patients with glioblastoma under compassionate use.
In an independent comment, Adilia Hormigo, MD, PhD, director of the neuro-oncology program at the Tisch Cancer Institute, Mount Sinai Health System, New York, noted that a clinical trial is needed to evaluate the device. “But this is an interesting idea, and we have to be open-minded in treating this fatal disease.”
Oscillating magnetic fields
The Oncomagnetic device consists of three oncoscillators that are attached to the outside of a helmet and are connected to a microprocessor-based electronic controller powered by a rechargeable battery.
It consists of a series of rotating magnets that produce oscillating magnetic fields that cover the entire brain, including the upper part of the brain stem. The device induces rapid apoptosis of glioblastoma cells, Dr. Baskin explained. Its mechanism of action involves disruption of the electron transport in the mitochondrial respiratory chain, causing an elevation of reactive oxygen species and caspase-dependent cancer cell death.
Dr. Baskin emphasized that the new Oncomagnetic device is very different from the Optune device (Novocare), which is already approved by the Food and Drug Administration and has been shown to increase survival among patients with glioblastoma. Optune uses tumor-treating fields (TTFs), which are electromagnetic waves that are delivered via an electric field generator through four transducer arrays that are placed on a shaved scalp. Preclinical studies indicated that the TTFs disrupt cell division by disrupting several steps in the mitotic process that are crucial for cell division.
Both of these devices “are using a type of external maneuver” rather than invasive intracranial approaches, said Dr. Hormingo. The experimental Oncomagnetic device may have an advantage in that it needs to be worn by the patient for fewer hours, she commented. A better understanding of the physics and underlying mechanism is needed, however. Clinical trials are an essential next step.
Most common brain cancer in adults
Glioblastoma is the most common malignant tumor of the brain in adults. Outcomes continue to be dismal. In more than 40 years, median survival has only modestly improved.
“We haven’t gotten very far with glioblastoma despite millions of dollars in research,” Dr. Baskin said. “With treatment, survival is about 15 months, and those are not very good months.”
Out of the box
Standard treatments for glioblastoma include surgery, radiotherapy, and chemotherapy, and many patients cannot tolerate some of these, Dr. Baskin noted. Hence, there is a great need for a different therapeutic approach that yields better outcomes with lower toxicity.
“We didn’t want to develop another chemotherapeutic agent that would help you live another 2 months,” he said in an interview. “We were trying to think out of the box.
“If you want to do something that will really make a difference in an aggressive tumor like glioblastoma, you have to attack something so basic that the tumor can’t evade it,” he said. “For example, with temozolomide, if it is unmethylated, the tumor can repair the DNA damage from the chemotherapy. Even if you’re sensitive to begin with, over time, the tumor will eventually become resistant.”
The new device stems from work by Dr. Baskin and colleagues on mitochondria, which he describes as the powerhouse of the cell. “Mitochondrial DNA can’t repair itself, so if you damage the mitochondria, you will damage the cell, and theoretically, it cannot repair itself,” he said.
In preclinical models, the oscillating magnetic fields generated by the new device were shown to kill patient-derived glioblastoma cells in cell culture without having cytotoxic effects on cortical neurons and normal human astrocytes. Animal studies also showed that it was effective and nontoxic, explained Dr. Baskin.
However, getting the device to human clinical trials has been slow going. “We wanted to start an early-phase trial for an investigational device, but the FDA is overwhelmed with COVID-related applications,” he said. “That has taken priority, and we understand that. So we were able to evaluate it on a patient through compassionate use via the [Food and Drug Administration]–approved Expanded Access Program.”
Exciting possibilities
The patient was a 53-year-old man who had undergone radiotherapy and chemotherapy, and the tumor was progressing. Imaging revealed the presence of leptomeningeal disease, which is associated with a poor outcome and a median survival of 3.5-3.9 months.
The patient was fitted with the helmet device and wore it under supervision for the first 3 days of treatment, during which time the strength of the oscillating magnetic fields was escalated. After this initial supervised phase, the treatment continued at home without supervision, using the same regimen as on the third day.
Treatment was first administered for 2 hours while under supervision and was then gradually increased to a maximum of 6 hours per day. The patient was evaluated clinically on days 7, 16, 30, and 44 after initiation of treatment. No serious adverse events were reported during treatment. The patient’s wife reported subjective improvement in speech and cognitive function.
Dr. Baskin noted that the patient had been experiencing falls for the past year and a half before treatment was initiated. “And then he tripped and fell and sustained a head injury that he subsequently died from,” he said.
Autopsy results confirmed the rapid response to treatment, and tumor shrinkage appeared to correlate with the treatment dose.
“Our results in the laboratory and with this patient open a new world of noninvasive and nontoxic therapy for brain cancer, with many exciting possibilities for the future,” Dr. Baskin commented.
He said his team has experimented with this approach with other tumor types in the laboratory, including triple-negative breast cancer and lung cancer. “We’ve only tried it in a culture so far, but it seems to melt the cancer cells,” he said.
The work was supported by a grant from the Translational Research Initiative of the Houston Methodist Research Institute and several foundations. Dr. Baskin and two coauthors are listed as inventors on a U.S. patent application filed by Houston Methodist Hospital for the device used in this report.
A version of this article first appeared on Medscape.com.
This is the first time that the wearable Oncomagnetic device was tried with a patient.
The patient had end-stage recurrent glioblastoma and had undergone all standard therapy options. He wore the device for 5 weeks but died from an unrelated injury, so the treatment period was cut short.
A brain scan showed a 31% reduction of contrast-enhanced tumor volume, and an autopsy of his brain confirmed the rapid response to the treatment.
The case study was published online on July 22, 2021, in Frontiers in Oncology.
“I believe that there is a great potential with this device,” said study author David S. Baskin, MD, director of the Kenneth R. Peak Center for Brain and Pituitary Tumor Treatment in the department of neurosurgery at Houston Methodist Hospital. “This is a very exciting time.”
The team is now treating several patients with glioblastoma under compassionate use.
In an independent comment, Adilia Hormigo, MD, PhD, director of the neuro-oncology program at the Tisch Cancer Institute, Mount Sinai Health System, New York, noted that a clinical trial is needed to evaluate the device. “But this is an interesting idea, and we have to be open-minded in treating this fatal disease.”
Oscillating magnetic fields
The Oncomagnetic device consists of three oncoscillators that are attached to the outside of a helmet and are connected to a microprocessor-based electronic controller powered by a rechargeable battery.
It consists of a series of rotating magnets that produce oscillating magnetic fields that cover the entire brain, including the upper part of the brain stem. The device induces rapid apoptosis of glioblastoma cells, Dr. Baskin explained. Its mechanism of action involves disruption of the electron transport in the mitochondrial respiratory chain, causing an elevation of reactive oxygen species and caspase-dependent cancer cell death.
Dr. Baskin emphasized that the new Oncomagnetic device is very different from the Optune device (Novocare), which is already approved by the Food and Drug Administration and has been shown to increase survival among patients with glioblastoma. Optune uses tumor-treating fields (TTFs), which are electromagnetic waves that are delivered via an electric field generator through four transducer arrays that are placed on a shaved scalp. Preclinical studies indicated that the TTFs disrupt cell division by disrupting several steps in the mitotic process that are crucial for cell division.
Both of these devices “are using a type of external maneuver” rather than invasive intracranial approaches, said Dr. Hormingo. The experimental Oncomagnetic device may have an advantage in that it needs to be worn by the patient for fewer hours, she commented. A better understanding of the physics and underlying mechanism is needed, however. Clinical trials are an essential next step.
Most common brain cancer in adults
Glioblastoma is the most common malignant tumor of the brain in adults. Outcomes continue to be dismal. In more than 40 years, median survival has only modestly improved.
“We haven’t gotten very far with glioblastoma despite millions of dollars in research,” Dr. Baskin said. “With treatment, survival is about 15 months, and those are not very good months.”
Out of the box
Standard treatments for glioblastoma include surgery, radiotherapy, and chemotherapy, and many patients cannot tolerate some of these, Dr. Baskin noted. Hence, there is a great need for a different therapeutic approach that yields better outcomes with lower toxicity.
“We didn’t want to develop another chemotherapeutic agent that would help you live another 2 months,” he said in an interview. “We were trying to think out of the box.
“If you want to do something that will really make a difference in an aggressive tumor like glioblastoma, you have to attack something so basic that the tumor can’t evade it,” he said. “For example, with temozolomide, if it is unmethylated, the tumor can repair the DNA damage from the chemotherapy. Even if you’re sensitive to begin with, over time, the tumor will eventually become resistant.”
The new device stems from work by Dr. Baskin and colleagues on mitochondria, which he describes as the powerhouse of the cell. “Mitochondrial DNA can’t repair itself, so if you damage the mitochondria, you will damage the cell, and theoretically, it cannot repair itself,” he said.
In preclinical models, the oscillating magnetic fields generated by the new device were shown to kill patient-derived glioblastoma cells in cell culture without having cytotoxic effects on cortical neurons and normal human astrocytes. Animal studies also showed that it was effective and nontoxic, explained Dr. Baskin.
However, getting the device to human clinical trials has been slow going. “We wanted to start an early-phase trial for an investigational device, but the FDA is overwhelmed with COVID-related applications,” he said. “That has taken priority, and we understand that. So we were able to evaluate it on a patient through compassionate use via the [Food and Drug Administration]–approved Expanded Access Program.”
Exciting possibilities
The patient was a 53-year-old man who had undergone radiotherapy and chemotherapy, and the tumor was progressing. Imaging revealed the presence of leptomeningeal disease, which is associated with a poor outcome and a median survival of 3.5-3.9 months.
The patient was fitted with the helmet device and wore it under supervision for the first 3 days of treatment, during which time the strength of the oscillating magnetic fields was escalated. After this initial supervised phase, the treatment continued at home without supervision, using the same regimen as on the third day.
Treatment was first administered for 2 hours while under supervision and was then gradually increased to a maximum of 6 hours per day. The patient was evaluated clinically on days 7, 16, 30, and 44 after initiation of treatment. No serious adverse events were reported during treatment. The patient’s wife reported subjective improvement in speech and cognitive function.
Dr. Baskin noted that the patient had been experiencing falls for the past year and a half before treatment was initiated. “And then he tripped and fell and sustained a head injury that he subsequently died from,” he said.
Autopsy results confirmed the rapid response to treatment, and tumor shrinkage appeared to correlate with the treatment dose.
“Our results in the laboratory and with this patient open a new world of noninvasive and nontoxic therapy for brain cancer, with many exciting possibilities for the future,” Dr. Baskin commented.
He said his team has experimented with this approach with other tumor types in the laboratory, including triple-negative breast cancer and lung cancer. “We’ve only tried it in a culture so far, but it seems to melt the cancer cells,” he said.
The work was supported by a grant from the Translational Research Initiative of the Houston Methodist Research Institute and several foundations. Dr. Baskin and two coauthors are listed as inventors on a U.S. patent application filed by Houston Methodist Hospital for the device used in this report.
A version of this article first appeared on Medscape.com.
This is the first time that the wearable Oncomagnetic device was tried with a patient.
The patient had end-stage recurrent glioblastoma and had undergone all standard therapy options. He wore the device for 5 weeks but died from an unrelated injury, so the treatment period was cut short.
A brain scan showed a 31% reduction of contrast-enhanced tumor volume, and an autopsy of his brain confirmed the rapid response to the treatment.
The case study was published online on July 22, 2021, in Frontiers in Oncology.
“I believe that there is a great potential with this device,” said study author David S. Baskin, MD, director of the Kenneth R. Peak Center for Brain and Pituitary Tumor Treatment in the department of neurosurgery at Houston Methodist Hospital. “This is a very exciting time.”
The team is now treating several patients with glioblastoma under compassionate use.
In an independent comment, Adilia Hormigo, MD, PhD, director of the neuro-oncology program at the Tisch Cancer Institute, Mount Sinai Health System, New York, noted that a clinical trial is needed to evaluate the device. “But this is an interesting idea, and we have to be open-minded in treating this fatal disease.”
Oscillating magnetic fields
The Oncomagnetic device consists of three oncoscillators that are attached to the outside of a helmet and are connected to a microprocessor-based electronic controller powered by a rechargeable battery.
It consists of a series of rotating magnets that produce oscillating magnetic fields that cover the entire brain, including the upper part of the brain stem. The device induces rapid apoptosis of glioblastoma cells, Dr. Baskin explained. Its mechanism of action involves disruption of the electron transport in the mitochondrial respiratory chain, causing an elevation of reactive oxygen species and caspase-dependent cancer cell death.
Dr. Baskin emphasized that the new Oncomagnetic device is very different from the Optune device (Novocare), which is already approved by the Food and Drug Administration and has been shown to increase survival among patients with glioblastoma. Optune uses tumor-treating fields (TTFs), which are electromagnetic waves that are delivered via an electric field generator through four transducer arrays that are placed on a shaved scalp. Preclinical studies indicated that the TTFs disrupt cell division by disrupting several steps in the mitotic process that are crucial for cell division.
Both of these devices “are using a type of external maneuver” rather than invasive intracranial approaches, said Dr. Hormingo. The experimental Oncomagnetic device may have an advantage in that it needs to be worn by the patient for fewer hours, she commented. A better understanding of the physics and underlying mechanism is needed, however. Clinical trials are an essential next step.
Most common brain cancer in adults
Glioblastoma is the most common malignant tumor of the brain in adults. Outcomes continue to be dismal. In more than 40 years, median survival has only modestly improved.
“We haven’t gotten very far with glioblastoma despite millions of dollars in research,” Dr. Baskin said. “With treatment, survival is about 15 months, and those are not very good months.”
Out of the box
Standard treatments for glioblastoma include surgery, radiotherapy, and chemotherapy, and many patients cannot tolerate some of these, Dr. Baskin noted. Hence, there is a great need for a different therapeutic approach that yields better outcomes with lower toxicity.
“We didn’t want to develop another chemotherapeutic agent that would help you live another 2 months,” he said in an interview. “We were trying to think out of the box.
“If you want to do something that will really make a difference in an aggressive tumor like glioblastoma, you have to attack something so basic that the tumor can’t evade it,” he said. “For example, with temozolomide, if it is unmethylated, the tumor can repair the DNA damage from the chemotherapy. Even if you’re sensitive to begin with, over time, the tumor will eventually become resistant.”
The new device stems from work by Dr. Baskin and colleagues on mitochondria, which he describes as the powerhouse of the cell. “Mitochondrial DNA can’t repair itself, so if you damage the mitochondria, you will damage the cell, and theoretically, it cannot repair itself,” he said.
In preclinical models, the oscillating magnetic fields generated by the new device were shown to kill patient-derived glioblastoma cells in cell culture without having cytotoxic effects on cortical neurons and normal human astrocytes. Animal studies also showed that it was effective and nontoxic, explained Dr. Baskin.
However, getting the device to human clinical trials has been slow going. “We wanted to start an early-phase trial for an investigational device, but the FDA is overwhelmed with COVID-related applications,” he said. “That has taken priority, and we understand that. So we were able to evaluate it on a patient through compassionate use via the [Food and Drug Administration]–approved Expanded Access Program.”
Exciting possibilities
The patient was a 53-year-old man who had undergone radiotherapy and chemotherapy, and the tumor was progressing. Imaging revealed the presence of leptomeningeal disease, which is associated with a poor outcome and a median survival of 3.5-3.9 months.
The patient was fitted with the helmet device and wore it under supervision for the first 3 days of treatment, during which time the strength of the oscillating magnetic fields was escalated. After this initial supervised phase, the treatment continued at home without supervision, using the same regimen as on the third day.
Treatment was first administered for 2 hours while under supervision and was then gradually increased to a maximum of 6 hours per day. The patient was evaluated clinically on days 7, 16, 30, and 44 after initiation of treatment. No serious adverse events were reported during treatment. The patient’s wife reported subjective improvement in speech and cognitive function.
Dr. Baskin noted that the patient had been experiencing falls for the past year and a half before treatment was initiated. “And then he tripped and fell and sustained a head injury that he subsequently died from,” he said.
Autopsy results confirmed the rapid response to treatment, and tumor shrinkage appeared to correlate with the treatment dose.
“Our results in the laboratory and with this patient open a new world of noninvasive and nontoxic therapy for brain cancer, with many exciting possibilities for the future,” Dr. Baskin commented.
He said his team has experimented with this approach with other tumor types in the laboratory, including triple-negative breast cancer and lung cancer. “We’ve only tried it in a culture so far, but it seems to melt the cancer cells,” he said.
The work was supported by a grant from the Translational Research Initiative of the Houston Methodist Research Institute and several foundations. Dr. Baskin and two coauthors are listed as inventors on a U.S. patent application filed by Houston Methodist Hospital for the device used in this report.
A version of this article first appeared on Medscape.com.
Federal Health Care Data Trends 2021
A Federal Practitioner Exclusive
Topics Include:

- Asthma
- Traumatic Brain Injury
- Dementia
- Post-Traumatic Stress Disorder
- Pain
- Migraine
- Suicide/Suicide Prevention
- Depression
- Anxiety
- Substance Use Disorder
- Tobacco
- Diabetes/Cardiovascular Disease
- Diabetic Retinopathy
- COVID-19
- Vaccination
- HIV
- Cancer Screening
To read the supplement click on the cover image or here
A Federal Practitioner Exclusive
Topics Include:
- Asthma
- Traumatic Brain Injury
- Dementia
- Post-Traumatic Stress Disorder
- Pain
- Migraine
- Suicide/Suicide Prevention
- Depression
- Anxiety
- Substance Use Disorder
- Tobacco
- Diabetes/Cardiovascular Disease
- Diabetic Retinopathy
- COVID-19
- Vaccination
- HIV
- Cancer Screening
To read the supplement click on the cover image or here
A Federal Practitioner Exclusive
Topics Include:
- Asthma
- Traumatic Brain Injury
- Dementia
- Post-Traumatic Stress Disorder
- Pain
- Migraine
- Suicide/Suicide Prevention
- Depression
- Anxiety
- Substance Use Disorder
- Tobacco
- Diabetes/Cardiovascular Disease
- Diabetic Retinopathy
- COVID-19
- Vaccination
- HIV
- Cancer Screening
To read the supplement click on the cover image or here

FDA warns of higher death risk with Pepaxto in multiple myeloma
participating in the ongoing OCEAN clinical trial.

The drug was granted accelerated approval in February 2021 for use in combination with dexamethasone in the treatment of adults with relapsed or refractory multiple myeloma who had received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
As a condition of the accelerated approval, the manufacturer, Oncopeptides, was required to conduct a confirmatory clinical trial and launched the OCEAN trial.
Enrollment in OCEAN as well as other ongoing trials of the drug have now been halted, according to the FDA alert.
The warning comes in the wake of OCEAN trial findings showing worse survival among patients in the experimental group, who were receiving melphalan plus low-dose dexamethasone, compared with patients in the control group, who were receiving pomalidomide plus low-dose dexamethasone (hazard ratio for overall survival, 1.104). Median overall survival in the treatment and control groups was 19.7 and 25.0 months, respectively.
Health care professionals should “review patients’ progress on Pepaxto and discuss the risks of continued administration with each patient in the context of other treatments,” and patients currently receiving the drug should discuss the risks and benefits with their health care professional, the FDA advises. “Patients receiving clinical benefit from Pepaxto may continue treatment in the OCEAN trial provided they are informed of the risks and sign a revised written informed consent.”
The FDA also hinted at “a future public meeting to discuss the safety findings and explore the continued marketing of Pepaxto,” which has a price tag of $19,000 per treatment course.
Accelerated approval data
Melphalan flufenamide was initially evaluated in combination with low-dose dexamethasone in the multicenter, single-arm HORIZON trial of adults with relapsed or refractory multiple myeloma who received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
Patients received melphalan flufenamide at a dose of 40 mg intravenously on day 1 along with oral dexamethasone at a dose of 40 mg (or 20 mg for those over age 75 years) on days 1, 8, 15, and 22 of each 28-day cycle until disease progression or unacceptable toxicity.
The most common adverse reactions, occurring in at least 20% of patients, were fatigue, nausea, diarrhea, pyrexia, and respiratory tract infection. The most common laboratory abnormalities, occurring in at least 50% of patients, were decreased leukocytes, platelets, lymphocytes, neutrophils, and hemoglobin, and increased creatinine.
Accelerated approval was granted after the HORIZON trial showed an overall response rate of 23.7% and median duration of response of 4.2 months. The application by Oncopeptides received priority review and orphan drug status.
Confirmatory trial data
The confirmatory OCEAN trial compared melphalan flufenamide plus low-dose dexamethasone to pomalidomide plus low-dose dexamethasone in patients with relapsed or refractory multiple myeloma following 2-4 lines of therapy and in patients who were resistant to lenalidomide in the last line of therapy.
The FDA conducted an efficacy and safety evaluation of the OCEAN trial using a data cutoff date of February 3, 2021. At a median follow-up of 19.1 months, 117 of 246 patients (48%) in the melphalan flufenamide group had died, compared with 108 of 249 patients (43%) in the pomalidomide control group.
“Patient safety is paramount to Oncopeptides,” the company said in a press statement, which also notes that “dialogue with the FDA” is ongoing and that updated information will be provided as it becomes available.
The company plans to submit complete data from the OCEAN study to the International Myeloma Workshop meeting in Vienna being held September 8-11, 2021.
Health care professionals and patients should report adverse events or quality issues experienced with melphalan flufenamide or any other medication to the FDA MedWatch Adverse Event Reporting program, either online or by downloading and completing a reporting form and submitting via fax at 1-800-FDA-0178.
A version of this article first appeared on Medscape.com.
participating in the ongoing OCEAN clinical trial.
The drug was granted accelerated approval in February 2021 for use in combination with dexamethasone in the treatment of adults with relapsed or refractory multiple myeloma who had received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
As a condition of the accelerated approval, the manufacturer, Oncopeptides, was required to conduct a confirmatory clinical trial and launched the OCEAN trial.
Enrollment in OCEAN as well as other ongoing trials of the drug have now been halted, according to the FDA alert.
The warning comes in the wake of OCEAN trial findings showing worse survival among patients in the experimental group, who were receiving melphalan plus low-dose dexamethasone, compared with patients in the control group, who were receiving pomalidomide plus low-dose dexamethasone (hazard ratio for overall survival, 1.104). Median overall survival in the treatment and control groups was 19.7 and 25.0 months, respectively.
Health care professionals should “review patients’ progress on Pepaxto and discuss the risks of continued administration with each patient in the context of other treatments,” and patients currently receiving the drug should discuss the risks and benefits with their health care professional, the FDA advises. “Patients receiving clinical benefit from Pepaxto may continue treatment in the OCEAN trial provided they are informed of the risks and sign a revised written informed consent.”
The FDA also hinted at “a future public meeting to discuss the safety findings and explore the continued marketing of Pepaxto,” which has a price tag of $19,000 per treatment course.
Accelerated approval data
Melphalan flufenamide was initially evaluated in combination with low-dose dexamethasone in the multicenter, single-arm HORIZON trial of adults with relapsed or refractory multiple myeloma who received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
Patients received melphalan flufenamide at a dose of 40 mg intravenously on day 1 along with oral dexamethasone at a dose of 40 mg (or 20 mg for those over age 75 years) on days 1, 8, 15, and 22 of each 28-day cycle until disease progression or unacceptable toxicity.
The most common adverse reactions, occurring in at least 20% of patients, were fatigue, nausea, diarrhea, pyrexia, and respiratory tract infection. The most common laboratory abnormalities, occurring in at least 50% of patients, were decreased leukocytes, platelets, lymphocytes, neutrophils, and hemoglobin, and increased creatinine.
Accelerated approval was granted after the HORIZON trial showed an overall response rate of 23.7% and median duration of response of 4.2 months. The application by Oncopeptides received priority review and orphan drug status.
Confirmatory trial data
The confirmatory OCEAN trial compared melphalan flufenamide plus low-dose dexamethasone to pomalidomide plus low-dose dexamethasone in patients with relapsed or refractory multiple myeloma following 2-4 lines of therapy and in patients who were resistant to lenalidomide in the last line of therapy.
The FDA conducted an efficacy and safety evaluation of the OCEAN trial using a data cutoff date of February 3, 2021. At a median follow-up of 19.1 months, 117 of 246 patients (48%) in the melphalan flufenamide group had died, compared with 108 of 249 patients (43%) in the pomalidomide control group.
“Patient safety is paramount to Oncopeptides,” the company said in a press statement, which also notes that “dialogue with the FDA” is ongoing and that updated information will be provided as it becomes available.
The company plans to submit complete data from the OCEAN study to the International Myeloma Workshop meeting in Vienna being held September 8-11, 2021.
Health care professionals and patients should report adverse events or quality issues experienced with melphalan flufenamide or any other medication to the FDA MedWatch Adverse Event Reporting program, either online or by downloading and completing a reporting form and submitting via fax at 1-800-FDA-0178.
A version of this article first appeared on Medscape.com.
participating in the ongoing OCEAN clinical trial.
The drug was granted accelerated approval in February 2021 for use in combination with dexamethasone in the treatment of adults with relapsed or refractory multiple myeloma who had received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
As a condition of the accelerated approval, the manufacturer, Oncopeptides, was required to conduct a confirmatory clinical trial and launched the OCEAN trial.
Enrollment in OCEAN as well as other ongoing trials of the drug have now been halted, according to the FDA alert.
The warning comes in the wake of OCEAN trial findings showing worse survival among patients in the experimental group, who were receiving melphalan plus low-dose dexamethasone, compared with patients in the control group, who were receiving pomalidomide plus low-dose dexamethasone (hazard ratio for overall survival, 1.104). Median overall survival in the treatment and control groups was 19.7 and 25.0 months, respectively.
Health care professionals should “review patients’ progress on Pepaxto and discuss the risks of continued administration with each patient in the context of other treatments,” and patients currently receiving the drug should discuss the risks and benefits with their health care professional, the FDA advises. “Patients receiving clinical benefit from Pepaxto may continue treatment in the OCEAN trial provided they are informed of the risks and sign a revised written informed consent.”
The FDA also hinted at “a future public meeting to discuss the safety findings and explore the continued marketing of Pepaxto,” which has a price tag of $19,000 per treatment course.
Accelerated approval data
Melphalan flufenamide was initially evaluated in combination with low-dose dexamethasone in the multicenter, single-arm HORIZON trial of adults with relapsed or refractory multiple myeloma who received at least four prior lines of therapy and whose disease was refractory to at least one proteasome inhibitor, one immunomodulator, and one CD38-directed monoclonal antibody.
Patients received melphalan flufenamide at a dose of 40 mg intravenously on day 1 along with oral dexamethasone at a dose of 40 mg (or 20 mg for those over age 75 years) on days 1, 8, 15, and 22 of each 28-day cycle until disease progression or unacceptable toxicity.
The most common adverse reactions, occurring in at least 20% of patients, were fatigue, nausea, diarrhea, pyrexia, and respiratory tract infection. The most common laboratory abnormalities, occurring in at least 50% of patients, were decreased leukocytes, platelets, lymphocytes, neutrophils, and hemoglobin, and increased creatinine.
Accelerated approval was granted after the HORIZON trial showed an overall response rate of 23.7% and median duration of response of 4.2 months. The application by Oncopeptides received priority review and orphan drug status.
Confirmatory trial data
The confirmatory OCEAN trial compared melphalan flufenamide plus low-dose dexamethasone to pomalidomide plus low-dose dexamethasone in patients with relapsed or refractory multiple myeloma following 2-4 lines of therapy and in patients who were resistant to lenalidomide in the last line of therapy.
The FDA conducted an efficacy and safety evaluation of the OCEAN trial using a data cutoff date of February 3, 2021. At a median follow-up of 19.1 months, 117 of 246 patients (48%) in the melphalan flufenamide group had died, compared with 108 of 249 patients (43%) in the pomalidomide control group.
“Patient safety is paramount to Oncopeptides,” the company said in a press statement, which also notes that “dialogue with the FDA” is ongoing and that updated information will be provided as it becomes available.
The company plans to submit complete data from the OCEAN study to the International Myeloma Workshop meeting in Vienna being held September 8-11, 2021.
Health care professionals and patients should report adverse events or quality issues experienced with melphalan flufenamide or any other medication to the FDA MedWatch Adverse Event Reporting program, either online or by downloading and completing a reporting form and submitting via fax at 1-800-FDA-0178.
A version of this article first appeared on Medscape.com.
Many pandemic-driven changes to cancer clinical trials should remain
Many of the changes to cancer clinical trials forced through by the COVID-19 pandemic should remain, as they have made trials “more patient centered and efficient,” according to a group of thought leaders in oncology.
Among the potential improvements were more efficient study enrollment through secure electronic platforms, direct shipment of oral drugs to patients, remote assessment of adverse events, and streamlined data collection.
These changes should be implemented on a permanent basis, the group argues in a commentary published online July 21, 2021, in Cancer Discovery, a journal of the American Association for Cancer Research.
“The ability to distribute oral investigational drugs by mail to patients at their home has probably been the single most impactful change to clinical trial conduct, linked with virtual visits with patients to assess side effects and symptoms,” commented lead author Keith Flaherty, MD, who is director of clinical research at Massachusetts General Hospital, a professor at Harvard Medical School, Boston, and a member of the AACR board of directors.
“This has made it more feasible for patients for whom participation in clinical trials poses a disruption of their ability to work or provide care for family members to participate in trials,” he added in a press statement issued by the AACR.
Pandemic halted many clinical trials
A survey of cancer programs in early 2020 showed that nearly 60% halted screening and/or enrollment for at least some trials because of COVID-19.
“In the spring of 2020, clinical trial conduct halted and then restarted focusing on the bare minimum procedures that first allowed patients continued access to their experimental therapies, and then allowed clinical trial sites and sponsors to collect information on the effects of the therapies,” the authors said.
“The COVID-19–induced changes to clinical trials were a big challenge, probably the largest change in clinical trial conduct since the start of modern oncology clinical testing,” they commented.
“But it also represents an opportunity to rethink the key aspects of clinical trial conduct that are strictly necessary to reach the goal of testing the effectiveness of cancer therapies, and which others are dispensable or provide only minor additional contributions,” they added.
As previously reported at the time by this news organization, efforts to find alternative approaches to conducting trials amid the pandemic led to the emergence of a few “silver linings.”
Key adaptations made to clinical trials and highlighted by the authors include:
- Uptake of remote consenting and telemedicine
- Use of alternative laboratories and imaging centers
- Delivery or administration of investigational drugs at patients’ homes or local clinics
- Commercial attainment of study drugs already approved for other indications
Indeed, the restrictions encountered during the pandemic underscore the importance of designing patient-centered trials versus study site–centered trials, added Antoni Ribas, MD, commentary coauthor and immediate past president of the AACR.
Many of the changes implemented during the pandemic could help increase access for patients living in underserved communities who are underrepresented in clinical trials, he explained.
Harnessing the lessons learned
The authors also recommended the following additional adaptations, which they believe will enhance efficiency and further expand access to clinical trials:
- Incorporating patient-reported outcomes and alternative endpoints in efficacy assessments
- Aiming for 100% remote drug infusions and monitoring
- Increasing funding for clinical trials conducted in underserved communities
- Expanding clinical trial eligibility to include patients with a wide range of comorbidities
- Reducing collection of low-grade adverse events and allowing minor protocol deviations
The group’s recommendations are based on discussions by the AACR COVID-19 and Cancer Task Force, in which they participated.
The American Society of Clinical Oncology is also working to leverage pandemic-related lessons to streamline care and trial planning.
ASCO’s “Road to Recovery” recommendations, published in December 2020, aim to “ensure lessons learned from the COVID-19 experience are used to craft a more equitable, accessible, and efficient clinical research system that protects patient safety, ensures scientific integrity, and maintains data quality,” the authors explained.
Dr. Flaherty and colleagues further underscore the importance of focusing on improvements going forward.
“Guided by lessons learned, many of the remote assessments and trial efficiencies deployed during the pandemic can be preserved and improved upon. We strongly encourage use of these streamlined procedures where appropriate in future prospectively designed cancer clinical trials,” they wrote.
Dr. Flaherty reported receiving personal fees from numerous pharmaceutical companies. Dr. Ribas reported receiving grants from Agilent and Bristol Myers Squibb.
A version of this article first appeared on Medscape.com.
Many of the changes to cancer clinical trials forced through by the COVID-19 pandemic should remain, as they have made trials “more patient centered and efficient,” according to a group of thought leaders in oncology.
Among the potential improvements were more efficient study enrollment through secure electronic platforms, direct shipment of oral drugs to patients, remote assessment of adverse events, and streamlined data collection.
These changes should be implemented on a permanent basis, the group argues in a commentary published online July 21, 2021, in Cancer Discovery, a journal of the American Association for Cancer Research.
“The ability to distribute oral investigational drugs by mail to patients at their home has probably been the single most impactful change to clinical trial conduct, linked with virtual visits with patients to assess side effects and symptoms,” commented lead author Keith Flaherty, MD, who is director of clinical research at Massachusetts General Hospital, a professor at Harvard Medical School, Boston, and a member of the AACR board of directors.
“This has made it more feasible for patients for whom participation in clinical trials poses a disruption of their ability to work or provide care for family members to participate in trials,” he added in a press statement issued by the AACR.
Pandemic halted many clinical trials
A survey of cancer programs in early 2020 showed that nearly 60% halted screening and/or enrollment for at least some trials because of COVID-19.
“In the spring of 2020, clinical trial conduct halted and then restarted focusing on the bare minimum procedures that first allowed patients continued access to their experimental therapies, and then allowed clinical trial sites and sponsors to collect information on the effects of the therapies,” the authors said.
“The COVID-19–induced changes to clinical trials were a big challenge, probably the largest change in clinical trial conduct since the start of modern oncology clinical testing,” they commented.
“But it also represents an opportunity to rethink the key aspects of clinical trial conduct that are strictly necessary to reach the goal of testing the effectiveness of cancer therapies, and which others are dispensable or provide only minor additional contributions,” they added.
As previously reported at the time by this news organization, efforts to find alternative approaches to conducting trials amid the pandemic led to the emergence of a few “silver linings.”
Key adaptations made to clinical trials and highlighted by the authors include:
- Uptake of remote consenting and telemedicine
- Use of alternative laboratories and imaging centers
- Delivery or administration of investigational drugs at patients’ homes or local clinics
- Commercial attainment of study drugs already approved for other indications
Indeed, the restrictions encountered during the pandemic underscore the importance of designing patient-centered trials versus study site–centered trials, added Antoni Ribas, MD, commentary coauthor and immediate past president of the AACR.
Many of the changes implemented during the pandemic could help increase access for patients living in underserved communities who are underrepresented in clinical trials, he explained.
Harnessing the lessons learned
The authors also recommended the following additional adaptations, which they believe will enhance efficiency and further expand access to clinical trials:
- Incorporating patient-reported outcomes and alternative endpoints in efficacy assessments
- Aiming for 100% remote drug infusions and monitoring
- Increasing funding for clinical trials conducted in underserved communities
- Expanding clinical trial eligibility to include patients with a wide range of comorbidities
- Reducing collection of low-grade adverse events and allowing minor protocol deviations
The group’s recommendations are based on discussions by the AACR COVID-19 and Cancer Task Force, in which they participated.
The American Society of Clinical Oncology is also working to leverage pandemic-related lessons to streamline care and trial planning.
ASCO’s “Road to Recovery” recommendations, published in December 2020, aim to “ensure lessons learned from the COVID-19 experience are used to craft a more equitable, accessible, and efficient clinical research system that protects patient safety, ensures scientific integrity, and maintains data quality,” the authors explained.
Dr. Flaherty and colleagues further underscore the importance of focusing on improvements going forward.
“Guided by lessons learned, many of the remote assessments and trial efficiencies deployed during the pandemic can be preserved and improved upon. We strongly encourage use of these streamlined procedures where appropriate in future prospectively designed cancer clinical trials,” they wrote.
Dr. Flaherty reported receiving personal fees from numerous pharmaceutical companies. Dr. Ribas reported receiving grants from Agilent and Bristol Myers Squibb.
A version of this article first appeared on Medscape.com.
Many of the changes to cancer clinical trials forced through by the COVID-19 pandemic should remain, as they have made trials “more patient centered and efficient,” according to a group of thought leaders in oncology.
Among the potential improvements were more efficient study enrollment through secure electronic platforms, direct shipment of oral drugs to patients, remote assessment of adverse events, and streamlined data collection.
These changes should be implemented on a permanent basis, the group argues in a commentary published online July 21, 2021, in Cancer Discovery, a journal of the American Association for Cancer Research.
“The ability to distribute oral investigational drugs by mail to patients at their home has probably been the single most impactful change to clinical trial conduct, linked with virtual visits with patients to assess side effects and symptoms,” commented lead author Keith Flaherty, MD, who is director of clinical research at Massachusetts General Hospital, a professor at Harvard Medical School, Boston, and a member of the AACR board of directors.
“This has made it more feasible for patients for whom participation in clinical trials poses a disruption of their ability to work or provide care for family members to participate in trials,” he added in a press statement issued by the AACR.
Pandemic halted many clinical trials
A survey of cancer programs in early 2020 showed that nearly 60% halted screening and/or enrollment for at least some trials because of COVID-19.
“In the spring of 2020, clinical trial conduct halted and then restarted focusing on the bare minimum procedures that first allowed patients continued access to their experimental therapies, and then allowed clinical trial sites and sponsors to collect information on the effects of the therapies,” the authors said.
“The COVID-19–induced changes to clinical trials were a big challenge, probably the largest change in clinical trial conduct since the start of modern oncology clinical testing,” they commented.
“But it also represents an opportunity to rethink the key aspects of clinical trial conduct that are strictly necessary to reach the goal of testing the effectiveness of cancer therapies, and which others are dispensable or provide only minor additional contributions,” they added.
As previously reported at the time by this news organization, efforts to find alternative approaches to conducting trials amid the pandemic led to the emergence of a few “silver linings.”
Key adaptations made to clinical trials and highlighted by the authors include:
- Uptake of remote consenting and telemedicine
- Use of alternative laboratories and imaging centers
- Delivery or administration of investigational drugs at patients’ homes or local clinics
- Commercial attainment of study drugs already approved for other indications
Indeed, the restrictions encountered during the pandemic underscore the importance of designing patient-centered trials versus study site–centered trials, added Antoni Ribas, MD, commentary coauthor and immediate past president of the AACR.
Many of the changes implemented during the pandemic could help increase access for patients living in underserved communities who are underrepresented in clinical trials, he explained.
Harnessing the lessons learned
The authors also recommended the following additional adaptations, which they believe will enhance efficiency and further expand access to clinical trials:
- Incorporating patient-reported outcomes and alternative endpoints in efficacy assessments
- Aiming for 100% remote drug infusions and monitoring
- Increasing funding for clinical trials conducted in underserved communities
- Expanding clinical trial eligibility to include patients with a wide range of comorbidities
- Reducing collection of low-grade adverse events and allowing minor protocol deviations
The group’s recommendations are based on discussions by the AACR COVID-19 and Cancer Task Force, in which they participated.
The American Society of Clinical Oncology is also working to leverage pandemic-related lessons to streamline care and trial planning.
ASCO’s “Road to Recovery” recommendations, published in December 2020, aim to “ensure lessons learned from the COVID-19 experience are used to craft a more equitable, accessible, and efficient clinical research system that protects patient safety, ensures scientific integrity, and maintains data quality,” the authors explained.
Dr. Flaherty and colleagues further underscore the importance of focusing on improvements going forward.
“Guided by lessons learned, many of the remote assessments and trial efficiencies deployed during the pandemic can be preserved and improved upon. We strongly encourage use of these streamlined procedures where appropriate in future prospectively designed cancer clinical trials,” they wrote.
Dr. Flaherty reported receiving personal fees from numerous pharmaceutical companies. Dr. Ribas reported receiving grants from Agilent and Bristol Myers Squibb.
A version of this article first appeared on Medscape.com.