User login
Diabetes Hub contains news and clinical review articles for physicians seeking the most up-to-date information on the rapidly evolving options for treating and preventing Type 2 Diabetes in at-risk patients. The Diabetes Hub is powered by Frontline Medical Communications.
FDA approves mobile medical app for real-time sharing of glucose data
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
The first set of mobile medical apps that permit secure, real-time data sharing by people who have diabetes and use a continuous glucose monitor were approved today by the Food and Drug Administration.
The Dexcom Share Direct Secondary Displays system allows caregivers to a person with diabetes to monitor that individual’s blood sugar levels remotely through an Apple mobile device such as an iPhone. Devices like the Dexcom Share were previously available through open source efforts but were not in compliance with regulatory requirements. The Dexcom Share system is the first legally marketed solution for real-time remote monitoring of a patient’s CGM data, according to a press release from the FDA.
“This innovative technology has been eagerly awaited by the diabetes community, especially caregivers of children with diabetes who want to monitor their glucose levels remotely,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “Today’s marketing permission paves the way for similar technologies to be marketed in the United States.”
The CGM is worn externally and includes a small, wire-like sensor inserted just under the wearer’s skin. The device monitors and continuously displays an estimate of blood glucose levels as well as the direction and rate of change of these estimates.
The app transmits real-time CGM data to a Web-based storage location. The app of the “follower” can then download the CGM data and display it in real-time.
The FDA has classified the device as low to moderate in risk and exempt from premarket submissions. In the future, manufacturers wishing to market devices like the Dexcom Share system will not need premarket clearance by the FDA prior to marketing. They will still need to register and list their device with the agency, as well as follow other applicable laws and regulations.
“Exempting devices from premarket review is part of the FDA’s effort to ensure these products provide accurate and reliable results while still encouraging the development of devices that meet the needs of people living with diabetes and their caregivers,” said Dr. Gutierrez.
The Dexcom Share system does not replace real-time continuous glucose monitoring or standard home blood glucose monitoring. CGM values alone are not approved to determine dosing of diabetes medications. CGMs must be calibrated by blood glucose meters, and treatment decisions, such as insulin dosing, should be based on readings from a blood glucose meter.The Dexcom Share system is manufactured by Dexcom, Inc., located in San Diego, Calif.
Lower diabetes risk seen in lupus patients using hydroxychloroquine
The risk for developing diabetes mellitus among patients with systemic lupus erythematosus declined significantly with increasing use of hydroxychloroquine in a retrospective cohort study of a national health insurance database in Taiwan.
This population-based cohort study is the first to demonstrate that hydroxychloroquine (HCQ) reduces the risk of diabetes in a dose-dependent manner in systemic lupus erythematosus (SLE) patients. In previous research, HCQ use has been shown to improve insulin sensitivity in SLE patients, and in rheumatoid arthritis patients, longer duration of HCQ treatment (> 4 years) has been shown before to have the greatest effect on lowering the incidence of diabetes, according to Dr. Der-Yuan Chen of Taichung (Taiwan) Veterans General Hospital and his associates.

The investigators analyzed the first year of medication use in 8,628 patients with SLE in the National Health Insurance Research Database of Taiwan during 2001-2008, excluding those with less than 3 years of follow-up data and comorbid diagnoses of RA, psoriasis, or diabetes (Rheumatology [Oxford] 2015 Jan. 12 [doi:10.1093/rheumatology/keu451]).
The patients’ mean age was 37 years, and 88% were female. The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion had taken glucocorticoids (92% vs. 73%). Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease.
The effect of HCQ on diabetes risk became apparent in patients with an average daily glucocorticoid dose equivalent to 10 mg prednisolone or greater. That glucocorticoid dose was associated with increased diabetes risk (hazard ratio, 2.29; 95% confidence interval, 1.34-3.93), but a cumulative dose of 129 g HCQ or greater (equal to 200 mg/day for 1.8 years) reduced the risk of diabetes (HR, 0.26; 95% CI, 0.18-0.37), compared with those who did not take the drug. There was no significant relationship between HCQ use and incident diabetes for those who had a cumulative dose of less than 129 g (HR, 1.13; 95% CI, 0.81-1.59).
Patients who took both an average daily glucocorticoid dose equivalent to at least 10 mg prednisolone and a cumulative dose of less than 129 g HCQ had a higher diabetes risk than did those taking a smaller prednisolone dose, whereas those who took at least 129 g HCQ had a reduced risk of diabetes despite taking high-dose glucocorticoids.
Hazard ratios for diabetes ranged from 0.10 to 0.41 among different groupings of patients who took at least 129 g HCQ, according to age, use of glucocorticoids or disease-modifying antirheumatic drugs, and presence of comorbidities.
The investigators noted that “there are substantial differences in the clinical characteristics of SLE patients with and without HCQ use ... [that] may reflect the discrepancy in lupus disease activity between the two groups.” The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion of them had taken glucocorticoids (92% vs. 73%).
Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease. HCQ users also took more methotrexate, sulfasalazine, and azathioprine, but less cyclophosphamide, than did those without HCQ use.
While the greater use of cyclophosphamide by HCQ nonusers may indicate more frequent lupus renal involvement and potentially bias them to increased risk of diabetes because of disease activity and chronic inflammation’s effect on insulin resistance, the greater cumulative glucocorticoid dose taken by HCQ users also can predispose to the development of diabetes, the authors said.
The study is limited in its ability to tease out the contributions of clinical characteristics between HCQ users and nonusers, because the patients’ lupus disease severity and laboratory data are unknown, and treatment data were recorded only from the first year. There was also a lack of data on lifestyle, body-mass index, and family history, the investigators said.
The study had no specific funding. The authors declared having no conflicts of interest.
The risk for developing diabetes mellitus among patients with systemic lupus erythematosus declined significantly with increasing use of hydroxychloroquine in a retrospective cohort study of a national health insurance database in Taiwan.
This population-based cohort study is the first to demonstrate that hydroxychloroquine (HCQ) reduces the risk of diabetes in a dose-dependent manner in systemic lupus erythematosus (SLE) patients. In previous research, HCQ use has been shown to improve insulin sensitivity in SLE patients, and in rheumatoid arthritis patients, longer duration of HCQ treatment (> 4 years) has been shown before to have the greatest effect on lowering the incidence of diabetes, according to Dr. Der-Yuan Chen of Taichung (Taiwan) Veterans General Hospital and his associates.
The investigators analyzed the first year of medication use in 8,628 patients with SLE in the National Health Insurance Research Database of Taiwan during 2001-2008, excluding those with less than 3 years of follow-up data and comorbid diagnoses of RA, psoriasis, or diabetes (Rheumatology [Oxford] 2015 Jan. 12 [doi:10.1093/rheumatology/keu451]).
The patients’ mean age was 37 years, and 88% were female. The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion had taken glucocorticoids (92% vs. 73%). Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease.
The effect of HCQ on diabetes risk became apparent in patients with an average daily glucocorticoid dose equivalent to 10 mg prednisolone or greater. That glucocorticoid dose was associated with increased diabetes risk (hazard ratio, 2.29; 95% confidence interval, 1.34-3.93), but a cumulative dose of 129 g HCQ or greater (equal to 200 mg/day for 1.8 years) reduced the risk of diabetes (HR, 0.26; 95% CI, 0.18-0.37), compared with those who did not take the drug. There was no significant relationship between HCQ use and incident diabetes for those who had a cumulative dose of less than 129 g (HR, 1.13; 95% CI, 0.81-1.59).
Patients who took both an average daily glucocorticoid dose equivalent to at least 10 mg prednisolone and a cumulative dose of less than 129 g HCQ had a higher diabetes risk than did those taking a smaller prednisolone dose, whereas those who took at least 129 g HCQ had a reduced risk of diabetes despite taking high-dose glucocorticoids.
Hazard ratios for diabetes ranged from 0.10 to 0.41 among different groupings of patients who took at least 129 g HCQ, according to age, use of glucocorticoids or disease-modifying antirheumatic drugs, and presence of comorbidities.
The investigators noted that “there are substantial differences in the clinical characteristics of SLE patients with and without HCQ use ... [that] may reflect the discrepancy in lupus disease activity between the two groups.” The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion of them had taken glucocorticoids (92% vs. 73%).
Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease. HCQ users also took more methotrexate, sulfasalazine, and azathioprine, but less cyclophosphamide, than did those without HCQ use.
While the greater use of cyclophosphamide by HCQ nonusers may indicate more frequent lupus renal involvement and potentially bias them to increased risk of diabetes because of disease activity and chronic inflammation’s effect on insulin resistance, the greater cumulative glucocorticoid dose taken by HCQ users also can predispose to the development of diabetes, the authors said.
The study is limited in its ability to tease out the contributions of clinical characteristics between HCQ users and nonusers, because the patients’ lupus disease severity and laboratory data are unknown, and treatment data were recorded only from the first year. There was also a lack of data on lifestyle, body-mass index, and family history, the investigators said.
The study had no specific funding. The authors declared having no conflicts of interest.
The risk for developing diabetes mellitus among patients with systemic lupus erythematosus declined significantly with increasing use of hydroxychloroquine in a retrospective cohort study of a national health insurance database in Taiwan.
This population-based cohort study is the first to demonstrate that hydroxychloroquine (HCQ) reduces the risk of diabetes in a dose-dependent manner in systemic lupus erythematosus (SLE) patients. In previous research, HCQ use has been shown to improve insulin sensitivity in SLE patients, and in rheumatoid arthritis patients, longer duration of HCQ treatment (> 4 years) has been shown before to have the greatest effect on lowering the incidence of diabetes, according to Dr. Der-Yuan Chen of Taichung (Taiwan) Veterans General Hospital and his associates.
The investigators analyzed the first year of medication use in 8,628 patients with SLE in the National Health Insurance Research Database of Taiwan during 2001-2008, excluding those with less than 3 years of follow-up data and comorbid diagnoses of RA, psoriasis, or diabetes (Rheumatology [Oxford] 2015 Jan. 12 [doi:10.1093/rheumatology/keu451]).
The patients’ mean age was 37 years, and 88% were female. The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion had taken glucocorticoids (92% vs. 73%). Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease.
The effect of HCQ on diabetes risk became apparent in patients with an average daily glucocorticoid dose equivalent to 10 mg prednisolone or greater. That glucocorticoid dose was associated with increased diabetes risk (hazard ratio, 2.29; 95% confidence interval, 1.34-3.93), but a cumulative dose of 129 g HCQ or greater (equal to 200 mg/day for 1.8 years) reduced the risk of diabetes (HR, 0.26; 95% CI, 0.18-0.37), compared with those who did not take the drug. There was no significant relationship between HCQ use and incident diabetes for those who had a cumulative dose of less than 129 g (HR, 1.13; 95% CI, 0.81-1.59).
Patients who took both an average daily glucocorticoid dose equivalent to at least 10 mg prednisolone and a cumulative dose of less than 129 g HCQ had a higher diabetes risk than did those taking a smaller prednisolone dose, whereas those who took at least 129 g HCQ had a reduced risk of diabetes despite taking high-dose glucocorticoids.
Hazard ratios for diabetes ranged from 0.10 to 0.41 among different groupings of patients who took at least 129 g HCQ, according to age, use of glucocorticoids or disease-modifying antirheumatic drugs, and presence of comorbidities.
The investigators noted that “there are substantial differences in the clinical characteristics of SLE patients with and without HCQ use ... [that] may reflect the discrepancy in lupus disease activity between the two groups.” The patients who had used HCQ were significantly older (49 years vs. 37 years) and a greater proportion of them had taken glucocorticoids (92% vs. 73%).
Those who had taken HCQ had significantly lower rates of hyperlipidemia, hypertension, stroke, and renal disease. HCQ users also took more methotrexate, sulfasalazine, and azathioprine, but less cyclophosphamide, than did those without HCQ use.
While the greater use of cyclophosphamide by HCQ nonusers may indicate more frequent lupus renal involvement and potentially bias them to increased risk of diabetes because of disease activity and chronic inflammation’s effect on insulin resistance, the greater cumulative glucocorticoid dose taken by HCQ users also can predispose to the development of diabetes, the authors said.
The study is limited in its ability to tease out the contributions of clinical characteristics between HCQ users and nonusers, because the patients’ lupus disease severity and laboratory data are unknown, and treatment data were recorded only from the first year. There was also a lack of data on lifestyle, body-mass index, and family history, the investigators said.
The study had no specific funding. The authors declared having no conflicts of interest.
FROM RHEUMATOLOGY
Key clinical point: Cumulative HCQ use of at least 129 g may be associated with lower risk of diabetes in patients with lupus and concomitant glucocorticoid use equivalent to 10 mg/day or more of prednisolone.
Major finding: A cumulative dose of 129 g HCQ or greater (equal to 200 mg/day for 1.8 years) reduced the risk of diabetes (HR, 0.26; 95% CI, 0.18-0.37), compared with those who did not take HCQ.
Data source: A retrospective cohort study of 8,628 patients with SLE in a national health insurance database in Taiwan.
Disclosures: The study had no specific funding. The authors declared having no conflicts of interest.

Zero coronary calcium means very low 10-year event risk
CHICAGO – Absence of coronary artery calcium upon imaging results in an impressively low cardiovascular event rate over the next 10 years regardless of an individual’s level of standard risk factors, according to prospective data from the MESA study.
In contrast, a coronary artery calcium (CAC) score of 1-10, often described as minimal CAC, nearly doubles the 10-year risk, compared with a baseline CAC score of 0.
Prior to these new 10-year data, many cardiologists considered a CAC score of 1-10 as tantamount to no CAC. Not so, Dr. Parag H. Joshi said at the American Heart Association scientific sessions.

“A CAC of 0 is presumably identifying someone without any atherosclerosis. Just the presence of minimal calcium suggests that atherosclerosis is building up. Our data suggest that among individuals with a CAC of 1-10, current smoking, elevated non-HDL cholesterol, and particularly hypertension should be treated aggressively,” said Dr. Joshi, a clinical fellow in cardiovascular diseases and prevention at Johns Hopkins University, Baltimore.
Prior studies totaling more than 50,000 subjects with a CAC score of 0 have shown very low cardiovascular event rates over 4-5 years of follow-up. However, current cardiovascular risk estimates focus on 10-year risk. This new analysis from MESA (Multi-Ethnic Study of Atherosclerosis) is the first study to provide prospective, 10-year events data, and those data are highly reassuring, he added.
MESA is a prospective, population-based cohort study. This analysis included 6,814 subjects aged 45-84 who were free of clinical cardiovascular disease at baseline, when their CAC score was determined. At that time, 3,415 participants had a CAC score of 0 and 508 had a score of 1-10.
During a median 10.3 years of follow-up, 123 cardiovascular events occurred, roughly one-third of which were nonfatal acute MIs and half of which were nonfatal strokes; the remainder were cardiovascular deaths.
The event rate was 2.9/1,000 person-years in subjects with a CAC of 0 and significantly greater at 5.5/1,000 person-years with a score of 1-10. However, since the cardiovascular risk factor profile of the zero CAC group was generally more favorable, Dr. Joshi and coinvestigators carried out a Cox proportional hazards analysis factoring in demographics, standard cardiovascular risk factors, body mass index, C-reactive protein level, and carotid intima media thickness. The adjusted 10-year event risk in the group with a CAC score of 1-10 was 1.9-fold greater than with a CAC of 0.
The highest 10-year event rate was noted in subjects with at least three of the following four risk factors at baseline: hypertension, current smoking, diabetes, and hyperlipidemia. The rate was 6.5/1,000 person-years in such individuals if they had a CAC of 0 and doubled at 13.1/1,000 person-years with a score of 1-10.
In a multivariate Cox analysis, age, smoking, and hypertension proved to be significant predictors of cardiovascular events in the group with a CAC of 0 as well as in those with a CAC of 1-10. But there was one important difference between the two groups: While the hazard ratio for cardiovascular events associated with hypertension versus no hypertension was 2.1 in subjects with a CAC of 0, the presence of hypertension in individuals with a CAC of 1-10 increased their event risk by 10.2-fold, or nearly five times greater than the risk increase associated with hypertension in persons with a CAC of 0, Dr. Joshi observed.
Non–HDL cholesterol level was predictive of cardiovascular risk in subjects with a CAC of 1-10 but not in those with a score of 0.
When actual event rates were compared with those predicted by the atherosclerotic cardiovascular disease (ASCVD) risk estimator introduced in the 2013 AHA/American College of Cardiology cholesterol guidelines, the event rate in subjects with an ASCVD 10-year risk estimate of 7.5%-15% but a CAC of 0 was just 4.4%.
Audience members noted that CAC scores didn’t do a very good job of stratifying stroke risk in MESA. That’s not surprising, since the score reflects coronary but not carotid artery calcium. But it is a limitation of CAC as a predictive tool, especially in light of the fact that strokes accounted for half of all cardiovascular events in the study.
Asked where he and his coinvestigators plan to go from here, Dr. Joshi said a randomized, controlled trial would be ideal, but to date funding isn’t available. However, the observational data from MESA and other studies suggest such a trial may not even be needed.
“Certainly the guidelines do allow for CAC scoring to be used in clinical decision making,” he noted.
The MESA study is funded by the National Heart, Lung, and Blood Institute. Dr. Joshi reported having no financial conflicts.
CHICAGO – Absence of coronary artery calcium upon imaging results in an impressively low cardiovascular event rate over the next 10 years regardless of an individual’s level of standard risk factors, according to prospective data from the MESA study.
In contrast, a coronary artery calcium (CAC) score of 1-10, often described as minimal CAC, nearly doubles the 10-year risk, compared with a baseline CAC score of 0.
Prior to these new 10-year data, many cardiologists considered a CAC score of 1-10 as tantamount to no CAC. Not so, Dr. Parag H. Joshi said at the American Heart Association scientific sessions.
“A CAC of 0 is presumably identifying someone without any atherosclerosis. Just the presence of minimal calcium suggests that atherosclerosis is building up. Our data suggest that among individuals with a CAC of 1-10, current smoking, elevated non-HDL cholesterol, and particularly hypertension should be treated aggressively,” said Dr. Joshi, a clinical fellow in cardiovascular diseases and prevention at Johns Hopkins University, Baltimore.
Prior studies totaling more than 50,000 subjects with a CAC score of 0 have shown very low cardiovascular event rates over 4-5 years of follow-up. However, current cardiovascular risk estimates focus on 10-year risk. This new analysis from MESA (Multi-Ethnic Study of Atherosclerosis) is the first study to provide prospective, 10-year events data, and those data are highly reassuring, he added.
MESA is a prospective, population-based cohort study. This analysis included 6,814 subjects aged 45-84 who were free of clinical cardiovascular disease at baseline, when their CAC score was determined. At that time, 3,415 participants had a CAC score of 0 and 508 had a score of 1-10.
During a median 10.3 years of follow-up, 123 cardiovascular events occurred, roughly one-third of which were nonfatal acute MIs and half of which were nonfatal strokes; the remainder were cardiovascular deaths.
The event rate was 2.9/1,000 person-years in subjects with a CAC of 0 and significantly greater at 5.5/1,000 person-years with a score of 1-10. However, since the cardiovascular risk factor profile of the zero CAC group was generally more favorable, Dr. Joshi and coinvestigators carried out a Cox proportional hazards analysis factoring in demographics, standard cardiovascular risk factors, body mass index, C-reactive protein level, and carotid intima media thickness. The adjusted 10-year event risk in the group with a CAC score of 1-10 was 1.9-fold greater than with a CAC of 0.
The highest 10-year event rate was noted in subjects with at least three of the following four risk factors at baseline: hypertension, current smoking, diabetes, and hyperlipidemia. The rate was 6.5/1,000 person-years in such individuals if they had a CAC of 0 and doubled at 13.1/1,000 person-years with a score of 1-10.
In a multivariate Cox analysis, age, smoking, and hypertension proved to be significant predictors of cardiovascular events in the group with a CAC of 0 as well as in those with a CAC of 1-10. But there was one important difference between the two groups: While the hazard ratio for cardiovascular events associated with hypertension versus no hypertension was 2.1 in subjects with a CAC of 0, the presence of hypertension in individuals with a CAC of 1-10 increased their event risk by 10.2-fold, or nearly five times greater than the risk increase associated with hypertension in persons with a CAC of 0, Dr. Joshi observed.
Non–HDL cholesterol level was predictive of cardiovascular risk in subjects with a CAC of 1-10 but not in those with a score of 0.
When actual event rates were compared with those predicted by the atherosclerotic cardiovascular disease (ASCVD) risk estimator introduced in the 2013 AHA/American College of Cardiology cholesterol guidelines, the event rate in subjects with an ASCVD 10-year risk estimate of 7.5%-15% but a CAC of 0 was just 4.4%.
Audience members noted that CAC scores didn’t do a very good job of stratifying stroke risk in MESA. That’s not surprising, since the score reflects coronary but not carotid artery calcium. But it is a limitation of CAC as a predictive tool, especially in light of the fact that strokes accounted for half of all cardiovascular events in the study.
Asked where he and his coinvestigators plan to go from here, Dr. Joshi said a randomized, controlled trial would be ideal, but to date funding isn’t available. However, the observational data from MESA and other studies suggest such a trial may not even be needed.
“Certainly the guidelines do allow for CAC scoring to be used in clinical decision making,” he noted.
The MESA study is funded by the National Heart, Lung, and Blood Institute. Dr. Joshi reported having no financial conflicts.
CHICAGO – Absence of coronary artery calcium upon imaging results in an impressively low cardiovascular event rate over the next 10 years regardless of an individual’s level of standard risk factors, according to prospective data from the MESA study.
In contrast, a coronary artery calcium (CAC) score of 1-10, often described as minimal CAC, nearly doubles the 10-year risk, compared with a baseline CAC score of 0.
Prior to these new 10-year data, many cardiologists considered a CAC score of 1-10 as tantamount to no CAC. Not so, Dr. Parag H. Joshi said at the American Heart Association scientific sessions.
“A CAC of 0 is presumably identifying someone without any atherosclerosis. Just the presence of minimal calcium suggests that atherosclerosis is building up. Our data suggest that among individuals with a CAC of 1-10, current smoking, elevated non-HDL cholesterol, and particularly hypertension should be treated aggressively,” said Dr. Joshi, a clinical fellow in cardiovascular diseases and prevention at Johns Hopkins University, Baltimore.
Prior studies totaling more than 50,000 subjects with a CAC score of 0 have shown very low cardiovascular event rates over 4-5 years of follow-up. However, current cardiovascular risk estimates focus on 10-year risk. This new analysis from MESA (Multi-Ethnic Study of Atherosclerosis) is the first study to provide prospective, 10-year events data, and those data are highly reassuring, he added.
MESA is a prospective, population-based cohort study. This analysis included 6,814 subjects aged 45-84 who were free of clinical cardiovascular disease at baseline, when their CAC score was determined. At that time, 3,415 participants had a CAC score of 0 and 508 had a score of 1-10.
During a median 10.3 years of follow-up, 123 cardiovascular events occurred, roughly one-third of which were nonfatal acute MIs and half of which were nonfatal strokes; the remainder were cardiovascular deaths.
The event rate was 2.9/1,000 person-years in subjects with a CAC of 0 and significantly greater at 5.5/1,000 person-years with a score of 1-10. However, since the cardiovascular risk factor profile of the zero CAC group was generally more favorable, Dr. Joshi and coinvestigators carried out a Cox proportional hazards analysis factoring in demographics, standard cardiovascular risk factors, body mass index, C-reactive protein level, and carotid intima media thickness. The adjusted 10-year event risk in the group with a CAC score of 1-10 was 1.9-fold greater than with a CAC of 0.
The highest 10-year event rate was noted in subjects with at least three of the following four risk factors at baseline: hypertension, current smoking, diabetes, and hyperlipidemia. The rate was 6.5/1,000 person-years in such individuals if they had a CAC of 0 and doubled at 13.1/1,000 person-years with a score of 1-10.
In a multivariate Cox analysis, age, smoking, and hypertension proved to be significant predictors of cardiovascular events in the group with a CAC of 0 as well as in those with a CAC of 1-10. But there was one important difference between the two groups: While the hazard ratio for cardiovascular events associated with hypertension versus no hypertension was 2.1 in subjects with a CAC of 0, the presence of hypertension in individuals with a CAC of 1-10 increased their event risk by 10.2-fold, or nearly five times greater than the risk increase associated with hypertension in persons with a CAC of 0, Dr. Joshi observed.
Non–HDL cholesterol level was predictive of cardiovascular risk in subjects with a CAC of 1-10 but not in those with a score of 0.
When actual event rates were compared with those predicted by the atherosclerotic cardiovascular disease (ASCVD) risk estimator introduced in the 2013 AHA/American College of Cardiology cholesterol guidelines, the event rate in subjects with an ASCVD 10-year risk estimate of 7.5%-15% but a CAC of 0 was just 4.4%.
Audience members noted that CAC scores didn’t do a very good job of stratifying stroke risk in MESA. That’s not surprising, since the score reflects coronary but not carotid artery calcium. But it is a limitation of CAC as a predictive tool, especially in light of the fact that strokes accounted for half of all cardiovascular events in the study.
Asked where he and his coinvestigators plan to go from here, Dr. Joshi said a randomized, controlled trial would be ideal, but to date funding isn’t available. However, the observational data from MESA and other studies suggest such a trial may not even be needed.
“Certainly the guidelines do allow for CAC scoring to be used in clinical decision making,” he noted.
The MESA study is funded by the National Heart, Lung, and Blood Institute. Dr. Joshi reported having no financial conflicts.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: A coronary artery calcium score of 0 appears to trump the 10-year atherosclerotic cardiovascular disease risk estimator introduced in the 2013 AHA/ACC cholesterol guidelines.
Major finding: The actual 10-year cardiovascular event rate in subjects with a coronary artery calcium score of 0 was just 4.4% – below the guideline-recommended threshold for statin therapy– even though their predicted risk using the AHA/ACC risk estimator was 7.5%-15%.
Data source: The Multi-Ethnic Study of Atherosclerosis is a prospective, population-based cohort study. This analysis included 6,814 subjects aged 45-84 who were free of clinical cardiovascular disease at baseline.
Disclosures: The MESA study is funded by the National Heart, Lung, and Blood Institute. The presenter reported having no financial conflicts.

HF: Glucose at ED presentation may predict mortality
Among adults who present to the emergency department with acute heart failure, blood glucose levels may predict 30-day mortality, regardless of whether or not the patients have preexisting diabetes.
At ED presentation, patients with acute heart failure syndrome “demonstrate a wide spectrum of physiological and metabolic perturbations.” Hyperglycemia occurs in up to 40% of them, irrespective of their diabetes status. “If blood glucose measurement is prognostically useful, it may be of broad potential utility because it is a rapid, readily available, and inexpensive test that can be used in the acute setting to allow rapid risk stratification for a wide range of potential outcomes,” according to Dr. Maneesh Sud of the University of Toronto and Toronto General Hospital and his associates.
To examine whether initial glucose level correlated with later outcomes, the investigators analyzed data from two large population-based cohorts of patients hospitalized for acute HF during a 3-year period. A total of 9,275 of the 16,524 patients (median age, 79 years) did not have preexisting diabetes (56%), and the remaining 44% did (Eur. Heart J. 2015 [doi:10.1093/eurheartj/ehu462]).
Among patients with diabetes, a blood glucose level exceeding 11.1 mmol/L was associated with significantly increased all-cause 30-day mortality, compared with normal glucose levels, with a hazard ratio (HR) of 1.48. Among patients without diabetes, a blood glucose level exceeding 6.1 mmol/L was associated with significantly increased all-cause 30-day mortality, with an HR of 1.26, and that risk rose with increasing glucose levels to 1.50 at the maximum level of 11.1 mmol/L.
The risk for cardiovascular death within 30 days increased as blood glucose levels rose for both groups of patients, as did the risk for cardiovascular hospitalization. In addition, both patients who had preexisting diabetes and those who did not were at significantly increased risk for diabetes-related hospitalization if their blood glucose level exceeded the normal range at presentation to the ED. And, in patients without diabetes who had elevated blood glucose levels, the risk of developing diabetes was significantly increased, in a dose-dependent fashion. These findings suggest that determining blood glucose levels at ED presentation “may serve as a screen to identify high-risk patients who warrant formal testing for diabetes, allowing for prompt referral to prevent further morbidity and mortality,” Dr. Sud and his associates said.
Among adults who present to the emergency department with acute heart failure, blood glucose levels may predict 30-day mortality, regardless of whether or not the patients have preexisting diabetes.
At ED presentation, patients with acute heart failure syndrome “demonstrate a wide spectrum of physiological and metabolic perturbations.” Hyperglycemia occurs in up to 40% of them, irrespective of their diabetes status. “If blood glucose measurement is prognostically useful, it may be of broad potential utility because it is a rapid, readily available, and inexpensive test that can be used in the acute setting to allow rapid risk stratification for a wide range of potential outcomes,” according to Dr. Maneesh Sud of the University of Toronto and Toronto General Hospital and his associates.
To examine whether initial glucose level correlated with later outcomes, the investigators analyzed data from two large population-based cohorts of patients hospitalized for acute HF during a 3-year period. A total of 9,275 of the 16,524 patients (median age, 79 years) did not have preexisting diabetes (56%), and the remaining 44% did (Eur. Heart J. 2015 [doi:10.1093/eurheartj/ehu462]).
Among patients with diabetes, a blood glucose level exceeding 11.1 mmol/L was associated with significantly increased all-cause 30-day mortality, compared with normal glucose levels, with a hazard ratio (HR) of 1.48. Among patients without diabetes, a blood glucose level exceeding 6.1 mmol/L was associated with significantly increased all-cause 30-day mortality, with an HR of 1.26, and that risk rose with increasing glucose levels to 1.50 at the maximum level of 11.1 mmol/L.
The risk for cardiovascular death within 30 days increased as blood glucose levels rose for both groups of patients, as did the risk for cardiovascular hospitalization. In addition, both patients who had preexisting diabetes and those who did not were at significantly increased risk for diabetes-related hospitalization if their blood glucose level exceeded the normal range at presentation to the ED. And, in patients without diabetes who had elevated blood glucose levels, the risk of developing diabetes was significantly increased, in a dose-dependent fashion. These findings suggest that determining blood glucose levels at ED presentation “may serve as a screen to identify high-risk patients who warrant formal testing for diabetes, allowing for prompt referral to prevent further morbidity and mortality,” Dr. Sud and his associates said.
Among adults who present to the emergency department with acute heart failure, blood glucose levels may predict 30-day mortality, regardless of whether or not the patients have preexisting diabetes.
At ED presentation, patients with acute heart failure syndrome “demonstrate a wide spectrum of physiological and metabolic perturbations.” Hyperglycemia occurs in up to 40% of them, irrespective of their diabetes status. “If blood glucose measurement is prognostically useful, it may be of broad potential utility because it is a rapid, readily available, and inexpensive test that can be used in the acute setting to allow rapid risk stratification for a wide range of potential outcomes,” according to Dr. Maneesh Sud of the University of Toronto and Toronto General Hospital and his associates.
To examine whether initial glucose level correlated with later outcomes, the investigators analyzed data from two large population-based cohorts of patients hospitalized for acute HF during a 3-year period. A total of 9,275 of the 16,524 patients (median age, 79 years) did not have preexisting diabetes (56%), and the remaining 44% did (Eur. Heart J. 2015 [doi:10.1093/eurheartj/ehu462]).
Among patients with diabetes, a blood glucose level exceeding 11.1 mmol/L was associated with significantly increased all-cause 30-day mortality, compared with normal glucose levels, with a hazard ratio (HR) of 1.48. Among patients without diabetes, a blood glucose level exceeding 6.1 mmol/L was associated with significantly increased all-cause 30-day mortality, with an HR of 1.26, and that risk rose with increasing glucose levels to 1.50 at the maximum level of 11.1 mmol/L.
The risk for cardiovascular death within 30 days increased as blood glucose levels rose for both groups of patients, as did the risk for cardiovascular hospitalization. In addition, both patients who had preexisting diabetes and those who did not were at significantly increased risk for diabetes-related hospitalization if their blood glucose level exceeded the normal range at presentation to the ED. And, in patients without diabetes who had elevated blood glucose levels, the risk of developing diabetes was significantly increased, in a dose-dependent fashion. These findings suggest that determining blood glucose levels at ED presentation “may serve as a screen to identify high-risk patients who warrant formal testing for diabetes, allowing for prompt referral to prevent further morbidity and mortality,” Dr. Sud and his associates said.
FROM EUROPEAN HEART JOURNAL
Key clinical point: Among patients who present to the ED with acute HF, blood glucose levels predict 30-day mortality and other outcomes.
Major finding: Patients with diabetes who had elevated blood glucose levels showed increased 30-day mortality with a hazard ratio of up to 1.48, while those without diabetes showed increased 30-day mortality with a hazard ratio of up to 1.50.
Data source: A secondary analysis of data from two population-based cohorts comprising 16,524 patients who presented to the ED with acute HF during a 3-year period.
Disclosures: This study was supported by the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Ontario. Dr. Sud and his associates reported having no relevant financial disclosures.
FDA approves vagal blocking device for obesity
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
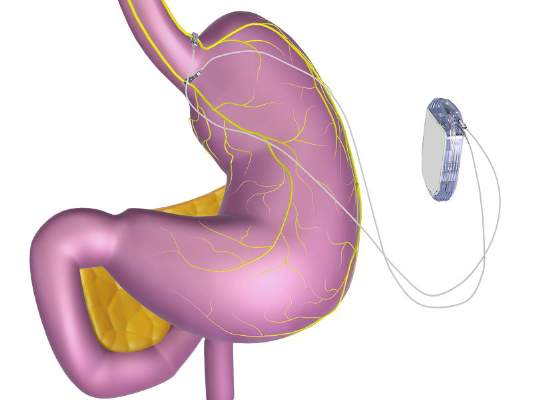
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
A novel implantable device that delivers electrical pulses to the intra-abdominal vagus nerve has been approved for treatment of obesity in adults, providing a less invasive alternative to bariatric surgery.
The Maestro Rechargeable System is “the first weight loss treatment device that targets the nerve pathway between the brain and the stomach that controls feelings of hunger and fullness,” according to the Food and Drug Administration statement released Jan. 14. The last device approved by the FDA for treatment of obesity was the Realize gastric band, in September 2007.
The device is approved for adults aged 18 and older with a body mass index of at least 40-45 kg/m2, or at least 35-39.9 kg/m2 with a related health condition such as high blood pressure or high cholesterol levels, who have tried to lose weight in a supervised weight management program within the past 5 years.
The system includes a rechargeable electrical pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure. “It works by sending intermittent electrical pulses to the trunks in the abdominal vagus nerve, which is involved in regulating stomach emptying and signaling to the brain that the stomach feels empty or full,” the FDA statement said, adding: “Although it is known that the electric stimulation blocks nerve activity between the brain and the stomach, the specific mechanisms for weight loss due to use of the device are unknown.”
The manufacturer, EnteroMedics, refers to the treatment as “VBLOC therapy,” delivered by the Maestro System. The company expects that the device will be available this year “on a limited basis” at select Bariatric Centers of Excellence in the United States, according to a statement issued by EnteroMedics on Jan. 14.
FDA approval was based on the results of the ReCharge study of 233 patients with a body mass index of at least 35 kg/m2; the device was activated in 157 patients, and the remaining patients had the device implanted but it was not activated and they served as controls.
After 12 months, those with the activated device lost 8.5% more excess weight than did the controls. Among those who had the device activated, almost 53% lost at least 20% of their excess weight and 38% lost at least 35% of their excess weight, according to the FDA.
The study did not meet the primary effectiveness endpoint, which was that those on active treatment would lose at least 10% more excess weight than would the controls. However, the majority of an FDA advisory panel that reviewed the data at a meeting in June 2014 supported approval, agreeing that the benefits outweighed the risks for the proposed indication. Panelists cited the fact that the study safety endpoint was met and that the device was effective in helping some people lose weight.
The FDA statement said the decision to approve the device was based on the panel’s recommendation, the study results, and an FDA survey of patient preferences for obesity devices, which found that “a group of patients would accept risks associated with this surgically implanted device for the amounts of weight loss expected to be provided by the device.”
As a condition for approval, EnteroMedics is required to conduct a 5-year postmarketing study that will collect safety and effectiveness data in at least 100 patients, including weight loss, adverse events, surgical revisions and explants and changes in obesity-related comorbidities, according to the FDA.
Serious adverse events in the ReCharge study were nausea, pain at the neuroregulator site, vomiting, and surgical complications; other adverse events were heartburn, problems swallowing, belching, mild nausea, and chest pain, the FDA noted.
The EnteroMedics statement says that contraindications for VBLOC therapy include liver cirrhosis, portal hypertension, esophageal varices or an uncorrectable, clinically significant hiatal hernia; patients for whom magnetic resonance imaging or diathermy use is planned; patients at high risk for surgical complications; and patients who have permanently implanted, electrically-powered medical devices or gastrointestinal devices or prostheses, such as pacemakers, implanted defibrillators, or neurostimulators.
FDA panel votes against approval of oral desmopressin for nocturia
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”

Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”
Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
HYATTSVILLE, MD. – The majority of a Food and Drug Administration advisory panel voted against approval of an orally disintegrating sublingual tablet formulation of desmopressin as a treatment for nocturia, citing uncertainties about the clinical benefits in the pivotal trials and concerns about the risk of hyponatremia.
At the meeting on Jan. 12, the FDA Endocrinologic and Metabolic Drugs Advisory Committee voted 10 to 5, with two abstentions, that the benefits of this desmopressin formulation in clinical trials did not outweigh the risks for the proposed indication, the treatment of nocturia due to nocturnal polyuria in adults who wake up two or more times each night to void. The indication proposed by Ferring Pharmaceuticals includes the statement that prior to treatment, “lifestyle changes and other treatable medical causes of nocturia should be addressed.”
Although the panel agreed that treatment was associated with statistically significant effects on the two primary endpoints in the two pivotal trials, those voting against approval said there were uncertainties about the clinical benefits, which several panelists said could be addressed in preapproval studies.
The panel generally agreed that the studies demonstrated a low risk of hyponatremia, the main safety concern associated with treatment. But they said that it was unclear whether the appropriate patients would be selected for treatment and would be properly monitored as recommended during treatment in real-world clinical situations. Examples included whether patients would remember to stop taking the medication when they developed an illness that could increase their risk of hyponatremia, or if they developed a disease or condition that affects sodium as they got older. There was also some uncertainty over the clinical significance of the small decreases in serum sodium in patients during the course of the 3-month studies, which evaluated two gender-specific doses that were lower than the higher doses previously studied that were associated with an increased risk of hyponatremia.
“The clinical benefit of the active medication relative to the substantial improvements seen in the placebo arm was relatively small,” said panelist Dr. Brendan Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston, who voted against approval. “The sponsor is on to something with respect to the time to the first nocturnal void being longer” among those on the drug, an exploratory endpoint that he recommended should be studied further. The company’s plan to address the outliers with substantial drops in sodium that are considered more dangerous was “reasonable,” he said, but added that he shared the concern of other committee members “that implementing that risk-mitigation strategy in the real world after approval would be difficult.”
The company developed lower doses to minimize the risk of hyponatremia and conducted two pivotal phase III confirmatory randomized, placebo-controlled studies of 268 women and almost 400 men with at least two voids per night (an average of 2.9 episodes), evaluating a 25-mcg dose in women and 50 mcg and 75 mcg in men over 3 months. Exclusion criteria included treatable causes of nocturia, such as diabetes insipidus and cardiac failure, and medical conditions that increased risk, such as hyponatremia and psychogenic polydipsia. (The company is not pursuing approval of the 75-mcg dose in men.)
Among the women, the number of voids per night over 3 months dropped by a mean of 1.46 among those on desmopressin vs. 1.24 among those on placebo, a 0.22 difference that was statistically significant, despite the large placebo effect. In the study of men, the number of voids per night dropped by a mean of 1.25 among those on the 50-mcg dose, vs. 0.88 among those on placebo, a difference of 0.37 that was also statistically significant.
In both studies, patients on desmopressin were almost twice as likely to achieve at least a 33% reduction in voids, the second primary endpoint. The large placebo effect in both studies, particularly among the women, is typically observed in studies of lower urinary tract dysfunction treatment, according to the company and several panelists.
An increase in the time to the first nocturnal void, a secondary endpoint, was increased by a mean of 49 minutes in women and by 39 minutes in men over placebo, which were both statistically significant differences.
There were two cases (1%) of hyponatremia among the women and three cases (3%) among the men on the 50-mcg dose; no patients on these two doses developed a serum sodium below 125 mmol/L (severe hyponatremia).
While the studies showed statistically significant decreases in the primary endpoints, the FDA reviewers questioned the magnitude of the clinical benefit. Other issues raised by FDA reviewers included the “urologically heterogeneous” population of patients in the two studies, which included, for example, patients with small-capacity bladders and those taking medications for overactive bladder or benign prostatic hypertrophy.
Currently, there is no FDA-approved treatment for nocturia or nocturnal polyuria. Desmopressin, in a tablet formulation, is approved in the United States for treating central diabetes insipidus and nocturnal enuresis in children and is occasionally used off-label for patients with nocturia. It is also approved for preventing bleeding in patients with hemophilia A or type 1 von Willebrand’s disease with a subcutaneous injection or nasal spray formulation.
The 25-mcg and the 50-mcg doses of the desmopressin orally disintegrating tablet are equivalent to the 42-mcg and 83-mcg doses of the desmopressin tablet, respectively, according to the FDA.
The orally disintegrating tablet formulation of desmopressin was approved in Canada in 2014 as a treatment for nocturia due to nocturnal polyuria at the same doses were proposed for approval in the United States. Higher doses have been approved in more than 65 countries).
The FDA usually follows the recommendations of its advisory panels.
In a statement issued after the meeting, Dr. Paul Korner, senior vice president for U.S. development at Ferring, said, “We look forward to working with the FDA to ensure it has the data needed to complete its evaluation of the safety and efficacy of this medication.”
The FDA is expected to make a decision during the first quarter of 2015, according to the company.
FDA panelists were cleared of conflicts.
AT AN FDA ADVISORY COMMITTEE MEETING

Diabetes substantially overtreated in older adults
Most older patients with diabetes who had complex or poor health status and limited life expectancy were still given insulin or sulfonylureas to achieve tight glycemic control, even though such intensive treatment likely causes more harm than good in this patient population, according to a report published online Jan. 12 in JAMA Internal Medicine.
Diabetes is highly prevalent among Americans aged 65 years and older, and tight glycemic control achieved through medication use poses significant threats to them – chiefly, hypoglycemia –without imparting the benefits seen in younger, healthier patients, said Dr. Kasia J. Lipska of Yale University, New Haven, Conn., and her associates. Hypoglycemia is associated with increased mortality, cardiovascular disease, falls and accidents, dementia, and a poor health-related quality of life in older adults.

To examine such overtreatment, the investigators analyzed National Health and Nutrition Examination Survey data during a 10-year period. They focused on 1,288 participants aged 65 years and older (mean age, 73 years) whose hemoglobin A1c levels indicated glycemic control that was categorized as poor (9% or higher), moderate (7%-8.9%), or tight (<7%); the latter category included those patients with very tight glycemic control (<6.5%).
Dr. Lipska and her associates found that 62% of the sample, representing nearly 4 million older Americans, had tight glycemic control, including 42%, representing 2.6 million older Americans, who had very tight glycemic control.
Most of those patients were taking insulin or sulfonylureas. That included the majority of patients who had complex health profiles or poor health because of such coexisting conditions as arthritis, heart failure, lung disease, chronic kidney disease, coronary heart disease, stroke, urinary incontinence, and functional impairments.
“Given incomplete information on all comorbidities in our study, it is likely that we underestimated the complexity of health status, and thus, true estimates of adults who are potentially overtreated may be even higher,” the investigators said (JAMA Intern. Med. 2015 Jan. 12 [doi:10.1001/jamainternmed.2014.7345]).
“Recognition of both the harms and the benefits of glycemic control is critical for patients and physicians and other health care professionals to make informed decisions about glucose-lowering treatment,” the investigators added.
A Pepper Center Career Development Award, the National Institute on Aging, the Yale Center for Investigation, the American Federation for Aging Research, the Paul B. Beeson Career Development Program, and the National Institutes of Health funded the study. Dr. Lipska reported having no financial conflicts. Her associates reported ties to Medtronic, Johnson & Johnson, and FAIR Health.
Most older patients with diabetes who had complex or poor health status and limited life expectancy were still given insulin or sulfonylureas to achieve tight glycemic control, even though such intensive treatment likely causes more harm than good in this patient population, according to a report published online Jan. 12 in JAMA Internal Medicine.
Diabetes is highly prevalent among Americans aged 65 years and older, and tight glycemic control achieved through medication use poses significant threats to them – chiefly, hypoglycemia –without imparting the benefits seen in younger, healthier patients, said Dr. Kasia J. Lipska of Yale University, New Haven, Conn., and her associates. Hypoglycemia is associated with increased mortality, cardiovascular disease, falls and accidents, dementia, and a poor health-related quality of life in older adults.
To examine such overtreatment, the investigators analyzed National Health and Nutrition Examination Survey data during a 10-year period. They focused on 1,288 participants aged 65 years and older (mean age, 73 years) whose hemoglobin A1c levels indicated glycemic control that was categorized as poor (9% or higher), moderate (7%-8.9%), or tight (<7%); the latter category included those patients with very tight glycemic control (<6.5%).
Dr. Lipska and her associates found that 62% of the sample, representing nearly 4 million older Americans, had tight glycemic control, including 42%, representing 2.6 million older Americans, who had very tight glycemic control.
Most of those patients were taking insulin or sulfonylureas. That included the majority of patients who had complex health profiles or poor health because of such coexisting conditions as arthritis, heart failure, lung disease, chronic kidney disease, coronary heart disease, stroke, urinary incontinence, and functional impairments.
“Given incomplete information on all comorbidities in our study, it is likely that we underestimated the complexity of health status, and thus, true estimates of adults who are potentially overtreated may be even higher,” the investigators said (JAMA Intern. Med. 2015 Jan. 12 [doi:10.1001/jamainternmed.2014.7345]).
“Recognition of both the harms and the benefits of glycemic control is critical for patients and physicians and other health care professionals to make informed decisions about glucose-lowering treatment,” the investigators added.
A Pepper Center Career Development Award, the National Institute on Aging, the Yale Center for Investigation, the American Federation for Aging Research, the Paul B. Beeson Career Development Program, and the National Institutes of Health funded the study. Dr. Lipska reported having no financial conflicts. Her associates reported ties to Medtronic, Johnson & Johnson, and FAIR Health.
Most older patients with diabetes who had complex or poor health status and limited life expectancy were still given insulin or sulfonylureas to achieve tight glycemic control, even though such intensive treatment likely causes more harm than good in this patient population, according to a report published online Jan. 12 in JAMA Internal Medicine.
Diabetes is highly prevalent among Americans aged 65 years and older, and tight glycemic control achieved through medication use poses significant threats to them – chiefly, hypoglycemia –without imparting the benefits seen in younger, healthier patients, said Dr. Kasia J. Lipska of Yale University, New Haven, Conn., and her associates. Hypoglycemia is associated with increased mortality, cardiovascular disease, falls and accidents, dementia, and a poor health-related quality of life in older adults.
To examine such overtreatment, the investigators analyzed National Health and Nutrition Examination Survey data during a 10-year period. They focused on 1,288 participants aged 65 years and older (mean age, 73 years) whose hemoglobin A1c levels indicated glycemic control that was categorized as poor (9% or higher), moderate (7%-8.9%), or tight (<7%); the latter category included those patients with very tight glycemic control (<6.5%).
Dr. Lipska and her associates found that 62% of the sample, representing nearly 4 million older Americans, had tight glycemic control, including 42%, representing 2.6 million older Americans, who had very tight glycemic control.
Most of those patients were taking insulin or sulfonylureas. That included the majority of patients who had complex health profiles or poor health because of such coexisting conditions as arthritis, heart failure, lung disease, chronic kidney disease, coronary heart disease, stroke, urinary incontinence, and functional impairments.
“Given incomplete information on all comorbidities in our study, it is likely that we underestimated the complexity of health status, and thus, true estimates of adults who are potentially overtreated may be even higher,” the investigators said (JAMA Intern. Med. 2015 Jan. 12 [doi:10.1001/jamainternmed.2014.7345]).
“Recognition of both the harms and the benefits of glycemic control is critical for patients and physicians and other health care professionals to make informed decisions about glucose-lowering treatment,” the investigators added.
A Pepper Center Career Development Award, the National Institute on Aging, the Yale Center for Investigation, the American Federation for Aging Research, the Paul B. Beeson Career Development Program, and the National Institutes of Health funded the study. Dr. Lipska reported having no financial conflicts. Her associates reported ties to Medtronic, Johnson & Johnson, and FAIR Health.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Most older patients with diabetes who are in poor health are given insulin or sulfonylureas to achieve tight glycemic control, even though such intensive control is more harmful than beneficial in that patient population.
Major finding: Of the sample, 62% (nearly 4 million older Americans) had tight glycemic control, including 42% (2.6 million older Americans) who had very tight glycemic control.
Data source: A cross-sectional analysis of diabetes treatment patterns in a nationally representative sample of 1,288 men and women aged 65 years and older.
Disclosures: A Pepper Center Career Development Award, the National Institute on Aging, the Yale Center for Investigation, the American Federation for Aging Research, the Paul B. Beeson Career Development Program, and the National Institutes of Health funded the study. Dr. Lipska reported having no financial conflicts. Her associates reported ties to Medtronic, Johnson & Johnson, and FAIR Health.
Unrecognized MI common with impaired fasting glucose
CHICAGO– Impaired fasting glucose was associated with a doubled risk of asymptomatic or unrecognized myocardial infarction during prospective follow-up of subjects without overt cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis.
The clinical relevance of this finding lies in previous work establishing that asymptomatic, unrecognized MIs are associated with a prognosis as serious as for symptomatic MIs.
“Because of the high prevalence of impaired fasting glucose, the implications of this finding may have ramifications for a large proportion of the adult population,” Dr. Richard B. Stacey observed at the American Heart Association Scientific Sessions.
The magnitude of the risk of unrecognized MI was associated with duration of exposure to abnormal blood glucose levels in the landmark population-based MESA study. Subjects with impaired fasting glucose (IFG) noted on at least three of five biannual examinations conducted over a 10-year period had an adjusted 1.97-fold greater risk of experiencing an unrecognized MI during that time, compared with MESA participants who had normal blood glucose, added Dr. Stacey of Wake Forest University in Winston-Salem, N.C.
He presented two new analyses from the MESA study. One was cross-sectional: Among 4,955 study participants with normal fasting glucose and 930 with IFG upon study enrollment, a baseline 12-lead ECG revealed a prior unrecognized MI in 1.4% of those with normal fasting glucose and 3.2% of those with IFG, a 2.3-fold increase. MESA participants were on average in their mid-60s, and none had clinical cardiovascular disease.
In a multivariate logistic regression analysis fully adjusted for demographics and the standard cardiovascular risk factors as well as body mass index and the use of antihypertensive and lipid-lowering medications, IFG was associated with a 1.63-fold increased risk of prior asymptomatic or unrecognized MI (P < .001).
The other, prospective, longitudinal analysis, included 872 subjects with baseline normal fasting glucose and 545 with IFG, none of whom had evidence of a prior unrecognized MI at entry. This analysis excluded individuals who developed diabetes mellitus, had a clinically recognized MI, or underwent coronary revascularization during follow-up. During 10 years of follow-up, 14.9% of the group who continued to have normal fasting glucose experienced an unrecognized MI, as did 23.8% of those with IFG at entry. The unrecognized MIs were detected either by the presence of pathologic Q waves or minor Q waves with ST-T abnormalities on ECG or by an MRI showing late gadolinium enhancement indicative of myocardial scar.
The MESA study is funded by the National Institutes of Health. Dr. Stacey reported having no financial conflicts.
CHICAGO– Impaired fasting glucose was associated with a doubled risk of asymptomatic or unrecognized myocardial infarction during prospective follow-up of subjects without overt cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis.
The clinical relevance of this finding lies in previous work establishing that asymptomatic, unrecognized MIs are associated with a prognosis as serious as for symptomatic MIs.
“Because of the high prevalence of impaired fasting glucose, the implications of this finding may have ramifications for a large proportion of the adult population,” Dr. Richard B. Stacey observed at the American Heart Association Scientific Sessions.
The magnitude of the risk of unrecognized MI was associated with duration of exposure to abnormal blood glucose levels in the landmark population-based MESA study. Subjects with impaired fasting glucose (IFG) noted on at least three of five biannual examinations conducted over a 10-year period had an adjusted 1.97-fold greater risk of experiencing an unrecognized MI during that time, compared with MESA participants who had normal blood glucose, added Dr. Stacey of Wake Forest University in Winston-Salem, N.C.
He presented two new analyses from the MESA study. One was cross-sectional: Among 4,955 study participants with normal fasting glucose and 930 with IFG upon study enrollment, a baseline 12-lead ECG revealed a prior unrecognized MI in 1.4% of those with normal fasting glucose and 3.2% of those with IFG, a 2.3-fold increase. MESA participants were on average in their mid-60s, and none had clinical cardiovascular disease.
In a multivariate logistic regression analysis fully adjusted for demographics and the standard cardiovascular risk factors as well as body mass index and the use of antihypertensive and lipid-lowering medications, IFG was associated with a 1.63-fold increased risk of prior asymptomatic or unrecognized MI (P < .001).
The other, prospective, longitudinal analysis, included 872 subjects with baseline normal fasting glucose and 545 with IFG, none of whom had evidence of a prior unrecognized MI at entry. This analysis excluded individuals who developed diabetes mellitus, had a clinically recognized MI, or underwent coronary revascularization during follow-up. During 10 years of follow-up, 14.9% of the group who continued to have normal fasting glucose experienced an unrecognized MI, as did 23.8% of those with IFG at entry. The unrecognized MIs were detected either by the presence of pathologic Q waves or minor Q waves with ST-T abnormalities on ECG or by an MRI showing late gadolinium enhancement indicative of myocardial scar.
The MESA study is funded by the National Institutes of Health. Dr. Stacey reported having no financial conflicts.
CHICAGO– Impaired fasting glucose was associated with a doubled risk of asymptomatic or unrecognized myocardial infarction during prospective follow-up of subjects without overt cardiovascular disease in the Multi-Ethnic Study of Atherosclerosis.
The clinical relevance of this finding lies in previous work establishing that asymptomatic, unrecognized MIs are associated with a prognosis as serious as for symptomatic MIs.
“Because of the high prevalence of impaired fasting glucose, the implications of this finding may have ramifications for a large proportion of the adult population,” Dr. Richard B. Stacey observed at the American Heart Association Scientific Sessions.
The magnitude of the risk of unrecognized MI was associated with duration of exposure to abnormal blood glucose levels in the landmark population-based MESA study. Subjects with impaired fasting glucose (IFG) noted on at least three of five biannual examinations conducted over a 10-year period had an adjusted 1.97-fold greater risk of experiencing an unrecognized MI during that time, compared with MESA participants who had normal blood glucose, added Dr. Stacey of Wake Forest University in Winston-Salem, N.C.
He presented two new analyses from the MESA study. One was cross-sectional: Among 4,955 study participants with normal fasting glucose and 930 with IFG upon study enrollment, a baseline 12-lead ECG revealed a prior unrecognized MI in 1.4% of those with normal fasting glucose and 3.2% of those with IFG, a 2.3-fold increase. MESA participants were on average in their mid-60s, and none had clinical cardiovascular disease.
In a multivariate logistic regression analysis fully adjusted for demographics and the standard cardiovascular risk factors as well as body mass index and the use of antihypertensive and lipid-lowering medications, IFG was associated with a 1.63-fold increased risk of prior asymptomatic or unrecognized MI (P < .001).
The other, prospective, longitudinal analysis, included 872 subjects with baseline normal fasting glucose and 545 with IFG, none of whom had evidence of a prior unrecognized MI at entry. This analysis excluded individuals who developed diabetes mellitus, had a clinically recognized MI, or underwent coronary revascularization during follow-up. During 10 years of follow-up, 14.9% of the group who continued to have normal fasting glucose experienced an unrecognized MI, as did 23.8% of those with IFG at entry. The unrecognized MIs were detected either by the presence of pathologic Q waves or minor Q waves with ST-T abnormalities on ECG or by an MRI showing late gadolinium enhancement indicative of myocardial scar.
The MESA study is funded by the National Institutes of Health. Dr. Stacey reported having no financial conflicts.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Consistent impaired fasting glucose may be associated with asymptomatic myocardial infarction.
Major finding: Individuals with impaired fasting glucose on at least three out of five structured clinic visits during 10 years of prospective follow-up were at twice the risk of experiencing an unrecognized MI, compared with those who had normal fasting glucose.
Data source: The Multi-Ethnic Study of Atherosclerosis, a prospective, longitudinal, population-based study.
Disclosures: MESA is funded by the National Institutes of Health. The presenter reported having no financial conflicts.
Elevated troponin present in 40% with T2D and stable heart disease
CHICAGO– Abnormal levels of high-sensitivity cardiac troponin T are present in 40% of type 2 diabetic patients with stable ischemic heart disease, and they do not bode well, according to a new secondary analysis of the BARI 2D study.
In BARI 2D, an abnormal high-sensitivity cardiac troponin T (hsTnT), defined as 14 ng/L or greater, a powerful marker of ongoing myocardial injury, was independently associated with a doubled 5-year risk of the composite endpoint of cardiovascular death, MI, or stroke. Moreover, and discouragingly so, prompt coronary revascularization did nothing to mitigate that risk, Dr. Brendan M. Everett reported at the American Heart Association Scientific Sessions.
Further, early coronary revascularization did not result in a reduction in abnormal hsTnT at 1 year of follow-up, said Dr. Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston.

“To better address the risk represented by an abnormal hsTnT, we need to gain an improved understanding of the biology of troponin release in this population,” he observed. “The fact that we saw an overall decrease of about 0.5% in hemoglobin A1c and an LDL reduction of 16 mg/dL at 1 year and still there was no change in hsTnT leaves me scratching my head. The abnormal hsTnT is clearly a marker of badness, but where is it coming from? Can we address it? Or are we just left to look at it and worry about our patients who have an abnormal hsTnT?”
The BARI 2D trial was designed to learn whether patients with type 2 diabetes and stable ischemic heart disease benefit from prompt coronary revascularization plus intensive medical therapy as compared with intensive medical therapy alone. As previously reported (N. Engl. J. Med. 2009;360:2503-15), this proved not to be the case; prompt revascularization conferred no outcome advantage.
The aim of Dr. Everett’s new secondary analysis of BARI 2D was to learn if the hsTnT assay can be used to identify a subgroup of patients with type 2 diabetes and stable ischemic heart disease who might benefit from prompt coronary revascularization. The rationale was that, in patients with acute coronary syndromes, it’s well established that an abnormal hsTnT is associated with poor prognosis, and such patients would benefit from early revascularization.
The secondary analysis included 2,285 type 2 diabetics with stable ischemic heart disease whose physicians first decided whether they were better candidates for percutaneous coronary intervention or CABG surgery. Patients were then randomized to prompt revascularization by the preferred method plus intensive medical therapy or to intensive medical therapy alone.
Forty percent of participants had an abnormal hsTnT at baseline. Their 5-year rate of the composite primary endpoint of cardiovascular death, MI, or stroke was 27.1%, compared with 12.9% in patients with a baseline hsTnT below 14 ng/mL. After adjusting in a multivariate analysis for various potential confounders – including age, race, and the standard cardiovascular risk factors – the group with an abnormal baseline hsTnT had a 2.09-fold increased risk of a major cardiovascular event.
Early revascularization, regardless of whether by percutaneous coronary intervention or coronary artery bypass graft surgery, provided no benefit no matter what the patient’s baseline hsTnT level. In patients with an hsTnT of 14 ng/L or greater, the 5-year rate of the composite outcome was 26.5% with early revascularization compared with 27.6% with intensive medical therapy. In those with an hsTnT below 14 ng/L, the rate was 11.8% in the early revascularization group and 14% with medical management, a trend favoring prompt revascularization that didn’t achieve statistical significance, according to Dr. Everett.
Of patients with an abnormal hsTnT at baseline, 77% still had an abnormal value at 1 year, regardless of whether they underwent prompt revascularization or intensive medical therapy alone.
Session moderator Dr. Mikhail N. Kosiborod commented that the new BARI 2D substudy highlights a dilemma: “We know that a large population of patients with diabetes, and to some extent those with prediabetes, have elevated hsTnT levels, and we know those patients don’t do well. What we don’t know is what to do about it.”
“What [Dr. Everett’s] study clearly demonstrates is that this does not appear to be driven by epicardial coronary artery disease. If we fix the epicardial CAD, it has absolutely no impact on the outcomes nor on the actual troponin level at follow-up. As far as I can tell, it doesn’t appear to be a glycemic control issue, either. It appears that this is a humoral issue. There are ‘evil humors’ – whatever they are – and we don’t really understand what they are or what to do about it,” said Dr. Kosiborod, professor of medicine at the University of Missouri, Kansas City.
“The truth of the matter is we have no idea what’s causing this low-grade myocardial necrosis, and it’s a hugely important thing,” he continued. “There is absolutely no question that elevated hsTnT, even at very low levels, has a huge impact on subsequent risk of heart failure. We know what the public health effects of heart failure are. And patients with diabetes and heart failure tend to do particularly poorly.”
The BARI 2D trial was funded by the National Institutes of Health. Dr. Everett’s secondary analysis was funded by Roche Diagnostics. He reported receiving research grants from Roche and Novartis.
CHICAGO– Abnormal levels of high-sensitivity cardiac troponin T are present in 40% of type 2 diabetic patients with stable ischemic heart disease, and they do not bode well, according to a new secondary analysis of the BARI 2D study.
In BARI 2D, an abnormal high-sensitivity cardiac troponin T (hsTnT), defined as 14 ng/L or greater, a powerful marker of ongoing myocardial injury, was independently associated with a doubled 5-year risk of the composite endpoint of cardiovascular death, MI, or stroke. Moreover, and discouragingly so, prompt coronary revascularization did nothing to mitigate that risk, Dr. Brendan M. Everett reported at the American Heart Association Scientific Sessions.
Further, early coronary revascularization did not result in a reduction in abnormal hsTnT at 1 year of follow-up, said Dr. Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston.
“To better address the risk represented by an abnormal hsTnT, we need to gain an improved understanding of the biology of troponin release in this population,” he observed. “The fact that we saw an overall decrease of about 0.5% in hemoglobin A1c and an LDL reduction of 16 mg/dL at 1 year and still there was no change in hsTnT leaves me scratching my head. The abnormal hsTnT is clearly a marker of badness, but where is it coming from? Can we address it? Or are we just left to look at it and worry about our patients who have an abnormal hsTnT?”
The BARI 2D trial was designed to learn whether patients with type 2 diabetes and stable ischemic heart disease benefit from prompt coronary revascularization plus intensive medical therapy as compared with intensive medical therapy alone. As previously reported (N. Engl. J. Med. 2009;360:2503-15), this proved not to be the case; prompt revascularization conferred no outcome advantage.
The aim of Dr. Everett’s new secondary analysis of BARI 2D was to learn if the hsTnT assay can be used to identify a subgroup of patients with type 2 diabetes and stable ischemic heart disease who might benefit from prompt coronary revascularization. The rationale was that, in patients with acute coronary syndromes, it’s well established that an abnormal hsTnT is associated with poor prognosis, and such patients would benefit from early revascularization.
The secondary analysis included 2,285 type 2 diabetics with stable ischemic heart disease whose physicians first decided whether they were better candidates for percutaneous coronary intervention or CABG surgery. Patients were then randomized to prompt revascularization by the preferred method plus intensive medical therapy or to intensive medical therapy alone.
Forty percent of participants had an abnormal hsTnT at baseline. Their 5-year rate of the composite primary endpoint of cardiovascular death, MI, or stroke was 27.1%, compared with 12.9% in patients with a baseline hsTnT below 14 ng/mL. After adjusting in a multivariate analysis for various potential confounders – including age, race, and the standard cardiovascular risk factors – the group with an abnormal baseline hsTnT had a 2.09-fold increased risk of a major cardiovascular event.
Early revascularization, regardless of whether by percutaneous coronary intervention or coronary artery bypass graft surgery, provided no benefit no matter what the patient’s baseline hsTnT level. In patients with an hsTnT of 14 ng/L or greater, the 5-year rate of the composite outcome was 26.5% with early revascularization compared with 27.6% with intensive medical therapy. In those with an hsTnT below 14 ng/L, the rate was 11.8% in the early revascularization group and 14% with medical management, a trend favoring prompt revascularization that didn’t achieve statistical significance, according to Dr. Everett.
Of patients with an abnormal hsTnT at baseline, 77% still had an abnormal value at 1 year, regardless of whether they underwent prompt revascularization or intensive medical therapy alone.
Session moderator Dr. Mikhail N. Kosiborod commented that the new BARI 2D substudy highlights a dilemma: “We know that a large population of patients with diabetes, and to some extent those with prediabetes, have elevated hsTnT levels, and we know those patients don’t do well. What we don’t know is what to do about it.”
“What [Dr. Everett’s] study clearly demonstrates is that this does not appear to be driven by epicardial coronary artery disease. If we fix the epicardial CAD, it has absolutely no impact on the outcomes nor on the actual troponin level at follow-up. As far as I can tell, it doesn’t appear to be a glycemic control issue, either. It appears that this is a humoral issue. There are ‘evil humors’ – whatever they are – and we don’t really understand what they are or what to do about it,” said Dr. Kosiborod, professor of medicine at the University of Missouri, Kansas City.
“The truth of the matter is we have no idea what’s causing this low-grade myocardial necrosis, and it’s a hugely important thing,” he continued. “There is absolutely no question that elevated hsTnT, even at very low levels, has a huge impact on subsequent risk of heart failure. We know what the public health effects of heart failure are. And patients with diabetes and heart failure tend to do particularly poorly.”
The BARI 2D trial was funded by the National Institutes of Health. Dr. Everett’s secondary analysis was funded by Roche Diagnostics. He reported receiving research grants from Roche and Novartis.
CHICAGO– Abnormal levels of high-sensitivity cardiac troponin T are present in 40% of type 2 diabetic patients with stable ischemic heart disease, and they do not bode well, according to a new secondary analysis of the BARI 2D study.
In BARI 2D, an abnormal high-sensitivity cardiac troponin T (hsTnT), defined as 14 ng/L or greater, a powerful marker of ongoing myocardial injury, was independently associated with a doubled 5-year risk of the composite endpoint of cardiovascular death, MI, or stroke. Moreover, and discouragingly so, prompt coronary revascularization did nothing to mitigate that risk, Dr. Brendan M. Everett reported at the American Heart Association Scientific Sessions.
Further, early coronary revascularization did not result in a reduction in abnormal hsTnT at 1 year of follow-up, said Dr. Everett, director of the general cardiology inpatient service at Brigham and Women’s Hospital, Boston.
“To better address the risk represented by an abnormal hsTnT, we need to gain an improved understanding of the biology of troponin release in this population,” he observed. “The fact that we saw an overall decrease of about 0.5% in hemoglobin A1c and an LDL reduction of 16 mg/dL at 1 year and still there was no change in hsTnT leaves me scratching my head. The abnormal hsTnT is clearly a marker of badness, but where is it coming from? Can we address it? Or are we just left to look at it and worry about our patients who have an abnormal hsTnT?”
The BARI 2D trial was designed to learn whether patients with type 2 diabetes and stable ischemic heart disease benefit from prompt coronary revascularization plus intensive medical therapy as compared with intensive medical therapy alone. As previously reported (N. Engl. J. Med. 2009;360:2503-15), this proved not to be the case; prompt revascularization conferred no outcome advantage.
The aim of Dr. Everett’s new secondary analysis of BARI 2D was to learn if the hsTnT assay can be used to identify a subgroup of patients with type 2 diabetes and stable ischemic heart disease who might benefit from prompt coronary revascularization. The rationale was that, in patients with acute coronary syndromes, it’s well established that an abnormal hsTnT is associated with poor prognosis, and such patients would benefit from early revascularization.
The secondary analysis included 2,285 type 2 diabetics with stable ischemic heart disease whose physicians first decided whether they were better candidates for percutaneous coronary intervention or CABG surgery. Patients were then randomized to prompt revascularization by the preferred method plus intensive medical therapy or to intensive medical therapy alone.
Forty percent of participants had an abnormal hsTnT at baseline. Their 5-year rate of the composite primary endpoint of cardiovascular death, MI, or stroke was 27.1%, compared with 12.9% in patients with a baseline hsTnT below 14 ng/mL. After adjusting in a multivariate analysis for various potential confounders – including age, race, and the standard cardiovascular risk factors – the group with an abnormal baseline hsTnT had a 2.09-fold increased risk of a major cardiovascular event.
Early revascularization, regardless of whether by percutaneous coronary intervention or coronary artery bypass graft surgery, provided no benefit no matter what the patient’s baseline hsTnT level. In patients with an hsTnT of 14 ng/L or greater, the 5-year rate of the composite outcome was 26.5% with early revascularization compared with 27.6% with intensive medical therapy. In those with an hsTnT below 14 ng/L, the rate was 11.8% in the early revascularization group and 14% with medical management, a trend favoring prompt revascularization that didn’t achieve statistical significance, according to Dr. Everett.
Of patients with an abnormal hsTnT at baseline, 77% still had an abnormal value at 1 year, regardless of whether they underwent prompt revascularization or intensive medical therapy alone.
Session moderator Dr. Mikhail N. Kosiborod commented that the new BARI 2D substudy highlights a dilemma: “We know that a large population of patients with diabetes, and to some extent those with prediabetes, have elevated hsTnT levels, and we know those patients don’t do well. What we don’t know is what to do about it.”
“What [Dr. Everett’s] study clearly demonstrates is that this does not appear to be driven by epicardial coronary artery disease. If we fix the epicardial CAD, it has absolutely no impact on the outcomes nor on the actual troponin level at follow-up. As far as I can tell, it doesn’t appear to be a glycemic control issue, either. It appears that this is a humoral issue. There are ‘evil humors’ – whatever they are – and we don’t really understand what they are or what to do about it,” said Dr. Kosiborod, professor of medicine at the University of Missouri, Kansas City.
“The truth of the matter is we have no idea what’s causing this low-grade myocardial necrosis, and it’s a hugely important thing,” he continued. “There is absolutely no question that elevated hsTnT, even at very low levels, has a huge impact on subsequent risk of heart failure. We know what the public health effects of heart failure are. And patients with diabetes and heart failure tend to do particularly poorly.”
The BARI 2D trial was funded by the National Institutes of Health. Dr. Everett’s secondary analysis was funded by Roche Diagnostics. He reported receiving research grants from Roche and Novartis.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Forty percent of patients with type 2 diabetes and stable ischemic heart disease are walking around with abnormal levels of hsTnT, placing them at substantial risk of a major cardiovascular event within 5 years.
Major finding: Prompt coronary revascularization does not reduce this risk, nor does it reduce the elevated troponin T level.
Data source: A secondary analysis of the randomized, prospective BARI 2D study involving 2,285 participants with type 2 diabetes and stable ischemic heart disease.
Disclosures: The BARI 2D study was funded by the National Institutes of Health. This new secondary analysis was funded by a research grant from Roche Diagnostics.

Insulin glargine shows cardiac safety in ORIGIN-ECHO
CHICAGO – Insulin glargine showed no effects on left ventricular mass or function during 3 years of follow-up in dysglycemic patients at high cardiovascular risk in the ORIGIN echocardiographic substudy.
This echocardiographic study of the ORIGIN (Outcome Reduction With an Initial Glargine Intervention) trial, the largest reported study of the effects of exogenous insulin on left ventricular mass and LV systolic and diastolic function, provides reassuring new evidence that insulin glargine is safe from a cardiac standpoint, Dr. Michelle Haroun said at the American Heart Association scientific sessions.
“The key here is that we didn’t see any signal whatsoever to suggest that insulin is putting patients at increased risk. We think that this finding is important. While you need to follow patients for a very long time to detect changes in clinical heart failure outcomes, we think we’d be able to detect subtle changes in endpoints like LV mass over a 3-year period if insulin was of harm to patients,” said Dr. Haroun of the Population Health Research Institute at McMaster University in Hamilton, Ont.
ORIGIN-ECHO involved 564 dysglycemic patients at high cardiovascular risk who were randomized to insulin glargine (Lantus) or standard therapy. All had echocardiograms at baseline and after 3 years of therapy. Participants had to have impaired fasting blood glucose, impaired glucose tolerance, or early type 2 diabetes managed with no more than one oral antiglycemic drug at baseline. This was a group at high cardiovascular risk: 32% had a prior MI, 84% had a history of hypertension, obesity was common, and the average age was 64. However, none of the participants had heart failure at baseline.
The study was undertaken because some of the medications used to treat hyperglycemia are associated with increased risk of heart failure. Regulatory agencies, physicians, and patients want to see evidence of cardiovascular safety, and until ORIGIN-ECHO, the effects of exogenous insulin on LV mass and function hadn’t been well studied.
Baseline LV mass and function values were within normal range and did not change significantly over 3 years of follow-up in either treatment arm. For example, left ventricular mass/height averaged 116 g/m at baseline and 115 g/m after 3 years on insulin glargine, and was comparable at 113 and 114 g/m, respectively, with standard therapy. This was an unexpected finding, according to Dr. Haroun.
“We thought patients with diabetes on standard therapy were going to develop left ventricular hypertrophy over a 3-year follow-up period, and they didn’t. That came as a bit of a surprise to us. We expected to see a lower rate of LVH in the patients on insulin glargine. This patient population was relatively early in their course of diabetes, and we believe our findings suggest that adequate management of cardiovascular risk factors – especially hypertension– and the use of cardioprotective drugs in this population may prevent or delay abnormalities in LV structure and function,” she said.
The primary outcomes of the full ORIGIN study involving more than 12,000 patients have previously been published (N. Engl. J. Med. 2012; 367:319-28). ORIGIN was sponsored by Sanofi. Dr. Haroun reported having no financial conflicts.
CHICAGO – Insulin glargine showed no effects on left ventricular mass or function during 3 years of follow-up in dysglycemic patients at high cardiovascular risk in the ORIGIN echocardiographic substudy.
This echocardiographic study of the ORIGIN (Outcome Reduction With an Initial Glargine Intervention) trial, the largest reported study of the effects of exogenous insulin on left ventricular mass and LV systolic and diastolic function, provides reassuring new evidence that insulin glargine is safe from a cardiac standpoint, Dr. Michelle Haroun said at the American Heart Association scientific sessions.
“The key here is that we didn’t see any signal whatsoever to suggest that insulin is putting patients at increased risk. We think that this finding is important. While you need to follow patients for a very long time to detect changes in clinical heart failure outcomes, we think we’d be able to detect subtle changes in endpoints like LV mass over a 3-year period if insulin was of harm to patients,” said Dr. Haroun of the Population Health Research Institute at McMaster University in Hamilton, Ont.
ORIGIN-ECHO involved 564 dysglycemic patients at high cardiovascular risk who were randomized to insulin glargine (Lantus) or standard therapy. All had echocardiograms at baseline and after 3 years of therapy. Participants had to have impaired fasting blood glucose, impaired glucose tolerance, or early type 2 diabetes managed with no more than one oral antiglycemic drug at baseline. This was a group at high cardiovascular risk: 32% had a prior MI, 84% had a history of hypertension, obesity was common, and the average age was 64. However, none of the participants had heart failure at baseline.
The study was undertaken because some of the medications used to treat hyperglycemia are associated with increased risk of heart failure. Regulatory agencies, physicians, and patients want to see evidence of cardiovascular safety, and until ORIGIN-ECHO, the effects of exogenous insulin on LV mass and function hadn’t been well studied.
Baseline LV mass and function values were within normal range and did not change significantly over 3 years of follow-up in either treatment arm. For example, left ventricular mass/height averaged 116 g/m at baseline and 115 g/m after 3 years on insulin glargine, and was comparable at 113 and 114 g/m, respectively, with standard therapy. This was an unexpected finding, according to Dr. Haroun.
“We thought patients with diabetes on standard therapy were going to develop left ventricular hypertrophy over a 3-year follow-up period, and they didn’t. That came as a bit of a surprise to us. We expected to see a lower rate of LVH in the patients on insulin glargine. This patient population was relatively early in their course of diabetes, and we believe our findings suggest that adequate management of cardiovascular risk factors – especially hypertension– and the use of cardioprotective drugs in this population may prevent or delay abnormalities in LV structure and function,” she said.
The primary outcomes of the full ORIGIN study involving more than 12,000 patients have previously been published (N. Engl. J. Med. 2012; 367:319-28). ORIGIN was sponsored by Sanofi. Dr. Haroun reported having no financial conflicts.
CHICAGO – Insulin glargine showed no effects on left ventricular mass or function during 3 years of follow-up in dysglycemic patients at high cardiovascular risk in the ORIGIN echocardiographic substudy.
This echocardiographic study of the ORIGIN (Outcome Reduction With an Initial Glargine Intervention) trial, the largest reported study of the effects of exogenous insulin on left ventricular mass and LV systolic and diastolic function, provides reassuring new evidence that insulin glargine is safe from a cardiac standpoint, Dr. Michelle Haroun said at the American Heart Association scientific sessions.
“The key here is that we didn’t see any signal whatsoever to suggest that insulin is putting patients at increased risk. We think that this finding is important. While you need to follow patients for a very long time to detect changes in clinical heart failure outcomes, we think we’d be able to detect subtle changes in endpoints like LV mass over a 3-year period if insulin was of harm to patients,” said Dr. Haroun of the Population Health Research Institute at McMaster University in Hamilton, Ont.
ORIGIN-ECHO involved 564 dysglycemic patients at high cardiovascular risk who were randomized to insulin glargine (Lantus) or standard therapy. All had echocardiograms at baseline and after 3 years of therapy. Participants had to have impaired fasting blood glucose, impaired glucose tolerance, or early type 2 diabetes managed with no more than one oral antiglycemic drug at baseline. This was a group at high cardiovascular risk: 32% had a prior MI, 84% had a history of hypertension, obesity was common, and the average age was 64. However, none of the participants had heart failure at baseline.
The study was undertaken because some of the medications used to treat hyperglycemia are associated with increased risk of heart failure. Regulatory agencies, physicians, and patients want to see evidence of cardiovascular safety, and until ORIGIN-ECHO, the effects of exogenous insulin on LV mass and function hadn’t been well studied.
Baseline LV mass and function values were within normal range and did not change significantly over 3 years of follow-up in either treatment arm. For example, left ventricular mass/height averaged 116 g/m at baseline and 115 g/m after 3 years on insulin glargine, and was comparable at 113 and 114 g/m, respectively, with standard therapy. This was an unexpected finding, according to Dr. Haroun.
“We thought patients with diabetes on standard therapy were going to develop left ventricular hypertrophy over a 3-year follow-up period, and they didn’t. That came as a bit of a surprise to us. We expected to see a lower rate of LVH in the patients on insulin glargine. This patient population was relatively early in their course of diabetes, and we believe our findings suggest that adequate management of cardiovascular risk factors – especially hypertension– and the use of cardioprotective drugs in this population may prevent or delay abnormalities in LV structure and function,” she said.
The primary outcomes of the full ORIGIN study involving more than 12,000 patients have previously been published (N. Engl. J. Med. 2012; 367:319-28). ORIGIN was sponsored by Sanofi. Dr. Haroun reported having no financial conflicts.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: The cardiac safety of insulin glargine in dysglycemic patients at high cardiovascular risk has received strong support from a 3-year echocardiographic study.
Major finding: Left ventricular mass over height was 116 g/m at baseline and 115 g/m after 3 years on insulin glargine.
Data source: The ORIGIN-ECHO substudy included 564 dysglycemic patients at high cardiovascular risk who were randomized to 3 years of insulin glargine or standard therapy.
Disclosures: The ORIGIN trial was sponsored by Sanofi. The presenter reported having no financial conflicts.