User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
The Growing Pains of Changing Times for Acne and Rosacea Pathophysiology: Where Will It All End Up?
It is interesting to observe the changes in dermatology that have occurred over the last 1 to 2 decades, especially as major advances in basic science research techniques have rapidly expanded our current understanding of the pathophysiology of many disease states—psoriasis, psoriatic arthritis, atopic dermatitis, alopecia areata, vitiligo, hidradenitis suppurativa, and lichen planus.1 Although acne vulgaris (AV) and rosacea do not make front-page news quite as often as some of these other aforementioned disease states in the pathophysiology arena, advances still have been made in understanding the pathophysiology, albeit slower and often less popularized in dermatology publications and other forms of media.2-4
If one looks at our fundamental understanding of AV, most of the discussion over multiple decades has been driven by new treatments and in some cases new formulations and packaging differences with topical agents. Although we understood that adrenarche, a subsequent increase in androgen synthesis, and the ensuing sebocyte development with formation of sebum were prerequisites for the development of AV, the absence of therapeutic options to address these vital components of AV—especially US Food and Drug Administration (FDA)–approved therapies—resulted in limited discussion about this specific area.5 Rather, the discussion was dominated by the notable role of Propionibacterium acnes (now called Cutibacterium acnes) in AV pathophysiology, as we had therapies such as benzoyl peroxide and antibiotics that improved AV in direct correlation with reductions in P acnes.6 This was soon coupled with an advanced understanding of how to reduce follicular hyperkeratinization with the development of topical tretinoin, followed by 3 other topical retinoids over time—adapalene, tazarotene, and trifarotene. Over subsequent years, slowly emerging basic science developments and collective data reviews added to our understanding of AV and how different therapies appear to work, including the role of toll-like receptors, anti-inflammatory properties of tetracyclines, and inflammasomes.7-9 Without a doubt, the availability of oral isotretinoin revolutionized AV therapy, especially in patients with severe refractory disease, with advanced formulations allowing for optimization of sustained remission without the need for high dietary fat intake.10-12
Progress in the pathophysiology of rosacea has been slower to develop, with the first true discussion of specific clinical presentations published after the new millennium.13 This was followed by more advanced basic science and clinical research, which led to an improved ability to understand modes of action of various therapies and to correlate treatment selection with specific visible manifestations of rosacea, including incorporation of physical devices.14-16 A newer perspective on evaluation and management of rosacea moved away from the “buckets” of rosacea subtypes to phenotypes observed at the time of clinical presentation.17,18
I could elaborate on research advancements with both diseases, but the bottom line is that information, developments, and current perspectives change over time. Keeping up is a challenge for all who study and practice dermatology. It is human nature to revert to what we already believe and do, which sometimes remains valid and other times is quite outdated and truly replaced by more optimal approaches. With AV and rosacea, progress is much slower in availability of newer agents. With AV, new agents have included topical dapsone, oral sarecycline, and topical clascoterone, with the latter being the first FDA-approved topical agent to mitigate the effects of androgens and sebum in both males and females. For rosacea, the 2 most recent FDA-approved therapies are minocycline foam and microencapsulated benzoyl peroxide. All of these therapies are proven to be effective for the modes of action and skin manifestations they specifically manage. Over the upcoming year, we are hoping to see the first triple-combination topical product come to market for AV, which will prompt our minds to consider if and how 3 established agents can work together to further augment treatment efficacy with favorable tolerability and safety.
Where will all of this end up? It is hard to say. We still have several other areas to tackle with both disease states, including establishing a well-substantiated understanding of the pathophysiologic role of the microbiome, sorting out the role of antibiotic use due to concerns about bacterial resistance, integration of FDA-approved physical devices in AV, and data on both diet and optimized skin care, to name a few.19-21
There is a lot on the plate to accomplish and digest. I have remained very involved in this subject matter for almost 3 decades and am still feeling the growing pains. Fortunately, the satisfaction of being part of a process so important to the lives of millions of patients makes this worth every moment. Stay tuned—more valuable information is to come.
- Wu J, Fang Z, Liu T, et al. Maximizing the utility of transcriptomics data in inflammatory skin diseases. Front Immunol. 2021;12:761890.
- Firlej E, Kowalska W, Szymaszek K, et al. The role of skin immune system in acne. J Clin Med. 2022;11:1579.
- Mias C, Mengeaud V, Bessou-Touya S, et al. Recent advances in understanding inflammatory acne: deciphering the relationship between Cutibacterium acnes and Th17 inflammatory pathway. J Eur Acad Dermatol Venereol. 2023;(37 suppl 2):3-11.
- Buddenkotte J, Steinhoff M. Recent advances in understanding and managing rosacea. F1000Res. 2018;7:F1000 Faculty Rev-1885. doi:10.12688/f1000research.16537.1
- Platsidaki E, Dessinioti C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Res. 2018;7:F1000 Faculty Rev-1953. doi:10.12688/f1000research.15659.1
- Leyden JJ. The evolving role of Propionibacterium acnes in acne. Semin Cutan Med Surg. 2001;20:139-143.
- Kim J. Review of the innate immune response in acne vulgaris: activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. Dermatology. 2005;211:193-198.
- Del Rosso JQ, Webster G, Weiss JS, et al. Nonantibiotic properties of tetracyclines in rosacea and their clinical implications. J Clin Aesthet Dermatol. 2021;14:14-21.
- Zhu W, Wang HL, Bu XL, et al. A narrative review of research progress on the role of NLRP3 inflammasome in acne vulgaris. Ann Transl Med. 2022;10:645.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(2 suppl):S3-S21.
- Webster GF, Leyden JJ, Gross JA. Comparative pharmacokinetic profiles of a novel isotretinoin formulation (isotretinoin-Lidose) and the innovator isotretinoin formulation: a randomized, treatment, crossover study. J Am Acad Dermatol. 2013;69:762-767.
- Del Rosso JQ, Stein Gold L, Seagal J, et al. An open-label, phase IV study evaluating Lidose-isotretinoin administered without food in patients with severe recalcitrant nodular acne: low relapse rates observed over the 104-week post-treatment period. J Clin Aesthet Dermatol. 2019;12:13-18.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Steinhoff M, Buddenkotte J, Aubert J, et al. Clinical, cellular, and molecular aspects in the pathophysiology of rosacea. J Investig Dermatol Symp Proc. 2011;15:2-11.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Tanghetti E, Del Rosso JQ, Thiboutot D, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 4: a status report on physical modalities and devices. Cutis. 2014;93:71-76.
- Del Rosso JQ, Gallo RL, Tanghetti E, et al. An evaluation of potential correlations between pathophysiologic mechanisms, clinical manifestations, and management of rosacea. Cutis. 2013;91(3 suppl):1-8.
- Schaller M, Almeida LMC, Bewley A, et al. Recommendations for rosacea diagnosis, classification and management: update from the global ROSacea COnsensus 2019 panel. Br J Dermatol. 2020;182:1269-1276.
- Xu H, Li H. Acne, the skin microbiome, and antibiotic treatment. Am J Clin Dermatol. 2019;20:335-344.
- Daou H, Paradiso M, Hennessy K. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Kayiran MA, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.
It is interesting to observe the changes in dermatology that have occurred over the last 1 to 2 decades, especially as major advances in basic science research techniques have rapidly expanded our current understanding of the pathophysiology of many disease states—psoriasis, psoriatic arthritis, atopic dermatitis, alopecia areata, vitiligo, hidradenitis suppurativa, and lichen planus.1 Although acne vulgaris (AV) and rosacea do not make front-page news quite as often as some of these other aforementioned disease states in the pathophysiology arena, advances still have been made in understanding the pathophysiology, albeit slower and often less popularized in dermatology publications and other forms of media.2-4
If one looks at our fundamental understanding of AV, most of the discussion over multiple decades has been driven by new treatments and in some cases new formulations and packaging differences with topical agents. Although we understood that adrenarche, a subsequent increase in androgen synthesis, and the ensuing sebocyte development with formation of sebum were prerequisites for the development of AV, the absence of therapeutic options to address these vital components of AV—especially US Food and Drug Administration (FDA)–approved therapies—resulted in limited discussion about this specific area.5 Rather, the discussion was dominated by the notable role of Propionibacterium acnes (now called Cutibacterium acnes) in AV pathophysiology, as we had therapies such as benzoyl peroxide and antibiotics that improved AV in direct correlation with reductions in P acnes.6 This was soon coupled with an advanced understanding of how to reduce follicular hyperkeratinization with the development of topical tretinoin, followed by 3 other topical retinoids over time—adapalene, tazarotene, and trifarotene. Over subsequent years, slowly emerging basic science developments and collective data reviews added to our understanding of AV and how different therapies appear to work, including the role of toll-like receptors, anti-inflammatory properties of tetracyclines, and inflammasomes.7-9 Without a doubt, the availability of oral isotretinoin revolutionized AV therapy, especially in patients with severe refractory disease, with advanced formulations allowing for optimization of sustained remission without the need for high dietary fat intake.10-12
Progress in the pathophysiology of rosacea has been slower to develop, with the first true discussion of specific clinical presentations published after the new millennium.13 This was followed by more advanced basic science and clinical research, which led to an improved ability to understand modes of action of various therapies and to correlate treatment selection with specific visible manifestations of rosacea, including incorporation of physical devices.14-16 A newer perspective on evaluation and management of rosacea moved away from the “buckets” of rosacea subtypes to phenotypes observed at the time of clinical presentation.17,18
I could elaborate on research advancements with both diseases, but the bottom line is that information, developments, and current perspectives change over time. Keeping up is a challenge for all who study and practice dermatology. It is human nature to revert to what we already believe and do, which sometimes remains valid and other times is quite outdated and truly replaced by more optimal approaches. With AV and rosacea, progress is much slower in availability of newer agents. With AV, new agents have included topical dapsone, oral sarecycline, and topical clascoterone, with the latter being the first FDA-approved topical agent to mitigate the effects of androgens and sebum in both males and females. For rosacea, the 2 most recent FDA-approved therapies are minocycline foam and microencapsulated benzoyl peroxide. All of these therapies are proven to be effective for the modes of action and skin manifestations they specifically manage. Over the upcoming year, we are hoping to see the first triple-combination topical product come to market for AV, which will prompt our minds to consider if and how 3 established agents can work together to further augment treatment efficacy with favorable tolerability and safety.
Where will all of this end up? It is hard to say. We still have several other areas to tackle with both disease states, including establishing a well-substantiated understanding of the pathophysiologic role of the microbiome, sorting out the role of antibiotic use due to concerns about bacterial resistance, integration of FDA-approved physical devices in AV, and data on both diet and optimized skin care, to name a few.19-21
There is a lot on the plate to accomplish and digest. I have remained very involved in this subject matter for almost 3 decades and am still feeling the growing pains. Fortunately, the satisfaction of being part of a process so important to the lives of millions of patients makes this worth every moment. Stay tuned—more valuable information is to come.
It is interesting to observe the changes in dermatology that have occurred over the last 1 to 2 decades, especially as major advances in basic science research techniques have rapidly expanded our current understanding of the pathophysiology of many disease states—psoriasis, psoriatic arthritis, atopic dermatitis, alopecia areata, vitiligo, hidradenitis suppurativa, and lichen planus.1 Although acne vulgaris (AV) and rosacea do not make front-page news quite as often as some of these other aforementioned disease states in the pathophysiology arena, advances still have been made in understanding the pathophysiology, albeit slower and often less popularized in dermatology publications and other forms of media.2-4
If one looks at our fundamental understanding of AV, most of the discussion over multiple decades has been driven by new treatments and in some cases new formulations and packaging differences with topical agents. Although we understood that adrenarche, a subsequent increase in androgen synthesis, and the ensuing sebocyte development with formation of sebum were prerequisites for the development of AV, the absence of therapeutic options to address these vital components of AV—especially US Food and Drug Administration (FDA)–approved therapies—resulted in limited discussion about this specific area.5 Rather, the discussion was dominated by the notable role of Propionibacterium acnes (now called Cutibacterium acnes) in AV pathophysiology, as we had therapies such as benzoyl peroxide and antibiotics that improved AV in direct correlation with reductions in P acnes.6 This was soon coupled with an advanced understanding of how to reduce follicular hyperkeratinization with the development of topical tretinoin, followed by 3 other topical retinoids over time—adapalene, tazarotene, and trifarotene. Over subsequent years, slowly emerging basic science developments and collective data reviews added to our understanding of AV and how different therapies appear to work, including the role of toll-like receptors, anti-inflammatory properties of tetracyclines, and inflammasomes.7-9 Without a doubt, the availability of oral isotretinoin revolutionized AV therapy, especially in patients with severe refractory disease, with advanced formulations allowing for optimization of sustained remission without the need for high dietary fat intake.10-12
Progress in the pathophysiology of rosacea has been slower to develop, with the first true discussion of specific clinical presentations published after the new millennium.13 This was followed by more advanced basic science and clinical research, which led to an improved ability to understand modes of action of various therapies and to correlate treatment selection with specific visible manifestations of rosacea, including incorporation of physical devices.14-16 A newer perspective on evaluation and management of rosacea moved away from the “buckets” of rosacea subtypes to phenotypes observed at the time of clinical presentation.17,18
I could elaborate on research advancements with both diseases, but the bottom line is that information, developments, and current perspectives change over time. Keeping up is a challenge for all who study and practice dermatology. It is human nature to revert to what we already believe and do, which sometimes remains valid and other times is quite outdated and truly replaced by more optimal approaches. With AV and rosacea, progress is much slower in availability of newer agents. With AV, new agents have included topical dapsone, oral sarecycline, and topical clascoterone, with the latter being the first FDA-approved topical agent to mitigate the effects of androgens and sebum in both males and females. For rosacea, the 2 most recent FDA-approved therapies are minocycline foam and microencapsulated benzoyl peroxide. All of these therapies are proven to be effective for the modes of action and skin manifestations they specifically manage. Over the upcoming year, we are hoping to see the first triple-combination topical product come to market for AV, which will prompt our minds to consider if and how 3 established agents can work together to further augment treatment efficacy with favorable tolerability and safety.
Where will all of this end up? It is hard to say. We still have several other areas to tackle with both disease states, including establishing a well-substantiated understanding of the pathophysiologic role of the microbiome, sorting out the role of antibiotic use due to concerns about bacterial resistance, integration of FDA-approved physical devices in AV, and data on both diet and optimized skin care, to name a few.19-21
There is a lot on the plate to accomplish and digest. I have remained very involved in this subject matter for almost 3 decades and am still feeling the growing pains. Fortunately, the satisfaction of being part of a process so important to the lives of millions of patients makes this worth every moment. Stay tuned—more valuable information is to come.
- Wu J, Fang Z, Liu T, et al. Maximizing the utility of transcriptomics data in inflammatory skin diseases. Front Immunol. 2021;12:761890.
- Firlej E, Kowalska W, Szymaszek K, et al. The role of skin immune system in acne. J Clin Med. 2022;11:1579.
- Mias C, Mengeaud V, Bessou-Touya S, et al. Recent advances in understanding inflammatory acne: deciphering the relationship between Cutibacterium acnes and Th17 inflammatory pathway. J Eur Acad Dermatol Venereol. 2023;(37 suppl 2):3-11.
- Buddenkotte J, Steinhoff M. Recent advances in understanding and managing rosacea. F1000Res. 2018;7:F1000 Faculty Rev-1885. doi:10.12688/f1000research.16537.1
- Platsidaki E, Dessinioti C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Res. 2018;7:F1000 Faculty Rev-1953. doi:10.12688/f1000research.15659.1
- Leyden JJ. The evolving role of Propionibacterium acnes in acne. Semin Cutan Med Surg. 2001;20:139-143.
- Kim J. Review of the innate immune response in acne vulgaris: activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. Dermatology. 2005;211:193-198.
- Del Rosso JQ, Webster G, Weiss JS, et al. Nonantibiotic properties of tetracyclines in rosacea and their clinical implications. J Clin Aesthet Dermatol. 2021;14:14-21.
- Zhu W, Wang HL, Bu XL, et al. A narrative review of research progress on the role of NLRP3 inflammasome in acne vulgaris. Ann Transl Med. 2022;10:645.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(2 suppl):S3-S21.
- Webster GF, Leyden JJ, Gross JA. Comparative pharmacokinetic profiles of a novel isotretinoin formulation (isotretinoin-Lidose) and the innovator isotretinoin formulation: a randomized, treatment, crossover study. J Am Acad Dermatol. 2013;69:762-767.
- Del Rosso JQ, Stein Gold L, Seagal J, et al. An open-label, phase IV study evaluating Lidose-isotretinoin administered without food in patients with severe recalcitrant nodular acne: low relapse rates observed over the 104-week post-treatment period. J Clin Aesthet Dermatol. 2019;12:13-18.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Steinhoff M, Buddenkotte J, Aubert J, et al. Clinical, cellular, and molecular aspects in the pathophysiology of rosacea. J Investig Dermatol Symp Proc. 2011;15:2-11.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Tanghetti E, Del Rosso JQ, Thiboutot D, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 4: a status report on physical modalities and devices. Cutis. 2014;93:71-76.
- Del Rosso JQ, Gallo RL, Tanghetti E, et al. An evaluation of potential correlations between pathophysiologic mechanisms, clinical manifestations, and management of rosacea. Cutis. 2013;91(3 suppl):1-8.
- Schaller M, Almeida LMC, Bewley A, et al. Recommendations for rosacea diagnosis, classification and management: update from the global ROSacea COnsensus 2019 panel. Br J Dermatol. 2020;182:1269-1276.
- Xu H, Li H. Acne, the skin microbiome, and antibiotic treatment. Am J Clin Dermatol. 2019;20:335-344.
- Daou H, Paradiso M, Hennessy K. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Kayiran MA, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.
- Wu J, Fang Z, Liu T, et al. Maximizing the utility of transcriptomics data in inflammatory skin diseases. Front Immunol. 2021;12:761890.
- Firlej E, Kowalska W, Szymaszek K, et al. The role of skin immune system in acne. J Clin Med. 2022;11:1579.
- Mias C, Mengeaud V, Bessou-Touya S, et al. Recent advances in understanding inflammatory acne: deciphering the relationship between Cutibacterium acnes and Th17 inflammatory pathway. J Eur Acad Dermatol Venereol. 2023;(37 suppl 2):3-11.
- Buddenkotte J, Steinhoff M. Recent advances in understanding and managing rosacea. F1000Res. 2018;7:F1000 Faculty Rev-1885. doi:10.12688/f1000research.16537.1
- Platsidaki E, Dessinioti C. Recent advances in understanding Propionibacterium acnes (Cutibacterium acnes) in acne. F1000Res. 2018;7:F1000 Faculty Rev-1953. doi:10.12688/f1000research.15659.1
- Leyden JJ. The evolving role of Propionibacterium acnes in acne. Semin Cutan Med Surg. 2001;20:139-143.
- Kim J. Review of the innate immune response in acne vulgaris: activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. Dermatology. 2005;211:193-198.
- Del Rosso JQ, Webster G, Weiss JS, et al. Nonantibiotic properties of tetracyclines in rosacea and their clinical implications. J Clin Aesthet Dermatol. 2021;14:14-21.
- Zhu W, Wang HL, Bu XL, et al. A narrative review of research progress on the role of NLRP3 inflammasome in acne vulgaris. Ann Transl Med. 2022;10:645.
- Leyden JJ, Del Rosso JQ, Baum EW. The use of isotretinoin in the treatment of acne vulgaris: clinical considerations and future directions. J Clin Aesthet Dermatol. 2014;7(2 suppl):S3-S21.
- Webster GF, Leyden JJ, Gross JA. Comparative pharmacokinetic profiles of a novel isotretinoin formulation (isotretinoin-Lidose) and the innovator isotretinoin formulation: a randomized, treatment, crossover study. J Am Acad Dermatol. 2013;69:762-767.
- Del Rosso JQ, Stein Gold L, Seagal J, et al. An open-label, phase IV study evaluating Lidose-isotretinoin administered without food in patients with severe recalcitrant nodular acne: low relapse rates observed over the 104-week post-treatment period. J Clin Aesthet Dermatol. 2019;12:13-18.
- Wilkin J, Dahl M, Detmar M, et al. Standard classification of rosacea: report of the National Rosacea Society Expert Committee on the classification and staging of rosacea. J Am Acad Dermatol. 2002;46:584-587.
- Steinhoff M, Buddenkotte J, Aubert J, et al. Clinical, cellular, and molecular aspects in the pathophysiology of rosacea. J Investig Dermatol Symp Proc. 2011;15:2-11.
- Yamasaki K, Gallo RL. The molecular pathology of rosacea. J Dermatol Sci. 2009;55:77-81.
- Tanghetti E, Del Rosso JQ, Thiboutot D, et al. Consensus recommendations from the American Acne & Rosacea Society on the management of rosacea, part 4: a status report on physical modalities and devices. Cutis. 2014;93:71-76.
- Del Rosso JQ, Gallo RL, Tanghetti E, et al. An evaluation of potential correlations between pathophysiologic mechanisms, clinical manifestations, and management of rosacea. Cutis. 2013;91(3 suppl):1-8.
- Schaller M, Almeida LMC, Bewley A, et al. Recommendations for rosacea diagnosis, classification and management: update from the global ROSacea COnsensus 2019 panel. Br J Dermatol. 2020;182:1269-1276.
- Xu H, Li H. Acne, the skin microbiome, and antibiotic treatment. Am J Clin Dermatol. 2019;20:335-344.
- Daou H, Paradiso M, Hennessy K. Rosacea and the microbiome: a systematic review. Dermatol Ther (Heidelb). 2021;11:1-12.
- Kayiran MA, Karadag AS, Al-Khuzaei S, et al. Antibiotic resistance in acne: mechanisms, complications and management. Am J Clin Dermatol. 2020;21:813-819.

Ulcerated Nodule on the Lip
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.

Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
.")
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.
Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
The Diagnosis: Cutaneous Metastasis
A shave biopsy of the lip revealed a diffuse cellular infiltrate filling the superficial and deep dermis (Figure 1A). Morphologically, the cells had abundant clear cytoplasm with eccentrically located, pleomorphic, hyperchromatic nuclei with occasional prominent nucleoli (Figure 1B). The cells stained positive for AE1/ AE3 on immunohistochemistry (Figure 2). A punch biopsy of the nodule in the right axillary vault revealed a morphologically similar proliferation of cells. A colonoscopy revealed a completely obstructing circumferential mass in the distal ascending colon. A biopsy of the mass confirmed invasive adenocarcinoma, supporting a diagnosis of cutaneous metastases from adenocarcinoma of the colon. The patient underwent resection of the lip tumor and started multiagent chemotherapy for his newly diagnosed stage IV adenocarcinoma of the colon. The patient died, despite therapy.
Cutaneous metastasis from solid malignancies is uncommon, as only 1.3% of them exhibit cutaneous manifestations at presentation.1 Cutaneous metastasis from signet ring cell adenocarcinoma (SRCA) of the colon is uncommon, and cutaneous metastasis of colorectal SRCA rarely precedes the diagnosis of the primary lesion.2 Among the colorectal cancers that metastasize to the skin, metastasis to the face occurs in only 0.5% of patients.3
Signet ring cell adenocarcinomas are poorly differentiated adenocarcinomas histologically characterized by the neoplastic cells’ circular to ovoid appearance with a flattened top.4,5 This distinctive shape is from the displacement of the nucleus to the periphery of the cell due to the accumulation of intracytoplasmic mucin.4 Classically, malignancies are characterized as an SRCA if more than 50% of the cells have a signet ring cell morphology; if the signet ring cells comprise less than 50% of the neoplasm, the tumor is designated as an adenocarcinoma with signet ring morphology.4 The most common cause of cutaneous metastasis with signet ring morphology is gastric cancer, while colorectal carcinoma is less common.1 Colorectal SRCAs usually are found in the right colon or the rectum in comparison to other colonic sites.6
Clinically, cutaneous metastasis can present in a variety of ways. The most common presentation is nodular lesions that may coalesce to become zosteriform in configuration or lesions that mimic inflammatory dermatoses.7 Cutaneous metastasis is more common in breast and lung cancer, and when it occurs secondary to colorectal cancer, cutaneous metastasis rarely predates the detection of the primary neoplasm.2
The clinical appearance of metastasis is not specific and can mimic many entities8; therefore, a high index of suspicion must be employed when managing patients, even those without a history of internal malignancy. In our patient, the smooth nodular lesion appeared similar to a basal cell carcinoma; however, basal cell carcinomas appear more pearly, and arborizing telangiectasia often is seen.9 Merkel cell carcinoma is common on sundamaged skin of the head and neck but clinically appears more violaceous than the lesion seen in our patient.10 Paracoccidioidomycosis may form ulcerated papulonodules or plaques, especially around the nose and mouth. In many of these cases, lesions develop from contiguous lesions of the oral mucosa; therefore, the presence of oral lesions will help distinguish this infectious entity from cutaneous metastasis. Multiple lesions usually are identified when there is hematogenous dissemination.11 Mycosis fungoides is a subtype of cutaneous T-cell lymphoma and is characterized by multiple patches, plaques, and nodules on sun-protected areas. Involvement of the head and neck is not common, except in the folliculotropic subtype, which has a separate and distinct clinical morphology.12
The development of signet ring morphology from an adenocarcinoma can be attributed to the activation of phosphatidylinositol 3-kinase (PI3K), which leads to downstream activation of mitogen-activated protein kinase (MAPK) and the subsequent loss of intercellular tight junctions. The mucin 4 gene, MUC4, also is upregulated by PI3K activation and possesses antiapoptotic and mitogenic effects in addition to its mucin secretory function.13
The neoplastic cells in SRCAs stain positive for mucicarmine, Alcian blue, and periodic acid–Schiff, which highlights the mucinous component of the cells.7 Immunohistochemical stains with CK7, CK20, AE1/AE3, and epithelial membrane antigen can be implemented to confirm an epithelial origin of the primary cancer.7,13 CK20 is a low-molecular-weight cytokeratin normally expressed by Merkel cells and by the epithelium of the gastrointestinal tract and urothelium, whereas CK7 expression typically is expressed in the lungs, ovaries, endometrium, and breasts, but not in the lower gastrointestinal tract.14 Differentiating primary cutaneous adenocarcinoma from cutaneous metastasis can be accomplished with a thorough clinical history; however, p63 positivity supports a primary cutaneous lesion and may be useful in certain situations.7 CDX2 stains can be utilized to aid in localizing the primary neoplasm when it is unknown, and when positive, it suggests a lower gastrointestinal tract origin. However, special AT-rich sequence-binding protein 2 (SATB2) recently has been proposed as a replacement immunohistochemical marker for CDX2, as it has greater specificity for SRCA of the lower gastrointestinal tract.15 Benign entities with signet ring cell morphology are difficult to distinguish from SRCA; however, malignant lesions are more likely to demonstrate an infiltrative growth pattern, frequent mitotic figures, and apoptosis. Immunohistochemistry also can be utilized to support the diagnosis of benign proliferation with signet ring morphology, as benign lesions often will demonstrate E-cadherin positivity and negativity for p53 and Ki-67.13
Cutaneous metastasis usually correlates to advanced disease and generally indicates a worse prognosis.13 Signet ring cell morphology in both gastric and colorectal cancer portends a poor prognosis, and there is a lower overall survival in patients with these malignancies compared to cancers of the same organ with non–signet ring cell morphology.4,8
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
- Mandzhieva B, Jalil A, Nadeem M, et al. Most common pathway of metastasis of rectal signet ring cell carcinoma to the skin: hematogenous. Cureus. 2020;12:E6890.
- Parente P, Ciardiello D, Reggiani Bonetti L, et al. Cutaneous metastasis from colorectal cancer: making light on an unusual and misdiagnosed event. Life. 2021;11:954.
- Picciariello A, Tomasicchio G, Lantone G, et al. Synchronous “skip” facial metastases from colorectal adenocarcinoma: a case report and review of literature. BMC Gastroenterol. 2022;22:68.
- Benesch MGK, Mathieson A. Epidemiology of signet ring cell adenocarcinomas. Cancers. 2020;12:1544.
- Xu Q, Karouji Y, Kobayashi M, et al. The PI 3-kinase-Rac-p38 MAP kinase pathway is involved in the formation of signet-ring cell carcinoma. Oncogene. 2003;22:5537-5544.
- Morales-Cruz M, Salgado-Nesme N, Trolle-Silva AM, et al. Signet ring cell carcinoma of the rectum: atypical metastatic presentation. BMJ Case Rep CP. 2019;12:E229135.
- Demirciog˘lu D, Öztürk Durmaz E, Demirkesen C, et al. Livedoid cutaneous metastasis of signet‐ring cell gastric carcinoma. J Cutan Pathol. 2021;48:785-788.
- Dong X, Sun G, Qu H, et al. Prognostic significance of signet-ring cell components in patients with gastric carcinoma of different stages. Front Surg. 2021;8:642468.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Nguyen AH, Tahseen AI, Vaudreuil AM, et al. Clinical features and treatment of vulvar Merkel cell carcinoma: a systematic review. Gynecol Oncol Res Pract. 2017;4:2.
- Marques, SA. Paracoccidioidomycosis. Clin Dermatol. 2012;30:610-615.
- Larocca C, Kupper T. Mycosis fungoides and Sézary syndrome. Hematol Oncol Clin. 2019;33:103-120.
- Gündüz Ö, Emeksiz MC, Atasoy P, et al. Signet-ring cells in the skin: a case of late-onset cutaneous metastasis of gastric carcinoma and a brief review of histological approach. Dermatol Rep. 2017;8:6819.
- Al-Taee A, Almukhtar R, Lai J, et al. Metastatic signet ring cell carcinoma of unknown primary origin: a case report and review of the literature. Ann Transl Med. 2016;4:283.
- Ma C, Lowenthal BM, Pai RK. SATB2 is superior to CDX2 in distinguishing signet ring cell carcinoma of the upper gastrointestinal tract and lower gastrointestinal tract. Am J Surg Pathol. 2018; 42:1715-1722.
A 79-year-old man with a medical history of type 2 diabetes mellitus, hypothyroidism, and atrial fibrillation presented with an enlarging lesion on the right side of the upper cutaneous lip of 5 weeks’ duration. He had no personal history of skin cancer or other malignancy and was up to date on all routine cancer screenings. He reported associated lip and oral cavity tenderness, weakness, and a 13.6-kg (30-lb) unintentional weight loss over the last 6 months. He had used over-the-counter bacitracin ointment on the lesion without relief. A full-body skin examination revealed a firm, mobile, flesh-colored, nondraining nodule in the right axillary vault.


Eccrine Porocarcinoma in 2 Patients
To the Editor:
Porocarcinoma is a rare malignancy of the eccrine sweat glands and is commonly misdiagnosed clinically. We present 2 cases of porocarcinoma and highlight key features of this uncommon disease.

A 65-year-old man presented to the emergency department with a chief concern of a bump on the head of 8 months' duration that gradually enlarged. The lesion recently became painful and contributed to frequent headaches. He reported a history of smoking 1 pack per day and denied trauma to the area or history of immunosuppression. He had no personal or family history of skin cancer. Physical examination revealed a 1.4-cm, heterochromic, pedunculated, keratotic tumor with crusting on the right temporal scalp (Figure 1). No lymphadenopathy was appreciated. The clinical differential diagnosis included irritated seborrheic keratosis, pyogenic granuloma, polypoid malignant melanoma, and nonmelanoma skin cancer. A biopsy of the lesion demonstrated a proliferation of cuboidal cells with focal ductular differentiation arranged in interanastamosing strands arising from the epidermis (Figure 2). Scattered mitotic figures, including atypical forms, cytologic atypia, and foci of necrosis, also were present. The findings were consistent with features of porocarcinoma. Contrast computed tomography of the neck showed no evidence of metastatic disease within the neck. A wide local excision was performed and yielded a tumor measuring 1.8×1.6×0.7 cm with a depth of 0.3 cm and uninvolved margins. No lymphovascular or perineural invasion was identified. At 4-month follow-up, the patient had a well-healed scar on the right scalp without evidence of recurrence or lymphadenopathy.
.")
A 32-year-old woman presented to dermatology with a chief concern of a mass on the back of 2 years’ duration that rapidly enlarged and became painful following irritation from her bra strap 2 months earlier. She had no relevant medical history. Physical examination revealed a firm, tender, heterochromic nodule measuring 3.0×2.8 cm on the left mid back inferior to the left scapula (Figure 3). The lesion expressed serosanguineous discharge. No lymphadenopathy was appreciated on examination. The clinical differential diagnosis included an inflamed cyst, nodular melanoma, cutaneous metastasis, and nonmelanoma skin cancer. The patient underwent an excisional biopsy, which demonstrated porocarcinoma with positive margins, microsatellitosis, and evidence of lymphovascular invasion. Carcinoembryonic antigen immunohistochemistry highlighted ducts within the tumor (Figure 4). The patient underwent re-excision with 2-cm margins, and no residual tumor was appreciated on pathology.

Positron emission tomography and computed tomography revealed a hypermetabolic left axillary lymph node. Ultrasound-guided fine-needle aspiration was positive for malignant cells consistent with metastatic carcinoma. Dissection of left axillary lymph nodes yielded metastatic porocarcinoma in 2 of 13 nodes. The largest tumor deposit measured 0.9 cm, and no extracapsular extension was identified. The patient continues to be monitored with semiannual full-body skin examinations as well as positron emission tomography and computed tomography scans, with no evidence of recurrence 2 years later.
.")
Porocarcinoma is a rare malignancy of the skin arising from the eccrine sweat glands1 with an incidence rate of 0.4 cases per 1 million person-years in the United States. These tumors represent 0.005% to 0.01% of all skin cancers.2 The mean age of onset is approximately 65 years with no predilection for sex. The mean time from initial presentation to treatment is 5.6 to 8.5 years.3-5
Eccrine sweat glands consist of a straight intradermal duct (syrinx); coiled intradermal duct; and spiral intraepidermal duct (acrosyringium), which opens onto the skin. Both eccrine poromas (solitary benign eccrine gland tumors) and eccrine porocarcinomas develop from the acrosyringium. Eccrine poromas most commonly are found in sites containing the highest density of eccrine glands such as the palms, soles, axillae, and forehead, whereas porocarcinomas most commonly are found on the head, neck, arms, and legs.1,3,4,6,7 A solitary painless nodule that may ulcerate or bleed is the most common presentation.1,3-5,7
The etiology of eccrine porocarcinoma is poorly understood, but it has been found to arise de novo or to develop from pre-existing poromas or even from nevus sebaceus of Jadassohn. Chronic sunlight exposure, irradiation, lymphedema, trauma, and immunosuppression (eg, Hodgkin disease, chronic lymphocytic leukemia, HIV) have been reported as potential predisposing factors.3,4,6,8,9
Eccrine porocarcinoma often is clinically misdiagnosed as nonmelanoma skin cancer, pyogenic granuloma, amelanotic melanoma, fibroma, verruca vulgaris, or metastatic carcinoma. Appropriate classification is essential, as metastasis is present in 25% to 31% of cases, and local recurrence occurs in 20% to 25% of cases.1,3-5,7
Microscopically, porocarcinomas are comprised of atypical basaloid epithelial cells with focal ductular differentiation. Typically, there is an extensive intraepidermal component that invades into the dermis in anastomosing ribbons and cords. The degree of nuclear atypia, mitotic activity, and invasive growth pattern, as well as the presence of necrosis, are useful histologic features to differentiate porocarcinoma from poroma, which may be present in the background. Given the sometimes-extensive squamous differentiation, porocarcinoma can be confused with squamous cell carcinoma. In these cases, immunohistochemical stains such as epithelial membrane antigen or carcinoembryonic antigen can be used to highlight the ductal differentiation.1,5,8,10
Poor histologic prognostic indicators include a high mitotic index (>14 mitoses per field), a tumor depth greater than 7 mm, and evidence of lymphovascular invasion. Positive lymph node involvement is associated with a 65% to 67% mortality rate.1,8
Because of its propensity to metastasize via the lymphatic system and the high mortality rate associated with such metastases, early identification and treatment are essential. Treatment is accomplished via Mohs micrographic surgery or wide local excision with negative margins. Lymphadenectomy is indicated if regional lymph nodes are involved. Radiation and chemotherapy have been used in patients with metastatic and recurrent disease with mixed results.1,3-5,7 There is no adequate standardized chemotherapy or drug regimen established for porocarcinoma.5 Tsunoda et al11 proposed that sentinel lymph node biopsy should be considered first-line management of eccrine porocarcinoma; however, this remains unproven on the basis of a limited case series. Others conclude that sentinel lymph node biopsy should be recommended for cases with poor histologic prognostic features.1,5
- Marone U, Caraco C, Anniciello AM, et al. Metastatic eccrine porocarcinoma: report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Salih AM, Kakamad FH, Baba HO, et al. Porocarcinoma; presentation and management, a meta-analysis of 453 cases. Ann Med Surg (Lond). 2017;20:74-79.
- Ritter AM, Graham RS, Amaker B, et al. Intracranial extension of an eccrine porocarcinoma. case report and review of the literature. J Neurosurg. 1999;90:138-140.
- Khaja M, Ashraf U, Mehershahi S, et al. Recurrent metastatic eccrine porocarcinoma: a case report and review of the literature. Am J Case Rep. 2019;20:179-183.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Lloyd MS, El-Muttardi N, Robson A. Eccrine porocarcinoma: a case report and review of the literature. Can J Plast Surg. 2003;11:153-156.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tarkhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413‐415.
- Prieto VG, Shea CR, Celebi JK, et al. Adnexal tumors. In: Busam KJ. Dermatopathology: A Volume in the Foundations in Diagnostic Pathology Series. 2nd ed. Elsevier; 2016:388-446.
- Tsunoda K, Onishi M, Maeda F, et al. Evaluation of sentinel lymph node biopsy for eccrine porocarcinoma. Acta Derm Venereol. 2019;99:691-692.
To the Editor:
Porocarcinoma is a rare malignancy of the eccrine sweat glands and is commonly misdiagnosed clinically. We present 2 cases of porocarcinoma and highlight key features of this uncommon disease.
A 65-year-old man presented to the emergency department with a chief concern of a bump on the head of 8 months' duration that gradually enlarged. The lesion recently became painful and contributed to frequent headaches. He reported a history of smoking 1 pack per day and denied trauma to the area or history of immunosuppression. He had no personal or family history of skin cancer. Physical examination revealed a 1.4-cm, heterochromic, pedunculated, keratotic tumor with crusting on the right temporal scalp (Figure 1). No lymphadenopathy was appreciated. The clinical differential diagnosis included irritated seborrheic keratosis, pyogenic granuloma, polypoid malignant melanoma, and nonmelanoma skin cancer. A biopsy of the lesion demonstrated a proliferation of cuboidal cells with focal ductular differentiation arranged in interanastamosing strands arising from the epidermis (Figure 2). Scattered mitotic figures, including atypical forms, cytologic atypia, and foci of necrosis, also were present. The findings were consistent with features of porocarcinoma. Contrast computed tomography of the neck showed no evidence of metastatic disease within the neck. A wide local excision was performed and yielded a tumor measuring 1.8×1.6×0.7 cm with a depth of 0.3 cm and uninvolved margins. No lymphovascular or perineural invasion was identified. At 4-month follow-up, the patient had a well-healed scar on the right scalp without evidence of recurrence or lymphadenopathy.
A 32-year-old woman presented to dermatology with a chief concern of a mass on the back of 2 years’ duration that rapidly enlarged and became painful following irritation from her bra strap 2 months earlier. She had no relevant medical history. Physical examination revealed a firm, tender, heterochromic nodule measuring 3.0×2.8 cm on the left mid back inferior to the left scapula (Figure 3). The lesion expressed serosanguineous discharge. No lymphadenopathy was appreciated on examination. The clinical differential diagnosis included an inflamed cyst, nodular melanoma, cutaneous metastasis, and nonmelanoma skin cancer. The patient underwent an excisional biopsy, which demonstrated porocarcinoma with positive margins, microsatellitosis, and evidence of lymphovascular invasion. Carcinoembryonic antigen immunohistochemistry highlighted ducts within the tumor (Figure 4). The patient underwent re-excision with 2-cm margins, and no residual tumor was appreciated on pathology.
Positron emission tomography and computed tomography revealed a hypermetabolic left axillary lymph node. Ultrasound-guided fine-needle aspiration was positive for malignant cells consistent with metastatic carcinoma. Dissection of left axillary lymph nodes yielded metastatic porocarcinoma in 2 of 13 nodes. The largest tumor deposit measured 0.9 cm, and no extracapsular extension was identified. The patient continues to be monitored with semiannual full-body skin examinations as well as positron emission tomography and computed tomography scans, with no evidence of recurrence 2 years later.
Porocarcinoma is a rare malignancy of the skin arising from the eccrine sweat glands1 with an incidence rate of 0.4 cases per 1 million person-years in the United States. These tumors represent 0.005% to 0.01% of all skin cancers.2 The mean age of onset is approximately 65 years with no predilection for sex. The mean time from initial presentation to treatment is 5.6 to 8.5 years.3-5
Eccrine sweat glands consist of a straight intradermal duct (syrinx); coiled intradermal duct; and spiral intraepidermal duct (acrosyringium), which opens onto the skin. Both eccrine poromas (solitary benign eccrine gland tumors) and eccrine porocarcinomas develop from the acrosyringium. Eccrine poromas most commonly are found in sites containing the highest density of eccrine glands such as the palms, soles, axillae, and forehead, whereas porocarcinomas most commonly are found on the head, neck, arms, and legs.1,3,4,6,7 A solitary painless nodule that may ulcerate or bleed is the most common presentation.1,3-5,7
The etiology of eccrine porocarcinoma is poorly understood, but it has been found to arise de novo or to develop from pre-existing poromas or even from nevus sebaceus of Jadassohn. Chronic sunlight exposure, irradiation, lymphedema, trauma, and immunosuppression (eg, Hodgkin disease, chronic lymphocytic leukemia, HIV) have been reported as potential predisposing factors.3,4,6,8,9
Eccrine porocarcinoma often is clinically misdiagnosed as nonmelanoma skin cancer, pyogenic granuloma, amelanotic melanoma, fibroma, verruca vulgaris, or metastatic carcinoma. Appropriate classification is essential, as metastasis is present in 25% to 31% of cases, and local recurrence occurs in 20% to 25% of cases.1,3-5,7
Microscopically, porocarcinomas are comprised of atypical basaloid epithelial cells with focal ductular differentiation. Typically, there is an extensive intraepidermal component that invades into the dermis in anastomosing ribbons and cords. The degree of nuclear atypia, mitotic activity, and invasive growth pattern, as well as the presence of necrosis, are useful histologic features to differentiate porocarcinoma from poroma, which may be present in the background. Given the sometimes-extensive squamous differentiation, porocarcinoma can be confused with squamous cell carcinoma. In these cases, immunohistochemical stains such as epithelial membrane antigen or carcinoembryonic antigen can be used to highlight the ductal differentiation.1,5,8,10
Poor histologic prognostic indicators include a high mitotic index (>14 mitoses per field), a tumor depth greater than 7 mm, and evidence of lymphovascular invasion. Positive lymph node involvement is associated with a 65% to 67% mortality rate.1,8
Because of its propensity to metastasize via the lymphatic system and the high mortality rate associated with such metastases, early identification and treatment are essential. Treatment is accomplished via Mohs micrographic surgery or wide local excision with negative margins. Lymphadenectomy is indicated if regional lymph nodes are involved. Radiation and chemotherapy have been used in patients with metastatic and recurrent disease with mixed results.1,3-5,7 There is no adequate standardized chemotherapy or drug regimen established for porocarcinoma.5 Tsunoda et al11 proposed that sentinel lymph node biopsy should be considered first-line management of eccrine porocarcinoma; however, this remains unproven on the basis of a limited case series. Others conclude that sentinel lymph node biopsy should be recommended for cases with poor histologic prognostic features.1,5
To the Editor:
Porocarcinoma is a rare malignancy of the eccrine sweat glands and is commonly misdiagnosed clinically. We present 2 cases of porocarcinoma and highlight key features of this uncommon disease.
A 65-year-old man presented to the emergency department with a chief concern of a bump on the head of 8 months' duration that gradually enlarged. The lesion recently became painful and contributed to frequent headaches. He reported a history of smoking 1 pack per day and denied trauma to the area or history of immunosuppression. He had no personal or family history of skin cancer. Physical examination revealed a 1.4-cm, heterochromic, pedunculated, keratotic tumor with crusting on the right temporal scalp (Figure 1). No lymphadenopathy was appreciated. The clinical differential diagnosis included irritated seborrheic keratosis, pyogenic granuloma, polypoid malignant melanoma, and nonmelanoma skin cancer. A biopsy of the lesion demonstrated a proliferation of cuboidal cells with focal ductular differentiation arranged in interanastamosing strands arising from the epidermis (Figure 2). Scattered mitotic figures, including atypical forms, cytologic atypia, and foci of necrosis, also were present. The findings were consistent with features of porocarcinoma. Contrast computed tomography of the neck showed no evidence of metastatic disease within the neck. A wide local excision was performed and yielded a tumor measuring 1.8×1.6×0.7 cm with a depth of 0.3 cm and uninvolved margins. No lymphovascular or perineural invasion was identified. At 4-month follow-up, the patient had a well-healed scar on the right scalp without evidence of recurrence or lymphadenopathy.
A 32-year-old woman presented to dermatology with a chief concern of a mass on the back of 2 years’ duration that rapidly enlarged and became painful following irritation from her bra strap 2 months earlier. She had no relevant medical history. Physical examination revealed a firm, tender, heterochromic nodule measuring 3.0×2.8 cm on the left mid back inferior to the left scapula (Figure 3). The lesion expressed serosanguineous discharge. No lymphadenopathy was appreciated on examination. The clinical differential diagnosis included an inflamed cyst, nodular melanoma, cutaneous metastasis, and nonmelanoma skin cancer. The patient underwent an excisional biopsy, which demonstrated porocarcinoma with positive margins, microsatellitosis, and evidence of lymphovascular invasion. Carcinoembryonic antigen immunohistochemistry highlighted ducts within the tumor (Figure 4). The patient underwent re-excision with 2-cm margins, and no residual tumor was appreciated on pathology.
Positron emission tomography and computed tomography revealed a hypermetabolic left axillary lymph node. Ultrasound-guided fine-needle aspiration was positive for malignant cells consistent with metastatic carcinoma. Dissection of left axillary lymph nodes yielded metastatic porocarcinoma in 2 of 13 nodes. The largest tumor deposit measured 0.9 cm, and no extracapsular extension was identified. The patient continues to be monitored with semiannual full-body skin examinations as well as positron emission tomography and computed tomography scans, with no evidence of recurrence 2 years later.
Porocarcinoma is a rare malignancy of the skin arising from the eccrine sweat glands1 with an incidence rate of 0.4 cases per 1 million person-years in the United States. These tumors represent 0.005% to 0.01% of all skin cancers.2 The mean age of onset is approximately 65 years with no predilection for sex. The mean time from initial presentation to treatment is 5.6 to 8.5 years.3-5
Eccrine sweat glands consist of a straight intradermal duct (syrinx); coiled intradermal duct; and spiral intraepidermal duct (acrosyringium), which opens onto the skin. Both eccrine poromas (solitary benign eccrine gland tumors) and eccrine porocarcinomas develop from the acrosyringium. Eccrine poromas most commonly are found in sites containing the highest density of eccrine glands such as the palms, soles, axillae, and forehead, whereas porocarcinomas most commonly are found on the head, neck, arms, and legs.1,3,4,6,7 A solitary painless nodule that may ulcerate or bleed is the most common presentation.1,3-5,7
The etiology of eccrine porocarcinoma is poorly understood, but it has been found to arise de novo or to develop from pre-existing poromas or even from nevus sebaceus of Jadassohn. Chronic sunlight exposure, irradiation, lymphedema, trauma, and immunosuppression (eg, Hodgkin disease, chronic lymphocytic leukemia, HIV) have been reported as potential predisposing factors.3,4,6,8,9
Eccrine porocarcinoma often is clinically misdiagnosed as nonmelanoma skin cancer, pyogenic granuloma, amelanotic melanoma, fibroma, verruca vulgaris, or metastatic carcinoma. Appropriate classification is essential, as metastasis is present in 25% to 31% of cases, and local recurrence occurs in 20% to 25% of cases.1,3-5,7
Microscopically, porocarcinomas are comprised of atypical basaloid epithelial cells with focal ductular differentiation. Typically, there is an extensive intraepidermal component that invades into the dermis in anastomosing ribbons and cords. The degree of nuclear atypia, mitotic activity, and invasive growth pattern, as well as the presence of necrosis, are useful histologic features to differentiate porocarcinoma from poroma, which may be present in the background. Given the sometimes-extensive squamous differentiation, porocarcinoma can be confused with squamous cell carcinoma. In these cases, immunohistochemical stains such as epithelial membrane antigen or carcinoembryonic antigen can be used to highlight the ductal differentiation.1,5,8,10
Poor histologic prognostic indicators include a high mitotic index (>14 mitoses per field), a tumor depth greater than 7 mm, and evidence of lymphovascular invasion. Positive lymph node involvement is associated with a 65% to 67% mortality rate.1,8
Because of its propensity to metastasize via the lymphatic system and the high mortality rate associated with such metastases, early identification and treatment are essential. Treatment is accomplished via Mohs micrographic surgery or wide local excision with negative margins. Lymphadenectomy is indicated if regional lymph nodes are involved. Radiation and chemotherapy have been used in patients with metastatic and recurrent disease with mixed results.1,3-5,7 There is no adequate standardized chemotherapy or drug regimen established for porocarcinoma.5 Tsunoda et al11 proposed that sentinel lymph node biopsy should be considered first-line management of eccrine porocarcinoma; however, this remains unproven on the basis of a limited case series. Others conclude that sentinel lymph node biopsy should be recommended for cases with poor histologic prognostic features.1,5
- Marone U, Caraco C, Anniciello AM, et al. Metastatic eccrine porocarcinoma: report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Salih AM, Kakamad FH, Baba HO, et al. Porocarcinoma; presentation and management, a meta-analysis of 453 cases. Ann Med Surg (Lond). 2017;20:74-79.
- Ritter AM, Graham RS, Amaker B, et al. Intracranial extension of an eccrine porocarcinoma. case report and review of the literature. J Neurosurg. 1999;90:138-140.
- Khaja M, Ashraf U, Mehershahi S, et al. Recurrent metastatic eccrine porocarcinoma: a case report and review of the literature. Am J Case Rep. 2019;20:179-183.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Lloyd MS, El-Muttardi N, Robson A. Eccrine porocarcinoma: a case report and review of the literature. Can J Plast Surg. 2003;11:153-156.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tarkhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413‐415.
- Prieto VG, Shea CR, Celebi JK, et al. Adnexal tumors. In: Busam KJ. Dermatopathology: A Volume in the Foundations in Diagnostic Pathology Series. 2nd ed. Elsevier; 2016:388-446.
- Tsunoda K, Onishi M, Maeda F, et al. Evaluation of sentinel lymph node biopsy for eccrine porocarcinoma. Acta Derm Venereol. 2019;99:691-692.
- Marone U, Caraco C, Anniciello AM, et al. Metastatic eccrine porocarcinoma: report of a case and review of the literature. World J Surg Oncol. 2011;9:32.
- Blake PW, Bradford PT, Devesa SS, et al. Cutaneous appendageal carcinoma incidence and survival patterns in the United States: a population-based study. Arch Dermatol. 2010;146:625-632.
- Salih AM, Kakamad FH, Baba HO, et al. Porocarcinoma; presentation and management, a meta-analysis of 453 cases. Ann Med Surg (Lond). 2017;20:74-79.
- Ritter AM, Graham RS, Amaker B, et al. Intracranial extension of an eccrine porocarcinoma. case report and review of the literature. J Neurosurg. 1999;90:138-140.
- Khaja M, Ashraf U, Mehershahi S, et al. Recurrent metastatic eccrine porocarcinoma: a case report and review of the literature. Am J Case Rep. 2019;20:179-183.
- Sawaya JL, Khachemoune A. Poroma: a review of eccrine, apocrine, and malignant forms. Int J Dermatol. 2014;53:1053-1061.
- Lloyd MS, El-Muttardi N, Robson A. Eccrine porocarcinoma: a case report and review of the literature. Can J Plast Surg. 2003;11:153-156.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tarkhan II, Domingo J. Metastasizing eccrine porocarcinoma developing in a sebaceous nevus of Jadassohn. report of a case. Arch Dermatol. 1985;121:413‐415.
- Prieto VG, Shea CR, Celebi JK, et al. Adnexal tumors. In: Busam KJ. Dermatopathology: A Volume in the Foundations in Diagnostic Pathology Series. 2nd ed. Elsevier; 2016:388-446.
- Tsunoda K, Onishi M, Maeda F, et al. Evaluation of sentinel lymph node biopsy for eccrine porocarcinoma. Acta Derm Venereol. 2019;99:691-692.
Practice Points
- Eccrine porocarcinoma is a rare malignancy that clinically mimics other cutaneous malignancies.
- Early histologic diagnosis is essential, as lymphatic metastasis is common and carries a 65% to 67% mortality rate.

Dermatology Author Gender Trends During the COVID-19 Pandemic
To the Editor:
Peer-reviewed publications are important determinants for promotions, academic leadership, and grants in dermatology.1 The impact of the COVID-19 pandemic on dermatology research productivity remains an area of investigation. We sought to determine authorship trends for males and females during the pandemic.
A cross-sectional retrospective study of the top 20 dermatology journals—determined by impact factor and Google Scholar H5-index—was conducted to identify manuscripts with submission date specified prepandemic (May 1, 2019–October 31, 2019) and during the pandemic (May 1, 2020–October 31, 2020). Submission date, first/last author name, sex, and affiliated country were extracted. Single authors were designated as first authors. Gender API (https://gender-api.com/en/) classified gender. A χ2 test (P<.05) compared differences in proportions of female first/last authors from 2019 to 2020.
Overall, 811 and 1061 articles submitted in 2019 and 2020, respectively, were included. There were 1517 articles submitted to clinical journals and 355 articles submitted to basic science journals (Table). For the 7 clinical journals included, there was a 7.7% decrease in the proportion of female last authors in 2020 vs 2019 (P=.002), with the largest decrease between August and September 2020. Although other comparisons did not yield statistically significant differences (P>.05 all)(Table), several trends were observed. For clinical journals, there was a 1.8% decrease in the proportion of female first authors. For the 4 basic science journals included, there was a 4.9% increase and a 0.3% decrease in percentages of female first and last authors, respectively, for 2020 vs 2019.

Our findings indicate that the COVID-19 pandemic may have impacted female authors’ productivity in clinical dermatology publications. In a survey-based study for 2010 to 2011, female physician-researchers (n=437) spent 8.5 more hours per week on domestic activities and childcare and were more likely to take time off for childcare if their partner worked full time compared with males (n=612)(42.6% vs 12.4%, respectively).2 Our observation that female last authors had a significant decrease in publications may suggest that this population had a disproportionate burden of domestic labor and childcare during the pandemic. It is possible that last authors, who generally are more senior researchers, may be more likely to have childcare, eldercare, and other types of domestic responsibilities. Similarly, in a study of surgery submissions (n=1068), there were 6%, 7%, and 4% decreases in percentages of female last, corresponding, and first authors, respectively, from 2019 to 2020.3Our study had limitations. Only 11 journals were analyzed because others did not have specified submission dates. Some journals only provided submission information for a subset of articles (eg, those published in the In Press section), which may have accounted for the large discrepancy in submission numbers for 2019 to 2020. Gender could not be determined for 1% of authors and was limited to female and male. Although our study submission time frame (May–October 2020) aimed at identifying research conducted during the height of the COVID-19 pandemic, some of these studies may have been conducted months or years before the pandemic. Future studies should focus on longer and more comprehensive time frames. Finally, estimated dates of stay-at-home orders fail to consider differences within countries.
The proportion of female US-affiliated first and last authors publishing in dermatology journals increased from 12% to 48% in 1976 and from 6% to 31% in 2006,4 which is encouraging. However, a gender gap persists, with one-third of National Institutes of Health grants in dermatology and one-fourth of research project grants in dermatology awarded to women.5 Consequences of the pandemic on academic productivity may include fewer women represented in higher academic ranks, lower compensation, and lower career satisfaction compared with men.1 We urge academic institutions and funding agencies to recognize and take action to mitigate long-term sequelae. Extended grant end dates and submission periods, funding opportunities dedicated to women, and prioritization of female-authored submissions are some strategies that can safeguard equitable career progression in dermatology research.
- Stewart C, Lipner SR. Gender and race trends in academic rank of dermatologists at top U.S. institutions: a cross-sectional study. Int J Womens Dermatol. 2020;6:283-285. doi:10.1016/j .ijwd.2020.04.010
- Jolly S, Griffith KA, DeCastro R, et al. Gender differences in time spent on parenting and domestic responsibilities by highachieving young physician-researchers. Ann Intern Med. 2014; 160:344-353. doi:10.7326/M13-0974
- Kibbe MR. Consequences of the COVID-19 pandemic on manuscript submissions by women. JAMA Surg. 2020;155:803-804. doi:10.1001/jamasurg.2020.3917
- Feramisco JD, Leitenberger JJ, Redfern SI, et al. A gender gap in the dermatology literature? cross-sectional analysis of manuscript authorship trends in dermatology journals during 3 decades. J Am Acad Dermatol. 2009;6:63-69. doi:10.1016/j.jaad.2008.06.044
- Cheng MY, Sukhov A, Sultani H, et al. Trends in national institutes of health funding of principal investigators in dermatology research by academic degree and sex. JAMA Dermatol. 2016;152:883-888. doi:10.1001/jamadermatol.2016.0271
To the Editor:
Peer-reviewed publications are important determinants for promotions, academic leadership, and grants in dermatology.1 The impact of the COVID-19 pandemic on dermatology research productivity remains an area of investigation. We sought to determine authorship trends for males and females during the pandemic.
A cross-sectional retrospective study of the top 20 dermatology journals—determined by impact factor and Google Scholar H5-index—was conducted to identify manuscripts with submission date specified prepandemic (May 1, 2019–October 31, 2019) and during the pandemic (May 1, 2020–October 31, 2020). Submission date, first/last author name, sex, and affiliated country were extracted. Single authors were designated as first authors. Gender API (https://gender-api.com/en/) classified gender. A χ2 test (P<.05) compared differences in proportions of female first/last authors from 2019 to 2020.
Overall, 811 and 1061 articles submitted in 2019 and 2020, respectively, were included. There were 1517 articles submitted to clinical journals and 355 articles submitted to basic science journals (Table). For the 7 clinical journals included, there was a 7.7% decrease in the proportion of female last authors in 2020 vs 2019 (P=.002), with the largest decrease between August and September 2020. Although other comparisons did not yield statistically significant differences (P>.05 all)(Table), several trends were observed. For clinical journals, there was a 1.8% decrease in the proportion of female first authors. For the 4 basic science journals included, there was a 4.9% increase and a 0.3% decrease in percentages of female first and last authors, respectively, for 2020 vs 2019.
Our findings indicate that the COVID-19 pandemic may have impacted female authors’ productivity in clinical dermatology publications. In a survey-based study for 2010 to 2011, female physician-researchers (n=437) spent 8.5 more hours per week on domestic activities and childcare and were more likely to take time off for childcare if their partner worked full time compared with males (n=612)(42.6% vs 12.4%, respectively).2 Our observation that female last authors had a significant decrease in publications may suggest that this population had a disproportionate burden of domestic labor and childcare during the pandemic. It is possible that last authors, who generally are more senior researchers, may be more likely to have childcare, eldercare, and other types of domestic responsibilities. Similarly, in a study of surgery submissions (n=1068), there were 6%, 7%, and 4% decreases in percentages of female last, corresponding, and first authors, respectively, from 2019 to 2020.3Our study had limitations. Only 11 journals were analyzed because others did not have specified submission dates. Some journals only provided submission information for a subset of articles (eg, those published in the In Press section), which may have accounted for the large discrepancy in submission numbers for 2019 to 2020. Gender could not be determined for 1% of authors and was limited to female and male. Although our study submission time frame (May–October 2020) aimed at identifying research conducted during the height of the COVID-19 pandemic, some of these studies may have been conducted months or years before the pandemic. Future studies should focus on longer and more comprehensive time frames. Finally, estimated dates of stay-at-home orders fail to consider differences within countries.
The proportion of female US-affiliated first and last authors publishing in dermatology journals increased from 12% to 48% in 1976 and from 6% to 31% in 2006,4 which is encouraging. However, a gender gap persists, with one-third of National Institutes of Health grants in dermatology and one-fourth of research project grants in dermatology awarded to women.5 Consequences of the pandemic on academic productivity may include fewer women represented in higher academic ranks, lower compensation, and lower career satisfaction compared with men.1 We urge academic institutions and funding agencies to recognize and take action to mitigate long-term sequelae. Extended grant end dates and submission periods, funding opportunities dedicated to women, and prioritization of female-authored submissions are some strategies that can safeguard equitable career progression in dermatology research.
To the Editor:
Peer-reviewed publications are important determinants for promotions, academic leadership, and grants in dermatology.1 The impact of the COVID-19 pandemic on dermatology research productivity remains an area of investigation. We sought to determine authorship trends for males and females during the pandemic.
A cross-sectional retrospective study of the top 20 dermatology journals—determined by impact factor and Google Scholar H5-index—was conducted to identify manuscripts with submission date specified prepandemic (May 1, 2019–October 31, 2019) and during the pandemic (May 1, 2020–October 31, 2020). Submission date, first/last author name, sex, and affiliated country were extracted. Single authors were designated as first authors. Gender API (https://gender-api.com/en/) classified gender. A χ2 test (P<.05) compared differences in proportions of female first/last authors from 2019 to 2020.
Overall, 811 and 1061 articles submitted in 2019 and 2020, respectively, were included. There were 1517 articles submitted to clinical journals and 355 articles submitted to basic science journals (Table). For the 7 clinical journals included, there was a 7.7% decrease in the proportion of female last authors in 2020 vs 2019 (P=.002), with the largest decrease between August and September 2020. Although other comparisons did not yield statistically significant differences (P>.05 all)(Table), several trends were observed. For clinical journals, there was a 1.8% decrease in the proportion of female first authors. For the 4 basic science journals included, there was a 4.9% increase and a 0.3% decrease in percentages of female first and last authors, respectively, for 2020 vs 2019.
Our findings indicate that the COVID-19 pandemic may have impacted female authors’ productivity in clinical dermatology publications. In a survey-based study for 2010 to 2011, female physician-researchers (n=437) spent 8.5 more hours per week on domestic activities and childcare and were more likely to take time off for childcare if their partner worked full time compared with males (n=612)(42.6% vs 12.4%, respectively).2 Our observation that female last authors had a significant decrease in publications may suggest that this population had a disproportionate burden of domestic labor and childcare during the pandemic. It is possible that last authors, who generally are more senior researchers, may be more likely to have childcare, eldercare, and other types of domestic responsibilities. Similarly, in a study of surgery submissions (n=1068), there were 6%, 7%, and 4% decreases in percentages of female last, corresponding, and first authors, respectively, from 2019 to 2020.3Our study had limitations. Only 11 journals were analyzed because others did not have specified submission dates. Some journals only provided submission information for a subset of articles (eg, those published in the In Press section), which may have accounted for the large discrepancy in submission numbers for 2019 to 2020. Gender could not be determined for 1% of authors and was limited to female and male. Although our study submission time frame (May–October 2020) aimed at identifying research conducted during the height of the COVID-19 pandemic, some of these studies may have been conducted months or years before the pandemic. Future studies should focus on longer and more comprehensive time frames. Finally, estimated dates of stay-at-home orders fail to consider differences within countries.
The proportion of female US-affiliated first and last authors publishing in dermatology journals increased from 12% to 48% in 1976 and from 6% to 31% in 2006,4 which is encouraging. However, a gender gap persists, with one-third of National Institutes of Health grants in dermatology and one-fourth of research project grants in dermatology awarded to women.5 Consequences of the pandemic on academic productivity may include fewer women represented in higher academic ranks, lower compensation, and lower career satisfaction compared with men.1 We urge academic institutions and funding agencies to recognize and take action to mitigate long-term sequelae. Extended grant end dates and submission periods, funding opportunities dedicated to women, and prioritization of female-authored submissions are some strategies that can safeguard equitable career progression in dermatology research.
- Stewart C, Lipner SR. Gender and race trends in academic rank of dermatologists at top U.S. institutions: a cross-sectional study. Int J Womens Dermatol. 2020;6:283-285. doi:10.1016/j .ijwd.2020.04.010
- Jolly S, Griffith KA, DeCastro R, et al. Gender differences in time spent on parenting and domestic responsibilities by highachieving young physician-researchers. Ann Intern Med. 2014; 160:344-353. doi:10.7326/M13-0974
- Kibbe MR. Consequences of the COVID-19 pandemic on manuscript submissions by women. JAMA Surg. 2020;155:803-804. doi:10.1001/jamasurg.2020.3917
- Feramisco JD, Leitenberger JJ, Redfern SI, et al. A gender gap in the dermatology literature? cross-sectional analysis of manuscript authorship trends in dermatology journals during 3 decades. J Am Acad Dermatol. 2009;6:63-69. doi:10.1016/j.jaad.2008.06.044
- Cheng MY, Sukhov A, Sultani H, et al. Trends in national institutes of health funding of principal investigators in dermatology research by academic degree and sex. JAMA Dermatol. 2016;152:883-888. doi:10.1001/jamadermatol.2016.0271
- Stewart C, Lipner SR. Gender and race trends in academic rank of dermatologists at top U.S. institutions: a cross-sectional study. Int J Womens Dermatol. 2020;6:283-285. doi:10.1016/j .ijwd.2020.04.010
- Jolly S, Griffith KA, DeCastro R, et al. Gender differences in time spent on parenting and domestic responsibilities by highachieving young physician-researchers. Ann Intern Med. 2014; 160:344-353. doi:10.7326/M13-0974
- Kibbe MR. Consequences of the COVID-19 pandemic on manuscript submissions by women. JAMA Surg. 2020;155:803-804. doi:10.1001/jamasurg.2020.3917
- Feramisco JD, Leitenberger JJ, Redfern SI, et al. A gender gap in the dermatology literature? cross-sectional analysis of manuscript authorship trends in dermatology journals during 3 decades. J Am Acad Dermatol. 2009;6:63-69. doi:10.1016/j.jaad.2008.06.044
- Cheng MY, Sukhov A, Sultani H, et al. Trends in national institutes of health funding of principal investigators in dermatology research by academic degree and sex. JAMA Dermatol. 2016;152:883-888. doi:10.1001/jamadermatol.2016.0271
Practice Points
- The academic productivity of female dermatologists as last authors in dermatology clinical journals has potentially been impacted by the COVID-19 pandemic.
- To potentially aid in the resurgence of female dermatologist authors impacted by the pandemic, academic institutions and funding agencies may consider implementing strategies such as extending grant end dates, providing dedicated funding opportunities, and prioritizing female-authored submissions in dermatology research.
Dupilumab-Associated Sweet Syndrome
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, was first described in 1964. 1 Since then, several subtypes of SS have been recognized, including classic or idiopathic, which typically follows an acute viral illness; cancer related, typically in the form of a paraneoplastic syndrome; and drug induced. 2 Drug-induced SS is defined by the following: (1) an abrupt onset of painful erythematous plaques or nodules; (2) histopathologic evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis; (3) pyrexia above 38 ° C; (4) temporal relationship between drug and clinical presentation or temporally related recurrence after rechallenge; and (5) temporally related resolution of lesions after drug withdrawal or treatment with systemic corticosteroids. 3 All 5 criteria must be met to make a diagnosis of drug-induced SS. Since these criteria were established by Walker and Cohen, 3 various drugs have been identified as causative agents, including antibiotics, antiepileptics, antiretrovirals, antineoplastic agents, antipsychotics, oral contraceptives, nonsteroidal anti-inflammatory agents, and retinoids. 4 W e present a rare case of SS caused by dupilumab, a monoclonal antibody therapy, used in the treatment of severe eosinophilic asthma and atopic dermatitis.
Case Report
A 53-year-old woman presented with painful skin lesions, arthralgia, fever, and leukocytosis following initiation of dupilumab. She had a history of adult-onset, severe, persistent eosinophilic asthma, as well as chronic rhinosinusitis with nasal polyps, plaque psoriasis, and hypertrophic cardiomyopathy. She started mepolizumab 3 years prior to the current presentation for persistently uncontrolled asthma with a baseline peripheral eosinophil count of 860 cells/µL. After 3 years of minimal response to mepolizumab, she was started on dupilumab. Within 2 weeks of the first dose of dupilumab, she started experiencing bilateral knee pain. She subsequently developed daily fevers (temperature, 38.3 °C to 39.4 °C), fatigue, and pain in the back of the neck and head. After the second dose of dupilumab, she started experiencing painful skin lesions on the bilateral knuckles, elbows, and abdomen (Figure 1). She had difficulty using her hands and walking secondary to intense arthralgia involving the bilateral finger joints, elbows, and knees. Her primary care physician obtained a laboratory evaluation, which revealed an elevated total white blood cell count of 20×103/mm3 (reference range, 4–11×103/mm3) with 27.5% neutrophils and severely elevated eosinophils above her baseline to 57.3% with an absolute eosinophil count of 11,700 cells/µL (reference range, <400 cells/µL). Further assessment revealed an elevated erythrocyte sedimentation rate of 64 mm/h (reference range, 0–30 mm/h) and C-reactive protein level of 34 mg/dL (reference range, ≤0.80 mg/dL), with negative antinuclear antibody, rheumatoid factor, antineutrophilic cytoplasmic antibody, and Lyme antibody. IgG, IgA, and IgM levels were within reference range, and the IgE level was not elevated above her baseline. She had normal serum tryptase, and a peripheral D816V c-KIT mutation was not detected. She was subsequently hospitalized for further evaluation, at which time there was no fever or localizing infectious signs or symptoms. An infectious evaluation including urinalysis; respiratory swab for adenovirus, coronaviruses, human metapneumovirus, rhinovirus/enterovirus, influenza A and B, parainfluenza viruses, respiratory syncytial virus, Chlamydophila pneumoniae, and Mycoplasma pneumoniae; Lyme serology; and a computed tomography (CT) scan of the chest, abdomen, and pelvis revealed no evidence of infection. A parasite evaluation was ordered but was not performed. There was no evidence of malignancy on CT of the chest, abdomen, and pelvis or CT of the head without contrast. A lumbar puncture was considered but was ultimately deferred.

At the current presentation, the patient was following up in the dermatology clinic shortly after discharge. The lesions on the fingers and arms were described by the dermatologist as deep, erythematous, 0.5-cm bullous papules. The differential diagnosis at this time included a cutaneous or systemic infection, vasculitis, drug eruption, or cutaneous manifestation of an autoimmune condition. A shave biopsy of a skin lesion on the right hand demonstrated epidermal necrosis with a dense dermal neutrophilic infiltrate consistent with a neutrophilic dermatosis (Figure 2). There was no evidence of leukocytoclastic vasculitis. The histologic differential diagnosis included cutaneous infection, neutrophilic dermatosis of the hand, and SS. Special stains for infectious organisms including Gram, Grocott methenamine-silver, and auramine-rhodamine stains were negative for bacterial, fungal, and mycobacterial organisms, ruling out cutaneous infection. A diagnosis of drug-induced SS was made based on the histologic findings, diffuse distribution of the lesions, negative infectious evaluation, lack of underlying malignancy or autoimmune conditions, and onset following initiation of dupilumab.
 and dense underlying inflammation. Neutrophils with associated karyorrhectic debris and eosinophils were seen")
Dupilumab was discontinued, and the patient was started on prednisone with rapid improvement in the symptoms. She underwent a slow taper of the prednisone over approximately 2 months with a slow downtrend of eosinophils. She was transitioned to a different biologic agent, benralizumab, with no further recurrence of the rash or arthralgia.
Comment
Dupilumab is a human monoclonal IgG4 antibody that inhibits IL-4 and IL-13 signaling by binding to the IL-4Rα subunit. By blocking IL-4Rα, dupilumab inhibits IL-4 and IL-13 cytokine-induced inflammatory responses, including the release of proinflammatory cytokines, chemokines, nitric oxide, and IgE. Currently, dupilumab is approved to treat refractory forms of moderate to severe asthma characterized by an eosinophilic phenotype or with corticosteroid-dependent asthma, moderate to severe atopic dermatitis, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis. The most common adverse events (incidence ≥1%) are injection-site reactions, oropharyngeal pain, and eosinophilia.5 Interestingly, our patient did exhibit a high degree of eosinophilia; however, she met all criteria for drug-induced SS, and the skin biopsy was not consistent with an eosinophilic process. Notably, the peripheral neutrophils were not elevated. Neutrophilia often is seen in classic SS but is not required for a diagnosis of drug-induced SS. Rare cases of dupilumab-associated arthritis and serum sickness–like reaction have been described,6-8 but our patient’s presentation was distinct, given other described signs, symptoms, and skin biopsy results. Histopathology results were not consistent with leukocytoclastic vasculitis, a potential mimicker of SS. Although the infectious and paraneoplastic evaluation was not exhaustive, the negative imaging from head to pelvis, the lack of recurrence of skin lesions, and the laboratory abnormalities after dupilumab discontinuation supported the conclusion that the culprit was not an infection or underlying malignancy. She had not started any other medications during this time frame, leaving dupilumab as the most likely offending agent. The mechanism for this reaction is not clear. It is possible that inhibition of IL-4 and IL-13 in the T helper 2 (TH2) cell pathway may have led to upregulated IL-17–mediated inflammation9 as well as a neutrophilic process in the skin, but this would not explain the concurrent peripheral eosinophilia that was noted. Further studies are needed to investigate the pathophysiology of SS.
Conclusion
We report a rare case of dupilumab-induced SS. Corticosteroids accompanied by cessation of the medication proved to be an effective treatment.
- Sweet RB. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Jackson K, Bahna SL. Hypersensitivity and adverse reactions to biologics for asthma and allergic diseases. Expert Rev Clin Immunol. 2020;16:311-319.
- Willsmore ZN, Woolf RT, Hughes C, et al. Development of inflammatory arthritis and enthesitis in patients on dupilumab: a case series. Br J Dermatol. 2019;181:1068-1070.
- de Wijs LEM, van der Waa JD, de Jong PHP, et al. Acute arthritis and arthralgia as an adverse drug reaction to dupilumab. Clin Exp Dermatol. 2020;45:262-263.
- Treudler R, Delaroque N, Puder M, et al. Dupilumab-induced serum sickness-like reaction: an unusual adverse effect in a patient with atopic eczema. J Eur Acad Dermatol Venereol. 2021;35:E30-E32.
- Guenova E, Skabytska Y, Hoetzenecker W, et al. IL-4 abrogates TH17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A. 2015;112:2163-2168.
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, was first described in 1964. 1 Since then, several subtypes of SS have been recognized, including classic or idiopathic, which typically follows an acute viral illness; cancer related, typically in the form of a paraneoplastic syndrome; and drug induced. 2 Drug-induced SS is defined by the following: (1) an abrupt onset of painful erythematous plaques or nodules; (2) histopathologic evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis; (3) pyrexia above 38 ° C; (4) temporal relationship between drug and clinical presentation or temporally related recurrence after rechallenge; and (5) temporally related resolution of lesions after drug withdrawal or treatment with systemic corticosteroids. 3 All 5 criteria must be met to make a diagnosis of drug-induced SS. Since these criteria were established by Walker and Cohen, 3 various drugs have been identified as causative agents, including antibiotics, antiepileptics, antiretrovirals, antineoplastic agents, antipsychotics, oral contraceptives, nonsteroidal anti-inflammatory agents, and retinoids. 4 W e present a rare case of SS caused by dupilumab, a monoclonal antibody therapy, used in the treatment of severe eosinophilic asthma and atopic dermatitis.
Case Report
A 53-year-old woman presented with painful skin lesions, arthralgia, fever, and leukocytosis following initiation of dupilumab. She had a history of adult-onset, severe, persistent eosinophilic asthma, as well as chronic rhinosinusitis with nasal polyps, plaque psoriasis, and hypertrophic cardiomyopathy. She started mepolizumab 3 years prior to the current presentation for persistently uncontrolled asthma with a baseline peripheral eosinophil count of 860 cells/µL. After 3 years of minimal response to mepolizumab, she was started on dupilumab. Within 2 weeks of the first dose of dupilumab, she started experiencing bilateral knee pain. She subsequently developed daily fevers (temperature, 38.3 °C to 39.4 °C), fatigue, and pain in the back of the neck and head. After the second dose of dupilumab, she started experiencing painful skin lesions on the bilateral knuckles, elbows, and abdomen (Figure 1). She had difficulty using her hands and walking secondary to intense arthralgia involving the bilateral finger joints, elbows, and knees. Her primary care physician obtained a laboratory evaluation, which revealed an elevated total white blood cell count of 20×103/mm3 (reference range, 4–11×103/mm3) with 27.5% neutrophils and severely elevated eosinophils above her baseline to 57.3% with an absolute eosinophil count of 11,700 cells/µL (reference range, <400 cells/µL). Further assessment revealed an elevated erythrocyte sedimentation rate of 64 mm/h (reference range, 0–30 mm/h) and C-reactive protein level of 34 mg/dL (reference range, ≤0.80 mg/dL), with negative antinuclear antibody, rheumatoid factor, antineutrophilic cytoplasmic antibody, and Lyme antibody. IgG, IgA, and IgM levels were within reference range, and the IgE level was not elevated above her baseline. She had normal serum tryptase, and a peripheral D816V c-KIT mutation was not detected. She was subsequently hospitalized for further evaluation, at which time there was no fever or localizing infectious signs or symptoms. An infectious evaluation including urinalysis; respiratory swab for adenovirus, coronaviruses, human metapneumovirus, rhinovirus/enterovirus, influenza A and B, parainfluenza viruses, respiratory syncytial virus, Chlamydophila pneumoniae, and Mycoplasma pneumoniae; Lyme serology; and a computed tomography (CT) scan of the chest, abdomen, and pelvis revealed no evidence of infection. A parasite evaluation was ordered but was not performed. There was no evidence of malignancy on CT of the chest, abdomen, and pelvis or CT of the head without contrast. A lumbar puncture was considered but was ultimately deferred.
At the current presentation, the patient was following up in the dermatology clinic shortly after discharge. The lesions on the fingers and arms were described by the dermatologist as deep, erythematous, 0.5-cm bullous papules. The differential diagnosis at this time included a cutaneous or systemic infection, vasculitis, drug eruption, or cutaneous manifestation of an autoimmune condition. A shave biopsy of a skin lesion on the right hand demonstrated epidermal necrosis with a dense dermal neutrophilic infiltrate consistent with a neutrophilic dermatosis (Figure 2). There was no evidence of leukocytoclastic vasculitis. The histologic differential diagnosis included cutaneous infection, neutrophilic dermatosis of the hand, and SS. Special stains for infectious organisms including Gram, Grocott methenamine-silver, and auramine-rhodamine stains were negative for bacterial, fungal, and mycobacterial organisms, ruling out cutaneous infection. A diagnosis of drug-induced SS was made based on the histologic findings, diffuse distribution of the lesions, negative infectious evaluation, lack of underlying malignancy or autoimmune conditions, and onset following initiation of dupilumab.
Dupilumab was discontinued, and the patient was started on prednisone with rapid improvement in the symptoms. She underwent a slow taper of the prednisone over approximately 2 months with a slow downtrend of eosinophils. She was transitioned to a different biologic agent, benralizumab, with no further recurrence of the rash or arthralgia.
Comment
Dupilumab is a human monoclonal IgG4 antibody that inhibits IL-4 and IL-13 signaling by binding to the IL-4Rα subunit. By blocking IL-4Rα, dupilumab inhibits IL-4 and IL-13 cytokine-induced inflammatory responses, including the release of proinflammatory cytokines, chemokines, nitric oxide, and IgE. Currently, dupilumab is approved to treat refractory forms of moderate to severe asthma characterized by an eosinophilic phenotype or with corticosteroid-dependent asthma, moderate to severe atopic dermatitis, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis. The most common adverse events (incidence ≥1%) are injection-site reactions, oropharyngeal pain, and eosinophilia.5 Interestingly, our patient did exhibit a high degree of eosinophilia; however, she met all criteria for drug-induced SS, and the skin biopsy was not consistent with an eosinophilic process. Notably, the peripheral neutrophils were not elevated. Neutrophilia often is seen in classic SS but is not required for a diagnosis of drug-induced SS. Rare cases of dupilumab-associated arthritis and serum sickness–like reaction have been described,6-8 but our patient’s presentation was distinct, given other described signs, symptoms, and skin biopsy results. Histopathology results were not consistent with leukocytoclastic vasculitis, a potential mimicker of SS. Although the infectious and paraneoplastic evaluation was not exhaustive, the negative imaging from head to pelvis, the lack of recurrence of skin lesions, and the laboratory abnormalities after dupilumab discontinuation supported the conclusion that the culprit was not an infection or underlying malignancy. She had not started any other medications during this time frame, leaving dupilumab as the most likely offending agent. The mechanism for this reaction is not clear. It is possible that inhibition of IL-4 and IL-13 in the T helper 2 (TH2) cell pathway may have led to upregulated IL-17–mediated inflammation9 as well as a neutrophilic process in the skin, but this would not explain the concurrent peripheral eosinophilia that was noted. Further studies are needed to investigate the pathophysiology of SS.
Conclusion
We report a rare case of dupilumab-induced SS. Corticosteroids accompanied by cessation of the medication proved to be an effective treatment.
Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, was first described in 1964. 1 Since then, several subtypes of SS have been recognized, including classic or idiopathic, which typically follows an acute viral illness; cancer related, typically in the form of a paraneoplastic syndrome; and drug induced. 2 Drug-induced SS is defined by the following: (1) an abrupt onset of painful erythematous plaques or nodules; (2) histopathologic evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis; (3) pyrexia above 38 ° C; (4) temporal relationship between drug and clinical presentation or temporally related recurrence after rechallenge; and (5) temporally related resolution of lesions after drug withdrawal or treatment with systemic corticosteroids. 3 All 5 criteria must be met to make a diagnosis of drug-induced SS. Since these criteria were established by Walker and Cohen, 3 various drugs have been identified as causative agents, including antibiotics, antiepileptics, antiretrovirals, antineoplastic agents, antipsychotics, oral contraceptives, nonsteroidal anti-inflammatory agents, and retinoids. 4 W e present a rare case of SS caused by dupilumab, a monoclonal antibody therapy, used in the treatment of severe eosinophilic asthma and atopic dermatitis.
Case Report
A 53-year-old woman presented with painful skin lesions, arthralgia, fever, and leukocytosis following initiation of dupilumab. She had a history of adult-onset, severe, persistent eosinophilic asthma, as well as chronic rhinosinusitis with nasal polyps, plaque psoriasis, and hypertrophic cardiomyopathy. She started mepolizumab 3 years prior to the current presentation for persistently uncontrolled asthma with a baseline peripheral eosinophil count of 860 cells/µL. After 3 years of minimal response to mepolizumab, she was started on dupilumab. Within 2 weeks of the first dose of dupilumab, she started experiencing bilateral knee pain. She subsequently developed daily fevers (temperature, 38.3 °C to 39.4 °C), fatigue, and pain in the back of the neck and head. After the second dose of dupilumab, she started experiencing painful skin lesions on the bilateral knuckles, elbows, and abdomen (Figure 1). She had difficulty using her hands and walking secondary to intense arthralgia involving the bilateral finger joints, elbows, and knees. Her primary care physician obtained a laboratory evaluation, which revealed an elevated total white blood cell count of 20×103/mm3 (reference range, 4–11×103/mm3) with 27.5% neutrophils and severely elevated eosinophils above her baseline to 57.3% with an absolute eosinophil count of 11,700 cells/µL (reference range, <400 cells/µL). Further assessment revealed an elevated erythrocyte sedimentation rate of 64 mm/h (reference range, 0–30 mm/h) and C-reactive protein level of 34 mg/dL (reference range, ≤0.80 mg/dL), with negative antinuclear antibody, rheumatoid factor, antineutrophilic cytoplasmic antibody, and Lyme antibody. IgG, IgA, and IgM levels were within reference range, and the IgE level was not elevated above her baseline. She had normal serum tryptase, and a peripheral D816V c-KIT mutation was not detected. She was subsequently hospitalized for further evaluation, at which time there was no fever or localizing infectious signs or symptoms. An infectious evaluation including urinalysis; respiratory swab for adenovirus, coronaviruses, human metapneumovirus, rhinovirus/enterovirus, influenza A and B, parainfluenza viruses, respiratory syncytial virus, Chlamydophila pneumoniae, and Mycoplasma pneumoniae; Lyme serology; and a computed tomography (CT) scan of the chest, abdomen, and pelvis revealed no evidence of infection. A parasite evaluation was ordered but was not performed. There was no evidence of malignancy on CT of the chest, abdomen, and pelvis or CT of the head without contrast. A lumbar puncture was considered but was ultimately deferred.
At the current presentation, the patient was following up in the dermatology clinic shortly after discharge. The lesions on the fingers and arms were described by the dermatologist as deep, erythematous, 0.5-cm bullous papules. The differential diagnosis at this time included a cutaneous or systemic infection, vasculitis, drug eruption, or cutaneous manifestation of an autoimmune condition. A shave biopsy of a skin lesion on the right hand demonstrated epidermal necrosis with a dense dermal neutrophilic infiltrate consistent with a neutrophilic dermatosis (Figure 2). There was no evidence of leukocytoclastic vasculitis. The histologic differential diagnosis included cutaneous infection, neutrophilic dermatosis of the hand, and SS. Special stains for infectious organisms including Gram, Grocott methenamine-silver, and auramine-rhodamine stains were negative for bacterial, fungal, and mycobacterial organisms, ruling out cutaneous infection. A diagnosis of drug-induced SS was made based on the histologic findings, diffuse distribution of the lesions, negative infectious evaluation, lack of underlying malignancy or autoimmune conditions, and onset following initiation of dupilumab.
Dupilumab was discontinued, and the patient was started on prednisone with rapid improvement in the symptoms. She underwent a slow taper of the prednisone over approximately 2 months with a slow downtrend of eosinophils. She was transitioned to a different biologic agent, benralizumab, with no further recurrence of the rash or arthralgia.
Comment
Dupilumab is a human monoclonal IgG4 antibody that inhibits IL-4 and IL-13 signaling by binding to the IL-4Rα subunit. By blocking IL-4Rα, dupilumab inhibits IL-4 and IL-13 cytokine-induced inflammatory responses, including the release of proinflammatory cytokines, chemokines, nitric oxide, and IgE. Currently, dupilumab is approved to treat refractory forms of moderate to severe asthma characterized by an eosinophilic phenotype or with corticosteroid-dependent asthma, moderate to severe atopic dermatitis, chronic rhinosinusitis with nasal polyposis, and eosinophilic esophagitis. The most common adverse events (incidence ≥1%) are injection-site reactions, oropharyngeal pain, and eosinophilia.5 Interestingly, our patient did exhibit a high degree of eosinophilia; however, she met all criteria for drug-induced SS, and the skin biopsy was not consistent with an eosinophilic process. Notably, the peripheral neutrophils were not elevated. Neutrophilia often is seen in classic SS but is not required for a diagnosis of drug-induced SS. Rare cases of dupilumab-associated arthritis and serum sickness–like reaction have been described,6-8 but our patient’s presentation was distinct, given other described signs, symptoms, and skin biopsy results. Histopathology results were not consistent with leukocytoclastic vasculitis, a potential mimicker of SS. Although the infectious and paraneoplastic evaluation was not exhaustive, the negative imaging from head to pelvis, the lack of recurrence of skin lesions, and the laboratory abnormalities after dupilumab discontinuation supported the conclusion that the culprit was not an infection or underlying malignancy. She had not started any other medications during this time frame, leaving dupilumab as the most likely offending agent. The mechanism for this reaction is not clear. It is possible that inhibition of IL-4 and IL-13 in the T helper 2 (TH2) cell pathway may have led to upregulated IL-17–mediated inflammation9 as well as a neutrophilic process in the skin, but this would not explain the concurrent peripheral eosinophilia that was noted. Further studies are needed to investigate the pathophysiology of SS.
Conclusion
We report a rare case of dupilumab-induced SS. Corticosteroids accompanied by cessation of the medication proved to be an effective treatment.
- Sweet RB. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Jackson K, Bahna SL. Hypersensitivity and adverse reactions to biologics for asthma and allergic diseases. Expert Rev Clin Immunol. 2020;16:311-319.
- Willsmore ZN, Woolf RT, Hughes C, et al. Development of inflammatory arthritis and enthesitis in patients on dupilumab: a case series. Br J Dermatol. 2019;181:1068-1070.
- de Wijs LEM, van der Waa JD, de Jong PHP, et al. Acute arthritis and arthralgia as an adverse drug reaction to dupilumab. Clin Exp Dermatol. 2020;45:262-263.
- Treudler R, Delaroque N, Puder M, et al. Dupilumab-induced serum sickness-like reaction: an unusual adverse effect in a patient with atopic eczema. J Eur Acad Dermatol Venereol. 2021;35:E30-E32.
- Guenova E, Skabytska Y, Hoetzenecker W, et al. IL-4 abrogates TH17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A. 2015;112:2163-2168.
- Sweet RB. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Jackson K, Bahna SL. Hypersensitivity and adverse reactions to biologics for asthma and allergic diseases. Expert Rev Clin Immunol. 2020;16:311-319.
- Willsmore ZN, Woolf RT, Hughes C, et al. Development of inflammatory arthritis and enthesitis in patients on dupilumab: a case series. Br J Dermatol. 2019;181:1068-1070.
- de Wijs LEM, van der Waa JD, de Jong PHP, et al. Acute arthritis and arthralgia as an adverse drug reaction to dupilumab. Clin Exp Dermatol. 2020;45:262-263.
- Treudler R, Delaroque N, Puder M, et al. Dupilumab-induced serum sickness-like reaction: an unusual adverse effect in a patient with atopic eczema. J Eur Acad Dermatol Venereol. 2021;35:E30-E32.
- Guenova E, Skabytska Y, Hoetzenecker W, et al. IL-4 abrogates TH17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci U S A. 2015;112:2163-2168.
Practice Points
- Prescribers of dupilumab should be aware that Sweet syndrome is a potential adverse reaction.
- Sweet syndrome should be suspected if there is abrupt onset of painful erythematous plaques or nodules accompanied by pyrexia following injection of dupilumab. Biopsy of the nodules should be obtained to confirm the diagnosis.
- Systemic corticosteroids with cessation of dupilumab are effective treatments.
- Following treatment, dupilumab should not be reinitiated, and alternative therapies should be used.

Dyshidroticlike Contact Dermatitis and Paronychia Resulting From a Dip Powder Manicure
To the Editor:
A 58-year-old woman presented to our dermatology clinic with a pruritic weeping eruption circumferentially on the distal digits of both hands of 5 weeks’ duration. The patient disclosed that she had been receiving dip powder manicures at a local nail salon approximately every 2 weeks over the last 3 to 6 months. She had received frequent acrylic nail extensions over the last 8 years prior to starting the dip powder manicures. Physical examination revealed well-demarcated eczematous plaques involving the lateral and proximal nail folds of the right thumb with an overlying serous crust and loss of the cuticle (Figure 1A). Erythematous plaques with firm deep-seated microvesicles also were present on the other digits, distributed distal to the distal interphalangeal joints (Figure 1B). She was diagnosed with dyshidroticlike contact dermatitis and paronychia. Treatment included phenol 1.5% colorless solution and clobetasol ointment 0.05% for twice-daily application to the affected areas. The patient also was advised to stop receiving manicures. At 1-month follow-up, the paronychia had resolved and the dermatitis had nearly resolved.

Dip powder manicures use a wet adhesive base coat with acrylic powder and an activator topcoat to initiate a chemical reaction that hardens and sets the nail polish. The colored powder typically is applied by dipping the digit up to the distal interphalangeal joint into a small container of loose powder and then brushing away the excess (Figure 2). Acrylate, a chemical present in dip powders, is a known allergen and has been associated with the development of allergic contact dermatitis and onychodystrophy in patients after receiving acrylic and UV-cured gel polish manicures.1,2 Inadequate sanitation practices at nail salons also have been associated with infection transmission.3,4 Additionally, the news media has covered the potential risk of infection due to contamination from reused dip manicure powder and the use of communal powder containers.5

To increase clinical awareness of the dip manicure technique, we describe the presentation and successful treatment of dyshidroticlike contact dermatitis and paronychia that occurred in a patient after she received a dip powder manicure. Dermatoses and infection limited to the distal phalanges will present in patients more frequently as dip powder manicures continue to increase in popularity and frequency.
- Baran R. Nail cosmetics: allergies and irritations. Am J Clin Dermatol. 2002;3:547-555.
- Chen AF, Chimento SM, Hu S, et al. Nail damage from gel polish manicure. J Cosmet Dermatol. 2012;11:27-29.
- Schmidt AN, Zic JA, Boyd AS. Pedicure-associated Mycobacterium chelonae infection in a hospitalized patient. J Am Acad Dermatol. 2014;71:E248-E250.
- Sniezek PJ, Graham BS, Busch HB, et al. Rapidly growing mycobacterial infections after pedicures. Arch Dermatol. 2003;139:629-634.
- Joseph T. You could be risking an infection with nail dipping. NBC Universal Media, LLC. Updated July 11, 2019. Accessed June 7, 2023. https://www.nbcmiami.com/news/local/You-Could-Be-Risking-an-Infection-with-Nail-Dipping-512550372.html
To the Editor:
A 58-year-old woman presented to our dermatology clinic with a pruritic weeping eruption circumferentially on the distal digits of both hands of 5 weeks’ duration. The patient disclosed that she had been receiving dip powder manicures at a local nail salon approximately every 2 weeks over the last 3 to 6 months. She had received frequent acrylic nail extensions over the last 8 years prior to starting the dip powder manicures. Physical examination revealed well-demarcated eczematous plaques involving the lateral and proximal nail folds of the right thumb with an overlying serous crust and loss of the cuticle (Figure 1A). Erythematous plaques with firm deep-seated microvesicles also were present on the other digits, distributed distal to the distal interphalangeal joints (Figure 1B). She was diagnosed with dyshidroticlike contact dermatitis and paronychia. Treatment included phenol 1.5% colorless solution and clobetasol ointment 0.05% for twice-daily application to the affected areas. The patient also was advised to stop receiving manicures. At 1-month follow-up, the paronychia had resolved and the dermatitis had nearly resolved.
Dip powder manicures use a wet adhesive base coat with acrylic powder and an activator topcoat to initiate a chemical reaction that hardens and sets the nail polish. The colored powder typically is applied by dipping the digit up to the distal interphalangeal joint into a small container of loose powder and then brushing away the excess (Figure 2). Acrylate, a chemical present in dip powders, is a known allergen and has been associated with the development of allergic contact dermatitis and onychodystrophy in patients after receiving acrylic and UV-cured gel polish manicures.1,2 Inadequate sanitation practices at nail salons also have been associated with infection transmission.3,4 Additionally, the news media has covered the potential risk of infection due to contamination from reused dip manicure powder and the use of communal powder containers.5
To increase clinical awareness of the dip manicure technique, we describe the presentation and successful treatment of dyshidroticlike contact dermatitis and paronychia that occurred in a patient after she received a dip powder manicure. Dermatoses and infection limited to the distal phalanges will present in patients more frequently as dip powder manicures continue to increase in popularity and frequency.
To the Editor:
A 58-year-old woman presented to our dermatology clinic with a pruritic weeping eruption circumferentially on the distal digits of both hands of 5 weeks’ duration. The patient disclosed that she had been receiving dip powder manicures at a local nail salon approximately every 2 weeks over the last 3 to 6 months. She had received frequent acrylic nail extensions over the last 8 years prior to starting the dip powder manicures. Physical examination revealed well-demarcated eczematous plaques involving the lateral and proximal nail folds of the right thumb with an overlying serous crust and loss of the cuticle (Figure 1A). Erythematous plaques with firm deep-seated microvesicles also were present on the other digits, distributed distal to the distal interphalangeal joints (Figure 1B). She was diagnosed with dyshidroticlike contact dermatitis and paronychia. Treatment included phenol 1.5% colorless solution and clobetasol ointment 0.05% for twice-daily application to the affected areas. The patient also was advised to stop receiving manicures. At 1-month follow-up, the paronychia had resolved and the dermatitis had nearly resolved.
Dip powder manicures use a wet adhesive base coat with acrylic powder and an activator topcoat to initiate a chemical reaction that hardens and sets the nail polish. The colored powder typically is applied by dipping the digit up to the distal interphalangeal joint into a small container of loose powder and then brushing away the excess (Figure 2). Acrylate, a chemical present in dip powders, is a known allergen and has been associated with the development of allergic contact dermatitis and onychodystrophy in patients after receiving acrylic and UV-cured gel polish manicures.1,2 Inadequate sanitation practices at nail salons also have been associated with infection transmission.3,4 Additionally, the news media has covered the potential risk of infection due to contamination from reused dip manicure powder and the use of communal powder containers.5
To increase clinical awareness of the dip manicure technique, we describe the presentation and successful treatment of dyshidroticlike contact dermatitis and paronychia that occurred in a patient after she received a dip powder manicure. Dermatoses and infection limited to the distal phalanges will present in patients more frequently as dip powder manicures continue to increase in popularity and frequency.
- Baran R. Nail cosmetics: allergies and irritations. Am J Clin Dermatol. 2002;3:547-555.
- Chen AF, Chimento SM, Hu S, et al. Nail damage from gel polish manicure. J Cosmet Dermatol. 2012;11:27-29.
- Schmidt AN, Zic JA, Boyd AS. Pedicure-associated Mycobacterium chelonae infection in a hospitalized patient. J Am Acad Dermatol. 2014;71:E248-E250.
- Sniezek PJ, Graham BS, Busch HB, et al. Rapidly growing mycobacterial infections after pedicures. Arch Dermatol. 2003;139:629-634.
- Joseph T. You could be risking an infection with nail dipping. NBC Universal Media, LLC. Updated July 11, 2019. Accessed June 7, 2023. https://www.nbcmiami.com/news/local/You-Could-Be-Risking-an-Infection-with-Nail-Dipping-512550372.html
- Baran R. Nail cosmetics: allergies and irritations. Am J Clin Dermatol. 2002;3:547-555.
- Chen AF, Chimento SM, Hu S, et al. Nail damage from gel polish manicure. J Cosmet Dermatol. 2012;11:27-29.
- Schmidt AN, Zic JA, Boyd AS. Pedicure-associated Mycobacterium chelonae infection in a hospitalized patient. J Am Acad Dermatol. 2014;71:E248-E250.
- Sniezek PJ, Graham BS, Busch HB, et al. Rapidly growing mycobacterial infections after pedicures. Arch Dermatol. 2003;139:629-634.
- Joseph T. You could be risking an infection with nail dipping. NBC Universal Media, LLC. Updated July 11, 2019. Accessed June 7, 2023. https://www.nbcmiami.com/news/local/You-Could-Be-Risking-an-Infection-with-Nail-Dipping-512550372.html
Practice Points
- Manicures performed at nail salons have been associated with the development of paronychia due to inadequate sanitation practices and contact dermatitis caused by acrylates present in nail polish.
- The dip powder manicure is a relatively new manicure technique. The distribution of dermatoses and infection limited to the distal phalanges will present in patients more frequently as dip powder manicures continue to increase in popularity and are performed more frequently.

White Spots on the Extremities
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
.")
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
.")
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
The Diagnosis: Hypopigmented Mycosis Fungoides
Histopathology showed an atypical lymphoid infiltrate with expanded cytoplasm and hyperchromatic nuclei of irregular contours in the dermoepidermal junction (Figure 1). Immunohistochemical stains of atypical lymphocytes demonstrated the presence of CD3, CD8, and CD5, as well as the absence of CD7 and CD4 lymphocytes (Figure 2). The T-cell γ rearrangement showed polyclonal lymphocytes with 5% tumor cells. The histologic and clinical findings along with our patient’s medical history led to a diagnosis of stage IA (<10% body surface area involvement) hypopigmented mycosis fungoides (hMF).1 Our patient was treated with triamcinolone cream 0.1%; she noted an improvement in her symptoms at 2-month follow-up.
Hypopigmented MF is an uncommon manifestation of MF with unknown prevalence and incidence rates. Mycosis fungoides is considered the most common subtype of cutaneous T-cell lymphoma that classically presents as a chronic, indolent, hypopigmented or depigmented macule or patch, commonly with scaling, in sunprotected areas such as the trunk and proximal arms and legs. It predominantly affects younger adults with darker skin tones and may be present in the pediatric population within the first decade of life.1 Classically, MF affects White patients aged 55 to 60 years. Disease progression is slow, with an incidence rate of 10% of tumor or extracutaneous involvement in the early stages of disease. A lack of specificity on the clinical and histopathologic findings in the initial stage often contributes to the diagnostic delay of hMF. As seen in our patient, this disease can be misdiagnosed as tinea versicolor, postinflammatory hypopigmentation, vitiligo, pityriasis alba, subcutaneous lupus erythematosus, or Hansen disease due to prolonged hypopigmented lesions.2 The clinical findings and histopathologic results including immunohistochemistry confirmed the diagnosis of hMF and ruled out pityriasis alba, postinflammatory hypopigmentation, subcutaneous lupus erythematosus, and vitiligo.
The etiology and pathophysiology of hMF are not fully understood; however, it is hypothesized that melanocyte degeneration, abnormal melanogenesis, and disturbance of melanosome transfer result from the clonal expansion of T helper memory cells. T-cell dyscrasia has been reported to evolve into hMF during etanercept therapy.3 Clinically, hMF presents as hypopigmented papulosquamous, eczematous, or erythrodermic patches, plaques, and tumors with poorly defined atrophied borders. Multiple biopsies of steroid-naive lesions are needed for the diagnosis, as the initial hMF histologic finding cannot be specific for diagnostic confirmation. Common histopathologic findings include a bandlike lymphocytic infiltrate with epidermotropism, intraepidermal nests of atypical cells, or cerebriform nuclei lymphocytes on hematoxylin and eosin staining. In comparison to classical MF epidermotropism, CD4− and CD8+ atypical cells aid in the diagnosis of hMF. Although hMF carries a good prognosis and a benign clinical course,4 full-body computed tomography or positron emission tomography/computed tomography as well as laboratory analysis for lactate dehydrogenase should be pursued if lymphadenopathy, systemic symptoms, or advancedstage hMF are present.
Treatment of hMF depends on the disease stage. Psoralen plus UVA and narrowband UVB can be utilized for the initial stages with a relatively fast response and remission of lesions as early as the first 2 months of treatment. In addition to phototherapy, stage IA to IIA mycosis fungoides with localized skin lesions can benefit from topical steroids, topical retinoids, imiquimod, nitrogen mustard, and carmustine. For advanced stages of mycosis fungoides, combination therapy consisting of psoralen plus UVA with an oral retinoid, interferon alfa, and systemic chemotherapy commonly are prescribed. Maintenance therapy is used for prolonging remission; however, long-term phototherapy is not recommended due to the risk for skin cancer. Unfortunately, hMF requires long-term treatment due to its waxing and waning course, and recurrence may occur after complete resolution.5
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
- Furlan FC, Sanches JA. Hypopigmented mycosis fungoides: a review of its clinical features and pathophysiology. An Bras Dermatol. 2013;88:954-960.
- Lambroza E, Cohen SR, Lebwohl M, et al. Hypopigmented variant of mycosis fungoides: demography, histopathology, and treatment of seven cases. J Am Acad Dermatol. 1995;32:987-993.
- Chuang GS, Wasserman DI, Byers HR, et al. Hypopigmented T-cell dyscrasia evolving to hypopigmented mycosis fungoides during etanercept therapy. J Am Acad Dermatol. 2008;59(5 suppl):S121-S122.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/ European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014; 70:223.e1-17; quiz 240-242.
A 52-year-old Black woman presented with self-described whitened spots on the arms and legs of 2 years’ duration. She experienced no improvement with ketoconazole cream and topical calcineurin inhibitors prescribed during a prior dermatology visit at an outside institution. She denied pain or pruritus. A review of systems as well as the patient’s medical history were noncontributory. A prior biopsy at an outside institution revealed an interface dermatitis suggestive of cutaneous lupus erythematosus. The patient noted social drinking and denied tobacco use. She had no known allergies to medications and currently was on tamoxifen for breast cancer following a right mastectomy. Physical examination showed hypopigmented macules and patches on the left upper arm and right proximal leg. The center of the lesions was not erythematous or scaly. Palpation did not reveal enlarged lymph nodes, and laboratory analyses ruled out low levels of red blood cells, white blood cells, or platelets. Punch biopsies from the left arm and right thigh were performed.


Erythematous Dermal Facial Plaques in a Neutropenic Patient
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
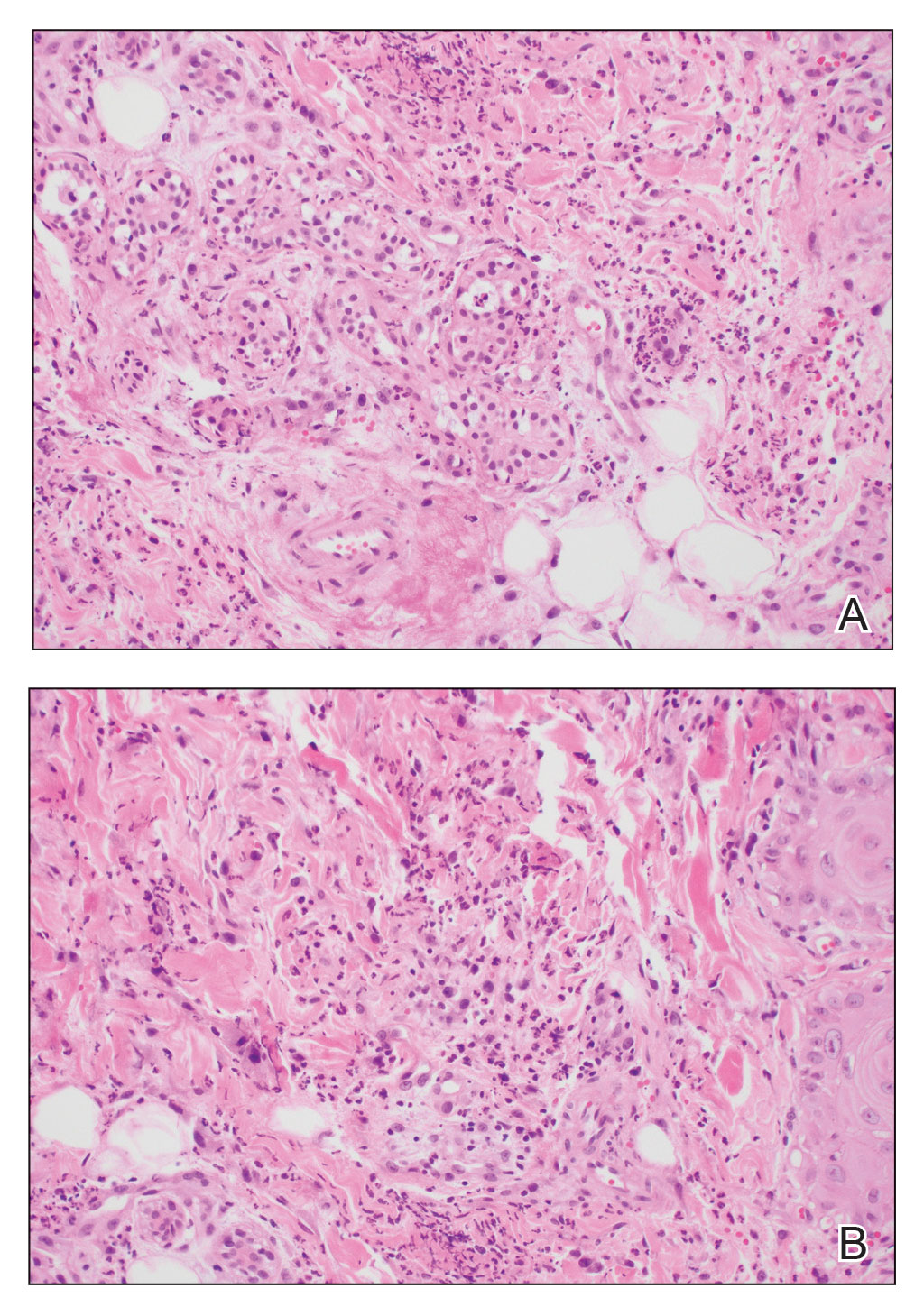
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
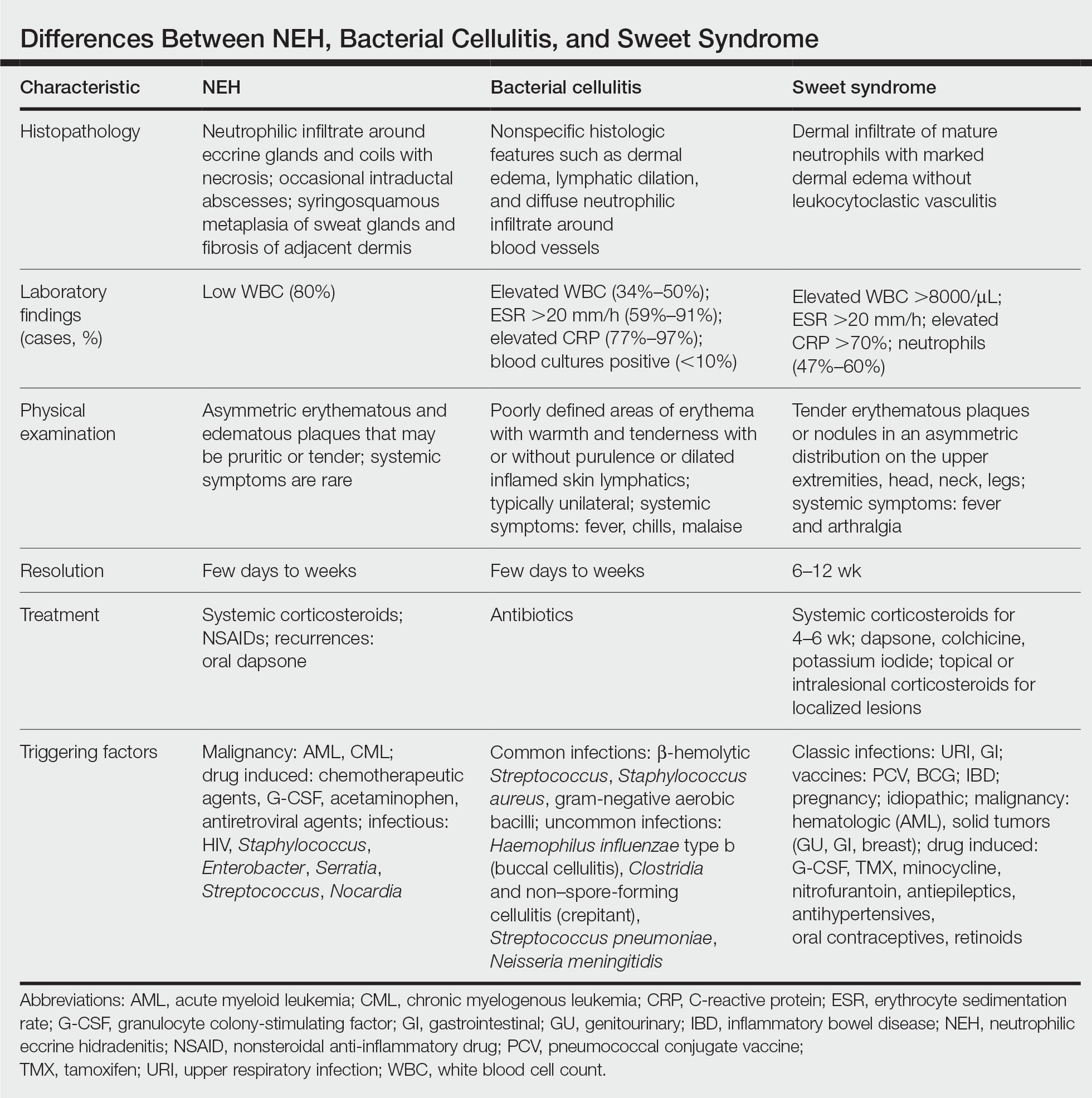
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
THE DIAGNOSIS: Neutrophilic Eccrine Hidradenitis
A biopsy from the left preauricular cheek demonstrated dermal neutrophilic inflammation around eccrine coils with focal necrosis (Figure). No notable diffuse dermal neutrophilic infiltrate was present, ruling out Sweet syndrome, and no notable interstitial neutrophilic infiltrate was present, making cellulitis and erysipelas less likely; panculture of tissue also was negative.1,2 Atypical cells in the deep dermis were positive for CD163 and negative for CD117, CD34, CD123, and myeloperoxidase, consistent with a diagnosis of neutrophilic eccrine hidradenitis (NEH) and reactive histiocytes.3 Treatment with oral prednisone resulted in rapid improvement of symptoms.
Neutrophilic eccrine hidradenitis is a rare reactive neutrophilic dermatosis characterized by eccrine gland involvement. This benign and self-limited condition presents as asymmetric erythematous papules and plaques.2 Among 8 granulocytopenic patients with neutrophilic dermatoses, 5 were diagnosed with NEH.4 Although first identified in 1982, NEH remains poorly understood.2 Initial theories suggested that NEH developed due to cytotoxic substances secreted in sweat glands causing necrosis and neutrophil chemotaxis; however, chemotherapy exposure cannot be linked to every case of NEH. Neutrophilic eccrine hidradenitis can be extremely difficult to differentiate clinically from conditions such as cellulitis and Sweet syndrome.
A patient history can be helpful in identifying triggering factors. Neutrophilic eccrine hidradenitis most commonly is associated with malignant, drug-induced, or infectious triggers, while Sweet syndrome has a broad range of associations including infections, vaccines, inflammatory bowel disease, pregnancy, malignancy, and drug-induced etiologies (Table).1 On average, NEH presents 10 days after chemotherapy induction, with 70% of cases presenting after the first chemotherapy cycle.5 Bacterial cellulitis or erysipelas have an infectious etiology, and patients may report symptoms such as fever, chills, or malaise. Immunosuppressed patients are at greater risk for infection; therefore, clinical signs of infection in a granulocytopenic patient should be addressed urgently.
Physical examination may have limited value in differentiating between these diagnoses, as neutrophilic dermatoses notoriously mimic infection. Cutaneous lesions can appear as pruritic or tender erythematous plaques, papules, or nodules in these conditions, though cellulitis and erysipelas tend to be unilateral and may have associated purulence or inflamed skin lymphatics. Given the potential for misdiagnosis, approaching patients with a broad differential can be helpful. In our patient, the differential diagnosis included Sweet syndrome, NEH, bacterial cellulitis, erysipelas, leukemia cutis, sarcoid, and eosinophilic cellulitis.
Leukemia cutis refers to the infiltration of neoplastic leukocytes in the skin and often occurs in patients with peripheral leukemia, most often acute myeloid leukemia or chronic lymphocytic leukemia. Patients with leukemia cutis often have a worse prognosis, as this finding signifies extramedullary spread of disease.6 Clinically, lesions can appear similar to those seen in our patient, though they typically are not symptomatic, can be nodular, tend to exhibit a violaceous hue, and occasionally may be hemorrhagic. Wells syndrome (also known as eosinophilic cellulitis) is an inflammatory dermatosis that presents as painful or pruritic, edematous and erythematous plaques.7,8 A green hue on resolution is present in some cases and may help clinicians differentiate this disease from mimickers.7 Often, eosinophilic cellulitis is misdiagnosed as bacterial cellulitis and treated with antibiotics. The presence of systemic symptoms such as fever or arthralgia is more typical of bacterial cellulitis, erysipelas, eosinophilic cellulitis, or Sweet syndrome than of NEH.1 Additionally, inflammatory markers (ie, C-reactive protein) and white blood cell counts tend to be elevated in bacterial cellulitis and Sweet syndrome, while leukopenia often is seen in NEH.
Histopathology is crucial in distinguishing these disease etiologies. Neutrophilic eccrine hidradenitis is diagnosed by the characteristic neutrophilic infiltrate and necrosis surrounding eccrine glands and coils. There also may be occasional intraductal abscesses and syringosquamous metaplasia of the sweat glands along with fibrosis of the adjacent dermis. In contrast, histologic sections of Sweet syndrome show numerous mature neutrophils infiltrating the dermis with marked papillary dermal edema. The histopathology of bacterial cellulitis and erysipelas is less specific, but common features include dermal edema, lymphatic dilation, and a diffuse neutrophilic infiltrate surrounding blood vessels. Pathogenic organisms may be seen on histopathology but are not required for the diagnosis of bacterial cellulitis or erysipelas.2 Additionally, blood and tissue culture can assist in identification of both the source of infection and the causative organism, but cultures may not always be positive.
Comparatively, the histopathologic features of eosinophilic cellulitis include dermal edema, eosinophilic infiltration, and flame figures that form when eosinophils degranulate and coat collagen fibers with major basic protein. Flame figures are characteristic but not pathognomonic for eosinophilic cellulitis.7 The histopathology of leukemia cutis varies based on the leukemia classification; generally, in acute myeloid leukemia the infiltrate is composed of neoplastic cells in the early stages of development that are positive for myeloid markers such as myeloperoxidase. Atypical and immature granulocytes within the leukocytic infiltrate differentiate this condition from the other diagnoses. Treatment may entail chemotherapy or radiotherapy, and this diagnosis generally carries the worst prognosis of all the conditions in the differential.6
Differentiating between these conditions is important in guiding treatment, especially in patients with febrile neutropenia. Unnecessary steroids in immunosuppressed patients can be dangerous, especially if the patient has an infection such as bacterial cellulitis. Furthermore, unwarranted antibiotic use for noninfectious conditions may expose patients to substantial side effects and not improve the condition. Neutrophilic eccrine hidradenitis typically is self-limited and treated symptomatically with systemic corticosteroids and nonsteroidal anti-inflammatory drugs.3 Sweet syndrome often requires a longer course of oral steroids. Leukemia cutis worsens as the leukemia advances, and treatment of the underlying malignancy is the most effective treatment.9
Early and accurate recognition of the diagnosis can prevent harmful diagnostic delay, unnecessary antibiotic use, or extended steroid taper in neutropenic patients. Appreciating the differences between these diagnoses can assist clinicians in investigating and tailoring a broad differential to specific patient presentations, which is especially critical when considering common mimickers for life-threatening conditions.
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
- Nelson CA, Stephen S, Ashchyan HJ, et al. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79:987-1006. doi:10.1016/j.jaad.2017.11.0642
- Srivastava M, Scharf S, Meehan SA, et al. Neutrophilic eccrine hidradenitis masquerading as facial cellulitis. J Am Acad Dermatol. 2007;56:693-696. doi:10.1016/j.jaad.2006.07.032
- Copaescu AM, Castilloux JF, Chababi-Atallah M, et al. A classic clinical case: neutrophilic eccrine hidradenitis. Case Rep Dermatol. 2013; 5:340-346. doi:10.1159/000356229
- Aractingi S, Mallet V, Pinquier L, et al. Neutrophilic dermatoses during granulocytopenia. Arch Dermatol. 1995;131:1141-1145.
- Cohen PR. Neutrophilic dermatoses occurring in oncology patients. Int J Dermatol. 2007;46:106-111. doi:10.1111/j.1365-4632.2006.02605.x
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol. 2006;5:908-911.
- Räßler F, Lukács J, Elsner P. Treatment of eosinophilic cellulitis (Wells syndrome): a systematic review. J Eur Acad Dermatol Venereol. 2016;30:1465-1479. doi:10.1111/jdv.13706
- Hobbs LK, Carr PC, Gru AA, et al. Case and review: cutaneous involvement by chronic neutrophilic leukemia vs Sweet syndrome: a diagnostic dilemma. J Cutan Pathol. 2021;48:644-649. doi:10.1111 /cup.13925
A 50-year-old woman undergoing cytarabine induction therapy for acute myeloid leukemia developed tender, erythematous, dermal plaques on the nasal dorsum, left medial eyebrow, left preauricular cheek, and right cheek. The rash erupted 7 days after receiving the cytarabine induction regimen. She had a fever (temperature, 39.9 °C [103.8 °F]) and also was neutropenic.
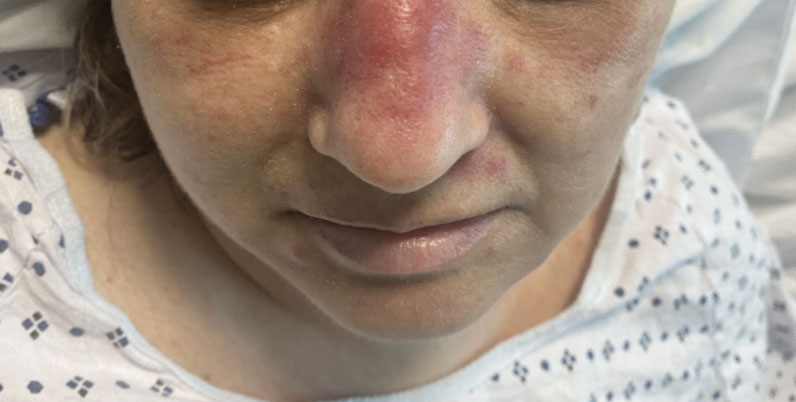
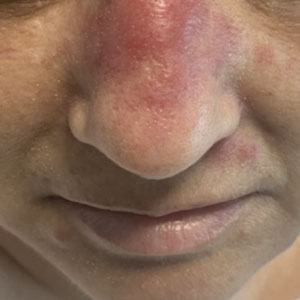
Long-term Remission of Pyoderma Gangrenosum, Acne, and Hidradenitis Suppurativa Syndrome
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.

Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.

After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.

The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.

Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.

PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
Pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS)(PASH) syndrome is a recently identified disease process within the spectrum of autoinflammatory diseases (AIDs), which are distinct from autoimmune, infectious, and allergic syndromes and are gaining increasing interest given their complex pathophysiology and therapeutic resistance.1 Autoinflammatory diseases are defined by a dysregulation of the innate immune system in the absence of typical autoimmune features, including autoantibodies and antigen-specific T lymphocytes.2 Mutations affecting proteins of the inflammasome or proteins involved in regulating inflammasome function have been associated with these AIDs.2
Many AIDs have cutaneous involvement, as seen in PASH syndrome. Pyoderma gangrenosum is a neutrophilic dermatosis presenting as skin ulcers with undermined, erythematous, violaceous borders. It can be isolated, syndromic, or associated with inflammatory conditions (eg, inflammatory bowel disease, rheumatologic disorders, hematologic disorders).1 Acne vulgaris develops because of chronic obstruction of hair follicles as a result of disordered keratinization and abnormal sebaceous stem cell differentiation.2 Propionibacterium acnes can reside and replicate within the biofilm community of the hair follicle and activate the inflammasome.2,3 Hidradenitis suppurativa, a chronic relapsing neutrophilic dermatosis, is a debilitating inflammatory disease of the hair follicles involving apocrine gland–bearing skin (ie, the axillary, inguinal, and anogenital regions).2 Onset often occurs between the ages of 20 and 40 years, with a 3-fold higher incidence in women compared to men.3 Patients experience painful, deep-seated nodules that drain into sinus tracts and abscesses. The condition can be isolated or associated with inflammatory conditions, such as inflammatory bowel disease.4
PASH syndrome has been described as a polygenic autoinflammatory condition that most commonly presents in young adults, with onset of acne beginning years prior to other manifestations. A study analyzing 5 patients with PASH syndrome reported an average age of 32.2 years at diagnosis with a disease duration of 3 to 7 years.5 Pathophysiology of this condition is not well understood, with many hypotheses calling upon dysregulation of the innate immune system, a commonality this syndrome may share with other AIDs. Given its poorly understood pathophysiology, treating PASH syndrome can be especially difficult. We report a novel case of disease remission lasting more than 4 years using adalimumab and cyclosporine. We also discuss prior treatment successes and hypotheses regarding etiologic factors in PASH syndrome.
Case Report
A 36-year-old woman presented for evaluation of open draining ulcerations on the back of 18 months’ duration. She had a 16-year history of scarring cystic acne of the face and HS of the groin. The patient’s family history was remarkable for severe cystic acne in her brother and son as well as HS in her mother and another brother. Her treatment history included isotretinoin, doxycycline, and topical steroids.
Physical examination revealed 2 ulcerations with violaceous borders involving the left upper back (greatest diameter, 5×7 cm)(Figure 1). Evidence of papular and cystic acne with residual scarring was noted on the cheeks. Scarring from HS was noted in the axillae and right groin. A biopsy from the edge of an ulceration on the back demonstrated epidermal spongiosis with acute and chronic inflammation and fibrosis (Figure 2). The clinicopathologic findings were most consistent with PG, and the patient was diagnosed with PASH syndrome, given the constellation of cutaneous lesions.
After treatment with topical and systemic antibiotics for acne and HS for more than 1 year failed, the patient was started on adalimumab. The initial dose was 160 mg subcutaneously, then 80 mg 2 weeks later, then 40 mg weekly thereafter. Doxycycline was continued for treatment of the acne and HS. After 6 weeks of adalimumab, the PG worsened and prednisone was added. She developed tender furuncles on the back, and cultures grew Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus that responded to ciprofloxacin and cephalexin.
Due to progression of PG on adalimumab, switching to an infliximab infusion or anakinra was considered, but these options were not covered by the patient’s health insurance. Three months after the initial presentation, the patient was started on cyclosporine 100 mg 3 times daily (5 mg/kg/d) while adalimumab was continued; the ulcers started to improve within 2.5 weeks. After 3 months (Figure 3), the cyclosporine was reduced to 100 mg twice daily, and adalimumab was continued. She had a slight flare of PG after 8 months of treatment when adalimumab was unavailable to her for 2 months. After 8 months on cyclosporine, the dosage was tapered to 100 mg/d and then completely discontinued after 12 months.
The patient has continued on adalimumab 40 mg weekly with excellent control of the PG (Figure 4), although she did have one HS flare in the left axilla 11 months after the initial treatment. The patient’s cystic acne has intermittently flared and has been managed with spironolactone 100 mg/d for 3 years. After 4 years of management, the patient’s PG and HS remain well controlled on adalimumab.
Comment
Our case represents a major step in refining long-term treatment approaches for PASH syndrome due to the 4-year remission. Prior cases have reported use of anakinra, anakinra-cyclosporine combination, prednisone, azathioprine, topical tacrolimus, etanercept, and dapsone without sustainable success.1-6 The case studies discussed below have achieved remission via alternative drug combinations.
Staub et al4 found greatest success with a combination of infliximab, dapsone, and cyclosporine, and their patient had been in remission for 20 months at time of publication. Their hypothesis proposed that multiple inflammatory signaling pathways are involved in PASH syndrome, and this is why combination therapy is required for remission.4 In 2018, Lamiaux et al7 demonstrated successful treatment with rifampicin and clindamycin. Their patient had been in remission for 22 months at the time of publication—this time frame included 12 months of combination therapy and 10 months without medication. The authors hypothesized that, because of the autoinflammatory nature of these antibiotics, this pharmacologic combination could eradicate pathogenic bacteria from host microbiota while also inhibiting neutrophil function and synthesis of chemokines and cytokines.7
More recently, reports have been published regarding the success of tildrakizumab, an IL-23 antagonist, and ixekizumab, an IL-17 antagonist, in the treatment of PASH syndrome.6,8 Ixekizumab was used in combination with doxycycline, and remission was achieved in 12 months.8 However, tildrakizumab was used alone and achieved greater than 75% improvement in disease manifestations within 2 months.
Marzano et al5 conducted protein arrays and enzyme-linked immunosorbent assay to analyze the expression of cytokine, chemokine, and effector molecule profiles in PASH syndrome. It was determined that serum analysis displayed a normal cytokine/chemokine profile, with the only abnormalities being anemia and elevated C-reactive protein. There were no statistically significant differences in serum levels of IL-1β, tumor necrosis factor (TNF) α, or IL-17 between PASH syndrome and healthy controls. However, cutaneous analysis revealed extensive cytokine and chemokine hyperactivity for IL-1β and IL-1β receptor; TNF-α; C-X-C motif ligands 1, 2, and 3; C-X-C motif ligand 16;
Ead et al3 presented a unique perspective focusing on cutaneous biofilm involvement in PASH syndrome. Microbes within these biofilms induce the migration and proliferation of inflammatory cells that consume factors normally utilized for tissue catabolism. These organisms deplete necessary biochemical cofactors used during healing. This lack of nutrients needed for healing not only slows the process but also promotes favorable conditions for the growth of anerobic species. In conjunction, biofilm formation restricts bacterial access to oxygen and nutrients, thus decreasing the bacterial metabolic rate and preventing the effects of antibiotic therapy. These features of biofilm communities contribute to inflammation and possibly the troubling resistance to many therapeutic options for PASH syndrome.
Each component of PASH syndrome has been associated with biofilm formation. As previously described, PG manifests in the skin as painful ulcerations, often with slough. This slough is hypothesized to be a consequence of increased vascular permeability and exudative byproducts that accompany the inflammatory nature of biofilms.3 Acne vulgaris has well-described associations with P acnes. Ead et al3 described P acnes as a component of the biofilm community within the microcomedone of hair follicles. This biofilm allows for antibiotic resistance occasionally seen in the treatment of acne and is potentially the pathogenic factor that both impedes healing and enhances the inflammatory state. Hidradenitis suppurativa has been associated with biofilm formation.3
In further pursuit of PASH syndrome pathophysiology, many experts have sought to uncover the relationship between PASH syndrome and the previously described pyogenic arthritis, PG, and acne (PAPA) syndrome, another entity within the AIDs spectrum (Table). This condition was first recognized in 1997 in a 3-generation family with 10 affected members.1 It is characterized by PG and acne, similar to PASH; however, PAPA syndrome includes PG arthritis and lacks HS. Pyogenic arthritis manifests as recurrent aseptic inflammation of the joints, mainly the elbows, knees, and ankles. Pyogenic arthritis commonly is the presenting symptom of PAPA syndrome, with onset in childhood.2 As patients age, the arthritic symptoms decrease, and skin manifestations become more prominent.
PAPA syndrome has autosomal-dominant inheritance with mutations on chromosome 15 in the proline-serine-threonine phosphatase interacting protein 1 (PSTPIP1) gene.1 This mutation induces hyperphosphorylation of PSTPIP1, allowing for increased binding affinity to pyrin. Both PSTPIP1 and pyrin are co-expressed as parts of the NLRP3 inflammasome in granulocytes and monocytes.1 As a result, pyrin is more highly bound and loses its inhibitory effect on the NLRP3 inflammasome pathway. This lack of inhibition allows for uninhibited cleavage of pro–IL-1β to active IL-1β by the inflammasome.1
Elevated concentrations of IL-1β in patients with PAPA syndrome result in a dysregulation of the innate immune system. IL-1β induces the release of proinflammatory cytokines, namely TNF-α; interferon γ; IL-8; and regulated on activation, normal T cell expressed and secreted (RANTES), all of which activate neutrophils and induce neutrophilic inflammation.2 IL-1β not only initiates this entire cascade but also acts as an antiapoptotic signal for neutrophils.2 When IL-1β reaches a critical threshold, it induces enough inflammation to cause severe tissue damage, thus causing joint and cutaneous disease in PAPA syndrome. IL-1 inhibitors (anakinra) or TNF-α inhibitors (etanercept, adalimumab, infliximab) have been used many times to successfully treat PAPA syndrome, with TNF-α inhibitors providing the most consistent results.
Another AIDs entity with similarities to both PAPA syndrome and PASH syndrome is pyogenic arthritis, PG, acne, and HS (PA-PASH) syndrome. First identified in 2012 by Bruzzese,9 genetic analyses revealed a p.E277D missense mutation in PSTPIP1 in PA-PASH syndrome. Research has suggested that the key molecular feature is neutrophil activation by TH17 cells and the TNF-α axis.9 This syndrome has not been further characterized, and little is known regarding adequate treatment for PA-PASH syndrome.
Although it is similar in phenotype to aspects of PAPA and PA-PASH syndromes, PASH syndrome has distinct genotypic and immunologic abnormalities. Genetic analysis of this condition has shown an increased number of CCTG repeats in proximity to the PSTPIP1 promoter. It is hypothesized that these additional repeats predispose patients to neutrophilic inflammation in a similar manner to a condition described in France, termed aseptic abscess syndrome.1,5 Other mutations have been identified, including those in IL-1N, PSMB8, MEFV, NOD2, NCSTN, and more.2,7 However, it has been determined that the majority of these variants have already been filed in the Single Nucleotide Polymorphism Database or in the Registry of Hereditary Auto-inflammatory Disorders Mutations.2 The question remains regarding the origin of inflammation seen in PASH syndrome; the potential role of biofilms; and the relationship between PASH, PAPA, and PA-PASH syndromes. Much work remains to be done in refining therapeutic options for PASH syndrome. Continued biochemical research is necessary, as well as collaboration among dermatologists worldwide who find success in treating this condition.
Conclusion
There are genotypic and phenotypic similarities between PASH, PAPA, and PA-PASH syndromes, with various mutations within or near the PSTPIP1 gene; however, their genetic discrepancies seem to play a major role in the pathophysiology of each syndrome. Much work remains to be done in PA-PASH syndrome, which has not yet been well described. Meanwhile, PAPA syndrome has been well characterized with mutations affecting proteins of the NLRP3 inflammasome, resulting in elevated IL-1β and excess neutrophilic inflammation. In PASH syndrome, the importance of increased repeats near the PSTPIP1 promoter is yet to be elucidated. It has been shown that these abnormalities predispose individuals to neutrophilic inflammation, but the mechanism by which they do so is unknown. In addition, consideration of biofilms and their predisposition to inflammation within the pathophysiology of PASH syndrome is a possibility that must be considered when discussing therapeutic options. Based on our case study and previous successes in treating PASH syndrome, it is clear that a multidrug approach is necessary for remission. It is likely that the etiology of PASH syndrome is multifaceted and involves hyperactivity in multiple arms of the innate immune system.
Patients with PASH syndrome have severely impaired quality of life and often experience social withdrawal due to the disfiguring sequelae and limited treatment options available. To improve patient outcomes, it is essential for physicians and scientists to report on successful treatment strategies and advances in immunologic understanding. Improved understanding of PASH syndrome calls for further genetic exploration into the role of additional genomic repeats and how these affect the PSTPIP1 gene and inflammasome activity. As medical advances improve understanding of the pathophysiology of this disease entity, it will likely become clear which mechanisms are most important in disease progression and how clinicians can best optimize treatment.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
- Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409-415.
- Cugno M, Borghi A, Marzano AV. PAPA, PASH and PAPASH syndromes: pathophysiology, presentation and treatment. Am J Clin Dermatol. 2017;18:555-562.
- Ead JK, Snyder RJ, Wise J, et al. Is PASH syndrome a biofilm disease?: a case series and review of the literature. Wounds. 2018;30:216-223.
- Staub J, Pfannschmidt N, Strohal R, et al. Successful treatment of PASH syndrome with infliximab, cyclosporine and dapsone. J Eur Acad Dermatol Venereol. 2015;29:2243-2247.
- Marzano AV, Ceccherini I, Gattorno M, et al. Association of pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) shares genetic and cytokine profiles with other autoinflammatory diseases. Medicine (Baltimore). 2014;93:E187.
- Kok Y, Nicolopoulos J, Varigos G, et al. Tildrakizumab in the treatment of PASH syndrome: a potential novel therapeutic target. Australas J Dermatol. 2020;61:E373-E374.
- Lamiaux M, Dabouz F, Wantz M, et al. Successful combined antibiotic therapy with oral clindamycin and oral rifampicin for pyoderma gangrenosum in patient with PASH syndrome. JAAD Case Rep. 2018;4:17-21.
- Gul MI, Singam V, Hanson C, et al. Remission of refractory PASH syndrome using ixekizumab and doxycycline. J Drugs Dermatol. 2020;19:1123.
- Bruzzese V. Pyoderma gangrenosum, acne conglobata, suppurative hidradenitis, and axial spondyloarthritis: efficacy of anti-tumor necrosis factor α therapy. J Clin Rheumatol. 2012;18:413-415.
Practice Points
- Despite phenotypic similarities among pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (PASH) syndrome; pyogenic arthritis, PG, and acne syndrome; and pyogenic arthritis–PASH syndrome, there are genotypic differences that contribute to unique inflammatory cytokine patterns and the need for distinct pharmacologic considerations within each entity.
- When formulating therapeutic regimens for patients with PASH syndrome, it is essential for dermatologists to consider the likelihood of hyperactivity in multiple pathways of the innate immune system and utilize a combination of multimodal antiinflammatory therapies.

Why Is There a Lack of Representation of Skin of Color in the COVID-19 Literature?
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
Throughout the COVID-19 pandemic, there has been a striking paucity of representations of patients with skin of color (SOC) in the dermatology literature. Was COVID-19 underdiagnosed in this patient population due to a lack of patient-centered resources and inadequate dermatology training; reduced access to care, resulting from social determinants of health and reduced skin-color concordance; or the absence of population-based prevalence studies?
Tan et al1 reviewed 51 articles describing skin findings secondary to COVID-19. Patients were stratified by country of origin, which yielded an increased prevalence of cutaneous manifestations among Americans and Europeans compared to Asians, but patients were not stratified by race.1 However, in one case series of 318 predominantly American patients, 89% were White and 0.7% were Black.2 This systematic review by Tan et al1 suggested that skin manifestations of COVID-19 were present in patients with SOC but less frequently than in White patients. However, case series are not a strong proxy for population-level prevalence.
More broadly, patients with SOC are underrepresented in Google image search results, as the medical resource websites (eg, DermNet [https://dermnetnz.org], MedicalNewsToday [www.medicalnewstoday.com], and Healthline [www.healthline.com]) are lacking these images.3 As a result, it is difficult for patients with SOC to recognize diseases presenting in darker skin types. This same tendency may exist for COVID-19 skin manifestations. A systematic review found that articles describing cutaneous manifestations of COVID-19 almost exclusively presented images of lighter skin and completely omitted darker skin.4 If images of patients with SOC are absent from online resources, it is increasingly unlikely for these patients to recognize if their skin lesions are associated with COVID-19, which may result in a decrease in the number of patients with SOC presenting with skin lesions secondary to COVID-19, thereby influencing the representation of patients with SOC in case studies.
The lack of representation of SOC in online resources mirrors the paucity of images in dermatology textbooks. According to a search of 7170 images in major dermatology textbooks, most images depicted light or white skin (80.6%), followed by medium or brown skin in 15.5% of images and dark or black skin in only 3.9%.5 Physicians rely on online and print resources for making diagnoses; inadequate resources highlight a component of a larger issue: inadequate training of dermatologists in SOC. In a survey of American dermatologists and dermatology residents (N=262), 47% thought that their medical education had not adequately trained them on skin conditions in Black patients.6
A lack of adequate training for dermatologists may decrease the rate of correct diagnosis of skin lesions secondary to COVID-19 in patients with SOC. A lack of trust in the health care system and social determinants of health may hinder patients with SOC from seeking medical help. Dermatology is the second least diverse of medical specialties; only 3% of dermatologists are Black.7 This is impactful: First, because minority physicians are increasingly likely to provide care for patients of the same race or background, and second, because race-concordant physician visits are associated with greater patient-reported positive affect.7 A lack of availability of race-concordant physicians or physicians with perceived cultural competence may deter patients with SOC from seeking help, which may be further prevalent in dermatologic practice.
Barriers at all levels of social determinants of health hinder access to health care. Patients with SOC experience greater housing insecurity, increased reliance on public transportation, more issues with health literacy, and limited English-language fluency.8 Combined, these factors equate to decreased access to health care resources and subsequently a lack of inclusion in case studies.
COVID-19 infection disproportionately affects patients with SOC,8 but there is a clear lack of representation of SOC in the COVID-19 dermatology literature. It is imperative to investigate factors that may contribute to this inequity. Recognizing skin manifestations can play a role in diagnosing COVID-19; increased awareness of its presentation in darker skin types may help bridge existing racial inequities. It is vital that physicians receive adequate resources and training to be able to recognize cutaneous manifestations of COVID-19 in all skin types. Finally, it is important to recognize that the lack of representation of SOC in the COVID-19 literature represents a larger trend that exists in dermatologic research that warrants further investigation and advocacy for inclusivity.
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815
- Tan SW, Tam YC, Oh CC. Skin manifestations of COVID-19: a worldwide review. JAAD Int. 2021;2:119-133. doi:10.1016/j.jdin.2020.12.003
- Freeman EE, McMahon DE, Lipoff JB, et al; American Academy of Dermatology Ad Hoc Task Force on COVID-19. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dematol. 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Fathy R, Lipoff JB. Lack of skin of color in Google image searches may reflect under-representation in all educational resources. J Am Acad Dermatol. 2022;86:E113-E114. doi:10.1016/j.jaad.2021.04.097
- Lester JC, Jia JL, Zhang L, et al. Absence of images of skin of colour in publications of COVID-19 skin manifestations. Br J Dermatol. 2020;183:593-595. doi:10.1111/bjd.19258
- Kamath P, Sundaram N, Morillo-Hernandez C, et al. Visual racism in internet searches and dermatology textbooks. J Am Acad Dermatol. 2021;85:1348-1349. doi:10.1016/j.jaad.2020.10.072
- Buster KJ, Stevens EI, Elmets CA. Dermatologic health disparities. Dermatol Clin. 2012;30:53-59,viii. doi:10.1016/j.det.2011.08.002
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587. doi:10.1016/j.jaad.2015.10.044
- Tai DBG, Shah A, Doubeni CA, et al. The disproportionate impact of COVID-19 on racial and ethnic minorities in the United States. Clin Infect Dis. 2021;72:703-706. doi:10.1093/cid/ciaa815