User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
What Is Your Diagnosis? Targetoid Hemosiderotic Hemangioma
A 45-year-old white woman presented with a solitary lesion on her lower back that had been present for approximately 2 years. The lesion changed monthly with menses. The patient described a brown papule that would enlarge, become deep purple in color, and develop a bruiselike rim with the onset of menses. Although the lesion was tender during menses, the tenderness and bruiselike rim would resolve following menses, and the lesion would revert back to a brown color. The patient was in good health and denied a history of previous trauma to the area or a history of abnormal vaginal bleeding or abdominal or pelvic pain. Physical examination revealed a 9x6 mm firm tan papule with a surrounding pale area and peripheral ecchymotic rim, resulting in a targetoid appearance. There were no other significant findings on cutaneous examination.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangiomas (THHs) were originally described in 1988 by Santa Cruz and Aronberg,1 who reported a series of patients with single vascular lesions with a distinctive targetoid appearance and characteristic histopathology. Despite some histopathologic similarities to malignant vascular neoplasms, these lesions followed a benign clinical course. Although now well recognized, a recent review of the literature revealed a total of only 46 reported cases of THHs.2 THHs appear most commonly on the trunk or lower extremities and are found in both genders equally. Most occur in young or middle-aged persons.2
The typical appearance of THH is that of a brown or violaceous papule or nodule surrounded initially by a pale area and later by an ecchymotic rim, resulting in a targetoid appearance. Over time, the ecchymotic rim expands peripherally and ultimately disappears, leaving the central papule.1 The lesion often is misdiagnosed clinically as a melanocytic nevus, hemangioma, or dermatofibroma; lesions in the process of evolution may even be mistaken for melanoma.
The histopathology of THH is variable and depends on the stage of evolution. A biphasic pattern is characteristic. Superficially, dilated vascular spaces lined by protuberant endothelial cells displaying a hobnail appearance are evident. Intraluminal papillary projections lined by a single layer of endothelial cells also may be present. Deep to this, a proliferation of narrow, angulated, and slitlike vascular spaces dissect between the collagen bundles.2 Fibrin thrombi, erythrocyte extravasation, and hemosiderin deposition are variable, as is a mild lymphocytic infiltrate. With maturity, the vascular spaces appear collapsed, and the stroma appears more fibrous with increased erythrocyte extravasation and hemosiderin deposition.
This histologic appearance of THH may be similar to that of early Kaposi sarcoma (KS). Features that may aid in distinguishing this lesion from KS include the prominent hemosiderin deposition and protuberant or “hobnailed” endothelial cells in dilated superficial vessels seen in THH versus the adnexocentricity seen in KS.3
The histopathologic differential diagnosis of THH includes hobnail hemangioma (HH), another recently described tumor. HH and THH share many similar features, including dilated vascular channels superficially and a collagen-dissecting pseudoangiosarcomatous pattern deeper, with the endothelial lining displaying hobnail cytomorphology. However, in HH a variable but often minimal amount of hemosiderin deposition is present. In addition, the HH lesion demonstrates a nontargetoid clinical appearance.4 One study reported a series of 62 cases with histology similar to that of THH. However, in each of these cases, the lesion had a nontargetoid clinical appearance.5 Despite the histologic similarity, the relationship between THH and HH remains unclear.
Trauma is thought to play a major role in the pathogenesis of THH.6 The ecchymotic rim of THH correlates to hemosiderin deposits, a defining histopathologic feature.2 Trauma to the vascular component of the central papule may result in extravasated erythrocytes and subsequent hemosiderin deposition. Trauma to a preexisting hemangioma resulting in thrombi also would explain histologic features such as intraluminal projections and fibrin thrombi.7
An interesting subset of THH consists of lesions with episodic and cyclic changes. These changes, though uncommon, have been reported to occur without preceding trauma,2,8 and the cause remains unknown. One report described a father and son with THH, both exhibiting cyclic changes with no apparent cause.8 Other cases occurring in women have been hypothesized to be mediated by hormonal factors. Carlson et al2 reported one case, similar to the one reported here, in which cyclic changes occurred during menses, suggesting the diagnosis of endometriosis. However, these changes may have been due to fluctuating estrogen levels. Estrogen has been demonstrated to promote vascular permeability and fragility,9 which may account for the extravasated erythrocytes and subsequent hemosiderin deposition characteristic of THH. A case termed targetoid hemangioma displayed similar clinical features, including cyclic changes in the ecchymotic rim occurring during menses.9 Although the histologic features varied from THH, the similar clinical presentation lends support to the diagnosis of THH. back to top
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Carlson JA, Daulat S, Goodheart H. Targetoid hemosiderotic hemangioma—a dynamic vascular tumor: report of 3 cases with episodic and cyclic changes and comparison with solitary angiokeratomas. J Am Acad Dermatol. 1999;41:215-224.
- Ho C, McCalmont TH. Targetoid hemosiderotic hemangioma: report of 24 cases, with emphasis on unusual features and comparison to early Kaposi’s sarcoma [abstract]. J Cutan Pathol. 1995;22:67A.
- Guillou L, Calonje E, Speight P, et al. Hobnail hemangioma: a pseudomalignant vascular lesion with a reappraisal of targetoid hemosiderotic hemangioma. Am J Surg Pathol. 1999;23:97-105.
- Mentzel TA, Partanen T, Kutzner H. Hobnail hemangioma (“targetoid hemosiderotic hemangioma”): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Rapini RP, Golitz LE. Targetoid hemosiderotic hemangioma. J Cutan Pathol. 1990;17:233-235.
- Requena L, Sangueza O. Cutaneous vascular proliferations, part II: hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997;37:887-919.
- Christenson L, VanBeek M, Davis D. Targetoid hemosiderotic hemangioma occurring in a father and son. Arch Dermatol. 2000;136:1571-1572.
- Morganroth GS, Tigelaar RE, Longley J, et al. Targetoid hemangioma associated with pregnancy and the menstrual cycle. J Am Acad Dermatol. 1955;32:282-284.
A 45-year-old white woman presented with a solitary lesion on her lower back that had been present for approximately 2 years. The lesion changed monthly with menses. The patient described a brown papule that would enlarge, become deep purple in color, and develop a bruiselike rim with the onset of menses. Although the lesion was tender during menses, the tenderness and bruiselike rim would resolve following menses, and the lesion would revert back to a brown color. The patient was in good health and denied a history of previous trauma to the area or a history of abnormal vaginal bleeding or abdominal or pelvic pain. Physical examination revealed a 9x6 mm firm tan papule with a surrounding pale area and peripheral ecchymotic rim, resulting in a targetoid appearance. There were no other significant findings on cutaneous examination.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangiomas (THHs) were originally described in 1988 by Santa Cruz and Aronberg,1 who reported a series of patients with single vascular lesions with a distinctive targetoid appearance and characteristic histopathology. Despite some histopathologic similarities to malignant vascular neoplasms, these lesions followed a benign clinical course. Although now well recognized, a recent review of the literature revealed a total of only 46 reported cases of THHs.2 THHs appear most commonly on the trunk or lower extremities and are found in both genders equally. Most occur in young or middle-aged persons.2
The typical appearance of THH is that of a brown or violaceous papule or nodule surrounded initially by a pale area and later by an ecchymotic rim, resulting in a targetoid appearance. Over time, the ecchymotic rim expands peripherally and ultimately disappears, leaving the central papule.1 The lesion often is misdiagnosed clinically as a melanocytic nevus, hemangioma, or dermatofibroma; lesions in the process of evolution may even be mistaken for melanoma.
The histopathology of THH is variable and depends on the stage of evolution. A biphasic pattern is characteristic. Superficially, dilated vascular spaces lined by protuberant endothelial cells displaying a hobnail appearance are evident. Intraluminal papillary projections lined by a single layer of endothelial cells also may be present. Deep to this, a proliferation of narrow, angulated, and slitlike vascular spaces dissect between the collagen bundles.2 Fibrin thrombi, erythrocyte extravasation, and hemosiderin deposition are variable, as is a mild lymphocytic infiltrate. With maturity, the vascular spaces appear collapsed, and the stroma appears more fibrous with increased erythrocyte extravasation and hemosiderin deposition.
This histologic appearance of THH may be similar to that of early Kaposi sarcoma (KS). Features that may aid in distinguishing this lesion from KS include the prominent hemosiderin deposition and protuberant or “hobnailed” endothelial cells in dilated superficial vessels seen in THH versus the adnexocentricity seen in KS.3
The histopathologic differential diagnosis of THH includes hobnail hemangioma (HH), another recently described tumor. HH and THH share many similar features, including dilated vascular channels superficially and a collagen-dissecting pseudoangiosarcomatous pattern deeper, with the endothelial lining displaying hobnail cytomorphology. However, in HH a variable but often minimal amount of hemosiderin deposition is present. In addition, the HH lesion demonstrates a nontargetoid clinical appearance.4 One study reported a series of 62 cases with histology similar to that of THH. However, in each of these cases, the lesion had a nontargetoid clinical appearance.5 Despite the histologic similarity, the relationship between THH and HH remains unclear.
Trauma is thought to play a major role in the pathogenesis of THH.6 The ecchymotic rim of THH correlates to hemosiderin deposits, a defining histopathologic feature.2 Trauma to the vascular component of the central papule may result in extravasated erythrocytes and subsequent hemosiderin deposition. Trauma to a preexisting hemangioma resulting in thrombi also would explain histologic features such as intraluminal projections and fibrin thrombi.7
An interesting subset of THH consists of lesions with episodic and cyclic changes. These changes, though uncommon, have been reported to occur without preceding trauma,2,8 and the cause remains unknown. One report described a father and son with THH, both exhibiting cyclic changes with no apparent cause.8 Other cases occurring in women have been hypothesized to be mediated by hormonal factors. Carlson et al2 reported one case, similar to the one reported here, in which cyclic changes occurred during menses, suggesting the diagnosis of endometriosis. However, these changes may have been due to fluctuating estrogen levels. Estrogen has been demonstrated to promote vascular permeability and fragility,9 which may account for the extravasated erythrocytes and subsequent hemosiderin deposition characteristic of THH. A case termed targetoid hemangioma displayed similar clinical features, including cyclic changes in the ecchymotic rim occurring during menses.9 Although the histologic features varied from THH, the similar clinical presentation lends support to the diagnosis of THH. back to top
A 45-year-old white woman presented with a solitary lesion on her lower back that had been present for approximately 2 years. The lesion changed monthly with menses. The patient described a brown papule that would enlarge, become deep purple in color, and develop a bruiselike rim with the onset of menses. Although the lesion was tender during menses, the tenderness and bruiselike rim would resolve following menses, and the lesion would revert back to a brown color. The patient was in good health and denied a history of previous trauma to the area or a history of abnormal vaginal bleeding or abdominal or pelvic pain. Physical examination revealed a 9x6 mm firm tan papule with a surrounding pale area and peripheral ecchymotic rim, resulting in a targetoid appearance. There were no other significant findings on cutaneous examination.
The Diagnosis: Targetoid Hemosiderotic Hemangioma
Targetoid hemosiderotic hemangiomas (THHs) were originally described in 1988 by Santa Cruz and Aronberg,1 who reported a series of patients with single vascular lesions with a distinctive targetoid appearance and characteristic histopathology. Despite some histopathologic similarities to malignant vascular neoplasms, these lesions followed a benign clinical course. Although now well recognized, a recent review of the literature revealed a total of only 46 reported cases of THHs.2 THHs appear most commonly on the trunk or lower extremities and are found in both genders equally. Most occur in young or middle-aged persons.2
The typical appearance of THH is that of a brown or violaceous papule or nodule surrounded initially by a pale area and later by an ecchymotic rim, resulting in a targetoid appearance. Over time, the ecchymotic rim expands peripherally and ultimately disappears, leaving the central papule.1 The lesion often is misdiagnosed clinically as a melanocytic nevus, hemangioma, or dermatofibroma; lesions in the process of evolution may even be mistaken for melanoma.
The histopathology of THH is variable and depends on the stage of evolution. A biphasic pattern is characteristic. Superficially, dilated vascular spaces lined by protuberant endothelial cells displaying a hobnail appearance are evident. Intraluminal papillary projections lined by a single layer of endothelial cells also may be present. Deep to this, a proliferation of narrow, angulated, and slitlike vascular spaces dissect between the collagen bundles.2 Fibrin thrombi, erythrocyte extravasation, and hemosiderin deposition are variable, as is a mild lymphocytic infiltrate. With maturity, the vascular spaces appear collapsed, and the stroma appears more fibrous with increased erythrocyte extravasation and hemosiderin deposition.
This histologic appearance of THH may be similar to that of early Kaposi sarcoma (KS). Features that may aid in distinguishing this lesion from KS include the prominent hemosiderin deposition and protuberant or “hobnailed” endothelial cells in dilated superficial vessels seen in THH versus the adnexocentricity seen in KS.3
The histopathologic differential diagnosis of THH includes hobnail hemangioma (HH), another recently described tumor. HH and THH share many similar features, including dilated vascular channels superficially and a collagen-dissecting pseudoangiosarcomatous pattern deeper, with the endothelial lining displaying hobnail cytomorphology. However, in HH a variable but often minimal amount of hemosiderin deposition is present. In addition, the HH lesion demonstrates a nontargetoid clinical appearance.4 One study reported a series of 62 cases with histology similar to that of THH. However, in each of these cases, the lesion had a nontargetoid clinical appearance.5 Despite the histologic similarity, the relationship between THH and HH remains unclear.
Trauma is thought to play a major role in the pathogenesis of THH.6 The ecchymotic rim of THH correlates to hemosiderin deposits, a defining histopathologic feature.2 Trauma to the vascular component of the central papule may result in extravasated erythrocytes and subsequent hemosiderin deposition. Trauma to a preexisting hemangioma resulting in thrombi also would explain histologic features such as intraluminal projections and fibrin thrombi.7
An interesting subset of THH consists of lesions with episodic and cyclic changes. These changes, though uncommon, have been reported to occur without preceding trauma,2,8 and the cause remains unknown. One report described a father and son with THH, both exhibiting cyclic changes with no apparent cause.8 Other cases occurring in women have been hypothesized to be mediated by hormonal factors. Carlson et al2 reported one case, similar to the one reported here, in which cyclic changes occurred during menses, suggesting the diagnosis of endometriosis. However, these changes may have been due to fluctuating estrogen levels. Estrogen has been demonstrated to promote vascular permeability and fragility,9 which may account for the extravasated erythrocytes and subsequent hemosiderin deposition characteristic of THH. A case termed targetoid hemangioma displayed similar clinical features, including cyclic changes in the ecchymotic rim occurring during menses.9 Although the histologic features varied from THH, the similar clinical presentation lends support to the diagnosis of THH. back to top
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Carlson JA, Daulat S, Goodheart H. Targetoid hemosiderotic hemangioma—a dynamic vascular tumor: report of 3 cases with episodic and cyclic changes and comparison with solitary angiokeratomas. J Am Acad Dermatol. 1999;41:215-224.
- Ho C, McCalmont TH. Targetoid hemosiderotic hemangioma: report of 24 cases, with emphasis on unusual features and comparison to early Kaposi’s sarcoma [abstract]. J Cutan Pathol. 1995;22:67A.
- Guillou L, Calonje E, Speight P, et al. Hobnail hemangioma: a pseudomalignant vascular lesion with a reappraisal of targetoid hemosiderotic hemangioma. Am J Surg Pathol. 1999;23:97-105.
- Mentzel TA, Partanen T, Kutzner H. Hobnail hemangioma (“targetoid hemosiderotic hemangioma”): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Rapini RP, Golitz LE. Targetoid hemosiderotic hemangioma. J Cutan Pathol. 1990;17:233-235.
- Requena L, Sangueza O. Cutaneous vascular proliferations, part II: hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997;37:887-919.
- Christenson L, VanBeek M, Davis D. Targetoid hemosiderotic hemangioma occurring in a father and son. Arch Dermatol. 2000;136:1571-1572.
- Morganroth GS, Tigelaar RE, Longley J, et al. Targetoid hemangioma associated with pregnancy and the menstrual cycle. J Am Acad Dermatol. 1955;32:282-284.
- Santa Cruz DJ, Aronberg J. Targetoid hemosiderotic hemangioma. J Am Acad Dermatol. 1988;19:550-558.
- Carlson JA, Daulat S, Goodheart H. Targetoid hemosiderotic hemangioma—a dynamic vascular tumor: report of 3 cases with episodic and cyclic changes and comparison with solitary angiokeratomas. J Am Acad Dermatol. 1999;41:215-224.
- Ho C, McCalmont TH. Targetoid hemosiderotic hemangioma: report of 24 cases, with emphasis on unusual features and comparison to early Kaposi’s sarcoma [abstract]. J Cutan Pathol. 1995;22:67A.
- Guillou L, Calonje E, Speight P, et al. Hobnail hemangioma: a pseudomalignant vascular lesion with a reappraisal of targetoid hemosiderotic hemangioma. Am J Surg Pathol. 1999;23:97-105.
- Mentzel TA, Partanen T, Kutzner H. Hobnail hemangioma (“targetoid hemosiderotic hemangioma”): clinicopathologic and immunohistochemical analysis of 62 cases. J Cutan Pathol. 1999;26:279-286.
- Rapini RP, Golitz LE. Targetoid hemosiderotic hemangioma. J Cutan Pathol. 1990;17:233-235.
- Requena L, Sangueza O. Cutaneous vascular proliferations, part II: hyperplasias and benign neoplasms. J Am Acad Dermatol. 1997;37:887-919.
- Christenson L, VanBeek M, Davis D. Targetoid hemosiderotic hemangioma occurring in a father and son. Arch Dermatol. 2000;136:1571-1572.
- Morganroth GS, Tigelaar RE, Longley J, et al. Targetoid hemangioma associated with pregnancy and the menstrual cycle. J Am Acad Dermatol. 1955;32:282-284.
Botanical Briefs: Capsicum Peppers
Clinical Information Chili peppers are well known among amateur chefs and kitchen workers for causing painful red hands and lips. Cases of painful red hands have been described in association with handling wet chili peppers for Asian cuisine (the so-called Hunan hand syndrome)1 and in individuals preparing Mexican cuisine who peel warm roasted peppers.2,3
Nonvesicular erythema on areas of contact—particularly the hands—has been reported.4 Burning of the lips and gastrointestinal mucosa is a well-known phenomenon to anyone who has eaten foods containing hot peppers. Those who handle chili peppers and accidentally touch their eyes also may experience burning of the eyes. These scenarios are believed to be irritant contact phenomena. Allergic contact dermatitis to Capsicum species appears to be rare.5-7 Patch testing can be done with 1% tincture of capsicum in alcohol.8 Red-pepper dermatitis has been reported in infants whose mothers ingested red peppers prior to breast feeding.9
Apart from the obvious avoidance of contact, reported treatments include topical application of lidocaine gel,1 immersion in 5% acetic acid,10 topical application of magnesium hydroxide aluminum hydroxide–simethicone suspension,11 topical application of vegetable oil,12 and a small amount of chlorine or ammonia in water.13 The burning mouth can be ameliorated by casein in dairy products or alcohol (eg, beer, vodka).13 back to top
Plant Information Capsicum peppers belong to the family Solanaceae, which also includes tomatoes, potatoes, tobacco, and the deadly nightshade.5 Nomenclature and classification of peppers has been fraught with confusion and change over the years.13-15 Terms like pepper, chile, chili, paprika, and capsicum are frequently used interchangeably. Currently, the Capsicum genus encompasses 5 well-described domesticated species and at least 20 wild species, as well as many hybrids and cultivars. The most common of these is Capsicum annuum, which includes the following pepper types: ancho/poblano, bell, cayenne, exotics, jalapeño, paprika, pimiento, piquin, serrano, and others. Capsicum frutescens is comprised of mainly the tabasco pepper but also includes malagueta and bird pepper varieties. The other domesticated species include Capsicum baccatum (ají, ají amarillo), Capsicum chinense (habañero, rica red), and Capsicum pubescens (rocoto, manzano).13,14 Capsicum species should not be confused with black or white pepper (Piper nigrum) or pimento (Pimento dioica).14
Capsicum species are perennial plants indigenous to tropical America that produce pungent fruits on a small spreading shrub. The Capsicum is oval to oblong with hollow berry-type fruit attached to pedicles and calyxes filled with seeds. The fruit has a wide range of sizes, shapes, and colors (Figure) and is well known for its pungency and often hot tast.13,14 Capsaicin is the dominant compound responsible for this pungency and heat. In the food industry, heat levels of peppers are expressed in Scoville units. These levels are based on multiple tests with high performance liquid chromatography and are intended for comparison purposes. The range is from 0 (mild bell) to 500,000 Scoville units (habañero, African “bird’s eye”). The common jalapeño or serrano peppers are rated at 5000 to 15,000 Scoville units.16
Capsicum have the earliest recorded culinary history, dating back to 7000 BC. The first known human contact with peppers was discovered in Mexico by archaeologist R.S. MacNeish, who found pepper seeds dating from approximately 7500 BC.14 Christopher Columbus and accompanying physician Diego Chanca were the first to describe Capsicum during Columbus’ second voyage in 1493. They described “ . . . bushes like rose bushes which make a fruit as long as cinnamon full of small grains as biting as pepper . . .”13 It appears that peppers were subsequently disseminated to Africa, India, the Middle East, the Far East, and Europe via post–Colombian trade routes.13 Today, peppers may be the most widely used spice worldwide.14,15 In the United States, demand for hot peppers exceeds supply, and many are imported annually. back to top
Medical Information Capsaicin, the active ingredient in Capsicum peppers, has been used as a topical medication for pain relief from arthritis,17 postherpetic neuralgia,18 diabetic neuropathy,19 and other painful phenomena.15,20 It also has been used in self-defense sprays.11
From a nutritional standpoint, peppers are rich in vitamin A, C, and B-complex and also may contain magnesium, iron, thiamin, riboflavin, and niacin.13 In herbal medicine, Capsicum are used internally to aid circulation, relieve gas and colic, aid digestion, and prevent infection. External uses include local analgesia and a remedy for cold feet.21
The chemical structure of capsaicin is 8-methyl-6-nonanoyl vanillylamide.15 Additional compounds subsequently identified include dihydrocapsaicin, nordihydrocapsaicin, homodihydrocapsaicin, and homocapsaicin.15 This should be distinguished from capsicum oleoresin, which is commonly used in over-the-counter topical pain relief products. Neither capsicum oleoresin nor the synthetic capsaicin appears to be as reliably neuropeptide-active as natural capsaicin itself.15 Capsaicin, when applied to the skin, induces release of substance P, producing erythema and pain. With repeated application, the substance P is depleted, and the sensory neuron stops producing substance P, leading to diminished pain. This is believed to be the mechanism by which management of certain neurogenic painful conditions (eg, postherpetic neuralgia, diabetic neuropathy) is achieved.15,20
Capsicum species have a rich history and flavor that adds spice and heat to our culinary environment. They extend beyond the kitchen to provide potential medical treatments for painful ailments. If not handled with care, they are responsible for painful red hands and lips. back to top
- Weinberg RB. Hunan hand. N Engl J Med. 1981;305:1020.
- Andrews J. Peppers: The Domesticated Capsicums. Austin, Tex: University of Texas Press; 1984.
- Jones LA, Tandberg D, Troutman WG. Household treatment for “chile burns” of the hands. J Toxicol Clin Toxicol. 1987;25:483-491.
- Williams SR, Clark RF, Dunford JV. Contact dermatitis associated with capsaicin: Hunan hand syndrome. Ann Emerg Med. 1995;25:713-715.
- Lovell CR. Plants and the Skin. London, England: Blackwell Scientific Publications; 1993.
- Cronin E. Dermatitis of the hands in caterers. Contact Dermatitis. 1987;17:265-269.
- Kanerva L, Estlander T, Jolanki R. Occupational allergic contact dermatitis from spices. Contact Dermatitis. 1996;35:157-162.
- Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis, 5th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001.
- Cooper RL, Cooper MM. Red pepper-induced dermatitis in breast-fed infants. Dermatology. 1996;193:61-62.
- Vogl TP. Treatment of Hunan hand. N Engl J Med. 1982;306:178.
- Herman LM, Kindschu MW, Shallash AJ. Treatment of mace dermatitis with topical antacid suspension. Am J Emerg Med. 1998;16:613-614.
- Burnett JW. Capsicum pepper dermatitis. Cutis. 1989;43:534.
- Andrews J. Red Hot Peppers. New York, NY: Macmillan Publishing Company; 1993.
- DeWitt D, Bosland PW. The Pepper Garden. Berkeley, Calif: Ten Speed Press; 1993.
- Cordell GA, Araujo OE. Capsaicin: identification, nomenclature, and pharmacotherapy. Ann Pharmacother. 1993;27:330-336.
- DeWitt D. The Chile Pepper Encyclopedia. New York, NY: William Morrow and Company, Inc; 1999.
- Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: double-blind trial. Clin Ther. 1991;13:383-395.
- Bernstein JE, Korman NJ, Bickers DR, et al. Topical capsaicin treatment of chronic postherpetic neuralgia. J Am Acad Dermatol. 1989;21:265-270.
- The Capsaicin Study Group. Treatment of painful diabetic neuropathy with topical capsaicin: a multicenter, double-blind, vehicle-controlled study. Arch Intern Med. 1991;151:2225-2229.
- Bernstein, JE. Capsaicin and substance P. Clin Dermatol. 1992;9:497-503.
- Chevallier A. The Encyclopedia of Medicinal Plants. New York, NY: DK Publishing, Inc; 1996.
Clinical Information Chili peppers are well known among amateur chefs and kitchen workers for causing painful red hands and lips. Cases of painful red hands have been described in association with handling wet chili peppers for Asian cuisine (the so-called Hunan hand syndrome)1 and in individuals preparing Mexican cuisine who peel warm roasted peppers.2,3
Nonvesicular erythema on areas of contact—particularly the hands—has been reported.4 Burning of the lips and gastrointestinal mucosa is a well-known phenomenon to anyone who has eaten foods containing hot peppers. Those who handle chili peppers and accidentally touch their eyes also may experience burning of the eyes. These scenarios are believed to be irritant contact phenomena. Allergic contact dermatitis to Capsicum species appears to be rare.5-7 Patch testing can be done with 1% tincture of capsicum in alcohol.8 Red-pepper dermatitis has been reported in infants whose mothers ingested red peppers prior to breast feeding.9
Apart from the obvious avoidance of contact, reported treatments include topical application of lidocaine gel,1 immersion in 5% acetic acid,10 topical application of magnesium hydroxide aluminum hydroxide–simethicone suspension,11 topical application of vegetable oil,12 and a small amount of chlorine or ammonia in water.13 The burning mouth can be ameliorated by casein in dairy products or alcohol (eg, beer, vodka).13 back to top
Plant Information Capsicum peppers belong to the family Solanaceae, which also includes tomatoes, potatoes, tobacco, and the deadly nightshade.5 Nomenclature and classification of peppers has been fraught with confusion and change over the years.13-15 Terms like pepper, chile, chili, paprika, and capsicum are frequently used interchangeably. Currently, the Capsicum genus encompasses 5 well-described domesticated species and at least 20 wild species, as well as many hybrids and cultivars. The most common of these is Capsicum annuum, which includes the following pepper types: ancho/poblano, bell, cayenne, exotics, jalapeño, paprika, pimiento, piquin, serrano, and others. Capsicum frutescens is comprised of mainly the tabasco pepper but also includes malagueta and bird pepper varieties. The other domesticated species include Capsicum baccatum (ají, ají amarillo), Capsicum chinense (habañero, rica red), and Capsicum pubescens (rocoto, manzano).13,14 Capsicum species should not be confused with black or white pepper (Piper nigrum) or pimento (Pimento dioica).14
Capsicum species are perennial plants indigenous to tropical America that produce pungent fruits on a small spreading shrub. The Capsicum is oval to oblong with hollow berry-type fruit attached to pedicles and calyxes filled with seeds. The fruit has a wide range of sizes, shapes, and colors (Figure) and is well known for its pungency and often hot tast.13,14 Capsaicin is the dominant compound responsible for this pungency and heat. In the food industry, heat levels of peppers are expressed in Scoville units. These levels are based on multiple tests with high performance liquid chromatography and are intended for comparison purposes. The range is from 0 (mild bell) to 500,000 Scoville units (habañero, African “bird’s eye”). The common jalapeño or serrano peppers are rated at 5000 to 15,000 Scoville units.16
Capsicum have the earliest recorded culinary history, dating back to 7000 BC. The first known human contact with peppers was discovered in Mexico by archaeologist R.S. MacNeish, who found pepper seeds dating from approximately 7500 BC.14 Christopher Columbus and accompanying physician Diego Chanca were the first to describe Capsicum during Columbus’ second voyage in 1493. They described “ . . . bushes like rose bushes which make a fruit as long as cinnamon full of small grains as biting as pepper . . .”13 It appears that peppers were subsequently disseminated to Africa, India, the Middle East, the Far East, and Europe via post–Colombian trade routes.13 Today, peppers may be the most widely used spice worldwide.14,15 In the United States, demand for hot peppers exceeds supply, and many are imported annually. back to top
Medical Information Capsaicin, the active ingredient in Capsicum peppers, has been used as a topical medication for pain relief from arthritis,17 postherpetic neuralgia,18 diabetic neuropathy,19 and other painful phenomena.15,20 It also has been used in self-defense sprays.11
From a nutritional standpoint, peppers are rich in vitamin A, C, and B-complex and also may contain magnesium, iron, thiamin, riboflavin, and niacin.13 In herbal medicine, Capsicum are used internally to aid circulation, relieve gas and colic, aid digestion, and prevent infection. External uses include local analgesia and a remedy for cold feet.21
The chemical structure of capsaicin is 8-methyl-6-nonanoyl vanillylamide.15 Additional compounds subsequently identified include dihydrocapsaicin, nordihydrocapsaicin, homodihydrocapsaicin, and homocapsaicin.15 This should be distinguished from capsicum oleoresin, which is commonly used in over-the-counter topical pain relief products. Neither capsicum oleoresin nor the synthetic capsaicin appears to be as reliably neuropeptide-active as natural capsaicin itself.15 Capsaicin, when applied to the skin, induces release of substance P, producing erythema and pain. With repeated application, the substance P is depleted, and the sensory neuron stops producing substance P, leading to diminished pain. This is believed to be the mechanism by which management of certain neurogenic painful conditions (eg, postherpetic neuralgia, diabetic neuropathy) is achieved.15,20
Capsicum species have a rich history and flavor that adds spice and heat to our culinary environment. They extend beyond the kitchen to provide potential medical treatments for painful ailments. If not handled with care, they are responsible for painful red hands and lips. back to top
Clinical Information Chili peppers are well known among amateur chefs and kitchen workers for causing painful red hands and lips. Cases of painful red hands have been described in association with handling wet chili peppers for Asian cuisine (the so-called Hunan hand syndrome)1 and in individuals preparing Mexican cuisine who peel warm roasted peppers.2,3
Nonvesicular erythema on areas of contact—particularly the hands—has been reported.4 Burning of the lips and gastrointestinal mucosa is a well-known phenomenon to anyone who has eaten foods containing hot peppers. Those who handle chili peppers and accidentally touch their eyes also may experience burning of the eyes. These scenarios are believed to be irritant contact phenomena. Allergic contact dermatitis to Capsicum species appears to be rare.5-7 Patch testing can be done with 1% tincture of capsicum in alcohol.8 Red-pepper dermatitis has been reported in infants whose mothers ingested red peppers prior to breast feeding.9
Apart from the obvious avoidance of contact, reported treatments include topical application of lidocaine gel,1 immersion in 5% acetic acid,10 topical application of magnesium hydroxide aluminum hydroxide–simethicone suspension,11 topical application of vegetable oil,12 and a small amount of chlorine or ammonia in water.13 The burning mouth can be ameliorated by casein in dairy products or alcohol (eg, beer, vodka).13 back to top
Plant Information Capsicum peppers belong to the family Solanaceae, which also includes tomatoes, potatoes, tobacco, and the deadly nightshade.5 Nomenclature and classification of peppers has been fraught with confusion and change over the years.13-15 Terms like pepper, chile, chili, paprika, and capsicum are frequently used interchangeably. Currently, the Capsicum genus encompasses 5 well-described domesticated species and at least 20 wild species, as well as many hybrids and cultivars. The most common of these is Capsicum annuum, which includes the following pepper types: ancho/poblano, bell, cayenne, exotics, jalapeño, paprika, pimiento, piquin, serrano, and others. Capsicum frutescens is comprised of mainly the tabasco pepper but also includes malagueta and bird pepper varieties. The other domesticated species include Capsicum baccatum (ají, ají amarillo), Capsicum chinense (habañero, rica red), and Capsicum pubescens (rocoto, manzano).13,14 Capsicum species should not be confused with black or white pepper (Piper nigrum) or pimento (Pimento dioica).14
Capsicum species are perennial plants indigenous to tropical America that produce pungent fruits on a small spreading shrub. The Capsicum is oval to oblong with hollow berry-type fruit attached to pedicles and calyxes filled with seeds. The fruit has a wide range of sizes, shapes, and colors (Figure) and is well known for its pungency and often hot tast.13,14 Capsaicin is the dominant compound responsible for this pungency and heat. In the food industry, heat levels of peppers are expressed in Scoville units. These levels are based on multiple tests with high performance liquid chromatography and are intended for comparison purposes. The range is from 0 (mild bell) to 500,000 Scoville units (habañero, African “bird’s eye”). The common jalapeño or serrano peppers are rated at 5000 to 15,000 Scoville units.16
Capsicum have the earliest recorded culinary history, dating back to 7000 BC. The first known human contact with peppers was discovered in Mexico by archaeologist R.S. MacNeish, who found pepper seeds dating from approximately 7500 BC.14 Christopher Columbus and accompanying physician Diego Chanca were the first to describe Capsicum during Columbus’ second voyage in 1493. They described “ . . . bushes like rose bushes which make a fruit as long as cinnamon full of small grains as biting as pepper . . .”13 It appears that peppers were subsequently disseminated to Africa, India, the Middle East, the Far East, and Europe via post–Colombian trade routes.13 Today, peppers may be the most widely used spice worldwide.14,15 In the United States, demand for hot peppers exceeds supply, and many are imported annually. back to top
Medical Information Capsaicin, the active ingredient in Capsicum peppers, has been used as a topical medication for pain relief from arthritis,17 postherpetic neuralgia,18 diabetic neuropathy,19 and other painful phenomena.15,20 It also has been used in self-defense sprays.11
From a nutritional standpoint, peppers are rich in vitamin A, C, and B-complex and also may contain magnesium, iron, thiamin, riboflavin, and niacin.13 In herbal medicine, Capsicum are used internally to aid circulation, relieve gas and colic, aid digestion, and prevent infection. External uses include local analgesia and a remedy for cold feet.21
The chemical structure of capsaicin is 8-methyl-6-nonanoyl vanillylamide.15 Additional compounds subsequently identified include dihydrocapsaicin, nordihydrocapsaicin, homodihydrocapsaicin, and homocapsaicin.15 This should be distinguished from capsicum oleoresin, which is commonly used in over-the-counter topical pain relief products. Neither capsicum oleoresin nor the synthetic capsaicin appears to be as reliably neuropeptide-active as natural capsaicin itself.15 Capsaicin, when applied to the skin, induces release of substance P, producing erythema and pain. With repeated application, the substance P is depleted, and the sensory neuron stops producing substance P, leading to diminished pain. This is believed to be the mechanism by which management of certain neurogenic painful conditions (eg, postherpetic neuralgia, diabetic neuropathy) is achieved.15,20
Capsicum species have a rich history and flavor that adds spice and heat to our culinary environment. They extend beyond the kitchen to provide potential medical treatments for painful ailments. If not handled with care, they are responsible for painful red hands and lips. back to top
- Weinberg RB. Hunan hand. N Engl J Med. 1981;305:1020.
- Andrews J. Peppers: The Domesticated Capsicums. Austin, Tex: University of Texas Press; 1984.
- Jones LA, Tandberg D, Troutman WG. Household treatment for “chile burns” of the hands. J Toxicol Clin Toxicol. 1987;25:483-491.
- Williams SR, Clark RF, Dunford JV. Contact dermatitis associated with capsaicin: Hunan hand syndrome. Ann Emerg Med. 1995;25:713-715.
- Lovell CR. Plants and the Skin. London, England: Blackwell Scientific Publications; 1993.
- Cronin E. Dermatitis of the hands in caterers. Contact Dermatitis. 1987;17:265-269.
- Kanerva L, Estlander T, Jolanki R. Occupational allergic contact dermatitis from spices. Contact Dermatitis. 1996;35:157-162.
- Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis, 5th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001.
- Cooper RL, Cooper MM. Red pepper-induced dermatitis in breast-fed infants. Dermatology. 1996;193:61-62.
- Vogl TP. Treatment of Hunan hand. N Engl J Med. 1982;306:178.
- Herman LM, Kindschu MW, Shallash AJ. Treatment of mace dermatitis with topical antacid suspension. Am J Emerg Med. 1998;16:613-614.
- Burnett JW. Capsicum pepper dermatitis. Cutis. 1989;43:534.
- Andrews J. Red Hot Peppers. New York, NY: Macmillan Publishing Company; 1993.
- DeWitt D, Bosland PW. The Pepper Garden. Berkeley, Calif: Ten Speed Press; 1993.
- Cordell GA, Araujo OE. Capsaicin: identification, nomenclature, and pharmacotherapy. Ann Pharmacother. 1993;27:330-336.
- DeWitt D. The Chile Pepper Encyclopedia. New York, NY: William Morrow and Company, Inc; 1999.
- Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: double-blind trial. Clin Ther. 1991;13:383-395.
- Bernstein JE, Korman NJ, Bickers DR, et al. Topical capsaicin treatment of chronic postherpetic neuralgia. J Am Acad Dermatol. 1989;21:265-270.
- The Capsaicin Study Group. Treatment of painful diabetic neuropathy with topical capsaicin: a multicenter, double-blind, vehicle-controlled study. Arch Intern Med. 1991;151:2225-2229.
- Bernstein, JE. Capsaicin and substance P. Clin Dermatol. 1992;9:497-503.
- Chevallier A. The Encyclopedia of Medicinal Plants. New York, NY: DK Publishing, Inc; 1996.
- Weinberg RB. Hunan hand. N Engl J Med. 1981;305:1020.
- Andrews J. Peppers: The Domesticated Capsicums. Austin, Tex: University of Texas Press; 1984.
- Jones LA, Tandberg D, Troutman WG. Household treatment for “chile burns” of the hands. J Toxicol Clin Toxicol. 1987;25:483-491.
- Williams SR, Clark RF, Dunford JV. Contact dermatitis associated with capsaicin: Hunan hand syndrome. Ann Emerg Med. 1995;25:713-715.
- Lovell CR. Plants and the Skin. London, England: Blackwell Scientific Publications; 1993.
- Cronin E. Dermatitis of the hands in caterers. Contact Dermatitis. 1987;17:265-269.
- Kanerva L, Estlander T, Jolanki R. Occupational allergic contact dermatitis from spices. Contact Dermatitis. 1996;35:157-162.
- Rietschel RL, Fowler JF. Fisher’s Contact Dermatitis, 5th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2001.
- Cooper RL, Cooper MM. Red pepper-induced dermatitis in breast-fed infants. Dermatology. 1996;193:61-62.
- Vogl TP. Treatment of Hunan hand. N Engl J Med. 1982;306:178.
- Herman LM, Kindschu MW, Shallash AJ. Treatment of mace dermatitis with topical antacid suspension. Am J Emerg Med. 1998;16:613-614.
- Burnett JW. Capsicum pepper dermatitis. Cutis. 1989;43:534.
- Andrews J. Red Hot Peppers. New York, NY: Macmillan Publishing Company; 1993.
- DeWitt D, Bosland PW. The Pepper Garden. Berkeley, Calif: Ten Speed Press; 1993.
- Cordell GA, Araujo OE. Capsaicin: identification, nomenclature, and pharmacotherapy. Ann Pharmacother. 1993;27:330-336.
- DeWitt D. The Chile Pepper Encyclopedia. New York, NY: William Morrow and Company, Inc; 1999.
- Deal CL, Schnitzer TJ, Lipstein E, et al. Treatment of arthritis with topical capsaicin: double-blind trial. Clin Ther. 1991;13:383-395.
- Bernstein JE, Korman NJ, Bickers DR, et al. Topical capsaicin treatment of chronic postherpetic neuralgia. J Am Acad Dermatol. 1989;21:265-270.
- The Capsaicin Study Group. Treatment of painful diabetic neuropathy with topical capsaicin: a multicenter, double-blind, vehicle-controlled study. Arch Intern Med. 1991;151:2225-2229.
- Bernstein, JE. Capsaicin and substance P. Clin Dermatol. 1992;9:497-503.
- Chevallier A. The Encyclopedia of Medicinal Plants. New York, NY: DK Publishing, Inc; 1996.
Bullous Eruption: A Manifestation of Lupus Erythematosus
Bullous systemic lupus erythematosus (BSLE) is a rare subset of systemic lupus erythematosus (SLE) associated with autoimmunity to type VII collagen.1 BSLE is an autoimmune-mediated, chronic, widespread, nonscarring, subepidermal blistering skin disease occurring in patients with SLE. In 23% of patients with SLE, cutaneous involvement is the initial manifestation. Approximately 76% of patients with SLE will have skin changes at some stage during the course of their disease. Among these patients, fewer than 5% will have chronic vesicobullous lesions.2 Generally, patients with BSLE meet the criteria for SLE as defined by the American College of Rheumatology (ACR) and have a widespread vesicobullous eruption that is generally unrelated to the severity of the SLE.3 Some patients have bullous eruptions related to lupus erythematosus (LE) but do not meet ACR criteria for SLE. We present such a patient and discuss the spectrum of bullous disease in patients with LE.
Case Report
A 17-year-old African American adolescent girl presented with a 2-day history of a blistering eruption with an abrupt onset. Physical examination revealed photodistributed tense bullae. Innumerable beadlike vesicles coalescing into larger bullae were noted on her face, with dramatic involvement of her lips and ears (Figures 1 and 2). Larger bullae on urticarial bases were found on her upper torso. Initially, no mucosal involvement was noted; however, within days, the patient developed oral and vaginal erosions. In the preceding 5 months, she had occasionally experienced a few blisters on her face but had otherwise been healthy. The patient was feeling well at the time of presentation and was not taking any medication except for a methylprednisolone dose pack prescribed during her visit to the emergency department the previous evening.
Results of a shave biopsy of an intact bulla revealed a neutrophil-rich subepidermal bulla with neutrophils stuffing the dermal papillae and lined up along the dermal-epidermal junction (DEJ) (Figure 3).
There was no leukocytoclastic vasculitis and no eosinophils were noted. Results of direct immunofluorescence (DIF) revealed IgG 4+ granular/continuous granular staining at the DEJ, trace IgM with 1+ staining of colloid bodies at the DEJ, 2+ granular/continuous granular C3 staining at the DEJ, and 3+ granular/continuous granular C1q staining at the DEJ (Figure 4).
The specimen was negative for IgA. Laboratory investigation revealed an antinuclear antibody of 1:160; anti-DNA of 1:265 (negative is <200); and negative ribonuclear protein antigen, Smith antigen, Sjögren syndrome A and B antigens, and lupus anticoagulant and anticardiolipin antibodies. Results of complete blood cell count (CBC), blood chemistry, and urinalysis were within reference range. No type VII collagen antibodies were found.
Treatment with oral steroids had begun prior to the patient presenting to dermatology, and no improvement was noted during a 1-week period. In anticipation of starting dapsone, a glucose-6-phosphate dehydrogenase level was ordered, and colchicine was begun at a dose of 0.6 mg 2 times a day. The bullous lesions showed some response within 2 days. The patient was subsequently switched to dapsone; however, colchicine was reinstated after she developed symptoms consistent with dapsone hypersensitivity, including a diffuse pruritic morbilliform eruption, nausea, and abdominal pain, with elevated liver enzyme levels—aspartate aminotransferase was 304 U/L (reference range, 0—37 U/L) and alanine aminotransferase was 360 U/L (reference range, 0–40 U/L). The eruption was eventually controlled with 0.6 mg of colchicine 3 times a day and prednisone. After multiple failed attempts to taper prednisone, 400 mg of hydroxychloroquine once a day was added. After 2 months, the patient was able to tolerate the steroid taper without a rebound flare of bullous lesions.
Comment
BSLE typically presents in the second or third decade of life. Patients with BSLE seldom have discoid lesions or annular erythema.4 Lesions may form large blisters on the trunk that resemble the lesions of bullous pemphigoid. Bullous skin lesions also may appear on flexural and extensor surfaces with a preference for sun-exposed areas. Bullae may form on an erythematous base or on normal skin. Some skin lesions present as herpetiform vesicles with clusters of ulcers. Because of the herpetiform grouping and dermatitis herpetiformlike histology, dermatitis herpetiformis should be included in the differential diagnosis, but can easily be ruled out with DIF. Oral manifestations, such as small blisters on the vermilion border of the lips, are seen in approximately 30% of cases.4
Epidermolysis bullosa acquisita (EBA) appears to share a common antigen with BSLE and has been noted in patients with LE.5 The 2 conditions may represent variants of the same condition. EBA typically presents in a patient’s fourth or fifth decade of life, with acrally distributed mechanobullous lesions or widespread inflammatory vesiculobullous lesions appearing like bullous pemphigoid.5 EBA is more likely than BSLE to result in scarring. Furthermore, mechanical skin fragility is not a common feature of BSLE, though it is a feature of EBA. BSLE lesions generally last for many weeks to months, can undergo remissions and exacerbations, and respond favorably to treatment with dapsone. Conversely, EBA often lasts for many years and is frequently treatment resistant.
Some patients with LE and bullous lesions do not meet ACR criteria for either BSLE or EBA. Our patient had serologic evidence of connective tissue disease, as well as DIF findings typical for LE. Her clinical lesions and response to treatment were similar to that of BSLE. These findings suggest that her condition represents part of a spectrum of connective tissue disease-related bullous dermatosis.
The underlying pathophysiology of BSLE and EBA relates to the structure of the DEJ. Anchoring complexes, which are specialized focal attachment sites within the DEJ, are structurally weakened by the binding of autoantibodies to its components.6 The components of the anchoring complexes, which contain type VII collagen, react with the autoantibodies in BSLE, compromising the integrity of the DEJ. This may lead to the formation of subepidermal blisters.6
The criteria for the diagnosis of BSLE proposed by Camisa and Sharma7 include a diagnosis of SLE based on the criteria of the ACR, vesicles, and bullae located on but not limited to sun-exposed skin; histopathologic findings similar to dermatitis herpetiformis; and deposition of IgG and/or IgM and often IgA at the basement membrane zone by DIF. Gammon and Briggaman5 classified BSLE into 2 distinct subtypes: patients with circulating antibodies to type VII collagen are designated as cases of BSLE-1, while patients designated as cases of BSLE-2 do not have these antibodies.
Some authors have suggested the current classification of BSLE be revised because some patients have autoantibodies bound to the epidermal side of 1 mol/L NaCl-split skin, which indicates involvement of DEJ components other than type VII collagen in BSLE.2,8 Patients also may test falsely negative for antibodies to type VII collagen, possibly because of degradation of the antibody during shipping or because they may possess antibodies to a different antigen. Failure to detect antibodies to type VII collagen does not rule out the possibility of BSLE, but data suggest that patients with lupus and bullous disease may represent a spectrum of related immunobullous disorders.
Histologically, the vesiculobullous eruption is typically characterized by dermal-epidermal separation with neutrophil-predominant inflammation in the upper dermis.7 In cases where the infiltrate concentrates in the dermal papillae as papillary microabscesses, the histologic picture is suggestive of dermatitis herpetiformis.4 Eosinophils also may be present, but are fewer in number. DIF studies characteristically show deposition of IgG, C3, IgA, and IgM at the DEJ in 2 types of patterns—granular and continuous granular—with an occasional mixed configuration.4 Circulating IgG antibodies to the DEJ have been detected in some, but not all, patients.
Ultrastructurally, electron microscopy localizes the blisters in the lamina densa region in most cases. Immunoelectron microscopic examination identified the deposition of the anti-DEJ antibodies on and beneath the lamina densa as in EBA, not in the lamina lucida as in bullous pemphigoid.4 These autoantibodies typically recognized the 290-kd and 145-kd antigens at the DEJ, with type VII collagen as the target. IgG autoantibodies to type VII collagen are believed to be pathogenic and contribute to the separation and blister formation both in BSLE and EBA.4 The production of these autoantibodies is regulated by the class II major histocompatibility complex.4
Unlike patients with EBA, most patients with BSLE respond dramatically to dapsone.5 Although dapsone has both antibiotic and anti-inflammatory properties, the anti-inflammatory mechanisms mediate the therapeutic effect in BSLE. Dapsone directly impairs the myeloperoxidase-hydrogen peroxide-halide system of polymorphonuclear neutrophils (PMNs), which prevents generation of proinflammatory oxygen intermediates caused by activation of neutrophils. Inhibition of PMN chemotaxis and mitogen-stimulated transformation of lymphocytes is another mechanism by which dapsone interferes with inflammation.9 Furthermore, dapsone prevents cyclooxygenase-mediated production of prostaglandin E2, further decreasing inflammation.
Patients with a glucose-6-phosphate dehydrogenase deficiency may experience severe hemolysis when taking dapsone; therefore, patients should be screened for this trait. Additionally, a baseline CBC and liver function test should be obtained and repeated weekly during the initial treatment period since dose-dependent hemolysis is the most common side effect of dapsone.10 Most patients will develop a 1- to 2-g drop in hemoglobin levels after initiation of treatment, which may be partially ameliorated by the concomitant use of 400 IU of vitamin E once a day.10 Other adverse reactions include methemoglobinemia, motor neuropathy, exfoliative dermatitis, hepatitis, headache, gastrointestinal upset, and rarely agranulocytosis.10 Dapsone also may induce a hypersensitivity syndrome with findings similar to those of infectious mononucleosis.9 The syndrome generally begins 4 to 6 weeks after initiation of treatment. Morbilliform eruptions with pruritus, fever, malaise, hepatitis, elevated erythrocyte sedimentation rate, lymphadenopathy, and lymphocytosis are common signs and symptoms associated with this syndrome.9 Immediate discontinuation of dapsone is recommended if symptoms arise. A good response to dapsone in the clearing of vesiculobullous lesions correlates with a better prognosis in BSLE; however, discontinuation of dapsone may allow new lesions to develop.4
Colchicine is a therapeutic option for treatment of neutrophil-mediated bullous diseases. Colchicine is known to interfere with PMN chemotaxis and the release of lysozymal enzymes by PMNs.11 Our patient achieved resolution of lesions with 0.6 mg of colchicine 3 times a day. Administered in low doses, colchicine has relatively few side effects. The most common are transient diarrhea and abdominal discomfort11; therefore, the dose requires titration to tolerance of these side effects. Other side effects of colchicine, such as neuropathy and bone marrow depression, are rare with low-dose therapy.11
Bullous lesions in patients with LE often fail to respond to treatment with systemic corticosteroids alone, and long-term corticosteroid treatment is associated with adverse metabolic effects and bone demineralization. To limit corticosteroid toxicity, adjuvant therapy with azathioprine, antimalarials, and cyclophosphamide have been reported to be useful in cases unresponsive or intolerant to dapsone.12,13 Patients initiating steroid therapy should receive a baseline weight and blood pressure measures, as well as an ophthalmologic examination. Pretreatment laboratory studies should include tests for CBC, electrolyte count, calcium level, alkaline phosphatase level, creatinine level, human immunodeficiency virus, tuberculosis, and bone densitometry. Weight, blood pressure, and blood glucose should be followed monthly until a response is established. The side effects of prolonged therapy with systemic steroids include: psychiatric disorders, sleep disturbances, cataracts, gastrointestinal upset, weight gain, peptic ulcer disease, hypertension, atherosclerosis, infection, growth failure, suppression of the hypothalmic-pituitary-adrenal axis, secondary amenorrhea, hyperglycemia, glucose intolerance, inhibition of wound healing, subcutaneous atrophy, acne, hirsutism, osteoporosis, and aseptic necrosis of bone.10 The initiation of bisphosphonate therapy when corticosteroid therapy is begun will prevent a significant loss of bone mineral density. Bisphosphonate therapy also can improve bone mineral density in patients with established bone loss due to corticosteroid therapy.
Prystowsky et al14 reported successful use of azathioprine to treat BSLE. By metabolizing to 6-thioguanine, azathioprine is incorporated into DNA yielding strand breaks secondary to blockage of DNA synthesis. Because azathioprine is metabolized by thiopurine methyltransferase, patients should be screened for activity of this enzyme prior to initiation of therapy.10,15 Individuals who are homozygous for the allele conferring low activity of this enzyme (0.3%) are at risk for profound myelosuppression with azathioprine. More commonly, patients have high levels of the enzyme and are at risk for undertreatment of their disease with inadequate doses. Long-term risks of azathioprine therapy include an increased incidence of malignancy such as lymphoma, leukemia, and squamous cell carcinoma. The prognosis in patients with SLE and bullous lesions is determined largely by the visceral manifestations of the SLE,5 yet the activity of the systemic and skin disease may not be linked.3,5 Our patient presented with immunobullous disease and serologic evidence of connective tissue disease. Her DIF finding was characteristic of LE. This case adds further evidence that there is a spectrum of related bullous disorders in patients with connective tissue disease.
- Fujii K, Fujimoto W, Ueda M, et al. Detection of anti-type VII collagen antibody in Sjögren's syndrome/lupus erythematosus overlap syndrome with transient bullous systemic lupus erythematosus. Br J Dermatol. 1998;139:302-306.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Cotell S, Robinson N, Lawrence C. Autoimmune blistering skin diseases. Am J Emerg Med. 2000;18:288-299.
- Weinberg MA, Insler MS, Campen RB. Mucocutaneous feature of autoimmune blistering diseases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:517-534.
- Gammon WR, Briggaman RA. Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus diseases of autoimmunity to type VII collagen. Dermatol Clin. 1993;11:535-547.
- Schmidt E, Zillikens D. Autoimmune and inherited subepidermal blistering diseases: advances in the clinic and the laboratory. Adv Dermatol. 2000;16:113-157.
- Camisa C, Sharma HM. Vesicobullous systems lupus erythematosus. report of two cases and review of the literature. J Am Acad Dermatol. 1983;9:924-933.
- Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135:569-573.
- Paniker U, Levine N. Dapsone and sulfapyridine. Dermatol Clin. 2001;19:79-86.
- Gleid M, Rico J. Treatment of autoimmune blistering diseases. Dermatol Clin. 1999;17:431-440.
- Cunningham B, Kirchmann T, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34:781-784.
- Mascaro JM, Herrero C, Hausmann G. Uncommon cutaneous manifestations of lupus erythematosus. Lupus. 1997;6:122-131.
- Yung A, Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol. 2000;41:234-237.
- Prystowsky JH, Finkel L, Tar L, et al. Bullous eruption in a woman with lupus erythematosus. Arch Dermatol. 1988;124:571, 574-575.
- Korman N. New and emerging therapies in the treatment of blistering diseases. Dermatol Clin. 2000;18:127-137.
- Pedro SD, Dahl MC. Direct immunofluorescence of bullous systemic lupus erythematosus. Arch Dermatol. 1973;107:118-120.
Bullous systemic lupus erythematosus (BSLE) is a rare subset of systemic lupus erythematosus (SLE) associated with autoimmunity to type VII collagen.1 BSLE is an autoimmune-mediated, chronic, widespread, nonscarring, subepidermal blistering skin disease occurring in patients with SLE. In 23% of patients with SLE, cutaneous involvement is the initial manifestation. Approximately 76% of patients with SLE will have skin changes at some stage during the course of their disease. Among these patients, fewer than 5% will have chronic vesicobullous lesions.2 Generally, patients with BSLE meet the criteria for SLE as defined by the American College of Rheumatology (ACR) and have a widespread vesicobullous eruption that is generally unrelated to the severity of the SLE.3 Some patients have bullous eruptions related to lupus erythematosus (LE) but do not meet ACR criteria for SLE. We present such a patient and discuss the spectrum of bullous disease in patients with LE.
Case Report
A 17-year-old African American adolescent girl presented with a 2-day history of a blistering eruption with an abrupt onset. Physical examination revealed photodistributed tense bullae. Innumerable beadlike vesicles coalescing into larger bullae were noted on her face, with dramatic involvement of her lips and ears (Figures 1 and 2). Larger bullae on urticarial bases were found on her upper torso. Initially, no mucosal involvement was noted; however, within days, the patient developed oral and vaginal erosions. In the preceding 5 months, she had occasionally experienced a few blisters on her face but had otherwise been healthy. The patient was feeling well at the time of presentation and was not taking any medication except for a methylprednisolone dose pack prescribed during her visit to the emergency department the previous evening.
Results of a shave biopsy of an intact bulla revealed a neutrophil-rich subepidermal bulla with neutrophils stuffing the dermal papillae and lined up along the dermal-epidermal junction (DEJ) (Figure 3).
There was no leukocytoclastic vasculitis and no eosinophils were noted. Results of direct immunofluorescence (DIF) revealed IgG 4+ granular/continuous granular staining at the DEJ, trace IgM with 1+ staining of colloid bodies at the DEJ, 2+ granular/continuous granular C3 staining at the DEJ, and 3+ granular/continuous granular C1q staining at the DEJ (Figure 4).
The specimen was negative for IgA. Laboratory investigation revealed an antinuclear antibody of 1:160; anti-DNA of 1:265 (negative is <200); and negative ribonuclear protein antigen, Smith antigen, Sjögren syndrome A and B antigens, and lupus anticoagulant and anticardiolipin antibodies. Results of complete blood cell count (CBC), blood chemistry, and urinalysis were within reference range. No type VII collagen antibodies were found.
Treatment with oral steroids had begun prior to the patient presenting to dermatology, and no improvement was noted during a 1-week period. In anticipation of starting dapsone, a glucose-6-phosphate dehydrogenase level was ordered, and colchicine was begun at a dose of 0.6 mg 2 times a day. The bullous lesions showed some response within 2 days. The patient was subsequently switched to dapsone; however, colchicine was reinstated after she developed symptoms consistent with dapsone hypersensitivity, including a diffuse pruritic morbilliform eruption, nausea, and abdominal pain, with elevated liver enzyme levels—aspartate aminotransferase was 304 U/L (reference range, 0—37 U/L) and alanine aminotransferase was 360 U/L (reference range, 0–40 U/L). The eruption was eventually controlled with 0.6 mg of colchicine 3 times a day and prednisone. After multiple failed attempts to taper prednisone, 400 mg of hydroxychloroquine once a day was added. After 2 months, the patient was able to tolerate the steroid taper without a rebound flare of bullous lesions.
Comment
BSLE typically presents in the second or third decade of life. Patients with BSLE seldom have discoid lesions or annular erythema.4 Lesions may form large blisters on the trunk that resemble the lesions of bullous pemphigoid. Bullous skin lesions also may appear on flexural and extensor surfaces with a preference for sun-exposed areas. Bullae may form on an erythematous base or on normal skin. Some skin lesions present as herpetiform vesicles with clusters of ulcers. Because of the herpetiform grouping and dermatitis herpetiformlike histology, dermatitis herpetiformis should be included in the differential diagnosis, but can easily be ruled out with DIF. Oral manifestations, such as small blisters on the vermilion border of the lips, are seen in approximately 30% of cases.4
Epidermolysis bullosa acquisita (EBA) appears to share a common antigen with BSLE and has been noted in patients with LE.5 The 2 conditions may represent variants of the same condition. EBA typically presents in a patient’s fourth or fifth decade of life, with acrally distributed mechanobullous lesions or widespread inflammatory vesiculobullous lesions appearing like bullous pemphigoid.5 EBA is more likely than BSLE to result in scarring. Furthermore, mechanical skin fragility is not a common feature of BSLE, though it is a feature of EBA. BSLE lesions generally last for many weeks to months, can undergo remissions and exacerbations, and respond favorably to treatment with dapsone. Conversely, EBA often lasts for many years and is frequently treatment resistant.
Some patients with LE and bullous lesions do not meet ACR criteria for either BSLE or EBA. Our patient had serologic evidence of connective tissue disease, as well as DIF findings typical for LE. Her clinical lesions and response to treatment were similar to that of BSLE. These findings suggest that her condition represents part of a spectrum of connective tissue disease-related bullous dermatosis.
The underlying pathophysiology of BSLE and EBA relates to the structure of the DEJ. Anchoring complexes, which are specialized focal attachment sites within the DEJ, are structurally weakened by the binding of autoantibodies to its components.6 The components of the anchoring complexes, which contain type VII collagen, react with the autoantibodies in BSLE, compromising the integrity of the DEJ. This may lead to the formation of subepidermal blisters.6
The criteria for the diagnosis of BSLE proposed by Camisa and Sharma7 include a diagnosis of SLE based on the criteria of the ACR, vesicles, and bullae located on but not limited to sun-exposed skin; histopathologic findings similar to dermatitis herpetiformis; and deposition of IgG and/or IgM and often IgA at the basement membrane zone by DIF. Gammon and Briggaman5 classified BSLE into 2 distinct subtypes: patients with circulating antibodies to type VII collagen are designated as cases of BSLE-1, while patients designated as cases of BSLE-2 do not have these antibodies.
Some authors have suggested the current classification of BSLE be revised because some patients have autoantibodies bound to the epidermal side of 1 mol/L NaCl-split skin, which indicates involvement of DEJ components other than type VII collagen in BSLE.2,8 Patients also may test falsely negative for antibodies to type VII collagen, possibly because of degradation of the antibody during shipping or because they may possess antibodies to a different antigen. Failure to detect antibodies to type VII collagen does not rule out the possibility of BSLE, but data suggest that patients with lupus and bullous disease may represent a spectrum of related immunobullous disorders.
Histologically, the vesiculobullous eruption is typically characterized by dermal-epidermal separation with neutrophil-predominant inflammation in the upper dermis.7 In cases where the infiltrate concentrates in the dermal papillae as papillary microabscesses, the histologic picture is suggestive of dermatitis herpetiformis.4 Eosinophils also may be present, but are fewer in number. DIF studies characteristically show deposition of IgG, C3, IgA, and IgM at the DEJ in 2 types of patterns—granular and continuous granular—with an occasional mixed configuration.4 Circulating IgG antibodies to the DEJ have been detected in some, but not all, patients.
Ultrastructurally, electron microscopy localizes the blisters in the lamina densa region in most cases. Immunoelectron microscopic examination identified the deposition of the anti-DEJ antibodies on and beneath the lamina densa as in EBA, not in the lamina lucida as in bullous pemphigoid.4 These autoantibodies typically recognized the 290-kd and 145-kd antigens at the DEJ, with type VII collagen as the target. IgG autoantibodies to type VII collagen are believed to be pathogenic and contribute to the separation and blister formation both in BSLE and EBA.4 The production of these autoantibodies is regulated by the class II major histocompatibility complex.4
Unlike patients with EBA, most patients with BSLE respond dramatically to dapsone.5 Although dapsone has both antibiotic and anti-inflammatory properties, the anti-inflammatory mechanisms mediate the therapeutic effect in BSLE. Dapsone directly impairs the myeloperoxidase-hydrogen peroxide-halide system of polymorphonuclear neutrophils (PMNs), which prevents generation of proinflammatory oxygen intermediates caused by activation of neutrophils. Inhibition of PMN chemotaxis and mitogen-stimulated transformation of lymphocytes is another mechanism by which dapsone interferes with inflammation.9 Furthermore, dapsone prevents cyclooxygenase-mediated production of prostaglandin E2, further decreasing inflammation.
Patients with a glucose-6-phosphate dehydrogenase deficiency may experience severe hemolysis when taking dapsone; therefore, patients should be screened for this trait. Additionally, a baseline CBC and liver function test should be obtained and repeated weekly during the initial treatment period since dose-dependent hemolysis is the most common side effect of dapsone.10 Most patients will develop a 1- to 2-g drop in hemoglobin levels after initiation of treatment, which may be partially ameliorated by the concomitant use of 400 IU of vitamin E once a day.10 Other adverse reactions include methemoglobinemia, motor neuropathy, exfoliative dermatitis, hepatitis, headache, gastrointestinal upset, and rarely agranulocytosis.10 Dapsone also may induce a hypersensitivity syndrome with findings similar to those of infectious mononucleosis.9 The syndrome generally begins 4 to 6 weeks after initiation of treatment. Morbilliform eruptions with pruritus, fever, malaise, hepatitis, elevated erythrocyte sedimentation rate, lymphadenopathy, and lymphocytosis are common signs and symptoms associated with this syndrome.9 Immediate discontinuation of dapsone is recommended if symptoms arise. A good response to dapsone in the clearing of vesiculobullous lesions correlates with a better prognosis in BSLE; however, discontinuation of dapsone may allow new lesions to develop.4
Colchicine is a therapeutic option for treatment of neutrophil-mediated bullous diseases. Colchicine is known to interfere with PMN chemotaxis and the release of lysozymal enzymes by PMNs.11 Our patient achieved resolution of lesions with 0.6 mg of colchicine 3 times a day. Administered in low doses, colchicine has relatively few side effects. The most common are transient diarrhea and abdominal discomfort11; therefore, the dose requires titration to tolerance of these side effects. Other side effects of colchicine, such as neuropathy and bone marrow depression, are rare with low-dose therapy.11
Bullous lesions in patients with LE often fail to respond to treatment with systemic corticosteroids alone, and long-term corticosteroid treatment is associated with adverse metabolic effects and bone demineralization. To limit corticosteroid toxicity, adjuvant therapy with azathioprine, antimalarials, and cyclophosphamide have been reported to be useful in cases unresponsive or intolerant to dapsone.12,13 Patients initiating steroid therapy should receive a baseline weight and blood pressure measures, as well as an ophthalmologic examination. Pretreatment laboratory studies should include tests for CBC, electrolyte count, calcium level, alkaline phosphatase level, creatinine level, human immunodeficiency virus, tuberculosis, and bone densitometry. Weight, blood pressure, and blood glucose should be followed monthly until a response is established. The side effects of prolonged therapy with systemic steroids include: psychiatric disorders, sleep disturbances, cataracts, gastrointestinal upset, weight gain, peptic ulcer disease, hypertension, atherosclerosis, infection, growth failure, suppression of the hypothalmic-pituitary-adrenal axis, secondary amenorrhea, hyperglycemia, glucose intolerance, inhibition of wound healing, subcutaneous atrophy, acne, hirsutism, osteoporosis, and aseptic necrosis of bone.10 The initiation of bisphosphonate therapy when corticosteroid therapy is begun will prevent a significant loss of bone mineral density. Bisphosphonate therapy also can improve bone mineral density in patients with established bone loss due to corticosteroid therapy.
Prystowsky et al14 reported successful use of azathioprine to treat BSLE. By metabolizing to 6-thioguanine, azathioprine is incorporated into DNA yielding strand breaks secondary to blockage of DNA synthesis. Because azathioprine is metabolized by thiopurine methyltransferase, patients should be screened for activity of this enzyme prior to initiation of therapy.10,15 Individuals who are homozygous for the allele conferring low activity of this enzyme (0.3%) are at risk for profound myelosuppression with azathioprine. More commonly, patients have high levels of the enzyme and are at risk for undertreatment of their disease with inadequate doses. Long-term risks of azathioprine therapy include an increased incidence of malignancy such as lymphoma, leukemia, and squamous cell carcinoma. The prognosis in patients with SLE and bullous lesions is determined largely by the visceral manifestations of the SLE,5 yet the activity of the systemic and skin disease may not be linked.3,5 Our patient presented with immunobullous disease and serologic evidence of connective tissue disease. Her DIF finding was characteristic of LE. This case adds further evidence that there is a spectrum of related bullous disorders in patients with connective tissue disease.
Bullous systemic lupus erythematosus (BSLE) is a rare subset of systemic lupus erythematosus (SLE) associated with autoimmunity to type VII collagen.1 BSLE is an autoimmune-mediated, chronic, widespread, nonscarring, subepidermal blistering skin disease occurring in patients with SLE. In 23% of patients with SLE, cutaneous involvement is the initial manifestation. Approximately 76% of patients with SLE will have skin changes at some stage during the course of their disease. Among these patients, fewer than 5% will have chronic vesicobullous lesions.2 Generally, patients with BSLE meet the criteria for SLE as defined by the American College of Rheumatology (ACR) and have a widespread vesicobullous eruption that is generally unrelated to the severity of the SLE.3 Some patients have bullous eruptions related to lupus erythematosus (LE) but do not meet ACR criteria for SLE. We present such a patient and discuss the spectrum of bullous disease in patients with LE.
Case Report
A 17-year-old African American adolescent girl presented with a 2-day history of a blistering eruption with an abrupt onset. Physical examination revealed photodistributed tense bullae. Innumerable beadlike vesicles coalescing into larger bullae were noted on her face, with dramatic involvement of her lips and ears (Figures 1 and 2). Larger bullae on urticarial bases were found on her upper torso. Initially, no mucosal involvement was noted; however, within days, the patient developed oral and vaginal erosions. In the preceding 5 months, she had occasionally experienced a few blisters on her face but had otherwise been healthy. The patient was feeling well at the time of presentation and was not taking any medication except for a methylprednisolone dose pack prescribed during her visit to the emergency department the previous evening.
Results of a shave biopsy of an intact bulla revealed a neutrophil-rich subepidermal bulla with neutrophils stuffing the dermal papillae and lined up along the dermal-epidermal junction (DEJ) (Figure 3).
There was no leukocytoclastic vasculitis and no eosinophils were noted. Results of direct immunofluorescence (DIF) revealed IgG 4+ granular/continuous granular staining at the DEJ, trace IgM with 1+ staining of colloid bodies at the DEJ, 2+ granular/continuous granular C3 staining at the DEJ, and 3+ granular/continuous granular C1q staining at the DEJ (Figure 4).
The specimen was negative for IgA. Laboratory investigation revealed an antinuclear antibody of 1:160; anti-DNA of 1:265 (negative is <200); and negative ribonuclear protein antigen, Smith antigen, Sjögren syndrome A and B antigens, and lupus anticoagulant and anticardiolipin antibodies. Results of complete blood cell count (CBC), blood chemistry, and urinalysis were within reference range. No type VII collagen antibodies were found.
Treatment with oral steroids had begun prior to the patient presenting to dermatology, and no improvement was noted during a 1-week period. In anticipation of starting dapsone, a glucose-6-phosphate dehydrogenase level was ordered, and colchicine was begun at a dose of 0.6 mg 2 times a day. The bullous lesions showed some response within 2 days. The patient was subsequently switched to dapsone; however, colchicine was reinstated after she developed symptoms consistent with dapsone hypersensitivity, including a diffuse pruritic morbilliform eruption, nausea, and abdominal pain, with elevated liver enzyme levels—aspartate aminotransferase was 304 U/L (reference range, 0—37 U/L) and alanine aminotransferase was 360 U/L (reference range, 0–40 U/L). The eruption was eventually controlled with 0.6 mg of colchicine 3 times a day and prednisone. After multiple failed attempts to taper prednisone, 400 mg of hydroxychloroquine once a day was added. After 2 months, the patient was able to tolerate the steroid taper without a rebound flare of bullous lesions.
Comment
BSLE typically presents in the second or third decade of life. Patients with BSLE seldom have discoid lesions or annular erythema.4 Lesions may form large blisters on the trunk that resemble the lesions of bullous pemphigoid. Bullous skin lesions also may appear on flexural and extensor surfaces with a preference for sun-exposed areas. Bullae may form on an erythematous base or on normal skin. Some skin lesions present as herpetiform vesicles with clusters of ulcers. Because of the herpetiform grouping and dermatitis herpetiformlike histology, dermatitis herpetiformis should be included in the differential diagnosis, but can easily be ruled out with DIF. Oral manifestations, such as small blisters on the vermilion border of the lips, are seen in approximately 30% of cases.4
Epidermolysis bullosa acquisita (EBA) appears to share a common antigen with BSLE and has been noted in patients with LE.5 The 2 conditions may represent variants of the same condition. EBA typically presents in a patient’s fourth or fifth decade of life, with acrally distributed mechanobullous lesions or widespread inflammatory vesiculobullous lesions appearing like bullous pemphigoid.5 EBA is more likely than BSLE to result in scarring. Furthermore, mechanical skin fragility is not a common feature of BSLE, though it is a feature of EBA. BSLE lesions generally last for many weeks to months, can undergo remissions and exacerbations, and respond favorably to treatment with dapsone. Conversely, EBA often lasts for many years and is frequently treatment resistant.
Some patients with LE and bullous lesions do not meet ACR criteria for either BSLE or EBA. Our patient had serologic evidence of connective tissue disease, as well as DIF findings typical for LE. Her clinical lesions and response to treatment were similar to that of BSLE. These findings suggest that her condition represents part of a spectrum of connective tissue disease-related bullous dermatosis.
The underlying pathophysiology of BSLE and EBA relates to the structure of the DEJ. Anchoring complexes, which are specialized focal attachment sites within the DEJ, are structurally weakened by the binding of autoantibodies to its components.6 The components of the anchoring complexes, which contain type VII collagen, react with the autoantibodies in BSLE, compromising the integrity of the DEJ. This may lead to the formation of subepidermal blisters.6
The criteria for the diagnosis of BSLE proposed by Camisa and Sharma7 include a diagnosis of SLE based on the criteria of the ACR, vesicles, and bullae located on but not limited to sun-exposed skin; histopathologic findings similar to dermatitis herpetiformis; and deposition of IgG and/or IgM and often IgA at the basement membrane zone by DIF. Gammon and Briggaman5 classified BSLE into 2 distinct subtypes: patients with circulating antibodies to type VII collagen are designated as cases of BSLE-1, while patients designated as cases of BSLE-2 do not have these antibodies.
Some authors have suggested the current classification of BSLE be revised because some patients have autoantibodies bound to the epidermal side of 1 mol/L NaCl-split skin, which indicates involvement of DEJ components other than type VII collagen in BSLE.2,8 Patients also may test falsely negative for antibodies to type VII collagen, possibly because of degradation of the antibody during shipping or because they may possess antibodies to a different antigen. Failure to detect antibodies to type VII collagen does not rule out the possibility of BSLE, but data suggest that patients with lupus and bullous disease may represent a spectrum of related immunobullous disorders.
Histologically, the vesiculobullous eruption is typically characterized by dermal-epidermal separation with neutrophil-predominant inflammation in the upper dermis.7 In cases where the infiltrate concentrates in the dermal papillae as papillary microabscesses, the histologic picture is suggestive of dermatitis herpetiformis.4 Eosinophils also may be present, but are fewer in number. DIF studies characteristically show deposition of IgG, C3, IgA, and IgM at the DEJ in 2 types of patterns—granular and continuous granular—with an occasional mixed configuration.4 Circulating IgG antibodies to the DEJ have been detected in some, but not all, patients.
Ultrastructurally, electron microscopy localizes the blisters in the lamina densa region in most cases. Immunoelectron microscopic examination identified the deposition of the anti-DEJ antibodies on and beneath the lamina densa as in EBA, not in the lamina lucida as in bullous pemphigoid.4 These autoantibodies typically recognized the 290-kd and 145-kd antigens at the DEJ, with type VII collagen as the target. IgG autoantibodies to type VII collagen are believed to be pathogenic and contribute to the separation and blister formation both in BSLE and EBA.4 The production of these autoantibodies is regulated by the class II major histocompatibility complex.4
Unlike patients with EBA, most patients with BSLE respond dramatically to dapsone.5 Although dapsone has both antibiotic and anti-inflammatory properties, the anti-inflammatory mechanisms mediate the therapeutic effect in BSLE. Dapsone directly impairs the myeloperoxidase-hydrogen peroxide-halide system of polymorphonuclear neutrophils (PMNs), which prevents generation of proinflammatory oxygen intermediates caused by activation of neutrophils. Inhibition of PMN chemotaxis and mitogen-stimulated transformation of lymphocytes is another mechanism by which dapsone interferes with inflammation.9 Furthermore, dapsone prevents cyclooxygenase-mediated production of prostaglandin E2, further decreasing inflammation.
Patients with a glucose-6-phosphate dehydrogenase deficiency may experience severe hemolysis when taking dapsone; therefore, patients should be screened for this trait. Additionally, a baseline CBC and liver function test should be obtained and repeated weekly during the initial treatment period since dose-dependent hemolysis is the most common side effect of dapsone.10 Most patients will develop a 1- to 2-g drop in hemoglobin levels after initiation of treatment, which may be partially ameliorated by the concomitant use of 400 IU of vitamin E once a day.10 Other adverse reactions include methemoglobinemia, motor neuropathy, exfoliative dermatitis, hepatitis, headache, gastrointestinal upset, and rarely agranulocytosis.10 Dapsone also may induce a hypersensitivity syndrome with findings similar to those of infectious mononucleosis.9 The syndrome generally begins 4 to 6 weeks after initiation of treatment. Morbilliform eruptions with pruritus, fever, malaise, hepatitis, elevated erythrocyte sedimentation rate, lymphadenopathy, and lymphocytosis are common signs and symptoms associated with this syndrome.9 Immediate discontinuation of dapsone is recommended if symptoms arise. A good response to dapsone in the clearing of vesiculobullous lesions correlates with a better prognosis in BSLE; however, discontinuation of dapsone may allow new lesions to develop.4
Colchicine is a therapeutic option for treatment of neutrophil-mediated bullous diseases. Colchicine is known to interfere with PMN chemotaxis and the release of lysozymal enzymes by PMNs.11 Our patient achieved resolution of lesions with 0.6 mg of colchicine 3 times a day. Administered in low doses, colchicine has relatively few side effects. The most common are transient diarrhea and abdominal discomfort11; therefore, the dose requires titration to tolerance of these side effects. Other side effects of colchicine, such as neuropathy and bone marrow depression, are rare with low-dose therapy.11
Bullous lesions in patients with LE often fail to respond to treatment with systemic corticosteroids alone, and long-term corticosteroid treatment is associated with adverse metabolic effects and bone demineralization. To limit corticosteroid toxicity, adjuvant therapy with azathioprine, antimalarials, and cyclophosphamide have been reported to be useful in cases unresponsive or intolerant to dapsone.12,13 Patients initiating steroid therapy should receive a baseline weight and blood pressure measures, as well as an ophthalmologic examination. Pretreatment laboratory studies should include tests for CBC, electrolyte count, calcium level, alkaline phosphatase level, creatinine level, human immunodeficiency virus, tuberculosis, and bone densitometry. Weight, blood pressure, and blood glucose should be followed monthly until a response is established. The side effects of prolonged therapy with systemic steroids include: psychiatric disorders, sleep disturbances, cataracts, gastrointestinal upset, weight gain, peptic ulcer disease, hypertension, atherosclerosis, infection, growth failure, suppression of the hypothalmic-pituitary-adrenal axis, secondary amenorrhea, hyperglycemia, glucose intolerance, inhibition of wound healing, subcutaneous atrophy, acne, hirsutism, osteoporosis, and aseptic necrosis of bone.10 The initiation of bisphosphonate therapy when corticosteroid therapy is begun will prevent a significant loss of bone mineral density. Bisphosphonate therapy also can improve bone mineral density in patients with established bone loss due to corticosteroid therapy.
Prystowsky et al14 reported successful use of azathioprine to treat BSLE. By metabolizing to 6-thioguanine, azathioprine is incorporated into DNA yielding strand breaks secondary to blockage of DNA synthesis. Because azathioprine is metabolized by thiopurine methyltransferase, patients should be screened for activity of this enzyme prior to initiation of therapy.10,15 Individuals who are homozygous for the allele conferring low activity of this enzyme (0.3%) are at risk for profound myelosuppression with azathioprine. More commonly, patients have high levels of the enzyme and are at risk for undertreatment of their disease with inadequate doses. Long-term risks of azathioprine therapy include an increased incidence of malignancy such as lymphoma, leukemia, and squamous cell carcinoma. The prognosis in patients with SLE and bullous lesions is determined largely by the visceral manifestations of the SLE,5 yet the activity of the systemic and skin disease may not be linked.3,5 Our patient presented with immunobullous disease and serologic evidence of connective tissue disease. Her DIF finding was characteristic of LE. This case adds further evidence that there is a spectrum of related bullous disorders in patients with connective tissue disease.
- Fujii K, Fujimoto W, Ueda M, et al. Detection of anti-type VII collagen antibody in Sjögren's syndrome/lupus erythematosus overlap syndrome with transient bullous systemic lupus erythematosus. Br J Dermatol. 1998;139:302-306.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Cotell S, Robinson N, Lawrence C. Autoimmune blistering skin diseases. Am J Emerg Med. 2000;18:288-299.
- Weinberg MA, Insler MS, Campen RB. Mucocutaneous feature of autoimmune blistering diseases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:517-534.
- Gammon WR, Briggaman RA. Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus diseases of autoimmunity to type VII collagen. Dermatol Clin. 1993;11:535-547.
- Schmidt E, Zillikens D. Autoimmune and inherited subepidermal blistering diseases: advances in the clinic and the laboratory. Adv Dermatol. 2000;16:113-157.
- Camisa C, Sharma HM. Vesicobullous systems lupus erythematosus. report of two cases and review of the literature. J Am Acad Dermatol. 1983;9:924-933.
- Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135:569-573.
- Paniker U, Levine N. Dapsone and sulfapyridine. Dermatol Clin. 2001;19:79-86.
- Gleid M, Rico J. Treatment of autoimmune blistering diseases. Dermatol Clin. 1999;17:431-440.
- Cunningham B, Kirchmann T, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34:781-784.
- Mascaro JM, Herrero C, Hausmann G. Uncommon cutaneous manifestations of lupus erythematosus. Lupus. 1997;6:122-131.
- Yung A, Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol. 2000;41:234-237.
- Prystowsky JH, Finkel L, Tar L, et al. Bullous eruption in a woman with lupus erythematosus. Arch Dermatol. 1988;124:571, 574-575.
- Korman N. New and emerging therapies in the treatment of blistering diseases. Dermatol Clin. 2000;18:127-137.
- Pedro SD, Dahl MC. Direct immunofluorescence of bullous systemic lupus erythematosus. Arch Dermatol. 1973;107:118-120.
- Fujii K, Fujimoto W, Ueda M, et al. Detection of anti-type VII collagen antibody in Sjögren's syndrome/lupus erythematosus overlap syndrome with transient bullous systemic lupus erythematosus. Br J Dermatol. 1998;139:302-306.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Cotell S, Robinson N, Lawrence C. Autoimmune blistering skin diseases. Am J Emerg Med. 2000;18:288-299.
- Weinberg MA, Insler MS, Campen RB. Mucocutaneous feature of autoimmune blistering diseases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:517-534.
- Gammon WR, Briggaman RA. Epidermolysis bullosa acquisita and bullous systemic lupus erythematosus diseases of autoimmunity to type VII collagen. Dermatol Clin. 1993;11:535-547.
- Schmidt E, Zillikens D. Autoimmune and inherited subepidermal blistering diseases: advances in the clinic and the laboratory. Adv Dermatol. 2000;16:113-157.
- Camisa C, Sharma HM. Vesicobullous systems lupus erythematosus. report of two cases and review of the literature. J Am Acad Dermatol. 1983;9:924-933.
- Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135:569-573.
- Paniker U, Levine N. Dapsone and sulfapyridine. Dermatol Clin. 2001;19:79-86.
- Gleid M, Rico J. Treatment of autoimmune blistering diseases. Dermatol Clin. 1999;17:431-440.
- Cunningham B, Kirchmann T, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34:781-784.
- Mascaro JM, Herrero C, Hausmann G. Uncommon cutaneous manifestations of lupus erythematosus. Lupus. 1997;6:122-131.
- Yung A, Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol. 2000;41:234-237.
- Prystowsky JH, Finkel L, Tar L, et al. Bullous eruption in a woman with lupus erythematosus. Arch Dermatol. 1988;124:571, 574-575.
- Korman N. New and emerging therapies in the treatment of blistering diseases. Dermatol Clin. 2000;18:127-137.
- Pedro SD, Dahl MC. Direct immunofluorescence of bullous systemic lupus erythematosus. Arch Dermatol. 1973;107:118-120.
Cutaneous T-Cell Lymphoma in a Patient With Neurofibromatosis Type 1
A variety of neoplasms may occur in association with neurofibromatosis type 1 (NF1). We describe a patient with NF1 and mycosis fungoides. Recommendations for the initial and long-term evaluations of patients with neurofibromatosis are presented.
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is an autosomal-dominant syndrome with no racial, gender, or ethnic predilection.1 It has an estimated incidence of 1 in 3000 to 1 in 7800 people.1,2 Although there are at least 7 types of neurofibromatosis, NF1 is the most common form. Numerous malignancies have been linked to NF1 including pheochromocytoma, thyroid carcinoma, leukemia, and melanoma.3,4 Over the past 30 years, there has been one case report of the coexistence of the leukemic phase of cutaneous T-cell lymphoma and NF1.5 We report the occurrence of the plaque variant of cutaneous T-cell lymphoma in a patient with NF1.
Case Report
A 72-year-old white man with a lifelong history of NF1 presented for evaluation of an erythematous plaque on his left hip. It had been present for 3 months and had slowly increased in size. The lesion had been treated with econazole cream without improvement. The patient's medical history included atrial fibrillation necessitating daily treatment with amiodarone and acetylsalicylic acid. His family history was significant for a mother with NF1. Although currently retired, he had worked as a professor for more than 20 years.
Physical examination revealed an erythematous scaly plaque 14x16 cm in diameter over his left hip extending to his left flank (Figure). The patient had hundreds of flesh-colored nodules varying in size from 0.5 to 3.0 cm distributed over his entire body. In addition, he had 2- to 3-mm hyperpigmented macules in the axilla and inguinal areas, scattered larger café-au-lait macules, and iris hamartomas (Lisch nodules).6 There was no palpable lymphadenopathy or organomegaly.
Laboratory data included a complete blood count, which consisted of a hemoglobin level of 16 g/dL; hematocrit level of 46.5%; and normal blood differential and white blood cell counts. The lymphocyte profile was remarkable for an absolute CD4 count of 340 mm3 (reference range, 359–1519 mm3) and the percentage of CD4+ lymphocytes equal to 26.2% (reference range, 30.8%–58.5%). His chest x-ray revealed moderate pulmonary fibrosis and changes consistent with chronic obstructive pulmonary disease, but no active lung disease. A computed tomographic scan of the abdomen and pelvis was unremarkable for any lymphadenopathy, but there was displacement of the left kidney and bowel into the left hemithorax and a foramen of Bochdalek hernia.
The results of a 3-mm punch skin biopsy of the plaque on the patient's left flank demonstrated psoriasiform acanthosis with a mild to moderate atypical epidermotropic lymphocytic infiltrate and a subjacent neurofibroma. Immunophenotyping revealed prominent epidermotropism of CD3+ lymphocytes. Fewer intraepidermal lymphocytes marked with CD4 and CD5 immunostains. No CD7 deletion was present, but a T-cell gamma gene rearrangement was identified. The patient was diagnosed with stage 1A cutaneous T-cell lymphoma (CTCL)(T1N0M0).
Comment
NF1 is the most common of the neurocutaneous disorders.3 At present, National Institutes of Health criteria for diagnosis include 2 or more of the following: 6 or more café-au-lait macules larger than 5 mm in greatest diameter in prepubertal patients and larger than 15 mm in greatest diameter in postpubertal patients; 2 or more neurofibromas of any type or one plexiform neurofibroma; freckling in the axillary or inguinal area; optic nerve glioma; 2 or more Lisch nodules; a distinctive osseous lesion such as sphenoid wing dysplasia or thinning of the bone cortex with or without pseudoarthrosis; a first-degree relative (parent, sibling, or offspring) with NF1 by the above criteria.1,2,6,7 Different criteria appear at various stages of the patient's development (ie, café-au-lait macules appear at birth and neurofibromas emerge at adolescence).8 Lisch nodules have been regarded as pathognomonic.4 Intertriginous freckling is highly suggestive of the diagnosis but also may be seen in Watson syndrome. The 5 subtypes of neurofibromas are cutaneous, subcutaneous, nodular, plexiform, and diffuse.4 Other associated findings include vision changes, cognitive disorders, growth problems, and musculoskeletal disorders.4 Our patient fulfilled the criteria for NF1 by having neurofibromas, café-au-lait macules, Lisch nodules, intertriginous freckling, and a positive family history.
The gene defect for NF1 has been mapped to chromosome 17. The NF gene acts as a tumor-suppressor gene that dampens the production of the ras proto-oncogene.9 Therefore, the mutation of this gene contributes to tumor progression.6,8
One in 4 patients with NF1 will develop a secondary tumor throughout his or her lifetime. Tumors found with NF1 include those of neural crest origin such as pheochromocytoma, neuroblastoma, and melanoma.10 Non-neural crest tumors include Wilms tumor, rhabdomyosarcoma, granular cell tumors, malignant nodular hidradenoma, and leukemia.11,12 Patients with both NF1 and juvenile xanthogranuloma appear to be at increased risk for leukemia. Malignant peripheral nerve sheath tumors are associated with NF1 and are a potential cause of mortality in adult patients with NF1. Typically, the malignancy will occur before age 38 years, arising in a longstanding neurofibroma. Multiple malignancies occur in 12% of patients.3 To our knowledge, plaque-stage mycosis fungoides has not been previously reported.
As is the case with NF1, mycosis fungoides has been associated with secondary malignancies, most commonly lymphoproliferative disorders. It is postulated that the increase in immune dysfunction and T-cell dysregulation fosters tumor growth in these other locations.10 The cancer incidence in patients with CTCL is deemed 2.4 times greater than the general population and 3.3 times greater than in white males alone.13 An excess of lung and colon cancer, as well as non-Hodgkin's lymphoma has been found in these patients.10
One study from Duke University revealed that 15.9% of patients with CTCL will have a second malignancy.9 Other studies have quoted risks as high as 74% following the first 5 years of diagnosis of CTCL.9,14,15 It appears that secondary malignancies are more likely if a CTCL patient has received chemotherapy and has a positive family history for other malignancies. The incidence of mycosis fungoides is 0.4/100,000 people. Based on currently accepted incidence data for mycosis fungoides and NF1, we would expect the combined incidence to be 0.4/25x107 to 0.4/78x107.
It is important that clinicians thoroughly investigate all suspicious lesions in patients with neurofibromatosis because cutaneous malignancies may be erroneously diagnosed as tinea corporis or contact dermatitis.
Acknowledgments—The authors would like to offer gratitude to Richard Marshall, MD, from the Department of Pathology at Touro Hospital in New Orleans, and to Alun Wang, MD, PhD, from the Department of Pathology and Dermatopathology at the Tulane University Medical Center, for their assistance and patience in corroborating this manuscript.
- Hager CM, Cohen PR, Tschen JA. Segmental neurofibromatosis: case reports and review. J Am Acad Dermatol. 1997;37:864-869.
- Zvulunov A, Esterly NR. Neurocutaneous syndromes associated with pigmentary skin lesions. J Am Acad Dermatol. 1995;32:915-935.
- Hope DG, Mulvihill JJ. Malignancies in neurofibromatosis. Adv Neurol. 1981;29:33-56.
- Eichenfield LF, Levy ML, Paller AS, et al. Guidelines of care for neurofibromatosis type 1. J Am Acad Dermatol. 1997;37:625-630.
- Trattner A, David M, Ingber A, et al. Coexistence of late-onset neurofibromatosis and cutaneous T cell lymphoma. J Am Acad Dermatol. 1990;23:932-934.
- Mulvihill JJ, Parry DM, Sherman JL, et al. NIH conference. neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990;113:39-52.
- Leroy K, Dumas V, Martin-Garcia N, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients. Arch Dermatol. 2001;137:908-913.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617-1627.
- Olsen EA, Delzell E, Jegasothy BV. Second malignancies in cutaneous T cell lymphoma. J Am Acad Dermatol. 1984;10:197-204.
- Knight WA III, Murphy WK, Gottlieb JA. Neurofibromatosis associated with malignant neurofibromas. Arch Dermatol. 1973;107:747-750.
- Mastrangelo MF, Goepp CE, Patel YA, et al. Cutaneous melanoma in a patient with neurofibromatosis. Arch Dermatol. 1979;115:864-865.
- Wu H, Elenitsas R. Malignant nodular hidradenoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Cutis. 2001;68:273-278.
- Otsuka F, Kawashima T, Imakado S. Lisch nodules and skin manifestations in neurofibromatosis type 1. Arch Dermatol. 2001;137:232-233.
- Duvic M. Treatment of cutaneous T-cell lymphoma from a dermatologist's perspective. Clin Lymphoma. 2000;1:515-520.
- Kantor AF, Curtis RE, Vonderheid EC, et al. Risk of secondary malignancy after cutaneous T-cell lymphoma. Cancer. 1989;63:1612-1615.
A variety of neoplasms may occur in association with neurofibromatosis type 1 (NF1). We describe a patient with NF1 and mycosis fungoides. Recommendations for the initial and long-term evaluations of patients with neurofibromatosis are presented.
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is an autosomal-dominant syndrome with no racial, gender, or ethnic predilection.1 It has an estimated incidence of 1 in 3000 to 1 in 7800 people.1,2 Although there are at least 7 types of neurofibromatosis, NF1 is the most common form. Numerous malignancies have been linked to NF1 including pheochromocytoma, thyroid carcinoma, leukemia, and melanoma.3,4 Over the past 30 years, there has been one case report of the coexistence of the leukemic phase of cutaneous T-cell lymphoma and NF1.5 We report the occurrence of the plaque variant of cutaneous T-cell lymphoma in a patient with NF1.
Case Report
A 72-year-old white man with a lifelong history of NF1 presented for evaluation of an erythematous plaque on his left hip. It had been present for 3 months and had slowly increased in size. The lesion had been treated with econazole cream without improvement. The patient's medical history included atrial fibrillation necessitating daily treatment with amiodarone and acetylsalicylic acid. His family history was significant for a mother with NF1. Although currently retired, he had worked as a professor for more than 20 years.
Physical examination revealed an erythematous scaly plaque 14x16 cm in diameter over his left hip extending to his left flank (Figure). The patient had hundreds of flesh-colored nodules varying in size from 0.5 to 3.0 cm distributed over his entire body. In addition, he had 2- to 3-mm hyperpigmented macules in the axilla and inguinal areas, scattered larger café-au-lait macules, and iris hamartomas (Lisch nodules).6 There was no palpable lymphadenopathy or organomegaly.
Laboratory data included a complete blood count, which consisted of a hemoglobin level of 16 g/dL; hematocrit level of 46.5%; and normal blood differential and white blood cell counts. The lymphocyte profile was remarkable for an absolute CD4 count of 340 mm3 (reference range, 359–1519 mm3) and the percentage of CD4+ lymphocytes equal to 26.2% (reference range, 30.8%–58.5%). His chest x-ray revealed moderate pulmonary fibrosis and changes consistent with chronic obstructive pulmonary disease, but no active lung disease. A computed tomographic scan of the abdomen and pelvis was unremarkable for any lymphadenopathy, but there was displacement of the left kidney and bowel into the left hemithorax and a foramen of Bochdalek hernia.
The results of a 3-mm punch skin biopsy of the plaque on the patient's left flank demonstrated psoriasiform acanthosis with a mild to moderate atypical epidermotropic lymphocytic infiltrate and a subjacent neurofibroma. Immunophenotyping revealed prominent epidermotropism of CD3+ lymphocytes. Fewer intraepidermal lymphocytes marked with CD4 and CD5 immunostains. No CD7 deletion was present, but a T-cell gamma gene rearrangement was identified. The patient was diagnosed with stage 1A cutaneous T-cell lymphoma (CTCL)(T1N0M0).
Comment
NF1 is the most common of the neurocutaneous disorders.3 At present, National Institutes of Health criteria for diagnosis include 2 or more of the following: 6 or more café-au-lait macules larger than 5 mm in greatest diameter in prepubertal patients and larger than 15 mm in greatest diameter in postpubertal patients; 2 or more neurofibromas of any type or one plexiform neurofibroma; freckling in the axillary or inguinal area; optic nerve glioma; 2 or more Lisch nodules; a distinctive osseous lesion such as sphenoid wing dysplasia or thinning of the bone cortex with or without pseudoarthrosis; a first-degree relative (parent, sibling, or offspring) with NF1 by the above criteria.1,2,6,7 Different criteria appear at various stages of the patient's development (ie, café-au-lait macules appear at birth and neurofibromas emerge at adolescence).8 Lisch nodules have been regarded as pathognomonic.4 Intertriginous freckling is highly suggestive of the diagnosis but also may be seen in Watson syndrome. The 5 subtypes of neurofibromas are cutaneous, subcutaneous, nodular, plexiform, and diffuse.4 Other associated findings include vision changes, cognitive disorders, growth problems, and musculoskeletal disorders.4 Our patient fulfilled the criteria for NF1 by having neurofibromas, café-au-lait macules, Lisch nodules, intertriginous freckling, and a positive family history.
The gene defect for NF1 has been mapped to chromosome 17. The NF gene acts as a tumor-suppressor gene that dampens the production of the ras proto-oncogene.9 Therefore, the mutation of this gene contributes to tumor progression.6,8
One in 4 patients with NF1 will develop a secondary tumor throughout his or her lifetime. Tumors found with NF1 include those of neural crest origin such as pheochromocytoma, neuroblastoma, and melanoma.10 Non-neural crest tumors include Wilms tumor, rhabdomyosarcoma, granular cell tumors, malignant nodular hidradenoma, and leukemia.11,12 Patients with both NF1 and juvenile xanthogranuloma appear to be at increased risk for leukemia. Malignant peripheral nerve sheath tumors are associated with NF1 and are a potential cause of mortality in adult patients with NF1. Typically, the malignancy will occur before age 38 years, arising in a longstanding neurofibroma. Multiple malignancies occur in 12% of patients.3 To our knowledge, plaque-stage mycosis fungoides has not been previously reported.
As is the case with NF1, mycosis fungoides has been associated with secondary malignancies, most commonly lymphoproliferative disorders. It is postulated that the increase in immune dysfunction and T-cell dysregulation fosters tumor growth in these other locations.10 The cancer incidence in patients with CTCL is deemed 2.4 times greater than the general population and 3.3 times greater than in white males alone.13 An excess of lung and colon cancer, as well as non-Hodgkin's lymphoma has been found in these patients.10
One study from Duke University revealed that 15.9% of patients with CTCL will have a second malignancy.9 Other studies have quoted risks as high as 74% following the first 5 years of diagnosis of CTCL.9,14,15 It appears that secondary malignancies are more likely if a CTCL patient has received chemotherapy and has a positive family history for other malignancies. The incidence of mycosis fungoides is 0.4/100,000 people. Based on currently accepted incidence data for mycosis fungoides and NF1, we would expect the combined incidence to be 0.4/25x107 to 0.4/78x107.
It is important that clinicians thoroughly investigate all suspicious lesions in patients with neurofibromatosis because cutaneous malignancies may be erroneously diagnosed as tinea corporis or contact dermatitis.
Acknowledgments—The authors would like to offer gratitude to Richard Marshall, MD, from the Department of Pathology at Touro Hospital in New Orleans, and to Alun Wang, MD, PhD, from the Department of Pathology and Dermatopathology at the Tulane University Medical Center, for their assistance and patience in corroborating this manuscript.
A variety of neoplasms may occur in association with neurofibromatosis type 1 (NF1). We describe a patient with NF1 and mycosis fungoides. Recommendations for the initial and long-term evaluations of patients with neurofibromatosis are presented.
Neurofibromatosis type 1 (NF1), also known as von Recklinghausen disease, is an autosomal-dominant syndrome with no racial, gender, or ethnic predilection.1 It has an estimated incidence of 1 in 3000 to 1 in 7800 people.1,2 Although there are at least 7 types of neurofibromatosis, NF1 is the most common form. Numerous malignancies have been linked to NF1 including pheochromocytoma, thyroid carcinoma, leukemia, and melanoma.3,4 Over the past 30 years, there has been one case report of the coexistence of the leukemic phase of cutaneous T-cell lymphoma and NF1.5 We report the occurrence of the plaque variant of cutaneous T-cell lymphoma in a patient with NF1.
Case Report
A 72-year-old white man with a lifelong history of NF1 presented for evaluation of an erythematous plaque on his left hip. It had been present for 3 months and had slowly increased in size. The lesion had been treated with econazole cream without improvement. The patient's medical history included atrial fibrillation necessitating daily treatment with amiodarone and acetylsalicylic acid. His family history was significant for a mother with NF1. Although currently retired, he had worked as a professor for more than 20 years.
Physical examination revealed an erythematous scaly plaque 14x16 cm in diameter over his left hip extending to his left flank (Figure). The patient had hundreds of flesh-colored nodules varying in size from 0.5 to 3.0 cm distributed over his entire body. In addition, he had 2- to 3-mm hyperpigmented macules in the axilla and inguinal areas, scattered larger café-au-lait macules, and iris hamartomas (Lisch nodules).6 There was no palpable lymphadenopathy or organomegaly.
Laboratory data included a complete blood count, which consisted of a hemoglobin level of 16 g/dL; hematocrit level of 46.5%; and normal blood differential and white blood cell counts. The lymphocyte profile was remarkable for an absolute CD4 count of 340 mm3 (reference range, 359–1519 mm3) and the percentage of CD4+ lymphocytes equal to 26.2% (reference range, 30.8%–58.5%). His chest x-ray revealed moderate pulmonary fibrosis and changes consistent with chronic obstructive pulmonary disease, but no active lung disease. A computed tomographic scan of the abdomen and pelvis was unremarkable for any lymphadenopathy, but there was displacement of the left kidney and bowel into the left hemithorax and a foramen of Bochdalek hernia.
The results of a 3-mm punch skin biopsy of the plaque on the patient's left flank demonstrated psoriasiform acanthosis with a mild to moderate atypical epidermotropic lymphocytic infiltrate and a subjacent neurofibroma. Immunophenotyping revealed prominent epidermotropism of CD3+ lymphocytes. Fewer intraepidermal lymphocytes marked with CD4 and CD5 immunostains. No CD7 deletion was present, but a T-cell gamma gene rearrangement was identified. The patient was diagnosed with stage 1A cutaneous T-cell lymphoma (CTCL)(T1N0M0).
Comment
NF1 is the most common of the neurocutaneous disorders.3 At present, National Institutes of Health criteria for diagnosis include 2 or more of the following: 6 or more café-au-lait macules larger than 5 mm in greatest diameter in prepubertal patients and larger than 15 mm in greatest diameter in postpubertal patients; 2 or more neurofibromas of any type or one plexiform neurofibroma; freckling in the axillary or inguinal area; optic nerve glioma; 2 or more Lisch nodules; a distinctive osseous lesion such as sphenoid wing dysplasia or thinning of the bone cortex with or without pseudoarthrosis; a first-degree relative (parent, sibling, or offspring) with NF1 by the above criteria.1,2,6,7 Different criteria appear at various stages of the patient's development (ie, café-au-lait macules appear at birth and neurofibromas emerge at adolescence).8 Lisch nodules have been regarded as pathognomonic.4 Intertriginous freckling is highly suggestive of the diagnosis but also may be seen in Watson syndrome. The 5 subtypes of neurofibromas are cutaneous, subcutaneous, nodular, plexiform, and diffuse.4 Other associated findings include vision changes, cognitive disorders, growth problems, and musculoskeletal disorders.4 Our patient fulfilled the criteria for NF1 by having neurofibromas, café-au-lait macules, Lisch nodules, intertriginous freckling, and a positive family history.
The gene defect for NF1 has been mapped to chromosome 17. The NF gene acts as a tumor-suppressor gene that dampens the production of the ras proto-oncogene.9 Therefore, the mutation of this gene contributes to tumor progression.6,8
One in 4 patients with NF1 will develop a secondary tumor throughout his or her lifetime. Tumors found with NF1 include those of neural crest origin such as pheochromocytoma, neuroblastoma, and melanoma.10 Non-neural crest tumors include Wilms tumor, rhabdomyosarcoma, granular cell tumors, malignant nodular hidradenoma, and leukemia.11,12 Patients with both NF1 and juvenile xanthogranuloma appear to be at increased risk for leukemia. Malignant peripheral nerve sheath tumors are associated with NF1 and are a potential cause of mortality in adult patients with NF1. Typically, the malignancy will occur before age 38 years, arising in a longstanding neurofibroma. Multiple malignancies occur in 12% of patients.3 To our knowledge, plaque-stage mycosis fungoides has not been previously reported.
As is the case with NF1, mycosis fungoides has been associated with secondary malignancies, most commonly lymphoproliferative disorders. It is postulated that the increase in immune dysfunction and T-cell dysregulation fosters tumor growth in these other locations.10 The cancer incidence in patients with CTCL is deemed 2.4 times greater than the general population and 3.3 times greater than in white males alone.13 An excess of lung and colon cancer, as well as non-Hodgkin's lymphoma has been found in these patients.10
One study from Duke University revealed that 15.9% of patients with CTCL will have a second malignancy.9 Other studies have quoted risks as high as 74% following the first 5 years of diagnosis of CTCL.9,14,15 It appears that secondary malignancies are more likely if a CTCL patient has received chemotherapy and has a positive family history for other malignancies. The incidence of mycosis fungoides is 0.4/100,000 people. Based on currently accepted incidence data for mycosis fungoides and NF1, we would expect the combined incidence to be 0.4/25x107 to 0.4/78x107.
It is important that clinicians thoroughly investigate all suspicious lesions in patients with neurofibromatosis because cutaneous malignancies may be erroneously diagnosed as tinea corporis or contact dermatitis.
Acknowledgments—The authors would like to offer gratitude to Richard Marshall, MD, from the Department of Pathology at Touro Hospital in New Orleans, and to Alun Wang, MD, PhD, from the Department of Pathology and Dermatopathology at the Tulane University Medical Center, for their assistance and patience in corroborating this manuscript.
- Hager CM, Cohen PR, Tschen JA. Segmental neurofibromatosis: case reports and review. J Am Acad Dermatol. 1997;37:864-869.
- Zvulunov A, Esterly NR. Neurocutaneous syndromes associated with pigmentary skin lesions. J Am Acad Dermatol. 1995;32:915-935.
- Hope DG, Mulvihill JJ. Malignancies in neurofibromatosis. Adv Neurol. 1981;29:33-56.
- Eichenfield LF, Levy ML, Paller AS, et al. Guidelines of care for neurofibromatosis type 1. J Am Acad Dermatol. 1997;37:625-630.
- Trattner A, David M, Ingber A, et al. Coexistence of late-onset neurofibromatosis and cutaneous T cell lymphoma. J Am Acad Dermatol. 1990;23:932-934.
- Mulvihill JJ, Parry DM, Sherman JL, et al. NIH conference. neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990;113:39-52.
- Leroy K, Dumas V, Martin-Garcia N, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients. Arch Dermatol. 2001;137:908-913.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617-1627.
- Olsen EA, Delzell E, Jegasothy BV. Second malignancies in cutaneous T cell lymphoma. J Am Acad Dermatol. 1984;10:197-204.
- Knight WA III, Murphy WK, Gottlieb JA. Neurofibromatosis associated with malignant neurofibromas. Arch Dermatol. 1973;107:747-750.
- Mastrangelo MF, Goepp CE, Patel YA, et al. Cutaneous melanoma in a patient with neurofibromatosis. Arch Dermatol. 1979;115:864-865.
- Wu H, Elenitsas R. Malignant nodular hidradenoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Cutis. 2001;68:273-278.
- Otsuka F, Kawashima T, Imakado S. Lisch nodules and skin manifestations in neurofibromatosis type 1. Arch Dermatol. 2001;137:232-233.
- Duvic M. Treatment of cutaneous T-cell lymphoma from a dermatologist's perspective. Clin Lymphoma. 2000;1:515-520.
- Kantor AF, Curtis RE, Vonderheid EC, et al. Risk of secondary malignancy after cutaneous T-cell lymphoma. Cancer. 1989;63:1612-1615.
- Hager CM, Cohen PR, Tschen JA. Segmental neurofibromatosis: case reports and review. J Am Acad Dermatol. 1997;37:864-869.
- Zvulunov A, Esterly NR. Neurocutaneous syndromes associated with pigmentary skin lesions. J Am Acad Dermatol. 1995;32:915-935.
- Hope DG, Mulvihill JJ. Malignancies in neurofibromatosis. Adv Neurol. 1981;29:33-56.
- Eichenfield LF, Levy ML, Paller AS, et al. Guidelines of care for neurofibromatosis type 1. J Am Acad Dermatol. 1997;37:625-630.
- Trattner A, David M, Ingber A, et al. Coexistence of late-onset neurofibromatosis and cutaneous T cell lymphoma. J Am Acad Dermatol. 1990;23:932-934.
- Mulvihill JJ, Parry DM, Sherman JL, et al. NIH conference. neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis): an update. Ann Intern Med. 1990;113:39-52.
- Leroy K, Dumas V, Martin-Garcia N, et al. Malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1: a clinicopathologic and molecular study of 17 patients. Arch Dermatol. 2001;137:908-913.
- Riccardi VM. Von Recklinghausen neurofibromatosis. N Engl J Med. 1981;305:1617-1627.
- Olsen EA, Delzell E, Jegasothy BV. Second malignancies in cutaneous T cell lymphoma. J Am Acad Dermatol. 1984;10:197-204.
- Knight WA III, Murphy WK, Gottlieb JA. Neurofibromatosis associated with malignant neurofibromas. Arch Dermatol. 1973;107:747-750.
- Mastrangelo MF, Goepp CE, Patel YA, et al. Cutaneous melanoma in a patient with neurofibromatosis. Arch Dermatol. 1979;115:864-865.
- Wu H, Elenitsas R. Malignant nodular hidradenoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Cutis. 2001;68:273-278.
- Otsuka F, Kawashima T, Imakado S. Lisch nodules and skin manifestations in neurofibromatosis type 1. Arch Dermatol. 2001;137:232-233.
- Duvic M. Treatment of cutaneous T-cell lymphoma from a dermatologist's perspective. Clin Lymphoma. 2000;1:515-520.
- Kantor AF, Curtis RE, Vonderheid EC, et al. Risk of secondary malignancy after cutaneous T-cell lymphoma. Cancer. 1989;63:1612-1615.
Lichen Planopilaris Presenting as Truncal Alopecia: A Case Presentation and Review of the Literature
Enoxaparin-Induced Generalized Exanthem
Enoxaparin and other low molecular weight heparins (LMWHs) are widely used to treat and prevent thromboembolic disorders. Rare adverse skin reactions to LMWH are largely confined to the localized sites of injections and include ecchymoses, erythematous plaques, and nodules.1-3 Previous reports of generalized skin rash due to high molecular weight heparin (HMWH) given intravenously and subcutaneously have been well described.4-7 Only one case of a generalized skin reaction to enoxaparin has been reported in Europe.8 We report the first case of a generalized maculopapular rash due to subcutaneous enoxaparin injections in the English literature. back to top
Case Report A 53-year-old white woman with morbid obesity and a history of deep venous thromboses, pulmonary emboli, and cor pulmonale developed a generalized pruritic rash on the fourth day of therapy with subcutaneous enoxaparin injections. The patient had been admitted to the hospital for a deep venous thrombosis in her leg. She initially was given intravenous heparin for 5 days, and then was switched to oral warfarin for anticoagulation. Warfarin was administered for one week but was discontinued because a Greenfield filter placement was being considered. She was then started on subcutaneous enoxaparin, 100 mg twice a day. By day 4 of enoxaparin administration, a rash appeared on the malar area of the face that quickly spread to the trunk and proximal extremities over the next few days. Tender lesions at injection sites on the abdomen also were noted. The patient had no known history of allergies to any medications prior to this episode. She denied any similar rash in the past. She had received heparin during previous hospitalizations without any adverse reactions. She denied receiving enoxaparin before this hospital stay. Her other medications included folic acid, metoprolol tartrate, amiodarone hydrochloride, acetaminophen with codeine, and albuterol inhaler, all of which she had been taking for many months to years. The patient was given albumin intravenously during the early part of her hospitalization for oliguria. No new medications were started prior to her admission.
On physical examination, there was a generalized blanching erythematous maculopapular rash, with marked accentuation on the nose and cheeks (Figures 1 and 2). There were tender ecchymotic nodules on the abdomen at the sites of previous enoxaparin injections. No mucous membrane lesions were seen.
| Figure not available. | Figure 1. Erythema with marked accentuation on the nose and cheeks. |
Results of laboratory studies, including complete blood cell count, blood chemistries, and transaminases, were within normal limits. There was no eosinophilia. Antinuclear antibodies were negative. Results of a skin biopsy of the maculopapular rash demonstrated a superficial perivascular infiltrate with lymphocytes and a few eosinophils (Figure 3). The clinical presentation and histologic findings were consistent with a drug eruption.
All medications were halted, and 60 mg of oral prednisone was started. The rash began to resolve 3 days later. Prednisone was tapered, and all of the patient’s long-standing medications were restarted. The patient also was restarted on warfarin for anticoagulation. There was no recurrence of the rash. A provocation test with enoxaparin was offered, but the patient declined. back to top
Comment Heparin-induced skin reactions associated with subcutaneous administration have been well described in the literature.1,9,10 Typical lesions are erythematous, tender nodules at injection sites. Reports of reactions with different preparations of HMWH and LMWH have implicated the heparin molecule rather than preservatives as the causative agent.2,11,12 Substitution of one heparin preparation for another does not prevent the recurrence of similar reactions in susceptible individuals.2,7,11,13
Enoxaparin is the first LMWH to be marketed in the United States. LMWHs are derived from unfractionated heparin, a heterogeneous mixture of polysaccharide chains. Unfractionated heparin is prepared from ox lung and bovine and porcine intestinal mucosa.11 Adverse effects are typically less common with LMWH than with unfractionated heparin, perhaps due to the smaller size of molecules, the greater homogeneity, and the exclusive porcine origin of LMWH.14
Localized adverse cutaneous reactions to LMWH have been reported. Ecchymoses and hematomas are thought to be directly related to the pharmacologic effects of the agent.15 Urticaria is probably due to local histamine release or is a classic type-I immediate hypersensitivity reaction. Early cases of urticaria with heparin were thought to be due to sensitivity to contaminant proteins.16 In recent years, reports of urticarial reactions have been rare.3 One case of generalized urticaria and angioedema has been attributed to enoxaparin.17 Skin necrosis from unfractionated heparin at injection sites typically occurs after 5 to 9 days of treatment.9 Lesions also have been noted at distant sites from subcutaneous and intravenous administration of unfractionated heparin.18,19 Similar cases of skin necrosis at injection sites have been reported with LMWH.15,20 Enoxaparin-induced skin necrosis distant from injection sites has been described in a patient with diabetes.21 Some patients may develop heparin-induced thrombocytopenia and present with erythematous plaques that rapidly evolve into necrosis.12 Occurring 6 to 10 days after the start of therapy, this condition is thought to be secondary to heparin-antiheparin IgG immune complexes that activate platelets and is more common in patients treated with unfractionated heparin than in patients on LMWH therapy.22
Delayed-type hypersensitivity skin reactions at injection sites have been reported with all types of LMWH.23,24 Lesions are well-circumscribed, erythematous, infiltrated, or vesicular plaques. Female sex, obesity, diabetes, and prolonged application of the drug are suspected risk factors for the development of sensitization to HMWH and LMWH.7,25
To our knowledge, this is the first case reported in the English literature of a generalized exanthem due to subcutaneous injection of enoxaparin. The temporal relationship of the medication and onset of rash, the patient’s erythematous nodules at enoxaparin injection sites, and the absence of recurrence after all other medications were restarted strongly implicate enoxaparin as the sole responsible agent for her generalized skin reaction. Incidentally, the patient was given albumin prior to the rash; however, the exceedingly rare, untoward reactions to albumin, such as nausea, fever, chills, or urticaria, usually disappear when the infusion is slowed or temporarily stopped. Our patient tolerated the albumin infusions without any adverse symptoms. In addition to being obese and female, our patient had been exposed to heparin multiple times in the past; thus, she shares the suspected risk factors that have been previously described in patients with localized hypersensitivity skin reactions to LMWH.
Lesions at the injection sites combined with the generalized rash that appeared 4 days after the onset of enoxaparin therapy suggest a type IV hypersensitivity reaction in this patient. A subcutaneous provocation test is considered the gold standard test for this condition.7 However, potential adverse effects such as anaphylactic shock or heparin-induced thrombocytopenia may occur.14 A provocation test was offered to the patient, but she declined.
There are several potential therapeutic alternatives to heparin therapy. Danaparoid sodium and pentosan polysulfate have polysaccharide chains with structures chemically different from heparin. Danaparoid, however, is a mucopolysaccharide derived from porcine intestinal mucosa and may share the same allergens with heparin. Allergic reactions with danaparoid and pentosan polysulfate have been reported.4,6,26 Hirudin is another anticoagulation agent that the patient may tolerate because recombinant hirudins do not cross-react with HMWH or LMWH and heparinoids.13,27 Unfortunately, they too can cause delayed-type hypersensitivity reactions.5 At present, our patient is on warfarin therapy.
Skin hypersensitivity reactions to enoxaparin are considered rare. However, given the widespread use of this and other LMWHs, it is important for the clinician to suspect this diagnosis when evaluating localized, as well as generalized, cutaneous eruptions.
Acknowledgments—The authors would like to thank Edward Heilman, MD, for his assistance with histopathology, and Leonard L. Gerschitz, MBA, RPh, for his pharmacologic expertise. back to top
- Manoharan A. Heparin-induced skin reaction with low molecular-weight heparin [letter]. Eur J Haematol. 1992;48:234.
- Phillips JK, Majumdar G, Hunt BJ, et al. Heparin-induced skin reaction due to two different preparations of low molecular weight heparin (LMWH). Br J Haematol. 1993;84:349-350.
- MacLean JA, Moscicki R, Bloch KJ. Adverse reactions to heparin. Ann Allergy. 1990;65:254-259.
- Koch P, Hindi S, Landwehr D. Delayed allergic skin reactions due to subcutaneous heparin-calcium, enoxaparin-sodium, pentosan polysulfate and acute skin lesions from systemic sodium-heparin. Contact Dermatitis. 1996;4:156-158.
- Schiffner R, Glassl A, Landthaler M, et al. Tolerance of desirudin in a patient with generalized eczema after intravenous challenge with heparin and a delayed-type skin reaction to high and low molecular weight heparins and heparinoids. Contact Dermatitis. 2000;42:49.
- Irion R, Gall H, Peter R. Delayed-type hypersensitivity to heparin with tolerance of its intravenous administration. Contact Dermatitis. 2000;3:249-250.
- Mendez J, Sanchis ME, de la Fuente R, et al. Delayed-type hypersensitivity to subcutaneous enoxaparin. Allergy. 1998;53:999-1003.
- Cordoba Lopez A, Bueno Alvarez-Arenas MI, Monterrubio Villar J, et al. Cutaneous hypersensitivity reaction to enoxaparin [in Spanish]. Med Clin (Barc). 2001;117:478-479.
- O’Toole RD. Letter: heparin: adverse reaction. Ann Intern Med. 1973;79:759.
- Klein GF, Kofler H, Wolf H, et al. Eczema-like, erythematous infiltrated plaques: a common side effect of subcutaneous heparin therapy. J Am Acad Dermatol. 1989;21:703-707.
- Ojukwu C, Jenkinson SD, Obeid D. Deep vein thrombosis in pregnancy and heparin hypersensitivity. Br J Obstet Gynaecol. 1996;13:934-936.
- Bircher A. The differential diagnosis of heparin-induced skin lesions. Br J Haematol. 1993;85:837-838.
- Grassegger A, Fritsch P, Reider N. Delayed-type hypersensitivity and cross-reactivity to heparin and danaparoid: a prospective study. Dermatol Surg. 2001;27:47-52.
- Wutschert R, Piletta P, Bounameaux H. Adverse skin reactions to low molecular weight heparins. Drug Safety. 1999;20:515-525.
- Tonn ME, Schaiff RA, Kollef MH. Enoxaparin-associated dermal necrosis: a consequence of cross-reactivity with heparin-mediated antibodies. Ann Pharmacother. 1997;31:323-326.
- Gaye A, Lerner C. Reactivity to preservative in heparin. [abstract]. J Allerg Clin Immunol. 1987;79:238.
- Odeh M, Oliven A. Urticaria and angioedema induced by low molecular weight heparin. Lancet. 1992;340:972-973.
- Levine LE, Bernstein JE, Soltani K, et al
Enoxaparin and other low molecular weight heparins (LMWHs) are widely used to treat and prevent thromboembolic disorders. Rare adverse skin reactions to LMWH are largely confined to the localized sites of injections and include ecchymoses, erythematous plaques, and nodules.1-3 Previous reports of generalized skin rash due to high molecular weight heparin (HMWH) given intravenously and subcutaneously have been well described.4-7 Only one case of a generalized skin reaction to enoxaparin has been reported in Europe.8 We report the first case of a generalized maculopapular rash due to subcutaneous enoxaparin injections in the English literature. back to top
Case Report A 53-year-old white woman with morbid obesity and a history of deep venous thromboses, pulmonary emboli, and cor pulmonale developed a generalized pruritic rash on the fourth day of therapy with subcutaneous enoxaparin injections. The patient had been admitted to the hospital for a deep venous thrombosis in her leg. She initially was given intravenous heparin for 5 days, and then was switched to oral warfarin for anticoagulation. Warfarin was administered for one week but was discontinued because a Greenfield filter placement was being considered. She was then started on subcutaneous enoxaparin, 100 mg twice a day. By day 4 of enoxaparin administration, a rash appeared on the malar area of the face that quickly spread to the trunk and proximal extremities over the next few days. Tender lesions at injection sites on the abdomen also were noted. The patient had no known history of allergies to any medications prior to this episode. She denied any similar rash in the past. She had received heparin during previous hospitalizations without any adverse reactions. She denied receiving enoxaparin before this hospital stay. Her other medications included folic acid, metoprolol tartrate, amiodarone hydrochloride, acetaminophen with codeine, and albuterol inhaler, all of which she had been taking for many months to years. The patient was given albumin intravenously during the early part of her hospitalization for oliguria. No new medications were started prior to her admission.
On physical examination, there was a generalized blanching erythematous maculopapular rash, with marked accentuation on the nose and cheeks (Figures 1 and 2). There were tender ecchymotic nodules on the abdomen at the sites of previous enoxaparin injections. No mucous membrane lesions were seen.
| Figure not available. | Figure 1. Erythema with marked accentuation on the nose and cheeks. |
Results of laboratory studies, including complete blood cell count, blood chemistries, and transaminases, were within normal limits. There was no eosinophilia. Antinuclear antibodies were negative. Results of a skin biopsy of the maculopapular rash demonstrated a superficial perivascular infiltrate with lymphocytes and a few eosinophils (Figure 3). The clinical presentation and histologic findings were consistent with a drug eruption.
All medications were halted, and 60 mg of oral prednisone was started. The rash began to resolve 3 days later. Prednisone was tapered, and all of the patient’s long-standing medications were restarted. The patient also was restarted on warfarin for anticoagulation. There was no recurrence of the rash. A provocation test with enoxaparin was offered, but the patient declined. back to top
Comment Heparin-induced skin reactions associated with subcutaneous administration have been well described in the literature.1,9,10 Typical lesions are erythematous, tender nodules at injection sites. Reports of reactions with different preparations of HMWH and LMWH have implicated the heparin molecule rather than preservatives as the causative agent.2,11,12 Substitution of one heparin preparation for another does not prevent the recurrence of similar reactions in susceptible individuals.2,7,11,13
Enoxaparin is the first LMWH to be marketed in the United States. LMWHs are derived from unfractionated heparin, a heterogeneous mixture of polysaccharide chains. Unfractionated heparin is prepared from ox lung and bovine and porcine intestinal mucosa.11 Adverse effects are typically less common with LMWH than with unfractionated heparin, perhaps due to the smaller size of molecules, the greater homogeneity, and the exclusive porcine origin of LMWH.14
Localized adverse cutaneous reactions to LMWH have been reported. Ecchymoses and hematomas are thought to be directly related to the pharmacologic effects of the agent.15 Urticaria is probably due to local histamine release or is a classic type-I immediate hypersensitivity reaction. Early cases of urticaria with heparin were thought to be due to sensitivity to contaminant proteins.16 In recent years, reports of urticarial reactions have been rare.3 One case of generalized urticaria and angioedema has been attributed to enoxaparin.17 Skin necrosis from unfractionated heparin at injection sites typically occurs after 5 to 9 days of treatment.9 Lesions also have been noted at distant sites from subcutaneous and intravenous administration of unfractionated heparin.18,19 Similar cases of skin necrosis at injection sites have been reported with LMWH.15,20 Enoxaparin-induced skin necrosis distant from injection sites has been described in a patient with diabetes.21 Some patients may develop heparin-induced thrombocytopenia and present with erythematous plaques that rapidly evolve into necrosis.12 Occurring 6 to 10 days after the start of therapy, this condition is thought to be secondary to heparin-antiheparin IgG immune complexes that activate platelets and is more common in patients treated with unfractionated heparin than in patients on LMWH therapy.22
Delayed-type hypersensitivity skin reactions at injection sites have been reported with all types of LMWH.23,24 Lesions are well-circumscribed, erythematous, infiltrated, or vesicular plaques. Female sex, obesity, diabetes, and prolonged application of the drug are suspected risk factors for the development of sensitization to HMWH and LMWH.7,25
To our knowledge, this is the first case reported in the English literature of a generalized exanthem due to subcutaneous injection of enoxaparin. The temporal relationship of the medication and onset of rash, the patient’s erythematous nodules at enoxaparin injection sites, and the absence of recurrence after all other medications were restarted strongly implicate enoxaparin as the sole responsible agent for her generalized skin reaction. Incidentally, the patient was given albumin prior to the rash; however, the exceedingly rare, untoward reactions to albumin, such as nausea, fever, chills, or urticaria, usually disappear when the infusion is slowed or temporarily stopped. Our patient tolerated the albumin infusions without any adverse symptoms. In addition to being obese and female, our patient had been exposed to heparin multiple times in the past; thus, she shares the suspected risk factors that have been previously described in patients with localized hypersensitivity skin reactions to LMWH.
Lesions at the injection sites combined with the generalized rash that appeared 4 days after the onset of enoxaparin therapy suggest a type IV hypersensitivity reaction in this patient. A subcutaneous provocation test is considered the gold standard test for this condition.7 However, potential adverse effects such as anaphylactic shock or heparin-induced thrombocytopenia may occur.14 A provocation test was offered to the patient, but she declined.
There are several potential therapeutic alternatives to heparin therapy. Danaparoid sodium and pentosan polysulfate have polysaccharide chains with structures chemically different from heparin. Danaparoid, however, is a mucopolysaccharide derived from porcine intestinal mucosa and may share the same allergens with heparin. Allergic reactions with danaparoid and pentosan polysulfate have been reported.4,6,26 Hirudin is another anticoagulation agent that the patient may tolerate because recombinant hirudins do not cross-react with HMWH or LMWH and heparinoids.13,27 Unfortunately, they too can cause delayed-type hypersensitivity reactions.5 At present, our patient is on warfarin therapy.
Skin hypersensitivity reactions to enoxaparin are considered rare. However, given the widespread use of this and other LMWHs, it is important for the clinician to suspect this diagnosis when evaluating localized, as well as generalized, cutaneous eruptions.
Acknowledgments—The authors would like to thank Edward Heilman, MD, for his assistance with histopathology, and Leonard L. Gerschitz, MBA, RPh, for his pharmacologic expertise. back to top
Enoxaparin and other low molecular weight heparins (LMWHs) are widely used to treat and prevent thromboembolic disorders. Rare adverse skin reactions to LMWH are largely confined to the localized sites of injections and include ecchymoses, erythematous plaques, and nodules.1-3 Previous reports of generalized skin rash due to high molecular weight heparin (HMWH) given intravenously and subcutaneously have been well described.4-7 Only one case of a generalized skin reaction to enoxaparin has been reported in Europe.8 We report the first case of a generalized maculopapular rash due to subcutaneous enoxaparin injections in the English literature. back to top
Case Report A 53-year-old white woman with morbid obesity and a history of deep venous thromboses, pulmonary emboli, and cor pulmonale developed a generalized pruritic rash on the fourth day of therapy with subcutaneous enoxaparin injections. The patient had been admitted to the hospital for a deep venous thrombosis in her leg. She initially was given intravenous heparin for 5 days, and then was switched to oral warfarin for anticoagulation. Warfarin was administered for one week but was discontinued because a Greenfield filter placement was being considered. She was then started on subcutaneous enoxaparin, 100 mg twice a day. By day 4 of enoxaparin administration, a rash appeared on the malar area of the face that quickly spread to the trunk and proximal extremities over the next few days. Tender lesions at injection sites on the abdomen also were noted. The patient had no known history of allergies to any medications prior to this episode. She denied any similar rash in the past. She had received heparin during previous hospitalizations without any adverse reactions. She denied receiving enoxaparin before this hospital stay. Her other medications included folic acid, metoprolol tartrate, amiodarone hydrochloride, acetaminophen with codeine, and albuterol inhaler, all of which she had been taking for many months to years. The patient was given albumin intravenously during the early part of her hospitalization for oliguria. No new medications were started prior to her admission.
On physical examination, there was a generalized blanching erythematous maculopapular rash, with marked accentuation on the nose and cheeks (Figures 1 and 2). There were tender ecchymotic nodules on the abdomen at the sites of previous enoxaparin injections. No mucous membrane lesions were seen.
| Figure not available. | Figure 1. Erythema with marked accentuation on the nose and cheeks. |
Results of laboratory studies, including complete blood cell count, blood chemistries, and transaminases, were within normal limits. There was no eosinophilia. Antinuclear antibodies were negative. Results of a skin biopsy of the maculopapular rash demonstrated a superficial perivascular infiltrate with lymphocytes and a few eosinophils (Figure 3). The clinical presentation and histologic findings were consistent with a drug eruption.
All medications were halted, and 60 mg of oral prednisone was started. The rash began to resolve 3 days later. Prednisone was tapered, and all of the patient’s long-standing medications were restarted. The patient also was restarted on warfarin for anticoagulation. There was no recurrence of the rash. A provocation test with enoxaparin was offered, but the patient declined. back to top
Comment Heparin-induced skin reactions associated with subcutaneous administration have been well described in the literature.1,9,10 Typical lesions are erythematous, tender nodules at injection sites. Reports of reactions with different preparations of HMWH and LMWH have implicated the heparin molecule rather than preservatives as the causative agent.2,11,12 Substitution of one heparin preparation for another does not prevent the recurrence of similar reactions in susceptible individuals.2,7,11,13
Enoxaparin is the first LMWH to be marketed in the United States. LMWHs are derived from unfractionated heparin, a heterogeneous mixture of polysaccharide chains. Unfractionated heparin is prepared from ox lung and bovine and porcine intestinal mucosa.11 Adverse effects are typically less common with LMWH than with unfractionated heparin, perhaps due to the smaller size of molecules, the greater homogeneity, and the exclusive porcine origin of LMWH.14
Localized adverse cutaneous reactions to LMWH have been reported. Ecchymoses and hematomas are thought to be directly related to the pharmacologic effects of the agent.15 Urticaria is probably due to local histamine release or is a classic type-I immediate hypersensitivity reaction. Early cases of urticaria with heparin were thought to be due to sensitivity to contaminant proteins.16 In recent years, reports of urticarial reactions have been rare.3 One case of generalized urticaria and angioedema has been attributed to enoxaparin.17 Skin necrosis from unfractionated heparin at injection sites typically occurs after 5 to 9 days of treatment.9 Lesions also have been noted at distant sites from subcutaneous and intravenous administration of unfractionated heparin.18,19 Similar cases of skin necrosis at injection sites have been reported with LMWH.15,20 Enoxaparin-induced skin necrosis distant from injection sites has been described in a patient with diabetes.21 Some patients may develop heparin-induced thrombocytopenia and present with erythematous plaques that rapidly evolve into necrosis.12 Occurring 6 to 10 days after the start of therapy, this condition is thought to be secondary to heparin-antiheparin IgG immune complexes that activate platelets and is more common in patients treated with unfractionated heparin than in patients on LMWH therapy.22
Delayed-type hypersensitivity skin reactions at injection sites have been reported with all types of LMWH.23,24 Lesions are well-circumscribed, erythematous, infiltrated, or vesicular plaques. Female sex, obesity, diabetes, and prolonged application of the drug are suspected risk factors for the development of sensitization to HMWH and LMWH.7,25
To our knowledge, this is the first case reported in the English literature of a generalized exanthem due to subcutaneous injection of enoxaparin. The temporal relationship of the medication and onset of rash, the patient’s erythematous nodules at enoxaparin injection sites, and the absence of recurrence after all other medications were restarted strongly implicate enoxaparin as the sole responsible agent for her generalized skin reaction. Incidentally, the patient was given albumin prior to the rash; however, the exceedingly rare, untoward reactions to albumin, such as nausea, fever, chills, or urticaria, usually disappear when the infusion is slowed or temporarily stopped. Our patient tolerated the albumin infusions without any adverse symptoms. In addition to being obese and female, our patient had been exposed to heparin multiple times in the past; thus, she shares the suspected risk factors that have been previously described in patients with localized hypersensitivity skin reactions to LMWH.
Lesions at the injection sites combined with the generalized rash that appeared 4 days after the onset of enoxaparin therapy suggest a type IV hypersensitivity reaction in this patient. A subcutaneous provocation test is considered the gold standard test for this condition.7 However, potential adverse effects such as anaphylactic shock or heparin-induced thrombocytopenia may occur.14 A provocation test was offered to the patient, but she declined.
There are several potential therapeutic alternatives to heparin therapy. Danaparoid sodium and pentosan polysulfate have polysaccharide chains with structures chemically different from heparin. Danaparoid, however, is a mucopolysaccharide derived from porcine intestinal mucosa and may share the same allergens with heparin. Allergic reactions with danaparoid and pentosan polysulfate have been reported.4,6,26 Hirudin is another anticoagulation agent that the patient may tolerate because recombinant hirudins do not cross-react with HMWH or LMWH and heparinoids.13,27 Unfortunately, they too can cause delayed-type hypersensitivity reactions.5 At present, our patient is on warfarin therapy.
Skin hypersensitivity reactions to enoxaparin are considered rare. However, given the widespread use of this and other LMWHs, it is important for the clinician to suspect this diagnosis when evaluating localized, as well as generalized, cutaneous eruptions.
Acknowledgments—The authors would like to thank Edward Heilman, MD, for his assistance with histopathology, and Leonard L. Gerschitz, MBA, RPh, for his pharmacologic expertise. back to top
- Manoharan A. Heparin-induced skin reaction with low molecular-weight heparin [letter]. Eur J Haematol. 1992;48:234.
- Phillips JK, Majumdar G, Hunt BJ, et al. Heparin-induced skin reaction due to two different preparations of low molecular weight heparin (LMWH). Br J Haematol. 1993;84:349-350.
- MacLean JA, Moscicki R, Bloch KJ. Adverse reactions to heparin. Ann Allergy. 1990;65:254-259.
- Koch P, Hindi S, Landwehr D. Delayed allergic skin reactions due to subcutaneous heparin-calcium, enoxaparin-sodium, pentosan polysulfate and acute skin lesions from systemic sodium-heparin. Contact Dermatitis. 1996;4:156-158.
- Schiffner R, Glassl A, Landthaler M, et al. Tolerance of desirudin in a patient with generalized eczema after intravenous challenge with heparin and a delayed-type skin reaction to high and low molecular weight heparins and heparinoids. Contact Dermatitis. 2000;42:49.
- Irion R, Gall H, Peter R. Delayed-type hypersensitivity to heparin with tolerance of its intravenous administration. Contact Dermatitis. 2000;3:249-250.
- Mendez J, Sanchis ME, de la Fuente R, et al. Delayed-type hypersensitivity to subcutaneous enoxaparin. Allergy. 1998;53:999-1003.
- Cordoba Lopez A, Bueno Alvarez-Arenas MI, Monterrubio Villar J, et al. Cutaneous hypersensitivity reaction to enoxaparin [in Spanish]. Med Clin (Barc). 2001;117:478-479.
- O’Toole RD. Letter: heparin: adverse reaction. Ann Intern Med. 1973;79:759.
- Klein GF, Kofler H, Wolf H, et al. Eczema-like, erythematous infiltrated plaques: a common side effect of subcutaneous heparin therapy. J Am Acad Dermatol. 1989;21:703-707.
- Ojukwu C, Jenkinson SD, Obeid D. Deep vein thrombosis in pregnancy and heparin hypersensitivity. Br J Obstet Gynaecol. 1996;13:934-936.
- Bircher A. The differential diagnosis of heparin-induced skin lesions. Br J Haematol. 1993;85:837-838.
- Grassegger A, Fritsch P, Reider N. Delayed-type hypersensitivity and cross-reactivity to heparin and danaparoid: a prospective study. Dermatol Surg. 2001;27:47-52.
- Wutschert R, Piletta P, Bounameaux H. Adverse skin reactions to low molecular weight heparins. Drug Safety. 1999;20:515-525.
- Tonn ME, Schaiff RA, Kollef MH. Enoxaparin-associated dermal necrosis: a consequence of cross-reactivity with heparin-mediated antibodies. Ann Pharmacother. 1997;31:323-326.
- Gaye A, Lerner C. Reactivity to preservative in heparin. [abstract]. J Allerg Clin Immunol. 1987;79:238.
- Odeh M, Oliven A. Urticaria and angioedema induced by low molecular weight heparin. Lancet. 1992;340:972-973.
- Levine LE, Bernstein JE, Soltani K, et al
- Manoharan A. Heparin-induced skin reaction with low molecular-weight heparin [letter]. Eur J Haematol. 1992;48:234.
- Phillips JK, Majumdar G, Hunt BJ, et al. Heparin-induced skin reaction due to two different preparations of low molecular weight heparin (LMWH). Br J Haematol. 1993;84:349-350.
- MacLean JA, Moscicki R, Bloch KJ. Adverse reactions to heparin. Ann Allergy. 1990;65:254-259.
- Koch P, Hindi S, Landwehr D. Delayed allergic skin reactions due to subcutaneous heparin-calcium, enoxaparin-sodium, pentosan polysulfate and acute skin lesions from systemic sodium-heparin. Contact Dermatitis. 1996;4:156-158.
- Schiffner R, Glassl A, Landthaler M, et al. Tolerance of desirudin in a patient with generalized eczema after intravenous challenge with heparin and a delayed-type skin reaction to high and low molecular weight heparins and heparinoids. Contact Dermatitis. 2000;42:49.
- Irion R, Gall H, Peter R. Delayed-type hypersensitivity to heparin with tolerance of its intravenous administration. Contact Dermatitis. 2000;3:249-250.
- Mendez J, Sanchis ME, de la Fuente R, et al. Delayed-type hypersensitivity to subcutaneous enoxaparin. Allergy. 1998;53:999-1003.
- Cordoba Lopez A, Bueno Alvarez-Arenas MI, Monterrubio Villar J, et al. Cutaneous hypersensitivity reaction to enoxaparin [in Spanish]. Med Clin (Barc). 2001;117:478-479.
- O’Toole RD. Letter: heparin: adverse reaction. Ann Intern Med. 1973;79:759.
- Klein GF, Kofler H, Wolf H, et al. Eczema-like, erythematous infiltrated plaques: a common side effect of subcutaneous heparin therapy. J Am Acad Dermatol. 1989;21:703-707.
- Ojukwu C, Jenkinson SD, Obeid D. Deep vein thrombosis in pregnancy and heparin hypersensitivity. Br J Obstet Gynaecol. 1996;13:934-936.
- Bircher A. The differential diagnosis of heparin-induced skin lesions. Br J Haematol. 1993;85:837-838.
- Grassegger A, Fritsch P, Reider N. Delayed-type hypersensitivity and cross-reactivity to heparin and danaparoid: a prospective study. Dermatol Surg. 2001;27:47-52.
- Wutschert R, Piletta P, Bounameaux H. Adverse skin reactions to low molecular weight heparins. Drug Safety. 1999;20:515-525.
- Tonn ME, Schaiff RA, Kollef MH. Enoxaparin-associated dermal necrosis: a consequence of cross-reactivity with heparin-mediated antibodies. Ann Pharmacother. 1997;31:323-326.
- Gaye A, Lerner C. Reactivity to preservative in heparin. [abstract]. J Allerg Clin Immunol. 1987;79:238.
- Odeh M, Oliven A. Urticaria and angioedema induced by low molecular weight heparin. Lancet. 1992;340:972-973.
- Levine LE, Bernstein JE, Soltani K, et al
Necrobiotic Xanthogranuloma Associated With a Benign Monoclonal Gammopathy
Skin Reaction Following Immunization With Smallpox Vaccine: A Personal Perspective
Smallpox is caused by the variola virus and is capable of causing serious complications. The disease is highly contagious and may present in several forms. The variola major form has a case fatality rate of about 30%.1 The smallpox vaccine was the first immunization used in modern medicine. The first inoculation was given in 1796 by Edward Jenner, MD, who immunized an 8-year-old boy with material taken from cowpox. The inoculation was shown to protect the child from smallpox when he was inoculated 6 weeks later with material that was taken from a smallpox pustule. Since then, mass immunization against smallpox has become a well-established method of fighting the disease. Immunization is based on epidermal inoculation of live vaccinia virus, another poxvirus that provokes immunization against smallpox but does not have the ability to cause the disease. The inoculation method includes putting a droplet of vaccine on the skin and then superficially poking the skin sufficiently to cause minimal blood drops. Disease protection following immunization lasts about 10 years.2
Because humans are the only natural host of the variola virus, the World Health Organization proclaimed smallpox the first disease to be eradicated worldwide. This was accomplished in 1977, when the last naturally occurring case in the world occurred in Somalia. The eradication of smallpox made it possible to stop the use of the vaccine without the threat of natural recurrence of the disease. In many places around the world, immunization was stopped before 1977 because many years had gone by without any documented cases of smallpox. In the United States, the last case was documented in 1949, and routine smallpox immunization was discontinued in 1972. In Israel, it was discontinued in 1980.
Since the events of September 11, 2001, there have been major concerns about the possibility of bioterrorism using the variola virus, which had been kept in laboratories and could be used as an unconventional weapon by countries or militant groups. Because many individuals are not immunized against smallpox, either naturally or artificially, the potential of a major epidemic occurring is significant.3
Because routine immunization against smallpox was discontinued in the United States and Israel, entire populations are vulnerable to the disease. The possibility that Israel will be involved in war became stronger when the United States and its allies targeted Iraq as a country that develops weapons of mass destruction. The use of smallpox as a biological weapon is a major possibility.4
To prepare against smallpox, the Israeli authorities have decided on a 2-step plan. In the first step, emergency personnel are to be vaccinated. These include medical and paramedical personnel, as well as people in other emergency services. The second stage of the plan is to immunize the entire population of Israel if one case of smallpox is diagnosed.
The immunization of healthcare workers in the first stage has 2 purposes. First, medical and other emergency personnel are expected to deal with infected or potentially infected people and thus must be protected against the disease. Second, in case of an epidemic, serious life-threatening disease could be aborted by variola immunoglobulin. All those immunized prior to a smallpox epidemic are the potential donors of human gamma globulin against the disease.
Immunization of medical personnel in Israel started in September 2002. The immunization was voluntary and thus included only a portion of the potential recipients. I was immunized on September 24, 2002, at my workplace. Assuming that many practitioners do not have experience with the vaccine, I decided to keep a diary and take pictures of the changes at the site of the inoculation.
Case Report
On day 1, 3 drops of dried blood (1 mm each) appeared. There was no itching or redness. Days 2 and 3 were the same as day 1, except that the puncture spots became black. A round, red, 4-mm papule appeared on day 4. There was some itching, and the black puncture spots at the area of inoculation remained the same. The papule became a pustule 3 to 4 mm in diameter surrounded by a 2- to 3-mm rim of red papule on day 5, and itching intensified. On day 6, the pustule became 5 to 6 mm, and the red rim around it became 2 to 3 mm. The area was not painful, except when touched. Itching became more obvious, and there was some tenderness of the axillary lymph nodes, with minimal enlargement of the glands. The pustule became 6 to 7 mm, and the red rim became 3 to 4 mm, with an additional 1- to 2-mm pink rim by day 7. Headache was present.
By day 8, the pustule was 10 mm, and the 3 dark 1-mm original puncture spots were still present inside the pustular area. The pustule felt hard when touched. The red rim was 2 to 3 mm, with an additional induration of skin 10 mm in diameter around the rim. The area was somewhat itchy, and headache was present. There was no change on day 9 except that the dark spots within the dry pustule were more obvious, increasing to 2 mm each. The size of the pustule was still the same on day 10, although it was less elevated. Central umbilication was obvious. On day 11, the pustule became dryer, and the black spots inside were 2 to 3 mm. Redness around the pustule was still 2 to 3 mm and was more intense after showering. Some induration of skin remained around the pustule and red area, and it was mildly painful when touched. Itching was lessened, and there was no pain at the axillary lymph nodes (Figure 1). The dry crust began to peel on day 12. The central area was confluent black, and the red rim was 2 to 3 mm, with some itching. On day 13, the crust had no yellow color of dried pus, and the roof of the crust had peeled off completely. The exposed base of the crust was dark black and hard. One to 2 mm of the red rim was still present, with less surrounding induration. There was mild itching but no pain or discharge. Minimal change was seen on day 14, except that the lesion began to shrink slightly.
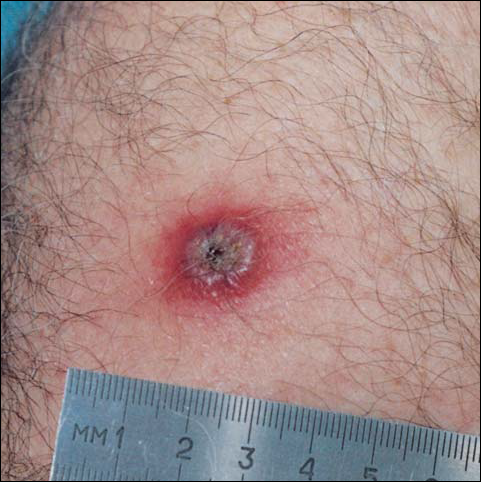
There was no pain on day 15, but the borders of the black crust began to separate from the red rim, and the red rim decreased to 1 to 2 mm. Itching became more intense, and the crust bled slightly at its border when touched. An adhesive bandage was put on the area. When the bandage was removed on day 16, it looked as if there was liquefaction of the crust, which had a yellowish color (Figure 2). The area was covered by gauze to enable air drying. On days 17 and 18, the crust was dry and brown, with no bleeding. It appeared smaller than the day before, the red rim had become pink, and there was mild itching (Figure 3). The skin above the pink rim began to peel on day 19, and there was no change in the scab. On one of the borders, there was a small discharge of serum. The scab fell off after showering (Figure 4). An ulcer 1- to 2-mm deep was obvious at the spot; the rim around the ulcer was red. The ulcer was smaller on day 20, and the rim around the ulcer was pink and still elevated above the surrounding normal skin. There was no discharge. On day 21, the base of the ulcer was dry and brown.
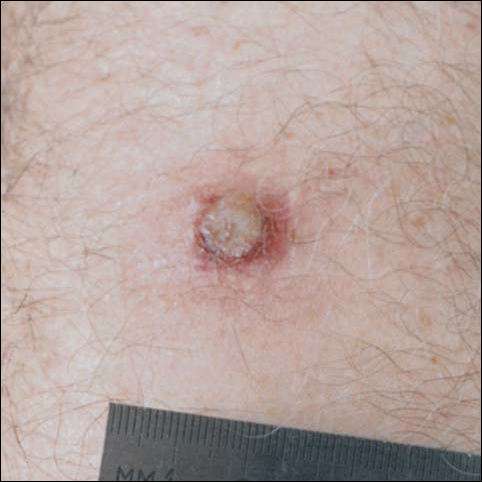


On day 22, the skin around the brown area was dry and pink, and the rim was 1 to 2 mm. Minimal changes were noted from days 23 to 27, and there was no itching. There was still no itching on day 28, and the red rim had disappeared. The central brown area peeled off, leaving a 4-mm pinkish red macule.
From days 29 to 75, the area gradually became pink. The skin looked thin and wrinkled, and there was no umbilication or elevation above the surrounding skin (Figure 5). No scar or induration was present, and symptoms had subsided.
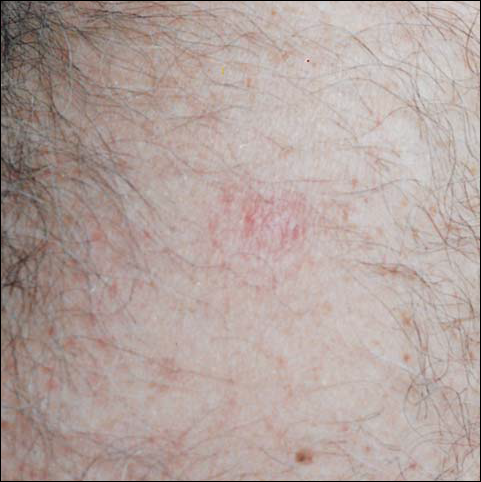
Comment
According to the US Centers for Disease Control and Prevention (CDC), if the smallpox vaccination is successful, a red and itchy bump will develop at the vaccine site in 3 to 4 days. In a week, the bump becomes a large blister, fills with pus, and begins to drain. During week 2, the blister begins to dry up, and a scab forms. The scab falls off during the third week, leaving a small scar.5 The described case follows the general description of the CDC. However, some differences should be mentioned. Itching at the site of vaccination was a major accompanying symptom. Swelling beyond the immediate site of skin reaction was obvious and lasted for more than a week. Axillary lymphadenopathy was noted. Discharge from the pustule was minimal. A scab was formed twice—once as the pus dried up and again after the first scab fell off and the drying of the wet base of the first scab dried up. At the end of the skin reaction, no scar remained.
Could the differences between the case described and the description by the CDC be explained by personal variation in skin reaction? Is the difference dependent on the skin care following the vaccination?
The instructions that were given to the Israeli recipients of the vaccine were to keep the area covered with loose gauze, change the gauze daily, not change regular skin washing habits, not touch the place of vaccination, and not put any topical preparation on the site. I followed the instructions except on day 15, when bleeding began as the scab started to peel off. At that point, a bandage was put over the area, thus limiting the ventilation and drying of the scab.
Complications following smallpox vaccination are relatively rare. Haim et al6 described an overall complication rate of 0.4 per 10,000 young army recruits. The complication rate was higher in primary recipients (those without previous exposure to the disease or vaccine).
Feery7 described an adverse skin reaction rate of 118 per 1 million recipients and a death rate of 1 to 5 per 1 million. The more serious skin reactions were eczema vaccinatum, progressive vaccinia, and neurologic and cardiac complications.
Monitoring the reaction to the vaccine is important in evaluating the intervention. Checking antibody titers postimmunization is possible. The micro–enzyme-linked immunosorbent assay technique was found to be more sensitive than plaque neutralization in finding a 4-fold increase in antibody titers.8 Postimmunization antibody checking is not done routinely, and it is not feasible when mass immunization is done over a short period as a crisis intervention.
As described, one of the reasons for the immunization of health workers in Israel is to build a pool of potential donors for variola immunoglobulin. The CDC description of a successful immunization includes the formation of a scar at the site of vaccination. Barghini9 has described an unusual reaction in an adult after revaccination against smallpox. In his case, there was a delay in the loss of the crust and a lack of cicatrix. In my case, crust appeared twice, and there is no remaining scar.
- Henderson DA, Inglesby TV, Bartlett JG, et al. Smallpox as a biological weapon: medical and public health management. JAMA. 1999;281:2127-2137.
- Katz L, Sagi R, Hurvitz A. Smallpox—past, present and future. Harefuah. 2002;141:43-50.
- Committee on infectious diseases. American Academy of Pediatrics. Smallpox vaccine. Pediatrics. 2002;110:841-845.
- Slater PE, Anis E, Leventhal A. Preparation for an outbreak of smallpox in Israel. Isr Med Assoc J. 2002;4:507-512.
- US Centers for Disease Control and Prevention. Smallpox vaccine overview.
Available at: http://www.bt.cdc.gov/agent/smallpox/vaccination/facts.asp
Accessed October 14, 2002. - Haim M, Gdalevich M, Mimouni D, et al. Adverse reactions to smallpox vaccine: the Israel defense force experience, 1991 to 1996. a comparison with previous surveys. Mil Med. 2000;165:287-289.
- Feery BJ. Adverse reaction after smallpox vaccination. Med J Aust. 1977;2:180-183.
- Lublin-Tennenbaum T, Katzenelson E, El-Ad B, et al. Correlation between cutaneous reaction in vaccines immunized against smallpox and antibody titer determined by plaque neutralization test and ELISA. Viral Immunol. 1990;3:19-25.
- Barghini G. Unusual reaction in an adult after revaccination against smallpox. Ann Sclavo. 1975;17:704-705.
Smallpox is caused by the variola virus and is capable of causing serious complications. The disease is highly contagious and may present in several forms. The variola major form has a case fatality rate of about 30%.1 The smallpox vaccine was the first immunization used in modern medicine. The first inoculation was given in 1796 by Edward Jenner, MD, who immunized an 8-year-old boy with material taken from cowpox. The inoculation was shown to protect the child from smallpox when he was inoculated 6 weeks later with material that was taken from a smallpox pustule. Since then, mass immunization against smallpox has become a well-established method of fighting the disease. Immunization is based on epidermal inoculation of live vaccinia virus, another poxvirus that provokes immunization against smallpox but does not have the ability to cause the disease. The inoculation method includes putting a droplet of vaccine on the skin and then superficially poking the skin sufficiently to cause minimal blood drops. Disease protection following immunization lasts about 10 years.2
Because humans are the only natural host of the variola virus, the World Health Organization proclaimed smallpox the first disease to be eradicated worldwide. This was accomplished in 1977, when the last naturally occurring case in the world occurred in Somalia. The eradication of smallpox made it possible to stop the use of the vaccine without the threat of natural recurrence of the disease. In many places around the world, immunization was stopped before 1977 because many years had gone by without any documented cases of smallpox. In the United States, the last case was documented in 1949, and routine smallpox immunization was discontinued in 1972. In Israel, it was discontinued in 1980.
Since the events of September 11, 2001, there have been major concerns about the possibility of bioterrorism using the variola virus, which had been kept in laboratories and could be used as an unconventional weapon by countries or militant groups. Because many individuals are not immunized against smallpox, either naturally or artificially, the potential of a major epidemic occurring is significant.3
Because routine immunization against smallpox was discontinued in the United States and Israel, entire populations are vulnerable to the disease. The possibility that Israel will be involved in war became stronger when the United States and its allies targeted Iraq as a country that develops weapons of mass destruction. The use of smallpox as a biological weapon is a major possibility.4
To prepare against smallpox, the Israeli authorities have decided on a 2-step plan. In the first step, emergency personnel are to be vaccinated. These include medical and paramedical personnel, as well as people in other emergency services. The second stage of the plan is to immunize the entire population of Israel if one case of smallpox is diagnosed.
The immunization of healthcare workers in the first stage has 2 purposes. First, medical and other emergency personnel are expected to deal with infected or potentially infected people and thus must be protected against the disease. Second, in case of an epidemic, serious life-threatening disease could be aborted by variola immunoglobulin. All those immunized prior to a smallpox epidemic are the potential donors of human gamma globulin against the disease.
Immunization of medical personnel in Israel started in September 2002. The immunization was voluntary and thus included only a portion of the potential recipients. I was immunized on September 24, 2002, at my workplace. Assuming that many practitioners do not have experience with the vaccine, I decided to keep a diary and take pictures of the changes at the site of the inoculation.
Case Report
On day 1, 3 drops of dried blood (1 mm each) appeared. There was no itching or redness. Days 2 and 3 were the same as day 1, except that the puncture spots became black. A round, red, 4-mm papule appeared on day 4. There was some itching, and the black puncture spots at the area of inoculation remained the same. The papule became a pustule 3 to 4 mm in diameter surrounded by a 2- to 3-mm rim of red papule on day 5, and itching intensified. On day 6, the pustule became 5 to 6 mm, and the red rim around it became 2 to 3 mm. The area was not painful, except when touched. Itching became more obvious, and there was some tenderness of the axillary lymph nodes, with minimal enlargement of the glands. The pustule became 6 to 7 mm, and the red rim became 3 to 4 mm, with an additional 1- to 2-mm pink rim by day 7. Headache was present.
By day 8, the pustule was 10 mm, and the 3 dark 1-mm original puncture spots were still present inside the pustular area. The pustule felt hard when touched. The red rim was 2 to 3 mm, with an additional induration of skin 10 mm in diameter around the rim. The area was somewhat itchy, and headache was present. There was no change on day 9 except that the dark spots within the dry pustule were more obvious, increasing to 2 mm each. The size of the pustule was still the same on day 10, although it was less elevated. Central umbilication was obvious. On day 11, the pustule became dryer, and the black spots inside were 2 to 3 mm. Redness around the pustule was still 2 to 3 mm and was more intense after showering. Some induration of skin remained around the pustule and red area, and it was mildly painful when touched. Itching was lessened, and there was no pain at the axillary lymph nodes (Figure 1). The dry crust began to peel on day 12. The central area was confluent black, and the red rim was 2 to 3 mm, with some itching. On day 13, the crust had no yellow color of dried pus, and the roof of the crust had peeled off completely. The exposed base of the crust was dark black and hard. One to 2 mm of the red rim was still present, with less surrounding induration. There was mild itching but no pain or discharge. Minimal change was seen on day 14, except that the lesion began to shrink slightly.
There was no pain on day 15, but the borders of the black crust began to separate from the red rim, and the red rim decreased to 1 to 2 mm. Itching became more intense, and the crust bled slightly at its border when touched. An adhesive bandage was put on the area. When the bandage was removed on day 16, it looked as if there was liquefaction of the crust, which had a yellowish color (Figure 2). The area was covered by gauze to enable air drying. On days 17 and 18, the crust was dry and brown, with no bleeding. It appeared smaller than the day before, the red rim had become pink, and there was mild itching (Figure 3). The skin above the pink rim began to peel on day 19, and there was no change in the scab. On one of the borders, there was a small discharge of serum. The scab fell off after showering (Figure 4). An ulcer 1- to 2-mm deep was obvious at the spot; the rim around the ulcer was red. The ulcer was smaller on day 20, and the rim around the ulcer was pink and still elevated above the surrounding normal skin. There was no discharge. On day 21, the base of the ulcer was dry and brown.
On day 22, the skin around the brown area was dry and pink, and the rim was 1 to 2 mm. Minimal changes were noted from days 23 to 27, and there was no itching. There was still no itching on day 28, and the red rim had disappeared. The central brown area peeled off, leaving a 4-mm pinkish red macule.
From days 29 to 75, the area gradually became pink. The skin looked thin and wrinkled, and there was no umbilication or elevation above the surrounding skin (Figure 5). No scar or induration was present, and symptoms had subsided.
Comment
According to the US Centers for Disease Control and Prevention (CDC), if the smallpox vaccination is successful, a red and itchy bump will develop at the vaccine site in 3 to 4 days. In a week, the bump becomes a large blister, fills with pus, and begins to drain. During week 2, the blister begins to dry up, and a scab forms. The scab falls off during the third week, leaving a small scar.5 The described case follows the general description of the CDC. However, some differences should be mentioned. Itching at the site of vaccination was a major accompanying symptom. Swelling beyond the immediate site of skin reaction was obvious and lasted for more than a week. Axillary lymphadenopathy was noted. Discharge from the pustule was minimal. A scab was formed twice—once as the pus dried up and again after the first scab fell off and the drying of the wet base of the first scab dried up. At the end of the skin reaction, no scar remained.
Could the differences between the case described and the description by the CDC be explained by personal variation in skin reaction? Is the difference dependent on the skin care following the vaccination?
The instructions that were given to the Israeli recipients of the vaccine were to keep the area covered with loose gauze, change the gauze daily, not change regular skin washing habits, not touch the place of vaccination, and not put any topical preparation on the site. I followed the instructions except on day 15, when bleeding began as the scab started to peel off. At that point, a bandage was put over the area, thus limiting the ventilation and drying of the scab.
Complications following smallpox vaccination are relatively rare. Haim et al6 described an overall complication rate of 0.4 per 10,000 young army recruits. The complication rate was higher in primary recipients (those without previous exposure to the disease or vaccine).
Feery7 described an adverse skin reaction rate of 118 per 1 million recipients and a death rate of 1 to 5 per 1 million. The more serious skin reactions were eczema vaccinatum, progressive vaccinia, and neurologic and cardiac complications.
Monitoring the reaction to the vaccine is important in evaluating the intervention. Checking antibody titers postimmunization is possible. The micro–enzyme-linked immunosorbent assay technique was found to be more sensitive than plaque neutralization in finding a 4-fold increase in antibody titers.8 Postimmunization antibody checking is not done routinely, and it is not feasible when mass immunization is done over a short period as a crisis intervention.
As described, one of the reasons for the immunization of health workers in Israel is to build a pool of potential donors for variola immunoglobulin. The CDC description of a successful immunization includes the formation of a scar at the site of vaccination. Barghini9 has described an unusual reaction in an adult after revaccination against smallpox. In his case, there was a delay in the loss of the crust and a lack of cicatrix. In my case, crust appeared twice, and there is no remaining scar.
Smallpox is caused by the variola virus and is capable of causing serious complications. The disease is highly contagious and may present in several forms. The variola major form has a case fatality rate of about 30%.1 The smallpox vaccine was the first immunization used in modern medicine. The first inoculation was given in 1796 by Edward Jenner, MD, who immunized an 8-year-old boy with material taken from cowpox. The inoculation was shown to protect the child from smallpox when he was inoculated 6 weeks later with material that was taken from a smallpox pustule. Since then, mass immunization against smallpox has become a well-established method of fighting the disease. Immunization is based on epidermal inoculation of live vaccinia virus, another poxvirus that provokes immunization against smallpox but does not have the ability to cause the disease. The inoculation method includes putting a droplet of vaccine on the skin and then superficially poking the skin sufficiently to cause minimal blood drops. Disease protection following immunization lasts about 10 years.2
Because humans are the only natural host of the variola virus, the World Health Organization proclaimed smallpox the first disease to be eradicated worldwide. This was accomplished in 1977, when the last naturally occurring case in the world occurred in Somalia. The eradication of smallpox made it possible to stop the use of the vaccine without the threat of natural recurrence of the disease. In many places around the world, immunization was stopped before 1977 because many years had gone by without any documented cases of smallpox. In the United States, the last case was documented in 1949, and routine smallpox immunization was discontinued in 1972. In Israel, it was discontinued in 1980.
Since the events of September 11, 2001, there have been major concerns about the possibility of bioterrorism using the variola virus, which had been kept in laboratories and could be used as an unconventional weapon by countries or militant groups. Because many individuals are not immunized against smallpox, either naturally or artificially, the potential of a major epidemic occurring is significant.3
Because routine immunization against smallpox was discontinued in the United States and Israel, entire populations are vulnerable to the disease. The possibility that Israel will be involved in war became stronger when the United States and its allies targeted Iraq as a country that develops weapons of mass destruction. The use of smallpox as a biological weapon is a major possibility.4
To prepare against smallpox, the Israeli authorities have decided on a 2-step plan. In the first step, emergency personnel are to be vaccinated. These include medical and paramedical personnel, as well as people in other emergency services. The second stage of the plan is to immunize the entire population of Israel if one case of smallpox is diagnosed.
The immunization of healthcare workers in the first stage has 2 purposes. First, medical and other emergency personnel are expected to deal with infected or potentially infected people and thus must be protected against the disease. Second, in case of an epidemic, serious life-threatening disease could be aborted by variola immunoglobulin. All those immunized prior to a smallpox epidemic are the potential donors of human gamma globulin against the disease.
Immunization of medical personnel in Israel started in September 2002. The immunization was voluntary and thus included only a portion of the potential recipients. I was immunized on September 24, 2002, at my workplace. Assuming that many practitioners do not have experience with the vaccine, I decided to keep a diary and take pictures of the changes at the site of the inoculation.
Case Report
On day 1, 3 drops of dried blood (1 mm each) appeared. There was no itching or redness. Days 2 and 3 were the same as day 1, except that the puncture spots became black. A round, red, 4-mm papule appeared on day 4. There was some itching, and the black puncture spots at the area of inoculation remained the same. The papule became a pustule 3 to 4 mm in diameter surrounded by a 2- to 3-mm rim of red papule on day 5, and itching intensified. On day 6, the pustule became 5 to 6 mm, and the red rim around it became 2 to 3 mm. The area was not painful, except when touched. Itching became more obvious, and there was some tenderness of the axillary lymph nodes, with minimal enlargement of the glands. The pustule became 6 to 7 mm, and the red rim became 3 to 4 mm, with an additional 1- to 2-mm pink rim by day 7. Headache was present.
By day 8, the pustule was 10 mm, and the 3 dark 1-mm original puncture spots were still present inside the pustular area. The pustule felt hard when touched. The red rim was 2 to 3 mm, with an additional induration of skin 10 mm in diameter around the rim. The area was somewhat itchy, and headache was present. There was no change on day 9 except that the dark spots within the dry pustule were more obvious, increasing to 2 mm each. The size of the pustule was still the same on day 10, although it was less elevated. Central umbilication was obvious. On day 11, the pustule became dryer, and the black spots inside were 2 to 3 mm. Redness around the pustule was still 2 to 3 mm and was more intense after showering. Some induration of skin remained around the pustule and red area, and it was mildly painful when touched. Itching was lessened, and there was no pain at the axillary lymph nodes (Figure 1). The dry crust began to peel on day 12. The central area was confluent black, and the red rim was 2 to 3 mm, with some itching. On day 13, the crust had no yellow color of dried pus, and the roof of the crust had peeled off completely. The exposed base of the crust was dark black and hard. One to 2 mm of the red rim was still present, with less surrounding induration. There was mild itching but no pain or discharge. Minimal change was seen on day 14, except that the lesion began to shrink slightly.
There was no pain on day 15, but the borders of the black crust began to separate from the red rim, and the red rim decreased to 1 to 2 mm. Itching became more intense, and the crust bled slightly at its border when touched. An adhesive bandage was put on the area. When the bandage was removed on day 16, it looked as if there was liquefaction of the crust, which had a yellowish color (Figure 2). The area was covered by gauze to enable air drying. On days 17 and 18, the crust was dry and brown, with no bleeding. It appeared smaller than the day before, the red rim had become pink, and there was mild itching (Figure 3). The skin above the pink rim began to peel on day 19, and there was no change in the scab. On one of the borders, there was a small discharge of serum. The scab fell off after showering (Figure 4). An ulcer 1- to 2-mm deep was obvious at the spot; the rim around the ulcer was red. The ulcer was smaller on day 20, and the rim around the ulcer was pink and still elevated above the surrounding normal skin. There was no discharge. On day 21, the base of the ulcer was dry and brown.
On day 22, the skin around the brown area was dry and pink, and the rim was 1 to 2 mm. Minimal changes were noted from days 23 to 27, and there was no itching. There was still no itching on day 28, and the red rim had disappeared. The central brown area peeled off, leaving a 4-mm pinkish red macule.
From days 29 to 75, the area gradually became pink. The skin looked thin and wrinkled, and there was no umbilication or elevation above the surrounding skin (Figure 5). No scar or induration was present, and symptoms had subsided.
Comment
According to the US Centers for Disease Control and Prevention (CDC), if the smallpox vaccination is successful, a red and itchy bump will develop at the vaccine site in 3 to 4 days. In a week, the bump becomes a large blister, fills with pus, and begins to drain. During week 2, the blister begins to dry up, and a scab forms. The scab falls off during the third week, leaving a small scar.5 The described case follows the general description of the CDC. However, some differences should be mentioned. Itching at the site of vaccination was a major accompanying symptom. Swelling beyond the immediate site of skin reaction was obvious and lasted for more than a week. Axillary lymphadenopathy was noted. Discharge from the pustule was minimal. A scab was formed twice—once as the pus dried up and again after the first scab fell off and the drying of the wet base of the first scab dried up. At the end of the skin reaction, no scar remained.
Could the differences between the case described and the description by the CDC be explained by personal variation in skin reaction? Is the difference dependent on the skin care following the vaccination?
The instructions that were given to the Israeli recipients of the vaccine were to keep the area covered with loose gauze, change the gauze daily, not change regular skin washing habits, not touch the place of vaccination, and not put any topical preparation on the site. I followed the instructions except on day 15, when bleeding began as the scab started to peel off. At that point, a bandage was put over the area, thus limiting the ventilation and drying of the scab.
Complications following smallpox vaccination are relatively rare. Haim et al6 described an overall complication rate of 0.4 per 10,000 young army recruits. The complication rate was higher in primary recipients (those without previous exposure to the disease or vaccine).
Feery7 described an adverse skin reaction rate of 118 per 1 million recipients and a death rate of 1 to 5 per 1 million. The more serious skin reactions were eczema vaccinatum, progressive vaccinia, and neurologic and cardiac complications.
Monitoring the reaction to the vaccine is important in evaluating the intervention. Checking antibody titers postimmunization is possible. The micro–enzyme-linked immunosorbent assay technique was found to be more sensitive than plaque neutralization in finding a 4-fold increase in antibody titers.8 Postimmunization antibody checking is not done routinely, and it is not feasible when mass immunization is done over a short period as a crisis intervention.
As described, one of the reasons for the immunization of health workers in Israel is to build a pool of potential donors for variola immunoglobulin. The CDC description of a successful immunization includes the formation of a scar at the site of vaccination. Barghini9 has described an unusual reaction in an adult after revaccination against smallpox. In his case, there was a delay in the loss of the crust and a lack of cicatrix. In my case, crust appeared twice, and there is no remaining scar.
- Henderson DA, Inglesby TV, Bartlett JG, et al. Smallpox as a biological weapon: medical and public health management. JAMA. 1999;281:2127-2137.
- Katz L, Sagi R, Hurvitz A. Smallpox—past, present and future. Harefuah. 2002;141:43-50.
- Committee on infectious diseases. American Academy of Pediatrics. Smallpox vaccine. Pediatrics. 2002;110:841-845.
- Slater PE, Anis E, Leventhal A. Preparation for an outbreak of smallpox in Israel. Isr Med Assoc J. 2002;4:507-512.
- US Centers for Disease Control and Prevention. Smallpox vaccine overview.
Available at: http://www.bt.cdc.gov/agent/smallpox/vaccination/facts.asp
Accessed October 14, 2002. - Haim M, Gdalevich M, Mimouni D, et al. Adverse reactions to smallpox vaccine: the Israel defense force experience, 1991 to 1996. a comparison with previous surveys. Mil Med. 2000;165:287-289.
- Feery BJ. Adverse reaction after smallpox vaccination. Med J Aust. 1977;2:180-183.
- Lublin-Tennenbaum T, Katzenelson E, El-Ad B, et al. Correlation between cutaneous reaction in vaccines immunized against smallpox and antibody titer determined by plaque neutralization test and ELISA. Viral Immunol. 1990;3:19-25.
- Barghini G. Unusual reaction in an adult after revaccination against smallpox. Ann Sclavo. 1975;17:704-705.
- Henderson DA, Inglesby TV, Bartlett JG, et al. Smallpox as a biological weapon: medical and public health management. JAMA. 1999;281:2127-2137.
- Katz L, Sagi R, Hurvitz A. Smallpox—past, present and future. Harefuah. 2002;141:43-50.
- Committee on infectious diseases. American Academy of Pediatrics. Smallpox vaccine. Pediatrics. 2002;110:841-845.
- Slater PE, Anis E, Leventhal A. Preparation for an outbreak of smallpox in Israel. Isr Med Assoc J. 2002;4:507-512.
- US Centers for Disease Control and Prevention. Smallpox vaccine overview.
Available at: http://www.bt.cdc.gov/agent/smallpox/vaccination/facts.asp
Accessed October 14, 2002. - Haim M, Gdalevich M, Mimouni D, et al. Adverse reactions to smallpox vaccine: the Israel defense force experience, 1991 to 1996. a comparison with previous surveys. Mil Med. 2000;165:287-289.
- Feery BJ. Adverse reaction after smallpox vaccination. Med J Aust. 1977;2:180-183.
- Lublin-Tennenbaum T, Katzenelson E, El-Ad B, et al. Correlation between cutaneous reaction in vaccines immunized against smallpox and antibody titer determined by plaque neutralization test and ELISA. Viral Immunol. 1990;3:19-25.
- Barghini G. Unusual reaction in an adult after revaccination against smallpox. Ann Sclavo. 1975;17:704-705.