User login
Timely Diagnosis of Lung Cancer in a Dedicated VA Referral Unit with Endobronchial Ultrasound Capability (FULL)
Lung cancer is the leading cause of cancer death in the US, with 154 050 deaths in 2018.1 There have been many attempts to reduce mortality of the disease through early diagnosis with use of computed tomography (CT). The National Lung Cancer Screening trial showed that screening high-risk populations with low-dose CT (LDCT) can reduce mortality.2 However, implementing LDCT screening in the clinical setting has proven challenging, as illustrated by the VA Lung Cancer Screening Demonstration Project (LCSDP).3 A lung cancer diagnosis typically comprises several steps that require different medical specialties; this can lead to delays. In the LCSDP, the mean time to diagnosis was 137 days.3 There are no federal standards for timeliness of lung cancer diagnosis.
The nonprofit RAND Corporation is the only American research organization that has published guidelines specifying acceptable intervals for the diagnosis and treatment of lung cancer. In Quality of Care for Oncologic Conditions and HIV, RAND Corporation researchers propose management quality indicators: lung cancer diagnosis within 2 months of an abnormal radiologic study and treatment within 6 weeks of diagnosis.4 The Swedish Lung Cancer Study5 and the Canadian Strategy for Cancer Control6 both recommended a standard of about 30 days—half the time recommended by the RAND Corporation.
Bukhari and colleagues at the Dayton US Department of Veterans Affairs (VA) Medical Center (VAMC) conducted a quality improvement study that examined lung cancer diagnosis and management.7 They found the time (SD) from abnormal chest imaging to diagnosis was 35.5 (31.6) days. Of those veterans who received a lung cancer diagnosis, 89.2% had the diagnosis made within the 60 days recommended by the RAND Corporation. Although these results surpass those of the LCSDP, they can be exceeded.
Beyond the potential emotional distress of awaiting the final diagnosis of a lung lesion, a delay in diagnosis and treatment may adversely affect outcomes. LDCT screening has been shown to reduce mortality, which implies a link between survival and time to intervention. There is no published evidence that time to diagnosis in advanced stage lung cancer affects outcome. The National Cancer Database (NCDB) contains informtion on about 70% of the cancers diagnosed each year in the US.8 An analysis of 4984 patients with stage IA squamous cell lung cancer undergoing lobectomy from NCDB showed that earlier surgery was associated with an absolute decrease in 5-year mortality of 5% to 8%. 9 Hence, at least in early-stage disease, reduced time from initial suspect imaging to definitive treatment may improve survival.
A system that coordinates the requisite diagnostic steps and avoids delays should provide a significant improvement in patient care. The results of such an approach that utilized nurse navigators has been previously published. 10 Here, we present the results of a dedicated VA referral clinic with priority access to pulmonary consultation and procedures in place that are designed to expedite the diagnosis of potential lung cancer.
Methods
The John L. McClellan Memorial Veterans Hospital (JLMMVH) in Little Rock, Arkansas institutional review board approved this study, which was performed in accordance with the Declaration of Helsinki. Requirement for informed consent was waived, and patient confidentiality was maintained throughout.
We have developed a plan of care specifically to facilitate diagnosis and treatment of the large number of veterans referred to the JLMMVH Diagnostic Clinic for abnormal results of chest imaging. The clinic has priority access to same-day imaging and subspecialty consultation services. In the clinic, medical students and residents perform evaluations and a registered nurse (RN) manager coordinates care.
A Diagnostic Clinic consult for abnormal thoracic imaging immediately triggers an e-consult to an interventional pulmonologist (Figure). The RN manager and pulmonologist perform a joint review of records/imaging prior to scheduling, and the pulmonologist triages the patient. Triage options include follow-up imaging, bronchoscopy with endobronchial ultrasound (EBUS), endoscopic ultrasound (EUS), and CT-guided biopsy.
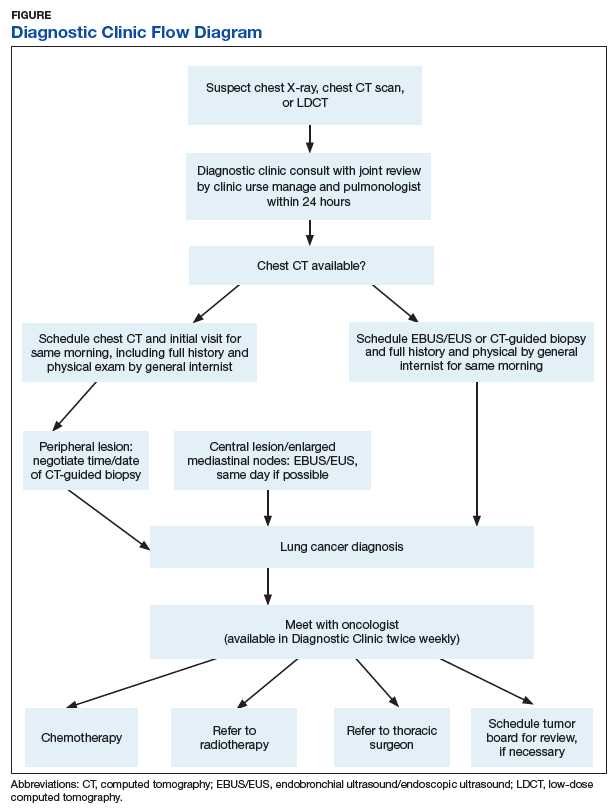
The RN manager then schedules a clinic visit that includes a medical evaluation by clinic staff and any indicated procedures on the same day. The interventional pulmonologist performs EBUS, EUS with the convex curvilinear bronchoscope, or both combined as indicated for diagnosis and staging. All procedures are performed in the JLMMVH bronchoscopy suite with standard conscious sedation using midazolam and fentanyl. Any other relevant procedures, such as pleural tap, also are performed at time of procedure. The pulmonologist and an attending pathologist interpret biopsies obtained in the bronchoscopy suite.
We performed a retrospective chart review of patients diagnosed with primary lung cancer through referral to the JLMMVH Diagnostic Clinic. The primary outcome was time from initial suspect chest imaging to cancer diagnosis. The study population consisted of patients referred for abnormal thoracic imaging between January 1, 2013 and December 31, 2016 and subsequently diagnosed with a primary lung cancer.
Subjects were excluded if (1) the patient was referred from outside our care network and a delay of > 10 days occurred between initial lesion imaging and referral; (2) the patient did not show up for appointments or chose to delay evaluation following referral; (3) biopsy demonstrated a nonlung primary cancer; and (4) serious intercurrent illness interrupted the diagnostic plan. In some cases, the radiologist or consulting pulmonologist had judged the lung lesion too small for immediate biopsy and recommended repeat imaging at a later date.
Patients were included in the study if the follow- up imaging led to a lung cancer diagnosis. However, because the interval between the initial imaging and the follow-up imaging in these patients did not represent a systems delay problem, the date of the scheduled follow-up abnormal imaging, which resulted in initiation of a potential cancer evaluation, served as the index suspect imaging date for this study.
Patient electronic medical records were reviewed and the following data were abstracted: date of the abnormal imaging that led to referral and time from abnormal chest X-ray to chest CT scan if applicable; date of referral and date of clinic visit; date of biopsy; date of lung cancer diagnosis; method of obtaining diagnostic specimen; lung cancer type and stage; type and date of treatment initiation or decision for supportive care only; and decision to seek further evaluation or care outside of our system.
All patients diagnosed with lung cancer during the study period were reviewed for inclusion, hence no required sample-size estimate was calculated. All outcomes were assessed as calendar days. The primary outcome was the time from the index suspect chest imaging study to the date of diagnosis of lung cancer. Prior to the initiation of our study, we chose this more stringent 30-day recommendation of the Canadian6 and Swedish5 studies as the comparator for our primary outcome, although data with respect to the 60-day Rand Corporation guidelines also are reported.4
Statistical Methods
The mean time to lung cancer diagnosis in our cohort was compared with this 30-day standard using a 2-sided Mann–Whitney U test. Normality of data distribution was determined using the Kolmogorov–Smirnov test. For statistical significance testing a P value of .05 was used. Statistical calculations were performed using R statistical software version 3.2.4. Secondary outcomes consisted of time from diagnosis to treatment; proportion of subjects diagnosed within 60 days; time from initial clinic visit to biopsy; and time from biopsy to diagnosis.
Results
Overall, 222 patients were diagnosed with a malignant lung lesion, of which 63 were excluded from analysis: 22 cancelled or did not appear for appointments, declined further evaluation, or completed evaluation outside of our network; 13 had the diagnosis made prior to Diagnostic Clinic visit; 13 proved to have a nonlung primary tumor presenting in the lung or mediastinal nodes; 12 were delayed > 10 days in referral from an outside network; and 3 had an intervening serious acute medical problem forcing delay in the diagnostic process.
Of the 159 included subjects, 154 (96.9%) were male, and the mean (SD) age was 67.6 (8.1) years. For 76 subjects, the abnormal chest X-ray and subsequent chest CT scan were performed the same day or the lung lesion had initially been noted on a CT scan. For 54 subjects, there was a delay of ≥ 1 week in obtaining a chest CT scan. The mean (SD) time from placement of the Diagnostic Clinic consultation by the primary care provider (PCP) or other provider and the initial Diagnostic Clinic visit was 6.3 (4.4) days. The mean (SD) time from suspect imaging to diagnosis (primary outcome) was 22.6(16.6) days.
The distribution of this outcome was nonnormal (Kolmogorov-Smirnov test P < .01). When compared with the standard of 30 days, the primary outcome of 22.6 days was significantly shorter (2-sided Mann–Whitney U test P < .01). Three-quarters (76.1%) of subjects were diagnosed within 30 days and 95.0% of subjects were diagnosed within 60 days of the initial imaging. For the 8 subjects diagnosed after 60 days, contributing factors included PCP delay in Diagnostic Clinic consultation, initial negative biopsy, delay in performance of chest CT scan prior to consultation, and outsourcing of positron emission tomography (PET) scans.
Overall, 57 (35.8%) of the subjects underwent biopsy on the day of their Diagnostic Clinic visit: 14 underwent CT-guided biopsy and 43 underwent EBUS/EUS. Within 2 days of the initial visit 106 subjects (66.7%) had undergone biopsy. The mean (SD) time from initial Diagnostic Clinic visit to biopsy was 6.3 (9.5) days. The mean (SD) interval was 1.8 (3.0) days for EBUS/ EUS and 11.3 (11.7) days for CT-guided biopsy. The mean (SD) interval from biopsy to diagnosis was 3.2 (6.2) days with 64 cases (40.3%) diagnosed the day of biopsy.
Excluding subjects whose treatment was delayed by patient choice or intercurrent illness, and those who left the VA system to seek treatment elsewhere (n = 21), 24 opted for palliative care, 5 died before treatment could be initiated, and 109 underwent treatment for their tumors (Table). The mean times (SD) from diagnosis to treatment were: chemotherapy alone 34.7 (25.3) days; chemoradiation 37.0 (22.8) days; surgery 44.3 (24.4) days; radiation therapy alone 47.9 (26.0) days. With respect to the RAND Corporation recommended diagnosis to treatment time, 60.9% of chemotherapy alone, 61.5% of chemoradiation, 66.7% of surgery, and 45.0% of radiation therapy alone treatments were initiated within the 6-week window.
Discussion
This retrospective case study demonstrates the effectiveness of a dedicated diagnostic clinic with priority EBUS/EUS access in diagnosing lung cancer within the VA system. Although there is no universally accepted quality standard for comparison, the RAND Corporation recommendation of 60 days from abnormal imaging to diagnosis and the Dayton VAMC published mean of 35.5 days are guideposts; however, the results from the Dayton VAMC may have been affected negatively by some subjects undergoing serial imaging for asymptomatic nodules. We chose a more stringent standard of 30 days as recommended by Swedish and Canadian task forces.
When diagnosing lung cancer, the overriding purpose of the Diagnostic Clinic is to minimize system delays. The method is to have as simple a task as possible for the PCP or other provider who identifies a lung nodule or mass and submits a single consultation request to the Diagnostic Clinic. Once this consultation is placed, the clinic RN manager oversees all further steps required for diagnosis and referral for treatment. The key factor in achieving a mean diagnosis time of 22.6 days is the cooperation between the RN manager and the interventional pulmonologist. When a consultation is received, the RN manager and pulmonologist review the data together and schedule the initial clinic visit; the goal is same-day biopsy, which is achieved in more than one-third of cases. Not all patients with a chest image suspected for lung cancer had it ordered by their PCP. For this reason, a Diagnostic Clinic consultation is available to all health care providers in our system. Many patients reach the clinic after the discovery of a suspect chest X-ray during an emergency department visit, a regularly scheduled subspecialty appointment, or during a preoperative evaluation.
The mean time from initial visit to biopsy was 1.8 days for EBUS/EUS compared with an interval of 11.3 days for CT-guided biopsy. This difference reflects the pulmonologist’s involvement in initial scheduling of Diagnostic Clinic patients. The ability of the pulmonologist to provide an accurate assessment of sample adequacy and a preliminary diagnosis at bedside, with concurrent confirmation by a staff pathologist, permitted the Diagnostic Clinic to inform 40.3% of patients of the finding of malignancy on the day of biopsy. A published comparison of the onsite review of biopsy material showed our pulmonologist and staff pathologists to be equally accurate in their interpretations.11
Sources of Delays
While this study documents the shortest intervals from suspect imaging to diagnosis reported to date, it also identifies sources of system delay in diagnosing lung cancer that JLMMVH could further optimize. The first is the time from initial abnormal chest X-ray imaging to performance of the chest CT scan. On occasion, the index lung lesion is identified unexpectedly on an outpatient or emergency department chest CT scan. With greater use of LDCT lung cancer screening, the initial detection of suspect lesions by CT scanning will increase in the future. However, the PCP most often investigates a patient complaint with a standard chest X-ray that reveals a suspect nodule or mass. When ordered by the PCP as an outpatient test, scheduling of the follow-up chest CT scan is not given priority. More than a third of subjects experienced a delay ≥ 1 week in obtaining a chest CT scan ordered by the PCP; for 29 subjects the delay was ≥ 3weeks. At JLMMVH, the Diagnostic Clinic is given priority in scheduling CT scans. Hence, for suspect lung lesions, the chest CT scan, if not already obtained, is generally performed on the morning of the clinic visit. Educating the PCP to refer the patient immediately to the Diagnostic Clinic rather than waiting to obtain an outpatient chest CT scan may remove this source of unnecessary delay.
Scheduling a CT-guided fine needle aspiration of a lung lesion is another source of system delay. When the chest CT scan is available at the time of the Diagnostic Clinic referral, the clinic visit is scheduled for the earliest day a required CT-guided biopsy can be performed. However, the mean time of 11.3 days from initial Diagnostic Clinic visit to CT-guided biopsy is indicative of the backlog faced by the interventional radiologists.
Although infrequent, PET scans that are required before biopsy can lead to substantial delays. PET scans are performed at our university affiliate, and the joint VA-university lung tumor board sometimes generates requests for such scans prior to tissue diagnosis, yet another source of delay.
The time from referral receipt to the Diagnostic Clinic visit averaged 6.3 days. This delay usually was determined by the availability of the CT-guided biopsy or the dedicated interventional pulmonologist. Although other interventional pulmonologists at JLMMVH may perform the requisite diagnostic procedures, they are not always available for immediate review of imaging studies of referred patients nor can their schedules flexibly accommodate the number of patients seen in our clinic for evaluation.
Lung Cancer Diagnosis
Prompt diagnosis in the setting of a worrisome chest X-ray may help decrease patient anxiety, but does the clinic improve lung cancer treatment outcomes? Such improvement has been demonstrated only in stage IA squamous cell lung cancer.9 Of our study population, 37.7% had squamous cell carcinoma, and 85.5% had non-small cell lung cancer. Of those with non-small cell lung cancer, 28.9% had a clinical stage I tumor. Stage I squamous cell carcinoma, the type of tumor most likely to benefit from early diagnosis and treatment, was diagnosed in 11.3% of patients. With the increased application of LDCT screening, the proportion of veterans identified with early stage lung cancer may rise. The Providence VAMC in Rhode Island reported its results from instituting LDCT screening.12 Prior to screening, 28% of patients diagnosed with lung cancer had a stage I tumor. Following the introduction of LDCT screening, 49% diagnosed by LDCT screening had a stage I tumor. Nearly a third of their patients diagnosed with lung cancer through LDCT screening had squamous cell tumor histology. Thus, we can anticipate an increasing number of veterans with early stage lung cancer who would benefit from timely diagnosis.
The JLMMVH is a referral center for the entire state of Arkansas. Quite a few of its referred patients come from a long distance, which may require overnight housing and other related travel expenses. Apart from any potential outcome benefit, the efficiencies of the system described herein include the minimization of extra trips, an inconvenience and cost to both patient and JLMMVH.
Although the primary task of the clinic is diagnosis, we also seek to facilitate timely treatment. Our lack of an on-site PET scanner and radiation therapy, resources present on-site at the Dayton VAMC, contribute to longer therapy wait times. The shortest mean wait time at JLMMVH is for chemotherapy alone (34.7 days), in part because the JLMMVH oncologists, performing initial consultations 2 to 3 times weekly in the Diagnostic Clinic, are more readily available than are our thoracic surgeons or radiation therapists. Yet overall, JLMMVH patients often face delay from the time of lung cancer diagnosis to initiation of treatment.
The Connecticut Veterans Affairs Healthcare System has published the results of changes in lung cancer management associated with a nurse navigator system.10 Prior to creating the position of cancer care coordinator, filled by an advanced practice RNs, the mean time from clinical suspicion of lung cancer to treatment was 117 days. After 4 years of such care navigation, this waiting time had decreased to 52.4 days. Associated with this dramatic improvement in overall waiting time were decreases in the turnaround time required for performance of CT and PET scans. With respect to this big picture view of lung cancer care, our Diagnostic Clinic serves as a model for the initial step of diagnosis. Coordination and streamlining of the various steps from diagnosis to definitive therapy shall require a more system-wide effort involving all the key players in cancer care.
Conclusion
We have developed a care pathway based in a dedicated diagnostic clinic and have been able to document the shortest interval from abnormality to diagnosis of lung cancer reported in the literature to date. Efficient functioning of this clinic is dependent upon the close cooperation between a full-time RN clinic manager and an interventional pulmonologist experienced in lung cancer management and able to interpret cytologic samples at the time of biopsy. Shortening the delay between diagnosis and definitive therapy remains a challenge and may benefit from the oncology nurse navigator model previously described within the VA system. 10
1. American Cancer Society. Cancer Facts & Figures. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf. Accessed July 13, 2019.
2. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Eng J Med. 2011;365(5):395-409.
3. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406.
4. Asch SM, Kerr EA, Hamilton EG, Reifel JL, McGlynn EA, eds. Quality of Care for Oncologic Conditions and HIV: A Review of the Literature and Quality Indicators. Santa Monica, CA: RAND Corporation; 2000.
5. Hillerdal G. [Recommendations from the Swedish Lung Cancer Study Group: Shorter waiting times are demanded for quality in diagnostic work-ups for lung care.] Swedish Med J 1999; 96: 4691.
6. Simunovic M, Gagliardi A, McCready D, Coates A, Levine M, DePetrillo D. A snapshot of waiting times for cancer surgery provided by surgeons affiliated with regional cancer centres in Ontario. CMAJ. 2001;165(4):421-425. [Canadian Strategy for Cancer Control]
7. Bukhari A, Kumar G, Rajsheker R, Markert R. Timeliness of lung cancer diagnosis and treatment. Fed Pract. 2017;34(suppl 1):24S-29S.
8. Bilimoria KY, Ko CY, Tomlinson JS, et al. Wait times for cancer surgery in the United States: trends and predictors of delays. Ann Surg. 2011;253(4):779-785.
9. Yang CJ, Wang H, Kumar A, et al. Impact of timing of lobectomy on survival for clinical stage IA lung squamous cell carcinoma. Chest. 2017;152(6):1239-1250.
10. Hunnibell LS, Rose MG, Connery DM, et al. Using nurse navigation to improve timeliness of lung cancer care at a veterans hospital. Clin J Oncol Nurs. 2012;16(1):29-36.
11. Meena N, Jeffus S, Massoll N, et al. Rapid onsite evaluation: a comparison of cytopathologist and pulmonologist performance. Cancer Cytopatho. 2016;124(4):279-84.
12. Okereke IC, Bates MF, Jankowich MD, et al. Effects of implementation of lung cancer screening at one Veterans Affairs Medical Center. Chest 2016;150(5):1023-1029.
Lung cancer is the leading cause of cancer death in the US, with 154 050 deaths in 2018.1 There have been many attempts to reduce mortality of the disease through early diagnosis with use of computed tomography (CT). The National Lung Cancer Screening trial showed that screening high-risk populations with low-dose CT (LDCT) can reduce mortality.2 However, implementing LDCT screening in the clinical setting has proven challenging, as illustrated by the VA Lung Cancer Screening Demonstration Project (LCSDP).3 A lung cancer diagnosis typically comprises several steps that require different medical specialties; this can lead to delays. In the LCSDP, the mean time to diagnosis was 137 days.3 There are no federal standards for timeliness of lung cancer diagnosis.
The nonprofit RAND Corporation is the only American research organization that has published guidelines specifying acceptable intervals for the diagnosis and treatment of lung cancer. In Quality of Care for Oncologic Conditions and HIV, RAND Corporation researchers propose management quality indicators: lung cancer diagnosis within 2 months of an abnormal radiologic study and treatment within 6 weeks of diagnosis.4 The Swedish Lung Cancer Study5 and the Canadian Strategy for Cancer Control6 both recommended a standard of about 30 days—half the time recommended by the RAND Corporation.
Bukhari and colleagues at the Dayton US Department of Veterans Affairs (VA) Medical Center (VAMC) conducted a quality improvement study that examined lung cancer diagnosis and management.7 They found the time (SD) from abnormal chest imaging to diagnosis was 35.5 (31.6) days. Of those veterans who received a lung cancer diagnosis, 89.2% had the diagnosis made within the 60 days recommended by the RAND Corporation. Although these results surpass those of the LCSDP, they can be exceeded.
Beyond the potential emotional distress of awaiting the final diagnosis of a lung lesion, a delay in diagnosis and treatment may adversely affect outcomes. LDCT screening has been shown to reduce mortality, which implies a link between survival and time to intervention. There is no published evidence that time to diagnosis in advanced stage lung cancer affects outcome. The National Cancer Database (NCDB) contains informtion on about 70% of the cancers diagnosed each year in the US.8 An analysis of 4984 patients with stage IA squamous cell lung cancer undergoing lobectomy from NCDB showed that earlier surgery was associated with an absolute decrease in 5-year mortality of 5% to 8%. 9 Hence, at least in early-stage disease, reduced time from initial suspect imaging to definitive treatment may improve survival.
A system that coordinates the requisite diagnostic steps and avoids delays should provide a significant improvement in patient care. The results of such an approach that utilized nurse navigators has been previously published. 10 Here, we present the results of a dedicated VA referral clinic with priority access to pulmonary consultation and procedures in place that are designed to expedite the diagnosis of potential lung cancer.
Methods
The John L. McClellan Memorial Veterans Hospital (JLMMVH) in Little Rock, Arkansas institutional review board approved this study, which was performed in accordance with the Declaration of Helsinki. Requirement for informed consent was waived, and patient confidentiality was maintained throughout.
We have developed a plan of care specifically to facilitate diagnosis and treatment of the large number of veterans referred to the JLMMVH Diagnostic Clinic for abnormal results of chest imaging. The clinic has priority access to same-day imaging and subspecialty consultation services. In the clinic, medical students and residents perform evaluations and a registered nurse (RN) manager coordinates care.
A Diagnostic Clinic consult for abnormal thoracic imaging immediately triggers an e-consult to an interventional pulmonologist (Figure). The RN manager and pulmonologist perform a joint review of records/imaging prior to scheduling, and the pulmonologist triages the patient. Triage options include follow-up imaging, bronchoscopy with endobronchial ultrasound (EBUS), endoscopic ultrasound (EUS), and CT-guided biopsy.
The RN manager then schedules a clinic visit that includes a medical evaluation by clinic staff and any indicated procedures on the same day. The interventional pulmonologist performs EBUS, EUS with the convex curvilinear bronchoscope, or both combined as indicated for diagnosis and staging. All procedures are performed in the JLMMVH bronchoscopy suite with standard conscious sedation using midazolam and fentanyl. Any other relevant procedures, such as pleural tap, also are performed at time of procedure. The pulmonologist and an attending pathologist interpret biopsies obtained in the bronchoscopy suite.
We performed a retrospective chart review of patients diagnosed with primary lung cancer through referral to the JLMMVH Diagnostic Clinic. The primary outcome was time from initial suspect chest imaging to cancer diagnosis. The study population consisted of patients referred for abnormal thoracic imaging between January 1, 2013 and December 31, 2016 and subsequently diagnosed with a primary lung cancer.
Subjects were excluded if (1) the patient was referred from outside our care network and a delay of > 10 days occurred between initial lesion imaging and referral; (2) the patient did not show up for appointments or chose to delay evaluation following referral; (3) biopsy demonstrated a nonlung primary cancer; and (4) serious intercurrent illness interrupted the diagnostic plan. In some cases, the radiologist or consulting pulmonologist had judged the lung lesion too small for immediate biopsy and recommended repeat imaging at a later date.
Patients were included in the study if the follow- up imaging led to a lung cancer diagnosis. However, because the interval between the initial imaging and the follow-up imaging in these patients did not represent a systems delay problem, the date of the scheduled follow-up abnormal imaging, which resulted in initiation of a potential cancer evaluation, served as the index suspect imaging date for this study.
Patient electronic medical records were reviewed and the following data were abstracted: date of the abnormal imaging that led to referral and time from abnormal chest X-ray to chest CT scan if applicable; date of referral and date of clinic visit; date of biopsy; date of lung cancer diagnosis; method of obtaining diagnostic specimen; lung cancer type and stage; type and date of treatment initiation or decision for supportive care only; and decision to seek further evaluation or care outside of our system.
All patients diagnosed with lung cancer during the study period were reviewed for inclusion, hence no required sample-size estimate was calculated. All outcomes were assessed as calendar days. The primary outcome was the time from the index suspect chest imaging study to the date of diagnosis of lung cancer. Prior to the initiation of our study, we chose this more stringent 30-day recommendation of the Canadian6 and Swedish5 studies as the comparator for our primary outcome, although data with respect to the 60-day Rand Corporation guidelines also are reported.4
Statistical Methods
The mean time to lung cancer diagnosis in our cohort was compared with this 30-day standard using a 2-sided Mann–Whitney U test. Normality of data distribution was determined using the Kolmogorov–Smirnov test. For statistical significance testing a P value of .05 was used. Statistical calculations were performed using R statistical software version 3.2.4. Secondary outcomes consisted of time from diagnosis to treatment; proportion of subjects diagnosed within 60 days; time from initial clinic visit to biopsy; and time from biopsy to diagnosis.
Results
Overall, 222 patients were diagnosed with a malignant lung lesion, of which 63 were excluded from analysis: 22 cancelled or did not appear for appointments, declined further evaluation, or completed evaluation outside of our network; 13 had the diagnosis made prior to Diagnostic Clinic visit; 13 proved to have a nonlung primary tumor presenting in the lung or mediastinal nodes; 12 were delayed > 10 days in referral from an outside network; and 3 had an intervening serious acute medical problem forcing delay in the diagnostic process.
Of the 159 included subjects, 154 (96.9%) were male, and the mean (SD) age was 67.6 (8.1) years. For 76 subjects, the abnormal chest X-ray and subsequent chest CT scan were performed the same day or the lung lesion had initially been noted on a CT scan. For 54 subjects, there was a delay of ≥ 1 week in obtaining a chest CT scan. The mean (SD) time from placement of the Diagnostic Clinic consultation by the primary care provider (PCP) or other provider and the initial Diagnostic Clinic visit was 6.3 (4.4) days. The mean (SD) time from suspect imaging to diagnosis (primary outcome) was 22.6(16.6) days.
The distribution of this outcome was nonnormal (Kolmogorov-Smirnov test P < .01). When compared with the standard of 30 days, the primary outcome of 22.6 days was significantly shorter (2-sided Mann–Whitney U test P < .01). Three-quarters (76.1%) of subjects were diagnosed within 30 days and 95.0% of subjects were diagnosed within 60 days of the initial imaging. For the 8 subjects diagnosed after 60 days, contributing factors included PCP delay in Diagnostic Clinic consultation, initial negative biopsy, delay in performance of chest CT scan prior to consultation, and outsourcing of positron emission tomography (PET) scans.
Overall, 57 (35.8%) of the subjects underwent biopsy on the day of their Diagnostic Clinic visit: 14 underwent CT-guided biopsy and 43 underwent EBUS/EUS. Within 2 days of the initial visit 106 subjects (66.7%) had undergone biopsy. The mean (SD) time from initial Diagnostic Clinic visit to biopsy was 6.3 (9.5) days. The mean (SD) interval was 1.8 (3.0) days for EBUS/ EUS and 11.3 (11.7) days for CT-guided biopsy. The mean (SD) interval from biopsy to diagnosis was 3.2 (6.2) days with 64 cases (40.3%) diagnosed the day of biopsy.
Excluding subjects whose treatment was delayed by patient choice or intercurrent illness, and those who left the VA system to seek treatment elsewhere (n = 21), 24 opted for palliative care, 5 died before treatment could be initiated, and 109 underwent treatment for their tumors (Table). The mean times (SD) from diagnosis to treatment were: chemotherapy alone 34.7 (25.3) days; chemoradiation 37.0 (22.8) days; surgery 44.3 (24.4) days; radiation therapy alone 47.9 (26.0) days. With respect to the RAND Corporation recommended diagnosis to treatment time, 60.9% of chemotherapy alone, 61.5% of chemoradiation, 66.7% of surgery, and 45.0% of radiation therapy alone treatments were initiated within the 6-week window.
Discussion
This retrospective case study demonstrates the effectiveness of a dedicated diagnostic clinic with priority EBUS/EUS access in diagnosing lung cancer within the VA system. Although there is no universally accepted quality standard for comparison, the RAND Corporation recommendation of 60 days from abnormal imaging to diagnosis and the Dayton VAMC published mean of 35.5 days are guideposts; however, the results from the Dayton VAMC may have been affected negatively by some subjects undergoing serial imaging for asymptomatic nodules. We chose a more stringent standard of 30 days as recommended by Swedish and Canadian task forces.
When diagnosing lung cancer, the overriding purpose of the Diagnostic Clinic is to minimize system delays. The method is to have as simple a task as possible for the PCP or other provider who identifies a lung nodule or mass and submits a single consultation request to the Diagnostic Clinic. Once this consultation is placed, the clinic RN manager oversees all further steps required for diagnosis and referral for treatment. The key factor in achieving a mean diagnosis time of 22.6 days is the cooperation between the RN manager and the interventional pulmonologist. When a consultation is received, the RN manager and pulmonologist review the data together and schedule the initial clinic visit; the goal is same-day biopsy, which is achieved in more than one-third of cases. Not all patients with a chest image suspected for lung cancer had it ordered by their PCP. For this reason, a Diagnostic Clinic consultation is available to all health care providers in our system. Many patients reach the clinic after the discovery of a suspect chest X-ray during an emergency department visit, a regularly scheduled subspecialty appointment, or during a preoperative evaluation.
The mean time from initial visit to biopsy was 1.8 days for EBUS/EUS compared with an interval of 11.3 days for CT-guided biopsy. This difference reflects the pulmonologist’s involvement in initial scheduling of Diagnostic Clinic patients. The ability of the pulmonologist to provide an accurate assessment of sample adequacy and a preliminary diagnosis at bedside, with concurrent confirmation by a staff pathologist, permitted the Diagnostic Clinic to inform 40.3% of patients of the finding of malignancy on the day of biopsy. A published comparison of the onsite review of biopsy material showed our pulmonologist and staff pathologists to be equally accurate in their interpretations.11
Sources of Delays
While this study documents the shortest intervals from suspect imaging to diagnosis reported to date, it also identifies sources of system delay in diagnosing lung cancer that JLMMVH could further optimize. The first is the time from initial abnormal chest X-ray imaging to performance of the chest CT scan. On occasion, the index lung lesion is identified unexpectedly on an outpatient or emergency department chest CT scan. With greater use of LDCT lung cancer screening, the initial detection of suspect lesions by CT scanning will increase in the future. However, the PCP most often investigates a patient complaint with a standard chest X-ray that reveals a suspect nodule or mass. When ordered by the PCP as an outpatient test, scheduling of the follow-up chest CT scan is not given priority. More than a third of subjects experienced a delay ≥ 1 week in obtaining a chest CT scan ordered by the PCP; for 29 subjects the delay was ≥ 3weeks. At JLMMVH, the Diagnostic Clinic is given priority in scheduling CT scans. Hence, for suspect lung lesions, the chest CT scan, if not already obtained, is generally performed on the morning of the clinic visit. Educating the PCP to refer the patient immediately to the Diagnostic Clinic rather than waiting to obtain an outpatient chest CT scan may remove this source of unnecessary delay.
Scheduling a CT-guided fine needle aspiration of a lung lesion is another source of system delay. When the chest CT scan is available at the time of the Diagnostic Clinic referral, the clinic visit is scheduled for the earliest day a required CT-guided biopsy can be performed. However, the mean time of 11.3 days from initial Diagnostic Clinic visit to CT-guided biopsy is indicative of the backlog faced by the interventional radiologists.
Although infrequent, PET scans that are required before biopsy can lead to substantial delays. PET scans are performed at our university affiliate, and the joint VA-university lung tumor board sometimes generates requests for such scans prior to tissue diagnosis, yet another source of delay.
The time from referral receipt to the Diagnostic Clinic visit averaged 6.3 days. This delay usually was determined by the availability of the CT-guided biopsy or the dedicated interventional pulmonologist. Although other interventional pulmonologists at JLMMVH may perform the requisite diagnostic procedures, they are not always available for immediate review of imaging studies of referred patients nor can their schedules flexibly accommodate the number of patients seen in our clinic for evaluation.
Lung Cancer Diagnosis
Prompt diagnosis in the setting of a worrisome chest X-ray may help decrease patient anxiety, but does the clinic improve lung cancer treatment outcomes? Such improvement has been demonstrated only in stage IA squamous cell lung cancer.9 Of our study population, 37.7% had squamous cell carcinoma, and 85.5% had non-small cell lung cancer. Of those with non-small cell lung cancer, 28.9% had a clinical stage I tumor. Stage I squamous cell carcinoma, the type of tumor most likely to benefit from early diagnosis and treatment, was diagnosed in 11.3% of patients. With the increased application of LDCT screening, the proportion of veterans identified with early stage lung cancer may rise. The Providence VAMC in Rhode Island reported its results from instituting LDCT screening.12 Prior to screening, 28% of patients diagnosed with lung cancer had a stage I tumor. Following the introduction of LDCT screening, 49% diagnosed by LDCT screening had a stage I tumor. Nearly a third of their patients diagnosed with lung cancer through LDCT screening had squamous cell tumor histology. Thus, we can anticipate an increasing number of veterans with early stage lung cancer who would benefit from timely diagnosis.
The JLMMVH is a referral center for the entire state of Arkansas. Quite a few of its referred patients come from a long distance, which may require overnight housing and other related travel expenses. Apart from any potential outcome benefit, the efficiencies of the system described herein include the minimization of extra trips, an inconvenience and cost to both patient and JLMMVH.
Although the primary task of the clinic is diagnosis, we also seek to facilitate timely treatment. Our lack of an on-site PET scanner and radiation therapy, resources present on-site at the Dayton VAMC, contribute to longer therapy wait times. The shortest mean wait time at JLMMVH is for chemotherapy alone (34.7 days), in part because the JLMMVH oncologists, performing initial consultations 2 to 3 times weekly in the Diagnostic Clinic, are more readily available than are our thoracic surgeons or radiation therapists. Yet overall, JLMMVH patients often face delay from the time of lung cancer diagnosis to initiation of treatment.
The Connecticut Veterans Affairs Healthcare System has published the results of changes in lung cancer management associated with a nurse navigator system.10 Prior to creating the position of cancer care coordinator, filled by an advanced practice RNs, the mean time from clinical suspicion of lung cancer to treatment was 117 days. After 4 years of such care navigation, this waiting time had decreased to 52.4 days. Associated with this dramatic improvement in overall waiting time were decreases in the turnaround time required for performance of CT and PET scans. With respect to this big picture view of lung cancer care, our Diagnostic Clinic serves as a model for the initial step of diagnosis. Coordination and streamlining of the various steps from diagnosis to definitive therapy shall require a more system-wide effort involving all the key players in cancer care.
Conclusion
We have developed a care pathway based in a dedicated diagnostic clinic and have been able to document the shortest interval from abnormality to diagnosis of lung cancer reported in the literature to date. Efficient functioning of this clinic is dependent upon the close cooperation between a full-time RN clinic manager and an interventional pulmonologist experienced in lung cancer management and able to interpret cytologic samples at the time of biopsy. Shortening the delay between diagnosis and definitive therapy remains a challenge and may benefit from the oncology nurse navigator model previously described within the VA system. 10
Lung cancer is the leading cause of cancer death in the US, with 154 050 deaths in 2018.1 There have been many attempts to reduce mortality of the disease through early diagnosis with use of computed tomography (CT). The National Lung Cancer Screening trial showed that screening high-risk populations with low-dose CT (LDCT) can reduce mortality.2 However, implementing LDCT screening in the clinical setting has proven challenging, as illustrated by the VA Lung Cancer Screening Demonstration Project (LCSDP).3 A lung cancer diagnosis typically comprises several steps that require different medical specialties; this can lead to delays. In the LCSDP, the mean time to diagnosis was 137 days.3 There are no federal standards for timeliness of lung cancer diagnosis.
The nonprofit RAND Corporation is the only American research organization that has published guidelines specifying acceptable intervals for the diagnosis and treatment of lung cancer. In Quality of Care for Oncologic Conditions and HIV, RAND Corporation researchers propose management quality indicators: lung cancer diagnosis within 2 months of an abnormal radiologic study and treatment within 6 weeks of diagnosis.4 The Swedish Lung Cancer Study5 and the Canadian Strategy for Cancer Control6 both recommended a standard of about 30 days—half the time recommended by the RAND Corporation.
Bukhari and colleagues at the Dayton US Department of Veterans Affairs (VA) Medical Center (VAMC) conducted a quality improvement study that examined lung cancer diagnosis and management.7 They found the time (SD) from abnormal chest imaging to diagnosis was 35.5 (31.6) days. Of those veterans who received a lung cancer diagnosis, 89.2% had the diagnosis made within the 60 days recommended by the RAND Corporation. Although these results surpass those of the LCSDP, they can be exceeded.
Beyond the potential emotional distress of awaiting the final diagnosis of a lung lesion, a delay in diagnosis and treatment may adversely affect outcomes. LDCT screening has been shown to reduce mortality, which implies a link between survival and time to intervention. There is no published evidence that time to diagnosis in advanced stage lung cancer affects outcome. The National Cancer Database (NCDB) contains informtion on about 70% of the cancers diagnosed each year in the US.8 An analysis of 4984 patients with stage IA squamous cell lung cancer undergoing lobectomy from NCDB showed that earlier surgery was associated with an absolute decrease in 5-year mortality of 5% to 8%. 9 Hence, at least in early-stage disease, reduced time from initial suspect imaging to definitive treatment may improve survival.
A system that coordinates the requisite diagnostic steps and avoids delays should provide a significant improvement in patient care. The results of such an approach that utilized nurse navigators has been previously published. 10 Here, we present the results of a dedicated VA referral clinic with priority access to pulmonary consultation and procedures in place that are designed to expedite the diagnosis of potential lung cancer.
Methods
The John L. McClellan Memorial Veterans Hospital (JLMMVH) in Little Rock, Arkansas institutional review board approved this study, which was performed in accordance with the Declaration of Helsinki. Requirement for informed consent was waived, and patient confidentiality was maintained throughout.
We have developed a plan of care specifically to facilitate diagnosis and treatment of the large number of veterans referred to the JLMMVH Diagnostic Clinic for abnormal results of chest imaging. The clinic has priority access to same-day imaging and subspecialty consultation services. In the clinic, medical students and residents perform evaluations and a registered nurse (RN) manager coordinates care.
A Diagnostic Clinic consult for abnormal thoracic imaging immediately triggers an e-consult to an interventional pulmonologist (Figure). The RN manager and pulmonologist perform a joint review of records/imaging prior to scheduling, and the pulmonologist triages the patient. Triage options include follow-up imaging, bronchoscopy with endobronchial ultrasound (EBUS), endoscopic ultrasound (EUS), and CT-guided biopsy.
The RN manager then schedules a clinic visit that includes a medical evaluation by clinic staff and any indicated procedures on the same day. The interventional pulmonologist performs EBUS, EUS with the convex curvilinear bronchoscope, or both combined as indicated for diagnosis and staging. All procedures are performed in the JLMMVH bronchoscopy suite with standard conscious sedation using midazolam and fentanyl. Any other relevant procedures, such as pleural tap, also are performed at time of procedure. The pulmonologist and an attending pathologist interpret biopsies obtained in the bronchoscopy suite.
We performed a retrospective chart review of patients diagnosed with primary lung cancer through referral to the JLMMVH Diagnostic Clinic. The primary outcome was time from initial suspect chest imaging to cancer diagnosis. The study population consisted of patients referred for abnormal thoracic imaging between January 1, 2013 and December 31, 2016 and subsequently diagnosed with a primary lung cancer.
Subjects were excluded if (1) the patient was referred from outside our care network and a delay of > 10 days occurred between initial lesion imaging and referral; (2) the patient did not show up for appointments or chose to delay evaluation following referral; (3) biopsy demonstrated a nonlung primary cancer; and (4) serious intercurrent illness interrupted the diagnostic plan. In some cases, the radiologist or consulting pulmonologist had judged the lung lesion too small for immediate biopsy and recommended repeat imaging at a later date.
Patients were included in the study if the follow- up imaging led to a lung cancer diagnosis. However, because the interval between the initial imaging and the follow-up imaging in these patients did not represent a systems delay problem, the date of the scheduled follow-up abnormal imaging, which resulted in initiation of a potential cancer evaluation, served as the index suspect imaging date for this study.
Patient electronic medical records were reviewed and the following data were abstracted: date of the abnormal imaging that led to referral and time from abnormal chest X-ray to chest CT scan if applicable; date of referral and date of clinic visit; date of biopsy; date of lung cancer diagnosis; method of obtaining diagnostic specimen; lung cancer type and stage; type and date of treatment initiation or decision for supportive care only; and decision to seek further evaluation or care outside of our system.
All patients diagnosed with lung cancer during the study period were reviewed for inclusion, hence no required sample-size estimate was calculated. All outcomes were assessed as calendar days. The primary outcome was the time from the index suspect chest imaging study to the date of diagnosis of lung cancer. Prior to the initiation of our study, we chose this more stringent 30-day recommendation of the Canadian6 and Swedish5 studies as the comparator for our primary outcome, although data with respect to the 60-day Rand Corporation guidelines also are reported.4
Statistical Methods
The mean time to lung cancer diagnosis in our cohort was compared with this 30-day standard using a 2-sided Mann–Whitney U test. Normality of data distribution was determined using the Kolmogorov–Smirnov test. For statistical significance testing a P value of .05 was used. Statistical calculations were performed using R statistical software version 3.2.4. Secondary outcomes consisted of time from diagnosis to treatment; proportion of subjects diagnosed within 60 days; time from initial clinic visit to biopsy; and time from biopsy to diagnosis.
Results
Overall, 222 patients were diagnosed with a malignant lung lesion, of which 63 were excluded from analysis: 22 cancelled or did not appear for appointments, declined further evaluation, or completed evaluation outside of our network; 13 had the diagnosis made prior to Diagnostic Clinic visit; 13 proved to have a nonlung primary tumor presenting in the lung or mediastinal nodes; 12 were delayed > 10 days in referral from an outside network; and 3 had an intervening serious acute medical problem forcing delay in the diagnostic process.
Of the 159 included subjects, 154 (96.9%) were male, and the mean (SD) age was 67.6 (8.1) years. For 76 subjects, the abnormal chest X-ray and subsequent chest CT scan were performed the same day or the lung lesion had initially been noted on a CT scan. For 54 subjects, there was a delay of ≥ 1 week in obtaining a chest CT scan. The mean (SD) time from placement of the Diagnostic Clinic consultation by the primary care provider (PCP) or other provider and the initial Diagnostic Clinic visit was 6.3 (4.4) days. The mean (SD) time from suspect imaging to diagnosis (primary outcome) was 22.6(16.6) days.
The distribution of this outcome was nonnormal (Kolmogorov-Smirnov test P < .01). When compared with the standard of 30 days, the primary outcome of 22.6 days was significantly shorter (2-sided Mann–Whitney U test P < .01). Three-quarters (76.1%) of subjects were diagnosed within 30 days and 95.0% of subjects were diagnosed within 60 days of the initial imaging. For the 8 subjects diagnosed after 60 days, contributing factors included PCP delay in Diagnostic Clinic consultation, initial negative biopsy, delay in performance of chest CT scan prior to consultation, and outsourcing of positron emission tomography (PET) scans.
Overall, 57 (35.8%) of the subjects underwent biopsy on the day of their Diagnostic Clinic visit: 14 underwent CT-guided biopsy and 43 underwent EBUS/EUS. Within 2 days of the initial visit 106 subjects (66.7%) had undergone biopsy. The mean (SD) time from initial Diagnostic Clinic visit to biopsy was 6.3 (9.5) days. The mean (SD) interval was 1.8 (3.0) days for EBUS/ EUS and 11.3 (11.7) days for CT-guided biopsy. The mean (SD) interval from biopsy to diagnosis was 3.2 (6.2) days with 64 cases (40.3%) diagnosed the day of biopsy.
Excluding subjects whose treatment was delayed by patient choice or intercurrent illness, and those who left the VA system to seek treatment elsewhere (n = 21), 24 opted for palliative care, 5 died before treatment could be initiated, and 109 underwent treatment for their tumors (Table). The mean times (SD) from diagnosis to treatment were: chemotherapy alone 34.7 (25.3) days; chemoradiation 37.0 (22.8) days; surgery 44.3 (24.4) days; radiation therapy alone 47.9 (26.0) days. With respect to the RAND Corporation recommended diagnosis to treatment time, 60.9% of chemotherapy alone, 61.5% of chemoradiation, 66.7% of surgery, and 45.0% of radiation therapy alone treatments were initiated within the 6-week window.
Discussion
This retrospective case study demonstrates the effectiveness of a dedicated diagnostic clinic with priority EBUS/EUS access in diagnosing lung cancer within the VA system. Although there is no universally accepted quality standard for comparison, the RAND Corporation recommendation of 60 days from abnormal imaging to diagnosis and the Dayton VAMC published mean of 35.5 days are guideposts; however, the results from the Dayton VAMC may have been affected negatively by some subjects undergoing serial imaging for asymptomatic nodules. We chose a more stringent standard of 30 days as recommended by Swedish and Canadian task forces.
When diagnosing lung cancer, the overriding purpose of the Diagnostic Clinic is to minimize system delays. The method is to have as simple a task as possible for the PCP or other provider who identifies a lung nodule or mass and submits a single consultation request to the Diagnostic Clinic. Once this consultation is placed, the clinic RN manager oversees all further steps required for diagnosis and referral for treatment. The key factor in achieving a mean diagnosis time of 22.6 days is the cooperation between the RN manager and the interventional pulmonologist. When a consultation is received, the RN manager and pulmonologist review the data together and schedule the initial clinic visit; the goal is same-day biopsy, which is achieved in more than one-third of cases. Not all patients with a chest image suspected for lung cancer had it ordered by their PCP. For this reason, a Diagnostic Clinic consultation is available to all health care providers in our system. Many patients reach the clinic after the discovery of a suspect chest X-ray during an emergency department visit, a regularly scheduled subspecialty appointment, or during a preoperative evaluation.
The mean time from initial visit to biopsy was 1.8 days for EBUS/EUS compared with an interval of 11.3 days for CT-guided biopsy. This difference reflects the pulmonologist’s involvement in initial scheduling of Diagnostic Clinic patients. The ability of the pulmonologist to provide an accurate assessment of sample adequacy and a preliminary diagnosis at bedside, with concurrent confirmation by a staff pathologist, permitted the Diagnostic Clinic to inform 40.3% of patients of the finding of malignancy on the day of biopsy. A published comparison of the onsite review of biopsy material showed our pulmonologist and staff pathologists to be equally accurate in their interpretations.11
Sources of Delays
While this study documents the shortest intervals from suspect imaging to diagnosis reported to date, it also identifies sources of system delay in diagnosing lung cancer that JLMMVH could further optimize. The first is the time from initial abnormal chest X-ray imaging to performance of the chest CT scan. On occasion, the index lung lesion is identified unexpectedly on an outpatient or emergency department chest CT scan. With greater use of LDCT lung cancer screening, the initial detection of suspect lesions by CT scanning will increase in the future. However, the PCP most often investigates a patient complaint with a standard chest X-ray that reveals a suspect nodule or mass. When ordered by the PCP as an outpatient test, scheduling of the follow-up chest CT scan is not given priority. More than a third of subjects experienced a delay ≥ 1 week in obtaining a chest CT scan ordered by the PCP; for 29 subjects the delay was ≥ 3weeks. At JLMMVH, the Diagnostic Clinic is given priority in scheduling CT scans. Hence, for suspect lung lesions, the chest CT scan, if not already obtained, is generally performed on the morning of the clinic visit. Educating the PCP to refer the patient immediately to the Diagnostic Clinic rather than waiting to obtain an outpatient chest CT scan may remove this source of unnecessary delay.
Scheduling a CT-guided fine needle aspiration of a lung lesion is another source of system delay. When the chest CT scan is available at the time of the Diagnostic Clinic referral, the clinic visit is scheduled for the earliest day a required CT-guided biopsy can be performed. However, the mean time of 11.3 days from initial Diagnostic Clinic visit to CT-guided biopsy is indicative of the backlog faced by the interventional radiologists.
Although infrequent, PET scans that are required before biopsy can lead to substantial delays. PET scans are performed at our university affiliate, and the joint VA-university lung tumor board sometimes generates requests for such scans prior to tissue diagnosis, yet another source of delay.
The time from referral receipt to the Diagnostic Clinic visit averaged 6.3 days. This delay usually was determined by the availability of the CT-guided biopsy or the dedicated interventional pulmonologist. Although other interventional pulmonologists at JLMMVH may perform the requisite diagnostic procedures, they are not always available for immediate review of imaging studies of referred patients nor can their schedules flexibly accommodate the number of patients seen in our clinic for evaluation.
Lung Cancer Diagnosis
Prompt diagnosis in the setting of a worrisome chest X-ray may help decrease patient anxiety, but does the clinic improve lung cancer treatment outcomes? Such improvement has been demonstrated only in stage IA squamous cell lung cancer.9 Of our study population, 37.7% had squamous cell carcinoma, and 85.5% had non-small cell lung cancer. Of those with non-small cell lung cancer, 28.9% had a clinical stage I tumor. Stage I squamous cell carcinoma, the type of tumor most likely to benefit from early diagnosis and treatment, was diagnosed in 11.3% of patients. With the increased application of LDCT screening, the proportion of veterans identified with early stage lung cancer may rise. The Providence VAMC in Rhode Island reported its results from instituting LDCT screening.12 Prior to screening, 28% of patients diagnosed with lung cancer had a stage I tumor. Following the introduction of LDCT screening, 49% diagnosed by LDCT screening had a stage I tumor. Nearly a third of their patients diagnosed with lung cancer through LDCT screening had squamous cell tumor histology. Thus, we can anticipate an increasing number of veterans with early stage lung cancer who would benefit from timely diagnosis.
The JLMMVH is a referral center for the entire state of Arkansas. Quite a few of its referred patients come from a long distance, which may require overnight housing and other related travel expenses. Apart from any potential outcome benefit, the efficiencies of the system described herein include the minimization of extra trips, an inconvenience and cost to both patient and JLMMVH.
Although the primary task of the clinic is diagnosis, we also seek to facilitate timely treatment. Our lack of an on-site PET scanner and radiation therapy, resources present on-site at the Dayton VAMC, contribute to longer therapy wait times. The shortest mean wait time at JLMMVH is for chemotherapy alone (34.7 days), in part because the JLMMVH oncologists, performing initial consultations 2 to 3 times weekly in the Diagnostic Clinic, are more readily available than are our thoracic surgeons or radiation therapists. Yet overall, JLMMVH patients often face delay from the time of lung cancer diagnosis to initiation of treatment.
The Connecticut Veterans Affairs Healthcare System has published the results of changes in lung cancer management associated with a nurse navigator system.10 Prior to creating the position of cancer care coordinator, filled by an advanced practice RNs, the mean time from clinical suspicion of lung cancer to treatment was 117 days. After 4 years of such care navigation, this waiting time had decreased to 52.4 days. Associated with this dramatic improvement in overall waiting time were decreases in the turnaround time required for performance of CT and PET scans. With respect to this big picture view of lung cancer care, our Diagnostic Clinic serves as a model for the initial step of diagnosis. Coordination and streamlining of the various steps from diagnosis to definitive therapy shall require a more system-wide effort involving all the key players in cancer care.
Conclusion
We have developed a care pathway based in a dedicated diagnostic clinic and have been able to document the shortest interval from abnormality to diagnosis of lung cancer reported in the literature to date. Efficient functioning of this clinic is dependent upon the close cooperation between a full-time RN clinic manager and an interventional pulmonologist experienced in lung cancer management and able to interpret cytologic samples at the time of biopsy. Shortening the delay between diagnosis and definitive therapy remains a challenge and may benefit from the oncology nurse navigator model previously described within the VA system. 10
1. American Cancer Society. Cancer Facts & Figures. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf. Accessed July 13, 2019.
2. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Eng J Med. 2011;365(5):395-409.
3. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406.
4. Asch SM, Kerr EA, Hamilton EG, Reifel JL, McGlynn EA, eds. Quality of Care for Oncologic Conditions and HIV: A Review of the Literature and Quality Indicators. Santa Monica, CA: RAND Corporation; 2000.
5. Hillerdal G. [Recommendations from the Swedish Lung Cancer Study Group: Shorter waiting times are demanded for quality in diagnostic work-ups for lung care.] Swedish Med J 1999; 96: 4691.
6. Simunovic M, Gagliardi A, McCready D, Coates A, Levine M, DePetrillo D. A snapshot of waiting times for cancer surgery provided by surgeons affiliated with regional cancer centres in Ontario. CMAJ. 2001;165(4):421-425. [Canadian Strategy for Cancer Control]
7. Bukhari A, Kumar G, Rajsheker R, Markert R. Timeliness of lung cancer diagnosis and treatment. Fed Pract. 2017;34(suppl 1):24S-29S.
8. Bilimoria KY, Ko CY, Tomlinson JS, et al. Wait times for cancer surgery in the United States: trends and predictors of delays. Ann Surg. 2011;253(4):779-785.
9. Yang CJ, Wang H, Kumar A, et al. Impact of timing of lobectomy on survival for clinical stage IA lung squamous cell carcinoma. Chest. 2017;152(6):1239-1250.
10. Hunnibell LS, Rose MG, Connery DM, et al. Using nurse navigation to improve timeliness of lung cancer care at a veterans hospital. Clin J Oncol Nurs. 2012;16(1):29-36.
11. Meena N, Jeffus S, Massoll N, et al. Rapid onsite evaluation: a comparison of cytopathologist and pulmonologist performance. Cancer Cytopatho. 2016;124(4):279-84.
12. Okereke IC, Bates MF, Jankowich MD, et al. Effects of implementation of lung cancer screening at one Veterans Affairs Medical Center. Chest 2016;150(5):1023-1029.
1. American Cancer Society. Cancer Facts & Figures. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf. Accessed July 13, 2019.
2. National Lung Screening Trial Research Team, Aberle DR, Adams AM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Eng J Med. 2011;365(5):395-409.
3. Kinsinger LS, Anderson C, Kim J, et al. Implementation of lung cancer screening in the Veterans Health Administration. JAMA Intern Med. 2017;177(3):399-406.
4. Asch SM, Kerr EA, Hamilton EG, Reifel JL, McGlynn EA, eds. Quality of Care for Oncologic Conditions and HIV: A Review of the Literature and Quality Indicators. Santa Monica, CA: RAND Corporation; 2000.
5. Hillerdal G. [Recommendations from the Swedish Lung Cancer Study Group: Shorter waiting times are demanded for quality in diagnostic work-ups for lung care.] Swedish Med J 1999; 96: 4691.
6. Simunovic M, Gagliardi A, McCready D, Coates A, Levine M, DePetrillo D. A snapshot of waiting times for cancer surgery provided by surgeons affiliated with regional cancer centres in Ontario. CMAJ. 2001;165(4):421-425. [Canadian Strategy for Cancer Control]
7. Bukhari A, Kumar G, Rajsheker R, Markert R. Timeliness of lung cancer diagnosis and treatment. Fed Pract. 2017;34(suppl 1):24S-29S.
8. Bilimoria KY, Ko CY, Tomlinson JS, et al. Wait times for cancer surgery in the United States: trends and predictors of delays. Ann Surg. 2011;253(4):779-785.
9. Yang CJ, Wang H, Kumar A, et al. Impact of timing of lobectomy on survival for clinical stage IA lung squamous cell carcinoma. Chest. 2017;152(6):1239-1250.
10. Hunnibell LS, Rose MG, Connery DM, et al. Using nurse navigation to improve timeliness of lung cancer care at a veterans hospital. Clin J Oncol Nurs. 2012;16(1):29-36.
11. Meena N, Jeffus S, Massoll N, et al. Rapid onsite evaluation: a comparison of cytopathologist and pulmonologist performance. Cancer Cytopatho. 2016;124(4):279-84.
12. Okereke IC, Bates MF, Jankowich MD, et al. Effects of implementation of lung cancer screening at one Veterans Affairs Medical Center. Chest 2016;150(5):1023-1029.
Pediatric, adolescent migraine treatment and prevention guidelines are updated
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
Two new guidelines on the treatment and prevention of migraines in children and adolescents have been released by the American Academy of Neurology and the American Headache Society.
This update to the previous guidelines released by the American Academy of Neurology in 2004 reflects the expansion in pharmacologic and nonpharmacologic approaches during the last 15 years, Andrew D. Hershey, MD, PhD, director of the division of neurology at Cincinnati Children’s Hospital and a fellow of the American Academy of Neurology, said in an interview.
“There has also been an increase in the number of randomized controlled studies, which have allowed for a more robust statement on acute and preventive treatments to be made,” said Dr. Hershey, who is also a senior author for both guidelines.
The two reports focused on separate issues: One guideline outlined the options for treatment of acute migraine, and the second guideline summarized the available studies on the effectiveness of preventive medications for migraine in children and adolescents.
The guidelines recommend a physical examination and history to establish a specific headache diagnosis and afford a treatment that provides fast and complete pain relief. Treatment should be initiated as soon as a patient realizes an attack is occurring. Patients with signs of secondary headache should be evaluated by a neurologist or a headache specialist.
Studies support the use of ibuprofen and acetaminophen for pain relief in cases of acute migraine, but only some triptans (such as almotriptan, rizatriptan, sumatriptan/naproxen, and zolmitriptan nasal spray) are approved for use in adolescents. Specifically, sumatriptan/naproxen was shown to be effective when compared with placebo in studies with adolescents, whose headache symptoms resolved within 2 hours.
It may be necessary to try more than one triptan, the guidelines noted, because patients respond differently to medications. A failure to respond to one triptan does not necessarily mean that treatment with another triptan will be unsuccessful.
The guidelines also focused on patient and family education to improve medication safety and adherence. Lifestyle modification, avoidance of migraine triggers, creating good sleep habits, and staying hydrated can help reduce migraines. While no medications improved associated symptoms of migraines such as nausea or vomiting, triptans did show a benefit in reducing phonophobia and photophobia.
Evidence for pharmacologic prevention of migraines in children and adolescents is limited, according to the guidelines. In the 15 studies included in a literature review, there was not sufficient evidence to show preventive treatments, such as divalproex, onabotulinumtoxinA, amitriptyline, nimodipine, and flunarizine, were more effective than placebo at reducing the frequency of headaches. There was some evidence to show propranolol in children and topiramate and cinnarizine in children and adolescents can reduce headache frequency. Children and adolescents who received cognitive-behavioral therapy together with amitriptyline were more likely to have reduced frequency of headaches than were those who received amitriptyline with patient education.
“The consensus conclusion was that a multidisciplinary approach that combines acute treatments, preventive treatments, and healthy habits is likely to have the best outcomes,” said Dr. Hershey.
Dr. Hershey acknowledged the many gaps between what is clinically observed and what the studies in the guidelines demonstrated.
“One of the biggest questions is how to minimize the expectation response in the controlled studies,” he said. “Additionally, we are moving toward a better recognition of the mechanism by which the various treatments work in a genetic-based disease that is polygenic in nature” with up to 38 different gene polymorphisms identified to date.
The guidelines also do not address newer treatments, such as calcitonin gene–related peptide (CGRP) antibodies, CGRP antagonists, serotonin antagonists, and devices because there are as yet no studies of their effectiveness in children and adolescents.
“They have been studied in adults, so will be prone to the expectation response; but given the large number of diverse therapies, one can hope that many of the gaps can be filled,” said Dr. Hershey.
The American Academy of Neurology provided funding for development of the guidelines and reimbursed authors who served as subcommittee members for travel expenses and in-person meetings. The authors reported personal and institutional relationships in the form of advisory board memberships, investigator appointments, speakers bureau positions, research support, grants, honorariums, consultancies, and publishing royalties for pharmaceutical companies and other organizations.
SOURCES: Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008095. Oskoui M et al. Neurology. 2019 Aug 14. doi: 10.1212/WNL.0000000000008105.
FROM NEUROLOGY
Two genetic variants modify risk of Alzheimer’s disease
according to research published online August 14 in Science Translational Medicine. The variants affect cerebrospinal fluid (CSF) concentrations of a soluble form of the TREM2 protein (sTREM2), which may be involved in Alzheimer’s disease pathology. “Increasing TREM2 or activating the TREM2 signaling pathway could offer a new therapeutic approach for treating Alzheimer’s disease,” wrote the researchers.

Yuetiva Deming, PhD, of the University of Wisconsin–Madison and colleagues conducted a genome-wide association study to identify genetic modifiers of CSF sTREM2. They analyzed CSF sTREM2 levels in 813 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Of this population, 172 participants had Alzheimer’s disease, 169 were cognitively normal, 183 had early mild cognitive impairment (MCI), 221 had late MCI, and 68 had significant memory concerns.
The rs1582763 single-nucleotide polymorphism (SNP) on chromosome 11 within the MS4A gene region was significantly associated with increased CSF levels of sTREM2. Conditional analyses of the MS4A locus indicated that rs6591561, a missense variant within MS4A4A, was associated with reduced CSF sTREM2. Analyzing 580 additional CSF sTREM2 samples, along with associated genetic data, from six other studies replicated these findings in an independent dataset.
Furthermore, Dr. Deming and colleagues found that rs1582763 was associated with reduced risk for Alzheimer’s disease and older age at Alzheimer’s disease onset. In addition, rs6591561 was associated with increased risk of Alzheimer’s disease and earlier onset of Alzheimer’s disease.
Subsequent analyses showed that rs1582763 modified the expression of the MS4A4A and MS4A6A genes in various tissues. This finding suggests that one or both of these genes are important for influencing the production of sTREM2, wrote Dr. Deming and colleagues. Using human macrophages as a proxy for microglia, the investigators observed that the MS4A4A and TREM2 proteins colocalized on lipid rafts at the plasma membrane. In addition, sTREM2 concentrations increased with MS4A4A overexpression, and silencing of MS4A4A reduced sTREM2 production.
These findings “provide a putative biological connection between the MS4A family, TREM2, and Alzheimer’s disease risk,” wrote the researchers. The data also suggest that MS4A4A is a potential therapeutic target in Alzheimer’s disease. Understanding the role of sTREM2 in Alzheimer’s disease will require additional research, but it may be involved in pathogenesis, wrote Dr. Deming and colleagues.
One of the study’s limitations is that the investigators included only common variants and thus could not determine the effect of genes that only harbor low-frequency or rare functional variants. Another limitation is that the data cannot support conclusions about whether other genes in the MS4A locus also modulate sTREM2, wrote Dr. Deming and colleagues.
Grants from the National Institutes of Health supported this study. The investigators disclosed consulting and other relationships with various pharmaceutical companies.
SOURCE: Deming Y et al. Sci Transl Med. 2019 Aug 14. doi: 10.1126/scitranslmed.aau2291.
according to research published online August 14 in Science Translational Medicine. The variants affect cerebrospinal fluid (CSF) concentrations of a soluble form of the TREM2 protein (sTREM2), which may be involved in Alzheimer’s disease pathology. “Increasing TREM2 or activating the TREM2 signaling pathway could offer a new therapeutic approach for treating Alzheimer’s disease,” wrote the researchers.
Yuetiva Deming, PhD, of the University of Wisconsin–Madison and colleagues conducted a genome-wide association study to identify genetic modifiers of CSF sTREM2. They analyzed CSF sTREM2 levels in 813 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Of this population, 172 participants had Alzheimer’s disease, 169 were cognitively normal, 183 had early mild cognitive impairment (MCI), 221 had late MCI, and 68 had significant memory concerns.
The rs1582763 single-nucleotide polymorphism (SNP) on chromosome 11 within the MS4A gene region was significantly associated with increased CSF levels of sTREM2. Conditional analyses of the MS4A locus indicated that rs6591561, a missense variant within MS4A4A, was associated with reduced CSF sTREM2. Analyzing 580 additional CSF sTREM2 samples, along with associated genetic data, from six other studies replicated these findings in an independent dataset.
Furthermore, Dr. Deming and colleagues found that rs1582763 was associated with reduced risk for Alzheimer’s disease and older age at Alzheimer’s disease onset. In addition, rs6591561 was associated with increased risk of Alzheimer’s disease and earlier onset of Alzheimer’s disease.
Subsequent analyses showed that rs1582763 modified the expression of the MS4A4A and MS4A6A genes in various tissues. This finding suggests that one or both of these genes are important for influencing the production of sTREM2, wrote Dr. Deming and colleagues. Using human macrophages as a proxy for microglia, the investigators observed that the MS4A4A and TREM2 proteins colocalized on lipid rafts at the plasma membrane. In addition, sTREM2 concentrations increased with MS4A4A overexpression, and silencing of MS4A4A reduced sTREM2 production.
These findings “provide a putative biological connection between the MS4A family, TREM2, and Alzheimer’s disease risk,” wrote the researchers. The data also suggest that MS4A4A is a potential therapeutic target in Alzheimer’s disease. Understanding the role of sTREM2 in Alzheimer’s disease will require additional research, but it may be involved in pathogenesis, wrote Dr. Deming and colleagues.
One of the study’s limitations is that the investigators included only common variants and thus could not determine the effect of genes that only harbor low-frequency or rare functional variants. Another limitation is that the data cannot support conclusions about whether other genes in the MS4A locus also modulate sTREM2, wrote Dr. Deming and colleagues.
Grants from the National Institutes of Health supported this study. The investigators disclosed consulting and other relationships with various pharmaceutical companies.
SOURCE: Deming Y et al. Sci Transl Med. 2019 Aug 14. doi: 10.1126/scitranslmed.aau2291.
according to research published online August 14 in Science Translational Medicine. The variants affect cerebrospinal fluid (CSF) concentrations of a soluble form of the TREM2 protein (sTREM2), which may be involved in Alzheimer’s disease pathology. “Increasing TREM2 or activating the TREM2 signaling pathway could offer a new therapeutic approach for treating Alzheimer’s disease,” wrote the researchers.
Yuetiva Deming, PhD, of the University of Wisconsin–Madison and colleagues conducted a genome-wide association study to identify genetic modifiers of CSF sTREM2. They analyzed CSF sTREM2 levels in 813 participants in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Of this population, 172 participants had Alzheimer’s disease, 169 were cognitively normal, 183 had early mild cognitive impairment (MCI), 221 had late MCI, and 68 had significant memory concerns.
The rs1582763 single-nucleotide polymorphism (SNP) on chromosome 11 within the MS4A gene region was significantly associated with increased CSF levels of sTREM2. Conditional analyses of the MS4A locus indicated that rs6591561, a missense variant within MS4A4A, was associated with reduced CSF sTREM2. Analyzing 580 additional CSF sTREM2 samples, along with associated genetic data, from six other studies replicated these findings in an independent dataset.
Furthermore, Dr. Deming and colleagues found that rs1582763 was associated with reduced risk for Alzheimer’s disease and older age at Alzheimer’s disease onset. In addition, rs6591561 was associated with increased risk of Alzheimer’s disease and earlier onset of Alzheimer’s disease.
Subsequent analyses showed that rs1582763 modified the expression of the MS4A4A and MS4A6A genes in various tissues. This finding suggests that one or both of these genes are important for influencing the production of sTREM2, wrote Dr. Deming and colleagues. Using human macrophages as a proxy for microglia, the investigators observed that the MS4A4A and TREM2 proteins colocalized on lipid rafts at the plasma membrane. In addition, sTREM2 concentrations increased with MS4A4A overexpression, and silencing of MS4A4A reduced sTREM2 production.
These findings “provide a putative biological connection between the MS4A family, TREM2, and Alzheimer’s disease risk,” wrote the researchers. The data also suggest that MS4A4A is a potential therapeutic target in Alzheimer’s disease. Understanding the role of sTREM2 in Alzheimer’s disease will require additional research, but it may be involved in pathogenesis, wrote Dr. Deming and colleagues.
One of the study’s limitations is that the investigators included only common variants and thus could not determine the effect of genes that only harbor low-frequency or rare functional variants. Another limitation is that the data cannot support conclusions about whether other genes in the MS4A locus also modulate sTREM2, wrote Dr. Deming and colleagues.
Grants from the National Institutes of Health supported this study. The investigators disclosed consulting and other relationships with various pharmaceutical companies.
SOURCE: Deming Y et al. Sci Transl Med. 2019 Aug 14. doi: 10.1126/scitranslmed.aau2291.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: Two variants of MS4A are associated with the risk of Alzheimer’s disease.
Major finding: The rs1582763 SNP is associated with reduced risk for Alzheimer’s disease, and rs6591561 is associated with increased risk of Alzheimer’s disease.
Study details: A genome-wide association study of 813 participants in the Alzheimer’s Disease Neuroimaging Initiative.
Disclosures: Grants from the National Institutes of Health supported this study. The investigators disclosed consulting and other relationships with various pharmaceutical companies.
Source: Deming Y et al. Sci Transl Med. 2019 Aug 14. doi: 10.1126/scitranslmed.aau2291.
Serum neurofilament light chain level may indicate MS disease activity
according to an investigation published online August 12 in JAMA Neurology. Furthermore, changes in sNfL levels are associated with disability worsening, and sNfL levels may be influenced by treatment. These data support the potential of sNfL as an objective surrogate of ongoing MS disease activity, according to the researchers.
Neuronal and axonal loss increase levels of NfL in cerebrospinal fluid (CSF) in patients with MS. Previous research indicated that sNfL levels are correlated with CSF levels of NfL and are associated with clinical and imaging measures of disease activity. For the purpose of repeated sampling, collecting blood from patients would be more practical than performing lumbar punctures, said the investigators. No long-term studies of sNfL concentrations and their associations with MS disease outcomes had been performed, however.
Ester Cantó, PhD, of the University of California, San Francisco (UCSF), and colleagues examined data from the prospective Expression, Proteomics, Imaging, Clinical (EPIC) study to assess sNfL as a biomarker of MS disease activity and progression. The ongoing EPIC study is being conducted at UCSF. Dr. Cantó and colleagues analyzed data collected from July 1, 2004, through August 31, 2017, for 607 patients with MS. Participants underwent clinical examinations and serum sample collections annually for 5 years, then at various time points for as long as 12 years. The median follow-up duration was 10 years. The researchers measured sNfL levels with a sensitive single-molecule array platform and compared them with clinical and MRI variables using univariable and multivariable analyses. Dr. Cantó and colleagues chose disability progression, defined as clinically significant worsening on the Expanded Disability Status Scale (EDSS) score, and brain fraction atrophy as their primary outcomes.
The population’s mean age was 42.5 years. About 70% of participants were women, and all were of non-Hispanic European descent. At baseline, sNfL levels were significantly associated with EDSS score, MS subtype, and treatment status.
Dr. Cantó and colleagues found a significant interaction between EDSS worsening and change in levels of sNfL over time. Baseline sNfL levels were associated with approximately 11.6% of the variance in participants’ brain fraction atrophy at year 10. When the investigators controlled for sex, age, and disease duration, they found that baseline sNfL levels were associated with 18% of the variance in brain fraction atrophy at year 10. After 5 years’ follow-up, active treatment was associated with lower levels of sNfL. High-efficacy treatments were associated with greater decreases in sNfL levels, compared with platform therapies.
More frequent sample acquisition could provide greater detail about changes in sNfL levels, wrote Dr. Cantó and colleagues. They acknowledged that their study had insufficient power for the researchers to assess the outcomes of individual MS therapies. Other limitations included the lack of data on NfL stability and the lack of a group of healthy controls.
“For an individual patient, the biomarker prognostic power of sNfL level for clinical and MRI outcomes was limited,” said the investigators. “Further prospective studies are necessary to assess the assay’s utility for decision making in individual patients.”
The National Institutes of Health and the U.S. National MS Society supported the study. Several of the investigators received compensation from Novartis, which provided funds for the reagents needed for the single-molecule array assay.
SOURCE: Cantó E et al. JAMA Neurol. 2019 Aug. 12. doi: 10.1001/jamaneurol.2019.2137.
according to an investigation published online August 12 in JAMA Neurology. Furthermore, changes in sNfL levels are associated with disability worsening, and sNfL levels may be influenced by treatment. These data support the potential of sNfL as an objective surrogate of ongoing MS disease activity, according to the researchers.
Neuronal and axonal loss increase levels of NfL in cerebrospinal fluid (CSF) in patients with MS. Previous research indicated that sNfL levels are correlated with CSF levels of NfL and are associated with clinical and imaging measures of disease activity. For the purpose of repeated sampling, collecting blood from patients would be more practical than performing lumbar punctures, said the investigators. No long-term studies of sNfL concentrations and their associations with MS disease outcomes had been performed, however.
Ester Cantó, PhD, of the University of California, San Francisco (UCSF), and colleagues examined data from the prospective Expression, Proteomics, Imaging, Clinical (EPIC) study to assess sNfL as a biomarker of MS disease activity and progression. The ongoing EPIC study is being conducted at UCSF. Dr. Cantó and colleagues analyzed data collected from July 1, 2004, through August 31, 2017, for 607 patients with MS. Participants underwent clinical examinations and serum sample collections annually for 5 years, then at various time points for as long as 12 years. The median follow-up duration was 10 years. The researchers measured sNfL levels with a sensitive single-molecule array platform and compared them with clinical and MRI variables using univariable and multivariable analyses. Dr. Cantó and colleagues chose disability progression, defined as clinically significant worsening on the Expanded Disability Status Scale (EDSS) score, and brain fraction atrophy as their primary outcomes.
The population’s mean age was 42.5 years. About 70% of participants were women, and all were of non-Hispanic European descent. At baseline, sNfL levels were significantly associated with EDSS score, MS subtype, and treatment status.
Dr. Cantó and colleagues found a significant interaction between EDSS worsening and change in levels of sNfL over time. Baseline sNfL levels were associated with approximately 11.6% of the variance in participants’ brain fraction atrophy at year 10. When the investigators controlled for sex, age, and disease duration, they found that baseline sNfL levels were associated with 18% of the variance in brain fraction atrophy at year 10. After 5 years’ follow-up, active treatment was associated with lower levels of sNfL. High-efficacy treatments were associated with greater decreases in sNfL levels, compared with platform therapies.
More frequent sample acquisition could provide greater detail about changes in sNfL levels, wrote Dr. Cantó and colleagues. They acknowledged that their study had insufficient power for the researchers to assess the outcomes of individual MS therapies. Other limitations included the lack of data on NfL stability and the lack of a group of healthy controls.
“For an individual patient, the biomarker prognostic power of sNfL level for clinical and MRI outcomes was limited,” said the investigators. “Further prospective studies are necessary to assess the assay’s utility for decision making in individual patients.”
The National Institutes of Health and the U.S. National MS Society supported the study. Several of the investigators received compensation from Novartis, which provided funds for the reagents needed for the single-molecule array assay.
SOURCE: Cantó E et al. JAMA Neurol. 2019 Aug. 12. doi: 10.1001/jamaneurol.2019.2137.
according to an investigation published online August 12 in JAMA Neurology. Furthermore, changes in sNfL levels are associated with disability worsening, and sNfL levels may be influenced by treatment. These data support the potential of sNfL as an objective surrogate of ongoing MS disease activity, according to the researchers.
Neuronal and axonal loss increase levels of NfL in cerebrospinal fluid (CSF) in patients with MS. Previous research indicated that sNfL levels are correlated with CSF levels of NfL and are associated with clinical and imaging measures of disease activity. For the purpose of repeated sampling, collecting blood from patients would be more practical than performing lumbar punctures, said the investigators. No long-term studies of sNfL concentrations and their associations with MS disease outcomes had been performed, however.
Ester Cantó, PhD, of the University of California, San Francisco (UCSF), and colleagues examined data from the prospective Expression, Proteomics, Imaging, Clinical (EPIC) study to assess sNfL as a biomarker of MS disease activity and progression. The ongoing EPIC study is being conducted at UCSF. Dr. Cantó and colleagues analyzed data collected from July 1, 2004, through August 31, 2017, for 607 patients with MS. Participants underwent clinical examinations and serum sample collections annually for 5 years, then at various time points for as long as 12 years. The median follow-up duration was 10 years. The researchers measured sNfL levels with a sensitive single-molecule array platform and compared them with clinical and MRI variables using univariable and multivariable analyses. Dr. Cantó and colleagues chose disability progression, defined as clinically significant worsening on the Expanded Disability Status Scale (EDSS) score, and brain fraction atrophy as their primary outcomes.
The population’s mean age was 42.5 years. About 70% of participants were women, and all were of non-Hispanic European descent. At baseline, sNfL levels were significantly associated with EDSS score, MS subtype, and treatment status.
Dr. Cantó and colleagues found a significant interaction between EDSS worsening and change in levels of sNfL over time. Baseline sNfL levels were associated with approximately 11.6% of the variance in participants’ brain fraction atrophy at year 10. When the investigators controlled for sex, age, and disease duration, they found that baseline sNfL levels were associated with 18% of the variance in brain fraction atrophy at year 10. After 5 years’ follow-up, active treatment was associated with lower levels of sNfL. High-efficacy treatments were associated with greater decreases in sNfL levels, compared with platform therapies.
More frequent sample acquisition could provide greater detail about changes in sNfL levels, wrote Dr. Cantó and colleagues. They acknowledged that their study had insufficient power for the researchers to assess the outcomes of individual MS therapies. Other limitations included the lack of data on NfL stability and the lack of a group of healthy controls.
“For an individual patient, the biomarker prognostic power of sNfL level for clinical and MRI outcomes was limited,” said the investigators. “Further prospective studies are necessary to assess the assay’s utility for decision making in individual patients.”
The National Institutes of Health and the U.S. National MS Society supported the study. Several of the investigators received compensation from Novartis, which provided funds for the reagents needed for the single-molecule array assay.
SOURCE: Cantó E et al. JAMA Neurol. 2019 Aug. 12. doi: 10.1001/jamaneurol.2019.2137.
FROM JAMA NEUROLOGY
Key clinical point: Serum neurofilament light chain level has potential as a surrogate of ongoing MS disease activity.
Major finding: Serum neurofilament light chain level is associated with brain fraction atrophy.
Study details: An ongoing, prospective, observational study of 607 patients with MS.
Disclosures: The National Institutes of Health and the U.S. National MS Society supported the study. Several of the investigators received compensation from Novartis, which provided funds for the reagents needed for the single-molecule array assay.
Source: Cantó E et al. JAMA Neurol. 2019 Aug 12. doi: 10.1001/jamaneurol.2019.2137.
Tamoxifen benefit in lower-risk breast cancer varies by intrinsic subtype
, finds a secondary analysis of the Stockholm Tamoxifen (STO-3) trial.
“Patients with estrogen receptor (ER)–positive breast cancer have a long-term risk for fatal disease. However, the tumor biological factors that influence the long-term risk and the benefit associated with endocrine therapy are not well understood,” noted the investigators, who conducted the research under senior investigator Linda Lindström, MSc, PhD, department of biosciences and nutrition, Karolinska Institutet, Stockholm.
The STO-3 trial spanned 1976 to 1990 and randomized postmenopausal patients with lymph node–negative breast cancer to receive at least 2 years of adjuvant tamoxifen or no endocrine therapy.
Dr. Lindström and coinvestigators used immunohistochemistry and Agilent microarrays to define tumor molecular subtype. Analyses were based on 462 patients with ER-positive disease: 336 with luminal A subtype tumors and 126 with luminal B subtype tumors.
Results reported in JAMA Oncology showed that the distant recurrence–free interval (DRFI) was significantly better with tamoxifen than with no endocrine therapy in both the luminal A group (P less than .001) and the luminal B group (P = .04).
Among patients given tamoxifen, the 25-year DRFI rate was 87% (95% confidence interval, 82%-93%) for those with luminal A tumors vs. 67% (95% CI, 56%-82%) for those with luminal B tumors. Among patients not given any endocrine therapy, it was 70% (95% CI, 62%-79%) vs. 54% (95% CI, 42%-70%), respectively.
Tamoxifen had a significant DRFI benefit for 15 years after diagnosis in the luminal A group (hazard ratio, 0.57; 95% CI, 0.35-0.94). In contrast, the benefit was significant for only 5 years in the luminal B group (HR, 0.38; 95% CI, 0.24-0.59).
“We conclude that tamoxifen appears to confer a long-term benefit for patients with lymph node–negative, ER-positive, luminal A subtype tumors, and a short-term benefit for patients with luminal B subtype tumors. Given that the risk of distant metastatic disease is low for patients with the luminal A subtype but persists in the long term, whereas the risk for patients with luminal B subtype is higher initially but decreases after 5 years, tamoxifen treatment is beneficial for patients with luminal A or luminal B subtype tumors,” Dr. Lindström and coinvestigators maintained.
“In patients with luminal B subtype, up-front chemotherapy should be discussed and endocrine therapy potentially extended for up to 10 years, particularly in those in the higher risk strata according to other tumor characteristics,” they recommended.
Dr. Lindström disclosed no conflicts of interest. The study was supported by the Swedish Research Council, FORTE, The Gösta Milton Donation Fund, the California Breast Cancer Research Program, The Iris, Stig och Gerry Castenbäcks Stiftelse för Cancerforskning, and Konung Gustaf V:s Jubileumsfond from Radiumhemmets Forskningsfonder.
SOURCE: Yu NY et al. JAMA Oncol. 2019 Aug 8. doi: 10.1001/jamaoncol.2019.1856.
, finds a secondary analysis of the Stockholm Tamoxifen (STO-3) trial.
“Patients with estrogen receptor (ER)–positive breast cancer have a long-term risk for fatal disease. However, the tumor biological factors that influence the long-term risk and the benefit associated with endocrine therapy are not well understood,” noted the investigators, who conducted the research under senior investigator Linda Lindström, MSc, PhD, department of biosciences and nutrition, Karolinska Institutet, Stockholm.
The STO-3 trial spanned 1976 to 1990 and randomized postmenopausal patients with lymph node–negative breast cancer to receive at least 2 years of adjuvant tamoxifen or no endocrine therapy.
Dr. Lindström and coinvestigators used immunohistochemistry and Agilent microarrays to define tumor molecular subtype. Analyses were based on 462 patients with ER-positive disease: 336 with luminal A subtype tumors and 126 with luminal B subtype tumors.
Results reported in JAMA Oncology showed that the distant recurrence–free interval (DRFI) was significantly better with tamoxifen than with no endocrine therapy in both the luminal A group (P less than .001) and the luminal B group (P = .04).
Among patients given tamoxifen, the 25-year DRFI rate was 87% (95% confidence interval, 82%-93%) for those with luminal A tumors vs. 67% (95% CI, 56%-82%) for those with luminal B tumors. Among patients not given any endocrine therapy, it was 70% (95% CI, 62%-79%) vs. 54% (95% CI, 42%-70%), respectively.
Tamoxifen had a significant DRFI benefit for 15 years after diagnosis in the luminal A group (hazard ratio, 0.57; 95% CI, 0.35-0.94). In contrast, the benefit was significant for only 5 years in the luminal B group (HR, 0.38; 95% CI, 0.24-0.59).
“We conclude that tamoxifen appears to confer a long-term benefit for patients with lymph node–negative, ER-positive, luminal A subtype tumors, and a short-term benefit for patients with luminal B subtype tumors. Given that the risk of distant metastatic disease is low for patients with the luminal A subtype but persists in the long term, whereas the risk for patients with luminal B subtype is higher initially but decreases after 5 years, tamoxifen treatment is beneficial for patients with luminal A or luminal B subtype tumors,” Dr. Lindström and coinvestigators maintained.
“In patients with luminal B subtype, up-front chemotherapy should be discussed and endocrine therapy potentially extended for up to 10 years, particularly in those in the higher risk strata according to other tumor characteristics,” they recommended.
Dr. Lindström disclosed no conflicts of interest. The study was supported by the Swedish Research Council, FORTE, The Gösta Milton Donation Fund, the California Breast Cancer Research Program, The Iris, Stig och Gerry Castenbäcks Stiftelse för Cancerforskning, and Konung Gustaf V:s Jubileumsfond from Radiumhemmets Forskningsfonder.
SOURCE: Yu NY et al. JAMA Oncol. 2019 Aug 8. doi: 10.1001/jamaoncol.2019.1856.
, finds a secondary analysis of the Stockholm Tamoxifen (STO-3) trial.
“Patients with estrogen receptor (ER)–positive breast cancer have a long-term risk for fatal disease. However, the tumor biological factors that influence the long-term risk and the benefit associated with endocrine therapy are not well understood,” noted the investigators, who conducted the research under senior investigator Linda Lindström, MSc, PhD, department of biosciences and nutrition, Karolinska Institutet, Stockholm.
The STO-3 trial spanned 1976 to 1990 and randomized postmenopausal patients with lymph node–negative breast cancer to receive at least 2 years of adjuvant tamoxifen or no endocrine therapy.
Dr. Lindström and coinvestigators used immunohistochemistry and Agilent microarrays to define tumor molecular subtype. Analyses were based on 462 patients with ER-positive disease: 336 with luminal A subtype tumors and 126 with luminal B subtype tumors.
Results reported in JAMA Oncology showed that the distant recurrence–free interval (DRFI) was significantly better with tamoxifen than with no endocrine therapy in both the luminal A group (P less than .001) and the luminal B group (P = .04).
Among patients given tamoxifen, the 25-year DRFI rate was 87% (95% confidence interval, 82%-93%) for those with luminal A tumors vs. 67% (95% CI, 56%-82%) for those with luminal B tumors. Among patients not given any endocrine therapy, it was 70% (95% CI, 62%-79%) vs. 54% (95% CI, 42%-70%), respectively.
Tamoxifen had a significant DRFI benefit for 15 years after diagnosis in the luminal A group (hazard ratio, 0.57; 95% CI, 0.35-0.94). In contrast, the benefit was significant for only 5 years in the luminal B group (HR, 0.38; 95% CI, 0.24-0.59).
“We conclude that tamoxifen appears to confer a long-term benefit for patients with lymph node–negative, ER-positive, luminal A subtype tumors, and a short-term benefit for patients with luminal B subtype tumors. Given that the risk of distant metastatic disease is low for patients with the luminal A subtype but persists in the long term, whereas the risk for patients with luminal B subtype is higher initially but decreases after 5 years, tamoxifen treatment is beneficial for patients with luminal A or luminal B subtype tumors,” Dr. Lindström and coinvestigators maintained.
“In patients with luminal B subtype, up-front chemotherapy should be discussed and endocrine therapy potentially extended for up to 10 years, particularly in those in the higher risk strata according to other tumor characteristics,” they recommended.
Dr. Lindström disclosed no conflicts of interest. The study was supported by the Swedish Research Council, FORTE, The Gösta Milton Donation Fund, the California Breast Cancer Research Program, The Iris, Stig och Gerry Castenbäcks Stiftelse för Cancerforskning, and Konung Gustaf V:s Jubileumsfond from Radiumhemmets Forskningsfonder.
SOURCE: Yu NY et al. JAMA Oncol. 2019 Aug 8. doi: 10.1001/jamaoncol.2019.1856.
FROM JAMA ONCOLOGY
Fatal Drug-Resistant Invasive Pulmonary Aspergillus fumigatus in a 56-Year-Old Immunosuppressed Man (FULL)
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1). 
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4). 
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
Historically, aspergillosis in patients with hematopoietic stem cell transplantation (HSCT) has carried a high mortality rate. However, recent data demonstrate a dramatic improvement in outcomes for patients with HSCT: 90-day survival increased from 22% before 2000 to 45% over the past 15 years.1 Improved outcomes coincide with changes in transplant immunosuppression practices, use of cross-sectional imaging for early disease identification, galactomannan screening, and the development of novel treatment options.
Voriconazole is an azole drug that blocks the synthesis of ergosterol, a vital component of the cellular membrane of fungi. Voriconazole was approved in 2002 after a clinical trial demonstrated an improvement in 50% of patients with invasive aspergillosis in the voriconazole arm vs 30% in the amphotericin B arm at 12 weeks.2 Amphotericin B is a polyene antifungal drug that binds with ergosterol, creating leaks in the cell membrane that lead to cellular demise. Voriconazole quickly became the first-line therapy for invasive aspergillosis and is recommended by both the Infectious Disease Society of American (IDSA) and the European Conference on Infections in Leukemia.3
Case Presentation
A 55-year-old man with high-risk chronic myelogenous leukemia (CML) underwent a 10 of 10 human leukocyte antigen allele and antigen-matched peripheral blood allogeneic HSCT with a myeloablative-conditioning regimen of busulfan and cyclophosphamide, along with prophylactic voriconazole, sulfamethoxazole/trimethoprim, and acyclovir. After successful engraftment (without significant neutropenia), his posttransplant course was complicated by grade 2 graft vs host disease (GVHD) of the skin, eyes, and liver, which responded well to steroids and tacrolimus. Voriconazole was continued for 5 months until immunosuppression was minimized (tacrolimus 1 mg twice daily). Two months later, the patient’s GVHD worsened, necessitating treatment at an outside hospital with high-dose prednisone (2 mg/kg/d) and cyclosporine (300 mg twice daily). Voriconazole prophylaxis was not reinitiated at that time.
One year later, at a routine follow-up appointment, the patient endorsed several weeks of malaise, weight loss, and nonproductive cough. The patient’s immunosuppression recently had been reduced to 1 mg/kg/d of prednisone and 100 mg of cyclosporine twice daily. A chest X-ray demonstrated multiple pulmonary nodules; follow-up chest computed tomography (CT) confirmed multiple nodular infiltrates with surrounding ground-glass opacities suspicious with a fungal infection (Figure 1).
Treatment with oral voriconazole (300 mg twice daily) was initiated for probable pulmonary aspergillosis. Cyclosporine (150 mg twice daily) and prednisone (1 mg/kg/d) were continued throughout treatment out of concern for hepatic GVHD. The patient’s symptoms improved over the next 10 days, and follow-up chest imaging demonstrated improvement.
Two weeks after initiation of voriconazole treatment, the patient developed a new productive cough and dyspnea, associated with fevers and chills. Repeat imaging revealed right lower-lobe pneumonia. The serum voriconazole trough level was checked and was 3.1 mg/L, suggesting therapeutic dosing. The patient subsequently developed acute respiratory distress syndrome and required intubation and mechanical ventilation. Repeat BAL sampling demonstrated multidrug-resistant Escherichia coli, a BAL galactomannan level of 2.0 ODI, and negative fungal cultures. The patient’s hospital course was complicated by profound hypoxemia, requiring prone positioning and neuromuscular blockade. He was treated with meropenem and voriconazole. His immunosuppression was reduced, but he rapidly developed acute liver injury from hepatic GVHD that resolved after reinitiation of cyclosporine and prednisone at 0.75 mg/kg/d.
The patient improved over the next 3 weeks and was successfully extubated. Repeat chest CT imaging demonstrated numerous pneumatoceles in the location of previous nodules, consistent with healing necrotic fungal disease, and a new right lower-lobe cavitary mass (Figure 2). Two days after transferring out of the intensive care unit, the patient again developed hypoxemia and fevers to 39° C. Bronchoscopy with BAL of the right lower lobe revealed positive A fumigatus and Rhizopus sp polymerase chain reaction (PCR) assays, although fungal cultures were positive only for A fumigatus. Liposomal amphotericin B (5 mg/kg) was added to voriconazole therapy to treat mucormycosis and to provide a second active agent against A fumigatus.
Unfortunately, the patient’s clinical status continued to deteriorate with signs of progressive respiratory failure and infection despite empiric, broad-spectrum antibiotics and dual antifungal therapy. His serum voriconazole level continued to be therapeutic at 1.9 mg/L. The patient declined reintubation and invasive mechanical ventilation, and he ultimately transitioned to comfort measures and died with his family at the bedside.
Autopsy demonstrated widely disseminated Aspergillus infection as the cause of death, with evidence of myocardial, neural, and vascular invasion of A fumigatus (Figures 3 and 4).
Discussion
This case of fatal, progressive, invasive, pulmonary aspergillosis demonstrates several important factors in the treatment of patients with this disease. Treatment failure usually relates to any of 4 possible factors: host immune status, severity or burden of disease, appropriate dosing of antifungal agents, and drug resistance. This patient’s immune system was heavily suppressed for a prolonged period. Attempts at reducing immunosuppression to the minimal required dosage to prevent a GVHD flare were unsuccessful and became an unmodifiable risk factor, a major contributor to his demise.
The risks of continuous high-dose immunosuppression in steroid-refractory GVHD is well understood and has been previously demonstrated to have up to 50% 4-year nonrelapse mortality, mainly due to overwhelming bacterial, viral, and fungal infections.4 All attempts should be made to cease or reduce immunosuppression in the setting of a severe infection, although this is sometimes impossible as in this case.
The patient’s disease burden was significant as evidenced by the bilateral, multifocal pulmonary nodules seen on chest imaging and the disseminated disease found at postmortem examination. His initial improvement in symptoms with voriconazole and the evolution of his images (with many of his initial pulmonary nodules becoming pneumatoceles) suggested a temporary positive immune response. The authors believe that the Rhizopus in his sputum represents noninvasive colonization of one of his pneumatoceles, because postmortem examination failed to reveal Rhizopus at any other location.
Voriconazole has excellent pulmonary and central nervous system penetration: In this patient serum levels were well within the therapeutic range. His peculiar drug resistance pattern (sensitivity to azoles and resistance to amphotericin) is unusual. Azole resistance in leukemia and patients with HSCT is more common than is amphotericin resistance, with current estimates of azole resistance close to 5%, ranging between 1% and 30%.5,6 Widespread use of antifungal prophylaxis with azoles likely selects for azole resistance.6
Despite this concern of azole resistance, current IDSA guidelines recommend against routine susceptibility testing of Aspergillus to azole therapy because of the current lack of consensus between the European Committee on Antibiotic Susceptibility Testing and Clinical and Laboratory Standards Institute on break points for resistance patterns.3,7 This is an area of emerging research, and proposed cut points for declaration of resistance do exist in the literature even if not globally agreed on.8
Combination antifungal therapy is an option for treatment in cases of possible drug resistance. Nonetheless, a recent randomized, double-blind, placebo-controlled, multicenter trial comparing voriconazole monotherapy with the combination of voriconazole and anidulafungin failed to demonstrate an overall mortality benefit in the primary analysis, although secondary analysis showed a mortality benefit with combination therapy in patients at highest risk for death.9
Despite the lack of unified standards with susceptibility testing, it may be reasonable to perform such tests in patients with demonstrating progressive disease. In this patient’s case, amphotericin B was added to treat the Rhizopus species found in his sputum, and while not the combination studied in the previously mentioned study, the drug should have provided an additional active agent for Aspergillus should this patient have had azole resistance.
Surprisingly, subsequent testing demonstrated the Aspergillus species to be resistant to amphotericin B. De novo amphotericin B-resistant A fumigates is extremely rare, with an expected incidence of 1% or less.10 The authors believe the patient may have demonstrated induction of amphotericin-B resistance through activation of fungal stress pathways by prior treatment with voriconazole. This has been demonstrated in vitro and should be considered should combination salvage therapy be required for the treatment of a refractory Aspergillus infection especially if patients have received prior treatment with voriconazole.11
Conclusion
This fatal case of invasive pulmonary aspergillosis illustrates the importance of considering the 4 main causes of treatment failure in an infection. Although the patient had a high burden of disease with a rare resistance pattern, he was treated with appropriate and well-dosed therapy. Ultimately, his unmodifiable immunosuppression was likely the driving factor leading to treatment failure and death. The indication for and number of bone marrow transplants continues to increase, thus exposure to and treatment of invasive fungal infections will increase accordingly. As such, providers should ensure that all causes of treatment failure are considered and addressed.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
1. Upton A, Kirby KA, Carpenter P, Boeckh M, Marr KA. Invasive aspergillosis following hematopoietic cell transplantation: outcomes and prognostic factors associated with mortality. Clin Infect Dis. 2007;44(4):531-540.
2. Herbrecht R, Denning DW, Patterson TF, et al; Invasive Fungal Infections Group of the European Organisation for Research and Treatment of Cancer and the Global Aspergillus Study Group. Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N Engl J Med. 2002;347(6):408-415.
3. Patterson TF, Thompson GR III, Denning DW, et al. Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the Infectious Disease Society of America. Clin Infect Dis. 2016;63(4):e1-e60.
4. García-Cadenas I, Rivera I, Martino R, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transplant. 2017;52(1):107-113.
5. Vermeulen E, Maertens J, De Bel A, et al. Nationwide surveillance of azole resistance in Aspergillus diseases. Antimicrob Agents Chemother. 2015;59(8):4569-4576.
6. Wiederhold NP, Patterson TF. Emergence of azole resistance in Aspergillus. Semin Respir Crit Care Med. 2015;36(5):673-680.
7. Cuenca-Estrella M, Moore CB, Barchiesi F, et al; AFST Subcommittee of the European Committee on Antimicrobial Susceptibility Testing. Multicenter evaluation of the reproducibility of the proposed antifungal susceptibility testing method for fermentative yeasts of the Antifungal Susceptibility Testing Subcommittee of the European Committee on Antimicrobial Susceptibility Testing (AFST-EUCAST). Clin Microbiol Infect. 2003;9(6):467-474.
8. Pfaller MA, Diekema DJ, Ghannoum MA, et al; Clinical and Laboratory Standards Institute Antifungal Testing Subcommittee. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by Clinical and Laboratory Standards Institute for broth microdilution methods. J Clin Microbiol. 2009;47(10):3142-3146.
9. Marr KA, Schlamm HT, Herbrecht R, et al. Combination antifungal therapy for invasive aspergillosis: a randomized trial. Ann Intern Med. 2015;162(2):81-89.
10. Tashiro M, Izumikawa K, Minematsu A, et al. Antifungal susceptibilities of Aspergillus fumigatus clinical isolates obtained in Nagasaki, Japan. Antimicrob Agents Chemother. 2012;56(1):584-587.
11. Rajendran R, Mowat E, Jones B, Williams C, Ramage G. Prior in vitro exposure to voriconazole confers resistance to amphotericin B in Aspergillus fumigatus biofilms. Int J Antimicrob Agents. 2015;46(3):342-345.
Interview with Andrew Pachner, MD, about the molecular processes of multiple sclerosis
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.

What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.
Andrew R. Pachner, MD is the Murray B. Bornstein professor of neurology at Geisel School of Medicine at Dartmouth and director of the Multiple Sclerosis Center at Dartmouth-Hitchcock Medical Center. We spoke to Dr. Pachner about his research into the molecular processes of multiple sclerosis (MS) and the potential impact on patient management.
What do we know about the molecular processes behind relapsing-remitting and progressive MS?
DR. PACHNER: The progress--in terms of molecules--has not been rapid in the field of MS. The only molecular biomarker we use in practice is oligoclonal bands or other measures of immunoglobulin production in the nervous system, and that biomarker was described in 1942. So, it has been a long time since we have seen a relevant molecule that we can use clinically.
But there has been a lot of progress in the general field of neuroinflammation. MS is one of a large number of diseases that results in neuroinflammation and demyelination.
One thing we have learned over time is that there are many different subtypes of MS. They probably have some shared molecular processes, but they also are likely to have divergent molecular processes.
Over the past 5 to 10 years, researchers have been interested in trying to dissect some of the molecular aspects of MS to identify biomarkers that can, in turn, differentiate subtypes of MS. This will help to identify different ways of treating MS that are optimal for individual patients. It is clear that each patient is quite different and unlikely to be standardized in the way they respond to treatment.
The degree to which relapsing-remitting and progressive MS are differentiated on the molecular level is dependent on how much influence there is of the immune system in the periphery. When MS first starts in a patient, the brain has either no or a very primitive immune system, and then over time it changes, and it becomes much more immune-oriented and populated by immune cells and molecules. So, there’s a trend over time of the central nervous system becoming increasingly populated by immune cells and able to make immune molecules.
What has your recent research on murine models representing these disease patterns shown?
DR. PACHNER: Even though in humans there is a continuum from relapsing remitting to progressive, it is not like they are completely separate. Frequently in the middle of relapsing-remitting disease there is some progression over time.
In mouse models, we like things to be very clear and separate. We try to make things as simple as possible because of the complexity of the nervous and immune systems.
The simple model for the relapsing-remitting disease is experimental autoimmune encephalomyelitis (EAE), the most commonly studied model of neuroinflammation.
For the progressive form of MS, we use the Theiler’s virus model, which is a type of virus called the picornavirus that is injected into the brain of mice resulting in a slowly progressive, chronic viral infection that looks very much like progressive MS.
In EAE, the disease is induced by presenting an antigen to the peripheral immune system, allowing cells from the peripheral immune system to enter into the central nervous system. It is a manifestation of inflammation and the immune response is in the periphery. In the Theiler’s model, it is a localized process within the central nervous system because the virus is injected directly into the brain.
We found that in EAE the pattern is very much dominated by what happens in the periphery and the injury is very transient. There are cells that enter the nervous system that cause inflammation and damage, but there are also processes that downregulate those cells and processes and eventually the animal improves--similar to an MS attack.
By contrast, in the Theiler’s model there is progressive injury that is dominated by two molecular processes in the central nervous system that we do not see in relapsing-remitting MS or in EAE, and that is the activation of Type 1 interferons and also a very pronounced immunoglobulin production along with all the molecules that help support plasma cells making immunoglobulin.
These are two different animal models that provide us insight into how the central nervous system can be injured in the course of neuroinflammation and they look to be very different in how they manifest themselves, both in the periphery and in the central nervous system.
How may these new findings impact the future management and treatment of MS?
DR. PACHNER: When I see a patient with MS, I tell them that we absolutely need to focus on your own disease and how it responds, rather than taking too much guidance from MS as a whole. Because each patient with MS is different.
One of the things that we have tried to do is to identify molecular markers that might help us in management and treatment. As an example, we have learned that some patients who present with their first episode of MS do very poorly. These patients have many more attacks and/or have very aggressive progression in terms of their disability so that they potentially could be in a wheelchair within a few years. Other patients have what we call a benign variant MS. These patients may have an initial episode that is not that different than the other patient, but this type of patient may not have anything else for the rest of their life.
We would like to have some differentiation of those two types of patients. In the first example you can try to be very aggressive and minimize the neuroinflammation with powerful immune-suppressing drugs that have a high risk of causing side effects, such as cancer or opportunistic infections, but on the other hand may have a high benefit in preventing future inflammatory events and progressive injury. But that would not be the correct treatment choice for the second patient example.
It would be nice to tailor treatment to a predictive biomarker. That is something we have been working very hard on. Based on some of the animal models, we have identified a molecular signature of inflammatory MS that is very predictive of future events and we are hoping that that will help us differentiate patients. In other words, not just treat every MS patient the same, but identify whether they need a very powerful immunosuppressant drug, or a mildly immunosuppressant drug, or no treatment at all.
If you have a patient who has one attack and never has any other problem with their MS, then they do not need to be on any treatment. Unfortunately, we do not have predictive value at this point for any molecule or any other attribute of the patient at this point in time. We are trying to remedy that.
That is one very practical aspect of our work in trying to understand the biology of the disease better--identifying molecules that are associated with future damage and inflammation and using those in a predictive manner in patients to guide treatment.
Another important aspect is the attempt to understand the biology of neuroinflammation and how it causes both demyelination and progressive injury to neurons.
References:
Pachner AR, DiSano K, Royce DB, Gilli F. Clinical utility of a molecular signature in inflammatory demyelinating diseases. Neurol Neuroimmunol Neuroinflamm.2019;6(1):e520.

Aspirin interacts with epigenetics to influence breast cancer mortality
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
This study offers new insights into the intersection of epigenetics, prediagnosis aspirin use, and breast cancer survival at a time when there is an urgent need to understand why some women respond differently to treatment and to find cost-effective therapies for the disease.
Epigenetics is a promising avenue of investigation because epigenetic shifts, such as DNA methylation, that impact the genes responsible for cell behavior and DNA damage and repair are known to contribute to and exacerbate cancer. These epigenetic signatures could act as biomarkers for risk in cancer and also aid with more effective treatment approaches. For example, aspirin is known to affect DNA methylation at certain sites in colon cancer, hence this study’s hypothesis that pre–cancer diagnosis aspirin use would interact with epigenetic signatures and influence breast cancer outcomes.
Kristen M. C. Malecki, PhD, is from the department of population health sciences in the School of Medicine and Public Health at the University of Wisconsin, Madison. The comments are adapted from an accompanying editorial (Cancer. 2019 Aug 12. doi: 10.1002/cncr.32365). Dr. Malecki declared support from the National Institutes of Health, National Institute for Environmental Health Sciences Breast Cancer, and the Environment Research Program.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
The impact of prediagnosis aspirin use on mortality in women with breast cancer is significantly tied to epigenetic changes in certain breast cancer-related genes, investigators reported.
While studies have shown aspirin reduces the risk of breast cancer development, there is limited and inconsistent data on the effect of aspirin on prognosis and mortality after a diagnosis of breast cancer, Tengteng Wang, PhD, from the department of epidemiology at the University of North Carolina at Chapel Hill and coauthors wrote in Cancer.
To address this, they analyzed data from 1,508 women who had a first diagnosis of primary breast cancer and were involved in the Long Island Breast Cancer Study Project; they then looked at the women’s methylation status, which is a mechanism of epigenetic change.
Around one in five participants reported ever using aspirin, and the analysis showed that ever use of aspirin was associated with an overall 13% decrease in breast cancer–specific mortality.
However researchers saw significant interactions between aspirin use and LINE-1 methylation status – which is a marker of methylation of genetic elements that play key roles in maintaining genomic stability – and breast cancer–specific genes.
They found that aspirin use in women with LINE-1 hypomethylation was associated with a risk of breast cancer–specific mortality that was 45% higher than that of nonusers (P = .05).
Compared with nonusers, aspirin users with methylated tumor BRCA1 promoter had significant 16% higher breast cancer mortality (P = .04) and 67% higher all-cause mortality (P = .02). However the study showed aspirin did not affect mortality in women with unmethylated BRCA1 promoter.
Among women with the PR breast cancer gene, aspirin use by those with methylation of the PR promoter was associated with a 63% higher breast cancer–specific mortality, but methylation showed no statistically significant effect on all-cause mortality, compared with nonusers.
The study found no significant change when they restricted the analysis to receptor-positive or invasive breast cancer, and the associations remained consistent even after adjusting for global methylation.
“Our findings suggest that the association between aspirin use and mortality after breast cancer may depend on methylation profiles and warrant further investigation,” the authors wrote. “These findings, if confirmed, may provide new biological insights into the association between aspirin use and breast cancer prognosis, may affect clinical decision making by identifying a subgroup of patients with breast cancer using epigenetic markers for whom prediagnosis aspirin use affects subsequent mortality, and may help refine risk-reduction strategies to improve survival among women with breast cancer.”
The study was partly supported by the National Institutes of Health. One author declared personal fees from the private sector outside the submitted work.
SOURCE: Wang T et al. Cancer. 2019 Aug 12. doi: 10.1002/cncr.32364.
FROM CANCER
Treatment Facility: An Important Prognostic Factor for Dedifferentiated Liposarcoma Survival (FULL)
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
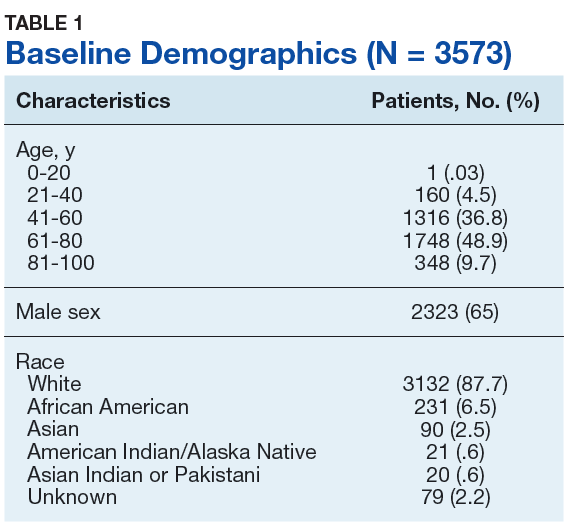
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
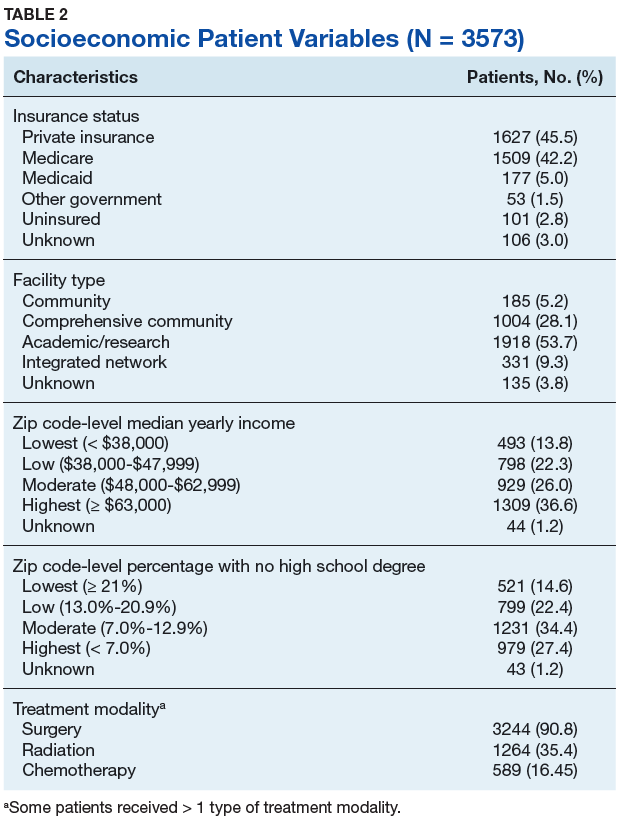
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
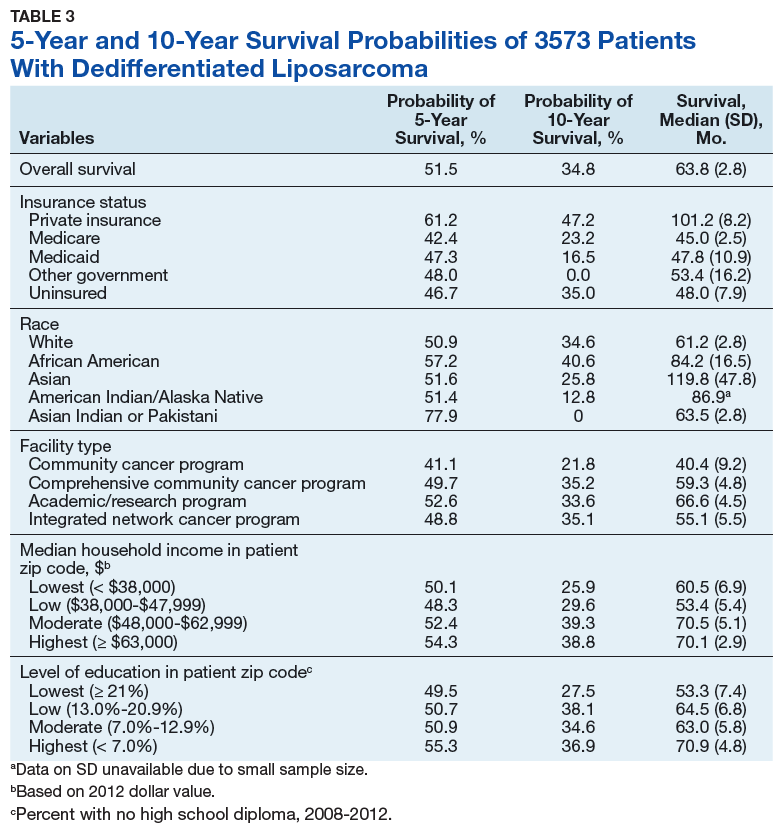
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
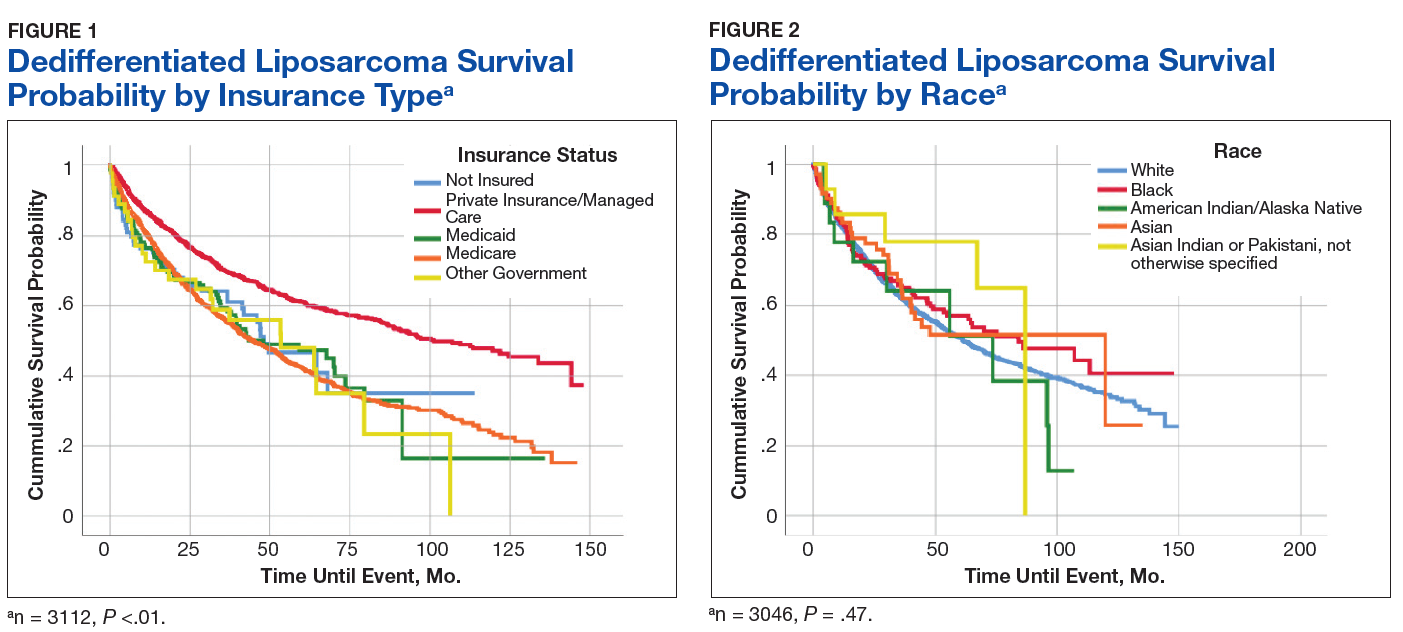
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
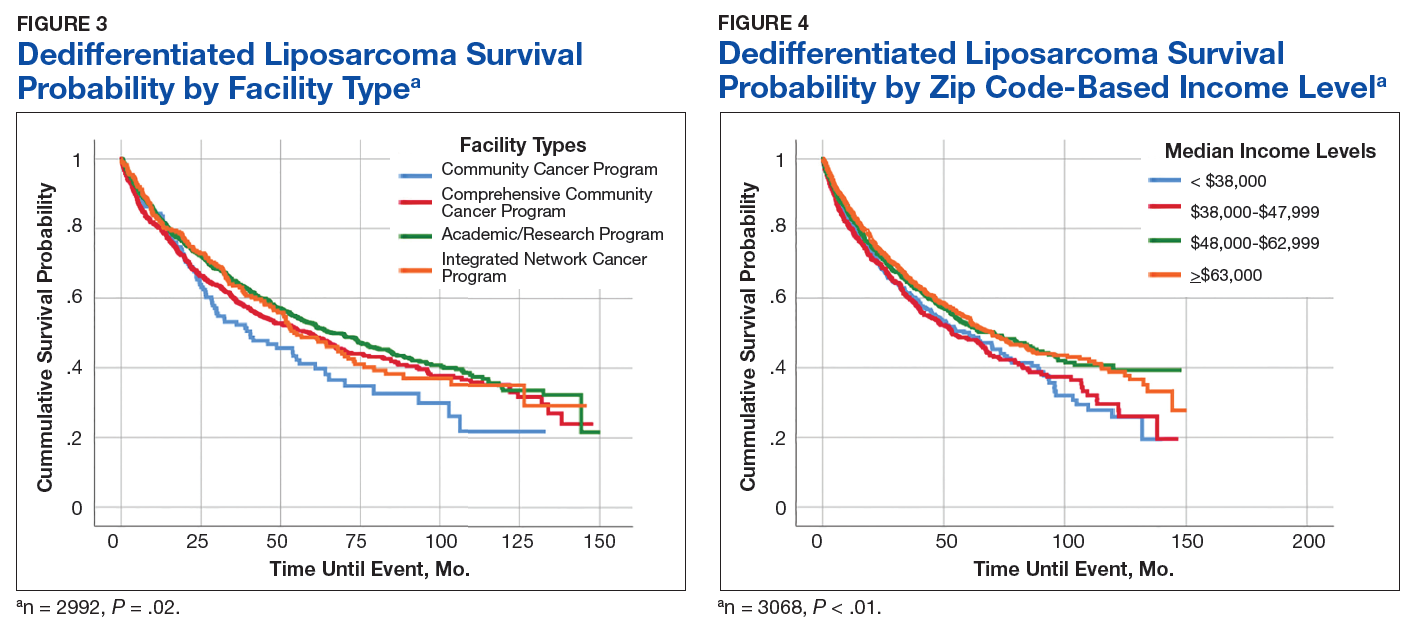
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
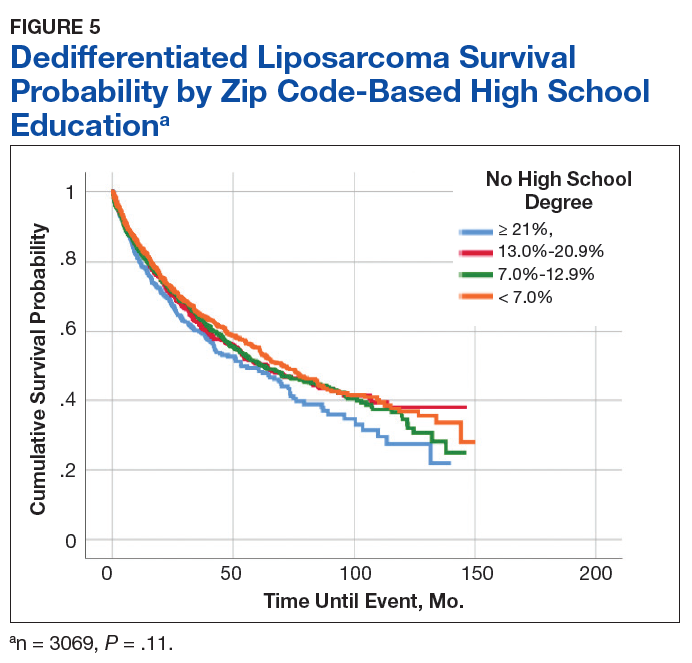
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
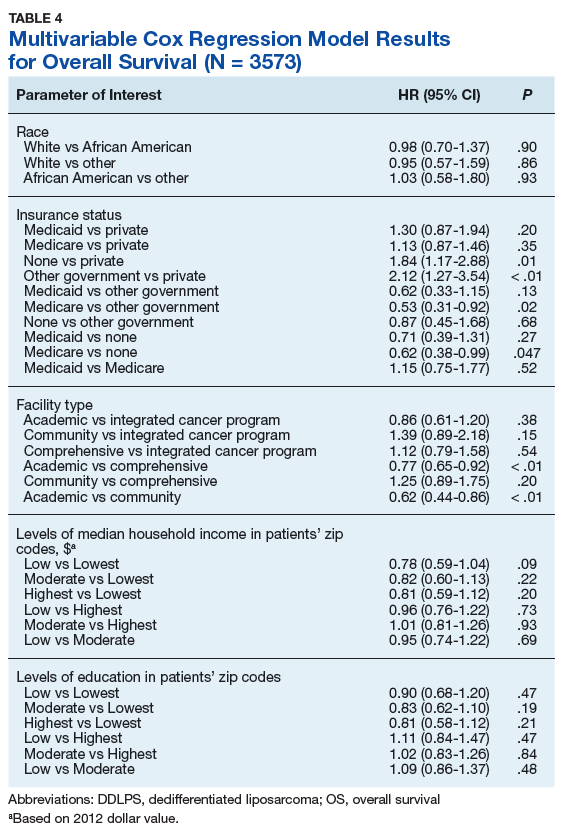
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Genomic Medicine and Genetic Counseling in the Department of Veterans Affairs and Department of Defense (FULL)
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.

We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.
We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.
Vickie Venne, MS. What is the Genomic Medicine Service (GMS) at the US Department of Veterans Affairs (VA)?
Renee Rider, JD, MS, LCGC. GMS is a telehealth service. We are part of central office and field stationed at the George E. Wahlen VA Medical Center (VAMC) in Salt Lake City, Utah. We provide care to about 90 VAMCs and their associated clinics. Veterans are referred to us by entering an interfacility consult in the VA Computerized Patient Record System (CPRS). We review the consult to determine whether the patient needs to be seen, whether we can answer with an e-consult, or whether we need more information. For the patients who need an appointment, the telehealth department at the veteran’s VA facility will contact the patient to arrange a visit with us. At the time of the appointment, the facility has a staff member available to seat the patient and connect them to us using video equipment.
We provide genetic care for all specialties, including cancer, women’s health, cardiology and neurology. In today’s discussion, we are focusing on cancer care.
Vickie Venne. What do patients do at facilities that don’t get care through GMS?
Renee Rider. There are a handful of facilities that provide their own genetic care in-house. For example, VA Boston Healthcare System in Massachusetts and the Michael E. DeBakey VAMC in Houston, Texas each have their own programs. For veterans who are not at a VA facility that has an agreement with GMS and do not have a different genetics program, their providers need to make referrals to community care.
Vickie Venne. How do patients get referred and what happens at their facility when the patients return to the specialty and primary care providers (PCP)? Ishta, who do you refer to GMS and how do you define them initially?
Ishta Thakar, MD, FACP. Referrals can come at a couple of points during a veteran’s journey at the VA. The VA covers obstetrics care for women veterans. Whenever a PCP or a women’s health provider is doing the initial history and physical on a new patient, if the female veteran has an extensive family history of breast, ovarian, colon, or endometrial cancer, then we take more history and we send a consult to GMS. The second instance would be if she tells us that she has had a personal history of breast, ovarian, or endometrial cancer and she has never had genetic testing. The third instance would be whenever we have a female veteran who is diagnosed with breast, ovarian, endometrial, or colon cancer. We would definitely talk to her about genetic counseling and send a referral to GMS. We would ask for a GMS consult for a patient with advanced maternal age, with exposure to some kind of teratogens, with an abnormal ultrasound, a family history of chromosomal disorders, or if she’s seeing an obstetrician who wants her to be tested. And finally, if a patient has a constellation of multiple cancers in the family and we don’t know what’s going on, we would also refer the patient to GMS.
Vickie Venne. That would be why GMS fields over 150 referrals every week. It is a large list. We also see veterans with personal or family histories of neurologic or cardiologic concerns as well.
Renee, as somebody who fields many of these referrals from unaffected individuals, what is the family history process?
Renee Rider. We don’t expect the referring provider to be a genetic expert. When a provider is seeing a constellation of several different cancers and he or she doesn’t know if there’s anything going on genetically or even if it’s possible, absolutely they should put in a referral to GMS. We have a triage counselor who reviews every consult that comes into our service within 24 hours.
Many cancers are due to exposures that are not concerning for a genetic etiology. We can let you know that it is not concerning, and the PCP can counsel the patient that it is very unlikely to be genetic in nature. We still give feedback even if it’s not someone who is appropriate for genetic counseling and testing. It is important to reach out to GMS even if you don’t know whether a cancer is genetic in nature.
It also is important to take your time when gathering family histories. We get a lot of patients who say, “There’s a lot of cancer in my family. I have no idea who had cancer, but I know a lot of people had cancer.” That’s not the day to put in a referral to GMS. At that point, providers should tell the patient to get as much information as they can about the family history and then reassess. It’s important for us to have accurate information. We’ve had several times where we receive a referral because the veteran says that their sister had ovarian cancer. And then when our staff calls, they later find out it was cervical cancer. That’s not a good use of the veteran’s time, and it’s not a good use of VA resources.
The other important thing about family histories is keeping the questions open-ended. Often a PCP or specialist will ask about a certain type of cancer: “Does anyone in your family have breast cancer, ovarian cancer?” Or if the veteran
is getting a colonoscopy, they ask, “Does anybody have colon cancer?” Where really, we need to be a little bit more open-ended. We prefer questions like, “Has anyone in your family
had cancer?” because that’s the question that prompts a response of, “Yes, 3 people in my family have had thyroid cancer.” That’s very important for us to know, too.
If you do get a positive response, probe a little bit more: what kind of cancer did someone have, how old were they when they had their cancer? And how are they related? Is this an aunt on your mom’s side or on your dad’s side? Those are the types of information that we need to figure out if that person needs a referral.
Vickie Venne. It’s a different story when people already have a cancer diagnosis. Which hematology or oncology patients are good referrals and why?
Lisa Arfons, MD. When patients come in with newly diagnosed cancer, breast for example, it is an emotional diagnosis and psychologicallydistressing. Oftentimes, they want to know why this happened to them. The issues surrounding
genetic testing also becomes very emotional. They want to know whether their children are at risk as well.
Genetic discussions take a long time. I rarely do that on the first visit. I always record for myself in my clinic note if something strikes me regarding the patient’s diagnosis. I quickly run through the National Comprehensive Cancer Network (NCCN) guidelines to remind myself of what I need to go over with the patient at our next meeting. Most patients don’t need to be referred to GMS, and most patients don’t need to be tested once they’re seen.
I often save the referral discussion for after I have established a rapport with a patient, we have a treatment plan, or they already have had their first surgery. Therefore, we are not making decisions about their first surgery based on the genetic medicine results.
If I’m considering a referral, I do a deeper dive with the patient. Is the patient older or younger than 45 years? I pull up NCCN guidelines and we go through the entire checklist.
We have male breast cancer patients at the VA—probably more than the community—so we refer those patients. At the Louis Stokes Cleveland VAMC in Ohio, we have had some in-depth discussions about referring male breast cancer patients for genetic testing and whether it was beneficial to older patients with male breast cancer. Ultimately, we decided that it was important for our male veterans to be tested because it empowered them to have better understanding of their medical conditions that may not just have effect on them but on their offspring, and that that can be a source of psychological and emotional support.
I don’t refer most people to GMS once I go through the checklist. I appreciate the action for an e-consult within the CPRS telemedicine consult itself, as Renee noted. If it is not necessary, GMS makes it an e-consult. I try to communicate that I don’t know whether it is necessary or not so that GMS understands where I’m coming from.
Vickie Venne. In the US Department of Defense (DoD) the process is quite different. Mauricio, can you explain the clinical referral process, who is referred, and how that works from a laboratory perspective?
Maj De Castro, MD, FACMG, USAF. The VA has led the way in demonstrating how to best provide for the medical genetic needs of a large, decentralized population distributed all over the country. Over the last 5 to 10 years, the DoD has made strides in recognizing the role genetics plays in the practice of everyday medicine and redoubling efforts to meet the needs of servicemembers.
The way that it traditionally has worked in the DoD is that military treatment facilities (MTFs) that have dedicated geneticists and genetic counselors: Kessler Medical Center in Mississippi, Walter Reed National Military Medical
Center in Maryland, Tripler Army Medical Center in Hawaii, Madigan Army Medical Center in Washington, Brooke Army Medical Center in Texas, Naval Medical Center San Diego in California, and Naval Medical Center Portsmouth in Virginia. A patient seeking genetic evaluation, counseling, or testing in those larger facilities would be referred to the genetics service by their primary care manager. Wait times vary, but it would usually be weeks, maybe months. However, the great majority of MTFs do not have dedicated genetics support. Most of the time, those patients would have to be referred to the local civilian community—there was no process for them to be seen in in the military healthcare system—with wait times that exceed 6 to 8 months in some cases. This is due to just not a military but a national shortage of genetics professionals (counselors and physicians).
Last year we started the telegenetics initiative, which is small compared to the VA—it is comprised of 2 geneticists and 1 genetic counselor—but with the full intent of growing it over time. Its purpose is to extend the resources we
had to other MTFs. Genetics professionals stationed state-side can provide care to remote facilities with limited access to local genetics support such as Cannon Air Force Base (AFB) or overseas facilities such as Spangdahlem AFB in Germany.
We recognize there are military-specific needs for the DoD regarding the genetic counseling process that have to take into account readiness, genetic discrimination, continued ability to serve and fitness for duty. For this important reason, we are seeking to expand our telegenetics initiative. The goal is to be able to provide 100% of all genetic counseling in-house, so to speak.
Currently, providers at the 4 pilot sites (Cannon AFB, Fort Bragg, Spangdahlem AFB, and Guantanamo Bay) send us referrals. We triage them and assign the patient to see a geneticist or a counselor depending on the indication.
On the laboratory side, it has been a very interesting experience. Because we provide comprehensive germline cancer testing at very little cost to the provider at any MTF, we have had high numbers of test requests over the years.
In addition to saving the DoD millions of dollars in testing, we have learned some interesting lessons in the process. For instance, we have worked closely with several different groups to better understand how to educate providers on the genetic counseling and testing process. This has allowed us to craft a thorough and inclusive consent form that addresses the needs of the DoD. We have also learned valuable lessons about population-based screening vs evidence-based testing, and lessons surrounding narrow-based testing (BRCA1 and BRCA2 only testing) vs ordering a more comprehensive panel that includes other genes supported by strong evidence (such as PALB2, CHEK2, or TP53).
For example, we have found that in a significant proportion of individuals with and without family history, there are clinically relevant variants in genes other than BRCA1 or BRCA2. And so, we have made part of our consent process,
a statement on secondary findings. If the patient consents, we will report pathogenic variants in other genes known to be associated with cancer (with strong evidence) even if the provider ordered a narrow panel such as BRCA1 and BRCA2 testing only. In about 1% to 4% of patients that would otherwise not meet NCCN guidelines, we’ve reported variants that were clinically actionable and changed the medical management of that patient.
We feel strongly that this is a conversation that we need to have in our field, and we realize it’s a complex issue, maybe we need to expand who gets testing. Guideline based testing is missing some patients out there that could benefit from it.
Vickie Venne. There certainly are many sides to the conversation of population-based vs evidence-based genetic testing. Genetic testing policies are changing rapidly. There are teams exploring comprehensive gene sequencing for
newborns and how that potential 1-time test can provide information will be reinterpreted as a person goes from cradle to grave. However, unlike the current DoD process, in the VA there are patients who we don’t see.
Renee Rider. I want to talk about money. When we order a genetic test, that test is paid for by the pathology department at the patient’s VAMC. Most of the pathology departments we work with are clear that they only can provide
genetic testing that is considered medically necessary. Thus, we review each test to make sure it meets established guidelines for testing. We don’t do population genetic screening as there isn’t evidence or guidelines to support offering it. We are strict about who does and does not get genetic testing, partly because we have a responsibility to pathology departments and to the taxpayers.
GMS focuses on conditions that are inherited, that is to say, we deal with germline genetics. Therefore, we discontinue referrals for somatic requests, such as when an OncotypeDX test is requested. It is my understanding that pharmacogenetic referrals may be sent to the new PHASeR initiative, which is a joint collaboration between the VA and Sanford Health and is headed by Deepak Voora, MD.
We generally don’t see patients who still are having diagnostic procedures done. For example, if a veteran has a suspicious breast mass, we recommend that the provider workup the mass before referring to GMS. Regardless of a genetic test result, a suspicious mass needs to be worked up. And, knowing if the mass is cancerous could change how we would proceed with the genetic workup. For example, if the mass were not cancerous, we may recommend that an affected relative have the first genetic evaluation. Furthermore, knowing if the patient has cancer changes how we interpret negative test results.
Another group of patients we don’t see are those who already had genetic testing done by the referring provider. It’s a VA directive that if you order a test, you’re the person who is responsible for giving the results. We agree with
this directive. If you don’t feel comfortable giving back test results, don’t order the test. Often, when a provider sends a patient to us after the test was done, we discover that the patient didn’t have appropriate pretest counseling. A test result, such as a variant of uncertain significance (VUS), should never be a surprise to either the provider or the patient.
Ishta Thakar. For newly diagnosed cancers, the first call is to the patient to inform them that they have cancer. We usually bring up genetic counseling or testing, if applicable, when they are ready to accept the diagnosis and have a conversation about it. All our consults are via telehealth, so none of our patients physically come to GMS in Salt Lake City. All the consults are done virtually.
For newly diagnosed patients, we would send a consult in within a couple of weeks. For patients who had a family history, the referral would not be urgent: They can be seen within about 3 months. The turnaround times for GMS are so much better than what we have available in the community where it’s often at least 6 months, as previously noted.
Vickie Venne. Thank you. We continue to work on that. One of the interesting things that we’ve done, which is the brainchild of Renee, is shared medical appointments.
Renee Rider. We have now created 4 group appointments for people who have concerns surrounding cancer. One group is for people who don’t have cancer but have family members who have cancer who may be the best testing candidate. For example, that might be a 30-year old who tells you that her mother had breast cancer at age 45 years. Her mother is still living, but she’s never had genetic testing. We would put her in a group where we discuss the importance of talking to the family members and encouraging them to go get that first genetic evaluation in the family.
Our second group is for people who don’t have cancer themselves, but have a family history of cancer and those affected relatives have passed away. The family needs a genetic evaluation, and the veteran is the best living testing candidate.
That group is geared towards education about the test and informed consent.
The third group is for people with cancer who qualify for genetic testing. We provide all of the information that they need to make an informed decision on having (or not having) genetic testing.
The final group is for people who have family histories of known genetic mutations in cancer genes. Again, we provide them with all of the information that they need to make an informed decision regarding genetic testing.
With the shared medical appointments, we have been able to greatly increase the number of patients that we can see. Our first 3 groups all meet once a week and can have 10 or 12 veterans. Our last group meets every other week and has a maximum of 6 veterans. Wait times for our groups are generally ≤ 2 weeks. All veterans can choose to have an individual appointment if they prefer. We regularly get unsolicited feedback from veterans that they learn a lot during our groups and appreciate it.
Our group appointments have lowered the wait time for the people in the groups. And, they’ve lowered the wait time for the people who are seen individually. They’ve allowed us to address the backlog of patients waiting to see us in a more timely manner. Our wait time for individual appointment had been approaching 6 months, and it is now about 1.5 months.
We also think that being in a group normalizes the experience. Most people don’t know anyone who has had genetic testing. Now, they are in a group with others going through the same experience. In one of my groups, a male veteran talked about his breast cancer being really rare. Another male in the group volunteer that he had breast cancer, too. They both seemed to appreciate not feeling alone.
Vickie Venne. I want to move to our final piece. What do the referring providers tell the patients about a genetics referral and what should they expect?
Lisa Arfons. First and foremost, I tell the patient that it is a discussion with a genetic counselor. I make it clear that they understand that it is a discussion. They then can agree or not agree to accept genetic testing if it’s recommended.
I talk in general terms about why I think it can be important for them to have the discussion, but that we don’t have great data for decisionmaking. We understand that there are more options for preventive measures but then it ultimately will be a discussion between the PCP, the patient, and their family members about how they proceed about the preventive measures. I want them to start thinking about how the genetic test results, regardless of if they are positive, negative, or a variant that is not yet understood, can impact their offspring.
Probably I am biased, as my mom had breast cancer and she underwent genetic testing. So, I have a bit of an offspring focus as well. I already mentioned that you must discuss about whether or not it’s worth screening or doing any preventive measures on contralateral breast, or screening for things like prostate cancer at age 75 years. And so I focus more on the family members.
I try to stay in my lane. I am extremely uncomfortable when I hear about someone in our facility sending off a blood test and then asking someone else to interpret the results and discuss it with the patient. Just because it’s a blood test and it’s easy to order doesn’t mean that it is easy to know what to do with it, and it needs to be respected as such.
Ishta Thakar. Our PCPs let the patients know that GMS will contact the patient to schedule a video appointment and that if they want to bring any family members along with them, they’re welcome to. We also explain that certain cancers are genetically based and that if they have a genetic mutation, it can be passed on to their offspring. I also explain that if they have certain mutations, then we would be more vigilant in screening them for other kinds of cancers. That’s the reason that we refer that they get counseled. After counseling if they’re ready for the testing, then the counselor orders the test and does the posttest discussion with the patient.
Vickie Venne. In the VA, people are invited to attend a genetic counseling session but can certainly decline. Does the the DoD have a different approach?
Maj De Castro. I would say that the great majority of active duty patients have limited knowledge of what to expect out of a genetics appointment. One of the main things we do is educate them on their rights and protections and the potential risks associated with performing genetic testing, in particular when it comes to their continued ability to serve. Genetic testing for clinical purposes is not mandatory in the DoD, patients can certainly decline testing. Because genetic testing has the potential to alter someone’s career, it is critical we have a very thorough and comprehensive pre- and posttest counseling sessions that includes everything from career implications to the Genetic Information Nondiscrimination Act (GINA) and genetic discrimination in the military, in addition to the standard of care medical information.
Scenarios in which a servicemember is negatively impacted by pursuing a genetic diagnosis are very rare. More than 90% of the time, genetic counseling and/or testing has no adverse career effect. When they do, it is out of concern for the safety and wellbeing of a servicemember. For instance, if we diagnosis a patient with a genetic form of some arrhythmogenic disorder, part of the treatment plan can be to limit that person’s level of exertion, because it could potentially lead to death. We don’t want to put someone in a situation that may trigger that.
Vickie Venne. We also have a certain number of veterans who ask us about their service disability pay and the impact of genetic testing on it. One example is veterans with prostate cancer who were exposed to Agent Orange, which has been associated with increased risk for developing prostate cancer. I have had men who have been referred for genetic evaluation ask, “Well, if I have an identifiable mutation, how will that impact my service disability?” So we discuss the carcinogenic process that may include an inherited component as well as the environmental risk factors. I think that’s a unique issue for a population we’re honored to be able to serve.
Renee Rider. When we are talking about how the population of veterans is unique, I think it is also important to acknowledge mental health. I’ve had several patients tell me that they have posttraumatic stress disorder or anxiety and the idea of getting an indeterminant test result, such as VUS, would really weigh on them.
In the community, a lot of providers order the biggest panel they can, but for these patients who are worried about getting those indeterminant test results, I’ve been able to work with them to limit the size of the panel. I order a small panel that only has genes that have implications for that veteran’s clinical management. For example, in a patient with ductal breast cancer, I remove the genes that cause lobular breast cancer. This takes a bit of knowledge and critical thinking that our VA genetic counselors have because they have experience with veterans and their needs.
As our time draws to a close, I have one final thought. This has been a heartwarming conversation today. It is really nice to hear that GMS services are appreciated. We in GMS want to partner with our referring providers. Help us help you! When you enter a referral, please let us know how we can help you. The more we understand why you are sending your veteran to GMS, the more we can help meet your needs. If there are any questions or problems, feel free to send us an email or pick up the phone and call us.