User login
New MS subtype shows absence of cerebral white matter demyelination
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
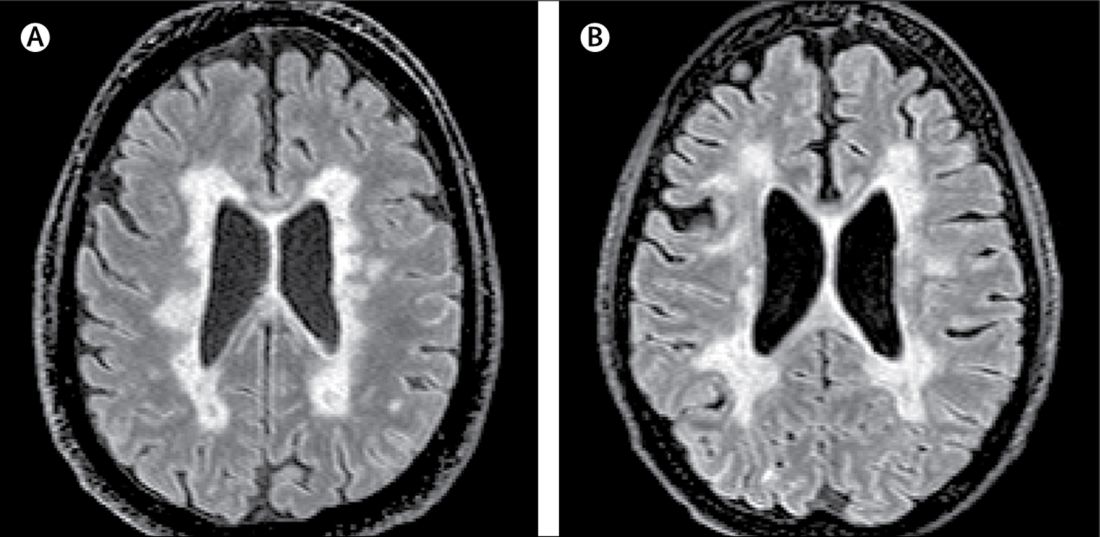
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
A new subtype of multiple sclerosis called myelocortical multiple sclerosis is characterized by demyelination only in the spinal cord and cerebral cortex and not in the cerebral white matter.
A paper published online Aug. 21 in Lancet Neurology presents the results of a study of the brains and spinal cords of 100 patients who died of multiple sclerosis.
Bruce D. Trapp, PhD, of the Lerner Research Institute at the Cleveland Clinic in Ohio, and his coauthors wrote that while the demyelination of cerebral white matter is a pathologic hallmark of multiple sclerosis, previous research has found only around half of cerebral T2-weighted hyperintense white matter lesions are demyelinated, and these lesions account for less than a third of variance in the rate of brain atrophy.
“In the absence of specific MRI metrics for demyelination, the relationship between cerebral white-matter demyelination and neurodegeneration remains speculative,” they wrote.
In this study, researchers scanned the brains with MRI before autopsy, then took centimeter-thick hemispheric slices to study the white-matter lesions. They identified 12 individuals as having what they describe as ‘myelocortical multiple sclerosis,’ characterized by the absence of areas of cerebral white-matter discoloration indicative of demyelinated lesions.
The authors then compared these individuals to 12 individuals with typical multiple sclerosis matched by age, sex, MRI protocol, multiple sclerosis disease subtype, disease duration, and Expanded Disability Status Scale.
They found that while individuals with myelocortical multiple sclerosis did not have demyelinated lesions in the cerebral white matter, they had similar areas of demyelinated lesions in the cerebral cortex to individuals with typical multiple sclerosis (median 4.45% vs. 9.74% respectively, P = .5512).
However, the individuals with myelocortical multiple sclerosis had a significantly smaller area of spinal cord demyelination (median 3.81% vs. 13.81%, P = .0083).
Individuals with myelocortical multiple sclerosis also had significantly lower mean cortical neuronal densities, compared with healthy control brains in layer III, layer V, and layer VI. But individuals with typical multiple sclerosis only had a lower cortical neuronal density in layer V when compared with controls.
Researchers also saw that in typical multiple sclerosis, neuronal density decreased as the area of brain white-matter demyelination increased. However, this negative linear correlation was not seen in myelocortical multiple sclerosis.
On MRI, researchers were still able to see abnormalities in the cerebral white matter in individuals with myelocortical multiple sclerosis, in T2-weighted, T1-weighted and magnetization transfer ratios (MTR) images.
They also found similar total T2-weighted and T1-weighted lesion volumes in individuals with myelocortical and with typical multiple sclerosis, although individuals with typical multiple sclerosis had significantly greater MTR lesion volumes.
“We propose that myelocortical multiple sclerosis is characterized by spinal cord demyelination, subpial cortical demyelination, and an absence of cerebral white-matter demyelination,” the authors wrote. “Our findings indicate that abnormal cerebral white-matter T2-T1-MTR regions of interest are not always demyelinated, and this pathological evidence suggests that cerebral white-matter demyelination and cortical neuronal degeneration can be independent events in myelocortical multiple sclerosis.”
The authors noted that their study may have been affected by selection bias, as all the patients in the study had died from complications of advanced multiple sclerosis. They suggested that it was therefore not appropriate to conclude that the prevalence of myelocortical multiple sclerosis seen in their sample would be similar across the multiple sclerosis population, nor were the findings likely to apply to people with earlier stage disease.
The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, and three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees, and non-financial support from pharmaceutical companies.
SOURCE: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
FROM LANCET NEUROLOGY
Key clinical point: Researchers have identified a new subtype of multiple sclerosis.
Major finding: Individuals with myelocortical multiple sclerosis show demyelination in the spinal cord and cortex only.
Study details: Post-mortem study of brains and spinal cords of 100 individuals with multiple sclerosis.
Disclosures: The study was funded by the U.S. National Institutes of Health and National Multiple Sclerosis Society. One author was an employee of Renovo Neural, three authors were employees of Biogen. One author declared a pending patent related to automated lesion segmentation from MRI images, and four authors declared funding, fees and non-financial support from pharmaceutical companies.
Source: Trapp B et al. Lancet Neurol. 2018 Aug 21. doi: 10.1016/ S1474-4422(18)30245-X.
Signal strength tied to potency of CAR T-cell therapy

Investigators found that chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model.
Intracellular signaling strength was a key determinant of T-cell fate in the study, which was published in Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
These findings suggest tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center in Seattle, Wash.
For this study, Salter and his colleagues used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T-cell signaling pathways, Salter said.
That overlap was somewhat surprising, according to Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor but the speed and strength of signaling, he added.
In particular, the CARs that evoked stronger signals also had increased T-cell dysfunction, decreasing their potency in the mouse model of lymphoma.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model.
By contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, Salter said.
These findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense.
“This is a modification that we think should be considered in future CAR design,” Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at efficacy, noted Stanley Riddell, MD, of Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said.
“But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics. He and Salter have filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
Investigators found that chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model.
Intracellular signaling strength was a key determinant of T-cell fate in the study, which was published in Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
These findings suggest tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center in Seattle, Wash.
For this study, Salter and his colleagues used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T-cell signaling pathways, Salter said.
That overlap was somewhat surprising, according to Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor but the speed and strength of signaling, he added.
In particular, the CARs that evoked stronger signals also had increased T-cell dysfunction, decreasing their potency in the mouse model of lymphoma.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model.
By contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, Salter said.
These findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense.
“This is a modification that we think should be considered in future CAR design,” Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at efficacy, noted Stanley Riddell, MD, of Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said.
“But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics. He and Salter have filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
Investigators found that chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model.
Intracellular signaling strength was a key determinant of T-cell fate in the study, which was published in Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
These findings suggest tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center in Seattle, Wash.
For this study, Salter and his colleagues used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T-cell signaling pathways, Salter said.
That overlap was somewhat surprising, according to Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor but the speed and strength of signaling, he added.
In particular, the CARs that evoked stronger signals also had increased T-cell dysfunction, decreasing their potency in the mouse model of lymphoma.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model.
By contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, Salter said.
These findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense.
“This is a modification that we think should be considered in future CAR design,” Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at efficacy, noted Stanley Riddell, MD, of Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said.
“But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics. He and Salter have filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
Signal strength may limit potency of CAR T-cell therapy
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
FROM SCIENCE SIGNALING
Key clinical point:
Major finding: T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in a mouse model of lymphoma, while the 4-1BB CAR signal led to T cells that better retained their function in vivo and had a longer median survival in the model.
Study details: Analysis of CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells using mass spectrometry, plus analysis of efficacy in a mouse model of lymphoma.
Disclosures: Study authors reported disclosures related to Juno therapeutics and a patent application related to use of mutant CD28 CARs for cellular therapy.
Source: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.

Role of SES in childhood cancer survival disparities

Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
CPI-613 receives orphan designation for PTCL
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
Meta-analysis supports rituximab maintenance in MCL
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
FROM HEMASPHERE
Key clinical point: Rituximab maintenance therapy improves outcomes in patients with MCL.
Major finding: Compared with observation or maintenance interferon-alfa, maintenance rituximab was associated with reduced risk of progression-free survival events (HR, 0.58) and increased risk of grade 3 or 4 neutropenia (RR, 2.02).
Study details: A meta-analysis of six randomized controlled trials including 858 patients with MCL who had a response to induction therapy.
Disclosures: Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
Source: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
Real-world bleeding risk with ibrutinib

The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
Latex Allergy From Biologic Injectable Devices


Phase 1 CAR T trial for NHL launches in Cleveland
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
Key clinical point: A phase 1 trial of CAR T therapy is enrolling adult patients with NHL who have not responded to standard therapies.
Major finding: The trial site has the ability to manufacture the cells on site, saving patients time.
Study details: A phase 1 trial to evaluate safety.
Disclosures: The study will be funded by University Hospitals Seidman Cancer Center.
First CAR T-cell therapy approvals bolster booming immunotherapy market
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.