User login
Spinal Muscular Atrophy Added to Recommended Uniform Screening Panel
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.

Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Screening will enable early detection, but the treatment’s exceptional cost could present a barrier to patients.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.
Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Spinal muscular atrophy (SMA) is now among the disorders officially included in the Recommended Uniform Screening Panel (RUSP), which state public health departments use to screen newborns for genetic disorders.
Secretary of the Department of Health and Human Services (HHS) Alex M. Azar II formally added SMA to the panel on July 2 on the recommendation of the Advisory Committee on Heritable Disorders in Newborns and Children.
“Adding SMA to the list will help ensure that babies born with SMA are identified, so that they have the opportunity to benefit from early treatment and intervention,” according to a statement from the Muscular Dystrophy Association about the decision. “This testing can also provide families with a genetic diagnosis—information that often is required to determine whether their child is eligible to participate in clinical trials.”
Adding SMA to the RUSP does not mean that states must screen newborns for the disorder. Each state’s public health apparatus decides independently whether to accept the recommendation and which disorders on the RUSP to screen for. Most states screen for most disorders on the RUSP. Evidence compiled by the advisory committee suggested a wide variation in resources, infrastructure, funding, and time to implementation among states.
Drug Approval Raised Ethical Questions
An estimated one in 11,000 newborns has SMA, a disorder caused by mutations in the survival motor neuron 1 (SMN1) gene. SMA affects motor neurons in the brainstem and spinal cord, thus leading to motor weakness and atrophy. The only treatment for SMA had been palliative care until the FDA approved nusinersen for the disorder in December 2016. The drug’s approval has raised ethical questions.1–3
After reviewing the evidence at its February 8 meeting, the advisory committee recommended adding SMA screening to the RUSP in a March 8 letter from committee chair Joseph A. Bocchini Jr, MD, Professor and Chair of Pediatrics at Louisiana State University Health in Shreveport.
Secretary Azar accepted the recommendation based on the evidence the committee provided; he also requested a follow-up report within two years “describing the status of implementing newborn screening for SMA and clinical outcomes of early treatment, including any potential harms, for infants diagnosed with SMA.”
The advisory committee makes its recommendations to HHS about which heritable disorders to include in the RUSP after it has assessed a systematic, evidence-based review conducted by an external, independent group. Alex R. Kemper, MD, MPH, Professor of Pediatrics at the Ohio State University and Division Chief of Ambulatory Pediatrics at Nationwide Children’s Hospital, both in Columbus, led the review group for SMA. Dr. Kemper is also deputy editor of Pediatrics and a member of the US Preventive Services Task Force.
According to Secretary Azar’s summary in his July 2 letter of acceptance, the evidence review suggested that “early screening and treatment can lead to decreased mortality for individuals with SMA and improved motor milestones.”
“SMA can be detected through newborn screening, and treatment is now available that can not only reduce the risk of death, but decrease the development of neurologic impairment,” he said in an interview. “As with adding any condition to newborn screening, public health laboratories will need to develop strategies to incorporate the screening test. The current FDA-approved treatment, nusinersen, is delivered by lumbar puncture into the spinal fluid. In addition, there are exciting advances in gene therapy leading to new treatment approaches.”
Symptom Onset Distinguishes the Types of SMA
Approximately 95% of SMA cases result from the deletion of exon 7 from both alleles of SMN1. Other, rarer cases are caused by mutations in different genes. Without the SMN protein produced by SMN1, a person gradually loses muscle function.
A similar gene, SMN2, also can produce the SMN protein, but in much lower amounts—typically less than 10% of what a person needs. People can, however, have multiple copies of SMN2, which can produce slightly more SMN protein and slow the disease process.
The five types of SMA are determined according to symptom onset, which directly correlates with disorder severity and prognosis. Approximately 54% of SMA cases are Type I, in which progressive weakness occurs over the first six months of life and results in early death. Only 18% of children with Type I live past age 4, and 68% die by age 2. Type 0 is rarer, but more severe, usually causing fetal loss or early infant death.
Type II represents 18% of SMA cases and causes progressive weakness by age 15 months. Most people with Type II survive to their 30s but later experience respiratory failure and rarely reach their 40s. Individuals with Types III and IV typically have a normal lifespan and only begin to see progressive muscle weakness after age 1 or in adulthood.
Dr. Kemper’s group focused on the three types diagnosed in infancy: Types I, II, and III. “It will be critical to make sure that infants diagnosed with SMA through newborn screening receive follow-up shortly afterward to determine whether they would benefit from nusinersen,” said Dr. Kemper. “More information is needed about the long-term outcomes of those infants who begin treatment following newborn screening, so we not only know about outcomes in later childhood and adolescence, but treatment approaches can be further refined and personalized.”
Long-Term Data on Nusinersen Are Lacking
Nusinersen alters the splicing of precursor messenger RNA in SMN2 so that the mRNA strands are longer, which increases the amount of SMN protein produced. Concerns about the medication, however, have included its cost—$750,000 in the first year and $375,000 every following year for life—and potential adverse events from repeated administration. Nusinersen is injected into the spinal canal four times in the first year and once annually thereafter, and the painful injections require patient immobilization. Potential adverse events include thrombocytopenia and nephrotoxicity, along with potential complications from repeated lumbar punctures over time.2
Other concerns about the drug include its limited evidence base, lack of long-term data, associated costs with administration (eg, travel costs), the potential for patients taking nusinersen to be excluded from future clinical trials on other treatments, and ensuring parents have enough information on the drug’s limitations and potential risks to provide adequate informed consent.2
Yet evidence to date is favorable in children with early onset SMA. Dr. Bocchini wrote in the letter to Secretary Azar that “limited data suggest that treatment effect is greater when the treatment is initiated before symptoms develop and when the individual has more copies of SMN2.”
Dr. Kemper’s group concluded that screening can detect SMA in newborns and that treatment can modify the disease course. “Grey literature suggests those with total disease duration less than or equal to 12 weeks before nusinersen treatment were more likely to have better outcomes than those with longer periods of disease duration.
“Presymptomatic treatment alters the natural history” of the disorder, the group found, although outcome data past age 1 are not yet available. Based on findings from a New York pilot program, they predicted that nationwide newborn screening would avert 33 deaths and 48 cases of children who were dependent on a ventilator among an annual cohort of four million births.
At the time of the evidence review, Massachusetts, Minnesota, Missouri, North Carolina, New York, Utah, and Wisconsin initiated pilot programs or whole-population mandated screening for SMA. Of the three states that reported costs, all reported costs of $1 or less per screen.
The research for the evidence review was funded by a Health Resources and Services Administration grant to Duke University in Durham, North Carolina. No disclosures were provided for evidence review group members.
—Tara Haelle
References
1. King NMP, Bishop CE. New treatments for serious conditions: ethical implications. Gene Ther. 2017;24(9):534-538.
2. Gerrity MS, Prasad V, Obley AJ. Concerns about the approval of nusinersen sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178(6):743-744.
3. Burgart AM, Magnus D, Tabor HK, et al. Ethical challenges confronted when providing nusinersen treatment for spinal muscular atrophy. JAMA Pediatr. 2018;172(2):188-192.
Does TBI Increase the Risk of Suicide?
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.

A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Compared with the general population, people who seek medical attention for TBI may have almost twice the risk of suicide.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.
A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
Residents of Denmark who seek medical attention for traumatic brain injury (TBI) have an increased risk of suicide, compared with the general Danish population without TBI, according to a study published in the August 14 issue of JAMA. “Additional analyses revealed that the risk of suicide was higher for individuals with severe TBI, numerous medical contacts, and longer hospital stays,” said lead author Trine Madsen, PhD. Individuals were at highest risk in the first six months after discharge, said Dr. Madsen, who is a postdoctoral fellow at the Danish Research Institute for Suicide Prevention in Hellerup.
A history of TBI previously has been associated with higher rates of self-harm, suicide, and death than are found in the general population. However, previous studies have been limited by methodological shortcomings, such as small sample sizes and low numbers of suicide cases with TBI. Dr. Madsen and colleagues conducted a retrospective cohort study using nationwide registers covering 7,418,391 individuals living in Denmark between 1980 and 2014 with 164,265,624 person-years’ follow-up. Of these people, 567,823 (7.6%) had a medical contact for TBI, which included mild TBI (ie, concussion), skull fracture without documented TBI, and severe TBI (ie, head injuries with evidence of structural brain injury).
Of 34,529 individuals who died by suicide, 3,536 (10.2%) had medical contact for TBI, including 2,701 for mild TBI, 174 for skull fracture without documented TBI, and 661 for severe TBI. The absolute suicide rate was 41 per 100,000 person-years among those with TBI versus 20 per 100,000 person-years among those with no diagnosis of TBI. After accounting for relevant covariates such as fractures not involving the skull, psychiatric diagnoses, and deliberate self-harm, the adjusted incidence ratio was 1.90.
This study “provides insights into the underappreciated relationship between TBI and suicide,” said Lee Goldstein, MD, PhD, and Ramon Diaz-Arrastia, MD, PhD, in an accompanying editorial. “The results … point to an important clinical triad—TBI history, recent injury (especially with long hospital stays), and more numerous postinjury medical contacts for TBI—that serves as a red flag for increased suicide risk,” said Dr. Goldstein, who is affiliated with Boston University School of Medicine, and Dr. Diaz-Arrastia, of the University of Pennsylvania’s Perelman School of Medicine in Philadelphia. The results “indicate that increased suicide risk is relevant across all TBI severity levels, including the far more common mild injuries. Clinicians, health care professionals, and mental health practitioners must take notice of this important information.”
—Glenn S. Williams
Suggested Reading
Goldstein L, Diaz-Arrastia R. Traumatic brain injury and risk of suicide. JAMA. 2018;320(6):554-556.
Madsen T, Erlangsen A, Orlovska S, et al. Association between traumatic brain injury and risk of suicide. JAMA. 2018;320(6):580-588.
NICE says CAR T-cell therapy isn’t cost-effective

The National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of axicabtagene ciloleucel (Yescarta) in England.
Axicabtagene ciloleucel is a chimeric antigen receptor (CAR) T-cell therapy that was just approved by the European Commission to treat patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal B-cell lymphoma (PMBCL) who have received two or more lines of systemic therapy.
However, NICE has said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy.
Additionally, the cost of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of National Health Service (NHS) resources.
NICE’s draft guidance points out that there is no standard treatment for patients with relapsed or refractory DLBCL or PMBCL who have received two or more systemic therapies. These patients receive best supportive care, which usually includes salvage chemotherapy.
Results from the ZUMA-1 trial suggest the majority of DLBCL/PMBCL patients given axicabtagene ciloleucel do respond to treatment.
However, there is no direct data comparing axicabtagene ciloleucel with salvage chemotherapy, so the benefit of the CAR T-cell therapy over chemotherapy is unknown.
The draft guidance also notes that axicabtagene ciloleucel meets NICE’s criteria to be considered a life-extending treatment at the end of life.
However, the CAR T-cell therapy cannot be considered a cost-effective use of NHS resources. The cost-effectiveness estimates for axicabtagene ciloleucel, compared with salvage chemotherapy, were above £50,000 per year of quality adjusted life gained, the upper limit of the specially extended range of cost-effectiveness for cancer treatments.
Furthermore, axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund. NICE said axicabtagene ciloleucel does not have the plausible potential to be cost effective, which would be necessary for inclusion in the fund while further evidence of the treatment’s longer-term benefits is collected.
“Although promising, there is still much more we need to know about CAR-T, and, unfortunately, in this case, we are not able to recommend axicabtagene ciloleucel for use in the NHS in England at the cost per patient set by Kite Pharma,” said Meindert Boysen, director of the centre for health technology evaluation at NICE.
The consultation period for the draft guidance runs until September 18, 2018.
The National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of axicabtagene ciloleucel (Yescarta) in England.
Axicabtagene ciloleucel is a chimeric antigen receptor (CAR) T-cell therapy that was just approved by the European Commission to treat patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal B-cell lymphoma (PMBCL) who have received two or more lines of systemic therapy.
However, NICE has said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy.
Additionally, the cost of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of National Health Service (NHS) resources.
NICE’s draft guidance points out that there is no standard treatment for patients with relapsed or refractory DLBCL or PMBCL who have received two or more systemic therapies. These patients receive best supportive care, which usually includes salvage chemotherapy.
Results from the ZUMA-1 trial suggest the majority of DLBCL/PMBCL patients given axicabtagene ciloleucel do respond to treatment.
However, there is no direct data comparing axicabtagene ciloleucel with salvage chemotherapy, so the benefit of the CAR T-cell therapy over chemotherapy is unknown.
The draft guidance also notes that axicabtagene ciloleucel meets NICE’s criteria to be considered a life-extending treatment at the end of life.
However, the CAR T-cell therapy cannot be considered a cost-effective use of NHS resources. The cost-effectiveness estimates for axicabtagene ciloleucel, compared with salvage chemotherapy, were above £50,000 per year of quality adjusted life gained, the upper limit of the specially extended range of cost-effectiveness for cancer treatments.
Furthermore, axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund. NICE said axicabtagene ciloleucel does not have the plausible potential to be cost effective, which would be necessary for inclusion in the fund while further evidence of the treatment’s longer-term benefits is collected.
“Although promising, there is still much more we need to know about CAR-T, and, unfortunately, in this case, we are not able to recommend axicabtagene ciloleucel for use in the NHS in England at the cost per patient set by Kite Pharma,” said Meindert Boysen, director of the centre for health technology evaluation at NICE.
The consultation period for the draft guidance runs until September 18, 2018.
The National Institute for Health and Care Excellence (NICE) has issued a draft guidance recommending against the use of axicabtagene ciloleucel (Yescarta) in England.
Axicabtagene ciloleucel is a chimeric antigen receptor (CAR) T-cell therapy that was just approved by the European Commission to treat patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) or primary mediastinal B-cell lymphoma (PMBCL) who have received two or more lines of systemic therapy.
However, NICE has said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy.
Additionally, the cost of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of National Health Service (NHS) resources.
NICE’s draft guidance points out that there is no standard treatment for patients with relapsed or refractory DLBCL or PMBCL who have received two or more systemic therapies. These patients receive best supportive care, which usually includes salvage chemotherapy.
Results from the ZUMA-1 trial suggest the majority of DLBCL/PMBCL patients given axicabtagene ciloleucel do respond to treatment.
However, there is no direct data comparing axicabtagene ciloleucel with salvage chemotherapy, so the benefit of the CAR T-cell therapy over chemotherapy is unknown.
The draft guidance also notes that axicabtagene ciloleucel meets NICE’s criteria to be considered a life-extending treatment at the end of life.
However, the CAR T-cell therapy cannot be considered a cost-effective use of NHS resources. The cost-effectiveness estimates for axicabtagene ciloleucel, compared with salvage chemotherapy, were above £50,000 per year of quality adjusted life gained, the upper limit of the specially extended range of cost-effectiveness for cancer treatments.
Furthermore, axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund. NICE said axicabtagene ciloleucel does not have the plausible potential to be cost effective, which would be necessary for inclusion in the fund while further evidence of the treatment’s longer-term benefits is collected.
“Although promising, there is still much more we need to know about CAR-T, and, unfortunately, in this case, we are not able to recommend axicabtagene ciloleucel for use in the NHS in England at the cost per patient set by Kite Pharma,” said Meindert Boysen, director of the centre for health technology evaluation at NICE.
The consultation period for the draft guidance runs until September 18, 2018.
European Commission approves first CAR T-cell therapies
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
The , two chimeric antigen receptor (CAR) T-cell therapies.
Tisagenlecleucel is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
Axicabtagene ciloleucel is approved for adults with relapsed/refractory DLBCL and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy. The treatment is marketed by Kite, a Gilead company.
The axicabtagene ciloleucel approval is based on results from the single arm, ZUMA-1 trial. During the study of 101 patients who received a single infusion, 72% responded to therapy and 51% achieved a complete response. At 1 year, median overall survival had not been reached.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
Updated results from JULIET were presented at the annual congress of the European Hematology Association in June 2018. The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so because of disease progression or clinical deterioration.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
Updated results from ELIANA were published in New England Journal of Medicine (2018;378:439-48).
The trial included 75 children and young adults with relapsed/refractory ALL. The overall remission rate was 81% (61/75), with 60% of patients (n = 45) achieving a complete remission (CR) and 21% (n = 16) achieving a CR with incomplete hematologic recovery (CRi).
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Serum Nf-L shows promise as biomarker for BMT response in MS
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.

Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.
Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
LOS ANGELES – Serum neurofilament light chain shows promise as a biomarker for disease severity and response to treatment with autologous bone marrow transplant in patients with multiple sclerosis, according to findings from an analysis of paired samples.
Serum neurofilament light chain (Nf-L) levels were found to be significantly elevated in both the serum and cerebrospinal fluid (CSF) of 23 patients with aggressive multiple sclerosis (MS), compared with samples from 5 controls with noninflammatory neurologic conditions such as chronic fatigue syndrome or migraine. The levels in the patients with MS were significantly reduced following autologous bone marrow transplant (BMT), Simon Thebault, MD, reported in a poster at the annual meeting of the American Academy of Neurology.
Nf-L has been shown to be a biomarker for neuronal damage and to have potential for denoting disease severity and treatment response in MS; for this analysis, samples from patients were collected at baseline and annually for 3 years after transplant. Samples from controls were obtained for comparison.
“Our objective, really, was [to determine if we could] detect a treatment response in neurofilament light chain, and secondly, did the degree of neurofilament light chain being high at baseline predict anything about subsequent disease outcome?” Dr. Thebault, a third-year resident at the University of Ottawa, reported in the poster.
Indeed, a treatment response was detected; baseline levels were high, and levels in both serum and CSF were down significantly (P = .05) at both 1 and 3 years following bone marrow transplant, and they stayed down. “In fact, they were so low, they were not significantly different from [the levels] in noninflammatory controls,” he said, noting that serum and CSF NfL levels were highly correlated (r = .833; P less than .001).
Study subjects were patients with aggressive MS, characterized by early disease onset, rapid progression, frequent relapses, and poor treatment responses. Such patients who have received autologous BMT represent an interesting group for examining treatment responses, he said.
At baseline, these patients presumably have a high burden of tissue injury, which would be representative of high levels of Nf-L; good, prospective data show these patients have no evidence of ongoing inflammatory disease following an intensive regimen that includes chemoablation and reinfusion of autologous stem cells, which is representative of a significant reduction of neurofilament levels, he explained.
Importantly, serum and CSF levels were correlated in this study, he said, noting that most prior work has involved CSF, but “the real clinical utility in neurofilament light chain is probably in the serum, because patients don’t like having lumbar punctures too often.”
With respect to the second question pertaining to disease outcomes, the study did show that patients with baseline Nf-L levels above the median for the group had worse T1 lesion volumes at baseline and after transplant (P = .0021 at 36 months), and also had worsening brain atrophy even after transplant (P = .0389 at 60 months).
The curves appeared to be diverging, suggesting that, in the absence of inflammatory disease activity, patients with high Nf-L levels at baseline continue to have ongoing atrophy at a differential rate, compared with those with low baseline levels, Dr. Thebault said.
Other findings included better Expanded Disability Status Scale outcomes after transplant in patients with lower baseline Nf-L, and a trend toward worsening outcomes among those with higher baseline Nf-L levels, although the study may have been underpowered for this. Additionally, lower baseline Nf-L predicted significantly lower T1 and T2 lesion volume, number of gadolinium-enhancing lesions, and a reduction in brain atrophy, whereas higher baseline Nf-L predicted the opposite, he noted, adding that N-acetylaspartate-to-creatinine ratios also tended to vary inversely with Nf-L levels.
The most important findings of the study are “the sustained reduction in Nf-L levels after BMT, and that higher baseline levels predicted worse MRI outcomes,” Dr. Thebault noted. “Together these data add to a growing body of evidence that suggests that serum Nf-L has a role in monitoring treatment responses and even a predictive value in determining disease severity.”
This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
SOURCE: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
REPORTING FROM AAN 2018
Key clinical point:
Major finding: Serum and cerebrospinal fluid Nf-L levels declines significantly after bone marrow transplant (P less than .05) and did not differ from the levels in controls.
Study details: An analysis of paired samples from 23 patients with multiple sclerosis and 5 controls.
Disclosures: This study was supported by Quanterix, which provided Nf-L assay kits. Dr. Thebault reported having no disclosures.
Source: Thebault S et al. Neurology. 2018 Apr;90(15 Suppl.):P5.036.
EC approves CAR T-cell therapy for DLBCL, PMBCL

The European Commission (EC) has approved the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®) to treat two types of lymphoma.
Axicabtagene ciloleucel is now approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy.
The approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival rate was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome.
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher cytokine release syndrome occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Commission (EC) has approved the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®) to treat two types of lymphoma.
Axicabtagene ciloleucel is now approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy.
The approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival rate was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome.
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher cytokine release syndrome occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
The European Commission (EC) has approved the chimeric antigen receptor (CAR) T-cell therapy axicabtagene ciloleucel (Yescarta®) to treat two types of lymphoma.
Axicabtagene ciloleucel is now approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) and primary mediastinal large B-cell lymphoma (PMBCL) after two or more lines of systemic therapy.
The approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
The EC’s approval of axicabtagene ciloleucel is supported by data from the ZUMA-1 trial.
Results from this phase 2 trial were presented at the 2017 ASH Annual Meeting and published simultaneously in NEJM.
The trial enrolled 111 patients with relapsed/refractory B-cell lymphomas. There were 101 patients who received axicabtagene ciloleucel—77 with DLBCL, 8 with PMBCL, and 16 with transformed follicular lymphoma (TFL).
Patients received conditioning with low-dose cyclophosphamide and fludarabine, followed by axicabtagene ciloleucel.
The objective response rate (ORR) was 82% (n=83), and the complete response (CR) rate was 54% (n=55).
Among the DLBCL patients, the ORR was 82% (63/77), and the CR rate was 49% (38/77). In the patients with PMBCL or TFL, the ORR was 83% (20/24), and the CR rate was 71% (17/24).
With a median follow-up of 15.4 months, 42% of patients retained their response, and 40% retained a CR.
At 18 months, the overall survival rate was 52%. Most deaths were due to disease progression.
However, 2 patients died of adverse events related to axicabtagene ciloleucel, both cytokine release syndrome.
The most common grade 3 or higher adverse events were neutropenia (78%), anemia (43%), thrombocytopenia (38%), and febrile neutropenia (31%).
Grade 3 or higher cytokine release syndrome occurred in 13% of patients, and grade 3 or higher neurologic events occurred in 28%.
ctDNA predicts early outcomes in DLBCL
Measurement of circulating tumor DNA (ctDNA) could be a new and useful tool for predicting survival outcomes and response to therapy in patients with diffuse large B-cell lymphoma (DLBCL), according to authors of a recent prospective study.
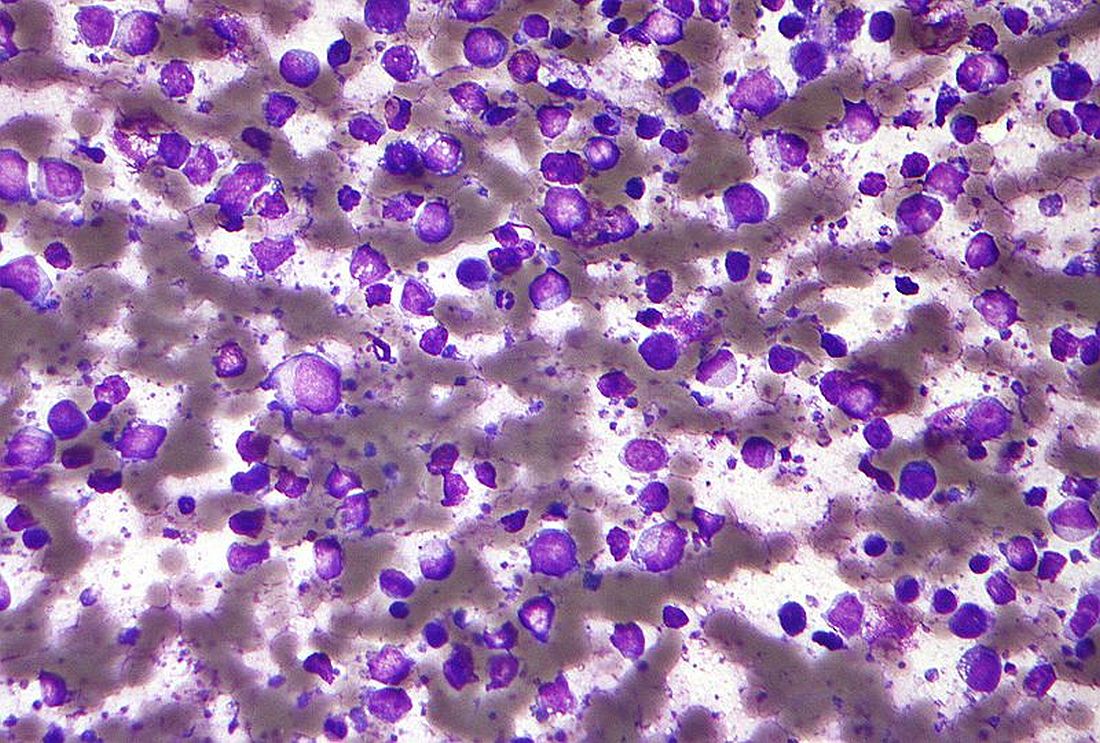
Pretreatment ctDNA levels predicted 24-month event-free survival – an important disease milestone in DLBCL – as well as overall survival in the study, which included more than 200 patients at six institutions in North America and Europe.
Changes in ctDNA during treatment were prognostic for outcomes as early as 21 days into therapy, according to corresponding author Ash A. Alizadeh, MD, PhD, of Stanford (Calif.) University and his coinvestigators.
“Our data suggest that both pretreatment and dynamic assessments of ctDNA are feasible and can add to established risk factors,” Dr. Alizadeh and his coauthors reported in the Journal of Clinical Oncology.
ctDNA was detected in 98% of the 217 patients evaluated, which demonstrated the “potentially universal applicability” of this approach, they wrote in the report.
In an evaluation of pretreatment ctDNA levels, investigators found a 2.5 log haploid genome equivalents per milliliter (hGE/mL) threshold stratified patient outcomes. Event-free survival was significantly inferior at 24 months in patients with ctDNA above that threshold, with hazard ratios of 2.6 (P = .007) for frontline treatment and 2.9 (P = 0.01) for salvage.
On-treatment ctDNA levels were favorably prognostic for outcomes in patients receiving frontline therapy, according to investigators. An early molecular response (EMR), defined as a 2-log decrease in ctDNA levels after one cycle of treatment, was associated with a 24-month event-free survival of 83% versus 50% for no EMR (P = .0015).
Major molecular response (MMR), defined as a 2.5-log drop in ctDNA after two cycles of treatment, was associated with a 24-month event-free survival of 82% versus 46% for no MMR in patients on frontline therapy (P less than .001).
In one cohort of patients receiving salvage therapy, EMR also predicted superior 24-month event-free survival, according to investigators.
The EMR measure was also favorably prognostic for overall survival in both the frontline and salvage settings.
The prognostic value of measuring ctDNA was independent of International Prognostic Index (IPI) and interim PET/CT studies, results of multivariable analysis showed.
Patients had “excellent outcomes” if they had both molecular response and favorable interim PET results, and conversely, patients were at “extremely high risk” for treatment failure if they had no molecular response and a positive PET scan.
“The identification of patients at exceptionally high risk (i.e., interim PET/CT positive and not achieving EMR/MMR) could provide an opportunity for early intervention with alternative treatments, including autologous bone marrow transplantation or chimeric antigen receptor T cells,” the researchers wrote.
Patients in the study were all treated with combination immunochemotherapy according to local standards.
Dr. Alizadeh reported disclosures related to CiberMed, Forty Seven, Janssen Oncology, Celgene, Roche/Genentech, and Gilead, as well as patent filings on ctDNA detection assigned to Stanford University.
SOURCE: Kurtz DM et al. J Clin Oncol. 2018 Aug 20. doi: 10.1200/JCO.2018.78.5246.
Measurement of circulating tumor DNA (ctDNA) could be a new and useful tool for predicting survival outcomes and response to therapy in patients with diffuse large B-cell lymphoma (DLBCL), according to authors of a recent prospective study.
Pretreatment ctDNA levels predicted 24-month event-free survival – an important disease milestone in DLBCL – as well as overall survival in the study, which included more than 200 patients at six institutions in North America and Europe.
Changes in ctDNA during treatment were prognostic for outcomes as early as 21 days into therapy, according to corresponding author Ash A. Alizadeh, MD, PhD, of Stanford (Calif.) University and his coinvestigators.
“Our data suggest that both pretreatment and dynamic assessments of ctDNA are feasible and can add to established risk factors,” Dr. Alizadeh and his coauthors reported in the Journal of Clinical Oncology.
ctDNA was detected in 98% of the 217 patients evaluated, which demonstrated the “potentially universal applicability” of this approach, they wrote in the report.
In an evaluation of pretreatment ctDNA levels, investigators found a 2.5 log haploid genome equivalents per milliliter (hGE/mL) threshold stratified patient outcomes. Event-free survival was significantly inferior at 24 months in patients with ctDNA above that threshold, with hazard ratios of 2.6 (P = .007) for frontline treatment and 2.9 (P = 0.01) for salvage.
On-treatment ctDNA levels were favorably prognostic for outcomes in patients receiving frontline therapy, according to investigators. An early molecular response (EMR), defined as a 2-log decrease in ctDNA levels after one cycle of treatment, was associated with a 24-month event-free survival of 83% versus 50% for no EMR (P = .0015).
Major molecular response (MMR), defined as a 2.5-log drop in ctDNA after two cycles of treatment, was associated with a 24-month event-free survival of 82% versus 46% for no MMR in patients on frontline therapy (P less than .001).
In one cohort of patients receiving salvage therapy, EMR also predicted superior 24-month event-free survival, according to investigators.
The EMR measure was also favorably prognostic for overall survival in both the frontline and salvage settings.
The prognostic value of measuring ctDNA was independent of International Prognostic Index (IPI) and interim PET/CT studies, results of multivariable analysis showed.
Patients had “excellent outcomes” if they had both molecular response and favorable interim PET results, and conversely, patients were at “extremely high risk” for treatment failure if they had no molecular response and a positive PET scan.
“The identification of patients at exceptionally high risk (i.e., interim PET/CT positive and not achieving EMR/MMR) could provide an opportunity for early intervention with alternative treatments, including autologous bone marrow transplantation or chimeric antigen receptor T cells,” the researchers wrote.
Patients in the study were all treated with combination immunochemotherapy according to local standards.
Dr. Alizadeh reported disclosures related to CiberMed, Forty Seven, Janssen Oncology, Celgene, Roche/Genentech, and Gilead, as well as patent filings on ctDNA detection assigned to Stanford University.
SOURCE: Kurtz DM et al. J Clin Oncol. 2018 Aug 20. doi: 10.1200/JCO.2018.78.5246.
Measurement of circulating tumor DNA (ctDNA) could be a new and useful tool for predicting survival outcomes and response to therapy in patients with diffuse large B-cell lymphoma (DLBCL), according to authors of a recent prospective study.
Pretreatment ctDNA levels predicted 24-month event-free survival – an important disease milestone in DLBCL – as well as overall survival in the study, which included more than 200 patients at six institutions in North America and Europe.
Changes in ctDNA during treatment were prognostic for outcomes as early as 21 days into therapy, according to corresponding author Ash A. Alizadeh, MD, PhD, of Stanford (Calif.) University and his coinvestigators.
“Our data suggest that both pretreatment and dynamic assessments of ctDNA are feasible and can add to established risk factors,” Dr. Alizadeh and his coauthors reported in the Journal of Clinical Oncology.
ctDNA was detected in 98% of the 217 patients evaluated, which demonstrated the “potentially universal applicability” of this approach, they wrote in the report.
In an evaluation of pretreatment ctDNA levels, investigators found a 2.5 log haploid genome equivalents per milliliter (hGE/mL) threshold stratified patient outcomes. Event-free survival was significantly inferior at 24 months in patients with ctDNA above that threshold, with hazard ratios of 2.6 (P = .007) for frontline treatment and 2.9 (P = 0.01) for salvage.
On-treatment ctDNA levels were favorably prognostic for outcomes in patients receiving frontline therapy, according to investigators. An early molecular response (EMR), defined as a 2-log decrease in ctDNA levels after one cycle of treatment, was associated with a 24-month event-free survival of 83% versus 50% for no EMR (P = .0015).
Major molecular response (MMR), defined as a 2.5-log drop in ctDNA after two cycles of treatment, was associated with a 24-month event-free survival of 82% versus 46% for no MMR in patients on frontline therapy (P less than .001).
In one cohort of patients receiving salvage therapy, EMR also predicted superior 24-month event-free survival, according to investigators.
The EMR measure was also favorably prognostic for overall survival in both the frontline and salvage settings.
The prognostic value of measuring ctDNA was independent of International Prognostic Index (IPI) and interim PET/CT studies, results of multivariable analysis showed.
Patients had “excellent outcomes” if they had both molecular response and favorable interim PET results, and conversely, patients were at “extremely high risk” for treatment failure if they had no molecular response and a positive PET scan.
“The identification of patients at exceptionally high risk (i.e., interim PET/CT positive and not achieving EMR/MMR) could provide an opportunity for early intervention with alternative treatments, including autologous bone marrow transplantation or chimeric antigen receptor T cells,” the researchers wrote.
Patients in the study were all treated with combination immunochemotherapy according to local standards.
Dr. Alizadeh reported disclosures related to CiberMed, Forty Seven, Janssen Oncology, Celgene, Roche/Genentech, and Gilead, as well as patent filings on ctDNA detection assigned to Stanford University.
SOURCE: Kurtz DM et al. J Clin Oncol. 2018 Aug 20. doi: 10.1200/JCO.2018.78.5246.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: Early molecular response (a 2-log decrease in ctDNA levels after one treatment cycle) was associated with a 24-month event-free survival of 83% versus 50% for no early molecular response (P = .0015).
Study details: Prospective analysis of pretreatment and dynamic on-treatment ctDNA levels in patients with DLBCL who received standard immunochemotherapy.
Disclosures: Study authors reported disclosures related to CiberMed, Forty Seven, Janssen Oncology, Celgene, Roche/Genentech, and Gilead, among others.
Source: Kurtz DM et al. J Clin Oncol. 2018 Aug 20. doi: 10.1200/JCO.2018.78.5246.
EC approves CAR T-cell therapy for ALL, DLBCL

The European Commission (EC) has granted approval for tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy targeting CD19.
Tisagenlecleucel (formerly CTL019) is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
The EC’s approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
JULIET trial
Updated results from JULIET were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached.
At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least one AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Commission (EC) has granted approval for tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy targeting CD19.
Tisagenlecleucel (formerly CTL019) is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
The EC’s approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
JULIET trial
Updated results from JULIET were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached.
At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least one AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
The European Commission (EC) has granted approval for tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy targeting CD19.
Tisagenlecleucel (formerly CTL019) is now approved for use in pediatric and young adult patients up to 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
Tisagenlecleucel is also approved to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
The EC’s approval extends to all member countries of the European Union, as well as Norway, Iceland, and Liechtenstein.
Novartis expects to launch tisagenlecleucel initially for pediatric ALL. The company said timing for tisagenlecleucel availability in each country will depend on multiple factors, including the onboarding of qualified treatment centers for the appropriate indications, as well as the completion of national reimbursement procedures.
The EC’s approval of tisagenlecleucel is based on results from the phase 2 JULIET and ELIANA trials.
JULIET trial
Updated results from JULIET were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
The trial enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Most of the patients who discontinued before dosing did so due to disease progression or clinical deterioration. The patients’ median age at baseline was 56 (range, 22-76).
Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The median time from infusion to data cutoff was 13.9 months.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached.
At the time of data cutoff, none of the responders had gone on to receive a stem cell transplant.
For all infused patients (n=111), the 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Fifteen percent of patients received tocilizumab for CRS, including 3% of patients with grade 2 CRS and 50% of patients with grade 3 CRS.
Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
ELIANA trial
Updated results from ELIANA were published in NEJM in February.
The trial included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All 75 patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi.
All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to transplant while in remission. At last follow-up, 4 were still in remission, and 4 had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
All patients experienced at least one AE, and 95% had AEs thought to be related to tisagenlecleucel. The rate of grade 3/4 AEs was 88%, and the rate of related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
FDA approves new use for ibrutinib in WM

The US Food and Drug Administration (FDA) has approved ibrutinib (Imbruvica®) for use in combination with rituximab to treat adults with Waldenström’s macroglobulinemia (WM).
This is the ninth FDA approval for ibrutinib and the second approval for the drug in WM.
The latest approval was supported by the phase 3 iNNOVATE trial, in which researchers compared ibrutinib plus rituximab to rituximab alone in patients with previously untreated or relapsed/refractory WM.
Results from iNNOVATE were presented at the 2018 ASCO Annual Meeting (abstract 8003) and simultaneously published in NEJM.
The trial enrolled 150 patients. They received rituximab at 375 mg/m2 with weekly infusions at weeks 1 to 4 and 17 to 20. They also received either ibrutinib (420 mg) or placebo once daily continuously until criteria for permanent discontinuation were met.
Overall response rates were significantly higher in the ibrutinib arm than the placebo arm—92% and 47%, respectively (P<0.0001). Complete response rates were 3% and 1%, respectively.
The median time to next treatment was not reached for the ibrutinib arm and was 18 months for the placebo arm (hazard ratio=0.096; P<0.0001). Of the patients randomized to ibrutinib plus rituximab, 75% continued on treatment at last follow-up.
The 30-month progression-free survival rates were 82% in the ibrutinib arm and 28% in the placebo arm. The median progression-free survival was not reached in the ibrutinib arm and was 20.3 months in the placebo arm (hazard ratio=0.20; P<0.0001).
The 30-month overall survival rates were 94% in the ibrutinib arm and 92% in the placebo arm.
Grade 3 or higher treatment-emergent adverse events (AEs) occurred in 60% of patients in the ibrutinib arm and 61% in the placebo arm. Serious AEs occurred in 43% and 33%, respectively.
There were no fatal AEs in the ibrutinib arm and 3 in the rituximab arm.
Grade 3 or higher AEs that occurred more frequently in the ibrutinib arm than the placebo arm included atrial fibrillation (12% vs 1%) and hypertension (13% vs 4%).
AEs that occurred less frequently in the ibrutinib arm than the placebo arm included grade 3 or higher infusion reactions (1% vs 16%) and any-grade IgM flare (8% vs 47%).
About ibrutinib
Ibrutinib is a Bruton’s tyrosine kinase inhibitor that is FDA-approved to treat chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), chronic graft-versus-host disease (cGVHD), and WM.
In November 2013, ibrutinib was approved to treat adults with MCL who have received at least one prior therapy.
In February 2014, ibrutinib was approved to treat adults with CLL who have received at least one prior therapy. In July 2014, ibrutinib received approval for adult CLL patients with 17p deletion, and, in March 2016, ibrutinib was approved as a frontline CLL treatment.
Ibrutinib was approved for use as a single agent in adults with WM in January 2015.
In May 2016, ibrutinib was approved in combination with bendamustine and rituximab for adults with previously treated CLL/SLL.
In January 2017, ibrutinib was approved for adults with MZL who require systemic therapy and have received at least one prior anti-CD20-based therapy.
In August 2017, ibrutinib was approved to treat adults with cGVHD that did not respond to one or more lines of systemic therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved ibrutinib (Imbruvica®) for use in combination with rituximab to treat adults with Waldenström’s macroglobulinemia (WM).
This is the ninth FDA approval for ibrutinib and the second approval for the drug in WM.
The latest approval was supported by the phase 3 iNNOVATE trial, in which researchers compared ibrutinib plus rituximab to rituximab alone in patients with previously untreated or relapsed/refractory WM.
Results from iNNOVATE were presented at the 2018 ASCO Annual Meeting (abstract 8003) and simultaneously published in NEJM.
The trial enrolled 150 patients. They received rituximab at 375 mg/m2 with weekly infusions at weeks 1 to 4 and 17 to 20. They also received either ibrutinib (420 mg) or placebo once daily continuously until criteria for permanent discontinuation were met.
Overall response rates were significantly higher in the ibrutinib arm than the placebo arm—92% and 47%, respectively (P<0.0001). Complete response rates were 3% and 1%, respectively.
The median time to next treatment was not reached for the ibrutinib arm and was 18 months for the placebo arm (hazard ratio=0.096; P<0.0001). Of the patients randomized to ibrutinib plus rituximab, 75% continued on treatment at last follow-up.
The 30-month progression-free survival rates were 82% in the ibrutinib arm and 28% in the placebo arm. The median progression-free survival was not reached in the ibrutinib arm and was 20.3 months in the placebo arm (hazard ratio=0.20; P<0.0001).
The 30-month overall survival rates were 94% in the ibrutinib arm and 92% in the placebo arm.
Grade 3 or higher treatment-emergent adverse events (AEs) occurred in 60% of patients in the ibrutinib arm and 61% in the placebo arm. Serious AEs occurred in 43% and 33%, respectively.
There were no fatal AEs in the ibrutinib arm and 3 in the rituximab arm.
Grade 3 or higher AEs that occurred more frequently in the ibrutinib arm than the placebo arm included atrial fibrillation (12% vs 1%) and hypertension (13% vs 4%).
AEs that occurred less frequently in the ibrutinib arm than the placebo arm included grade 3 or higher infusion reactions (1% vs 16%) and any-grade IgM flare (8% vs 47%).
About ibrutinib
Ibrutinib is a Bruton’s tyrosine kinase inhibitor that is FDA-approved to treat chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), chronic graft-versus-host disease (cGVHD), and WM.
In November 2013, ibrutinib was approved to treat adults with MCL who have received at least one prior therapy.
In February 2014, ibrutinib was approved to treat adults with CLL who have received at least one prior therapy. In July 2014, ibrutinib received approval for adult CLL patients with 17p deletion, and, in March 2016, ibrutinib was approved as a frontline CLL treatment.
Ibrutinib was approved for use as a single agent in adults with WM in January 2015.
In May 2016, ibrutinib was approved in combination with bendamustine and rituximab for adults with previously treated CLL/SLL.
In January 2017, ibrutinib was approved for adults with MZL who require systemic therapy and have received at least one prior anti-CD20-based therapy.
In August 2017, ibrutinib was approved to treat adults with cGVHD that did not respond to one or more lines of systemic therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved ibrutinib (Imbruvica®) for use in combination with rituximab to treat adults with Waldenström’s macroglobulinemia (WM).
This is the ninth FDA approval for ibrutinib and the second approval for the drug in WM.
The latest approval was supported by the phase 3 iNNOVATE trial, in which researchers compared ibrutinib plus rituximab to rituximab alone in patients with previously untreated or relapsed/refractory WM.
Results from iNNOVATE were presented at the 2018 ASCO Annual Meeting (abstract 8003) and simultaneously published in NEJM.
The trial enrolled 150 patients. They received rituximab at 375 mg/m2 with weekly infusions at weeks 1 to 4 and 17 to 20. They also received either ibrutinib (420 mg) or placebo once daily continuously until criteria for permanent discontinuation were met.
Overall response rates were significantly higher in the ibrutinib arm than the placebo arm—92% and 47%, respectively (P<0.0001). Complete response rates were 3% and 1%, respectively.
The median time to next treatment was not reached for the ibrutinib arm and was 18 months for the placebo arm (hazard ratio=0.096; P<0.0001). Of the patients randomized to ibrutinib plus rituximab, 75% continued on treatment at last follow-up.
The 30-month progression-free survival rates were 82% in the ibrutinib arm and 28% in the placebo arm. The median progression-free survival was not reached in the ibrutinib arm and was 20.3 months in the placebo arm (hazard ratio=0.20; P<0.0001).
The 30-month overall survival rates were 94% in the ibrutinib arm and 92% in the placebo arm.
Grade 3 or higher treatment-emergent adverse events (AEs) occurred in 60% of patients in the ibrutinib arm and 61% in the placebo arm. Serious AEs occurred in 43% and 33%, respectively.
There were no fatal AEs in the ibrutinib arm and 3 in the rituximab arm.
Grade 3 or higher AEs that occurred more frequently in the ibrutinib arm than the placebo arm included atrial fibrillation (12% vs 1%) and hypertension (13% vs 4%).
AEs that occurred less frequently in the ibrutinib arm than the placebo arm included grade 3 or higher infusion reactions (1% vs 16%) and any-grade IgM flare (8% vs 47%).
About ibrutinib
Ibrutinib is a Bruton’s tyrosine kinase inhibitor that is FDA-approved to treat chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), mantle cell lymphoma (MCL), marginal zone lymphoma (MZL), chronic graft-versus-host disease (cGVHD), and WM.
In November 2013, ibrutinib was approved to treat adults with MCL who have received at least one prior therapy.
In February 2014, ibrutinib was approved to treat adults with CLL who have received at least one prior therapy. In July 2014, ibrutinib received approval for adult CLL patients with 17p deletion, and, in March 2016, ibrutinib was approved as a frontline CLL treatment.
Ibrutinib was approved for use as a single agent in adults with WM in January 2015.
In May 2016, ibrutinib was approved in combination with bendamustine and rituximab for adults with previously treated CLL/SLL.
In January 2017, ibrutinib was approved for adults with MZL who require systemic therapy and have received at least one prior anti-CD20-based therapy.
In August 2017, ibrutinib was approved to treat adults with cGVHD that did not respond to one or more lines of systemic therapy.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Combo treatment yields MRD-negative remissions in CLL
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
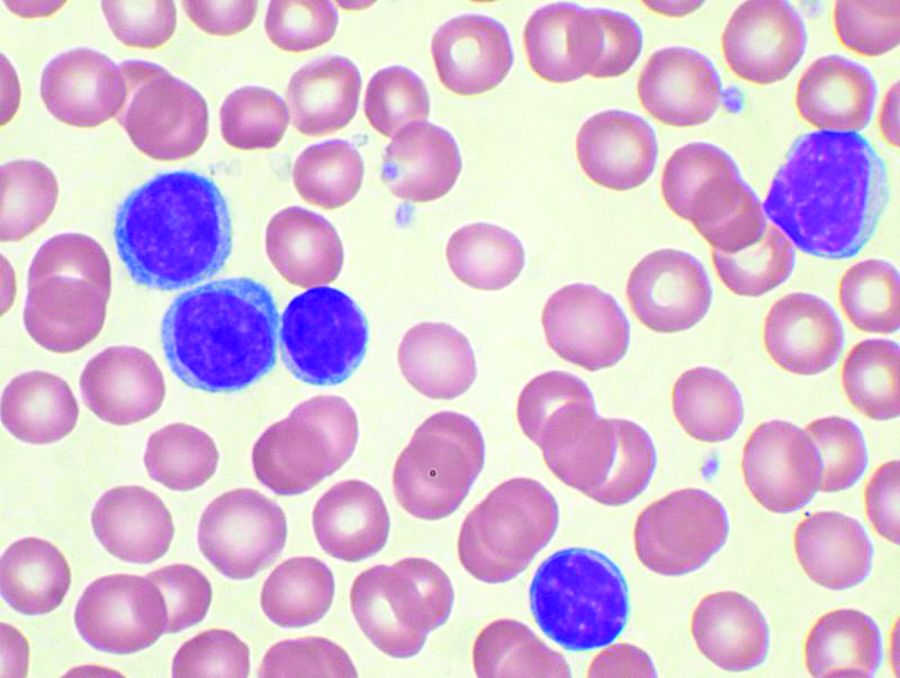
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
The combination of the anti-CD20 antibody obinutuzumab and venetoclax in chronic lymphocytic leukemia shows a high overall response rate and compares favorably with established therapies, according to a new report.
The ongoing, open-label, phase 2 study examined the outcomes of six induction cycles, followed by up to 24 months of maintenance treatment with obinutuzumab and venetoclax, in 66 patients with chronic lymphocytic leukemia (CLL). Of the 63 patients included in the efficacy analysis, 34 (54%) had treatment-naive and 29 (46%) had relapsed or refractory disease.
After an initial debulking with two cycles of bendamustine, followed by the obinutuzumab and venetoclax treatment, researchers observed an overall response rate of 95%. By the end of the induction phase, all the treatment-naive patients responded, as did 90% of the relapsed or refractory patients. Five patients had achieved complete remission and 55 patients had a partial response, the researchers reported in Lancet Oncology.
By 15 months, both progression-free and overall survival was 100% among treatment-naive patients, while progression-free survival was 83% and overall survival was 90% among the relapsed or refractory patients at this point.
Researchers observed minimal residual disease (MRD) negativity in the peripheral blood of 91% of treatment-naive patients and 83% of relapsed or refractory patients.
The combination of venetoclax and obinutuzumab was chosen based on earlier trial data, which suggested a synergy between venetoclax and the less-potent anti-CD20 antibody rituximab.
Paula Cramer, MD, from the German CLL Study Group at University Hospital, Cologne, and her coauthors described the efficacy of the venetoclax and obinutuzumab combination as “encouraging.”
“The combination of venetoclax and obinutuzumab yields fast responses with MRD-negative remissions in most patients,” they wrote. “Based on the experience with venetoclax combined with rituximab in another trial and with venetoclax and obinutuzumab in this and another study, these deep, MRD-negative remissions seem to last for a substantial time after treatment termination.”
Of the 677 adverse events, 427 (63%) were deemed to be related to the study treatment, and 69 of these were serious adverse events.
The most common of these were infections, experienced by four patients during the debulking with bendamustine, and 18 cases in 11 patients during the induction treatment. This included pneumonia, sepsis and cytomegalovirus infection, as well as neutropenia and thrombocytopenia.
Six patients also experienced infusion-related reactions, four had coronary artery disorder – one during debulking and three during induction – and there were three cases of neoplasms.
Five patients in the relapsed or refractory group died; three of sepsis related to study treatment, and two from unrelated Richter’s transformation.
“With three deaths from sepsis in 66 enrolled patients, the treatment-related mortality seems high; however, in cases of low patient numbers, a few patients can have a substantial effect on the overall results,” the researchers wrote.
The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria and other support from the pharmaceutical industry, including from the study sponsors.
SOURCE: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.
FROM LANCET ONCOLOGY
Key clinical point:
Major finding: The overall response rate for obinutuzumab plus venetoclax in CLL was 95%.
Study details: An ongoing, phase 2, open-label trial in 66 patients with chronic lymphocytic leukemia.
Disclosures: The study was funded by F Hoffmann-La Roche and AbbVie. Several authors reported research funding, grants, honoraria, and other support from the pharmaceutical industry, including from the study sponsors.
Source: Cramer P et al. Lancet Oncol. 2018 Aug 13. doi: 10.1016/S1470-2045(18)30414-5.