User login
An Unusual Case of Sporadic Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
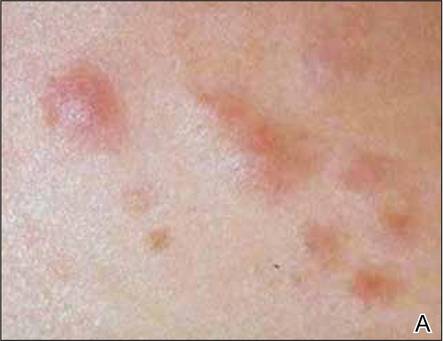

Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.

The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
|
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
|
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.