User login
Sildenafil associated with persistent pulmonary hypertension in neonates with early IUGR
LAS VEGAS – Increased rates of persistent neonatal pulmonary hypertension in neonates put the brakes on STRIDER, an international placebo-controlled study looking at sildenafil as a treatment for early-onset intrauterine growth restriction (IUGR).
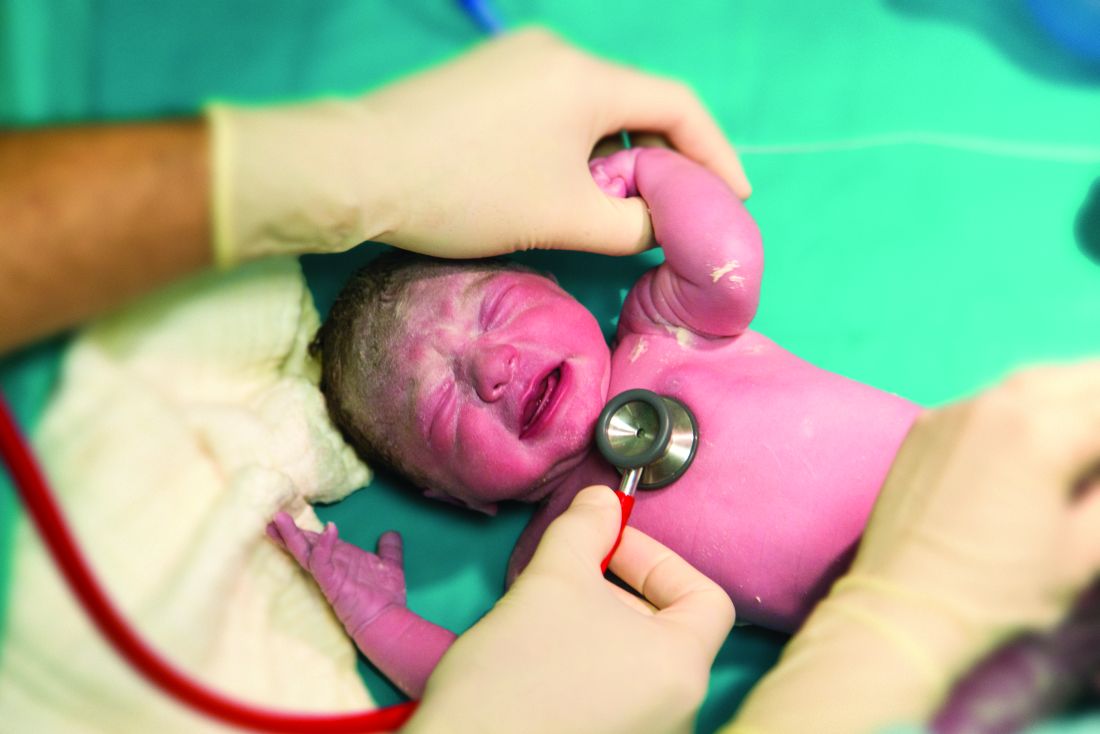
The study’s independent data safety monitoring board halted STRIDER (Sildenafil Therapy in Dismal Prognosis Early-Onset Fetal Growth Restriction) last July, after an interim safety analysis identified possible fetal harm and no signal of benefit over placebo, Dr. Anouk Pels reported at the annual meeting of the Society for Maternal-Fetal Medicine. The late-breaking presentation at the meeting revealed the first outcome data details.
The board had “serious concerns that sildenafil may cause harm to newborn children. … Given the results , it is extremely unlikely that any benefit could be shown on the primary endpoint if the trial is continued to its completion,” said Dr. Pels of the University of Amsterdam. “Our recommendation is not to use sildenafil for this indication in pregnant women.”
Although the link remains as-yet unproven, pulmonary hypertension among sildenafil-exposed neonates is biologically plausible, she said. It could have been a symptomatic rebound response to the discontinuation of constant intrauterine sildenafil exposure – or it could have been a hint of something more profound. Like the genital vasculature, pulmonary vasculature is a target of the drug. Intrauterine exposure to sildenafil could theoretically alter its development.
“It’s possible that sildenafil may be causing structural changes in the pulmonary vasculature of fetuses. This needs to be explored further, and we will do so by performing additional analyses on autopsy data and placental histology.”
STRIDER involved 261 pregnant women diagnosed with severe early-onset fetal growth restriction. They were randomized to sildenafil 25 mg or placebo three times daily until delivery or 32 weeks’ gestation. A safety analysis was conducted after every 50 patients were enrolled. The preplanned interim analysis was conducted after half of the cohort had been enrolled and received at least one dose of the study medication.
The primary outcome was a composite measure of neonatal mortality or major neonatal morbidity at hospital discharge.
Gestational age at baseline was about 24.6 weeks, and the estimated fetal weight by ultrasound, 465 g. About 45% of pregnancies had evidence of a notching in the uterine artery. In about 42%, the pulsatility index of the umbilical artery was above the 95th percentile; the pulsatility index of the middle cerebral artery was below the 5% percentile in about 70% of cases.
About a quarter of the women had a diagnosis of pregnancy-induced hypertension, and another quarter, preeclampsia. Women used the study medication for a mean of 22 days.
There were no significant between-group differences in maternal outcomes. Median gestational age at delivery was 28 weeks in both groups. About 10% in each group experienced new-onset pregnancy-induced hypertension; About a quarter of each group developed new-onset preeclampsia or HELLP (hemolysis, elevated liver enzymes, low platelet count). There were no between-group differences in the number of maternal antihypertensives prescribed.
The primary combined outcome of neonatal mortality or major neonatal morbidity occurred in 66 of the sildenafil-exposed neonates and 58 of the placebo-exposed infants (61% vs. 54%) – not a significant difference. Fetal death occurred in 23 and 29 pregnancies, respectively (21% and 27%); neonatal death occurred in 21 and 11, respectively (19% and 10%). Overall, fetal/neonatal mortality was similar between the sildenafil and placebo groups (41% and 37%, respectively).
Of the 64 sildenafil-exposed neonates who survived to hospital discharge, 22 exhibited clinically relevant morbidity. Of the 67 in the placebo-treated group who survived to hospital discharge, 18 had clinically relevant morbidity. Overall, 42 in the sildenafil group and 49 of the placebo group survived to hospital discharge without relevant morbidity.
There were a number of secondary outcomes, none exhibiting any significant between-group differences. These included the median weights of those who experienced intrauterine death (425 g and 350 g), median live birth weight (725 g and 783 g), intraventricular hemorrhage of grade II or IV (3% and 2%), periventricular hemorrhage grade II or higher (0%, both groups), necrotizing enterocolitis grade II or higher (7% and 8%), and at least one culture-proven or clinical infection (41% and 33%).
The significantly higher rates of pulmonary hypertension in the sildenafil-exposed neonates was the showstopper. Almost half of the 21 who died had proven pulmonary hypertension (10), as did 6 of the 64 who survived – an excess of 16 cases. Of proven cases, 11 were persistent pulmonary hypertension. There were also two cases of sepsis-associated pulmonary hypertension and four cases of bronchopulmonary dysplasia associated with the disorder. In the placebo-exposed group, pulmonary hypertension occurred in 3 of the 11 deaths and 1 of the 67 who survived. Of proven cases, two were persistent pulmonary hypertension. There was no sepsis-associated pulmonary hypertension, but there were three cases of bronchopulmonary dysplasia associated with the disorder. Some children had both persistent pulmonary hypertension and bronchopulmonary dysplasia.
“While there was no difference in the primary outcomes or in overall mortality, there were more cases of pulmonary hypertension in the sildenafil group,” Dr. Pels said. “We can speculate on the cause, whether it was related to sildenafil and why, or whether it was simply chance. This is the reason we need to conduct more in-depth analyses of these data.”
Funding came from federal health agencies and universities in the countries where STRIDER was conducted, including New Zealand, Australia, the United Kingdom, Ireland, and the Netherlands. Dr. Pels had no relevant financial disclosures.
SOURCE: Pels A et al. The Pregnancy Meeting, Late-Breaker 2.
LAS VEGAS – Increased rates of persistent neonatal pulmonary hypertension in neonates put the brakes on STRIDER, an international placebo-controlled study looking at sildenafil as a treatment for early-onset intrauterine growth restriction (IUGR).
The study’s independent data safety monitoring board halted STRIDER (Sildenafil Therapy in Dismal Prognosis Early-Onset Fetal Growth Restriction) last July, after an interim safety analysis identified possible fetal harm and no signal of benefit over placebo, Dr. Anouk Pels reported at the annual meeting of the Society for Maternal-Fetal Medicine. The late-breaking presentation at the meeting revealed the first outcome data details.
The board had “serious concerns that sildenafil may cause harm to newborn children. … Given the results , it is extremely unlikely that any benefit could be shown on the primary endpoint if the trial is continued to its completion,” said Dr. Pels of the University of Amsterdam. “Our recommendation is not to use sildenafil for this indication in pregnant women.”
Although the link remains as-yet unproven, pulmonary hypertension among sildenafil-exposed neonates is biologically plausible, she said. It could have been a symptomatic rebound response to the discontinuation of constant intrauterine sildenafil exposure – or it could have been a hint of something more profound. Like the genital vasculature, pulmonary vasculature is a target of the drug. Intrauterine exposure to sildenafil could theoretically alter its development.
“It’s possible that sildenafil may be causing structural changes in the pulmonary vasculature of fetuses. This needs to be explored further, and we will do so by performing additional analyses on autopsy data and placental histology.”
STRIDER involved 261 pregnant women diagnosed with severe early-onset fetal growth restriction. They were randomized to sildenafil 25 mg or placebo three times daily until delivery or 32 weeks’ gestation. A safety analysis was conducted after every 50 patients were enrolled. The preplanned interim analysis was conducted after half of the cohort had been enrolled and received at least one dose of the study medication.
The primary outcome was a composite measure of neonatal mortality or major neonatal morbidity at hospital discharge.
Gestational age at baseline was about 24.6 weeks, and the estimated fetal weight by ultrasound, 465 g. About 45% of pregnancies had evidence of a notching in the uterine artery. In about 42%, the pulsatility index of the umbilical artery was above the 95th percentile; the pulsatility index of the middle cerebral artery was below the 5% percentile in about 70% of cases.
About a quarter of the women had a diagnosis of pregnancy-induced hypertension, and another quarter, preeclampsia. Women used the study medication for a mean of 22 days.
There were no significant between-group differences in maternal outcomes. Median gestational age at delivery was 28 weeks in both groups. About 10% in each group experienced new-onset pregnancy-induced hypertension; About a quarter of each group developed new-onset preeclampsia or HELLP (hemolysis, elevated liver enzymes, low platelet count). There were no between-group differences in the number of maternal antihypertensives prescribed.
The primary combined outcome of neonatal mortality or major neonatal morbidity occurred in 66 of the sildenafil-exposed neonates and 58 of the placebo-exposed infants (61% vs. 54%) – not a significant difference. Fetal death occurred in 23 and 29 pregnancies, respectively (21% and 27%); neonatal death occurred in 21 and 11, respectively (19% and 10%). Overall, fetal/neonatal mortality was similar between the sildenafil and placebo groups (41% and 37%, respectively).
Of the 64 sildenafil-exposed neonates who survived to hospital discharge, 22 exhibited clinically relevant morbidity. Of the 67 in the placebo-treated group who survived to hospital discharge, 18 had clinically relevant morbidity. Overall, 42 in the sildenafil group and 49 of the placebo group survived to hospital discharge without relevant morbidity.
There were a number of secondary outcomes, none exhibiting any significant between-group differences. These included the median weights of those who experienced intrauterine death (425 g and 350 g), median live birth weight (725 g and 783 g), intraventricular hemorrhage of grade II or IV (3% and 2%), periventricular hemorrhage grade II or higher (0%, both groups), necrotizing enterocolitis grade II or higher (7% and 8%), and at least one culture-proven or clinical infection (41% and 33%).
The significantly higher rates of pulmonary hypertension in the sildenafil-exposed neonates was the showstopper. Almost half of the 21 who died had proven pulmonary hypertension (10), as did 6 of the 64 who survived – an excess of 16 cases. Of proven cases, 11 were persistent pulmonary hypertension. There were also two cases of sepsis-associated pulmonary hypertension and four cases of bronchopulmonary dysplasia associated with the disorder. In the placebo-exposed group, pulmonary hypertension occurred in 3 of the 11 deaths and 1 of the 67 who survived. Of proven cases, two were persistent pulmonary hypertension. There was no sepsis-associated pulmonary hypertension, but there were three cases of bronchopulmonary dysplasia associated with the disorder. Some children had both persistent pulmonary hypertension and bronchopulmonary dysplasia.
“While there was no difference in the primary outcomes or in overall mortality, there were more cases of pulmonary hypertension in the sildenafil group,” Dr. Pels said. “We can speculate on the cause, whether it was related to sildenafil and why, or whether it was simply chance. This is the reason we need to conduct more in-depth analyses of these data.”
Funding came from federal health agencies and universities in the countries where STRIDER was conducted, including New Zealand, Australia, the United Kingdom, Ireland, and the Netherlands. Dr. Pels had no relevant financial disclosures.
SOURCE: Pels A et al. The Pregnancy Meeting, Late-Breaker 2.
LAS VEGAS – Increased rates of persistent neonatal pulmonary hypertension in neonates put the brakes on STRIDER, an international placebo-controlled study looking at sildenafil as a treatment for early-onset intrauterine growth restriction (IUGR).
The study’s independent data safety monitoring board halted STRIDER (Sildenafil Therapy in Dismal Prognosis Early-Onset Fetal Growth Restriction) last July, after an interim safety analysis identified possible fetal harm and no signal of benefit over placebo, Dr. Anouk Pels reported at the annual meeting of the Society for Maternal-Fetal Medicine. The late-breaking presentation at the meeting revealed the first outcome data details.
The board had “serious concerns that sildenafil may cause harm to newborn children. … Given the results , it is extremely unlikely that any benefit could be shown on the primary endpoint if the trial is continued to its completion,” said Dr. Pels of the University of Amsterdam. “Our recommendation is not to use sildenafil for this indication in pregnant women.”
Although the link remains as-yet unproven, pulmonary hypertension among sildenafil-exposed neonates is biologically plausible, she said. It could have been a symptomatic rebound response to the discontinuation of constant intrauterine sildenafil exposure – or it could have been a hint of something more profound. Like the genital vasculature, pulmonary vasculature is a target of the drug. Intrauterine exposure to sildenafil could theoretically alter its development.
“It’s possible that sildenafil may be causing structural changes in the pulmonary vasculature of fetuses. This needs to be explored further, and we will do so by performing additional analyses on autopsy data and placental histology.”
STRIDER involved 261 pregnant women diagnosed with severe early-onset fetal growth restriction. They were randomized to sildenafil 25 mg or placebo three times daily until delivery or 32 weeks’ gestation. A safety analysis was conducted after every 50 patients were enrolled. The preplanned interim analysis was conducted after half of the cohort had been enrolled and received at least one dose of the study medication.
The primary outcome was a composite measure of neonatal mortality or major neonatal morbidity at hospital discharge.
Gestational age at baseline was about 24.6 weeks, and the estimated fetal weight by ultrasound, 465 g. About 45% of pregnancies had evidence of a notching in the uterine artery. In about 42%, the pulsatility index of the umbilical artery was above the 95th percentile; the pulsatility index of the middle cerebral artery was below the 5% percentile in about 70% of cases.
About a quarter of the women had a diagnosis of pregnancy-induced hypertension, and another quarter, preeclampsia. Women used the study medication for a mean of 22 days.
There were no significant between-group differences in maternal outcomes. Median gestational age at delivery was 28 weeks in both groups. About 10% in each group experienced new-onset pregnancy-induced hypertension; About a quarter of each group developed new-onset preeclampsia or HELLP (hemolysis, elevated liver enzymes, low platelet count). There were no between-group differences in the number of maternal antihypertensives prescribed.
The primary combined outcome of neonatal mortality or major neonatal morbidity occurred in 66 of the sildenafil-exposed neonates and 58 of the placebo-exposed infants (61% vs. 54%) – not a significant difference. Fetal death occurred in 23 and 29 pregnancies, respectively (21% and 27%); neonatal death occurred in 21 and 11, respectively (19% and 10%). Overall, fetal/neonatal mortality was similar between the sildenafil and placebo groups (41% and 37%, respectively).
Of the 64 sildenafil-exposed neonates who survived to hospital discharge, 22 exhibited clinically relevant morbidity. Of the 67 in the placebo-treated group who survived to hospital discharge, 18 had clinically relevant morbidity. Overall, 42 in the sildenafil group and 49 of the placebo group survived to hospital discharge without relevant morbidity.
There were a number of secondary outcomes, none exhibiting any significant between-group differences. These included the median weights of those who experienced intrauterine death (425 g and 350 g), median live birth weight (725 g and 783 g), intraventricular hemorrhage of grade II or IV (3% and 2%), periventricular hemorrhage grade II or higher (0%, both groups), necrotizing enterocolitis grade II or higher (7% and 8%), and at least one culture-proven or clinical infection (41% and 33%).
The significantly higher rates of pulmonary hypertension in the sildenafil-exposed neonates was the showstopper. Almost half of the 21 who died had proven pulmonary hypertension (10), as did 6 of the 64 who survived – an excess of 16 cases. Of proven cases, 11 were persistent pulmonary hypertension. There were also two cases of sepsis-associated pulmonary hypertension and four cases of bronchopulmonary dysplasia associated with the disorder. In the placebo-exposed group, pulmonary hypertension occurred in 3 of the 11 deaths and 1 of the 67 who survived. Of proven cases, two were persistent pulmonary hypertension. There was no sepsis-associated pulmonary hypertension, but there were three cases of bronchopulmonary dysplasia associated with the disorder. Some children had both persistent pulmonary hypertension and bronchopulmonary dysplasia.
“While there was no difference in the primary outcomes or in overall mortality, there were more cases of pulmonary hypertension in the sildenafil group,” Dr. Pels said. “We can speculate on the cause, whether it was related to sildenafil and why, or whether it was simply chance. This is the reason we need to conduct more in-depth analyses of these data.”
Funding came from federal health agencies and universities in the countries where STRIDER was conducted, including New Zealand, Australia, the United Kingdom, Ireland, and the Netherlands. Dr. Pels had no relevant financial disclosures.
SOURCE: Pels A et al. The Pregnancy Meeting, Late-Breaker 2.
REPORTING FROM THE PREGNANCY MEETING
Key clinical point:
Major finding: There were 16 cases of persistent pulmonary hypertension in the treated group and four in the placebo group.
Study details: The randomized study involved 261 pregnant women treated with sildenafil for early-onset intrauterine growth restriction.
Disclosures: Funding agencies and universities in the countries involved in the trial contributed to funding. Dr. Pels had no relevant financial disclosures.
Source: Pels A et al. The Pregnancy Meeting, Late-Breaker 2.
Umbilical cord milking tied to severe IVH in very premature neonates
Delayed cord clamping and cutting is safer
LAS VEGAS – Umbilical cord milking can cause severe intraventricular hemorrhage (IVH) in very premature neonates and should not be performed on these cerebrovascularly fragile premature babies.

Just six of these procedures would be needed to cause a case of severe IVH in neonates born at 23-27 weeks’ gestation, Michael W. Varner, MD, said at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“Centers practicing umbilical cord milking should consider discontinuing this practice in infants 23-27 weeks’ gestation,” said Dr. Varner of the University of Utah, Salt Lake City.
The damage to the brains of very young preemies appears to be a direct result of the fluid overload caused by milking, he said. “From a mechanistic perspective, we can intuit that these findings are consistent with cord milking. This causes increasing venous return to the right atrium where it enters the foramen ovale and aorta. These very premature babies have more pulmonary vasoconstriction, which shunts more blood toward the brain. This results in fluctuations in flow in an immature brain with fragile germinal matrices and perhaps further compromised by chorioamnionitis inflammation, resulting in IVH.”
Premature Infants Receiving Milking or Delayed Cord Clamping (PREMOD2) was a noninferiority trial of umbilical cord milking compared to delayed cord clamping and cutting in preterm infants. Conducted at 11 sites in the United States and Europe, the study was halted prematurely when the data safety monitoring board determined that cord milking increased the risk of IVH in younger preemies and was no better than delayed cutting in the older preemies. The analysis presented at the meeting is the first public discussion of the data details.
The trial involved 474 premature neonates. They were randomized to placental transfusion via a 60-second delay in cord clamping and cutting or to umbilical cord milking, which involved grasping the cord and manually pushing the cord blood toward the infant four times before clamping. All participating sites received a video demonstrating the proper procedure. The cohort also was divided by gestational age: 23-27 weeks and 28-31 weeks.
The primary endpoint was a combination of severe IVH (grade 3 or higher) and neonatal death. Overall, the primary endpoint occurred in 29 of those randomized to cord milking (12%) and 20 randomized to delayed clamping (8%) – a significant difference.
This finding was largely driven by the treatment differences in the 23-27 week group, Dr. Varner said. Severe IVH occurred in 20 (22%) of those randomized to cord milking and five (6%) of those randomized to delayed clamping – a highly statistically significant difference with a P value of 0.0019.
In the 28-31 week group, there were no cases of severe IVH in the cord milking group, and three cases in the delayed clamping group; the difference was not statistically significant.
Overall, deaths were similar between the cord milking and cord clamping groups (17 and 15, respectively). Most of these deaths occurred in the younger group (14 in the cord milking group and 13 in the clamping group). There were five deaths in the older group: three in the cord milking group and two in the clamping group. None of these differences were statistically significant.
After seeing these data in a preplanned interim safety analysis, the Data Safety Monitoring Board stopped the study, saying that the intervention appeared dangerous for the younger babies, and no better than the delayed cutting and clamping for the older group, Dr. Varner said.
Since the trial was halted, investigators have been dissecting the data to identify any other intracranial hemorrhage risks particular to the infants. They found no significant differences in maternal characteristics at baseline, and – other than age and randomization– nothing significantly different between the infant groups. Severe persistent IVH occurred in almost 70% of the infants born at 23 weeks’ gestation but in only 7% in the delayed cord clamping group. The risks declined rapidly with increasing gestational age, although they were at all times greater than the risk of IVH in the cord clamping group.
“Looking at the data by gestational age, it’s clear that the majority of the severe IVH occurrences were in the 23 weekers, and also occurred in the first 7 days of life,” Dr. Varner said.
The cohort will be followed for at least another year, he added, as investigators track neurodevelopmental outcomes.
Investigators are particularly interested in differences in motor and language skills, as well as general cognitive development.
The study was sponsored by theEunice Kennedy Shriver National Institute of Child Health and Development. Neither Dr. Varner nor any of the coauthors had any financial declarations.
SOURCE: Katheria AC et al. The Pregnancy Meeting, late breaking abstract 1.
Delayed cord clamping and cutting is safer
Delayed cord clamping and cutting is safer
LAS VEGAS – Umbilical cord milking can cause severe intraventricular hemorrhage (IVH) in very premature neonates and should not be performed on these cerebrovascularly fragile premature babies.
Just six of these procedures would be needed to cause a case of severe IVH in neonates born at 23-27 weeks’ gestation, Michael W. Varner, MD, said at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“Centers practicing umbilical cord milking should consider discontinuing this practice in infants 23-27 weeks’ gestation,” said Dr. Varner of the University of Utah, Salt Lake City.
The damage to the brains of very young preemies appears to be a direct result of the fluid overload caused by milking, he said. “From a mechanistic perspective, we can intuit that these findings are consistent with cord milking. This causes increasing venous return to the right atrium where it enters the foramen ovale and aorta. These very premature babies have more pulmonary vasoconstriction, which shunts more blood toward the brain. This results in fluctuations in flow in an immature brain with fragile germinal matrices and perhaps further compromised by chorioamnionitis inflammation, resulting in IVH.”
Premature Infants Receiving Milking or Delayed Cord Clamping (PREMOD2) was a noninferiority trial of umbilical cord milking compared to delayed cord clamping and cutting in preterm infants. Conducted at 11 sites in the United States and Europe, the study was halted prematurely when the data safety monitoring board determined that cord milking increased the risk of IVH in younger preemies and was no better than delayed cutting in the older preemies. The analysis presented at the meeting is the first public discussion of the data details.
The trial involved 474 premature neonates. They were randomized to placental transfusion via a 60-second delay in cord clamping and cutting or to umbilical cord milking, which involved grasping the cord and manually pushing the cord blood toward the infant four times before clamping. All participating sites received a video demonstrating the proper procedure. The cohort also was divided by gestational age: 23-27 weeks and 28-31 weeks.
The primary endpoint was a combination of severe IVH (grade 3 or higher) and neonatal death. Overall, the primary endpoint occurred in 29 of those randomized to cord milking (12%) and 20 randomized to delayed clamping (8%) – a significant difference.
This finding was largely driven by the treatment differences in the 23-27 week group, Dr. Varner said. Severe IVH occurred in 20 (22%) of those randomized to cord milking and five (6%) of those randomized to delayed clamping – a highly statistically significant difference with a P value of 0.0019.
In the 28-31 week group, there were no cases of severe IVH in the cord milking group, and three cases in the delayed clamping group; the difference was not statistically significant.
Overall, deaths were similar between the cord milking and cord clamping groups (17 and 15, respectively). Most of these deaths occurred in the younger group (14 in the cord milking group and 13 in the clamping group). There were five deaths in the older group: three in the cord milking group and two in the clamping group. None of these differences were statistically significant.
After seeing these data in a preplanned interim safety analysis, the Data Safety Monitoring Board stopped the study, saying that the intervention appeared dangerous for the younger babies, and no better than the delayed cutting and clamping for the older group, Dr. Varner said.
Since the trial was halted, investigators have been dissecting the data to identify any other intracranial hemorrhage risks particular to the infants. They found no significant differences in maternal characteristics at baseline, and – other than age and randomization– nothing significantly different between the infant groups. Severe persistent IVH occurred in almost 70% of the infants born at 23 weeks’ gestation but in only 7% in the delayed cord clamping group. The risks declined rapidly with increasing gestational age, although they were at all times greater than the risk of IVH in the cord clamping group.
“Looking at the data by gestational age, it’s clear that the majority of the severe IVH occurrences were in the 23 weekers, and also occurred in the first 7 days of life,” Dr. Varner said.
The cohort will be followed for at least another year, he added, as investigators track neurodevelopmental outcomes.
Investigators are particularly interested in differences in motor and language skills, as well as general cognitive development.
The study was sponsored by theEunice Kennedy Shriver National Institute of Child Health and Development. Neither Dr. Varner nor any of the coauthors had any financial declarations.
SOURCE: Katheria AC et al. The Pregnancy Meeting, late breaking abstract 1.
LAS VEGAS – Umbilical cord milking can cause severe intraventricular hemorrhage (IVH) in very premature neonates and should not be performed on these cerebrovascularly fragile premature babies.
Just six of these procedures would be needed to cause a case of severe IVH in neonates born at 23-27 weeks’ gestation, Michael W. Varner, MD, said at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“Centers practicing umbilical cord milking should consider discontinuing this practice in infants 23-27 weeks’ gestation,” said Dr. Varner of the University of Utah, Salt Lake City.
The damage to the brains of very young preemies appears to be a direct result of the fluid overload caused by milking, he said. “From a mechanistic perspective, we can intuit that these findings are consistent with cord milking. This causes increasing venous return to the right atrium where it enters the foramen ovale and aorta. These very premature babies have more pulmonary vasoconstriction, which shunts more blood toward the brain. This results in fluctuations in flow in an immature brain with fragile germinal matrices and perhaps further compromised by chorioamnionitis inflammation, resulting in IVH.”
Premature Infants Receiving Milking or Delayed Cord Clamping (PREMOD2) was a noninferiority trial of umbilical cord milking compared to delayed cord clamping and cutting in preterm infants. Conducted at 11 sites in the United States and Europe, the study was halted prematurely when the data safety monitoring board determined that cord milking increased the risk of IVH in younger preemies and was no better than delayed cutting in the older preemies. The analysis presented at the meeting is the first public discussion of the data details.
The trial involved 474 premature neonates. They were randomized to placental transfusion via a 60-second delay in cord clamping and cutting or to umbilical cord milking, which involved grasping the cord and manually pushing the cord blood toward the infant four times before clamping. All participating sites received a video demonstrating the proper procedure. The cohort also was divided by gestational age: 23-27 weeks and 28-31 weeks.
The primary endpoint was a combination of severe IVH (grade 3 or higher) and neonatal death. Overall, the primary endpoint occurred in 29 of those randomized to cord milking (12%) and 20 randomized to delayed clamping (8%) – a significant difference.
This finding was largely driven by the treatment differences in the 23-27 week group, Dr. Varner said. Severe IVH occurred in 20 (22%) of those randomized to cord milking and five (6%) of those randomized to delayed clamping – a highly statistically significant difference with a P value of 0.0019.
In the 28-31 week group, there were no cases of severe IVH in the cord milking group, and three cases in the delayed clamping group; the difference was not statistically significant.
Overall, deaths were similar between the cord milking and cord clamping groups (17 and 15, respectively). Most of these deaths occurred in the younger group (14 in the cord milking group and 13 in the clamping group). There were five deaths in the older group: three in the cord milking group and two in the clamping group. None of these differences were statistically significant.
After seeing these data in a preplanned interim safety analysis, the Data Safety Monitoring Board stopped the study, saying that the intervention appeared dangerous for the younger babies, and no better than the delayed cutting and clamping for the older group, Dr. Varner said.
Since the trial was halted, investigators have been dissecting the data to identify any other intracranial hemorrhage risks particular to the infants. They found no significant differences in maternal characteristics at baseline, and – other than age and randomization– nothing significantly different between the infant groups. Severe persistent IVH occurred in almost 70% of the infants born at 23 weeks’ gestation but in only 7% in the delayed cord clamping group. The risks declined rapidly with increasing gestational age, although they were at all times greater than the risk of IVH in the cord clamping group.
“Looking at the data by gestational age, it’s clear that the majority of the severe IVH occurrences were in the 23 weekers, and also occurred in the first 7 days of life,” Dr. Varner said.
The cohort will be followed for at least another year, he added, as investigators track neurodevelopmental outcomes.
Investigators are particularly interested in differences in motor and language skills, as well as general cognitive development.
The study was sponsored by theEunice Kennedy Shriver National Institute of Child Health and Development. Neither Dr. Varner nor any of the coauthors had any financial declarations.
SOURCE: Katheria AC et al. The Pregnancy Meeting, late breaking abstract 1.
REPORTING FROM THE PREGNANCY MEETING
New atopic dermatitis agents expand treatment options
GRAND CAYMAN, CAYMAN ISLANDS –
Moisturizers that confer skin barrier protection, lipid-replenishing topicals, and some biologics are available now or will soon be available for patients with AD, Joseph Fowler Jr., MD, said at the Caribbean Dermatology Symposium provided by Global Academy for Medical Education.
With the approval of dupilumab for moderate to severe disease in 2017, a biologic finally became available for treating AD, said Dr. Fowler of the University of Louisville (Ky.). While it’s not a cure and may take as long as 6 months to really kick in, “I think almost everyone gets some benefit from it. And although it’s not approved yet for anyone under 18, I’m sure it will be.”
He provided a brief rundown of dupilumab; crisaborole, another relatively new agent for AD; and some agents that are being investigated.
- Dupilumab. For AD, dupilumab, which inhibits interleukin-4 and interleukin-13 signaling, is usually started at 600 mg, then tapered to 300 mg subcutaneously every 2 weeks. Its pivotal data showed a mean 70% decrease in Eczema Area and Severity Index (EASI) scores over 16 weeks at that dose.*
“Again, I would say most patients do get benefit from this, but they might not see it for more than 3 months, and even up to 6 months. I’m not sure why, but some develop eye symptoms – I think these are more severe cases who also have respiratory atopy. I would also be interested to see if dupilumab might work on patients with chronic hand eczema,” he said.
- Crisaborole ointment 2%. A nonsteroidal topical phosphodiesterase 4 (PDE4) inhibitor approved in 2016 for mild to moderate AD in people aged 2 and older, crisaborole (Eucrisa) blocks the release of cyclic adenosine monophosphate (cAMP), which is elevated in AD. Lower cAMP levels lead to lower levels of inflammatory cytokines. In its pivotal phase 3 study, about 35% of patients achieved clinical success – an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, or at least a two-grade improvement over baseline.
“In my opinion, it’s similar or slightly better than topical corticosteroids, and safer as well, especially in our younger patients, or when the face or intertriginous areas are involved,” Dr. Fowler said. “There is often some application site stinging and burning. If you put it in the fridge and get it good and cold when it goes on, that seems to moderate the sensation. It’s a good steroid-sparing option.”
Crisaborole is now being investigated for use in infants aged 3-24 months with mild to moderate AD.
- Tofacitinib ointment. This topical form of tofacitinib, an inhibitor of Janus kinase 1 and 3, is being evaluated in a placebo-controlled trial in adults with mild to moderate AD. There are also a few reports of oral tofacitinib improving AD, including a case report (Clin Exp Dermatol. 2017 Dec;42[8]:942-4). Dr. Fowler noted a small series of six adults with moderate to severe AD uncontrolled with methotrexate or azathioprine. The patients received oral tofacitinib 5 mg twice a day for 8-29 weeks; there was a mean 67% improvement in the Scoring Atopic Dermatitis (SCORAD) index.
- Ustekinumab. The interleukin-12 and -23 antagonist indicated for moderate to severe psoriasis has also made an appearance in the AD literature, including an Austrian report of three patients with severe AD who received 45 mg of ustekinumab (Stelara) subcutaneously at 0, 4 and 12 weeks. By week 16, all of them experienced a 50% reduction in their EASI score, with a marked reduction in interleukin-22 markers (J Am Acad Dermatol. 2017 Jan;76[1]:91-7.e3).
But no matter which therapy is chosen, regular moisturizing is critically important, Dr. Fowler remarked. Expensive prescription moisturizers are available, but he questioned whether they offer any cost-worthy extra benefit over a good nonprescription moisturizer.
“Do these super-moisturizers protect the skin barrier any more than petrolatum? I can’t answer that. They promise better results, but each patient and doc have to make the decision. If your patient can afford it, maybe some will be better for their skin, but really, it’s not as important as some of the other medications. So, I tell them, if cost is an issue, don’t worry about the fancy moisturizers.”
Dr. Fowler disclosed relationships with multiple pharmaceutical companies.
Global Academy and this news organization are owned by the same parent company.
Correction, 2/15/19: An earlier version of this article mischaracterized the chemical action of dupilumab.
GRAND CAYMAN, CAYMAN ISLANDS –
Moisturizers that confer skin barrier protection, lipid-replenishing topicals, and some biologics are available now or will soon be available for patients with AD, Joseph Fowler Jr., MD, said at the Caribbean Dermatology Symposium provided by Global Academy for Medical Education.
With the approval of dupilumab for moderate to severe disease in 2017, a biologic finally became available for treating AD, said Dr. Fowler of the University of Louisville (Ky.). While it’s not a cure and may take as long as 6 months to really kick in, “I think almost everyone gets some benefit from it. And although it’s not approved yet for anyone under 18, I’m sure it will be.”
He provided a brief rundown of dupilumab; crisaborole, another relatively new agent for AD; and some agents that are being investigated.
- Dupilumab. For AD, dupilumab, which inhibits interleukin-4 and interleukin-13 signaling, is usually started at 600 mg, then tapered to 300 mg subcutaneously every 2 weeks. Its pivotal data showed a mean 70% decrease in Eczema Area and Severity Index (EASI) scores over 16 weeks at that dose.*
“Again, I would say most patients do get benefit from this, but they might not see it for more than 3 months, and even up to 6 months. I’m not sure why, but some develop eye symptoms – I think these are more severe cases who also have respiratory atopy. I would also be interested to see if dupilumab might work on patients with chronic hand eczema,” he said.
- Crisaborole ointment 2%. A nonsteroidal topical phosphodiesterase 4 (PDE4) inhibitor approved in 2016 for mild to moderate AD in people aged 2 and older, crisaborole (Eucrisa) blocks the release of cyclic adenosine monophosphate (cAMP), which is elevated in AD. Lower cAMP levels lead to lower levels of inflammatory cytokines. In its pivotal phase 3 study, about 35% of patients achieved clinical success – an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, or at least a two-grade improvement over baseline.
“In my opinion, it’s similar or slightly better than topical corticosteroids, and safer as well, especially in our younger patients, or when the face or intertriginous areas are involved,” Dr. Fowler said. “There is often some application site stinging and burning. If you put it in the fridge and get it good and cold when it goes on, that seems to moderate the sensation. It’s a good steroid-sparing option.”
Crisaborole is now being investigated for use in infants aged 3-24 months with mild to moderate AD.
- Tofacitinib ointment. This topical form of tofacitinib, an inhibitor of Janus kinase 1 and 3, is being evaluated in a placebo-controlled trial in adults with mild to moderate AD. There are also a few reports of oral tofacitinib improving AD, including a case report (Clin Exp Dermatol. 2017 Dec;42[8]:942-4). Dr. Fowler noted a small series of six adults with moderate to severe AD uncontrolled with methotrexate or azathioprine. The patients received oral tofacitinib 5 mg twice a day for 8-29 weeks; there was a mean 67% improvement in the Scoring Atopic Dermatitis (SCORAD) index.
- Ustekinumab. The interleukin-12 and -23 antagonist indicated for moderate to severe psoriasis has also made an appearance in the AD literature, including an Austrian report of three patients with severe AD who received 45 mg of ustekinumab (Stelara) subcutaneously at 0, 4 and 12 weeks. By week 16, all of them experienced a 50% reduction in their EASI score, with a marked reduction in interleukin-22 markers (J Am Acad Dermatol. 2017 Jan;76[1]:91-7.e3).
But no matter which therapy is chosen, regular moisturizing is critically important, Dr. Fowler remarked. Expensive prescription moisturizers are available, but he questioned whether they offer any cost-worthy extra benefit over a good nonprescription moisturizer.
“Do these super-moisturizers protect the skin barrier any more than petrolatum? I can’t answer that. They promise better results, but each patient and doc have to make the decision. If your patient can afford it, maybe some will be better for their skin, but really, it’s not as important as some of the other medications. So, I tell them, if cost is an issue, don’t worry about the fancy moisturizers.”
Dr. Fowler disclosed relationships with multiple pharmaceutical companies.
Global Academy and this news organization are owned by the same parent company.
Correction, 2/15/19: An earlier version of this article mischaracterized the chemical action of dupilumab.
GRAND CAYMAN, CAYMAN ISLANDS –
Moisturizers that confer skin barrier protection, lipid-replenishing topicals, and some biologics are available now or will soon be available for patients with AD, Joseph Fowler Jr., MD, said at the Caribbean Dermatology Symposium provided by Global Academy for Medical Education.
With the approval of dupilumab for moderate to severe disease in 2017, a biologic finally became available for treating AD, said Dr. Fowler of the University of Louisville (Ky.). While it’s not a cure and may take as long as 6 months to really kick in, “I think almost everyone gets some benefit from it. And although it’s not approved yet for anyone under 18, I’m sure it will be.”
He provided a brief rundown of dupilumab; crisaborole, another relatively new agent for AD; and some agents that are being investigated.
- Dupilumab. For AD, dupilumab, which inhibits interleukin-4 and interleukin-13 signaling, is usually started at 600 mg, then tapered to 300 mg subcutaneously every 2 weeks. Its pivotal data showed a mean 70% decrease in Eczema Area and Severity Index (EASI) scores over 16 weeks at that dose.*
“Again, I would say most patients do get benefit from this, but they might not see it for more than 3 months, and even up to 6 months. I’m not sure why, but some develop eye symptoms – I think these are more severe cases who also have respiratory atopy. I would also be interested to see if dupilumab might work on patients with chronic hand eczema,” he said.
- Crisaborole ointment 2%. A nonsteroidal topical phosphodiesterase 4 (PDE4) inhibitor approved in 2016 for mild to moderate AD in people aged 2 and older, crisaborole (Eucrisa) blocks the release of cyclic adenosine monophosphate (cAMP), which is elevated in AD. Lower cAMP levels lead to lower levels of inflammatory cytokines. In its pivotal phase 3 study, about 35% of patients achieved clinical success – an Investigator’s Static Global Assessment (ISGA) score of 0 or 1, or at least a two-grade improvement over baseline.
“In my opinion, it’s similar or slightly better than topical corticosteroids, and safer as well, especially in our younger patients, or when the face or intertriginous areas are involved,” Dr. Fowler said. “There is often some application site stinging and burning. If you put it in the fridge and get it good and cold when it goes on, that seems to moderate the sensation. It’s a good steroid-sparing option.”
Crisaborole is now being investigated for use in infants aged 3-24 months with mild to moderate AD.
- Tofacitinib ointment. This topical form of tofacitinib, an inhibitor of Janus kinase 1 and 3, is being evaluated in a placebo-controlled trial in adults with mild to moderate AD. There are also a few reports of oral tofacitinib improving AD, including a case report (Clin Exp Dermatol. 2017 Dec;42[8]:942-4). Dr. Fowler noted a small series of six adults with moderate to severe AD uncontrolled with methotrexate or azathioprine. The patients received oral tofacitinib 5 mg twice a day for 8-29 weeks; there was a mean 67% improvement in the Scoring Atopic Dermatitis (SCORAD) index.
- Ustekinumab. The interleukin-12 and -23 antagonist indicated for moderate to severe psoriasis has also made an appearance in the AD literature, including an Austrian report of three patients with severe AD who received 45 mg of ustekinumab (Stelara) subcutaneously at 0, 4 and 12 weeks. By week 16, all of them experienced a 50% reduction in their EASI score, with a marked reduction in interleukin-22 markers (J Am Acad Dermatol. 2017 Jan;76[1]:91-7.e3).
But no matter which therapy is chosen, regular moisturizing is critically important, Dr. Fowler remarked. Expensive prescription moisturizers are available, but he questioned whether they offer any cost-worthy extra benefit over a good nonprescription moisturizer.
“Do these super-moisturizers protect the skin barrier any more than petrolatum? I can’t answer that. They promise better results, but each patient and doc have to make the decision. If your patient can afford it, maybe some will be better for their skin, but really, it’s not as important as some of the other medications. So, I tell them, if cost is an issue, don’t worry about the fancy moisturizers.”
Dr. Fowler disclosed relationships with multiple pharmaceutical companies.
Global Academy and this news organization are owned by the same parent company.
Correction, 2/15/19: An earlier version of this article mischaracterized the chemical action of dupilumab.
REPORTING FROM THE CARIBBEAN DERMATOLOGY SYMPOSIUM
FDA panels back intranasal esketamine for refractory depression
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.

Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.
Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.
Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
Hidradenitis suppurativa linked to increased lymphoma risk
Lymphomas appear to be up to four times more likely in patients with hidradenitis suppurativa than among the general population, Rachel Tannenbaum and her colleagues reported in a Research Letter in JAMA Dermatology.
The risks of Hodgkin (HL), non-Hodgkin (NHL), and cutaneous T-cell lymphoma (CTCL) all were significantly higher among patients with HS, wrote Ms. Tannenbaum, Andrew Strunk, and Amit Garg, MD. Males and older patients carried higher risks than females and younger patients, they found.
The team members, of Hofstra University, Hempstead, N.Y., conducted a health care database study comprising 55 million patients included in 27 integrated U.S. health care systems. All the subjects were at least 18 years old; records indicated active HS during the study period of 2013-2018. A regression analysis controlled for age and sex.
The database contained 62,690 patients with HS. The majority (74%) were female and were aged 44 years or younger (57%).
All three lymphomas were more common among HS patients than patients without HS, including non-Hodgkin lymphoma (0.40% vs. 0.35%,) Hodgkin lymphoma (0.17% vs. 0.09%), and cutaneous T-cell lymphoma (0.06% vs. 0.02%).
The multivariate analysis determined that HS patients were twice as likely to develop both non-Hodgkin and Hodgkin lymphoma (odds ratio, 2.0 and 2.21, respectively). They were four times more likely to develop cutaneous T-cell lymphoma (OR, 4.31).
All three lymphomas were more common among males than females: NHL, 0.62% vs. 0.32%; HL, 0.28% vs. 0.13%; and CTCL, 0.09% vs. 0.04%. This translated into significantly increased HS-associated risks, Ms. Tannenbaum and her coauthors noted. “For example, the [odds ratios] for the association between HS and HL were higher in males (OR, 2.97; 95% confidence interval, 2.22-3.99) than in females (OR, 1.86; 95% CI, 1.44-2.39) (P = .02),” they wrote.
Lymphomas were more common among HS patients in every age group. Patients with HS aged 45-64 years were 38% more likely to develop NHL, and those older than 65, about twice as likely (OR, 1.99).
“To our knowledge, this is the first investigation to systematically evaluate this association in a U.S. population of patients with HS,” the research team concluded.
The study was supported by a grant from AbbVie. Ms. Tannenbaum and Mr. Strunk reported no disclosures. Dr. Garg reported financial relationships with AbbVie and several other pharmaceutical companies.
SOURCE: Tannenbaum R et al. JAMA Dermatol. 2019 Jan 30. doi: 10.1001/jamadermatol.2018.5230.
Lymphomas appear to be up to four times more likely in patients with hidradenitis suppurativa than among the general population, Rachel Tannenbaum and her colleagues reported in a Research Letter in JAMA Dermatology.
The risks of Hodgkin (HL), non-Hodgkin (NHL), and cutaneous T-cell lymphoma (CTCL) all were significantly higher among patients with HS, wrote Ms. Tannenbaum, Andrew Strunk, and Amit Garg, MD. Males and older patients carried higher risks than females and younger patients, they found.
The team members, of Hofstra University, Hempstead, N.Y., conducted a health care database study comprising 55 million patients included in 27 integrated U.S. health care systems. All the subjects were at least 18 years old; records indicated active HS during the study period of 2013-2018. A regression analysis controlled for age and sex.
The database contained 62,690 patients with HS. The majority (74%) were female and were aged 44 years or younger (57%).
All three lymphomas were more common among HS patients than patients without HS, including non-Hodgkin lymphoma (0.40% vs. 0.35%,) Hodgkin lymphoma (0.17% vs. 0.09%), and cutaneous T-cell lymphoma (0.06% vs. 0.02%).
The multivariate analysis determined that HS patients were twice as likely to develop both non-Hodgkin and Hodgkin lymphoma (odds ratio, 2.0 and 2.21, respectively). They were four times more likely to develop cutaneous T-cell lymphoma (OR, 4.31).
All three lymphomas were more common among males than females: NHL, 0.62% vs. 0.32%; HL, 0.28% vs. 0.13%; and CTCL, 0.09% vs. 0.04%. This translated into significantly increased HS-associated risks, Ms. Tannenbaum and her coauthors noted. “For example, the [odds ratios] for the association between HS and HL were higher in males (OR, 2.97; 95% confidence interval, 2.22-3.99) than in females (OR, 1.86; 95% CI, 1.44-2.39) (P = .02),” they wrote.
Lymphomas were more common among HS patients in every age group. Patients with HS aged 45-64 years were 38% more likely to develop NHL, and those older than 65, about twice as likely (OR, 1.99).
“To our knowledge, this is the first investigation to systematically evaluate this association in a U.S. population of patients with HS,” the research team concluded.
The study was supported by a grant from AbbVie. Ms. Tannenbaum and Mr. Strunk reported no disclosures. Dr. Garg reported financial relationships with AbbVie and several other pharmaceutical companies.
SOURCE: Tannenbaum R et al. JAMA Dermatol. 2019 Jan 30. doi: 10.1001/jamadermatol.2018.5230.
Lymphomas appear to be up to four times more likely in patients with hidradenitis suppurativa than among the general population, Rachel Tannenbaum and her colleagues reported in a Research Letter in JAMA Dermatology.
The risks of Hodgkin (HL), non-Hodgkin (NHL), and cutaneous T-cell lymphoma (CTCL) all were significantly higher among patients with HS, wrote Ms. Tannenbaum, Andrew Strunk, and Amit Garg, MD. Males and older patients carried higher risks than females and younger patients, they found.
The team members, of Hofstra University, Hempstead, N.Y., conducted a health care database study comprising 55 million patients included in 27 integrated U.S. health care systems. All the subjects were at least 18 years old; records indicated active HS during the study period of 2013-2018. A regression analysis controlled for age and sex.
The database contained 62,690 patients with HS. The majority (74%) were female and were aged 44 years or younger (57%).
All three lymphomas were more common among HS patients than patients without HS, including non-Hodgkin lymphoma (0.40% vs. 0.35%,) Hodgkin lymphoma (0.17% vs. 0.09%), and cutaneous T-cell lymphoma (0.06% vs. 0.02%).
The multivariate analysis determined that HS patients were twice as likely to develop both non-Hodgkin and Hodgkin lymphoma (odds ratio, 2.0 and 2.21, respectively). They were four times more likely to develop cutaneous T-cell lymphoma (OR, 4.31).
All three lymphomas were more common among males than females: NHL, 0.62% vs. 0.32%; HL, 0.28% vs. 0.13%; and CTCL, 0.09% vs. 0.04%. This translated into significantly increased HS-associated risks, Ms. Tannenbaum and her coauthors noted. “For example, the [odds ratios] for the association between HS and HL were higher in males (OR, 2.97; 95% confidence interval, 2.22-3.99) than in females (OR, 1.86; 95% CI, 1.44-2.39) (P = .02),” they wrote.
Lymphomas were more common among HS patients in every age group. Patients with HS aged 45-64 years were 38% more likely to develop NHL, and those older than 65, about twice as likely (OR, 1.99).
“To our knowledge, this is the first investigation to systematically evaluate this association in a U.S. population of patients with HS,” the research team concluded.
The study was supported by a grant from AbbVie. Ms. Tannenbaum and Mr. Strunk reported no disclosures. Dr. Garg reported financial relationships with AbbVie and several other pharmaceutical companies.
SOURCE: Tannenbaum R et al. JAMA Dermatol. 2019 Jan 30. doi: 10.1001/jamadermatol.2018.5230.
FROM JAMA DERMATOLOGY
Key clinical point: Hidradenitis suppurativa appears to increase the risk of cutaneous T-cell lymphoma, Hodgkin, and non-Hodgkin lymphomas.
Major finding: Lymphomas are up to four times more common among patients with hidradenitis suppurativa than those without the chronic inflammatory disorder.
Study details: The database review comprised more than 55 million patients in 27 linked health care systems.
Disclosures: This study was supported by a grant from AbbVie. Ms. Tannenbaum and Mr. Strunk reported no disclosures. Dr. Garg reported financial relationships with AbbVie and several other pharmaceutical companies.
Source: Tannenbaum R et al. JAMA Dermatol. 2019 Jan 30. doi: 10.1001/jamadermatol.2018.5230.
Case report: Longstanding actinic keratosis responds to kanuka honey
GRAND CAYMAN, CAYMAN ISLANDS – Not all honeys are created equal, Theodore Rosen, MD, said at the meeting provided by Global Academy for Medical Education.

“It seems that kanuka is the new manuka,” said Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston. These lesser-known New Zealand bush honeys may be something to watch because research and case reports continue to provide intriguing hints of how these honeys exert their immunomodulatory effects on skin, he commented, describing a recent case report describing the elimination of a large, long-standing actinic keratosis (AK) with application of kanuka honey.
Manuka (Leptospermum scoparium) is a large bush native to both Australia and New Zealand. Kanuka (Kunzea ericoides) is quite similar in size and appearance, but native only to New Zealand. Honey made from the flowers of these bushes possesses some unique properties that make it an attractive addition to wound healing regimens, according to a 2014 study (Int J Gen Med. 2014;7:149-58).
The study examined samples of manuka, kanuka, a manuka/kanuka blend, and clover honey. The investigators found that kanuka honey, and to a lesser extent manuka honey, exerted a potent anti-inflammatory effect in human embryonic kidney cells. The honeys interfered with toll-like receptor 1 and 2 signaling, which would reduce the production of proinflammatory cytokines.
Kanuka’s potency seems directly related to its unusually high level of arabinogalactan, according to Saras Mane, MD, primary author of the AK case report (Case Rep Dermatol Med. 2018 May 31;2018:4628971). Dr. Mane is with the Medical Research Institute of New Zealand in Wellington.
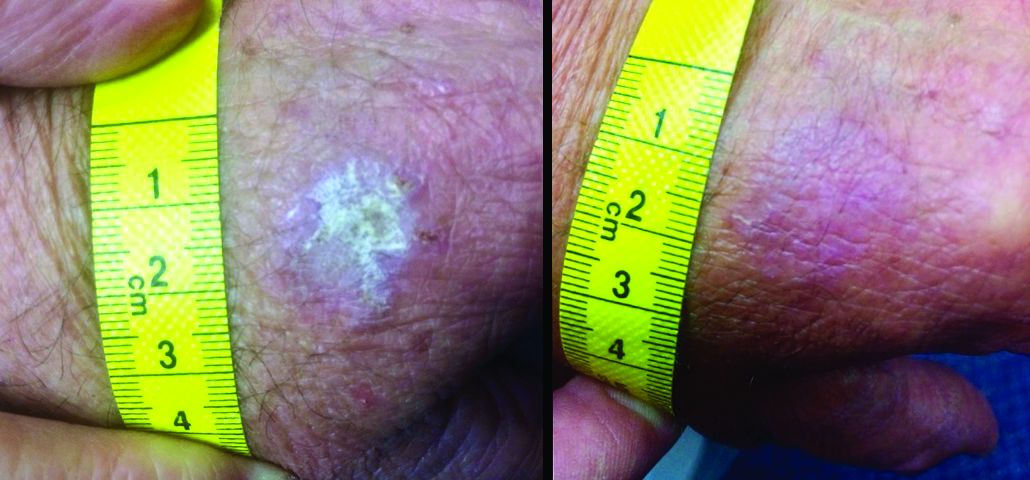
“The immunomodulatory properties of kanuka honey in particular are thought to be more potent than other New Zealand honeys due to the relatively high concentrations of arabinogalactan proteins present,” Dr. Mane and his coauthors wrote in the case report. “These proteins have been shown to stimulate release of TNF-alpha from monocytic cell lines in vitro.”
The report involved a 66-year-old man who was enrolled in a randomized trial of a commercialized medical-grade kanuka honey ointment (Honevo, 90% kanuka honey, 10% glycerin; Honeylab NZ) for rosacea.
The patient also had multiple AKs, including a raised, crusted, scaly lesion measuring 20 mm by 21 mm with marginal erythema on the back of one hand. The lesion had been present and dormant for a number of years, but it had recently begun to grow.

“This gentleman decided he’d just try the honey on his AK, too,” Dr. Rosen said. The man reported applying a small amount to the lesion and erythematous area once a day, leaving it on for about 30 to 60 minutes. After 5 days, he stopped because the lesion became tender. During the next two days, the patient reported “picking at” the lesion, which was softening. He repeated this cycle of treatment for 3 months with no other therapy to the lesion.
“The lesion gradually reduced in size with an initial rapid reduction in its dry, crusted nature,” the authors reported. “After 3 months, residual appearance of the lesion was a 20 mm by 17 mm area of pink skin with no elements of hypertrophy, crusting, or loss of skin integrity,” they noted. “At 6 months, there were no signs of recurrence. At 9 months, the appearance of the skin had fully returned to normal. A telephone follow-up was conducted at 2 years after treatment, and the patient reported that his skin in the area was still completely normal and that there were no signs of recurrence.”
Dr. Mane noted that they had only clinical evidence, and no histology of the lesion either before or after its change. “The AK was diagnosed and treated in primary care, where it is not usual for AKs to be biopsied, and the decision to write up the case was made after the course of treatment had finished,” they said.
“Immunomodulatory topical agents are already widely used in the treatment of AK as an immune component is evident in its etiology,” they wrote. “Immunocompromised patients have 250 times the risk of developing an AK than the general population.”
Dr. Rosen said that kanuka honey is also being investigated in psoriasis, eczema, acne, herpes simplex virus, and diaper dermatitis. It is also being studied for rosacea.
Dr. Mane declared no conflicts of interest. Some coauthors disclosed that they have previously received funding from HoneyLab NZ. Dr. Rosen has no commercial interest in HoneyLab.
The meeting was sponsored by Global Academy for Medical Education; Global Academy and this news organization are owned by the same parent company.
GRAND CAYMAN, CAYMAN ISLANDS – Not all honeys are created equal, Theodore Rosen, MD, said at the meeting provided by Global Academy for Medical Education.
“It seems that kanuka is the new manuka,” said Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston. These lesser-known New Zealand bush honeys may be something to watch because research and case reports continue to provide intriguing hints of how these honeys exert their immunomodulatory effects on skin, he commented, describing a recent case report describing the elimination of a large, long-standing actinic keratosis (AK) with application of kanuka honey.
Manuka (Leptospermum scoparium) is a large bush native to both Australia and New Zealand. Kanuka (Kunzea ericoides) is quite similar in size and appearance, but native only to New Zealand. Honey made from the flowers of these bushes possesses some unique properties that make it an attractive addition to wound healing regimens, according to a 2014 study (Int J Gen Med. 2014;7:149-58).
The study examined samples of manuka, kanuka, a manuka/kanuka blend, and clover honey. The investigators found that kanuka honey, and to a lesser extent manuka honey, exerted a potent anti-inflammatory effect in human embryonic kidney cells. The honeys interfered with toll-like receptor 1 and 2 signaling, which would reduce the production of proinflammatory cytokines.
Kanuka’s potency seems directly related to its unusually high level of arabinogalactan, according to Saras Mane, MD, primary author of the AK case report (Case Rep Dermatol Med. 2018 May 31;2018:4628971). Dr. Mane is with the Medical Research Institute of New Zealand in Wellington.
“The immunomodulatory properties of kanuka honey in particular are thought to be more potent than other New Zealand honeys due to the relatively high concentrations of arabinogalactan proteins present,” Dr. Mane and his coauthors wrote in the case report. “These proteins have been shown to stimulate release of TNF-alpha from monocytic cell lines in vitro.”
The report involved a 66-year-old man who was enrolled in a randomized trial of a commercialized medical-grade kanuka honey ointment (Honevo, 90% kanuka honey, 10% glycerin; Honeylab NZ) for rosacea.
The patient also had multiple AKs, including a raised, crusted, scaly lesion measuring 20 mm by 21 mm with marginal erythema on the back of one hand. The lesion had been present and dormant for a number of years, but it had recently begun to grow.
“This gentleman decided he’d just try the honey on his AK, too,” Dr. Rosen said. The man reported applying a small amount to the lesion and erythematous area once a day, leaving it on for about 30 to 60 minutes. After 5 days, he stopped because the lesion became tender. During the next two days, the patient reported “picking at” the lesion, which was softening. He repeated this cycle of treatment for 3 months with no other therapy to the lesion.
“The lesion gradually reduced in size with an initial rapid reduction in its dry, crusted nature,” the authors reported. “After 3 months, residual appearance of the lesion was a 20 mm by 17 mm area of pink skin with no elements of hypertrophy, crusting, or loss of skin integrity,” they noted. “At 6 months, there were no signs of recurrence. At 9 months, the appearance of the skin had fully returned to normal. A telephone follow-up was conducted at 2 years after treatment, and the patient reported that his skin in the area was still completely normal and that there were no signs of recurrence.”
Dr. Mane noted that they had only clinical evidence, and no histology of the lesion either before or after its change. “The AK was diagnosed and treated in primary care, where it is not usual for AKs to be biopsied, and the decision to write up the case was made after the course of treatment had finished,” they said.
“Immunomodulatory topical agents are already widely used in the treatment of AK as an immune component is evident in its etiology,” they wrote. “Immunocompromised patients have 250 times the risk of developing an AK than the general population.”
Dr. Rosen said that kanuka honey is also being investigated in psoriasis, eczema, acne, herpes simplex virus, and diaper dermatitis. It is also being studied for rosacea.
Dr. Mane declared no conflicts of interest. Some coauthors disclosed that they have previously received funding from HoneyLab NZ. Dr. Rosen has no commercial interest in HoneyLab.
The meeting was sponsored by Global Academy for Medical Education; Global Academy and this news organization are owned by the same parent company.
GRAND CAYMAN, CAYMAN ISLANDS – Not all honeys are created equal, Theodore Rosen, MD, said at the meeting provided by Global Academy for Medical Education.
“It seems that kanuka is the new manuka,” said Dr. Rosen, professor of dermatology at Baylor College of Medicine, Houston. These lesser-known New Zealand bush honeys may be something to watch because research and case reports continue to provide intriguing hints of how these honeys exert their immunomodulatory effects on skin, he commented, describing a recent case report describing the elimination of a large, long-standing actinic keratosis (AK) with application of kanuka honey.
Manuka (Leptospermum scoparium) is a large bush native to both Australia and New Zealand. Kanuka (Kunzea ericoides) is quite similar in size and appearance, but native only to New Zealand. Honey made from the flowers of these bushes possesses some unique properties that make it an attractive addition to wound healing regimens, according to a 2014 study (Int J Gen Med. 2014;7:149-58).
The study examined samples of manuka, kanuka, a manuka/kanuka blend, and clover honey. The investigators found that kanuka honey, and to a lesser extent manuka honey, exerted a potent anti-inflammatory effect in human embryonic kidney cells. The honeys interfered with toll-like receptor 1 and 2 signaling, which would reduce the production of proinflammatory cytokines.
Kanuka’s potency seems directly related to its unusually high level of arabinogalactan, according to Saras Mane, MD, primary author of the AK case report (Case Rep Dermatol Med. 2018 May 31;2018:4628971). Dr. Mane is with the Medical Research Institute of New Zealand in Wellington.
“The immunomodulatory properties of kanuka honey in particular are thought to be more potent than other New Zealand honeys due to the relatively high concentrations of arabinogalactan proteins present,” Dr. Mane and his coauthors wrote in the case report. “These proteins have been shown to stimulate release of TNF-alpha from monocytic cell lines in vitro.”
The report involved a 66-year-old man who was enrolled in a randomized trial of a commercialized medical-grade kanuka honey ointment (Honevo, 90% kanuka honey, 10% glycerin; Honeylab NZ) for rosacea.
The patient also had multiple AKs, including a raised, crusted, scaly lesion measuring 20 mm by 21 mm with marginal erythema on the back of one hand. The lesion had been present and dormant for a number of years, but it had recently begun to grow.
“This gentleman decided he’d just try the honey on his AK, too,” Dr. Rosen said. The man reported applying a small amount to the lesion and erythematous area once a day, leaving it on for about 30 to 60 minutes. After 5 days, he stopped because the lesion became tender. During the next two days, the patient reported “picking at” the lesion, which was softening. He repeated this cycle of treatment for 3 months with no other therapy to the lesion.
“The lesion gradually reduced in size with an initial rapid reduction in its dry, crusted nature,” the authors reported. “After 3 months, residual appearance of the lesion was a 20 mm by 17 mm area of pink skin with no elements of hypertrophy, crusting, or loss of skin integrity,” they noted. “At 6 months, there were no signs of recurrence. At 9 months, the appearance of the skin had fully returned to normal. A telephone follow-up was conducted at 2 years after treatment, and the patient reported that his skin in the area was still completely normal and that there were no signs of recurrence.”
Dr. Mane noted that they had only clinical evidence, and no histology of the lesion either before or after its change. “The AK was diagnosed and treated in primary care, where it is not usual for AKs to be biopsied, and the decision to write up the case was made after the course of treatment had finished,” they said.
“Immunomodulatory topical agents are already widely used in the treatment of AK as an immune component is evident in its etiology,” they wrote. “Immunocompromised patients have 250 times the risk of developing an AK than the general population.”
Dr. Rosen said that kanuka honey is also being investigated in psoriasis, eczema, acne, herpes simplex virus, and diaper dermatitis. It is also being studied for rosacea.
Dr. Mane declared no conflicts of interest. Some coauthors disclosed that they have previously received funding from HoneyLab NZ. Dr. Rosen has no commercial interest in HoneyLab.
The meeting was sponsored by Global Academy for Medical Education; Global Academy and this news organization are owned by the same parent company.
REPORTING FROM THE ANNUAL CARIBBEAN DERMATOLOGY SYMPOSIUM

Phase 3 studies of antiamyloid Alzheimer’s drug crenezumab stopped
After a disappointing interim analysis, Roche and its collaborator AC Immune are halting two phase 3 trials of the antiamyloid antibody crenezumab.

CREAD 1 and CREAD 2 enrolled patients with prodromal-to-mild sporadic Alzheimer’s disease. The preplanned interim safety and efficacy analysis determined that neither study was likely to meet the primary endpoint of change from baseline on the Clinical Dementia Rating-sum of boxes score.
There were no unexpected safety signals associated with the drug, despite a quadrupling of the phase 3 dose from that used in phase 2. The company in its press release said that it will continue to conduct the Autosomal Dominant Alzheimer’s Disease (ADAD) trial as part of the Alzheimer’s Prevention Initiative (API). ADAD is a large South American trial of crenezumab in Colombian families with familial Alzheimer’s caused by mutations in the presenilin-1 gene (PSEN1).
Roche did not release any data but said the trial results will be discussed at an upcoming scientific meeting.
“While the results with crenezumab are disappointing, they meaningfully contribute to our understanding of Alzheimer’s disease,” Sandra Horning, MD, Roche’s chief medical officer and executive vice president for global development, said in an interview. “We gratefully acknowledge the participants in the CREAD trials and the efforts of everyone involved in this important program.”
The decision was not a surprise to researchers who have followed the antibody’s development. It advanced into phase 3 with lackluster phase 2 cognitive, imaging, and biomarker data. Its selection as the therapeutic agent for the ADAD trial was a key driver in its continued development, securing Roche $100 million in federal funds to help launch ADAD, the first-ever Alzheimer’s primary prevention study.
Despite its failure in sporadic Alzheimer’s, there is still some hope that crenezumab might benefit people with the PSEN1 mutation, said Richard Caselli, MD, professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.
“The Colombian trial is aimed at dominantly-inherited AD due to a PSEN1 mutation, so it is different enough to imagine it still might make a difference in patients in whom amyloid metabolism is actually defective due to functionally altered amyloid precursor protein or gamma secretase,” he said in an interview. “Possibly some might argue that many of the patients in the crenezumab trial likely had additional pathologies so that even if the AD component responded, the overall clinical picture might not reflect it due to the other components. That would be interesting if proven and could even argue against equating young-onset with late-onset AD, at least for clinical purposes, as is currently envisioned.”
Michael Wolfe, PhD, had a different take on the matter.
“Although amyloid-beta [Abeta] production is not necessarily altered in sporadic AD, there is essentially the same pathology, presentation, and progression with familial and sporadic AD, suggesting a common molecular mechanism,” said Dr. Wolfe, who is the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence. “It’s hard to say Abeta is the pathogenic species in familial but not sporadic AD.

“To me, the failures of the antiamyloid approaches are because the drugs are given too late, are targeting the wrong form of Abeta, or are targeting an enzyme [for example, beta secretase1] that has other important functions. Most likely it’s a combination of these reasons. One could argue that even if some form of Abeta is the pathogenic entity, it is not a practical target because intervention may need to be initiated many years before the onset of symptoms.”
Despite the long string of failed antiamyloid antibodies, it’s not yet time to give up on the approach, said James Kupiec, MD, chief medical officer at ProMIS Neurosciences of Toronto.
“I understand where the pessimism [around antiamyloid antibodies] is coming from, and I also understand the enthusiasm from these companies to pursue them,” said Dr. Kupiec, who formerly headed Pfizer’s neuroscience research unit. “Targeting plaque is clearly not going to do the job. But in my opinion, the deeper pathophysiologic questions have not been adequately addressed. I’m not willing to throw in the towel. The correct molecular species [of amyloid] has not been appropriately or adequately tested in studies with monoclonal antibodies.”
The antibodies that have been failing for 5 years now were designed in the early 2000s, Dr. Kupiec pointed out, when knowledge of the various amyloid species was still immature. Newer candidates can target specific conformations of the protein – monomers and oligomers – before they aggregate into insoluble sheets. “Solanezumab was the first of these, paving the way for this new generation of antibodies,” Dr. Kupiec said.
Because they target soluble Abeta, not amyloid plaques, these domain-specific antibodies are less likely to elicit ARIA (amyloid-related imaging abnormalities), the inflammatory reaction that’s been associated with plaque dissolution in other antibody trials. ARIA has been a dose-limiting step for antiamyloid antibodies – one that conformationally targeted antibodies could avoid, Dr. Kupiec said.
“There may be some limited success with the these, and there may be enough of a treatment effect to secure approval,” he said. “The question is: Can we generate a higher effect size with an antibody that is more selective to the toxic forms of Abeta?”
PMN310 is ProMIS’ attempt to thread this needle. In preclinical studies, the antibody did not bind to amyloid monomers, plaques, or vascular Abeta aggregates. The company expects to take this antibody into phase 1 trials later this year.
“If we have a molecule that doesn’t bind to monomers or to plaques, but only to the toxic oligomer, then that is an something well worth testing in the clinic,” he said.
Dr. Caselli and Dr. Wolfe have no financial disclosures.
On behalf of the millions of people living with Alzheimer’s disease and their families that we serve and represent, the Alzheimer’s Association is disappointed to learn that these trials have been stopped.

We learn something from every Alzheimer’s clinical trial. The Alzheimer’s Association looks forward to hearing details of these studies at an upcoming scientific meeting.
More important, we must redouble our efforts to better understand the causes of the disease, and to discover additional therapeutic targets. No stone can be left unturned in the pursuit of better treatments and effective preventions.
The Alzheimer’s Association is investing in research looking at a variety of novel targets for treatment and prevention, including brain inflammation, the life and death cycle of brain cells, how brain cells use different energy sources, and the impact of lifestyle.
• Lifestyle interventions include leading the U.S. POINTER Study.
• To further the study of blood pressure control on reducing risk of mild cognitive impairment and dementia, the Alzheimer’s Association recently announced seed funding of SPRINT MIND 2.0.
• Part The Cloud Translational Research program fills a gap in Alzheimer’s drug development by supporting more than 30 early phase clinical studies.
• The Association is also funding research into the causes of the disease.
The emotional and financial cost of Alzheimer’s is enormous. At the Alzheimer’s Association, we will not stop. We will not slow down in our fight against this terrible disease.
Maria Carrillo, PhD , is the Alzheimer’s Association’s chief science officer.
On behalf of the millions of people living with Alzheimer’s disease and their families that we serve and represent, the Alzheimer’s Association is disappointed to learn that these trials have been stopped.
We learn something from every Alzheimer’s clinical trial. The Alzheimer’s Association looks forward to hearing details of these studies at an upcoming scientific meeting.
More important, we must redouble our efforts to better understand the causes of the disease, and to discover additional therapeutic targets. No stone can be left unturned in the pursuit of better treatments and effective preventions.
The Alzheimer’s Association is investing in research looking at a variety of novel targets for treatment and prevention, including brain inflammation, the life and death cycle of brain cells, how brain cells use different energy sources, and the impact of lifestyle.
• Lifestyle interventions include leading the U.S. POINTER Study.
• To further the study of blood pressure control on reducing risk of mild cognitive impairment and dementia, the Alzheimer’s Association recently announced seed funding of SPRINT MIND 2.0.
• Part The Cloud Translational Research program fills a gap in Alzheimer’s drug development by supporting more than 30 early phase clinical studies.
• The Association is also funding research into the causes of the disease.
The emotional and financial cost of Alzheimer’s is enormous. At the Alzheimer’s Association, we will not stop. We will not slow down in our fight against this terrible disease.
Maria Carrillo, PhD , is the Alzheimer’s Association’s chief science officer.
On behalf of the millions of people living with Alzheimer’s disease and their families that we serve and represent, the Alzheimer’s Association is disappointed to learn that these trials have been stopped.
We learn something from every Alzheimer’s clinical trial. The Alzheimer’s Association looks forward to hearing details of these studies at an upcoming scientific meeting.
More important, we must redouble our efforts to better understand the causes of the disease, and to discover additional therapeutic targets. No stone can be left unturned in the pursuit of better treatments and effective preventions.
The Alzheimer’s Association is investing in research looking at a variety of novel targets for treatment and prevention, including brain inflammation, the life and death cycle of brain cells, how brain cells use different energy sources, and the impact of lifestyle.
• Lifestyle interventions include leading the U.S. POINTER Study.
• To further the study of blood pressure control on reducing risk of mild cognitive impairment and dementia, the Alzheimer’s Association recently announced seed funding of SPRINT MIND 2.0.
• Part The Cloud Translational Research program fills a gap in Alzheimer’s drug development by supporting more than 30 early phase clinical studies.
• The Association is also funding research into the causes of the disease.
The emotional and financial cost of Alzheimer’s is enormous. At the Alzheimer’s Association, we will not stop. We will not slow down in our fight against this terrible disease.
Maria Carrillo, PhD , is the Alzheimer’s Association’s chief science officer.
After a disappointing interim analysis, Roche and its collaborator AC Immune are halting two phase 3 trials of the antiamyloid antibody crenezumab.
CREAD 1 and CREAD 2 enrolled patients with prodromal-to-mild sporadic Alzheimer’s disease. The preplanned interim safety and efficacy analysis determined that neither study was likely to meet the primary endpoint of change from baseline on the Clinical Dementia Rating-sum of boxes score.
There were no unexpected safety signals associated with the drug, despite a quadrupling of the phase 3 dose from that used in phase 2. The company in its press release said that it will continue to conduct the Autosomal Dominant Alzheimer’s Disease (ADAD) trial as part of the Alzheimer’s Prevention Initiative (API). ADAD is a large South American trial of crenezumab in Colombian families with familial Alzheimer’s caused by mutations in the presenilin-1 gene (PSEN1).
Roche did not release any data but said the trial results will be discussed at an upcoming scientific meeting.
“While the results with crenezumab are disappointing, they meaningfully contribute to our understanding of Alzheimer’s disease,” Sandra Horning, MD, Roche’s chief medical officer and executive vice president for global development, said in an interview. “We gratefully acknowledge the participants in the CREAD trials and the efforts of everyone involved in this important program.”
The decision was not a surprise to researchers who have followed the antibody’s development. It advanced into phase 3 with lackluster phase 2 cognitive, imaging, and biomarker data. Its selection as the therapeutic agent for the ADAD trial was a key driver in its continued development, securing Roche $100 million in federal funds to help launch ADAD, the first-ever Alzheimer’s primary prevention study.
Despite its failure in sporadic Alzheimer’s, there is still some hope that crenezumab might benefit people with the PSEN1 mutation, said Richard Caselli, MD, professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.
“The Colombian trial is aimed at dominantly-inherited AD due to a PSEN1 mutation, so it is different enough to imagine it still might make a difference in patients in whom amyloid metabolism is actually defective due to functionally altered amyloid precursor protein or gamma secretase,” he said in an interview. “Possibly some might argue that many of the patients in the crenezumab trial likely had additional pathologies so that even if the AD component responded, the overall clinical picture might not reflect it due to the other components. That would be interesting if proven and could even argue against equating young-onset with late-onset AD, at least for clinical purposes, as is currently envisioned.”
Michael Wolfe, PhD, had a different take on the matter.
“Although amyloid-beta [Abeta] production is not necessarily altered in sporadic AD, there is essentially the same pathology, presentation, and progression with familial and sporadic AD, suggesting a common molecular mechanism,” said Dr. Wolfe, who is the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence. “It’s hard to say Abeta is the pathogenic species in familial but not sporadic AD.
“To me, the failures of the antiamyloid approaches are because the drugs are given too late, are targeting the wrong form of Abeta, or are targeting an enzyme [for example, beta secretase1] that has other important functions. Most likely it’s a combination of these reasons. One could argue that even if some form of Abeta is the pathogenic entity, it is not a practical target because intervention may need to be initiated many years before the onset of symptoms.”
Despite the long string of failed antiamyloid antibodies, it’s not yet time to give up on the approach, said James Kupiec, MD, chief medical officer at ProMIS Neurosciences of Toronto.
“I understand where the pessimism [around antiamyloid antibodies] is coming from, and I also understand the enthusiasm from these companies to pursue them,” said Dr. Kupiec, who formerly headed Pfizer’s neuroscience research unit. “Targeting plaque is clearly not going to do the job. But in my opinion, the deeper pathophysiologic questions have not been adequately addressed. I’m not willing to throw in the towel. The correct molecular species [of amyloid] has not been appropriately or adequately tested in studies with monoclonal antibodies.”
The antibodies that have been failing for 5 years now were designed in the early 2000s, Dr. Kupiec pointed out, when knowledge of the various amyloid species was still immature. Newer candidates can target specific conformations of the protein – monomers and oligomers – before they aggregate into insoluble sheets. “Solanezumab was the first of these, paving the way for this new generation of antibodies,” Dr. Kupiec said.
Because they target soluble Abeta, not amyloid plaques, these domain-specific antibodies are less likely to elicit ARIA (amyloid-related imaging abnormalities), the inflammatory reaction that’s been associated with plaque dissolution in other antibody trials. ARIA has been a dose-limiting step for antiamyloid antibodies – one that conformationally targeted antibodies could avoid, Dr. Kupiec said.
“There may be some limited success with the these, and there may be enough of a treatment effect to secure approval,” he said. “The question is: Can we generate a higher effect size with an antibody that is more selective to the toxic forms of Abeta?”
PMN310 is ProMIS’ attempt to thread this needle. In preclinical studies, the antibody did not bind to amyloid monomers, plaques, or vascular Abeta aggregates. The company expects to take this antibody into phase 1 trials later this year.
“If we have a molecule that doesn’t bind to monomers or to plaques, but only to the toxic oligomer, then that is an something well worth testing in the clinic,” he said.
Dr. Caselli and Dr. Wolfe have no financial disclosures.
After a disappointing interim analysis, Roche and its collaborator AC Immune are halting two phase 3 trials of the antiamyloid antibody crenezumab.
CREAD 1 and CREAD 2 enrolled patients with prodromal-to-mild sporadic Alzheimer’s disease. The preplanned interim safety and efficacy analysis determined that neither study was likely to meet the primary endpoint of change from baseline on the Clinical Dementia Rating-sum of boxes score.
There were no unexpected safety signals associated with the drug, despite a quadrupling of the phase 3 dose from that used in phase 2. The company in its press release said that it will continue to conduct the Autosomal Dominant Alzheimer’s Disease (ADAD) trial as part of the Alzheimer’s Prevention Initiative (API). ADAD is a large South American trial of crenezumab in Colombian families with familial Alzheimer’s caused by mutations in the presenilin-1 gene (PSEN1).
Roche did not release any data but said the trial results will be discussed at an upcoming scientific meeting.
“While the results with crenezumab are disappointing, they meaningfully contribute to our understanding of Alzheimer’s disease,” Sandra Horning, MD, Roche’s chief medical officer and executive vice president for global development, said in an interview. “We gratefully acknowledge the participants in the CREAD trials and the efforts of everyone involved in this important program.”
The decision was not a surprise to researchers who have followed the antibody’s development. It advanced into phase 3 with lackluster phase 2 cognitive, imaging, and biomarker data. Its selection as the therapeutic agent for the ADAD trial was a key driver in its continued development, securing Roche $100 million in federal funds to help launch ADAD, the first-ever Alzheimer’s primary prevention study.
Despite its failure in sporadic Alzheimer’s, there is still some hope that crenezumab might benefit people with the PSEN1 mutation, said Richard Caselli, MD, professor of neurology at the Mayo Clinic Arizona in Scottsdale and associate director and clinical core director of the Arizona Alzheimer’s Disease Center.
“The Colombian trial is aimed at dominantly-inherited AD due to a PSEN1 mutation, so it is different enough to imagine it still might make a difference in patients in whom amyloid metabolism is actually defective due to functionally altered amyloid precursor protein or gamma secretase,” he said in an interview. “Possibly some might argue that many of the patients in the crenezumab trial likely had additional pathologies so that even if the AD component responded, the overall clinical picture might not reflect it due to the other components. That would be interesting if proven and could even argue against equating young-onset with late-onset AD, at least for clinical purposes, as is currently envisioned.”
Michael Wolfe, PhD, had a different take on the matter.
“Although amyloid-beta [Abeta] production is not necessarily altered in sporadic AD, there is essentially the same pathology, presentation, and progression with familial and sporadic AD, suggesting a common molecular mechanism,” said Dr. Wolfe, who is the Mathias P. Mertes Professor of Medicinal Chemistry at the University of Kansas, Lawrence. “It’s hard to say Abeta is the pathogenic species in familial but not sporadic AD.
“To me, the failures of the antiamyloid approaches are because the drugs are given too late, are targeting the wrong form of Abeta, or are targeting an enzyme [for example, beta secretase1] that has other important functions. Most likely it’s a combination of these reasons. One could argue that even if some form of Abeta is the pathogenic entity, it is not a practical target because intervention may need to be initiated many years before the onset of symptoms.”
Despite the long string of failed antiamyloid antibodies, it’s not yet time to give up on the approach, said James Kupiec, MD, chief medical officer at ProMIS Neurosciences of Toronto.
“I understand where the pessimism [around antiamyloid antibodies] is coming from, and I also understand the enthusiasm from these companies to pursue them,” said Dr. Kupiec, who formerly headed Pfizer’s neuroscience research unit. “Targeting plaque is clearly not going to do the job. But in my opinion, the deeper pathophysiologic questions have not been adequately addressed. I’m not willing to throw in the towel. The correct molecular species [of amyloid] has not been appropriately or adequately tested in studies with monoclonal antibodies.”
The antibodies that have been failing for 5 years now were designed in the early 2000s, Dr. Kupiec pointed out, when knowledge of the various amyloid species was still immature. Newer candidates can target specific conformations of the protein – monomers and oligomers – before they aggregate into insoluble sheets. “Solanezumab was the first of these, paving the way for this new generation of antibodies,” Dr. Kupiec said.
Because they target soluble Abeta, not amyloid plaques, these domain-specific antibodies are less likely to elicit ARIA (amyloid-related imaging abnormalities), the inflammatory reaction that’s been associated with plaque dissolution in other antibody trials. ARIA has been a dose-limiting step for antiamyloid antibodies – one that conformationally targeted antibodies could avoid, Dr. Kupiec said.
“There may be some limited success with the these, and there may be enough of a treatment effect to secure approval,” he said. “The question is: Can we generate a higher effect size with an antibody that is more selective to the toxic forms of Abeta?”
PMN310 is ProMIS’ attempt to thread this needle. In preclinical studies, the antibody did not bind to amyloid monomers, plaques, or vascular Abeta aggregates. The company expects to take this antibody into phase 1 trials later this year.
“If we have a molecule that doesn’t bind to monomers or to plaques, but only to the toxic oligomer, then that is an something well worth testing in the clinic,” he said.
Dr. Caselli and Dr. Wolfe have no financial disclosures.
Guselkumab tops secukinumab over 48 weeks for plaque psoriasis
GRAND CAYMAN, CAYMAN ISLANDS – Guselkumab bested secukinumab in a 48-week-long study of plaque psoriasis, with 84.5% of patients on the interleukin (IL)-23 blocker hitting at least a 90% improvement in their Psoriasis Area Severity Index (PASI), compared with 70% of those taking secukinumab, which blocks IL-17.

The drug was numerically, but not significantly, better than secukinumab in the PASI 75 response at weeks 12 and 48 (84.6% for guselkumab at both time points vs. 80.2% for secukinumab at both time points). This finding on the primary secondary endpoint knocked the P values of the other five into “nominally significant” ranges. But the responses were still good enough for researchers to tag guselkumab as noninferior to its competitor, Jeffrey M. Sobell, MD, said at the meeting provided by Global Academy for Medical Education.
The difference also speaks to the difference in the drugs’ onset of action and its peak efficacy, said Dr. Sobell, of the department of dermatology at Tufts University, Boston.
“In both groups, the PASI 90 increased similarly in the first month,” to about 20%, he commented. “But at week 12 and after, it was consistently higher in guselkumab, peaking around week 28. Secukinumab peaked around weeks 16 to 20 and then slowly declined.”
Despite not being statistically significant, the other secondary efficacy endpoints were certainly enough to pique the audience’s attention. At week 48, guselkumab topped secukinumab in both PASI 100 (58.2% vs. 48.4%, respectively) and Investigator’s Global Assessment (IGA) scores of 0 (62.2% vs. 50.4%) and 0-1 (85% vs. 74.9%).
ECLIPSE randomized 1,048 patients with moderate to severe plaque psoriasis to 100-mg subcutaneous guselkumab at weeks 0, 4, and 12, followed by dosing every 8 weeks, or to 300-mg subcutaneous secukinumab administered by two subcutaneous injections of 150 mg at weeks 0, 1, 2, 3, and 4, followed by dosing every 4 weeks. The primary endpoint of the study was the proportion of patients achieving a PASI 90 response at week 48. Secondary endpoints were assessed at weeks 12 and 48, with safety monitoring through week 56.
The mean baseline Body Surface Area score was 24, and the mean PASI score was 20. Patients had already been treated with phototherapy (51.8%), nonbiologic systemic medications (53.7%), and biologics (29%). About 37% were naive to both nonbiologics and biologics.
Both drugs were well tolerated, with no unanticipated adverse events. Through week 44, the discontinuation rates were 5% for guselkumab and 9% for secukinumab. Adverse events were common in both arms (77.9% and 81.6%, respectively). Serious adverse events occurred in 6.2% and 7.2%, respectively. These included serious infections in six patients taking guselkumab and five taking secukinumab. Superficial Candida infections occurred in 2% of the guselkumab group and 5.7% of the secukinumab group; Tinea infections occurred in 1.7% and 4.5%, respectively.
The session was sponsored by Janssen, the manufacturer of guselkumab (Tremfya). Dr. Sobell is a consultant for Janssen and also disclosed relationships with AbbVie, Amgen, Celgene, Eli Lilly, Merck, Novartis, Regeneron, and Sun Pharma. Secukinumab is marketed as Cosentyx.
Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
GRAND CAYMAN, CAYMAN ISLANDS – Guselkumab bested secukinumab in a 48-week-long study of plaque psoriasis, with 84.5% of patients on the interleukin (IL)-23 blocker hitting at least a 90% improvement in their Psoriasis Area Severity Index (PASI), compared with 70% of those taking secukinumab, which blocks IL-17.
The drug was numerically, but not significantly, better than secukinumab in the PASI 75 response at weeks 12 and 48 (84.6% for guselkumab at both time points vs. 80.2% for secukinumab at both time points). This finding on the primary secondary endpoint knocked the P values of the other five into “nominally significant” ranges. But the responses were still good enough for researchers to tag guselkumab as noninferior to its competitor, Jeffrey M. Sobell, MD, said at the meeting provided by Global Academy for Medical Education.
The difference also speaks to the difference in the drugs’ onset of action and its peak efficacy, said Dr. Sobell, of the department of dermatology at Tufts University, Boston.
“In both groups, the PASI 90 increased similarly in the first month,” to about 20%, he commented. “But at week 12 and after, it was consistently higher in guselkumab, peaking around week 28. Secukinumab peaked around weeks 16 to 20 and then slowly declined.”
Despite not being statistically significant, the other secondary efficacy endpoints were certainly enough to pique the audience’s attention. At week 48, guselkumab topped secukinumab in both PASI 100 (58.2% vs. 48.4%, respectively) and Investigator’s Global Assessment (IGA) scores of 0 (62.2% vs. 50.4%) and 0-1 (85% vs. 74.9%).
ECLIPSE randomized 1,048 patients with moderate to severe plaque psoriasis to 100-mg subcutaneous guselkumab at weeks 0, 4, and 12, followed by dosing every 8 weeks, or to 300-mg subcutaneous secukinumab administered by two subcutaneous injections of 150 mg at weeks 0, 1, 2, 3, and 4, followed by dosing every 4 weeks. The primary endpoint of the study was the proportion of patients achieving a PASI 90 response at week 48. Secondary endpoints were assessed at weeks 12 and 48, with safety monitoring through week 56.
The mean baseline Body Surface Area score was 24, and the mean PASI score was 20. Patients had already been treated with phototherapy (51.8%), nonbiologic systemic medications (53.7%), and biologics (29%). About 37% were naive to both nonbiologics and biologics.
Both drugs were well tolerated, with no unanticipated adverse events. Through week 44, the discontinuation rates were 5% for guselkumab and 9% for secukinumab. Adverse events were common in both arms (77.9% and 81.6%, respectively). Serious adverse events occurred in 6.2% and 7.2%, respectively. These included serious infections in six patients taking guselkumab and five taking secukinumab. Superficial Candida infections occurred in 2% of the guselkumab group and 5.7% of the secukinumab group; Tinea infections occurred in 1.7% and 4.5%, respectively.
The session was sponsored by Janssen, the manufacturer of guselkumab (Tremfya). Dr. Sobell is a consultant for Janssen and also disclosed relationships with AbbVie, Amgen, Celgene, Eli Lilly, Merck, Novartis, Regeneron, and Sun Pharma. Secukinumab is marketed as Cosentyx.
Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
GRAND CAYMAN, CAYMAN ISLANDS – Guselkumab bested secukinumab in a 48-week-long study of plaque psoriasis, with 84.5% of patients on the interleukin (IL)-23 blocker hitting at least a 90% improvement in their Psoriasis Area Severity Index (PASI), compared with 70% of those taking secukinumab, which blocks IL-17.
The drug was numerically, but not significantly, better than secukinumab in the PASI 75 response at weeks 12 and 48 (84.6% for guselkumab at both time points vs. 80.2% for secukinumab at both time points). This finding on the primary secondary endpoint knocked the P values of the other five into “nominally significant” ranges. But the responses were still good enough for researchers to tag guselkumab as noninferior to its competitor, Jeffrey M. Sobell, MD, said at the meeting provided by Global Academy for Medical Education.
The difference also speaks to the difference in the drugs’ onset of action and its peak efficacy, said Dr. Sobell, of the department of dermatology at Tufts University, Boston.
“In both groups, the PASI 90 increased similarly in the first month,” to about 20%, he commented. “But at week 12 and after, it was consistently higher in guselkumab, peaking around week 28. Secukinumab peaked around weeks 16 to 20 and then slowly declined.”
Despite not being statistically significant, the other secondary efficacy endpoints were certainly enough to pique the audience’s attention. At week 48, guselkumab topped secukinumab in both PASI 100 (58.2% vs. 48.4%, respectively) and Investigator’s Global Assessment (IGA) scores of 0 (62.2% vs. 50.4%) and 0-1 (85% vs. 74.9%).
ECLIPSE randomized 1,048 patients with moderate to severe plaque psoriasis to 100-mg subcutaneous guselkumab at weeks 0, 4, and 12, followed by dosing every 8 weeks, or to 300-mg subcutaneous secukinumab administered by two subcutaneous injections of 150 mg at weeks 0, 1, 2, 3, and 4, followed by dosing every 4 weeks. The primary endpoint of the study was the proportion of patients achieving a PASI 90 response at week 48. Secondary endpoints were assessed at weeks 12 and 48, with safety monitoring through week 56.
The mean baseline Body Surface Area score was 24, and the mean PASI score was 20. Patients had already been treated with phototherapy (51.8%), nonbiologic systemic medications (53.7%), and biologics (29%). About 37% were naive to both nonbiologics and biologics.
Both drugs were well tolerated, with no unanticipated adverse events. Through week 44, the discontinuation rates were 5% for guselkumab and 9% for secukinumab. Adverse events were common in both arms (77.9% and 81.6%, respectively). Serious adverse events occurred in 6.2% and 7.2%, respectively. These included serious infections in six patients taking guselkumab and five taking secukinumab. Superficial Candida infections occurred in 2% of the guselkumab group and 5.7% of the secukinumab group; Tinea infections occurred in 1.7% and 4.5%, respectively.
The session was sponsored by Janssen, the manufacturer of guselkumab (Tremfya). Dr. Sobell is a consultant for Janssen and also disclosed relationships with AbbVie, Amgen, Celgene, Eli Lilly, Merck, Novartis, Regeneron, and Sun Pharma. Secukinumab is marketed as Cosentyx.
Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
REPORTING FROM THE CARIBBEAN DERMATOLOGY SYMPOSIUM
Key clinical point: Guselkumab outperformed secukinumab for patients with moderate to severe plaque psoriasis.
Major finding: PASI 90 was achieved in 84.5% of patients on guselkumab and 70% on secukinumab.
Study details: The phase 3 study randomized 1,048 patients to guselkumab or secukinumab.
Disclosures: The session was sponsored by Janssen, the manufacturer of guselkumab (Tremfya). Dr. Sobell is a consultant for Janssen and also disclosed relationships with AbbVie, Amgen, Celgene, Eli Lilly, Merck, Novartis, Regeneron, and Sun Pharma.
SPRINT MIND published: Extension trial to add 2 years’ follow-up
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.
SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.

The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.

Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.
Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
SPRINT MIND offers hope that a very achievable blood pressure goal can dramatically alter the trajectory from mild cognitive impairment to dementia, Kristine Yaffe, MD, wrote in an accompanying editorial. But at this point, it’s impossible to make specific clinical recommendations.
Additionally it is not possible, right now, to know which hypertension treatment regimens were most effective in improved cognitive outcomes.
“Information necessary to compare the effects of classes of antihypertensive agents on cognitive outcomes is also not provided. SPRINT used a quasi-pragmatic approach with suggestions for treatment choice, but practitioners approached SBP control individually, and most participants were taking multiple drugs.”
Nevertheless, the positive secondary findings and the encouraging trajectory on dementia risk should fix blood pressure management squarely into a cornerstone of dementia prevention algorithms.
“The SPRINT MIND study may not be the final approach for prevention of AD or other cognitive impairment, but it represents a major leap forward in what has emerged as a marathon journey.”
Dr. Kristine Yaffe is professor of psychiatry, neurology and epidemiology and the Roy and Marie Scola Endowed Chair at the University of California, San Francisco.
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.
SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.
The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
A new iteration of the SPRINT MIND hypertension trial will seek to prove conclusively the original study’s tantalizing suggestion: that intensive blood pressure control decreases the risk of developing mild cognitive impairment (MCI) and, eventually, dementia.
SPRINT MIND 2.0 will re-recruit SPRINT MIND subjects and enable another follow-up cognitive assessment and other clinical tests as they remain on their standard of care blood pressure regimen. It is largely funded by an $800,000 grant from the Alzheimer’s Association.
Initially released last July at the Alzheimer’s Association International Conference, the results of the SPRINT MIND have now appeared online in JAMA. Although it failed to meet its primary endpoint of reducing dementia incidence, the study did score on two secondary endpoints. Patients who reduced their systolic blood pressure to less than 120 mm Hg were 19% less likely to develop MCI and 17% less likely to be diagnosed with all-cause dementia than were those who achieved a hypertension target of less than 140 mm Hg.
The secondary results, and positive movement in the primary results, sparked excitement in the dementia research community last summer. They have suggested that the median 5-year follow-up just wasn’t long enough to show any significant effects on dementia, which can take years to fully manifest. Adding 2 more years with SPRINT MIND 2.0 should be long enough to discern those benefits, if indeed they exist.
“SPRINT MIND 2.0 and the work leading up to it offers genuine, concrete hope,” Maria C. Carrillo, PhD, chief science officer for the Alzheimer’s Association, said in a press statement. “MCI is a known risk factor for dementia, and everyone who experiences dementia passes through MCI. When you prevent new cases of MCI, you are preventing new cases of dementia. The Alzheimer’s Association finds these data to be compelling and is committed to getting clarity and certainty on the dementia outcome by following participants for a longer period of time.”
The study strengthens the new and energetic push to find ways to prevent dementia, which has proven itself intractable in every drug study to date.
“This study is in line with where the field of dementia research is going: preventing memory loss earlier,” said Laurie Ryan, PhD, chief of the dementias of aging branch in the National Institute on Aging. “Much like we have research-based interventions for heart health and cancer prevention, we hope to have guidance based on this and subsequent studies that will more definitively show how to slow or even stop dementia well before symptoms appear.”
NIA director Richard J. Hodes, MD, agreed.
“Dementia continues to be a large public health challenge, and based on the primary results of this study, we still have yet to find an intervention strategy proven to reduce the risk of dementia,” he said in a press statement. “Nevertheless, the secondary results showing that intensive lowering of blood pressure may reduce risk for MCI, a known risk factor for dementia, gives us additional avenues to explore on the path to prevention.”
SPRINT MIND was a substudy of the Systolic Blood Pressure Intervention Trial (SPRINT). It compared two strategies for managing hypertension in older adults. The intensive strategy had a target of less than 120 mm Hg, while standard care had a target of less than 140 mm Hg. SPRINT showed that more intensive blood pressure control produced a 25% reduction in the composite primary composite endpoint of cardiovascular events, stroke, and cardiovascular death. The intensive arm was so successful that SPRINT helped inform the 2017 high blood pressure clinical guidelines from the American Heart Association and American College of Cardiology.
The SPRINT MIND substudy, headed by Jeff D. Williamson, MD, of Wake Forest University, Winston-Salem, NC, asked whether intensive management had any effect on probable all-cause dementia or MCI, as well as imaging evidence of changes in white matter lesions and brain volume. It followed patients for up to 7 years and comprised 9,361 SPRINT subjects at least 50 years old (mean, 68 years) with at least one cardiovascular risk factor. Nearly a third (30%) were black, and 10% Hispanic. The primary outcome was incident probable dementia. Secondary outcomes were MCI and a composite of MCI and/or probable dementia. About a third had a SBP of 132 mm Hg or less, another third had a systolic pressure of 132-145 mm Hg, and the remainder had a systolic pressure greater than 145 mm Hg.
Physicians could use their choice of antihypertensive treatments. The study protocol encouraged, but did not mandate, thiazide-type diuretics as a first-line agent, followed by loop diuretics and beta-adrenergic blockers. Chlorthalidone was encouraged as the primary thiazide-type diuretic, and amlodipine as the preferred calcium-channel blocker.
The interventions did successfully control blood pressure, with a significant difference between the treatment groups. The mean SBP was 121.6 mm Hg in the intensive therapy group and 134.8 mm Hg in the standard group – a statistically significant difference of 13.3 mm Hg.
Dementia developed in 149 in the aggressive control group and 176 in the standard group – a nonsignificant difference of 17% (hazard ratio, 0.83). MCI developed in 287 in the intensive group and 353 in the standard treatment group. This amounted to a statistically significant 19% reduction. There was also a significant 15% reduction in the composite outcome of MCI or probable dementia in favor of intensive treatment.
As evidenced by the Alzheimer’s Association grant, dementia researchers chose to focus on SPRINT MIND’s positive secondary endpoints. At the AAIC meeting, Dr. Williamson even suggested that antihypertensive medications could be seen as disease-modifying agents for cognitive decline. Data support his claim: No dementia intervention yet tested has approached this level of success.
“I think we can say this is the first disease-modifying strategy to reduce the risk of MCI,” Dr. Williamson said during a press briefing. And although the primary endpoint – the 17% relative risk reduction for probable all-cause dementia – didn’t meet statistical significance, “It’s comforting to see that the benefit went in the same direction and was of the same magnitude..”
SOURCE: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
FROM JAMA
Key clinical point: Keeping systolic blood pressure lower than 120 mm Hg did not significantly reduce the risk of all-cause dementia in patients with hypertension, but it did lower the risk of mild cognitive impairment and probable dementia.
Major finding: The intensively treated group had a nonsignificant 17% lower risk of dementia, and significant reductions in the risk of MCI (19%) and probable dementia (15%).
Study details: SPRINT MIND was a substudy of the SPRINT antihypertension trial.
Source: Williamson JD et al. JAMA 2019 Jan 28. doi:10.1001/jama.2018.21442.
Isotretinoin treatment reorganizes dermal microbiome in acne patients
GRAND CAYMAN, CAYMAN ISLANDS – Isotretinoin, the go-to guy for severe acne, may not be so much a local cop as a community organizer, Kenneth B. Gordon, MD, said at the meeting provided by Global Academy for Medical Education.

“It now appears that with and that the microbial community is replenished with the types associated with healthy skin,” said Dr. Gordon, professor and chair of dermatology at the Medical College of Wisconsin, Milwaukee. When these new bacteria move in, they push pathogenic species out of the neighborhood “and create a new skin microbial community. Maybe this is the real reason our patients tend to stay better, once we get them better with isotretinoin.”
Dr. Gordon discussed new data published last October in the Journal of Investigative Dermatology (J Invest Dermatol. 2018 Oct 24. doi: 10.1016/j.jid.2018.09.023). In a letter to the editor, William H. McCoy, IV, MD, PhD, of Washington University, St. Louis, and his associates suggest that isotretinoin induces a “sebaceous drought,” which shifts the skin microbiome from pathogenic to normophysiological.
Isotretinoin is the gold standard treatment for severe acne, but its method of action has never been fully elucidated, Dr. Gordon said. It clearly targets the sebaceous gland – decreasing sebocyte proliferation and suppressing sebum production – but an emerging body of research suggests that the drug also markedly affects dermal microbial colonization.
The entire concept of a skin microbiome is nearly as new as this new concept of isotretinoin’s effect upon it. Only in the last few years have researchers begun to characterize the complex microbial film that keeps skin healthy and resistant to infection. Dermal dysbiosis has now been associated with acne, psoriasis and psoriatic arthritis, and atopic dermatitis.
The 2-year pilot study compared the dermal microbiome of isotretinoin-treated acne patients with that of patients with untreated acne and normal skin. Skin samples underwent genomic analysis before isotretinoin treatment, at several periods during treatment, and about 5 months after treatment stopped. Untreated controls were evaluated at baseline and at 2, 5, and 10 months.
Not surprisingly, before treatment the microbiome was similar in both acne groups, but markedly different from that seen in normal skin. As isotretinoin’s “oil drought” dragged on, levels of Cutibacterium acnes (the new appellation for P. acnes) declined. Staphylococcus species initially increased, but then declined as well. Simultaneously, four new taxa (Rothia, Flavobacterium, Enterobacter, and Micrococcus) increased. Most patients had a restructuring of their Propionibacterium community, populated largely by the less-pathogenic strains found on normal skin.
“We suggest that isotretinoin creates a Propionibacterium ‘population bottleneck’ that selects for ‘healthy’ Propionibacterium communities and other sebaceous skin taxa that persist after treatment, resulting in long-term acne remission [i.e., normal skin],” the investigators wrote.
This is a new and very exciting finding, Dr. Gordon commented. “It appears that the reason our isotretinoin patients stay better once they get better is not from targeting the sebaceous gland itself, but by repairing the skin’s microbiome and getting it back to normal.”
Dr. Gordon reported financial relationships with numerous pharmaceutical companies. Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
GRAND CAYMAN, CAYMAN ISLANDS – Isotretinoin, the go-to guy for severe acne, may not be so much a local cop as a community organizer, Kenneth B. Gordon, MD, said at the meeting provided by Global Academy for Medical Education.
“It now appears that with and that the microbial community is replenished with the types associated with healthy skin,” said Dr. Gordon, professor and chair of dermatology at the Medical College of Wisconsin, Milwaukee. When these new bacteria move in, they push pathogenic species out of the neighborhood “and create a new skin microbial community. Maybe this is the real reason our patients tend to stay better, once we get them better with isotretinoin.”
Dr. Gordon discussed new data published last October in the Journal of Investigative Dermatology (J Invest Dermatol. 2018 Oct 24. doi: 10.1016/j.jid.2018.09.023). In a letter to the editor, William H. McCoy, IV, MD, PhD, of Washington University, St. Louis, and his associates suggest that isotretinoin induces a “sebaceous drought,” which shifts the skin microbiome from pathogenic to normophysiological.
Isotretinoin is the gold standard treatment for severe acne, but its method of action has never been fully elucidated, Dr. Gordon said. It clearly targets the sebaceous gland – decreasing sebocyte proliferation and suppressing sebum production – but an emerging body of research suggests that the drug also markedly affects dermal microbial colonization.
The entire concept of a skin microbiome is nearly as new as this new concept of isotretinoin’s effect upon it. Only in the last few years have researchers begun to characterize the complex microbial film that keeps skin healthy and resistant to infection. Dermal dysbiosis has now been associated with acne, psoriasis and psoriatic arthritis, and atopic dermatitis.
The 2-year pilot study compared the dermal microbiome of isotretinoin-treated acne patients with that of patients with untreated acne and normal skin. Skin samples underwent genomic analysis before isotretinoin treatment, at several periods during treatment, and about 5 months after treatment stopped. Untreated controls were evaluated at baseline and at 2, 5, and 10 months.
Not surprisingly, before treatment the microbiome was similar in both acne groups, but markedly different from that seen in normal skin. As isotretinoin’s “oil drought” dragged on, levels of Cutibacterium acnes (the new appellation for P. acnes) declined. Staphylococcus species initially increased, but then declined as well. Simultaneously, four new taxa (Rothia, Flavobacterium, Enterobacter, and Micrococcus) increased. Most patients had a restructuring of their Propionibacterium community, populated largely by the less-pathogenic strains found on normal skin.
“We suggest that isotretinoin creates a Propionibacterium ‘population bottleneck’ that selects for ‘healthy’ Propionibacterium communities and other sebaceous skin taxa that persist after treatment, resulting in long-term acne remission [i.e., normal skin],” the investigators wrote.
This is a new and very exciting finding, Dr. Gordon commented. “It appears that the reason our isotretinoin patients stay better once they get better is not from targeting the sebaceous gland itself, but by repairing the skin’s microbiome and getting it back to normal.”
Dr. Gordon reported financial relationships with numerous pharmaceutical companies. Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
GRAND CAYMAN, CAYMAN ISLANDS – Isotretinoin, the go-to guy for severe acne, may not be so much a local cop as a community organizer, Kenneth B. Gordon, MD, said at the meeting provided by Global Academy for Medical Education.
“It now appears that with and that the microbial community is replenished with the types associated with healthy skin,” said Dr. Gordon, professor and chair of dermatology at the Medical College of Wisconsin, Milwaukee. When these new bacteria move in, they push pathogenic species out of the neighborhood “and create a new skin microbial community. Maybe this is the real reason our patients tend to stay better, once we get them better with isotretinoin.”
Dr. Gordon discussed new data published last October in the Journal of Investigative Dermatology (J Invest Dermatol. 2018 Oct 24. doi: 10.1016/j.jid.2018.09.023). In a letter to the editor, William H. McCoy, IV, MD, PhD, of Washington University, St. Louis, and his associates suggest that isotretinoin induces a “sebaceous drought,” which shifts the skin microbiome from pathogenic to normophysiological.
Isotretinoin is the gold standard treatment for severe acne, but its method of action has never been fully elucidated, Dr. Gordon said. It clearly targets the sebaceous gland – decreasing sebocyte proliferation and suppressing sebum production – but an emerging body of research suggests that the drug also markedly affects dermal microbial colonization.
The entire concept of a skin microbiome is nearly as new as this new concept of isotretinoin’s effect upon it. Only in the last few years have researchers begun to characterize the complex microbial film that keeps skin healthy and resistant to infection. Dermal dysbiosis has now been associated with acne, psoriasis and psoriatic arthritis, and atopic dermatitis.
The 2-year pilot study compared the dermal microbiome of isotretinoin-treated acne patients with that of patients with untreated acne and normal skin. Skin samples underwent genomic analysis before isotretinoin treatment, at several periods during treatment, and about 5 months after treatment stopped. Untreated controls were evaluated at baseline and at 2, 5, and 10 months.
Not surprisingly, before treatment the microbiome was similar in both acne groups, but markedly different from that seen in normal skin. As isotretinoin’s “oil drought” dragged on, levels of Cutibacterium acnes (the new appellation for P. acnes) declined. Staphylococcus species initially increased, but then declined as well. Simultaneously, four new taxa (Rothia, Flavobacterium, Enterobacter, and Micrococcus) increased. Most patients had a restructuring of their Propionibacterium community, populated largely by the less-pathogenic strains found on normal skin.
“We suggest that isotretinoin creates a Propionibacterium ‘population bottleneck’ that selects for ‘healthy’ Propionibacterium communities and other sebaceous skin taxa that persist after treatment, resulting in long-term acne remission [i.e., normal skin],” the investigators wrote.
This is a new and very exciting finding, Dr. Gordon commented. “It appears that the reason our isotretinoin patients stay better once they get better is not from targeting the sebaceous gland itself, but by repairing the skin’s microbiome and getting it back to normal.”
Dr. Gordon reported financial relationships with numerous pharmaceutical companies. Global Academy and this news organization are owned by the same parent company.
This article was updated 2/1/19.
REPORTING FROM THE CARIBBEAN DERMATOLOGY SYMPOSIUM