User login
Tool may help predict persistent postconcussion symptoms
A new tool – a clinical risk score – may help identify which children and adolescents who recently sustained a head injury are at risk for persistent postconcussion symptoms, according to a report published online March 7 in JAMA.
Approximately one-third of pediatric patients with concussion will have ongoing somatic, cognitive, psychological, and/or behavioral symptoms at 28 days, and at present, there are no tools to help predict which patients will be affected. The 5P (Preventing Postconcussive Problems in Pediatrics) study was performed to develop and validate a clinical risk score for this purpose, said Dr. Roger Zemek of Children’s Hospital of Eastern Ontario Research Institute, Ottawa, and his associates (JAMA 2016 Mar 8;315[10]:1014-25).

This prospective cohort study involved patients aged 5-17 years (median age, 12 years) who presented to one of nine Canadian pediatric emergency departments within 48 hours of sustaining a concussion.
In the derivation cohort, 510 of 1,701 participants (30%) met the criteria for persistent postconcussion symptoms (PPCS). A total of 47 possible predictive variables were assessed for their usefulness in predicting PPCS in this cohort. They were collected from demographic data, patient history, injury characteristics, physical examination, results on the Acute Concussion Evaluation Inventory and the Postconcussion Symptom Inventory, and patient/parent responses to weekly follow-up surveys during the month following the injury.
The investigators devised a clinical risk score using the nine predictors they found to be most accurate: patient age, patient gender, the presence or absence of prior concussion, migraine history, the presence or absence of current headache, sensitivity to noise, fatigue, slow responses to questions, and an abnormal tandem stance. They then selected three cutoff points to delineate PPCS risk: 0-3 points indicated low risk, 4-8 points indicated intermediate risk, and 9 or more points indicated high risk.
Treating physicians also were asked to predict the likelihood of PPCS.
In the validation cohort, 291 of 883 participants (33%) met the criteria for PPCS.
For low-risk patients, the sensitivity of the clinical risk score was 94%, the specificity was 18%, the negative predictive value was 85%, and the positive predictive value was 36%. For high-risk patients, the sensitivity of the clinical risk score was 20%, the specificity was 94%, the negative predictive value was 70%, and the positive predictive value was 60%.
In both sets of patients, the clinical risk score was significantly better than physician judgment in predicting PPCS. However, in its present form, it is only modestly accurate at distinguishing who will and who will not have the disorder. This tool could be further refined, perhaps by adding information regarding biomarkers, genetic susceptibility, or advanced neuroimaging, Dr. Zemek and his associates wrote.
“Before this score is adopted in clinical practice, further research is needed for external validation, assessment of accuracy in an office setting, and determination of clinical utility,” they concluded.
This work was supported by the Canadian Institutes of Health Research, the Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team, and the Alberta Children’s Hospital Foundation. Dr. Zemek and his associates reported having no relevant financial disclosures.
The clinical risk score devised by Zemek et al. may facilitate selection of patients who are at highest risk of long-term impairment, both for more intensive monitoring and treatment in the clinical setting and for inclusion in much-needed interventional trials in the research setting. It also may support clinicians in reassuring low-risk patients and their families of the likelihood of full recovery.
However, this tool first must be validated in other settings where children and adolescents are assessed for head injury, including general emergency departments; urgent care centers; and primary care, orthopedic, and sports medicine practices. Its performance should also be evaluated when used in conjunction with bedside vestibular ocular measures, serum biomarkers, genetic factors, and advanced neuroimaging measures. And determining its usefulness in other patient groups excluded from this trial also is warranted, including children younger than age 5, those with multiple trauma, and those found to have structural abnormalities on neuroimaging tests.
Dr. Lynn Babcock is in the division of pediatric emergency medicine at the University of Cincinnati and at the Cincinnati Children’s Hospital Medical Center. Dr. Brad G. Kurowski is in the division of physical medicine and rehabilitation in the department of pediatrics at Cincinnati Children’s Hospital Medical Center. Dr. Kurowski reported receiving grants from the National Institutes of Health and the Centers for Disease Control and Prevention. Dr. Babcock and Dr. Kurowski made these remarks in an editorial accompanying Dr. Zemek’s report (JAMA 2016 Mar 8;315[10]:987-8).
The clinical risk score devised by Zemek et al. may facilitate selection of patients who are at highest risk of long-term impairment, both for more intensive monitoring and treatment in the clinical setting and for inclusion in much-needed interventional trials in the research setting. It also may support clinicians in reassuring low-risk patients and their families of the likelihood of full recovery.
However, this tool first must be validated in other settings where children and adolescents are assessed for head injury, including general emergency departments; urgent care centers; and primary care, orthopedic, and sports medicine practices. Its performance should also be evaluated when used in conjunction with bedside vestibular ocular measures, serum biomarkers, genetic factors, and advanced neuroimaging measures. And determining its usefulness in other patient groups excluded from this trial also is warranted, including children younger than age 5, those with multiple trauma, and those found to have structural abnormalities on neuroimaging tests.
Dr. Lynn Babcock is in the division of pediatric emergency medicine at the University of Cincinnati and at the Cincinnati Children’s Hospital Medical Center. Dr. Brad G. Kurowski is in the division of physical medicine and rehabilitation in the department of pediatrics at Cincinnati Children’s Hospital Medical Center. Dr. Kurowski reported receiving grants from the National Institutes of Health and the Centers for Disease Control and Prevention. Dr. Babcock and Dr. Kurowski made these remarks in an editorial accompanying Dr. Zemek’s report (JAMA 2016 Mar 8;315[10]:987-8).
The clinical risk score devised by Zemek et al. may facilitate selection of patients who are at highest risk of long-term impairment, both for more intensive monitoring and treatment in the clinical setting and for inclusion in much-needed interventional trials in the research setting. It also may support clinicians in reassuring low-risk patients and their families of the likelihood of full recovery.
However, this tool first must be validated in other settings where children and adolescents are assessed for head injury, including general emergency departments; urgent care centers; and primary care, orthopedic, and sports medicine practices. Its performance should also be evaluated when used in conjunction with bedside vestibular ocular measures, serum biomarkers, genetic factors, and advanced neuroimaging measures. And determining its usefulness in other patient groups excluded from this trial also is warranted, including children younger than age 5, those with multiple trauma, and those found to have structural abnormalities on neuroimaging tests.
Dr. Lynn Babcock is in the division of pediatric emergency medicine at the University of Cincinnati and at the Cincinnati Children’s Hospital Medical Center. Dr. Brad G. Kurowski is in the division of physical medicine and rehabilitation in the department of pediatrics at Cincinnati Children’s Hospital Medical Center. Dr. Kurowski reported receiving grants from the National Institutes of Health and the Centers for Disease Control and Prevention. Dr. Babcock and Dr. Kurowski made these remarks in an editorial accompanying Dr. Zemek’s report (JAMA 2016 Mar 8;315[10]:987-8).
A new tool – a clinical risk score – may help identify which children and adolescents who recently sustained a head injury are at risk for persistent postconcussion symptoms, according to a report published online March 7 in JAMA.
Approximately one-third of pediatric patients with concussion will have ongoing somatic, cognitive, psychological, and/or behavioral symptoms at 28 days, and at present, there are no tools to help predict which patients will be affected. The 5P (Preventing Postconcussive Problems in Pediatrics) study was performed to develop and validate a clinical risk score for this purpose, said Dr. Roger Zemek of Children’s Hospital of Eastern Ontario Research Institute, Ottawa, and his associates (JAMA 2016 Mar 8;315[10]:1014-25).
This prospective cohort study involved patients aged 5-17 years (median age, 12 years) who presented to one of nine Canadian pediatric emergency departments within 48 hours of sustaining a concussion.
In the derivation cohort, 510 of 1,701 participants (30%) met the criteria for persistent postconcussion symptoms (PPCS). A total of 47 possible predictive variables were assessed for their usefulness in predicting PPCS in this cohort. They were collected from demographic data, patient history, injury characteristics, physical examination, results on the Acute Concussion Evaluation Inventory and the Postconcussion Symptom Inventory, and patient/parent responses to weekly follow-up surveys during the month following the injury.
The investigators devised a clinical risk score using the nine predictors they found to be most accurate: patient age, patient gender, the presence or absence of prior concussion, migraine history, the presence or absence of current headache, sensitivity to noise, fatigue, slow responses to questions, and an abnormal tandem stance. They then selected three cutoff points to delineate PPCS risk: 0-3 points indicated low risk, 4-8 points indicated intermediate risk, and 9 or more points indicated high risk.
Treating physicians also were asked to predict the likelihood of PPCS.
In the validation cohort, 291 of 883 participants (33%) met the criteria for PPCS.
For low-risk patients, the sensitivity of the clinical risk score was 94%, the specificity was 18%, the negative predictive value was 85%, and the positive predictive value was 36%. For high-risk patients, the sensitivity of the clinical risk score was 20%, the specificity was 94%, the negative predictive value was 70%, and the positive predictive value was 60%.
In both sets of patients, the clinical risk score was significantly better than physician judgment in predicting PPCS. However, in its present form, it is only modestly accurate at distinguishing who will and who will not have the disorder. This tool could be further refined, perhaps by adding information regarding biomarkers, genetic susceptibility, or advanced neuroimaging, Dr. Zemek and his associates wrote.
“Before this score is adopted in clinical practice, further research is needed for external validation, assessment of accuracy in an office setting, and determination of clinical utility,” they concluded.
This work was supported by the Canadian Institutes of Health Research, the Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team, and the Alberta Children’s Hospital Foundation. Dr. Zemek and his associates reported having no relevant financial disclosures.
A new tool – a clinical risk score – may help identify which children and adolescents who recently sustained a head injury are at risk for persistent postconcussion symptoms, according to a report published online March 7 in JAMA.
Approximately one-third of pediatric patients with concussion will have ongoing somatic, cognitive, psychological, and/or behavioral symptoms at 28 days, and at present, there are no tools to help predict which patients will be affected. The 5P (Preventing Postconcussive Problems in Pediatrics) study was performed to develop and validate a clinical risk score for this purpose, said Dr. Roger Zemek of Children’s Hospital of Eastern Ontario Research Institute, Ottawa, and his associates (JAMA 2016 Mar 8;315[10]:1014-25).
This prospective cohort study involved patients aged 5-17 years (median age, 12 years) who presented to one of nine Canadian pediatric emergency departments within 48 hours of sustaining a concussion.
In the derivation cohort, 510 of 1,701 participants (30%) met the criteria for persistent postconcussion symptoms (PPCS). A total of 47 possible predictive variables were assessed for their usefulness in predicting PPCS in this cohort. They were collected from demographic data, patient history, injury characteristics, physical examination, results on the Acute Concussion Evaluation Inventory and the Postconcussion Symptom Inventory, and patient/parent responses to weekly follow-up surveys during the month following the injury.
The investigators devised a clinical risk score using the nine predictors they found to be most accurate: patient age, patient gender, the presence or absence of prior concussion, migraine history, the presence or absence of current headache, sensitivity to noise, fatigue, slow responses to questions, and an abnormal tandem stance. They then selected three cutoff points to delineate PPCS risk: 0-3 points indicated low risk, 4-8 points indicated intermediate risk, and 9 or more points indicated high risk.
Treating physicians also were asked to predict the likelihood of PPCS.
In the validation cohort, 291 of 883 participants (33%) met the criteria for PPCS.
For low-risk patients, the sensitivity of the clinical risk score was 94%, the specificity was 18%, the negative predictive value was 85%, and the positive predictive value was 36%. For high-risk patients, the sensitivity of the clinical risk score was 20%, the specificity was 94%, the negative predictive value was 70%, and the positive predictive value was 60%.
In both sets of patients, the clinical risk score was significantly better than physician judgment in predicting PPCS. However, in its present form, it is only modestly accurate at distinguishing who will and who will not have the disorder. This tool could be further refined, perhaps by adding information regarding biomarkers, genetic susceptibility, or advanced neuroimaging, Dr. Zemek and his associates wrote.
“Before this score is adopted in clinical practice, further research is needed for external validation, assessment of accuracy in an office setting, and determination of clinical utility,” they concluded.
This work was supported by the Canadian Institutes of Health Research, the Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team, and the Alberta Children’s Hospital Foundation. Dr. Zemek and his associates reported having no relevant financial disclosures.
FROM JAMA
Key clinical point: A new tool has been developed to help identify children and adolescents at risk for persistent postconcussion symptoms.
Major finding: For high-risk patients, the sensitivity of the clinical risk score was 20%, the specificity was 93%, the negative predictive value was 70%, and the positive predictive value was 60%.
Data source: A prospective, multicenter cohort study involving 1,701 pediatric patients to develop a clinical risk score, and a validation study involving 883 to test the performance of that tool.
Disclosures: This work was supported by the Canadian Institutes of Health Research, the Ontario Neurotrauma Foundation Mild Traumatic Brain Injury Team, and the Alberta Children’s Hospital Foundation. Dr. Zemek and his associates reported having no relevant financial disclosures.

No LIGHT shed on CV safety of naltrexone-bupropion
The cardiovascular risks of the weight-loss combination drug naltrexone-bupropion are still uncertain, because the FDA-mandated safety trial, LIGHT, was terminated prematurely due to the sponsoring drug company’s breach of confidentiality. Other irregularities also were discovered, according to a report published online March 8 in JAMA.
Orexigen violated its agreement with FDA not to disclose the findings of an interim analysis performed after only 25% of expected CV events accrued. These findings appeared highly favorable for the drug, prompting the sponsor to publicly, and prematurely, claim naltrexone-bupropion reduced CV risk by 41% independently of its weight-loss effect. This favorable outcome was later found to be “driven almost exclusively by a 12-to-1 imbalance in death favoring naltrexone-bupropion among patients who were no longer taking the study drug,” said Dr. Steven E. Nissen of the Cleveland Clinic Center for Cardiovascular Research and his associates.

They now report the results of a second interim analysis of LIGHT (Cardiovascular Outcomes Study of Naltrexone SR/Bupropion SR in Overweight and Obese Subjects With Cardiovascular Risk Factors) performed after 50% of expected CV events accrued, as well as those of a final analysis performed when the trial was halted and 64% of expected CV events accrued. These findings are much less favorable, showing no significant difference between naltrexone-bupropion and placebo in the rate of major adverse cardiovascular events (MACE). In addition, extremely poor adherence to both active treatment and placebo over time, together with the complex statistical issues involved, call into question any interpretation of the results and preclude “any reliable conclusions about the long-term safety of naltrexone-bupropion,” the investigators said.
“The cardiovascular safety of this treatment remains uncertain and will require evaluation in a new, adequately powered outcome trial.” The results of such a study, should it be undertaken, wouldn’t be available for at least 3-4 years, they noted.
In their report, the investigators described in detail why the FDA required a CV safety study even though the agency had already approved naltrexone-bupropion as a weight-loss treatment.
The study was a phase IIIb randomized, double-blind, placebo-controlled trial involving 8,910 overweight/obese patients aged 45 or older (mean age, 61 years) who were at risk of adverse CV outcomes due to preexisting CV disease; exercise-induced angina; an unfavorable ankle/brachial index; stenosis of coronary, carotid, or lower extremity arteries; type 2 diabetes; hypertension; dyslipidemia; or smoking. These participants received daily tablets of either 32 mg naltrexone plus 360 mg bupropion or a matching placebo and were to be followed for 4 years at 266 U.S. medical centers.
The analysis performed after 50% of expected CV events accrued showed that MACE – the primary safety outcome – occurred in 2.0% of the naltrexone-bupropion group and 2.3% of the placebo group, a nonsignificant difference. Differences between the two study groups also were nonsignificant for the individual components of the primary outcome, including cardiovascular death (0.4% vs. 0.8%), nonfatal stroke (0.5% vs. 0.4%), and nonfatal MI (1.2% vs. 1.2%). These results were confirmed in a sensitivity analysis based on the final data collection, when 64% of expected CV events accrued: MACE occurred in 2.7% of the naltrexone-bupropion group and 2.8% of the placebo group, another nonsignificant difference.
The active treatment also was no better than placebo regarding all secondary outcomes, including all-cause mortality, CV hospitalization, and coronary revascularization, Dr. Nissen and his associates said (JAMA. 2016 Mar 8. doi: 10.1001/jama.2016.1558). Treatment adherence was low. At 16 weeks, only 64% of the naltrexone-bupropion group and 72% of the placebo group were still taking the study treatment. By 2 years, only 27% of the naltrexone-bupropion group and 17% of the placebo group were. The mean duration of treatment was only 18.4 weeks for naltrexone-bupropion and 16.3 weeks for placebo. Most treatment discontinuations were due to a failure to lose weight, but a substantial proportion occurred because of treatment-related increases in blood pressure or heart rate.
The active treatment’s ability to reduce body weight was deemed “modest.” When the trial was halted, the mean decrease in weight was 3.9 kg with naltrexone-bupropion, representing a 3.6% reduction in total weight, and was 1.2 kg with placebo, representing a 1.1% reduction.
Adverse effects developed in 28.1% of patients taking naltrexone-bupropion, which was a significantly greater proportion than the 8.7% rate in the placebo group. The most common adverse events leading to discontinuation of the study drug affected the gastrointestinal system (nausea, constipation, vomiting), the CNS (tremor, dizziness, headache), and mental/emotional problems (insomnia, anxiety, hallucinations, depression).
The sponsor of this trial, Orexigen Therapeutics, disregarded the multiple harms caused by breaches of confidentiality, treated the academic investigators unfairly, ignored the trial’s data monitoring committee, and defied the FDA. Their statements about the naltrexone-bupropion’s cardiovascular efficacy were highly misleading, and there were obvious signs that the supporting data were not reliable. For example, nearly all of the CV mortality in the first interim analysis occurred well after participants had stopped taking their medication.
Yet the drug company was not sanctioned, and the naltrexone-bupropion retains FDA approval. At a minimum, the FDA should withhold approval until a viable replacement study is conducted, or it should require Risk Evaluation and Mitigation Strategies to counter the misinformation disseminated by the study sponsor, or it should restrict use of the drug outright.
Clinicians should be aware of these research improprieties and should know that the purported weight-loss benefit of naltrexone-bupropion is only 2.7 kg more than that achieved with placebo. How does this modest benefit balance against an unknown cardiovascular risk? In addition, the drug’s lack of efficacy clearly contributed to the very poor adherence rate in this trial: At 1 year, only 37.5% were still taking naltrexone-bupropion and 26.3% were still taking placebo.
Dr. Joshua M. Sharfstein is in the department of health policy and management at Johns Hopkins Bloomberg School of Public Health, Baltimore. Dr. Bruce M. Psaty is in the Cardiovascular Health Research Unit and the departments of medicine, epidemiology, and health services at the University of Washington, Seattle. They reported that this work was supported by the National Heart, Lung, and Blood Institute. Dr. Sharfstein reported serving as principal deputy commissioner of the FDA in 2009-2011. Dr. Psaty reported serving on the data monitoring committee of a clinical trial funded by Zoll LifeCor, on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson, and on the FDA Science Board. Dr. Sharfstein and Dr. Psaty made these remarks in an editorial accompanying Dr. Nissen’s report (JAMA. 2016;315:984-6).
The sponsor of this trial, Orexigen Therapeutics, disregarded the multiple harms caused by breaches of confidentiality, treated the academic investigators unfairly, ignored the trial’s data monitoring committee, and defied the FDA. Their statements about the naltrexone-bupropion’s cardiovascular efficacy were highly misleading, and there were obvious signs that the supporting data were not reliable. For example, nearly all of the CV mortality in the first interim analysis occurred well after participants had stopped taking their medication.
Yet the drug company was not sanctioned, and the naltrexone-bupropion retains FDA approval. At a minimum, the FDA should withhold approval until a viable replacement study is conducted, or it should require Risk Evaluation and Mitigation Strategies to counter the misinformation disseminated by the study sponsor, or it should restrict use of the drug outright.
Clinicians should be aware of these research improprieties and should know that the purported weight-loss benefit of naltrexone-bupropion is only 2.7 kg more than that achieved with placebo. How does this modest benefit balance against an unknown cardiovascular risk? In addition, the drug’s lack of efficacy clearly contributed to the very poor adherence rate in this trial: At 1 year, only 37.5% were still taking naltrexone-bupropion and 26.3% were still taking placebo.
Dr. Joshua M. Sharfstein is in the department of health policy and management at Johns Hopkins Bloomberg School of Public Health, Baltimore. Dr. Bruce M. Psaty is in the Cardiovascular Health Research Unit and the departments of medicine, epidemiology, and health services at the University of Washington, Seattle. They reported that this work was supported by the National Heart, Lung, and Blood Institute. Dr. Sharfstein reported serving as principal deputy commissioner of the FDA in 2009-2011. Dr. Psaty reported serving on the data monitoring committee of a clinical trial funded by Zoll LifeCor, on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson, and on the FDA Science Board. Dr. Sharfstein and Dr. Psaty made these remarks in an editorial accompanying Dr. Nissen’s report (JAMA. 2016;315:984-6).
The sponsor of this trial, Orexigen Therapeutics, disregarded the multiple harms caused by breaches of confidentiality, treated the academic investigators unfairly, ignored the trial’s data monitoring committee, and defied the FDA. Their statements about the naltrexone-bupropion’s cardiovascular efficacy were highly misleading, and there were obvious signs that the supporting data were not reliable. For example, nearly all of the CV mortality in the first interim analysis occurred well after participants had stopped taking their medication.
Yet the drug company was not sanctioned, and the naltrexone-bupropion retains FDA approval. At a minimum, the FDA should withhold approval until a viable replacement study is conducted, or it should require Risk Evaluation and Mitigation Strategies to counter the misinformation disseminated by the study sponsor, or it should restrict use of the drug outright.
Clinicians should be aware of these research improprieties and should know that the purported weight-loss benefit of naltrexone-bupropion is only 2.7 kg more than that achieved with placebo. How does this modest benefit balance against an unknown cardiovascular risk? In addition, the drug’s lack of efficacy clearly contributed to the very poor adherence rate in this trial: At 1 year, only 37.5% were still taking naltrexone-bupropion and 26.3% were still taking placebo.
Dr. Joshua M. Sharfstein is in the department of health policy and management at Johns Hopkins Bloomberg School of Public Health, Baltimore. Dr. Bruce M. Psaty is in the Cardiovascular Health Research Unit and the departments of medicine, epidemiology, and health services at the University of Washington, Seattle. They reported that this work was supported by the National Heart, Lung, and Blood Institute. Dr. Sharfstein reported serving as principal deputy commissioner of the FDA in 2009-2011. Dr. Psaty reported serving on the data monitoring committee of a clinical trial funded by Zoll LifeCor, on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson, and on the FDA Science Board. Dr. Sharfstein and Dr. Psaty made these remarks in an editorial accompanying Dr. Nissen’s report (JAMA. 2016;315:984-6).
The cardiovascular risks of the weight-loss combination drug naltrexone-bupropion are still uncertain, because the FDA-mandated safety trial, LIGHT, was terminated prematurely due to the sponsoring drug company’s breach of confidentiality. Other irregularities also were discovered, according to a report published online March 8 in JAMA.
Orexigen violated its agreement with FDA not to disclose the findings of an interim analysis performed after only 25% of expected CV events accrued. These findings appeared highly favorable for the drug, prompting the sponsor to publicly, and prematurely, claim naltrexone-bupropion reduced CV risk by 41% independently of its weight-loss effect. This favorable outcome was later found to be “driven almost exclusively by a 12-to-1 imbalance in death favoring naltrexone-bupropion among patients who were no longer taking the study drug,” said Dr. Steven E. Nissen of the Cleveland Clinic Center for Cardiovascular Research and his associates.
They now report the results of a second interim analysis of LIGHT (Cardiovascular Outcomes Study of Naltrexone SR/Bupropion SR in Overweight and Obese Subjects With Cardiovascular Risk Factors) performed after 50% of expected CV events accrued, as well as those of a final analysis performed when the trial was halted and 64% of expected CV events accrued. These findings are much less favorable, showing no significant difference between naltrexone-bupropion and placebo in the rate of major adverse cardiovascular events (MACE). In addition, extremely poor adherence to both active treatment and placebo over time, together with the complex statistical issues involved, call into question any interpretation of the results and preclude “any reliable conclusions about the long-term safety of naltrexone-bupropion,” the investigators said.
“The cardiovascular safety of this treatment remains uncertain and will require evaluation in a new, adequately powered outcome trial.” The results of such a study, should it be undertaken, wouldn’t be available for at least 3-4 years, they noted.
In their report, the investigators described in detail why the FDA required a CV safety study even though the agency had already approved naltrexone-bupropion as a weight-loss treatment.
The study was a phase IIIb randomized, double-blind, placebo-controlled trial involving 8,910 overweight/obese patients aged 45 or older (mean age, 61 years) who were at risk of adverse CV outcomes due to preexisting CV disease; exercise-induced angina; an unfavorable ankle/brachial index; stenosis of coronary, carotid, or lower extremity arteries; type 2 diabetes; hypertension; dyslipidemia; or smoking. These participants received daily tablets of either 32 mg naltrexone plus 360 mg bupropion or a matching placebo and were to be followed for 4 years at 266 U.S. medical centers.
The analysis performed after 50% of expected CV events accrued showed that MACE – the primary safety outcome – occurred in 2.0% of the naltrexone-bupropion group and 2.3% of the placebo group, a nonsignificant difference. Differences between the two study groups also were nonsignificant for the individual components of the primary outcome, including cardiovascular death (0.4% vs. 0.8%), nonfatal stroke (0.5% vs. 0.4%), and nonfatal MI (1.2% vs. 1.2%). These results were confirmed in a sensitivity analysis based on the final data collection, when 64% of expected CV events accrued: MACE occurred in 2.7% of the naltrexone-bupropion group and 2.8% of the placebo group, another nonsignificant difference.
The active treatment also was no better than placebo regarding all secondary outcomes, including all-cause mortality, CV hospitalization, and coronary revascularization, Dr. Nissen and his associates said (JAMA. 2016 Mar 8. doi: 10.1001/jama.2016.1558). Treatment adherence was low. At 16 weeks, only 64% of the naltrexone-bupropion group and 72% of the placebo group were still taking the study treatment. By 2 years, only 27% of the naltrexone-bupropion group and 17% of the placebo group were. The mean duration of treatment was only 18.4 weeks for naltrexone-bupropion and 16.3 weeks for placebo. Most treatment discontinuations were due to a failure to lose weight, but a substantial proportion occurred because of treatment-related increases in blood pressure or heart rate.
The active treatment’s ability to reduce body weight was deemed “modest.” When the trial was halted, the mean decrease in weight was 3.9 kg with naltrexone-bupropion, representing a 3.6% reduction in total weight, and was 1.2 kg with placebo, representing a 1.1% reduction.
Adverse effects developed in 28.1% of patients taking naltrexone-bupropion, which was a significantly greater proportion than the 8.7% rate in the placebo group. The most common adverse events leading to discontinuation of the study drug affected the gastrointestinal system (nausea, constipation, vomiting), the CNS (tremor, dizziness, headache), and mental/emotional problems (insomnia, anxiety, hallucinations, depression).
The cardiovascular risks of the weight-loss combination drug naltrexone-bupropion are still uncertain, because the FDA-mandated safety trial, LIGHT, was terminated prematurely due to the sponsoring drug company’s breach of confidentiality. Other irregularities also were discovered, according to a report published online March 8 in JAMA.
Orexigen violated its agreement with FDA not to disclose the findings of an interim analysis performed after only 25% of expected CV events accrued. These findings appeared highly favorable for the drug, prompting the sponsor to publicly, and prematurely, claim naltrexone-bupropion reduced CV risk by 41% independently of its weight-loss effect. This favorable outcome was later found to be “driven almost exclusively by a 12-to-1 imbalance in death favoring naltrexone-bupropion among patients who were no longer taking the study drug,” said Dr. Steven E. Nissen of the Cleveland Clinic Center for Cardiovascular Research and his associates.
They now report the results of a second interim analysis of LIGHT (Cardiovascular Outcomes Study of Naltrexone SR/Bupropion SR in Overweight and Obese Subjects With Cardiovascular Risk Factors) performed after 50% of expected CV events accrued, as well as those of a final analysis performed when the trial was halted and 64% of expected CV events accrued. These findings are much less favorable, showing no significant difference between naltrexone-bupropion and placebo in the rate of major adverse cardiovascular events (MACE). In addition, extremely poor adherence to both active treatment and placebo over time, together with the complex statistical issues involved, call into question any interpretation of the results and preclude “any reliable conclusions about the long-term safety of naltrexone-bupropion,” the investigators said.
“The cardiovascular safety of this treatment remains uncertain and will require evaluation in a new, adequately powered outcome trial.” The results of such a study, should it be undertaken, wouldn’t be available for at least 3-4 years, they noted.
In their report, the investigators described in detail why the FDA required a CV safety study even though the agency had already approved naltrexone-bupropion as a weight-loss treatment.
The study was a phase IIIb randomized, double-blind, placebo-controlled trial involving 8,910 overweight/obese patients aged 45 or older (mean age, 61 years) who were at risk of adverse CV outcomes due to preexisting CV disease; exercise-induced angina; an unfavorable ankle/brachial index; stenosis of coronary, carotid, or lower extremity arteries; type 2 diabetes; hypertension; dyslipidemia; or smoking. These participants received daily tablets of either 32 mg naltrexone plus 360 mg bupropion or a matching placebo and were to be followed for 4 years at 266 U.S. medical centers.
The analysis performed after 50% of expected CV events accrued showed that MACE – the primary safety outcome – occurred in 2.0% of the naltrexone-bupropion group and 2.3% of the placebo group, a nonsignificant difference. Differences between the two study groups also were nonsignificant for the individual components of the primary outcome, including cardiovascular death (0.4% vs. 0.8%), nonfatal stroke (0.5% vs. 0.4%), and nonfatal MI (1.2% vs. 1.2%). These results were confirmed in a sensitivity analysis based on the final data collection, when 64% of expected CV events accrued: MACE occurred in 2.7% of the naltrexone-bupropion group and 2.8% of the placebo group, another nonsignificant difference.
The active treatment also was no better than placebo regarding all secondary outcomes, including all-cause mortality, CV hospitalization, and coronary revascularization, Dr. Nissen and his associates said (JAMA. 2016 Mar 8. doi: 10.1001/jama.2016.1558). Treatment adherence was low. At 16 weeks, only 64% of the naltrexone-bupropion group and 72% of the placebo group were still taking the study treatment. By 2 years, only 27% of the naltrexone-bupropion group and 17% of the placebo group were. The mean duration of treatment was only 18.4 weeks for naltrexone-bupropion and 16.3 weeks for placebo. Most treatment discontinuations were due to a failure to lose weight, but a substantial proportion occurred because of treatment-related increases in blood pressure or heart rate.
The active treatment’s ability to reduce body weight was deemed “modest.” When the trial was halted, the mean decrease in weight was 3.9 kg with naltrexone-bupropion, representing a 3.6% reduction in total weight, and was 1.2 kg with placebo, representing a 1.1% reduction.
Adverse effects developed in 28.1% of patients taking naltrexone-bupropion, which was a significantly greater proportion than the 8.7% rate in the placebo group. The most common adverse events leading to discontinuation of the study drug affected the gastrointestinal system (nausea, constipation, vomiting), the CNS (tremor, dizziness, headache), and mental/emotional problems (insomnia, anxiety, hallucinations, depression).
FROM JAMA
Key clinical point: The cardiovascular risks of the weight-loss combination drug naltrexone-bupropion are still uncertain.
Major finding: The primary safety outcome, major adverse cardiovascular events, occurred in 2.0% of the naltrexone-bupropion group and 2.3% of the placebo group, a nonsignificant difference.
Data source: LIGHT, a multicenter randomized placebo-controlled double-blind noninferiority trial involving 8,910 overweight/obese patients.
Disclosures: This trial was sponsored by Orexigen Therapeutics and Takeda Pharmaceuticals. Dr. Nissen reported receiving grants from The Medicines Company, Amgen, Pfizer, AstraZeneca, Esperion Therapeutics, Eli Lilly, and Cerenis, and consulting for numerous drug companies that pay his fees directly to charities. His associates reported ties to numerous industry sources.

Ixekizumab for plaque psoriasis improves work productivity
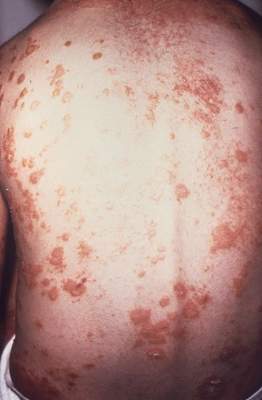
Ixekizumab improved work productivity among patients with moderate to severe plaque psoriasis in three manufacturer-sponsored phase III clinical trials, according to a report published online March 7 in JAMA Dermatology.
In previous studies, many adults with plaque psoriasis have reported that their skin condition forces them to miss work, reduces their productivity at work, requires them to take early retirement, and restricts them to part-time work when they would prefer to work full time. This is associated with substantial economic consequences for the patient and his or her family, for employers, and for society at large.
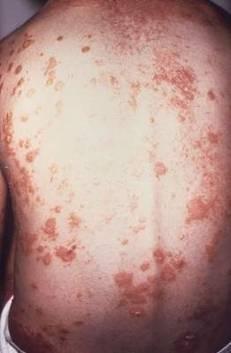
“A recent systematic review estimated the annual indirect economic burden of psoriasis in the United States to be $23.9 billion to $35.4 billion from productivity losses due to absenteeism and presenteeism,” said Dr. April W. Armstrong of the University of Southern California, Los Angeles, and her associates.
They assessed whether ixekizumab improved work productivity by analyzing data regarding that secondary endpoint in the three randomized double-blind UNCOVER trials, which compared the agent against either a matching placebo or etanercept in 3,866 patients with chronic plaque psoriasis involving at least 10% of their body surface area, sPGA (static Physician Global Assessment) scores of 3 or higher, and PASI (Psoriasis Area Severity Index) scores of 12 or higher. The study participants were treated for 12 weeks with either the active drug or a comparator; in two of the studies, those who responded to ixekizumab could then continue on maintenance therapy for up to a further 60 weeks.
Work productivity was measured using the WPAI-PSO (Work Productivity and Activity Impairment-Psoriasis) questionnaire, which assesses psoriasis-associated absenteeism (time missed from work), presenteeism (impairment and reduced effectiveness at work), and overall loss of work productivity.
Across all three trials, 12 weeks of ixekizumab was significantly better than placebo or etanercept was at increasing work productivity and reducing impairment at work. These benefits were sustained in patients who continued long-term treatment, while patients who discontinued treatment lost these benefits and also showed significant worsening in absenteeism, Dr. Armstrong and her associates said (JAMA Dermatol. 2016 Mar 7. doi: 10.1001/jamadermatol.2016.0269).
The improvements in work productivity paralleled improvements in patients’ PASI scores while taking ixekizumab.
Ixekizumab improved work productivity among patients with moderate to severe plaque psoriasis in three manufacturer-sponsored phase III clinical trials, according to a report published online March 7 in JAMA Dermatology.
In previous studies, many adults with plaque psoriasis have reported that their skin condition forces them to miss work, reduces their productivity at work, requires them to take early retirement, and restricts them to part-time work when they would prefer to work full time. This is associated with substantial economic consequences for the patient and his or her family, for employers, and for society at large.
“A recent systematic review estimated the annual indirect economic burden of psoriasis in the United States to be $23.9 billion to $35.4 billion from productivity losses due to absenteeism and presenteeism,” said Dr. April W. Armstrong of the University of Southern California, Los Angeles, and her associates.
They assessed whether ixekizumab improved work productivity by analyzing data regarding that secondary endpoint in the three randomized double-blind UNCOVER trials, which compared the agent against either a matching placebo or etanercept in 3,866 patients with chronic plaque psoriasis involving at least 10% of their body surface area, sPGA (static Physician Global Assessment) scores of 3 or higher, and PASI (Psoriasis Area Severity Index) scores of 12 or higher. The study participants were treated for 12 weeks with either the active drug or a comparator; in two of the studies, those who responded to ixekizumab could then continue on maintenance therapy for up to a further 60 weeks.
Work productivity was measured using the WPAI-PSO (Work Productivity and Activity Impairment-Psoriasis) questionnaire, which assesses psoriasis-associated absenteeism (time missed from work), presenteeism (impairment and reduced effectiveness at work), and overall loss of work productivity.
Across all three trials, 12 weeks of ixekizumab was significantly better than placebo or etanercept was at increasing work productivity and reducing impairment at work. These benefits were sustained in patients who continued long-term treatment, while patients who discontinued treatment lost these benefits and also showed significant worsening in absenteeism, Dr. Armstrong and her associates said (JAMA Dermatol. 2016 Mar 7. doi: 10.1001/jamadermatol.2016.0269).
The improvements in work productivity paralleled improvements in patients’ PASI scores while taking ixekizumab.
Ixekizumab improved work productivity among patients with moderate to severe plaque psoriasis in three manufacturer-sponsored phase III clinical trials, according to a report published online March 7 in JAMA Dermatology.
In previous studies, many adults with plaque psoriasis have reported that their skin condition forces them to miss work, reduces their productivity at work, requires them to take early retirement, and restricts them to part-time work when they would prefer to work full time. This is associated with substantial economic consequences for the patient and his or her family, for employers, and for society at large.
“A recent systematic review estimated the annual indirect economic burden of psoriasis in the United States to be $23.9 billion to $35.4 billion from productivity losses due to absenteeism and presenteeism,” said Dr. April W. Armstrong of the University of Southern California, Los Angeles, and her associates.
They assessed whether ixekizumab improved work productivity by analyzing data regarding that secondary endpoint in the three randomized double-blind UNCOVER trials, which compared the agent against either a matching placebo or etanercept in 3,866 patients with chronic plaque psoriasis involving at least 10% of their body surface area, sPGA (static Physician Global Assessment) scores of 3 or higher, and PASI (Psoriasis Area Severity Index) scores of 12 or higher. The study participants were treated for 12 weeks with either the active drug or a comparator; in two of the studies, those who responded to ixekizumab could then continue on maintenance therapy for up to a further 60 weeks.
Work productivity was measured using the WPAI-PSO (Work Productivity and Activity Impairment-Psoriasis) questionnaire, which assesses psoriasis-associated absenteeism (time missed from work), presenteeism (impairment and reduced effectiveness at work), and overall loss of work productivity.
Across all three trials, 12 weeks of ixekizumab was significantly better than placebo or etanercept was at increasing work productivity and reducing impairment at work. These benefits were sustained in patients who continued long-term treatment, while patients who discontinued treatment lost these benefits and also showed significant worsening in absenteeism, Dr. Armstrong and her associates said (JAMA Dermatol. 2016 Mar 7. doi: 10.1001/jamadermatol.2016.0269).
The improvements in work productivity paralleled improvements in patients’ PASI scores while taking ixekizumab.
FROM JAMA DERMATOLOGY
Key clinical point: Ixekizumab improved work productivity among patients with moderate to severe plaque psoriasis in three manufacturer-sponsored phase III clinical trials.
Major finding: Across all three trials, 12 weeks of ixekizumab was significantly better than placebo or etanercept was at increasing work productivity and reducing impairment at work.
Data source: An analysis of the secondary endpoint of work productivity in three international randomized, controlled phase III trials involving 3,866 patients.
Disclosures: The three UNCOVER trials were supported by Eli Lilly, maker of ixekizumab. Dr. Armstrong reported being a consultant for, receiving honoraria from, and receiving grant support from Eli Lilly and additional pharmaceutical companies. Her associates reported ties to numerous industry sources.
Induction at 39 Weeks Doesn’t Affect C-section Rate in Older Women
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.

|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.
|
|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.
|
|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE

Induction at 39 weeks doesn’t affect C-section rate in older women
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.
|
|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.
|
|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
The findings of Walker et al. suggest that a fundamental belief that guides our decisions about the timing of delivery – namely, that induction of labor at term increases the risk of cesarean delivery – may not be true after all.
|
|
Dr. William A, Grobman |
However, the authors note that larger studies of the issue are needed to confirm these findings and to further explore the effects of induction on maternal and neonatal outcomes, especially stillbirth and other uncommon adverse outcomes. I am the principal investigator of such a trial (NCT01990612), which is currently underway within the Maternal–Fetal Medicine Units Network of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
This trial, which has a targeted enrollment of 6,000 women, is designed to identify differences in perinatal outcomes among nulliparous women with uncomplicated singleton pregnancies who are randomly assigned to induction between 39 weeks 0 days and 39 weeks 4 days of gestation or to expectant management. The trial is more than halfway complete.
Dr. William A. Grobman is at Northwestern University, Chicago. He reported having no relevant financial disclosures. These remarks are adapted from an accompanying editorial (N Engl J Med. 2016 March 2. doi: 10.1056/NEJMe1516461).
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
For women aged 35 years and older, induction of labor at 39 weeks neither raised nor lowered the rate of cesarean delivery, compared with expectant management, in a randomized clinical trial in the United Kingdom.
Labor induction at or before the due date may be beneficial in older women “because the gestational age at delivery that is associated with the lowest cumulative risk of perinatal death is 38 weeks.” However, induction in general is associated with an increased rate of cesarean delivery.
Previous studies of the relative benefits and harms of labor induction in this age group haven’t shed much light on the issue; they were small, observational in design, and were performed in the 1970s, so their findings may not be applicable to modern obstetric practice, wrote Dr. Kate F. Walker of University of Nottingham, England, and her associates.
They tested the hypothesis that labor induction at 39 weeks would reduce the rate of cesarean delivery in nulliparous women of advanced maternal age in the 35/39 Trial. It involved 619 women aged 35 years and older with singleton, uncomplicated pregnancies who were treated during a 2.5-year period at 39 secondary- and tertiary-care medical centers across the United Kingdom. The study participants were randomly assigned late in their pregnancies to either labor induction at 39 weeks (304 women) or expectant management (314 women).
The study hypothesis was not proven: there was no significant difference in the frequency of cesarean section between the induction group (32%) and the expectant management group (33%), for a relative risk of 0.99.
There also were no significant differences between the two study groups in maternal outcomes or neonatal outcomes (N Engl J Med. 2016 Mar 2;374:813-22. doi: 10.1056/NEJMoa1509117).
The trial did not address whether induction at 39 weeks’ gestation could prevent stillbirth, the researchers reported, but it provides support for the safety of performing a larger trial to test the effects of induction on stillbirths and other adverse neonatal outcomes.
The women assigned to labor induction did not differ from those assigned to expectant management in their subjective assessments of satisfaction with the childbirth experience. However, all the study participants had agreed to randomization. Women who had strong preferences about induction or expectant management, or strong feelings about their involvement in treatment decisions – and who may therefore have had a different take on their childbirth experience if labor were induced – were excluded from the study.
The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Among women aged 35 years and older, labor induction at 39 weeks didn’t affect the rate of cesarean delivery.
Major finding: There was no significant difference in the frequency of C-section between the induction group (32%) and the expectant management group (33%).
Data source: A multicenter, randomized trial involving 619 older pregnant women in the United Kingdom.
Disclosures: The study was supported by the National Institute for Health Research in the United Kingdom. Dr. Walker reported having no relevant financial disclosures; one of her associates reported ties to Roche Diagnostics, GlaxoSmithKline, General Electric, Chiesi, and Action Medical Research.

Unintended pregnancies fall to 30-year low
The rate of unintended pregnancies in the United States declined 18% between 2008 and 2011 after a long interval of minimal change, and it is now at the lowest level in 30 years, according to a report published online March 2 in the New England Journal of Medicine.
This “substantial” reduction occurred across all ages, ethnicities, income levels, education levels, and religious affiliations. However, large disparities among demographic groups were still present in 2011, the most recent year for which national data are available for analysis.

“In particular, poor, black, and Hispanic women and girls continued to have much higher rates of unintended pregnancy than did whites and those with higher incomes. Much more progress can be made in eliminating these disparities,” wrote Lawrence B. Finer, Ph.D., and Mia R. Zolna of the Guttmacher Institute, New York.
National rates of unintended pregnancies haven’t been examined since 2008. To update these figures, the investigators analyzed information from the National Center for Health Statistics, the National Survey of Family Growth, and others.
They calculated that there were 6.1 million pregnancies in the United States in 2011, of which 45% (2.8 million) were unintended. The rate was 45 unintended pregnancies for every 1,000 women of childbearing age in 2011, compared with 54/1,000 in 2008, which corresponds to an 18% decline (N Engl J Med.2016;374:843-52. doi: 10.1056/NEJMsa1506575).
“This was the first substantial decline since at least 1981,” the investigators wrote.
Abortion rates remained steady during the study period. In 2011, the percentage of unintended pregnancies that ended in abortion was 42%, compared with 40% in 2008.
Although the study was not designed to determine the reasons for the decline, several possible factors deserve consideration, according to the investigators. Changes in sexual behavior are unlikely to have driven this reduction, since the incidence of sexual activity tends to remain static over time; changes in the composition of the population also aren’t likely causes, since the demographic subgroups who have higher rates of unintended pregnancy actually increased during this period, the investigators said.
Changes in the desire for pregnancy may have contributed a small amount to the decline, as surveys showed that many women said they would reduce or delay their childbearing during the recent economic recession.
The most likely explanation for the decline, the investigators wrote, is a change in the frequency and type of contraceptive use. Several studies have reported that women at high risk of unintended pregnancy increased both their use of any contraception and their use of highly effective long-acting methods, particularly IUDs.
This study was supported chiefly by the Susan Thompson Buffet Foundation, with additional support from the Guttmacher Center through a grant from the National Institutes of Health. The investigators reported having no other relevant financial disclosures.
The rate of unintended pregnancies in the United States declined 18% between 2008 and 2011 after a long interval of minimal change, and it is now at the lowest level in 30 years, according to a report published online March 2 in the New England Journal of Medicine.
This “substantial” reduction occurred across all ages, ethnicities, income levels, education levels, and religious affiliations. However, large disparities among demographic groups were still present in 2011, the most recent year for which national data are available for analysis.
“In particular, poor, black, and Hispanic women and girls continued to have much higher rates of unintended pregnancy than did whites and those with higher incomes. Much more progress can be made in eliminating these disparities,” wrote Lawrence B. Finer, Ph.D., and Mia R. Zolna of the Guttmacher Institute, New York.
National rates of unintended pregnancies haven’t been examined since 2008. To update these figures, the investigators analyzed information from the National Center for Health Statistics, the National Survey of Family Growth, and others.
They calculated that there were 6.1 million pregnancies in the United States in 2011, of which 45% (2.8 million) were unintended. The rate was 45 unintended pregnancies for every 1,000 women of childbearing age in 2011, compared with 54/1,000 in 2008, which corresponds to an 18% decline (N Engl J Med.2016;374:843-52. doi: 10.1056/NEJMsa1506575).
“This was the first substantial decline since at least 1981,” the investigators wrote.
Abortion rates remained steady during the study period. In 2011, the percentage of unintended pregnancies that ended in abortion was 42%, compared with 40% in 2008.
Although the study was not designed to determine the reasons for the decline, several possible factors deserve consideration, according to the investigators. Changes in sexual behavior are unlikely to have driven this reduction, since the incidence of sexual activity tends to remain static over time; changes in the composition of the population also aren’t likely causes, since the demographic subgroups who have higher rates of unintended pregnancy actually increased during this period, the investigators said.
Changes in the desire for pregnancy may have contributed a small amount to the decline, as surveys showed that many women said they would reduce or delay their childbearing during the recent economic recession.
The most likely explanation for the decline, the investigators wrote, is a change in the frequency and type of contraceptive use. Several studies have reported that women at high risk of unintended pregnancy increased both their use of any contraception and their use of highly effective long-acting methods, particularly IUDs.
This study was supported chiefly by the Susan Thompson Buffet Foundation, with additional support from the Guttmacher Center through a grant from the National Institutes of Health. The investigators reported having no other relevant financial disclosures.
The rate of unintended pregnancies in the United States declined 18% between 2008 and 2011 after a long interval of minimal change, and it is now at the lowest level in 30 years, according to a report published online March 2 in the New England Journal of Medicine.
This “substantial” reduction occurred across all ages, ethnicities, income levels, education levels, and religious affiliations. However, large disparities among demographic groups were still present in 2011, the most recent year for which national data are available for analysis.
“In particular, poor, black, and Hispanic women and girls continued to have much higher rates of unintended pregnancy than did whites and those with higher incomes. Much more progress can be made in eliminating these disparities,” wrote Lawrence B. Finer, Ph.D., and Mia R. Zolna of the Guttmacher Institute, New York.
National rates of unintended pregnancies haven’t been examined since 2008. To update these figures, the investigators analyzed information from the National Center for Health Statistics, the National Survey of Family Growth, and others.
They calculated that there were 6.1 million pregnancies in the United States in 2011, of which 45% (2.8 million) were unintended. The rate was 45 unintended pregnancies for every 1,000 women of childbearing age in 2011, compared with 54/1,000 in 2008, which corresponds to an 18% decline (N Engl J Med.2016;374:843-52. doi: 10.1056/NEJMsa1506575).
“This was the first substantial decline since at least 1981,” the investigators wrote.
Abortion rates remained steady during the study period. In 2011, the percentage of unintended pregnancies that ended in abortion was 42%, compared with 40% in 2008.
Although the study was not designed to determine the reasons for the decline, several possible factors deserve consideration, according to the investigators. Changes in sexual behavior are unlikely to have driven this reduction, since the incidence of sexual activity tends to remain static over time; changes in the composition of the population also aren’t likely causes, since the demographic subgroups who have higher rates of unintended pregnancy actually increased during this period, the investigators said.
Changes in the desire for pregnancy may have contributed a small amount to the decline, as surveys showed that many women said they would reduce or delay their childbearing during the recent economic recession.
The most likely explanation for the decline, the investigators wrote, is a change in the frequency and type of contraceptive use. Several studies have reported that women at high risk of unintended pregnancy increased both their use of any contraception and their use of highly effective long-acting methods, particularly IUDs.
This study was supported chiefly by the Susan Thompson Buffet Foundation, with additional support from the Guttmacher Center through a grant from the National Institutes of Health. The investigators reported having no other relevant financial disclosures.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Unintended pregnancies fell by 18% between 2008 and 2011.
Major finding: The rate of unintended pregnancies was 45 for every 1,000 women of childbearing age in 2011, compared with 54/1,000 in 2008.
Data source: An analysis of data from numerous nationally representative surveys and databases regarding pregnancy intentions and outcomes in 2011.
Disclosures: The study was supported chiefly by the Susan Thompson Buffet Foundation, with additional support from the Guttmacher Center through a grant from the National Institutes of Health. The investigators reported having no other relevant financial disclosures.

Pro basketball players’ hearts: LV keeps growing, aortic root doesn’t
For the first time, cardiologists have characterized the adaptive cardiac remodeling in a large cohort of National Basketball Association players, which establishes a normative database and allows physicians to distinguish it from occult pathologic changes that may precipitate sudden cardiac death, according to an imaging study.
“We hope that the present data will help to focus decision making and improve clinical acumen for the purpose of primary prevention of cardiac emergencies in U.S. basketball players and in the athletic community at large,” said Dr. David J. Engel and his associates of Columbia University, New York.

Until now, most of the literature concerning the structural features of the athletic heart has been based on European studies, where comprehensive cardiac screening of all elite athletes is mandatory. The typical sports activities and the demographics of athletes in the U.S. are different, and their cardiologic profiles have not been well studied because detailed cardiac examinations are not compulsory. But the NBA recently mandated that all athletes undergo annual preseason medical evaluations including stress echocardiograms, and allowed the division of cardiology at Columbia to assess the results each year.
“A detailed understanding of normal and expected cardiac remodeling in U.S. basketball players has significant clinical importance given that the incidence of sports-related sudden cardiac death in the U.S. is highest among basketball players and that the most common cause ... in this population is hypertrophic cardiomyopathy,” the investigators noted.
Their analysis of all 526 ECGs performed on NBA players during a 1-year period “will provide an invaluable frame of reference to enhance player safety for the large group of U.S. basketball players in training at all skill levels, and in the athletic community at large,” they said.
The study participants were aged 18-39 years (mean age, 25.7 years). Roughly 77% were African American, 20% were white, 2% were Hispanic, and 1% were Asian or other ethnicities. The mean height was 200.2 cm (6’7”).
Left ventricular cavity size was larger than that in the general population, but LV size was proportional to the athletes’ large body size. “Scaling LV size to body size is vitally important in the cardiac evaluation of basketball players, whose heights extend to 218 cm and body surface areas to 2.8 m2,” Dr. Engel and his associates said (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0252).
Left ventricular hypertrophy (LVH) was identified in only 27% of the athletes. African Americans had increased indices of LVH, compared with whites, and had a higher incidence of nondilated concentric hypertrophy, while whites showed predominantly eccentric dilated hypertrophy. These findings should help clinicians recognize genuine hypertrophic cardiomyopathy, which is a contraindication to participating in all but the most low-intensity competitive sports.
Most of the participants had a normal left ventricular ejection fraction, and all showed normal augmentation of LV systolic function with exercise.
Aortic root diameter was larger than that in the general population but similar to that in other elite athletes. Surprisingly, aortic root diameter increased with increasing body size only to a certain point, reaching a plateau at 31-35 mm. Fewer than 5% of the participants had an aortic root diameter of 40 mm or more, and the maximal diameter was 42 mm. “These data have important implications in the evaluation of exceptionally large athletes and question the applicability in individuals with significantly increased biometrics of the traditional formula to estimate aortic root diameter that assumes a linear association between [it] and body surface area,” they noted.
“We hope that the results of this study will assist recognition of cardiac pathologic change and provide a framework to help avoid unnecessary exclusions of athletes from competition. We believe that these data have additional applicability to other sports that preselect for athletes with height, such as volleyball, rowing, and track and field,” Dr. Engel and his associates added.
This study was supported by the National Basketball Association as part of a medical services agreement with Columbia University. Dr. Engel and his associates reported having no relevant financial disclosures.
The most interesting finding of this study was that despite the immense body size of the athletes, aortic root diameter exceeded 40 mm in less than 5%, and when dilation did occur it was of a very small magnitude, with a maximal diameter of 42 mm.
This important finding confirms that only mild aortic dilation should be considered physiologic among athletes, and that even athletes at the extreme end of the height spectrum should not be expected to show proportionally extreme aortic dilation.
Unlike ventricular size, which increases proportionally with body size, aortic dilation has an upper limit. Athletes with aortic dimensions that exceed this limit should be considered at risk for aortopathy and either prohibited from competitive sports or closely monitored if they do participate.
Dr. Aaron L. Baggish of the Cardiovascular Performance Program at Massachusetts General Hospital, Boston, made these remarks in an accompanying editorial (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0289). He reported having no relevant financial conflicts of interest.
The most interesting finding of this study was that despite the immense body size of the athletes, aortic root diameter exceeded 40 mm in less than 5%, and when dilation did occur it was of a very small magnitude, with a maximal diameter of 42 mm.
This important finding confirms that only mild aortic dilation should be considered physiologic among athletes, and that even athletes at the extreme end of the height spectrum should not be expected to show proportionally extreme aortic dilation.
Unlike ventricular size, which increases proportionally with body size, aortic dilation has an upper limit. Athletes with aortic dimensions that exceed this limit should be considered at risk for aortopathy and either prohibited from competitive sports or closely monitored if they do participate.
Dr. Aaron L. Baggish of the Cardiovascular Performance Program at Massachusetts General Hospital, Boston, made these remarks in an accompanying editorial (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0289). He reported having no relevant financial conflicts of interest.
The most interesting finding of this study was that despite the immense body size of the athletes, aortic root diameter exceeded 40 mm in less than 5%, and when dilation did occur it was of a very small magnitude, with a maximal diameter of 42 mm.
This important finding confirms that only mild aortic dilation should be considered physiologic among athletes, and that even athletes at the extreme end of the height spectrum should not be expected to show proportionally extreme aortic dilation.
Unlike ventricular size, which increases proportionally with body size, aortic dilation has an upper limit. Athletes with aortic dimensions that exceed this limit should be considered at risk for aortopathy and either prohibited from competitive sports or closely monitored if they do participate.
Dr. Aaron L. Baggish of the Cardiovascular Performance Program at Massachusetts General Hospital, Boston, made these remarks in an accompanying editorial (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0289). He reported having no relevant financial conflicts of interest.
For the first time, cardiologists have characterized the adaptive cardiac remodeling in a large cohort of National Basketball Association players, which establishes a normative database and allows physicians to distinguish it from occult pathologic changes that may precipitate sudden cardiac death, according to an imaging study.
“We hope that the present data will help to focus decision making and improve clinical acumen for the purpose of primary prevention of cardiac emergencies in U.S. basketball players and in the athletic community at large,” said Dr. David J. Engel and his associates of Columbia University, New York.
Until now, most of the literature concerning the structural features of the athletic heart has been based on European studies, where comprehensive cardiac screening of all elite athletes is mandatory. The typical sports activities and the demographics of athletes in the U.S. are different, and their cardiologic profiles have not been well studied because detailed cardiac examinations are not compulsory. But the NBA recently mandated that all athletes undergo annual preseason medical evaluations including stress echocardiograms, and allowed the division of cardiology at Columbia to assess the results each year.
“A detailed understanding of normal and expected cardiac remodeling in U.S. basketball players has significant clinical importance given that the incidence of sports-related sudden cardiac death in the U.S. is highest among basketball players and that the most common cause ... in this population is hypertrophic cardiomyopathy,” the investigators noted.
Their analysis of all 526 ECGs performed on NBA players during a 1-year period “will provide an invaluable frame of reference to enhance player safety for the large group of U.S. basketball players in training at all skill levels, and in the athletic community at large,” they said.
The study participants were aged 18-39 years (mean age, 25.7 years). Roughly 77% were African American, 20% were white, 2% were Hispanic, and 1% were Asian or other ethnicities. The mean height was 200.2 cm (6’7”).
Left ventricular cavity size was larger than that in the general population, but LV size was proportional to the athletes’ large body size. “Scaling LV size to body size is vitally important in the cardiac evaluation of basketball players, whose heights extend to 218 cm and body surface areas to 2.8 m2,” Dr. Engel and his associates said (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0252).
Left ventricular hypertrophy (LVH) was identified in only 27% of the athletes. African Americans had increased indices of LVH, compared with whites, and had a higher incidence of nondilated concentric hypertrophy, while whites showed predominantly eccentric dilated hypertrophy. These findings should help clinicians recognize genuine hypertrophic cardiomyopathy, which is a contraindication to participating in all but the most low-intensity competitive sports.
Most of the participants had a normal left ventricular ejection fraction, and all showed normal augmentation of LV systolic function with exercise.
Aortic root diameter was larger than that in the general population but similar to that in other elite athletes. Surprisingly, aortic root diameter increased with increasing body size only to a certain point, reaching a plateau at 31-35 mm. Fewer than 5% of the participants had an aortic root diameter of 40 mm or more, and the maximal diameter was 42 mm. “These data have important implications in the evaluation of exceptionally large athletes and question the applicability in individuals with significantly increased biometrics of the traditional formula to estimate aortic root diameter that assumes a linear association between [it] and body surface area,” they noted.
“We hope that the results of this study will assist recognition of cardiac pathologic change and provide a framework to help avoid unnecessary exclusions of athletes from competition. We believe that these data have additional applicability to other sports that preselect for athletes with height, such as volleyball, rowing, and track and field,” Dr. Engel and his associates added.
This study was supported by the National Basketball Association as part of a medical services agreement with Columbia University. Dr. Engel and his associates reported having no relevant financial disclosures.
For the first time, cardiologists have characterized the adaptive cardiac remodeling in a large cohort of National Basketball Association players, which establishes a normative database and allows physicians to distinguish it from occult pathologic changes that may precipitate sudden cardiac death, according to an imaging study.
“We hope that the present data will help to focus decision making and improve clinical acumen for the purpose of primary prevention of cardiac emergencies in U.S. basketball players and in the athletic community at large,” said Dr. David J. Engel and his associates of Columbia University, New York.
Until now, most of the literature concerning the structural features of the athletic heart has been based on European studies, where comprehensive cardiac screening of all elite athletes is mandatory. The typical sports activities and the demographics of athletes in the U.S. are different, and their cardiologic profiles have not been well studied because detailed cardiac examinations are not compulsory. But the NBA recently mandated that all athletes undergo annual preseason medical evaluations including stress echocardiograms, and allowed the division of cardiology at Columbia to assess the results each year.
“A detailed understanding of normal and expected cardiac remodeling in U.S. basketball players has significant clinical importance given that the incidence of sports-related sudden cardiac death in the U.S. is highest among basketball players and that the most common cause ... in this population is hypertrophic cardiomyopathy,” the investigators noted.
Their analysis of all 526 ECGs performed on NBA players during a 1-year period “will provide an invaluable frame of reference to enhance player safety for the large group of U.S. basketball players in training at all skill levels, and in the athletic community at large,” they said.
The study participants were aged 18-39 years (mean age, 25.7 years). Roughly 77% were African American, 20% were white, 2% were Hispanic, and 1% were Asian or other ethnicities. The mean height was 200.2 cm (6’7”).
Left ventricular cavity size was larger than that in the general population, but LV size was proportional to the athletes’ large body size. “Scaling LV size to body size is vitally important in the cardiac evaluation of basketball players, whose heights extend to 218 cm and body surface areas to 2.8 m2,” Dr. Engel and his associates said (JAMA Cardiol. 2016 Feb 24. doi: 10.1001/jamacardio.2015.0252).
Left ventricular hypertrophy (LVH) was identified in only 27% of the athletes. African Americans had increased indices of LVH, compared with whites, and had a higher incidence of nondilated concentric hypertrophy, while whites showed predominantly eccentric dilated hypertrophy. These findings should help clinicians recognize genuine hypertrophic cardiomyopathy, which is a contraindication to participating in all but the most low-intensity competitive sports.
Most of the participants had a normal left ventricular ejection fraction, and all showed normal augmentation of LV systolic function with exercise.
Aortic root diameter was larger than that in the general population but similar to that in other elite athletes. Surprisingly, aortic root diameter increased with increasing body size only to a certain point, reaching a plateau at 31-35 mm. Fewer than 5% of the participants had an aortic root diameter of 40 mm or more, and the maximal diameter was 42 mm. “These data have important implications in the evaluation of exceptionally large athletes and question the applicability in individuals with significantly increased biometrics of the traditional formula to estimate aortic root diameter that assumes a linear association between [it] and body surface area,” they noted.
“We hope that the results of this study will assist recognition of cardiac pathologic change and provide a framework to help avoid unnecessary exclusions of athletes from competition. We believe that these data have additional applicability to other sports that preselect for athletes with height, such as volleyball, rowing, and track and field,” Dr. Engel and his associates added.
This study was supported by the National Basketball Association as part of a medical services agreement with Columbia University. Dr. Engel and his associates reported having no relevant financial disclosures.
FROM JAMA CARDIOLOGY
Key clinical point: Cardiologists characterized normal, adaptive cardiac remodeling in NBA players, allowing physicians to distinguish it from occult pathologic changes that may precipitate sudden cardiac death.
Major finding: Aortic root diameter increased with increasing body size only to a certain point, reaching a plateau at 31-35 mm.
Data source: An observational cohort study in which echocardiograms of 526 professional athletes were analyzed.
Disclosures: This study was supported by the National Basketball Association as part of a medical services agreement with Columbia University. Dr. Engel and his associates reported having no relevant financial disclosures.

Degludec/liraglutide Noninferior to Uptitrated Glargine
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.

After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.
After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.
After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
FROM JAMA

Degludec/liraglutide noninferior to uptitrated glargine
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.

After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.
After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
For adults whose type 2 diabetes is not controlled by glargine plus metformin, switching from glargine to combination degludec/liraglutide lowered hemoglobin A1c levels to a noninferior degree, compared with continued uptitrating of glargine, according to a report published online March 1 in JAMA.
In DUAL V, an international, randomized, phase III clinical trial sponsored and conducted by Novo Nordisk, maker of degludec/liraglutide, 557 patients (mean age, 59 years) on a regimen of glargine plus metformin were randomly assigned to switch from glargine to combination degludec/liraglutide or to uptitrate their glargine dose to a target HbA1c level, said Dr. Ildiko Lingvay of the departments of internal medicine and clinical sciences, University of Texas, Dallas, and her associates.
After 26 weeks, HbA1c levels decreased to 6.6% with the study intervention and to 7.1% with the increased glargine. This nonsignificant difference in the primary efficacy endpoint demonstrates the noninferiority of degludec/liraglutide. Combination degludec/liraglutide also was associated with a 1.4-kg reduction in mean body weight to 86.9 kg, while increasing glargine was associated with a 1.8-kg increase in body weight to 89.1 kg.
In addition, hypoglycemic episodes occurred in 28.4% of the degludec/liraglutide group, significantly fewer than the 49.1% of the glargine group who experienced hypoglycemic episodes. The respective rates of hypoglycemic episodes were 2.23 vs. 5.05 per patient-year of exposure to the drugs.
Patients taking degludec/liraglutide reported improved scores on measures of physical health, physical functioning, bodily pain, and the impact of diabetes treatment on daily living, while those taking glargine reported worsening of these scores. Patients in the intervention group also reported higher satisfaction with treatment than did those in the control group, Dr. Lingvay and her associates said (JAMA. 2016 Mar 1. doi: 10.101/jama.2016.1252). The rate of adverse events was 343.3 per 100 person-years of exposure for degludec/liraglutide and 286.4 per 100 person-years for glargine. The respective rates of serious adverse events were 3.9 and 6.7 per 100 person-years of exposure.
“As a once-daily, single injection that is effective, is associated with weight loss, and [carries] a low risk of hypoglycemia, degludec/liraglutide may overcome many of the barriers to treatment intensification in patients treated with basal insulin,” the investigators said.
DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.
FROM JAMA
Key clinical point: Switching to combination degludec/liraglutide produced noninferior reductions in HbA1c levels in adults with uncontrolled type 2 diabetes, compared with continued uptitrating of glargine.
Major finding: After 26 weeks, mean HbA1c levels decreased to 6.6% with degludec/liraglutide and to 7.1% with increased glargine, a nonsignificant difference.
Data source: DUAL V, an international, randomized, phase III noninferiority trial in 557 adults with uncontrolled type 2 diabetes treated for 26 weeks.
Disclosures: DUAL V was sponsored by Novo Nordisk, maker of degludec/liraglutide, which also participated in the study design and conduct; the collection, analysis, and interpretation of the data; and the writing of the report. Dr. Lingvay reported ties to Novo Nordisk, Pfizer/Merck, GI Dynamics, AstraZeneca, Boehringer-Ingelheim, Janssen, and Sanofi; her associates reported ties to numerous industry sources.

DTC genomic tests: Some patients dissatisfied with physician interpretation
Twenty-seven percent of patients who purchased direct-to-consumer personal genomic testing in a large cohort study shared their results with their primary care providers, often looking for him or her to translate the complex and vague findings into simple, concrete health care advice, according to a report published online Feb. 29 in Annals of Internal Medicine.
A significant portion – 18% – of these patients came away feeling very dissatisfied with the provider’s response. They reported that the clinician’s interpretation of the results didn’t accord with the company’s interpretation, that the clinician didn’t adequately understand genetics, or that the clinician was unwilling to discuss the results or apply them to the medical care, said Cathelijne H. van der Wouden of Harvard University, Boston, and the University of Utrecht (the Netherlands), and her associates.

“All primary-care providers should have adequate clinical skills to at least engage in a discussion about genetic testing that describes its benefits and limitations and provides an account of why further action is or is not recommended,” they said.
The investigators performed what they described as the first large, longitudinal study to describe patients’ experience with commercially available genomic testing: the Impact of Personal Genomics (PGen) study. They characterized it as an “academia-industry partnership” among researchers at Harvard; Brigham and Women’s Hospital; the University of Michigan School of Public Health; 23andMe; Pathway Genomics; and SoundRocket, a scientific survey group. They focused on 1,026 adults who underwent genomic analysis, which included genetic testing for approximately 25-30 disease-risk and pharmacologic risk factors, during a 5-month period. These patients reported their experience in detailed surveys at baseline and 6 months after receiving their genomic test results.
According to these tests, the participants were found to be “at elevated genetic risk” for a mean of 20% of the 25-30 medical conditions assessed. Approximately 20%-24% of the time, participants were predicted to have an “atypical” response to 8-9 medications assessed.
Most patients were “highly satisfied” with their decision to undergo genomic testing.
A total of 278 respondents (27%) reported that they shared their results with their primary care providers and another 78 (8%) did so with another health care provider such as a physician assistant/nurse, ob.gyn., cardiologist, oncologist, neurologist, psychiatrist/psychologist, rheumatologist, endocrinologist, or immunologist. Thirty-five percent of these respondents said they were very satisfied with this discussion, while 18% said they were “not at all satisfied.” Some of the dissatisfied patients said their clinicians were unwilling or “afraid” to even look at the test results, Ms. Van der Wouden and her associates said (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M15-0995).Another reason for dissatisfaction may be that primary care providers didn’t act on a result patients felt was important, “even though such inaction may have been medically appropriate (for example, in the case of an increased risk for Alzheimer’s disease, bipolar disorder, or leukemia). Some patient dissatisfaction may stem from the disconnect between how companies market (and their customers perceive) the utility of [personal genomic testing] and how the medical community describes (and its members perceive) this utility,” the investigators added.
Physicians should not have to spend valuable time during clinical encounters deflating the misguided marketing message of companies that sell personal genomic testing to a public that has unrealistic expectations.
However, such testing isn’t the first “health” product to generate false hopes. And the health system is probably the best source of authoritative advice to counter the deceptive appeal of genetic risk prediction.
Dr. Wylie Burke and Susan Brown Trinidad are in the department of bioethics and humanities at the University of Washington, Seattle. They reported having no conflicts of interest. Dr. Burke and Ms. Trinidad made these remarks in an editorial accompanying Ms. Van der Wouden’s report (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M16-0257).
Physicians should not have to spend valuable time during clinical encounters deflating the misguided marketing message of companies that sell personal genomic testing to a public that has unrealistic expectations.
However, such testing isn’t the first “health” product to generate false hopes. And the health system is probably the best source of authoritative advice to counter the deceptive appeal of genetic risk prediction.
Dr. Wylie Burke and Susan Brown Trinidad are in the department of bioethics and humanities at the University of Washington, Seattle. They reported having no conflicts of interest. Dr. Burke and Ms. Trinidad made these remarks in an editorial accompanying Ms. Van der Wouden’s report (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M16-0257).
Physicians should not have to spend valuable time during clinical encounters deflating the misguided marketing message of companies that sell personal genomic testing to a public that has unrealistic expectations.
However, such testing isn’t the first “health” product to generate false hopes. And the health system is probably the best source of authoritative advice to counter the deceptive appeal of genetic risk prediction.
Dr. Wylie Burke and Susan Brown Trinidad are in the department of bioethics and humanities at the University of Washington, Seattle. They reported having no conflicts of interest. Dr. Burke and Ms. Trinidad made these remarks in an editorial accompanying Ms. Van der Wouden’s report (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M16-0257).
Twenty-seven percent of patients who purchased direct-to-consumer personal genomic testing in a large cohort study shared their results with their primary care providers, often looking for him or her to translate the complex and vague findings into simple, concrete health care advice, according to a report published online Feb. 29 in Annals of Internal Medicine.
A significant portion – 18% – of these patients came away feeling very dissatisfied with the provider’s response. They reported that the clinician’s interpretation of the results didn’t accord with the company’s interpretation, that the clinician didn’t adequately understand genetics, or that the clinician was unwilling to discuss the results or apply them to the medical care, said Cathelijne H. van der Wouden of Harvard University, Boston, and the University of Utrecht (the Netherlands), and her associates.
“All primary-care providers should have adequate clinical skills to at least engage in a discussion about genetic testing that describes its benefits and limitations and provides an account of why further action is or is not recommended,” they said.
The investigators performed what they described as the first large, longitudinal study to describe patients’ experience with commercially available genomic testing: the Impact of Personal Genomics (PGen) study. They characterized it as an “academia-industry partnership” among researchers at Harvard; Brigham and Women’s Hospital; the University of Michigan School of Public Health; 23andMe; Pathway Genomics; and SoundRocket, a scientific survey group. They focused on 1,026 adults who underwent genomic analysis, which included genetic testing for approximately 25-30 disease-risk and pharmacologic risk factors, during a 5-month period. These patients reported their experience in detailed surveys at baseline and 6 months after receiving their genomic test results.
According to these tests, the participants were found to be “at elevated genetic risk” for a mean of 20% of the 25-30 medical conditions assessed. Approximately 20%-24% of the time, participants were predicted to have an “atypical” response to 8-9 medications assessed.
Most patients were “highly satisfied” with their decision to undergo genomic testing.
A total of 278 respondents (27%) reported that they shared their results with their primary care providers and another 78 (8%) did so with another health care provider such as a physician assistant/nurse, ob.gyn., cardiologist, oncologist, neurologist, psychiatrist/psychologist, rheumatologist, endocrinologist, or immunologist. Thirty-five percent of these respondents said they were very satisfied with this discussion, while 18% said they were “not at all satisfied.” Some of the dissatisfied patients said their clinicians were unwilling or “afraid” to even look at the test results, Ms. Van der Wouden and her associates said (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M15-0995).Another reason for dissatisfaction may be that primary care providers didn’t act on a result patients felt was important, “even though such inaction may have been medically appropriate (for example, in the case of an increased risk for Alzheimer’s disease, bipolar disorder, or leukemia). Some patient dissatisfaction may stem from the disconnect between how companies market (and their customers perceive) the utility of [personal genomic testing] and how the medical community describes (and its members perceive) this utility,” the investigators added.
Twenty-seven percent of patients who purchased direct-to-consumer personal genomic testing in a large cohort study shared their results with their primary care providers, often looking for him or her to translate the complex and vague findings into simple, concrete health care advice, according to a report published online Feb. 29 in Annals of Internal Medicine.
A significant portion – 18% – of these patients came away feeling very dissatisfied with the provider’s response. They reported that the clinician’s interpretation of the results didn’t accord with the company’s interpretation, that the clinician didn’t adequately understand genetics, or that the clinician was unwilling to discuss the results or apply them to the medical care, said Cathelijne H. van der Wouden of Harvard University, Boston, and the University of Utrecht (the Netherlands), and her associates.
“All primary-care providers should have adequate clinical skills to at least engage in a discussion about genetic testing that describes its benefits and limitations and provides an account of why further action is or is not recommended,” they said.
The investigators performed what they described as the first large, longitudinal study to describe patients’ experience with commercially available genomic testing: the Impact of Personal Genomics (PGen) study. They characterized it as an “academia-industry partnership” among researchers at Harvard; Brigham and Women’s Hospital; the University of Michigan School of Public Health; 23andMe; Pathway Genomics; and SoundRocket, a scientific survey group. They focused on 1,026 adults who underwent genomic analysis, which included genetic testing for approximately 25-30 disease-risk and pharmacologic risk factors, during a 5-month period. These patients reported their experience in detailed surveys at baseline and 6 months after receiving their genomic test results.
According to these tests, the participants were found to be “at elevated genetic risk” for a mean of 20% of the 25-30 medical conditions assessed. Approximately 20%-24% of the time, participants were predicted to have an “atypical” response to 8-9 medications assessed.
Most patients were “highly satisfied” with their decision to undergo genomic testing.
A total of 278 respondents (27%) reported that they shared their results with their primary care providers and another 78 (8%) did so with another health care provider such as a physician assistant/nurse, ob.gyn., cardiologist, oncologist, neurologist, psychiatrist/psychologist, rheumatologist, endocrinologist, or immunologist. Thirty-five percent of these respondents said they were very satisfied with this discussion, while 18% said they were “not at all satisfied.” Some of the dissatisfied patients said their clinicians were unwilling or “afraid” to even look at the test results, Ms. Van der Wouden and her associates said (Ann Intern Med. 2016 Feb 29. doi: 10.7326/M15-0995).Another reason for dissatisfaction may be that primary care providers didn’t act on a result patients felt was important, “even though such inaction may have been medically appropriate (for example, in the case of an increased risk for Alzheimer’s disease, bipolar disorder, or leukemia). Some patient dissatisfaction may stem from the disconnect between how companies market (and their customers perceive) the utility of [personal genomic testing] and how the medical community describes (and its members perceive) this utility,” the investigators added.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point: Twenty-seven percent of patients who purchased direct-to-consumer personal genomic testing shared their results with their primary care physicians.
Major finding: Patients were found to be “at elevated genetic risk” for a mean of 20% of the 25-30 medical conditions assessed, and were predicted to have an “atypical” response to 8-9 medications 20%-24% of the time.
Data source: A prospective longitudinal cohort study involving 1,026 adults who underwent commercially available personal genomic testing during a 5-month period.
Disclosures: The study was supported by academia and industry, including the National Human Genomic Research Institute, the De Koninklijke Nederlandse Maatschappij Pharmacie Stipendiafonds, and the National Institutes of Health. Ms. van der Wouden reported having no relevant disclosures while her associates reported ties to industry.
