User login
Biosimilars in Psoriasis: The Future or Not?
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
Practice Points
- Three biosimilars have been approved by the US Food and Drug Administration to treat adult patients with plaque psoriasis and psoriatic arthritis.
- By virtue of their production, biosimilars are not identical to their reference products, and we must ensure that their safety is comparable.
New Biologics in Psoriasis: An Update on IL-23 and IL-17 Inhibitors
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
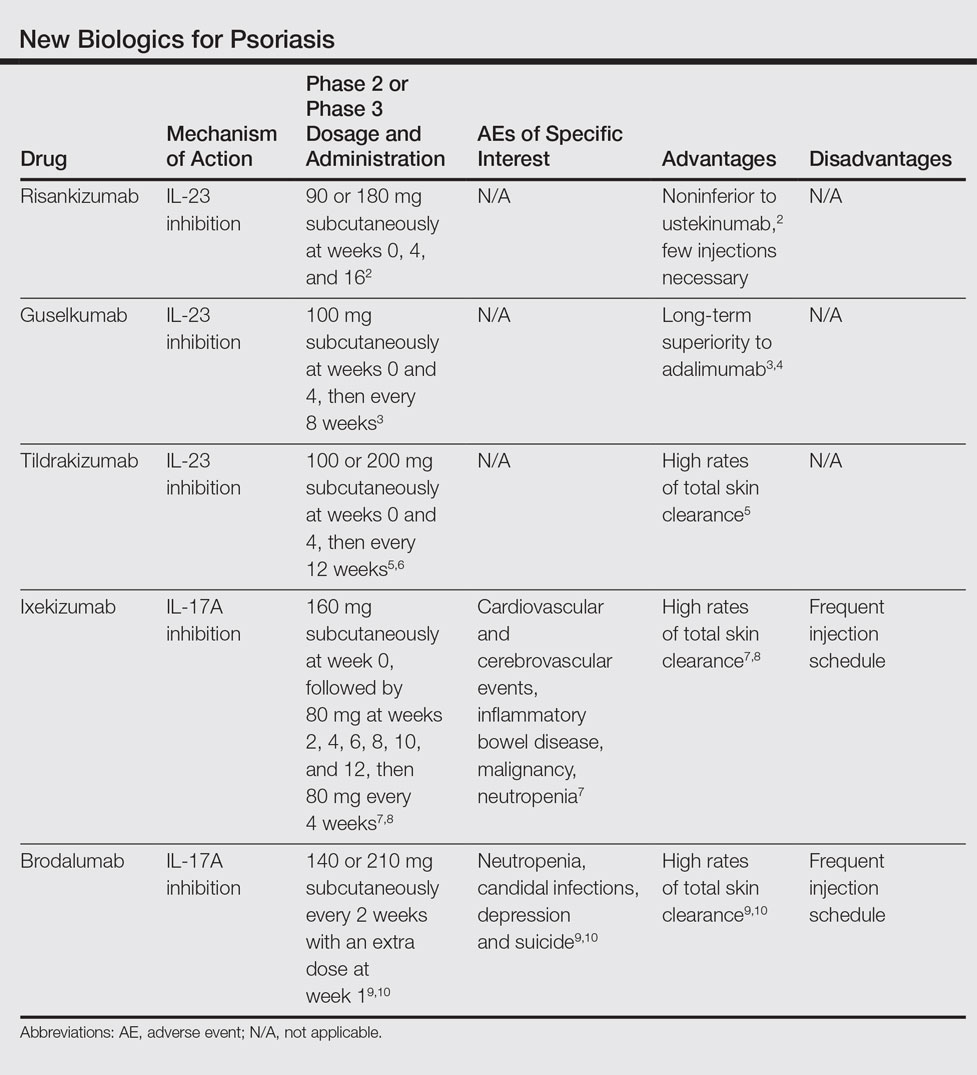
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
Practice Points
- The newest biologics for treatment of moderate to severe plaque psoriasis are IL-23 and IL-17 inhibitors with unprecedented efficacy of complete skin clearance compared to older biologics.
- Risankizumab, guselkumab, and tildrakizumab are new IL-23 inhibitors currently in phase 3 trials with promising early efficacy and safety results.
- Ixekizumab, which recently was approved, and brodalumab, which is pending US Food and Drug Administration review, are new IL-17 inhibitors that achieved total skin clearance in more than one-quarter of phase 3 participants after 12 weeks of treatment.

Advances in Minimally Invasive and Noninvasive Treatments for Submental Fat
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
Submental fat (SMF) accumulation within the subcutaneous (preplatysmal) or subplatysmal fat compartment of the cervical anatomy results in an obtuse cervicomental angle and loss of mandibular and cervical contours. It is a common cosmetic concern due to its aesthetic association with weight gain and aging.1 Minimally invasive or noninvasive submental lipolytic agents and techniques are sought for patients who are not candidates for surgery or prefer more conservative cosmetic treatments. These methods typically are only effective in addressing preplatysmal SMF, as subplatysmal SMF requires more surgical methods due to its less-accessible location. The pathology of SMF should initially be assessed by clinical examination or ultrasonography. In this article, we review the most relevant clinical and safety data on minimally invasive and noninvasive treatments for SMF, including laser-assisted lipolysis (LAL), radiofrequency (RF)–assisted lipolysis, deoxycholic acid (DCA), and cryolipolysis.
MINIMALLY INVASIVE MODALITIES
Traditional, or tumescent, liposuction is still widely considered the most effective method for removal of large masses of adiposity. Laser- and RF-assisted adjuncts have been more recently developed to improve patient side effects and recovery time and reduce the manual effort of surgeons. Of note, these adjuncts, with some exceptions, still require the same invasiveness as traditional liposuction, involving submental stab incisions of up to 2.4 mm.
Laser-Assisted Lipolysis
Laser-assisted lipolysis produces a similar effect as suction-assisted lipoplasty by focusing pulses of laser energy through a 1-mm wide fiber optic cannula and inducing thermally mediated adipolysis. The directed laser results in adipocyte rupturing with added benefits of skin retraction and small vessel coagulation, thus lessening intraoperative blood loss.2 This technique typically requires smaller incisions than traditional liposuction. The most common laser lipolysis systems used in cosmetic dermatology are the 920- to 980-nm diode lasers and 1064- to 1440-nm Nd:YAG lasers. The 924-nm diode, 1064-nm Nd:YAG, and 1064/1320-nm Nd:YAG have been best characterized in clinical trials, as reviewed by Fakhouri et al,3 with demonstrated efficacy in reducing SMF density.
The first randomized prospective trial comparing LAL (using 1064-nm Nd:YAG) and traditional liposuction in various anatomical areas on 25 patients showed no difference in cosmetic results, ecchymoses, edema, or retraction, and significantly lower postoperative pain ratings (P<.0001) in LAL.4 A more recent prospective randomized comparison of LAL (980-nm diode laser; 6–8 W) and traditional liposuction of the submental area in 40 female patients showed greater reduction in SMF thickness in the LAL group compared to the liposuction group at 2-month follow-up (6.2 vs 8.22 unspecified units; P<.001) with significant improvement from baseline in both groups (P<.001).5 However, the cosmetic benefit of LAL over traditional liposuction remains controversial and has not been unequivocally established in the literature.
Common adverse events (AEs) are postoperative swelling, ecchymoses, and pain, and complications of interest are nodularity, skin infections, burns, and nerve damage.6 In one retrospective investigation (N=537), these complications occurred at a rate of less than 1% (4 burns and 1 skin infection).6 Patients treated with LAL may report fewer AEs, especially pain and bleeding, compared to liposuction-treated patients.3
RF-Assisted Lipolysis
Radiofrequency-assisted lipolysis is one of the newest technologies in lipocontouring. NeckTite (Invasix Aesthetic Solutions) is effective for treatment of preplatysmal adiposity and cervicomental lipocontouring; a 2.4-mm bipolar probe that is inserted into the subdermal space and connected with an external electrode emits RF energy and simultaneously coagulates and aspirates adipose tissue. NeckTite also may be used in conjunction with FaceTite (Invasix Aesthetic Solutions), which promotes fibroseptal network remodeling and dermal contraction.2
In the first published investigation of the efficacy and safety of NeckTite, 47 of 55 patients received treatment of slight to moderate SMF (average body mass index [BMI], 25 kg/m2) with NeckTite and FaceTite or NeckTite alone.7 At 6-month follow-up, 87% (48/55) of patients subjectively rated treatment efficacy as satisfactory, and 2 independent physicians rated the improvement between before-and-after frontal and lateral photographs of the submental area as moderate to excellent in 95% (52/55) of all cases. Reported complications in this study were full-thickness burns resulting in minor scarring (2/55 [4%]), neck tissue hardness that resolved with daily massage after 3 months (5/55 [9%]), and transient facial nerve paresis of the mandibular branch that resolved after 2 months (1/55 [2%]).7
NONINVASIVE MODALITIES
RF-Assisted Contouring
Another exciting development in RF technology is truSculpt (Cutera), a noninvasive contouring device that is placed over the epidermis and emits RF energy that preferentially heats fat more than other tissue types. In a single-center prospective trial of efficacy and safety in the treatment of SMF, 17 patients received 2 treatments with truSculpt administered 1 month apart.8 At 1- and 6-month follow-up, 82.3% (14/17) and 52.9% (9/17) of patients showed improvement on physician assessment. Submental circumference and ultrasonographic fat thickness reductions at 1-month follow-up were 1.4 cm (5.7% of pretreatment circumference [P<.001]) and 5.4 mm (9.7% of pretreatment fat thickness [P=.005]), respectively. At further longer-term follow-up to 6 months, submental circumference was 0.9 cm (3.8% of pretreatment circumference [P<.001]) and ultrasonographic fat reduction was 6.8 mm (10.5% of pretreatment fat thickness [P=.006]). Commonly reported AEs were pain (rate not given), erythema (8/17 [47%]), edema (1/17 [6%]), and vesicle formation (1/17 [6%]); all were self-resolving. Erythema usually subsided within 6 hours posttreatment. No other AEs or complications were reported.8
Deoxycholic Acid
Deoxycholic acid (DCA)(formerly ATX-101) is an injectable liquid formulation of synthetic DCA that was approved by the US Food and Drug Administration (FDA) in 2015 for moderate to severe SMF. Deoxycholic acid exists endogenously as a bile salt emulsifier and has been shown to cause dose-dependent adipocyte lysis, necrosis, disruption and dissolution of fat architecture, and inflammatory targeting of adipocytes by immune cells.9,10 Thus, DCA causes targeted adipocytolysis and is a novel medical agent in the treatment of SMF. Supplied in 2-mL vials, clinicians may inject 10 mL at each treatment for up to 6 treatments administered 1 month apart.11
Efficacy
REFINE-1, a pivotal North American–based phase 3 trial, investigated the efficacy and safety of DCA.12 A total of 506 participants with scores of 2 (moderate) or 3 (severe) on the Clinician-Reported Submental Fat Rating Scale (CR-SMFRS) and a mean BMI of 29 kg/m2 were randomized to receive preplatysmal fat injections of 2 mg/cm2 of DCA (n=256) or placebo (n=250). Participants received up to 10 mL of product (mean total of 25 mL of DCA across all visits) at each treatment session for up to 6 sessions depending on individual efficacy, with approximately 28 days between sessions. Sixty-four percent of the treatment group received all 6 treatments. At 12-week follow-up after the last treatment session, 70% of DCA-treated participants versus 18.6% of placebo-treated participants (P<.001) improved by 1 grade or more on the CR-SMFRS and 13.4% versus 0% (P<.001) improved by 2 grades or more. Skin laxity was unchanged or improved in 92.7% of the DCA group and 87.6% of the placebo group.12
REFINE-2, the second of the North American phase 3 trials, had parallel inclusionary criteria and study design and established efficacy of 2 mg/cm2 DCA over placebo in 516 participants (randomized 1:1).13 At 12 weeks posttreatment, 66.5% of DCA-treated participants versus 22.2% of placebo-treated participants improved by 1 grade or more according to the CR-SMFRS (P<.001) and 18.6% versus 3% improved by 2 grades or more in SMF (P<.001). Magnetic resonance imaging analysis of participants in the DCA (n=113) and placebo groups (n=112) showed that 40.2% versus 5.2% (P<.001) exhibited 10% or more reduction in submental volume, with similar comparative rates of SMF thickness reduction via caliper measurements.13
Safety
Safety data from REFINE-1 showed higher rates of treatment-related AEs in DCA-treated participants compared to placebo, including hematoma (70% vs 67.3%), anesthesia (66.9% vs 4.4%), pain (65.4% vs 23.4%), edema (52.9% vs 21.8%), induration (18.3% vs 1.6%), paresthesia (12.8% vs 3.2%), nodule formation (12.5% vs 0.8%), and pruritus (8.6% vs 3.6%).12 In this trial, 11 of 258 cases (4.3%) of marginal mandibular nerve paresis and asymmetric smile occurred, all in DCA-treated participants and with a median duration of 31 days. Dysphagia resolving in a median duration of 4 days occurred in 1.6% (4/258) of DCA-treated participants.12 REFINE-2 exhibited similar rates of common AEs. Complications of note were 14 cases of marginal mandibular nerve paresis (11 in DCA group, 3 in placebo group) attributed to injection technique, 1 case of skin ulceration possibly related to accidental injection into dermis, and 6 cases of dysphagia in DCA participants attributed to higher volume treatment sessions and postinjection swelling. Dysphagia lasted a median of 2.5 days and resolved without sequelae.13
Overall, DCA demonstrated high rates of minor injection-site AEs that resolved without sequelae and could be mitigated by comfort therapies (eg, lidocaine, nonsteroidal anti-inflammatory drugs) as well as understanding the anatomy of the submental region. Adverse effects of particular interest included marginal mandibular nerve palsy, skin ulceration, and dysphagia.12,13
Cryolipolysis
Cryolipolysis is an advancement that utilizes the application of noninvasive cooling temperatures to the skin’s surface to destroy underlying adipocytes based on the concept that lipid-filled cells are more susceptible to cold-induced injury than water-filled cells. Thus, cryolipolysis selectively targets adipose tissue, leading to cell death without harm to surrounding cells and without the need for surgery or injections.14
Cryolipolysis typically is delivered via a vacuum applicator (CoolMini, Zeltiq Aesthetics Inc), which applies temperatures of –10°C (14°F) to the skin in cycles of 60 minutes each. Initially approved by the FDA for treatment of flank adiposity in 2010, cryolipolysis has since been approved for treatment of the abdomen, thighs, and submental area.14 An advantage of cryolipolysis is that it does not require frequent treatment sessions for maximal efficacy.
Efficacy
The efficacy of cryolipolysis in the treatment of SMF was established in a multicenter device investigation resulting in its FDA approval for the submental region.15 Sixty participants with a mean BMI of 31.8 kg/m2 received 1 (1/60) or 2 (59/60) treatment sessions of the submental area administered 6 weeks apart. Primary efficacy assessments included analysis by 3 blinded reviewers who viewed photographs of each participant at baseline, immediately posttreatment, 6 weeks posttreatment, and 12 weeks posttreatment; ultrasonographic measurements of SMF thickness; and a 12-point patient satisfaction questionnaire. Blinded reviewers correctly identified baseline images in 91.4% (55/60) of cases. Ultrasonography confirmed a mean reduction in SMF of 2 mm (P<.0001) or 20% of fat thickness at 12 weeks posttreatment. On subjective patient satisfaction surveys, 83% (50/60) of participants were satisfied with the procedure and 77% (46/60) reported a visible reduction in fat and perceived an improvement in appearance.15
Safety
The most common immediate posttreatment AEs were erythema/purpura (100%), numbness (90%), edema (62%), tingling (30%), blanching (25%), and bruising (3%) at the site of cryolipolysis with resolution within 1 week posttreatment, except for numbness.15 At 6-week follow-up, all AEs had resolved, except continued numbness in 4 participants that resolved by 12-week follow-up. A further event of note was fullness in the throat in 1 participant that was attributed to swelling and resolved at 40 days posttreatment without incident. No serious AEs were reported in this trial.15
A particularly concerning but rare complication that is increasing in awareness is paradoxical adipose hyperplasia following cryolipolysis. Patients may develop firm painless areas of soft tissue enlargements in the area of cryolipolysis typically 3 to 6 months posttreatment.16 The largest published report recorded an incidence rate of 0.46% (n=2, all males) at a single-center institution of 422 cryolipolysis treatments.16 Other incidence rates reported are 0.0051% and 0.78%.17 Causes and associations are not known, though male gender is speculated to increase risk.
Conclusion
This article highlights the available information on advances in minimally invasive and noninvasive treatments for SMF accumulation. The efficacy and safety trials varied in quality and in different methods of end point analysis of SMF reduction. Further, few trials have featured head-to-head comparisons of treatments.
Although liposuction and adjuncts remain the gold standard in large-mass lipid removal, these procedures are invasive and exhibit typical risks of surgery. Given its sensitive location, the submental area may require the use of more delicate therapeutic methods, including completely noninvasive devices such as truSculpt and cryolipolysis. Regardless of the chosen treatment, the most important factors in yielding patient satisfaction and SMF improvement are proper patient selection and an understanding of the anatomical source of adiposity to be addressed with the therapeutic modalities.
[polldaddy:9711250]
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
- Hatef DA, Koshy JC, Sandoval SE, et al. The submental fat compartment of the neck. Semin Plast Surg. 2009;23:288-291.
- Mulholland RS. Nonexcisional, minimally invasive rejuvenation of the neck. Clin Plast Surg. 2014;41:11-31.
- Fakhouri TM, El Tal AK, Abrou AE, et al. Laser-assisted lipolysis: a review. Dermatol Surg. 2012;38:155-169.
- Prado A, Andrades P, Danilla S, et al. A prospective, randomized, double-blind, controlled clinical trial comparing laser-assisted lipoplasty with suction-assisted lipoplasty. Plast Reconstr Surg. 2006;118:1032-1045.
- Valizadeh N, Jalaly NY, Zarghampour M, et al. Evaluation of safety and efficacy of 980-nm diode laser-assisted lipolysis versus traditional liposuction for submental rejuvenation: a randomized clinical trial. J Cosmet Laser Ther. 2016;18:41-45.
- Katz B, McBean J. Laser-assisted lipolysis: a report on complications. J Cosmet Laser Ther. 2008;10:231-233.
- Keramidas E, Rodopoulou S. Radiofrequency-assisted liposuction for neck and lower face adipodermal remodeling and contouring. Plast Reconstr Surg Glob Open. 2016;4:e850.
- Park JH, Kim JI, Park HJ, et al. Evaluation of safety and efficacy of noninvasive radiofrequency technology for submental rejuvenation [published online July 12, 2016]. Lasers Med Sci. 2016;31:1599-1605.
- Yagima Odo ME, Cucé LC, Odo LM, et al. Action of sodium deoxycholate on subcutaneous human tissue: local and systemic effects. Dermatol Surg. 2007;33:178-188; discussion 188-189.
- Rotunda AM, Suzuki H, Moy RL, et al. Detergent effects of sodium deoxycholate are a major feature of an injectable phosphatidylcholine formulation used for localized fat dissolution. Dermatol Surg. 2004;30:1001-1008.
- Kybella [package insert]. Westlake Village, CA: Kythera Biopharmaceuticals, Inc; 2015.
- Jones DH, Carruthers J, Joseph JH, et al. REFINE-1, a multicenter, randomized, double-blind, placebo-controlled, phase 3 trial with ATX-101, an injectable drug for submental fat reduction. Dermatol Surg. 2016;42:38-49.
- Humphrey S, Sykes J, Kantor J, et al. ATX-101 for reduction of submental fat: a phase III randomized controlled trial [published online July 16, 2016]. J Am Acad Dermatol. 2016;75:788-797.e7.
- Manstein D, Laubach H, Watanabe K, et al. Selective cryolysis: a novel method of non-invasive fat removal. Lasers Surg Med. 2008;40:595-604.
- Kilmer SL, Burns AJ, Zelickson BD. Safety and efficacy of cryolipolysis for non-invasive reduction of submental fat. Lasers Surg Med. 2016;48:3-13.
- Singh SM, Geddes ER, Boutrous SG, et al. Paradoxical adipose hyperplasia secondary to cryolipolysis: an underreported entity? Lasers Surg Med. 2015;47:476-478.
- Kelly E, Rodriguez-Feliz J, Kelly ME. Paradoxical adipose hyperplasia after cryolipolysis: a report on incidence and common factors identified in 510 patients. Plast Reconst Surg. 2016;137:639e-640e.
Practice Points
- New developments in minimally invasive techniques for treating submental adiposity include laser-assisted and radiofrequency-assisted lipoplasty with demonstrated clinical benefit and acceptable safety.
- Noninvasive treatments for submental adiposity include radiofrequency-assisted contouring devices, deoxycholic acid, and cryolipolysis, which offer an alternative to more invasive procedures such as lipoplasty.
- There are no comparative studies to date to suggest noninferiority of these noninvasive treatments compared to lipoplasty.
Facial Rejuvenation With Fractional Laser Resurfacing

Timed Sequential Therapy for Actinic Keratosis: Report From the Mount Sinai Winter Symposium
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Efficacy and Safety of New Dermal Fillers
Facial aging is the result of the interplay between loss of skin elasticity, changes in subcutaneous fat and other soft-tissue layers, and skeletal remodeling with chronological age.1 Dermal fillers are effective for the treatment of rhytides, facial scars, and lipoatrophy, as well as facial contouring and augmentation. Given that multiple filler options exist, updated reviews are necessary to inform clinicians of the choices that are available. We provide a detailed review of the clinical efficacy and safety of the dermal fillers with the most recent approvals by the US Food and Drug Administration (FDA).
Polymethylmethacrylate
Polymethylmethacrylate (PMMA) microspheres suspended in bovine collagen and lidocaine 0.3% were approved in 2006 for use in nasolabial folds (NLFs) and in 2014 for acne scars. Now branded as Bellafill (Suneva Medical, Inc), it is the only permanent injectable filler currently available. Once injected, the particles are not reabsorbed and can only be removed by procedural extraction (eg, liposuction of the surrounding fat); however, the permanence of PMMA does not extend to facial rejuvenation, which can last up to 5 years. Prior to use, skin testing for bovine collagen reaction is necessary. In a clinical trial of 147 patients with moderate to severe acne scarring, patients were randomized to receive PMMA in collagen (n=97) or saline (n=50).2 Injections were administered using a linear threading or serial puncture technique, and patients were reevaluated after 4 weeks for touch-up injections. After 6 months, 64% of patients treated with PMMA in collagen achieved improvement in acne scars by 2 points or more on the acne scar rating scale versus 33% of the control group (P=.0005).2
Treatment-related adverse events (AEs) include injection-site pain, bruising, swelling, erythema, and more rarely pruritus and lumps/granulomas.3 A 5-year longitudinal safety investigation of 871 patients initially treated with PMMA in collagen for NLF correction revealed that 17 patients (2.0%) had biopsy-confirmed granulomas with half of these retained at study end.4 Fifteen of these patients were treated with intralesional corticosteroids alone or in combination with intralesional 5-fluorouracil, oral antibiotics, or topical calcineurin inhibitors; 1 patient was untreated and another used topical corticosteroids. The authors noted no correlation between treatment method and granuloma response.4 Polymethylmethacrylate in collagen is contraindicated in patients with lidocaine or bovine collagen sensitivity and is not indicated for use in lip augmentation due to high rates of nodule formation.3
Hyaluronic Acid
Hyaluronic acid (HA) is a naturally occurring glycosaminoglycan polymer found in the extracellular matrix of the dermis. Hyaluronic acid fillers are bacteria derived and come in gel form. A useful advantage of HA fillers compared to other dermal fillers is the commercial availability of hyaluronidase to correct injections. Preinjection skin testing is not necessary.5
This category of nonpermanent dermal fillers has the most robust market choices. Older HA dermal fillers with reliable and proven efficacy are Restylane (Galderma Laboratories, LP)(facial rhytides, lip augmentation), Juvéderm (Ultra/Ultra XC/Ultra Plus/Ultra Plus XC [Allergan, Inc])(facial rhytides, lip augmentation), Hydrelle (Anika Therapeutics, Inc)(facial rhytides), and Prevelle Silk (Mentor Corporation)(facial rhytides); they will not be reviewed here. Newer agents include Belotero Balance (Merz Aesthetics), Juvéderm Voluma XC (Allergan, Inc), Restylane Silk (Galderma Laboratories, LP), and Restylane Lyft (Galderma Laboratories, LP).
Belotero Balance
Belotero Balance is used to treat fine lines and wrinkles, especially NLFs.6 The initial pivotal studies that led to FDA approval in 2011 demonstrated noninferiority and superiority to bovine collagen for use in the treatment of NLFs.7,8 One hundred eighteen patients with bilateral NLFs that were rated as 2 (moderate) or 3 (severe) on the wrinkle severity rating scale (WSRS) were randomized to split-face injection of Belotero Balance in one NLF and bovine collagen in the contralateral NLF.7 An additional injection at week 2 was allowed for optimal correction. Belotero Balance was noninferior to bovine collagen at week 2, with mean improvement in WSRS of 1.52 versus 1.57 (P=.50). Belotero Balance was superior to bovine collagen in mean WSRS improvement at weeks 12 (1.25 vs 0.26; P<.001), 16 (1.09 vs 0.66; P<.001), and 24 (1.08 vs 0.50; P<.001).7 In a subsequent open-label extension study, which included 95 of 118 patients who received Belotero Balance injections in both NLFs at week 24, 80.2% of patients showed sustained improvement in WSRS from baseline for 48 weeks without further injection.8
The first comparative study of Belotero Balance with other established HA fillers at the time—Restylane and Juvéderm Ultra 3/Ultra Plus XC—to treat NLFs demonstrated noninferiority.9 Forty patients with bilateral, moderate to severe NLFs (rated 3 or 4 on the Merz severity scale) were randomized to split-face groups of Belotero Balance versus Restylane or Belotero Balance versus Juvéderm. At 12 months, NLF severity improved from 2.3 to 1.5 in the Restylane group and from 2.3 to 1.6 in the Juvéderm group.9
Belotero Balance has been compared to Juvéderm Ultra XC for use in perioral lines.10 The study included 136 patients with moderate to severe perioral lines, according to the perioral lines severity scale, who were randomized (1:1 ratio) to receive injections of Belotero Balance or Juvéderm Ultra XC to correct upper and lower perioral lines, with assessment at week 2 for optimization. After 6 months, 87% of Juvéderm-treated patients compared to 72% of Belotero Balance–treated patients had 1-point improvement in perioral lines (P<.04). Juvéderm-treated patients also reported significantly less pain than Belotero Balance–treated patients (P<.001).10
Treatment-related AEs are described in the Table, with the majority occurring at lower rates compared to a collagen control group and self-resolving within 2 weeks.7
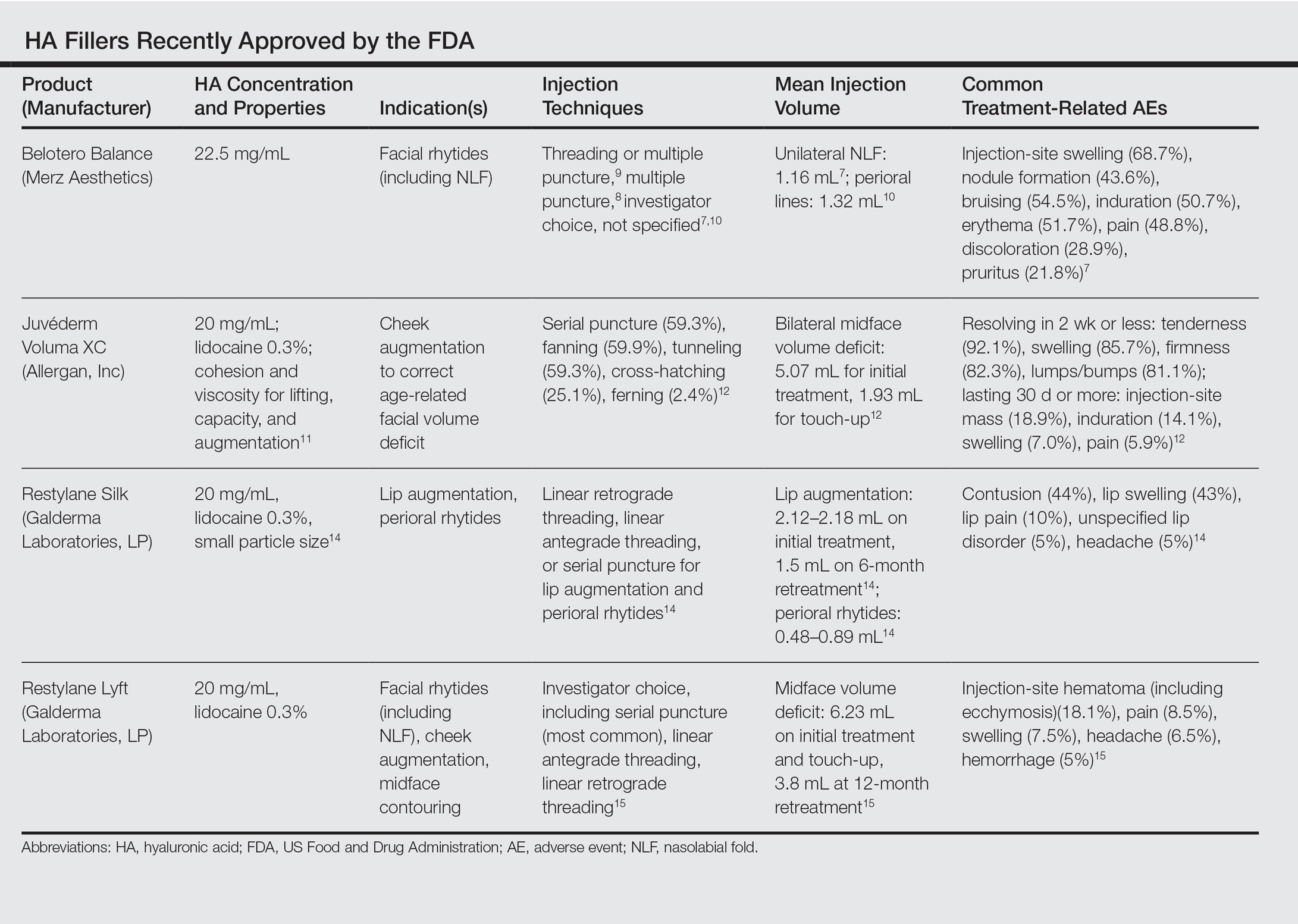
Juvéderm Voluma XC
Juvéderm Voluma XC was FDA approved in 2013 for cheek augmentation to correct age-related volume deficit restoration by subcutaneous or subperiosteal injections. In its landmark multicenter investigation, 282 patients with moderate to severe midface (eg, zygomaticomalar, anteromedial cheek, submalar regions) volume deficit measured on a validated midface volume deficit scale (MFVDS) were treated with Juvéderm Voluma XC (n=235) or control (n=47).11 Patients were reevaluated at 30 days and 81.9% received touch-up injections. At a 6-month primary evaluation, 86% of the Juvéderm-treated patients versus 39% of the control patients showed 1-point improvement on the MFVDS (P<.001). At 24-months’ follow-up, 44.6% of patients sustained efficacy.11 Of these aforementioned patients, 167 received repeat treatment due to lost correction or patient request and 91.1% improved by 1 point or more on the MFVDS on evaluation 12 months after repeat treatment.12 For this same population of patients, a 2-year extended follow-up of patient-reported outcomes revealed that 49% of patients felt fulfilled in their treatment goals 2 years after treatment and 79% of patients rated improvement from baseline based on the global aesthetic improvement scale.13 Efficacy studies involving Juvéderm Voluma XC are currently ongoing for facial temporal aging (registered at www.clinicaltrials.gov with the identifier NCT02437903) and recruiting for mandibular hypoplasia (NCT02330016).
Common treatment-related AEs are detailed in the Table. Two patients required treatment with hyaluronidase for chronic lumpiness and nodularity following non–treatment-related cellulitis.11 The product is contraindicated in patients with allergy to lidocaine.
Restylane Silk
Restylane Silk was approved in 2014 for lip augmentation and perioral rhytides. Efficacy and safety was demonstrated in a large multicenter randomized investigation in which 221 patients seeking lip augmentation received either Restylane Silk (n=177) injected submucosally for treatment of the upper and lower lips and/or intradermally for perioral rhytides or no treatment (n=44).14 Restylane treatment group patients optionally received touch-up at 2 weeks for optimization. All patients, including the control group, received injections at 6 months. At the 2-month primary end point, 80.2% of the treatment group exhibited at least 1-point improvement in upper lip fullness on the Medicis lip fullness scale compared to 11.9% (P<.001) of the control group; response rates for the lower lips were 84.2% versus 18.4% (P<.001). Patients in the treatment group receiving injections for perioral rhytides showed significant improvement in perioral rhytides through week 24 compared to patients treated for lip augmentation only (P<.001).14 Restylane Silk currently is undergoing investigation for cheek rejuvenation (NCT02636894, NCT02679924) and treatment of hand photoaging (NCT02780258).
The most common AEs are listed in the Table. No lip disorders were considered clinically concerning on evaluation. Concomitant lip augmentation and treatment of perioral rhytides yielded similar rates of AEs.14 Restylane Silk is not to be used in patients with known lidocaine allergy.
Restylane Lyft
Restylane Lyft (formerly known as Perlane-L) was approved in 2010 for use in facial rhytides, including NLFs, and gained approval in 2015 for use in cheek augmentation and midface contouring. Only its efficacy and safety for the more recent indication will be reviewed here.
In an evaluator-blinded investigation of 200 patients with mild to substantial bilateral midface deficiency based on the Medicis midface volume scale (MMVS), patients were randomized to receive supraperiosteal and subcutaneous treatment with Restylane Lyft (n=150) or no treatment (n=50).15 Touch-up injections at week 2 or month 12 were available to treatment group patients and all patients were given either an initial treatment or retreatment at 12 months. Primary end point evaluation at week 8 showed that 89% of treatment group patients had at least 1 grade MMVS improvement compared to 16% of the control group (P<.001). Although the percentage of these MMVS responders in the treatment group decreased with each follow-up period to 54.3% at month 12, retreatment was effective in reproducing a similar MMVS response rate as with initial treatment.15 Restylane Lyft is under ongoing investigation for dorsal hand rejuvenation (NCT02650921).
In addition to the common treatment-related AEs listed in the Table, 2 patients reported serious AEs, including bilateral implant-site inflammation and unilateral implant-site hematoma and infection (organism not described), all of which resolved with unspecified treatment.15 Lidocaine allergies are contraindications for use.
Conclusion
Several new options in dermal fillers have been approved in recent years and have demonstrated efficacy and acceptable safety in various cosmetic rejuvenation applications. Restylane Silk and Restylane Lyft are undergoing further studies to evaluate use in hand rejuvenation, an area that currently has few cosmetic filler treatment options. As technology continues to progress and new formulations of dermal fillers with varied properties and benefits are available, clinicians should expect multiple options for use in rhytides, volume deficits, and contouring.
ADDENDUM
After the manuscript was accepted for publication, Juvéderm Volbella XC (Allergan, Inc) was approved by the FDA for use in lip augmentation and thus is not included in this review.
- Fitzgerald R, Graivier MH, Kane M, et al. Update on facial aging. Aesthet Surg J. 2010;30(suppl):S11-S24.
- Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
- Bellafill [package insert]. San Diego, CA: Suneva Medical, Inc; 2015.
- Cohen S, Dover J, Monheit G, et al. Five-year safety and satisfaction study of PMMA-collagen in the correction of nasolabial folds. Dermatol Surg. 2015;41(suppl 1):S302-S313.
- Greene JJ, Sidle DM. The hyaluronic acid fillers: current understanding of the tissue device interface. Facial Plast Surg Clin North Am. 2015;23:423-432.
- Lorenc ZP, Fagien S, Flynn TC, et al. Review of key Belotero Balance safety and efficacy trials. Plast Reconstr Surg. 2013;132(4, suppl 2):33S-40S.
- Narins RS, Coleman W, Donofrio L, et al. Nonanimal sourced hyaluronic acid–based dermal filler using a cohesive polydensified matrix technology is superior to bovine collagen in the correction of moderate to severe nasolabial folds: results from a 6-month, randomized, blinded, controlled, multicenter study. Dermatol Surg. 2010;36(suppl 1):730-740.
- Narins RS, Coleman WP 3rd, Donofrio LM, et al. Improvement in nasolabial folds with a hyaluronic acid filler using a cohesive polydensified matrix technology: results from an 18-month open-label extension trial. Dermatol Surg. 2010;36(suppl 3):1800-1808.
- Prager W, Wissmueller E, Havermann I, et al. A prospective, split-face, randomized, comparative study of safety and 12-month longevity of three formulations of hyaluronic acid dermal filler for treatment of nasolabial folds. Dermatol Surg. 2012;38(7, pt 2):1143-1150.
- Butterwick K, Marmur E, Narurkar V, et al. HYC-24L demonstrates greater effectiveness with less pain than CPM-22.5 for treatment of perioral lines in a randomized controlled trial. Dermatol Surg. 2015;41:1351-1360.
- Jones D, Murphy DK. Volumizing hyaluronic acid filler for midface volume deficit: 2-year results from a pivotal single-blind randomized controlled study. Dermatol Surg. 2013;39:1602-1612.
- Baumann L, Narins RS, Beer K, et al. Volumizing hyaluronic acid filler for midface volume deficit: results after repeat treatment. Dermatol Surg. 2015;41(suppl 1):S284-S292.
- Few J, Cox SE, Paradkar-Mitragotri D, et al. A multicenter, single-blind randomized, controlled study of a volumizing hyaluronic acid filler for midface volume deficit: patient-reported outcomes at 2 years. Aesthet Surg J. 2015;35:589-599.
- Beer K, Glogau RG, Dover JS, et al. A randomized, evaluator-blinded, controlled study of effectiveness and safety of small particle hyaluronic acid plus lidocaine for lip augmentation and perioral rhytides. Dermatol Surg. 2015;41(suppl 1):S127-S136.
- Weiss RA, Moradi A, Bank D, et al. Effectiveness and safety of large gel particle hyaluronic acid with lidocaine for correction of midface volume deficit or contour deficiency. Dermatol Surg. 2016;42:699-709.
Facial aging is the result of the interplay between loss of skin elasticity, changes in subcutaneous fat and other soft-tissue layers, and skeletal remodeling with chronological age.1 Dermal fillers are effective for the treatment of rhytides, facial scars, and lipoatrophy, as well as facial contouring and augmentation. Given that multiple filler options exist, updated reviews are necessary to inform clinicians of the choices that are available. We provide a detailed review of the clinical efficacy and safety of the dermal fillers with the most recent approvals by the US Food and Drug Administration (FDA).
Polymethylmethacrylate
Polymethylmethacrylate (PMMA) microspheres suspended in bovine collagen and lidocaine 0.3% were approved in 2006 for use in nasolabial folds (NLFs) and in 2014 for acne scars. Now branded as Bellafill (Suneva Medical, Inc), it is the only permanent injectable filler currently available. Once injected, the particles are not reabsorbed and can only be removed by procedural extraction (eg, liposuction of the surrounding fat); however, the permanence of PMMA does not extend to facial rejuvenation, which can last up to 5 years. Prior to use, skin testing for bovine collagen reaction is necessary. In a clinical trial of 147 patients with moderate to severe acne scarring, patients were randomized to receive PMMA in collagen (n=97) or saline (n=50).2 Injections were administered using a linear threading or serial puncture technique, and patients were reevaluated after 4 weeks for touch-up injections. After 6 months, 64% of patients treated with PMMA in collagen achieved improvement in acne scars by 2 points or more on the acne scar rating scale versus 33% of the control group (P=.0005).2
Treatment-related adverse events (AEs) include injection-site pain, bruising, swelling, erythema, and more rarely pruritus and lumps/granulomas.3 A 5-year longitudinal safety investigation of 871 patients initially treated with PMMA in collagen for NLF correction revealed that 17 patients (2.0%) had biopsy-confirmed granulomas with half of these retained at study end.4 Fifteen of these patients were treated with intralesional corticosteroids alone or in combination with intralesional 5-fluorouracil, oral antibiotics, or topical calcineurin inhibitors; 1 patient was untreated and another used topical corticosteroids. The authors noted no correlation between treatment method and granuloma response.4 Polymethylmethacrylate in collagen is contraindicated in patients with lidocaine or bovine collagen sensitivity and is not indicated for use in lip augmentation due to high rates of nodule formation.3
Hyaluronic Acid
Hyaluronic acid (HA) is a naturally occurring glycosaminoglycan polymer found in the extracellular matrix of the dermis. Hyaluronic acid fillers are bacteria derived and come in gel form. A useful advantage of HA fillers compared to other dermal fillers is the commercial availability of hyaluronidase to correct injections. Preinjection skin testing is not necessary.5
This category of nonpermanent dermal fillers has the most robust market choices. Older HA dermal fillers with reliable and proven efficacy are Restylane (Galderma Laboratories, LP)(facial rhytides, lip augmentation), Juvéderm (Ultra/Ultra XC/Ultra Plus/Ultra Plus XC [Allergan, Inc])(facial rhytides, lip augmentation), Hydrelle (Anika Therapeutics, Inc)(facial rhytides), and Prevelle Silk (Mentor Corporation)(facial rhytides); they will not be reviewed here. Newer agents include Belotero Balance (Merz Aesthetics), Juvéderm Voluma XC (Allergan, Inc), Restylane Silk (Galderma Laboratories, LP), and Restylane Lyft (Galderma Laboratories, LP).
Belotero Balance
Belotero Balance is used to treat fine lines and wrinkles, especially NLFs.6 The initial pivotal studies that led to FDA approval in 2011 demonstrated noninferiority and superiority to bovine collagen for use in the treatment of NLFs.7,8 One hundred eighteen patients with bilateral NLFs that were rated as 2 (moderate) or 3 (severe) on the wrinkle severity rating scale (WSRS) were randomized to split-face injection of Belotero Balance in one NLF and bovine collagen in the contralateral NLF.7 An additional injection at week 2 was allowed for optimal correction. Belotero Balance was noninferior to bovine collagen at week 2, with mean improvement in WSRS of 1.52 versus 1.57 (P=.50). Belotero Balance was superior to bovine collagen in mean WSRS improvement at weeks 12 (1.25 vs 0.26; P<.001), 16 (1.09 vs 0.66; P<.001), and 24 (1.08 vs 0.50; P<.001).7 In a subsequent open-label extension study, which included 95 of 118 patients who received Belotero Balance injections in both NLFs at week 24, 80.2% of patients showed sustained improvement in WSRS from baseline for 48 weeks without further injection.8
The first comparative study of Belotero Balance with other established HA fillers at the time—Restylane and Juvéderm Ultra 3/Ultra Plus XC—to treat NLFs demonstrated noninferiority.9 Forty patients with bilateral, moderate to severe NLFs (rated 3 or 4 on the Merz severity scale) were randomized to split-face groups of Belotero Balance versus Restylane or Belotero Balance versus Juvéderm. At 12 months, NLF severity improved from 2.3 to 1.5 in the Restylane group and from 2.3 to 1.6 in the Juvéderm group.9
Belotero Balance has been compared to Juvéderm Ultra XC for use in perioral lines.10 The study included 136 patients with moderate to severe perioral lines, according to the perioral lines severity scale, who were randomized (1:1 ratio) to receive injections of Belotero Balance or Juvéderm Ultra XC to correct upper and lower perioral lines, with assessment at week 2 for optimization. After 6 months, 87% of Juvéderm-treated patients compared to 72% of Belotero Balance–treated patients had 1-point improvement in perioral lines (P<.04). Juvéderm-treated patients also reported significantly less pain than Belotero Balance–treated patients (P<.001).10
Treatment-related AEs are described in the Table, with the majority occurring at lower rates compared to a collagen control group and self-resolving within 2 weeks.7
Juvéderm Voluma XC
Juvéderm Voluma XC was FDA approved in 2013 for cheek augmentation to correct age-related volume deficit restoration by subcutaneous or subperiosteal injections. In its landmark multicenter investigation, 282 patients with moderate to severe midface (eg, zygomaticomalar, anteromedial cheek, submalar regions) volume deficit measured on a validated midface volume deficit scale (MFVDS) were treated with Juvéderm Voluma XC (n=235) or control (n=47).11 Patients were reevaluated at 30 days and 81.9% received touch-up injections. At a 6-month primary evaluation, 86% of the Juvéderm-treated patients versus 39% of the control patients showed 1-point improvement on the MFVDS (P<.001). At 24-months’ follow-up, 44.6% of patients sustained efficacy.11 Of these aforementioned patients, 167 received repeat treatment due to lost correction or patient request and 91.1% improved by 1 point or more on the MFVDS on evaluation 12 months after repeat treatment.12 For this same population of patients, a 2-year extended follow-up of patient-reported outcomes revealed that 49% of patients felt fulfilled in their treatment goals 2 years after treatment and 79% of patients rated improvement from baseline based on the global aesthetic improvement scale.13 Efficacy studies involving Juvéderm Voluma XC are currently ongoing for facial temporal aging (registered at www.clinicaltrials.gov with the identifier NCT02437903) and recruiting for mandibular hypoplasia (NCT02330016).
Common treatment-related AEs are detailed in the Table. Two patients required treatment with hyaluronidase for chronic lumpiness and nodularity following non–treatment-related cellulitis.11 The product is contraindicated in patients with allergy to lidocaine.
Restylane Silk
Restylane Silk was approved in 2014 for lip augmentation and perioral rhytides. Efficacy and safety was demonstrated in a large multicenter randomized investigation in which 221 patients seeking lip augmentation received either Restylane Silk (n=177) injected submucosally for treatment of the upper and lower lips and/or intradermally for perioral rhytides or no treatment (n=44).14 Restylane treatment group patients optionally received touch-up at 2 weeks for optimization. All patients, including the control group, received injections at 6 months. At the 2-month primary end point, 80.2% of the treatment group exhibited at least 1-point improvement in upper lip fullness on the Medicis lip fullness scale compared to 11.9% (P<.001) of the control group; response rates for the lower lips were 84.2% versus 18.4% (P<.001). Patients in the treatment group receiving injections for perioral rhytides showed significant improvement in perioral rhytides through week 24 compared to patients treated for lip augmentation only (P<.001).14 Restylane Silk currently is undergoing investigation for cheek rejuvenation (NCT02636894, NCT02679924) and treatment of hand photoaging (NCT02780258).
The most common AEs are listed in the Table. No lip disorders were considered clinically concerning on evaluation. Concomitant lip augmentation and treatment of perioral rhytides yielded similar rates of AEs.14 Restylane Silk is not to be used in patients with known lidocaine allergy.
Restylane Lyft
Restylane Lyft (formerly known as Perlane-L) was approved in 2010 for use in facial rhytides, including NLFs, and gained approval in 2015 for use in cheek augmentation and midface contouring. Only its efficacy and safety for the more recent indication will be reviewed here.
In an evaluator-blinded investigation of 200 patients with mild to substantial bilateral midface deficiency based on the Medicis midface volume scale (MMVS), patients were randomized to receive supraperiosteal and subcutaneous treatment with Restylane Lyft (n=150) or no treatment (n=50).15 Touch-up injections at week 2 or month 12 were available to treatment group patients and all patients were given either an initial treatment or retreatment at 12 months. Primary end point evaluation at week 8 showed that 89% of treatment group patients had at least 1 grade MMVS improvement compared to 16% of the control group (P<.001). Although the percentage of these MMVS responders in the treatment group decreased with each follow-up period to 54.3% at month 12, retreatment was effective in reproducing a similar MMVS response rate as with initial treatment.15 Restylane Lyft is under ongoing investigation for dorsal hand rejuvenation (NCT02650921).
In addition to the common treatment-related AEs listed in the Table, 2 patients reported serious AEs, including bilateral implant-site inflammation and unilateral implant-site hematoma and infection (organism not described), all of which resolved with unspecified treatment.15 Lidocaine allergies are contraindications for use.
Conclusion
Several new options in dermal fillers have been approved in recent years and have demonstrated efficacy and acceptable safety in various cosmetic rejuvenation applications. Restylane Silk and Restylane Lyft are undergoing further studies to evaluate use in hand rejuvenation, an area that currently has few cosmetic filler treatment options. As technology continues to progress and new formulations of dermal fillers with varied properties and benefits are available, clinicians should expect multiple options for use in rhytides, volume deficits, and contouring.
ADDENDUM
After the manuscript was accepted for publication, Juvéderm Volbella XC (Allergan, Inc) was approved by the FDA for use in lip augmentation and thus is not included in this review.
Facial aging is the result of the interplay between loss of skin elasticity, changes in subcutaneous fat and other soft-tissue layers, and skeletal remodeling with chronological age.1 Dermal fillers are effective for the treatment of rhytides, facial scars, and lipoatrophy, as well as facial contouring and augmentation. Given that multiple filler options exist, updated reviews are necessary to inform clinicians of the choices that are available. We provide a detailed review of the clinical efficacy and safety of the dermal fillers with the most recent approvals by the US Food and Drug Administration (FDA).
Polymethylmethacrylate
Polymethylmethacrylate (PMMA) microspheres suspended in bovine collagen and lidocaine 0.3% were approved in 2006 for use in nasolabial folds (NLFs) and in 2014 for acne scars. Now branded as Bellafill (Suneva Medical, Inc), it is the only permanent injectable filler currently available. Once injected, the particles are not reabsorbed and can only be removed by procedural extraction (eg, liposuction of the surrounding fat); however, the permanence of PMMA does not extend to facial rejuvenation, which can last up to 5 years. Prior to use, skin testing for bovine collagen reaction is necessary. In a clinical trial of 147 patients with moderate to severe acne scarring, patients were randomized to receive PMMA in collagen (n=97) or saline (n=50).2 Injections were administered using a linear threading or serial puncture technique, and patients were reevaluated after 4 weeks for touch-up injections. After 6 months, 64% of patients treated with PMMA in collagen achieved improvement in acne scars by 2 points or more on the acne scar rating scale versus 33% of the control group (P=.0005).2
Treatment-related adverse events (AEs) include injection-site pain, bruising, swelling, erythema, and more rarely pruritus and lumps/granulomas.3 A 5-year longitudinal safety investigation of 871 patients initially treated with PMMA in collagen for NLF correction revealed that 17 patients (2.0%) had biopsy-confirmed granulomas with half of these retained at study end.4 Fifteen of these patients were treated with intralesional corticosteroids alone or in combination with intralesional 5-fluorouracil, oral antibiotics, or topical calcineurin inhibitors; 1 patient was untreated and another used topical corticosteroids. The authors noted no correlation between treatment method and granuloma response.4 Polymethylmethacrylate in collagen is contraindicated in patients with lidocaine or bovine collagen sensitivity and is not indicated for use in lip augmentation due to high rates of nodule formation.3
Hyaluronic Acid
Hyaluronic acid (HA) is a naturally occurring glycosaminoglycan polymer found in the extracellular matrix of the dermis. Hyaluronic acid fillers are bacteria derived and come in gel form. A useful advantage of HA fillers compared to other dermal fillers is the commercial availability of hyaluronidase to correct injections. Preinjection skin testing is not necessary.5
This category of nonpermanent dermal fillers has the most robust market choices. Older HA dermal fillers with reliable and proven efficacy are Restylane (Galderma Laboratories, LP)(facial rhytides, lip augmentation), Juvéderm (Ultra/Ultra XC/Ultra Plus/Ultra Plus XC [Allergan, Inc])(facial rhytides, lip augmentation), Hydrelle (Anika Therapeutics, Inc)(facial rhytides), and Prevelle Silk (Mentor Corporation)(facial rhytides); they will not be reviewed here. Newer agents include Belotero Balance (Merz Aesthetics), Juvéderm Voluma XC (Allergan, Inc), Restylane Silk (Galderma Laboratories, LP), and Restylane Lyft (Galderma Laboratories, LP).
Belotero Balance
Belotero Balance is used to treat fine lines and wrinkles, especially NLFs.6 The initial pivotal studies that led to FDA approval in 2011 demonstrated noninferiority and superiority to bovine collagen for use in the treatment of NLFs.7,8 One hundred eighteen patients with bilateral NLFs that were rated as 2 (moderate) or 3 (severe) on the wrinkle severity rating scale (WSRS) were randomized to split-face injection of Belotero Balance in one NLF and bovine collagen in the contralateral NLF.7 An additional injection at week 2 was allowed for optimal correction. Belotero Balance was noninferior to bovine collagen at week 2, with mean improvement in WSRS of 1.52 versus 1.57 (P=.50). Belotero Balance was superior to bovine collagen in mean WSRS improvement at weeks 12 (1.25 vs 0.26; P<.001), 16 (1.09 vs 0.66; P<.001), and 24 (1.08 vs 0.50; P<.001).7 In a subsequent open-label extension study, which included 95 of 118 patients who received Belotero Balance injections in both NLFs at week 24, 80.2% of patients showed sustained improvement in WSRS from baseline for 48 weeks without further injection.8
The first comparative study of Belotero Balance with other established HA fillers at the time—Restylane and Juvéderm Ultra 3/Ultra Plus XC—to treat NLFs demonstrated noninferiority.9 Forty patients with bilateral, moderate to severe NLFs (rated 3 or 4 on the Merz severity scale) were randomized to split-face groups of Belotero Balance versus Restylane or Belotero Balance versus Juvéderm. At 12 months, NLF severity improved from 2.3 to 1.5 in the Restylane group and from 2.3 to 1.6 in the Juvéderm group.9
Belotero Balance has been compared to Juvéderm Ultra XC for use in perioral lines.10 The study included 136 patients with moderate to severe perioral lines, according to the perioral lines severity scale, who were randomized (1:1 ratio) to receive injections of Belotero Balance or Juvéderm Ultra XC to correct upper and lower perioral lines, with assessment at week 2 for optimization. After 6 months, 87% of Juvéderm-treated patients compared to 72% of Belotero Balance–treated patients had 1-point improvement in perioral lines (P<.04). Juvéderm-treated patients also reported significantly less pain than Belotero Balance–treated patients (P<.001).10
Treatment-related AEs are described in the Table, with the majority occurring at lower rates compared to a collagen control group and self-resolving within 2 weeks.7
Juvéderm Voluma XC
Juvéderm Voluma XC was FDA approved in 2013 for cheek augmentation to correct age-related volume deficit restoration by subcutaneous or subperiosteal injections. In its landmark multicenter investigation, 282 patients with moderate to severe midface (eg, zygomaticomalar, anteromedial cheek, submalar regions) volume deficit measured on a validated midface volume deficit scale (MFVDS) were treated with Juvéderm Voluma XC (n=235) or control (n=47).11 Patients were reevaluated at 30 days and 81.9% received touch-up injections. At a 6-month primary evaluation, 86% of the Juvéderm-treated patients versus 39% of the control patients showed 1-point improvement on the MFVDS (P<.001). At 24-months’ follow-up, 44.6% of patients sustained efficacy.11 Of these aforementioned patients, 167 received repeat treatment due to lost correction or patient request and 91.1% improved by 1 point or more on the MFVDS on evaluation 12 months after repeat treatment.12 For this same population of patients, a 2-year extended follow-up of patient-reported outcomes revealed that 49% of patients felt fulfilled in their treatment goals 2 years after treatment and 79% of patients rated improvement from baseline based on the global aesthetic improvement scale.13 Efficacy studies involving Juvéderm Voluma XC are currently ongoing for facial temporal aging (registered at www.clinicaltrials.gov with the identifier NCT02437903) and recruiting for mandibular hypoplasia (NCT02330016).
Common treatment-related AEs are detailed in the Table. Two patients required treatment with hyaluronidase for chronic lumpiness and nodularity following non–treatment-related cellulitis.11 The product is contraindicated in patients with allergy to lidocaine.
Restylane Silk
Restylane Silk was approved in 2014 for lip augmentation and perioral rhytides. Efficacy and safety was demonstrated in a large multicenter randomized investigation in which 221 patients seeking lip augmentation received either Restylane Silk (n=177) injected submucosally for treatment of the upper and lower lips and/or intradermally for perioral rhytides or no treatment (n=44).14 Restylane treatment group patients optionally received touch-up at 2 weeks for optimization. All patients, including the control group, received injections at 6 months. At the 2-month primary end point, 80.2% of the treatment group exhibited at least 1-point improvement in upper lip fullness on the Medicis lip fullness scale compared to 11.9% (P<.001) of the control group; response rates for the lower lips were 84.2% versus 18.4% (P<.001). Patients in the treatment group receiving injections for perioral rhytides showed significant improvement in perioral rhytides through week 24 compared to patients treated for lip augmentation only (P<.001).14 Restylane Silk currently is undergoing investigation for cheek rejuvenation (NCT02636894, NCT02679924) and treatment of hand photoaging (NCT02780258).
The most common AEs are listed in the Table. No lip disorders were considered clinically concerning on evaluation. Concomitant lip augmentation and treatment of perioral rhytides yielded similar rates of AEs.14 Restylane Silk is not to be used in patients with known lidocaine allergy.
Restylane Lyft
Restylane Lyft (formerly known as Perlane-L) was approved in 2010 for use in facial rhytides, including NLFs, and gained approval in 2015 for use in cheek augmentation and midface contouring. Only its efficacy and safety for the more recent indication will be reviewed here.
In an evaluator-blinded investigation of 200 patients with mild to substantial bilateral midface deficiency based on the Medicis midface volume scale (MMVS), patients were randomized to receive supraperiosteal and subcutaneous treatment with Restylane Lyft (n=150) or no treatment (n=50).15 Touch-up injections at week 2 or month 12 were available to treatment group patients and all patients were given either an initial treatment or retreatment at 12 months. Primary end point evaluation at week 8 showed that 89% of treatment group patients had at least 1 grade MMVS improvement compared to 16% of the control group (P<.001). Although the percentage of these MMVS responders in the treatment group decreased with each follow-up period to 54.3% at month 12, retreatment was effective in reproducing a similar MMVS response rate as with initial treatment.15 Restylane Lyft is under ongoing investigation for dorsal hand rejuvenation (NCT02650921).
In addition to the common treatment-related AEs listed in the Table, 2 patients reported serious AEs, including bilateral implant-site inflammation and unilateral implant-site hematoma and infection (organism not described), all of which resolved with unspecified treatment.15 Lidocaine allergies are contraindications for use.
Conclusion
Several new options in dermal fillers have been approved in recent years and have demonstrated efficacy and acceptable safety in various cosmetic rejuvenation applications. Restylane Silk and Restylane Lyft are undergoing further studies to evaluate use in hand rejuvenation, an area that currently has few cosmetic filler treatment options. As technology continues to progress and new formulations of dermal fillers with varied properties and benefits are available, clinicians should expect multiple options for use in rhytides, volume deficits, and contouring.
ADDENDUM
After the manuscript was accepted for publication, Juvéderm Volbella XC (Allergan, Inc) was approved by the FDA for use in lip augmentation and thus is not included in this review.
- Fitzgerald R, Graivier MH, Kane M, et al. Update on facial aging. Aesthet Surg J. 2010;30(suppl):S11-S24.
- Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
- Bellafill [package insert]. San Diego, CA: Suneva Medical, Inc; 2015.
- Cohen S, Dover J, Monheit G, et al. Five-year safety and satisfaction study of PMMA-collagen in the correction of nasolabial folds. Dermatol Surg. 2015;41(suppl 1):S302-S313.
- Greene JJ, Sidle DM. The hyaluronic acid fillers: current understanding of the tissue device interface. Facial Plast Surg Clin North Am. 2015;23:423-432.
- Lorenc ZP, Fagien S, Flynn TC, et al. Review of key Belotero Balance safety and efficacy trials. Plast Reconstr Surg. 2013;132(4, suppl 2):33S-40S.
- Narins RS, Coleman W, Donofrio L, et al. Nonanimal sourced hyaluronic acid–based dermal filler using a cohesive polydensified matrix technology is superior to bovine collagen in the correction of moderate to severe nasolabial folds: results from a 6-month, randomized, blinded, controlled, multicenter study. Dermatol Surg. 2010;36(suppl 1):730-740.
- Narins RS, Coleman WP 3rd, Donofrio LM, et al. Improvement in nasolabial folds with a hyaluronic acid filler using a cohesive polydensified matrix technology: results from an 18-month open-label extension trial. Dermatol Surg. 2010;36(suppl 3):1800-1808.
- Prager W, Wissmueller E, Havermann I, et al. A prospective, split-face, randomized, comparative study of safety and 12-month longevity of three formulations of hyaluronic acid dermal filler for treatment of nasolabial folds. Dermatol Surg. 2012;38(7, pt 2):1143-1150.
- Butterwick K, Marmur E, Narurkar V, et al. HYC-24L demonstrates greater effectiveness with less pain than CPM-22.5 for treatment of perioral lines in a randomized controlled trial. Dermatol Surg. 2015;41:1351-1360.
- Jones D, Murphy DK. Volumizing hyaluronic acid filler for midface volume deficit: 2-year results from a pivotal single-blind randomized controlled study. Dermatol Surg. 2013;39:1602-1612.
- Baumann L, Narins RS, Beer K, et al. Volumizing hyaluronic acid filler for midface volume deficit: results after repeat treatment. Dermatol Surg. 2015;41(suppl 1):S284-S292.
- Few J, Cox SE, Paradkar-Mitragotri D, et al. A multicenter, single-blind randomized, controlled study of a volumizing hyaluronic acid filler for midface volume deficit: patient-reported outcomes at 2 years. Aesthet Surg J. 2015;35:589-599.
- Beer K, Glogau RG, Dover JS, et al. A randomized, evaluator-blinded, controlled study of effectiveness and safety of small particle hyaluronic acid plus lidocaine for lip augmentation and perioral rhytides. Dermatol Surg. 2015;41(suppl 1):S127-S136.
- Weiss RA, Moradi A, Bank D, et al. Effectiveness and safety of large gel particle hyaluronic acid with lidocaine for correction of midface volume deficit or contour deficiency. Dermatol Surg. 2016;42:699-709.
- Fitzgerald R, Graivier MH, Kane M, et al. Update on facial aging. Aesthet Surg J. 2010;30(suppl):S11-S24.
- Karnik J, Baumann L, Bruce S, et al. A double-blind, randomized, multicenter, controlled trial of suspended polymethylmethacrylate microspheres for the correction of atrophic facial acne scars. J Am Acad Dermatol. 2014;71:77-83.
- Bellafill [package insert]. San Diego, CA: Suneva Medical, Inc; 2015.
- Cohen S, Dover J, Monheit G, et al. Five-year safety and satisfaction study of PMMA-collagen in the correction of nasolabial folds. Dermatol Surg. 2015;41(suppl 1):S302-S313.
- Greene JJ, Sidle DM. The hyaluronic acid fillers: current understanding of the tissue device interface. Facial Plast Surg Clin North Am. 2015;23:423-432.
- Lorenc ZP, Fagien S, Flynn TC, et al. Review of key Belotero Balance safety and efficacy trials. Plast Reconstr Surg. 2013;132(4, suppl 2):33S-40S.
- Narins RS, Coleman W, Donofrio L, et al. Nonanimal sourced hyaluronic acid–based dermal filler using a cohesive polydensified matrix technology is superior to bovine collagen in the correction of moderate to severe nasolabial folds: results from a 6-month, randomized, blinded, controlled, multicenter study. Dermatol Surg. 2010;36(suppl 1):730-740.
- Narins RS, Coleman WP 3rd, Donofrio LM, et al. Improvement in nasolabial folds with a hyaluronic acid filler using a cohesive polydensified matrix technology: results from an 18-month open-label extension trial. Dermatol Surg. 2010;36(suppl 3):1800-1808.
- Prager W, Wissmueller E, Havermann I, et al. A prospective, split-face, randomized, comparative study of safety and 12-month longevity of three formulations of hyaluronic acid dermal filler for treatment of nasolabial folds. Dermatol Surg. 2012;38(7, pt 2):1143-1150.
- Butterwick K, Marmur E, Narurkar V, et al. HYC-24L demonstrates greater effectiveness with less pain than CPM-22.5 for treatment of perioral lines in a randomized controlled trial. Dermatol Surg. 2015;41:1351-1360.
- Jones D, Murphy DK. Volumizing hyaluronic acid filler for midface volume deficit: 2-year results from a pivotal single-blind randomized controlled study. Dermatol Surg. 2013;39:1602-1612.
- Baumann L, Narins RS, Beer K, et al. Volumizing hyaluronic acid filler for midface volume deficit: results after repeat treatment. Dermatol Surg. 2015;41(suppl 1):S284-S292.
- Few J, Cox SE, Paradkar-Mitragotri D, et al. A multicenter, single-blind randomized, controlled study of a volumizing hyaluronic acid filler for midface volume deficit: patient-reported outcomes at 2 years. Aesthet Surg J. 2015;35:589-599.
- Beer K, Glogau RG, Dover JS, et al. A randomized, evaluator-blinded, controlled study of effectiveness and safety of small particle hyaluronic acid plus lidocaine for lip augmentation and perioral rhytides. Dermatol Surg. 2015;41(suppl 1):S127-S136.
- Weiss RA, Moradi A, Bank D, et al. Effectiveness and safety of large gel particle hyaluronic acid with lidocaine for correction of midface volume deficit or contour deficiency. Dermatol Surg. 2016;42:699-709.
Practice Points
- The merits of new dermal fillers approved by the US Food and Drug Administration should be weighed with an understanding of aesthetic indications of use, duration of efficacy, and common adverse effects, in line with patient preference.
- The most common adverse effects are injection-site contusion, swelling, and pain, usually self-resolving within days to 2 weeks. Patient quality of care can be improved with forewarning and emphasis on alleviating symptoms.
An Update on Neurotoxin Products and Administration Methods
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
The first botulinum neurotoxin (BoNT) approved by the US Food and Drug Administration (FDA) was onabotulinumtoxinA in 1989 for the treatment of strabismus and blepharospasm. It was not until 1992, however, that the aesthetic benefits of BoNT were first reported in the medical literature by Carruthers and Carruthers,1 and a cosmetic indication was not approved by the FDA until 2002. Since that time, the popularity of BoNT products has grown rapidly with a nearly 6500% increase in popularity from 1997 to 2015 in addition to the introduction of a variety of new BoNT formulations to the market.2 It is estimated by the American Society for Aesthetic Plastic Surgery that there were at least 4,000,000 BoNT injections performed in 2015 alone, making it the most popular nonsurgical aesthetic procedure available.2 As the demand for minimally invasive cosmetic procedures continues to increase, we will continue to see the introduction of additional formulations of BoNT products as well as novel administration techniques and delivery devices. In this article, we provide an update on current and upcoming BoNT products and also review the literature on novel administration methods based on studies published from January 1, 2014, to December 31, 2015.
Current Products
To date, there are only 4 FDA-approved formulations of BoNT available for clinical use (eg, cervical dystonia, strabismus, blepharospasm, headache, urinary incontinence) in the United States: abobotulinumtoxinA, incobotulinumtoxinA, onabotulinumtoxinA, and rimabotulinumtoxinB.The FDA-approved dermatologic indications (eg, moderate to severe glabellar or canthal lines, severe axillary hyperhidrosis) for these products are provided in the Table. On a global scale, there are several other commonly utilized formulations of BoNT, including a Korean serotype resembling onabotulinumtoxinA and a Chinese botulinum toxin type A.3 Although there is some evidence to demonstrate comparable efficacy and safety of these latter products, the literature is relatively lacking in comparison to the FDA-approved products.4,5
Upcoming Products
Currently, there are several new BoNT formulations being studied for clinical use. RT 002 (Revance Therapeutics, Inc) is a novel injectable formulation of onabotulinumtoxinA that consists of the purified neurotoxin in combination with patented TransMTS peptides that have been shown to provide high-binding avidity for the neurotoxin, and thus the product is designed to reduce diffusion to adjacent muscles and diminish unwanted effects. With a reduced level of neurotoxin dissemination, it is theorized that a higher administration of targeted doses can be injected, which may lead to a longer duration of desired effects.6 A clinical pilot study done to establish the safety and efficacy of RT 002 for treatment of moderate to severe glabellar lines evaluated 4 equally sized cohorts of 12 participants, each receiving single-dose administration of RT 002 ranging in potency equivalent to 25 U, 50 U, 75 U, and 100 U of abobotulinumtoxinA as determined by the gelatin phosphate method.6 It was concluded that RT 002 is both safe and efficacious with an extended duration of action, with a median duration of effect of 7 months observed in the highest dose group (dose equivalent to 100 U of abobotulinumtoxinA). Notably, 80% of all 48 participants maintained a minimum 1-point improvement in investigator-determined glabellar line severity scores at the 6-month time point and 60% achieved wrinkle scores of none or mild at 6 months posttreatment.6
DWP 450 (Daewoong Pharmaceutical Co, Ltd) is derived from the wild-type Clostridium botulinum and is reported to be of higher purity than standard onabotulinumtoxinA. An initial 16-week pilot study demonstrated that 20 U of DWP 450 is noninferior and of comparable efficacy and safety to 20 U of onabotulinumtoxinA in the treatment of glabellar lines.7
NTC (Botulax [Hugel, Inc]) is the name of the toxin derived from the C botulinum strain CBFC26, which has already been approved in many Asian, European, and Latin American countries for the treatment of blepharospasm. This formulation has demonstrated noninferiority to onabotulinumtoxinA at equivalent 20-U doses for the treatment of moderate to severe glabellar lines in a double-blind, randomized, multicenter, phase 3 trial of 272 participants with a 16-week follow-up.8
MT 10109L (Medytox Inc) is a unique product in that it is distributed as a liquid type A botulinum toxin rather than the standard freeze-dried formulation; thus, a major advantage of this product is its convenience, as it does not need reconstitution or dilution prior to administration. In a double-blind, randomized, active drug–controlled, phase 3 study of 168 participants, it was determined that MT 10109L (20 U) is comparable in efficacy to onabotulinumtoxinA (20 U) for the treatment of moderate to severe glabellar lines. No significant difference was seen between the 2 treatment groups when glabellar lines were assessed at rest at 4 and 16 weeks after treatment, but a significantly greater improvement in glabellar lines was seen at maximum frown in the MT 10109L group at the 16-week follow-up (P=.0064).9
Administration Techniques
With regard to safe and effective BoNT product administration techniques, a variety of studies have provided insight into optimal practice methods. A 2015 expert consensus statement formed by an American Society for Dermatologic Surgery task force reviewed data from 42 papers and unanimously determined that for all current type A BoNT products available in the United States, a vial of BoNT reconstituted appropriately for the purpose of facial injections can be reconstituted at least 4 weeks prior to administration without contamination risk or decrease in efficacy and that multiple patients can be treated with the same vial.Although the statement was not explicit on whether or not preserved or unpreserved saline is to be used, it is considered routine practice to use preservative-containing saline to reconstitute BoNT, as it has been shown to reduce patient discomfort and is not associated with adverse effects.10
Pain Minimization
With respect to minimizing the pain associated with BoNT injections, several studies have assessed administration techniques to minimize patient discomfort. A split-face, double-blind study of 20 participants demonstrated that the use of a 32-gauge needle has a significantly greater chance of reducing clinically significant levels of pain as compared to a 30-gauge needle when performing facial injections (P=.04). Overall, however, injections of the face and arms were on average only nominally and not significantly more painful with 30-gauge needles compared to 32-gauge needles.11
Another technique that has been found to reduce patient discomfort is the application of cold packs prior to injection. A study of patients with chronic facial palsy observed a significant reduction in pain with the administration of a cold (3°C–5°C) gel pack for 1 minute compared to a room temperature (20°C) gel pack prior to the administration of onabotulinumtoxinA into the platysma (P<.001).12 In the matter of injection with rimabotulinumtoxinB, which has been shown to be considerably more painful to receive than its more popularly administered counterpart onabotulinumtoxinA, a split-face pilot study examined the effect of increasing the pH of rimabotulinumtoxinB to 7.5 with sodium bicarbonate from the usual pH of 5.6.13,14 Pain was reported to be considerably less in the higher pH group and no reduction of efficacy was seen over the 10-week follow-up period.14
Delivery Methods
Several preliminary studies also have examined novel delivery techniques to identify minimally painful yet effective methods for administering BoNT. It has been reported that standard BoNT formulations are not effective as topical agents in a comparison study in which onabotulinumtoxinA injection was compared to topically applied onabotulinumtoxinA.15 However, a follow-up prospective study by the same authors has demonstrated efficacy of topical onabotulinumtoxinA following pretreatment with a fractional ablative CO2 laser for treatment of crow’s-feet. In this randomized, split-face, controlled trial (N=10), participants were first pretreated with topical lidocaine 30% before receiving a single pass of fractional ablative CO2 laser with no overlap and a pulse energy of 100 mJ. Within 60 seconds of laser treatment, participants then received either 100 U of abobotulinumtoxinA diluted in 0.1 mL of saline or simple normal saline applied topically. A clinically significant improvement in periorbital wrinkles was seen both at 1-week and 1-month posttreatment in the laser and onabotulinumtoxinA–treated group compared to the laser and topical saline–treated group (P<.02).15
Another unique administration method studied, albeit with less successful results, involves the use of iontophoresis to deliver BoNT painlessly in a transdermal fashion with the assistance of an electrical current.16 This prospective, randomized, assessor-blinded, split-axilla, controlled trial of 11 participants compared the effectiveness of administering onabotulinumtoxinA via iontophoresis to traditional injection with onabotulinumtoxinA (250 U). Iontophoresis was accomplished with a single electrode pad soaked with 250 U of onabotulinumtoxinA applied directly to the axilla and a second electrode pad soaked in 0.9% saline applied to the hand to complete the circuit. An alternating electrical current was slowly increased for 30 minutes to a maximum current of 15 mA with a voltage of 12 V. Among the 11 participants recruited, the side treated with traditional injection showed a significantly greater percentage reduction in baseline sweating at the 1-week, 1-month, and 6-month posttreatment evaluations compared to iontophoresis (84%, 76%, and 50%, respectively vs 73%, 22%, and 32%, respectively)(P<.05). Despite being less efficacious than standard injection therapy, participants reported that iontophoresis delivery was significantly less painful (P<.05).16
A high-pressure oxygen delivery device, which utilizes a powerful jet of microdroplets containing water, the drug, air, and oxygen to deliver medication onto the skin surface, also has been studied as a means of delivery of BoNT in a minimally painful manner. In this study, the device was used to assess the efficacy of transdermal delivery of BoNT via jet nebulization in the treatment of primary palmar, plantar, and axillary hyperhidrosis.17 The 20 participants included in the study were randomized to receive either a combination of lidocaine and onabotulinumtoxinA (50 U) administered through the device or lidocaine delivered through the device followed by multiple transcutaneous injections of onabotulinumtoxinA (100 U). Both treatments significantly reduced sweating compared to baseline as measured by a visual analogue scale at 3-month follow-up (P<.001), but the combination delivery of lidocaine and onabotulinumtoxinA via the device resulted in significantly less procedure-related pain and sweating (P<.001) as well as significantly greater patient satisfaction (P<.001).17
Optimizing Aesthetic Outcomes
A frequent concern of patients receiving BoNT for cosmetic purposes is a desire to avoid a “frozen” or expressionless look. As such, many clinicians have attempted a variety of techniques to achieve more natural aesthetic results. One such method is known as the multipoint and multilevel injection technique, which consists of utilizing multiple injection sites at varying depths (intramuscular, subcutaneous, or intradermal) and doses (2–6 U) depending on the degree of contractility of the targeted muscle. In a preliminary study of 223 participants using this technique with a total dose of 125 U of abobotulinumtoxinA, good and natural results were reported with perseveration of facial emotion in all participants in addition to a mean overall satisfaction rate of 6.4 of 7 on the Facial Line Treatment Satisfaction Questionnaire with the maximum satisfaction rating possible reported in 66% of cases.18 It also has been postulated that injection depth of BoNT can affect brow elevation whereupon deeper injection depths can result in inactivation of the brow depressors and allow for increased elevation of the eyebrows. This technique has been applied in attempts to correct brow height asymmetry. However, a prospective, split-face study of 23 women suggested that this method is not effective.19 Participants received 64 U of onabotulinumtoxinA via 16 injection sites in the glabella, forehead, and lateral canthal area with either all deep or all shallow injections depending on the side treated and whether brow-lift was desired. Results at 4 weeks posttreatment showed no significant difference in brow height, and it was concluded that eyebrow depressor muscles cannot be accurately targeted with deep injection into the muscle belly for correction of eyebrow height discrepancies.19 Conversely, a 5-year retrospective, nonrandomized study of 227 patients with 563 treatments utilizing a “microdroplet” technique reported success at selectively targeting the eyebrow depressors while leaving the brow elevators unaffected.20 Here, a total dose of 33 U of onabotulinumtoxinA was administered via microdroplets of 10 to 20 μL, each with more than 60 to 100 injections into the brow, glabella, and crow’s-feet area. This method of injection resulted in a statistically significant improvement of forehead lines and brow ptosis and furrowing at follow-up between 10 and 45 days after treatment (P<.0001). Additionally, average brow height was significantly increased from 24.6 mm to 25 mm after treatment (P=.02).20
Conclusion
The use of BoNT products for both on- and off-label cosmetic and medical indications has rapidly grown over the past 2 decades. As demonstrated in this review, a variety of promising new products and delivery techniques are being developed. Given the rise in popularity of BoNT products among both physicians and consumers, clinicians should be aware of the current data and ongoing research.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
- Carruthers JD, Carruthers JA. Treatment of glabellar frown lines with C. botulinum-A exotoxin. J Dermatol Surg Oncol. 1992;18:17-21.
- American Society for Aesthetic Plastic Surgery. Cosmetic Surgery National Data Bank statistics. American Society for Aesthetic Plastic Surgery website. http://www.surgery.org/sites/default/files/ASAPS-Stats2015.pdf. Accessed June 12, 2016.
- Walker TJ, Dayan SH. Comparison and overview of currently available neurotoxins. J Clin Aesthet Dermatol. 2014;7:31-39.
- Feng Z, Sun Q, He L, et al. Optimal dosage of botulinum toxin type A for treatment of glabellar frown lines: efficacy and safety in a clinical trial. Dermatol Surg. 2015;41(suppl 1):S56-S63.
- Jiang HY, Chen S, Zhou J, et al. Diffusion of two botulinum toxins type A on the forehead: double-blinded, randomized, controlled study. Dermatol Surg. 2014;40:184-192.
- Garcia-Murray E, Velasco Villasenor ML, Acevedo B, et al. Safety and efficacy of RT002, an injectable botulinum toxin type A, for treating glabellar lines: results of a phase 1/2, open-label, sequential dose-escalation study. Dermatol Surg. 2015;41(suppl 1):S47-S55.
- Won CH, Kim HK, Kim BJ, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol. 2015;54:227-234.
- Kim BJ, Kwon HH, Park SY, et al. Double-blind, randomized non-inferiority trial of a novel botulinum toxin A processed from the strain CBFC26, compared with onabotulinumtoxin A in the treatment of glabellar lines. J Eur Acad Dermatol Venereol. 2014;28:1761-1767.
- Kim JE, Song EJ, Choi GS, et al. The efficacy and safety of liquid-type botulinum toxin type A for the management of moderate to severe glabellar frown lines. Plast Reconstr Surg. 2015;135:732-741.
- Alam M, Bolotin D, Carruthers J, et al. Consensus statement regarding storage and reuse of previously reconstituted neuromodulators. Dermatol Surg. 2015;41:321-326.
- Alam M, Geisler A, Sadhwani D, et al. Effect of needle size on pain perception in patients treated with botulinum toxin type A injections: a randomized clinical trial. JAMA Dermatol. 2015;151:1194-1199.
- Pucks N, Thomas A, Hallam MJ, et al. Cutaneous cooling to manage botulinum toxin injection-associated pain in patients with facial palsy: a randomised controlled trial. J Plast Reconstr Aesthet Surg. 2015;68:1701-1705.
- Kranz G, Sycha T, Voller B, et al. Pain sensation during intradermal injections of three different botulinum toxin preparations in different doses and dilutions. Dermatol Surg. 2006;32:886-890.
- Lowe PL, Lowe NJ. Botulinum toxin type B: pH change reduces injection pain, retains efficacy. Dermatol Surg. 2014;40:1328-1333.
- Mahmoud BH, Burnett C, Ozog D. Prospective randomized controlled study to determine the effect of topical application of botulinum toxin A for crow’s feet after treatment with ablative fractional CO2 laser. Dermatol Surg. 2015;41(suppl 1):S75-S81.
- Montaser-Kouhsari L, Zartab H, Fanian F, et al. Comparison of intradermal injection with iontophoresis of abo-botulinum toxin A for the treatment of primary axillary hyperhidrosis: a randomized, controlled trial. J Dermatolog Treat. 2014;25:337-341.
- Iannitti T, Palmieri B, Aspiro A, et al. A preliminary study of painless and effective transdermal botulinum toxin A delivery by jet nebulization for treatment of primary hyperhidrosis. Drug Des Devel Ther. 2014;8:931-935.
- Iozzo I, Tengattini V, Antonucci VA. Multipoint and multilevel injection technique of botulinum toxin A in facial aesthetics. J Cosmet Dermatol. 2014;13:135-142.
- Sneath J, Humphrey S, Carruthers A, et al. Injecting botulinum toxin at different depths is not effective for the correction of eyebrow asymmetry. Dermatol Surg. 2015;41(suppl 1):S82-S87.
- Steinsapir KD, Rootman D, Wulc A, et al. Cosmetic microdroplet botulinum toxin A forehead lift: a new treatment paradigm. Ophthal Plast Reconstr Surg. 2015;31:263-268.
Practice Points
- Botulinum neurotoxin (BoNT) injection is the most popular nonsurgical aesthetic procedure available of which there are currently 4 products approved by the US Food and Drug Administration.
- A variety of new BoNT products with unique properties and formulations are currently being studied, some of which are already available for clinical use in foreign markets.
- Administration technique and novel product delivery methods also can be utilized to minimize pain and maximize aesthetic outcomes.
Oral Therapies for Psoriasis: Report From the AAD Meeting
Patients with psoriasis now have several treatment options to help control their disease. Among them are oral therapies. Dr. Gary Goldenberg reviews clearance data on approved therapies and ones on the horizon.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Patients with psoriasis now have several treatment options to help control their disease. Among them are oral therapies. Dr. Gary Goldenberg reviews clearance data on approved therapies and ones on the horizon.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Patients with psoriasis now have several treatment options to help control their disease. Among them are oral therapies. Dr. Gary Goldenberg reviews clearance data on approved therapies and ones on the horizon.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Enlarged Facial Pores: An Update on Treatments
Enlarged facial pores are superficial skin structures that are visualized as small openings on the skin corresponding to the openings of the pilosebaceous apparatus. These openings may be impacted with horny follicular plugs consisting of sebaceous debris that appear as open comedones.1 Skin pores is a lay term that is poorly defined in the medical literature and often is categorized in terms of arbitrary circular diameters determined through cosmetic skin analyzers.2 The term refers to pilosebaceous follicular enlargements (with or without open comedonal horny impactions) that can be visualized by the naked eye, most commonly occurring on the face and scalp. These enlarged pores remain a pervasive cosmetic concern that impacts patient quality of life. Enlarged pores are difficult to treat, in part due to lack of knowledge of the pathophysiology; thus, we review the currently proposed causes of enlarged pilosebaceous openings and the treatments in the scope of this pathogenesis with a focus on therapeutic efficacy.
Pathogenesis of Enlarged Facial Pores
It is now thought that seborrhea, loss of skin elasticity and tension, and hair follicle size are most clinically relevant to the pathogenesis of enlarged pores.2 Other potential associated and causative factors include genetic predisposition, acne, comedogenic xenobiotics, chronic photodamage, chronic radiodermatitis, and vitamin A deficiency.1,3
The direct relationship between sebum output and pore size has been well established, particularly in men who generally have higher sebum output levels than women, which likely is testosterone driven.4,5 However, there are contradictory data on whether sex affects pore size, as females also exhibit contributory hormonal factors. Sebum output and pore size increase substantially during the ovulation phase of the female menstrual cycle, likely secondary to increased progesterone affecting sebaceous gland activity.2,4 The presence of acne also is associated with enlarged facial pores, though the extent of seborrhea as a confounding factor is unclear. Furthermore, acne severity does not correlate with increased pore size.5 However, the processes of acne and facial pores are interlinked, given the frequent occurrence of open comedones within the pores.
Skin elasticity and tensile strength when defined visually and mechanically has shown a negative correlation with facial pore size and density.5 It is well known that cutaneous aging and chronic photodamage cause perturbation in the collagen and elastin framework that allows for the skin to maintain its resilient properties.6 Aged and photodamaged skin also demonstrates decreased expression of microfibril-associated glycoprotein-1 (MAGP-1), a crucial component in elastic fiber assembly and skin elasticity in the dermis and perifollicular/pore areas.7
Pore density and size appears to range diversely across ethnicities, though Chinese women exhibit notably lower pore size and density across all ages as compared to other ethnicities.8 Black individuals have aberrant epidermal architecture, defined as the presence of stalagmitelike structures at the dermoepidermal junction, correlating with enlarged pore size compared to other ethnicities.2,8
Treating Enlarged Facial Pores
Treatments for enlarged facial pores primarily aim to decrease sebum production, rejuvenate skin, remove hair, and/or decrease follicular size. Evidence-based studies are limited, and many currently used therapies have not been studied with enlarged facial pores as a primary investigative outcome. Here, we include studies that report efficacy in decreasing pore size specifically. It is important to note the lack of a uniform and objective modality with which to report skin pore size. Studies use a wide range of techniques including patient self-reporting, physician observation, and software image analyzers.
Topical Therapies
Topical retinoids are vitamin A derivatives, and they are first-line therapies in reversing the aberrant collagen and elastin-associated epidermal and dermal changes that occur with chronological aging and photoaging. Tretinoin, isotretinoin, and tazarotene have shown efficacy in multiple parameters of skin rejuvenation, including facial pores, skin wrinkling, hyperpigmentation, skin laxity, and sebum production.9 However, it is important to note that retinoids treat keratinocyte atypia in acne, and efficacy in facial pores is confounded by improvement in follicular keratinization. Because studies have not distinctly uncoupled this association, it is erroneous to conclude that retinoids reduce facial pore size and density irrespective of concomitant acne vulgaris.
Tazarotene has been evaluated for use in reducing facial pore size. In one investigation, 568 patients with moderate wrinkling or hyperpigmentation were randomized to receive tazarotene cream 0.1% or placebo once daily for 24 weeks and were evaluated for enlarged facial pores as a secondary outcome using a double-blinded physician 5-point scale.10 At week 24, 42% of tazarotene-treated patients achieved improvement of at least 1 point compared to 20% of placebo-treated patients (P<.001). Adverse events were dermatitic, as can be expected of retinoids, leading to a 4% discontinuation rate in the tazarotene group compared to 1% in the placebo group.10
Tretinoin has long been used off label for antiaging treatments but has only recently shown efficacy for facial pores. In one study, 60 women who had previously sought antiaging procedures were treated with tretinoin cream 0.025% once daily and no other antiaging products or procedures for 90 days.11 Facial pore evaluations were determined by a modified dermatoscope with a polarized analyzer for clinical scoring using a photonumeric scale. Patients improved from a baseline average score of 3.2 in facial pores to a posttreatment average score of 2.0 (P<.05) at day 84. This improvement was sustained from day 28 of treatment and corresponded to patient self-perception. Adverse events included xerosis, desquamation, burning, and erythema, which led to 3 premature discontinuations.11
Various chemical peel formulations are used in skin rejuvenation and have shown application in enlarged facial pores. Chemical peels act at the epidermal or dermal level to induce temporary breakdown and regeneration of healthier cells and improved skin matrix.12 Twenty-two Japanese women applied glycolic acid (30% solution) every 2 weeks for a total of 5 treatments and exhibited reduced appearance of conspicuous, open, and dark pores, defined by surface area and shading as determined through dermatoscopic and software analysis, with mean improvement rates of 34.6%, 11%, and 34.3%, respectively. More than 70% of participants exhibited improvement in enlarged facial pores.13 A study involving a 40% glycolic acid and vitamin C formulation demonstrated significant improvement in facial pores (28.3%; P<.001).14
The newest topical therapies studied for use in minimizing facial pilosebaceous openings are natural plant-derived copper chlorophyllin complex sodium salt (CHLcu) and tetra-hydro-jasmonic acid (LR2412). Clinical trials of these botanicals are limited with small sample sizes but are included here as novel treatments requiring further investigation.
Chlorophyllin copper complex sodium salt is derived from chlorophyll, a green pigment found in plants, and has been investigated as a topical gel in liposomal dispersions for application in photodamaged and aged skin. Chlorophyllin copper complex sodium salt exerts in vitro hyaluronidase inhibitory activity to maintain hyaluronic acid in the extracellular matrix and counteract the structural breakdown of cutaneous aging.15 Two small single-center pilot trials enrolled 10 participants each in a 3-week study of CHLcu 0.1% twice daily and an 8-week study of CHLcu 0.066% twice daily.16,17 After 3 weeks, patients treated with CHLcu 0.1% exhibited a 22.2% improvement in facial pores by clinical assessment grading, though this improvement was not significant on software imaging analysis. Patients improved the most on parameters of facial seborrhea by clinical assessment.16 After 8 weeks, patients treated with CHLcu 0.066% exhibited 25.3% improvement in facial pores by clinical assessment grading.17 Treatments were reported to be well tolerated without noted adverse events in both studies.
Tetra-hydro-jasmonic acid is an analogue of jasmonic acid, a plant hormone derived from linoleic acid. Due to its favorable safety profile and bioavailability, penetration into epidermal and dermal layers, and potential effects in rejuvenating desquamation, LR2412 is currently being assessed for treatment of skin wrinkles, texture, and pores.18 Its effect is thought to relate to stimulation of laminin-5, collagen IV, and fibrillin deposition at the dermoepidermal junction.19 In an open-label trial of a topical preparation of LR2412, 15 participants were treated twice daily for 6 weeks and assessed through investigator clinical assessment scoring.20 Investigator scoring of pores improved by 25.2% from baseline (P<.05) after 6 weeks of treatment. Improvement in pores was seen as early as days 1 and 3. No serious adverse events were reported, though 2 participants developed acne on follow-up.20
Tetra-hydro-jasmonic acid also is formulated with retinol (retinol 0.2%/LR2412 2.0%) and demonstrated cosmetic efficacy in a noninferiority trial with tretinoin cream 0.025%.11 Sixty patients each were randomized to retinol/LR2412 or tretinoin at bedtime and treated for 90 days. At day 84, participants in the retinol/LR2412 group exhibited an improvement in investigator clinical assessment scoring from a baseline of 3.6 to 2.5 (P<.05). There were no significant differences in investigator-assessed efficacy between the treatment arms. Participants reported similar or better results and fewer side effects with retinol/LR2412 on self-questionnaires. Eight participants treated with retinol/LR2412 and 15 participants treated with tretinoin reported various incidences of skin irritation, burning, and desquamation.11
Oral Therapies
The most commonly used oral therapies for enlarged pores are antiandrogens, such as combined oral contraceptives, spironolactone, and cyproterone acetate, which modulate sebum production due to the presence of androgen receptors within sebaceous glands.21 Forty-four white women in an open-label, phase 4 study were treated with combined oral contraceptives containing chlormadinone acetate–ethinyl estradiol for 6 menstrual cycles, with standardized photography taken before and after the treatment period for software analysis. After 6 treatment cycles, 9.1% (4/44) of participants had visibly enlarged pores of the forehead and cheeks compared to 43.2% (19/44) of participants at baseline (P<.0001).22 The effects of other antiandrogens on facial pores have not been studied in this capacity.
Lasers, Radiofrequency, and Ultrasound Devices
The development of various devices that can deliver targeted thermal or ultrasound energy to the skin offers the newest and most robust modality in cosmetic therapy. The mechanism of their efficacy may be due to a combination of induced remodeling of collagen fibers near pilosebaceous openings to increase skin elasticity and decrease sebum production.2,23
Devices with established antiaging effects have been extensively reviewed and include the gold particle 800-nm diode laser, 1450-nm diode laser, microneedle apparatuses, fractional radiofrequency devices, 2790-nm erbium:YAG laser, nonablative 1410-nm fractionated erbium-doped fiber laser, and nonablative 1440-nm fractional laser.2
Literature on the use of these devices for minimizing facial pore size is limited. One treatment of intense focused ultrasound using a 3-mm transducer successfully improved overall pore appearance in 91% of sites at 6-week follow-up on a clinical grading scale.24 Three sessions of nonablative 1410-nm fractionated erbium-doped fiber laser treatments yielded facial skin pore minimization of greater than 51% in 14 of 15 participants.25
The nonablative 1440-nm diode fractional laser received 510(k) clearance by the US Food and Drug Administration in 2011 for aesthetic use in chronologically aged and photoaged skin. Twenty participants treated for 2 weeks and a total of 6 facial treatments with this laser system showed a 17% average improvement in facial pore score on software analysis (P≤.002). Adverse events were mild and included erythema and xerosis.26
Conclusion
The reliability of available literature on efficacy of various treatments in diminishing facial skin pores has been challenging given that most studies are low in power, lack control groups, use nonuniform methods of reporting outcomes, and do not report complete adverse events. Thus, all results should be interpreted with caution.
Overall, it is clear that the pathogenesis of enlarged facial pores is multifactorial and complex, necessitating a similar approach to therapeutics. Topical treatments offer a range of diverse therapies with proven benefit in facial pore reduction. The advent of lasers and devices offers constantly evolving therapeutic options with diffuse antiaging effects. Despite the numerous topical, oral, and device-oriented options, enlarged facial pores remain a challenging cosmetic concern. More robust efficacy studies on new treatments are necessary.
- Uhoda E, Pierard-Franchimont C, Petit L, et al. The conundrum of skin pores in dermocosmetology. Dermatology. 2005;210:3-7.
- Lee SJ, Seok J, Jeong SY, et al. Facial pores: definition, causes, and treatment options. Dermatol Surg. 2016;42:277-285.
- Pierard GE, Pierard-Franchimont C, Marks R, et al. EEMCO guidance for the in vivo assessment of skin greasiness. The EEMCO Group. Skin Pharmacol Appl Skin Physiol. 2000;13:372-389.
- Roh M, Han M, Kim D, et al. Sebum output as a factor contributing to the size of facial pores. Br J Dermatol. 2006;155:890-894.
- Kim BY, Choi JW, Park KC, et al. Sebum, acne, skin elasticity, and gender difference-which is the major influencing factor for facial pores? Skin Res Technol. 2013;19:E45-E53.
- Uitto J. The role of elastin and collagen in cutaneous aging: intrinsic aging versus photoexposure. J Drugs Dermatol. 2008;7(2 suppl):S12-S16.
- Zheng Q, Chen S, Chen Y, et al. Investigation of age-related decline of microfibril-associated glycoprotein-1 in human skin through immunohistochemistry study. Clin Cosmet Investig Dermatol. 2013;6:317-323.
- Sugiyama-Nakagiri Y, Sugata K, Hachiya A, et al. Ethnic differences in the structural properties of facial skin. J Dermatol Sci. 2009;53:135-139.
- Mukherjee S, Date A, Patravale V, et al. Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety. Clin Interv Aging. 2006;1:327-348.
- Kang S, Krueger GG, Tanghetti EA, et al; Tazarotene Cream in Photodamage Study Group. A multicenter, randomized, double-blind trial of tazarotene 0.1% cream in the treatment of photodamage. J Am Acad Dermatol. 2005;52:268-274.
- Bouloc A, Vergnanini AL, Issa MC. A double-blind randomized study comparing the association of retinol and LR2412 with tretinoin 0.025% in photoaged skin. J Cosmet Dermatol. 2015;14:40-46.
- Fischer TC, Perosino E, Poli F, et al. Chemical peels in aesthetic dermatology: an update 2009 [published online September 8, 2009]. J Eur Acad Dermatol Venereol. 2010;24:281-292.
- Kakudo N, Kushida S, Tanaka N, et al. A novel method to measure conspicuous facial pores using computer analysis of digital-camera-captured images: the effect of glycolic acid chemical peeling. Skin Res Technol. 2011;17:427-433.
- Kim WS. Efficacy and safety of a new superficial chemical peel using alpha-hydroxy acid, vitamin C and oxygen for melasma. J Cosmet Laser Ther. 2013;15:21-24.
- McCook JP, Dorogi PL, Vasily DB, et al. In vitro inhibition of hyaluronidase by sodium copper chlorophyllin complex and chlorophyllin analogs. Clin Cosmet Investig Dermatol. 2015;8:443-448.
- Stephens TJ, McCook JP, Herndon JH Jr. Pilot study of topical copper chlorophyllin complex in subjects with facial acne and large pores. J Drugs Dermatol. 2015;14:589-592.
- Sigler ML, Stephens TJ. Assessment of the safety and efficacy of topical copper chlorophyllin in women with photodamaged facial skin. J Drugs Dermatol. 2015;14:401-404.
- Alexiades M. Jasmonates and tetrahydrojasmonic acid: a novel class of anti-aging molecules. J Drugs Dermatol. 2016;15:206-207.
- Tran C, Michelet JF, Simonetti L, et al. In vitro and in vivo studies with tetra-hydro-jasmonic acid (LR2412) reveal its potential to correct signs of skin ageing. J Eur Acad Dermatol Venereol. 2014;28:415-423.
- Alexiades M. Clinical assessment of a novel jasmonate cosmeceutical, LR2412-Cx, for the treatment of skin aging. J Drugs Dermatol. 2016;15:209-215.
- Lam C, Zaenglein AL. Contraceptive use in acne. Clin Dermatol. 2014;32:502-515.
- Kerscher M, Reuther T, Bayrhammer J, et al. Effects of an oral contraceptive containing chlormadinone and ethinylestradiol on acne-prone skin of women of different age groups: an open-label, single-centre, phase IV study. Clin Drug Investig. 2008;28:703-711.
- Schmults CD, Phelps R, Goldberg DJ. Nonablative facial remodeling: erythema reduction and histologic evidence of new collagen formation using a 300-microsecond 1064-nm Nd:YAG laser. Arch Dermatol. 2004;140:1373-1376.
- Lee HJ, Lee KR, Park JY, et al. The efficacy and safety of intense focused ultrasound in the treatment of enlarged facial pores in Asian skin. J Dermatolog Treat. 2015;26:73-77.
- Suh DH, Chang KY, Lee SJ, et al. Treatment of dilated pores with 1410-nm fractional erbium-doped fiber laser. Lasers Med Sci. 2015;30:1135-1139.
- Saedi N, Petrell K, Arndt K, et al. Evaluating facial pores and skin texture after low-energy nonablative fractional 1440-nm laser treatments. J Am Acad Dermatol. 2013;68:113-118.
Enlarged facial pores are superficial skin structures that are visualized as small openings on the skin corresponding to the openings of the pilosebaceous apparatus. These openings may be impacted with horny follicular plugs consisting of sebaceous debris that appear as open comedones.1 Skin pores is a lay term that is poorly defined in the medical literature and often is categorized in terms of arbitrary circular diameters determined through cosmetic skin analyzers.2 The term refers to pilosebaceous follicular enlargements (with or without open comedonal horny impactions) that can be visualized by the naked eye, most commonly occurring on the face and scalp. These enlarged pores remain a pervasive cosmetic concern that impacts patient quality of life. Enlarged pores are difficult to treat, in part due to lack of knowledge of the pathophysiology; thus, we review the currently proposed causes of enlarged pilosebaceous openings and the treatments in the scope of this pathogenesis with a focus on therapeutic efficacy.
Pathogenesis of Enlarged Facial Pores
It is now thought that seborrhea, loss of skin elasticity and tension, and hair follicle size are most clinically relevant to the pathogenesis of enlarged pores.2 Other potential associated and causative factors include genetic predisposition, acne, comedogenic xenobiotics, chronic photodamage, chronic radiodermatitis, and vitamin A deficiency.1,3
The direct relationship between sebum output and pore size has been well established, particularly in men who generally have higher sebum output levels than women, which likely is testosterone driven.4,5 However, there are contradictory data on whether sex affects pore size, as females also exhibit contributory hormonal factors. Sebum output and pore size increase substantially during the ovulation phase of the female menstrual cycle, likely secondary to increased progesterone affecting sebaceous gland activity.2,4 The presence of acne also is associated with enlarged facial pores, though the extent of seborrhea as a confounding factor is unclear. Furthermore, acne severity does not correlate with increased pore size.5 However, the processes of acne and facial pores are interlinked, given the frequent occurrence of open comedones within the pores.
Skin elasticity and tensile strength when defined visually and mechanically has shown a negative correlation with facial pore size and density.5 It is well known that cutaneous aging and chronic photodamage cause perturbation in the collagen and elastin framework that allows for the skin to maintain its resilient properties.6 Aged and photodamaged skin also demonstrates decreased expression of microfibril-associated glycoprotein-1 (MAGP-1), a crucial component in elastic fiber assembly and skin elasticity in the dermis and perifollicular/pore areas.7
Pore density and size appears to range diversely across ethnicities, though Chinese women exhibit notably lower pore size and density across all ages as compared to other ethnicities.8 Black individuals have aberrant epidermal architecture, defined as the presence of stalagmitelike structures at the dermoepidermal junction, correlating with enlarged pore size compared to other ethnicities.2,8
Treating Enlarged Facial Pores
Treatments for enlarged facial pores primarily aim to decrease sebum production, rejuvenate skin, remove hair, and/or decrease follicular size. Evidence-based studies are limited, and many currently used therapies have not been studied with enlarged facial pores as a primary investigative outcome. Here, we include studies that report efficacy in decreasing pore size specifically. It is important to note the lack of a uniform and objective modality with which to report skin pore size. Studies use a wide range of techniques including patient self-reporting, physician observation, and software image analyzers.
Topical Therapies
Topical retinoids are vitamin A derivatives, and they are first-line therapies in reversing the aberrant collagen and elastin-associated epidermal and dermal changes that occur with chronological aging and photoaging. Tretinoin, isotretinoin, and tazarotene have shown efficacy in multiple parameters of skin rejuvenation, including facial pores, skin wrinkling, hyperpigmentation, skin laxity, and sebum production.9 However, it is important to note that retinoids treat keratinocyte atypia in acne, and efficacy in facial pores is confounded by improvement in follicular keratinization. Because studies have not distinctly uncoupled this association, it is erroneous to conclude that retinoids reduce facial pore size and density irrespective of concomitant acne vulgaris.
Tazarotene has been evaluated for use in reducing facial pore size. In one investigation, 568 patients with moderate wrinkling or hyperpigmentation were randomized to receive tazarotene cream 0.1% or placebo once daily for 24 weeks and were evaluated for enlarged facial pores as a secondary outcome using a double-blinded physician 5-point scale.10 At week 24, 42% of tazarotene-treated patients achieved improvement of at least 1 point compared to 20% of placebo-treated patients (P<.001). Adverse events were dermatitic, as can be expected of retinoids, leading to a 4% discontinuation rate in the tazarotene group compared to 1% in the placebo group.10
Tretinoin has long been used off label for antiaging treatments but has only recently shown efficacy for facial pores. In one study, 60 women who had previously sought antiaging procedures were treated with tretinoin cream 0.025% once daily and no other antiaging products or procedures for 90 days.11 Facial pore evaluations were determined by a modified dermatoscope with a polarized analyzer for clinical scoring using a photonumeric scale. Patients improved from a baseline average score of 3.2 in facial pores to a posttreatment average score of 2.0 (P<.05) at day 84. This improvement was sustained from day 28 of treatment and corresponded to patient self-perception. Adverse events included xerosis, desquamation, burning, and erythema, which led to 3 premature discontinuations.11
Various chemical peel formulations are used in skin rejuvenation and have shown application in enlarged facial pores. Chemical peels act at the epidermal or dermal level to induce temporary breakdown and regeneration of healthier cells and improved skin matrix.12 Twenty-two Japanese women applied glycolic acid (30% solution) every 2 weeks for a total of 5 treatments and exhibited reduced appearance of conspicuous, open, and dark pores, defined by surface area and shading as determined through dermatoscopic and software analysis, with mean improvement rates of 34.6%, 11%, and 34.3%, respectively. More than 70% of participants exhibited improvement in enlarged facial pores.13 A study involving a 40% glycolic acid and vitamin C formulation demonstrated significant improvement in facial pores (28.3%; P<.001).14
The newest topical therapies studied for use in minimizing facial pilosebaceous openings are natural plant-derived copper chlorophyllin complex sodium salt (CHLcu) and tetra-hydro-jasmonic acid (LR2412). Clinical trials of these botanicals are limited with small sample sizes but are included here as novel treatments requiring further investigation.
Chlorophyllin copper complex sodium salt is derived from chlorophyll, a green pigment found in plants, and has been investigated as a topical gel in liposomal dispersions for application in photodamaged and aged skin. Chlorophyllin copper complex sodium salt exerts in vitro hyaluronidase inhibitory activity to maintain hyaluronic acid in the extracellular matrix and counteract the structural breakdown of cutaneous aging.15 Two small single-center pilot trials enrolled 10 participants each in a 3-week study of CHLcu 0.1% twice daily and an 8-week study of CHLcu 0.066% twice daily.16,17 After 3 weeks, patients treated with CHLcu 0.1% exhibited a 22.2% improvement in facial pores by clinical assessment grading, though this improvement was not significant on software imaging analysis. Patients improved the most on parameters of facial seborrhea by clinical assessment.16 After 8 weeks, patients treated with CHLcu 0.066% exhibited 25.3% improvement in facial pores by clinical assessment grading.17 Treatments were reported to be well tolerated without noted adverse events in both studies.
Tetra-hydro-jasmonic acid is an analogue of jasmonic acid, a plant hormone derived from linoleic acid. Due to its favorable safety profile and bioavailability, penetration into epidermal and dermal layers, and potential effects in rejuvenating desquamation, LR2412 is currently being assessed for treatment of skin wrinkles, texture, and pores.18 Its effect is thought to relate to stimulation of laminin-5, collagen IV, and fibrillin deposition at the dermoepidermal junction.19 In an open-label trial of a topical preparation of LR2412, 15 participants were treated twice daily for 6 weeks and assessed through investigator clinical assessment scoring.20 Investigator scoring of pores improved by 25.2% from baseline (P<.05) after 6 weeks of treatment. Improvement in pores was seen as early as days 1 and 3. No serious adverse events were reported, though 2 participants developed acne on follow-up.20
Tetra-hydro-jasmonic acid also is formulated with retinol (retinol 0.2%/LR2412 2.0%) and demonstrated cosmetic efficacy in a noninferiority trial with tretinoin cream 0.025%.11 Sixty patients each were randomized to retinol/LR2412 or tretinoin at bedtime and treated for 90 days. At day 84, participants in the retinol/LR2412 group exhibited an improvement in investigator clinical assessment scoring from a baseline of 3.6 to 2.5 (P<.05). There were no significant differences in investigator-assessed efficacy between the treatment arms. Participants reported similar or better results and fewer side effects with retinol/LR2412 on self-questionnaires. Eight participants treated with retinol/LR2412 and 15 participants treated with tretinoin reported various incidences of skin irritation, burning, and desquamation.11
Oral Therapies
The most commonly used oral therapies for enlarged pores are antiandrogens, such as combined oral contraceptives, spironolactone, and cyproterone acetate, which modulate sebum production due to the presence of androgen receptors within sebaceous glands.21 Forty-four white women in an open-label, phase 4 study were treated with combined oral contraceptives containing chlormadinone acetate–ethinyl estradiol for 6 menstrual cycles, with standardized photography taken before and after the treatment period for software analysis. After 6 treatment cycles, 9.1% (4/44) of participants had visibly enlarged pores of the forehead and cheeks compared to 43.2% (19/44) of participants at baseline (P<.0001).22 The effects of other antiandrogens on facial pores have not been studied in this capacity.
Lasers, Radiofrequency, and Ultrasound Devices
The development of various devices that can deliver targeted thermal or ultrasound energy to the skin offers the newest and most robust modality in cosmetic therapy. The mechanism of their efficacy may be due to a combination of induced remodeling of collagen fibers near pilosebaceous openings to increase skin elasticity and decrease sebum production.2,23
Devices with established antiaging effects have been extensively reviewed and include the gold particle 800-nm diode laser, 1450-nm diode laser, microneedle apparatuses, fractional radiofrequency devices, 2790-nm erbium:YAG laser, nonablative 1410-nm fractionated erbium-doped fiber laser, and nonablative 1440-nm fractional laser.2
Literature on the use of these devices for minimizing facial pore size is limited. One treatment of intense focused ultrasound using a 3-mm transducer successfully improved overall pore appearance in 91% of sites at 6-week follow-up on a clinical grading scale.24 Three sessions of nonablative 1410-nm fractionated erbium-doped fiber laser treatments yielded facial skin pore minimization of greater than 51% in 14 of 15 participants.25
The nonablative 1440-nm diode fractional laser received 510(k) clearance by the US Food and Drug Administration in 2011 for aesthetic use in chronologically aged and photoaged skin. Twenty participants treated for 2 weeks and a total of 6 facial treatments with this laser system showed a 17% average improvement in facial pore score on software analysis (P≤.002). Adverse events were mild and included erythema and xerosis.26
Conclusion
The reliability of available literature on efficacy of various treatments in diminishing facial skin pores has been challenging given that most studies are low in power, lack control groups, use nonuniform methods of reporting outcomes, and do not report complete adverse events. Thus, all results should be interpreted with caution.
Overall, it is clear that the pathogenesis of enlarged facial pores is multifactorial and complex, necessitating a similar approach to therapeutics. Topical treatments offer a range of diverse therapies with proven benefit in facial pore reduction. The advent of lasers and devices offers constantly evolving therapeutic options with diffuse antiaging effects. Despite the numerous topical, oral, and device-oriented options, enlarged facial pores remain a challenging cosmetic concern. More robust efficacy studies on new treatments are necessary.
Enlarged facial pores are superficial skin structures that are visualized as small openings on the skin corresponding to the openings of the pilosebaceous apparatus. These openings may be impacted with horny follicular plugs consisting of sebaceous debris that appear as open comedones.1 Skin pores is a lay term that is poorly defined in the medical literature and often is categorized in terms of arbitrary circular diameters determined through cosmetic skin analyzers.2 The term refers to pilosebaceous follicular enlargements (with or without open comedonal horny impactions) that can be visualized by the naked eye, most commonly occurring on the face and scalp. These enlarged pores remain a pervasive cosmetic concern that impacts patient quality of life. Enlarged pores are difficult to treat, in part due to lack of knowledge of the pathophysiology; thus, we review the currently proposed causes of enlarged pilosebaceous openings and the treatments in the scope of this pathogenesis with a focus on therapeutic efficacy.
Pathogenesis of Enlarged Facial Pores
It is now thought that seborrhea, loss of skin elasticity and tension, and hair follicle size are most clinically relevant to the pathogenesis of enlarged pores.2 Other potential associated and causative factors include genetic predisposition, acne, comedogenic xenobiotics, chronic photodamage, chronic radiodermatitis, and vitamin A deficiency.1,3
The direct relationship between sebum output and pore size has been well established, particularly in men who generally have higher sebum output levels than women, which likely is testosterone driven.4,5 However, there are contradictory data on whether sex affects pore size, as females also exhibit contributory hormonal factors. Sebum output and pore size increase substantially during the ovulation phase of the female menstrual cycle, likely secondary to increased progesterone affecting sebaceous gland activity.2,4 The presence of acne also is associated with enlarged facial pores, though the extent of seborrhea as a confounding factor is unclear. Furthermore, acne severity does not correlate with increased pore size.5 However, the processes of acne and facial pores are interlinked, given the frequent occurrence of open comedones within the pores.
Skin elasticity and tensile strength when defined visually and mechanically has shown a negative correlation with facial pore size and density.5 It is well known that cutaneous aging and chronic photodamage cause perturbation in the collagen and elastin framework that allows for the skin to maintain its resilient properties.6 Aged and photodamaged skin also demonstrates decreased expression of microfibril-associated glycoprotein-1 (MAGP-1), a crucial component in elastic fiber assembly and skin elasticity in the dermis and perifollicular/pore areas.7
Pore density and size appears to range diversely across ethnicities, though Chinese women exhibit notably lower pore size and density across all ages as compared to other ethnicities.8 Black individuals have aberrant epidermal architecture, defined as the presence of stalagmitelike structures at the dermoepidermal junction, correlating with enlarged pore size compared to other ethnicities.2,8
Treating Enlarged Facial Pores
Treatments for enlarged facial pores primarily aim to decrease sebum production, rejuvenate skin, remove hair, and/or decrease follicular size. Evidence-based studies are limited, and many currently used therapies have not been studied with enlarged facial pores as a primary investigative outcome. Here, we include studies that report efficacy in decreasing pore size specifically. It is important to note the lack of a uniform and objective modality with which to report skin pore size. Studies use a wide range of techniques including patient self-reporting, physician observation, and software image analyzers.
Topical Therapies
Topical retinoids are vitamin A derivatives, and they are first-line therapies in reversing the aberrant collagen and elastin-associated epidermal and dermal changes that occur with chronological aging and photoaging. Tretinoin, isotretinoin, and tazarotene have shown efficacy in multiple parameters of skin rejuvenation, including facial pores, skin wrinkling, hyperpigmentation, skin laxity, and sebum production.9 However, it is important to note that retinoids treat keratinocyte atypia in acne, and efficacy in facial pores is confounded by improvement in follicular keratinization. Because studies have not distinctly uncoupled this association, it is erroneous to conclude that retinoids reduce facial pore size and density irrespective of concomitant acne vulgaris.
Tazarotene has been evaluated for use in reducing facial pore size. In one investigation, 568 patients with moderate wrinkling or hyperpigmentation were randomized to receive tazarotene cream 0.1% or placebo once daily for 24 weeks and were evaluated for enlarged facial pores as a secondary outcome using a double-blinded physician 5-point scale.10 At week 24, 42% of tazarotene-treated patients achieved improvement of at least 1 point compared to 20% of placebo-treated patients (P<.001). Adverse events were dermatitic, as can be expected of retinoids, leading to a 4% discontinuation rate in the tazarotene group compared to 1% in the placebo group.10
Tretinoin has long been used off label for antiaging treatments but has only recently shown efficacy for facial pores. In one study, 60 women who had previously sought antiaging procedures were treated with tretinoin cream 0.025% once daily and no other antiaging products or procedures for 90 days.11 Facial pore evaluations were determined by a modified dermatoscope with a polarized analyzer for clinical scoring using a photonumeric scale. Patients improved from a baseline average score of 3.2 in facial pores to a posttreatment average score of 2.0 (P<.05) at day 84. This improvement was sustained from day 28 of treatment and corresponded to patient self-perception. Adverse events included xerosis, desquamation, burning, and erythema, which led to 3 premature discontinuations.11
Various chemical peel formulations are used in skin rejuvenation and have shown application in enlarged facial pores. Chemical peels act at the epidermal or dermal level to induce temporary breakdown and regeneration of healthier cells and improved skin matrix.12 Twenty-two Japanese women applied glycolic acid (30% solution) every 2 weeks for a total of 5 treatments and exhibited reduced appearance of conspicuous, open, and dark pores, defined by surface area and shading as determined through dermatoscopic and software analysis, with mean improvement rates of 34.6%, 11%, and 34.3%, respectively. More than 70% of participants exhibited improvement in enlarged facial pores.13 A study involving a 40% glycolic acid and vitamin C formulation demonstrated significant improvement in facial pores (28.3%; P<.001).14
The newest topical therapies studied for use in minimizing facial pilosebaceous openings are natural plant-derived copper chlorophyllin complex sodium salt (CHLcu) and tetra-hydro-jasmonic acid (LR2412). Clinical trials of these botanicals are limited with small sample sizes but are included here as novel treatments requiring further investigation.
Chlorophyllin copper complex sodium salt is derived from chlorophyll, a green pigment found in plants, and has been investigated as a topical gel in liposomal dispersions for application in photodamaged and aged skin. Chlorophyllin copper complex sodium salt exerts in vitro hyaluronidase inhibitory activity to maintain hyaluronic acid in the extracellular matrix and counteract the structural breakdown of cutaneous aging.15 Two small single-center pilot trials enrolled 10 participants each in a 3-week study of CHLcu 0.1% twice daily and an 8-week study of CHLcu 0.066% twice daily.16,17 After 3 weeks, patients treated with CHLcu 0.1% exhibited a 22.2% improvement in facial pores by clinical assessment grading, though this improvement was not significant on software imaging analysis. Patients improved the most on parameters of facial seborrhea by clinical assessment.16 After 8 weeks, patients treated with CHLcu 0.066% exhibited 25.3% improvement in facial pores by clinical assessment grading.17 Treatments were reported to be well tolerated without noted adverse events in both studies.
Tetra-hydro-jasmonic acid is an analogue of jasmonic acid, a plant hormone derived from linoleic acid. Due to its favorable safety profile and bioavailability, penetration into epidermal and dermal layers, and potential effects in rejuvenating desquamation, LR2412 is currently being assessed for treatment of skin wrinkles, texture, and pores.18 Its effect is thought to relate to stimulation of laminin-5, collagen IV, and fibrillin deposition at the dermoepidermal junction.19 In an open-label trial of a topical preparation of LR2412, 15 participants were treated twice daily for 6 weeks and assessed through investigator clinical assessment scoring.20 Investigator scoring of pores improved by 25.2% from baseline (P<.05) after 6 weeks of treatment. Improvement in pores was seen as early as days 1 and 3. No serious adverse events were reported, though 2 participants developed acne on follow-up.20
Tetra-hydro-jasmonic acid also is formulated with retinol (retinol 0.2%/LR2412 2.0%) and demonstrated cosmetic efficacy in a noninferiority trial with tretinoin cream 0.025%.11 Sixty patients each were randomized to retinol/LR2412 or tretinoin at bedtime and treated for 90 days. At day 84, participants in the retinol/LR2412 group exhibited an improvement in investigator clinical assessment scoring from a baseline of 3.6 to 2.5 (P<.05). There were no significant differences in investigator-assessed efficacy between the treatment arms. Participants reported similar or better results and fewer side effects with retinol/LR2412 on self-questionnaires. Eight participants treated with retinol/LR2412 and 15 participants treated with tretinoin reported various incidences of skin irritation, burning, and desquamation.11
Oral Therapies
The most commonly used oral therapies for enlarged pores are antiandrogens, such as combined oral contraceptives, spironolactone, and cyproterone acetate, which modulate sebum production due to the presence of androgen receptors within sebaceous glands.21 Forty-four white women in an open-label, phase 4 study were treated with combined oral contraceptives containing chlormadinone acetate–ethinyl estradiol for 6 menstrual cycles, with standardized photography taken before and after the treatment period for software analysis. After 6 treatment cycles, 9.1% (4/44) of participants had visibly enlarged pores of the forehead and cheeks compared to 43.2% (19/44) of participants at baseline (P<.0001).22 The effects of other antiandrogens on facial pores have not been studied in this capacity.
Lasers, Radiofrequency, and Ultrasound Devices
The development of various devices that can deliver targeted thermal or ultrasound energy to the skin offers the newest and most robust modality in cosmetic therapy. The mechanism of their efficacy may be due to a combination of induced remodeling of collagen fibers near pilosebaceous openings to increase skin elasticity and decrease sebum production.2,23
Devices with established antiaging effects have been extensively reviewed and include the gold particle 800-nm diode laser, 1450-nm diode laser, microneedle apparatuses, fractional radiofrequency devices, 2790-nm erbium:YAG laser, nonablative 1410-nm fractionated erbium-doped fiber laser, and nonablative 1440-nm fractional laser.2
Literature on the use of these devices for minimizing facial pore size is limited. One treatment of intense focused ultrasound using a 3-mm transducer successfully improved overall pore appearance in 91% of sites at 6-week follow-up on a clinical grading scale.24 Three sessions of nonablative 1410-nm fractionated erbium-doped fiber laser treatments yielded facial skin pore minimization of greater than 51% in 14 of 15 participants.25
The nonablative 1440-nm diode fractional laser received 510(k) clearance by the US Food and Drug Administration in 2011 for aesthetic use in chronologically aged and photoaged skin. Twenty participants treated for 2 weeks and a total of 6 facial treatments with this laser system showed a 17% average improvement in facial pore score on software analysis (P≤.002). Adverse events were mild and included erythema and xerosis.26
Conclusion
The reliability of available literature on efficacy of various treatments in diminishing facial skin pores has been challenging given that most studies are low in power, lack control groups, use nonuniform methods of reporting outcomes, and do not report complete adverse events. Thus, all results should be interpreted with caution.
Overall, it is clear that the pathogenesis of enlarged facial pores is multifactorial and complex, necessitating a similar approach to therapeutics. Topical treatments offer a range of diverse therapies with proven benefit in facial pore reduction. The advent of lasers and devices offers constantly evolving therapeutic options with diffuse antiaging effects. Despite the numerous topical, oral, and device-oriented options, enlarged facial pores remain a challenging cosmetic concern. More robust efficacy studies on new treatments are necessary.
- Uhoda E, Pierard-Franchimont C, Petit L, et al. The conundrum of skin pores in dermocosmetology. Dermatology. 2005;210:3-7.
- Lee SJ, Seok J, Jeong SY, et al. Facial pores: definition, causes, and treatment options. Dermatol Surg. 2016;42:277-285.
- Pierard GE, Pierard-Franchimont C, Marks R, et al. EEMCO guidance for the in vivo assessment of skin greasiness. The EEMCO Group. Skin Pharmacol Appl Skin Physiol. 2000;13:372-389.
- Roh M, Han M, Kim D, et al. Sebum output as a factor contributing to the size of facial pores. Br J Dermatol. 2006;155:890-894.
- Kim BY, Choi JW, Park KC, et al. Sebum, acne, skin elasticity, and gender difference-which is the major influencing factor for facial pores? Skin Res Technol. 2013;19:E45-E53.
- Uitto J. The role of elastin and collagen in cutaneous aging: intrinsic aging versus photoexposure. J Drugs Dermatol. 2008;7(2 suppl):S12-S16.
- Zheng Q, Chen S, Chen Y, et al. Investigation of age-related decline of microfibril-associated glycoprotein-1 in human skin through immunohistochemistry study. Clin Cosmet Investig Dermatol. 2013;6:317-323.
- Sugiyama-Nakagiri Y, Sugata K, Hachiya A, et al. Ethnic differences in the structural properties of facial skin. J Dermatol Sci. 2009;53:135-139.
- Mukherjee S, Date A, Patravale V, et al. Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety. Clin Interv Aging. 2006;1:327-348.
- Kang S, Krueger GG, Tanghetti EA, et al; Tazarotene Cream in Photodamage Study Group. A multicenter, randomized, double-blind trial of tazarotene 0.1% cream in the treatment of photodamage. J Am Acad Dermatol. 2005;52:268-274.
- Bouloc A, Vergnanini AL, Issa MC. A double-blind randomized study comparing the association of retinol and LR2412 with tretinoin 0.025% in photoaged skin. J Cosmet Dermatol. 2015;14:40-46.
- Fischer TC, Perosino E, Poli F, et al. Chemical peels in aesthetic dermatology: an update 2009 [published online September 8, 2009]. J Eur Acad Dermatol Venereol. 2010;24:281-292.
- Kakudo N, Kushida S, Tanaka N, et al. A novel method to measure conspicuous facial pores using computer analysis of digital-camera-captured images: the effect of glycolic acid chemical peeling. Skin Res Technol. 2011;17:427-433.
- Kim WS. Efficacy and safety of a new superficial chemical peel using alpha-hydroxy acid, vitamin C and oxygen for melasma. J Cosmet Laser Ther. 2013;15:21-24.
- McCook JP, Dorogi PL, Vasily DB, et al. In vitro inhibition of hyaluronidase by sodium copper chlorophyllin complex and chlorophyllin analogs. Clin Cosmet Investig Dermatol. 2015;8:443-448.
- Stephens TJ, McCook JP, Herndon JH Jr. Pilot study of topical copper chlorophyllin complex in subjects with facial acne and large pores. J Drugs Dermatol. 2015;14:589-592.
- Sigler ML, Stephens TJ. Assessment of the safety and efficacy of topical copper chlorophyllin in women with photodamaged facial skin. J Drugs Dermatol. 2015;14:401-404.
- Alexiades M. Jasmonates and tetrahydrojasmonic acid: a novel class of anti-aging molecules. J Drugs Dermatol. 2016;15:206-207.
- Tran C, Michelet JF, Simonetti L, et al. In vitro and in vivo studies with tetra-hydro-jasmonic acid (LR2412) reveal its potential to correct signs of skin ageing. J Eur Acad Dermatol Venereol. 2014;28:415-423.
- Alexiades M. Clinical assessment of a novel jasmonate cosmeceutical, LR2412-Cx, for the treatment of skin aging. J Drugs Dermatol. 2016;15:209-215.
- Lam C, Zaenglein AL. Contraceptive use in acne. Clin Dermatol. 2014;32:502-515.
- Kerscher M, Reuther T, Bayrhammer J, et al. Effects of an oral contraceptive containing chlormadinone and ethinylestradiol on acne-prone skin of women of different age groups: an open-label, single-centre, phase IV study. Clin Drug Investig. 2008;28:703-711.
- Schmults CD, Phelps R, Goldberg DJ. Nonablative facial remodeling: erythema reduction and histologic evidence of new collagen formation using a 300-microsecond 1064-nm Nd:YAG laser. Arch Dermatol. 2004;140:1373-1376.
- Lee HJ, Lee KR, Park JY, et al. The efficacy and safety of intense focused ultrasound in the treatment of enlarged facial pores in Asian skin. J Dermatolog Treat. 2015;26:73-77.
- Suh DH, Chang KY, Lee SJ, et al. Treatment of dilated pores with 1410-nm fractional erbium-doped fiber laser. Lasers Med Sci. 2015;30:1135-1139.
- Saedi N, Petrell K, Arndt K, et al. Evaluating facial pores and skin texture after low-energy nonablative fractional 1440-nm laser treatments. J Am Acad Dermatol. 2013;68:113-118.
- Uhoda E, Pierard-Franchimont C, Petit L, et al. The conundrum of skin pores in dermocosmetology. Dermatology. 2005;210:3-7.
- Lee SJ, Seok J, Jeong SY, et al. Facial pores: definition, causes, and treatment options. Dermatol Surg. 2016;42:277-285.
- Pierard GE, Pierard-Franchimont C, Marks R, et al. EEMCO guidance for the in vivo assessment of skin greasiness. The EEMCO Group. Skin Pharmacol Appl Skin Physiol. 2000;13:372-389.
- Roh M, Han M, Kim D, et al. Sebum output as a factor contributing to the size of facial pores. Br J Dermatol. 2006;155:890-894.
- Kim BY, Choi JW, Park KC, et al. Sebum, acne, skin elasticity, and gender difference-which is the major influencing factor for facial pores? Skin Res Technol. 2013;19:E45-E53.
- Uitto J. The role of elastin and collagen in cutaneous aging: intrinsic aging versus photoexposure. J Drugs Dermatol. 2008;7(2 suppl):S12-S16.
- Zheng Q, Chen S, Chen Y, et al. Investigation of age-related decline of microfibril-associated glycoprotein-1 in human skin through immunohistochemistry study. Clin Cosmet Investig Dermatol. 2013;6:317-323.
- Sugiyama-Nakagiri Y, Sugata K, Hachiya A, et al. Ethnic differences in the structural properties of facial skin. J Dermatol Sci. 2009;53:135-139.
- Mukherjee S, Date A, Patravale V, et al. Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety. Clin Interv Aging. 2006;1:327-348.
- Kang S, Krueger GG, Tanghetti EA, et al; Tazarotene Cream in Photodamage Study Group. A multicenter, randomized, double-blind trial of tazarotene 0.1% cream in the treatment of photodamage. J Am Acad Dermatol. 2005;52:268-274.
- Bouloc A, Vergnanini AL, Issa MC. A double-blind randomized study comparing the association of retinol and LR2412 with tretinoin 0.025% in photoaged skin. J Cosmet Dermatol. 2015;14:40-46.
- Fischer TC, Perosino E, Poli F, et al. Chemical peels in aesthetic dermatology: an update 2009 [published online September 8, 2009]. J Eur Acad Dermatol Venereol. 2010;24:281-292.
- Kakudo N, Kushida S, Tanaka N, et al. A novel method to measure conspicuous facial pores using computer analysis of digital-camera-captured images: the effect of glycolic acid chemical peeling. Skin Res Technol. 2011;17:427-433.
- Kim WS. Efficacy and safety of a new superficial chemical peel using alpha-hydroxy acid, vitamin C and oxygen for melasma. J Cosmet Laser Ther. 2013;15:21-24.
- McCook JP, Dorogi PL, Vasily DB, et al. In vitro inhibition of hyaluronidase by sodium copper chlorophyllin complex and chlorophyllin analogs. Clin Cosmet Investig Dermatol. 2015;8:443-448.
- Stephens TJ, McCook JP, Herndon JH Jr. Pilot study of topical copper chlorophyllin complex in subjects with facial acne and large pores. J Drugs Dermatol. 2015;14:589-592.
- Sigler ML, Stephens TJ. Assessment of the safety and efficacy of topical copper chlorophyllin in women with photodamaged facial skin. J Drugs Dermatol. 2015;14:401-404.
- Alexiades M. Jasmonates and tetrahydrojasmonic acid: a novel class of anti-aging molecules. J Drugs Dermatol. 2016;15:206-207.
- Tran C, Michelet JF, Simonetti L, et al. In vitro and in vivo studies with tetra-hydro-jasmonic acid (LR2412) reveal its potential to correct signs of skin ageing. J Eur Acad Dermatol Venereol. 2014;28:415-423.
- Alexiades M. Clinical assessment of a novel jasmonate cosmeceutical, LR2412-Cx, for the treatment of skin aging. J Drugs Dermatol. 2016;15:209-215.
- Lam C, Zaenglein AL. Contraceptive use in acne. Clin Dermatol. 2014;32:502-515.
- Kerscher M, Reuther T, Bayrhammer J, et al. Effects of an oral contraceptive containing chlormadinone and ethinylestradiol on acne-prone skin of women of different age groups: an open-label, single-centre, phase IV study. Clin Drug Investig. 2008;28:703-711.
- Schmults CD, Phelps R, Goldberg DJ. Nonablative facial remodeling: erythema reduction and histologic evidence of new collagen formation using a 300-microsecond 1064-nm Nd:YAG laser. Arch Dermatol. 2004;140:1373-1376.
- Lee HJ, Lee KR, Park JY, et al. The efficacy and safety of intense focused ultrasound in the treatment of enlarged facial pores in Asian skin. J Dermatolog Treat. 2015;26:73-77.
- Suh DH, Chang KY, Lee SJ, et al. Treatment of dilated pores with 1410-nm fractional erbium-doped fiber laser. Lasers Med Sci. 2015;30:1135-1139.
- Saedi N, Petrell K, Arndt K, et al. Evaluating facial pores and skin texture after low-energy nonablative fractional 1440-nm laser treatments. J Am Acad Dermatol. 2013;68:113-118.
Practice Points
- The pathogenesis of enlarged facial pores is speculated to be associated with sebum production, skin aging and photodamage, and hair follicle size, among other factors.
- Current treatment modalities for enlarged facial pores target these factors and include topical retinoids, chemical peels, oral antiandrogens, lasers, radiofrequency, and ultrasound devices, with the latter devices offering the most novel and robust choices.
- New botanically derived topical treatments, specifically copper chlorophyllin complex sodium salt and tetra-hydro-jasmonic acid, are in development with initial positive results, though studies are still limited.
Actinic Keratosis as a Marker of Field Cancerization in Excision Specimens of Cutaneous Malignancies
The concept of field cancerization was first proposed in 1953 by Slaughter et al1 in their study of oral squamous carcinomas. Their findings of multifocal patches of premalignant disease, abnormal tissue surrounding tumors, multiple localized primary tumors, and tumor recurrence following surgical resection was suggestive of a field of dysplastic cells with malignant potential.1 Since then, modern molecular techniques have been used to establish a genetic basis for this model in many different types of cancer, including cutaneous malignancies.2-4 The field begins from a singular stem cell, which undergoes one or more genetic changes that allow for a growth advantage compared to surrounding cells. The stem cell then divides, forming a patch of clonal daughter cells that displace the surrounding normal epithelium. Growth of this patch eventually leads to a dysplastic field of monoclonal cells, which notably does not yet show invasive growth or metastatic behavior. Over time, continued carcinogenic exposure results in additional genetic alterations among different cells in the field, which leads to new subclonal proliferations that share common clonal origin but exhibit unique genetic changes. Eventually, transformative events may occur, resulting in cells with invasive and metastatic properties, thus forming a carcinoma.5
In the case of cutaneous malignancies, UV radiation in the form of UVA and UVB rays is the most common source of carcinogenesis. It is well established that UV radiation has numerous effects on the body, including but not limited to local and systemic immunosuppression, alteration of signal transduction pathways, and the development of mutations in DNA via direct damage by UVB or indirect damage by free radical formation with UVA.6,7 Normally, DNA is protected from UV radiation–induced genetic alteration by the p53 gene, TP53. As such, damage to this gene is highly associated with cancer induction. One study found that more than 90% of squamous cell carcinomas (SCCs) and more than 50% of basal cell carcinomas (BCCs) contain UV-like mutations in TP53.8 The concept of field cancerization suggests that because the skin surrounding cutaneous malignancies has been exposed to the same chronic UV light as the initial lesion, it is at an increased risk for genetic abnormalities and thus possible malignant transformation.
Actinic keratoses (AKs) are common neoplasms of the skin that generally are regarded as precancerous lesions or may be considered to be the earliest stage of SCC in situ.9 Actinic keratoses usually develop as a consequence of chronic exposure to UV radiation and often are clinically apparent as erythematous scaly papules in sun-exposed areas (Figure 1).10 They also are identified histologically as atypical keratinocytes along the basal layer of the epidermis with possible enlargement, hyperchromatic nuclei, lack of maturation, mitotic figures, inflammatory infiltrate, and/or hyperkeratosis.10 Furthermore, the genetic changes associated with AKs are well documented and are strongly associated with changes to p53.11 Given these characteristics, AKs serve as good markers of genetic damage with potential for malignancy. In this study, we used histologically identified AKs to assess the presence of field damage in the tissue immediately surrounding excision specimens of SCCs, BCCs, and malignant melanomas (MMs).
Methods
This study was approved by the Program for the Protection of Human Subjects at the Icahn School of Medicine at Mount Sinai (New York, New York) prior to initiation. All cutaneous specimens submitted to the dermatopathology service for consultation between April 2013 and June 2013 were reviewed for inclusion in this study. Data collection was extended for MMs to include all specimens from January 2013 to June 2013 given the limited number of cases in the original data collection period.
Initial screening for this study was done electronically and assessed for a diagnosis of SCC (Figure 2), BCC (Figure 3), or MM (Figure 4) as determined by a board-certified dermatopathologist (G.G.). The resulting pool of specimens was then screened to include only excision specimens and to exclude curettage specimens and superficial specimens that lacked dermis. In this study, we chose to look at reexcisions rather than initial biopsies so that there was a greater likelihood of having an intact epidermis surrounding a malignancy that could be assessed for the presence of AKs as markers for field cancerization. Specimens were examined in full via serial transverse cross-sections at 3-mm intervals. Additional step sections were obtained at smaller intervals when margins were close or unclear.
Selected cases were reassessed by a board-certified dermatopathologist (G.G.) to confirm the diagnosis and to assess for the presence of at least 1 AK within the specimen sample that was separated from the original malignancy by histologically normal-appearing cells. Samples were also assessed for the presence of an AK within 0.1 mm of the distal lateral margins of the tissue sample. Information regarding patient age, gender, lesion location, lesion type, and specimen size was collected for each sample. In accordance with institutional review board protocol, research data were collected without any protected health information. All analyses and results were deidentified and stored on a password-protected computer database. Statistical analysis was performed using SPSS software. When applicable, P<.05 was considered to indicate statistical significance.
Results
There were 205 cases that passed the initial screening filters, of which 56 were excluded due to the presence of curettage or lack of a sufficient tissue sample. Of the remaining 149 cases, the distribution by malignancy type was tabulated along with the percentage of observed AKs. If an AK was observed, the percentage that had an AK at the lateral margins (marginal AK) was determined (Table 1). A χ2 analysis determined that AKs were observed significantly more often in SCC specimens (57% [35/61]) than BCC (33% [21/64]) or malignant melanoma (25% [6/24]) specimens (P=.0125).
Statistics regarding patient age and gender as well as specimen size were stratified by malignancy type (Table 2). Using a receiver operating characteristic curve and the Youden index, an optimal cutoff of older than 67 years was determined to increase probability of observing an AK (P=.077) with sensitivity of 0.531 and specificity of 0.529. The distribution of specimen excision location for each malignancy type is shown in Table 3.
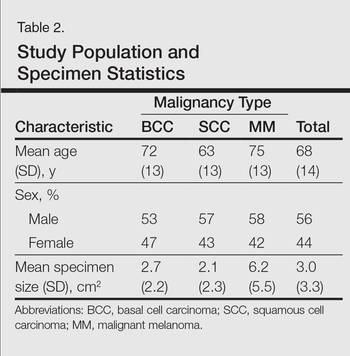
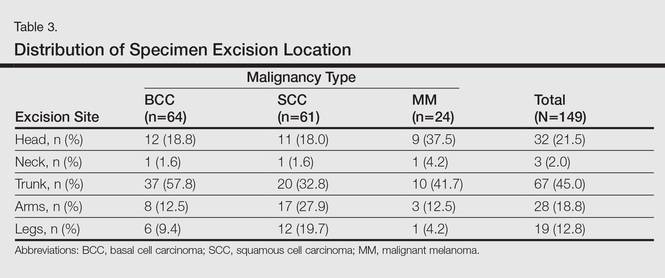
A multivariate analysis was performed to determine if the variables of patient age, gender, biopsy size, malignancy type (SCC, BCC, or MM), or cancer location (head, neck, trunk, arms, or legs) were independently useful in predicting whether an AK would be observed in the excision specimen. The significance of variables in the logistic regression model was assessed using a backward stepwise regression selection procedure entering variables if P<.15 and excluding variables if P>.25. Significant variables in predicting the occurrence of AK were SCC malignancy type (P=.007; odds ratio [OR], 2.61) and location on the head (P=.044; OR, 2.39) and arms (P=.042; OR, 2.55).
Comment
The χ2 analysis of our data showed that SCC specimens were significantly more likely to have an associated AK than either BCCs or MMs (P=.0125), which is not surprising given that AKs are considered by many to be early-stage SCCs.12 It is important to note, however, that BCCs and MMs both had nonnegligible rates of associated AKs. Although BCC and MM do not arise from the same background of genetic changes as SCC, this finding is noteworthy because it demonstrates definitive field damage with malignant potential in the area surrounding these cutaneous malignancies.
Our data also showed that there was a significantly greater association of AKs in malignancies located on the head (P=.044) and arms (P=.042), possibly because these 2 areas tend to be the most sun exposed and thus are more likely to have sustained field damage as evidenced by the higher percentage of AKs. A study by Jonason et al13 described a similar finding in which sun-exposed skin exhibited significantly more frequent (P=.04) and larger (P=.02) clonal patches of mutated p53 keratinocytes than sun-protected skin.
It is likely that the field damage surrounding the cutaneous lesions in our study is actually greater than what we reported because the AK was present at the margin of the excision specimens the majority of the time (56%), which suggests that there likely may have been more AKs found if a wider area surrounding the malignancy had been studied given that AKs often are at the periphery of the lesion and may be missed by a small excision. Fewer marginal AKs were observed with MM cases, possibly because the excision specimens were more than double the size of SCC or BCC excisions. Furthermore, there likely is to be more damage than what can be appreciated by visual changes alone.
Kanjilal et al14 used polymerase chain reaction and DNA sequencing to demonstrate numerous p53 mutations in nonmalignant-appearing skin surrounding BCCs and SCCs. Brennan et al15 found p53 mutations in surgical margins of excised SCCs considered to be tumor free by histopathologic analysis in more than half of the specimens studied. Notably, tumor recurrence was significantly more likely in areas where mutations were found and no tumor recurrence was seen in areas free of p53 mutations (P=.02).15 Tabor et al4 similarly found genetically altered fields in histologically clear surgical margins of SCCs but also showed that local tumor recurrence following excision had more molecular markers in common with the nonresected premalignant field than it did with the primary tumor. Thus, these studies provide a genetic basis for field damage that can exist even in histologically benign-appearing cells.
We believe our findings are clinically relevant, as they provide additional evidence for the theory of field cancerization as demonstrated by the nonnegligible rates of AKs and thus field damage with malignant potential in the skin immediately surrounding cutaneous malignancies. The limitations of our study, however, include a small sample size; no consideration of the effects of prior topical, field, or systemic treatments; and lack of a control group. Nevertheless, our findings emphasize the importance of assessing the extent of field damage when determining treatment strategies. Clinicians treating cutaneous malignancies should consider the need for field therapy, especially in sun-exposed regions, to avoid additional primary tumors.16 Further research is needed, however, to identify optimal methods for quantifying field damage clinically and determining the most effective treatment strategies.
- Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Braakhuis B, Tabor M, Kummer J, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Stern R, Bolshakov S, Nataraj A, et al. p53 Mutation in nonmelanoma skin cancers occurring in psoralen ultraviolet A-treated patients: evidence for heterogeneity and field cancerization. J Invest Dermatol. 2002;119:522-526.
- Tabor M, Brakenhoff R, van Houten VM, et al. Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001;7:1523-1532.
- Torezan L. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Ullrich S, Kripke M, Ananthaswamy H. Mechanisms underlying UV-induced immune suppression: implications for sunscreen design. Exp Dermatol. 2002;11:1-4.
- de Gruijl FR. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 2000;319:359-366.
- Brash DE, Ziegler A, Jonason AS, et al. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1996;1:136-142.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Rossi R, Mori M, Lotti T. Actinic keratosis. Int J Dermatol. 2007;46:895-904.
- Ziegler A, Jonason AS, Leffel DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773-776.
- Cockerell C. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42:11-17.
- Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci. 1996;93:14025-14029.
- Kanjilal S, Strom SS, Clayman GL, et al. p53 Mutations in nonmelanoma skin cancer of the head and neck: molecular evidence for field cancerization. Cancer Res. 1995;55:3604-3609.
- Brennan JA, Mao L, Hruban RH, et al. Molecular assessment of histopathological staging in squamous cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braathen LR, Morton CA, Basset-Seguin N, et al. Photodynamic therapy for skin field cancerization: an international consensus. International Society for Photodynamic Therapy in Dermatology. J Eur Acad Dermatol Venereol. 2012;26:1063-1066.
The concept of field cancerization was first proposed in 1953 by Slaughter et al1 in their study of oral squamous carcinomas. Their findings of multifocal patches of premalignant disease, abnormal tissue surrounding tumors, multiple localized primary tumors, and tumor recurrence following surgical resection was suggestive of a field of dysplastic cells with malignant potential.1 Since then, modern molecular techniques have been used to establish a genetic basis for this model in many different types of cancer, including cutaneous malignancies.2-4 The field begins from a singular stem cell, which undergoes one or more genetic changes that allow for a growth advantage compared to surrounding cells. The stem cell then divides, forming a patch of clonal daughter cells that displace the surrounding normal epithelium. Growth of this patch eventually leads to a dysplastic field of monoclonal cells, which notably does not yet show invasive growth or metastatic behavior. Over time, continued carcinogenic exposure results in additional genetic alterations among different cells in the field, which leads to new subclonal proliferations that share common clonal origin but exhibit unique genetic changes. Eventually, transformative events may occur, resulting in cells with invasive and metastatic properties, thus forming a carcinoma.5
In the case of cutaneous malignancies, UV radiation in the form of UVA and UVB rays is the most common source of carcinogenesis. It is well established that UV radiation has numerous effects on the body, including but not limited to local and systemic immunosuppression, alteration of signal transduction pathways, and the development of mutations in DNA via direct damage by UVB or indirect damage by free radical formation with UVA.6,7 Normally, DNA is protected from UV radiation–induced genetic alteration by the p53 gene, TP53. As such, damage to this gene is highly associated with cancer induction. One study found that more than 90% of squamous cell carcinomas (SCCs) and more than 50% of basal cell carcinomas (BCCs) contain UV-like mutations in TP53.8 The concept of field cancerization suggests that because the skin surrounding cutaneous malignancies has been exposed to the same chronic UV light as the initial lesion, it is at an increased risk for genetic abnormalities and thus possible malignant transformation.
Actinic keratoses (AKs) are common neoplasms of the skin that generally are regarded as precancerous lesions or may be considered to be the earliest stage of SCC in situ.9 Actinic keratoses usually develop as a consequence of chronic exposure to UV radiation and often are clinically apparent as erythematous scaly papules in sun-exposed areas (Figure 1).10 They also are identified histologically as atypical keratinocytes along the basal layer of the epidermis with possible enlargement, hyperchromatic nuclei, lack of maturation, mitotic figures, inflammatory infiltrate, and/or hyperkeratosis.10 Furthermore, the genetic changes associated with AKs are well documented and are strongly associated with changes to p53.11 Given these characteristics, AKs serve as good markers of genetic damage with potential for malignancy. In this study, we used histologically identified AKs to assess the presence of field damage in the tissue immediately surrounding excision specimens of SCCs, BCCs, and malignant melanomas (MMs).
Methods
This study was approved by the Program for the Protection of Human Subjects at the Icahn School of Medicine at Mount Sinai (New York, New York) prior to initiation. All cutaneous specimens submitted to the dermatopathology service for consultation between April 2013 and June 2013 were reviewed for inclusion in this study. Data collection was extended for MMs to include all specimens from January 2013 to June 2013 given the limited number of cases in the original data collection period.
Initial screening for this study was done electronically and assessed for a diagnosis of SCC (Figure 2), BCC (Figure 3), or MM (Figure 4) as determined by a board-certified dermatopathologist (G.G.). The resulting pool of specimens was then screened to include only excision specimens and to exclude curettage specimens and superficial specimens that lacked dermis. In this study, we chose to look at reexcisions rather than initial biopsies so that there was a greater likelihood of having an intact epidermis surrounding a malignancy that could be assessed for the presence of AKs as markers for field cancerization. Specimens were examined in full via serial transverse cross-sections at 3-mm intervals. Additional step sections were obtained at smaller intervals when margins were close or unclear.
Selected cases were reassessed by a board-certified dermatopathologist (G.G.) to confirm the diagnosis and to assess for the presence of at least 1 AK within the specimen sample that was separated from the original malignancy by histologically normal-appearing cells. Samples were also assessed for the presence of an AK within 0.1 mm of the distal lateral margins of the tissue sample. Information regarding patient age, gender, lesion location, lesion type, and specimen size was collected for each sample. In accordance with institutional review board protocol, research data were collected without any protected health information. All analyses and results were deidentified and stored on a password-protected computer database. Statistical analysis was performed using SPSS software. When applicable, P<.05 was considered to indicate statistical significance.
Results
There were 205 cases that passed the initial screening filters, of which 56 were excluded due to the presence of curettage or lack of a sufficient tissue sample. Of the remaining 149 cases, the distribution by malignancy type was tabulated along with the percentage of observed AKs. If an AK was observed, the percentage that had an AK at the lateral margins (marginal AK) was determined (Table 1). A χ2 analysis determined that AKs were observed significantly more often in SCC specimens (57% [35/61]) than BCC (33% [21/64]) or malignant melanoma (25% [6/24]) specimens (P=.0125).
Statistics regarding patient age and gender as well as specimen size were stratified by malignancy type (Table 2). Using a receiver operating characteristic curve and the Youden index, an optimal cutoff of older than 67 years was determined to increase probability of observing an AK (P=.077) with sensitivity of 0.531 and specificity of 0.529. The distribution of specimen excision location for each malignancy type is shown in Table 3.
A multivariate analysis was performed to determine if the variables of patient age, gender, biopsy size, malignancy type (SCC, BCC, or MM), or cancer location (head, neck, trunk, arms, or legs) were independently useful in predicting whether an AK would be observed in the excision specimen. The significance of variables in the logistic regression model was assessed using a backward stepwise regression selection procedure entering variables if P<.15 and excluding variables if P>.25. Significant variables in predicting the occurrence of AK were SCC malignancy type (P=.007; odds ratio [OR], 2.61) and location on the head (P=.044; OR, 2.39) and arms (P=.042; OR, 2.55).
Comment
The χ2 analysis of our data showed that SCC specimens were significantly more likely to have an associated AK than either BCCs or MMs (P=.0125), which is not surprising given that AKs are considered by many to be early-stage SCCs.12 It is important to note, however, that BCCs and MMs both had nonnegligible rates of associated AKs. Although BCC and MM do not arise from the same background of genetic changes as SCC, this finding is noteworthy because it demonstrates definitive field damage with malignant potential in the area surrounding these cutaneous malignancies.
Our data also showed that there was a significantly greater association of AKs in malignancies located on the head (P=.044) and arms (P=.042), possibly because these 2 areas tend to be the most sun exposed and thus are more likely to have sustained field damage as evidenced by the higher percentage of AKs. A study by Jonason et al13 described a similar finding in which sun-exposed skin exhibited significantly more frequent (P=.04) and larger (P=.02) clonal patches of mutated p53 keratinocytes than sun-protected skin.
It is likely that the field damage surrounding the cutaneous lesions in our study is actually greater than what we reported because the AK was present at the margin of the excision specimens the majority of the time (56%), which suggests that there likely may have been more AKs found if a wider area surrounding the malignancy had been studied given that AKs often are at the periphery of the lesion and may be missed by a small excision. Fewer marginal AKs were observed with MM cases, possibly because the excision specimens were more than double the size of SCC or BCC excisions. Furthermore, there likely is to be more damage than what can be appreciated by visual changes alone.
Kanjilal et al14 used polymerase chain reaction and DNA sequencing to demonstrate numerous p53 mutations in nonmalignant-appearing skin surrounding BCCs and SCCs. Brennan et al15 found p53 mutations in surgical margins of excised SCCs considered to be tumor free by histopathologic analysis in more than half of the specimens studied. Notably, tumor recurrence was significantly more likely in areas where mutations were found and no tumor recurrence was seen in areas free of p53 mutations (P=.02).15 Tabor et al4 similarly found genetically altered fields in histologically clear surgical margins of SCCs but also showed that local tumor recurrence following excision had more molecular markers in common with the nonresected premalignant field than it did with the primary tumor. Thus, these studies provide a genetic basis for field damage that can exist even in histologically benign-appearing cells.
We believe our findings are clinically relevant, as they provide additional evidence for the theory of field cancerization as demonstrated by the nonnegligible rates of AKs and thus field damage with malignant potential in the skin immediately surrounding cutaneous malignancies. The limitations of our study, however, include a small sample size; no consideration of the effects of prior topical, field, or systemic treatments; and lack of a control group. Nevertheless, our findings emphasize the importance of assessing the extent of field damage when determining treatment strategies. Clinicians treating cutaneous malignancies should consider the need for field therapy, especially in sun-exposed regions, to avoid additional primary tumors.16 Further research is needed, however, to identify optimal methods for quantifying field damage clinically and determining the most effective treatment strategies.
The concept of field cancerization was first proposed in 1953 by Slaughter et al1 in their study of oral squamous carcinomas. Their findings of multifocal patches of premalignant disease, abnormal tissue surrounding tumors, multiple localized primary tumors, and tumor recurrence following surgical resection was suggestive of a field of dysplastic cells with malignant potential.1 Since then, modern molecular techniques have been used to establish a genetic basis for this model in many different types of cancer, including cutaneous malignancies.2-4 The field begins from a singular stem cell, which undergoes one or more genetic changes that allow for a growth advantage compared to surrounding cells. The stem cell then divides, forming a patch of clonal daughter cells that displace the surrounding normal epithelium. Growth of this patch eventually leads to a dysplastic field of monoclonal cells, which notably does not yet show invasive growth or metastatic behavior. Over time, continued carcinogenic exposure results in additional genetic alterations among different cells in the field, which leads to new subclonal proliferations that share common clonal origin but exhibit unique genetic changes. Eventually, transformative events may occur, resulting in cells with invasive and metastatic properties, thus forming a carcinoma.5
In the case of cutaneous malignancies, UV radiation in the form of UVA and UVB rays is the most common source of carcinogenesis. It is well established that UV radiation has numerous effects on the body, including but not limited to local and systemic immunosuppression, alteration of signal transduction pathways, and the development of mutations in DNA via direct damage by UVB or indirect damage by free radical formation with UVA.6,7 Normally, DNA is protected from UV radiation–induced genetic alteration by the p53 gene, TP53. As such, damage to this gene is highly associated with cancer induction. One study found that more than 90% of squamous cell carcinomas (SCCs) and more than 50% of basal cell carcinomas (BCCs) contain UV-like mutations in TP53.8 The concept of field cancerization suggests that because the skin surrounding cutaneous malignancies has been exposed to the same chronic UV light as the initial lesion, it is at an increased risk for genetic abnormalities and thus possible malignant transformation.
Actinic keratoses (AKs) are common neoplasms of the skin that generally are regarded as precancerous lesions or may be considered to be the earliest stage of SCC in situ.9 Actinic keratoses usually develop as a consequence of chronic exposure to UV radiation and often are clinically apparent as erythematous scaly papules in sun-exposed areas (Figure 1).10 They also are identified histologically as atypical keratinocytes along the basal layer of the epidermis with possible enlargement, hyperchromatic nuclei, lack of maturation, mitotic figures, inflammatory infiltrate, and/or hyperkeratosis.10 Furthermore, the genetic changes associated with AKs are well documented and are strongly associated with changes to p53.11 Given these characteristics, AKs serve as good markers of genetic damage with potential for malignancy. In this study, we used histologically identified AKs to assess the presence of field damage in the tissue immediately surrounding excision specimens of SCCs, BCCs, and malignant melanomas (MMs).
Methods
This study was approved by the Program for the Protection of Human Subjects at the Icahn School of Medicine at Mount Sinai (New York, New York) prior to initiation. All cutaneous specimens submitted to the dermatopathology service for consultation between April 2013 and June 2013 were reviewed for inclusion in this study. Data collection was extended for MMs to include all specimens from January 2013 to June 2013 given the limited number of cases in the original data collection period.
Initial screening for this study was done electronically and assessed for a diagnosis of SCC (Figure 2), BCC (Figure 3), or MM (Figure 4) as determined by a board-certified dermatopathologist (G.G.). The resulting pool of specimens was then screened to include only excision specimens and to exclude curettage specimens and superficial specimens that lacked dermis. In this study, we chose to look at reexcisions rather than initial biopsies so that there was a greater likelihood of having an intact epidermis surrounding a malignancy that could be assessed for the presence of AKs as markers for field cancerization. Specimens were examined in full via serial transverse cross-sections at 3-mm intervals. Additional step sections were obtained at smaller intervals when margins were close or unclear.
Selected cases were reassessed by a board-certified dermatopathologist (G.G.) to confirm the diagnosis and to assess for the presence of at least 1 AK within the specimen sample that was separated from the original malignancy by histologically normal-appearing cells. Samples were also assessed for the presence of an AK within 0.1 mm of the distal lateral margins of the tissue sample. Information regarding patient age, gender, lesion location, lesion type, and specimen size was collected for each sample. In accordance with institutional review board protocol, research data were collected without any protected health information. All analyses and results were deidentified and stored on a password-protected computer database. Statistical analysis was performed using SPSS software. When applicable, P<.05 was considered to indicate statistical significance.
Results
There were 205 cases that passed the initial screening filters, of which 56 were excluded due to the presence of curettage or lack of a sufficient tissue sample. Of the remaining 149 cases, the distribution by malignancy type was tabulated along with the percentage of observed AKs. If an AK was observed, the percentage that had an AK at the lateral margins (marginal AK) was determined (Table 1). A χ2 analysis determined that AKs were observed significantly more often in SCC specimens (57% [35/61]) than BCC (33% [21/64]) or malignant melanoma (25% [6/24]) specimens (P=.0125).
Statistics regarding patient age and gender as well as specimen size were stratified by malignancy type (Table 2). Using a receiver operating characteristic curve and the Youden index, an optimal cutoff of older than 67 years was determined to increase probability of observing an AK (P=.077) with sensitivity of 0.531 and specificity of 0.529. The distribution of specimen excision location for each malignancy type is shown in Table 3.
A multivariate analysis was performed to determine if the variables of patient age, gender, biopsy size, malignancy type (SCC, BCC, or MM), or cancer location (head, neck, trunk, arms, or legs) were independently useful in predicting whether an AK would be observed in the excision specimen. The significance of variables in the logistic regression model was assessed using a backward stepwise regression selection procedure entering variables if P<.15 and excluding variables if P>.25. Significant variables in predicting the occurrence of AK were SCC malignancy type (P=.007; odds ratio [OR], 2.61) and location on the head (P=.044; OR, 2.39) and arms (P=.042; OR, 2.55).
Comment
The χ2 analysis of our data showed that SCC specimens were significantly more likely to have an associated AK than either BCCs or MMs (P=.0125), which is not surprising given that AKs are considered by many to be early-stage SCCs.12 It is important to note, however, that BCCs and MMs both had nonnegligible rates of associated AKs. Although BCC and MM do not arise from the same background of genetic changes as SCC, this finding is noteworthy because it demonstrates definitive field damage with malignant potential in the area surrounding these cutaneous malignancies.
Our data also showed that there was a significantly greater association of AKs in malignancies located on the head (P=.044) and arms (P=.042), possibly because these 2 areas tend to be the most sun exposed and thus are more likely to have sustained field damage as evidenced by the higher percentage of AKs. A study by Jonason et al13 described a similar finding in which sun-exposed skin exhibited significantly more frequent (P=.04) and larger (P=.02) clonal patches of mutated p53 keratinocytes than sun-protected skin.
It is likely that the field damage surrounding the cutaneous lesions in our study is actually greater than what we reported because the AK was present at the margin of the excision specimens the majority of the time (56%), which suggests that there likely may have been more AKs found if a wider area surrounding the malignancy had been studied given that AKs often are at the periphery of the lesion and may be missed by a small excision. Fewer marginal AKs were observed with MM cases, possibly because the excision specimens were more than double the size of SCC or BCC excisions. Furthermore, there likely is to be more damage than what can be appreciated by visual changes alone.
Kanjilal et al14 used polymerase chain reaction and DNA sequencing to demonstrate numerous p53 mutations in nonmalignant-appearing skin surrounding BCCs and SCCs. Brennan et al15 found p53 mutations in surgical margins of excised SCCs considered to be tumor free by histopathologic analysis in more than half of the specimens studied. Notably, tumor recurrence was significantly more likely in areas where mutations were found and no tumor recurrence was seen in areas free of p53 mutations (P=.02).15 Tabor et al4 similarly found genetically altered fields in histologically clear surgical margins of SCCs but also showed that local tumor recurrence following excision had more molecular markers in common with the nonresected premalignant field than it did with the primary tumor. Thus, these studies provide a genetic basis for field damage that can exist even in histologically benign-appearing cells.
We believe our findings are clinically relevant, as they provide additional evidence for the theory of field cancerization as demonstrated by the nonnegligible rates of AKs and thus field damage with malignant potential in the skin immediately surrounding cutaneous malignancies. The limitations of our study, however, include a small sample size; no consideration of the effects of prior topical, field, or systemic treatments; and lack of a control group. Nevertheless, our findings emphasize the importance of assessing the extent of field damage when determining treatment strategies. Clinicians treating cutaneous malignancies should consider the need for field therapy, especially in sun-exposed regions, to avoid additional primary tumors.16 Further research is needed, however, to identify optimal methods for quantifying field damage clinically and determining the most effective treatment strategies.
- Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Braakhuis B, Tabor M, Kummer J, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Stern R, Bolshakov S, Nataraj A, et al. p53 Mutation in nonmelanoma skin cancers occurring in psoralen ultraviolet A-treated patients: evidence for heterogeneity and field cancerization. J Invest Dermatol. 2002;119:522-526.
- Tabor M, Brakenhoff R, van Houten VM, et al. Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001;7:1523-1532.
- Torezan L. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Ullrich S, Kripke M, Ananthaswamy H. Mechanisms underlying UV-induced immune suppression: implications for sunscreen design. Exp Dermatol. 2002;11:1-4.
- de Gruijl FR. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 2000;319:359-366.
- Brash DE, Ziegler A, Jonason AS, et al. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1996;1:136-142.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Rossi R, Mori M, Lotti T. Actinic keratosis. Int J Dermatol. 2007;46:895-904.
- Ziegler A, Jonason AS, Leffel DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773-776.
- Cockerell C. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42:11-17.
- Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci. 1996;93:14025-14029.
- Kanjilal S, Strom SS, Clayman GL, et al. p53 Mutations in nonmelanoma skin cancer of the head and neck: molecular evidence for field cancerization. Cancer Res. 1995;55:3604-3609.
- Brennan JA, Mao L, Hruban RH, et al. Molecular assessment of histopathological staging in squamous cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braathen LR, Morton CA, Basset-Seguin N, et al. Photodynamic therapy for skin field cancerization: an international consensus. International Society for Photodynamic Therapy in Dermatology. J Eur Acad Dermatol Venereol. 2012;26:1063-1066.
- Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6:963-968.
- Braakhuis B, Tabor M, Kummer J, et al. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63:1727-1730.
- Stern R, Bolshakov S, Nataraj A, et al. p53 Mutation in nonmelanoma skin cancers occurring in psoralen ultraviolet A-treated patients: evidence for heterogeneity and field cancerization. J Invest Dermatol. 2002;119:522-526.
- Tabor M, Brakenhoff R, van Houten VM, et al. Persistence of genetically altered fields in head and neck cancer patients: biological and clinical implications. Clin Cancer Res. 2001;7:1523-1532.
- Torezan L. Cutaneous field cancerization: clinical, histopathological and therapeutic aspects. An Bras Dermatol. 2013;88:775-786.
- Ullrich S, Kripke M, Ananthaswamy H. Mechanisms underlying UV-induced immune suppression: implications for sunscreen design. Exp Dermatol. 2002;11:1-4.
- de Gruijl FR. Photocarcinogenesis: UVA vs UVB. Methods Enzymol. 2000;319:359-366.
- Brash DE, Ziegler A, Jonason AS, et al. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1996;1:136-142.
- Ackerman AB, Mones JM. Solar (actinic) keratosis is squamous cell carcinoma. Br J Dermatol. 2006;155:9-22.
- Rossi R, Mori M, Lotti T. Actinic keratosis. Int J Dermatol. 2007;46:895-904.
- Ziegler A, Jonason AS, Leffel DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773-776.
- Cockerell C. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42:11-17.
- Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci. 1996;93:14025-14029.
- Kanjilal S, Strom SS, Clayman GL, et al. p53 Mutations in nonmelanoma skin cancer of the head and neck: molecular evidence for field cancerization. Cancer Res. 1995;55:3604-3609.
- Brennan JA, Mao L, Hruban RH, et al. Molecular assessment of histopathological staging in squamous cell carcinoma of the head and neck. N Engl J Med. 1995;332:429-435.
- Braathen LR, Morton CA, Basset-Seguin N, et al. Photodynamic therapy for skin field cancerization: an international consensus. International Society for Photodynamic Therapy in Dermatology. J Eur Acad Dermatol Venereol. 2012;26:1063-1066.
Practice Points
- Clinically apparent and subclinical actinic keratoses usually are present in patients, a concept known as field cancerization, and it is important to treat both types of lesions.
- Actinic keratoses are present in the field of cutaneous malignancies, including basal cell carcinoma, squamous cell carcinoma, and melanoma.