User login
Testosterone gel increases LV mass in older men
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.

“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.
“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
PHILADELPHIA – Testosterone gel for treatment of hypogonadism in older men boosted their left ventricular mass by 3.5% in a single year in the multicenter, double-blind, placebo-controlled Testosterone Cardiovascular Trial, although the clinical implications of this impressive increase remain unclear, Elizabeth Hutchins, MD, reported at the American Heart Association scientific sessions.
“I do think these results should be considered as part of the safety profile for testosterone gel and also represent an interesting and understudied area for future research,” said Dr. Hutchins, a hospitalist affiliated with the Los Angeles Biomedical Research Center at Harbor-UCLA Medical Center.
The Testosterone Cardiovascular Trial was one of seven coordinated placebo-controlled, double-blind clinical trials of the impact of raising serum testosterone levels in older men with low testosterone. Some results of what are known as the TTrials have previously been reported (Endocr Rev. 2018 Jun 1;39[3]:369-86).
Dr. Hutchins presented new findings on the effect of treatment with 1% topical testosterone gel on body surface area–indexed left ventricular mass. The trial utilized a widely prescribed, commercially available product known as AndroGel. The study included 123 men over age 65 with low serum testosterone and coronary CT angiography images obtained at baseline and again after 1 year of double-blind testosterone gel or placebo. More than 80% of the men were above age 75, half were obese, more than two-thirds had hypertension, and 30% had diabetes.
The men initially applied 5 g of the testosterone gel daily, providing 15 mg/day of testosterone, with subsequent dosing adjustments as needed based on serum testosterone levels measured at a central laboratory. Participants were evaluated in office visits with serum testosterone measurements every 3 months. Testosterone levels in the men assigned to active treatment quickly rose to normal range and stayed there for the full 12 months, while the placebo-treated controls continued to have below-normal testosterone throughout the trial.
The key study finding was that LV mass indexed to body surface area rose significantly in the testosterone gel group, from an average of 71.5 g/m2 at baseline to 74.8 g/m2 at 1 year. That’s a statistically significant 3.5% increase. In contrast, LV mass remained flat across the year in controls: 73.8 g/m2 at baseline and 73.3 g/m2 at 12 months.
There was, however, no change over time in left or right atrial or ventricular chamber volumes in the testosterone gel recipients, nor in the controls.
Session comoderator Eric D. Peterson, MD, professor of medicine and a cardiologist at Duke University in Durham, N.C., said that “this is a very important topic,” then posed a provocative question to Dr. Hutchins: “If the intervention had been running instead of testosterone gel, would the results have looked similar, and would you be concluding that there should be a warning around the use of running?”
Dr. Hutchins replied that she’s given that question much thought.
“Of course, exercise leads to LV hypertrophy and we consider that to be good muscle, and high blood pressure leads to LV hypertrophy and we consider that bad muscle. So which one is it in this case? From what I can find in the literature, it seems that incremental increases in LV mass in the absence of being an athlete are deleterious. But I think we would need outcomes-based research to really answer that question,” she said.
Dr. Hutchins noted that this was the first-ever randomized controlled trial to measure the effect of testosterone therapy on LV mass in humans. The documented increase achieved with 1 year of testosterone gel doesn’t come close to reaching the threshold of LV hypertrophy, which is about 125 g/m2 for men. But evidence from animal and observational human studies suggests that even in the absence of LV hypertrophy, increases in LV mass are associated with increased mortality, she added.
She reported having no financial conflicts regarding her study, sponsored by the National Institutes of Health.
SOURCE: Hutchins E. AHA 2019, Session FS.AOS.04.
REPORTING FROM AHA 2019
SPRINT-type BP control provides up to 3 years of additional life
PHILADELPHIA – in age-dependent fashion, compared with the older target standard BP, according to a novel analysis of data from the landmark SPRINT trial.

SPRINT randomized 9,361 hypertensive patients aged 50 years or older with at least one additional cardiovascular risk factor to intensive control with a target systolic BP of less than 120 mm Hg or to the then-standard target of less than 140 mm Hg. The trial was stopped early for ethical reasons when an interim analysis showed intensive control was associated with a 27% reduction in mortality. But that 27% reduction in mortality risk is a tough concept for many patients to interpret in practical terms, Muthiah Vaduganathan, MD, observed at the American Heart Association scientific sessions.
So he and his coinvestigators sliced and diced the mountainous SPRINT data in a novel way, using an actuarial statistical analysis.
“These actuarial data from SPRINT support the survival benefits of intensive blood pressure control, especially when initiated in middle-aged, high-risk adults. Our analysis really reaffirms the original SPRINT trial results [N Engl J Med. 2015 Nov 26;373(22):2103-16] and helps present them in an alternative format that can potentially be more easily communicated to clinicians, patients, and the public at large,” explained Dr. Vaduganathan, a cardiologist at Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
The impact of intensive BP control on residual survival was magnified in patients who were younger, since they intrinsically have a longer expected survival and will apply the antihypertensive regimen over a longer period. For example, the actuarial analysis concluded that the mean survival benefit of starting intensive BP lowering, rather than settling for a target systolic BP of less than 140 mm Hg starting at age 50 years, was 3 additional years of life, as compared with 1.1 additional years in 65-year-olds and 0.5 years in patients aged 85 years or older. The same approach can be applied to patients at any individual age from 50 to 95 years at the time of enrollment.
“This is very helpful in conveying messages to individual patients. Often if you tell a patient: ‘Your risk is going to go down by 27%,’ it’s tough for them to recognize what the baseline is and if that actually applied to them. So this may personalize that decision-making conversation,” according to the cardiologist.
One audience member commented that this SPRINT analysis might actually underestimate the true survival advantage of intensive BP lowering. He noted that SPRINT, which was halted after an average of 3.3 years, didn’t show a significant benefit for intensive BP lowering in terms of stroke reduction, whereas the ACCORD trial did, but that benefit didn’t occur until after 3 years into the study (Hypertension. 2018 Aug;72[2]:323-30).
Dr. Vaduganathan conceded that’s a limitation of his analysis.
“The assumption we’ve used is that long-term cardiovascular benefits are going to be as seen in the trial, but since SPRINT was stopped early, some benefits may be exaggerated and some may not have been observed yet,” he agreed.
Another audience member observed, “I think a lot of patients will think: ‘Okay, you’re tacking on a year at the end, when I’m going to be 89 and demented.’ The National Institute on Aging is focusing a lot more now on nondisabled life expectancy or healthy life expectancy.’”
Dr. Vaduganathan offered a degree of reassurance on this score. Because of time limitations, he said, he only presented the life expectancy results. But he and his coworkers have performed the same actuarial analysis of the SPRINT data for other endpoints related to freedom from various forms of disease or disability and found a consistent effect: Intensive BP control was associated with a longer time to onset of morbidity.
SPRINT was sponsored primarily by the National Heart, Lung, and Blood Institute. Dr. Vaduganathan reported that he receives research support from/and or serves on advisory boards for Amgen, AstraZeneca, Baxter Healthcare, Bayer, Boehringer Ingelheim, and Relypsa.
SOURCE: Vaduganathan M et al. AHA 2019, Abstract MDP233.
PHILADELPHIA – in age-dependent fashion, compared with the older target standard BP, according to a novel analysis of data from the landmark SPRINT trial.
SPRINT randomized 9,361 hypertensive patients aged 50 years or older with at least one additional cardiovascular risk factor to intensive control with a target systolic BP of less than 120 mm Hg or to the then-standard target of less than 140 mm Hg. The trial was stopped early for ethical reasons when an interim analysis showed intensive control was associated with a 27% reduction in mortality. But that 27% reduction in mortality risk is a tough concept for many patients to interpret in practical terms, Muthiah Vaduganathan, MD, observed at the American Heart Association scientific sessions.
So he and his coinvestigators sliced and diced the mountainous SPRINT data in a novel way, using an actuarial statistical analysis.
“These actuarial data from SPRINT support the survival benefits of intensive blood pressure control, especially when initiated in middle-aged, high-risk adults. Our analysis really reaffirms the original SPRINT trial results [N Engl J Med. 2015 Nov 26;373(22):2103-16] and helps present them in an alternative format that can potentially be more easily communicated to clinicians, patients, and the public at large,” explained Dr. Vaduganathan, a cardiologist at Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
The impact of intensive BP control on residual survival was magnified in patients who were younger, since they intrinsically have a longer expected survival and will apply the antihypertensive regimen over a longer period. For example, the actuarial analysis concluded that the mean survival benefit of starting intensive BP lowering, rather than settling for a target systolic BP of less than 140 mm Hg starting at age 50 years, was 3 additional years of life, as compared with 1.1 additional years in 65-year-olds and 0.5 years in patients aged 85 years or older. The same approach can be applied to patients at any individual age from 50 to 95 years at the time of enrollment.
“This is very helpful in conveying messages to individual patients. Often if you tell a patient: ‘Your risk is going to go down by 27%,’ it’s tough for them to recognize what the baseline is and if that actually applied to them. So this may personalize that decision-making conversation,” according to the cardiologist.
One audience member commented that this SPRINT analysis might actually underestimate the true survival advantage of intensive BP lowering. He noted that SPRINT, which was halted after an average of 3.3 years, didn’t show a significant benefit for intensive BP lowering in terms of stroke reduction, whereas the ACCORD trial did, but that benefit didn’t occur until after 3 years into the study (Hypertension. 2018 Aug;72[2]:323-30).
Dr. Vaduganathan conceded that’s a limitation of his analysis.
“The assumption we’ve used is that long-term cardiovascular benefits are going to be as seen in the trial, but since SPRINT was stopped early, some benefits may be exaggerated and some may not have been observed yet,” he agreed.
Another audience member observed, “I think a lot of patients will think: ‘Okay, you’re tacking on a year at the end, when I’m going to be 89 and demented.’ The National Institute on Aging is focusing a lot more now on nondisabled life expectancy or healthy life expectancy.’”
Dr. Vaduganathan offered a degree of reassurance on this score. Because of time limitations, he said, he only presented the life expectancy results. But he and his coworkers have performed the same actuarial analysis of the SPRINT data for other endpoints related to freedom from various forms of disease or disability and found a consistent effect: Intensive BP control was associated with a longer time to onset of morbidity.
SPRINT was sponsored primarily by the National Heart, Lung, and Blood Institute. Dr. Vaduganathan reported that he receives research support from/and or serves on advisory boards for Amgen, AstraZeneca, Baxter Healthcare, Bayer, Boehringer Ingelheim, and Relypsa.
SOURCE: Vaduganathan M et al. AHA 2019, Abstract MDP233.
PHILADELPHIA – in age-dependent fashion, compared with the older target standard BP, according to a novel analysis of data from the landmark SPRINT trial.
SPRINT randomized 9,361 hypertensive patients aged 50 years or older with at least one additional cardiovascular risk factor to intensive control with a target systolic BP of less than 120 mm Hg or to the then-standard target of less than 140 mm Hg. The trial was stopped early for ethical reasons when an interim analysis showed intensive control was associated with a 27% reduction in mortality. But that 27% reduction in mortality risk is a tough concept for many patients to interpret in practical terms, Muthiah Vaduganathan, MD, observed at the American Heart Association scientific sessions.
So he and his coinvestigators sliced and diced the mountainous SPRINT data in a novel way, using an actuarial statistical analysis.
“These actuarial data from SPRINT support the survival benefits of intensive blood pressure control, especially when initiated in middle-aged, high-risk adults. Our analysis really reaffirms the original SPRINT trial results [N Engl J Med. 2015 Nov 26;373(22):2103-16] and helps present them in an alternative format that can potentially be more easily communicated to clinicians, patients, and the public at large,” explained Dr. Vaduganathan, a cardiologist at Brigham and Women’s Hospital and Harvard Medical School, both in Boston.
The impact of intensive BP control on residual survival was magnified in patients who were younger, since they intrinsically have a longer expected survival and will apply the antihypertensive regimen over a longer period. For example, the actuarial analysis concluded that the mean survival benefit of starting intensive BP lowering, rather than settling for a target systolic BP of less than 140 mm Hg starting at age 50 years, was 3 additional years of life, as compared with 1.1 additional years in 65-year-olds and 0.5 years in patients aged 85 years or older. The same approach can be applied to patients at any individual age from 50 to 95 years at the time of enrollment.
“This is very helpful in conveying messages to individual patients. Often if you tell a patient: ‘Your risk is going to go down by 27%,’ it’s tough for them to recognize what the baseline is and if that actually applied to them. So this may personalize that decision-making conversation,” according to the cardiologist.
One audience member commented that this SPRINT analysis might actually underestimate the true survival advantage of intensive BP lowering. He noted that SPRINT, which was halted after an average of 3.3 years, didn’t show a significant benefit for intensive BP lowering in terms of stroke reduction, whereas the ACCORD trial did, but that benefit didn’t occur until after 3 years into the study (Hypertension. 2018 Aug;72[2]:323-30).
Dr. Vaduganathan conceded that’s a limitation of his analysis.
“The assumption we’ve used is that long-term cardiovascular benefits are going to be as seen in the trial, but since SPRINT was stopped early, some benefits may be exaggerated and some may not have been observed yet,” he agreed.
Another audience member observed, “I think a lot of patients will think: ‘Okay, you’re tacking on a year at the end, when I’m going to be 89 and demented.’ The National Institute on Aging is focusing a lot more now on nondisabled life expectancy or healthy life expectancy.’”
Dr. Vaduganathan offered a degree of reassurance on this score. Because of time limitations, he said, he only presented the life expectancy results. But he and his coworkers have performed the same actuarial analysis of the SPRINT data for other endpoints related to freedom from various forms of disease or disability and found a consistent effect: Intensive BP control was associated with a longer time to onset of morbidity.
SPRINT was sponsored primarily by the National Heart, Lung, and Blood Institute. Dr. Vaduganathan reported that he receives research support from/and or serves on advisory boards for Amgen, AstraZeneca, Baxter Healthcare, Bayer, Boehringer Ingelheim, and Relypsa.
SOURCE: Vaduganathan M et al. AHA 2019, Abstract MDP233.
REPORTING FROM AHA 2019
COACT: No benefit of immediate PCI for non–ST-elevation cardiac arrest at 1 year
PHILADELPHIA – Immediate coronary angiography following restoration of spontaneous circulation after out-of-hospital cardiac arrest without ST-elevation MI (STEMI) offered no survival benefit at 1 year, compared with a strategy of delaying angiography until after neurologic recovery, in the landmark COACT trial, Jorrit Lemkes, MD, reported at the American Heart Association scientific sessions.

Nor was immediate coronary angiography advantageous in terms of any of the numerous secondary long-term endpoints, including rates of MI, revascularization, hospitalization for heart failure, implantable cardioverter defibrillator (ICD) shocks, or quality of life measures (see graphic), according to Dr. Lemkes, an interventional cardiologist at Amsterdam University Medical Center.
COACT, a 552-patient randomized trial conducted at 19 Dutch centers, was the first-ever randomized trial to evaluate the role of immediate coronary angiography in patients resuscitated from out-of-hospital cardiac arrest with no signs of ST elevation on ECG. The study hypothesis was that this practice would result in improved survival and other outcomes. But such was not the case in the previously reported 90-day analysis (N Engl J Med. 2019 Apr 11;380[15]:1397-407). Nonetheless, the new 1-year results were eagerly awaited because observational data had suggested that immediate angiography after cardiac arrest without STEMI might provide a survival advantage that manifests late.
It now appears the observational studies were misleading. The 1-year survival rate was 61.4% in the immediate angioplasty group and similar at 64% with delayed angioplasty.
By way of background, Dr. Lemkes noted that both European and American guidelines give a class 1 recommendation to immediate coronary angiography with percutaneous coronary intervention (PCI) in patients who present with STEMI and cardiac arrest. It’s an endorsement grounded in compelling clinical trials evidence demonstrating that this practice reduces mortality and recurrent ischemia, salvages myocardium, and restores left ventricular function. In contrast, current guidelines offer only a tepid recommendation for immediate PCI in patients with cardiac arrest without STEMI because the only supporting evidence has been observational, with its inherent susceptibility to bias.
Discussant Joaquin E. Cigarroa, MD, said that the 1-year outcomes shouldn’t be surprising, since the 90-day results failed to show any between-group differences in myocardial injury or ischemia.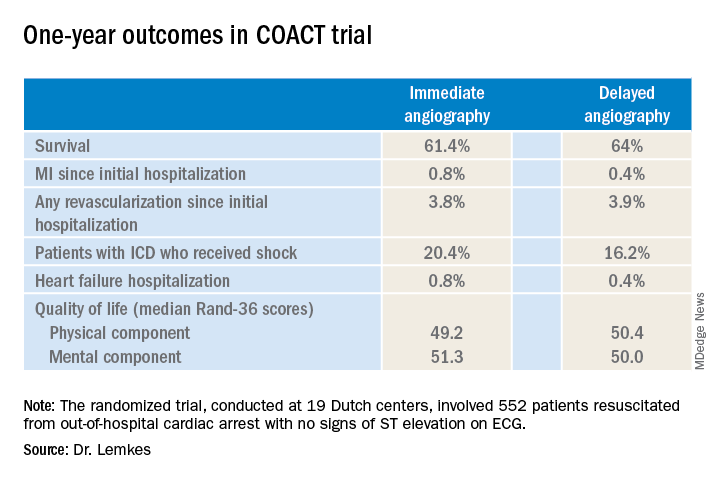
“At present, the results of COACT with regards to primary and secondary outcomes should guide practitioners that angiography remains essential, but that early angiography does not improve outcomes, compared to delayed angiography,” declared Dr. Cigarroa, chief of cardiology and professor of medicine at Oregon Health & Science University, Portland.
His choice of the words “at present” was key, as COACT won’t be the last word on the subject. Dr. Cigarroa believes that it’s critically important to understand the relatively narrow profile of the patients included in the study, because the results may or may not prove to be generalizable to the broader population of out-of-hospital cardiac arrest patients encountered in clinical practice. Nearly 80% of COACT participants had a witnessed arrest, the median time to basic life support was just 2 minutes, and the median time to return of spontaneous circulation was 15 minutes.
He urged his colleagues to stay tuned for reports from ongoing randomized trials examining the potential role of immediate angiography in broader populations of patients with out-of-hospital cardiac arrest without STEMI, including the Swedish DISCO (Direct or Subacute Coronary Angiography in Out-of-Hospital Cardiac Arrest) trial and the smaller University of Minnesota–sponsored ACCESS trial.
Dr. Lemkes reported having no financial conflicts regarding the COACT trial, funded by the Netherlands Heart Institute and unrestricted research grants from Biotronik and AstraZeneca.
PHILADELPHIA – Immediate coronary angiography following restoration of spontaneous circulation after out-of-hospital cardiac arrest without ST-elevation MI (STEMI) offered no survival benefit at 1 year, compared with a strategy of delaying angiography until after neurologic recovery, in the landmark COACT trial, Jorrit Lemkes, MD, reported at the American Heart Association scientific sessions.
Nor was immediate coronary angiography advantageous in terms of any of the numerous secondary long-term endpoints, including rates of MI, revascularization, hospitalization for heart failure, implantable cardioverter defibrillator (ICD) shocks, or quality of life measures (see graphic), according to Dr. Lemkes, an interventional cardiologist at Amsterdam University Medical Center.
COACT, a 552-patient randomized trial conducted at 19 Dutch centers, was the first-ever randomized trial to evaluate the role of immediate coronary angiography in patients resuscitated from out-of-hospital cardiac arrest with no signs of ST elevation on ECG. The study hypothesis was that this practice would result in improved survival and other outcomes. But such was not the case in the previously reported 90-day analysis (N Engl J Med. 2019 Apr 11;380[15]:1397-407). Nonetheless, the new 1-year results were eagerly awaited because observational data had suggested that immediate angiography after cardiac arrest without STEMI might provide a survival advantage that manifests late.
It now appears the observational studies were misleading. The 1-year survival rate was 61.4% in the immediate angioplasty group and similar at 64% with delayed angioplasty.
By way of background, Dr. Lemkes noted that both European and American guidelines give a class 1 recommendation to immediate coronary angiography with percutaneous coronary intervention (PCI) in patients who present with STEMI and cardiac arrest. It’s an endorsement grounded in compelling clinical trials evidence demonstrating that this practice reduces mortality and recurrent ischemia, salvages myocardium, and restores left ventricular function. In contrast, current guidelines offer only a tepid recommendation for immediate PCI in patients with cardiac arrest without STEMI because the only supporting evidence has been observational, with its inherent susceptibility to bias.
Discussant Joaquin E. Cigarroa, MD, said that the 1-year outcomes shouldn’t be surprising, since the 90-day results failed to show any between-group differences in myocardial injury or ischemia.
“At present, the results of COACT with regards to primary and secondary outcomes should guide practitioners that angiography remains essential, but that early angiography does not improve outcomes, compared to delayed angiography,” declared Dr. Cigarroa, chief of cardiology and professor of medicine at Oregon Health & Science University, Portland.
His choice of the words “at present” was key, as COACT won’t be the last word on the subject. Dr. Cigarroa believes that it’s critically important to understand the relatively narrow profile of the patients included in the study, because the results may or may not prove to be generalizable to the broader population of out-of-hospital cardiac arrest patients encountered in clinical practice. Nearly 80% of COACT participants had a witnessed arrest, the median time to basic life support was just 2 minutes, and the median time to return of spontaneous circulation was 15 minutes.
He urged his colleagues to stay tuned for reports from ongoing randomized trials examining the potential role of immediate angiography in broader populations of patients with out-of-hospital cardiac arrest without STEMI, including the Swedish DISCO (Direct or Subacute Coronary Angiography in Out-of-Hospital Cardiac Arrest) trial and the smaller University of Minnesota–sponsored ACCESS trial.
Dr. Lemkes reported having no financial conflicts regarding the COACT trial, funded by the Netherlands Heart Institute and unrestricted research grants from Biotronik and AstraZeneca.
PHILADELPHIA – Immediate coronary angiography following restoration of spontaneous circulation after out-of-hospital cardiac arrest without ST-elevation MI (STEMI) offered no survival benefit at 1 year, compared with a strategy of delaying angiography until after neurologic recovery, in the landmark COACT trial, Jorrit Lemkes, MD, reported at the American Heart Association scientific sessions.
Nor was immediate coronary angiography advantageous in terms of any of the numerous secondary long-term endpoints, including rates of MI, revascularization, hospitalization for heart failure, implantable cardioverter defibrillator (ICD) shocks, or quality of life measures (see graphic), according to Dr. Lemkes, an interventional cardiologist at Amsterdam University Medical Center.
COACT, a 552-patient randomized trial conducted at 19 Dutch centers, was the first-ever randomized trial to evaluate the role of immediate coronary angiography in patients resuscitated from out-of-hospital cardiac arrest with no signs of ST elevation on ECG. The study hypothesis was that this practice would result in improved survival and other outcomes. But such was not the case in the previously reported 90-day analysis (N Engl J Med. 2019 Apr 11;380[15]:1397-407). Nonetheless, the new 1-year results were eagerly awaited because observational data had suggested that immediate angiography after cardiac arrest without STEMI might provide a survival advantage that manifests late.
It now appears the observational studies were misleading. The 1-year survival rate was 61.4% in the immediate angioplasty group and similar at 64% with delayed angioplasty.
By way of background, Dr. Lemkes noted that both European and American guidelines give a class 1 recommendation to immediate coronary angiography with percutaneous coronary intervention (PCI) in patients who present with STEMI and cardiac arrest. It’s an endorsement grounded in compelling clinical trials evidence demonstrating that this practice reduces mortality and recurrent ischemia, salvages myocardium, and restores left ventricular function. In contrast, current guidelines offer only a tepid recommendation for immediate PCI in patients with cardiac arrest without STEMI because the only supporting evidence has been observational, with its inherent susceptibility to bias.
Discussant Joaquin E. Cigarroa, MD, said that the 1-year outcomes shouldn’t be surprising, since the 90-day results failed to show any between-group differences in myocardial injury or ischemia.
“At present, the results of COACT with regards to primary and secondary outcomes should guide practitioners that angiography remains essential, but that early angiography does not improve outcomes, compared to delayed angiography,” declared Dr. Cigarroa, chief of cardiology and professor of medicine at Oregon Health & Science University, Portland.
His choice of the words “at present” was key, as COACT won’t be the last word on the subject. Dr. Cigarroa believes that it’s critically important to understand the relatively narrow profile of the patients included in the study, because the results may or may not prove to be generalizable to the broader population of out-of-hospital cardiac arrest patients encountered in clinical practice. Nearly 80% of COACT participants had a witnessed arrest, the median time to basic life support was just 2 minutes, and the median time to return of spontaneous circulation was 15 minutes.
He urged his colleagues to stay tuned for reports from ongoing randomized trials examining the potential role of immediate angiography in broader populations of patients with out-of-hospital cardiac arrest without STEMI, including the Swedish DISCO (Direct or Subacute Coronary Angiography in Out-of-Hospital Cardiac Arrest) trial and the smaller University of Minnesota–sponsored ACCESS trial.
Dr. Lemkes reported having no financial conflicts regarding the COACT trial, funded by the Netherlands Heart Institute and unrestricted research grants from Biotronik and AstraZeneca.
REPORTING FROM AHA 2019
Pharmacist BP telemonitoring cut cardiovascular events, turned profit
PHILADELPHIA – A home blood pressure telemonitoring program featuring pharmacist management of patients with uncontrolled hypertension reduced cardiovascular events by half and was cost saving over the course of 5 years, even though the intervention ended after year 1, Karen L. Margolis, MD, reported at the American Heart Association scientific sessions.

“The return on investment was 126%. That means that for every dollar spent on the intervention, that dollar was recouped by $1.00 plus another $1.26,” explained Dr. Margolis, a general internist who serves as executive director for research at the HealthPartners Institute in Bloomington, Minn., and professor of medicine at the University of Minnesota, Minneapolis.
She presented 5-year follow-up data from the Hyperlink (Home Blood Pressure Telemonitoring and Case Management to Control Hypertension) study, a cluster randomized controlled trial involving 16 primary care clinics. Half of the clinics were randomized to the intervention, which entailed home blood pressure telemonitoring and pharmacist-led case management in collaboration with the primary care team. The other eight clinics provided usual care. The intervention portion of the trial, which lasted for 12 months, included 450 adults with uncontrolled hypertension as defined by repeated on-treatment blood pressure readings of 140/90 mm Hg or more. Participants’ baseline mean blood pressure was 148/85 mm Hg while on an average of one and a half antihypertensive drug classes. On average, pharmacists ended up adding one additional drug from a different antihypertensive drug class to achieve improved blood pressure control.
The details of the intervention and the short-term blood pressure results have previously been reported (JAMA. 2013 Jul 3;310[1]:46-56). Briefly, 6 months into the study, patients in the intervention arm averaged 11/6 mm Hg lower blood pressure than did the usual care controls. At 12 months – when the intervention ended – the between-group difference was similar at 10/5 mm Hg. At 18 months, the difference, while attenuated, remained significant at 7/3 mm Hg in favor of the intervention group. However, at 54 months, the intervention group’s advantage – a 3–mm Hg lower SBP and a 1–mm Hg lower DBP than in controls – was no longer significant.
The exciting new findings Dr. Margolis presented at the AHA scientific sessions focused on 5-year outcomes. Since HealthPartners is an integrated health care system, follow-up was essentially complete.
“None of the other telemetry studies I’m aware of have published anything on cardiovascular events. And we were somewhat surprised when we looked at our data to see fairly substantial differences in our primary outcome,” she noted.
That outcome was a composite of MI, stroke, heart failure, or cardiovascular death occurring over 5 years. The rate was 4.4% in the intervention group and nearly double at 8.6% in controls. That translated to a 51% relative risk reduction. The biggest difference was in stroke: 4 cases in the intervention arm, 12 in usual care controls.
The 5-year coronary revascularization rate was 5.3% in the intervention arm and 10.4% in controls, for a 52% relative risk reduction.
A major caveat regarding the Hyperlink trial was that, even at 450 patients and 5 years of follow-up, the study was underpowered to show significant differences in event rates, with P =.09 for the primary endpoint.
That being said, the financial results were striking. The intervention cost $1,511 per patient in 2017 U.S. dollars. The cost of treatment for major adverse cardiovascular events totaled $758,000 in the intervention group and $1,538,000 in usual care controls. That works out to $3,420 less per patient in the intervention arm. Offset by the cost of the intervention, that spells a net savings of $1,908 per patient achieved by implementing the year-long intervention. It’s a rare instance in health care of an intervention that actually makes money.
These results were unusual enough that Dr. Margolis and her coinvestigators decided to feed their wealth of SBP readings into a microsimulation model, which they ran 1,000 times. The model predicted – in light of the fact that patients in the intervention group were on average 2 years older than the controls were – that the expected reduction in the primary endpoint was 12% rather than the observed 51% relative risk reduction.
How to explain the discrepancy? The Hyperlink results could have been due to chance. Or it could be, Dr. Margolis surmised, that the pharmacists helped accomplish improvements in other cardiovascular risk factors, such as hyperlipidemia, smoking, or sedentary behavior. That’s unknown, since the investigators focused on changes in blood pressure only. Future studies of home telemonitoring and pharmacist case management of uncontrolled hypertension should be powered to detect significant differences in cardiovascular events and should track additional risk factors, she concluded.
She reported having no financial conflicts regarding the study.
SOURCE: Margolis KL. AHA 2019. Abstract MDP232.
PHILADELPHIA – A home blood pressure telemonitoring program featuring pharmacist management of patients with uncontrolled hypertension reduced cardiovascular events by half and was cost saving over the course of 5 years, even though the intervention ended after year 1, Karen L. Margolis, MD, reported at the American Heart Association scientific sessions.
“The return on investment was 126%. That means that for every dollar spent on the intervention, that dollar was recouped by $1.00 plus another $1.26,” explained Dr. Margolis, a general internist who serves as executive director for research at the HealthPartners Institute in Bloomington, Minn., and professor of medicine at the University of Minnesota, Minneapolis.
She presented 5-year follow-up data from the Hyperlink (Home Blood Pressure Telemonitoring and Case Management to Control Hypertension) study, a cluster randomized controlled trial involving 16 primary care clinics. Half of the clinics were randomized to the intervention, which entailed home blood pressure telemonitoring and pharmacist-led case management in collaboration with the primary care team. The other eight clinics provided usual care. The intervention portion of the trial, which lasted for 12 months, included 450 adults with uncontrolled hypertension as defined by repeated on-treatment blood pressure readings of 140/90 mm Hg or more. Participants’ baseline mean blood pressure was 148/85 mm Hg while on an average of one and a half antihypertensive drug classes. On average, pharmacists ended up adding one additional drug from a different antihypertensive drug class to achieve improved blood pressure control.
The details of the intervention and the short-term blood pressure results have previously been reported (JAMA. 2013 Jul 3;310[1]:46-56). Briefly, 6 months into the study, patients in the intervention arm averaged 11/6 mm Hg lower blood pressure than did the usual care controls. At 12 months – when the intervention ended – the between-group difference was similar at 10/5 mm Hg. At 18 months, the difference, while attenuated, remained significant at 7/3 mm Hg in favor of the intervention group. However, at 54 months, the intervention group’s advantage – a 3–mm Hg lower SBP and a 1–mm Hg lower DBP than in controls – was no longer significant.
The exciting new findings Dr. Margolis presented at the AHA scientific sessions focused on 5-year outcomes. Since HealthPartners is an integrated health care system, follow-up was essentially complete.
“None of the other telemetry studies I’m aware of have published anything on cardiovascular events. And we were somewhat surprised when we looked at our data to see fairly substantial differences in our primary outcome,” she noted.
That outcome was a composite of MI, stroke, heart failure, or cardiovascular death occurring over 5 years. The rate was 4.4% in the intervention group and nearly double at 8.6% in controls. That translated to a 51% relative risk reduction. The biggest difference was in stroke: 4 cases in the intervention arm, 12 in usual care controls.
The 5-year coronary revascularization rate was 5.3% in the intervention arm and 10.4% in controls, for a 52% relative risk reduction.
A major caveat regarding the Hyperlink trial was that, even at 450 patients and 5 years of follow-up, the study was underpowered to show significant differences in event rates, with P =.09 for the primary endpoint.
That being said, the financial results were striking. The intervention cost $1,511 per patient in 2017 U.S. dollars. The cost of treatment for major adverse cardiovascular events totaled $758,000 in the intervention group and $1,538,000 in usual care controls. That works out to $3,420 less per patient in the intervention arm. Offset by the cost of the intervention, that spells a net savings of $1,908 per patient achieved by implementing the year-long intervention. It’s a rare instance in health care of an intervention that actually makes money.
These results were unusual enough that Dr. Margolis and her coinvestigators decided to feed their wealth of SBP readings into a microsimulation model, which they ran 1,000 times. The model predicted – in light of the fact that patients in the intervention group were on average 2 years older than the controls were – that the expected reduction in the primary endpoint was 12% rather than the observed 51% relative risk reduction.
How to explain the discrepancy? The Hyperlink results could have been due to chance. Or it could be, Dr. Margolis surmised, that the pharmacists helped accomplish improvements in other cardiovascular risk factors, such as hyperlipidemia, smoking, or sedentary behavior. That’s unknown, since the investigators focused on changes in blood pressure only. Future studies of home telemonitoring and pharmacist case management of uncontrolled hypertension should be powered to detect significant differences in cardiovascular events and should track additional risk factors, she concluded.
She reported having no financial conflicts regarding the study.
SOURCE: Margolis KL. AHA 2019. Abstract MDP232.
PHILADELPHIA – A home blood pressure telemonitoring program featuring pharmacist management of patients with uncontrolled hypertension reduced cardiovascular events by half and was cost saving over the course of 5 years, even though the intervention ended after year 1, Karen L. Margolis, MD, reported at the American Heart Association scientific sessions.
“The return on investment was 126%. That means that for every dollar spent on the intervention, that dollar was recouped by $1.00 plus another $1.26,” explained Dr. Margolis, a general internist who serves as executive director for research at the HealthPartners Institute in Bloomington, Minn., and professor of medicine at the University of Minnesota, Minneapolis.
She presented 5-year follow-up data from the Hyperlink (Home Blood Pressure Telemonitoring and Case Management to Control Hypertension) study, a cluster randomized controlled trial involving 16 primary care clinics. Half of the clinics were randomized to the intervention, which entailed home blood pressure telemonitoring and pharmacist-led case management in collaboration with the primary care team. The other eight clinics provided usual care. The intervention portion of the trial, which lasted for 12 months, included 450 adults with uncontrolled hypertension as defined by repeated on-treatment blood pressure readings of 140/90 mm Hg or more. Participants’ baseline mean blood pressure was 148/85 mm Hg while on an average of one and a half antihypertensive drug classes. On average, pharmacists ended up adding one additional drug from a different antihypertensive drug class to achieve improved blood pressure control.
The details of the intervention and the short-term blood pressure results have previously been reported (JAMA. 2013 Jul 3;310[1]:46-56). Briefly, 6 months into the study, patients in the intervention arm averaged 11/6 mm Hg lower blood pressure than did the usual care controls. At 12 months – when the intervention ended – the between-group difference was similar at 10/5 mm Hg. At 18 months, the difference, while attenuated, remained significant at 7/3 mm Hg in favor of the intervention group. However, at 54 months, the intervention group’s advantage – a 3–mm Hg lower SBP and a 1–mm Hg lower DBP than in controls – was no longer significant.
The exciting new findings Dr. Margolis presented at the AHA scientific sessions focused on 5-year outcomes. Since HealthPartners is an integrated health care system, follow-up was essentially complete.
“None of the other telemetry studies I’m aware of have published anything on cardiovascular events. And we were somewhat surprised when we looked at our data to see fairly substantial differences in our primary outcome,” she noted.
That outcome was a composite of MI, stroke, heart failure, or cardiovascular death occurring over 5 years. The rate was 4.4% in the intervention group and nearly double at 8.6% in controls. That translated to a 51% relative risk reduction. The biggest difference was in stroke: 4 cases in the intervention arm, 12 in usual care controls.
The 5-year coronary revascularization rate was 5.3% in the intervention arm and 10.4% in controls, for a 52% relative risk reduction.
A major caveat regarding the Hyperlink trial was that, even at 450 patients and 5 years of follow-up, the study was underpowered to show significant differences in event rates, with P =.09 for the primary endpoint.
That being said, the financial results were striking. The intervention cost $1,511 per patient in 2017 U.S. dollars. The cost of treatment for major adverse cardiovascular events totaled $758,000 in the intervention group and $1,538,000 in usual care controls. That works out to $3,420 less per patient in the intervention arm. Offset by the cost of the intervention, that spells a net savings of $1,908 per patient achieved by implementing the year-long intervention. It’s a rare instance in health care of an intervention that actually makes money.
These results were unusual enough that Dr. Margolis and her coinvestigators decided to feed their wealth of SBP readings into a microsimulation model, which they ran 1,000 times. The model predicted – in light of the fact that patients in the intervention group were on average 2 years older than the controls were – that the expected reduction in the primary endpoint was 12% rather than the observed 51% relative risk reduction.
How to explain the discrepancy? The Hyperlink results could have been due to chance. Or it could be, Dr. Margolis surmised, that the pharmacists helped accomplish improvements in other cardiovascular risk factors, such as hyperlipidemia, smoking, or sedentary behavior. That’s unknown, since the investigators focused on changes in blood pressure only. Future studies of home telemonitoring and pharmacist case management of uncontrolled hypertension should be powered to detect significant differences in cardiovascular events and should track additional risk factors, she concluded.
She reported having no financial conflicts regarding the study.
SOURCE: Margolis KL. AHA 2019. Abstract MDP232.
REPORTING FROM THE AHA SCIENTIFIC SESSIONS
Consider hyperbaric oxygen for inflammatory ileal pouchitis
SAN ANTONIO – , Hamna Fahad, MD, reported at the annual meeting of the American College of Gastroenterology.

Dr. Fahad, of the Cleveland Clinic, presented a retrospective case series of 21 consecutive clinic patients who presented with inflammatory bowel disease, a surgically created ileal pouch–anal anastomosis, and medically refractory pouchitis. All patients received 30 hyperbaric oxygen treatment sessions, each an hour long, over the course of 2 months. This intensive regimen worked out to 3-5 sessions per week involving 100% oxygen pressurized to 2.4-3.0 ATA.
Overall, 19 of 21 patients experienced improvement in their modified Pouchitis Disease Activity Index (mPDAI) score. The mean total mPDAI at baseline was 8.71, improving significantly to 5 post treatment. The mPDAI symptoms subscore also showed significant improvement in response to a course of hyperbaric oxygen therapy, decreasing from 4 points to 2. The cuff subscore fell from 3 to 0, and the pouch body subscore improved from 3 to 2.
Thirteen of 21 patients reported subjective symptomatic improvement in stool frequency, bleeding, urgency, and fevers, including 6 with complete symptomatic remission. Seventeen patients demonstrated significant endoscopic improvement upon blinded assessment. Seven of 9 patients with fistulae experienced healing of the fistula tract.
The treatment entailed no side effects. However, the benefits weren’t uniformly durable. Several patients underwent a second 30-session round of hyperbaric oxygen therapy within a year because of recurrent pouchitis symptoms refractory to corticosteroids, biologics, and other medications.
Dr. Fahad said the mechanism of benefit for hyperbaric oxygen in the treatment of chronic inflammatory pouchitis is probably severalfold: reversal of a disordered microbiome through inhibition of the growth of anaerobes, reduced production of tumor necrosis factor–alpha and other inflammatory cytokines, and increased plasma oxygen, which reduces ischemia at the tissue level, thereby promoting tissue healing.
Audience members had a practical question: How can they get this treatment paid for? One gastroenterologist said she has encountered considerable payer resistance when she has sought coverage of hyperbaric oxygen for patients with ulcerative colitis and fistulae, even though there is already published evidence of benefit. But Dr. Fahad’s groundbreaking study provides the first such evidence in pouchitis. So how did she and her coworkers do it? Eighty percent of the pouchitis patients obtained payer approval only upon appeal, which was readily granted, she explained.
Dr. Fahad reported having no financial conflicts regarding her study, conducted without commercial support.
SOURCE: Fahad H. ACG 2019 Abstract 38.
SAN ANTONIO – , Hamna Fahad, MD, reported at the annual meeting of the American College of Gastroenterology.
Dr. Fahad, of the Cleveland Clinic, presented a retrospective case series of 21 consecutive clinic patients who presented with inflammatory bowel disease, a surgically created ileal pouch–anal anastomosis, and medically refractory pouchitis. All patients received 30 hyperbaric oxygen treatment sessions, each an hour long, over the course of 2 months. This intensive regimen worked out to 3-5 sessions per week involving 100% oxygen pressurized to 2.4-3.0 ATA.
Overall, 19 of 21 patients experienced improvement in their modified Pouchitis Disease Activity Index (mPDAI) score. The mean total mPDAI at baseline was 8.71, improving significantly to 5 post treatment. The mPDAI symptoms subscore also showed significant improvement in response to a course of hyperbaric oxygen therapy, decreasing from 4 points to 2. The cuff subscore fell from 3 to 0, and the pouch body subscore improved from 3 to 2.
Thirteen of 21 patients reported subjective symptomatic improvement in stool frequency, bleeding, urgency, and fevers, including 6 with complete symptomatic remission. Seventeen patients demonstrated significant endoscopic improvement upon blinded assessment. Seven of 9 patients with fistulae experienced healing of the fistula tract.
The treatment entailed no side effects. However, the benefits weren’t uniformly durable. Several patients underwent a second 30-session round of hyperbaric oxygen therapy within a year because of recurrent pouchitis symptoms refractory to corticosteroids, biologics, and other medications.
Dr. Fahad said the mechanism of benefit for hyperbaric oxygen in the treatment of chronic inflammatory pouchitis is probably severalfold: reversal of a disordered microbiome through inhibition of the growth of anaerobes, reduced production of tumor necrosis factor–alpha and other inflammatory cytokines, and increased plasma oxygen, which reduces ischemia at the tissue level, thereby promoting tissue healing.
Audience members had a practical question: How can they get this treatment paid for? One gastroenterologist said she has encountered considerable payer resistance when she has sought coverage of hyperbaric oxygen for patients with ulcerative colitis and fistulae, even though there is already published evidence of benefit. But Dr. Fahad’s groundbreaking study provides the first such evidence in pouchitis. So how did she and her coworkers do it? Eighty percent of the pouchitis patients obtained payer approval only upon appeal, which was readily granted, she explained.
Dr. Fahad reported having no financial conflicts regarding her study, conducted without commercial support.
SOURCE: Fahad H. ACG 2019 Abstract 38.
SAN ANTONIO – , Hamna Fahad, MD, reported at the annual meeting of the American College of Gastroenterology.
Dr. Fahad, of the Cleveland Clinic, presented a retrospective case series of 21 consecutive clinic patients who presented with inflammatory bowel disease, a surgically created ileal pouch–anal anastomosis, and medically refractory pouchitis. All patients received 30 hyperbaric oxygen treatment sessions, each an hour long, over the course of 2 months. This intensive regimen worked out to 3-5 sessions per week involving 100% oxygen pressurized to 2.4-3.0 ATA.
Overall, 19 of 21 patients experienced improvement in their modified Pouchitis Disease Activity Index (mPDAI) score. The mean total mPDAI at baseline was 8.71, improving significantly to 5 post treatment. The mPDAI symptoms subscore also showed significant improvement in response to a course of hyperbaric oxygen therapy, decreasing from 4 points to 2. The cuff subscore fell from 3 to 0, and the pouch body subscore improved from 3 to 2.
Thirteen of 21 patients reported subjective symptomatic improvement in stool frequency, bleeding, urgency, and fevers, including 6 with complete symptomatic remission. Seventeen patients demonstrated significant endoscopic improvement upon blinded assessment. Seven of 9 patients with fistulae experienced healing of the fistula tract.
The treatment entailed no side effects. However, the benefits weren’t uniformly durable. Several patients underwent a second 30-session round of hyperbaric oxygen therapy within a year because of recurrent pouchitis symptoms refractory to corticosteroids, biologics, and other medications.
Dr. Fahad said the mechanism of benefit for hyperbaric oxygen in the treatment of chronic inflammatory pouchitis is probably severalfold: reversal of a disordered microbiome through inhibition of the growth of anaerobes, reduced production of tumor necrosis factor–alpha and other inflammatory cytokines, and increased plasma oxygen, which reduces ischemia at the tissue level, thereby promoting tissue healing.
Audience members had a practical question: How can they get this treatment paid for? One gastroenterologist said she has encountered considerable payer resistance when she has sought coverage of hyperbaric oxygen for patients with ulcerative colitis and fistulae, even though there is already published evidence of benefit. But Dr. Fahad’s groundbreaking study provides the first such evidence in pouchitis. So how did she and her coworkers do it? Eighty percent of the pouchitis patients obtained payer approval only upon appeal, which was readily granted, she explained.
Dr. Fahad reported having no financial conflicts regarding her study, conducted without commercial support.
SOURCE: Fahad H. ACG 2019 Abstract 38.
REPORTING FROM ACG 2019
Recognize early window of opportunity in hidradenitis suppurativa
MADRID – Antonio Martorell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.

This distinction is critical in recognizing a window of opportunity in the management of hidradenitis suppurativa (HS): The period early in the disease course when medical therapy alone can be life-changing.
“The window of opportunity is an old concept in gastroenterology, but it’s a new idea in dermatology: It’s the moment in which the patient can have the best results with medical control of inflammation, before progression to tissue scarring has occurred,” explained Dr. Martorell, a dermatologist at the Hospital of Manises in Valencia, Spain, and coauthor of recent HS treatment recommendations by an international expert panel (J Eur Acad Dermatol Venereol. 2019 Jan;33[1]:19-31).
Symptoms in patients with HS can be caused by either dynamic or static lesions. Dynamic lesions arise directly from acute inflammation that can be treated with antibiotics and immunomodulatory therapy. Static lesions are associated with tissue scarring secondary to inflammatory activity and generally benefit only from surgery.
Dynamic lesions consist of nodules, abscesses, and some but not all fistulae. Although the traditional view among dermatologists has been that fistulae simply don’t respond to medical therapy and must be treated surgically, Dr. Martorell and coinvestigators have recently demonstrated in a retrospective study of 117 fistulae in 40 patients that ultrasound was useful in distinguishing four fistular subtypes, two of which responded reasonably well to medical management.
What the investigators call Type A or dermal fistulae are by definition not connected to tunnels. In Dr. Martorell’s study, they had a 95% complete resolution rate after 6 months of various medications. Dermoepidermal fistulae tunnel through the dermis to the epidermis; they had a 65% complete resolution rate. In contrast, Type C or complex fistulae, identified by the ultrasound finding of multiple tunnels extending through the dermis into underlying fat tissue, had no significant response to medical management. Neither did Type D fistulae, which are essentially Type C lesions with scarring (Dermatol Surg. 2019 Oct;45[10]:1237-44).
Thus, ultrasound can have an important impact on patient management and the decision to opt for a combined medical/surgical approach.
“It’s important to apply the HS severity scores and complete the clinical exam, but it’s also important to use ultrasound or another imaging technique,” Dr. Martorell concluded.
MADRID – Antonio Martorell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
This distinction is critical in recognizing a window of opportunity in the management of hidradenitis suppurativa (HS): The period early in the disease course when medical therapy alone can be life-changing.
“The window of opportunity is an old concept in gastroenterology, but it’s a new idea in dermatology: It’s the moment in which the patient can have the best results with medical control of inflammation, before progression to tissue scarring has occurred,” explained Dr. Martorell, a dermatologist at the Hospital of Manises in Valencia, Spain, and coauthor of recent HS treatment recommendations by an international expert panel (J Eur Acad Dermatol Venereol. 2019 Jan;33[1]:19-31).
Symptoms in patients with HS can be caused by either dynamic or static lesions. Dynamic lesions arise directly from acute inflammation that can be treated with antibiotics and immunomodulatory therapy. Static lesions are associated with tissue scarring secondary to inflammatory activity and generally benefit only from surgery.
Dynamic lesions consist of nodules, abscesses, and some but not all fistulae. Although the traditional view among dermatologists has been that fistulae simply don’t respond to medical therapy and must be treated surgically, Dr. Martorell and coinvestigators have recently demonstrated in a retrospective study of 117 fistulae in 40 patients that ultrasound was useful in distinguishing four fistular subtypes, two of which responded reasonably well to medical management.
What the investigators call Type A or dermal fistulae are by definition not connected to tunnels. In Dr. Martorell’s study, they had a 95% complete resolution rate after 6 months of various medications. Dermoepidermal fistulae tunnel through the dermis to the epidermis; they had a 65% complete resolution rate. In contrast, Type C or complex fistulae, identified by the ultrasound finding of multiple tunnels extending through the dermis into underlying fat tissue, had no significant response to medical management. Neither did Type D fistulae, which are essentially Type C lesions with scarring (Dermatol Surg. 2019 Oct;45[10]:1237-44).
Thus, ultrasound can have an important impact on patient management and the decision to opt for a combined medical/surgical approach.
“It’s important to apply the HS severity scores and complete the clinical exam, but it’s also important to use ultrasound or another imaging technique,” Dr. Martorell concluded.
MADRID – Antonio Martorell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
This distinction is critical in recognizing a window of opportunity in the management of hidradenitis suppurativa (HS): The period early in the disease course when medical therapy alone can be life-changing.
“The window of opportunity is an old concept in gastroenterology, but it’s a new idea in dermatology: It’s the moment in which the patient can have the best results with medical control of inflammation, before progression to tissue scarring has occurred,” explained Dr. Martorell, a dermatologist at the Hospital of Manises in Valencia, Spain, and coauthor of recent HS treatment recommendations by an international expert panel (J Eur Acad Dermatol Venereol. 2019 Jan;33[1]:19-31).
Symptoms in patients with HS can be caused by either dynamic or static lesions. Dynamic lesions arise directly from acute inflammation that can be treated with antibiotics and immunomodulatory therapy. Static lesions are associated with tissue scarring secondary to inflammatory activity and generally benefit only from surgery.
Dynamic lesions consist of nodules, abscesses, and some but not all fistulae. Although the traditional view among dermatologists has been that fistulae simply don’t respond to medical therapy and must be treated surgically, Dr. Martorell and coinvestigators have recently demonstrated in a retrospective study of 117 fistulae in 40 patients that ultrasound was useful in distinguishing four fistular subtypes, two of which responded reasonably well to medical management.
What the investigators call Type A or dermal fistulae are by definition not connected to tunnels. In Dr. Martorell’s study, they had a 95% complete resolution rate after 6 months of various medications. Dermoepidermal fistulae tunnel through the dermis to the epidermis; they had a 65% complete resolution rate. In contrast, Type C or complex fistulae, identified by the ultrasound finding of multiple tunnels extending through the dermis into underlying fat tissue, had no significant response to medical management. Neither did Type D fistulae, which are essentially Type C lesions with scarring (Dermatol Surg. 2019 Oct;45[10]:1237-44).
Thus, ultrasound can have an important impact on patient management and the decision to opt for a combined medical/surgical approach.
“It’s important to apply the HS severity scores and complete the clinical exam, but it’s also important to use ultrasound or another imaging technique,” Dr. Martorell concluded.
EXPERT ANALYSIS FROM THE EADV CONGRESS
Bimekizumab elevates psoriasis therapy
MADRID – Renowned dermatologic clinical trialist Kim A. Papp, MD, PhD, is known to pick his words carefully, and the word he uses to describe the quality of life improvement documented in psoriasis patients treated with the novel investigational humanized monoclonal antibody bimekizumab is “phenomenal.”

Dr. Papp was lead investigator in the previously reported phase 2b multicenter BE ABLE 1 trial, in which 250 patients with moderate to severe chronic plaque psoriasis were randomized double-blind to various doses of bimekizumab or placebo every 4 weeks for 12 weeks (J Am Acad Dermatol. 2018 Aug;79[2]:277-86.e10. doi: 10.1016/j.jaad.2018.03.037). He was also lead investigator in the 48-week phase 2b BE ABLE 2 extension study. He presented the 60-week quality-of-life BE ABLE 2 results for the first time at the annual congress of the European Academy of Dermatology and Venereology.
“Small numbers, but the results are nonetheless very compelling,” said Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
Bimekizumab is unique in that it selectively neutralizes both interleukin-17A and -17F, two closely related proinflammatory cytokines which, when upregulated, synergize with other proinflammatory cytokines to drive psoriasis and other immune-mediated inflammatory diseases. In contrast, secukinumab (Cosentyx) and ixekizumab (Taltz) specifically inhibit only IL-17A, and brodalumab (Siliq) targets the IL-17 receptor A. The bimekizumab clinical trials program – a work in progress – aims to demonstrate that dual neutralization of IL-17A and -17F provides a more complete therapeutic approach in psoriasis, with greater efficacy and fewer safety concerns than with current biologics, the dermatologist explained.
In BE ABLE 1, the primary endpoint of at least a 90% reduction in Psoriasis Area and Severity Index (PASI90) response was achieved at week 12 in 46%-79% of patients randomized to bimekizumab in dose-dependent fashion. Those PASI90 responses were maintained with additional treatment out to week 60 in BE ABLE 2 in 80%-100% of patients.
Dr. Papp’s focus at EADV 2019 was on the quality-of-life improvement achieved in bimekizumab-treated patients, a benefit not captured by PASI scores. For this purpose, he and coinvestigators turned to the Dermatology Life Quality Index (DLQI), measured in structured fashion every 4 weeks out to week 60.
“We often forget that even though we’re looking at the patient from the outside, what’s really important is how well they respond to our treatments internally. The DLQI is not a perfect tool, but it’s the best tool we have available. It gives us a fairly good survey of the various domains that affect patients’ day-to-day living,” he said.
In BE ABLE 1, the proportion of week-12 PASI90 responders achieving a DLQI of 0 or 1 – indicative of essentially no disease impact on quality of life – increased rapidly up until week 8. At week 12, 70%-100% of the PASI90 responders in the various treatment arms had a DLQI of 0 or 1. This quality-of-life improvement, like the PASI90 response, proved durable: When the week-12 PASI90 responders were assessed at week 60 in BE ABLE 2, 76%-93% of them had a DLQI of 0 or 1.
The improvements in quality of life correlated with clinical response. BE ABLE enrollees had an average PASI score of 19 at baseline. Overall, 79% of those with an absolute PASI score of 0 at week 12 had a DLQI of 0 or 1 at that time, as did 95% of those with a PASI of 0 at week 60. A PASI of 1 was associated with a 77% likelihood of a DLQI of 0 or 1 at week 12 and an 82% rate at week 60. In contrast, patients with an absolute PASI of 2-4 at week 12 had a 46% rate of DLQI 0/1, and those with a PASI 2-4 at week 60 had a 50% chance of having a DLQI of 0/1.
Phase 3 clinical trials of bimekizumab totaling several thousand psoriasis patients are ongoing.
The BE ABLE trials were sponsored by UCB Pharma. Dr. Papp reported serving as a consultant to and/or recipient of research grants from UCB and dozens of other pharmaceutical companies.
SOURCE: Papp KA. EADV 2019 Abstract FC02.02.
MADRID – Renowned dermatologic clinical trialist Kim A. Papp, MD, PhD, is known to pick his words carefully, and the word he uses to describe the quality of life improvement documented in psoriasis patients treated with the novel investigational humanized monoclonal antibody bimekizumab is “phenomenal.”
Dr. Papp was lead investigator in the previously reported phase 2b multicenter BE ABLE 1 trial, in which 250 patients with moderate to severe chronic plaque psoriasis were randomized double-blind to various doses of bimekizumab or placebo every 4 weeks for 12 weeks (J Am Acad Dermatol. 2018 Aug;79[2]:277-86.e10. doi: 10.1016/j.jaad.2018.03.037). He was also lead investigator in the 48-week phase 2b BE ABLE 2 extension study. He presented the 60-week quality-of-life BE ABLE 2 results for the first time at the annual congress of the European Academy of Dermatology and Venereology.
“Small numbers, but the results are nonetheless very compelling,” said Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
Bimekizumab is unique in that it selectively neutralizes both interleukin-17A and -17F, two closely related proinflammatory cytokines which, when upregulated, synergize with other proinflammatory cytokines to drive psoriasis and other immune-mediated inflammatory diseases. In contrast, secukinumab (Cosentyx) and ixekizumab (Taltz) specifically inhibit only IL-17A, and brodalumab (Siliq) targets the IL-17 receptor A. The bimekizumab clinical trials program – a work in progress – aims to demonstrate that dual neutralization of IL-17A and -17F provides a more complete therapeutic approach in psoriasis, with greater efficacy and fewer safety concerns than with current biologics, the dermatologist explained.
In BE ABLE 1, the primary endpoint of at least a 90% reduction in Psoriasis Area and Severity Index (PASI90) response was achieved at week 12 in 46%-79% of patients randomized to bimekizumab in dose-dependent fashion. Those PASI90 responses were maintained with additional treatment out to week 60 in BE ABLE 2 in 80%-100% of patients.
Dr. Papp’s focus at EADV 2019 was on the quality-of-life improvement achieved in bimekizumab-treated patients, a benefit not captured by PASI scores. For this purpose, he and coinvestigators turned to the Dermatology Life Quality Index (DLQI), measured in structured fashion every 4 weeks out to week 60.
“We often forget that even though we’re looking at the patient from the outside, what’s really important is how well they respond to our treatments internally. The DLQI is not a perfect tool, but it’s the best tool we have available. It gives us a fairly good survey of the various domains that affect patients’ day-to-day living,” he said.
In BE ABLE 1, the proportion of week-12 PASI90 responders achieving a DLQI of 0 or 1 – indicative of essentially no disease impact on quality of life – increased rapidly up until week 8. At week 12, 70%-100% of the PASI90 responders in the various treatment arms had a DLQI of 0 or 1. This quality-of-life improvement, like the PASI90 response, proved durable: When the week-12 PASI90 responders were assessed at week 60 in BE ABLE 2, 76%-93% of them had a DLQI of 0 or 1.
The improvements in quality of life correlated with clinical response. BE ABLE enrollees had an average PASI score of 19 at baseline. Overall, 79% of those with an absolute PASI score of 0 at week 12 had a DLQI of 0 or 1 at that time, as did 95% of those with a PASI of 0 at week 60. A PASI of 1 was associated with a 77% likelihood of a DLQI of 0 or 1 at week 12 and an 82% rate at week 60. In contrast, patients with an absolute PASI of 2-4 at week 12 had a 46% rate of DLQI 0/1, and those with a PASI 2-4 at week 60 had a 50% chance of having a DLQI of 0/1.
Phase 3 clinical trials of bimekizumab totaling several thousand psoriasis patients are ongoing.
The BE ABLE trials were sponsored by UCB Pharma. Dr. Papp reported serving as a consultant to and/or recipient of research grants from UCB and dozens of other pharmaceutical companies.
SOURCE: Papp KA. EADV 2019 Abstract FC02.02.
MADRID – Renowned dermatologic clinical trialist Kim A. Papp, MD, PhD, is known to pick his words carefully, and the word he uses to describe the quality of life improvement documented in psoriasis patients treated with the novel investigational humanized monoclonal antibody bimekizumab is “phenomenal.”
Dr. Papp was lead investigator in the previously reported phase 2b multicenter BE ABLE 1 trial, in which 250 patients with moderate to severe chronic plaque psoriasis were randomized double-blind to various doses of bimekizumab or placebo every 4 weeks for 12 weeks (J Am Acad Dermatol. 2018 Aug;79[2]:277-86.e10. doi: 10.1016/j.jaad.2018.03.037). He was also lead investigator in the 48-week phase 2b BE ABLE 2 extension study. He presented the 60-week quality-of-life BE ABLE 2 results for the first time at the annual congress of the European Academy of Dermatology and Venereology.
“Small numbers, but the results are nonetheless very compelling,” said Dr. Papp, president and founder of Probity Medical Research in Waterloo, Ont.
Bimekizumab is unique in that it selectively neutralizes both interleukin-17A and -17F, two closely related proinflammatory cytokines which, when upregulated, synergize with other proinflammatory cytokines to drive psoriasis and other immune-mediated inflammatory diseases. In contrast, secukinumab (Cosentyx) and ixekizumab (Taltz) specifically inhibit only IL-17A, and brodalumab (Siliq) targets the IL-17 receptor A. The bimekizumab clinical trials program – a work in progress – aims to demonstrate that dual neutralization of IL-17A and -17F provides a more complete therapeutic approach in psoriasis, with greater efficacy and fewer safety concerns than with current biologics, the dermatologist explained.
In BE ABLE 1, the primary endpoint of at least a 90% reduction in Psoriasis Area and Severity Index (PASI90) response was achieved at week 12 in 46%-79% of patients randomized to bimekizumab in dose-dependent fashion. Those PASI90 responses were maintained with additional treatment out to week 60 in BE ABLE 2 in 80%-100% of patients.
Dr. Papp’s focus at EADV 2019 was on the quality-of-life improvement achieved in bimekizumab-treated patients, a benefit not captured by PASI scores. For this purpose, he and coinvestigators turned to the Dermatology Life Quality Index (DLQI), measured in structured fashion every 4 weeks out to week 60.
“We often forget that even though we’re looking at the patient from the outside, what’s really important is how well they respond to our treatments internally. The DLQI is not a perfect tool, but it’s the best tool we have available. It gives us a fairly good survey of the various domains that affect patients’ day-to-day living,” he said.
In BE ABLE 1, the proportion of week-12 PASI90 responders achieving a DLQI of 0 or 1 – indicative of essentially no disease impact on quality of life – increased rapidly up until week 8. At week 12, 70%-100% of the PASI90 responders in the various treatment arms had a DLQI of 0 or 1. This quality-of-life improvement, like the PASI90 response, proved durable: When the week-12 PASI90 responders were assessed at week 60 in BE ABLE 2, 76%-93% of them had a DLQI of 0 or 1.
The improvements in quality of life correlated with clinical response. BE ABLE enrollees had an average PASI score of 19 at baseline. Overall, 79% of those with an absolute PASI score of 0 at week 12 had a DLQI of 0 or 1 at that time, as did 95% of those with a PASI of 0 at week 60. A PASI of 1 was associated with a 77% likelihood of a DLQI of 0 or 1 at week 12 and an 82% rate at week 60. In contrast, patients with an absolute PASI of 2-4 at week 12 had a 46% rate of DLQI 0/1, and those with a PASI 2-4 at week 60 had a 50% chance of having a DLQI of 0/1.
Phase 3 clinical trials of bimekizumab totaling several thousand psoriasis patients are ongoing.
The BE ABLE trials were sponsored by UCB Pharma. Dr. Papp reported serving as a consultant to and/or recipient of research grants from UCB and dozens of other pharmaceutical companies.
SOURCE: Papp KA. EADV 2019 Abstract FC02.02.
REPORTING FROM THE EADV CONGRESS
Experts in Europe issue guidance on atopic dermatitis in pregnancy
MADRID – European atopic dermatitis experts have issued formal guidance on a seriously neglected topic: treatment of the disease during pregnancy, breastfeeding, and in men planning to father children.

The impetus for the project was clear: “,” Christian Vestergaard, MD, PhD, declared at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of the task force’s position paper on the topic, for which he served as first author. The group’s recommendations are based on expert opinion, since randomized clinical trial literature in this area is nonexistent because of ethical concerns. But the task force, comprising a who’s who in European dermatology, drew on a wealth of collective clinical experience in this area.
“We have all of Europe involved in doing this position statement. It’s meant as what we think is proper treatment and what we can say about the different drugs,” explained Dr. Vestergaard, a dermatologist at the University of Aarhus (Denmark).
Most nonobstetricians are intimidated by atopic dermatitis (AD) in pregnancy, and are concerned about the potential for treatment-related harm to the fetus. As a consequence, they are reluctant to recommend anything beyond weak class I topical corticosteroids and emollients. That’s clearly insufficient in light of the vast scope of need, he asserted. After all, AD affects 15%-20% of all children and persists or reappears in adulthood in one out of five of them. Half of those adults are women, many of whom will at some point wish to become pregnant. And many men with AD will eventually want to father children.
A key message from the task force is that untreated AD in pregnancy potentially places the mother and fetus at risk of serious complications, including Staphylococcus aureus infection and eczema herpeticum.
“If you take one thing away from our position paper, it’s that you can use class II or III topical corticosteroids in pregnant women as first-line therapy,” Dr. Vestergaard said.
This stance contradicts a longstanding widely held concern that topical steroids in pregnancy might increase the risk of facial cleft in the offspring, a worry that has been convincingly debunked in a Cochrane systematic review of 14 studies including more than 1.6 million pregnancies. The report concluded there was no association between topical corticosteroids of any potency with preterm delivery, birth defects, or low Apgar scores (Cochrane Database Syst Rev. 2015 Oct 26. doi: 10.1002/14651858.CD007346.pub3).
The task force recommends that if class II or III topical corticosteroid use in pregnancy exceeds 200 g/month, it’s worth considering add-on UV therapy, with narrow band UVB-311 nm as the regimen of choice; it can be used liberally. UV therapy with psoralens is not advised because of a theoretical risk of mutagenicity.
Product labeling for the topical calcineurin inhibitors declares that the agents should not be used during pregnancy. However, the European task force position paper takes issue with that and declares that topical tacrolimus (Protopic) can be considered an off-label first-line therapy in pregnant women with an insufficient response to liberal use of emollients. The same holds true for breastfeeding patients with AD. Just as when topical corticosteroids are used in the nipple area, topical tacrolimus should be applied after nursing, and the nipple area should be gently cleaned before nursing.
The rationale behind recommending topical tacrolimus as a first-line treatment is that systemic absorption of the drug is trivial. Plus, observational studies of oral tacrolimus in pregnant women who have received a solid organ transplant have shown no increase in congenital malformations.
The task force recommends against the use of topical pimecrolimus (Elidel) or crisaborole (Eucrisa) in pregnancy or lactation due to lack of clinical experience in these settings, Dr. Vestergaard continued.
The task force position is that chlorhexidine and other topical antiseptics – with the notable exception of triclosan – can be used in pregnancy to prevent recurrent skin infections. Aminoglycosides should be avoided, but topical fusidic acid is a reasonable antibiotic for treatment of small areas of clinically infected atopic dermatitis in pregnancy.
Systemic therapies
If disease control is insufficient with topical therapy, it’s appropriate to engage in shared decision-making with the patient regarding systemic treatment. She needs to understand up front that the worldwide overall background stillbirth rate in the general population is about 3%, and that severe congenital malformations are present in up to 6% of all live births.
“You need to inform them that they can have systemic therapy and give birth to a child with congenital defects which have nothing to do with the medication,” noted Dr. Vestergaard.
That said, the task force recommends cyclosporine as the off-label, first-line systemic therapy in pregnancy and lactation when long-term treatment is required. This guidance is based largely upon reassuring evidence in solid organ transplant recipients.
The recommended second-line therapy is systemic corticosteroids, but it’s a qualified recommendation. Dr. Vestergaard and colleagues find that systemic corticosteroid therapy is only rarely needed in pregnant AD patients, and the task force recommendation is to limit the use to less than 2-3 weeks and no more than 0.5 mg/kg per day of prednisone. Dexamethasone is not recommended.
Azathioprine should not be started in pregnancy, according to the task force, but when no other options are available, it may be continued in women already on the drug, albeit at half of the prepregnancy dose.
Dupilumab (Dupixent) is to be avoided in pregnant women with AD until more clinical experience becomes available.
Treatment of prospective fathers with AD
The European task force recommends that topical therapies can be prescribed in prospective fathers without any special concerns. The same is true for systemic corticosteroids. Methotrexate should be halted 3 months before planned pregnancy, as is the case for mycophenolate mofetil (CellCept). Azathioprine is recommended when other options have failed. Cyclosporine is deemed a reasonable option in the treatment of men with severe AD at the time of conception if other treatments have failed; of note, neither the Food and Drug Administration nor the European regulatory agency have issued contraindications for the use of the drug in men who wish to become fathers.
Mycophenolate mofetil carries a theoretical risk of teratogenicity. The European task force recommends that men should use condoms while on the drug and for at least 90 days afterward.
Unplanned pregnancy in women on systemic therapy
The recommended course of action is to immediately stop systemic therapy, intensify appropriate topical therapy in anticipation of worsening AD, and refer the patient to an obstetrician and a teratology information center for an individualized risk assessment. Methotrexate and mycophenolate mofetil are known teratogens.
The full 16-page task force position paper was published shortly before EADV 2019 (J Eur Acad Dermatol Venereol. 2019 Sep;33[9]:1644-59).
The report was developed without commercial sponsorship. Dr. Vestergaard indicated he has received research grants from and/or serves as a consultant to eight pharmaceutical companies.
Jenny Murase, MD, is with the department of dermatology, University of California, San Francisco, and is the director of medical consultative dermatology at the Palo Alto Foundation Medical Group, Mountain View, Calif. She has served on advisory boards for Dermira, Sanofi, and UCB; performed dermatologic consulting for UpToDate and Ferndale, and given nonbranded lectures for disease state management awareness for Regeneron and UCB.
Jenny Murase, MD, is with the department of dermatology, University of California, San Francisco, and is the director of medical consultative dermatology at the Palo Alto Foundation Medical Group, Mountain View, Calif. She has served on advisory boards for Dermira, Sanofi, and UCB; performed dermatologic consulting for UpToDate and Ferndale, and given nonbranded lectures for disease state management awareness for Regeneron and UCB.
Jenny Murase, MD, is with the department of dermatology, University of California, San Francisco, and is the director of medical consultative dermatology at the Palo Alto Foundation Medical Group, Mountain View, Calif. She has served on advisory boards for Dermira, Sanofi, and UCB; performed dermatologic consulting for UpToDate and Ferndale, and given nonbranded lectures for disease state management awareness for Regeneron and UCB.
MADRID – European atopic dermatitis experts have issued formal guidance on a seriously neglected topic: treatment of the disease during pregnancy, breastfeeding, and in men planning to father children.
The impetus for the project was clear: “,” Christian Vestergaard, MD, PhD, declared at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of the task force’s position paper on the topic, for which he served as first author. The group’s recommendations are based on expert opinion, since randomized clinical trial literature in this area is nonexistent because of ethical concerns. But the task force, comprising a who’s who in European dermatology, drew on a wealth of collective clinical experience in this area.
“We have all of Europe involved in doing this position statement. It’s meant as what we think is proper treatment and what we can say about the different drugs,” explained Dr. Vestergaard, a dermatologist at the University of Aarhus (Denmark).
Most nonobstetricians are intimidated by atopic dermatitis (AD) in pregnancy, and are concerned about the potential for treatment-related harm to the fetus. As a consequence, they are reluctant to recommend anything beyond weak class I topical corticosteroids and emollients. That’s clearly insufficient in light of the vast scope of need, he asserted. After all, AD affects 15%-20% of all children and persists or reappears in adulthood in one out of five of them. Half of those adults are women, many of whom will at some point wish to become pregnant. And many men with AD will eventually want to father children.
A key message from the task force is that untreated AD in pregnancy potentially places the mother and fetus at risk of serious complications, including Staphylococcus aureus infection and eczema herpeticum.
“If you take one thing away from our position paper, it’s that you can use class II or III topical corticosteroids in pregnant women as first-line therapy,” Dr. Vestergaard said.
This stance contradicts a longstanding widely held concern that topical steroids in pregnancy might increase the risk of facial cleft in the offspring, a worry that has been convincingly debunked in a Cochrane systematic review of 14 studies including more than 1.6 million pregnancies. The report concluded there was no association between topical corticosteroids of any potency with preterm delivery, birth defects, or low Apgar scores (Cochrane Database Syst Rev. 2015 Oct 26. doi: 10.1002/14651858.CD007346.pub3).
The task force recommends that if class II or III topical corticosteroid use in pregnancy exceeds 200 g/month, it’s worth considering add-on UV therapy, with narrow band UVB-311 nm as the regimen of choice; it can be used liberally. UV therapy with psoralens is not advised because of a theoretical risk of mutagenicity.
Product labeling for the topical calcineurin inhibitors declares that the agents should not be used during pregnancy. However, the European task force position paper takes issue with that and declares that topical tacrolimus (Protopic) can be considered an off-label first-line therapy in pregnant women with an insufficient response to liberal use of emollients. The same holds true for breastfeeding patients with AD. Just as when topical corticosteroids are used in the nipple area, topical tacrolimus should be applied after nursing, and the nipple area should be gently cleaned before nursing.
The rationale behind recommending topical tacrolimus as a first-line treatment is that systemic absorption of the drug is trivial. Plus, observational studies of oral tacrolimus in pregnant women who have received a solid organ transplant have shown no increase in congenital malformations.
The task force recommends against the use of topical pimecrolimus (Elidel) or crisaborole (Eucrisa) in pregnancy or lactation due to lack of clinical experience in these settings, Dr. Vestergaard continued.
The task force position is that chlorhexidine and other topical antiseptics – with the notable exception of triclosan – can be used in pregnancy to prevent recurrent skin infections. Aminoglycosides should be avoided, but topical fusidic acid is a reasonable antibiotic for treatment of small areas of clinically infected atopic dermatitis in pregnancy.
Systemic therapies
If disease control is insufficient with topical therapy, it’s appropriate to engage in shared decision-making with the patient regarding systemic treatment. She needs to understand up front that the worldwide overall background stillbirth rate in the general population is about 3%, and that severe congenital malformations are present in up to 6% of all live births.
“You need to inform them that they can have systemic therapy and give birth to a child with congenital defects which have nothing to do with the medication,” noted Dr. Vestergaard.
That said, the task force recommends cyclosporine as the off-label, first-line systemic therapy in pregnancy and lactation when long-term treatment is required. This guidance is based largely upon reassuring evidence in solid organ transplant recipients.
The recommended second-line therapy is systemic corticosteroids, but it’s a qualified recommendation. Dr. Vestergaard and colleagues find that systemic corticosteroid therapy is only rarely needed in pregnant AD patients, and the task force recommendation is to limit the use to less than 2-3 weeks and no more than 0.5 mg/kg per day of prednisone. Dexamethasone is not recommended.
Azathioprine should not be started in pregnancy, according to the task force, but when no other options are available, it may be continued in women already on the drug, albeit at half of the prepregnancy dose.
Dupilumab (Dupixent) is to be avoided in pregnant women with AD until more clinical experience becomes available.
Treatment of prospective fathers with AD
The European task force recommends that topical therapies can be prescribed in prospective fathers without any special concerns. The same is true for systemic corticosteroids. Methotrexate should be halted 3 months before planned pregnancy, as is the case for mycophenolate mofetil (CellCept). Azathioprine is recommended when other options have failed. Cyclosporine is deemed a reasonable option in the treatment of men with severe AD at the time of conception if other treatments have failed; of note, neither the Food and Drug Administration nor the European regulatory agency have issued contraindications for the use of the drug in men who wish to become fathers.
Mycophenolate mofetil carries a theoretical risk of teratogenicity. The European task force recommends that men should use condoms while on the drug and for at least 90 days afterward.
Unplanned pregnancy in women on systemic therapy
The recommended course of action is to immediately stop systemic therapy, intensify appropriate topical therapy in anticipation of worsening AD, and refer the patient to an obstetrician and a teratology information center for an individualized risk assessment. Methotrexate and mycophenolate mofetil are known teratogens.
The full 16-page task force position paper was published shortly before EADV 2019 (J Eur Acad Dermatol Venereol. 2019 Sep;33[9]:1644-59).
The report was developed without commercial sponsorship. Dr. Vestergaard indicated he has received research grants from and/or serves as a consultant to eight pharmaceutical companies.
MADRID – European atopic dermatitis experts have issued formal guidance on a seriously neglected topic: treatment of the disease during pregnancy, breastfeeding, and in men planning to father children.
The impetus for the project was clear: “,” Christian Vestergaard, MD, PhD, declared at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
He presented highlights of the task force’s position paper on the topic, for which he served as first author. The group’s recommendations are based on expert opinion, since randomized clinical trial literature in this area is nonexistent because of ethical concerns. But the task force, comprising a who’s who in European dermatology, drew on a wealth of collective clinical experience in this area.
“We have all of Europe involved in doing this position statement. It’s meant as what we think is proper treatment and what we can say about the different drugs,” explained Dr. Vestergaard, a dermatologist at the University of Aarhus (Denmark).
Most nonobstetricians are intimidated by atopic dermatitis (AD) in pregnancy, and are concerned about the potential for treatment-related harm to the fetus. As a consequence, they are reluctant to recommend anything beyond weak class I topical corticosteroids and emollients. That’s clearly insufficient in light of the vast scope of need, he asserted. After all, AD affects 15%-20% of all children and persists or reappears in adulthood in one out of five of them. Half of those adults are women, many of whom will at some point wish to become pregnant. And many men with AD will eventually want to father children.
A key message from the task force is that untreated AD in pregnancy potentially places the mother and fetus at risk of serious complications, including Staphylococcus aureus infection and eczema herpeticum.
“If you take one thing away from our position paper, it’s that you can use class II or III topical corticosteroids in pregnant women as first-line therapy,” Dr. Vestergaard said.
This stance contradicts a longstanding widely held concern that topical steroids in pregnancy might increase the risk of facial cleft in the offspring, a worry that has been convincingly debunked in a Cochrane systematic review of 14 studies including more than 1.6 million pregnancies. The report concluded there was no association between topical corticosteroids of any potency with preterm delivery, birth defects, or low Apgar scores (Cochrane Database Syst Rev. 2015 Oct 26. doi: 10.1002/14651858.CD007346.pub3).
The task force recommends that if class II or III topical corticosteroid use in pregnancy exceeds 200 g/month, it’s worth considering add-on UV therapy, with narrow band UVB-311 nm as the regimen of choice; it can be used liberally. UV therapy with psoralens is not advised because of a theoretical risk of mutagenicity.
Product labeling for the topical calcineurin inhibitors declares that the agents should not be used during pregnancy. However, the European task force position paper takes issue with that and declares that topical tacrolimus (Protopic) can be considered an off-label first-line therapy in pregnant women with an insufficient response to liberal use of emollients. The same holds true for breastfeeding patients with AD. Just as when topical corticosteroids are used in the nipple area, topical tacrolimus should be applied after nursing, and the nipple area should be gently cleaned before nursing.
The rationale behind recommending topical tacrolimus as a first-line treatment is that systemic absorption of the drug is trivial. Plus, observational studies of oral tacrolimus in pregnant women who have received a solid organ transplant have shown no increase in congenital malformations.
The task force recommends against the use of topical pimecrolimus (Elidel) or crisaborole (Eucrisa) in pregnancy or lactation due to lack of clinical experience in these settings, Dr. Vestergaard continued.
The task force position is that chlorhexidine and other topical antiseptics – with the notable exception of triclosan – can be used in pregnancy to prevent recurrent skin infections. Aminoglycosides should be avoided, but topical fusidic acid is a reasonable antibiotic for treatment of small areas of clinically infected atopic dermatitis in pregnancy.
Systemic therapies
If disease control is insufficient with topical therapy, it’s appropriate to engage in shared decision-making with the patient regarding systemic treatment. She needs to understand up front that the worldwide overall background stillbirth rate in the general population is about 3%, and that severe congenital malformations are present in up to 6% of all live births.
“You need to inform them that they can have systemic therapy and give birth to a child with congenital defects which have nothing to do with the medication,” noted Dr. Vestergaard.
That said, the task force recommends cyclosporine as the off-label, first-line systemic therapy in pregnancy and lactation when long-term treatment is required. This guidance is based largely upon reassuring evidence in solid organ transplant recipients.
The recommended second-line therapy is systemic corticosteroids, but it’s a qualified recommendation. Dr. Vestergaard and colleagues find that systemic corticosteroid therapy is only rarely needed in pregnant AD patients, and the task force recommendation is to limit the use to less than 2-3 weeks and no more than 0.5 mg/kg per day of prednisone. Dexamethasone is not recommended.
Azathioprine should not be started in pregnancy, according to the task force, but when no other options are available, it may be continued in women already on the drug, albeit at half of the prepregnancy dose.
Dupilumab (Dupixent) is to be avoided in pregnant women with AD until more clinical experience becomes available.
Treatment of prospective fathers with AD
The European task force recommends that topical therapies can be prescribed in prospective fathers without any special concerns. The same is true for systemic corticosteroids. Methotrexate should be halted 3 months before planned pregnancy, as is the case for mycophenolate mofetil (CellCept). Azathioprine is recommended when other options have failed. Cyclosporine is deemed a reasonable option in the treatment of men with severe AD at the time of conception if other treatments have failed; of note, neither the Food and Drug Administration nor the European regulatory agency have issued contraindications for the use of the drug in men who wish to become fathers.
Mycophenolate mofetil carries a theoretical risk of teratogenicity. The European task force recommends that men should use condoms while on the drug and for at least 90 days afterward.
Unplanned pregnancy in women on systemic therapy
The recommended course of action is to immediately stop systemic therapy, intensify appropriate topical therapy in anticipation of worsening AD, and refer the patient to an obstetrician and a teratology information center for an individualized risk assessment. Methotrexate and mycophenolate mofetil are known teratogens.
The full 16-page task force position paper was published shortly before EADV 2019 (J Eur Acad Dermatol Venereol. 2019 Sep;33[9]:1644-59).
The report was developed without commercial sponsorship. Dr. Vestergaard indicated he has received research grants from and/or serves as a consultant to eight pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Eluxadoline effective for IBS in loperamide nonresponders
SAN ANTONIO – Darren M. Brenner, MD, reported at the annual meeting of the American College of Gastroenterology.

“From the totality of the clinical trials data we have now, we believe that eluxadoline can be effective both in patients who are naive to other treatments and in patients who have failed loperamide therapy,” concluded Dr. Brenner, a gastroenterologist at Northwestern University, Chicago.
Eluxadoline (Viberzi) is a novel mixed mu- and kappa-opioid receptor agonist and delta-opioid receptor antagonist approved by the Food and Drug Administration for treatment of irritable bowel syndrome with diarrhea (IBS-D) in adults. In contrast, loperamide, a mu-opioid receptor agonist, is not approved for that indication. Yet loperamide is widely prescribed for this purpose, despite the fact that both the Canadian Association of Gastroenterology and the ACG now recommend against this practice.
“There is a lack of conclusive evidence to support the use of loperamide for the relief of global IBS-D symptoms. It works on the stool symptoms – stool frequency and texture – but has never been shown to be beneficial for the abdominal pain symptoms or discomfort or bloating. That being said, as practitioners we continue to see loperamide used as a first-line agent,” Dr. Brenner noted.
RELIEF was a multicenter, prospective, double-blind study which randomized 346 patients with moderate to severe IBS-D to eluxadoline at 100 mg twice daily or placebo for 12 weeks. All participants were required to have an intact gallbladder as per the drug’s labeling guidance, and all had a self-reported recent inadequate response to loperamide.
The primary composite endpoint in the RELIEF trial was a 40% or greater improvement from baseline in the 11-point Daily Worst Abdominal Pain score plus a Bristol Stool Form score below 5 on the same day for at least 50% of study days. At baseline, participants had an average Worst Abdominal Pain score of 6.2 on the 0-10 scale and a Bristol score of 6.2. The primary endpoint was achieved at week 12 in 23% of the eluxadoline group, significantly better than the 10% rate in controls. The eluxadoline group also showed significantly greater improvement on the many secondary endpoints having to do with urgency-free days, stool consistency, bowel movement frequency, abdominal discomfort, bloating, and the experience of adequate relief of symptoms.
The safety profile of eluxadoline mirrored that of placebo, with no serious adverse events recorded in either study arm and a 2.9% study discontinuation rate because of treatment-emergent adverse events in the eluxadoline group. Asked why he thinks eluxadoline was effective in improving the full range of IBS-D symptoms when loperamide wasn’t, even though both drugs are mu-opioid receptor agonists, Dr. Brenner replied, “The problem is mu receptors line the entire GI tract, so you can actually push somebody from diarrhea to opioid-induced constipation – and that’s not the goal. What delta does is alleviate some of the adverse events by binding to the receptor, which results in increased transit time, reduced secretion, and increased absorption. Delta brings things back towards the center. We also believe antagonism of delta potentiates analgesic effects at the mu receptor, improves the pain component, gut symptoms, and stool symptoms.”
Dr. Brenner reported serving as a consultant to and member of a speaker’s bureau for Allergan, which markets eluxadoline and sponsored the RELIEF trial.
SAN ANTONIO – Darren M. Brenner, MD, reported at the annual meeting of the American College of Gastroenterology.
“From the totality of the clinical trials data we have now, we believe that eluxadoline can be effective both in patients who are naive to other treatments and in patients who have failed loperamide therapy,” concluded Dr. Brenner, a gastroenterologist at Northwestern University, Chicago.
Eluxadoline (Viberzi) is a novel mixed mu- and kappa-opioid receptor agonist and delta-opioid receptor antagonist approved by the Food and Drug Administration for treatment of irritable bowel syndrome with diarrhea (IBS-D) in adults. In contrast, loperamide, a mu-opioid receptor agonist, is not approved for that indication. Yet loperamide is widely prescribed for this purpose, despite the fact that both the Canadian Association of Gastroenterology and the ACG now recommend against this practice.
“There is a lack of conclusive evidence to support the use of loperamide for the relief of global IBS-D symptoms. It works on the stool symptoms – stool frequency and texture – but has never been shown to be beneficial for the abdominal pain symptoms or discomfort or bloating. That being said, as practitioners we continue to see loperamide used as a first-line agent,” Dr. Brenner noted.
RELIEF was a multicenter, prospective, double-blind study which randomized 346 patients with moderate to severe IBS-D to eluxadoline at 100 mg twice daily or placebo for 12 weeks. All participants were required to have an intact gallbladder as per the drug’s labeling guidance, and all had a self-reported recent inadequate response to loperamide.
The primary composite endpoint in the RELIEF trial was a 40% or greater improvement from baseline in the 11-point Daily Worst Abdominal Pain score plus a Bristol Stool Form score below 5 on the same day for at least 50% of study days. At baseline, participants had an average Worst Abdominal Pain score of 6.2 on the 0-10 scale and a Bristol score of 6.2. The primary endpoint was achieved at week 12 in 23% of the eluxadoline group, significantly better than the 10% rate in controls. The eluxadoline group also showed significantly greater improvement on the many secondary endpoints having to do with urgency-free days, stool consistency, bowel movement frequency, abdominal discomfort, bloating, and the experience of adequate relief of symptoms.
The safety profile of eluxadoline mirrored that of placebo, with no serious adverse events recorded in either study arm and a 2.9% study discontinuation rate because of treatment-emergent adverse events in the eluxadoline group. Asked why he thinks eluxadoline was effective in improving the full range of IBS-D symptoms when loperamide wasn’t, even though both drugs are mu-opioid receptor agonists, Dr. Brenner replied, “The problem is mu receptors line the entire GI tract, so you can actually push somebody from diarrhea to opioid-induced constipation – and that’s not the goal. What delta does is alleviate some of the adverse events by binding to the receptor, which results in increased transit time, reduced secretion, and increased absorption. Delta brings things back towards the center. We also believe antagonism of delta potentiates analgesic effects at the mu receptor, improves the pain component, gut symptoms, and stool symptoms.”
Dr. Brenner reported serving as a consultant to and member of a speaker’s bureau for Allergan, which markets eluxadoline and sponsored the RELIEF trial.
SAN ANTONIO – Darren M. Brenner, MD, reported at the annual meeting of the American College of Gastroenterology.
“From the totality of the clinical trials data we have now, we believe that eluxadoline can be effective both in patients who are naive to other treatments and in patients who have failed loperamide therapy,” concluded Dr. Brenner, a gastroenterologist at Northwestern University, Chicago.
Eluxadoline (Viberzi) is a novel mixed mu- and kappa-opioid receptor agonist and delta-opioid receptor antagonist approved by the Food and Drug Administration for treatment of irritable bowel syndrome with diarrhea (IBS-D) in adults. In contrast, loperamide, a mu-opioid receptor agonist, is not approved for that indication. Yet loperamide is widely prescribed for this purpose, despite the fact that both the Canadian Association of Gastroenterology and the ACG now recommend against this practice.
“There is a lack of conclusive evidence to support the use of loperamide for the relief of global IBS-D symptoms. It works on the stool symptoms – stool frequency and texture – but has never been shown to be beneficial for the abdominal pain symptoms or discomfort or bloating. That being said, as practitioners we continue to see loperamide used as a first-line agent,” Dr. Brenner noted.
RELIEF was a multicenter, prospective, double-blind study which randomized 346 patients with moderate to severe IBS-D to eluxadoline at 100 mg twice daily or placebo for 12 weeks. All participants were required to have an intact gallbladder as per the drug’s labeling guidance, and all had a self-reported recent inadequate response to loperamide.
The primary composite endpoint in the RELIEF trial was a 40% or greater improvement from baseline in the 11-point Daily Worst Abdominal Pain score plus a Bristol Stool Form score below 5 on the same day for at least 50% of study days. At baseline, participants had an average Worst Abdominal Pain score of 6.2 on the 0-10 scale and a Bristol score of 6.2. The primary endpoint was achieved at week 12 in 23% of the eluxadoline group, significantly better than the 10% rate in controls. The eluxadoline group also showed significantly greater improvement on the many secondary endpoints having to do with urgency-free days, stool consistency, bowel movement frequency, abdominal discomfort, bloating, and the experience of adequate relief of symptoms.
The safety profile of eluxadoline mirrored that of placebo, with no serious adverse events recorded in either study arm and a 2.9% study discontinuation rate because of treatment-emergent adverse events in the eluxadoline group. Asked why he thinks eluxadoline was effective in improving the full range of IBS-D symptoms when loperamide wasn’t, even though both drugs are mu-opioid receptor agonists, Dr. Brenner replied, “The problem is mu receptors line the entire GI tract, so you can actually push somebody from diarrhea to opioid-induced constipation – and that’s not the goal. What delta does is alleviate some of the adverse events by binding to the receptor, which results in increased transit time, reduced secretion, and increased absorption. Delta brings things back towards the center. We also believe antagonism of delta potentiates analgesic effects at the mu receptor, improves the pain component, gut symptoms, and stool symptoms.”
Dr. Brenner reported serving as a consultant to and member of a speaker’s bureau for Allergan, which markets eluxadoline and sponsored the RELIEF trial.
REPORTING FROM ACG 2019
Dupilumab-induced head and neck erythema described in atopic dermatitis patients
MADRID – A growing recognition that from their background skin disease emerged as a hot topic of discussion at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.

“During treatment with dupilumab, we saw something that is really different from the classic eczema that patients experienced prior to dupilumab, with no or minimal scaling, itch, or burning sensation. We do not believe this is a delayed effect of dupilumab on that specific region. We think this is a dupilumab-induced entity that we’re looking at. You should take home, in my opinion, that this is a common side effect that’s underreported in daily practice at this moment, and it’s not reported in clinical trials at all,” said Linde de Wijs, MD, of the department of dermatology, Erasmus University Medical Center in Rotterdam, the Netherlands.
She presented a detailed case series of seven affected patients which included histologic examination of lesional skin biopsies. The biopsies were characterized by a perivascular lymphohistiocytic infiltrate, an increase in ectatic capillaries in the papillary dermis, and a notable dearth of spongiosis, eosinophils, and neutrophils. Four patients had bulbous elongated rete ridges evocative of a psoriasiform dermatitis. The overall histologic picture was suggestive of a drug-induced skin reaction.
A striking finding was that, even though AD patients typically place high importance on achieving total clearing of disease on the head and neck, these seven closely studied patients nonetheless rated their treatment satisfaction as 9 out of a possible 10 points. Dr. de Wijs interpreted this as testimony to dupilumab’s potent efficacy and comparatively acceptable safety profile, especially the apparent side effect’s absence of scaling and itch.
“Remember, these are patients with really severe atopic dermatitis who’ve been treated with a lot of immunosuppressants prior to dupilumab,” she said.
Once the investigators began to suspect the existence of a novel dupilumab-induced skin reaction, they conducted a retrospective chart review of more than 150 patients treated with dupilumab (Dupixent) and determined that roughly 30% had developed this distinctive sharply demarcated patchy erythema on the head and neck characterized by absence of itch. The sequence involved clearance of the AD in response to dupilumab, followed by gradual development of the head and neck erythema 10-39 weeks after the start of treatment.
The erythema proved treatment refractory. Dr. de Wijs and her colleagues tried topical corticosteroids, including potent ones, as well as topical tacrolimus, antifungals, antibiotics, emollients, oral steroids, and antihistamines, to no avail. Patch testing to investigate allergic contact dermatitis as a possible etiology was unremarkable.
She hypothesized that, since dupilumab blocks the key signaling pathways for Th2 T-cell differentiation by targeting the interleukin-4 receptor alpha, it’s possible that the biologic promotes a shift towards activation of the Th17 pathway, which might explain the observed histologic findings.
The fact that this erythema wasn’t reported in the major randomized clinical trials of dupilumab underscores the enormous value of clinical practice registries, she said.
“We are not the only ones observing this phenomenon,” noted Dr. de Wijs, citing recently published reports by other investigators (J Am Acad Dermatol. 2020 Jan;82[1]:230-2; JAMA Dermatol. 2019 Jul 1;155[7]:850-2).
Indeed, her talk was immediately followed by a presentation by Sebastien Barbarot, MD, PhD, who reported on a French national retrospective study of head and neck dermatitis arising in patients on dupilumab that was conducted by the French Atopic Dermatitis Network using the organization’s GREAT database. Among 1,000 adult patients with AD treated with the biologic at 29 French centers, 10 developed a de novo head and neck dermatitis, and 32 others experienced more than 50% worsening of eczema signs on the head and neck from baseline beginning about 2 months after starting on dupilumab.
This 4.2% incidence is probably an underestimate, since dermatologists weren’t aware of the phenomenon and didn’t specifically ask patients about it, observed Dr. Barbarot, a dermatologist at the University of Nantes (France).
Among the key findings: No differences in clinical characteristics were found between the de novo and exacerbation groups, nearly half of affected patients had concomitant conjunctivitis, and seven patients discontinued dupilumab because of an intolerable burning sensation on the head/neck.
“I think this condition is quite different from rosacea,” Dr. Barbarot emphasized.
French dermatologists generally turned to topical corticosteroids or topical tacrolimus to treat the face and neck dermatitis, with mixed results; 22 of the 42 patients showed improvement and 8 worsened.

Marjolein de Bruin-Weller, MD, PhD, a dermatologist at Utrecht (the Netherlands) University and head of the Dutch National Eczema Expertise Center, said she and her colleagues have also encountered this dupilumab-related head and neck erythema and are convinced that a subset of affected patients have Malassezia-induced dermatitis with neutrophils present on lesional biopsies. “It responds very well to treatment. I think it’s very important to try itraconazole because sometimes it works,” she said.
Dr. de Wijs replied that she and her coworkers tried 2 weeks of itraconazole in several patients, with no effect. And none of their seven biopsied patients had an increase in neutrophils.
“It might be a very heterogenous polyform entity that we’re now observing,” she commented. Dr. de Bruin-Weller concurred.
Dr. Barbarot said he’d be interested in a formal study of antifungal therapy in patients with dupilumab-related head and neck dermatitis. Mechanistically, it seems plausible that dupilumab-induced activation of the TH17 pathway might lead to proliferation of Malassezia fungus.
Dr. de Wijs and Dr. Barbarot reported having no financial conflicts regarding their respective studies, which were conducted free of commercial sponsorship.
MADRID – A growing recognition that from their background skin disease emerged as a hot topic of discussion at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“During treatment with dupilumab, we saw something that is really different from the classic eczema that patients experienced prior to dupilumab, with no or minimal scaling, itch, or burning sensation. We do not believe this is a delayed effect of dupilumab on that specific region. We think this is a dupilumab-induced entity that we’re looking at. You should take home, in my opinion, that this is a common side effect that’s underreported in daily practice at this moment, and it’s not reported in clinical trials at all,” said Linde de Wijs, MD, of the department of dermatology, Erasmus University Medical Center in Rotterdam, the Netherlands.
She presented a detailed case series of seven affected patients which included histologic examination of lesional skin biopsies. The biopsies were characterized by a perivascular lymphohistiocytic infiltrate, an increase in ectatic capillaries in the papillary dermis, and a notable dearth of spongiosis, eosinophils, and neutrophils. Four patients had bulbous elongated rete ridges evocative of a psoriasiform dermatitis. The overall histologic picture was suggestive of a drug-induced skin reaction.
A striking finding was that, even though AD patients typically place high importance on achieving total clearing of disease on the head and neck, these seven closely studied patients nonetheless rated their treatment satisfaction as 9 out of a possible 10 points. Dr. de Wijs interpreted this as testimony to dupilumab’s potent efficacy and comparatively acceptable safety profile, especially the apparent side effect’s absence of scaling and itch.
“Remember, these are patients with really severe atopic dermatitis who’ve been treated with a lot of immunosuppressants prior to dupilumab,” she said.
Once the investigators began to suspect the existence of a novel dupilumab-induced skin reaction, they conducted a retrospective chart review of more than 150 patients treated with dupilumab (Dupixent) and determined that roughly 30% had developed this distinctive sharply demarcated patchy erythema on the head and neck characterized by absence of itch. The sequence involved clearance of the AD in response to dupilumab, followed by gradual development of the head and neck erythema 10-39 weeks after the start of treatment.
The erythema proved treatment refractory. Dr. de Wijs and her colleagues tried topical corticosteroids, including potent ones, as well as topical tacrolimus, antifungals, antibiotics, emollients, oral steroids, and antihistamines, to no avail. Patch testing to investigate allergic contact dermatitis as a possible etiology was unremarkable.
She hypothesized that, since dupilumab blocks the key signaling pathways for Th2 T-cell differentiation by targeting the interleukin-4 receptor alpha, it’s possible that the biologic promotes a shift towards activation of the Th17 pathway, which might explain the observed histologic findings.
The fact that this erythema wasn’t reported in the major randomized clinical trials of dupilumab underscores the enormous value of clinical practice registries, she said.
“We are not the only ones observing this phenomenon,” noted Dr. de Wijs, citing recently published reports by other investigators (J Am Acad Dermatol. 2020 Jan;82[1]:230-2; JAMA Dermatol. 2019 Jul 1;155[7]:850-2).
Indeed, her talk was immediately followed by a presentation by Sebastien Barbarot, MD, PhD, who reported on a French national retrospective study of head and neck dermatitis arising in patients on dupilumab that was conducted by the French Atopic Dermatitis Network using the organization’s GREAT database. Among 1,000 adult patients with AD treated with the biologic at 29 French centers, 10 developed a de novo head and neck dermatitis, and 32 others experienced more than 50% worsening of eczema signs on the head and neck from baseline beginning about 2 months after starting on dupilumab.
This 4.2% incidence is probably an underestimate, since dermatologists weren’t aware of the phenomenon and didn’t specifically ask patients about it, observed Dr. Barbarot, a dermatologist at the University of Nantes (France).
Among the key findings: No differences in clinical characteristics were found between the de novo and exacerbation groups, nearly half of affected patients had concomitant conjunctivitis, and seven patients discontinued dupilumab because of an intolerable burning sensation on the head/neck.
“I think this condition is quite different from rosacea,” Dr. Barbarot emphasized.
French dermatologists generally turned to topical corticosteroids or topical tacrolimus to treat the face and neck dermatitis, with mixed results; 22 of the 42 patients showed improvement and 8 worsened.
Marjolein de Bruin-Weller, MD, PhD, a dermatologist at Utrecht (the Netherlands) University and head of the Dutch National Eczema Expertise Center, said she and her colleagues have also encountered this dupilumab-related head and neck erythema and are convinced that a subset of affected patients have Malassezia-induced dermatitis with neutrophils present on lesional biopsies. “It responds very well to treatment. I think it’s very important to try itraconazole because sometimes it works,” she said.
Dr. de Wijs replied that she and her coworkers tried 2 weeks of itraconazole in several patients, with no effect. And none of their seven biopsied patients had an increase in neutrophils.
“It might be a very heterogenous polyform entity that we’re now observing,” she commented. Dr. de Bruin-Weller concurred.
Dr. Barbarot said he’d be interested in a formal study of antifungal therapy in patients with dupilumab-related head and neck dermatitis. Mechanistically, it seems plausible that dupilumab-induced activation of the TH17 pathway might lead to proliferation of Malassezia fungus.
Dr. de Wijs and Dr. Barbarot reported having no financial conflicts regarding their respective studies, which were conducted free of commercial sponsorship.
MADRID – A growing recognition that from their background skin disease emerged as a hot topic of discussion at a meeting of the European Task Force on Atopic Dermatitis held in conjunction with the annual congress of the European Academy of Dermatology and Venereology.
“During treatment with dupilumab, we saw something that is really different from the classic eczema that patients experienced prior to dupilumab, with no or minimal scaling, itch, or burning sensation. We do not believe this is a delayed effect of dupilumab on that specific region. We think this is a dupilumab-induced entity that we’re looking at. You should take home, in my opinion, that this is a common side effect that’s underreported in daily practice at this moment, and it’s not reported in clinical trials at all,” said Linde de Wijs, MD, of the department of dermatology, Erasmus University Medical Center in Rotterdam, the Netherlands.
She presented a detailed case series of seven affected patients which included histologic examination of lesional skin biopsies. The biopsies were characterized by a perivascular lymphohistiocytic infiltrate, an increase in ectatic capillaries in the papillary dermis, and a notable dearth of spongiosis, eosinophils, and neutrophils. Four patients had bulbous elongated rete ridges evocative of a psoriasiform dermatitis. The overall histologic picture was suggestive of a drug-induced skin reaction.
A striking finding was that, even though AD patients typically place high importance on achieving total clearing of disease on the head and neck, these seven closely studied patients nonetheless rated their treatment satisfaction as 9 out of a possible 10 points. Dr. de Wijs interpreted this as testimony to dupilumab’s potent efficacy and comparatively acceptable safety profile, especially the apparent side effect’s absence of scaling and itch.
“Remember, these are patients with really severe atopic dermatitis who’ve been treated with a lot of immunosuppressants prior to dupilumab,” she said.
Once the investigators began to suspect the existence of a novel dupilumab-induced skin reaction, they conducted a retrospective chart review of more than 150 patients treated with dupilumab (Dupixent) and determined that roughly 30% had developed this distinctive sharply demarcated patchy erythema on the head and neck characterized by absence of itch. The sequence involved clearance of the AD in response to dupilumab, followed by gradual development of the head and neck erythema 10-39 weeks after the start of treatment.
The erythema proved treatment refractory. Dr. de Wijs and her colleagues tried topical corticosteroids, including potent ones, as well as topical tacrolimus, antifungals, antibiotics, emollients, oral steroids, and antihistamines, to no avail. Patch testing to investigate allergic contact dermatitis as a possible etiology was unremarkable.
She hypothesized that, since dupilumab blocks the key signaling pathways for Th2 T-cell differentiation by targeting the interleukin-4 receptor alpha, it’s possible that the biologic promotes a shift towards activation of the Th17 pathway, which might explain the observed histologic findings.
The fact that this erythema wasn’t reported in the major randomized clinical trials of dupilumab underscores the enormous value of clinical practice registries, she said.
“We are not the only ones observing this phenomenon,” noted Dr. de Wijs, citing recently published reports by other investigators (J Am Acad Dermatol. 2020 Jan;82[1]:230-2; JAMA Dermatol. 2019 Jul 1;155[7]:850-2).
Indeed, her talk was immediately followed by a presentation by Sebastien Barbarot, MD, PhD, who reported on a French national retrospective study of head and neck dermatitis arising in patients on dupilumab that was conducted by the French Atopic Dermatitis Network using the organization’s GREAT database. Among 1,000 adult patients with AD treated with the biologic at 29 French centers, 10 developed a de novo head and neck dermatitis, and 32 others experienced more than 50% worsening of eczema signs on the head and neck from baseline beginning about 2 months after starting on dupilumab.
This 4.2% incidence is probably an underestimate, since dermatologists weren’t aware of the phenomenon and didn’t specifically ask patients about it, observed Dr. Barbarot, a dermatologist at the University of Nantes (France).
Among the key findings: No differences in clinical characteristics were found between the de novo and exacerbation groups, nearly half of affected patients had concomitant conjunctivitis, and seven patients discontinued dupilumab because of an intolerable burning sensation on the head/neck.
“I think this condition is quite different from rosacea,” Dr. Barbarot emphasized.
French dermatologists generally turned to topical corticosteroids or topical tacrolimus to treat the face and neck dermatitis, with mixed results; 22 of the 42 patients showed improvement and 8 worsened.
Marjolein de Bruin-Weller, MD, PhD, a dermatologist at Utrecht (the Netherlands) University and head of the Dutch National Eczema Expertise Center, said she and her colleagues have also encountered this dupilumab-related head and neck erythema and are convinced that a subset of affected patients have Malassezia-induced dermatitis with neutrophils present on lesional biopsies. “It responds very well to treatment. I think it’s very important to try itraconazole because sometimes it works,” she said.
Dr. de Wijs replied that she and her coworkers tried 2 weeks of itraconazole in several patients, with no effect. And none of their seven biopsied patients had an increase in neutrophils.
“It might be a very heterogenous polyform entity that we’re now observing,” she commented. Dr. de Bruin-Weller concurred.
Dr. Barbarot said he’d be interested in a formal study of antifungal therapy in patients with dupilumab-related head and neck dermatitis. Mechanistically, it seems plausible that dupilumab-induced activation of the TH17 pathway might lead to proliferation of Malassezia fungus.
Dr. de Wijs and Dr. Barbarot reported having no financial conflicts regarding their respective studies, which were conducted free of commercial sponsorship.
REPORTING FROM THE EADV CONGRESS