User login
Measles Resurgence: A Dermatologist’s Guide
Measles Resurgence: A Dermatologist’s Guide
Measles, also known as rubeola, is a highly contagious paramyxovirus that has neared elimination in the United States since 2000 due to widespread adoption of the measles vaccine; however, measles recently has made a comeback, with outbreaks reported in more than 60 countries. In the United States, vaccine hesitancy coupled with decreasing vaccination rates, international travel to endemic areas, and decreased funding and resources for monitoring and immunization programs likely led to a re-emergence of measles cases.1,2 The resurgence of measles is troubling given its infectiousness and potential severity in at-risk populations. Since measles has a basic reproduction number of 12 to 18 (ie, 1 infected individual will on average infect 12 to 18 others3), it has the capacity to spread quickly. This is why, prior to the development of the measles vaccine in the 1960s, it was responsible for millions of deaths across the globe.
Prior to the introduction of the measles vaccine, both physicians and the public generally were aware of the signs and symptoms of measles due to its prevalence; however, since there have been so few cases in recent decades, images and descriptions of patients presenting with measles can be found only in textbooks, and many physicians are ill-prepared to diagnose the disease.4 In response to the recent surge in measles cases, dermatologists—who often are among the first medical professionals to encounter febrile patients with rashes—must be prepared to bridge this divide. Herein, we review the clinical signs, diagnostic approach, operational precautions, and public health responsibilities that dermatologists must relearn amid the current measles outbreak.
Background
Measles is primarily transmitted via respiratory droplets and may remain airborne for up to 2 hours.5 It also can be transmitted through direct contact with secretions such as mucus. Indirect transmission via fomites, while certainly plausible, is thought to be the least effective mechanism of transmission.6 Following exposure, the incubation period ranges from 7 to 21 days, during which the virus replicates asymptomatically before causing clinical disease.7 Herd immunity for measles requires 93% immunity in the population; public health agencies typically target greater than 95% immunity.8 Humans are the only reservoir for the measles virus, making eradication possible.
The road to eradication began with the introduction of the measles vaccine in 1963 and subsequent development of the combined measles-mumps-rubella (MMR) vaccine in 1971. As MMR is a live vaccine, 2 doses confer approximately 97% protection.9 The first dose is given at 12 to 15 months of age, and the second dose is given at 4 to 6 years of age. Immunity is considered lifelong, and the Centers for Disease Control and Prevention and the World Health Organization do not recommend routine measles boosters for individuals who have completed the primary 2-dose series.10,11
Widespread vaccination led to a dramatic reduction in incidence, with many countries eliminating measles infections.7 The United States declared measles eliminated in 2000, with confirmed cases between 2000 and 2020 ranging from 37 to 1282.12 Vaccination progress stalled in the late 1990s due to vaccine hesitancy resulting from (subsequently debunked) reports of an association between the MMR vaccine and autism.13 Despite efforts to correct this misinformation, many patients continue to espouse these concerns.
Recognizing Measles: Clinical Presentation
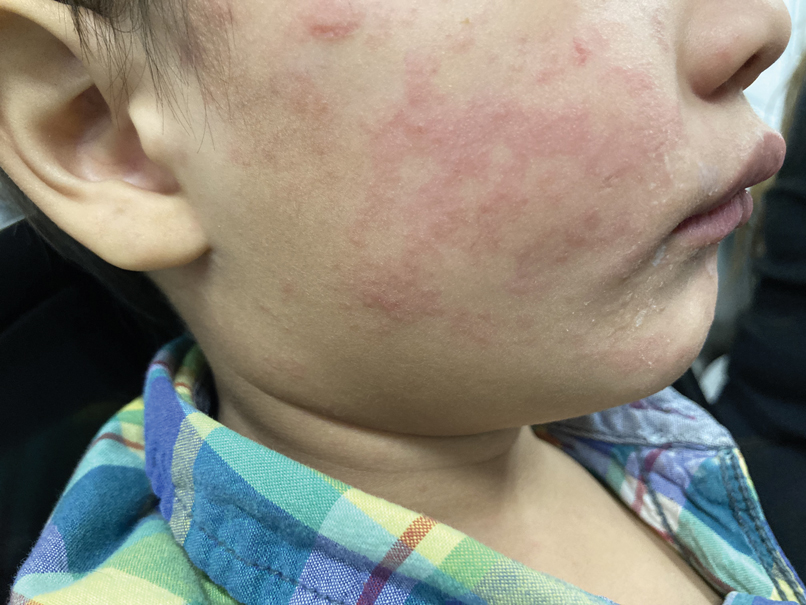
Measles, which most often manifests in childhood but also can occur in adults, follows a distinctive clinical course. The prodromal phase is characterized by high fever, cough, coryza (nasal congestion), and conjunctivitis— conjunctivitis—the 3 “Cs” that serve as early warning signs of the disease. Patients may develop small white macules on the buccal mucosa known as Koplik spots (phonetically the fourth “C”), which appear just before the rash. Three to 5 days after the onset of systemic symptoms, patients will develop a classic morbilliform exanthem. In some cases, the exanthem manifests on the head and neck (Figure 1)—first behind the ears and along the hairline, then spreading caudally to the trunk and extremities. The lesions may become confluent, with patients presenting with diffuse erythema. The exanthem fades over several days to weeks, often accompanied by superficial desquamation.14
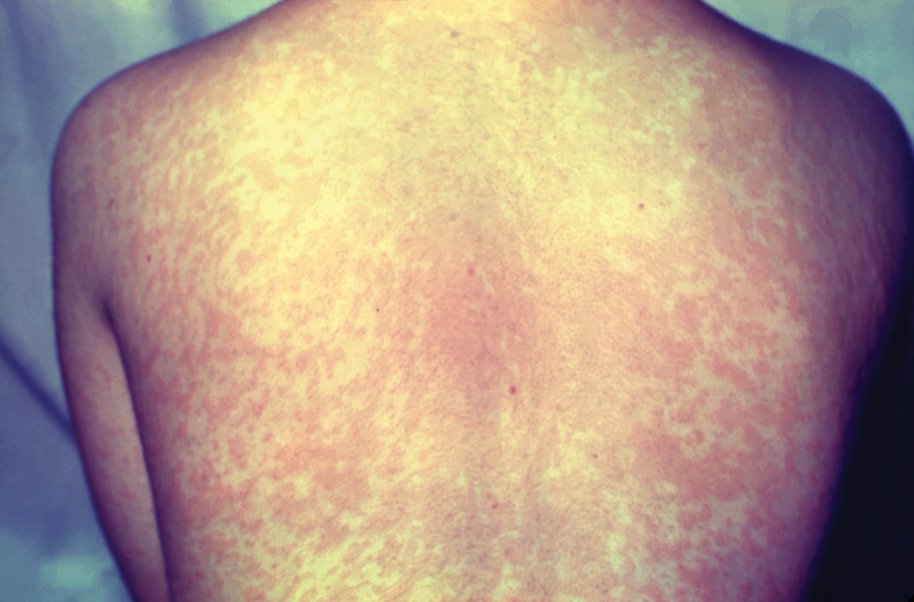
Given the nonspecificity of the early symptoms of measles, a high index of suspicion is needed for patients presenting with a febrile illness and a morbilliform eruption (Figure 2). Consideration of MMR vaccination status, exposure history, and local outbreak patterns can help guide risk stratification and the need for testing. Immunocompromised individuals, including those receiving immunosuppressive therapies for dermatologic conditions, may present atypically, lacking the prototypical exanthem or displaying milder signs and further complicating the diagnosis.15 The differential diagnosis for measles includes a drug reaction or other viral exanthem, and a detailed history may help elucidate the culprit.
Evaluation and Diagnosis
Definitive diagnosis of measles relies on both molecular and serologic testing. Nasopharyngeal swabs for measles polymerase chain reaction testing are obtained using synthetic (noncotton) swabs placed in a viral transport medium. Serum samples also should be collected for measles IgM and IgG antibody testing. Importantly, measles is a reportable illness, and testing may be coordinated with local departments of health.
Determining a patient’s immune status may be important for certain populations. Patients with documented 2-dose MMR vaccination, positive measles IgG serology, or a prior confirmed measles infection are considered immune. While a positive measles IgG indicates immunity, a negative result in an exposed patient should prompt consideration of postexposure prophylaxis with intravenous immunoglobulin.
Many patients, specifically those presenting to dermatology, are taking immunomodulatory or immunosuppressive medications—a contraindication for vaccination with the live MMR vaccine. At the time of publication, there was a single reported case of a patient taking a tumor necrosis factor α inhibitor for rheumatoid arthritis who had acquired measles.16 While the benefits of titer assessment in patients who are starting or continuing immunomodulatory therapy are not known and currently it is not recommended by the Centers for Disease Control and Prevention, dermatologists might consider checking MMR titers and vaccinating (or referring for vaccination) nonimmune patients.17
Infection Control
Early identification of a suspected measles case is paramount. Patients in whom measles is a possibility should be isolated as quickly as possible, and the patient and accompanying caregivers should be masked. Clinical staff should don appropriate personal protective equipment, including an N95 mask. Coordination with the local department of health must occur as soon as measles is suspected.
If testing is an option in the outpatient setting, a nasopharyngeal viral swab and serologic titers can be obtained. If testing is not available on site, patients should be sent to appropriate care facilities; prenotification is critical to prevent nosocomial outbreaks. Patients should be encouraged to isolate and avoid public spaces and/or public transport for 4 days following development of an exanthem.18 Offices should develop clinical protocols for suspected measles cases with training for clinical and office staff.
Final Thoughts
As measles outbreaks become more prevalent, it is incumbent upon physicians to remind ourselves of the signs and symptoms of this largely eliminated disease so that we may pursue early detection and intervention strategies. The primary cutaneous manifestations of measles make dermatologists critical to early recognition and containment efforts. Dermatologists should prepare for the arrival of patients with measles by maintaining vigilance for the classic signs of the disease, implementing stringent isolation protocols, verifying patient immunity when appropriate, and partnering closely with public health authorities.
More broadly, efforts to contain and re-establish a paradigm for eliminating measles outbreaks must be pursued. Encouraging vaccination and developing programs to help combat misinformation surrounding vaccines are critical to this effort. In an era of vaccine hesitancy, measles is a multidisciplinary public health emergency. Dermatologists must remain ready.
- Bedford H, Elliman D. Measles rates are rising again. BMJ. 2024;384.
- Harris E. Measles outbreaks grow amid declining vaccination rates. JAMA. 2023;330:2242.
- Guerra FM, Bolotin S, Lim G, et al. The basic reproduction number (R0) of measles: a systematic review. Lancet Infect Dis. 2017;17:E420-E428.
- Swartz MK. Measles: public and professional education. J Pediatr Health Care. 2019;33:367-368.
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for measles in healthcare settings. Accessed April 27, 2025. https://www.cdc.gov/infection-control/hcp/measles/
- Moss WJ, Griffin DE, Feinstone WH. Measles. In: Vaccines for Biodefense and Emerging and Neglected Diseases. Elsevier; 2009: 551-565.
- Moss WJ. Measles. Lancet. 2017;390:2490-2502.
- Maintain the vaccination coverage level of 2 doses of the MMR vaccine for children in kindergarten— IID04. Healthy People 2030 website. Accessed May 6, 2025. https://odphp.health.gov/healthypeople/objectives-and-data/browse-objectives/vaccination/maintain-vaccination-coverage-level-2-doses-mmr-vaccine-children-kindergarten-iid-04
- Franconeri L, Antona D, Cauchemez S, et al. Two-dose measles vaccine effectiveness remains high over time: a French observational study, 2017–2019. Vaccine. 2023;41:5797-5804.
- World Health Organization. Measles. Accessed May 8, 2025. https:// www.who.int/news-room/fact-sheets/detail/measles
- Centers for Disease Control and Prevention. Measles vaccine recommendations. Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/vaccine-considerations/index.html
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Accessed May 6, 2025. https://www.cdc.gov/measles/cases-outbreaks.html
- Dyer C. Lancet retracts Wakefield’s MMR paper. BMJ. 2010;340.
- Alves Graber EM, Andrade FJ, Bost W, et al. An update and review of measles for emergency physicians. J Emerg Med. 2020;58:610-615.
- Kaplan LJ, Daum RS, Smaron M, et al. Severe measles in immunocompromised patients. JAMA. 1992;267:1237-1241.
- Takahashi E, Kurosaka D, Yoshida K, et al. Onset of modified measles after etanercept treatment in rheumatoid arthritis. Japanese J Clin Immunol. 2010;33:37-41.
- Worth A, Waldman RA, Dieckhaus K, et al. Art of prevention: our approach to the measles-mumps-rubella vaccine in adult patients vaccinated against measles before 1968 on biologic therapy for the treatment of psoriasis. Int J Womens Dermatol. 2019;6:94.
- Centers for Disease Control and Prevention. Clinical overview of measles (rubeola). Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/clinical-overview/index.html
Measles, also known as rubeola, is a highly contagious paramyxovirus that has neared elimination in the United States since 2000 due to widespread adoption of the measles vaccine; however, measles recently has made a comeback, with outbreaks reported in more than 60 countries. In the United States, vaccine hesitancy coupled with decreasing vaccination rates, international travel to endemic areas, and decreased funding and resources for monitoring and immunization programs likely led to a re-emergence of measles cases.1,2 The resurgence of measles is troubling given its infectiousness and potential severity in at-risk populations. Since measles has a basic reproduction number of 12 to 18 (ie, 1 infected individual will on average infect 12 to 18 others3), it has the capacity to spread quickly. This is why, prior to the development of the measles vaccine in the 1960s, it was responsible for millions of deaths across the globe.
Prior to the introduction of the measles vaccine, both physicians and the public generally were aware of the signs and symptoms of measles due to its prevalence; however, since there have been so few cases in recent decades, images and descriptions of patients presenting with measles can be found only in textbooks, and many physicians are ill-prepared to diagnose the disease.4 In response to the recent surge in measles cases, dermatologists—who often are among the first medical professionals to encounter febrile patients with rashes—must be prepared to bridge this divide. Herein, we review the clinical signs, diagnostic approach, operational precautions, and public health responsibilities that dermatologists must relearn amid the current measles outbreak.
Background
Measles is primarily transmitted via respiratory droplets and may remain airborne for up to 2 hours.5 It also can be transmitted through direct contact with secretions such as mucus. Indirect transmission via fomites, while certainly plausible, is thought to be the least effective mechanism of transmission.6 Following exposure, the incubation period ranges from 7 to 21 days, during which the virus replicates asymptomatically before causing clinical disease.7 Herd immunity for measles requires 93% immunity in the population; public health agencies typically target greater than 95% immunity.8 Humans are the only reservoir for the measles virus, making eradication possible.
The road to eradication began with the introduction of the measles vaccine in 1963 and subsequent development of the combined measles-mumps-rubella (MMR) vaccine in 1971. As MMR is a live vaccine, 2 doses confer approximately 97% protection.9 The first dose is given at 12 to 15 months of age, and the second dose is given at 4 to 6 years of age. Immunity is considered lifelong, and the Centers for Disease Control and Prevention and the World Health Organization do not recommend routine measles boosters for individuals who have completed the primary 2-dose series.10,11
Widespread vaccination led to a dramatic reduction in incidence, with many countries eliminating measles infections.7 The United States declared measles eliminated in 2000, with confirmed cases between 2000 and 2020 ranging from 37 to 1282.12 Vaccination progress stalled in the late 1990s due to vaccine hesitancy resulting from (subsequently debunked) reports of an association between the MMR vaccine and autism.13 Despite efforts to correct this misinformation, many patients continue to espouse these concerns.
Recognizing Measles: Clinical Presentation
Measles, which most often manifests in childhood but also can occur in adults, follows a distinctive clinical course. The prodromal phase is characterized by high fever, cough, coryza (nasal congestion), and conjunctivitis— conjunctivitis—the 3 “Cs” that serve as early warning signs of the disease. Patients may develop small white macules on the buccal mucosa known as Koplik spots (phonetically the fourth “C”), which appear just before the rash. Three to 5 days after the onset of systemic symptoms, patients will develop a classic morbilliform exanthem. In some cases, the exanthem manifests on the head and neck (Figure 1)—first behind the ears and along the hairline, then spreading caudally to the trunk and extremities. The lesions may become confluent, with patients presenting with diffuse erythema. The exanthem fades over several days to weeks, often accompanied by superficial desquamation.14
Given the nonspecificity of the early symptoms of measles, a high index of suspicion is needed for patients presenting with a febrile illness and a morbilliform eruption (Figure 2). Consideration of MMR vaccination status, exposure history, and local outbreak patterns can help guide risk stratification and the need for testing. Immunocompromised individuals, including those receiving immunosuppressive therapies for dermatologic conditions, may present atypically, lacking the prototypical exanthem or displaying milder signs and further complicating the diagnosis.15 The differential diagnosis for measles includes a drug reaction or other viral exanthem, and a detailed history may help elucidate the culprit.
Evaluation and Diagnosis
Definitive diagnosis of measles relies on both molecular and serologic testing. Nasopharyngeal swabs for measles polymerase chain reaction testing are obtained using synthetic (noncotton) swabs placed in a viral transport medium. Serum samples also should be collected for measles IgM and IgG antibody testing. Importantly, measles is a reportable illness, and testing may be coordinated with local departments of health.
Determining a patient’s immune status may be important for certain populations. Patients with documented 2-dose MMR vaccination, positive measles IgG serology, or a prior confirmed measles infection are considered immune. While a positive measles IgG indicates immunity, a negative result in an exposed patient should prompt consideration of postexposure prophylaxis with intravenous immunoglobulin.
Many patients, specifically those presenting to dermatology, are taking immunomodulatory or immunosuppressive medications—a contraindication for vaccination with the live MMR vaccine. At the time of publication, there was a single reported case of a patient taking a tumor necrosis factor α inhibitor for rheumatoid arthritis who had acquired measles.16 While the benefits of titer assessment in patients who are starting or continuing immunomodulatory therapy are not known and currently it is not recommended by the Centers for Disease Control and Prevention, dermatologists might consider checking MMR titers and vaccinating (or referring for vaccination) nonimmune patients.17
Infection Control
Early identification of a suspected measles case is paramount. Patients in whom measles is a possibility should be isolated as quickly as possible, and the patient and accompanying caregivers should be masked. Clinical staff should don appropriate personal protective equipment, including an N95 mask. Coordination with the local department of health must occur as soon as measles is suspected.
If testing is an option in the outpatient setting, a nasopharyngeal viral swab and serologic titers can be obtained. If testing is not available on site, patients should be sent to appropriate care facilities; prenotification is critical to prevent nosocomial outbreaks. Patients should be encouraged to isolate and avoid public spaces and/or public transport for 4 days following development of an exanthem.18 Offices should develop clinical protocols for suspected measles cases with training for clinical and office staff.
Final Thoughts
As measles outbreaks become more prevalent, it is incumbent upon physicians to remind ourselves of the signs and symptoms of this largely eliminated disease so that we may pursue early detection and intervention strategies. The primary cutaneous manifestations of measles make dermatologists critical to early recognition and containment efforts. Dermatologists should prepare for the arrival of patients with measles by maintaining vigilance for the classic signs of the disease, implementing stringent isolation protocols, verifying patient immunity when appropriate, and partnering closely with public health authorities.
More broadly, efforts to contain and re-establish a paradigm for eliminating measles outbreaks must be pursued. Encouraging vaccination and developing programs to help combat misinformation surrounding vaccines are critical to this effort. In an era of vaccine hesitancy, measles is a multidisciplinary public health emergency. Dermatologists must remain ready.
Measles, also known as rubeola, is a highly contagious paramyxovirus that has neared elimination in the United States since 2000 due to widespread adoption of the measles vaccine; however, measles recently has made a comeback, with outbreaks reported in more than 60 countries. In the United States, vaccine hesitancy coupled with decreasing vaccination rates, international travel to endemic areas, and decreased funding and resources for monitoring and immunization programs likely led to a re-emergence of measles cases.1,2 The resurgence of measles is troubling given its infectiousness and potential severity in at-risk populations. Since measles has a basic reproduction number of 12 to 18 (ie, 1 infected individual will on average infect 12 to 18 others3), it has the capacity to spread quickly. This is why, prior to the development of the measles vaccine in the 1960s, it was responsible for millions of deaths across the globe.
Prior to the introduction of the measles vaccine, both physicians and the public generally were aware of the signs and symptoms of measles due to its prevalence; however, since there have been so few cases in recent decades, images and descriptions of patients presenting with measles can be found only in textbooks, and many physicians are ill-prepared to diagnose the disease.4 In response to the recent surge in measles cases, dermatologists—who often are among the first medical professionals to encounter febrile patients with rashes—must be prepared to bridge this divide. Herein, we review the clinical signs, diagnostic approach, operational precautions, and public health responsibilities that dermatologists must relearn amid the current measles outbreak.
Background
Measles is primarily transmitted via respiratory droplets and may remain airborne for up to 2 hours.5 It also can be transmitted through direct contact with secretions such as mucus. Indirect transmission via fomites, while certainly plausible, is thought to be the least effective mechanism of transmission.6 Following exposure, the incubation period ranges from 7 to 21 days, during which the virus replicates asymptomatically before causing clinical disease.7 Herd immunity for measles requires 93% immunity in the population; public health agencies typically target greater than 95% immunity.8 Humans are the only reservoir for the measles virus, making eradication possible.
The road to eradication began with the introduction of the measles vaccine in 1963 and subsequent development of the combined measles-mumps-rubella (MMR) vaccine in 1971. As MMR is a live vaccine, 2 doses confer approximately 97% protection.9 The first dose is given at 12 to 15 months of age, and the second dose is given at 4 to 6 years of age. Immunity is considered lifelong, and the Centers for Disease Control and Prevention and the World Health Organization do not recommend routine measles boosters for individuals who have completed the primary 2-dose series.10,11
Widespread vaccination led to a dramatic reduction in incidence, with many countries eliminating measles infections.7 The United States declared measles eliminated in 2000, with confirmed cases between 2000 and 2020 ranging from 37 to 1282.12 Vaccination progress stalled in the late 1990s due to vaccine hesitancy resulting from (subsequently debunked) reports of an association between the MMR vaccine and autism.13 Despite efforts to correct this misinformation, many patients continue to espouse these concerns.
Recognizing Measles: Clinical Presentation
Measles, which most often manifests in childhood but also can occur in adults, follows a distinctive clinical course. The prodromal phase is characterized by high fever, cough, coryza (nasal congestion), and conjunctivitis— conjunctivitis—the 3 “Cs” that serve as early warning signs of the disease. Patients may develop small white macules on the buccal mucosa known as Koplik spots (phonetically the fourth “C”), which appear just before the rash. Three to 5 days after the onset of systemic symptoms, patients will develop a classic morbilliform exanthem. In some cases, the exanthem manifests on the head and neck (Figure 1)—first behind the ears and along the hairline, then spreading caudally to the trunk and extremities. The lesions may become confluent, with patients presenting with diffuse erythema. The exanthem fades over several days to weeks, often accompanied by superficial desquamation.14
Given the nonspecificity of the early symptoms of measles, a high index of suspicion is needed for patients presenting with a febrile illness and a morbilliform eruption (Figure 2). Consideration of MMR vaccination status, exposure history, and local outbreak patterns can help guide risk stratification and the need for testing. Immunocompromised individuals, including those receiving immunosuppressive therapies for dermatologic conditions, may present atypically, lacking the prototypical exanthem or displaying milder signs and further complicating the diagnosis.15 The differential diagnosis for measles includes a drug reaction or other viral exanthem, and a detailed history may help elucidate the culprit.
Evaluation and Diagnosis
Definitive diagnosis of measles relies on both molecular and serologic testing. Nasopharyngeal swabs for measles polymerase chain reaction testing are obtained using synthetic (noncotton) swabs placed in a viral transport medium. Serum samples also should be collected for measles IgM and IgG antibody testing. Importantly, measles is a reportable illness, and testing may be coordinated with local departments of health.
Determining a patient’s immune status may be important for certain populations. Patients with documented 2-dose MMR vaccination, positive measles IgG serology, or a prior confirmed measles infection are considered immune. While a positive measles IgG indicates immunity, a negative result in an exposed patient should prompt consideration of postexposure prophylaxis with intravenous immunoglobulin.
Many patients, specifically those presenting to dermatology, are taking immunomodulatory or immunosuppressive medications—a contraindication for vaccination with the live MMR vaccine. At the time of publication, there was a single reported case of a patient taking a tumor necrosis factor α inhibitor for rheumatoid arthritis who had acquired measles.16 While the benefits of titer assessment in patients who are starting or continuing immunomodulatory therapy are not known and currently it is not recommended by the Centers for Disease Control and Prevention, dermatologists might consider checking MMR titers and vaccinating (or referring for vaccination) nonimmune patients.17
Infection Control
Early identification of a suspected measles case is paramount. Patients in whom measles is a possibility should be isolated as quickly as possible, and the patient and accompanying caregivers should be masked. Clinical staff should don appropriate personal protective equipment, including an N95 mask. Coordination with the local department of health must occur as soon as measles is suspected.
If testing is an option in the outpatient setting, a nasopharyngeal viral swab and serologic titers can be obtained. If testing is not available on site, patients should be sent to appropriate care facilities; prenotification is critical to prevent nosocomial outbreaks. Patients should be encouraged to isolate and avoid public spaces and/or public transport for 4 days following development of an exanthem.18 Offices should develop clinical protocols for suspected measles cases with training for clinical and office staff.
Final Thoughts
As measles outbreaks become more prevalent, it is incumbent upon physicians to remind ourselves of the signs and symptoms of this largely eliminated disease so that we may pursue early detection and intervention strategies. The primary cutaneous manifestations of measles make dermatologists critical to early recognition and containment efforts. Dermatologists should prepare for the arrival of patients with measles by maintaining vigilance for the classic signs of the disease, implementing stringent isolation protocols, verifying patient immunity when appropriate, and partnering closely with public health authorities.
More broadly, efforts to contain and re-establish a paradigm for eliminating measles outbreaks must be pursued. Encouraging vaccination and developing programs to help combat misinformation surrounding vaccines are critical to this effort. In an era of vaccine hesitancy, measles is a multidisciplinary public health emergency. Dermatologists must remain ready.
- Bedford H, Elliman D. Measles rates are rising again. BMJ. 2024;384.
- Harris E. Measles outbreaks grow amid declining vaccination rates. JAMA. 2023;330:2242.
- Guerra FM, Bolotin S, Lim G, et al. The basic reproduction number (R0) of measles: a systematic review. Lancet Infect Dis. 2017;17:E420-E428.
- Swartz MK. Measles: public and professional education. J Pediatr Health Care. 2019;33:367-368.
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for measles in healthcare settings. Accessed April 27, 2025. https://www.cdc.gov/infection-control/hcp/measles/
- Moss WJ, Griffin DE, Feinstone WH. Measles. In: Vaccines for Biodefense and Emerging and Neglected Diseases. Elsevier; 2009: 551-565.
- Moss WJ. Measles. Lancet. 2017;390:2490-2502.
- Maintain the vaccination coverage level of 2 doses of the MMR vaccine for children in kindergarten— IID04. Healthy People 2030 website. Accessed May 6, 2025. https://odphp.health.gov/healthypeople/objectives-and-data/browse-objectives/vaccination/maintain-vaccination-coverage-level-2-doses-mmr-vaccine-children-kindergarten-iid-04
- Franconeri L, Antona D, Cauchemez S, et al. Two-dose measles vaccine effectiveness remains high over time: a French observational study, 2017–2019. Vaccine. 2023;41:5797-5804.
- World Health Organization. Measles. Accessed May 8, 2025. https:// www.who.int/news-room/fact-sheets/detail/measles
- Centers for Disease Control and Prevention. Measles vaccine recommendations. Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/vaccine-considerations/index.html
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Accessed May 6, 2025. https://www.cdc.gov/measles/cases-outbreaks.html
- Dyer C. Lancet retracts Wakefield’s MMR paper. BMJ. 2010;340.
- Alves Graber EM, Andrade FJ, Bost W, et al. An update and review of measles for emergency physicians. J Emerg Med. 2020;58:610-615.
- Kaplan LJ, Daum RS, Smaron M, et al. Severe measles in immunocompromised patients. JAMA. 1992;267:1237-1241.
- Takahashi E, Kurosaka D, Yoshida K, et al. Onset of modified measles after etanercept treatment in rheumatoid arthritis. Japanese J Clin Immunol. 2010;33:37-41.
- Worth A, Waldman RA, Dieckhaus K, et al. Art of prevention: our approach to the measles-mumps-rubella vaccine in adult patients vaccinated against measles before 1968 on biologic therapy for the treatment of psoriasis. Int J Womens Dermatol. 2019;6:94.
- Centers for Disease Control and Prevention. Clinical overview of measles (rubeola). Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/clinical-overview/index.html
- Bedford H, Elliman D. Measles rates are rising again. BMJ. 2024;384.
- Harris E. Measles outbreaks grow amid declining vaccination rates. JAMA. 2023;330:2242.
- Guerra FM, Bolotin S, Lim G, et al. The basic reproduction number (R0) of measles: a systematic review. Lancet Infect Dis. 2017;17:E420-E428.
- Swartz MK. Measles: public and professional education. J Pediatr Health Care. 2019;33:367-368.
- Centers for Disease Control and Prevention. Interim infection prevention and control recommendations for measles in healthcare settings. Accessed April 27, 2025. https://www.cdc.gov/infection-control/hcp/measles/
- Moss WJ, Griffin DE, Feinstone WH. Measles. In: Vaccines for Biodefense and Emerging and Neglected Diseases. Elsevier; 2009: 551-565.
- Moss WJ. Measles. Lancet. 2017;390:2490-2502.
- Maintain the vaccination coverage level of 2 doses of the MMR vaccine for children in kindergarten— IID04. Healthy People 2030 website. Accessed May 6, 2025. https://odphp.health.gov/healthypeople/objectives-and-data/browse-objectives/vaccination/maintain-vaccination-coverage-level-2-doses-mmr-vaccine-children-kindergarten-iid-04
- Franconeri L, Antona D, Cauchemez S, et al. Two-dose measles vaccine effectiveness remains high over time: a French observational study, 2017–2019. Vaccine. 2023;41:5797-5804.
- World Health Organization. Measles. Accessed May 8, 2025. https:// www.who.int/news-room/fact-sheets/detail/measles
- Centers for Disease Control and Prevention. Measles vaccine recommendations. Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/vaccine-considerations/index.html
- Centers for Disease Control and Prevention. Measles cases and outbreaks. Accessed May 6, 2025. https://www.cdc.gov/measles/cases-outbreaks.html
- Dyer C. Lancet retracts Wakefield’s MMR paper. BMJ. 2010;340.
- Alves Graber EM, Andrade FJ, Bost W, et al. An update and review of measles for emergency physicians. J Emerg Med. 2020;58:610-615.
- Kaplan LJ, Daum RS, Smaron M, et al. Severe measles in immunocompromised patients. JAMA. 1992;267:1237-1241.
- Takahashi E, Kurosaka D, Yoshida K, et al. Onset of modified measles after etanercept treatment in rheumatoid arthritis. Japanese J Clin Immunol. 2010;33:37-41.
- Worth A, Waldman RA, Dieckhaus K, et al. Art of prevention: our approach to the measles-mumps-rubella vaccine in adult patients vaccinated against measles before 1968 on biologic therapy for the treatment of psoriasis. Int J Womens Dermatol. 2019;6:94.
- Centers for Disease Control and Prevention. Clinical overview of measles (rubeola). Accessed May 8, 2025. https://www.cdc.gov/measles/hcp/clinical-overview/index.html
Measles Resurgence: A Dermatologist’s Guide
Measles Resurgence: A Dermatologist’s Guide

Immune Responses and Health Disparities Warrant Scabies Vaccine Development
Immune Responses and Health Disparities Warrant Scabies Vaccine Development
The scabies mite, originally known as Acarus scabiei,1 now is considered an arthropod of the class Arachnida, order Astigmata, and family Sarcoptidae.2 Scabies mites are able to adhere to the surface of human skin.3 The mites burrow and lay eggs in the top layer of the epidermis; most patients have 10 to 15 mites.3 The patient’s immune system incites an allergic reaction to the mite protein and feces in the skin, causing itching and rash.4
Scabies is common in indigenous populations and in low-income areas of developing countries.5 It is most prevalent in Africa, South America, Australia, and Southeast Asia, in part due to poverty, poor nutritional status, homelessness, and inadequate hygiene.2 In 2009, the World Health Organization declared scabies a neglected skin disease2; however, in 2010, 1.5 million disability adjusted life-years were attributed to scabies,6 and it is estimated that 200 million people worldwide have scabies at any given time. Children and elderly individuals in resource-poor communities are the most at risk. In fact, 5% to 50% of children in low-income areas have scabies.4
The purpose of this article is to provide background on scabies and its effect on the human immune system. We also discuss manipulation of the immune response for the purposes of creating a potential scabies vaccine.
Life Cycle and Transmission
The life cycle of Sarcoptes scabiei consists of 4 stages. The first is the egg. As female scabies mites burrow under the skin, they lay 2 to 3 ovular eggs per day.3 The second stage is the larva. When the egg hatches, the larva has 3 pairs of legs and travels to the surface of the skin where it burrows into the stratum corneum, creating short, nearly invisible burrows called molting pouches. After 3 to 4 days, the larva molts into a nymph, which is the third stage. The nymph has 4 pairs of legs and will continue to grow before molting into an adult, which is the fourth stage. Both the larva and nymph may be found in hair follicles or molting pouches. The fourth stage is the adult, which is round and saclike and does not have eyes. Adult females are 0.30 mm to 0.45 mm long and 0.25 mm to 0.35 mm wide, which is half the size of adult males.3 On warm skin, the female mite can crawl at a rate of 2.5 cm per minute.7
Scabies mites mate via an active male penetrating the molting pouch of a female. This only occurs once but leaves the female fertile for the rest of her life. Once a female is pregnant, she leaves her molting pouch and travels along the surface of the skin looking for a place to make her permanent burrow.3 The most common sites for scabies burrows are the axillae, umbilicus, interdigital spaces, beltline, buttocks, flexor surfaces of the wrists, female nipples, and male penile shaft.5 Once she finds an acceptable location, the female scabies mite will create a serpentine burrow and lay her eggs. Once she burrows, she will stay there and continue to lay eggs for the rest of her life, lengthening the burrow as needed.3 Female mites lay their eggs in the superficial epidermis, and the eggs take approximately 2 to 3 weeks to hatch. Female mites die 30 to 60 days later.2
Scabies infestations typically spread via the transfer of pregnant adult females during skin-to-skin contact, but they also can spread via fomites.3 During all stages of their life cycle, scabies mites can secrete enzymes that allow them to penetrate the intact epidermis in less than 30 minutes; in fact, an otherwise healthy patient with scabies must have 15 to 20 minutes of close skin-to-skin contact with an infected individual for the disease to be transmitted.7 Because scabies mites can survive for more than 3 days outside the human body, it is thought that fomites also may be involved in transmission. Scabies mites also have been collected from clothing, bedding, and furniture, which further supports the idea that fomites are involved in disease transmission.7
Clinical Manifestation of Scabies
Scabies symptoms include severe pruritus as well as linear burrows and vesicles in the interdigital spaces on the hands, wrists, arms and legs, and lower abdomen. Infants and young children also can develop a rash on the palms, soles, ankles, and scalp. Men can develop inflammatory scabies nodules on the penis and scrotum, while women can develop these nodules on the nipple.4 Type I and type IV hypersensitivity reactions contribute to the rash and itching associated with scabies infestation via host allergic and inflammatory reactions to the mites and their byproducts. Patients with scabies typically are infested with fewer than 15 mites,6 but just a few can cause substantial pruritus and scratching, leading to hyperkeratosis.8
Additionally, when patients with scabies scratch the skin, they become vulnerable to bacterial infections.4 Scabies lesions can be coinfected with group A streptococci and Staphylococcus aureus,8 potentially leading to abscesses and septicemia. These secondary infections also can cause renal and cardiac complications; in fact, in tropical areas, scabies infections are considered a risk factor for kidney disease and rheumatic heart disease.4
The 2 main forms of scabies infestations are ordinary and crusted. The most common form is ordinary scabies, which typically manifests with fewer than 15 mites per patient; crusted scabies (CS) is the more rare and extreme form.6 Cases of CS present with thousands to millions of mites per patient, leading to more widespread and severe symptoms.4 Because of the large increase in the number of mites, CS is more contagious than ordinary scabies.6
Patients with CS typically present with hyperkeratotic skin disease, as evidenced by thick scaly crusts with large numbers of mites, which can lead to permanent skin disfiguration. Patients with CS also can develop deep fissuring of the crusts, within which other microbes can gain entry to the body and lead to secondary infection and possibly sepsis and death. Also, because of the increased number of mites as well as the crusted skin, patients with CS are contagious for longer. As it is more difficult to eradicate, reinfestation is common with CS.6
Patients with compromised immune systems are predisposed to CS. Specifically, patients with HIV or human T-lymphotropic virus 1 or those undergoing organ transplantation are thought to be the most at risk for CS.6 Crusted scabies also has been identified in large numbers in patients with Down syndrome and in Aboriginal Australians; however, the reasoning for this is poorly understood.6
Immune Response
The inflammatory reaction associated with scabies infestations occurs 4 to 6 weeks after initial exposure. It is hypothesized that scabies can alter parts of the host immune system, which contributes to the delayed onset of symptoms. Scabies mites also produce inactivated protease paralogues and serpins, which help to protect the mites from the host immune system by inhibiting the complement system.6
The complement system is part of the innate immune response and is the first line of defense against pathogens. Specifically with scabies infestations, C3 and C4 complement components have been found in skin lesions.6 C3a and C4a fragments cause local inflammation, while C3a and C5a activate mast cells to release histamine and tumor necrosis factor (TNF) α, further amplifying the inflammatory response; however, CS lesions show low C3 and C4, which can indicate immunodeficiency in patients with CS. It also can be due to the sheer number of mites in a CS infection causing the host immune system to be overloaded.6
Innate effector immune cells also are an important part of the innate immune response to scabies; for example, eosinophilia is seen in scabies infections. Specifically, in CS, eosinophils help modulate and sustain the T-helper (Th) 2 inflammatory response. One cytokine secreted by Th2 cells is IL-5, which is closely associated with the attraction, maturation, and survival of eosinophils.6 Eosinophils also can influence the Th1 inflammatory response in that they produce IL-12, interferon (IFN) γ, and several Toll-like receptors. Furthermore, eosinophilic expression of IL-2 can lead to expansion of regulatory T cells, while eosinophilic expression of IL-10 and transforming growth factor (TGF) Β also can suppress local inflammation by influencing regulatory T cells.6
Additionally, mast cells and basophils are important in the IgE-mediated allergic reaction as well as the host immune response to parasites. When activated, basophils and mast cells produce TNF-α, IL-6, Il-4, IL-5, and IL-13, which contribute to the Th2 inflammatory response; however, the role of mast cells and basophils in scabies infections still is poorly understood.6
Macrophages, neutrophils, and dendritic cells (DCs) contribute to phagocytosis, antigen presentation, and differentiation of T cells, which also contribute to the inflammatory and allergic reactions associated with parasitic infections.6 Macrophages have been found in low numbers in scabies infestation, possibly due to immune-modulating molecules secreted by scabies mites. Early in an infestation, the mites secrete immune-modulating molecules, which inhibit macrophage migration to the site of inflammation, allowing the mites to grow.6 Neutrophils and DCs also are involved in the host immune response to scabies. Neutrophils are the predominant inflammatory cell infiltrate in scabies lesions. The scabies protein SMSB4 inhibits neutrophil opsonization and phagocytosis, thus suppressing bacterial killing.6 Some of the first antigen-presenting cells encountered by the antigen are DCs. They are involved in preparing the antigens for presentation to effector T cells, which leads to T-cell differentiation and activation.6
Cytokines are another important factor in the innate immune response. The host immune response to ordinary scabies is Th1-cell mediated, during which CD4+ and CD8+ T cells secrete IFN-γ, TNF-α, and IL-2.6 Therefore, IFN- γ and TNF-α are elevated in the serum of patients with ordinary scabies. Conversely, the host immune response to CS is Th2-cell mediated. T-helper 2 cells are needed in IgE-mediated hypersensitivity reactions, and they secrete IL-4, IL-5, and IL-13. In the serum of patients with CS, IL-l4, IL-5, and IL-13 are elevated while IFN-γ is decreased.6 Additionally, IL-6, TGF-Β, IL-23, IL-1Β, or IL-18 can induce Th17 cells to generate and secrete IL-17, which enhances the inflammatory response by inducing further expression of TNF-α, IL-1Β, IL-6, keratinocytes, and fibroblasts. T-helper 17 and IL-17 also are involved in psoriasis and atopic dermatitis, as well as Leishmania major and Schistosoma japonicum.6
Regulatory T cells Tregs secrete TGF-Β and IL-10, which suppress pathologic inflammation, and IL-10 is substantially reduced in patients with CS compared to those with ordinary scabies and uninfected control patients. Additionally, IL-10 can inhibit the synthesis of TNF-γ and IFN-α. Reduced IL-10 expression can lead to proliferation of IL-17 secretion, resulting in a regulatory T cell/Th17 dysfunctional immune response.6
Immunoglobulins are antibodies that are involved in the host’s adaptive immune response. The first antibody to appear in response to an antigen is IgM, and IgM bound to scabies antigens is present in 74%6 of patients with ordinary scabies. Because IgM is the first antibody to appear in response to a scabies infection, detection of serum IgM may allow for earlier detection of scabies; however, IgM has a high cross-reactivity between scabies mites and dust mites, which can hinder scabies diagnosis via IgM detection.6
Both patients with ordinary scabies and CS also show an increased circulatory IgG concentration compared to control groups; patients with CS have higher concentrations. Increased IgG also can be in part due to concurrent bacterial infections.6 When IgG or IgM antibodies bind to a pathogen, they activate the complement cascade, which further enhances the activity of these antibodies.9
Additionally, IgA is important in mucosal immune function. In both patients with ordinary scabies and CS, there is increased IgA binding to recombinant scabies mite antigens.6 Sarcoptes scabiei proteases that are localized in the mite’s gut and scybala suggest their involvement in mite digestion and burrowing. The increased secretion of these proteases into the host skin may contribute to the increased IgA,9 and these increased IgA levels have been shown to be positively correlated with severity of scabies infection.6
Also essential in allergic and parasitic inflammation, IgE is observed at higher levels in secondary infections of scabies compared to primary infections.6 Additionally, T-cell infiltrates are implicated in adaptive immune response to scabies. CD4+ T cells are the most prevalent T cells in ordinary scabies skin lesions; however, CD4+ T cells are minimal and CD8+ T cells are elevated in CS skin lesions. The increased CD8+ T cells may cause apoptosis of keratinocytes, leading to epidermal hyperproliferation. The apoptotic keratinocytes can secrete cytokines, which can lead to tissue damage.6 These T cells also may be involved in the failure of the skin’s immune system to mount an effective response to the parasite infestation, leading to uncontrolled parasitic growth. Because patients with AIDS who are infected with scabies mites often develop CS, it is also thought that CD4+ T cells are essential in the immune response to scabies.6
Diagnosis and Current Treatment Options
Current diagnosis of scabies is based on mites, eggs, and fecal matter from the host’s skin. Dermoscopy and fluorescent dermoscopy can be helpful in identifying the mites, eggs, and feces on the patient’s skin. Scabies treatment sometimes may be based solely on symptoms without any positive tests.8
Acaricides are the current method of treatment for scabies infestations.5 Acaricides can be expensive and toxic to the environment and food sources,10 and some agents have been associated with neurotoxicity5 in children or the development of certain cancers.11 Although topical acaricides are the standard form of treatment, oral ivermectin also can be used. Ivermectin is not associated with selective fetal toxicity, but there are limited safety data in pregnant women and in children weighing less than 15 kg (33 lb). Additionally, because symptoms typically are not present during an early infection, treating everyone in the household and those who had close contact with the patient can help prevent reinfection.4
Although these drugs have been shown to be effective at treating scabies, scabies mites are becoming increasingly resistant to acaricides.5 There are 4 main proposed mechanisms for why this occurs.12 The first is through voltage-gated sodium channels, which are involved in the normal functioning of neurons and myocytes. Permethrin, a type of acaricide, binds to voltage-gated sodium channels when it is in an open or active state and prevents it from closing. This creates repetitive neuron firing and hyperactivity, which ultimately kills the scabies mite. Some mites have mutated to close this channel, which reduces the binding potential of permethrin. Glutathione S-transferase is another mechanism of resistance. It catalyzes a bond that tags drugs for elimination. Increased activity or expressivity of glutathione S-transferase by scabies mites can lead to drug resistance.12 Adenosine triphosphate– binding cassette (ABC) transporters also may contribute to this resistance. The ABC transporters use adenosine triphosphate to facilitate the import or export of molecules. Scabies mites express a protein called the multidrug-resistant protein, which is an ABC transporter that is associated with drug resistance and is present in scabies mites.12 Lastly, ligand-gated chloride channels have been implicated in scabies resistance to acaricides. Ligand-gated chloride channels also are important in normal functioning of neurons and myocytes. Some antiparasitic drugs act on these channels, leading to a continuous influx of chloride, but some scabies mites have mutated this pathway.12
Pesticides and the Risk for Cancer
Pesticides commonly are used to treat scabies; however, a link between pesticide exposure and leukemia and lymphoma has been seen through epidemiologic studies, and there also is increasing biological evidence to suggest this.11 For example, the pesticide permethrin, which works by paralyzing the nervous system of insects,13 has been associated with an increased risk for leukemia and lymphoma in humans. Permethrin is a pyrethroid and, compared to control patients, children with leukemia had higher levels of pyrethroid metabolites in their blood.14 Numerical and structural chromosomal aberrations that give rise to gene fusions are the most common abnormalities seen in leukemia, and permethrin has been shown to induce DNA breaks, chromosome aberrations, and sister chromatid exchanges.14 Permethrin also has been associated with an increased risk for multiple myeloma.13
Furthermore, in utero exposure to pesticides has been associated with an increased risk for childhood leukemia.15 Pesticide exposure shortly before conception, during pregnancy, and after birth is associated with an increased risk for acute lymphocytic leukemia.16 In fact, the children of mothers who were exposed to pesticides 3 months before conception have been found to be at least twice as likely to be diagnosed with acute lymphocytic leukemia within the first year of life compared with children whose mothers were not exposed to pesticides.17 It is hypothesized that permethrin can cross the placenta and alter the hematopoietic precursor cells in the fetus, resulting in leukemogenesis.18 Pyrethroid metabolites also have been detected in umbilical cord blood samples and breast milk.15
In contrast to the research demonstrating a link between permethrin and cancer, other studies have found no association between permethrin19 and leukemia20; non-Hodgkin lymphoma19; or cancers of the colon, rectum, pancreas, lungs, skin, female breast, prostate, and urinary bladder.20 Because of conflicting research on the link between permethrin and cancer, more research is needed.,20
Importance of a Scabies Vaccine
Because scabies mites are developing increasing treatment resistance, more radical approaches such as vaccines are becoming important. While a scabies vaccine is still aspirational, animals that have been infected for a second time with scabies demonstrate a milder response to the second infection compared to the first infection, which could mean there is a potential for disease prevention through a vaccine.21 While educating patients and physicians, reporting cases of infection, and improving drug supply and access can help decrease scabies infestations, these are costly and difficult to implement. Scabies already is most prevalent in low-income areas, so costly interventions are even less feasible. An effective, one-dose vaccine would cost less than these efforts and therefore could be implemented more easily.9
In older adults, scabies more often manifests atypically and is more likely to progress to CS. Aged care centers are prone to institutional outbreaks, even in developed countries, so a vaccine also would greatly help this population. Additionally, the number of children attending day care centers, which also are prone to scabies outbreaks, is increasing. When a child contracts scabies, all close contacts need to be treated, so a preventive vaccine can be useful.9
One potential candidate for a scabies vaccine is total mite extract. Studies show that rabbits immunized with a total mite extract induce antibodies to more antigens than rabbits naturally infested with scabies mites; however, the mites cannot be cultured in vitro, which makes obtaining a large amount of their total extract difficult. Therefore, recombinant vaccines also have been proposed, as they are more easily available.22 One recombinant vaccine candidate is recombinant S scabiei serpin (rSs-serpin). Immunization with rSs-serpin has strong immunogenicity and produced immune protection in rabbits.22
Two other recombinant vaccine candidates are the rSs chitinaselike protein (CLP) 12 and the rSsCLP5. Chitinaselike proteins are very similar to chitinases; however, they are unable to degrade chitin. They are involved in immune reactions to infections, and CLPs from scabies mites have been shown to induce the host immune response.22 For example, in a particular rabbit study, rSsCLP5 demonstrated high immunoreactivity and immunogenicity. In fact, after exposure to S scabiei, 74.3% of rabbits who were vaccinated with rSsCLP5 had no detectable lesions.5 Also, after immunization with rSsCLP5 and rSsCLP12, there were increased levels of specific IgG and IgE antibodies produced and decreased numbers of infesting mites.22 Weight loss also is associated with severe scabies infection. Rabbits vaccinated with rSsCLP5 and exposed to the parasite gained weight, indicating protection via rSsCLP5. Even rabbits who did develop symptoms of scabies after immunization with rSsCLP5 and exposure to S scabiei showed less serious manifestations.5
A combination vaccine cocktail of rSs-serpin, rSsCLP12, and rSsCLP5 also has been proposed by Shen et al.22 Four test groups and a control group (n=12 per group) were included in a vaccine trial. Between 83.33% and 91.67% of rabbits vaccinated with this mixed recombinant cocktail vaccine had no detectable skin lesions from scabies. After immunization with the cocktail vaccine, the specific serum IgG and IgE antibodies also increased. For both IgG and IgE, increased levels were first detected at 1 week postimmunization and peaked at 2 weeks postimmunization.22 A multiepitope vaccine derived from these 3 recombinant proteins also was explored by Shen et al22; fewer rabbits vaccinated with it had no detectable scabies skin lesions compared to those treated with the vaccine cocktail. Although the multiepitope vaccine yielded less immume protection, it was associated with a slower disease course and milder symptoms compared with no vaccination.22
Two more proposed scabies recombinant vaccine candidates are derived from the antigens Ssag1 and Ssag2; however, rabbits vaccinated with Ssag1 or Ssag2 showed no immune protection or mite burden reduction.22 The lack of protection could be due to denaturation or degradation of the protective antigens. It also can be due to the low abundance of these antigens, meaning they may not be vital for the mite’s survival—survival—a potential avenue for future research. The antigens also could have lost their native structure and immunogenic properties during the purification and production process. Therefore, more research is needed to investigate how to purify these vaccines to keep the peptides more structurally similar to their native makeups.10 More research also is needed to better understand the antigen or antigens and their mechanisms that elicit a protective immune response.9
Final Thoughts
Scabies causes severe pruritus in mild cases but also can lead to severe disfigurement, sepsis, and even death. Scabies infestations are seen disproportionately more often in low-income and resource-poor communities, and the current treatment options are less accessible to these populations. Scabies infestations induce a complex immune response that involves multiple aspects of both the innate and adaptive immune systems and can be targeted to create a scabies vaccine. Development of a scabies vaccine is crucial considering the growing resistance to current standard treatments. Acaricides potentially are associated with an increased risk for malignancy, which further amplifies the need for a scabies vaccine. There currently are multiple promising scabies vaccine candidates; however, more research is needed to better understand the host’s immune response to scabies as well as how to more accurately and efficiently produce the vaccine. The development of a safe, effective, economical vaccine that can be mass distributed would be beneficial in the treatment of scabies, especially in resource-poor communities.
- Arlian LG, Morgan MS. A review of Sarcoptes scabiei: past, present and future. Parasit Vectors. 2017;10:297. doi:10.1186/s13071-017-2234-1
- Murray RL, Crane JS. Scabies. In: StatPearls. StatPearls Publishing. Updated July 31, 2023.
- Centers for Disease Control and Prevention. CDC—scabies—biology. November 2, 2010. https://www.cdc.gov/dpdx/scabies/index.html
- World Health Organization. Scabies. May 31, 2023. Accessed May 8, 2025. https://www.who.int/news-room/fact-sheets/detail/scabies
- Shen N, Zhang H, Ren Y, et al. A chitinase-like protein from Sarcoptes scabiei as a candidate anti-mite vaccine that contributes to immune protection in rabbits. Parasit Vectors. 2018;11:599. doi:10.1186/s13071- 018-3184-y
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Hicks MI, Elston DM. Scabies. Dermatolog Ther. 2009;22:279-292. doi:10.1111/j.1529-8019.2009.01243.x
- Morgan MS, Arlian LG, Rider SD, et al. A proteomic analysis of Sarcoptes scabiei (acari: Sarcoptidae). J Med Entomol. 2016;53:553-561. doi:10.1093/jme/tjv247
- Liu X, Walton S, Mounsey K. Vaccine against scabies: necessity and possibility. Parasitology. 2014;141:725-732. doi:10.1017 /s0031182013002047
- Casais R, Granda V, Balseiro A, et al. Vaccination of rabbits with immunodominant antigens from Sarcoptes scabiei induced high levels of humoral responses and pro-inflammatory cytokines but confers limited protection. Parasit Vectors. 2016;9:435. doi:10.1186 /s13071-016-1717-9?
- Navarrete-Meneses MP, Pedraza-Meléndez AI, Salas-Labadía C, et al. Low concentrations of permethrin and malathion induce numerical and structural abnormalities in KMT2A and IGH genes in vitro. J Appl Toxicol. 2018;38:1262-1270. doi:10.1002/jat.3638
- Khalil S, Abbas O, Kibbi AG, et al. Scabies in the age of increasing drug resistance. PLoS Negl Trop Dis. 2017;11:E0005920. doi:10.1371 /journal.pntd.0005920
- Rusiecki JA, Patel R, Koutros S, et al. Cancer incidence among pesticide applicators exposed to permethrin in the Agricultural Health Study. Environ Health Perspect. 2009;117:581-586. doi:10.1289 /ehp.11318
- Navarrete-Meneses MP, Salas-Labadía C, Sanabrais-Jiménez M, et al. Exposure to the insecticides permethrin and malathion induces leukemia and lymphoma-associated gene aberrations in vitro. Toxicol In Vitro. 2017;44:17-26. doi:10.1016/j.tiv.2017.06.013
- Navarrete-Meneses MDP, Pérez-Vera P. Pyrethroid pesticide exposure and hematological cancer: epidemiological, biological and molecular evidence. Rev Environ Health. 2019;34:197-210. doi:10.1515 /reveh-2018-0070
- Madrigal JM, Jones RR, Gunier RB, et al. Residential exposure to carbamate, organophosphate, and pyrethroid insecticides in house dust and risk of childhood acute lymphoblastic leukemia. Environ Res. 2021;201:111501. doi:10.1016/j.envres.2021.111501
- Ferreira JD, Couto AC, Pombo-de-Oliveira MS, et al. In utero pesticide exposure and leukemia in Brazilian children <2 years of age. Environ Health Perspect. 2013;121:269-275. doi:10.1289/ehp.1103942
- Borkhardt A, Wilda M, Fuchs U, et al. Congenital leukaemia after heavy abuse of permethrin during pregnancy. Arch Dis Child Fetal Neonatal Ed. 2003;88:F436-F437. doi:10.1136/fn.88.5.f436
- De Roos AJ, Schinasi LH, Miligi L, et al. Occupational insecticide exposure and risk of non]Hodgkin lymphoma: a pooled case]control study from the InterLymph consortium. Int J Cancer. 2021;149:1768-1786. doi:10.1002/ijc.33740
- Boffett, P, Desai V. Exposure to permethrin and cancer risk: a systematic review. Crit Rev Toxicol. 2018;48:433-442. doi:10.1080/1040 8444.2018.1439449
- Adji A, Rumokoy LJM, Salaki CL. Scabies vaccine as a new breakthrough for the challenge of acaricides resistance. Adv Biolog Sci Res. 2020;8:208-213. doi:10.2991/absr.k.200513.036
- Shen N, Wei W, Chen Y, et al. Vaccination with a cocktail vaccine elicits significant protection against Sarcoptes scabiei in rabbits, whereas the multi-epitope vaccine offers limited protection. Exp Parasitol. 2023;245:108442. doi:10.1016/j.exppara.2022.108442
The scabies mite, originally known as Acarus scabiei,1 now is considered an arthropod of the class Arachnida, order Astigmata, and family Sarcoptidae.2 Scabies mites are able to adhere to the surface of human skin.3 The mites burrow and lay eggs in the top layer of the epidermis; most patients have 10 to 15 mites.3 The patient’s immune system incites an allergic reaction to the mite protein and feces in the skin, causing itching and rash.4
Scabies is common in indigenous populations and in low-income areas of developing countries.5 It is most prevalent in Africa, South America, Australia, and Southeast Asia, in part due to poverty, poor nutritional status, homelessness, and inadequate hygiene.2 In 2009, the World Health Organization declared scabies a neglected skin disease2; however, in 2010, 1.5 million disability adjusted life-years were attributed to scabies,6 and it is estimated that 200 million people worldwide have scabies at any given time. Children and elderly individuals in resource-poor communities are the most at risk. In fact, 5% to 50% of children in low-income areas have scabies.4
The purpose of this article is to provide background on scabies and its effect on the human immune system. We also discuss manipulation of the immune response for the purposes of creating a potential scabies vaccine.
Life Cycle and Transmission
The life cycle of Sarcoptes scabiei consists of 4 stages. The first is the egg. As female scabies mites burrow under the skin, they lay 2 to 3 ovular eggs per day.3 The second stage is the larva. When the egg hatches, the larva has 3 pairs of legs and travels to the surface of the skin where it burrows into the stratum corneum, creating short, nearly invisible burrows called molting pouches. After 3 to 4 days, the larva molts into a nymph, which is the third stage. The nymph has 4 pairs of legs and will continue to grow before molting into an adult, which is the fourth stage. Both the larva and nymph may be found in hair follicles or molting pouches. The fourth stage is the adult, which is round and saclike and does not have eyes. Adult females are 0.30 mm to 0.45 mm long and 0.25 mm to 0.35 mm wide, which is half the size of adult males.3 On warm skin, the female mite can crawl at a rate of 2.5 cm per minute.7
Scabies mites mate via an active male penetrating the molting pouch of a female. This only occurs once but leaves the female fertile for the rest of her life. Once a female is pregnant, she leaves her molting pouch and travels along the surface of the skin looking for a place to make her permanent burrow.3 The most common sites for scabies burrows are the axillae, umbilicus, interdigital spaces, beltline, buttocks, flexor surfaces of the wrists, female nipples, and male penile shaft.5 Once she finds an acceptable location, the female scabies mite will create a serpentine burrow and lay her eggs. Once she burrows, she will stay there and continue to lay eggs for the rest of her life, lengthening the burrow as needed.3 Female mites lay their eggs in the superficial epidermis, and the eggs take approximately 2 to 3 weeks to hatch. Female mites die 30 to 60 days later.2
Scabies infestations typically spread via the transfer of pregnant adult females during skin-to-skin contact, but they also can spread via fomites.3 During all stages of their life cycle, scabies mites can secrete enzymes that allow them to penetrate the intact epidermis in less than 30 minutes; in fact, an otherwise healthy patient with scabies must have 15 to 20 minutes of close skin-to-skin contact with an infected individual for the disease to be transmitted.7 Because scabies mites can survive for more than 3 days outside the human body, it is thought that fomites also may be involved in transmission. Scabies mites also have been collected from clothing, bedding, and furniture, which further supports the idea that fomites are involved in disease transmission.7
Clinical Manifestation of Scabies
Scabies symptoms include severe pruritus as well as linear burrows and vesicles in the interdigital spaces on the hands, wrists, arms and legs, and lower abdomen. Infants and young children also can develop a rash on the palms, soles, ankles, and scalp. Men can develop inflammatory scabies nodules on the penis and scrotum, while women can develop these nodules on the nipple.4 Type I and type IV hypersensitivity reactions contribute to the rash and itching associated with scabies infestation via host allergic and inflammatory reactions to the mites and their byproducts. Patients with scabies typically are infested with fewer than 15 mites,6 but just a few can cause substantial pruritus and scratching, leading to hyperkeratosis.8
Additionally, when patients with scabies scratch the skin, they become vulnerable to bacterial infections.4 Scabies lesions can be coinfected with group A streptococci and Staphylococcus aureus,8 potentially leading to abscesses and septicemia. These secondary infections also can cause renal and cardiac complications; in fact, in tropical areas, scabies infections are considered a risk factor for kidney disease and rheumatic heart disease.4
The 2 main forms of scabies infestations are ordinary and crusted. The most common form is ordinary scabies, which typically manifests with fewer than 15 mites per patient; crusted scabies (CS) is the more rare and extreme form.6 Cases of CS present with thousands to millions of mites per patient, leading to more widespread and severe symptoms.4 Because of the large increase in the number of mites, CS is more contagious than ordinary scabies.6
Patients with CS typically present with hyperkeratotic skin disease, as evidenced by thick scaly crusts with large numbers of mites, which can lead to permanent skin disfiguration. Patients with CS also can develop deep fissuring of the crusts, within which other microbes can gain entry to the body and lead to secondary infection and possibly sepsis and death. Also, because of the increased number of mites as well as the crusted skin, patients with CS are contagious for longer. As it is more difficult to eradicate, reinfestation is common with CS.6
Patients with compromised immune systems are predisposed to CS. Specifically, patients with HIV or human T-lymphotropic virus 1 or those undergoing organ transplantation are thought to be the most at risk for CS.6 Crusted scabies also has been identified in large numbers in patients with Down syndrome and in Aboriginal Australians; however, the reasoning for this is poorly understood.6
Immune Response
The inflammatory reaction associated with scabies infestations occurs 4 to 6 weeks after initial exposure. It is hypothesized that scabies can alter parts of the host immune system, which contributes to the delayed onset of symptoms. Scabies mites also produce inactivated protease paralogues and serpins, which help to protect the mites from the host immune system by inhibiting the complement system.6
The complement system is part of the innate immune response and is the first line of defense against pathogens. Specifically with scabies infestations, C3 and C4 complement components have been found in skin lesions.6 C3a and C4a fragments cause local inflammation, while C3a and C5a activate mast cells to release histamine and tumor necrosis factor (TNF) α, further amplifying the inflammatory response; however, CS lesions show low C3 and C4, which can indicate immunodeficiency in patients with CS. It also can be due to the sheer number of mites in a CS infection causing the host immune system to be overloaded.6
Innate effector immune cells also are an important part of the innate immune response to scabies; for example, eosinophilia is seen in scabies infections. Specifically, in CS, eosinophils help modulate and sustain the T-helper (Th) 2 inflammatory response. One cytokine secreted by Th2 cells is IL-5, which is closely associated with the attraction, maturation, and survival of eosinophils.6 Eosinophils also can influence the Th1 inflammatory response in that they produce IL-12, interferon (IFN) γ, and several Toll-like receptors. Furthermore, eosinophilic expression of IL-2 can lead to expansion of regulatory T cells, while eosinophilic expression of IL-10 and transforming growth factor (TGF) Β also can suppress local inflammation by influencing regulatory T cells.6
Additionally, mast cells and basophils are important in the IgE-mediated allergic reaction as well as the host immune response to parasites. When activated, basophils and mast cells produce TNF-α, IL-6, Il-4, IL-5, and IL-13, which contribute to the Th2 inflammatory response; however, the role of mast cells and basophils in scabies infections still is poorly understood.6
Macrophages, neutrophils, and dendritic cells (DCs) contribute to phagocytosis, antigen presentation, and differentiation of T cells, which also contribute to the inflammatory and allergic reactions associated with parasitic infections.6 Macrophages have been found in low numbers in scabies infestation, possibly due to immune-modulating molecules secreted by scabies mites. Early in an infestation, the mites secrete immune-modulating molecules, which inhibit macrophage migration to the site of inflammation, allowing the mites to grow.6 Neutrophils and DCs also are involved in the host immune response to scabies. Neutrophils are the predominant inflammatory cell infiltrate in scabies lesions. The scabies protein SMSB4 inhibits neutrophil opsonization and phagocytosis, thus suppressing bacterial killing.6 Some of the first antigen-presenting cells encountered by the antigen are DCs. They are involved in preparing the antigens for presentation to effector T cells, which leads to T-cell differentiation and activation.6
Cytokines are another important factor in the innate immune response. The host immune response to ordinary scabies is Th1-cell mediated, during which CD4+ and CD8+ T cells secrete IFN-γ, TNF-α, and IL-2.6 Therefore, IFN- γ and TNF-α are elevated in the serum of patients with ordinary scabies. Conversely, the host immune response to CS is Th2-cell mediated. T-helper 2 cells are needed in IgE-mediated hypersensitivity reactions, and they secrete IL-4, IL-5, and IL-13. In the serum of patients with CS, IL-l4, IL-5, and IL-13 are elevated while IFN-γ is decreased.6 Additionally, IL-6, TGF-Β, IL-23, IL-1Β, or IL-18 can induce Th17 cells to generate and secrete IL-17, which enhances the inflammatory response by inducing further expression of TNF-α, IL-1Β, IL-6, keratinocytes, and fibroblasts. T-helper 17 and IL-17 also are involved in psoriasis and atopic dermatitis, as well as Leishmania major and Schistosoma japonicum.6
Regulatory T cells Tregs secrete TGF-Β and IL-10, which suppress pathologic inflammation, and IL-10 is substantially reduced in patients with CS compared to those with ordinary scabies and uninfected control patients. Additionally, IL-10 can inhibit the synthesis of TNF-γ and IFN-α. Reduced IL-10 expression can lead to proliferation of IL-17 secretion, resulting in a regulatory T cell/Th17 dysfunctional immune response.6
Immunoglobulins are antibodies that are involved in the host’s adaptive immune response. The first antibody to appear in response to an antigen is IgM, and IgM bound to scabies antigens is present in 74%6 of patients with ordinary scabies. Because IgM is the first antibody to appear in response to a scabies infection, detection of serum IgM may allow for earlier detection of scabies; however, IgM has a high cross-reactivity between scabies mites and dust mites, which can hinder scabies diagnosis via IgM detection.6
Both patients with ordinary scabies and CS also show an increased circulatory IgG concentration compared to control groups; patients with CS have higher concentrations. Increased IgG also can be in part due to concurrent bacterial infections.6 When IgG or IgM antibodies bind to a pathogen, they activate the complement cascade, which further enhances the activity of these antibodies.9
Additionally, IgA is important in mucosal immune function. In both patients with ordinary scabies and CS, there is increased IgA binding to recombinant scabies mite antigens.6 Sarcoptes scabiei proteases that are localized in the mite’s gut and scybala suggest their involvement in mite digestion and burrowing. The increased secretion of these proteases into the host skin may contribute to the increased IgA,9 and these increased IgA levels have been shown to be positively correlated with severity of scabies infection.6
Also essential in allergic and parasitic inflammation, IgE is observed at higher levels in secondary infections of scabies compared to primary infections.6 Additionally, T-cell infiltrates are implicated in adaptive immune response to scabies. CD4+ T cells are the most prevalent T cells in ordinary scabies skin lesions; however, CD4+ T cells are minimal and CD8+ T cells are elevated in CS skin lesions. The increased CD8+ T cells may cause apoptosis of keratinocytes, leading to epidermal hyperproliferation. The apoptotic keratinocytes can secrete cytokines, which can lead to tissue damage.6 These T cells also may be involved in the failure of the skin’s immune system to mount an effective response to the parasite infestation, leading to uncontrolled parasitic growth. Because patients with AIDS who are infected with scabies mites often develop CS, it is also thought that CD4+ T cells are essential in the immune response to scabies.6
Diagnosis and Current Treatment Options
Current diagnosis of scabies is based on mites, eggs, and fecal matter from the host’s skin. Dermoscopy and fluorescent dermoscopy can be helpful in identifying the mites, eggs, and feces on the patient’s skin. Scabies treatment sometimes may be based solely on symptoms without any positive tests.8
Acaricides are the current method of treatment for scabies infestations.5 Acaricides can be expensive and toxic to the environment and food sources,10 and some agents have been associated with neurotoxicity5 in children or the development of certain cancers.11 Although topical acaricides are the standard form of treatment, oral ivermectin also can be used. Ivermectin is not associated with selective fetal toxicity, but there are limited safety data in pregnant women and in children weighing less than 15 kg (33 lb). Additionally, because symptoms typically are not present during an early infection, treating everyone in the household and those who had close contact with the patient can help prevent reinfection.4
Although these drugs have been shown to be effective at treating scabies, scabies mites are becoming increasingly resistant to acaricides.5 There are 4 main proposed mechanisms for why this occurs.12 The first is through voltage-gated sodium channels, which are involved in the normal functioning of neurons and myocytes. Permethrin, a type of acaricide, binds to voltage-gated sodium channels when it is in an open or active state and prevents it from closing. This creates repetitive neuron firing and hyperactivity, which ultimately kills the scabies mite. Some mites have mutated to close this channel, which reduces the binding potential of permethrin. Glutathione S-transferase is another mechanism of resistance. It catalyzes a bond that tags drugs for elimination. Increased activity or expressivity of glutathione S-transferase by scabies mites can lead to drug resistance.12 Adenosine triphosphate– binding cassette (ABC) transporters also may contribute to this resistance. The ABC transporters use adenosine triphosphate to facilitate the import or export of molecules. Scabies mites express a protein called the multidrug-resistant protein, which is an ABC transporter that is associated with drug resistance and is present in scabies mites.12 Lastly, ligand-gated chloride channels have been implicated in scabies resistance to acaricides. Ligand-gated chloride channels also are important in normal functioning of neurons and myocytes. Some antiparasitic drugs act on these channels, leading to a continuous influx of chloride, but some scabies mites have mutated this pathway.12
Pesticides and the Risk for Cancer
Pesticides commonly are used to treat scabies; however, a link between pesticide exposure and leukemia and lymphoma has been seen through epidemiologic studies, and there also is increasing biological evidence to suggest this.11 For example, the pesticide permethrin, which works by paralyzing the nervous system of insects,13 has been associated with an increased risk for leukemia and lymphoma in humans. Permethrin is a pyrethroid and, compared to control patients, children with leukemia had higher levels of pyrethroid metabolites in their blood.14 Numerical and structural chromosomal aberrations that give rise to gene fusions are the most common abnormalities seen in leukemia, and permethrin has been shown to induce DNA breaks, chromosome aberrations, and sister chromatid exchanges.14 Permethrin also has been associated with an increased risk for multiple myeloma.13
Furthermore, in utero exposure to pesticides has been associated with an increased risk for childhood leukemia.15 Pesticide exposure shortly before conception, during pregnancy, and after birth is associated with an increased risk for acute lymphocytic leukemia.16 In fact, the children of mothers who were exposed to pesticides 3 months before conception have been found to be at least twice as likely to be diagnosed with acute lymphocytic leukemia within the first year of life compared with children whose mothers were not exposed to pesticides.17 It is hypothesized that permethrin can cross the placenta and alter the hematopoietic precursor cells in the fetus, resulting in leukemogenesis.18 Pyrethroid metabolites also have been detected in umbilical cord blood samples and breast milk.15
In contrast to the research demonstrating a link between permethrin and cancer, other studies have found no association between permethrin19 and leukemia20; non-Hodgkin lymphoma19; or cancers of the colon, rectum, pancreas, lungs, skin, female breast, prostate, and urinary bladder.20 Because of conflicting research on the link between permethrin and cancer, more research is needed.,20
Importance of a Scabies Vaccine
Because scabies mites are developing increasing treatment resistance, more radical approaches such as vaccines are becoming important. While a scabies vaccine is still aspirational, animals that have been infected for a second time with scabies demonstrate a milder response to the second infection compared to the first infection, which could mean there is a potential for disease prevention through a vaccine.21 While educating patients and physicians, reporting cases of infection, and improving drug supply and access can help decrease scabies infestations, these are costly and difficult to implement. Scabies already is most prevalent in low-income areas, so costly interventions are even less feasible. An effective, one-dose vaccine would cost less than these efforts and therefore could be implemented more easily.9
In older adults, scabies more often manifests atypically and is more likely to progress to CS. Aged care centers are prone to institutional outbreaks, even in developed countries, so a vaccine also would greatly help this population. Additionally, the number of children attending day care centers, which also are prone to scabies outbreaks, is increasing. When a child contracts scabies, all close contacts need to be treated, so a preventive vaccine can be useful.9
One potential candidate for a scabies vaccine is total mite extract. Studies show that rabbits immunized with a total mite extract induce antibodies to more antigens than rabbits naturally infested with scabies mites; however, the mites cannot be cultured in vitro, which makes obtaining a large amount of their total extract difficult. Therefore, recombinant vaccines also have been proposed, as they are more easily available.22 One recombinant vaccine candidate is recombinant S scabiei serpin (rSs-serpin). Immunization with rSs-serpin has strong immunogenicity and produced immune protection in rabbits.22
Two other recombinant vaccine candidates are the rSs chitinaselike protein (CLP) 12 and the rSsCLP5. Chitinaselike proteins are very similar to chitinases; however, they are unable to degrade chitin. They are involved in immune reactions to infections, and CLPs from scabies mites have been shown to induce the host immune response.22 For example, in a particular rabbit study, rSsCLP5 demonstrated high immunoreactivity and immunogenicity. In fact, after exposure to S scabiei, 74.3% of rabbits who were vaccinated with rSsCLP5 had no detectable lesions.5 Also, after immunization with rSsCLP5 and rSsCLP12, there were increased levels of specific IgG and IgE antibodies produced and decreased numbers of infesting mites.22 Weight loss also is associated with severe scabies infection. Rabbits vaccinated with rSsCLP5 and exposed to the parasite gained weight, indicating protection via rSsCLP5. Even rabbits who did develop symptoms of scabies after immunization with rSsCLP5 and exposure to S scabiei showed less serious manifestations.5
A combination vaccine cocktail of rSs-serpin, rSsCLP12, and rSsCLP5 also has been proposed by Shen et al.22 Four test groups and a control group (n=12 per group) were included in a vaccine trial. Between 83.33% and 91.67% of rabbits vaccinated with this mixed recombinant cocktail vaccine had no detectable skin lesions from scabies. After immunization with the cocktail vaccine, the specific serum IgG and IgE antibodies also increased. For both IgG and IgE, increased levels were first detected at 1 week postimmunization and peaked at 2 weeks postimmunization.22 A multiepitope vaccine derived from these 3 recombinant proteins also was explored by Shen et al22; fewer rabbits vaccinated with it had no detectable scabies skin lesions compared to those treated with the vaccine cocktail. Although the multiepitope vaccine yielded less immume protection, it was associated with a slower disease course and milder symptoms compared with no vaccination.22
Two more proposed scabies recombinant vaccine candidates are derived from the antigens Ssag1 and Ssag2; however, rabbits vaccinated with Ssag1 or Ssag2 showed no immune protection or mite burden reduction.22 The lack of protection could be due to denaturation or degradation of the protective antigens. It also can be due to the low abundance of these antigens, meaning they may not be vital for the mite’s survival—survival—a potential avenue for future research. The antigens also could have lost their native structure and immunogenic properties during the purification and production process. Therefore, more research is needed to investigate how to purify these vaccines to keep the peptides more structurally similar to their native makeups.10 More research also is needed to better understand the antigen or antigens and their mechanisms that elicit a protective immune response.9
Final Thoughts
Scabies causes severe pruritus in mild cases but also can lead to severe disfigurement, sepsis, and even death. Scabies infestations are seen disproportionately more often in low-income and resource-poor communities, and the current treatment options are less accessible to these populations. Scabies infestations induce a complex immune response that involves multiple aspects of both the innate and adaptive immune systems and can be targeted to create a scabies vaccine. Development of a scabies vaccine is crucial considering the growing resistance to current standard treatments. Acaricides potentially are associated with an increased risk for malignancy, which further amplifies the need for a scabies vaccine. There currently are multiple promising scabies vaccine candidates; however, more research is needed to better understand the host’s immune response to scabies as well as how to more accurately and efficiently produce the vaccine. The development of a safe, effective, economical vaccine that can be mass distributed would be beneficial in the treatment of scabies, especially in resource-poor communities.
The scabies mite, originally known as Acarus scabiei,1 now is considered an arthropod of the class Arachnida, order Astigmata, and family Sarcoptidae.2 Scabies mites are able to adhere to the surface of human skin.3 The mites burrow and lay eggs in the top layer of the epidermis; most patients have 10 to 15 mites.3 The patient’s immune system incites an allergic reaction to the mite protein and feces in the skin, causing itching and rash.4
Scabies is common in indigenous populations and in low-income areas of developing countries.5 It is most prevalent in Africa, South America, Australia, and Southeast Asia, in part due to poverty, poor nutritional status, homelessness, and inadequate hygiene.2 In 2009, the World Health Organization declared scabies a neglected skin disease2; however, in 2010, 1.5 million disability adjusted life-years were attributed to scabies,6 and it is estimated that 200 million people worldwide have scabies at any given time. Children and elderly individuals in resource-poor communities are the most at risk. In fact, 5% to 50% of children in low-income areas have scabies.4
The purpose of this article is to provide background on scabies and its effect on the human immune system. We also discuss manipulation of the immune response for the purposes of creating a potential scabies vaccine.
Life Cycle and Transmission
The life cycle of Sarcoptes scabiei consists of 4 stages. The first is the egg. As female scabies mites burrow under the skin, they lay 2 to 3 ovular eggs per day.3 The second stage is the larva. When the egg hatches, the larva has 3 pairs of legs and travels to the surface of the skin where it burrows into the stratum corneum, creating short, nearly invisible burrows called molting pouches. After 3 to 4 days, the larva molts into a nymph, which is the third stage. The nymph has 4 pairs of legs and will continue to grow before molting into an adult, which is the fourth stage. Both the larva and nymph may be found in hair follicles or molting pouches. The fourth stage is the adult, which is round and saclike and does not have eyes. Adult females are 0.30 mm to 0.45 mm long and 0.25 mm to 0.35 mm wide, which is half the size of adult males.3 On warm skin, the female mite can crawl at a rate of 2.5 cm per minute.7
Scabies mites mate via an active male penetrating the molting pouch of a female. This only occurs once but leaves the female fertile for the rest of her life. Once a female is pregnant, she leaves her molting pouch and travels along the surface of the skin looking for a place to make her permanent burrow.3 The most common sites for scabies burrows are the axillae, umbilicus, interdigital spaces, beltline, buttocks, flexor surfaces of the wrists, female nipples, and male penile shaft.5 Once she finds an acceptable location, the female scabies mite will create a serpentine burrow and lay her eggs. Once she burrows, she will stay there and continue to lay eggs for the rest of her life, lengthening the burrow as needed.3 Female mites lay their eggs in the superficial epidermis, and the eggs take approximately 2 to 3 weeks to hatch. Female mites die 30 to 60 days later.2
Scabies infestations typically spread via the transfer of pregnant adult females during skin-to-skin contact, but they also can spread via fomites.3 During all stages of their life cycle, scabies mites can secrete enzymes that allow them to penetrate the intact epidermis in less than 30 minutes; in fact, an otherwise healthy patient with scabies must have 15 to 20 minutes of close skin-to-skin contact with an infected individual for the disease to be transmitted.7 Because scabies mites can survive for more than 3 days outside the human body, it is thought that fomites also may be involved in transmission. Scabies mites also have been collected from clothing, bedding, and furniture, which further supports the idea that fomites are involved in disease transmission.7
Clinical Manifestation of Scabies
Scabies symptoms include severe pruritus as well as linear burrows and vesicles in the interdigital spaces on the hands, wrists, arms and legs, and lower abdomen. Infants and young children also can develop a rash on the palms, soles, ankles, and scalp. Men can develop inflammatory scabies nodules on the penis and scrotum, while women can develop these nodules on the nipple.4 Type I and type IV hypersensitivity reactions contribute to the rash and itching associated with scabies infestation via host allergic and inflammatory reactions to the mites and their byproducts. Patients with scabies typically are infested with fewer than 15 mites,6 but just a few can cause substantial pruritus and scratching, leading to hyperkeratosis.8
Additionally, when patients with scabies scratch the skin, they become vulnerable to bacterial infections.4 Scabies lesions can be coinfected with group A streptococci and Staphylococcus aureus,8 potentially leading to abscesses and septicemia. These secondary infections also can cause renal and cardiac complications; in fact, in tropical areas, scabies infections are considered a risk factor for kidney disease and rheumatic heart disease.4
The 2 main forms of scabies infestations are ordinary and crusted. The most common form is ordinary scabies, which typically manifests with fewer than 15 mites per patient; crusted scabies (CS) is the more rare and extreme form.6 Cases of CS present with thousands to millions of mites per patient, leading to more widespread and severe symptoms.4 Because of the large increase in the number of mites, CS is more contagious than ordinary scabies.6
Patients with CS typically present with hyperkeratotic skin disease, as evidenced by thick scaly crusts with large numbers of mites, which can lead to permanent skin disfiguration. Patients with CS also can develop deep fissuring of the crusts, within which other microbes can gain entry to the body and lead to secondary infection and possibly sepsis and death. Also, because of the increased number of mites as well as the crusted skin, patients with CS are contagious for longer. As it is more difficult to eradicate, reinfestation is common with CS.6
Patients with compromised immune systems are predisposed to CS. Specifically, patients with HIV or human T-lymphotropic virus 1 or those undergoing organ transplantation are thought to be the most at risk for CS.6 Crusted scabies also has been identified in large numbers in patients with Down syndrome and in Aboriginal Australians; however, the reasoning for this is poorly understood.6
Immune Response
The inflammatory reaction associated with scabies infestations occurs 4 to 6 weeks after initial exposure. It is hypothesized that scabies can alter parts of the host immune system, which contributes to the delayed onset of symptoms. Scabies mites also produce inactivated protease paralogues and serpins, which help to protect the mites from the host immune system by inhibiting the complement system.6
The complement system is part of the innate immune response and is the first line of defense against pathogens. Specifically with scabies infestations, C3 and C4 complement components have been found in skin lesions.6 C3a and C4a fragments cause local inflammation, while C3a and C5a activate mast cells to release histamine and tumor necrosis factor (TNF) α, further amplifying the inflammatory response; however, CS lesions show low C3 and C4, which can indicate immunodeficiency in patients with CS. It also can be due to the sheer number of mites in a CS infection causing the host immune system to be overloaded.6
Innate effector immune cells also are an important part of the innate immune response to scabies; for example, eosinophilia is seen in scabies infections. Specifically, in CS, eosinophils help modulate and sustain the T-helper (Th) 2 inflammatory response. One cytokine secreted by Th2 cells is IL-5, which is closely associated with the attraction, maturation, and survival of eosinophils.6 Eosinophils also can influence the Th1 inflammatory response in that they produce IL-12, interferon (IFN) γ, and several Toll-like receptors. Furthermore, eosinophilic expression of IL-2 can lead to expansion of regulatory T cells, while eosinophilic expression of IL-10 and transforming growth factor (TGF) Β also can suppress local inflammation by influencing regulatory T cells.6
Additionally, mast cells and basophils are important in the IgE-mediated allergic reaction as well as the host immune response to parasites. When activated, basophils and mast cells produce TNF-α, IL-6, Il-4, IL-5, and IL-13, which contribute to the Th2 inflammatory response; however, the role of mast cells and basophils in scabies infections still is poorly understood.6
Macrophages, neutrophils, and dendritic cells (DCs) contribute to phagocytosis, antigen presentation, and differentiation of T cells, which also contribute to the inflammatory and allergic reactions associated with parasitic infections.6 Macrophages have been found in low numbers in scabies infestation, possibly due to immune-modulating molecules secreted by scabies mites. Early in an infestation, the mites secrete immune-modulating molecules, which inhibit macrophage migration to the site of inflammation, allowing the mites to grow.6 Neutrophils and DCs also are involved in the host immune response to scabies. Neutrophils are the predominant inflammatory cell infiltrate in scabies lesions. The scabies protein SMSB4 inhibits neutrophil opsonization and phagocytosis, thus suppressing bacterial killing.6 Some of the first antigen-presenting cells encountered by the antigen are DCs. They are involved in preparing the antigens for presentation to effector T cells, which leads to T-cell differentiation and activation.6
Cytokines are another important factor in the innate immune response. The host immune response to ordinary scabies is Th1-cell mediated, during which CD4+ and CD8+ T cells secrete IFN-γ, TNF-α, and IL-2.6 Therefore, IFN- γ and TNF-α are elevated in the serum of patients with ordinary scabies. Conversely, the host immune response to CS is Th2-cell mediated. T-helper 2 cells are needed in IgE-mediated hypersensitivity reactions, and they secrete IL-4, IL-5, and IL-13. In the serum of patients with CS, IL-l4, IL-5, and IL-13 are elevated while IFN-γ is decreased.6 Additionally, IL-6, TGF-Β, IL-23, IL-1Β, or IL-18 can induce Th17 cells to generate and secrete IL-17, which enhances the inflammatory response by inducing further expression of TNF-α, IL-1Β, IL-6, keratinocytes, and fibroblasts. T-helper 17 and IL-17 also are involved in psoriasis and atopic dermatitis, as well as Leishmania major and Schistosoma japonicum.6
Regulatory T cells Tregs secrete TGF-Β and IL-10, which suppress pathologic inflammation, and IL-10 is substantially reduced in patients with CS compared to those with ordinary scabies and uninfected control patients. Additionally, IL-10 can inhibit the synthesis of TNF-γ and IFN-α. Reduced IL-10 expression can lead to proliferation of IL-17 secretion, resulting in a regulatory T cell/Th17 dysfunctional immune response.6
Immunoglobulins are antibodies that are involved in the host’s adaptive immune response. The first antibody to appear in response to an antigen is IgM, and IgM bound to scabies antigens is present in 74%6 of patients with ordinary scabies. Because IgM is the first antibody to appear in response to a scabies infection, detection of serum IgM may allow for earlier detection of scabies; however, IgM has a high cross-reactivity between scabies mites and dust mites, which can hinder scabies diagnosis via IgM detection.6
Both patients with ordinary scabies and CS also show an increased circulatory IgG concentration compared to control groups; patients with CS have higher concentrations. Increased IgG also can be in part due to concurrent bacterial infections.6 When IgG or IgM antibodies bind to a pathogen, they activate the complement cascade, which further enhances the activity of these antibodies.9
Additionally, IgA is important in mucosal immune function. In both patients with ordinary scabies and CS, there is increased IgA binding to recombinant scabies mite antigens.6 Sarcoptes scabiei proteases that are localized in the mite’s gut and scybala suggest their involvement in mite digestion and burrowing. The increased secretion of these proteases into the host skin may contribute to the increased IgA,9 and these increased IgA levels have been shown to be positively correlated with severity of scabies infection.6
Also essential in allergic and parasitic inflammation, IgE is observed at higher levels in secondary infections of scabies compared to primary infections.6 Additionally, T-cell infiltrates are implicated in adaptive immune response to scabies. CD4+ T cells are the most prevalent T cells in ordinary scabies skin lesions; however, CD4+ T cells are minimal and CD8+ T cells are elevated in CS skin lesions. The increased CD8+ T cells may cause apoptosis of keratinocytes, leading to epidermal hyperproliferation. The apoptotic keratinocytes can secrete cytokines, which can lead to tissue damage.6 These T cells also may be involved in the failure of the skin’s immune system to mount an effective response to the parasite infestation, leading to uncontrolled parasitic growth. Because patients with AIDS who are infected with scabies mites often develop CS, it is also thought that CD4+ T cells are essential in the immune response to scabies.6
Diagnosis and Current Treatment Options
Current diagnosis of scabies is based on mites, eggs, and fecal matter from the host’s skin. Dermoscopy and fluorescent dermoscopy can be helpful in identifying the mites, eggs, and feces on the patient’s skin. Scabies treatment sometimes may be based solely on symptoms without any positive tests.8
Acaricides are the current method of treatment for scabies infestations.5 Acaricides can be expensive and toxic to the environment and food sources,10 and some agents have been associated with neurotoxicity5 in children or the development of certain cancers.11 Although topical acaricides are the standard form of treatment, oral ivermectin also can be used. Ivermectin is not associated with selective fetal toxicity, but there are limited safety data in pregnant women and in children weighing less than 15 kg (33 lb). Additionally, because symptoms typically are not present during an early infection, treating everyone in the household and those who had close contact with the patient can help prevent reinfection.4
Although these drugs have been shown to be effective at treating scabies, scabies mites are becoming increasingly resistant to acaricides.5 There are 4 main proposed mechanisms for why this occurs.12 The first is through voltage-gated sodium channels, which are involved in the normal functioning of neurons and myocytes. Permethrin, a type of acaricide, binds to voltage-gated sodium channels when it is in an open or active state and prevents it from closing. This creates repetitive neuron firing and hyperactivity, which ultimately kills the scabies mite. Some mites have mutated to close this channel, which reduces the binding potential of permethrin. Glutathione S-transferase is another mechanism of resistance. It catalyzes a bond that tags drugs for elimination. Increased activity or expressivity of glutathione S-transferase by scabies mites can lead to drug resistance.12 Adenosine triphosphate– binding cassette (ABC) transporters also may contribute to this resistance. The ABC transporters use adenosine triphosphate to facilitate the import or export of molecules. Scabies mites express a protein called the multidrug-resistant protein, which is an ABC transporter that is associated with drug resistance and is present in scabies mites.12 Lastly, ligand-gated chloride channels have been implicated in scabies resistance to acaricides. Ligand-gated chloride channels also are important in normal functioning of neurons and myocytes. Some antiparasitic drugs act on these channels, leading to a continuous influx of chloride, but some scabies mites have mutated this pathway.12
Pesticides and the Risk for Cancer
Pesticides commonly are used to treat scabies; however, a link between pesticide exposure and leukemia and lymphoma has been seen through epidemiologic studies, and there also is increasing biological evidence to suggest this.11 For example, the pesticide permethrin, which works by paralyzing the nervous system of insects,13 has been associated with an increased risk for leukemia and lymphoma in humans. Permethrin is a pyrethroid and, compared to control patients, children with leukemia had higher levels of pyrethroid metabolites in their blood.14 Numerical and structural chromosomal aberrations that give rise to gene fusions are the most common abnormalities seen in leukemia, and permethrin has been shown to induce DNA breaks, chromosome aberrations, and sister chromatid exchanges.14 Permethrin also has been associated with an increased risk for multiple myeloma.13
Furthermore, in utero exposure to pesticides has been associated with an increased risk for childhood leukemia.15 Pesticide exposure shortly before conception, during pregnancy, and after birth is associated with an increased risk for acute lymphocytic leukemia.16 In fact, the children of mothers who were exposed to pesticides 3 months before conception have been found to be at least twice as likely to be diagnosed with acute lymphocytic leukemia within the first year of life compared with children whose mothers were not exposed to pesticides.17 It is hypothesized that permethrin can cross the placenta and alter the hematopoietic precursor cells in the fetus, resulting in leukemogenesis.18 Pyrethroid metabolites also have been detected in umbilical cord blood samples and breast milk.15
In contrast to the research demonstrating a link between permethrin and cancer, other studies have found no association between permethrin19 and leukemia20; non-Hodgkin lymphoma19; or cancers of the colon, rectum, pancreas, lungs, skin, female breast, prostate, and urinary bladder.20 Because of conflicting research on the link between permethrin and cancer, more research is needed.,20
Importance of a Scabies Vaccine
Because scabies mites are developing increasing treatment resistance, more radical approaches such as vaccines are becoming important. While a scabies vaccine is still aspirational, animals that have been infected for a second time with scabies demonstrate a milder response to the second infection compared to the first infection, which could mean there is a potential for disease prevention through a vaccine.21 While educating patients and physicians, reporting cases of infection, and improving drug supply and access can help decrease scabies infestations, these are costly and difficult to implement. Scabies already is most prevalent in low-income areas, so costly interventions are even less feasible. An effective, one-dose vaccine would cost less than these efforts and therefore could be implemented more easily.9
In older adults, scabies more often manifests atypically and is more likely to progress to CS. Aged care centers are prone to institutional outbreaks, even in developed countries, so a vaccine also would greatly help this population. Additionally, the number of children attending day care centers, which also are prone to scabies outbreaks, is increasing. When a child contracts scabies, all close contacts need to be treated, so a preventive vaccine can be useful.9
One potential candidate for a scabies vaccine is total mite extract. Studies show that rabbits immunized with a total mite extract induce antibodies to more antigens than rabbits naturally infested with scabies mites; however, the mites cannot be cultured in vitro, which makes obtaining a large amount of their total extract difficult. Therefore, recombinant vaccines also have been proposed, as they are more easily available.22 One recombinant vaccine candidate is recombinant S scabiei serpin (rSs-serpin). Immunization with rSs-serpin has strong immunogenicity and produced immune protection in rabbits.22
Two other recombinant vaccine candidates are the rSs chitinaselike protein (CLP) 12 and the rSsCLP5. Chitinaselike proteins are very similar to chitinases; however, they are unable to degrade chitin. They are involved in immune reactions to infections, and CLPs from scabies mites have been shown to induce the host immune response.22 For example, in a particular rabbit study, rSsCLP5 demonstrated high immunoreactivity and immunogenicity. In fact, after exposure to S scabiei, 74.3% of rabbits who were vaccinated with rSsCLP5 had no detectable lesions.5 Also, after immunization with rSsCLP5 and rSsCLP12, there were increased levels of specific IgG and IgE antibodies produced and decreased numbers of infesting mites.22 Weight loss also is associated with severe scabies infection. Rabbits vaccinated with rSsCLP5 and exposed to the parasite gained weight, indicating protection via rSsCLP5. Even rabbits who did develop symptoms of scabies after immunization with rSsCLP5 and exposure to S scabiei showed less serious manifestations.5
A combination vaccine cocktail of rSs-serpin, rSsCLP12, and rSsCLP5 also has been proposed by Shen et al.22 Four test groups and a control group (n=12 per group) were included in a vaccine trial. Between 83.33% and 91.67% of rabbits vaccinated with this mixed recombinant cocktail vaccine had no detectable skin lesions from scabies. After immunization with the cocktail vaccine, the specific serum IgG and IgE antibodies also increased. For both IgG and IgE, increased levels were first detected at 1 week postimmunization and peaked at 2 weeks postimmunization.22 A multiepitope vaccine derived from these 3 recombinant proteins also was explored by Shen et al22; fewer rabbits vaccinated with it had no detectable scabies skin lesions compared to those treated with the vaccine cocktail. Although the multiepitope vaccine yielded less immume protection, it was associated with a slower disease course and milder symptoms compared with no vaccination.22
Two more proposed scabies recombinant vaccine candidates are derived from the antigens Ssag1 and Ssag2; however, rabbits vaccinated with Ssag1 or Ssag2 showed no immune protection or mite burden reduction.22 The lack of protection could be due to denaturation or degradation of the protective antigens. It also can be due to the low abundance of these antigens, meaning they may not be vital for the mite’s survival—survival—a potential avenue for future research. The antigens also could have lost their native structure and immunogenic properties during the purification and production process. Therefore, more research is needed to investigate how to purify these vaccines to keep the peptides more structurally similar to their native makeups.10 More research also is needed to better understand the antigen or antigens and their mechanisms that elicit a protective immune response.9
Final Thoughts
Scabies causes severe pruritus in mild cases but also can lead to severe disfigurement, sepsis, and even death. Scabies infestations are seen disproportionately more often in low-income and resource-poor communities, and the current treatment options are less accessible to these populations. Scabies infestations induce a complex immune response that involves multiple aspects of both the innate and adaptive immune systems and can be targeted to create a scabies vaccine. Development of a scabies vaccine is crucial considering the growing resistance to current standard treatments. Acaricides potentially are associated with an increased risk for malignancy, which further amplifies the need for a scabies vaccine. There currently are multiple promising scabies vaccine candidates; however, more research is needed to better understand the host’s immune response to scabies as well as how to more accurately and efficiently produce the vaccine. The development of a safe, effective, economical vaccine that can be mass distributed would be beneficial in the treatment of scabies, especially in resource-poor communities.
- Arlian LG, Morgan MS. A review of Sarcoptes scabiei: past, present and future. Parasit Vectors. 2017;10:297. doi:10.1186/s13071-017-2234-1
- Murray RL, Crane JS. Scabies. In: StatPearls. StatPearls Publishing. Updated July 31, 2023.
- Centers for Disease Control and Prevention. CDC—scabies—biology. November 2, 2010. https://www.cdc.gov/dpdx/scabies/index.html
- World Health Organization. Scabies. May 31, 2023. Accessed May 8, 2025. https://www.who.int/news-room/fact-sheets/detail/scabies
- Shen N, Zhang H, Ren Y, et al. A chitinase-like protein from Sarcoptes scabiei as a candidate anti-mite vaccine that contributes to immune protection in rabbits. Parasit Vectors. 2018;11:599. doi:10.1186/s13071- 018-3184-y
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Hicks MI, Elston DM. Scabies. Dermatolog Ther. 2009;22:279-292. doi:10.1111/j.1529-8019.2009.01243.x
- Morgan MS, Arlian LG, Rider SD, et al. A proteomic analysis of Sarcoptes scabiei (acari: Sarcoptidae). J Med Entomol. 2016;53:553-561. doi:10.1093/jme/tjv247
- Liu X, Walton S, Mounsey K. Vaccine against scabies: necessity and possibility. Parasitology. 2014;141:725-732. doi:10.1017 /s0031182013002047
- Casais R, Granda V, Balseiro A, et al. Vaccination of rabbits with immunodominant antigens from Sarcoptes scabiei induced high levels of humoral responses and pro-inflammatory cytokines but confers limited protection. Parasit Vectors. 2016;9:435. doi:10.1186 /s13071-016-1717-9?
- Navarrete-Meneses MP, Pedraza-Meléndez AI, Salas-Labadía C, et al. Low concentrations of permethrin and malathion induce numerical and structural abnormalities in KMT2A and IGH genes in vitro. J Appl Toxicol. 2018;38:1262-1270. doi:10.1002/jat.3638
- Khalil S, Abbas O, Kibbi AG, et al. Scabies in the age of increasing drug resistance. PLoS Negl Trop Dis. 2017;11:E0005920. doi:10.1371 /journal.pntd.0005920
- Rusiecki JA, Patel R, Koutros S, et al. Cancer incidence among pesticide applicators exposed to permethrin in the Agricultural Health Study. Environ Health Perspect. 2009;117:581-586. doi:10.1289 /ehp.11318
- Navarrete-Meneses MP, Salas-Labadía C, Sanabrais-Jiménez M, et al. Exposure to the insecticides permethrin and malathion induces leukemia and lymphoma-associated gene aberrations in vitro. Toxicol In Vitro. 2017;44:17-26. doi:10.1016/j.tiv.2017.06.013
- Navarrete-Meneses MDP, Pérez-Vera P. Pyrethroid pesticide exposure and hematological cancer: epidemiological, biological and molecular evidence. Rev Environ Health. 2019;34:197-210. doi:10.1515 /reveh-2018-0070
- Madrigal JM, Jones RR, Gunier RB, et al. Residential exposure to carbamate, organophosphate, and pyrethroid insecticides in house dust and risk of childhood acute lymphoblastic leukemia. Environ Res. 2021;201:111501. doi:10.1016/j.envres.2021.111501
- Ferreira JD, Couto AC, Pombo-de-Oliveira MS, et al. In utero pesticide exposure and leukemia in Brazilian children <2 years of age. Environ Health Perspect. 2013;121:269-275. doi:10.1289/ehp.1103942
- Borkhardt A, Wilda M, Fuchs U, et al. Congenital leukaemia after heavy abuse of permethrin during pregnancy. Arch Dis Child Fetal Neonatal Ed. 2003;88:F436-F437. doi:10.1136/fn.88.5.f436
- De Roos AJ, Schinasi LH, Miligi L, et al. Occupational insecticide exposure and risk of non]Hodgkin lymphoma: a pooled case]control study from the InterLymph consortium. Int J Cancer. 2021;149:1768-1786. doi:10.1002/ijc.33740
- Boffett, P, Desai V. Exposure to permethrin and cancer risk: a systematic review. Crit Rev Toxicol. 2018;48:433-442. doi:10.1080/1040 8444.2018.1439449
- Adji A, Rumokoy LJM, Salaki CL. Scabies vaccine as a new breakthrough for the challenge of acaricides resistance. Adv Biolog Sci Res. 2020;8:208-213. doi:10.2991/absr.k.200513.036
- Shen N, Wei W, Chen Y, et al. Vaccination with a cocktail vaccine elicits significant protection against Sarcoptes scabiei in rabbits, whereas the multi-epitope vaccine offers limited protection. Exp Parasitol. 2023;245:108442. doi:10.1016/j.exppara.2022.108442
- Arlian LG, Morgan MS. A review of Sarcoptes scabiei: past, present and future. Parasit Vectors. 2017;10:297. doi:10.1186/s13071-017-2234-1
- Murray RL, Crane JS. Scabies. In: StatPearls. StatPearls Publishing. Updated July 31, 2023.
- Centers for Disease Control and Prevention. CDC—scabies—biology. November 2, 2010. https://www.cdc.gov/dpdx/scabies/index.html
- World Health Organization. Scabies. May 31, 2023. Accessed May 8, 2025. https://www.who.int/news-room/fact-sheets/detail/scabies
- Shen N, Zhang H, Ren Y, et al. A chitinase-like protein from Sarcoptes scabiei as a candidate anti-mite vaccine that contributes to immune protection in rabbits. Parasit Vectors. 2018;11:599. doi:10.1186/s13071- 018-3184-y
- Bhat SA, Mounsey KE, Liu X, et al. Host immune responses to the itch mite, Sarcoptes scabiei, in humans. Parasit Vectors. 2017;10:385. doi:10.1186/s13071-017-2320-4
- Hicks MI, Elston DM. Scabies. Dermatolog Ther. 2009;22:279-292. doi:10.1111/j.1529-8019.2009.01243.x
- Morgan MS, Arlian LG, Rider SD, et al. A proteomic analysis of Sarcoptes scabiei (acari: Sarcoptidae). J Med Entomol. 2016;53:553-561. doi:10.1093/jme/tjv247
- Liu X, Walton S, Mounsey K. Vaccine against scabies: necessity and possibility. Parasitology. 2014;141:725-732. doi:10.1017 /s0031182013002047
- Casais R, Granda V, Balseiro A, et al. Vaccination of rabbits with immunodominant antigens from Sarcoptes scabiei induced high levels of humoral responses and pro-inflammatory cytokines but confers limited protection. Parasit Vectors. 2016;9:435. doi:10.1186 /s13071-016-1717-9?
- Navarrete-Meneses MP, Pedraza-Meléndez AI, Salas-Labadía C, et al. Low concentrations of permethrin and malathion induce numerical and structural abnormalities in KMT2A and IGH genes in vitro. J Appl Toxicol. 2018;38:1262-1270. doi:10.1002/jat.3638
- Khalil S, Abbas O, Kibbi AG, et al. Scabies in the age of increasing drug resistance. PLoS Negl Trop Dis. 2017;11:E0005920. doi:10.1371 /journal.pntd.0005920
- Rusiecki JA, Patel R, Koutros S, et al. Cancer incidence among pesticide applicators exposed to permethrin in the Agricultural Health Study. Environ Health Perspect. 2009;117:581-586. doi:10.1289 /ehp.11318
- Navarrete-Meneses MP, Salas-Labadía C, Sanabrais-Jiménez M, et al. Exposure to the insecticides permethrin and malathion induces leukemia and lymphoma-associated gene aberrations in vitro. Toxicol In Vitro. 2017;44:17-26. doi:10.1016/j.tiv.2017.06.013
- Navarrete-Meneses MDP, Pérez-Vera P. Pyrethroid pesticide exposure and hematological cancer: epidemiological, biological and molecular evidence. Rev Environ Health. 2019;34:197-210. doi:10.1515 /reveh-2018-0070
- Madrigal JM, Jones RR, Gunier RB, et al. Residential exposure to carbamate, organophosphate, and pyrethroid insecticides in house dust and risk of childhood acute lymphoblastic leukemia. Environ Res. 2021;201:111501. doi:10.1016/j.envres.2021.111501
- Ferreira JD, Couto AC, Pombo-de-Oliveira MS, et al. In utero pesticide exposure and leukemia in Brazilian children <2 years of age. Environ Health Perspect. 2013;121:269-275. doi:10.1289/ehp.1103942
- Borkhardt A, Wilda M, Fuchs U, et al. Congenital leukaemia after heavy abuse of permethrin during pregnancy. Arch Dis Child Fetal Neonatal Ed. 2003;88:F436-F437. doi:10.1136/fn.88.5.f436
- De Roos AJ, Schinasi LH, Miligi L, et al. Occupational insecticide exposure and risk of non]Hodgkin lymphoma: a pooled case]control study from the InterLymph consortium. Int J Cancer. 2021;149:1768-1786. doi:10.1002/ijc.33740
- Boffett, P, Desai V. Exposure to permethrin and cancer risk: a systematic review. Crit Rev Toxicol. 2018;48:433-442. doi:10.1080/1040 8444.2018.1439449
- Adji A, Rumokoy LJM, Salaki CL. Scabies vaccine as a new breakthrough for the challenge of acaricides resistance. Adv Biolog Sci Res. 2020;8:208-213. doi:10.2991/absr.k.200513.036
- Shen N, Wei W, Chen Y, et al. Vaccination with a cocktail vaccine elicits significant protection against Sarcoptes scabiei in rabbits, whereas the multi-epitope vaccine offers limited protection. Exp Parasitol. 2023;245:108442. doi:10.1016/j.exppara.2022.108442
Immune Responses and Health Disparities Warrant Scabies Vaccine Development
Immune Responses and Health Disparities Warrant Scabies Vaccine Development
PRACTICE POINTS
- Dermatologists should be aware of the impact scabies has on patients, especially on those in lower socioeconomic groups.
- Physicians and patients should be educated on scabies prevention and treatment to help decrease the spread of scabies infections.
Managing Seromas Following Skin Graft Placement in Dermatologic Surgery
Managing Seromas Following Skin Graft Placement in Dermatologic Surgery
A seroma is a collection of serous lymphatic fluid that forms in an anatomic or surgically created dead space—a void left between tissue layers, such as between the skin and underlying tissue, where fluid can accumulate. Seromas represent possible postoperative complications in many types of procedures, including general, oncologic, reconstructive, and dermatologic surgeries.1-3 While seroma formation following dermatologic surgery generally is uncommon, associated procedures include placement of split- or full-thickness skin grafts or liposuction.4,5 Many seromas follow a self-limited course. In some cases, seromas may cause discomfort, recur, or possibly become infected. Surgical techniques for prevention of seroma formation have been described in the dermatologic literature, but discussion of seroma management, particularly in dermatology, is not well documented. In this article, we describe a management approach for primary, recurrent, or late-stage seromas following placement of split- and full-thickness skin grafts in dermatologic surgery.
Practice Gap
To minimize the risk for seroma formation, attention should be paid to reducing dead space during graft placement. Small slits may be created in the skin graft after placement if the graft is larger than 2 to 3 cm in diameter to facilitate fluid drainage.6 Additionally, a tie-over bolster dressing that provides sustained even pressure over the entire graft should be applied and left in place for 1 week.7 Adjunctive measures, such as the use of fibrin sealants or quilting sutures, may further reduce the likelihood of fluid accumulation.7,8 Factors such as obesity, smoking, limited mobility, and inadequate elevation of the extremities undergoing surgery also should be addressed preoperatively to optimize outcomes of skin grafts.
Although these preventive strategies can be used during skin graft placement, seromas still can occur. Seromas typically manifest during the postoperative period after the removal of the protective dressings, including the bolster. The characteristic finding is the formation of a fluid-filled bulla under the graft. The associated serous lymphatic collection usually is yellow-tinged but may appear violaceous if bleeding has occurred beneath the graft. If the patient presents within 24 to 48 hours of seroma formation, the bulla may be tense or slightly tense; however, if days to weeks have passed since the seroma formed, the lesion may undergo fibrosis with thickening of the overlying tissue. If untreated, fibrosis may progress for several weeks, eventually resulting in nodule formation. Chronic seromas with retained fluid will persist for months. Seromas are more likely to develop under larger skin grafts (typically those exceeding 5-10 cm in diameter) or grafts placed in dependent positions, such as areas below the level of the heart where fluid pooling is more likely, especially on the arms and legs with associated movement.
The Technique
Our approach to seroma management is based on the timeline at presentation and whether the seroma is primary or recurrent or demonstrates late fibrosis. Successful management of primary seromas is centered on prompt drainage. Complete drainage using a #11 surgical blade may be accomplished with a single puncture to create a 2- to 3-mm opening for smaller seromas. Larger or multiple seromas under larger skin grafts may require creating multiple small punctures or small slits (ie, 5-10 mm) to allow for adequate drainage and reduce the incidence of seroma reaccumulation. Once successful drainage has occurred, a pressure dressing consisting of a thin layer of petroleum based ointment, a nonadherent dressing, gauze, and secure tape can help reduce the risk for reaccumulation.
Infrequently, seromas will reaccumulate under a skin graft. If this occurs, the graft may appear fibrous with lumps and loculations of seroma fluid separated by intact graft tissue, resulting in a “bound down” appearance (eFigure 1). This may require creating adequate slits for drainage in the graft. Multiple slits should be created if the seroma is larger (typically more than 3-4 cm in diameter) or loculations are present. If the fluid continues to reaccumulate and the drainage slits reseal, the next step is to cut a small hole in the graft to allow for uninterrupted drainage (eFigure 2). Manual digital pressure with moist gauze can assist in decompressing the seroma and removing residual fluid and gelatinous contents, promoting continuous drainage and preventing further fluid buildup (eFigure 3). These openings heal by secondary intention (eFigure 4). Local care during this time also is achieved with a thin layer of ointment, a nonstick pad, gauze, and secure tape. Dressings should be changed every 1 to 2 days until healing is complete.
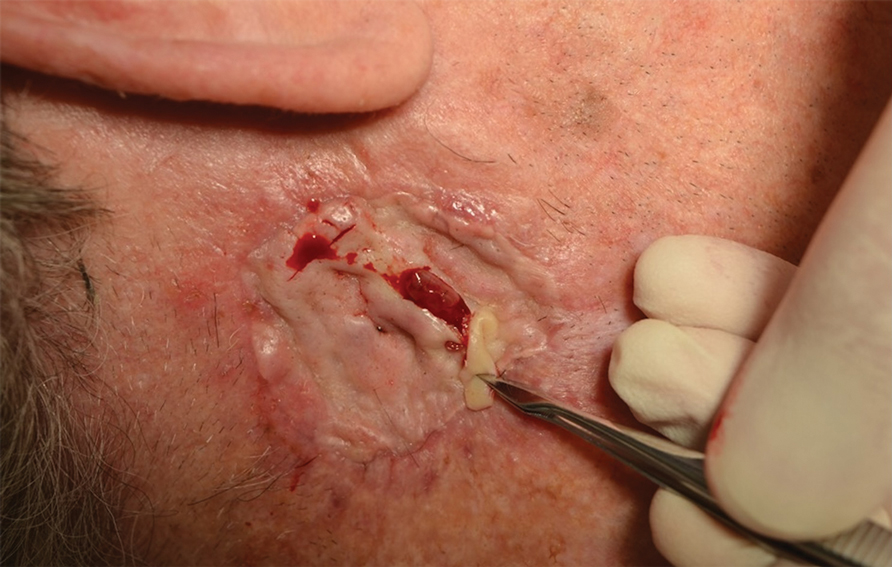
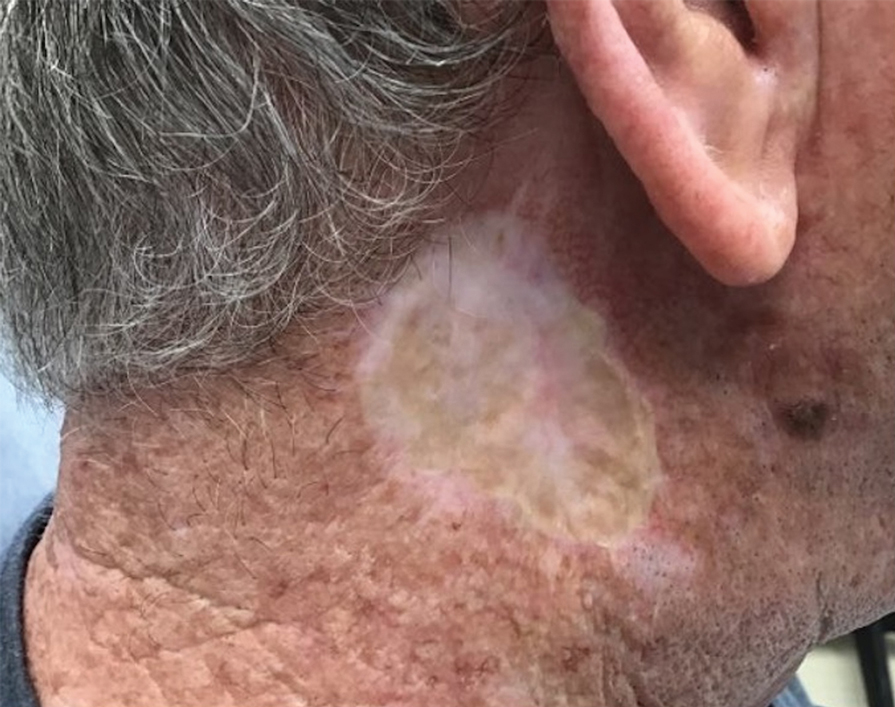
Seromas that lead to fibrotic nodule formation—typically occurring within several weeks to months if untreated—require additional steps for resolution. Once fibrosis occurs, these nodules can be managed by (1) placing adequate local anesthesia, (2) tangentially excising the nodules using either a skin biopsy blade or a #10 or #15 surgical blade, (3) using a handheld heat cautery or electrocautery device to achieve hemostasis, and (4) performing local care, as with any shave or tangential biopsy, until healing is complete. Typically, this requires a single treatment.
Practice Implications
While conservative management with continued compression dressings can be considered for postoperative seroma formation, interventional management sometimes is required. The size and duration of the seroma often guide management. For small seromas (typically less than 2-3 cm in diameter), a small slit incision with a #11 surgical blade may be performed at the dependent point of the seroma. Gentle pressure with a cotton-tipped applicator or moist gauze can be useful to express serous fluid; however, care should be taken not to disrupt adherence of the graft. Recurrent seromas or those with late fibrosis benefit from creation of a surgical window to allow uninterrupted drainage and removal of fibrous components, then can be left to heal by secondary intention with conservative local care.
- DeWitt C, Norris I, Fischer A, et al. A dermatologic approach to a recurrent auricular seroma. Dermatol Surg. 2018;44:1033-1035. doi:10.1097/DSS.0000000000001390
- Woodworth PA, McBoyle MF, Helmer SD, et al. Seroma formation after breast cancer surgery: incidence and predicting factors. Am Surg. 2000;66:444-451.
- Salari N, Fatahi B, Bartina Y, et al. The global prevalence of seroma after abdominoplasty: a systematic review and meta-analysis. Aesthetic Plast Surg. 2021;45:2821-2836. doi:10.1007/s00266-021-02365-6
- Bolognia J, Cerroni L, Schaffer JV. Dermatology. Elsevier; 2018.
- Taha AA, Wahba MM, Tahseen H. Liposuction: drains, are they adequate? Plast Reconstr Surg Glob Open. 2020;8:E2677. doi:10.1097/ GOX.0000000000002677
- Ishii N, Sakai S, Kishi K. A simple and safe method to create a drainage hole for thick skin grafts. Eplasty. 2017;17:ic27.
- Davis M, Baird D, Hill D, et al. Management of full-thickness skin grafts. Proc (Bayl Univ Med Cent). 2021;34:683-686. doi:10.1080 /08998280.2021.1953867
- Mittermayr R, Wassermann E, Thurnher M, et al. Skin graft fixation by slow clotting fibrin sealant applied as a thin layer. Burns. 2006; 32:305-311. doi:10.1016/j.burns.2005.10.010
A seroma is a collection of serous lymphatic fluid that forms in an anatomic or surgically created dead space—a void left between tissue layers, such as between the skin and underlying tissue, where fluid can accumulate. Seromas represent possible postoperative complications in many types of procedures, including general, oncologic, reconstructive, and dermatologic surgeries.1-3 While seroma formation following dermatologic surgery generally is uncommon, associated procedures include placement of split- or full-thickness skin grafts or liposuction.4,5 Many seromas follow a self-limited course. In some cases, seromas may cause discomfort, recur, or possibly become infected. Surgical techniques for prevention of seroma formation have been described in the dermatologic literature, but discussion of seroma management, particularly in dermatology, is not well documented. In this article, we describe a management approach for primary, recurrent, or late-stage seromas following placement of split- and full-thickness skin grafts in dermatologic surgery.
Practice Gap
To minimize the risk for seroma formation, attention should be paid to reducing dead space during graft placement. Small slits may be created in the skin graft after placement if the graft is larger than 2 to 3 cm in diameter to facilitate fluid drainage.6 Additionally, a tie-over bolster dressing that provides sustained even pressure over the entire graft should be applied and left in place for 1 week.7 Adjunctive measures, such as the use of fibrin sealants or quilting sutures, may further reduce the likelihood of fluid accumulation.7,8 Factors such as obesity, smoking, limited mobility, and inadequate elevation of the extremities undergoing surgery also should be addressed preoperatively to optimize outcomes of skin grafts.
Although these preventive strategies can be used during skin graft placement, seromas still can occur. Seromas typically manifest during the postoperative period after the removal of the protective dressings, including the bolster. The characteristic finding is the formation of a fluid-filled bulla under the graft. The associated serous lymphatic collection usually is yellow-tinged but may appear violaceous if bleeding has occurred beneath the graft. If the patient presents within 24 to 48 hours of seroma formation, the bulla may be tense or slightly tense; however, if days to weeks have passed since the seroma formed, the lesion may undergo fibrosis with thickening of the overlying tissue. If untreated, fibrosis may progress for several weeks, eventually resulting in nodule formation. Chronic seromas with retained fluid will persist for months. Seromas are more likely to develop under larger skin grafts (typically those exceeding 5-10 cm in diameter) or grafts placed in dependent positions, such as areas below the level of the heart where fluid pooling is more likely, especially on the arms and legs with associated movement.
The Technique
Our approach to seroma management is based on the timeline at presentation and whether the seroma is primary or recurrent or demonstrates late fibrosis. Successful management of primary seromas is centered on prompt drainage. Complete drainage using a #11 surgical blade may be accomplished with a single puncture to create a 2- to 3-mm opening for smaller seromas. Larger or multiple seromas under larger skin grafts may require creating multiple small punctures or small slits (ie, 5-10 mm) to allow for adequate drainage and reduce the incidence of seroma reaccumulation. Once successful drainage has occurred, a pressure dressing consisting of a thin layer of petroleum based ointment, a nonadherent dressing, gauze, and secure tape can help reduce the risk for reaccumulation.
Infrequently, seromas will reaccumulate under a skin graft. If this occurs, the graft may appear fibrous with lumps and loculations of seroma fluid separated by intact graft tissue, resulting in a “bound down” appearance (eFigure 1). This may require creating adequate slits for drainage in the graft. Multiple slits should be created if the seroma is larger (typically more than 3-4 cm in diameter) or loculations are present. If the fluid continues to reaccumulate and the drainage slits reseal, the next step is to cut a small hole in the graft to allow for uninterrupted drainage (eFigure 2). Manual digital pressure with moist gauze can assist in decompressing the seroma and removing residual fluid and gelatinous contents, promoting continuous drainage and preventing further fluid buildup (eFigure 3). These openings heal by secondary intention (eFigure 4). Local care during this time also is achieved with a thin layer of ointment, a nonstick pad, gauze, and secure tape. Dressings should be changed every 1 to 2 days until healing is complete.
Seromas that lead to fibrotic nodule formation—typically occurring within several weeks to months if untreated—require additional steps for resolution. Once fibrosis occurs, these nodules can be managed by (1) placing adequate local anesthesia, (2) tangentially excising the nodules using either a skin biopsy blade or a #10 or #15 surgical blade, (3) using a handheld heat cautery or electrocautery device to achieve hemostasis, and (4) performing local care, as with any shave or tangential biopsy, until healing is complete. Typically, this requires a single treatment.
Practice Implications
While conservative management with continued compression dressings can be considered for postoperative seroma formation, interventional management sometimes is required. The size and duration of the seroma often guide management. For small seromas (typically less than 2-3 cm in diameter), a small slit incision with a #11 surgical blade may be performed at the dependent point of the seroma. Gentle pressure with a cotton-tipped applicator or moist gauze can be useful to express serous fluid; however, care should be taken not to disrupt adherence of the graft. Recurrent seromas or those with late fibrosis benefit from creation of a surgical window to allow uninterrupted drainage and removal of fibrous components, then can be left to heal by secondary intention with conservative local care.
A seroma is a collection of serous lymphatic fluid that forms in an anatomic or surgically created dead space—a void left between tissue layers, such as between the skin and underlying tissue, where fluid can accumulate. Seromas represent possible postoperative complications in many types of procedures, including general, oncologic, reconstructive, and dermatologic surgeries.1-3 While seroma formation following dermatologic surgery generally is uncommon, associated procedures include placement of split- or full-thickness skin grafts or liposuction.4,5 Many seromas follow a self-limited course. In some cases, seromas may cause discomfort, recur, or possibly become infected. Surgical techniques for prevention of seroma formation have been described in the dermatologic literature, but discussion of seroma management, particularly in dermatology, is not well documented. In this article, we describe a management approach for primary, recurrent, or late-stage seromas following placement of split- and full-thickness skin grafts in dermatologic surgery.
Practice Gap
To minimize the risk for seroma formation, attention should be paid to reducing dead space during graft placement. Small slits may be created in the skin graft after placement if the graft is larger than 2 to 3 cm in diameter to facilitate fluid drainage.6 Additionally, a tie-over bolster dressing that provides sustained even pressure over the entire graft should be applied and left in place for 1 week.7 Adjunctive measures, such as the use of fibrin sealants or quilting sutures, may further reduce the likelihood of fluid accumulation.7,8 Factors such as obesity, smoking, limited mobility, and inadequate elevation of the extremities undergoing surgery also should be addressed preoperatively to optimize outcomes of skin grafts.
Although these preventive strategies can be used during skin graft placement, seromas still can occur. Seromas typically manifest during the postoperative period after the removal of the protective dressings, including the bolster. The characteristic finding is the formation of a fluid-filled bulla under the graft. The associated serous lymphatic collection usually is yellow-tinged but may appear violaceous if bleeding has occurred beneath the graft. If the patient presents within 24 to 48 hours of seroma formation, the bulla may be tense or slightly tense; however, if days to weeks have passed since the seroma formed, the lesion may undergo fibrosis with thickening of the overlying tissue. If untreated, fibrosis may progress for several weeks, eventually resulting in nodule formation. Chronic seromas with retained fluid will persist for months. Seromas are more likely to develop under larger skin grafts (typically those exceeding 5-10 cm in diameter) or grafts placed in dependent positions, such as areas below the level of the heart where fluid pooling is more likely, especially on the arms and legs with associated movement.
The Technique
Our approach to seroma management is based on the timeline at presentation and whether the seroma is primary or recurrent or demonstrates late fibrosis. Successful management of primary seromas is centered on prompt drainage. Complete drainage using a #11 surgical blade may be accomplished with a single puncture to create a 2- to 3-mm opening for smaller seromas. Larger or multiple seromas under larger skin grafts may require creating multiple small punctures or small slits (ie, 5-10 mm) to allow for adequate drainage and reduce the incidence of seroma reaccumulation. Once successful drainage has occurred, a pressure dressing consisting of a thin layer of petroleum based ointment, a nonadherent dressing, gauze, and secure tape can help reduce the risk for reaccumulation.
Infrequently, seromas will reaccumulate under a skin graft. If this occurs, the graft may appear fibrous with lumps and loculations of seroma fluid separated by intact graft tissue, resulting in a “bound down” appearance (eFigure 1). This may require creating adequate slits for drainage in the graft. Multiple slits should be created if the seroma is larger (typically more than 3-4 cm in diameter) or loculations are present. If the fluid continues to reaccumulate and the drainage slits reseal, the next step is to cut a small hole in the graft to allow for uninterrupted drainage (eFigure 2). Manual digital pressure with moist gauze can assist in decompressing the seroma and removing residual fluid and gelatinous contents, promoting continuous drainage and preventing further fluid buildup (eFigure 3). These openings heal by secondary intention (eFigure 4). Local care during this time also is achieved with a thin layer of ointment, a nonstick pad, gauze, and secure tape. Dressings should be changed every 1 to 2 days until healing is complete.
Seromas that lead to fibrotic nodule formation—typically occurring within several weeks to months if untreated—require additional steps for resolution. Once fibrosis occurs, these nodules can be managed by (1) placing adequate local anesthesia, (2) tangentially excising the nodules using either a skin biopsy blade or a #10 or #15 surgical blade, (3) using a handheld heat cautery or electrocautery device to achieve hemostasis, and (4) performing local care, as with any shave or tangential biopsy, until healing is complete. Typically, this requires a single treatment.
Practice Implications
While conservative management with continued compression dressings can be considered for postoperative seroma formation, interventional management sometimes is required. The size and duration of the seroma often guide management. For small seromas (typically less than 2-3 cm in diameter), a small slit incision with a #11 surgical blade may be performed at the dependent point of the seroma. Gentle pressure with a cotton-tipped applicator or moist gauze can be useful to express serous fluid; however, care should be taken not to disrupt adherence of the graft. Recurrent seromas or those with late fibrosis benefit from creation of a surgical window to allow uninterrupted drainage and removal of fibrous components, then can be left to heal by secondary intention with conservative local care.
- DeWitt C, Norris I, Fischer A, et al. A dermatologic approach to a recurrent auricular seroma. Dermatol Surg. 2018;44:1033-1035. doi:10.1097/DSS.0000000000001390
- Woodworth PA, McBoyle MF, Helmer SD, et al. Seroma formation after breast cancer surgery: incidence and predicting factors. Am Surg. 2000;66:444-451.
- Salari N, Fatahi B, Bartina Y, et al. The global prevalence of seroma after abdominoplasty: a systematic review and meta-analysis. Aesthetic Plast Surg. 2021;45:2821-2836. doi:10.1007/s00266-021-02365-6
- Bolognia J, Cerroni L, Schaffer JV. Dermatology. Elsevier; 2018.
- Taha AA, Wahba MM, Tahseen H. Liposuction: drains, are they adequate? Plast Reconstr Surg Glob Open. 2020;8:E2677. doi:10.1097/ GOX.0000000000002677
- Ishii N, Sakai S, Kishi K. A simple and safe method to create a drainage hole for thick skin grafts. Eplasty. 2017;17:ic27.
- Davis M, Baird D, Hill D, et al. Management of full-thickness skin grafts. Proc (Bayl Univ Med Cent). 2021;34:683-686. doi:10.1080 /08998280.2021.1953867
- Mittermayr R, Wassermann E, Thurnher M, et al. Skin graft fixation by slow clotting fibrin sealant applied as a thin layer. Burns. 2006; 32:305-311. doi:10.1016/j.burns.2005.10.010
- DeWitt C, Norris I, Fischer A, et al. A dermatologic approach to a recurrent auricular seroma. Dermatol Surg. 2018;44:1033-1035. doi:10.1097/DSS.0000000000001390
- Woodworth PA, McBoyle MF, Helmer SD, et al. Seroma formation after breast cancer surgery: incidence and predicting factors. Am Surg. 2000;66:444-451.
- Salari N, Fatahi B, Bartina Y, et al. The global prevalence of seroma after abdominoplasty: a systematic review and meta-analysis. Aesthetic Plast Surg. 2021;45:2821-2836. doi:10.1007/s00266-021-02365-6
- Bolognia J, Cerroni L, Schaffer JV. Dermatology. Elsevier; 2018.
- Taha AA, Wahba MM, Tahseen H. Liposuction: drains, are they adequate? Plast Reconstr Surg Glob Open. 2020;8:E2677. doi:10.1097/ GOX.0000000000002677
- Ishii N, Sakai S, Kishi K. A simple and safe method to create a drainage hole for thick skin grafts. Eplasty. 2017;17:ic27.
- Davis M, Baird D, Hill D, et al. Management of full-thickness skin grafts. Proc (Bayl Univ Med Cent). 2021;34:683-686. doi:10.1080 /08998280.2021.1953867
- Mittermayr R, Wassermann E, Thurnher M, et al. Skin graft fixation by slow clotting fibrin sealant applied as a thin layer. Burns. 2006; 32:305-311. doi:10.1016/j.burns.2005.10.010
Managing Seromas Following Skin Graft Placement in Dermatologic Surgery
Managing Seromas Following Skin Graft Placement in Dermatologic Surgery
PRACTICE POINTS
- If seromas are identified early (within 24 to 48 hours postoperatively), prompt drainage with a small incision can prevent complications, such as fibrosis or nodule formation, and improve patient comfort.
- For larger or recurrent seromas, multiple small slits or a surgical window should be created to ensure continuous drainage and prevent reaccumulation. Manual compression with moist gauze also can aid in fluid removal.
- If fibrosis develops and leads to nodule formation, early excision of the fibrotic tissue with local anesthesia is essential for resolution. This approach typically requires a single treatment, with secondary intention healing.

Advocacy and Compliance Issues Impacting Dermatology in 2025
Advocacy and Compliance Issues Impacting Dermatology in 2025
The US health care system presents major administrative burdens—particularly in coding, billing, and reimbursement—that impact clinical efficiency and patient access. Dermatologists have experienced disproportionate reimbursement declines. A longitudinal review of 20 dermatologic service codes found a 10% average decline in Medicare reimbursement between 2000 and 2020.1 A recent cross-sectional study showed a 4.7% average decline in reimbursement rates from 2007 to 2021 for commonly performed dermatologic procedures, with variation across procedure categories.2 These reductions threaten practice sustainability and highlight the urgent need for comprehensive, long-term payment reform to preserve access to high-quality dermatologic care.
In dermatopathology, policy changes to reimbursement and laboratory oversight directly impact practice operations. Specialty-specific advocacy remains vital in driving policy changes. In this article, we highlight a recent advocacy win—the reversal of immunohistochemistry (IHC) stain denials—and provide updates on a new position statement on IHC guidance. We also outline regulatory changes to the Clinical Laboratory Improvement Amendments (CLIA) of 1988 and College of American Pathologists (CAP) laboratory director requirements and emphasize the importance of continued legislative advocacy.
Reversal of Reimbursement Denials for IHC Stains
EviCore, a medical benefits management company serving over one-third of insured individuals in the United States, is hired by an extensive network of insurance companies to develop clinical and laboratory guidelines and utilization and payment integrity programs.3 EviCore’s laboratory management guidelines for 2024 denied IHC stains (Current Procedural Terminology codes 88341 and 88342) as not medically necessary when associated with specific International Statistical Classification of Diseases, Tenth Revision, skin lesion codes (eTable 1).3-5 These policies caused major disruption to dermatopathology services nationwide, impacting both academic and private laboratories (eTable 2).5 The implementation of such blanket denials interferes with clinical decision-making, compromising diagnostic quality by restricting medically necessary and essential laboratory and pathology services. The American Academy of Dermatology Association (AADA) and CAP leadership formally objected to the policy, citing how these reimbursement denials fail to account for the importance of clinical judgment and diagnostic nuance.6
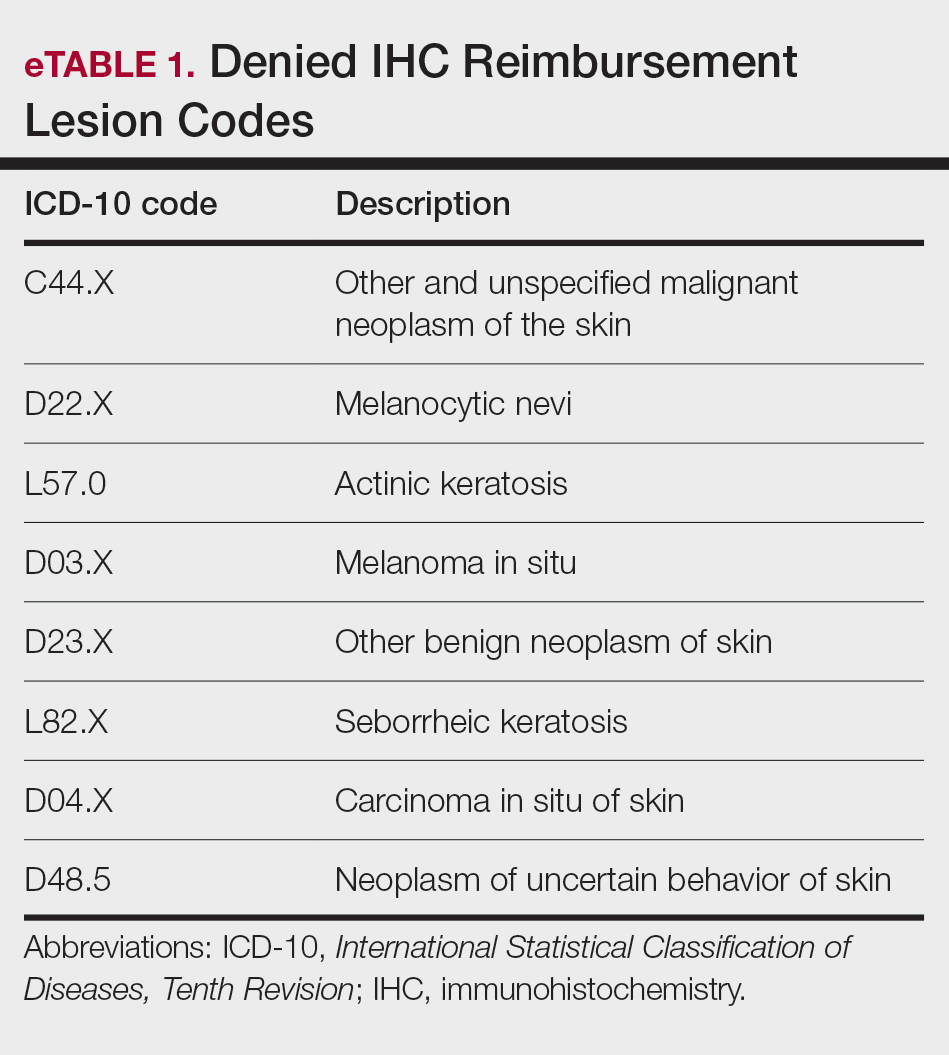

Thanks to broad advocacy efforts, EviCore updated its guidelines effective January 1, 2025. The skin-related International Statistical Classification of Diseases, Tenth Revision, codes were removed from IHC coverage restrictions, with automatic payment reinstated retroactive to March 15, 2024. EviCore also rescinded language denying reimbursement if a diagnosis could be made without the use of IHC stains.7 While this reversal is a notable achievement, ongoing monitoring of emerging trends in claim denials remains crucial. Continued advocacy, proper documentation, and adherence to American Society of Dermatopathology (ASDP) Appropriate Use Criteria is essential to protecting clinical autonomy.
The AADA’s Dermatopathology Committee developed a new position statement on IHC utilization supporting the advocacy efforts with payers, who recently have tried to implement restrictive limitations.8 Immunohistochemistry is considered a valuable tool for dermatopathology diagnosis, and its utility aids in the confirmation, exclusion, or change in diagnosis.9 By clearly outlining the clinical value of IHC in dermatopathology, this statement reinforces the need to advocate against restrictive payer policies to preserve physician autonomy and promote appropriate, evidence-based use of IHC stains.8
In addition, the ASDP Standards of Practice Committee is working with the Johns Hopkins–Global Appropriateness Measures data-powered analytics platform to develop physician-led IHC benchmarks. The ASDP Appropriate Use Criteria mobile application is a valuable clinical tool for dermatopathologists, general pathologists, dermatologists, and other providers, offering case-based recommendations for test utilization grounded in current evidence.9
Legislative Advocacy: Support for H.R. 879
Physician payment cuts have reached a critical tipping point. Since 2001, physicians have experienced a 33% average reduction in Medicare reimbursement, unadjusted for inflation or rising overhead.10 In January 2025, the Centers for Medicare & Medicaid Services (CMS) imposed a further 2.83% cut, despite projecting a 3.5% increase in the Medicare Economic Index.11,12 Dermatologists and other physician groups cannot continue to absorb these reductions, as they have several consequences, including the inability to maintain practices, forcing some physicians out of business, driving health care consolidation, and limiting patient access.
The Medicare Patient Access and Practice Stabilization Act (H.R. 879)13 is bipartisan legislation that seeks to stop the 2.8% Medicare physician payment cut that went into effect in January 2025, provide physicians with an additional 2% inflation-adjusted payment increase for 2025, and help stabilize Medicare reimbursement rates.13,14 As the impact of continued cuts threatens both patient access and practice viability, member engagement is essential to advancing federal physician payment reform. To support sustainable payment reform and protect access to care, visit the AADA Advocacy Action Center online.14
2025 CLIA and CAP Laboratory Director Requirements: What’s Changing?
As of December 28, 2024, updated CLIA regulations took effect for all laboratories performing moderate- or high-complexity testing. These revisions aim to modernize outdated requirements and update regulations to incorporate technological advancements such as automation and artificial intelligence.15 New CLIA standards require laboratory directors with Doctor of Medicine or Doctor of Osteopathy degrees to be certified in anatomic and/or clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology.15 For physicians who do not hold these board-certified qualifications, there are alternative pathways to becoming a laboratory director based on experience and education for physicians licensed to practice in the jurisdiction where the laboratory is located. For high-complexity laboratories, individuals need at least 2 years of experience directing or supervising high-complexity testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities. For moderate-complexity laboratories, individuals need at least 1 year of experience supervising nonwaived laboratory testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities.16
If the current laboratory director is not board certified in pathology, the new regulation will permit the grandfathering of current laboratory directors if existing laboratory directors have remained continuously employed in their current role since December 28, 2024.16 Therefore, individuals who were already employed in qualifying positions as of December 28, 2024, will be grandfathered in and will not need to meet the new educational requirements if they remain employed without interruption. All individuals qualifying after December 28, 2024, will be required to do so under the new provisions stated earlier.
The CMS updated laboratory personnel requirements, thereby impacting all CLIA-certified laboratories and those seeking CLIA certification. Likewise, laboratories seeking accreditation by the CAP must meet the new laboratory personnel requirements.17 In some cases, CAP requirements are more stringent than the CLIA regulations (CAP accreditation is more stringent in areas of quality control, personnel qualifications, proficiency testing, and in oversight of laboratory developed tests).15-17 If more stringent state or local regulations are in place for personnel qualifications, including requirements for state licensure, they must be followed.
The AADA formed an ad hoc workgroup to address the CLIA laboratory director requirements and is actively engaging CMS to amend these requirements immediately. Formal objections have been submitted, and direct dialogue with CMS leadership is under way in collaboration with the American Board of Dermatology and leading dermatology and pathology societies.
Final Thoughts
Advocacy remains essential to the future of dermatology. From payer policy reversals to laboratory compliance reforms and federal payment advocacy, physicians must remain engaged. Whether it is safeguarding diagnostic autonomy or securing financial sustainability, we must continue to put “skin in the game.”
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Mazmudar RS, Sheth A, Tripathi R, et al. Inflation-adjusted trends in Medicare reimbursement for common dermatologic procedures, 2007-2021. JAMA Dermatol. 2021;157:1355-1358.
- Miller TC, Rucker P, Armstrong D. “Not medically necessary”: inside the company helping America’s biggest health insurers deny coverage for care. ProPublica. October 23, 2024. Accessed April 23, 2025. https://www.propublica.org/article/evicore-health-insurance-denials-cigna-unitedhealthcare-aetna-prior-authorizations
- EviCore healthcare. Immunohistochemistry (IHC). Lab Management Guidelines v2.0.2024. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/MOL.CS_.104.A_Immunohistochemistry%20%28IHC%29_V2.0.2024_eff11.01.2024_pub12.31.2024.pdf
- EviCore. Laboratory management. Accessed April 23, 2025. https://www.evicore.com/provider/clinical-guidelines-details?solution=laboratory%20management
- Saad AJ. College of American Pathologists. December 12, 2023. Accessed April 23, 2025. https://documents.cap.org/documents /Wellmark-Letter- https://documents.cap.org/documents/wellmarkcap-letter2023.pdf
- EviCore healthcare. Clinical Guidelines: Lab Management Program. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/Cigna_LabMgmt_V1.0.2025_eff01.01.2025_pub08.22.2024_0.pdf
- American Academy of Dermatology Association. Position statement on immunohistochemistry utilization. Accessed May 9, 2024. https://server.aad.org/forms/policies/Uploads/PS/PS-Immunohistochemistry%20Utilization.pdf
- Naert KA, Trotter MJ. Utilization and utility of immunohistochemistry in dermatopathology. Am J Dermatopathol. 2013;35:74-77.
- American Medical Association. Medicare physician payment continues to fall further behind practice cost inflation. Accessed April 23, 2025. https:// www.ama-assn.org/system/files/2025-medicare-updates-inflation-chart.pdf
- Centers for Medicare & Medicaid Services. Calendar year (CY) 2025 Medicare Physician Fee Schedule final rule. Accessed April 23, 2025. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2025-medicare-physician-fee-schedule-final-rule
- American Medical Association. The Medicare Economic Index. Accessed April 23 2025. https://www.ama-assn.org/system/files/medicare-basics-medicare-economic-index.pdf
- Medicare Patient Access and Practice Stabilization Act, HR 879, 119th Cong (2025). Accessed April 23, 2025. https://www.congress.gov/bill/119th-congress/house-bill/879
- American Academy of Dermatology Association. AADA advocacy action center. Accessed April 23, 2025. https://www.aad.org/member/advocacy/take-action
- Department of Health and Human Services. Centers for Medicare & Medicaid Services. Clinical Laboratory Improvement Amendments of 1988 (CLIA) fees; histocompatibility, personnel, and alternative sanctions for certificate of waiver laboratories. Fed Regist. 2023;88:89976-90044.
- College of American Pathologists. CAP accreditation checklists – 2024 edition. Accessed April 23, 2025. https://documents.cap.org/documents/2024-Checklist-Summary.pdf?_gl=1*1b4rei9*_ga*NDc0NjYwNjM5LjE3NDQ3NTI4NjA.*_ga_97ZFJSQQ0X*MTc0NDc2OTc3My40LjEuMTc0NDc2OTgyOC4wLjAuMA
- Bennett SA, Conn CM, Gill HE, et al. Regulatory requirements for laboratory developed tests in the United States. J Immunol Methods. 2025;537:113813.
The US health care system presents major administrative burdens—particularly in coding, billing, and reimbursement—that impact clinical efficiency and patient access. Dermatologists have experienced disproportionate reimbursement declines. A longitudinal review of 20 dermatologic service codes found a 10% average decline in Medicare reimbursement between 2000 and 2020.1 A recent cross-sectional study showed a 4.7% average decline in reimbursement rates from 2007 to 2021 for commonly performed dermatologic procedures, with variation across procedure categories.2 These reductions threaten practice sustainability and highlight the urgent need for comprehensive, long-term payment reform to preserve access to high-quality dermatologic care.
In dermatopathology, policy changes to reimbursement and laboratory oversight directly impact practice operations. Specialty-specific advocacy remains vital in driving policy changes. In this article, we highlight a recent advocacy win—the reversal of immunohistochemistry (IHC) stain denials—and provide updates on a new position statement on IHC guidance. We also outline regulatory changes to the Clinical Laboratory Improvement Amendments (CLIA) of 1988 and College of American Pathologists (CAP) laboratory director requirements and emphasize the importance of continued legislative advocacy.
Reversal of Reimbursement Denials for IHC Stains
EviCore, a medical benefits management company serving over one-third of insured individuals in the United States, is hired by an extensive network of insurance companies to develop clinical and laboratory guidelines and utilization and payment integrity programs.3 EviCore’s laboratory management guidelines for 2024 denied IHC stains (Current Procedural Terminology codes 88341 and 88342) as not medically necessary when associated with specific International Statistical Classification of Diseases, Tenth Revision, skin lesion codes (eTable 1).3-5 These policies caused major disruption to dermatopathology services nationwide, impacting both academic and private laboratories (eTable 2).5 The implementation of such blanket denials interferes with clinical decision-making, compromising diagnostic quality by restricting medically necessary and essential laboratory and pathology services. The American Academy of Dermatology Association (AADA) and CAP leadership formally objected to the policy, citing how these reimbursement denials fail to account for the importance of clinical judgment and diagnostic nuance.6
Thanks to broad advocacy efforts, EviCore updated its guidelines effective January 1, 2025. The skin-related International Statistical Classification of Diseases, Tenth Revision, codes were removed from IHC coverage restrictions, with automatic payment reinstated retroactive to March 15, 2024. EviCore also rescinded language denying reimbursement if a diagnosis could be made without the use of IHC stains.7 While this reversal is a notable achievement, ongoing monitoring of emerging trends in claim denials remains crucial. Continued advocacy, proper documentation, and adherence to American Society of Dermatopathology (ASDP) Appropriate Use Criteria is essential to protecting clinical autonomy.
The AADA’s Dermatopathology Committee developed a new position statement on IHC utilization supporting the advocacy efforts with payers, who recently have tried to implement restrictive limitations.8 Immunohistochemistry is considered a valuable tool for dermatopathology diagnosis, and its utility aids in the confirmation, exclusion, or change in diagnosis.9 By clearly outlining the clinical value of IHC in dermatopathology, this statement reinforces the need to advocate against restrictive payer policies to preserve physician autonomy and promote appropriate, evidence-based use of IHC stains.8
In addition, the ASDP Standards of Practice Committee is working with the Johns Hopkins–Global Appropriateness Measures data-powered analytics platform to develop physician-led IHC benchmarks. The ASDP Appropriate Use Criteria mobile application is a valuable clinical tool for dermatopathologists, general pathologists, dermatologists, and other providers, offering case-based recommendations for test utilization grounded in current evidence.9
Legislative Advocacy: Support for H.R. 879
Physician payment cuts have reached a critical tipping point. Since 2001, physicians have experienced a 33% average reduction in Medicare reimbursement, unadjusted for inflation or rising overhead.10 In January 2025, the Centers for Medicare & Medicaid Services (CMS) imposed a further 2.83% cut, despite projecting a 3.5% increase in the Medicare Economic Index.11,12 Dermatologists and other physician groups cannot continue to absorb these reductions, as they have several consequences, including the inability to maintain practices, forcing some physicians out of business, driving health care consolidation, and limiting patient access.
The Medicare Patient Access and Practice Stabilization Act (H.R. 879)13 is bipartisan legislation that seeks to stop the 2.8% Medicare physician payment cut that went into effect in January 2025, provide physicians with an additional 2% inflation-adjusted payment increase for 2025, and help stabilize Medicare reimbursement rates.13,14 As the impact of continued cuts threatens both patient access and practice viability, member engagement is essential to advancing federal physician payment reform. To support sustainable payment reform and protect access to care, visit the AADA Advocacy Action Center online.14
2025 CLIA and CAP Laboratory Director Requirements: What’s Changing?
As of December 28, 2024, updated CLIA regulations took effect for all laboratories performing moderate- or high-complexity testing. These revisions aim to modernize outdated requirements and update regulations to incorporate technological advancements such as automation and artificial intelligence.15 New CLIA standards require laboratory directors with Doctor of Medicine or Doctor of Osteopathy degrees to be certified in anatomic and/or clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology.15 For physicians who do not hold these board-certified qualifications, there are alternative pathways to becoming a laboratory director based on experience and education for physicians licensed to practice in the jurisdiction where the laboratory is located. For high-complexity laboratories, individuals need at least 2 years of experience directing or supervising high-complexity testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities. For moderate-complexity laboratories, individuals need at least 1 year of experience supervising nonwaived laboratory testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities.16
If the current laboratory director is not board certified in pathology, the new regulation will permit the grandfathering of current laboratory directors if existing laboratory directors have remained continuously employed in their current role since December 28, 2024.16 Therefore, individuals who were already employed in qualifying positions as of December 28, 2024, will be grandfathered in and will not need to meet the new educational requirements if they remain employed without interruption. All individuals qualifying after December 28, 2024, will be required to do so under the new provisions stated earlier.
The CMS updated laboratory personnel requirements, thereby impacting all CLIA-certified laboratories and those seeking CLIA certification. Likewise, laboratories seeking accreditation by the CAP must meet the new laboratory personnel requirements.17 In some cases, CAP requirements are more stringent than the CLIA regulations (CAP accreditation is more stringent in areas of quality control, personnel qualifications, proficiency testing, and in oversight of laboratory developed tests).15-17 If more stringent state or local regulations are in place for personnel qualifications, including requirements for state licensure, they must be followed.
The AADA formed an ad hoc workgroup to address the CLIA laboratory director requirements and is actively engaging CMS to amend these requirements immediately. Formal objections have been submitted, and direct dialogue with CMS leadership is under way in collaboration with the American Board of Dermatology and leading dermatology and pathology societies.
Final Thoughts
Advocacy remains essential to the future of dermatology. From payer policy reversals to laboratory compliance reforms and federal payment advocacy, physicians must remain engaged. Whether it is safeguarding diagnostic autonomy or securing financial sustainability, we must continue to put “skin in the game.”
The US health care system presents major administrative burdens—particularly in coding, billing, and reimbursement—that impact clinical efficiency and patient access. Dermatologists have experienced disproportionate reimbursement declines. A longitudinal review of 20 dermatologic service codes found a 10% average decline in Medicare reimbursement between 2000 and 2020.1 A recent cross-sectional study showed a 4.7% average decline in reimbursement rates from 2007 to 2021 for commonly performed dermatologic procedures, with variation across procedure categories.2 These reductions threaten practice sustainability and highlight the urgent need for comprehensive, long-term payment reform to preserve access to high-quality dermatologic care.
In dermatopathology, policy changes to reimbursement and laboratory oversight directly impact practice operations. Specialty-specific advocacy remains vital in driving policy changes. In this article, we highlight a recent advocacy win—the reversal of immunohistochemistry (IHC) stain denials—and provide updates on a new position statement on IHC guidance. We also outline regulatory changes to the Clinical Laboratory Improvement Amendments (CLIA) of 1988 and College of American Pathologists (CAP) laboratory director requirements and emphasize the importance of continued legislative advocacy.
Reversal of Reimbursement Denials for IHC Stains
EviCore, a medical benefits management company serving over one-third of insured individuals in the United States, is hired by an extensive network of insurance companies to develop clinical and laboratory guidelines and utilization and payment integrity programs.3 EviCore’s laboratory management guidelines for 2024 denied IHC stains (Current Procedural Terminology codes 88341 and 88342) as not medically necessary when associated with specific International Statistical Classification of Diseases, Tenth Revision, skin lesion codes (eTable 1).3-5 These policies caused major disruption to dermatopathology services nationwide, impacting both academic and private laboratories (eTable 2).5 The implementation of such blanket denials interferes with clinical decision-making, compromising diagnostic quality by restricting medically necessary and essential laboratory and pathology services. The American Academy of Dermatology Association (AADA) and CAP leadership formally objected to the policy, citing how these reimbursement denials fail to account for the importance of clinical judgment and diagnostic nuance.6
Thanks to broad advocacy efforts, EviCore updated its guidelines effective January 1, 2025. The skin-related International Statistical Classification of Diseases, Tenth Revision, codes were removed from IHC coverage restrictions, with automatic payment reinstated retroactive to March 15, 2024. EviCore also rescinded language denying reimbursement if a diagnosis could be made without the use of IHC stains.7 While this reversal is a notable achievement, ongoing monitoring of emerging trends in claim denials remains crucial. Continued advocacy, proper documentation, and adherence to American Society of Dermatopathology (ASDP) Appropriate Use Criteria is essential to protecting clinical autonomy.
The AADA’s Dermatopathology Committee developed a new position statement on IHC utilization supporting the advocacy efforts with payers, who recently have tried to implement restrictive limitations.8 Immunohistochemistry is considered a valuable tool for dermatopathology diagnosis, and its utility aids in the confirmation, exclusion, or change in diagnosis.9 By clearly outlining the clinical value of IHC in dermatopathology, this statement reinforces the need to advocate against restrictive payer policies to preserve physician autonomy and promote appropriate, evidence-based use of IHC stains.8
In addition, the ASDP Standards of Practice Committee is working with the Johns Hopkins–Global Appropriateness Measures data-powered analytics platform to develop physician-led IHC benchmarks. The ASDP Appropriate Use Criteria mobile application is a valuable clinical tool for dermatopathologists, general pathologists, dermatologists, and other providers, offering case-based recommendations for test utilization grounded in current evidence.9
Legislative Advocacy: Support for H.R. 879
Physician payment cuts have reached a critical tipping point. Since 2001, physicians have experienced a 33% average reduction in Medicare reimbursement, unadjusted for inflation or rising overhead.10 In January 2025, the Centers for Medicare & Medicaid Services (CMS) imposed a further 2.83% cut, despite projecting a 3.5% increase in the Medicare Economic Index.11,12 Dermatologists and other physician groups cannot continue to absorb these reductions, as they have several consequences, including the inability to maintain practices, forcing some physicians out of business, driving health care consolidation, and limiting patient access.
The Medicare Patient Access and Practice Stabilization Act (H.R. 879)13 is bipartisan legislation that seeks to stop the 2.8% Medicare physician payment cut that went into effect in January 2025, provide physicians with an additional 2% inflation-adjusted payment increase for 2025, and help stabilize Medicare reimbursement rates.13,14 As the impact of continued cuts threatens both patient access and practice viability, member engagement is essential to advancing federal physician payment reform. To support sustainable payment reform and protect access to care, visit the AADA Advocacy Action Center online.14
2025 CLIA and CAP Laboratory Director Requirements: What’s Changing?
As of December 28, 2024, updated CLIA regulations took effect for all laboratories performing moderate- or high-complexity testing. These revisions aim to modernize outdated requirements and update regulations to incorporate technological advancements such as automation and artificial intelligence.15 New CLIA standards require laboratory directors with Doctor of Medicine or Doctor of Osteopathy degrees to be certified in anatomic and/or clinical pathology by the American Board of Pathology or the American Osteopathic Board of Pathology.15 For physicians who do not hold these board-certified qualifications, there are alternative pathways to becoming a laboratory director based on experience and education for physicians licensed to practice in the jurisdiction where the laboratory is located. For high-complexity laboratories, individuals need at least 2 years of experience directing or supervising high-complexity testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities. For moderate-complexity laboratories, individuals need at least 1 year of experience supervising nonwaived laboratory testing and at least 20 continuing education credit hours in laboratory practice that cover director responsibilities.16
If the current laboratory director is not board certified in pathology, the new regulation will permit the grandfathering of current laboratory directors if existing laboratory directors have remained continuously employed in their current role since December 28, 2024.16 Therefore, individuals who were already employed in qualifying positions as of December 28, 2024, will be grandfathered in and will not need to meet the new educational requirements if they remain employed without interruption. All individuals qualifying after December 28, 2024, will be required to do so under the new provisions stated earlier.
The CMS updated laboratory personnel requirements, thereby impacting all CLIA-certified laboratories and those seeking CLIA certification. Likewise, laboratories seeking accreditation by the CAP must meet the new laboratory personnel requirements.17 In some cases, CAP requirements are more stringent than the CLIA regulations (CAP accreditation is more stringent in areas of quality control, personnel qualifications, proficiency testing, and in oversight of laboratory developed tests).15-17 If more stringent state or local regulations are in place for personnel qualifications, including requirements for state licensure, they must be followed.
The AADA formed an ad hoc workgroup to address the CLIA laboratory director requirements and is actively engaging CMS to amend these requirements immediately. Formal objections have been submitted, and direct dialogue with CMS leadership is under way in collaboration with the American Board of Dermatology and leading dermatology and pathology societies.
Final Thoughts
Advocacy remains essential to the future of dermatology. From payer policy reversals to laboratory compliance reforms and federal payment advocacy, physicians must remain engaged. Whether it is safeguarding diagnostic autonomy or securing financial sustainability, we must continue to put “skin in the game.”
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Mazmudar RS, Sheth A, Tripathi R, et al. Inflation-adjusted trends in Medicare reimbursement for common dermatologic procedures, 2007-2021. JAMA Dermatol. 2021;157:1355-1358.
- Miller TC, Rucker P, Armstrong D. “Not medically necessary”: inside the company helping America’s biggest health insurers deny coverage for care. ProPublica. October 23, 2024. Accessed April 23, 2025. https://www.propublica.org/article/evicore-health-insurance-denials-cigna-unitedhealthcare-aetna-prior-authorizations
- EviCore healthcare. Immunohistochemistry (IHC). Lab Management Guidelines v2.0.2024. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/MOL.CS_.104.A_Immunohistochemistry%20%28IHC%29_V2.0.2024_eff11.01.2024_pub12.31.2024.pdf
- EviCore. Laboratory management. Accessed April 23, 2025. https://www.evicore.com/provider/clinical-guidelines-details?solution=laboratory%20management
- Saad AJ. College of American Pathologists. December 12, 2023. Accessed April 23, 2025. https://documents.cap.org/documents /Wellmark-Letter- https://documents.cap.org/documents/wellmarkcap-letter2023.pdf
- EviCore healthcare. Clinical Guidelines: Lab Management Program. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/Cigna_LabMgmt_V1.0.2025_eff01.01.2025_pub08.22.2024_0.pdf
- American Academy of Dermatology Association. Position statement on immunohistochemistry utilization. Accessed May 9, 2024. https://server.aad.org/forms/policies/Uploads/PS/PS-Immunohistochemistry%20Utilization.pdf
- Naert KA, Trotter MJ. Utilization and utility of immunohistochemistry in dermatopathology. Am J Dermatopathol. 2013;35:74-77.
- American Medical Association. Medicare physician payment continues to fall further behind practice cost inflation. Accessed April 23, 2025. https:// www.ama-assn.org/system/files/2025-medicare-updates-inflation-chart.pdf
- Centers for Medicare & Medicaid Services. Calendar year (CY) 2025 Medicare Physician Fee Schedule final rule. Accessed April 23, 2025. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2025-medicare-physician-fee-schedule-final-rule
- American Medical Association. The Medicare Economic Index. Accessed April 23 2025. https://www.ama-assn.org/system/files/medicare-basics-medicare-economic-index.pdf
- Medicare Patient Access and Practice Stabilization Act, HR 879, 119th Cong (2025). Accessed April 23, 2025. https://www.congress.gov/bill/119th-congress/house-bill/879
- American Academy of Dermatology Association. AADA advocacy action center. Accessed April 23, 2025. https://www.aad.org/member/advocacy/take-action
- Department of Health and Human Services. Centers for Medicare & Medicaid Services. Clinical Laboratory Improvement Amendments of 1988 (CLIA) fees; histocompatibility, personnel, and alternative sanctions for certificate of waiver laboratories. Fed Regist. 2023;88:89976-90044.
- College of American Pathologists. CAP accreditation checklists – 2024 edition. Accessed April 23, 2025. https://documents.cap.org/documents/2024-Checklist-Summary.pdf?_gl=1*1b4rei9*_ga*NDc0NjYwNjM5LjE3NDQ3NTI4NjA.*_ga_97ZFJSQQ0X*MTc0NDc2OTc3My40LjEuMTc0NDc2OTgyOC4wLjAuMA
- Bennett SA, Conn CM, Gill HE, et al. Regulatory requirements for laboratory developed tests in the United States. J Immunol Methods. 2025;537:113813.
- Pollock JR, Chen JY, Dorius DA, et al. Decreasing physician Medicare reimbursement for dermatology services. J Am Acad Dermatol. 2022;86:1154-1156.
- Mazmudar RS, Sheth A, Tripathi R, et al. Inflation-adjusted trends in Medicare reimbursement for common dermatologic procedures, 2007-2021. JAMA Dermatol. 2021;157:1355-1358.
- Miller TC, Rucker P, Armstrong D. “Not medically necessary”: inside the company helping America’s biggest health insurers deny coverage for care. ProPublica. October 23, 2024. Accessed April 23, 2025. https://www.propublica.org/article/evicore-health-insurance-denials-cigna-unitedhealthcare-aetna-prior-authorizations
- EviCore healthcare. Immunohistochemistry (IHC). Lab Management Guidelines v2.0.2024. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/MOL.CS_.104.A_Immunohistochemistry%20%28IHC%29_V2.0.2024_eff11.01.2024_pub12.31.2024.pdf
- EviCore. Laboratory management. Accessed April 23, 2025. https://www.evicore.com/provider/clinical-guidelines-details?solution=laboratory%20management
- Saad AJ. College of American Pathologists. December 12, 2023. Accessed April 23, 2025. https://documents.cap.org/documents /Wellmark-Letter- https://documents.cap.org/documents/wellmarkcap-letter2023.pdf
- EviCore healthcare. Clinical Guidelines: Lab Management Program. Accessed April 23, 2025. https://www.evicore.com/sites/default/files/clinical-guidelines/2024-08/Cigna_LabMgmt_V1.0.2025_eff01.01.2025_pub08.22.2024_0.pdf
- American Academy of Dermatology Association. Position statement on immunohistochemistry utilization. Accessed May 9, 2024. https://server.aad.org/forms/policies/Uploads/PS/PS-Immunohistochemistry%20Utilization.pdf
- Naert KA, Trotter MJ. Utilization and utility of immunohistochemistry in dermatopathology. Am J Dermatopathol. 2013;35:74-77.
- American Medical Association. Medicare physician payment continues to fall further behind practice cost inflation. Accessed April 23, 2025. https:// www.ama-assn.org/system/files/2025-medicare-updates-inflation-chart.pdf
- Centers for Medicare & Medicaid Services. Calendar year (CY) 2025 Medicare Physician Fee Schedule final rule. Accessed April 23, 2025. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2025-medicare-physician-fee-schedule-final-rule
- American Medical Association. The Medicare Economic Index. Accessed April 23 2025. https://www.ama-assn.org/system/files/medicare-basics-medicare-economic-index.pdf
- Medicare Patient Access and Practice Stabilization Act, HR 879, 119th Cong (2025). Accessed April 23, 2025. https://www.congress.gov/bill/119th-congress/house-bill/879
- American Academy of Dermatology Association. AADA advocacy action center. Accessed April 23, 2025. https://www.aad.org/member/advocacy/take-action
- Department of Health and Human Services. Centers for Medicare & Medicaid Services. Clinical Laboratory Improvement Amendments of 1988 (CLIA) fees; histocompatibility, personnel, and alternative sanctions for certificate of waiver laboratories. Fed Regist. 2023;88:89976-90044.
- College of American Pathologists. CAP accreditation checklists – 2024 edition. Accessed April 23, 2025. https://documents.cap.org/documents/2024-Checklist-Summary.pdf?_gl=1*1b4rei9*_ga*NDc0NjYwNjM5LjE3NDQ3NTI4NjA.*_ga_97ZFJSQQ0X*MTc0NDc2OTc3My40LjEuMTc0NDc2OTgyOC4wLjAuMA
- Bennett SA, Conn CM, Gill HE, et al. Regulatory requirements for laboratory developed tests in the United States. J Immunol Methods. 2025;537:113813.
Advocacy and Compliance Issues Impacting Dermatology in 2025
Advocacy and Compliance Issues Impacting Dermatology in 2025
PRACTICE POINTS
- Recent advocacy efforts have led to the reversal of widespread insurer denials for immunohistochemistry stains; however, continued vigilance is necessary, as restrictive coverage policies may re-emerge.
- Laboratory directors must comply with updated Clinical Laboratory Improvement Amendments of 1988 and College of American Pathologists personnel requirements effective December 28, 2024, including stricter board certification and 2 years of laboratory training or experience and 20 hours of continuing education requirements.
- The American Society of Dermatopathology Appropriate Use Criteria mobile application provides physicians with evidence-based guidance for test selection in dermatopathology.
Scurvy in Hospitalized Patients
Scurvy in Hospitalized Patients
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
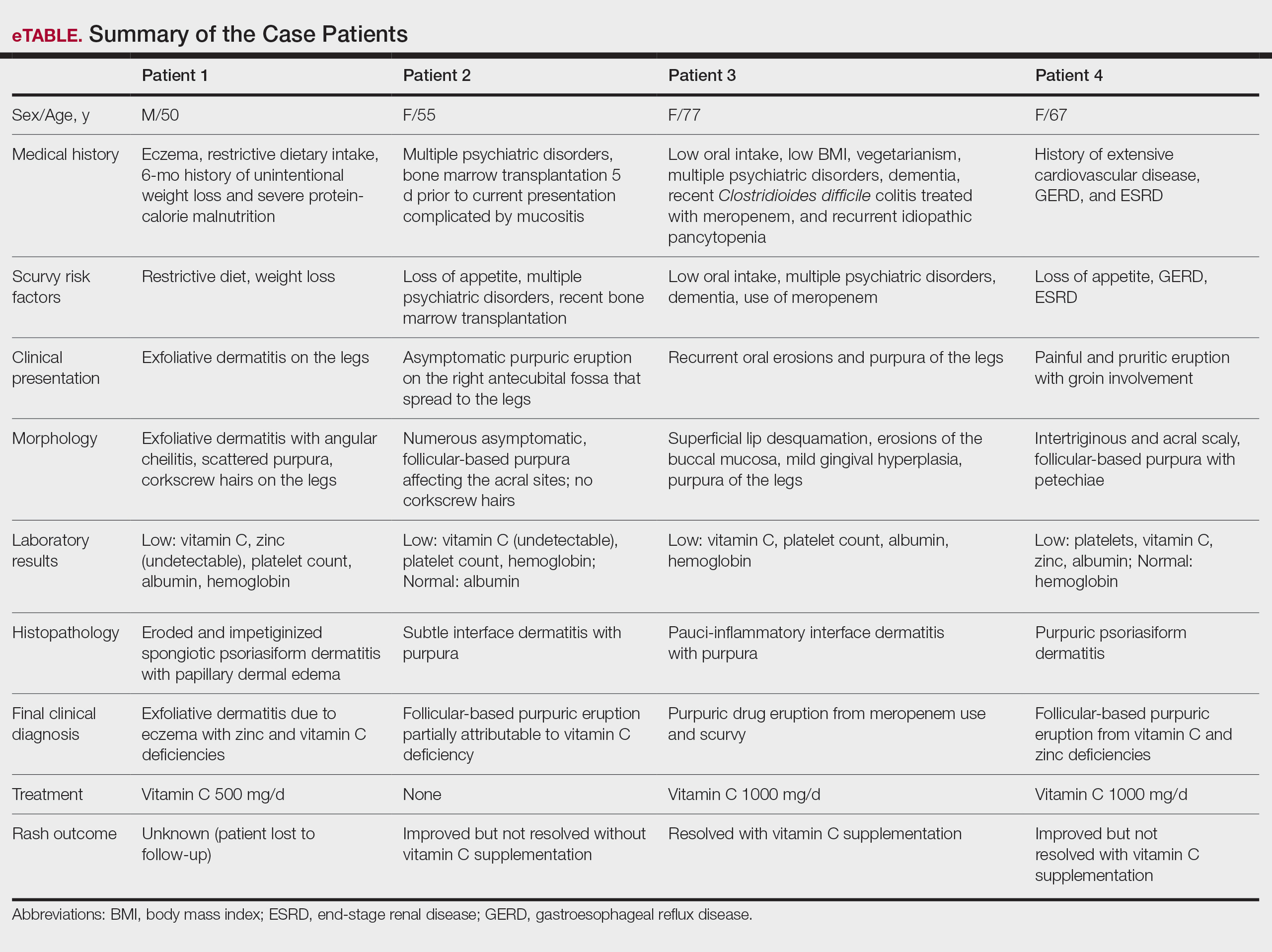
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
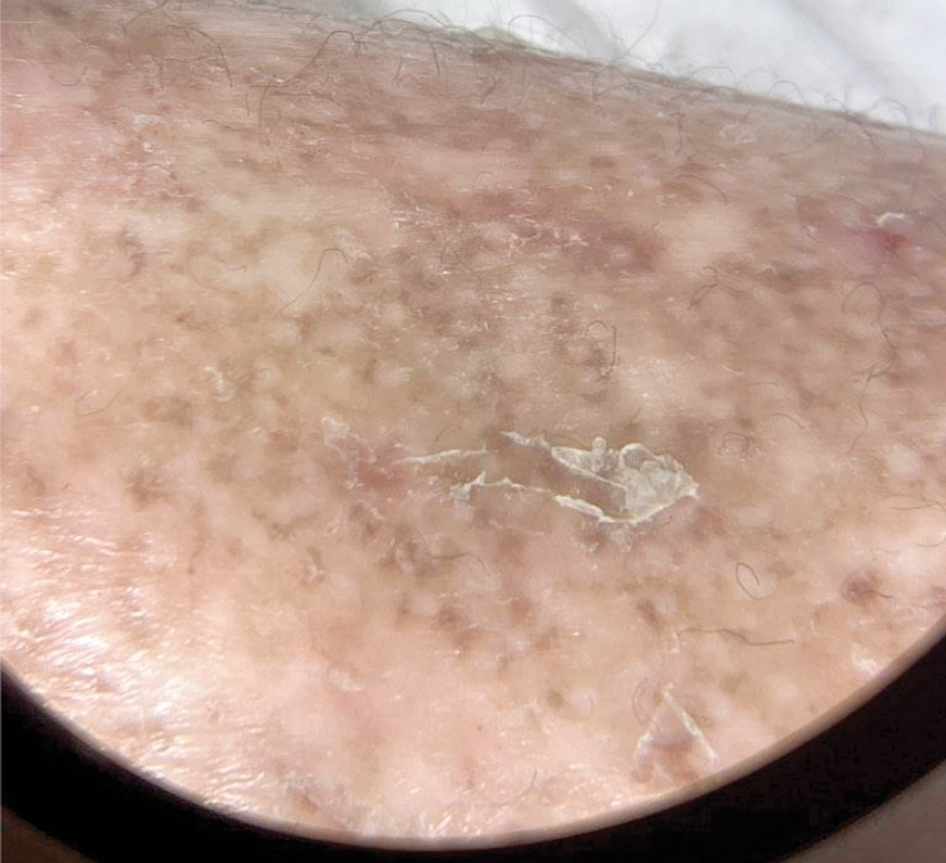
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
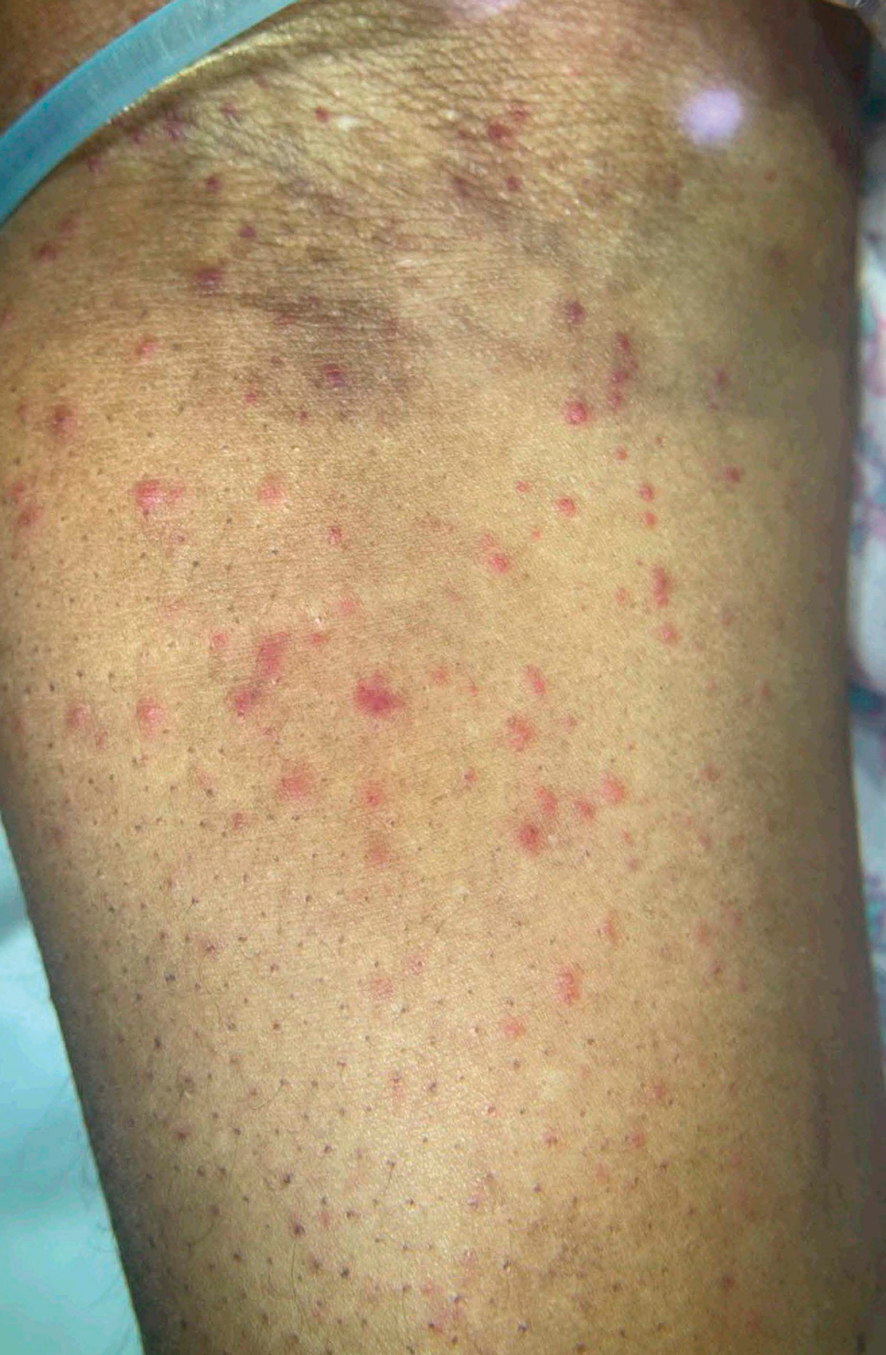
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
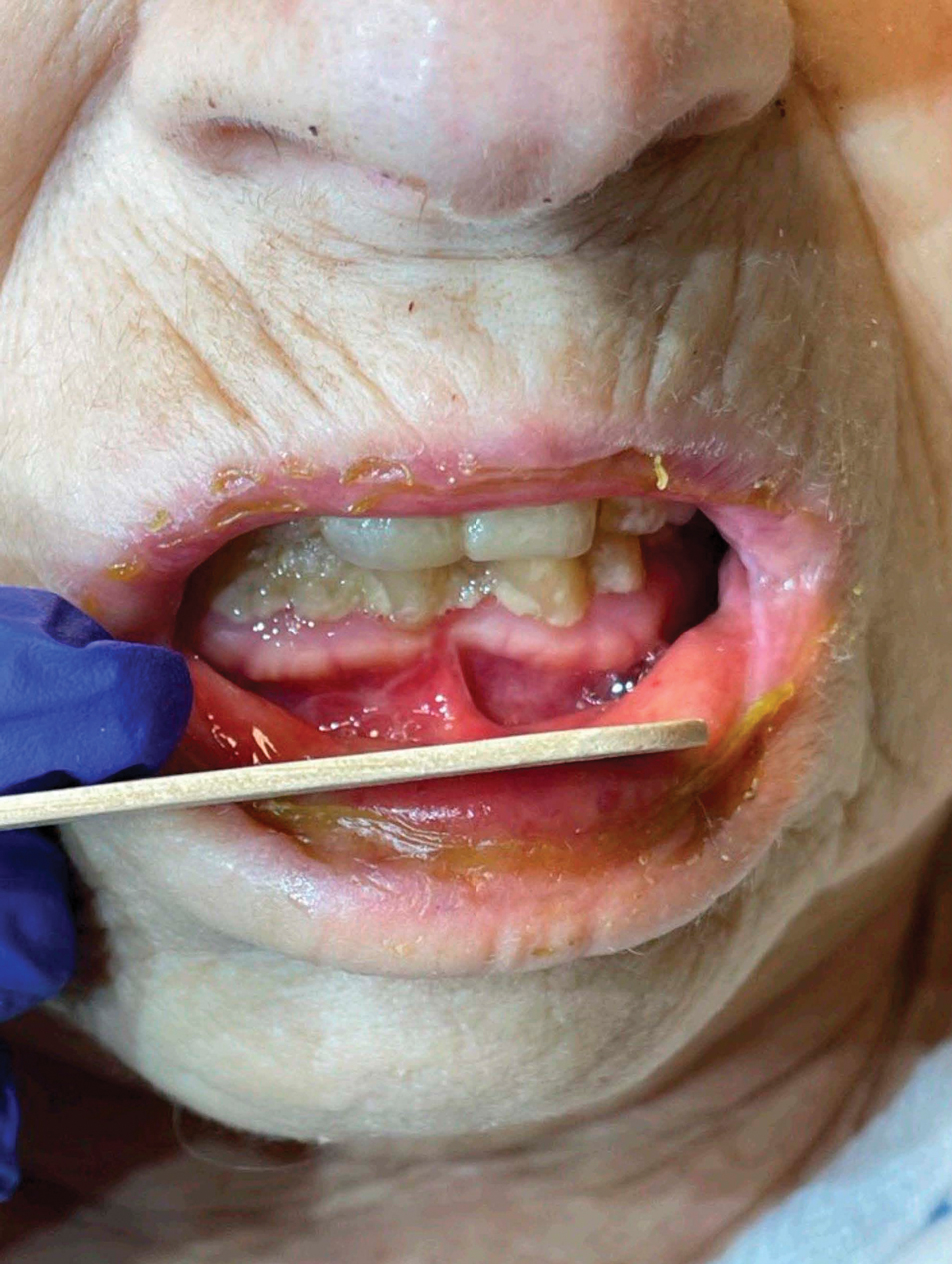
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
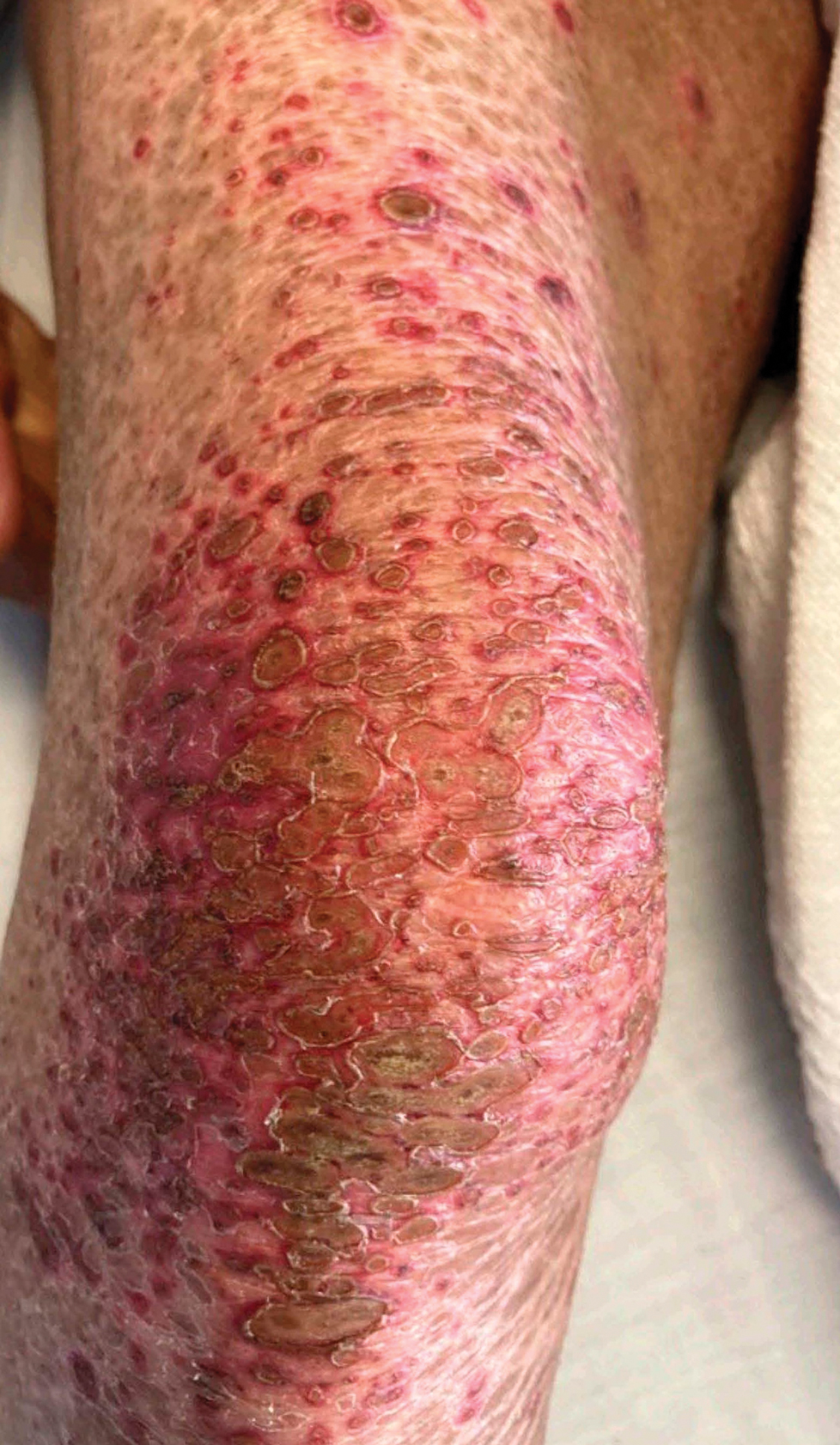
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
Scurvy, caused by vitamin C or ascorbic acid deficiency, historically has been associated primarily with developing nations and famine; however, specific populations in industrialized nations remain at an increased risk, particularly individuals with a history of smoking, alcohol use, restrictive diet, poor oral intake, psychiatric disorders, dementia, bone marrow transplantation, gastroesophageal reflux disease, end-stage renal disease, and hospitalization.1 Micronutrient deficiency– associated dermatoses have been linked to poor clinical outcomes in hospitalized patients.2 In this case series, we report 4 hospitalized patients with scurvy, each presenting with unique comorbidities and risk factors for vitamin C deficiency (eTable).
Case Reports
Patient 1—A 50-year-old man with a 6-month history of eczema and restrictive dietary intake was admitted to the hospital for septic shock attributed to a left foot infection of 5-days’ duration. The patient had experienced unintentional weight loss with severe protein-calorie malnutrition. His dietary history was notable for selective eating behaviors, intermittent meal skipping, and vegetarianism. Mucocutaneous examination by the dermatology consult team showed exfoliative dermatitis with angular cheilitis, corkscrew hairs on the legs (eFigure 1), and scattered purpura throughout the body. The differential diagnosis included eczema exacerbation, cutaneous T-cell lymphoma/Sézary syndrome, and malnutrition-related dermatosis. Punch biopsies of the left medial knee and right lateral arm revealed impetiginized, spongiotic, psoriasiform dermatitis with papillary dermal edema. The histologic changes were consistent with malnutrition-related dermatosis. Laboratory results included low vitamin C levels (0.1 mg/dL [reference range, 0.2-2.1 mg/dL]), undetectable zinc levels (<10 μg/dL [reference range ,60-130 μg/dL]), a low platelet count (21 kμ/L [reference range, 150-400 k/μL]),low albumin levels (0.9 mg/dL (13.0 g/dL [reference range, 14.0-17.4 g/dL]). The final diagnosis was exfoliative dermatitis due to eczema and multiple nutrient deficiencies (vitamin C and zinc). The patient was treated with vitamin C 500 mg/d and was started on mirtazapine to improve his appetite. Following a 3-month hospitalization, the patient was lost to follow-up after discharge.
Patient 2—A 55-year-old woman with a history of multiple psychiatric disorders presented to the dermatology consult service with an asymptomatic purpuric eruption on the right antecubital fossa of 2 days’ duration that spread to the proximal thighs. Five days prior to presentation, she had received an allogeneic bone marrow transplant complicated by mucositis. She also reported a 4-month history of decreased appetite. At the current presentation, numerous acral, follicular based, purpuric macules and papules without associated corkscrew hairs were observed (eFigure 2). The differential diagnosis included a purpuric drug reaction, viral exanthem, acute graft-vs-host disease, neutrophilic dermatoses, and vitamin C deficiency–related dermatosis. Laboratory results revealed undetectable vitamin C levels (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (8 k/μL [reference range, 150-400 k/μL]), normal albumin levels (3.7 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (7.8 g/dL [reference range, 14.0-17.4 g/dL]). Based on the histopathologic finding of subtle interface dermatitis with purpura from a punch biopsy of the right forearm, the eruption was attributed to scurvy. Although dermatology recommended supplementation with vitamin C 1000 mg/d, the decision was deferred by the primary team and the purpura improved without it—suggesting the purpura was only partly attributable to low vitamin C.
Patient 3—A 77-year-old woman with a history of low oral intake, a low body mass index (18.15 kg/m2 [reference range, 18.5-24.9]), vegetarianism, multiple psychiatric disorders, dementia, recent Clostridioides difficile colitis treated with meropenem, and recurrent idiopathic pancytopenia presented to the hospital with recurrent oral erosions and purpura of the legs for an unknown period. Physical examination by the dermatology consult team revealed superficial lip desquamation; erosions of the buccal mucosa with no involvement of the inner lip or gingiva; mild gingival hyperplasia (eFigure 3); and scaly, purpuric, follicular macules and papules on the legs. The arms and legs were devoid of hair. Laboratory results were notable for low vitamin C levels (0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), a low platelet count (28 k/μL [reference range, 150-500 k/μL]), low albumin levels (2.9 g/dL [reference range, 3.5-5.0 g/dL]), and low hemoglobin (8.8 g/dL [reference range, 12.0-16.0 g/ dL]). A punch biopsy from the left thigh revealed pauci-inflammatory interface dermatitis with purpura. Based on the clinical and histologic findings, a final diagnosis of purpuric drug eruption (from the meropenem) and scurvy was made. Nutritional support included supplementation with vitamin C 1000 mg/d. The patient’s oral erosions and purpura gradually resolved with treatment throughout her 1.5-month hospitalization.
Patient 4—A 67-year-old woman with a history of extensive cardiovascular disease, gastroesophageal reflux disease without esophagitis, end-stage renal disease not requiring hemodialysis, and loss of appetite presented with a painful pruritic eruption on the legs with groin involvement of 2 months’ duration. The patient was admitted to the hospital for worsening mental status and weakness accompanied by dark stools, hematuria, and a productive cough with red-tinged sputum. Physical examination by the dermatology consult team showed a scaly, follicular, purpuric eruption affecting the acral and intertriginous sites (eFigure 4). The patient had sparse leg hair, making it difficult to assess for hair tortuosity. A punch biopsy of the left posterior knee revealed purpuric psoriasiform dermatitis, which was consistent with nutritional deficiency– associated dermatosis. Laboratory results included low vitamin C (<0.1 mg/dL [reference range, 0.3-2.7 mg/dL]), zinc, (58 μg/dL [reference range, 60-130 μg/dL]), and albumin levels (3.3 g/dL [reference range, 3.5-5.0 g/dL]) and a low platelet count (67 k/μL [reference range, 150- 500 k/μL]). The patient was started on supplementation with vitamin C 1000 mg/d with improvement of the purpura.
Comment
Micronutrient deficiencies may be common in hospitalized patients due to an increased prevalence of predisposing risk factors including infection, malnutrition, malabsorptive conditions, psychiatric diseases, and chronic illnesses.3 Acute-phase response in hospitalized patients also has been strongly associated with decreased plasma vitamin C levels.4 This phenomenon is postulated to be due to the increase in ascorbic acid uptake by circulating granulocytes in acute disease5; however because low vitamin C levels during the acute-phase response may not always accurately reflect total body stores, other clinical features should be assessed. Previously reported social history risk factors include smoking, alcohol consumption, marijuana use, restrictive diets, vegetarianism, and living alone.6,7
The unifying clinical clues for scurvy in our 4 patients were a history of poor oral intake and purpura. While purpura is nonspecific and can appear after traumatic injury to the skin in elderly patients with photodamage and coagulation disorders, it also is associated with vitamin C deficiency, even with a normal platelet count, circulating von Willebrand factor levels, and prothrombin time/partial thromboplastin time.8 This is because vitamin C is vital in forming the collagen and extracellular matrix. Specifically, it is a cofactor for lysine and proline hydroxylase enzymes needed for the á-helix crosslinks in collagen, which are essential for its structural integrity.9 Collagen is a structural protein that maintains the blood vessel walls, skin, and the basement membrane. A deficiency in vitamin C leads to impairment in collagen synthesis, and insufficient collagen results in compromised connective tissue, blood vessels, and hair strength, which may lead to purpura. All of our patients had thrombocytopenia, and similarly, consideration for scurvy in hospitalized patients with risk factors for micronutrient deficiency is a must. Additional findings such as a follicular-based pattern of the purpura, hair tortuosity, restrictive dietary history, histopathology reports consistent with nutritional dermatoses, serum vitamin C levels, and improvement with vitamin C supplementation are more specific for scurvy. All of these factors can assist the clinician in detecting and confirming these micronutrient deficiencies.
Although there are no established therapeutic guidelines for scurvy, the mainstay of treatment is vitamin C repletion, either orally or parenterally. In hospitalized patients, one suggested regimen is 1000 mg of intravenous ascorbic acid daily for 3 days, followed by further supplementation with a dose of 250 to 500 mg twice daily for 1 month as needed after discharge.10 Symptom improvement occurs about 72 hours after vitamin replacement.8 We recommended 500 to 1000 mg of daily vitamin C supplementation for our patients.
Final Thoughts
This case series highlights the importance of maintaining a high index of suspicion for scurvy in hospitalized patients presenting with purpura, especially in a follicular-based pattern, who have multiple medical comorbidities and risk factors for vitamin C deficiency. The manifestations of scurvy are heterogeneous, necessitating a comprehensive mucocutaneous examination. The diagnosis of scurvy requires correlation of the findings from the patient history, clinical examination, laboratory results, and histopathology.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999; 41:895-910.
- Marsh RL, Trinidad J, Shearer S, et al. Association between micronutrient deficiency dermatoses and clinical outcomes in hospitalized patients. J Am Acad Dermatol. 2020;82:1226-1228.
- Hoffman M, Micheletti RG, Shields BE. Nutritional dermatoses in the hospitalized patient. Cutis. 2020;105:296-302, 308, E1-E5.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Moser U, Weber F. Uptake of ascorbic acid by human granulocytes. Int J Vitam Nutr Res. 1984;54:47-53.
- Swanson AM, Hughey LC. Acute inpatient presentation of scurvy. Cutis. 2010;86:205-207.
- Christopher KL, Menachof KK, Fathi R. Scurvy masquerading as reactive arthritis. Cutis. 2019;103:E21-E23.
- Antonelli M, Burzo ML, Pecorini G, et al. Scurvy as cause of purpura in the XXI century: a review on this “ancient” disease. Eur Rev Med Pharmacol Sci. 2018;22:4355-4358.
- Maxfield L, Daley SF, Crane JS. Vitamin C deficiency. StatPearls [Internet]. Updated November 12, 2023. Accessed September 6, 2024. https://www.ncbi.nlm.nih.gov/books/NBK493187/
- Gandhi M, Elfeky O, Ertugrul H, et al. Scurvy: rediscovering a forgotten disease. Diseases. 2023;11:78.
Scurvy in Hospitalized Patients
Scurvy in Hospitalized Patients
PRACTICE POINTS
- Clinicians should maintain a high index of suspicion for vitamin C deficiency/scurvy in hospitalized patients with purpura who have multiple medical comorbidities and risk factors.
- A low platelet count may mask underlying vitamin C deficiency, and patients may have concurrent deficiencies in other nutrients such as zinc.

Comparing the Quality of Patient Guidance on Dermatologic Care Generated by ChatGPT vs Reddit
Comparing the Quality of Patient Guidance on Dermatologic Care Generated by ChatGPT vs Reddit
To the Editor:
Online resources that are convenient and affordable play a crucial role in mitigating health inequality and improving patient access to health care information; however, the benefits are limited by the quality of information available, as medical misinformation can lead to patients engaging in harmful practices, making dangerous decisions, and even avoiding safe and effective treatments. In this study, we aimed to assess and compare the quality of patient guidance on dermatologic care generated by ChatGPT vs Reddit based on accuracy, appropriateness, and safety. It is essential to assess the quality and reliability of online health information to support patients in making informed decisions about their health.
The emergence and advancement of artificial intelligence and large language models such as ChatGPT present a new method for patients to access health care advice. ChatGPT can engage in conversation by accessing information from existing publicly available data on the internet, including books and websites, up to the year 2023 and providing humanlike responses with context.1 ChatGPT’s access to a breadth of online evidence-based literature ensures the dissemination of quality information that is quick and without inherent bias, offering the potential to more closely align with health care professionals. ChatGPT’s use in dermatology by patients has shown efficacy, with a 98.87% approval rate by dermatologists scoring its ability to recommend appropriate medication for common dermatologic conditions.2 However, ChatGPT has limitations when providing health care advice and has been observed to misunderstand health care standards, lack personalization, and offer incorrect references; currently, the latest publicly available version (ChatGPT 3.5) also is unable to analyze clinical images.3,4
Reddit is an online social media forum that allows users to post questions and photographs to which anyone can reply and offer advice. Patients may find comfort in online communities where they can connect with others facing similar challenges related to their diagnosis. Within these communities, the responses often share users’ own lived experiences and offer support based on what has and has not worked for them. Prior research found that users intentionally seeking health information via Reddit are likely to implement the advice they receive even without verification of its credibility, suggesting a trust and receptibility to ideas offered on the platform.5 Furthermore, a study analyzing the dermatologic content of 17 dermatology related subreddits that had 1000 or more subscribers found that 70.6% of posts fell under the category of “seeking health/cosmetic advice.”6 Reddit users thus are vulnerable to receiving advice based on personal bias and exposing their health information to the public.
We hypothesized that ChatGPT would provide users with guidance that was more closely aligned with typical dermatologists’ advice due to its thorough analysis and compilation of diverse sources and recommendations available on the internet. We expected Reddit to yield recommendations of lesser quality and a diminished safety score, primarily due to the absence of credibility-vetting mechanisms and the influence of personal biases within the advice shared.
User-submitted posts to large dermatologic community Reddit forums representing a few of the most common skin conditions (r/eczema, r/acne, r/Folliculitis, r/SebDerm, r/Hidradenitis, r/keratosis, and r/Psoriasis) were retrospectively reviewed from January 2024 to March 2024. The most popular posts that did not include photographs were included in our study. Posts with photographs were excluded, as clinical images were not able to be uploaded to the publicly available ChatGPT 3.5. We collected real user questions about common skin conditions from Reddit forums and then asked ChatGPT to answer those same questions. We compared ChatGPT’s responses to the most upvoted Reddit comments to see how they matched up (eTable).

Each ChatGPT response and the top-rated Reddit comment were independently evaluated by a board certified dermatologist (S.A.) and a dermatology resident (A.H.K.). The quality of the ChatGPT and Reddit responses were determined by scoring the accuracy, appropriateness, safety consideration, and specificity on a 5-point Likert scale (1=low, 5=high). The 2 evaluators’ mean scores for each of the 4 categories were calculated based on adequate interrater reliability, which was tested using Cohen’s κ coefficient. Related-samples sign tests were used to compare ChatGPT and Reddit responses for each of the 4 categories. Analysis was completed using SPSS statistics software version 29.0 (IBM). The evaluators also were asked to provide qualitative feedback on the strengths and weaknesses of each response.
Our retrospective review yielded 20 total questions: 5 (25%) on atopic dermatitis, 4 (20%) on acne, 4 (20%) on hidradenitis suppurativa, 4 (20%) on psoriasis, 1 (5%) on folliculitis, 1 (5%) on keratosis pilaris, and 1 (5%) on seborrheic dermatitis. The number of posts was limited to 20 due to the extensive time required for grading each response. These 20 questions were selected from a larger pool of eligible posts based on factors such as clarity and relevance to common skin conditions. With regard to the types of questions that were asked, 6 (30%) were related to general management of a diagnosis, 5 (25%) were on treatment recommendations for symptom relief, 3 (15%) were on optimal utilization of current treatment regimens, 2 (10%) were on prescription side effects, 2 (10%) were on diagnosis presentation, 1 (5%) was on potential triggers of the diagnosis, and 1 (5%) was on natural treatment recommendations.
Mean (SD) evaluator scores for accuracy were significantly higher among ChatGPT responses compared with Reddit (4.63 [0.60] vs 2.60 [0.98])(P<.001). ChatGPT responses also were significantly higher for appropriateness compared with Reddit (4.55 [0.71] vs 2.58 [1.02])(P<.001) and safety consideration (4.88 [0.56] vs 2.80[0.97])(P <.001). There was no significant difference in mean specificity scores between ChatGPT and Reddit (4.25[1.02] vs 3.80 [0.70])(P=.096)(Figure).
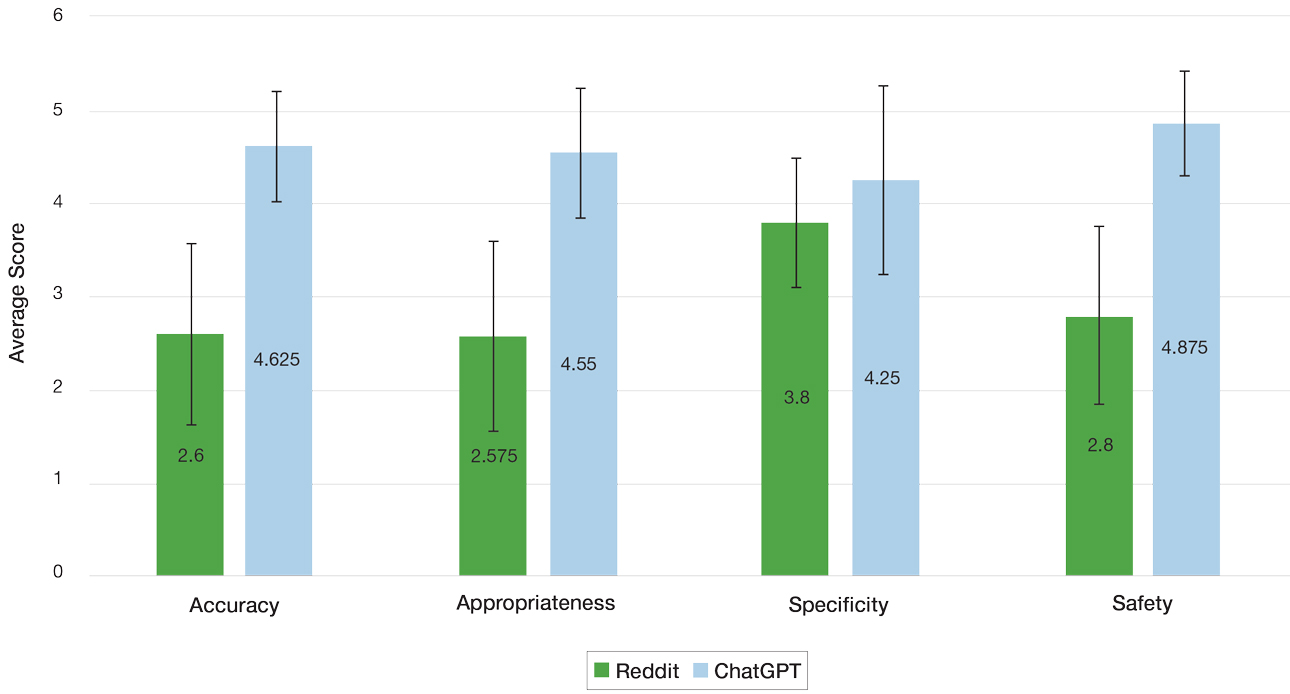
For the Reddit responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.200 (95% CI, –.089 to .489) for accuracy, 0.255 (95% CI, .014-.497) for appropriateness, 0.385 (95% CI, .176-.594) for safety consideration, and –0.024 (95% CI, –.177 to .129) for specificity. For the ChatGPT responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.426 (95% CI, .122-.730) for accuracy, 0.571 (95% CI, .294-.849) for appropriateness, 0.655 (95% CI, .632-.678) for safety consideration, and 0.313 (95% CI, .043-.584) for specificity.
The strengths and weaknesses of the responses also were qualitatively analyzed. One commonly observed strength was ChatGPT’s frequent and appropriate recommendation for users to consult a dermatologist. In the case of atopic dermatitis—one of the more frequently asked about conditions—ChatGPT consistently emphasized evidence-based strategies such as gentle skin care and moisturization, reflecting alignment with clinical guidelines. Additionally, a common weakness of both ChatGPT and Reddit responses generally was the lack of personalized guidance and comprehensive discussion of the risks and benefits of specific treatments. It also was noted that neither platform consistently explored differential diagnoses—for example, distinguishing atopic dermatitis from conditions such as allergic contact dermatitis—limiting the diagnostic depth of the responses.
ChatGPT and Reddit can provide patients with quick and accessible health information for various dermatologic concerns. The results of our study demonstrated a significantly higher level of accuracy, appropriateness, and safety of responses generated by ChatGPT compared with human-generated responses on Reddit (P<.001). Both platforms offered similarly specific responses to user inquiries, demonstrating ChatGPT’s ability to comprehend user questions and draw from publicly available texts and Reddit users’ contributing insights based on their own first-hand experiences.
Reddit’s dermatologic forums often feature personal anecdotes and unique treatments described by individual users. Although specific to particular dermatologic concerns, such advice lacks an evidence-based standard of care. With the noted inherent trust of patients seeking guidance within Reddit communities, patients may follow unhelpful or potentially dangerous medical advice.5 A study examining 300 user-submitted posts on popular Reddit dermatology forums during the COVID-19 pandemic found that the mean scores for top-rated comments’ potential to be misleading or dangerous was 2.33 out of 5 on a Likert scale (95% CI, 2.18- 2.48).7 Dermatologists should be aware of the potential risks associated with dermatologic advice offered on Reddit and should caution patients against relying solely on this information without consulting a qualified dermatologist first.
Reddit’s open-forum design provides licensed dermatologists with the opportunity to disseminate evidence based information regarding dermatologic conditions. Currently, there is a subreddit (r/AskDocs) that allows users to post medical questions that can be answered by moderator-verified physicians. Participation from dermatologists in online communities such as this can improve the quality of dermatologic information shared online, combat misinformation, and promote safe skin care practices.
ChatGPT offers more accurate, appropriate, and safe information compared to Reddit responses, but its answers lack personalization. In a clinical setting, a personalized treatment plan from a physician can be tailored with a comprehensive discussion of the risks and benefits. Further, clinical settings allow for diagnosis and confirmation via biopsy and meticulous history taking to ensure that the diagnosis and treatment plan are accurate. While ChatGPT may be an option for seeking basic advice on dermatologic conditions, a licensed dermatologist should always be consulted for proper medical advice. Services such as telehealth may be another option to for patients with limited access to care.
Since ChatGPT 3.5 does not support the ability to upload images, our study acknowledges a limitation regarding the inclusion of Reddit posts containing photographs. Images can improve the response quality from both Reddit users and ChatGPT. While the updated ChatGPT 4o is capable of processing images, it requires a monthly subscription fee. The free version was chosen for use in this study, as this may reflect the most likely version that patients of low socioeconomic status would utilize to access dermatologic care; however, there is potential for growth and improvement of ChatGPT’s capability in providing medical advice.
This study compared the strengths and limitations of ChatGPT’s and Reddit’s responses to common dermatologic inquiries. ChatGPT and Reddit both show potential to be helpful sources of dermatologic health information; however, their current versions have many limitations and require caution and careful examination by patients of the guidance provided. Clinicians should be aware of these limitations when advising patients and emphasize the importance of consulting a licensed dermatologist for personalized, evidence-based care. For the best medical advice, it is always advisable to consult with a licensed dermatologist.
- Roumeliotis KI, Tselikas ND. ChatGPT and open-AI models: a preliminary review. Future Internet. 2023;15:192. doi:10.3390/fi15060192
- Iqbal U, Lee LTJ, Rahmanti AR, et al. Can large language models provide secondary reliable opinion on treatment options for dermatological diseases? J Am Med Inform Assoc. 2024;31:1341-1347. doi:10.1093/jamia/ocae067
- Whiles BB, Bird VG, Canales BK, et al. Caution! AI bot has entered the patient chat: ChatGPT has limitations in providing accurate urologic healthcare advice. Urology. 2023;180:278-284. doi:10.1016/j.urology.2023.07.010
- Nastasi AJ, Courtright KR, Halpern SD, et al. A vignette-based evaluation of ChatGPT’s ability to provide appropriate and equitable medical advice across care contexts. Sci Rep. 2023;13:17885. doi:10.1038/s41598-023-45223-y
- Record RA, Silberman WR, Santiago JE, et al. I sought it, I Reddit: examining health information engagement behaviors among Reddit users. J Health Commun. 2018;23:470-476. doi:10.1080/1081073 0.2018.1465493
- Buntinx-Krieg T, Caravaglio J, Domozych R, et al. Dermatology on Reddit: elucidating trends in dermatologic communications on the world wide web. Dermatol Online J. 2017;23:13030/qt9dr1f7x6.
- Aboul-Fettouh N, Lee KP, Kash N, et al. Social media and dermatology during the COVID-19 pandemic: analyzing usersubmitted posts seeking dermatologic advice on Reddit. Cureus. 2023;15:E33720. doi:10.7759/cureus.33720
To the Editor:
Online resources that are convenient and affordable play a crucial role in mitigating health inequality and improving patient access to health care information; however, the benefits are limited by the quality of information available, as medical misinformation can lead to patients engaging in harmful practices, making dangerous decisions, and even avoiding safe and effective treatments. In this study, we aimed to assess and compare the quality of patient guidance on dermatologic care generated by ChatGPT vs Reddit based on accuracy, appropriateness, and safety. It is essential to assess the quality and reliability of online health information to support patients in making informed decisions about their health.
The emergence and advancement of artificial intelligence and large language models such as ChatGPT present a new method for patients to access health care advice. ChatGPT can engage in conversation by accessing information from existing publicly available data on the internet, including books and websites, up to the year 2023 and providing humanlike responses with context.1 ChatGPT’s access to a breadth of online evidence-based literature ensures the dissemination of quality information that is quick and without inherent bias, offering the potential to more closely align with health care professionals. ChatGPT’s use in dermatology by patients has shown efficacy, with a 98.87% approval rate by dermatologists scoring its ability to recommend appropriate medication for common dermatologic conditions.2 However, ChatGPT has limitations when providing health care advice and has been observed to misunderstand health care standards, lack personalization, and offer incorrect references; currently, the latest publicly available version (ChatGPT 3.5) also is unable to analyze clinical images.3,4
Reddit is an online social media forum that allows users to post questions and photographs to which anyone can reply and offer advice. Patients may find comfort in online communities where they can connect with others facing similar challenges related to their diagnosis. Within these communities, the responses often share users’ own lived experiences and offer support based on what has and has not worked for them. Prior research found that users intentionally seeking health information via Reddit are likely to implement the advice they receive even without verification of its credibility, suggesting a trust and receptibility to ideas offered on the platform.5 Furthermore, a study analyzing the dermatologic content of 17 dermatology related subreddits that had 1000 or more subscribers found that 70.6% of posts fell under the category of “seeking health/cosmetic advice.”6 Reddit users thus are vulnerable to receiving advice based on personal bias and exposing their health information to the public.
We hypothesized that ChatGPT would provide users with guidance that was more closely aligned with typical dermatologists’ advice due to its thorough analysis and compilation of diverse sources and recommendations available on the internet. We expected Reddit to yield recommendations of lesser quality and a diminished safety score, primarily due to the absence of credibility-vetting mechanisms and the influence of personal biases within the advice shared.
User-submitted posts to large dermatologic community Reddit forums representing a few of the most common skin conditions (r/eczema, r/acne, r/Folliculitis, r/SebDerm, r/Hidradenitis, r/keratosis, and r/Psoriasis) were retrospectively reviewed from January 2024 to March 2024. The most popular posts that did not include photographs were included in our study. Posts with photographs were excluded, as clinical images were not able to be uploaded to the publicly available ChatGPT 3.5. We collected real user questions about common skin conditions from Reddit forums and then asked ChatGPT to answer those same questions. We compared ChatGPT’s responses to the most upvoted Reddit comments to see how they matched up (eTable).
Each ChatGPT response and the top-rated Reddit comment were independently evaluated by a board certified dermatologist (S.A.) and a dermatology resident (A.H.K.). The quality of the ChatGPT and Reddit responses were determined by scoring the accuracy, appropriateness, safety consideration, and specificity on a 5-point Likert scale (1=low, 5=high). The 2 evaluators’ mean scores for each of the 4 categories were calculated based on adequate interrater reliability, which was tested using Cohen’s κ coefficient. Related-samples sign tests were used to compare ChatGPT and Reddit responses for each of the 4 categories. Analysis was completed using SPSS statistics software version 29.0 (IBM). The evaluators also were asked to provide qualitative feedback on the strengths and weaknesses of each response.
Our retrospective review yielded 20 total questions: 5 (25%) on atopic dermatitis, 4 (20%) on acne, 4 (20%) on hidradenitis suppurativa, 4 (20%) on psoriasis, 1 (5%) on folliculitis, 1 (5%) on keratosis pilaris, and 1 (5%) on seborrheic dermatitis. The number of posts was limited to 20 due to the extensive time required for grading each response. These 20 questions were selected from a larger pool of eligible posts based on factors such as clarity and relevance to common skin conditions. With regard to the types of questions that were asked, 6 (30%) were related to general management of a diagnosis, 5 (25%) were on treatment recommendations for symptom relief, 3 (15%) were on optimal utilization of current treatment regimens, 2 (10%) were on prescription side effects, 2 (10%) were on diagnosis presentation, 1 (5%) was on potential triggers of the diagnosis, and 1 (5%) was on natural treatment recommendations.
Mean (SD) evaluator scores for accuracy were significantly higher among ChatGPT responses compared with Reddit (4.63 [0.60] vs 2.60 [0.98])(P<.001). ChatGPT responses also were significantly higher for appropriateness compared with Reddit (4.55 [0.71] vs 2.58 [1.02])(P<.001) and safety consideration (4.88 [0.56] vs 2.80[0.97])(P <.001). There was no significant difference in mean specificity scores between ChatGPT and Reddit (4.25[1.02] vs 3.80 [0.70])(P=.096)(Figure).
For the Reddit responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.200 (95% CI, –.089 to .489) for accuracy, 0.255 (95% CI, .014-.497) for appropriateness, 0.385 (95% CI, .176-.594) for safety consideration, and –0.024 (95% CI, –.177 to .129) for specificity. For the ChatGPT responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.426 (95% CI, .122-.730) for accuracy, 0.571 (95% CI, .294-.849) for appropriateness, 0.655 (95% CI, .632-.678) for safety consideration, and 0.313 (95% CI, .043-.584) for specificity.
The strengths and weaknesses of the responses also were qualitatively analyzed. One commonly observed strength was ChatGPT’s frequent and appropriate recommendation for users to consult a dermatologist. In the case of atopic dermatitis—one of the more frequently asked about conditions—ChatGPT consistently emphasized evidence-based strategies such as gentle skin care and moisturization, reflecting alignment with clinical guidelines. Additionally, a common weakness of both ChatGPT and Reddit responses generally was the lack of personalized guidance and comprehensive discussion of the risks and benefits of specific treatments. It also was noted that neither platform consistently explored differential diagnoses—for example, distinguishing atopic dermatitis from conditions such as allergic contact dermatitis—limiting the diagnostic depth of the responses.
ChatGPT and Reddit can provide patients with quick and accessible health information for various dermatologic concerns. The results of our study demonstrated a significantly higher level of accuracy, appropriateness, and safety of responses generated by ChatGPT compared with human-generated responses on Reddit (P<.001). Both platforms offered similarly specific responses to user inquiries, demonstrating ChatGPT’s ability to comprehend user questions and draw from publicly available texts and Reddit users’ contributing insights based on their own first-hand experiences.
Reddit’s dermatologic forums often feature personal anecdotes and unique treatments described by individual users. Although specific to particular dermatologic concerns, such advice lacks an evidence-based standard of care. With the noted inherent trust of patients seeking guidance within Reddit communities, patients may follow unhelpful or potentially dangerous medical advice.5 A study examining 300 user-submitted posts on popular Reddit dermatology forums during the COVID-19 pandemic found that the mean scores for top-rated comments’ potential to be misleading or dangerous was 2.33 out of 5 on a Likert scale (95% CI, 2.18- 2.48).7 Dermatologists should be aware of the potential risks associated with dermatologic advice offered on Reddit and should caution patients against relying solely on this information without consulting a qualified dermatologist first.
Reddit’s open-forum design provides licensed dermatologists with the opportunity to disseminate evidence based information regarding dermatologic conditions. Currently, there is a subreddit (r/AskDocs) that allows users to post medical questions that can be answered by moderator-verified physicians. Participation from dermatologists in online communities such as this can improve the quality of dermatologic information shared online, combat misinformation, and promote safe skin care practices.
ChatGPT offers more accurate, appropriate, and safe information compared to Reddit responses, but its answers lack personalization. In a clinical setting, a personalized treatment plan from a physician can be tailored with a comprehensive discussion of the risks and benefits. Further, clinical settings allow for diagnosis and confirmation via biopsy and meticulous history taking to ensure that the diagnosis and treatment plan are accurate. While ChatGPT may be an option for seeking basic advice on dermatologic conditions, a licensed dermatologist should always be consulted for proper medical advice. Services such as telehealth may be another option to for patients with limited access to care.
Since ChatGPT 3.5 does not support the ability to upload images, our study acknowledges a limitation regarding the inclusion of Reddit posts containing photographs. Images can improve the response quality from both Reddit users and ChatGPT. While the updated ChatGPT 4o is capable of processing images, it requires a monthly subscription fee. The free version was chosen for use in this study, as this may reflect the most likely version that patients of low socioeconomic status would utilize to access dermatologic care; however, there is potential for growth and improvement of ChatGPT’s capability in providing medical advice.
This study compared the strengths and limitations of ChatGPT’s and Reddit’s responses to common dermatologic inquiries. ChatGPT and Reddit both show potential to be helpful sources of dermatologic health information; however, their current versions have many limitations and require caution and careful examination by patients of the guidance provided. Clinicians should be aware of these limitations when advising patients and emphasize the importance of consulting a licensed dermatologist for personalized, evidence-based care. For the best medical advice, it is always advisable to consult with a licensed dermatologist.
To the Editor:
Online resources that are convenient and affordable play a crucial role in mitigating health inequality and improving patient access to health care information; however, the benefits are limited by the quality of information available, as medical misinformation can lead to patients engaging in harmful practices, making dangerous decisions, and even avoiding safe and effective treatments. In this study, we aimed to assess and compare the quality of patient guidance on dermatologic care generated by ChatGPT vs Reddit based on accuracy, appropriateness, and safety. It is essential to assess the quality and reliability of online health information to support patients in making informed decisions about their health.
The emergence and advancement of artificial intelligence and large language models such as ChatGPT present a new method for patients to access health care advice. ChatGPT can engage in conversation by accessing information from existing publicly available data on the internet, including books and websites, up to the year 2023 and providing humanlike responses with context.1 ChatGPT’s access to a breadth of online evidence-based literature ensures the dissemination of quality information that is quick and without inherent bias, offering the potential to more closely align with health care professionals. ChatGPT’s use in dermatology by patients has shown efficacy, with a 98.87% approval rate by dermatologists scoring its ability to recommend appropriate medication for common dermatologic conditions.2 However, ChatGPT has limitations when providing health care advice and has been observed to misunderstand health care standards, lack personalization, and offer incorrect references; currently, the latest publicly available version (ChatGPT 3.5) also is unable to analyze clinical images.3,4
Reddit is an online social media forum that allows users to post questions and photographs to which anyone can reply and offer advice. Patients may find comfort in online communities where they can connect with others facing similar challenges related to their diagnosis. Within these communities, the responses often share users’ own lived experiences and offer support based on what has and has not worked for them. Prior research found that users intentionally seeking health information via Reddit are likely to implement the advice they receive even without verification of its credibility, suggesting a trust and receptibility to ideas offered on the platform.5 Furthermore, a study analyzing the dermatologic content of 17 dermatology related subreddits that had 1000 or more subscribers found that 70.6% of posts fell under the category of “seeking health/cosmetic advice.”6 Reddit users thus are vulnerable to receiving advice based on personal bias and exposing their health information to the public.
We hypothesized that ChatGPT would provide users with guidance that was more closely aligned with typical dermatologists’ advice due to its thorough analysis and compilation of diverse sources and recommendations available on the internet. We expected Reddit to yield recommendations of lesser quality and a diminished safety score, primarily due to the absence of credibility-vetting mechanisms and the influence of personal biases within the advice shared.
User-submitted posts to large dermatologic community Reddit forums representing a few of the most common skin conditions (r/eczema, r/acne, r/Folliculitis, r/SebDerm, r/Hidradenitis, r/keratosis, and r/Psoriasis) were retrospectively reviewed from January 2024 to March 2024. The most popular posts that did not include photographs were included in our study. Posts with photographs were excluded, as clinical images were not able to be uploaded to the publicly available ChatGPT 3.5. We collected real user questions about common skin conditions from Reddit forums and then asked ChatGPT to answer those same questions. We compared ChatGPT’s responses to the most upvoted Reddit comments to see how they matched up (eTable).
Each ChatGPT response and the top-rated Reddit comment were independently evaluated by a board certified dermatologist (S.A.) and a dermatology resident (A.H.K.). The quality of the ChatGPT and Reddit responses were determined by scoring the accuracy, appropriateness, safety consideration, and specificity on a 5-point Likert scale (1=low, 5=high). The 2 evaluators’ mean scores for each of the 4 categories were calculated based on adequate interrater reliability, which was tested using Cohen’s κ coefficient. Related-samples sign tests were used to compare ChatGPT and Reddit responses for each of the 4 categories. Analysis was completed using SPSS statistics software version 29.0 (IBM). The evaluators also were asked to provide qualitative feedback on the strengths and weaknesses of each response.
Our retrospective review yielded 20 total questions: 5 (25%) on atopic dermatitis, 4 (20%) on acne, 4 (20%) on hidradenitis suppurativa, 4 (20%) on psoriasis, 1 (5%) on folliculitis, 1 (5%) on keratosis pilaris, and 1 (5%) on seborrheic dermatitis. The number of posts was limited to 20 due to the extensive time required for grading each response. These 20 questions were selected from a larger pool of eligible posts based on factors such as clarity and relevance to common skin conditions. With regard to the types of questions that were asked, 6 (30%) were related to general management of a diagnosis, 5 (25%) were on treatment recommendations for symptom relief, 3 (15%) were on optimal utilization of current treatment regimens, 2 (10%) were on prescription side effects, 2 (10%) were on diagnosis presentation, 1 (5%) was on potential triggers of the diagnosis, and 1 (5%) was on natural treatment recommendations.
Mean (SD) evaluator scores for accuracy were significantly higher among ChatGPT responses compared with Reddit (4.63 [0.60] vs 2.60 [0.98])(P<.001). ChatGPT responses also were significantly higher for appropriateness compared with Reddit (4.55 [0.71] vs 2.58 [1.02])(P<.001) and safety consideration (4.88 [0.56] vs 2.80[0.97])(P <.001). There was no significant difference in mean specificity scores between ChatGPT and Reddit (4.25[1.02] vs 3.80 [0.70])(P=.096)(Figure).
For the Reddit responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.200 (95% CI, –.089 to .489) for accuracy, 0.255 (95% CI, .014-.497) for appropriateness, 0.385 (95% CI, .176-.594) for safety consideration, and –0.024 (95% CI, –.177 to .129) for specificity. For the ChatGPT responses, the weighted Cohen’s κ coefficient between the 2 evaluators was 0.426 (95% CI, .122-.730) for accuracy, 0.571 (95% CI, .294-.849) for appropriateness, 0.655 (95% CI, .632-.678) for safety consideration, and 0.313 (95% CI, .043-.584) for specificity.
The strengths and weaknesses of the responses also were qualitatively analyzed. One commonly observed strength was ChatGPT’s frequent and appropriate recommendation for users to consult a dermatologist. In the case of atopic dermatitis—one of the more frequently asked about conditions—ChatGPT consistently emphasized evidence-based strategies such as gentle skin care and moisturization, reflecting alignment with clinical guidelines. Additionally, a common weakness of both ChatGPT and Reddit responses generally was the lack of personalized guidance and comprehensive discussion of the risks and benefits of specific treatments. It also was noted that neither platform consistently explored differential diagnoses—for example, distinguishing atopic dermatitis from conditions such as allergic contact dermatitis—limiting the diagnostic depth of the responses.
ChatGPT and Reddit can provide patients with quick and accessible health information for various dermatologic concerns. The results of our study demonstrated a significantly higher level of accuracy, appropriateness, and safety of responses generated by ChatGPT compared with human-generated responses on Reddit (P<.001). Both platforms offered similarly specific responses to user inquiries, demonstrating ChatGPT’s ability to comprehend user questions and draw from publicly available texts and Reddit users’ contributing insights based on their own first-hand experiences.
Reddit’s dermatologic forums often feature personal anecdotes and unique treatments described by individual users. Although specific to particular dermatologic concerns, such advice lacks an evidence-based standard of care. With the noted inherent trust of patients seeking guidance within Reddit communities, patients may follow unhelpful or potentially dangerous medical advice.5 A study examining 300 user-submitted posts on popular Reddit dermatology forums during the COVID-19 pandemic found that the mean scores for top-rated comments’ potential to be misleading or dangerous was 2.33 out of 5 on a Likert scale (95% CI, 2.18- 2.48).7 Dermatologists should be aware of the potential risks associated with dermatologic advice offered on Reddit and should caution patients against relying solely on this information without consulting a qualified dermatologist first.
Reddit’s open-forum design provides licensed dermatologists with the opportunity to disseminate evidence based information regarding dermatologic conditions. Currently, there is a subreddit (r/AskDocs) that allows users to post medical questions that can be answered by moderator-verified physicians. Participation from dermatologists in online communities such as this can improve the quality of dermatologic information shared online, combat misinformation, and promote safe skin care practices.
ChatGPT offers more accurate, appropriate, and safe information compared to Reddit responses, but its answers lack personalization. In a clinical setting, a personalized treatment plan from a physician can be tailored with a comprehensive discussion of the risks and benefits. Further, clinical settings allow for diagnosis and confirmation via biopsy and meticulous history taking to ensure that the diagnosis and treatment plan are accurate. While ChatGPT may be an option for seeking basic advice on dermatologic conditions, a licensed dermatologist should always be consulted for proper medical advice. Services such as telehealth may be another option to for patients with limited access to care.
Since ChatGPT 3.5 does not support the ability to upload images, our study acknowledges a limitation regarding the inclusion of Reddit posts containing photographs. Images can improve the response quality from both Reddit users and ChatGPT. While the updated ChatGPT 4o is capable of processing images, it requires a monthly subscription fee. The free version was chosen for use in this study, as this may reflect the most likely version that patients of low socioeconomic status would utilize to access dermatologic care; however, there is potential for growth and improvement of ChatGPT’s capability in providing medical advice.
This study compared the strengths and limitations of ChatGPT’s and Reddit’s responses to common dermatologic inquiries. ChatGPT and Reddit both show potential to be helpful sources of dermatologic health information; however, their current versions have many limitations and require caution and careful examination by patients of the guidance provided. Clinicians should be aware of these limitations when advising patients and emphasize the importance of consulting a licensed dermatologist for personalized, evidence-based care. For the best medical advice, it is always advisable to consult with a licensed dermatologist.
- Roumeliotis KI, Tselikas ND. ChatGPT and open-AI models: a preliminary review. Future Internet. 2023;15:192. doi:10.3390/fi15060192
- Iqbal U, Lee LTJ, Rahmanti AR, et al. Can large language models provide secondary reliable opinion on treatment options for dermatological diseases? J Am Med Inform Assoc. 2024;31:1341-1347. doi:10.1093/jamia/ocae067
- Whiles BB, Bird VG, Canales BK, et al. Caution! AI bot has entered the patient chat: ChatGPT has limitations in providing accurate urologic healthcare advice. Urology. 2023;180:278-284. doi:10.1016/j.urology.2023.07.010
- Nastasi AJ, Courtright KR, Halpern SD, et al. A vignette-based evaluation of ChatGPT’s ability to provide appropriate and equitable medical advice across care contexts. Sci Rep. 2023;13:17885. doi:10.1038/s41598-023-45223-y
- Record RA, Silberman WR, Santiago JE, et al. I sought it, I Reddit: examining health information engagement behaviors among Reddit users. J Health Commun. 2018;23:470-476. doi:10.1080/1081073 0.2018.1465493
- Buntinx-Krieg T, Caravaglio J, Domozych R, et al. Dermatology on Reddit: elucidating trends in dermatologic communications on the world wide web. Dermatol Online J. 2017;23:13030/qt9dr1f7x6.
- Aboul-Fettouh N, Lee KP, Kash N, et al. Social media and dermatology during the COVID-19 pandemic: analyzing usersubmitted posts seeking dermatologic advice on Reddit. Cureus. 2023;15:E33720. doi:10.7759/cureus.33720
- Roumeliotis KI, Tselikas ND. ChatGPT and open-AI models: a preliminary review. Future Internet. 2023;15:192. doi:10.3390/fi15060192
- Iqbal U, Lee LTJ, Rahmanti AR, et al. Can large language models provide secondary reliable opinion on treatment options for dermatological diseases? J Am Med Inform Assoc. 2024;31:1341-1347. doi:10.1093/jamia/ocae067
- Whiles BB, Bird VG, Canales BK, et al. Caution! AI bot has entered the patient chat: ChatGPT has limitations in providing accurate urologic healthcare advice. Urology. 2023;180:278-284. doi:10.1016/j.urology.2023.07.010
- Nastasi AJ, Courtright KR, Halpern SD, et al. A vignette-based evaluation of ChatGPT’s ability to provide appropriate and equitable medical advice across care contexts. Sci Rep. 2023;13:17885. doi:10.1038/s41598-023-45223-y
- Record RA, Silberman WR, Santiago JE, et al. I sought it, I Reddit: examining health information engagement behaviors among Reddit users. J Health Commun. 2018;23:470-476. doi:10.1080/1081073 0.2018.1465493
- Buntinx-Krieg T, Caravaglio J, Domozych R, et al. Dermatology on Reddit: elucidating trends in dermatologic communications on the world wide web. Dermatol Online J. 2017;23:13030/qt9dr1f7x6.
- Aboul-Fettouh N, Lee KP, Kash N, et al. Social media and dermatology during the COVID-19 pandemic: analyzing usersubmitted posts seeking dermatologic advice on Reddit. Cureus. 2023;15:E33720. doi:10.7759/cureus.33720
Comparing the Quality of Patient Guidance on Dermatologic Care Generated by ChatGPT vs Reddit
Comparing the Quality of Patient Guidance on Dermatologic Care Generated by ChatGPT vs Reddit
PRACTICE POINTS
- ChatGPT and Reddit are free, convenient, and accessible online resources that patients may use for guidance on dermatologic care.
- Dermatologists should be aware of the potential risks associated with obtaining medical guidance from ChatGPT and Reddit and caution patients on them.
- An increasing presence of dermatologists on online public forums can increase the dissemination of reliable health care information.
Remarkable Response to Vismodegib in a Locally Advanced Basal Cell Carcinoma on the Nose
Remarkable Response to Vismodegib in a Locally Advanced Basal Cell Carcinoma on the Nose
A 90-year-old man presented for evaluation of a large basal cell carcinoma (BCC) involving the nasal region. The lesion was a 7×4-cm pink, crusted, verrucous plaque covering the majority of the nose and extending onto the malar cheeks that originally had been biopsied 26 years prior, and repeat biopsy was performed 3 years prior. Results from both biopsies were consistent with BCC. The patient had avoided treatment for many years due to fear of losing his nose.
Given the size and location of the tumor, surgical intervention posed major challenges for both functional and cosmetic outcomes. After careful consideration and discussion of treatment options, which included Mohs micrographic surgery (MMS), wide local excision, radiation therapy, and systemic therapy, the decision was made to start the patient on vismodegib 150 mg once daily as well as L-carnitine 330 mg twice daily to help with muscle cramps. A baseline complete metabolic panel with an estimated glomerular filtration rate was unremarkable.
By the patient’s first follow-up visit after 2 months of therapy, he had experienced marked clinical improvement with notable regression of the tumor (Figure 1). He reported no adverse effects (eg, muscle cramps, dysgeusia, hair loss, nausea, vomiting, diarrhea). At subsequent follow-up visits, the patient continued to demonstrate clinical improvement. His only adverse effect was a 6-kg weight loss over the prior 6 months of initiating therapy despite no changes in taste or appetite. His dose of vismodegib was decreased to an alternative regimen of 150 mg daily for the first 2 weeks of each month with a drug holiday the rest of the month. Since that time, his weight has stabilized and he has continued with treatment.
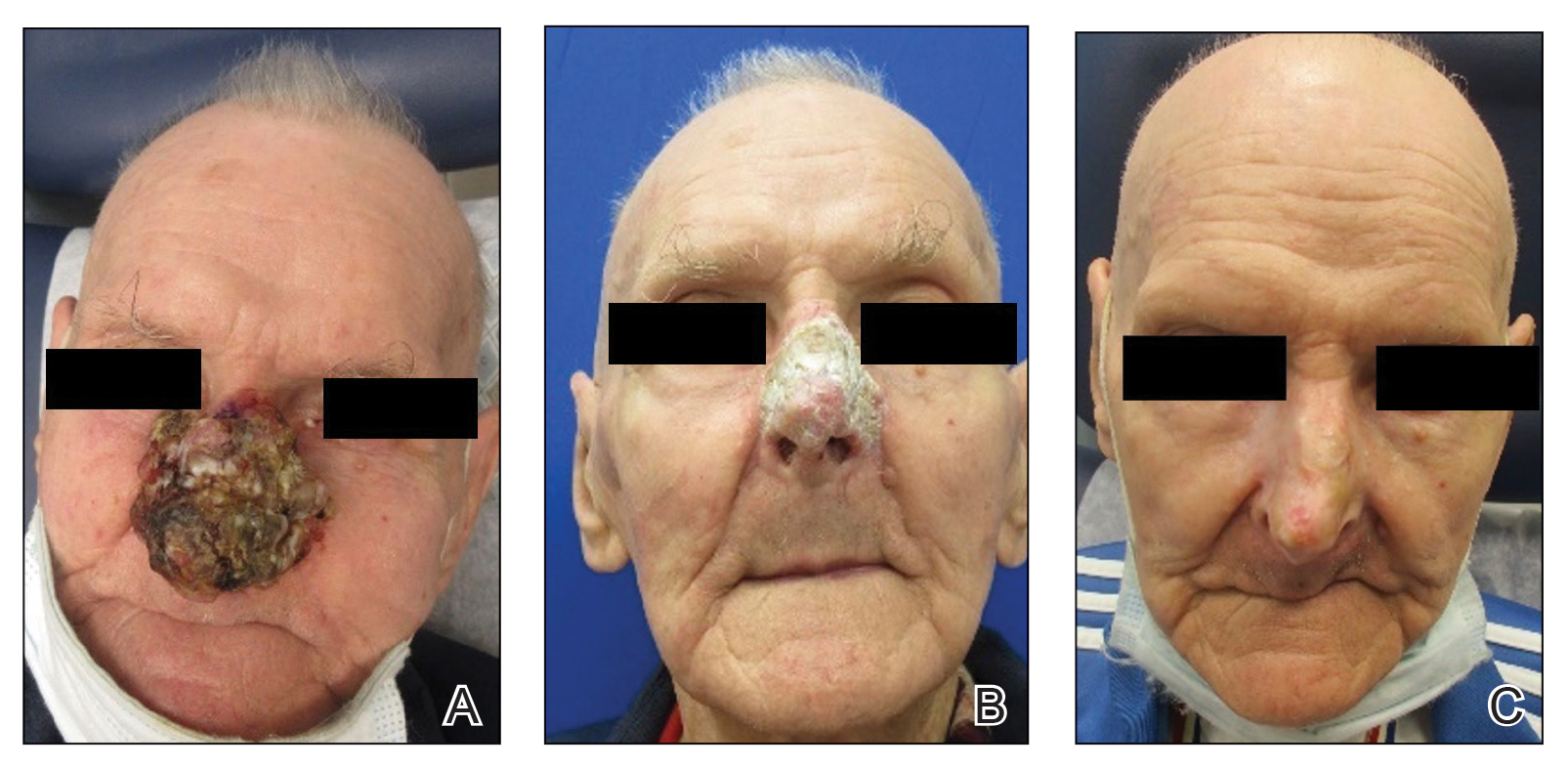
Comment
Vismodegib was the first Hedgehog (Hh) inhibitor approved by the US Food and Drug Administration for management of selected locally advanced and metastatic BCC in adults.1,2 Genetic alterations in the Hh signaling pathway resulting in proliferation of basal cells are present in nearly all BCCs.2 In normal function, when the Hh ligand is absent at the patched (PTCH1) receptor, smoothened (SMO) is inhibited. When Hh ligand binds PTCH1, SMO is activated with downstream effects of triggering cell survival and proliferation in the nucleus via GLI. Loss of function mutations at the PTCH1 receptor or SMO-activating mutations lead to the same downstream effects, even when Hh ligand is absent.1 This allows for unregulated tumor growth.
Vismodegib is a small-molecule SMO inhibitor that blocks aberrant activation of the Hh signaling pathway, thereby slowing the growth of BCCs (Figure 2).3,4 Vismodegib and sonidegib have been used to treat patients with basal cell nevus syndrome as well as metastatic or locally advanced BCCs. At least 50% of advanced BCCs develop resistance to vismodegib, commonly via acquiring mutations in SMO.4
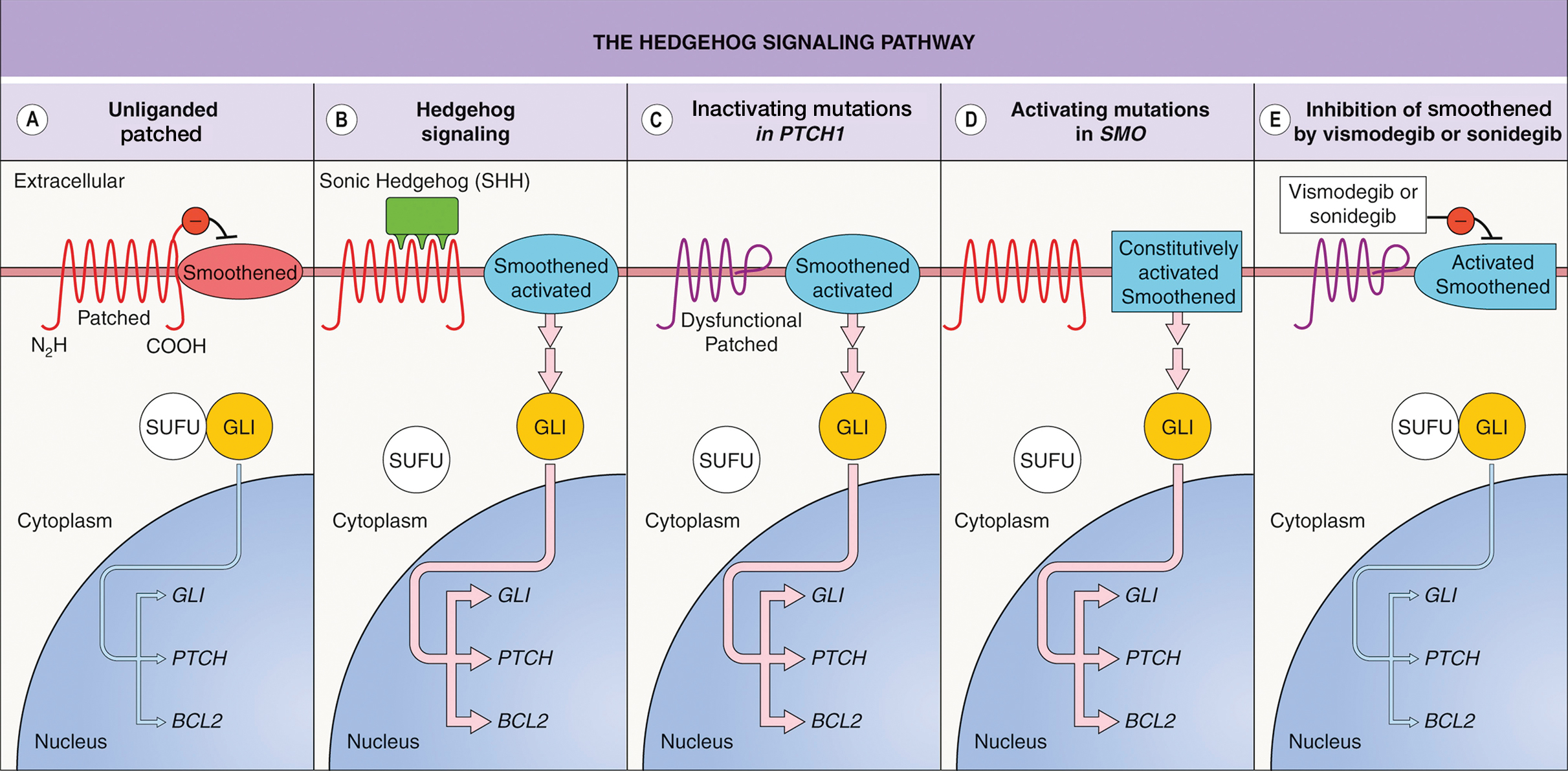
Basal cell carcinoma can be classified as low or high risk based on risk for recurrence. First-line treatments for low-risk BCC are surgical excision, electrodessication and curettage, and MMS.4 Second-line treatment includes radiation therapy. High-risk tumors include those involving anatomic locations of Area H near the eyelids, nose, ears, hands, feet, or genitals in addition to tumors with an aggressive histologic subtype.4,5 First-line treatments for high-risk BCC are MMS or surgical excision. Second-line treatments are radiation therapy or systemic therapy, such as vismodegib.4
Although Hh inhibitors are not a first-line treatment, our case highlights vismodegib’s effectiveness in the management of a large unresectable BCC on the nose of an elderly patient. Our patient opted out of surgical first-line options due to functional and cosmetic concerns.4 He also declined radiation treatment due to financial cost and difficulty with transportation. The patient chose to pursue systemic vismodegib therapy through shared decision-making with dermatology. Vismodegib treatment alone granted our patient a highly remarkable result.
There are limited clinical data on the effectiveness and safety profile of vismodegib in elderly patients, even though this is a high-risk population for BCC.6 In a study that categorized responses to vismodegib in 13 patients with canthal BCC, 5 experienced a complete clinical response (defined as complete regression of the tumor), and 8 achieved partial clinical response (defined as regression but not to the extent of a complete response).7 Our patient’s successful response is notable, as it reinforces vismodegib’s effectiveness as a treatment option for BCC in a sensitive facial area. In addition, our patient’s minimal adverse effect profile is evidence in support of establishing visogemib’s role as a viable treatment option in advanced BCC in the elderly.
Alternative dosing regimens of vismodegib involve the use of drug holidays.8 Utilizing a regimen of 1 week with and 3 weeks without vismodegib for 5 to 14 cycles has led to the resolution of BCC with decreased adverse effects.8 Furthermore, the MIKIE study demonstrated the efficacy of 2 dosing regimens: 12 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 12 weeks of vismodegib 150 mg and 24 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 8 weeks of vismodegib 150 mg.9 Both regimens appeared viable to treat BCC in patients who were at risk for treatment discontinuation due to adverse effects.10
One adverse effect associated with vismodegib is muscle cramps, which are a potential cause of treatment discontinuation. The mechanism by which vismodegib causes cramps is not fully understood but is attributed to contractions from Ca2+ influx into muscle cells and a lack of adenosine triphosphate to allow muscle relaxation.11 This is due to vismodegib’s inhibition of the SMO signaling pathway and activation of the SMO–Ca2+/ AMP-related kinase axis.12 L-carnitine can be used as an adjuvant with vismodegib to address this adverse effect. L-carnitine is found in muscle cells, where its role is to produce energy by utilizing fatty acids.13 It is hypothesized that L-carnitine helps prevent cramps through production of adenosine triphosphate via fatty acid Β-oxidation that aids in stabilizing the sarcolemma and promoting muscle relaxation in skeletal muscle.13,14 Evidence suggests that making L-carnitine a common adjuvant to vismodegib can aid in preventing this adverse effect.
Vismodegib can be an effective treatment option for large nasal BCCs that are difficult to resect. Our case demonstrates both clinical efficacy and a favorable safety profile in an elderly patient. Further studies and long-term follow-up are warranted to establish the role of vismodegib in the evolving landscape of BCC management.
- Peris K, Fargnoli MC, Garbe C, et al. European Dermatology Forum (EDF), the European Association of Dermato-Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur J Cancer. 2019;118:10-34. doi:10.1016/j.ejca.2019.06.003
- Alkeraye SS, Alhammad GA, Binkhonain FK. Vismodegib for basal cell carcinoma and beyond: what dermatologists need to know. Cutis. 2022;110:155-158. doi:10.12788/cutis.0601
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: contemporary approaches to diagnosis, treatment, and prevention. J Am Acad Dermatol. 2019;80:321-339. doi:10.1016/j.jaad.2018.02.083
- Wolf IH, Soyer P, McMeniman EK, et al. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Dermatology. 5th ed. Elsevier; 2024:1888-1910. doi:10.1016/B978-0-7020-8225-2.00108-6
- National Comprehensive Cancer Network. Guidelines for patients: basal cell carcinoma. 2025. Accessed April 7, 2025. https://www.nccn.org/patients/guidelines/content/PDF/basal-cell-patient-guideline.pdf
- Ad Hoc Task Force; Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550. doi:10.1016/j .jaad.2012.06.009
- Passarelli A, Galdo G, Aieta M, et al. Vismodegib experience in elderly patients with basal cell carcinoma: case reports and review of the literature. Int J Mol Sci. 2020;21:8596. doi:10.3390/ijms21228596
- Oliphant H, Laybourne J, Chan K, et al. Vismodegib for periocular basal cell carcinoma: an international multicentre case series. Eye (Lond). 2020;34:2076-2081. doi:10.1038/s41433-020-0778-3
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322. doi:10.1001 /jamadermatol.2016.5058
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basalcell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412. doi:10.1016 /S1470-2045(17)30072-4
- Svoboda SA, Johnson NM, Phillips MA. Systemic targeted treatments for basal cell carcinoma. Cutis. 2022;109:E25-E31. doi:10.12788/cutis.0560
- Nakanishi H, Kurosaki M, Tsuchiya K, et al. L-carnitine reduces muscle cramps in patients with cirrhosis. Clin Gastroenterol Hepatol. 2015;13:1540-1543. doi:10.1016/j.cgh.2014.12.005
- Teperino R, Amann S, Bayer M, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414-426. doi:10.1016/j.cell.2012.09.021
- Dinehart M, McMurray S, Dinehart SM, et al. L-carnitine reduces muscle cramps in patients taking vismodegib. SKIN J Cutan Med. 2018;2:90-95. doi:10.25251/skin.2.2.1
A 90-year-old man presented for evaluation of a large basal cell carcinoma (BCC) involving the nasal region. The lesion was a 7×4-cm pink, crusted, verrucous plaque covering the majority of the nose and extending onto the malar cheeks that originally had been biopsied 26 years prior, and repeat biopsy was performed 3 years prior. Results from both biopsies were consistent with BCC. The patient had avoided treatment for many years due to fear of losing his nose.
Given the size and location of the tumor, surgical intervention posed major challenges for both functional and cosmetic outcomes. After careful consideration and discussion of treatment options, which included Mohs micrographic surgery (MMS), wide local excision, radiation therapy, and systemic therapy, the decision was made to start the patient on vismodegib 150 mg once daily as well as L-carnitine 330 mg twice daily to help with muscle cramps. A baseline complete metabolic panel with an estimated glomerular filtration rate was unremarkable.
By the patient’s first follow-up visit after 2 months of therapy, he had experienced marked clinical improvement with notable regression of the tumor (Figure 1). He reported no adverse effects (eg, muscle cramps, dysgeusia, hair loss, nausea, vomiting, diarrhea). At subsequent follow-up visits, the patient continued to demonstrate clinical improvement. His only adverse effect was a 6-kg weight loss over the prior 6 months of initiating therapy despite no changes in taste or appetite. His dose of vismodegib was decreased to an alternative regimen of 150 mg daily for the first 2 weeks of each month with a drug holiday the rest of the month. Since that time, his weight has stabilized and he has continued with treatment.
Comment
Vismodegib was the first Hedgehog (Hh) inhibitor approved by the US Food and Drug Administration for management of selected locally advanced and metastatic BCC in adults.1,2 Genetic alterations in the Hh signaling pathway resulting in proliferation of basal cells are present in nearly all BCCs.2 In normal function, when the Hh ligand is absent at the patched (PTCH1) receptor, smoothened (SMO) is inhibited. When Hh ligand binds PTCH1, SMO is activated with downstream effects of triggering cell survival and proliferation in the nucleus via GLI. Loss of function mutations at the PTCH1 receptor or SMO-activating mutations lead to the same downstream effects, even when Hh ligand is absent.1 This allows for unregulated tumor growth.
Vismodegib is a small-molecule SMO inhibitor that blocks aberrant activation of the Hh signaling pathway, thereby slowing the growth of BCCs (Figure 2).3,4 Vismodegib and sonidegib have been used to treat patients with basal cell nevus syndrome as well as metastatic or locally advanced BCCs. At least 50% of advanced BCCs develop resistance to vismodegib, commonly via acquiring mutations in SMO.4
Basal cell carcinoma can be classified as low or high risk based on risk for recurrence. First-line treatments for low-risk BCC are surgical excision, electrodessication and curettage, and MMS.4 Second-line treatment includes radiation therapy. High-risk tumors include those involving anatomic locations of Area H near the eyelids, nose, ears, hands, feet, or genitals in addition to tumors with an aggressive histologic subtype.4,5 First-line treatments for high-risk BCC are MMS or surgical excision. Second-line treatments are radiation therapy or systemic therapy, such as vismodegib.4
Although Hh inhibitors are not a first-line treatment, our case highlights vismodegib’s effectiveness in the management of a large unresectable BCC on the nose of an elderly patient. Our patient opted out of surgical first-line options due to functional and cosmetic concerns.4 He also declined radiation treatment due to financial cost and difficulty with transportation. The patient chose to pursue systemic vismodegib therapy through shared decision-making with dermatology. Vismodegib treatment alone granted our patient a highly remarkable result.
There are limited clinical data on the effectiveness and safety profile of vismodegib in elderly patients, even though this is a high-risk population for BCC.6 In a study that categorized responses to vismodegib in 13 patients with canthal BCC, 5 experienced a complete clinical response (defined as complete regression of the tumor), and 8 achieved partial clinical response (defined as regression but not to the extent of a complete response).7 Our patient’s successful response is notable, as it reinforces vismodegib’s effectiveness as a treatment option for BCC in a sensitive facial area. In addition, our patient’s minimal adverse effect profile is evidence in support of establishing visogemib’s role as a viable treatment option in advanced BCC in the elderly.
Alternative dosing regimens of vismodegib involve the use of drug holidays.8 Utilizing a regimen of 1 week with and 3 weeks without vismodegib for 5 to 14 cycles has led to the resolution of BCC with decreased adverse effects.8 Furthermore, the MIKIE study demonstrated the efficacy of 2 dosing regimens: 12 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 12 weeks of vismodegib 150 mg and 24 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 8 weeks of vismodegib 150 mg.9 Both regimens appeared viable to treat BCC in patients who were at risk for treatment discontinuation due to adverse effects.10
One adverse effect associated with vismodegib is muscle cramps, which are a potential cause of treatment discontinuation. The mechanism by which vismodegib causes cramps is not fully understood but is attributed to contractions from Ca2+ influx into muscle cells and a lack of adenosine triphosphate to allow muscle relaxation.11 This is due to vismodegib’s inhibition of the SMO signaling pathway and activation of the SMO–Ca2+/ AMP-related kinase axis.12 L-carnitine can be used as an adjuvant with vismodegib to address this adverse effect. L-carnitine is found in muscle cells, where its role is to produce energy by utilizing fatty acids.13 It is hypothesized that L-carnitine helps prevent cramps through production of adenosine triphosphate via fatty acid Β-oxidation that aids in stabilizing the sarcolemma and promoting muscle relaxation in skeletal muscle.13,14 Evidence suggests that making L-carnitine a common adjuvant to vismodegib can aid in preventing this adverse effect.
Vismodegib can be an effective treatment option for large nasal BCCs that are difficult to resect. Our case demonstrates both clinical efficacy and a favorable safety profile in an elderly patient. Further studies and long-term follow-up are warranted to establish the role of vismodegib in the evolving landscape of BCC management.
A 90-year-old man presented for evaluation of a large basal cell carcinoma (BCC) involving the nasal region. The lesion was a 7×4-cm pink, crusted, verrucous plaque covering the majority of the nose and extending onto the malar cheeks that originally had been biopsied 26 years prior, and repeat biopsy was performed 3 years prior. Results from both biopsies were consistent with BCC. The patient had avoided treatment for many years due to fear of losing his nose.
Given the size and location of the tumor, surgical intervention posed major challenges for both functional and cosmetic outcomes. After careful consideration and discussion of treatment options, which included Mohs micrographic surgery (MMS), wide local excision, radiation therapy, and systemic therapy, the decision was made to start the patient on vismodegib 150 mg once daily as well as L-carnitine 330 mg twice daily to help with muscle cramps. A baseline complete metabolic panel with an estimated glomerular filtration rate was unremarkable.
By the patient’s first follow-up visit after 2 months of therapy, he had experienced marked clinical improvement with notable regression of the tumor (Figure 1). He reported no adverse effects (eg, muscle cramps, dysgeusia, hair loss, nausea, vomiting, diarrhea). At subsequent follow-up visits, the patient continued to demonstrate clinical improvement. His only adverse effect was a 6-kg weight loss over the prior 6 months of initiating therapy despite no changes in taste or appetite. His dose of vismodegib was decreased to an alternative regimen of 150 mg daily for the first 2 weeks of each month with a drug holiday the rest of the month. Since that time, his weight has stabilized and he has continued with treatment.
Comment
Vismodegib was the first Hedgehog (Hh) inhibitor approved by the US Food and Drug Administration for management of selected locally advanced and metastatic BCC in adults.1,2 Genetic alterations in the Hh signaling pathway resulting in proliferation of basal cells are present in nearly all BCCs.2 In normal function, when the Hh ligand is absent at the patched (PTCH1) receptor, smoothened (SMO) is inhibited. When Hh ligand binds PTCH1, SMO is activated with downstream effects of triggering cell survival and proliferation in the nucleus via GLI. Loss of function mutations at the PTCH1 receptor or SMO-activating mutations lead to the same downstream effects, even when Hh ligand is absent.1 This allows for unregulated tumor growth.
Vismodegib is a small-molecule SMO inhibitor that blocks aberrant activation of the Hh signaling pathway, thereby slowing the growth of BCCs (Figure 2).3,4 Vismodegib and sonidegib have been used to treat patients with basal cell nevus syndrome as well as metastatic or locally advanced BCCs. At least 50% of advanced BCCs develop resistance to vismodegib, commonly via acquiring mutations in SMO.4
Basal cell carcinoma can be classified as low or high risk based on risk for recurrence. First-line treatments for low-risk BCC are surgical excision, electrodessication and curettage, and MMS.4 Second-line treatment includes radiation therapy. High-risk tumors include those involving anatomic locations of Area H near the eyelids, nose, ears, hands, feet, or genitals in addition to tumors with an aggressive histologic subtype.4,5 First-line treatments for high-risk BCC are MMS or surgical excision. Second-line treatments are radiation therapy or systemic therapy, such as vismodegib.4
Although Hh inhibitors are not a first-line treatment, our case highlights vismodegib’s effectiveness in the management of a large unresectable BCC on the nose of an elderly patient. Our patient opted out of surgical first-line options due to functional and cosmetic concerns.4 He also declined radiation treatment due to financial cost and difficulty with transportation. The patient chose to pursue systemic vismodegib therapy through shared decision-making with dermatology. Vismodegib treatment alone granted our patient a highly remarkable result.
There are limited clinical data on the effectiveness and safety profile of vismodegib in elderly patients, even though this is a high-risk population for BCC.6 In a study that categorized responses to vismodegib in 13 patients with canthal BCC, 5 experienced a complete clinical response (defined as complete regression of the tumor), and 8 achieved partial clinical response (defined as regression but not to the extent of a complete response).7 Our patient’s successful response is notable, as it reinforces vismodegib’s effectiveness as a treatment option for BCC in a sensitive facial area. In addition, our patient’s minimal adverse effect profile is evidence in support of establishing visogemib’s role as a viable treatment option in advanced BCC in the elderly.
Alternative dosing regimens of vismodegib involve the use of drug holidays.8 Utilizing a regimen of 1 week with and 3 weeks without vismodegib for 5 to 14 cycles has led to the resolution of BCC with decreased adverse effects.8 Furthermore, the MIKIE study demonstrated the efficacy of 2 dosing regimens: 12 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 12 weeks of vismodegib 150 mg and 24 weeks of vismodegib 150 mg followed by 3 cycles of 8 placebo weeks and 8 weeks of vismodegib 150 mg.9 Both regimens appeared viable to treat BCC in patients who were at risk for treatment discontinuation due to adverse effects.10
One adverse effect associated with vismodegib is muscle cramps, which are a potential cause of treatment discontinuation. The mechanism by which vismodegib causes cramps is not fully understood but is attributed to contractions from Ca2+ influx into muscle cells and a lack of adenosine triphosphate to allow muscle relaxation.11 This is due to vismodegib’s inhibition of the SMO signaling pathway and activation of the SMO–Ca2+/ AMP-related kinase axis.12 L-carnitine can be used as an adjuvant with vismodegib to address this adverse effect. L-carnitine is found in muscle cells, where its role is to produce energy by utilizing fatty acids.13 It is hypothesized that L-carnitine helps prevent cramps through production of adenosine triphosphate via fatty acid Β-oxidation that aids in stabilizing the sarcolemma and promoting muscle relaxation in skeletal muscle.13,14 Evidence suggests that making L-carnitine a common adjuvant to vismodegib can aid in preventing this adverse effect.
Vismodegib can be an effective treatment option for large nasal BCCs that are difficult to resect. Our case demonstrates both clinical efficacy and a favorable safety profile in an elderly patient. Further studies and long-term follow-up are warranted to establish the role of vismodegib in the evolving landscape of BCC management.
- Peris K, Fargnoli MC, Garbe C, et al. European Dermatology Forum (EDF), the European Association of Dermato-Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur J Cancer. 2019;118:10-34. doi:10.1016/j.ejca.2019.06.003
- Alkeraye SS, Alhammad GA, Binkhonain FK. Vismodegib for basal cell carcinoma and beyond: what dermatologists need to know. Cutis. 2022;110:155-158. doi:10.12788/cutis.0601
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: contemporary approaches to diagnosis, treatment, and prevention. J Am Acad Dermatol. 2019;80:321-339. doi:10.1016/j.jaad.2018.02.083
- Wolf IH, Soyer P, McMeniman EK, et al. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Dermatology. 5th ed. Elsevier; 2024:1888-1910. doi:10.1016/B978-0-7020-8225-2.00108-6
- National Comprehensive Cancer Network. Guidelines for patients: basal cell carcinoma. 2025. Accessed April 7, 2025. https://www.nccn.org/patients/guidelines/content/PDF/basal-cell-patient-guideline.pdf
- Ad Hoc Task Force; Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550. doi:10.1016/j .jaad.2012.06.009
- Passarelli A, Galdo G, Aieta M, et al. Vismodegib experience in elderly patients with basal cell carcinoma: case reports and review of the literature. Int J Mol Sci. 2020;21:8596. doi:10.3390/ijms21228596
- Oliphant H, Laybourne J, Chan K, et al. Vismodegib for periocular basal cell carcinoma: an international multicentre case series. Eye (Lond). 2020;34:2076-2081. doi:10.1038/s41433-020-0778-3
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322. doi:10.1001 /jamadermatol.2016.5058
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basalcell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412. doi:10.1016 /S1470-2045(17)30072-4
- Svoboda SA, Johnson NM, Phillips MA. Systemic targeted treatments for basal cell carcinoma. Cutis. 2022;109:E25-E31. doi:10.12788/cutis.0560
- Nakanishi H, Kurosaki M, Tsuchiya K, et al. L-carnitine reduces muscle cramps in patients with cirrhosis. Clin Gastroenterol Hepatol. 2015;13:1540-1543. doi:10.1016/j.cgh.2014.12.005
- Teperino R, Amann S, Bayer M, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414-426. doi:10.1016/j.cell.2012.09.021
- Dinehart M, McMurray S, Dinehart SM, et al. L-carnitine reduces muscle cramps in patients taking vismodegib. SKIN J Cutan Med. 2018;2:90-95. doi:10.25251/skin.2.2.1
- Peris K, Fargnoli MC, Garbe C, et al. European Dermatology Forum (EDF), the European Association of Dermato-Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). Diagnosis and treatment of basal cell carcinoma: European consensus-based interdisciplinary guidelines. Eur J Cancer. 2019;118:10-34. doi:10.1016/j.ejca.2019.06.003
- Alkeraye SS, Alhammad GA, Binkhonain FK. Vismodegib for basal cell carcinoma and beyond: what dermatologists need to know. Cutis. 2022;110:155-158. doi:10.12788/cutis.0601
- Cameron MC, Lee E, Hibler BP, et al. Basal cell carcinoma: contemporary approaches to diagnosis, treatment, and prevention. J Am Acad Dermatol. 2019;80:321-339. doi:10.1016/j.jaad.2018.02.083
- Wolf IH, Soyer P, McMeniman EK, et al. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Dermatology. 5th ed. Elsevier; 2024:1888-1910. doi:10.1016/B978-0-7020-8225-2.00108-6
- National Comprehensive Cancer Network. Guidelines for patients: basal cell carcinoma. 2025. Accessed April 7, 2025. https://www.nccn.org/patients/guidelines/content/PDF/basal-cell-patient-guideline.pdf
- Ad Hoc Task Force; Connolly SM, Baker DR, Coldiron BM, et al. AAD/ACMS/ASDSA/ASMS 2012 appropriate use criteria for Mohs micrographic surgery: a report of the American Academy of Dermatology, American College of Mohs Surgery, American Society for Dermatologic Surgery Association, and the American Society for Mohs Surgery. J Am Acad Dermatol. 2012;67:531-550. doi:10.1016/j .jaad.2012.06.009
- Passarelli A, Galdo G, Aieta M, et al. Vismodegib experience in elderly patients with basal cell carcinoma: case reports and review of the literature. Int J Mol Sci. 2020;21:8596. doi:10.3390/ijms21228596
- Oliphant H, Laybourne J, Chan K, et al. Vismodegib for periocular basal cell carcinoma: an international multicentre case series. Eye (Lond). 2020;34:2076-2081. doi:10.1038/s41433-020-0778-3
- Becker LR, Aakhus AE, Reich HC, et al. A novel alternate dosing of vismodegib for treatment of patients with advanced basal cell carcinomas. JAMA Dermatol. 2017;153:321-322. doi:10.1001 /jamadermatol.2016.5058
- Dréno B, Kunstfeld R, Hauschild A, et al. Two intermittent vismodegib dosing regimens in patients with multiple basalcell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017;18:404-412. doi:10.1016 /S1470-2045(17)30072-4
- Svoboda SA, Johnson NM, Phillips MA. Systemic targeted treatments for basal cell carcinoma. Cutis. 2022;109:E25-E31. doi:10.12788/cutis.0560
- Nakanishi H, Kurosaki M, Tsuchiya K, et al. L-carnitine reduces muscle cramps in patients with cirrhosis. Clin Gastroenterol Hepatol. 2015;13:1540-1543. doi:10.1016/j.cgh.2014.12.005
- Teperino R, Amann S, Bayer M, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414-426. doi:10.1016/j.cell.2012.09.021
- Dinehart M, McMurray S, Dinehart SM, et al. L-carnitine reduces muscle cramps in patients taking vismodegib. SKIN J Cutan Med. 2018;2:90-95. doi:10.25251/skin.2.2.1
Remarkable Response to Vismodegib in a Locally Advanced Basal Cell Carcinoma on the Nose
Remarkable Response to Vismodegib in a Locally Advanced Basal Cell Carcinoma on the Nose
PRACTICE POINTS
- Dermatologists should consider using vismodegib for treatment of unresectable basal cell carcinoma.
- Vismodegib dosing regimens can vary; drug holidays can be used to mitigate adverse effects while maintaining desirable treatment outcomes.

Nonhealing Ulcer on the Lower Lip
Nonhealing Ulcer on the Lower Lip
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
THE DIAGNOSIS: Syphilis
The differential diagnosis of oral lesions can be complex; in our patient, we considered conditions such as pyogenic granuloma, herpes simplex virus, and syphilis, despite the presence of pain. Immunohistochemical staining for spirochete antigens was positive, and serologic confirmation through a positive rapid plasma reagin (RPR) test confirmed the diagnosis of primary syphilis. The patient was promptly referred back to the primary care physician for treatment with intramuscular penicillin, leading to resolution of the lesion. At 3 months’ follow-up in our clinic, the lesion was fully resolved.
A primary syphilitic chancre is the initial lesion caused by Treponema pallidum, typically manifesting as a painless ulcer at the infection site, usually in the genital area; however, chancres also may manifest in other locations (eg, the anus or oral cavity) due to direct contact with infectious lesions on another individual. Our case represents an atypical presentation of an oral syphilitic chancre.
Syphilis is a sexually transmitted infection with various clinical manifestations. It is crucial to consider syphilis in the differential diagnosis of ulcerative lesions even when pain is present, especially in high-risk individuals such as those who engage in unprotected sex.1,2 Oral syphilitic chancres have been documented in the medical literature for more than a century, underscoring the importance of maintaining a high index of suspicion for diagnosis and a low threshold for obtaining an RPR test to facilitate early detection and treatment.2,3 Notably, the prevalence of syphilis is higher in men who have sex with men, particularly among those who engage in unprotected oral and anal sex. Increased screening and early treatment are essential to control the spread of disease within all populations. Doxycycline postexposure prophylaxis (doxyPEP) is used as a preventive measure for syphilis, chlamydia, and gonorrhea.4 This regimen consists of 200 mg of doxycycline taken within 24 hours but no later than 72 hours after unprotected anal, vaginal, or oral sex.
Our case highlights the importance of considering the differential diagnosis of oral ulcers, particularly in high-risk populations such as men who have sex with men. Prompt diagnosis, effective treatment, and preventive strategies such as doxyPEP are essential for controlling syphilis. Comprehensive patient education and regular follow-up appointments are critical components of successful management.
The United States has experienced a considerable rise in primary and congenital syphilis cases, with an 80% increase between 2018 and 2022.6 Serologic testing is the primary method for diagnosing, staging, and managing syphilis. Sexually active patients with suspected syphilis or unexplained symptoms should undergo testing. Prompt diagnosis and treatment can prevent systemic complications, including ocular involvement and permanent blindness.
Syphilis is transmitted through direct contact with a syphilitic ulcer or saliva or blood from an infected individual. Oral syphilitic ulcers can develop on the lips, tongue, oral mucosa, and tonsils. Chancres can range from a few millimeters to several centimeters, with an incubation period of 10 to 90 days (average, 21 days). The chancre lasts 3 to 6 weeks and heals spontaneously. Without treatment, primary syphilis can progress to secondary syphilis, characterized by a papulosquamous eruption and mucosal involvement, and potentially tertiary syphilis, which can affect the central nervous system, heart, bones, and skin.7
Immunocompromised patients, especially those diagnosed with HIV, face increased risks including altered clinical presentations (eg, multiple or deep chancres), delayed healing, overlapping stages of disease, and increased severity of organ involvement. All sexually active individuals should be screened for syphilis every 3 to 6 months, particularly those with unexplained oral ulcers.
Serologic testing is fundamental for syphilis diagnosis and management. Nontreponemal tests such as RPR and treponemal tests such as the fluorescent treponemal antibody absorption test provide comprehensive diagnostic information. Early diagnosis and empiric treatment are crucial in suspected cases. Ocular screening is recommended for suspected or confirmed syphilis cases.7
Management of syphilis includes treating all sexual partners and providing thorough patient education on the disease. Monitoring for the Jarisch-Herxheimer reaction—an acute febrile reaction following penicillin therapy—is important, especially in pregnant patients.5 Serologic evaluation at 6 and 12 months posttreatment is recommended, with more frequent evaluations if follow-up is uncertain, particularly for those with inconsistent access to health care or in whom reinfection is suspected. Guidelines from the Centers for Disease Control and Prevention advocate for intramuscular penicillin G benzathine as the preferred treatment, with specific dosing for adults and children.7 Due to the ongoing bicillin shortage, alternatives such as extencilline have temporarily been allowed for use in the United States.8
The rising incidence of syphilis in the United States underscores the critical need for enhanced public health initiatives focusing on education, screening, and early intervention. Comprehensive sexual education that includes information about syphilis and other sexually transmitted infections, proper use of prophylactic measures such as condoms, and the benefits of doxyPEP can considerably reduce transmission rates. Health care providers should routinely discuss these preventive measures with their patients, especially those in high-risk groups.
Our case highlights the importance of considering syphilis in the differential diagnosis of oral ulcers, particularly in high-risk populations. Timely diagnosis, effective treatment, and preventive measures such as doxyPEP are essential for managing and controlling syphilis. The rising incidence of syphilis in the United States warrants increased screening, patient education, and public health interventions to address this notable health challenge. The syphilis crisis calls for coordinated efforts from health care providers, public health officials, and community leaders to curb the spread of this infection and protect public health.
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
- Mayer KH, Traeger M, Marcus JL. Doxycycline postexposure prophylaxis and sexually transmitted infections. JAMA. 2023;330:1381-1382. doi:10.1001/jama.2023.16416
- Cossman JP, Fournier JB. Frequency of syphilis diagnoses by dermatologists. JAMA Dermatol. 2017;153:718-719. doi:10.1001 /jamadermatol.2017.0460
- Porterfield C, Brodell D, Dolohanty L, et al. Primary syphilis presenting as a chronic lip ulcer. Cureus. 2020;12:E7086. doi:10.7759 /cureus.7086
- Schamberg JF. An epidemic of chancres of the lip from kissing. JAMA. 1911;LVII:783-784. doi:10.1001/jama.1911.04260090005002
- Farmer TW. Jarisch-Herxheimer reaction in early syphilis. JAMA. 1948;138:480–485. doi:10.1001/jama.1948.02900070012003
- Winney A. Why is syphilis spiking in the U.S.? Johns Hopkins Bloomberg School of Public Health. Johns Hopkins Bloomberg School of Public Health. Published March 13, 2024. Accessed April 30, 2025. https://publichealth.jhu.edu/why-is-syphilis-spiking-in-the-us
- Koundanya VV, Tripathy K. Syphilis ocular manifestations. StatPearls Publishing; 2021. Updated August 25, 2023. Accessed May 6, 2025. https://www.ncbi.nlm.nih.gov/books/NBK558957/
- CDC. FDA announcement on availability of extencilline. National Center for HIV, Viral Hepatitis, STD, and Tuberculosis Prevention. Published July 19, 2024. Accessed April 30, 2025. https://www.cdc.gov/nchhstp/director-letters/extencilline-during-bicillin-l-a-shortage.html
Nonhealing Ulcer on the Lower Lip
Nonhealing Ulcer on the Lower Lip
A 54-year-old HIV-negative man with a history of having sex with men presented to his primary care physician with an ulcer on the lower lip of 3 weeks’ duration. The patient reported that the lesion had appeared as a typical cold sore with pain in the area. A 9-day course of oral valacyclovir prescribed by the primary care physician provided no relief or improvement. A 2-mm punch biopsy was performed.
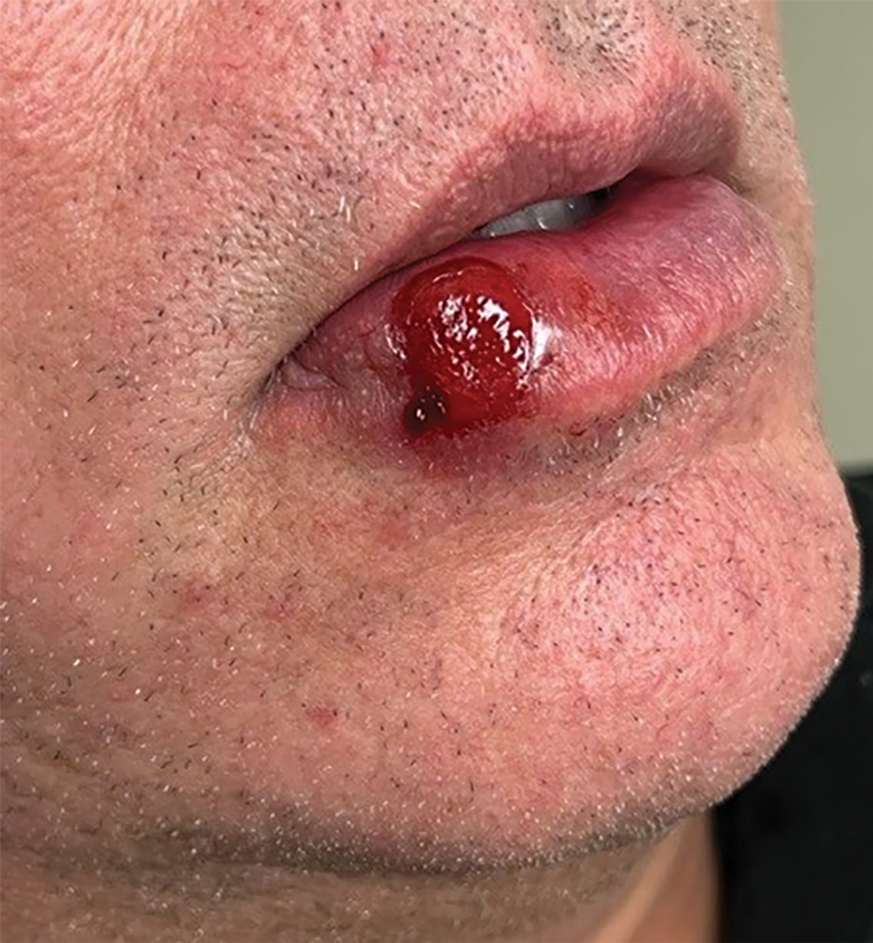

Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
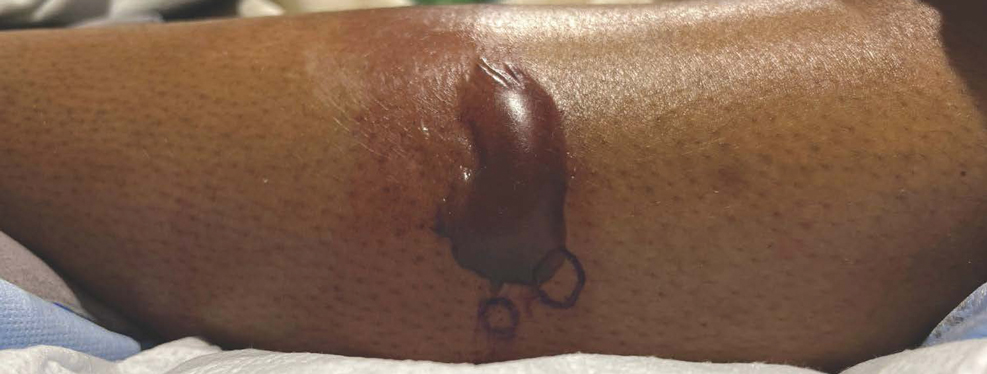
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
THE DIAGNOSIS: Bullous Hemorrhagic Dermatosis
Biopsy results showed an intraepidermal blister with a floor composed of maturing epidermis. The roof of the blister was composed of necrotic keratinocytes with overlying orthokeratosis, and the cavity was filled with a moderate amount of fibrin and dead cells with neutrophils. Direct immunofluorescence (DIF) using specific antihuman IgG, IgM, IgA, C3, and fibrin was negative. Aerobic, anaerobic, and fungal cultures also were negative. With these histopathologic findings, medication exposure, and timing of bullae onset, our patient was diagnosed with bullous hemorrhagic dermatosis (BHD) secondary to enoxaparin administration. Enoxaparin was continued due to increased risk for coagulopathy, and there was complete resolution of the bullae after 5 weeks with no residual symptoms.
Bullous hemorrhagic dermatosis is a rare eruption that can occur after administration of heparin and low-molecular-weight heparin, with enoxaparin being the most commonly implicated drug.1 The lesions typically are seen in elderly men in the seventh decade of life and appear within a median of 7 days after drug exposure. The time course for the postexposure eruption can vary from 2 to 21 days, with reports of skin lesions appearing up to 4 months after exposure.1,2 hemorrhagic bullae (Figure) typically on the arms and legs, though lesions also can develop on the trunk. The lesions can occur in distant areas from the injection site, suggesting BHD may be a systemic reaction, although the etiology is poorly understood.1
Another heparin reaction that can manifest similarly to BHD is heparin-induced skin necrosis.3 Patients with this condition also may have associated heparin-induced thrombocytopenia upon laboratory investigation and have a more aggressive clinical course than BHD. Biopsy can help differentiate BHD and early heparin-induced skin necrosis if the clinical manifestation is unclear. Histopathologically, BHD typically has intraepidermal bullae filled with blood, whereas heparin-induced skin necrosis has dermal thrombi.1,4 Treatment of both conditions differs in whether to discontinue anticoagulants: heparin-induced skin necrosis requires discontinuation of the medication, while BHD does not.2,3
In patients with BHD, the lesions are self-resolving, and treatment is supportive, although whether enoxaparin is discontinued varies among physicians.2 Lesions typically resolve within 2 weeks of onset, although it is unclear whether continuing anticoagulants delays resolution.1 Discontinuing anticoagulants in certain patients can be life-threatening due to complex comorbidities (eg, risk for venous thromboembolism or pulmonary embolism from prolonged hospitalization or severe trauma) and is not necessary for the resolution of BHD.
In addition to BHD and heparin-induced skin necrosis, our differential diagnosis included bullous pemphigoid, coma blisters, and Vibrio vulnificus infection. Although bullous pemphigoid can manifest with tense bullae that are pauci-inflammatory on histology, DIF would show linear IgG and C3 deposition at the dermal-epidermal junction. In our patient, DIF was negative and favored another etiology for the lesions. Coma blisters can occur in areas of sustained pressure and typically develop in patients with a prolonged hospitalization or those who are sedentary for long periods of time. The distribution of bullae on our patient’s bilateral pretibial shins made this diagnosis unlikely. Vibrio vulnificus infection can manifest as hemorrhagic bullae, though typically after a break in the skin exposed to brackish water. Vibrio vulnificus infection can be life-threatening, resulting in septicemia and increased mortality, and a thorough patient history is important for diagnosis.5
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
- Russo A, Curtis S, Balbuena-Merle R, et al. Bullous hemorrhagic dermatosis is an under-recognized side effect of full dose lowmolecular weight heparin: a case report and review of the literature. Exp Hematol Oncol. 2018;7:15. doi:10.1186/s40164-018-0108-7
- Dhattarwal N, Gurjar R. Bullous hemorrhagic dermatosis: a rare cutaneous reaction of heparin. J Postgrad Med. 2023;69:97-98. doi:10.4103/jpgm.jpgm_282_22
- Maldonado Cid P, Alonso de Celada RM, Noguera Morel L, et al. Cutaneous adverse events associated with heparin. Clin Exp Dermatol. 2012;37:707-711. doi:10.1111/j.1365-2230.2012.04395.x
- Handschin AE, Trentz O, Kock HJ, et al. Low molecular weight heparininduced skin necrosis-a systematic review. Langenbecks Arch Surg. 2005;390:249-254. doi:10.1007/s00423-004-0522-7
- Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723-1733. doi:10.1128/IAI.01046-08
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
Large Bullae on the Legs in a Hospitalized Patient Following a Gunshot Wound
A 19-year-old man developed fluid-filled blisters on both legs within 1 month of a prolonged hospitalization following a gunshot wound that resulted in complete paralysis of the legs. His medical history was otherwise unremarkable. Medications started during hospitalization included moxifloxacin, levetiracetam, and prophylactic subcutaneous enoxaparin. Physical examination by dermatology revealed tense blood-filled bullae measuring several centimeters with well-demarcated, pink to red, irregularly shaped patches on both legs. A biopsy of a blister was taken.
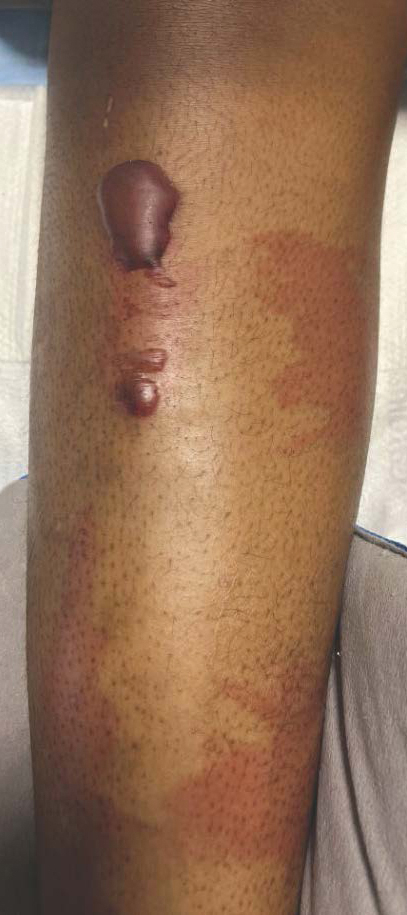

Anaphylaxis Preparedness Starts in Your Office: Insights on Caregiver Readiness and Epinephrine Device Selection

Click here to read more.
Click here to read more.