User login
EHR default order slashes unnecessary imaging during palliative RT
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
Simply adding a default order to the electronic health record that automatically opts patients out of commonly used but unnecessary radiation oncology procedures can dramatically curtail their use, suggests a stepped-wedge, cluster-randomized, controlled trial.
Daily x-ray or CT imaging is often used to better reproducibly position patients during curative radiotherapy, but guidelines consider it unnecessary during palliative radiotherapy because of limited clinical benefit, according to the investigators, led by Sonam Sharma, MD, of the Icahn School of Medicine at Mount Sinai, New York, and the Abramson Cancer Center at the University of Pennsylvania, Philadelphia. “Unnecessary imaging can increase treatment time and expense for patients in distress,” they noted.
The investigators conducted a 2-year trial among 21 radiation oncologists from five practices (one university, four community) in which they added to the EHR a default order that specified no daily imaging during palliative radiation therapy. (Radiation oncologists could select another imaging frequency if they preferred.) The default order was first rolled out in the university practice and subsequently in the community practices.
Study analyses were based on 1,019 adult patients with bone, soft tissue, or brain metastases who received 1,188 courses of palliative three-dimensional conformal radiotherapy during the trial.
Results reported in a JAMA Oncology research letter showed that the proportion of patients receiving daily imaging during their palliative radiotherapy (imaging during 80% or more of treatments) fell from 68.2% during the combined preintervention periods to 32.4% during the combined intervention periods.
After potential confounders were taken into account, implementation of the default order in the EHR was associated with a more than halving of the odds of daily imaging during palliative radiotherapy (adjusted odds ratio, 0.37; P = .003), with an adjusted percentage point reduction of –18.8.
Findings were similar in the university practice alone (aOR, 0.33; P = .01; –22.3 percentage points) and in the community practices alone (aOR, 0.45; P = .02; –27.5 percentage points).
“In a network of five radiation oncology practices, introducing a default order in the EHR reduced unnecessary daily imaging during palliative radiotherapy,” Dr. Sharma and colleagues concluded. “Our findings suggest that simple nudges, such as setting default orders, can meaningfully reduce unnecessary care.”
Dr. Sharma reported that she had no relevant conflicts of interest. The study was funded in part by the National Cancer Institute and the University of Pennsylvania Health System through the Penn Medicine Nudge Unit and the department of radiation oncology.
SOURCE: Sharma S et al. JAMA Oncol. 2019 Jun 27. doi: 10.1001/jamaoncol.2019.1432.
FROM JAMA ONCOLOGY
Sarcoma—rare, but not insignificant
This year, progress in treating rare cancers has been named the advance of the year by the American Society of Clinical Oncology (ASCO). Advancements in treating desmoid tumors, a subtype of sarcoma, was highlighted as one of the prominent breakthroughs for a rare cancer. While sarcoma is statistically rare, the impact of the disease is great, particularly on patients and families. ASCO’s recognition of rare cancer advancements demonstrates what the sarcoma community has long known: that “rare” shouldn’t mean unimportant or overlooked. In fact, the contributions of families, patients, caregivers, clinicians, researchers, foundations, organizations, and agencies in bringing sarcoma to the forefront and giving it prominence—spending time, effort, and energy in finding effective treatments—is of utmost importance, despite the disease’s rarity.
The Sarcoma Foundation of America (SFA) is leading the race to cure sarcoma, and it is doing so through research, advocacy, and education. Since its founding in 2001, donors to the foundation have funded over $9 million in research, with almost $2 million to be invested in research projects this year alone. The SFA supports research focused on discovering and developing new and effective therapies to treat and eradicate sarcoma—often highrisk, high-reward projects that would not likely be funded by the government or commercial interests. Driving the research agenda are members of its Medical Advisory Board—some of the brightest scientific minds in the world today, several of whom also serve on the Editorial Advisory Board of this, the SFA’s official journal. We are thankful for their dedication. Together, their efforts will continue to make a difference in the lives of those impacted by sarcoma.
The Sarcoma Foundation of America
CureSarcoma.org
This year, progress in treating rare cancers has been named the advance of the year by the American Society of Clinical Oncology (ASCO). Advancements in treating desmoid tumors, a subtype of sarcoma, was highlighted as one of the prominent breakthroughs for a rare cancer. While sarcoma is statistically rare, the impact of the disease is great, particularly on patients and families. ASCO’s recognition of rare cancer advancements demonstrates what the sarcoma community has long known: that “rare” shouldn’t mean unimportant or overlooked. In fact, the contributions of families, patients, caregivers, clinicians, researchers, foundations, organizations, and agencies in bringing sarcoma to the forefront and giving it prominence—spending time, effort, and energy in finding effective treatments—is of utmost importance, despite the disease’s rarity.
The Sarcoma Foundation of America (SFA) is leading the race to cure sarcoma, and it is doing so through research, advocacy, and education. Since its founding in 2001, donors to the foundation have funded over $9 million in research, with almost $2 million to be invested in research projects this year alone. The SFA supports research focused on discovering and developing new and effective therapies to treat and eradicate sarcoma—often highrisk, high-reward projects that would not likely be funded by the government or commercial interests. Driving the research agenda are members of its Medical Advisory Board—some of the brightest scientific minds in the world today, several of whom also serve on the Editorial Advisory Board of this, the SFA’s official journal. We are thankful for their dedication. Together, their efforts will continue to make a difference in the lives of those impacted by sarcoma.
The Sarcoma Foundation of America
CureSarcoma.org
This year, progress in treating rare cancers has been named the advance of the year by the American Society of Clinical Oncology (ASCO). Advancements in treating desmoid tumors, a subtype of sarcoma, was highlighted as one of the prominent breakthroughs for a rare cancer. While sarcoma is statistically rare, the impact of the disease is great, particularly on patients and families. ASCO’s recognition of rare cancer advancements demonstrates what the sarcoma community has long known: that “rare” shouldn’t mean unimportant or overlooked. In fact, the contributions of families, patients, caregivers, clinicians, researchers, foundations, organizations, and agencies in bringing sarcoma to the forefront and giving it prominence—spending time, effort, and energy in finding effective treatments—is of utmost importance, despite the disease’s rarity.
The Sarcoma Foundation of America (SFA) is leading the race to cure sarcoma, and it is doing so through research, advocacy, and education. Since its founding in 2001, donors to the foundation have funded over $9 million in research, with almost $2 million to be invested in research projects this year alone. The SFA supports research focused on discovering and developing new and effective therapies to treat and eradicate sarcoma—often highrisk, high-reward projects that would not likely be funded by the government or commercial interests. Driving the research agenda are members of its Medical Advisory Board—some of the brightest scientific minds in the world today, several of whom also serve on the Editorial Advisory Board of this, the SFA’s official journal. We are thankful for their dedication. Together, their efforts will continue to make a difference in the lives of those impacted by sarcoma.
The Sarcoma Foundation of America
CureSarcoma.org
From the journals: sarcoma around the world
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
EWING SARCOMA IN NEPAL: Investigators reported what they believe to be the first prospective clinical trial providing state-of-the-art chemotherapy to patients with Ewing sarcoma in Nepal. They treated 20 newly diagnosed patients with combination chemotherapy, including a course of etoposide and ifosfamide during external-beam radiotherapy. Radiotherapy was the only available treatment modality for local tumor control because advanced tumor-orthopedic services are not available in Nepal.
The 11 females and 9 males enrolled ranged in age from 6 to 37 years.
The treatment protocol—based on the Nepali-Norwegian Ewing Sarcoma Study treatment initiative— consisted of:
- Cyclophosphamide (1,200 mg/m2 as a 30-minute intravenous [IV] infusion)
- Doxorubicin (40 mg/m2/d as a 4-hour IV infusion on days 1 and 2; total dose, 80 mg/m2 in 2 days; total cumulative dose, 400 mg/m2)
- Etoposide (150 mg/m2/d as a 2-hour IV infusion; total dose, 450 mg/m2 in 3 days)
- Ifosfamide (3,000 mg/m2 over 21 to 24 hours as a 3-day continuous IV infusion; total dose, 9,000 mg/m2 in 3 days)
- Vincristine (1.5 mg/m2 IV push; maximum, 2 mg)
Patients received 5 courses of chemotherapy, then radiotherapy twice daily for 4 weeks for a total accumulated 54-Gy dose with a course of etoposide and ifosfamide, followed by 6 additional courses of chemotherapy.
Patients had primary tumors in the following sites: femur (n = 4), pubic bone (n = 1), fibula (n = 1), thoracic wall or costae (n = 4), clavicle (n = 1), craniofacial bone (n = 3), humerus (n = 3), forearm (n = 1), musculus sartorius with invasion into adjacent femur (n = 1), and uterine cervix (n = 1).
Eleven patients completed the entire treatment regimen, 6 of whom had no evidence of disease at a median follow-up of 2.3 years (range, 1.3 to 3.1 years). Four of them died of metastatic disease, and 1 experienced a recurrence 6 months later.
Three patients died due to chemotherapy- related toxicity, and 6 patients did not complete the treatment protocol, 4 of whom experienced progressive disease, were lost to follow-up, and presumed dead.
The investigators concluded that radiotherapy as the sole local treatment modality in combination with chemotherapy is feasible. They observed no fractures among the 15 patients who received radiotherapy.
SOURCE: Jha AK, Neupane P, Pradhan M, et al. Ewing sarcoma in Nepal treated with combined chemotherapy and definitive radiotherapy. J Glob Oncol. 2019;5:1-10.
PEDIATRIC SOFT TISSUE AND BONE SARCOMAS IN TANZANIA: In this retrospective review, investigators documented the epidemiologic and clinical features of pediatric sarcomas in the largest pediatric oncology center in Tanzania—Muhimbili National Hospital. Their objective in collecting the data was to compare the results with those of other countries and ultimately prioritize treatment protocols and resources for the more common pediatric sarcomas in Tanzania. Prior to this study, no data existed on the frequency and types most commonly seen in the country.
Between 2011 and 2016, the investigators collected information on 135 pediatric cases seen at the hospital. Eighty-nine cases (66%) were soft tissue sarcomas (STS) and 46 (34%) were bone sarcomas. Most patients, they reported, presented with a painless swelling.
Investigators found that, as in other countries, embryonal rhabdomyosarcoma accounted for the majority (75%) of all sarcomas seen in this study and osteosarcoma accounted for most (87%) bone sarcomas. However, unlike pediatric sarcomas in other countries, few cases of Ewing sarcoma were diagnosed during the study period.
An important disparity between Tanzania and other countries is that most patients in Tanzania present with advanced- stage disease, when the possibility of curative therapy is vastly reduced. Investigators found the lung to be the most common site of distant metastasis.
Other clinical and tumor characteristics reported in this study included:
- Slight female predominance (51%)
- Mean age, 6.3 years
- 42% of STS patients were younger than 5 years (n = 37)
- 46% of bone sarcoma patients were 10 to 15 years old (n = 21)
- Head and neck were the most common sites for STS
- Extremities were the most common sites for bone sarcomas
- Most patients presented with large tumors (>5 cm for STS and >8 cm for bone sarcomas).
The investigators believe these findings and others they reported will help them adapt treatment protocols used in Europe and America so that they will be most appropriate for their patients.
SOURCE: Siwillis EM, Dharse NJ, Scanlan T, et al. Pediatric soft tissue and bone sarcomas in Tanzania: Epidemiology and clinical features. J Glob Oncol. 2019;5:1-6.
PEDIATRIC OSTEOSARCOMA IN LEBANON: Investigators at a single institution in Lebanon reported a similar survival rate for newly diagnosed patients with pediatric osteosarcoma treated at their center as for those treated in more developed countries. In a retrospective review of the medical records of 38 patients treated at the American University of Beirut Medical Center between August 2001 and May 2012, they determined the 5-year overall survival (OS) for all patients to be 74% and the event-free survival (EFS), 62%. Patients with localized disease had a 5-year OS of 81% and an EFS of 68%. Patients with metastatic disease had OS and EFS rates of about 42%.
All patients with localized disease received chemotherapy according to the Pediatric Oncology Group 9351 protocol, which consisted of cisplatin, doxorubicin, and methotrexate. If patients had metastatic disease or tumor necrosis less than 90%, they also received ifosfamide and etoposide.
Patients were a mean age of 12.9 years at diagnosis and there were an equal number of male and female patients. Most patients (n=34) had a primary tumor site affecting the long bones around the knee.
Six patients had metastatic disease to the lungs, and 3 patients had multifocal bone disease with lung metastases.
Thirty-three patients (86.8%) underwent surgical resection after 2 courses of induction chemotherapy. Twenty-two (66.7%) of these patients had a delay in local tumor control of more than 4 weeks. And 12 patients (31.5%) had tumor necrosis of less than 90%.
The investigators analyzed the prognostic importance of age, sex, metastatic disease, tumor site, delay in local control, and degree of tumor necrosis. Bivariate analysis revealed that only the degree of tumor necrosis was a statistically significant adverse prognostic factor for EFS (P=.001) and OS (P=.002).
SOURCE: Abou Ali B, Salman M, Ghanem KM, et al. Clinical prognostic factors and outcome in pediatric osteosarcoma: Effect of delay in local control and degree of necrosis in a multidisciplinary setting in Lebanon. J Glob Oncol. 2019;5:1-8.
Wolf in sheep’s clothing: metatarsal osteosarcoma
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
Case Presentation and Summary
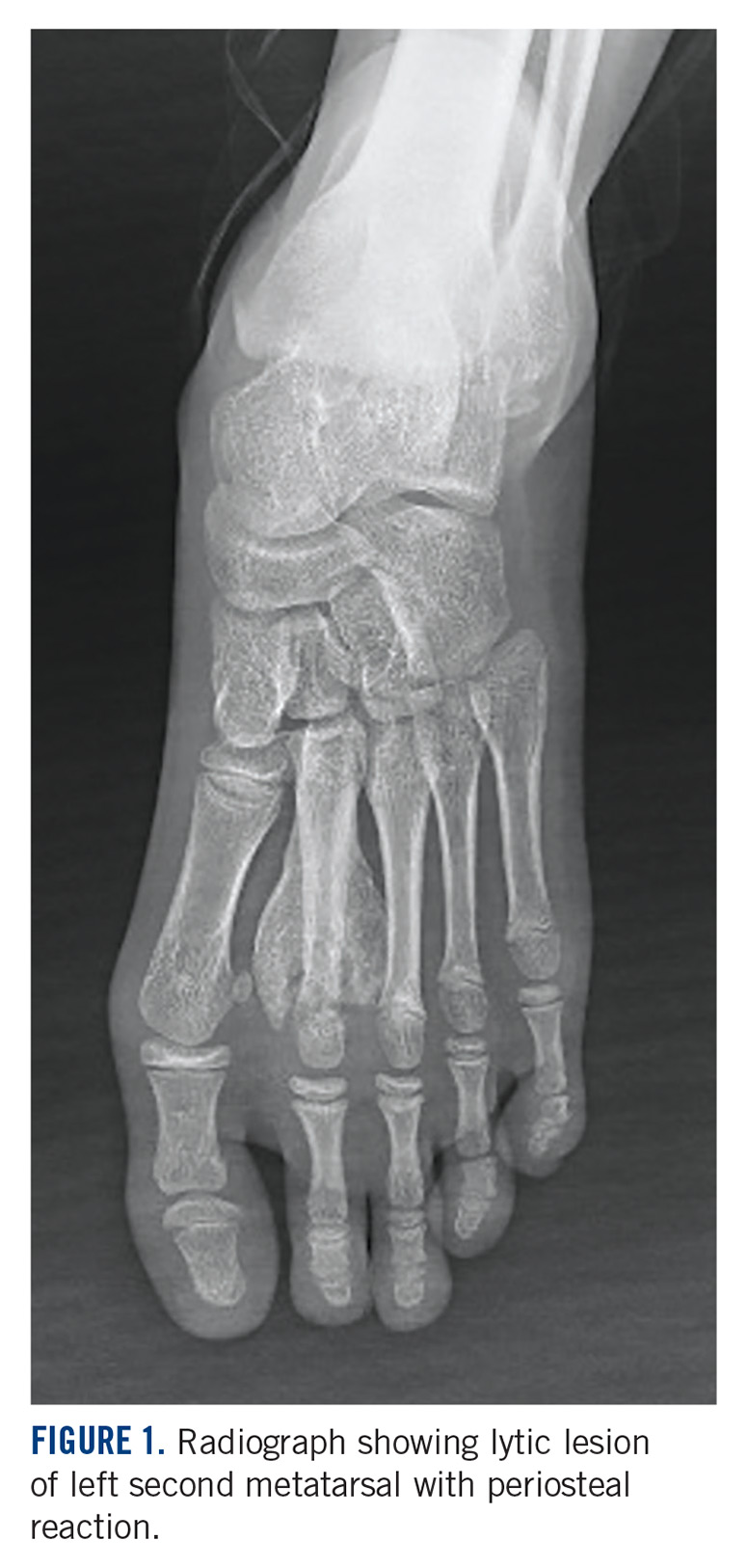
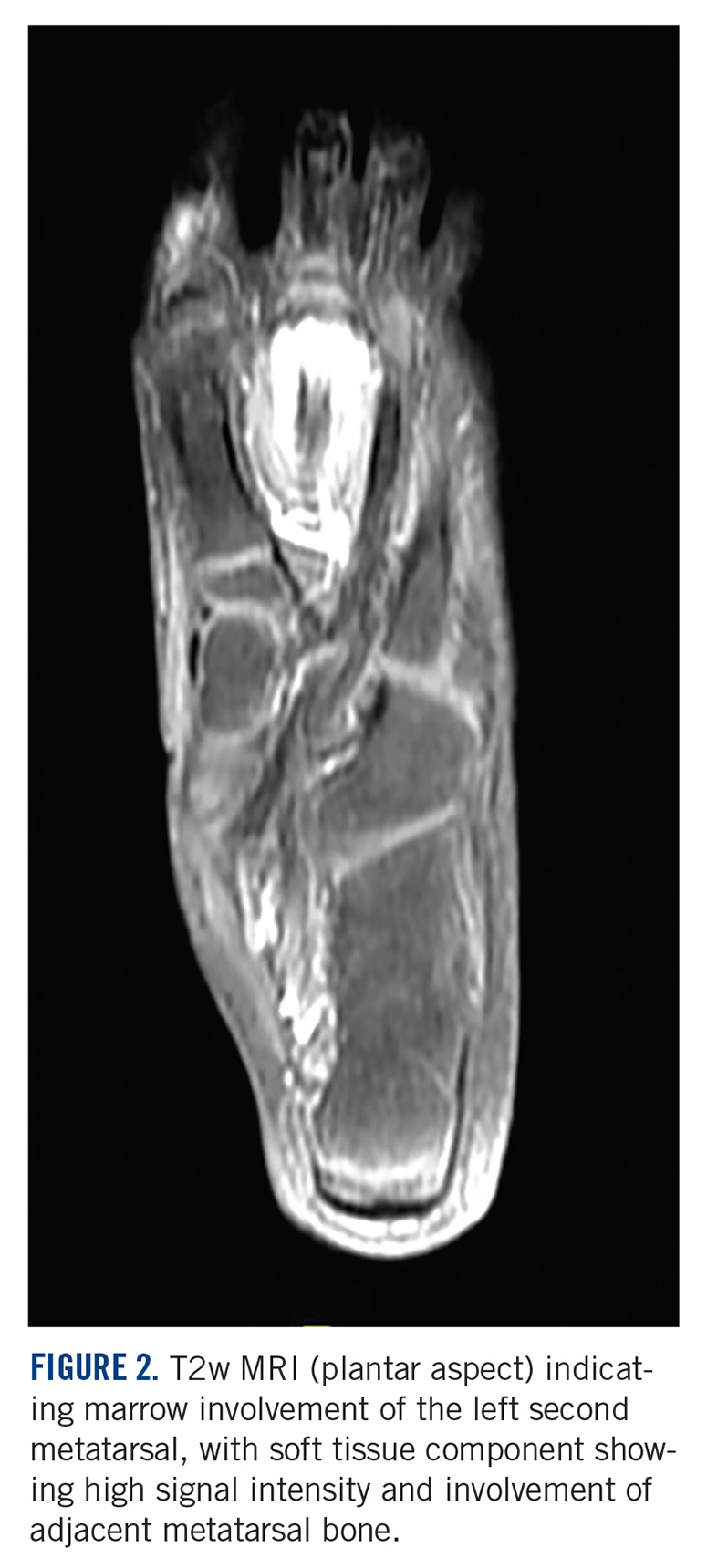
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
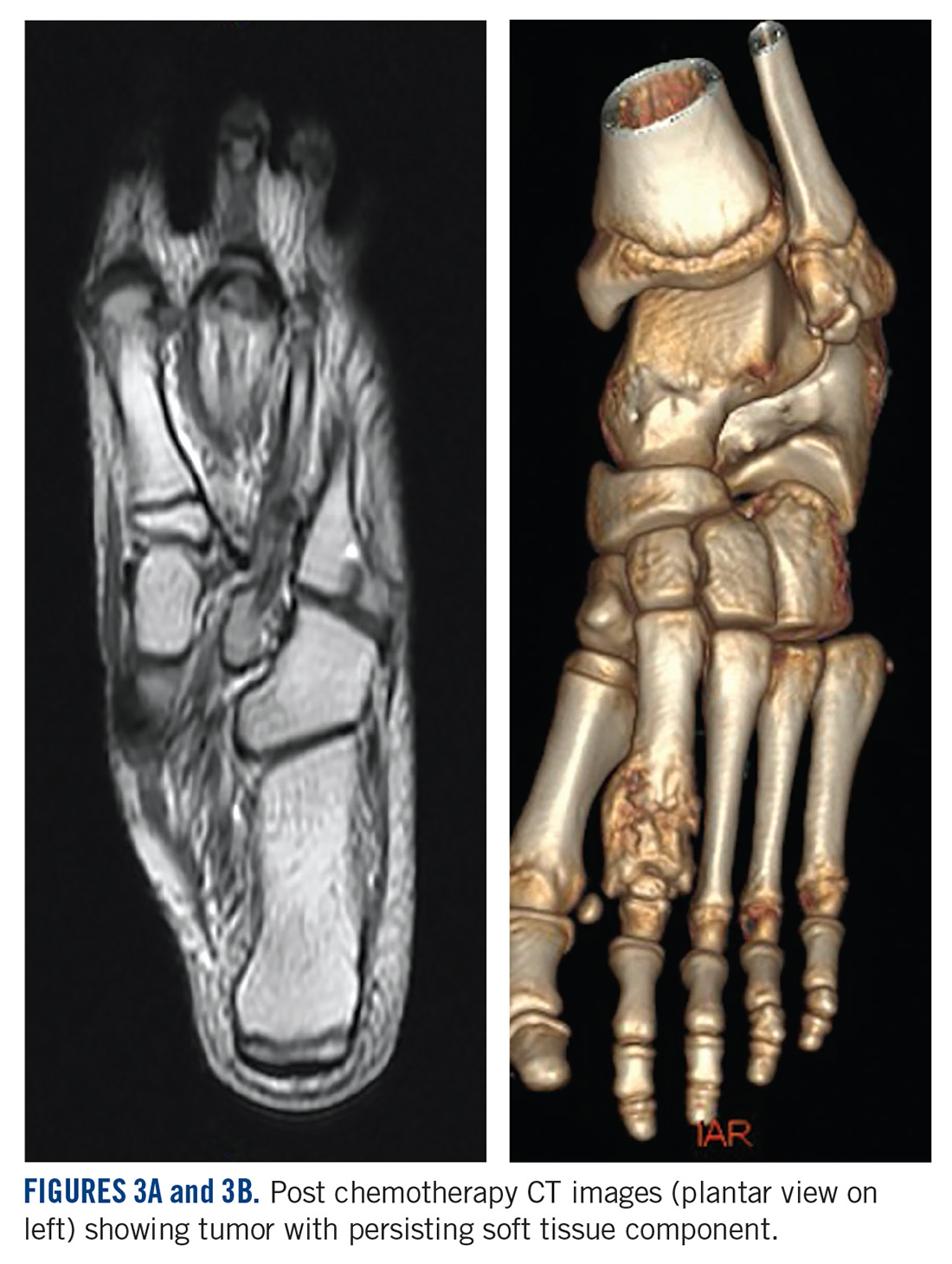
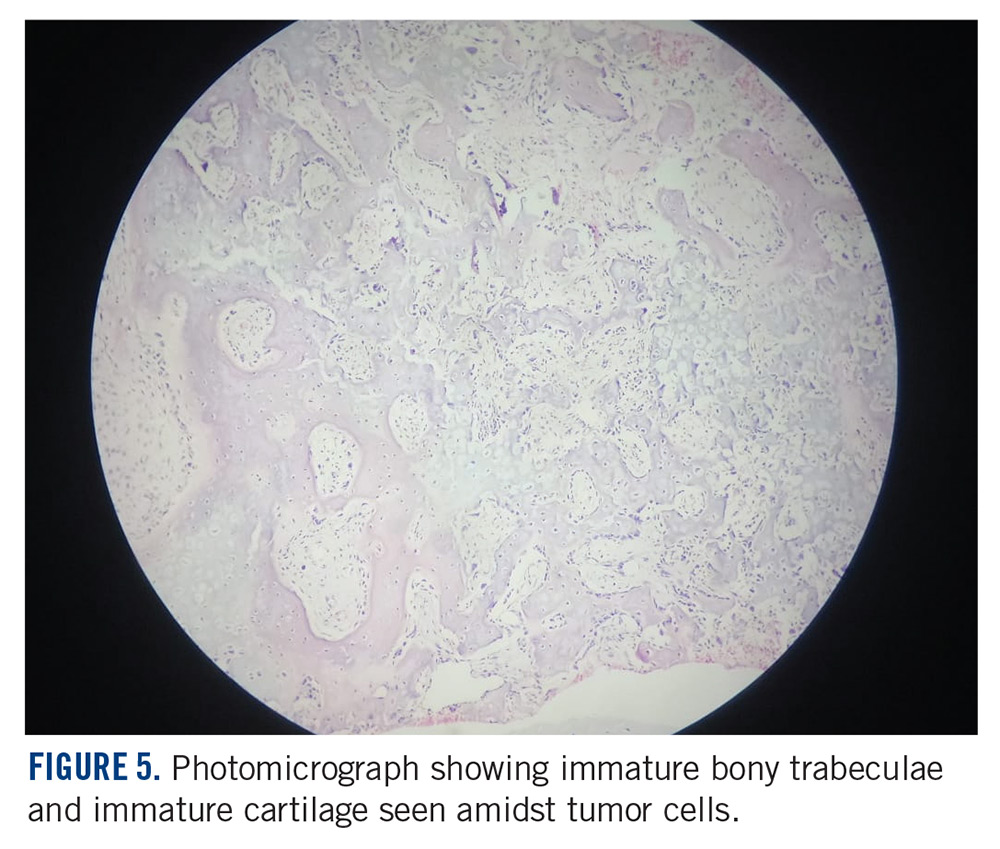
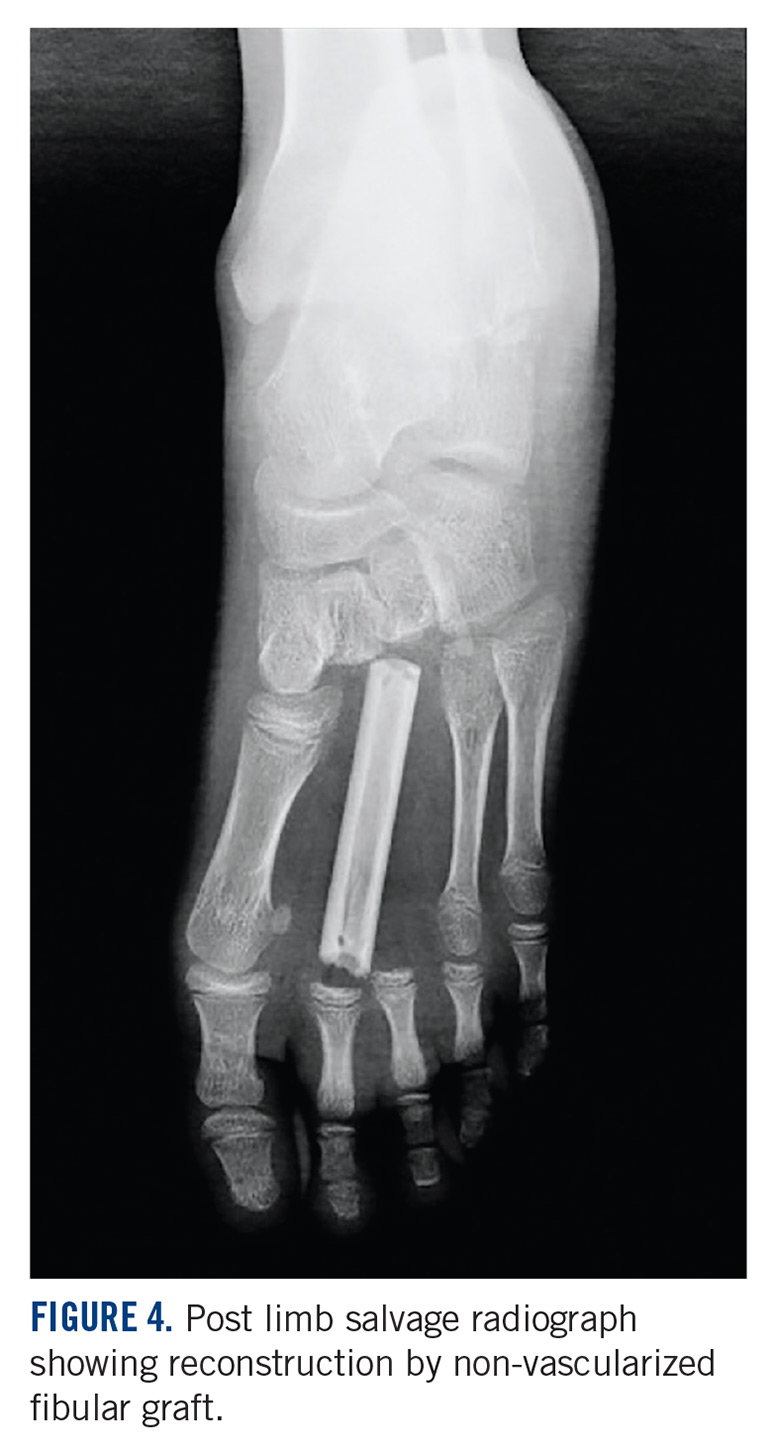
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.

A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
Case Presentation and Summary
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.
A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
Metatarsal bones are an unusual subsite for small bone involvement in osteosarcomas. This subgroup is often misdiagnosed and hence associated with significant treatment delays. The standard treatment of metatarsal osteosarcomas remains the same as for those treated at other sites, namely neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy. Limb salvage surgery or metatarsectomy in the foot is often a challenge owing to the poor compartmentalization of the disease. We hereby describe the case of a young girl with a metatarsal osteosarcoma who was managed with neoadjuvant chemotherapy and limb salvage surgery.
Introduction
Osteosarcomas are the most common primary malignant bone tumor in children and adolescents. Although predominantly occurring in pediatric and adolescent age groups, bimodal distribution (with a second incidence peak occurring in the sixth and seventh decades) is not uncommon.1 Osteosarcomas of the foot and small bones represent a rare and distinct clinical entity. This must have been a well-known observation for years that led to Watson-Jones stating, “Sarcoma of this [metatarsal] bone has not yet been reported in thousands of years in any country.”2 The incidence of osteosarcomas of the foot is estimated to be from 0.2% to 2%.3
These tumors, owing to their rarity, often lead to diagnostic dilemmas and hence treatment delays.4 They are usually mistaken for inflammatory conditions and often treated with—but not limited to—curettages and drainage procedures.5 The following case of osteosarcoma of the metatarsal bone in a young girl highlights the importance of having a high index of clinical suspicion prior to treatment.
Case Presentation and Summary
A 10-year-old girl visited our outpatient clinic with a painful progressive swelling on the dorsum of the left foot of 2 months’ duration. There was no history of antecedent trauma or fever. Physical examination revealed a bony hard swelling measuring around 5 x 6 cm on the dorsum of the left foot around the region of the second metatarsal. There was no regional lymphadenopathy or distal neurovascular deficit. She was evaluated with a plain radiograph that demonstrated a lytic lesion in the left second metatarsal associated with cortical destruction and periosteal reaction (Figure 1). A subsequent magnetic resonance image (MRI) revealed a bony lesion destroying part of the left second metatarsal with cortical destruction and marrow involvement and affecting the soft tissue around the adjacent third metatarsal (Figure 2). Needle biopsy showed chondroblastic osteosarcoma. Computed tomography (CT) of the thorax and bone scan were both negative for distant metastases.
She received 3 cycles of a MAP (highdose methotrexate, doxorubicin, and cisplatin) regimen as neoadjuvant chemotherapy. Response assessment scans showed partial response (Figures 3A and B). We performed a wide excision of the second and third metatarsal with reconstruction using a segment of non-vascularized fibular graft as rigid fixation (Figure 4). The postoperative period was uneventful. She was able to begin partial weight bearing on the fourth postoperative day and her sutures were removed on the twelfth postoperative day. She received adjuvant chemotherapy following surgery. The final histopathology report showed residual disease with Huvos grade III response (>90% necrosis) with all margins negative for malignancy (Figure 5). At present, the child is disease-free at 5 months of treatment completion and is undergoing regular follow-up visits.
Discussion
Metatarsal involvement amongst smallbone osteosarcomas is uncommon.3 There are about 32 cases of osteosarcomas reported in the literature from 1940 to 2018 involving the metatarsal bones (Table 1). According to a review article from the Mayo Clinic, the most common bone of the foot involved is the calcaneum.6 While the incidence of osteosarcomas of the foot as a whole is around 0.2% to 2%,3 metatarsal involvement is documented in 0.5% of these patients.7 However, a recent study depicted metatarsal involvement in 33% of all osteosarcomas of the foot.8
Osteosarcomas at conventional sites tend to have a bimodal age distribution with respect to disease affliction.9 Metatarsal osteosarcomas, however, are more common in an older age group.4,10 Our patient is probably the second youngest reported case of metatarsal osteosarcoma in the literature.11
Biscaglia et al propounded that osteosarcomas of the metatarsal were a distinct subgroup due to the rarity of occurrence, anatomical location, and prognosis.4 This often led to misdiagnosis and subsequent inadequate or inappropriate surgery. In six out of the ten cases (60%) described in Table 1, an incorrect pretreatment diagnosis was made that led to treatment delay. None, except one patient, received neoadjuvant chemotherapy, which is currently the standard of care. The average duration from symptom onset to diagnosis was found to be 2 years.4 However, in our case, the duration of symptoms was approximately 2 months.
Surgery for metatarsal osteosarcomas can be challenging, as the compartments of the foot are narrow spaces with poor demarcation. Limb salvage surgery in the form of metatarsectomy needs proper preoperative planning and execution. Neoadjuvant chemotherapy will serve to downstage the tumor within the fascial barriers of the metatarsal compartment.It has also been postulated that osteosarcoma of the foot may have a better prognosis and survival compared to other osteosarcoma subsites.10 This can be extrapolated from the fact that the majority are found to be low grade, and despite a long delay in treatment, there was no rapid increase in size and/or metastatic spread. However, tumor grade remains an important factor affecting survival— patients with higher grade tumors have worse survival.8
A number of differentials, including benign tumors, are to be kept in mind when diagnosing and treating such patients (Table 2). The most common benign tumors affecting the metatarsal are giant cell tumors (GCT) followed by chondromyxoid fibroma. Osteosarcomas and Ewing sarcomas constitute the malignant tumors.12 Occasionally, infections like osteomyelitis of the small bones may mimic malignancy. The absence of an extensive soft tissue component and/or calcifications with the presence of bony changes (like sequestrum) favors a diagnosis of infection/osteomyelitis. In addition, clinical findings like fever, skin redness, and presence of a painful swelling (especially after onset of fever) point to an inflammatory pathology rather than malignancy. Stress fractures rarely simulate tumors. MRI showing marrow and soft tissue edema with a visible fracture line points to the diagnosis.
A plane radiograph showing cortical bone destruction with a soft tissue component and calcification should be considered suspicious and must be thoroughly evaluated prior to surgical treatment.13 In a young patient such as ours, the important differentials that need to be considered include Ewing sarcoma, chronic osteomyelitis, and eosinophilic granuloma, which can radiologically mimic osteosarcoma at this location.
Conclusions
Osteosarcoma of the metatarsal is rare. Our case remains unique as it reports the second youngest patient in the literature. Erroneous or delayed diagnosis resulting in inadequate tumor excision and limb loss (amputation) often occurs in a majority of the cases. Proper pretreatment radiological imaging becomes imperative, and when clinical suspicion is high, a needle biopsy must follow in those cases. Early diagnosis with administration of neoadjuvant chemotherapy may allow us to perform limb salvage surgery or wide excision in these cases.
Acknowledgement
We would like to thank Dr. Sithara Aravind, Associate Professor, Department of Pathology, Malabar Cancer Center, for the photomicrographs.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
1. Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
2. Watson-Jones R. Fractures and Joint Injuries. Vol. I, 4th ed. Edinburgh and London: E & S Livingstone Ltd.1960:347.
3. Wu KK. Osteogenic sarcoma of the tarsal navicular bone. J Foot Surg. 1989;28(4):363-369.
4. Biscaglia R, Gasbarrini A, Böhling T, Bacchini P, Bertoni F, Picci P. Osteosarcoma of the bones of the foot: an easily misdiagnosed malignant tumour. Mayo Clin Proc. 1998;73(9):842-847.
5. Kundu ZS, Gupta V, Sangwan SS, Rana P. Curettage of benign bone tumors and tumor like lesions: A retrospective analysis. Indian J Orthop. 2013;47(3):295-301.
6. Choong PFM, Qureshil AA, Sim FH, Unni KK. Osteosarcoma of the foot. A review of 52 patients at the Mayo Clinic. Acta Orthop Scand. 1999;70(4):361-364.
7. Sneppen O, Dissing I, Heerfordt J, Schiödt T. Osteosarcoma of the metatarsal bones: Review of the literature and report of a case. Acta Orthop Scand. 1978;49(2):220-223.
8. Anninga JK, Picci P, Fiocco M, et al. Osteosarcoma of the hands and feet: a distinct clinico-pathological subgroup. Virchows Arch. 2013;462(1):109-120.
9. Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology and End Results Program. Cancer.
2009;115(7):1531-1543.
10. Wang CW, Chen CY, Yang RS. Talar osteosarcoma treated with limb sparing surgery. J Bone Joint Surg Am. 2011;93:e22.
11. Aycan OE, Vanel D, Righi A, Arikan Y, Manfrini M. Chondroblastoma-like osteosarcoma:
a case report and review. Skeletal Radiol. 2015;44(6):869-873.
12. Jarkiewicz-Kochman E, Gołebiowski M, Swiatkowski J, Pacholec E, Rajewski R. Tumours of the metatarsus. Ortop Traumatol Rehabil. 2007;9(3):319-330.
13. Schatz J, Soper J, McCormack S, Healy M, Deady L, Brown W. Imaging of tumours in the ankle and foot. Top Magn Reson Imaging. 2010;21(1):37-50.
14. Fukuda K, Ushigome S, Nikaidou T, Asanuma K, Masui F. Osteosarcoma of the metatarsal. Skeletal Radiol. 1999;28(5):294-297.
15. Parsa R, Marcus M, Orlando R, Parsa C. Low-grade central osteosarcoma of the second metatarsal in a 72 year old male. Internet J Orthop Surg. 2013;21(2): 1-8.
16. Lee EY, Seeger LL, Nelson SD, Eckardt JJ. Primary osteosarcoma of a metatarsal bone. Skeletal Radiol. 2000;29(8):474-476.
17. Padhy D, Madhuri V, Pulimood SA, Danda S, Walter NM, Wang LL. Metatarsal osteosarcoma in Rothmund-Thomson syndrome: a case report. J Bone Joint
Surg Am. 2010;92(3):726-730.
18. Mohammadi A, Porghasem J, Noroozinia F, Ilkhanizadeh B, Ghasemi-Rad M, Khenari S. Periosteal osteosarcoma of the fifth metatarsal: A rare pedal tumor. J Foot Ankle Surg. 2011;50(5):620-622.
19. Nishio J, Iwasaki H, Takagi S, et al. Low-grade central osteosarcoma of the metatarsal bone: A clinicopathological, immunohistochemical, cytogenetic and molecular cytogenetic analysis. Anticancer Res. 2012;32(12):5429-5435.
An unusual presentation of low-grade clavicle osteosarcoma: a case report and literature review
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
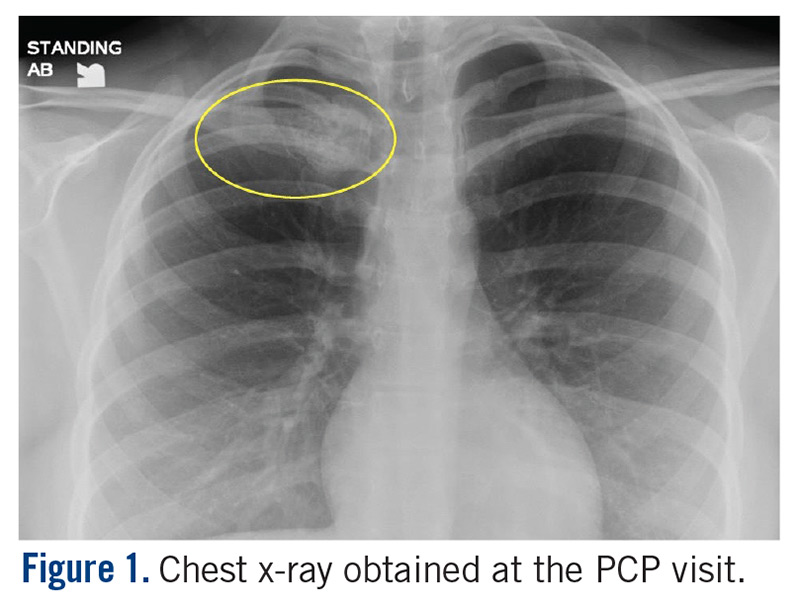
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
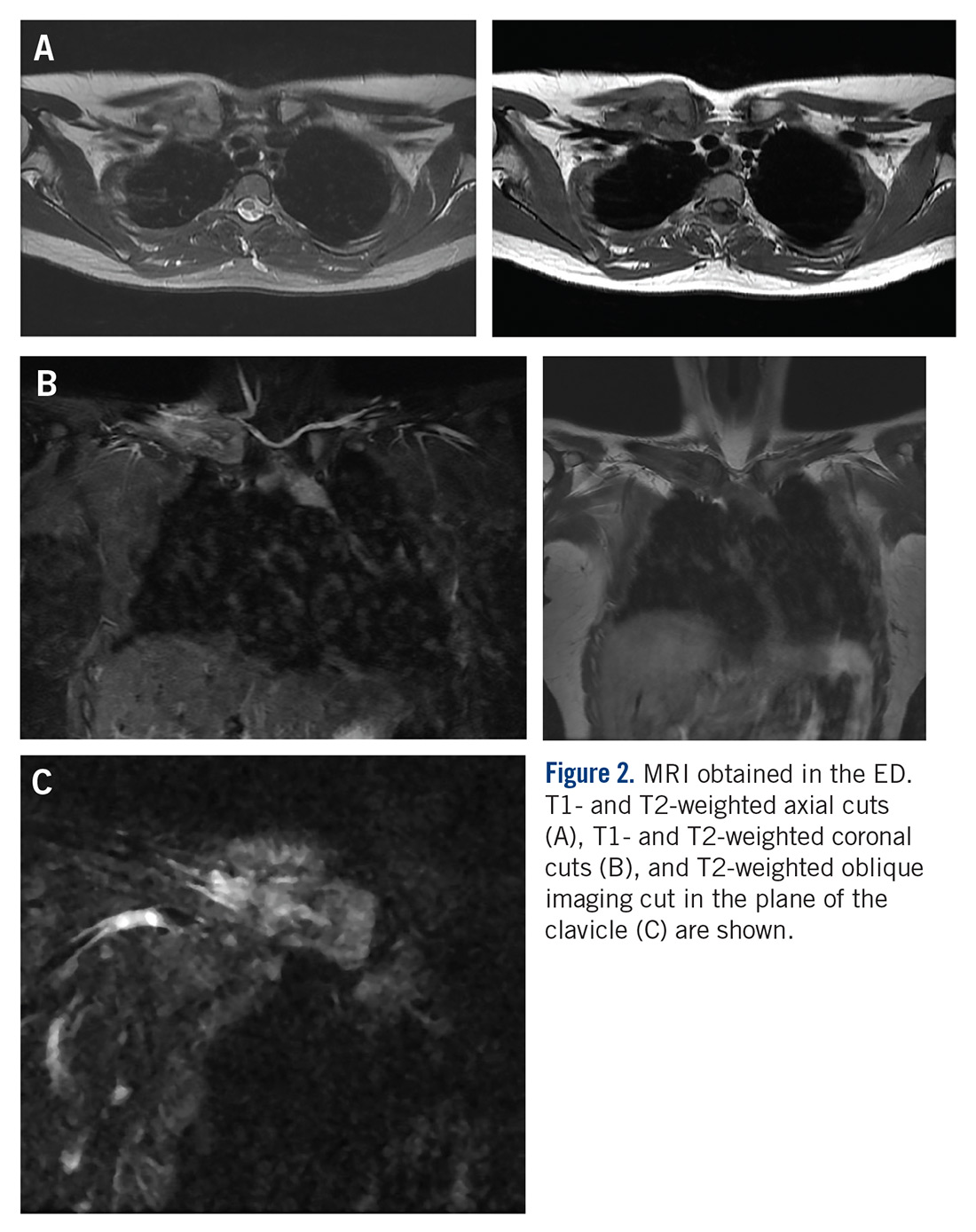
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
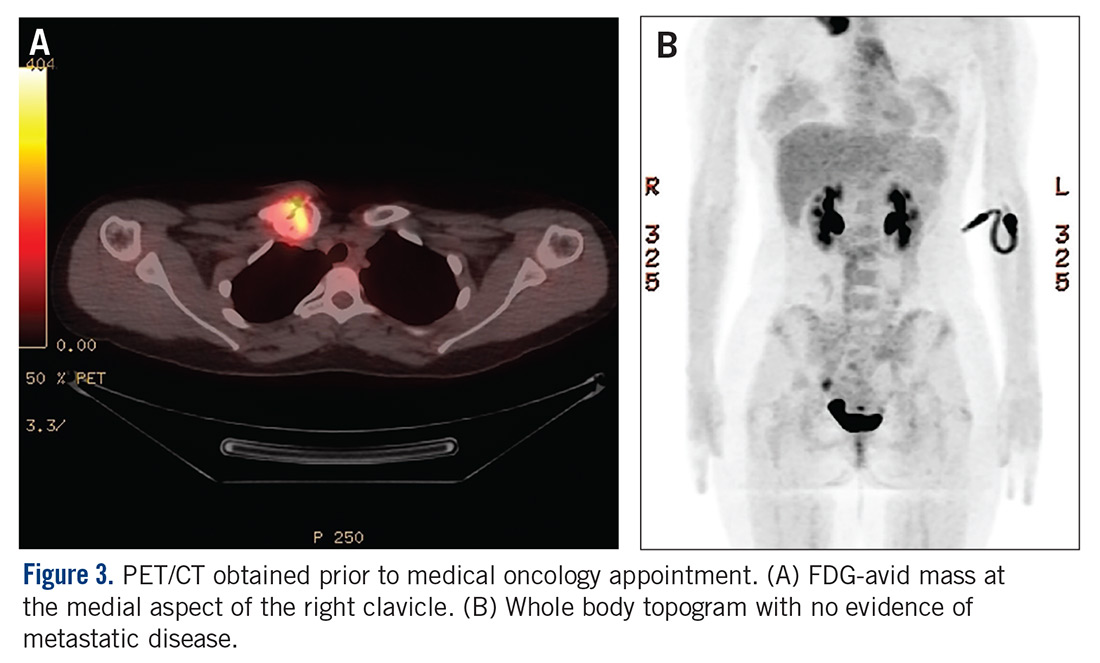
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).

Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
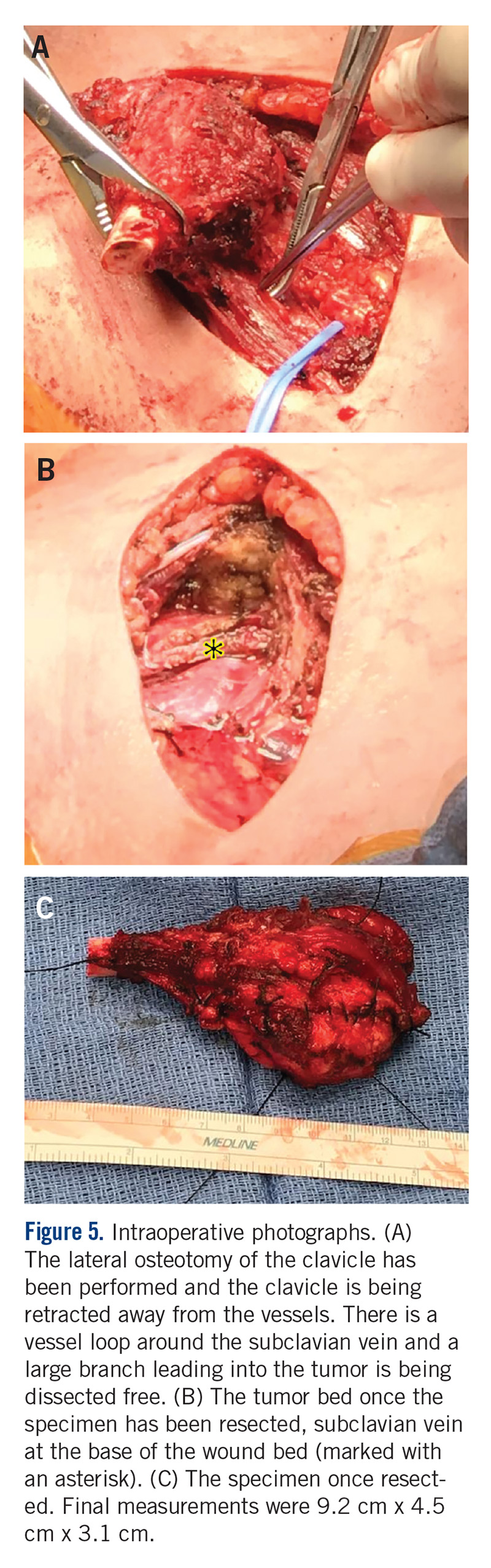
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).
Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
Osteosarcoma (OS) is a rare disease with approximately 800- 900 newly diagnosed cases each year in the United States. Of those, the majority occur about the knee. The distal femur is the most common site, followed by the proximal tibia, with the proximal humerus being a distant third. OS of the clavicle has been reported, with the earliest case report dating from 1975.1 Since then, additional case reports of high-grade OS of the clavicle have been published.2,3 We describe the case of a 16-year-old female who presented with a mass on her right medial clavicle, which was confirmed to be a low-grade central OS.
Case Presentation
The patient is a 16-year-old female who presented to the Emergency Department (ED) for evaluation of a mass on her right clavicle, after being evaluated by her primary care physician (PCP). She noted an enlarging mass over the previous 2 months but stated that it had been asymptomatic until 4 days prior to presentation to her PCP, at which time she had developed tenderness to palpation and pain with range of motion of the right arm. X-rays were obtained at the PCP’s office and she was referred to the ED for further evaluation. She denied constitutional symptoms.
At the ED visit, she was noted to have an area of erythema and tenderness over the medial aspect of the right clavicle with increased bony prominence. A chest x-ray demonstrated medial clavicle enlargement with periosteal reaction and sclerosis (Figure 1).
MRI demonstrated a 6-cm x 3.8-cm x 4.1-cm mass arising from the right medial clavicle with cortical destruction and concomitant displacement of the right subclavian and brachiocephalic veins (Figure 2). A CT-guided biopsy was performed 1 week later and demonstrated low-grade OS. The pathologist was concerned about the possibility of sampling error and the presence of a higher-grade component, as low-grade OS of the clavicle had not been reported.
The patient was evaluated by a pediatric hematologist/oncologist 2 weeks later after having obtained the biopsy and a PET/CT scan. At that time, the PET/CT showed an FDG-avid mass at the clavicle without evidence of pulmonary metastatic disease (Figure 3). She was subsequently evaluated by orthopedic oncology, at which time a discussion was had regarding further treatment. There was essentially no literature to guide the surgical and medical teams, as low-grade clavicular OS is unknown. Based on the evidence of localized, low-grade disease, the determination was made to proceedwith surgical resection. In the event that high-grade disease was identified at the time of final pathological evaluation, the pediatric hematology/oncology team felt that administering all of the patient’s chemotherapy postoperatively would be acceptable and not affect her long-term prognosis. CT and CT angiogram were obtained for further operative planning (Figure 4).
Given the intimacy of the mass to the subclavian vessels, she was also seen preoperatively by pediatric general and cardiothoracic surgeons. The plan was formulated to have them in the operating room for mobilization of the subclavian vessels and in the event that a sternotomy was required for proximal control of the vessels. Following this visit, the case was discussed at the multidisciplinary pediatric tumor board and the consensus was to proceed with surgical resection.
Surgical Technique
General endotracheal anesthesia was administered without complication. The patient was positioned supine with a soft bump under her shoulders to place her neck in slight extension and thus facilitate access to the clavicle and great vessels. A 14-cm oblique incision was made over the subcutaneous clavicle extending to the contralateral sternoclavicular joint. Dissection was carried down to the fascia and the biopsy site was excised with the skin paddle. Dissection was carried through the sternocleidomastoid superiorly and the pectoralis major inferiorly, to 8 cm lateral from the right sternoclavicular joint. The clavicle was osteotomized well lateral of the palpable tumor and a marrow margin was sent for frozen section, which was found to be negative.
Dissection was continued circumferentially. Assistance from pediatric general and cardiothoracic surgery was required at the inferior aspect of the mass to assist with exposure and control of the subclavian vein (Figure 5A). A large branch of the subclavian vein near its junction with the internal jugular vein was found to be involved with the tumor and thus required suture ligation. The subclavian vein was noted to be intimate with the mass and somewhat friable. With the vein mobilized, a cuff of normal tissue was obtained inferiorly and superiorly to the mass. Medially, the sternoclavicular joint was disarticulated (Figure 5B). At this point, the specimen was delivered from the operative field and tagged in the usual fashion (Figure 5C). A medial soft tissue margin from the sternal side of the sternoclavicular joint was also sent and found to be negative for tumor. The wound was closed in layered fashion over a ¼” Penrose drain. A soft dressing was placed, and the patient was successfully extubated and transferred to the post-anesthesia care unit in stable condition.
Postoperative Course
The patient was found to be neurologically and vascularly intact on postoperative exam and was discharged on postoperative day 1.
She was seen 14 days postoperatively and was doing well at that time, with full range of motion of the shoulder, elbow, wrist, and hand. Final pathology confirmed a low-grade OS with extraosseous extension. All margins were negative except the medial (sternoclavicular joint) margin and the inferior margin adjacent to the subclavian vein. The intraoperative frozen section from the medial margin was negative for tumor.
The pediatric hematology/oncology team determined that, as no high-grade areas were identified, chemotherapy should be deferred. The positive margins were also discussed with the patient and her family specifically regarding further possible treatments. The findings from the pathology were discussed in a multidisciplinary tumor board and it was felt that, given the low-grade nature of the lesion as well as the high morbidity and risk of mortality with further surgery, additional surgery would be potentially more harmful than helpful. Additionally, low-grade OS is extremely resistant to radiotherapy. The plan remains to monitor her for local recurrence as well as metastases with serial imaging.
Discussion
The clavicle is one of the first bones in the body to ossify but one of the last to have final physeal closure. Its unique characteristics have led to various descriptions, such as a “short tubular bone” versus a “flat bone.”4,5 Of note are its paucity of a true intramedullary space and scanty red marrow, which make it an unlikely site for a primarily intramedullary- based neoplasm to arise.4 However, it has also been noted that malignant lesions are more common in the clavicle than benign lesions, and special attention should be paid to aggressiveappearing lesions in the clavicle.
Radiographs can be misleading as well. Prior studies have demonstrated that low-grade central OS can be readily misdiagnosed as fibrous dysplasia, desmoplastic fibroma, nonossifying fibroma, osteoblastoma, and aneurysmal bone cyst.6 Findings found in low-grade OS can include evidence of cortical interruption, local soft tissue mass development, intramedullary involvement, cortical destruction, and poor margination; however, low-grade OS is typically sclerotic and highly trabeculated. Cross-sectional imaging can help differentiate between OS and other more benign pathologies and should be considered in the clavicle where biopsy may be perilous.5
The difficulty of clavicular biopsy has been reported. Not only does clavicular anatomy make biopsy hazardous, but also the potential for sampling error does exist. In a case report of one patient with a highgrade lesion, fine needle aspiration biopsy was initially diagnosed as an aneurysmal bone cyst but was ultimately found to be osteosarcoma.2 Histology of low-grade lesions usually demonstrates minimal cytological atypia, rare mitotic activity, and variable osteoid production.5 Lower mitotic indices typically make wide resection curative for these patients, without the need for chemotherapy.
In this case, wide resection was carried out with the subclavian vein as the posterior-inferior margin and the sternoclavicular joint as the medial margin. Though the intra-operative medial margin was clear of disease, final pathology demonstrated focal (microscopic) involvement of the posterior and medial margins. A study of soft tissue sarcoma evaluated positive margins and concluded that the imperative of preservation of vital structures supersedes the need for negative margins.7,8 The rate of metastasis and overall survival was similar to surgical resections with positive margins. In the case of our patient, further resection would have carried significant morbidity and possibly mortality, including sacrifice of the major vessels to the arm below and entering into the sternum and thoracic cavity. The likely disability as well as the hazards of surgery were deemed to be too great to justify further excision. Frequent cross-sectional imaging will be necessary to evaluate the presence of recurrent or metastatic disease. To our knowledge, this is the first documented case of low-grade clavicle OS. This report demonstrates the need for multidisciplinary sarcoma care at a center of excellence, particularly in instances of unusual diagnoses.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
1. Zinghi G. Osteosarcoma of the clavicle (description of a case) [in Italian]. Chir Organi Mov. 1975;62(6):671-674.
2. Cundy WJ, Carter C, Dhatrak D, Clayer M. Primary osteosarcoma of the clavicle and the perils of bone biopsy. BMJ Case Rep. 2015;2015:bcr2014208859.
3. Greenspan A, Unni KK, Mann J. Case report 804: Chondroblastic osteosarcoma grade 3 of the left clavicle. Skeletal Radiol. 1993;22(6):469-471.
4. Rossi B, Fabbriciani C, Chalidis BE, Visci F, Maccauro G. Primary malignant clavicular tumours: a clinicopathological analysis of six cases and evaluation of surgical management. Arch Orthop Trauma Surg. 2011;131(7):935-939.
5. Andresen KJ, Sundaram M, Unni KK, Sim FH. Imaging features of low-grade central osteosarcoma of the long bones and pelvis. Skeletal Radiol. 2004;33(7):373-379.
6. Malhas AM, Sumathi VP, James SL, et al. Low-grade central osteosarcoma: A difficult condition to diagnose. Sarcoma. 2012; 2012:764796.
7. O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866-2875.
8. Kawaguchi N, Ahmed AR, Matsumoto S, Manabe J, Matsushita Y. The concept of curative margin in surgery for bone and soft tissue sarcoma. Clin Orthop Relat Res. 2004;419:165-172.
Trial matches pediatric cancer patients to targeted therapies
Researchers have found they can screen pediatric cancer patients for genetic alterations and match those patients to appropriate targeted therapies.
Thus far, 24% of the patients screened have been matched and assigned to a treatment, and 10% have been enrolled on treatment protocols.
The patients were screened and matched as part of the National Cancer Institute–Children’s Oncology Group Pediatric MATCH (Molecular Analysis for Therapy Choice) trial.
Results from this trial are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Donald Williams Parsons, MD, PhD, of Baylor College of Medicine in Houston, Tex., presented some results at a press briefing in advance of the meeting. “[T]he last 10 years have been an incredible time in terms of learning more about the genetics and underlying molecular basis of both adult and pediatric cancers,” Dr. Parsons said.
He pointed out, however, that it is not yet known if this information will be useful in guiding the treatment of pediatric cancers. Specifically, how many pediatric patients can be matched to targeted therapies, and how effective will those therapies be?
The Pediatric MATCH trial (NCT03155620) was developed to answer these questions. Researchers plan to enroll at least 1,000 patients in this trial. Patients are eligible if they are 1-21 years of age and have refractory or recurrent solid tumors, non-Hodgkin lymphomas, or histiocytic disorders.
After patients are enrolled in the trial, their tumor samples undergo DNA and RNA sequencing, and the results are used to match each patient to a targeted therapy. At present, the trial can match patients to one of 10 drugs:
- larotrectinib (targeting NTRK fusions).
- erdafitinib (targeting FGFR1/2/3/4).
- tazemetostat (targeting EZH2 or members of the SWI/SNF complex).
- LY3023414 (targeting the PI3K/MTOR pathway).
- selumetinib (targeting the MAPK pathway).
- ensartinib (targeting ALK or ROS1).
- vemurafenib (targeting BRAF V600 mutations).
- olaparib (targeting defects in DNA damage repair).
- palbociclib (targeting alterations in cell cycle genes).
- ulixertinib (targeting MAPK pathway mutations).
Early results
From July 2017 through December 2018, 422 patients were enrolled in the trial. The patients had more than 60 different diagnoses, including brain tumors, sarcomas, neuroblastoma, renal and liver cancers, and other malignancies.
The researchers received tumor samples from 390 patients, attempted sequencing of 370 samples (95%), and completed sequencing of 357 samples (92%).
A treatment target was found in 112 (29%) patients, 95 (24%) of those patients were assigned to a treatment, and 39 (10%) were enrolled in a protocol. The median turnaround time from sample receipt to treatment assignment was 15 days.
“In addition to the sequencing being successful, the patients are being matched to the different treatments,” Dr. Parsons said. He added that the study is ongoing, so more of the matched and assigned patients will be enrolled in protocols in the future.
Dr. Parsons also presented results by tumor type. A targetable alteration was identified in 26% (67/255) of all non–central nervous system solid tumors, 13% (10/75) of osteosarcomas, 50% (18/36) of rhabdomyosarcomas, 21% (7/33) of Ewing sarcomas, 25% (9/36) of other sarcomas, 19% (5/26) of renal cancers, 16% (3/19) of carcinomas, 44% (8/18) of neuroblastomas, 43% (3/7) of liver cancers, and 29% (4/14) of “other” tumors.
Drilling down further, Dr. Parsons presented details on specific alterations in one cancer type: astrocytomas. Targetable alterations were found in 74% (29/39) of astrocytomas. This includes NF1 mutations (18%), BRAF V600E (15%), FGFR1 fusions/mutations (10%), BRAF fusions (10%), PIK3CA mutations (8%), NRAS/KRAS mutations (5%), and other alterations.
“Pretty remarkably, in this one diagnosis, there are patients who have been matched to nine of the ten different treatment arms,” Dr. Parsons said. “This study is allowing us to evaluate targeted therapies – specific types of investigational drugs – in patients with many different cancer types, some common, some very rare. So, hopefully, we can study these agents and identify signals of activity where some of these drugs may work for our patients.”
The Pediatric MATCH trial is sponsored by the National Cancer Institute. Dr. Parsons has patents, royalties, and other intellectual property related to genes discovered through sequencing of several adult cancer types.
SOURCE: Parsons DW et al. ASCO 2019, Abstract 10011.
Researchers have found they can screen pediatric cancer patients for genetic alterations and match those patients to appropriate targeted therapies.
Thus far, 24% of the patients screened have been matched and assigned to a treatment, and 10% have been enrolled on treatment protocols.
The patients were screened and matched as part of the National Cancer Institute–Children’s Oncology Group Pediatric MATCH (Molecular Analysis for Therapy Choice) trial.
Results from this trial are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Donald Williams Parsons, MD, PhD, of Baylor College of Medicine in Houston, Tex., presented some results at a press briefing in advance of the meeting. “[T]he last 10 years have been an incredible time in terms of learning more about the genetics and underlying molecular basis of both adult and pediatric cancers,” Dr. Parsons said.
He pointed out, however, that it is not yet known if this information will be useful in guiding the treatment of pediatric cancers. Specifically, how many pediatric patients can be matched to targeted therapies, and how effective will those therapies be?
The Pediatric MATCH trial (NCT03155620) was developed to answer these questions. Researchers plan to enroll at least 1,000 patients in this trial. Patients are eligible if they are 1-21 years of age and have refractory or recurrent solid tumors, non-Hodgkin lymphomas, or histiocytic disorders.
After patients are enrolled in the trial, their tumor samples undergo DNA and RNA sequencing, and the results are used to match each patient to a targeted therapy. At present, the trial can match patients to one of 10 drugs:
- larotrectinib (targeting NTRK fusions).
- erdafitinib (targeting FGFR1/2/3/4).
- tazemetostat (targeting EZH2 or members of the SWI/SNF complex).
- LY3023414 (targeting the PI3K/MTOR pathway).
- selumetinib (targeting the MAPK pathway).
- ensartinib (targeting ALK or ROS1).
- vemurafenib (targeting BRAF V600 mutations).
- olaparib (targeting defects in DNA damage repair).
- palbociclib (targeting alterations in cell cycle genes).
- ulixertinib (targeting MAPK pathway mutations).
Early results
From July 2017 through December 2018, 422 patients were enrolled in the trial. The patients had more than 60 different diagnoses, including brain tumors, sarcomas, neuroblastoma, renal and liver cancers, and other malignancies.
The researchers received tumor samples from 390 patients, attempted sequencing of 370 samples (95%), and completed sequencing of 357 samples (92%).
A treatment target was found in 112 (29%) patients, 95 (24%) of those patients were assigned to a treatment, and 39 (10%) were enrolled in a protocol. The median turnaround time from sample receipt to treatment assignment was 15 days.
“In addition to the sequencing being successful, the patients are being matched to the different treatments,” Dr. Parsons said. He added that the study is ongoing, so more of the matched and assigned patients will be enrolled in protocols in the future.
Dr. Parsons also presented results by tumor type. A targetable alteration was identified in 26% (67/255) of all non–central nervous system solid tumors, 13% (10/75) of osteosarcomas, 50% (18/36) of rhabdomyosarcomas, 21% (7/33) of Ewing sarcomas, 25% (9/36) of other sarcomas, 19% (5/26) of renal cancers, 16% (3/19) of carcinomas, 44% (8/18) of neuroblastomas, 43% (3/7) of liver cancers, and 29% (4/14) of “other” tumors.
Drilling down further, Dr. Parsons presented details on specific alterations in one cancer type: astrocytomas. Targetable alterations were found in 74% (29/39) of astrocytomas. This includes NF1 mutations (18%), BRAF V600E (15%), FGFR1 fusions/mutations (10%), BRAF fusions (10%), PIK3CA mutations (8%), NRAS/KRAS mutations (5%), and other alterations.
“Pretty remarkably, in this one diagnosis, there are patients who have been matched to nine of the ten different treatment arms,” Dr. Parsons said. “This study is allowing us to evaluate targeted therapies – specific types of investigational drugs – in patients with many different cancer types, some common, some very rare. So, hopefully, we can study these agents and identify signals of activity where some of these drugs may work for our patients.”
The Pediatric MATCH trial is sponsored by the National Cancer Institute. Dr. Parsons has patents, royalties, and other intellectual property related to genes discovered through sequencing of several adult cancer types.
SOURCE: Parsons DW et al. ASCO 2019, Abstract 10011.
Researchers have found they can screen pediatric cancer patients for genetic alterations and match those patients to appropriate targeted therapies.
Thus far, 24% of the patients screened have been matched and assigned to a treatment, and 10% have been enrolled on treatment protocols.
The patients were screened and matched as part of the National Cancer Institute–Children’s Oncology Group Pediatric MATCH (Molecular Analysis for Therapy Choice) trial.
Results from this trial are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Donald Williams Parsons, MD, PhD, of Baylor College of Medicine in Houston, Tex., presented some results at a press briefing in advance of the meeting. “[T]he last 10 years have been an incredible time in terms of learning more about the genetics and underlying molecular basis of both adult and pediatric cancers,” Dr. Parsons said.
He pointed out, however, that it is not yet known if this information will be useful in guiding the treatment of pediatric cancers. Specifically, how many pediatric patients can be matched to targeted therapies, and how effective will those therapies be?
The Pediatric MATCH trial (NCT03155620) was developed to answer these questions. Researchers plan to enroll at least 1,000 patients in this trial. Patients are eligible if they are 1-21 years of age and have refractory or recurrent solid tumors, non-Hodgkin lymphomas, or histiocytic disorders.
After patients are enrolled in the trial, their tumor samples undergo DNA and RNA sequencing, and the results are used to match each patient to a targeted therapy. At present, the trial can match patients to one of 10 drugs:
- larotrectinib (targeting NTRK fusions).
- erdafitinib (targeting FGFR1/2/3/4).
- tazemetostat (targeting EZH2 or members of the SWI/SNF complex).
- LY3023414 (targeting the PI3K/MTOR pathway).
- selumetinib (targeting the MAPK pathway).
- ensartinib (targeting ALK or ROS1).
- vemurafenib (targeting BRAF V600 mutations).
- olaparib (targeting defects in DNA damage repair).
- palbociclib (targeting alterations in cell cycle genes).
- ulixertinib (targeting MAPK pathway mutations).
Early results
From July 2017 through December 2018, 422 patients were enrolled in the trial. The patients had more than 60 different diagnoses, including brain tumors, sarcomas, neuroblastoma, renal and liver cancers, and other malignancies.
The researchers received tumor samples from 390 patients, attempted sequencing of 370 samples (95%), and completed sequencing of 357 samples (92%).
A treatment target was found in 112 (29%) patients, 95 (24%) of those patients were assigned to a treatment, and 39 (10%) were enrolled in a protocol. The median turnaround time from sample receipt to treatment assignment was 15 days.
“In addition to the sequencing being successful, the patients are being matched to the different treatments,” Dr. Parsons said. He added that the study is ongoing, so more of the matched and assigned patients will be enrolled in protocols in the future.
Dr. Parsons also presented results by tumor type. A targetable alteration was identified in 26% (67/255) of all non–central nervous system solid tumors, 13% (10/75) of osteosarcomas, 50% (18/36) of rhabdomyosarcomas, 21% (7/33) of Ewing sarcomas, 25% (9/36) of other sarcomas, 19% (5/26) of renal cancers, 16% (3/19) of carcinomas, 44% (8/18) of neuroblastomas, 43% (3/7) of liver cancers, and 29% (4/14) of “other” tumors.
Drilling down further, Dr. Parsons presented details on specific alterations in one cancer type: astrocytomas. Targetable alterations were found in 74% (29/39) of astrocytomas. This includes NF1 mutations (18%), BRAF V600E (15%), FGFR1 fusions/mutations (10%), BRAF fusions (10%), PIK3CA mutations (8%), NRAS/KRAS mutations (5%), and other alterations.
“Pretty remarkably, in this one diagnosis, there are patients who have been matched to nine of the ten different treatment arms,” Dr. Parsons said. “This study is allowing us to evaluate targeted therapies – specific types of investigational drugs – in patients with many different cancer types, some common, some very rare. So, hopefully, we can study these agents and identify signals of activity where some of these drugs may work for our patients.”
The Pediatric MATCH trial is sponsored by the National Cancer Institute. Dr. Parsons has patents, royalties, and other intellectual property related to genes discovered through sequencing of several adult cancer types.
SOURCE: Parsons DW et al. ASCO 2019, Abstract 10011.
REPORTING FROM ASCO 2019
Entrectinib exhibits activity in children with solid tumors
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
Entrectinib demonstrated “very promising” antitumor activity in children and adolescents with recurrent or refractory solid tumors, according to an investigator involved in a phase 1/1b trial.
Twelve of 29 patients enrolled in the trial have responded to entrectinib. All responders had fusions in genes targeted by the drug – NTRK1/2/3 (TRKA/B/C), ROS1, or ALK – or an ALK mutation.
Details of this study are scheduled to be presented at the annual meeting of the American Society of Clinical Oncology.
Giles W. Robinson, MD, of St. Jude Children’s Research Hospital in Memphis, Tenn., discussed the study during a press briefing in advance of the meeting.
“Entrectinib is an oral and potent inhibitor of the TRKA/B/C, ROS1, and ALK proteins, but it also penetrates into the brain to reach tumors in the brain and spine, which can be a hard area to get drugs to,” Dr. Robinson explained.
“Promising clinical activity was initially seen in the adult solid tumor patients with target rearrangements, and it was encouraging to see these patients also had responses when the tumors were located in their brains. And what got us really excited as pediatric oncologists was that a variety of pediatric cancers harbor these fusions and mutations within certain tumors.”
With this in mind, Dr. Robinson and colleagues conducted a phase 1/1b study (NCT02650401) of entrectinib in 29 patients with recurrent or refractory solid tumors, including central nervous system (CNS) tumors.
The patients’ median age was 7 years (range, 0-20 years), and roughly half of them were male (n = 15). Patients were diagnosed with neuroblastoma (n = 16), high-grade glioma (n = 5), inflammatory myofibroblastic tumors (n = 3), infantile fibrosarcoma (n = 2), CNS embryonal tumor (n = 1), melanoma (n = 1), and synovial sarcoma (n = 1).
In the dose-finding portion of the trial, patients received entrectinib at 250 mg/m2 (n = 3), 400 mg/m2 (n = 3), 550 mg/m2 (n = 7), or 750 mg/m2 (n = 3).
In the phase 1b portion, patients received entrectinib at 550 mg/m2 (n = 7) – the recommended dose – or 400 mg/m2 (n = 6) if they were unable to swallow intact capsules.
Dr. Robinson said entrectinib was “quite well tolerated” overall, but he did not present any data on adverse events. He did say dose-limiting toxicities included fatigue, elevated creatinine levels, dysgeusia resulting in loss of taste, weight gain, and, in one patient, pulmonary edema.
“Entrectinib produced striking, rapid, and durable responses in all children with refractory CNS and solid tumors that actually harbored these fusions in NTRK1/2/3, ROS1, or ALK,” Dr. Robinson said. “It also produced a significant response in one ALK-mutated neuroblastoma patient. [N]o responses were seen in tumors lacking aberrations in the target kinases.”
In all, 12 patients responded. The three complete responders had an ALK F1174L mutation, an ALK fusion, and an NTRK fusion, respectively. Five partial responders had NTRK fusions, three had ROS1 fusions, and one had an ALK fusion.
Three responders discontinued treatment. Ten patients were still receiving entrectinib at last follow-up, and 11 patients had died.
Progression-free survival was significantly longer among patients who had fusions than among those who did not (P less than .0001).
“To sum up, entrectinib really is very promising,” Dr. Robinson said. “It has very promising antitumor activity and progression-free survival but [only] in patients with target gene fusions.”
Dr. Robinson said this trial is ongoing, but it is now limited to patients with fusions targeted by entrectinib.
The trial is sponsored by Hoffman-La Roche Ltd. and supported by Alex’s Lemonade Stand Center of Excellence. Dr. Robinson has relationships with Lilly, Genentech/Roche, and Novartis.
SOURCE: Robinson GW et al. ASCO 2019. Abstract 10009.
REPORTING FROM ASCO 2019
Pexidartinib gets ODAC nod for tenosynovial giant cell tumor treatment
The Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted to support approval of the small molecule kinase inhibitor pexidartinib for the treatment of adults with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and is not amenable to improvement with surgery.
The drug was favored by a 12-3 margin (no abstentions), with the majority of panel members agreeing that it offers clinical benefits that outweigh significant risk for elevated liver enzymes and small but real potential for serious or even fatal liver injury.
The FDA usually follows the recommendation of advisory committees in deciding final approval. Daiichi Sankyo plans to market the drug under the trade name Turalio.
Final approval and marketing of the drug will hinge on a mandatory Risk Evaluation and Mitigation Strategy that will require certification of prescribers, patient participation in education about the need for frequent liver function testing and the signs and symptoms of liver injury, and distribution of the drug only to certified pharmacies.
Both the ODAC panel members and pexidartinib’s manufacturer, Daiichi Sankyo, agreed that the drug is effective, but opinions about the degree of clinical benefit and the risk-benefit ratio differed.
TGCT is a rare, nonmalignant, and nonlethal tumor of the synovium, bursae, or tendon sheath that can be locally aggressive, and for some patients completely disabling. Surgery is the primary mode of treatment, but less than 10% of patients have disease that is not amenable to resection; for these patients treatment options are limited, because there are no approved systemic therapies for the disease.
ENLIVEN
Evidence submitted to support the application comes from the phase 3 ENLIVEN trial, in which patients with TGCT not amenable to surgery were randomly assigned to receive pexidartinib or placebo. The trial was designed to enroll 126 patients to provide 90% power to detect a difference in objective response rate at a two-sided alpha level of 0.05, assuming an overall response rate of 10% with placebo, and 35% with pexidartinib.
The actual trial enrollment, however, fell a little short, with a total of 120 patients randomized.
The ORR at week 25 as assessed by blinded independent central reviewers, the primary endpoint, was 39% for the 61 patients in the pexidartinib group, compared with 0% for the 59 patients in the placebo group (P less than .0001).
There were also statistically significant improvements in the pexidartinib arm at week 25 in the secondary endpoints of mean change in range of motion (ROM), ORR by tumor-volume score at week 25, mean change from baseline in the Patient-Reported Outcomes Measurement Information physical function scale, and mean change in the Worst Stiffness numeric rating scale item. There were no significant differences between the groups for worst pain, however.
The FDA briefing document notes that “interpretation of the results of the secondary endpoints should be viewed with caution as there was a high proportion of missing data at week 25 for ROM, physical function, and worst stiffness (27%, 43% 43%, respectively); the proportion of patients with missing data was similar across study arms.”
Risk and benefits
The major issues before the ODAC included the validity of clinical outcome assessment given the large chunks of missing data and the major adverse event of liver injury, with a majority of patients on pexidartinib experiencing transaminase elevations. The potential for liver injury may be exacerbated by chronic use of the drug or by drugs used to treat comorbidities such as diabetes or cardiovascular disease, several panel members noted.
During the clinical development program for the drug, 2 of 768 patients who received the drug developed irreversible liver injury leading to liver transplant in 1 patient and death in the other.
Many of the panelists who voted to support approval did so with some reluctance about the adverse events. For example, Karin Anton Calis, PharmD, of the National Institutes of Health voted yes because of the efficacy of the drug in a disabling condition.
“Hopefully it will be used in a restricted system so that there can be adequate monitoring, but I’m still concerned about those patients that may have this unpredictable liver toxicity, “ he said.
Victor M. Villalobos MD, PhD, from the University of Colorado at Denver, Aurora, also voted yes.
“This is an ultra-rare disease with no good therapies available to patients and it can be highly morbid, and I feel that getting real-world data on how we can use this drug in a safe and effective manner will be really important to the academic community going forward,” he said.
Other panel members who also voted to support approval expressed concerns about the hepatotoxicity, but noted that the drug has the potential to change lives, as attested by TCGT patients who spoke during the public comment portion of the meeting.
However, panelist Doris Strader, MD, from the University of Vermont, Burlington, said that she voted no because, while she was sensitive to the potential benefits of the drug for some patients, “I was concerned about the missing data and was not convinced that there was clinically meaningful benefit. Likewise, while I understand that the hepatic injury is not liver failure, I am concerned that this may be persistent for a long time, and I worry that there is not enough to suggest that there is going to be rigorous monitoring of patients over their lifetimes.”
The Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted to support approval of the small molecule kinase inhibitor pexidartinib for the treatment of adults with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and is not amenable to improvement with surgery.
The drug was favored by a 12-3 margin (no abstentions), with the majority of panel members agreeing that it offers clinical benefits that outweigh significant risk for elevated liver enzymes and small but real potential for serious or even fatal liver injury.
The FDA usually follows the recommendation of advisory committees in deciding final approval. Daiichi Sankyo plans to market the drug under the trade name Turalio.
Final approval and marketing of the drug will hinge on a mandatory Risk Evaluation and Mitigation Strategy that will require certification of prescribers, patient participation in education about the need for frequent liver function testing and the signs and symptoms of liver injury, and distribution of the drug only to certified pharmacies.
Both the ODAC panel members and pexidartinib’s manufacturer, Daiichi Sankyo, agreed that the drug is effective, but opinions about the degree of clinical benefit and the risk-benefit ratio differed.
TGCT is a rare, nonmalignant, and nonlethal tumor of the synovium, bursae, or tendon sheath that can be locally aggressive, and for some patients completely disabling. Surgery is the primary mode of treatment, but less than 10% of patients have disease that is not amenable to resection; for these patients treatment options are limited, because there are no approved systemic therapies for the disease.
ENLIVEN
Evidence submitted to support the application comes from the phase 3 ENLIVEN trial, in which patients with TGCT not amenable to surgery were randomly assigned to receive pexidartinib or placebo. The trial was designed to enroll 126 patients to provide 90% power to detect a difference in objective response rate at a two-sided alpha level of 0.05, assuming an overall response rate of 10% with placebo, and 35% with pexidartinib.
The actual trial enrollment, however, fell a little short, with a total of 120 patients randomized.
The ORR at week 25 as assessed by blinded independent central reviewers, the primary endpoint, was 39% for the 61 patients in the pexidartinib group, compared with 0% for the 59 patients in the placebo group (P less than .0001).
There were also statistically significant improvements in the pexidartinib arm at week 25 in the secondary endpoints of mean change in range of motion (ROM), ORR by tumor-volume score at week 25, mean change from baseline in the Patient-Reported Outcomes Measurement Information physical function scale, and mean change in the Worst Stiffness numeric rating scale item. There were no significant differences between the groups for worst pain, however.
The FDA briefing document notes that “interpretation of the results of the secondary endpoints should be viewed with caution as there was a high proportion of missing data at week 25 for ROM, physical function, and worst stiffness (27%, 43% 43%, respectively); the proportion of patients with missing data was similar across study arms.”
Risk and benefits
The major issues before the ODAC included the validity of clinical outcome assessment given the large chunks of missing data and the major adverse event of liver injury, with a majority of patients on pexidartinib experiencing transaminase elevations. The potential for liver injury may be exacerbated by chronic use of the drug or by drugs used to treat comorbidities such as diabetes or cardiovascular disease, several panel members noted.
During the clinical development program for the drug, 2 of 768 patients who received the drug developed irreversible liver injury leading to liver transplant in 1 patient and death in the other.
Many of the panelists who voted to support approval did so with some reluctance about the adverse events. For example, Karin Anton Calis, PharmD, of the National Institutes of Health voted yes because of the efficacy of the drug in a disabling condition.
“Hopefully it will be used in a restricted system so that there can be adequate monitoring, but I’m still concerned about those patients that may have this unpredictable liver toxicity, “ he said.
Victor M. Villalobos MD, PhD, from the University of Colorado at Denver, Aurora, also voted yes.
“This is an ultra-rare disease with no good therapies available to patients and it can be highly morbid, and I feel that getting real-world data on how we can use this drug in a safe and effective manner will be really important to the academic community going forward,” he said.
Other panel members who also voted to support approval expressed concerns about the hepatotoxicity, but noted that the drug has the potential to change lives, as attested by TCGT patients who spoke during the public comment portion of the meeting.
However, panelist Doris Strader, MD, from the University of Vermont, Burlington, said that she voted no because, while she was sensitive to the potential benefits of the drug for some patients, “I was concerned about the missing data and was not convinced that there was clinically meaningful benefit. Likewise, while I understand that the hepatic injury is not liver failure, I am concerned that this may be persistent for a long time, and I worry that there is not enough to suggest that there is going to be rigorous monitoring of patients over their lifetimes.”
The Oncologic Drugs Advisory Committee (ODAC) of the Food and Drug Administration voted to support approval of the small molecule kinase inhibitor pexidartinib for the treatment of adults with symptomatic tenosynovial giant cell tumor (TGCT) associated with severe morbidity or functional limitations and is not amenable to improvement with surgery.
The drug was favored by a 12-3 margin (no abstentions), with the majority of panel members agreeing that it offers clinical benefits that outweigh significant risk for elevated liver enzymes and small but real potential for serious or even fatal liver injury.
The FDA usually follows the recommendation of advisory committees in deciding final approval. Daiichi Sankyo plans to market the drug under the trade name Turalio.
Final approval and marketing of the drug will hinge on a mandatory Risk Evaluation and Mitigation Strategy that will require certification of prescribers, patient participation in education about the need for frequent liver function testing and the signs and symptoms of liver injury, and distribution of the drug only to certified pharmacies.
Both the ODAC panel members and pexidartinib’s manufacturer, Daiichi Sankyo, agreed that the drug is effective, but opinions about the degree of clinical benefit and the risk-benefit ratio differed.
TGCT is a rare, nonmalignant, and nonlethal tumor of the synovium, bursae, or tendon sheath that can be locally aggressive, and for some patients completely disabling. Surgery is the primary mode of treatment, but less than 10% of patients have disease that is not amenable to resection; for these patients treatment options are limited, because there are no approved systemic therapies for the disease.
ENLIVEN
Evidence submitted to support the application comes from the phase 3 ENLIVEN trial, in which patients with TGCT not amenable to surgery were randomly assigned to receive pexidartinib or placebo. The trial was designed to enroll 126 patients to provide 90% power to detect a difference in objective response rate at a two-sided alpha level of 0.05, assuming an overall response rate of 10% with placebo, and 35% with pexidartinib.
The actual trial enrollment, however, fell a little short, with a total of 120 patients randomized.
The ORR at week 25 as assessed by blinded independent central reviewers, the primary endpoint, was 39% for the 61 patients in the pexidartinib group, compared with 0% for the 59 patients in the placebo group (P less than .0001).
There were also statistically significant improvements in the pexidartinib arm at week 25 in the secondary endpoints of mean change in range of motion (ROM), ORR by tumor-volume score at week 25, mean change from baseline in the Patient-Reported Outcomes Measurement Information physical function scale, and mean change in the Worst Stiffness numeric rating scale item. There were no significant differences between the groups for worst pain, however.
The FDA briefing document notes that “interpretation of the results of the secondary endpoints should be viewed with caution as there was a high proportion of missing data at week 25 for ROM, physical function, and worst stiffness (27%, 43% 43%, respectively); the proportion of patients with missing data was similar across study arms.”
Risk and benefits
The major issues before the ODAC included the validity of clinical outcome assessment given the large chunks of missing data and the major adverse event of liver injury, with a majority of patients on pexidartinib experiencing transaminase elevations. The potential for liver injury may be exacerbated by chronic use of the drug or by drugs used to treat comorbidities such as diabetes or cardiovascular disease, several panel members noted.
During the clinical development program for the drug, 2 of 768 patients who received the drug developed irreversible liver injury leading to liver transplant in 1 patient and death in the other.
Many of the panelists who voted to support approval did so with some reluctance about the adverse events. For example, Karin Anton Calis, PharmD, of the National Institutes of Health voted yes because of the efficacy of the drug in a disabling condition.
“Hopefully it will be used in a restricted system so that there can be adequate monitoring, but I’m still concerned about those patients that may have this unpredictable liver toxicity, “ he said.
Victor M. Villalobos MD, PhD, from the University of Colorado at Denver, Aurora, also voted yes.
“This is an ultra-rare disease with no good therapies available to patients and it can be highly morbid, and I feel that getting real-world data on how we can use this drug in a safe and effective manner will be really important to the academic community going forward,” he said.
Other panel members who also voted to support approval expressed concerns about the hepatotoxicity, but noted that the drug has the potential to change lives, as attested by TCGT patients who spoke during the public comment portion of the meeting.
However, panelist Doris Strader, MD, from the University of Vermont, Burlington, said that she voted no because, while she was sensitive to the potential benefits of the drug for some patients, “I was concerned about the missing data and was not convinced that there was clinically meaningful benefit. Likewise, while I understand that the hepatic injury is not liver failure, I am concerned that this may be persistent for a long time, and I worry that there is not enough to suggest that there is going to be rigorous monitoring of patients over their lifetimes.”
CAR T cells home in on HER2 in advanced sarcomas
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.

One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.
One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
ATLANTA – A novel chimeric antigen receptor (CAR) T-cell construct centered on HER2 as the target antigen was safe and showed early promise in the treatment of advanced sarcomas of bone and soft tissues in a phase I trial.
One patient, a 16-year-old girl with advanced osteosarcoma metastatic to her lungs, had a complete response to the therapy that is ongoing out to nearly 3 years, reported Shoba A. Navai, MD, from Baylor College of Medicine in Houston.
A second patient, an 8-year-old boy with rhabdomyosarcoma metastatic to bone marrow, had a complete response lasting 12 months. Upon relapse he was re-enrolled, received additional CAR T-cell infusions, and had a second complete response that has been ongoing for 17 months.
“HER2 CAR T cells can induce objective clinical responses in some patients with sarcoma, and engagement of endogenous immunity may aid in generation of tumor responses. We are currently working to validate these findings in other patients who were treated,” she said at a briefing at the annual meeting of the American Association for Cancer Research.
HER2 is a member of the human epidermal growth factor receptor family that is primarily expressed on the surface of tumor cells but is largely absent from nonmalignant tissues. HER2 can be expressed in a variety of sarcomas, including osteosarcoma, and HER2 expression in osteosarcoma correlates with worse overall survival.
Unlike HER2-positive breast cancers, however, HER2 expression levels in osteosarcoma are too low to be effectively targeted by anti-HER2 agents such as trastuzumab (Hereceptin).
But as Dr. Navai and colleagues have found, HER2 appears to be a valid target for CAR T-cell therapy in otherwise antigenically “cold” tumors – that is, tumors with few targetable antigens.
Old target, new weapon
They have developed a CAR T-cell construct using a HER2-directed antibody coupled with CD28 as the costimulatory molecule. As with other CAR T therapies, the patient’s T cells or selected T cell subsets are collected, transfected to express the antigen, and are then expanded and returned to the patient following lymphodepletion with either fludarabine alone or with cyclophosphamide.
Each patient received up to three infusions of autologous CAR T cells at a dose of 1 x 108 cells/m2, and eligible patients received up to five additional infusions without additional lymphodepletion.
Dr. Navai presented data on 10 patients treated to date, including the two mentioned before; the boy with rhabdomyosarcoma was counted as two separate patients for the purpose of the efficacy analysis.
All patients had metastatic disease, including five with osteosarcoma, three with rhabdomyosarcoma, one with Ewing sarcoma, and one with synovial sarcoma.
The lymphodepletion regimens did their job, inducing neutropenia (defined as an absolute neutrophil count less than 500 per milliliter ) for up to 14 days.
Eight patients developed grade 1 or 2 cytokine release syndrome within 24 hours of CAR T-cell infusion, and all cases completely resolved with supportive care within 5 days of onset.
In nine patients, T cells were successfully expanded, with a median peak expansion on day 7.
In all 10 patients, CAR T cells were detected by quantitative polymerase chain reaction 6 weeks after infusion.
In addition to the two patients with complete remissions already described, three patients had stable disease. The remaining patients had disease progression. At the most recent analysis, five patients were still alive, and five had died.
The infusions were safe, with no dose-limiting toxicities reported. No patient required a transfusion, and there were no opportunistic, infections, no neurotoxicities, and no lasting pulmonary or cardiac toxicities, Dr. Navai reported.
Some fare better than others
Nilofer S. Azad, MD, of the Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, who moderated the briefing, commented that the study had “very small numbers, but is still very exciting.”
She noted that the patients who benefited most from the therapy either had minimal residual disease or bone marrow disease without visceral disease; she asked Dr. Navai how this could be addressed going forward.
“The patients who seemed to have had responses both in this trial, as well as in our previous trial without lymphodepletion, tended to have less disease or more accessible disease. So we hypothesized that disease that’s in the bone marrow because it’s more accessible, or in the lungs, where also CAR T cells go after they are first infused, may be more amenable to treatment,” Dr. Navai said.
In contrast, larger tumors and more invasive disease may emit immune inhibitory signals that dampen the efficacy of CAR T cells, she added.
Development of the CAR T-cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai and Dr. Azad reported having no disclosures relevant to the work.
SOURCE: Navai SA et al. AACR 2019, Abstract LB-147.
REPORTING FROM AACR 2019
CAR T cells target HER2 expression in advanced sarcomas
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
ATLANTA – Sarcomas of bone and soft tissues are considered to be “antigenically cold” tumors, with few identifiable mutations that may be susceptible to targeted therapies.
Some sarcoma subtypes such as osteosarcoma and rhabomyosarcoma, however, frequently express the human epidermal growth factor receptor 2 on tumor surfaces. Although HER2 expression in these tumors is at too low a level for HER2-targeted therapies such as trastuzumab (Herceptin), HER2 appears to be an opportunistic target for chimeric antigen receptor (CAR) T-cell therapy, according to Shoba Navai, MD, from Baylor College of Medicine, Houston.
In a video interview at the 2019 annual meeting of the American Association for Cancer Research, Dr. Navai describes her team’s early experience using a HER2-targeted CAR-T cell construct and preinfusion lymphodepletion in patients with advanced sarcomas.
Development of the CAR-T cell construct is supported by the Cancer Prevention & Research Institute of Texas, Stand Up to Cancer, the St. Baldrick’s Foundation, Cookies for Kids’ Cancer, Alex’s Lemonade Stand, and a grant from the National Institutes of Health. Dr. Navai reported having no disclosures.
REPORTING FROM AACR 2019
