User login
Adolescent and young adult (AYA) survival trends
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
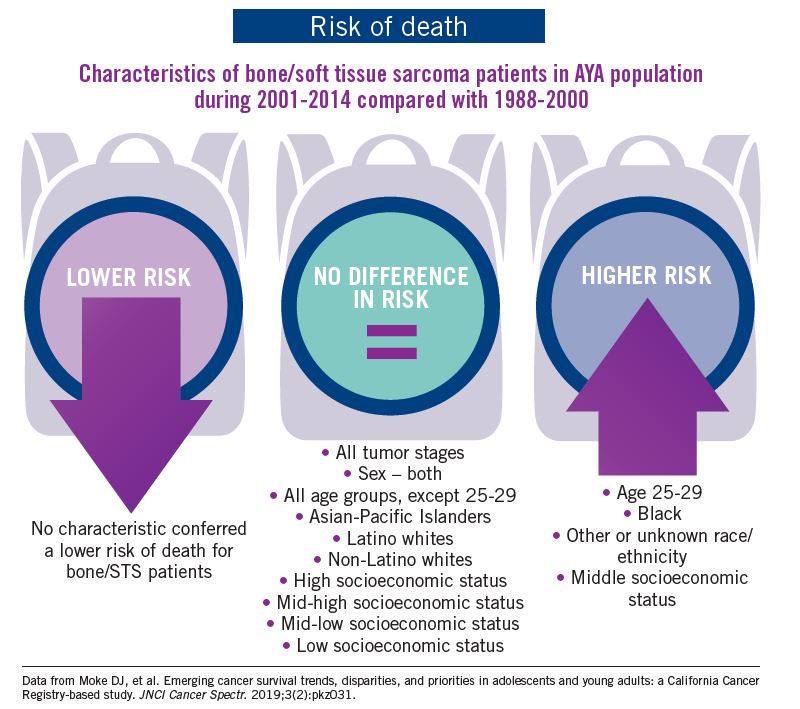
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
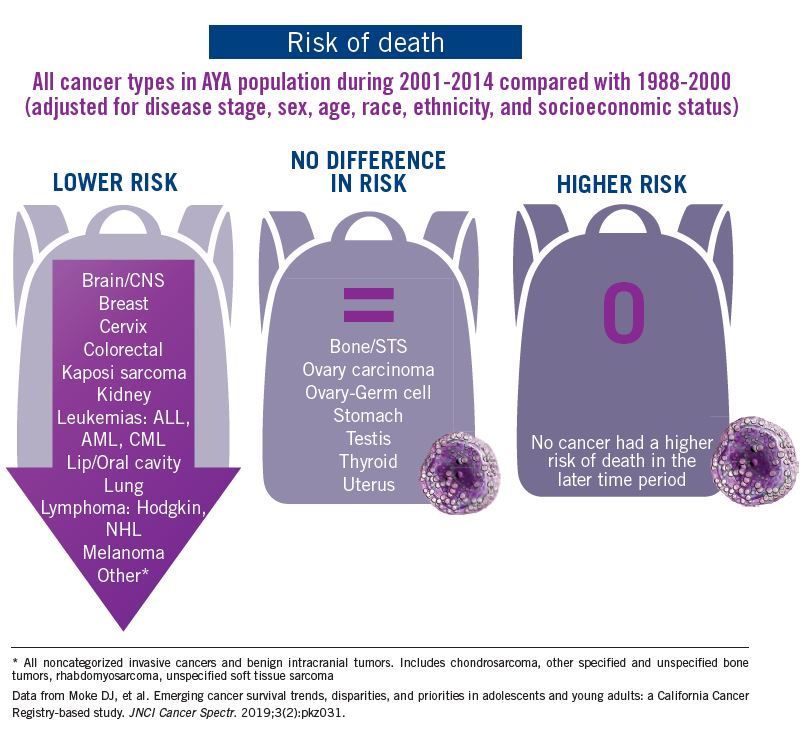
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
The good news: AYA survival improvement was at least as large as in younger children and older adults comparing deaths in two time periods, 1988-2000 and 2001-2014, in a California Cancer Registry.
The bad news: There was no statistically significant difference in survival between time periods for patients with bone and soft tissue sarcoma.
Conference Coverage: ASCO 2019
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
Behind Olaratumab's Phase 3 Disappointment
ANNOUNCE, the phase 3 trial designed to confirm the clinical benefit of olaratumab in patients with advanced soft tissue sarcoma (STS), failed to meet its primary endpoint of overall survival (OS) in all STS histologies and the leiomyosarcoma population. The previous phase 1b/2 signal-finding study of olaratumab had achieved an unprecedented improvement in OS, and the US Food and Drug Administration (FDA) awarded olaratumab accelerated approval in October 2016. By December 2018, olaratumab received additional accelerated, conditional, and full approvals in more than 40 countries worldwide. William D. Tap, MD, chief of the Sarcoma Medical Oncology Service at Memorial Sloan Kettering Cancer Center in New York, presented the phase 3 results and provided some explanations for the findings during the plenary session at ASCO.
ANNOUNCE (NCT02451943), which was designed and enrolled prior to olaratumab receiving accelerated approval, opened in September 2015 and completed accrual 10 months later in July 2016. Investigators randomized and treated 509 patients with advanced STS not amenable to curative therapy, 258 patients in the olaratumab-doxorubicin arm and 251 in the placebo-doxorubicin arm. Most patients (46%) had leiomyosarcoma, followed by liposarcoma (18%), pleomorphic sarcoma (13%), and 24% of the patient population had 26 unique histologies. Three-quarters of the patients had no prior systemic therapy.
Results
As of the data cutoff on December 5, 2018, there were no survival differences in the intention-to-treat population, in the total STS population nor in the leiomyosarcoma subpopulation, with olaratumab-doxorubicin compared to placebo-doxorubicin. For the total STS population, median OS with olaratumab- doxorubicin was 20.4 months and with placebo-doxorubicin 19.7 months. “This is the highest survival rate described to date in any phase 3 sarcoma study,” Dr. Tap said. “It is of particular interest as ANNOUNCE did not mandate treatment in the first line.” In the leiomyosarcoma population, median OS was 21.6 months with olaratumab and 21.9 months with placebo. The secondary endpoints of progression-free survival (PFS), overall response rate, and disease control rate did not favor olaratumab either.
Investigators are examining the relationship between PDGFRα expression and OS in ANNOUNCE. PDGFRα-positive tumors tended to do worse with olaratumab than PDGFRα-negative tumors. The investigators noticed a 6-month difference in OS between these populations favoring PDGFRα-negative tumors. Additional biomarker analyses are ongoing.
A large and concerted effort is underway, Dr. Tap said, to understand the results of the ANNOUNCE study alone and in context with the phase 1b/2 study. “There are no noted discrepancies in study conduct or data integrity which could explain these findings or the differences between the two studies.”
Possible explanations
The designs of the phase 1b/2 and phase 3 studies had some important differences. The phase 1b/2 study was a small, open-label, US-centric study (10 sites) that did not include a placebo or subtype- specified analyses. Its primary endpoint was PFS, it did not have a loading dose of olaratumab, and it specified the timing of dexrazoxane administration after 300 mg/m2 of doxorubicin.
ANNOUNCE, on the other hand, was a large (n=509), international (110 study sites), double-blind, placebo-controlled trial that had outcomes evaluated in STS and leiomyosarcoma. Its primary endpoint was OS, it had a loading dose of olaratumab of 20 mg/kg, and there was no restriction as to the timing of dexrazoxane administration.
Dr. Tap pointed out that in ANNOUNCE it was difficult to predict or control for factors that may have had an unanticipated influence on outcomes, such as albumin levels as a surrogate for disease burden and behavior of PDGFRα status. It is possible, he said, that olaratumab has no activity in STS and that the phase 1b/2 results were due to, among other things, the small sample size, numerous represented histologies with disparate clinical behavior, and the effect of subtype-specific therapies on overall survival, given subsequently or even by chance. On the other hand, it is also possible, he said, that olaratumab has some activity in STS, with outcomes being affected by the heterogeneity of the study populations, differences in trial design, and the performance of the ANNOUNCE control arm. Whatever the case, he said, accelerated approval allowed patients to have access to a potentially life-prolonging drug with little added toxicity.
Discussion
In the expert discussion following the presentation, Jaap Verweij, MD, PhD, of Erasmus University Medical Center in Rotterdam, The Netherlands, congratulated the investigators for performing the study at an unprecedented pace. He commented that lumping STS subtypes together is problematic, as different histological subtypes behave as though they are different diseases. Small numbers of each tumor subtype and subtypes with slow tumor growth can impact trial outcomes. In the phase 1b/2 and phase 3 trials, 26 different subtypes were represented in each study. Dr. Verweij pointed out this could have made a big difference in the phase 1b/2 study, in which there were only 66 patients in each arm.
It is striking to note, he said, that without exception, phase 2 randomized studies in STS involving doxorubicin consistently overestimated and wrongly predicted PFS in the subsequent phase 3 studies. And the situation is similar for OS. The results of the ANNOUNCE study are no exception, he added. “Taken together, these studies indicate that phase 2 studies in soft tissue sarcomas, certainly those involving additions of drugs to doxorubicin, even if randomized, should be interpreted with great caution,” he said.
SOURCE: Tap WD, et al. J Clin Oncol 37, 2019 (suppl; abstr LBA3)
The study was sponsored by Eli Lilly and Company.
Dr. Tap reported research funding from Lilly and Dr. Verweij had nothing to report related to this study. Abstract coauthors disclosed numerous financial relationships, including consulting/advisory roles and/or research funding from Lilly, and several were employed by Lilly.
Addition of Temozolomide May Improve Outcomes in RMS
Investigators from the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG) found that the addition of temozolomide (T) to vincristine and irinotecan (VI) may improve outcomes in adults and children with relapsed or refractory rhabdomyosarcoma (RMS). Principal investigator of the study, Anne Sophie Defachelles, MD, pediatric oncologist at the Centre Oscar Lambret in Lille, France, presented the results on behalf of the EpSSG.
The primary objective of the study was to evaluate the efficacy of VI and VIT regimens, defined as objective response (OR)—complete response (CR) plus partial response (PR)—after 2 cycles. Secondary objectives were progression-free survival (PFS), overall survival (OS), and safety in each arm, and the relative treatment effect of VIT compared to VI in terms of OR, survival, and safety.
The international, randomized (1:1), open-label, phase 2 trial (VIT-0910; NCT01355445) was conducted at 37 centers in 5 countries. Patients ages 6 months to 50 years with RMS were eligible. They could not have had prior irinotecan or temozolomide. A 2015 protocol amendment limited enrollment to patients at relapse and increased the enrollment goal by 40 patients. After the 2015 amendment, patients with refractory disease were no longer eligible.
From January 2012 to April 2018, investigators enrolled 120 patients, 60 on each arm. Two patients in the VI arm were not treated. Patients were a median age of 10.5 years in the VI arm and 12 years in the VIT arm, 92% (VI) and 87% (VIT) had relapsed disease, 8% (VI) and 13% (VIT) had refractory disease, and 55% (VI) and 68% (VIT) had metastatic disease at study entry.
Results
Patients achieved an OR rate of 44% (VIT) and 31% (VI) for the whole population, one-sided P value <.0001. The adjusted odds ratio for the whole population was 0.50, P=.09. PFS was 4.7 months (VIT) and 3.2 months (VI), “a nearly significant reduction in the risk of progression,” Dr. Defachelles noted. Median OS was 15.0 months (VIT) and 10.3 months (VI), which amounted to “a large and significant reduction in the risk of death,” she said. The adjusted hazard ratio was 0.55, P=.006.
Adverse events of grade 3 or higher were more frequent in the VIT arm, with hematologic toxicity the most frequent (81% for VIT, 59% for VI), followed by gastrointestinal adverse events. “VIT was significantly more toxic than VI,” Dr. Defachelles observed, “but the toxicity was manageable.”
“VIT is now the standard treatment in Europe for relapsed rhabdomyosarcoma and will be the control arm in the multiarm, multistage RMS study for relapsed patients,” she said.
In a discussion following the presentation, Lars M. Wagner, MD, of Cincinnati Children’s Hospital, pointed out that the study was not powered for the PFS and OS assessments. These were secondary objectives that should be considered exploratory. Therefore, he said, the outcome data is not conclusive. The role of temozolomide in RMS is also unclear, given recent negative results in patients with newly diagnosed metastatic RMS (Malempati et al, Cancer 2019). And he said it’s uncertain how these results apply to patients who received irinotecan upfront for RMS.
SOURCE: Defachelles AS, et al. J Clin Oncol 37, 2019 (suppl; abstr 10000)
The study was sponsored by Centre Oscar Lambret and SFCE (Société Française de Lutte contre les Cancers et Lucémies de l’Enfant et de l’Adolescent) served as collaborator.
Drs. Defachelles and Wagner had no relationships to disclose. A few coauthors had advisory/consulting or speaker roles for various commercial interests, including two for Merck (temozolomide).
Pazopanib Increases Pathologic Necrosis Rates in STS
Pazopanib added to a regimen of preoperative chemoradiation in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) significantly increased the rate of near-complete pathologic response in both children and adults with intermediate or high-risk disease. Pazopanib, a multitargeted receptor tyrosine kinase inhibitor, works in multiple signaling pathways involved in tumorigenesis— VEGFR-1, -2, -3, PDGFRα/β, and c-kit. A phase 3 study demonstrated significant improvement in progression-free survival (PFS) in advanced STS patients and was the basis for its approval in the US and elsewhere for treatment of this patient population. Preclinical data suggest synergy between pazopanib and cytotoxic chemotherapy, forming the rationale for the current trial with neoadjuvant pazopanib added to chemoradiation.
According to the investigators, the trial (ARST1321) is the first ever collaborative study codeveloped, written, and conducted by pediatric (Children’s Oncology Group) and adult (NRG Oncology) cancer cooperative groups (NCT02180867). Aaron R. Weiss, MD, of the Maine Medical Center in Portland and study cochair, presented the data for the chemotherapy arms at ASCO. The primary objectives of the study were to determine the feasibility of preoperative chemoradiation with or without pazopanib and to compare the rates of complete pathologic response in patients receiving radiation or chemoradiation with or without pazopanib. Pathologic necrosis rates of 90% or better have been found to be predictive of outcome in STS.
Patients with metastatic or non-metastatic NRSTS were eligible to enroll if they had initially unresectable extremity or trunk tumors with the expectation that they would be resectable after therapy. Patients had to be 2 years or older— there was no upper age limit—and had to be able to swallow a tablet whole. The dose-finding phase of the study determined the pediatric dose to be 350 mg/m2 and the adult dose to be 600 mg/m2, both taken orally and once daily. Patients in the chemotherapy cohort were then randomized to receive chemotherapy—ifosfamide and doxorubicin—with or without pazopanib. At 4 weeks, patients in both arms received preoperative radiotherapy (45 Gy in 25 fractions), and at week 13, surgery of the primary site if they did not have progressive disease. After surgery, patients received continuation therapy with or without pazopanib according to their randomization arm. Upon completion and recovery from the continuation therapy, patients could receive surgery/radiotherapy of their metastatic sites.
Results
As of the June 30, 2018, cutoff, 81 patients were enrolled on the chemotherapy arms: 42 in the pazopanib plus chemoradiation arm and 39 in the chemoradiation-only arm. Sixty-one percent of all patients were 18 years or older, and the median age was 20.3 years. Most patients (73%) did not have metastatic disease, and the major histologies represented were synovial sarcoma (49%) and undifferentiated pleomorphic sarcoma (25%).
At week 13, patients in the pazopanib arm showed significant improvement, with 14 (58%) of those evaluated having pathologic necrosis of at least 90%, compared with 4 (22%) in the chemoradiation-only arm (P=.02). The study was closed to further accrual.
Eighteen patients were not evaluable for pathologic response and 21 were pending pathologic evaluation at week 13. Radiographic response rates were not statistically significant on either arm. No complete responses (CR) were achieved in the pazopanib arm, but 14 patients (52%) achieved a partial response (PR) and 12 (44%) had stable disease (SD). In the chemoradiation-only arm, 2 patients (8%) achieved a CR, 12 (50%) a PR, and 8 (33%) SD. Fifteen patients in each arm were not evaluated for radiographic response.
The pazopanib arm experienced more febrile neutropenia and myelotoxicity during induction and continuation phases than the chemoradiation-only arm. In general, investigators indicated pazopanib combined with chemoradiation was well tolerated and no unexpected toxicities arose during the trial.
In the post-presentation discussion, Dr. Raphael E. Pollock, MD, PhD, of The Ohio State University, called it a tremendous challenge to interdigitate primary local therapies in systemic approaches, particularly in the neoadjuvant context. He pointed out that in an earlier study, a 95% to 100% necrosis level was needed to achieve a significant positive impact on outcomes and perhaps a subsequent prospective trial could determine the best level. He questioned whether the availability of only 60% of patient responses could affect the conclusions and whether the high number of toxicities (73.8% grade 3/4 with pazopanib) might be too high to consider the treatment for most patients, given the intensity of the regimen.
SOURCE: Weiss AR, et al. J Clin Oncol 37, 2019 (suppl; abstr 11002)
The study was sponsored by the National Cancer Institute.
Drs. Weiss and Pollock had no relationships with commercial interests to disclose. A few investigators disclosed advisory, consulting, or research roles with pharmaceutical companies, including one who received institutional research funding from Novartis (pazopanib).
Gemcitabine Plus Pazopanib a Potential Alternative in STS
In a phase 2 study of gemcitabine with pazopanib (G+P) or gemcitabine with docetaxel (G+D), investigators concluded the combination with pazopanib can be considered an alternative to that with docetaxel in select patients with advanced soft tissue sarcoma (STS). They reported similar progression-free survival (PFS) and rate of toxicity for the two regimens. Neeta Somaiah, MD, of the University of Texas MD Anderson Cancer Center in Houston, presented the findings of the investigator-initiated effort (NCT01593748) at ASCO.
The objective of the study, conducted at 10 centers across the United States, was to examine the activity of pazopanib when combined with gemcitabine as an alternative to the commonly used gemcitabine plus docetaxel regimen. Pazopanib is a multi-tyrosine kinase inhibitor with efficacy in non-adipocytic STS. Adult patients with metastatic or locally advanced non-adipocytic STS with ECOG performance of 0 or 1 were eligible. Patients had to have received prior anthracycline exposure unless it was contraindicated. The 1:1 randomization included stratification for pelvic radiation and leiomyosarcoma histology, which was felt to have a higher response rate with the pazopanib regimen.
The investigators enrolled 90 patients, 45 in each arm. Patients were a mean age of 56 years, and there was no difference in age or gender distribution between the arms. Patients with leiomyosarcoma (31% overall) or prior pelvic radiation (11% overall) were similar between the arms. The overall response rate using RECIST 1.1 criteria was partial response (PR) in 8 of 44 evaluable patients (18%) in the G+D arm and 5 of 43 evaluable patients (12%) in the G+P arm. Stable disease (SD) was observed in 21 patients (48%) in the G+D arm and 24 patients (56%) in the G+P arm. This amounted to a clinical benefit rate (PR + SD) of 66% and 68% for the G+D and G+P arms, respectively (Fisher’s exact test, P>.99). The median PFS was 4.1 months on both arms and the difference in median overall survival— 15.9 months in the G+D arm and 12.4 months in the G+P arm—was not statistically significant.
Adverse events (AEs) of grade 3 or higher occurred in 19.9% of patients on G+D and 20.6% on G+P. Serious AEs occurred in 33% (G+D) and 22% (G+P). Dose reductions were necessary in 80% of patients on G+P and doses were held in 93%. Dr. Somaiah explained that this may have been because the starting dose of gemcitabine and pazopanib (1000 mg/m2 of gemcitabine on days 1 and 8 and 800 mg of pazopanib) was “probably higher than what we should have started at.” The rate of doses held was also higher in the pazopanib arm (93%) compared with the docetaxel arm (58%). This was likely because pazopanib was a daily dosing, so if there was a toxicity it was more likely to be held than docetaxel, she observed. Grade 3 or higher toxicities occurring in 5% or more of patients in either arm consisted generally of cytopenias and fatigue. The G+P arm experienced a high amount of neutropenia, most likely because this arm did not receive granulocyte-colony stimulating factor (GCSF) support, as opposed to the G+D arm.
Dr. Somaiah pointed out that the 12% response rate for the G+P combination is similar to what has been previously presented and higher than single-agent gemcitabine or pazopanib, but not higher than the G+D combination. The PFS of 4.1 months was less than anticipated, she added, but it was similar on both arms. The investigators believe the G+P combination warrants further exploration.
SOURCE: Somaiah N, et al. J Clin Oncol 37, 2019 (suppl; abstr 11008)
The study was sponsored by the Medical University of South Carolina, with Novartis as collaborator.
Dr. Somaiah disclosed Advisory Board roles for Blueprint, Deciphera, and Bayer. Abstract coauthors disclosed advisory/consulting roles or research funding from various commercial interests, including Novartis (pazopanib) and Pfizer (gemcitabine).
rEECur Trial Finding Optimal Chemotherapy Regimen for Ewing Sarcoma
Interim results of the first and largest randomized trial in patients with refractory or recurrent Ewing sarcoma (ES), the rEECur trial, are guiding the way to finding the optimal chemotherapy regimen to treat the disease. Until now, there has been little prospective evidence and no randomized data to guide treatment choices in relapsed or refractory patients, and hence no real standard of care, according to the presentation at ASCO. Several molecularly targeted therapies are emerging, and they require a standardized chemotherapy backbone against which they can be tested.
The rEECur trial (ISRCTN36453794) is a multi-arm, multistage phase 2/3 “drop-a-loser” randomized trial designed to find the standard of care. The trial compares 4 chemotherapy regimens to each other and drops the least effective one after 50 patients per arm are enrolled and evaluated. The 3 remaining regimens continue until at least 75 patients on each arm are enrolled and evaluated, and then another arm would be dropped. The 2 remaining regimens continue to phase 3 evaluation. Four regimens are being tested at 8 centers in 17 countries: topotecan/ cyclophosphamide (TC), irinotecan/temozolomide (IT), gemcitabine/docetaxel (GD), and ifosfamide (IFOS). The primary objective is to identify the optimal regimen based on a balance between efficacy and toxicity. Martin G. McCabe, MB BChir, PhD, of the University of Manchester in the United Kingdom, presented the results on behalf of the investigators of the rEECur trial.
Results
Two hundred twenty patients 4 years or older and younger than 50 years with recurrent or refractory histologically confirmed ES of bone or soft tissue were randomized to receive GD (n=72) or TC, IT, or IFOS (n=148). Sixty-two GD patients and 123 TC/IT/IFOS patients were included in the primary outcome analysis. Patients were predominantly male (70%), with a median age of 19 years (range, 4 to 49). About two-thirds (67.3%) were post-pubertal. Most patients (85%) were primary refractory or experienced their first disease recurrence, and 89% had measurable disease.
Investigators assessed the primary outcome of objective response after 4 cycles of therapy and found 11% of patients treated with GD responded compared to 24% in the other 3 arms combined. When they subjected the data to Bayesian analysis, there was a 25% chance that the response rate in the GD arm was better than the response in Arm A, a 2% chance that it was better than Arm B, and a 3% chance that it was better than Arm C. Because this study was still blinded at the time of the presentation, investigators didn’t know which regimen constituted which arm. The probability that response favored GD, however, was low.
The investigators observed no surprising safety findings. Eighty-five percent of all patients experienced at least 1 adverse event. Most frequent grade 3‐5 events consisted of pneumonitis (50%, 60%), neutropenic fever (17%, 25%), and diarrhea (0, 12%) in GD and the combined 3 arms, respectively. Grade 3 events in the GD arm were lower than in the other 3 arms combined. There was 1 toxic death attributed to neutropenic sepsis in 1 of the 3 blinded arms.
Median progression-free survival (PFS) for all patients was approximately 5 months. Bayesian analysis suggested there was a low probability that GD was more effective than the other 3 arms: a 22% chance that GD was better than Arm A, a 3% chance that it was better than Arm B, and a 7% chance that it was better than Arm C. Bayesian analysis also suggested there was a probability that OS favored GD. Because the trial directs only the first 4 or 6 cycles of treatment and the patients receive more treatment after trial-directed therapy, investigators were not fully able to interpret this.
Data suggested GD is a less effective regimen than the other 3 regimens both by objective response rate and PFS, so GD has been dropped from the study. Investigators already had more than 75 evaluable patients in each of the 3 arms for the second interim analysis to take place. In a discussion following the presentation, Jayesh Desai, FRACP, of Peter MacCallum Cancer Centre in Melbourne, Australia, called this study a potentially practice-changing trial at this early stage, noting that the GD combination will be de-prioritized in practice based on these results.
SOURCE: McCabe MG, et al. J Clin Oncol 37, 2019 (suppl; abstr 11007)
The rEECur trial is sponsored by the University of Birmingham (UK) and received funding from the European Union’s Seventh Framework Programme under a grant agreement.
Dr. McCabe disclosed no conflicts of interest. Other authors disclosed consulting, advisory roles, or research funding from numerous pharmaceutical companies, including Lilly (gemcitabine) and Pfizer (irinotecan). Dr. Desai disclosed a consulting/advisory role and institutional research funding from Lilly.
Abemaciclib Meets Primary Endpoint in Phase 2 Trial of DDLS
The newer and more potent CDK4 inhibitor, abemaciclib, met its primary endpoint in the investigator-initiated, single-center, single-arm, phase 2 trial in patients with advanced progressive dedifferentiated liposarcoma (DDLS). Twenty-two patients (76%) achieved progression-free survival (PFS) at 12 weeks for a median PFS of 30 weeks. A subset of patients experienced prolonged clinical benefit, remaining on study with stable disease for over 900 days. The study (NCT02846987) was conducted at Memorial Sloan Kettering Cancer Center (MSKCC) in New York and Mark A. Dickson, MD, presented the results at ASCO.
Of three agents in the clinic with the potential to target CDK4 and CDK6—palbociclib, ribociclib, and abemaciclib— abemaciclib is more selective for CDK4 than CDK6. CDK4 amplification occurs in more than 90% of well-differentiated and dedifferentiated liposarcomas. Abemaciclib also has a different side effect profile, with less hematologic toxicity than the other 2 agents. The current study was considered positive if 15 patients or more of a 30-patient sample size were progression- free at 12 weeks.
Results
Thirty patients, 29 evaluable, with metastatic or recurrent DDLS were enrolled and treated with abemaciclib 200 mg orally twice daily between August 2016 and October 2018. Data cutoff for the presentation was the first week of May 2019. Patients were a median of 62 years, 60% were male, and half had no prior systemic treatment. Prior systemic treatments for those previously treated included doxorubicin, olaratumab, gemcitabine, docetaxel, ifosfamide, eribulin, and trabectedin. For 87%, the primary tumor was in their abdomen or retroperitoneum.
Toxicity was as expected with this class of agent, according to the investigators. The most common grades 2 and 3 toxicities, respectively, possibly related to the study drug, occurring in more than 1 patient included anemia (70%, 37%), thrombocytopenia (13%, 13%), neutropenia (43%, 17%), and lymphocyte count decreased (23%, 23%). Very few of these adverse events were grade 4—none for anemia, and 3% each for thrombocytopenia, neutropenia, and lymphocyte count decreased. Diarrhea of grades 2 and 3 occurred in 27% and 7% of patients, respectively, and was managed well with loperamide.
In addition to reaching the primary endpoint of 15 patients or more achieving PFS at 12 weeks, 1 patient had a confirmed partial response (PR) and another an unconfirmed PR. At data cutoff, 11 patients remained on study with stable disease or PR. The investigators conducted correlative studies that indicated all patients had CDK4 and MDM2 amplification with no loss of retinoblastoma tumor suppressor. They observed an inverse correlation between CDK4 amplification and PFS—the higher the level of CDK4 amplification, the shorter the PFS. They also found additional genomic alterations, including JUN, GLI1, ARID1A, TERT, and ATRX. TERT amplification was also associated with shorter PFS. Based on these findings, the investigators believe a phase 3 study of abemaciclib in DDLS is warranted.
Winette van der Graaf, MD, PhD, of the Netherlands Cancer Institute in Amsterdam, in the discussion following the presentation, concurred that it is certainly time for a multicenter phase 3 study of CDK4 inhibitors in DDLS, and a strong international collaboration is key to conducting such studies, particularly in rare cancers. On a critical note, Dr. van der Graaf expressed concern that no patient-reported outcomes were measured after 120 patients, including those in previous studies, were treated on palbociclib and abemaciclib. Given that the toxicities of the CDK4 inhibitors are quite different, she recommended including patient-reported outcomes in future studies using validated health-related quality-of-life instruments.
SOURCE: Dickson MA, et al. J Clin Oncol 37, 2019 (suppl; abstr 11004)
The study was sponsored by Memorial Sloan Kettering Cancer Center, with the study collaborator, Eli Lilly and Company.
Dr. Dickson disclosed research funding from Lilly, the company that provided the study drug. Dr. van der Graaf had no relevant relationships to disclose. Abstract coauthors had consulting/advisory roles or research funding from various companies, including Lilly.
nab-Sirolimus Provides Benefits in Advanced Malignant PEComa
In a prospective phase 2 study of nab-sirolimus in advanced malignant perivascular epithelioid cell tumor (PEComa), the mTOR inhibitor achieved an objective response rate (ORR) of 42% with an acceptable safety profile, despite using relatively high doses of nab-sirolimus compared to other mTOR inhibitors. Activation of the mTOR pathway is common in PEComa, and earlier case reports had indicated substantial clinical benefit with mTOR inhibitor treatment. nab-Sirolimus (ABI-009) is a novel intravenous mTOR inhibitor consisting of nanoparticles of albumin-bound sirolimus. It has significantly higher anti-tumor activity than oral mTOR inhibitors and greater mTOR target suppression at an equal dose. Andrew J. Wagner, MD, PhD, of the Dana-Farber Cancer Institute in Boston, presented the findings of AMPECT (NCT02494570)—Advanced Malignant PEComa Trial—at ASCO.
Investigators enrolled 34 patients 18 years or older with histologically confirmed malignant PEComa. Patients could not have had prior mTOR inhibitors. They received infusions of 100 mg/m2 nab-sirolimus on days 1 and 8 every 21 days until progression or unacceptable toxicity. Patients were a median age of 60 years and 44% were 65 or older; 82% were women, which is typical of the disease. Most patients (88%) had no prior systemic therapy for advanced PEComa.
Results
The drug was well tolerated, with toxicities similar to those of oral mTOR inhibitors. Treatment-related adverse events (TRAEs) occurring in 25% or more of patients were mostly grade 1 or 2 toxicities. Hematologic TRAEs included anemia (47%) and thrombocytopenia (32%) of any grade. Nonhematologic events of any grade included stomatitis/ mucositis (74%), dermatitis/rash (65%), fatigue (59%), nausea (47%), and diarrhea (38%), among others. A few grade 3 events occurred on study, most notably stomatitis/mucositis (18%). Severe adverse events (SAEs) were also uncommon, occurring in 7 of 34 patients (21%). Pneumonitis is common in orally administered mTOR inhibitors; 6 patients (18%) treated with nab-sirolimus had grade 1 or 2 pneumonitis.
Of the 31 evaluable patients, 13 (42%) had an objective response, all of which were partial responses (PR). Eleven (35%) had stable disease and 7 (23%) had progressive disease. The disease control rate, consisting of PR and stable disease, was 77%. The median duration of response had not been reached as of the data cutoff on May 10, 2019. At that time, it was 6.2 months (range, 1.5 to 27.7+). The median time to response was 1.4 months and the median progression-free survival (PFS) was 8.4 months. The PFS rate at 6 months was 61%. Three patients had received treatment for over a year and another 3 patients for more than 2 years.
Correlation with biomarkers
Of the 25 patients who had tissue suitable for next-generation sequencing, 9 had TSC2 mutations, 5 had TSC1 mutations, and 11 had neither mutation. Strikingly, 9 of 9 patients with TSC2 mutations developed a PR, while only 1 with a TSC1 mutation responded. One patient with no TSC1/2 mutation also responded and 2 patients with unknown mutational status responded. The investigators also analyzed pS6 status by immunohistochemistry—pS6 is a marker of mTOR hyperactivity. Twenty- five patient samples were available for analysis. Eight of 8 patients who were negative for pS6 staining did not have a response, while 10 of 17 (59%) who were pS6-positive had a PR.
In the discussion that followed, Winette van der Graaf, MD, of the Netherlands Cancer Institute in Amsterdam, noted that this study showed that biomarkers can be used for patient selection, although TSC2 mutations are not uniquely linked with response. She indicated a comparator with sirolimus would have been of great interest.
SOURCE: Wagner AJ, et al. J Clin Oncol 37, 2019 (suppl; abstr 11005).
The study was sponsored by Aadi Bioscience, Inc., and funded in part by a grant from the FDA Office of Orphan Products Development (OOPD).
Disclosures relevant to this presentation include contininstitutional research funding from Aadi Bioscience for Dr. Wagner and a few other abstract coauthors. Several coauthors are employed by Aadi Bioscience and have stock or other ownership interests. Dr. van der Graaf had nothing to disclose.
Cabozantinib Achieves Disease Control in GIST
The phase 2 EORTC 1317 trial, known as CaboGIST (NCT02216578), met its primary endpoint of progression-free survival (PFS) at 12 weeks in patients with metastatic gastrointestinal stromal tumor (GIST) treated with the tyrosine kinase inhibitor (TKI) cabozantinib. Twenty-four (58.5%) of the 41 patients in the primary study population, and 30 (60%) of the entire 50-patient population, were progression-free at 12 weeks. The study needed 21 patients to be progression- free for cabozantinib to warrant further exploration in GIST patients.
Cabozantinib is a multitargeted TKI inhibiting KIT, MET, AXL, and VEGFR2, which are potentially relevant targets in GIST. In patient-derived xenografts of GIST, cabozantinib demonstrated activity in imatinib-sensitive and -resistant models and inhibited tumor growth, proliferation, and angiogenesis. Additional preclinical experience suggested that cabozantinib could potentially be used as a potent MET inhibitor, overcoming upregulation of MET signaling that occurs with imatinib treatment of GIST, known as the kinase switch.
This investigator-initiated study had as its primary objective assessment of the safety and activity of cabozantinib in patients with metastatic GIST who had progressed on imatinib and sunitinib. The patients could not have been exposed to other KIT- or PDGFR-directed TKIs, such as regorafenib. Secondary objectives included the assessment of cabozantinib in different mutational subtypes of GIST. Patients received cabozantinib tablets once daily until they experienced no further clinical benefit or became intolerant to the drug or chose to discontinue therapy. Fifty patients started treatment between February 2017 and August 2018. All were evaluable for the primary endpoint, and one-third of patients contininstitutional cabozantinib treatment as of the database cutoff in January 2019.
Results
Patients were a median age of 63 years. Virtually all patients (92%) had prior surgery and only 8% had prior radiotherapy. The daily cabozantinib dose was a median 47.2 mg and duration of treatment was a median 20.4 weeks. No patient discontinued treatment due to toxicity, but 88% discontinued due to disease progression.
Safety signals were the same as for other indications in which cabozantinib is used. Almost all patients (94%) had at least 1 treatment-related adverse event of grades 1‐4, including diarrhea (74%), palmar-plantar erythrodysesthesia (58%), fatigue (46%), and hypertension (46%), which are typical of treatment with cabozantinib. Hematologic toxicities in this trial were clinically irrelevant, according to the investigators, consisting of small numbers of grades 2‐3 anemia, lymphopenia, white blood cell count abnormality, and neutropenia. Biochemical abnormalities included grades 3 and 4 hypophosphatemia, increased grades 3 and 4 gamma-glutamyl transferase, grade 3 hyponatremia, and grade 3 hypokalemia, in 8% or more of patients.
Overall survival was a median 14.4 months, with 16 patients still on treatment at the time of data cutoff. Twenty- four patients were progression-free at week 12, satisfying the study decision rule for clinical benefit. Median duration of PFS was 6.0 months. Seven patients (14%) achieved a confirmed partial response (PR) and 33 (66%) achieved stable disease (SD). Nine patients had progressive disease as their best response, 3 of whom had some clinical benefit. Forty patients (80%) experienced a clinical benefit of disease control (PR + SD).
An analysis of the relationship of genotype, duration, and RECIST response showed objective responses in patients with primary exon 11 mutations, with exon 9 mutations, and with exon 17 mutations, and in 2 patients without any known mutational information at the time of the presentation. Patients with stable disease were spread across all mutational subsets in the trial. The investigators suggested the definitive role of MET and AXL inhibition in GIST be assessed further in future clinical trials.
SOURCE: Schöffski P, et al. J Clin Oncol 37, 2019 (suppl; abstr 11006).
The study was sponsored by the European Organization for Research and Treatment of Cancer (EORTC).
Presenting author, Patrick Schöffski, MD, of KU Leuven and Leuven Cancer Institute in Belgium, disclosed institutional relationships with multiple pharmaceutical companies for consulting and research funding, including research funding from Exelixis, the developer of cabozantinib. No other abstract coauthor disclosed a relationship with Exelixis.
Larotectinib Effective in TRK Fusion Cancers
Pediatric patients with tropomyosin receptor kinase (TRK) fusions involving NTRK1, NTRK2, and NTRK3 genes had a high response rate with durable responses and a favorable safety profile when treated with larotrectinib, according to a presentation at ASCO. In this pediatric subset of children and adolescents from the SCOUT and NAVIGATE studies, the overall response rate (ORR) was 94%, with a 35% complete response (CR), 59% partial response (PR), and 6% stable disease as of the data cutoff at the end of July 2018.
TRK fusion cancer is a rare malignancy seen in a wide variety of adult and childhood tumor types. Among pediatric malignancies, infantile fibrosarcoma and congenital mesoblastic nephroma are rare, but have high NTRK gene fusion frequency. Other sarcomas and pediatric high-grade gliomas, for example, are less rare but have low NTRK gene fusion frequency. Larotrectinib, a first-in-class and the only selective TRK inhibitor, has high potency against the 3 NTRK genes that encode the neurotrophin receptors. It is highly selective and has limited inhibition of the other kinases. The US Food and Drug Administration approved larotrectinib for the treatment of patients with solid tumors harboring NTRK fusions. Cornelis Martinus van Tilburg, MD, of the Hopp Children’s Cancer Center, Heidelberg University Hospital, and German Cancer Research Center in Heidelberg, Germany, presented the findings.
Investigators enrolled 38 children and adolescents younger than 18 years from the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) studies of larotrectinib who had non-central nervous system (CNS) TRK fusion cancers. Not all patients had the recommended phase 2 dose, Dr. van Tilburg pointed out, but most did. Hence, 29 of the 38 patients received the 100 mg/m2 twice-daily, phase 2 dose until progression, withdrawal, or unacceptable toxicity.
Patients were young, with a median age of 2.3 years (range, 0.1 to 14.0 years). Almost two-thirds (61%) had prior surgery, 11% had prior radiotherapy, and 68% had prior systemic therapy. For 12 patients, larotrectinib was their first systemic therapy. The predominant tumor types were infantile fibrosarcoma (47%) and other soft tissue sarcoma (42%). And 47% of patients had NTRK3 fusions with ETV6, most of which were infantile fibrosarcoma.
Efficacy
Thirty-four patients were evaluable, and 32 had a reduction in tumor size, for an ORR of 94%, CR of 35%, and PR of 59%. Two patients with infantile fibrosarcoma had pathologic CRs—after treatment, no fibroid tissue in the tumors could be found. Median time to response was 1.8 months, median duration of treatment was 10.24 months, and 33 of 38 patients (87%) remained on treatment or underwent surgery with curative intent. As of the data cutoff of July 30, 2018, the secondary endpoints were not yet reached. However, 84% of responders were estimated to have a response duration of a year or more, and progression-free and overall survival looked very promising, according to Dr. van Tilburg.
Adverse events were primarily grades 1 and 2. The grades 3 and 4 treatment-related adverse events were quite few and consisted of increased alanine aminotransferase, decreased neutrophil count, and nausea. Longer follow-up of the patient safety profile is required, particularly since NTRK has multiple roles in neurodevelopment. The investigators recommended that routine testing for NTRK gene fusions in pediatric patients with cancer be conducted in appropriate clinical contexts.
In a discussion after the presentation, Daniel Alexander Morgenstern, MB BChir, PhD, of Great Ormond Street Hospital, London, UK, said that in many ways, the NTRK inhibitors have become the new poster child for precision oncology in pediatrics because of “these really spectacular results” with larotrectinib [and entrectinib]. One of the questions he raised regarding larotrectinib was the issue of CNS penetration, since patients with CNS cancer were not enrolled in the trial and preclinical data suggest limited CNS penetration for larotrectinib.
SOURCE: van Tilburg CM, et al. J Clin Oncol 37, 2019 (suppl; abstr 10010).
The studies were funded by Loxo Oncology, Inc., and Bayer AG.
Disclosures relevant to this presentation include consulting or advisory roles for Bayer for Drs. van Tilburg and Morgenstern. A few coauthors also had consulting/advisory roles or research funding from various companies, including Loxo and Bayer.
SFA awards grants to 15 researchers
Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant

Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant
Since its inception, the Sarcoma Foundation of America (SFA) has awarded research grants for the best, most promising research to cure sarcoma. This year SFA awarded $750,000 in research funds to 15 scientists as part of its 2019 SFA Research Grant program. The grants, worth $50,000 each, explore numerous sarcoma subtypes, multiple strategies, and different approaches to find effective treatments for many forms of the disease. Research projects are listed below alphabetically by investigator last name. More details are available on the SFA’s grant pages, available at https://www.curesarcoma.org/grant
Significant clinical response induced by vismodegib in advanced sarcoma: Hedgehog pathway inhibition
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
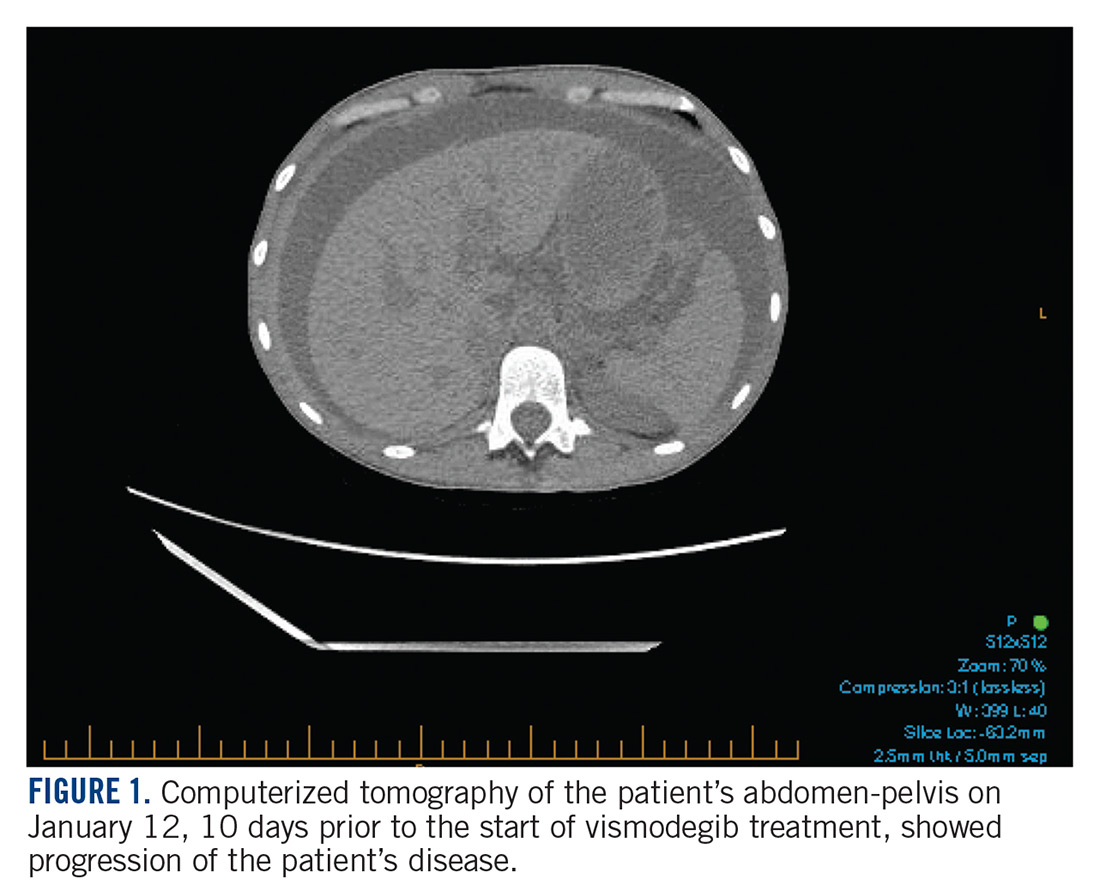
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
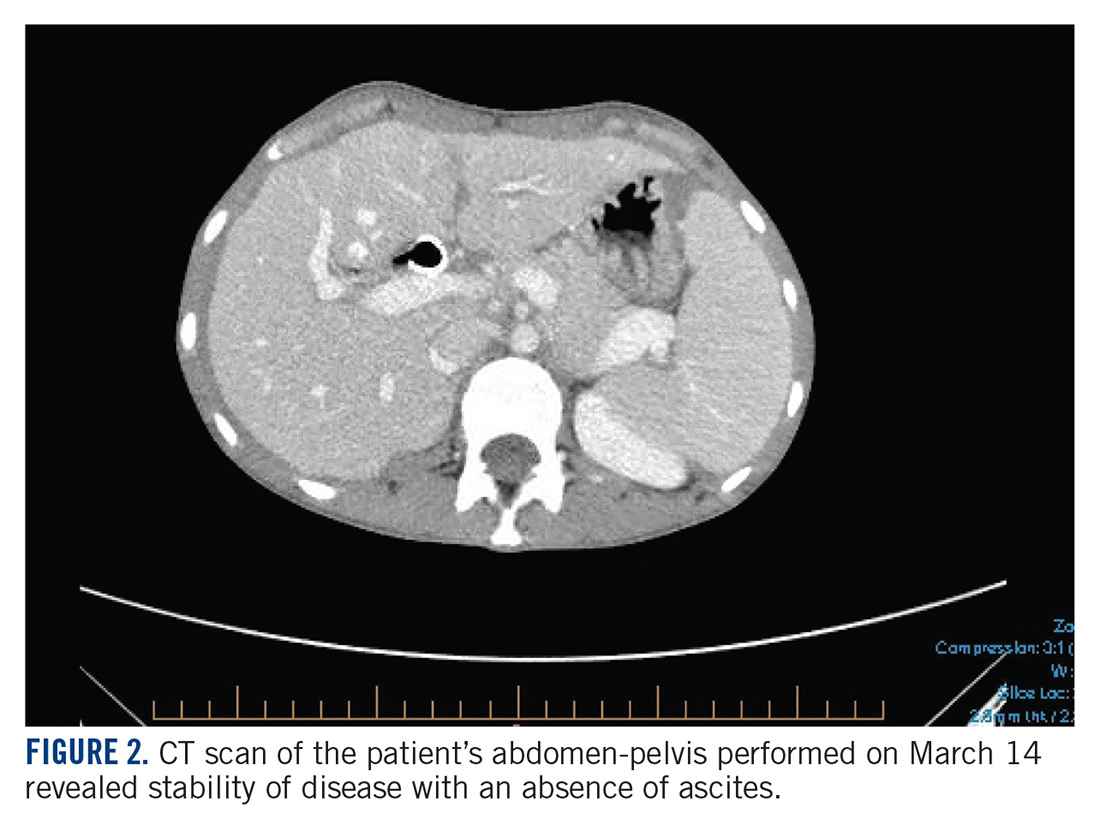
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
Spindle cell sarcomas are part of a rare, heterogeneous family of connective tissue tumors. These tumors are primarily treated with surgery and have a high risk of recurrence and distant metastasis with elevated mortality rates.1 Other than the evidence for first-line therapy with doxorubicin in advanced soft tissue sarcoma, little evidence exists to point to an optimal second-line therapy. This is due to the diversity of soft tissue sarcomas, which encompass approximately 70 different histologic subtypes that can each respond differently to treatment.2 As such, newer strategies, including immunotherapy and targeted molecular drugs, are being developed.
Quiescent in most adult tissues, the Hedgehog signaling pathway, when inappropriately activated, has been implicated in the development of multiple types of cancers, including basal cell, breast, prostate, hepatocellular, pancreatic, and brain cancer.3 The Hedgehog signaling pathway is an important regulator of cell growth and differentiation in early development, but when inappropriately activated can lead to cell proliferation and increased angiogenic factors, decreased apoptosis, and breakdown of tight junctions promoting cancer growth and metastasis.4 Recent data reveal that the Hedgehog pathway plays a specific role in activation of satellite cells, proliferation of myoblasts, and differentiation of skeletal muscle.5 Activation of this embryonic pathway has been implicated in embryonal rhabdoymyosarcoma, osteosarcoma, and chondrosarcoma.5-7
This pathway has recently been recognized as a therapeutic target, with the development of vismodegib, a targeted Hedgehog pathway inhibitor. This novel agent is in active use for treatment of advanced basal cell carcinoma and is currently undergoing trials for various other malignancies. Recently, a phase 2a basket study, called MyPathway, evaluated the use of targeted therapies in 35 different advanced refractory solid tumors harboring specific molecular alterations. Out of 21 patients with mutations in the Hedgehog pathway, 3 had a partial response to vismodegib—one had an unknown primary tumor, another a squamous skin cancer, and the third a salivary gland cancer.8 Vismodegib (GDC-0449) was also evaluated in a phase 2 multicenter clinical trial in patients with progressive advanced chondrosarcoma.7 Although the study did not meet its primary endpoint, the proportion of patients with non-progressive disease was 25.6% at 6 months. Investigators observed that the benefit occurred in the subset of patients with overexpression of the Hedgehog ligand. Genomic studies for mutations in SMO and PTCH genes were available for only 28 and 26 patients, respectively, of the 45 patients enrolled on the trial. While there were no mutations identified, expression data revealed that overexpression of the Hedgehog ligand was present in 65% of cases tested (13 out of 20 patients). In patients with stable disease at 6 months, all had overexpression of the Hedgehog ligand.7 These studies point to the potential use of vismodegib in both bone and soft tissue sarcomas, and more specifically, to the importance of genomic testing in these cases.
Case Presentation and Summary
This report describes the novel use of vismodegib, an oral Hedgehog signaling pathway inhibitor, in the treatment of a patient with metastatic soft tissue sarcoma.
An 18-year-old female with no particular previous illnesses was initially diagnosed with superficial soft tissue sarcoma overlying the right hip in 2013. Due to the complexity of pathology, a second opinion was requested and revealed atypical cellular spindle and epithelioid cells, morphologically and immunohistochemically suggestive of spindle cell sarcoma, not otherwise specified. She underwent negative-margin resection in January 2014. Her course was complicated by two recurrences in the right inguinal lymph nodes in July 2014 and July 2015. She was treated with lymph node dissection in 2014, followed by numerous right lymph node dissections and adjuvant radiation in 2015.
A routine computerized tomography (CT) scan of the thorax-abdomen and pelvis in August 2016 revealed recurrence of disease, with multiple lung nodules as well as metastases in the retroperitoneum. She received 6 cycles of gemcitabine and docetaxel with stability of disease. The patient was then started on a PI3K inhibitor as part of a clinical trial, as genotypic analysis of the tumor revealed an activating mutation of the PI3K gene. The patient’s course was complicated by acute obstructive renal failure requiring a double J stent for right-sided hydronephrosis.
Repeat imaging revealed disease progression, and the patient was then switched to liposomal doxorubicin alone for 4 months and then in combination with olaratumab. She received the combined treatment for a total of 3 months, which was then stopped when she was found to have new peritoneal implants and worsening ascites. At this time, tissue was sent for FoundationOne® next generation sequencing (NGS)-based genomic testing, and the patient received one dose of nivolumab.
In January 2018, 2 days after receiving her first dose of nivolumab, the patient required admission for worsening abdominal pain secondary to progression of her disease (FIGURE 1). She was found to have acute kidney injury on top of chronic kidney disease due to hydronephrosis requiring a left-sided double J stent. She also had transaminitis resulting from a common bile duct stricture treated with a biliary stent and worsening ascites requiring regular paracentesis. This was all in the context of new or growing metastatic implants.
At this time, the result of the FoundationOne genomic testing revealed PTCH1 loss of exons 1-24 and CDKN2A/B loss. Mutation of tumor suppressor gene PTCH1 leads to Hedgehog pathway activation and therefore the patient was started on vismodegib on January 22, 2018. She was discharged from the hospital in stable condition a day later, on January 23.
The patient’s clinical status subsequently improved, with significant reduction in her chronic abdominal pain and very minimal side effects. Clinically, the patient’s acute kidney injury resolved (from a creatinine of 272 μmol/L at discharge to 85 μmol/L after a week of treatment) and her liver enzymes normalized (from an alkaline phosphatase of 301 U/L to 83 U/L, and alanine transaminase of 111 U/L to 38 U/L). CT scan of her chest and abdomen, which was performed 1 month post treatment, revealed stability of disease with absence of ascites (FIGURE 2). The patient continued to have a good response to treatment for 6 months, with no recurrence of pain or ascites.
Six months later, in July 2018, the patient developed increasing pain and a CT scan revealed worsening of abdominopelvic carcinomatosis. In this context, vismodegib was discontinued on July 17. In the next 5 months, she went on to receive carboplatin and paclitaxel, gemcitabine, and nivolumab consecutively with no response. She was admitted to hospital on December 30 for a pain crisis. She passed away on January 9, 2019, from fecal peritonitis.
Discussion
To the best of our knowledge, this is the first patient with metastatic sarcoma to receive vismodegib, a Hedgehog signaling pathway inhibitor. She achieved an excellent clinical response with progression- free disease for approximately 6 months after starting treatment.
There is no current standard second- line treatment for metastatic soft tissue sarcoma. The choice of systemic therapy is histology-driven and therefore treatment is individualized for each patient. The future of oncology is heading towards an even more personalized approach with molecular profiling. Our case report highlights the relevance of genomic testing and targeted therapies, especially in cases of diverse clinical and biological disease behavior.
Molecular targeting is even more necessary in patients with advanced cancer who have failed multiple lines of treatment. As in our study, these patients can obtain a significant response with meaningful improvement in their quality of life. Future research is currently focusing on identifying new molecular targets in patients with advanced refractory cancers. Further studies will need to be done to determine whether these molecular targeting agents, such as vismodegib, lead to significant outcome changes in these patients.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
1. Collini P, Sorensen PHB, Patel S, et al. Sarcomas with spindle cell morphology. Semin Oncol. 2009;36(4):324-337.
2. Frezza AM, Stacchiotti S, Gronchi A. Systemic treatment in advanced soft tissue sarcoma: what is standard, what is new. BMC Med. 2017;15(1):109.
3. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properties and tumor microenvironment. Mol Cancer. 2016;15:24.
4. Abidi A. Hedgehog signaling pathway: a novel target for cancer therapy: vismodegib, a promising therapeutic option in treatment of basal cell carcinomas. Indian J Pharmacol. 2014;46(1): 3-12.
5. Belyea B, Kephart JG, Blum J, Kirsch DG, Linardic CM. Embryonic signaling pathways and rhabdomyosarcoma: contributions to cancer development and opportunities for therapeutic targeting. Sarcoma. 2012;2012:13.
6. Yao Z, Han L, Chen Y, et al. Hedgehog signalling in the tumourigenesis and metastasis of osteosarcoma, and its potential value in the clinical therapy of osteosarcoma. Cell Death Dis. 2018;9(6):701.
7. Italiano A, Le Cesne A, Bellera C, et al. GDC- 0449 in patients with advanced chondrosarcomas: a French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann Oncol. 2013;24(11):2922-2926.
8. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6): 536-542.
Clinical trials in sarcoma bring hope and promise
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
In this issue of The Sarcoma Journal, we highlight research and developments presented at the 2019 ASCO annual meeting. Despite the rarity of sarcoma, it was not lost among the thousands of abstracts, posters, and talks presented during the four-and-a-half days of the meeting.
In the past 5 years, there has been a resurgence of phase 3 clinical trials in sarcoma, including several large first-line studies comparing combination therapies to doxorubicin—the gold standard since the mid-1970s. None have shown superiority. Despite this, there has been a gradual improvement in overall survival. This is attributed to advances in the multidisciplinary management of sarcomas and available supportive care services as well as a better understanding of and emergent therapies for individual sarcoma subtypes.
In the United States, we have seen the approval of several agents in sarcoma: pazopanib, with a 3-month improvement in progression-free survival (PFS) over placebo; trabectedin in liposarcoma and leiomyosarcoma, with a 2.7-month improvement in PFS over dacarbazine; and eribulin, based on a liposarcoma subgroup analysis that showed a 7-month improvement in overall survival over dacarbazine.None of these therapies are approved in the US in the front-line setting; rather, all after a patient generally receives a doxorubicin- based therapy.
We have learned a great deal from these studies. They highlight some of the challenges in designing clinical trials in a rare and heterogeneous group of malignancies. The sarcoma community is very much focused on overcoming these challenges by designing clinical trials appropriate to the disease and the therapy that is being studied. This includes the incorporation of novel endpoints, imaging modalities, patient-reported outcome measures, and statistical methodologies to best serve the patient and to determine whether and how the therapy is helping them.
There is tremendous hope and promise in sarcoma due to significant advancements in our understanding of mesenchymal biology and of the genetic diversity in these diseases. This has led to an influx of promising agents and trials, many of which have transformed treatment paradigms on specific sarcoma subtypes. This issue provides a glimpse into the progress being made.
From Dr. Tap’s plenary presentation at ASCO 2019
The Sarcoma Journal Author Guidelines
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to [email protected]. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact [email protected] to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to [email protected]
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to [email protected]. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact [email protected] to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to [email protected]
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Submission
Quick Links
Guidelines for Submitting:
Review and State-of-the-Art Papers
Manuscript Submission
We require electronic submission of manuscripts. Please submit your manuscript and tables as a Microsoft Word file attached to an e-mail to [email protected]. We will confirm successful receipt of your manuscript by e-mail.
Cover Letter: The cover letter should include the name, address, telephone numbers, and e-mail address of the corresponding author. The letter should make it clear that the manuscript has not been published in another journal and is not under consideration by another journal, and that the final manuscript has been seen and approved by all authors.
Conflict of Interest: The authors should disclose in the cover letter any affiliations or financial arrangements with any company whose product appears prominently in the manuscript or with any company making a competing product. Such information will be kept in confidence while the paper is under review. If the article is accepted for publication, full disclosure will be required.
Third-party Support/Assistance: TSJ does not accept articles that have been developed by or written with financial support from a commercial entity (eg, pharmaceutical company or medical device manufacturer) or whose authors have received writing assistance from a commercially sponsored third party, such as a medical education company or a publication planner. Authors who have received such support or funding (and entities that have supported such articles) should contact [email protected] to explore opportunities to publish sponsored supplements to TSJ.
Original Research Exception. TSJ does accept original research articles in which the study was funded by a pharmaceutical company or in which the author(s) are employed by one, provided that all funding and affiliations are fully disclosed. Authors may not, however, receive any form of writing assistance. (See above).
The Peer Review Process: All TSJ articles undergo peer review to determine whether the submission meets the needs of TSJ readers and thus is suitable for publication.
Questions about this policy should be directed to [email protected]
Manuscript Style and Format
Manuscripts should conform to the International Committee of Medical Journal Editors (ICMJE) Recommendations, which are available at www.icmje.org. For questions about style, consult the American Medical Association Manual of Style: A Guide for Authors and Editors. 10th ed. New York, NY.: Oxford University Press, 2007. Keep abbreviations and acronyms to a minimum and spell out on first reference, eg, Sarcoma Foundation of America (SFA). Regarding medications, generic drug names should generally be used. When proprietary brands are used in research, include the brand name in parentheses in the Methods section.
The title page should include from top to bottom: article title; name, degree, and institutional affiliation of each author; previous presentation of the work, if any; name and address/fax/e-mail of the corresponding author; and manuscript word count, excluding tables, figures, and abstract. Pages should be numbered consecutively in the upper right corner beginning with the title page.
Please see the links, above, for detailed information on specific article types and departments.
Acknowledgements: This section is optional and may be used to acknowledge substantial contributions to the research or preparation of the manuscript made by individuals other than the authors.
Conflict of Interest: Each article should have a statement acknowledging any potential conflict of interest. If there is none, please state "No conflict of interest."
Permissions: Contributors to TSJ may be asked to obtain permission from the author and publisher for the use of quotes, illustrations, tables, and other materials taken from previously published works not in the public domain. The original source should be mentioned in the table footnote.
Please note the following:
- Papers that exceed the stipulated word counts will be returned to the author(s) for editing before the paper is sent out for review.
- Papers in which the references do not follow style will also be returned to the author for revision.
Original Research Reports
These are reports on randomized trials, interventional studies, cohort studies, case-control studies, epidemiologic assessments, other observational studies, surveys, cost-effectiveness analyses, and studies of screening and diagnostic tests as they pertain to sarcoma.
Original research reports will:
- Be no more than 4,500 words (including a structured abstract, references, and figure titles and legends).
- Have a structured abstract of no more than 250 words (AMA Manual chapter 2.5.1).
- Have a title (headline) of no more than 100 characters.
- Have no more than 5 tables and/or figures (AMA Manual chapter 4).
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; structured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 50 or fewer references, which will be in AMA style (AMA Manual chapter 3).
- Begin page numbering with the title page.
- Either provide sex-specific data (when appropriate) in describing outcomes of epidemiologic analyses or clinical trials, or specifically state that no gender-based differences were present.
Review and State-of-the-Art Papers
The editors will consider invited and uninvited review papers. These manuscripts gather and summarize information from current literature and data sources on clinical topics. They should do the following:
- Focus on novel approaches and cutting-edge therapies, as well as diagnoses, prognoses, and management.
- Include critical assessments thereof.
- Explore their potential for changing treatment.
Review articles are often used as guides in the practice setting, and therefore they must be systematic, must include relevant data, and must not be influenced by the authors’ opinions or biases (AMA Manual chapter 1.2).
The search and selection processes for research sources, such as databases, should be described in the manuscript. The research sources should be as current as possible, preferably with the search having been conducted within a few months of submission. Authors should detail in their cover letters how their review differs from existing reviews on the subject.
Review and state-of-the-art manuscripts will:
- Be no more than 5,000 words (including an unstructured abstract, reference list, tables, and figure titles and legends).
- Have an unstructured abstract of 250 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Have no more than 4 tables and/or figures, which should be submitted as separate files.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Limit figures, clinical images, and tables to those necessary to highlight key data.
- Be arranged as follows: title page; unstructured abstract and key words; abbreviations list; text; acknowledgments (if applicable); references; figure titles and legends; and tables.
- Have 30 or fewer references.
- Begin page numbering with the title page.
Case Reports
These reports usually describe a step-by-step approach to clinical decision making in the diagnosis and treatment of a patient who has an unusual or complicated presentation or diagnosis. They can be accompanied by a brief review of pertinent, current literature.
A case report will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the case details are presented.
- Have no more than 4 tables and/or figures.
- Include figures that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Adequately de-identify all patient information. If identifying information or figures are included, express written permission from the patient(s) must be provided at the time of manuscript submission.
- Begin page numbering with the title page.
Research Letters
New or preliminary research findings may be considered for publication as research letters.
A research letter will:
- Be limited to 2,000 words (including references, tables, and figure titles and legends).
- Have an unstructured abstract of 50 or fewer words (AMA Manual chapter 2.5.2-2.5.3).
- Have a title (headline) of no more than 100 characters.
- Begin with a brief summary before the details are presented.
- Have no more than 2 tables and/or figures.
- Include figures (if any) that are submitted as separate, high-resolution files.
- Have no more than 10 references.
- Begin page numbering with the title page.
Commentaries
Succinct opinion pieces will also be considered. These can address any current topic that has a bearing on clinical practice: research findings, health policy and/or law, ethics, or practice economics. The arguments should be focused and succinctly presented.
A commentary will:
- Have up to 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 1,200 words long.
- Have no more than 8 references (AMA Manual chapter 3).
- Have a title of 7 or fewer words.
- Begin page numbering with the title page.
Letters to the Editor
Letters to the editor should focus on a specific article that has been published in The Sarcoma Journal.
A letter to the editor will:
- Have no more than 3 authors, and will provide the full name, academic degrees, and a single institutional affiliation for each author.
- Provide disclosures, if relevant to the topic of the letter, for each letter author.
- Provide the e-mail address for the corresponding letter author.
- Be no more than 400 words long.
- Have no more than 5 references.
- Have a title of 5-7 words.
- Begin page numbering with the title page.
Letters will be sent for response to the authors of the original article. This response may be published or sent directly to the commentator at the discretion of the editor. Letters will be published at the discretion of the editors and are subject to abridgement and editing for style and content. Questions or comments that could be addressed directly to authors of the original article (including complaints about missed citations) should be sent directly to those authors.
Submission Checklist
Before you send in your manuscript for review, please check the following:
- Cover letter: Is a cover letter included with your manuscript submission?
- Title page: Is the title page presented as outlined?
- Corresponding author: Have you designated a corresponding author and provided current, correct contact information in the format described in AMA Manual chapter 2.10.4?
- Article authors: Have you provided first and last names and highest degrees for each author, according to the formats shown in AMA Manual chapter 2.2.1–2.2.4?
- Author affiliations: Have you included affiliations for each author according to AMA Manual chapter 2.3.3., as well as their current e-mail addresses?
- Word count: Does the word count include abstract, main running text, references, and tables, and does it appear on the title page of the manuscript?
- Formatting: Is your manuscript double spaced, and have you ensured that it is minimally formatted?
- Abstract: Have you included a structured or unstructured abstract (as stipulated by your article type) that has been formatted according to AMA Manual chapter 2.5 and the specific requirements of The Sarcoma Journal?
- Reference citations (“callouts”) in running text/tables: Per AMA Manual chapter 3, are the callouts in superscripts and in numerical order, and does each one match the corresponding reference in the reference list?
- Reference list: Are all references in the reference list complete, accurate, numerically ordered to match the callouts, and formatted according to AMA Manual chapter 3?
- Tables: Have all tables been prepared according to AMA Manual chapter 4.1?
- Figures: Do all figures meet the stated quality requirements to ensure best possible print reproduction? Are their titles and legends formatted according to AMA Manual chapter 4.2? Have they been prepared and uploaded as separate files that are labeled with the correct naming convention?
- Permissions: Have you obtained permission for use of copyrighted material from other sources (including the Web), and have all appropriate forms been completed and included with the submission, according to AMA Manual chapter 5.6?
- Final read-through: Have you checked the spelling and grammar within your manuscript? Does its outline match its content?
Rozlytrek approved for ROS1-positive metastatic NSCLC, cancers with NTRK gene fusion defects
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.

Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.
Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
Rozlytrek (entrectinib) has been approved to treat cancers with neurotrophic tyrosine receptor kinase (NTRK) gene fusion defects in adults and adolescents for whom there are no effective treatments, the Food and Drug Administration announced in a press release.
Entrectinib was also approved for the treatment of adults with metastatic non–small cell lung cancers that are ROS1-positive.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue-agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” FDA Acting Commissioner Ned Sharpless, MD, said in the release.
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than on the original tumor’s location. The previous tissue-agnostic indications approved by the FDA were pembrolizumab (Keytruda) for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib (Vitrakvi) for NTRK gene fusion tumors in 2018.
The approval of entrectinib was granted to Genentech. “Rozlytrek is the first FDA-approved treatment that selectively targets both ROS1 and NTRK fusions, and, importantly, has also shown responses in these rare cancer types that have spread to the brain,” Sandra Horning, MD, chief medical officer and head of global product development for Genentech, said in a separate press release.
Foundation Medicine will submit Foundation One CDx to the FDA for approval as a companion diagnostic for entrectinib, according to the Genentech release; an FDA-approved companion diagnostic for entrectinib is not available at this time.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK fusion–positive tumors by relying on efficacy information obtained primarily in adults,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
Entrectinib was evaluated in four clinical trials that included 54 adults with NTRK fusion–positive tumors. The overall response rate was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for 9 months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid, and colon/rectum.
Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
The most serious side effects of entrectinib are heart failure, central nervous system effects, changes in sleep pattern, skeletal fractures, hepatotoxicity, hyperuricemia, QT prolongation, and vision disorders. Females of reproductive age and males with a female partner of reproductive potential are advised to use effective contraception during treatment; the drug may cause harm to a developing fetus or newborn baby.
Genentech must provide additional clinical trial data to the FDA as a condition of the approval.
Gastrointestinal Stromal Tumors: Management of Localized Disease
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17 KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18 PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.

When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17 KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18 PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumor of the gastrointestinal (GI) tract and arise from the interstitial cells of Cajal of the myenteric plexus. These tumors are rare, with about 1 case per 100,000 persons diagnosed in the United States annually, but may be incidentally discovered in up to 1 in 5 autopsy specimens of older adults.1,2 Epidemiologic risk factors include increasing age, with a peak incidence between age 60 and 65 years, male gender, black race, and non-Hispanic white ethnicity. Germline predisposition can also increase the risk of developing GISTs; molecular drivers of GIST include gain-of-function mutations in the KIT proto-oncogene and platelet-derived growth factor receptor α (PDGFRA) gene, which both encode structurally similar tyrosine kinase receptors; germline mutations of succinate dehydrogenase (SDH) subunit genes; and mutations associated with neurofibromatosis type 1.
GISTs most commonly involve the stomach, followed by the small intestine, but can arise anywhere within the GI tract (esophagus, colon, rectum, and anus). They can also develop outside the GI tract, arising from the mesentery, omentum, and retroperitoneum. The majority of cases are localized or locoregional, whereas about 20% are metastatic at presentation.1 GISTs can occur in children, adolescents, and young adults. Pediatric GISTs represent a distinct subset marked by female predominance and gastric origin, are often multifocal, can sometimes have lymph node involvement, and typically lack mutations in the KIT and PDGFRA genes.
This review is the first of 2 articles focusing on the diagnosis and management of GISTs. Here, we review the evaluation and diagnosis of GISTs along with management of localized disease. Management of advanced disease is reviewed in a separate article.
Case Presentation
A 64-year-old African American man with progressive iron deficiency and abdominal discomfort undergoes upper and lower endoscopy and is found to have a bulging mass within his abdominal cavity. He undergoes a computed tomography (CT) evaluation of the chest, abdomen, and pelvis with contrast, which reveals the presence of a 10-cm gastric mass, with no other lesions identified. He undergoes surgical resection of the mass and presents for review of his pathology and to discuss his treatment plan.
What histopathologic features are consistent with GIST?
What factors are used for risk stratification and to predict likelihood of recurrence?
Clinical Presentation and Diagnosis
Most patients present with symptoms of overt or occult GI bleeding or abdominal discomfort, but a significant proportion of GISTs are discovered incidentally. Lymph node involvement is not typical, except for GISTs occurring in children and/or with rare syndromes. Most syndromic GISTs are multifocal and multicentric. After surgical resection, GISTs usually recur or metastasize within the abdominal cavity, including the omentum, peritoneum, or liver. These tumors rarely spread to the lungs, brain, or bones; when tumor spread does occur, it tends to be in heavily pre-treated patients with advanced disease who have been on multiple lines of therapy for a long duration of time.
The diagnosis usually can be made by histopathology. Specimens can be obtained by endoscopic ultrasound (EUS)– or CT-guided methods, the latter of which carries a very small risk of contamination from percutaneous biopsy. In terms of morphology, GISTs can be spindle cell, epithelioid, or mixed neoplasms. Epithelioid tumors are more commonly seen in the stomach and are often PDGFRA-mutated or SDH-deficient. The differential diagnosis includes other soft-tissue GI wall tumors such as leiomyosarcomas/leiomyomas, germ cell tumors, lymphomas, fibromatosis, and neuroendocrine and neurogenic tumors. A unique feature of GISTs that differentiates them from leiomyomas is near universal expression of CD117 by immunohistochemistry (IHC); this characteristic has allowed pathologists and providers to accurately distinguish true GISTs from other GI mesenchymal tumors.3 Recently, DOG1 (discovered on GIST1) immunoreactivity has been found to be helpful in identifying patients with CD117-negative GISTs. Initially identified through gene expression analysis of GISTs, DOG1 IHC can identify the common mutant c-Kit-driven CD117-positive GISTs as well as the rare CD117-negative GISTs, which are often driven by mutated PDGFRA.4 Importantly, IHC for KIT and DOG1 are not surrogates for mutational status, nor are they predictive of tyrosine kinase inhibitor (TKI) sensitivity. If IHC of a tumor specimen is CD117- and DOG1-negative, the specimen can be sent for KIT and PDGFRA mutational analysis to confirm the diagnosis. If analysis reveals that these genes are wild-type, then IHC staining for SDH B (SDHB) should follow to assess for an SDH-deficient GIST (negative staining).
Risk Stratification for Recurrence
The clinical behavior of GISTs can be variable. Some are indolent, while others behave more aggressively, with a greater malignant potential and a higher propensity to recur and metastasize. Clinical and pathologic features can provide important prognostic information that allows providers to risk-stratify patients. Various institutions have assessed prognostic variables for GISTs. In 2001, the National Institutes of Health (NIH) held a GIST workshop that proposed an approach to estimating metastatic risk based on tumor size and mitotic index (NIH or Fletcher criteria).5 Joensuu et al later proposed a modification of the NIH risk classification to include tumor location and tumor rupture (modified NIH criteria or Joensuu criteria).6-8 Similarly, the Armed Forces Institute of Pathology (AFIP) identified tumor site as a prognostic factor, with gastric GISTs having the best prognosis (AFIP-Miettinen criteria).9-11 Tabular schemes were designed which stratified patients into discrete groups with ranges for mitotic rate and tumor size. Nomograms for ease of use were then constructed utilizing a bimodal mitotic rate and included tumor site and size.12 Finally, contour maps were developed, which have the advantage of evaluating mitotic rate and tumor size as continuous nonlinear variables and also include tumor site and rupture (associated with a high risk of peritoneal metastasis) separately, further improving risk assessment. These contour maps have been validated against pooled data from 10 series (2560 patients).13 High-risk features identified from these studies include tumor location, size, mitotic rate and tumor rupture and are now used for deciding on the use of adjuvant imatinib and as requirements to enter clinical trials assessing adjuvant therapy for resected GISTs.
Case Continued
The patient’s operative and pathology reports indicate that the tumor is a spindle cell neoplasm of the stomach that is positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. Resection margins are negative. There are 10 mitoses per 50 high-power fields (HPF). Per the operative report, there was no intraoperative or intraperitoneal tumor rupture. Thus, while his GIST was gastric, which generally has a more favorable prognosis, the tumor harbors high-risk features based on its size and mitotic index.
What further testing should be requested?
Molecular Alterations
It is recommended that a mutational analysis be performed as part of the diagnostic work-up of all GISTs.14 Mutational analysis can provide prognostic and predictive information for sensitivity to imatinib and should be considered standard of care. It may also be useful for confirming a GIST diagnosis, or, if negative, lead to further evaluation with an IHC stain for SDHB. The c-Kit receptor is a member of the tyrosine kinase family and, through direct interactions with stem cell factor (SCF), can upregulate the PI3K/AKT/mTOR, Ras/Raf/MEK/ERK, and JAK-STAT pathways, resulting in transcription and translation of genes that enhance cell growth and survival.15 The cell of origin of GISTs, the interstitial cells of Cajal, are dependent on the SCF–c-Kit interaction for development.16 Likewise, the large majority of GISTs (about 70%) are driven by upregulation and constitutive activation of c-Kit, which is normally autoinhibited. About 80% of KIT mutations involve exon 11; these GISTs are most often associated with a gastric location and are associated with a favorable recurrence-free survival (RFS) rate with surgery alone.17 KIT exon 9 mutations are much less common, encompassing only about 10% of GIST KIT mutations, and GISTs with these mutations are more likely to arise from the small bowel.17
About 8% of GISTs harbor gain-of-function PDGFRA driver mutations rendering constitutively active PDGFRA.18 PDGFRA mutations are mutually exclusive from KIT mutations, and PDGFRA-mutated tumors most often occur in the stomach. PDGFRA mutations generally are associated with a lower mitotic rate and gastric location. Identification of the PDGFRA D842V mutation on exon 18, which is the most common, is important, as it is associated with imatinib resistance, and these patients should not be offered imatinib.19
Several other mutations associated with GISTs outside of the KIT and PDGFRA spectrum have been identified. About 10% of GISTs are wildtype for KIT and PDGFRA, and not all KIT/PDGFRA-wildtype GISTs are imatinib-sensitive and/or respond to other TKIs.18 These tumors may harbor aberrations in SDH and NF1, or less commonly, BRAF V600E, FGFR, and NTRK.20,21 SDH subunits B, C and D play a role in the Krebs cycle and electron transport chain. Germline mutations in these SDH subunits can result in the Carney-Stratakis syndrome characterized by the dyad of multifocal GISTs and multicentric paragangliomas.22 This syndrome is most likely to manifest in the pediatric or young adult population. In contradistinction is the Carney triad, which is associated with acquired loss of function of the SDHC gene due to promoter hypermethylation. This syndrome classically occurs in young women and is characterized by an indolent-behaving triad of multicentric GISTs, non-adrenal paragangliomas, and pulmonary chondromas.23 Like PDGFRA D842V–mutated GISTs, SDH-deficient and NF1-associated GISTs are considered imatinib resistant, and these patients should not be offered imatinib therapy.14
Case Continued
The patient’s GIST is found to harbor a KIT exon 11 single codon deletion. He appears anxious and asks to have everything done to prevent his GIST from coming back and to improve his lifespan.
What are the next steps in the management of this patient?
Management
A multidisciplinary team approach to the management of all GISTs is essential and includes input from radiology, gastroenterology, pathology, medical and surgical oncology, nuclear medicine, and nursing.
Surgical Resection
Small esophagogastric and duodenal GISTs ≤ 2 cm can be asymptomatic and managed with serial endoscopic surveillance, typically every 6 to 12 months, with biopsies if the tumors increase in size. GISTs larger than 2 cm require surgical resection, with resection of the full pseudocapsule and an R0 resection, if possible, since larger GISTs carry a higher risk of growth and recurrence. If an R0 resection would lead to significant morbidity or functional sequelae, an R1 may suffice. Rectal GISTs are an exception, where microscopic margins have been shown to be associated with an increased risk of local failure.24 It is important to explore the abdomen thoroughly for peritoneal, rectovaginal, and vesicular implants and metastasis to the liver. A formal lymph node dissection is not necessary because lymph nodes are rarely involved and should only be removed when clinically suspicious. Tumor rupture must be avoided. A laparoscopic approach should only be considered for smaller tumors, since there is a risk of tumor rupture with larger tumors.14
When is adjuvant imatinib indicated?
Adjuvant Imatinib
Among patients with local or locally advanced GISTs, the risk of death from recurrence with surgery alone can be high, with a historical 5-year overall survival (OS) of about 35%.25 As a result, multiple studies have assessed the benefit of adjuvant imatinib, which is now considered standard of care for patients with imatinib-sensitive, high-risk GISTs. In addition to inhibiting BCR-ABL, imatinib mesylate inhibits multiple other receptor tyrosine kinases, including PDGFR, SCF and c-Kit. As a result, imatinib has demonstrated in vitro inhibition of cell proliferation and apoptosis and clinical activity against GISTs expressing CD117.26 Importantly, adjuvant imatinib should only be offered to patients with imatinib-sensitive mutations, such as KIT exon 11 and KIT exon 9 mutations. Adjuvant imatinib should not be offered to patients with imatinib-insensitive mutations such as PDGFR 842V, NF1, or BRAF-related or SDH-deficient GISTs.
The ACOSOG Z9000 was the first study of adjuvant imatinib in patients with resected GISTs.25 This was a single-arm, phase 2 study involving 106 patients with surgically resected GISTs deemed high-risk for recurrence, defined as size > 10 cm, tumor rupture, or up to 4 peritoneal implants. Patients were treated with imatinib 400 mg daily for 1 year. The primary and secondary endpoints were OS and RFS, respectively. Long-term follow-up of this study demonstrated 1-, 3-, and 5-year OS of 99%, 97%, and 83%, and 1-, 3-, and 5-year RFS of 96%, 60%, and 40%, which compared favorably with historical controls. In a multivariable analysis, increasing tumor size, small bowel location, KIT exon 9 mutation, high mitotic rate, and older age were independent risk factors for a poor RFS.25 It is important to note that the benefit of adjuvant imatinib waned after discontinuation of therapy, creating a rationale to study adjuvant imatinib for longer periods of time.
As a result of the promising phase 2 data, ACOSOG opened a phase 3 randomized trial (Z9001) comparing 1 year of adjuvant imatinib to placebo among patients with surgically resected GISTs that were > 3 cm in size and that stained positive for CD117 on pathology. The trial accrued 713 patients and was stopped early at a planned interim analysis, which revealed a 1-year RFS of 98% for imatinib versus 83% for placebo (hazard ratio [HR], 0.35; P < 0.001). The 1-year OS did not differ between the 2 arms (92.2% vs 99.7%; HR, 0.66; P = 0.47).27 When comparing the 2 arms, imatinib was associated with a higher RFS among patients with a KIT exon 11 deletion, but not among patients with other KIT mutation types, PDGFRA mutations, or who were KIT/PDGFRA wildtype.28 Imatinib was granted approval by the US Food and Drug Administration (FDA) for the adjuvant treatment of high-risk GISTs based on the results of the ACOSOG Z9001 trial.
The EORTC 62024 study was a randomized placebo-controlled trial assessing the benefit of 2 years of adjuvant imatinib.29 Patients had to be considered intermediate or high risk per the 2002 NIH consensus classification to be eligible. The trial enrolled 918 patients. The 5-year OS rate, the original primary endpoint, did not differ between the 2 groups (100% vs 99%). The 3-year and 5-year RFS rates, secondary endpoints, were significantly longer among patients treated with imatinib (84% vs 66% and 69% vs 63%, respectively). Again, it was noted that the benefit of imatinib waned over time after treatment discontinuation.
The Scandinavian Sarcoma Group (SSG XVIII) trial was a prospective randomized phase 3 trial that compared 3 years versus 1 year of adjuvant imatinib.30 Patients had to be enrolled within 12 weeks of the postoperative period and had to have GISTs that were CD117-positive and with a high estimated risk of recurrence, per the modified NIH consensus criteria (size > 10 cm, > 10 mitoses per 50 HPF, diameter > 5 cm with mitotic count > 5, or tumor rupture before or at surgery). Three years of adjuvant imatinib was associated with a 54% reduction in the hazard for recurrence at 5 years (65.6% vs 47.9%; HR, 0.46; P < 0.001) and a 55% reduction in the hazard for death at 5 years (OS 92% vs 81.7%; HR, 0.45; P = 0.02). Based on the results of this study, the FDA granted approval for the use of 3 years of adjuvant imatinib in patients with high-risk resected GISTs.
The observation that a longer duration of adjuvant imatinib was associated with superior RFS and OS led to studies to further explore longer durations of adjuvant imatinib. The PERSIST-5 (Postresection Evaluation of Recurrence-free Survival for Gastrointestinal Stromal Tumors With 5 Years of Adjuvant Imatinib) was a multicenter, single-arm, phase 2 prospective study of adjuvant imatinib with a primary endpoint of RFS after 5 years.31 Patients had to have an intermediate or high risk of recurrence, which included GISTs at any site > 2 cm with > 5 mitoses per 50 HPF or nongastric GISTs that were ≥ 5 cm. With 91 patients enrolled, the estimated 5-year RFS was 90% and the OS was 95%. Of note, about half of the patients stopped treatment early due to a variety of reasons, including patient choice or adverse events. Importantly, there were no recurrences in patients with imatinib-sensitive mutations while on therapy. We know that in patients at high risk of relapse, adjuvant imatinib delays recurrence and improves survival, but whether any patients are cured, or their survival curves are just shifted to the right, is unknown. Only longer follow-up of existing studies, and the results of newer trials utilizing longer durations of adjuvant treatment, will help to determine the real value of adjuvant therapy for GIST patients.32 Based on this study, it would be reasonable to discuss a longer duration of imatinib with patients deemed to be at very high risk of recurrence and who are tolerating therapy well. We are awaiting the data from the randomized phase 3 Scandinavian Sarcoma Group XII trial comparing 5 years versus 3 years of adjuvant imatinib therapy, and from the French ImadGIST trial of adjuvant imatinib for 3 versus 6 years. A summary of the aforementioned key adjuvant trials is shown in the Table.
When imatinib is commenced, careful monitoring for treatment toxicities and drug interactions should ensue in order to improve compliance. Dose density should be maintained if possible, as retrospective studies suggest suboptimal plasma levels are associated with a worse outcome.33
When should neoadjuvant imatinib be considered?
Neoadjuvant Imatinib
Neoadjuvant imatinib should be considered for patients requiring total gastrectomy, esophagectomy, or abdominoperineal resection of the rectum in order to reduce tumor size, limit subsequent surgical morbidity, mitigate tumor bleeding and rupture, and aid with organ preservation. Patients with rectal GISTs that may otherwise warrant an abdominoperineal resection should be offered a trial of imatinib in the neoadjuvant setting. There is no evidence for the use of any other TKI aside from imatinib in the neoadjuvant or adjuvant setting. With neoadjuvant imatinib, it is difficult to accurately assess the mitotic rate in the resected tumor specimen.
The RTOG 0132/ACRIN 6665 trial was a prospective phase 2 study evaluating the efficacy of imatinib 600 mg daily in the perioperative setting.34 The trial enrolled 50 patients, 30 with primary GISTs (group A) and 22 with recurrent metastatic GISTs (group B). Based on data from the metastatic setting revealing a time to treatment response of about 2.5 months, patients were treated with 8 to 12 weeks of preoperative imatinib followed by 2 years of adjuvant imatinib. Imatinib was stopped 24 hours preoperatively and resumed as soon as possible postoperatively. In group A, 7% of patients achieved a partial response (PR), 83% achieved stable disease, and 2-year progression-free survival (PFS) and OS were 83% and 93%, respectively. In group B, 4.5% of patients achieved a PR, 91% achieved stable disease, and 4.5% experienced progressive disease in the preoperative period; the 2-year PFS and OS were 77% and 91%, respectively. The results of this trial demonstrated the feasibility of using perioperative imatinib with minimal effects on surgical outcomes and set the rationale to use neoadjuvant imatinib in select patients with borderline resectable or rectal GISTs. Another EORTC pooled analysis from 10 sarcoma centers revealed that after a median of 10 months of neoadjuvant imatinib, 83.2% of patients achieved an R0 resection and only 1% progressed during treatment.35 After a median follow-up of 46 months, the 5-year disease-free survival and OS were 65% and 87%, respectively.
Mutational testing should be performed beforehand to ensure the tumor is imatinib-sensitive. If a KIT exon 9 mutation is identified, then 400 mg twice daily should be considered (given the benefit seen with 800 mg imatinib for advanced GIST patients), although there are no studies to confirm this practice. Neoadjuvant imatinib is recommended for a total of 6 to 12 months to ensure maximal tumor debulking, but with very close monitoring and surgical input for disease resistance and growth.14 Imatinib should be stopped 1 to 2 days preoperatively and resumed once the patient has recovered from surgery for a total of 3 years (pre-/postoperatively combined). Neoadjuvant therapy has been shown to be safe and effective, but there have been no randomized trials to assess survival.
What is appropriate surveillance for resected GISTs?
Surveillance
There have been no randomized studies to guide the management of surveillance after surgical resection and adjuvant therapy. There is no known optimal follow-up schedule, but several have been proposed.13,36 Among high-risk patients, it is suggested to image every 3 to 6 months during adjuvant therapy, followed by every 3 months for 2 years after discontinuing therapy, then every 6 months for another 3 years and annually thereafter for an additional 5 years. High-risk patients usually relapse within 1 to 3 years after finishing adjuvant therapy, while low-risk patients can relapse later given that their disease can be slower growing. It has been recommended that low-risk patients undergo imaging every 6 months for 5 years, with follow-up individualized thereafter. Very-low-risk patients may not require more than annual imaging. Because most relapses occur within the peritoneum or liver, imaging should encompass the abdomen and pelvis. Surveillance imaging usually consists of CT scans of the abdomen and pelvis. MRI scans can be utilized for patients at lower risk or who are out several years in order to avoid excess radiation exposure. MRI is also specifically helpful for rectal and esophageal lesions. Chest CT or chest radiograph and bone scan are not routinely required for follow-up.
Case Conclusion
The patient receives adjuvant imatinib and experiences grade 2 myalgias, periorbital edema, and macrocytic anemia, which result in imatinib discontinuation after 3 years of treatment. He is seen every 3 to 6 months and a contrast CT abdomen and pelvis is obtained every 6 months for 5 years. During this 5-year follow-up period, he does not have any clinical or radiographic evidence of disease recurrence.
Further follow-up of this patient is presented in the second article in this 2-part review of management of GISTs.
Key Points
- GISTs are the most common mesenchymal neoplasms of the GI tract and can occasionally occur in extragastrointestinal locations as well.
- GISTs encompass a heterogeneous family of tumor subsets with different natural histories, mutations, and TKI responsiveness.
- Surgery is the mainstay of treatment for localized GISTs, with cure rates greater than 50%.
- For very small (< 2 cm) esophagogastric GISTs, endoscopic ultrasound evaluation and follow-up is recommended.
- For tumors ≥ 2 cm, biopsy and excision is the standard approach.
- For localized GISTs, complete surgical resection (R0) is standard treatment, with no lymphadenectomy for clinically negative lymph nodes.
- Mutational analysis should be considered standard of practice. It can be helpful for confirming the diagnosis and can be predictive and prognostic in determining specific TKI therapy and dose.
- Adjuvant imatinib at a dose of 400 mg for 3 years is standard of care for GISTs that are at high risk of relapse and are imatinib-sensitive, and it is the only TKI approved for adjuvant therapy. Patients with PDGFRA D842V, NF1, BRAF or SDH-deficient GISTs should not receive adjuvant imatinib therapy.
- Neoadjuvant therapy can be utilized for sites where extensive resection would lead to significant morbidity. It should be given for 6 to 12 months, but patients need to be monitored closely for tumor growth.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
3. Miettinen M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
4. West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutational status. Am J Pathol. 2004;165:107-113.
5. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81-89.
6. Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39:1411-1419.
7. Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumor. Br J Surg. 2010;97:1854-1859.
8. Holmenbakk T, Bjerkehagen B, Boye K, et al. Definition and clinical significance of tumor rupture in gastrointestinal stromal tumours of the small intestine. Br J Surg. 2016;103:684-691.
9. Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82-87.
10. Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477-489.
11. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29:52-68.
12. Gold JS, Gonen M, Gutierrez A, et al. Development and validation of a prognostic nomogram for recurrence-free survival after complete surgical resection of localized primary gastrointestinal stromal tumour: a retrospective analysis. Lancet Oncol. 2009;10:1045-1052.
13. Joensuu H, Vehtari A, Rihimaki J et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265-274.
14. Casali PG, Abecassis N, Bauer S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow up. Ann Oncol. 2018;29(Supplement_4): iv267.
15. Jing L, Yan-Ling W, Bing-Jia C, et al. The c-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
16. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
17. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33:634-642.
18. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813-3825.
19. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342-4349.
20. Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206-214.
21. Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339.
22. Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132-139.
23. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543-552.
24. Jakob J, Mussi C, Ronellenfitsch U, et al. Gastrointestinal stromal tumor of the rectum: results of surgical and multimodality therapy in the era of imatinib. Ann Surg Oncol. 2013;20:586-592.
25. DeMatteo RP, Ballman KV, Antonescu CR, et al. Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor (GIST): ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Ann Surg. 2013;258:422-429.
26. Gleevac (imatinib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2016.
27. DeMatteo RP, Ballman KV, Antonescu CR, et al. Placebo-controlled randomized trial of adjuvant imatinib mesylate following the resection of localized, primary gastrointestinal stromal tumor (GIST). Lancet. 2009;373:1097-1104.
28. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32:1563-1570.
29. Casali PG, Le Cesne A, Poveda Velasco A, et al. Imatinib failure-free survival (IFS) in patients with localized gastrointestinal stromal tumors (GIST) treated with adjuvant imatinib (IM): the EORTC/AGITG/FSG/GEIS/ISG randomized controlled phase III trial. J Clin Oncol. 2013;31. Abstract 10500.
30. Joensuu H, Eriksson M, Sundby HK, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307:1265-1272.
31. Raut CP, Espat NJ, Maki RG, et al. Efficacy and tolerability of 5-year adjuvant imatinib treatment for patients with resected intermediate- or high-risk primary gastrointestinal stromal tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018: e184060.
32. Benjamin RS, Casali PG. Adjuvant imatinib for GI stromal tumors: when and for how long? J Clin Oncol. 2016;34:215-218.
33. Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141-3147.
34. Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99:42-47.
35. Rutkowski P, Gronchi A, Hohenberger P, et al. Neoadjuvant imatinib in locally advanced gastrointestinal stromal tumors (GIST): the EORTC STBSG experience. Ann Surg Oncol. 2013;20:2937-2943.
36. Joensuu H, Martin-Broto J, Nishida T, et al. Follow-up strategies for patients with gastrointestinal stromal tumour treated with or without adjuvant imatinib after surgery. Eur J Cancer. 2015;51:1611-1617.
Gastrointestinal Stromal Tumors: Management of Advanced Disease
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.

A summary of FDA-approved drugs for treating GISTs is provided in Table 2.

Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.

Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.
A summary of FDA-approved drugs for treating GISTs is provided in Table 2.
Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.
Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
Most advanced gastrointestinal stromal tumors (GISTs) are due to a recurrence of localized disease, with only a small minority presenting with metastatic disease.1 Compared with chemotherapy, tyrosine kinase inhibitors (TKIs) have significantly improved the natural history of the disease, with median overall survival (OS) increasing from less than 1 year to about 5 years and approximately 1 in 5 patients achieving long-term survival.2 In addition, newer drugs in development and in clinical trials appear promising and have the potential to improve outcomes even further. This article reviews current evidence on options for treating metastatic or recurrent GISTs and GISTs that have progressed following initial therapy. The evaluation and diagnosis of GIST along with management of localized disease are reviewed in a separate article.
Case Presentation
A 64-year-old African American man underwent surgical resection of a 10-cm gastric mass, which pathology reported was positive for CD117, DOG1, and CD34 and negative for smooth muscle actin and S-100, consistent with a diagnosis of GIST. There were 10 mitoses per 50 HPF, and there was no intraoperative or intraperitoneal tumor rupture. The patient was treated with adjuvant imatinib, which was discontinued after 3 years due to grade 2 myalgias, periorbital edema, and macrocytic anemia. Surveillance included office visits every 3 to 6 months and a contrast CT abdomen and pelvis every 6 months. For the past 5 years, he has not had any clinical or radiographic evidence of disease recurrence. New imaging reveals multiple liver metastases and peritoneal implants. He feels fatigued and has lost about 10 lb since his last visit. He is 5 years out from his initial diagnosis and 2 years out from last receiving imatinib. His original tumor harbored a KIT exon 11 deletion.
What treatment should you recommend now?
Imatinib for Advanced GISTs
Before the first report of the efficacy of imatinib for metastatic GISTs in 2002, patients with advanced unresectable or metastatic GISTs were routinely treated with doxorubicin-based chemotherapy regimens, which were largely ineffective, with response rates (RRs) of around 5% and a median overall survival (OS) of less than 1 year.3,4 In 2002 a landmark phase 2 study revealed imatinib’s significant efficacy profile in advanced or metastatic GISTs, resulting in its approval by the US Food and Drug Administration (FDA).5 In this study, 147 patients with CD117-positive GISTs were randomly assigned to receive daily imatinib 400 mg or 600 mg for up to 36 months. The RRs were similar between the 2 groups (68.5% vs 67.6%), with a median time to response of 12 weeks and median duration of response of 118 days. Results of this study were much more favorable when compared to doxorubicin, rendering imatinib the new standard of care for advanced GISTs. A long-term follow-up of this study after a median of 63 months confirmed near identical RRs, progression-free survival (PFS), and median survival of 57 months among the 2 groups.6
Imatinib Daily Dosing
Although 400 mg of daily imatinib proved to be efficacious, it was unclear if a dose-response relationship existed for imatinib. An EORTC phase 2 study demonstrated a benefit of using a higher dose of imatinib at 400 mg twice daily, producing a RR of 71% (4% complete , 67% partial) and 1-year PFS of 73%, which appeared favorable compared with once-daily dosing and set the framework for larger phase 3 studies.7 Two phase 3 studies compared imatinib 400 mg once daily versus twice daily (until disease progression or unacceptable toxicity) among patients with CD117-positive advanced or metastatic GISTs. These studies were eventually combined into a meta-analysis (metaGIST) to compare RR, PFS and OS between the treatment groups. Both studies allowed cross-over to the 800 mg dose for patients who progressed on 400 mg daily.
The first study, conducted jointly by the EORTC, Italian Sarcoma Group, and Australasian Gastro-Intestinal Trials Group (EU-AUS),8 randomly assigned 946 patients to 400 mg once daily or twice daily. There were no differences in response rates between the groups, but the twice-daily group had a predicted 18% reduction in the hazard for progression compared with the once-daily group (estimated HR, 0.82; P = 0.026), which came at the expense of greater toxicities warranting dose reductions (60%) and treatment interruptions (64%). Cross-over to high-dose imatinib was feasible and safe, producing a partial response in 2%, stable disease in 27%, and a median PFS of 81 days. The second study was an intergroup study conducted jointly by SWOG, CALGB, NCI-C, and ECOG (S0033, US-CDN), with a nearly identical study design as the EU-AUS trial.9 The trial enrolled 746 patients. After a median follow up of 4.5 years, the median PFS and OS were not statistically different (18 vs 20 months and 55 vs 51 months, respectively). There were also no differences in response rates. One third of patients initially placed on the once-daily arm who crossed over after progression achieved a treatment response or stable disease.
The combined EU-AUS and US-CDN analysis (metaGIST) included 1640 patients with a median age of 60 years and 58% of whom were men; 818 and 822 patients were assigned to the 400 mg and 800 mg total daily doses, respectively.10 The median follow-up was 37.5 months. There were no differences in OS (49 vs 48.7 months), median PFS (18.9 vs 23.2 months), or overall response rates (51.4% vs 53.9%). Patients who had crossed over (n = 347) to the 800 mg total daily dose arm had a 7.7-month average PFS while on the higher daily dose. An analysis was performed on 377 patients in the EU-AUS trial assessing the impact of mutational status on clinical outcomes among imatinib-treated patients. KIT exon 9 activating mutations were found to be a significant independent prognostic factor for death when compared with KIT exon 11 mutations. However, the adverse prognostic value of KIT exon 9 mutations was partially overcome with higher doses of imatinib, as those who received 800 mg total had a significantly better PFS, with a 61% relative risk reduction, than those who received 400 mg. Altogether, it was concluded that imatinib 400 mg once daily should be the standard-of-care first-line treatment for advanced or metastatic GISTs, unless a KIT exon 9 mutation is present, in which case imatinib 800 mg should be considered, if 400 mg is well tolerated. In addition, patients treated with frontline imatinib at 400 mg once daily, if tolerated well, should be considered for imatinib 800 mg upon progression of disease.
Despite there being problems with secondary resistance, significant progress has occurred in the treatment of metastatic disease over a short period of time. Prior to 2000, median OS for patients with metastatic GISTs was 9 months. With the introduction of imatinib and other TKIs, the median OS has increased to 5 years, with an estimated 10-year OS rate of approximately 20%.2
Imatinib Interruption
Since at this point, imatinib was a well-established standard of care for advanced GISTs, it was questioned whether imatinib therapy could be interrupted. At this time, treatment interruption in a stop-and-go fashion was deemed feasible in other metastatic solid tumors such as colorectal cancer (OPTIMOX1).11 The BFR French trial showed that stopping imatinib therapy in patients who had a response or stable disease after 1, 3, or 5 years was generally followed by relatively rapid tumor progression (approximately 50% of patients within 6 months), even when tumors were previously removed.12 Therefore, it is recommended that treatment in the metastatic setting should be continued indefinitely, unless there is disease progression. Hence, unlike with colorectal cancer or chronic myelogenous leukemia, as of now there is no role for imatinib interruption in metastatic GISTs.
Case Continued
The patient is started on imatinib 400 mg daily, and overall he tolerates therapy well. Interval CT imaging reveals a treatment response. Two years later, imaging reveals an increase in the tumor size and density with a new nodule present within a preexisting mass. There are no clinical trials in the area.
What defines tumor progression?
Disease Progression
When GISTs are responding to treatment, on imaging the tumors can become more cystic and less dense but with an increase in size. In addition, tumor progression may not always be associated with increased size—increased density of the tumor or a nodule within a mass that may indicate progression. If CT imaging is equivocal for progression, positron emission tomography (PET) can play a role in identifying true progression. It is critically important that tumor size and density are carefully assessed when performing interval imaging. Of note, radiofrequency ablation, cryotherapy, or chemoembolization can be used for symptomatic liver metastases or oligometastatic disease. When evaluating for progression, one needs to ask patients about compliance (ie, maintaining dose intensity related to side effects of therapy as well as the financial burden of treatment—copay toxicity).
What are mechanisms of secondary imatinib resistance?
Imatinib resistance can be subtle in patients with GISTs, manifesting with new nodular, enhancing foci enclosed within a preexisting mass (resistant clonal nodule), or can be clinically or radiographically overt.13 Imatinib resistance occurs through multiple mechanisms including acquisition of secondary activating KIT mutations in the intracellular ATP-binding domain (exons 13 and 14) and the activation loop (exons 17 and 18).14
What are the treatment options for this patient?
Second-line Therapy
Sunitinib malate is a multitargeted TKI that not only targets c-Kit and PDGFRA, but also has anti-angiogenic activity through inhibition of vascular endothelial growth factor receptors (VEGFR). Sunitinib gained FDA approval for the second-line treatment of advanced GISTs based on an international double-blind trial that randomized 312 patients with imatinib-resistant metastatic GISTs in a 2:1 fashion to receive sunitinib 50 mg daily for 4 weeks on and 2 weeks off or placebo.15,16 The trial was unblinded early at the planned interim analysis, which revealed a marked benefit, producing a 66% reduction in the hazard risk of progression (27.3 vs 6.4 weeks, HR, 0.33; P < 0.001). The most common treatment-related adverse events were fatigue, diarrhea, skin discoloration, nausea, and hand-foot syndrome. Another open-label phase 2 study assessed a continuous dosing schema of sunitinib 37.5 mg daily, which has been shown to be effective with less toxicity.17 Among the 60 patients enrolled, the primary endpoint of clinical benefit rate at 24 weeks was reached in 53%, which consisted of 13% partial responses and 40% stable disease. Most toxicities were grade 1 or 2 and easily manageable through standard interventions. This has been recommended as an alternative to the initial scheduled regimen.18 Part of sunitinib’s success is its activity against GISTs harboring secondary KIT exon 13 and 14 mutations, and possibly its anti-angiogenic activity.19 Sunitinib is particularly efficacious among GISTs harboring KIT exon 9 mutations.
Third-line Therapy
Patients who have progressed on prior imatinib and sunitinib can receive third-line regorafenib, a multi-TKI that differs chemically from sorafenib by a fluorouracil group (fluoro-sorafenib). FDA approval of regorafenib was based on the phase 3 GRID (GIST Regorafenib In progressive Disease) multicenter international trial.20 This trial randomly assigned 199 patients in a 2:1 fashion to receive regorafenib 160 mg daily for 21 days out of 28-day cycles plus best supportive care (BSC) versus placebo plus BSC. Cross-over was allowed. Regorafenib significantly reduced the hazard risk of progression by 73% compared with placebo (4.8 vs 0.9 months; HR, 0.27; P < 0.001). There was no difference in OS, which may be because of cross-over (median OS, 17.4 months in both arms). As a result, regorafenib is now considered standard third-line treatment for patients with metastatic GISTs. It has a less favorable toxicity profile than imatinib, with hand-foot syndrome, transaminitis, hypertension and fatigue being the most common treatment toxicities. In order to avoid noncompliance, it is recommended to start at 80 mg and carefully titrate upwards to the 160 mg dose.
A list of landmark studies for advanced GISTs is provided in Table 1.
A summary of FDA-approved drugs for treating GISTs is provided in Table 2.
Clinical Trials
Clinical trial enrollment should be considered for all patients with advanced or unresectable GISTs throughout their treatment continuum. Owing to significant advances in genomic profiling through next-generation sequencing, multiple driver mutations have recently been identified, and targeted therapies are being explored in clinical trials.21 For example, the neurotrophic receptor tyrosine kinase (NTRK) gene appears to be mutated in a small number of advanced GISTs, and these can respond to the highly selective TRK inhibitor larotrectinib.22 Additionally, ongoing studies are assessing immunotherapies for sporadic GISTs and treatment for familial GISTs (Table 3). Some notable studies include those assessing the efficacy of agents that target KIT and PDGFR secondary mutations, including avapritinib (BLU-285) and DCC-2618, MEK inhibitors, and the multi-kinase inhibitor crenolanib for GISTs harboring the imatinib-resistant PDGFRA D842V mutation. There are also studies utilizing checkpoint inhibitors alone or in combination with imatinib.
Case Conclusion
Given the patient’s progression on imatinib, he is started on second-line sunitinib malate. He experiences grade 1 fatigue and hand-foot syndrome, which are managed supportively. After he has been on sunitinib for approximately 8 months, his disease progresses. He subsequently undergoes genomic profiling of his tumor and starts BLU-285 on a clinical trial.
Key Points
- For advanced and metastatic disease, TKIs have substantially improved the prognosis of KIT mutated GISTs, with 3 FDA-approved drugs: imatinib, sunitinib, and regorafenib. Imatinib 400 mg is the standard-of-care frontline therapy for locally advanced, unresectable, or metastatic imatinib-sensitive GISTs. If a patient has a KIT exon 9 mutation and 400 mg is well-tolerated, increasing to 800 mg is recommended. Imatinib should be continued indefinitely unless there is intolerance, a specific patient request for interruption, or progression of disease.
- When there is progression of disease in a patient with a sensitive mutation on 400 mg of imatinib, the dose can be increased to 800 mg.
- For patients who are imatinib-intolerant or have progression, standard second line is sunitinib.
- For patients who further progress or are sunitinib-intolerant, regorafenib is the standard third-line treatment.
- There needs to be close attention to side effects, drug and food interactions, and patient copay costs in order to maintain patient compliance while on TKI therapy.
- There are still major limitations in the systemic treatment of GISTs marked by their inherent genetic heterogeneity and secondary resistance. Continued translational and clinical research is needed in order to improve treatment for patients who develop secondary resistance or who have less common primary resistant mutations. Patients are encouraged to participate in clinical trials of new therapies.
Summary
GISTs are the most common mesenchymal tumors of the GI tract. They comprise an expanding landscape of tumors that are heterogenous in terms of natural history, mutations, and response to systemic treatments. The mainstay of treatment for localized GISTs is surgical resection followed by at least 3-years of adjuvant imatinib for patients with high-risk features who are imatinib-sensitive. Patients with GISTs harboring resistance mutations such as PDGFRA D842V or with SDH-deficient or NF1-associated GISTs should not receive adjuvant imatinib. Patients with more advanced GISTs and/or in difficult to resect sites harboring a sensitive mutation can be considered for neoadjuvant imatinib. Those with metastatic GISTs can receive first-, second-, and third-line imatinib, sunitinib, or regorafenib, respectively. Clinical trial enrollment should be encouraged for patients whose GISTs harbor primary imatinib-resistant mutations, and those with advanced or unresectable GISTs with secondary resistance.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
1. Ma GL, Murphy JD, Martinez ME et al. Epidemiology of gastrointestinal stromal tumors in the era of histology codes: results of a population-based study. Cancer Epidemiol Biomarkers Prev. 2015;24:298-302.
2. Heinrich MC, Rankin C, Blanke CD, et al. Correlation of long-term results of imatinib in advanced gastrointestinal stromal tumors with next-generation sequencing results: analysis of phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017;3:944-952.
3. DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51-58.
4. Goss GA, Merriam P, Manola J, et al. Clinical and pathological characteristics of gastrointestinal stromal tumors (GIST). Prog Proc Am Soc Clin Oncol. 2000;19:599a.
5. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002; 347:472-480.
6. Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase ii trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620-625.
7. Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevac) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39:2006-2011.
8. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364:1127-1134.
9. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626-632.
10. Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247-1253.
11. Tournigand C, Cervantes A, Figer A, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer –a GERCOR study. J Clin Oncol. 2006;24:394-400.
12. Blay JV, Cesne AL, Ray-Coquard I, et al. Prospective multicentric randomized phase iii study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: The French Sarcoma Group. J Clin Oncol. 2007;25:1107-1113.
13. Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1): 5398-5405.
14. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510-7518.
15. Sutent (sunitinib malate) [package insert]. New York, NY: Pfizer Labs; 2017.
16. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomized controlled trial. Lancet. 2006;368:1329-1338.
17. George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45:1959-1968.
18. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcomas. Switzerland: Springer International Publishing; 2013.
19. Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumors. J Clin Oncol. 2008;26:5352-5359.
20. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295-302.
21. Wilky BA, Villalobos VM. Emerging role for precision therapy through next-generation sequencing for sarcomas. JCO Precision Oncology. 2018;2:1-4.
22. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in trk fusion-positive cancers in adults and children. N Engl J Med. 2018;378:731-739.
FDA approves Turalio for symptomatic tenosynovial giant cell tumor
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.