User login
Report details financial burden of blood cancers
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
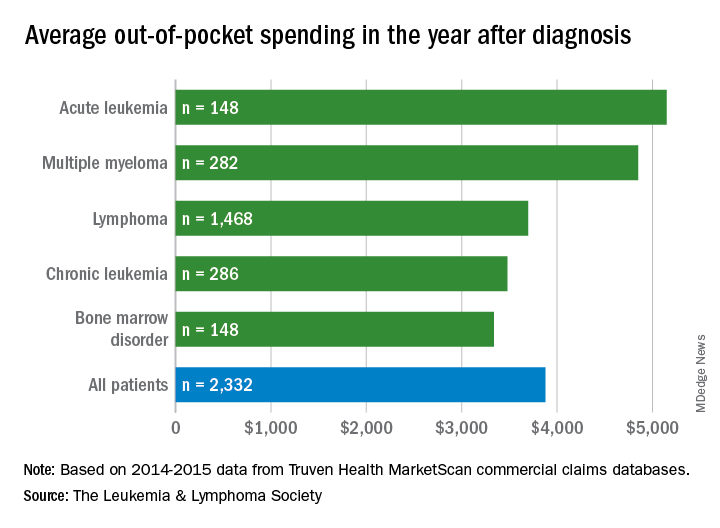
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
Molecule enhances PI activity in multiple myeloma
Researchers say they have identified a new class of protein disulfide isomerase (PDI) inhibitors that sensitize multiple myeloma (MM) cells to proteasome inhibitors (PIs).
The investigators screened approximately 20,000 compounds spanning multiple chemical libraries and found the compound E61 to be a “striking hit,” with a six-fold increase in bortezomib cytotoxicity and the ability to re-sensitize PI activity at low micromolar concentrations.
The researchers then synthesized and evaluated 150 E61 derivatives and discovered the lead candidate, E64FC26, which was highly synergistic with PIs at concentrations as low as 200 nM.
They reported that E64FC26 has “several advantages over previously reported PDI inhibitors, including superior potency and a pan-style mode of inhibition.”
“PDI is an attractive target in oncology, but good PDI inhibitors have been hard to find,” said Nathan G. Dolloff, PhD, of the Medical University of South Carolina (MUSC) in Charleston.
“The compounds we discovered have a lot of advantages, including high potency and good drug-like properties. We hope that those strengths translate into an effective new drug that can ultimately help patients.”
Dr. Dolloff and his colleagues reported their discovery in Leukemia.
The investigators detected the synergistic effects of E61 in combination with next-generation PIs, including carfilzomib, ixazomib, and oprozomib in both PI-sensitive and -resistant MM cell lines.
On the other hand, E61 had no effect on dexamethasone activity in dexamethasone-resistant cells. And E61 did not affect lenalidomide or doxorubicin cytotoxicity in PI-resistant cells.
The researchers also determined that E61 was only active in MM cells. E61 did not enhance the cytotoxic effects of PIs in normal cells.
This selective toxicity suggests that E61 may have “a wide therapeutic index in vivo,” the investigators wrote.
In vivo activity
To investigate the anti-MM activity and tolerability of E61 in vivo, the researchers treated a NOD-SCID IL2Rγ−/− mouse model with E61 at a continuous dose of 50 mg/kg/day.
E61 prolonged survival by 11 days in the treated mice (P=0.0007), and four of the 11 treated mice survived to the experiment’s end.
In another experiment, two of eight mice achieved a complete response.
After continuous dosing for 40 or more days, E61 was well tolerated, the investigators reported. The mice showed no overt signs of distress and did not lose weight.
Molecular target of E61
Using click (Cu(I)-catalyzed azide-alkyne cycloaddition) chemistry and a proteomics approach, the researchers then confirmed that PDI family members are the molecular target of E61.
Functional studies indicated that E61 inhibited PDI reductase activity in vitro. E61 also enhanced the accumulation of ubiquitinylated proteins and produced strong endoplasmic reticulum (ER) and oxidative stress responses when combined with PIs.
Anti-MM activity of E64FC26
The investigators used a structure activity relationship program to narrow the candidate molecules down to E64FC26.
E64FC26 demonstrated pan-inhibition in that it inhibited all members of the PDI family tested, including PDIA3, PDIA4, TXNDC5, and PDIA6.
E64FC26 also had greater in vitro potency against PDIA1 and the other PDI isoforms. It was the only compound to sensitize MM cells to PIs, with an average increase in PI sensitivity ranging from six- to seven-fold.
The researchers also noted that E64FC26 was superior to other PDI inhibitors they tested in activating ER stress.
The investigators tested the activity of E64FC26 in vivo using Vk*MYC transgenic mice, a model that closely resembles human MM.
Mice treated with E64FC26 had an immediate anti-MM response. Serum M-protein decreased in all mice by an average of 33 ± 7.9% (P=0.0135).
The investigators observed similar effects in a human xenotransplant MM model.
These mice were randomized to receive treatment with vehicle, E64FC26 (2 mg/kg for 3 days/week), bortezomib (0.25 mg/kg for 2 days/week), or a combination of the two agents.
E64FC26 increased median survival by 2 weeks compared with vehicle-treated mice (P<0.0001). By day 36, no vehicle-treated mouse survived, compared with 100% of the E64FC26-treated mice.
Single-agent bortezomib increased survival by 6 days (P=0.0007).
And the combination produced the greatest improvement in median survival, increasing it by 20 days (P<0.0001).
The investigators reported no overt toxicity or body weight fluctuation for mice treated with E64FC26 or the combination.
“These results provide preclinical proof of concept for the strategy of targeting PDI with this new class of compound for the treatment of MM,” the researchers concluded.
“One of the strengths of this study is that we spanned almost the entire drug discovery process,” Dr. Dolloff said. “We screened thousands of compounds, found an exciting molecule, deconvoluted what its binding target was, synthesized hundreds of derivatives to make it better, and then conducted animal studies.”
“The study has everything from biochemistry and cell biology to medicinal chemistry and animal pharmacology in it. There is still a lot of work to be done before this drug is ready for clinical trials in humans, but it has been a rewarding project, and I’m looking forward to the next steps.”
Dr. Dolloff is founder of Leukogene Therapeutics, Inc., which has licensed patents from MUSC, and a second study author is an inventor on patents. The other authors declared no conflicts of interest.
The research was supported by the National Institutes of Health/National Cancer Institute, the South Carolina Clinical & Translational Research Institute, the MUSC Hollings Cancer Center, and by the Hollings Cancer Center T32 Ruth L. Kirschstein National Research Service Award Training Program.
Researchers say they have identified a new class of protein disulfide isomerase (PDI) inhibitors that sensitize multiple myeloma (MM) cells to proteasome inhibitors (PIs).
The investigators screened approximately 20,000 compounds spanning multiple chemical libraries and found the compound E61 to be a “striking hit,” with a six-fold increase in bortezomib cytotoxicity and the ability to re-sensitize PI activity at low micromolar concentrations.
The researchers then synthesized and evaluated 150 E61 derivatives and discovered the lead candidate, E64FC26, which was highly synergistic with PIs at concentrations as low as 200 nM.
They reported that E64FC26 has “several advantages over previously reported PDI inhibitors, including superior potency and a pan-style mode of inhibition.”
“PDI is an attractive target in oncology, but good PDI inhibitors have been hard to find,” said Nathan G. Dolloff, PhD, of the Medical University of South Carolina (MUSC) in Charleston.
“The compounds we discovered have a lot of advantages, including high potency and good drug-like properties. We hope that those strengths translate into an effective new drug that can ultimately help patients.”
Dr. Dolloff and his colleagues reported their discovery in Leukemia.
The investigators detected the synergistic effects of E61 in combination with next-generation PIs, including carfilzomib, ixazomib, and oprozomib in both PI-sensitive and -resistant MM cell lines.
On the other hand, E61 had no effect on dexamethasone activity in dexamethasone-resistant cells. And E61 did not affect lenalidomide or doxorubicin cytotoxicity in PI-resistant cells.
The researchers also determined that E61 was only active in MM cells. E61 did not enhance the cytotoxic effects of PIs in normal cells.
This selective toxicity suggests that E61 may have “a wide therapeutic index in vivo,” the investigators wrote.
In vivo activity
To investigate the anti-MM activity and tolerability of E61 in vivo, the researchers treated a NOD-SCID IL2Rγ−/− mouse model with E61 at a continuous dose of 50 mg/kg/day.
E61 prolonged survival by 11 days in the treated mice (P=0.0007), and four of the 11 treated mice survived to the experiment’s end.
In another experiment, two of eight mice achieved a complete response.
After continuous dosing for 40 or more days, E61 was well tolerated, the investigators reported. The mice showed no overt signs of distress and did not lose weight.
Molecular target of E61
Using click (Cu(I)-catalyzed azide-alkyne cycloaddition) chemistry and a proteomics approach, the researchers then confirmed that PDI family members are the molecular target of E61.
Functional studies indicated that E61 inhibited PDI reductase activity in vitro. E61 also enhanced the accumulation of ubiquitinylated proteins and produced strong endoplasmic reticulum (ER) and oxidative stress responses when combined with PIs.
Anti-MM activity of E64FC26
The investigators used a structure activity relationship program to narrow the candidate molecules down to E64FC26.
E64FC26 demonstrated pan-inhibition in that it inhibited all members of the PDI family tested, including PDIA3, PDIA4, TXNDC5, and PDIA6.
E64FC26 also had greater in vitro potency against PDIA1 and the other PDI isoforms. It was the only compound to sensitize MM cells to PIs, with an average increase in PI sensitivity ranging from six- to seven-fold.
The researchers also noted that E64FC26 was superior to other PDI inhibitors they tested in activating ER stress.
The investigators tested the activity of E64FC26 in vivo using Vk*MYC transgenic mice, a model that closely resembles human MM.
Mice treated with E64FC26 had an immediate anti-MM response. Serum M-protein decreased in all mice by an average of 33 ± 7.9% (P=0.0135).
The investigators observed similar effects in a human xenotransplant MM model.
These mice were randomized to receive treatment with vehicle, E64FC26 (2 mg/kg for 3 days/week), bortezomib (0.25 mg/kg for 2 days/week), or a combination of the two agents.
E64FC26 increased median survival by 2 weeks compared with vehicle-treated mice (P<0.0001). By day 36, no vehicle-treated mouse survived, compared with 100% of the E64FC26-treated mice.
Single-agent bortezomib increased survival by 6 days (P=0.0007).
And the combination produced the greatest improvement in median survival, increasing it by 20 days (P<0.0001).
The investigators reported no overt toxicity or body weight fluctuation for mice treated with E64FC26 or the combination.
“These results provide preclinical proof of concept for the strategy of targeting PDI with this new class of compound for the treatment of MM,” the researchers concluded.
“One of the strengths of this study is that we spanned almost the entire drug discovery process,” Dr. Dolloff said. “We screened thousands of compounds, found an exciting molecule, deconvoluted what its binding target was, synthesized hundreds of derivatives to make it better, and then conducted animal studies.”
“The study has everything from biochemistry and cell biology to medicinal chemistry and animal pharmacology in it. There is still a lot of work to be done before this drug is ready for clinical trials in humans, but it has been a rewarding project, and I’m looking forward to the next steps.”
Dr. Dolloff is founder of Leukogene Therapeutics, Inc., which has licensed patents from MUSC, and a second study author is an inventor on patents. The other authors declared no conflicts of interest.
The research was supported by the National Institutes of Health/National Cancer Institute, the South Carolina Clinical & Translational Research Institute, the MUSC Hollings Cancer Center, and by the Hollings Cancer Center T32 Ruth L. Kirschstein National Research Service Award Training Program.
Researchers say they have identified a new class of protein disulfide isomerase (PDI) inhibitors that sensitize multiple myeloma (MM) cells to proteasome inhibitors (PIs).
The investigators screened approximately 20,000 compounds spanning multiple chemical libraries and found the compound E61 to be a “striking hit,” with a six-fold increase in bortezomib cytotoxicity and the ability to re-sensitize PI activity at low micromolar concentrations.
The researchers then synthesized and evaluated 150 E61 derivatives and discovered the lead candidate, E64FC26, which was highly synergistic with PIs at concentrations as low as 200 nM.
They reported that E64FC26 has “several advantages over previously reported PDI inhibitors, including superior potency and a pan-style mode of inhibition.”
“PDI is an attractive target in oncology, but good PDI inhibitors have been hard to find,” said Nathan G. Dolloff, PhD, of the Medical University of South Carolina (MUSC) in Charleston.
“The compounds we discovered have a lot of advantages, including high potency and good drug-like properties. We hope that those strengths translate into an effective new drug that can ultimately help patients.”
Dr. Dolloff and his colleagues reported their discovery in Leukemia.
The investigators detected the synergistic effects of E61 in combination with next-generation PIs, including carfilzomib, ixazomib, and oprozomib in both PI-sensitive and -resistant MM cell lines.
On the other hand, E61 had no effect on dexamethasone activity in dexamethasone-resistant cells. And E61 did not affect lenalidomide or doxorubicin cytotoxicity in PI-resistant cells.
The researchers also determined that E61 was only active in MM cells. E61 did not enhance the cytotoxic effects of PIs in normal cells.
This selective toxicity suggests that E61 may have “a wide therapeutic index in vivo,” the investigators wrote.
In vivo activity
To investigate the anti-MM activity and tolerability of E61 in vivo, the researchers treated a NOD-SCID IL2Rγ−/− mouse model with E61 at a continuous dose of 50 mg/kg/day.
E61 prolonged survival by 11 days in the treated mice (P=0.0007), and four of the 11 treated mice survived to the experiment’s end.
In another experiment, two of eight mice achieved a complete response.
After continuous dosing for 40 or more days, E61 was well tolerated, the investigators reported. The mice showed no overt signs of distress and did not lose weight.
Molecular target of E61
Using click (Cu(I)-catalyzed azide-alkyne cycloaddition) chemistry and a proteomics approach, the researchers then confirmed that PDI family members are the molecular target of E61.
Functional studies indicated that E61 inhibited PDI reductase activity in vitro. E61 also enhanced the accumulation of ubiquitinylated proteins and produced strong endoplasmic reticulum (ER) and oxidative stress responses when combined with PIs.
Anti-MM activity of E64FC26
The investigators used a structure activity relationship program to narrow the candidate molecules down to E64FC26.
E64FC26 demonstrated pan-inhibition in that it inhibited all members of the PDI family tested, including PDIA3, PDIA4, TXNDC5, and PDIA6.
E64FC26 also had greater in vitro potency against PDIA1 and the other PDI isoforms. It was the only compound to sensitize MM cells to PIs, with an average increase in PI sensitivity ranging from six- to seven-fold.
The researchers also noted that E64FC26 was superior to other PDI inhibitors they tested in activating ER stress.
The investigators tested the activity of E64FC26 in vivo using Vk*MYC transgenic mice, a model that closely resembles human MM.
Mice treated with E64FC26 had an immediate anti-MM response. Serum M-protein decreased in all mice by an average of 33 ± 7.9% (P=0.0135).
The investigators observed similar effects in a human xenotransplant MM model.
These mice were randomized to receive treatment with vehicle, E64FC26 (2 mg/kg for 3 days/week), bortezomib (0.25 mg/kg for 2 days/week), or a combination of the two agents.
E64FC26 increased median survival by 2 weeks compared with vehicle-treated mice (P<0.0001). By day 36, no vehicle-treated mouse survived, compared with 100% of the E64FC26-treated mice.
Single-agent bortezomib increased survival by 6 days (P=0.0007).
And the combination produced the greatest improvement in median survival, increasing it by 20 days (P<0.0001).
The investigators reported no overt toxicity or body weight fluctuation for mice treated with E64FC26 or the combination.
“These results provide preclinical proof of concept for the strategy of targeting PDI with this new class of compound for the treatment of MM,” the researchers concluded.
“One of the strengths of this study is that we spanned almost the entire drug discovery process,” Dr. Dolloff said. “We screened thousands of compounds, found an exciting molecule, deconvoluted what its binding target was, synthesized hundreds of derivatives to make it better, and then conducted animal studies.”
“The study has everything from biochemistry and cell biology to medicinal chemistry and animal pharmacology in it. There is still a lot of work to be done before this drug is ready for clinical trials in humans, but it has been a rewarding project, and I’m looking forward to the next steps.”
Dr. Dolloff is founder of Leukogene Therapeutics, Inc., which has licensed patents from MUSC, and a second study author is an inventor on patents. The other authors declared no conflicts of interest.
The research was supported by the National Institutes of Health/National Cancer Institute, the South Carolina Clinical & Translational Research Institute, the MUSC Hollings Cancer Center, and by the Hollings Cancer Center T32 Ruth L. Kirschstein National Research Service Award Training Program.
FDA clears portable hematology analyzer

The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
The U.S. Food and Drug Administration (FDA) has granted 510(k) clearance for PixCell Medical’s HemoScreen™.
This portable hematology analyzer is used to perform a complete blood count at the point of care.
HemoScreen requires a single drop of blood and uses disposable cartridges that provide automatic sample preparation.
HemoScreen can analyze 20 standard complete blood count parameters and produces results within 5 minutes.
Study results suggested that HemoScreen provides results comparable to those of another hematology analyzer, Sysmex XE-2100. This study was published in the Journal of Clinical Pathology in 2016.
“The HemoScreen delivers lab-accurate results,” said Avishay Bransky, PhD, chief executive officer of PixCell Medical.
He added that HemoScreen “would be especially useful” in physicians’ offices, emergency rooms, intensive care units, oncology clinics, and remote locations.
HemoScreen makes use of a technology called viscoelastic focusing, which employs microfluidics and machine vision algorithms to analyze cells.
ICYMI: Elotuzumab reduces progression risk in lenalidomide-refractory multiple myeloma
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA approves elotuzumab with pom/dex in refractory myeloma
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.

Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
FDA approves elotuzumab combo for rel/ref MM
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Haploidentical transplantation holds promise in relapsed myeloma
, investigators have reported.
The rate of non-relapse mortality at one year was 21% in the retrospective analysis of 96 patients, recently reported in the journal Biology of Blood and Marrow Transplantation.
Haploidentical allogeneic hematopoietic stem cell transplant (allo-HCT) is currently limited in use due to a high rate of relapse, but may hold potential promise for future applications, according to Firoozeh Sahebi, MD, a hematologist with the City of Hope Medical Center, Duarte, Calif., and colleagues. “Our results demonstrate that haploidentical allo-HCT can be safely performed in appropriate patients with MM who lack on HLA-matched sibling or unrelated donor.”
“The allo-HCT platform can be used in the context of other post-transplantation immune-based strategies, such as donor-derived chimeric antigen receptor T cells and natural killer cell infusions, newer immunomodulatory drugs or proteasome inhibitors, bispecific T cell engagers, and bispecific killer cell engagers, to further enhance antitumor effects and ultimately improve survival in an appropriate patient population,” Dr. Sahebi and colleagues said in their report.
The investigators reported results of a retrospective analysis including 96 patients with relapsed multiple myeloma who had failed at least one previous autologous HCT. They underwent haploidentical allo-HCT at European Society for Blood and Marrow Transplantation/Center for International Blood and Marrow Transplant Research centers between 2008 and 2016.
Median follow-up in the analysis was 24 months. Almost all patients (97%) achieved neutrophil engraftment by day 28, while 75% had recovery of platelets by day 60, Dr. Sahebi and co-investigators reported.
The 1-year nonrelapse mortality rate was 21%, but the cumulative risk of relapse and progression at 2 years was 56%, according to the study results. Two-year progression-free survival was reported to be 17%, while overall survival was 48%.
Acute graft-versus-host-disease (GVHD) of grades II-IV occurred in 39% by 100 days, while chronic GVHD was seen in 46% at 2 years, the report shows.
Factors linked to improved overall survival at 2 years included use of bone marrow as the source of stem cells, and the use of cyclophosphamide after transplantation, according to Dr. Sahebi and co-authors.
By contrast, factors that had no impact on overall survival, progression-free survival, or non-relapse mortality included disease status (ie, degree of response), gender, conditioning regimen intensity, presence of cytomegalovirus in the blood, or donor-recipient sex mismatch.
This analysis was conducted in part due to the limited availability of matched donors, along with the promising results of allo-HCT in other malignancies, according to investigators.
There were no conflicts of interest to report related to this research, Dr. Sahebi and colleagues reported in the journal.
SOURCE: Sahebi F, et al. Biol Blood Marrow Transplant. 2018 Sep 20. pii: S1083-8791(18)30575-5.
, investigators have reported.
The rate of non-relapse mortality at one year was 21% in the retrospective analysis of 96 patients, recently reported in the journal Biology of Blood and Marrow Transplantation.
Haploidentical allogeneic hematopoietic stem cell transplant (allo-HCT) is currently limited in use due to a high rate of relapse, but may hold potential promise for future applications, according to Firoozeh Sahebi, MD, a hematologist with the City of Hope Medical Center, Duarte, Calif., and colleagues. “Our results demonstrate that haploidentical allo-HCT can be safely performed in appropriate patients with MM who lack on HLA-matched sibling or unrelated donor.”
“The allo-HCT platform can be used in the context of other post-transplantation immune-based strategies, such as donor-derived chimeric antigen receptor T cells and natural killer cell infusions, newer immunomodulatory drugs or proteasome inhibitors, bispecific T cell engagers, and bispecific killer cell engagers, to further enhance antitumor effects and ultimately improve survival in an appropriate patient population,” Dr. Sahebi and colleagues said in their report.
The investigators reported results of a retrospective analysis including 96 patients with relapsed multiple myeloma who had failed at least one previous autologous HCT. They underwent haploidentical allo-HCT at European Society for Blood and Marrow Transplantation/Center for International Blood and Marrow Transplant Research centers between 2008 and 2016.
Median follow-up in the analysis was 24 months. Almost all patients (97%) achieved neutrophil engraftment by day 28, while 75% had recovery of platelets by day 60, Dr. Sahebi and co-investigators reported.
The 1-year nonrelapse mortality rate was 21%, but the cumulative risk of relapse and progression at 2 years was 56%, according to the study results. Two-year progression-free survival was reported to be 17%, while overall survival was 48%.
Acute graft-versus-host-disease (GVHD) of grades II-IV occurred in 39% by 100 days, while chronic GVHD was seen in 46% at 2 years, the report shows.
Factors linked to improved overall survival at 2 years included use of bone marrow as the source of stem cells, and the use of cyclophosphamide after transplantation, according to Dr. Sahebi and co-authors.
By contrast, factors that had no impact on overall survival, progression-free survival, or non-relapse mortality included disease status (ie, degree of response), gender, conditioning regimen intensity, presence of cytomegalovirus in the blood, or donor-recipient sex mismatch.
This analysis was conducted in part due to the limited availability of matched donors, along with the promising results of allo-HCT in other malignancies, according to investigators.
There were no conflicts of interest to report related to this research, Dr. Sahebi and colleagues reported in the journal.
SOURCE: Sahebi F, et al. Biol Blood Marrow Transplant. 2018 Sep 20. pii: S1083-8791(18)30575-5.
, investigators have reported.
The rate of non-relapse mortality at one year was 21% in the retrospective analysis of 96 patients, recently reported in the journal Biology of Blood and Marrow Transplantation.
Haploidentical allogeneic hematopoietic stem cell transplant (allo-HCT) is currently limited in use due to a high rate of relapse, but may hold potential promise for future applications, according to Firoozeh Sahebi, MD, a hematologist with the City of Hope Medical Center, Duarte, Calif., and colleagues. “Our results demonstrate that haploidentical allo-HCT can be safely performed in appropriate patients with MM who lack on HLA-matched sibling or unrelated donor.”
“The allo-HCT platform can be used in the context of other post-transplantation immune-based strategies, such as donor-derived chimeric antigen receptor T cells and natural killer cell infusions, newer immunomodulatory drugs or proteasome inhibitors, bispecific T cell engagers, and bispecific killer cell engagers, to further enhance antitumor effects and ultimately improve survival in an appropriate patient population,” Dr. Sahebi and colleagues said in their report.
The investigators reported results of a retrospective analysis including 96 patients with relapsed multiple myeloma who had failed at least one previous autologous HCT. They underwent haploidentical allo-HCT at European Society for Blood and Marrow Transplantation/Center for International Blood and Marrow Transplant Research centers between 2008 and 2016.
Median follow-up in the analysis was 24 months. Almost all patients (97%) achieved neutrophil engraftment by day 28, while 75% had recovery of platelets by day 60, Dr. Sahebi and co-investigators reported.
The 1-year nonrelapse mortality rate was 21%, but the cumulative risk of relapse and progression at 2 years was 56%, according to the study results. Two-year progression-free survival was reported to be 17%, while overall survival was 48%.
Acute graft-versus-host-disease (GVHD) of grades II-IV occurred in 39% by 100 days, while chronic GVHD was seen in 46% at 2 years, the report shows.
Factors linked to improved overall survival at 2 years included use of bone marrow as the source of stem cells, and the use of cyclophosphamide after transplantation, according to Dr. Sahebi and co-authors.
By contrast, factors that had no impact on overall survival, progression-free survival, or non-relapse mortality included disease status (ie, degree of response), gender, conditioning regimen intensity, presence of cytomegalovirus in the blood, or donor-recipient sex mismatch.
This analysis was conducted in part due to the limited availability of matched donors, along with the promising results of allo-HCT in other malignancies, according to investigators.
There were no conflicts of interest to report related to this research, Dr. Sahebi and colleagues reported in the journal.
SOURCE: Sahebi F, et al. Biol Blood Marrow Transplant. 2018 Sep 20. pii: S1083-8791(18)30575-5.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Key clinical point: Haploidentical allogeneic transplantation is feasible and had an acceptable rate of non-relapse mortality, setting the stage for its use in future combination strategies.
Major finding: The cumulative risk of relapse and progression at 2 years was 56%, and the 1-year nonrelapse mortality was 21%.
Study details: A retrospective analysis including 96 patients who underwent haploidentical allogeneic hematopoietic stem cell transplantation between 2008 and 2016.
Disclosures: Authors reported no conflicts of interest.
Source: Sahebi F, et al. Biol Blood Marrow Transplant. 2018 Sep 20. pii: S1083-8791(18)30575-5.
Genomic abnormalities shed light on racial disparity in myeloma
Researchers say they may have determined why African Americans have a two- to threefold increased risk of multiple myeloma (MM), compared with European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes – t(11;14), t(14;16), and t(14;20) – that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes, compared with individuals with African ancestry of less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
Previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias, Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minn., and his colleagues reported in Blood Cancer Journal.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” Dr. Rajkumar said in a statement.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 years. More samples were from men (54.3%) than women (45.7%). Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) odds ratio, 1.06; 95% confidence interval, 1.02-1.11; P = .05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations – individuals with 80% or greater African ancestry and individuals with less than 0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P = .008), compared with the European cohort.
The differences emerged in only the highest and lowest cohorts, they noted. Most patients (60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff for African ancestry was greater than 50%.
The research was supported by the National Cancer Institute and the Mayo Clinic. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
SOURCE: Baughn LB et al. Blood Cancer J. 2018 Oct 10;8(10):96.
Researchers say they may have determined why African Americans have a two- to threefold increased risk of multiple myeloma (MM), compared with European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes – t(11;14), t(14;16), and t(14;20) – that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes, compared with individuals with African ancestry of less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
Previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias, Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minn., and his colleagues reported in Blood Cancer Journal.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” Dr. Rajkumar said in a statement.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 years. More samples were from men (54.3%) than women (45.7%). Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) odds ratio, 1.06; 95% confidence interval, 1.02-1.11; P = .05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations – individuals with 80% or greater African ancestry and individuals with less than 0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P = .008), compared with the European cohort.
The differences emerged in only the highest and lowest cohorts, they noted. Most patients (60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff for African ancestry was greater than 50%.
The research was supported by the National Cancer Institute and the Mayo Clinic. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
SOURCE: Baughn LB et al. Blood Cancer J. 2018 Oct 10;8(10):96.
Researchers say they may have determined why African Americans have a two- to threefold increased risk of multiple myeloma (MM), compared with European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes – t(11;14), t(14;16), and t(14;20) – that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes, compared with individuals with African ancestry of less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
Previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias, Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minn., and his colleagues reported in Blood Cancer Journal.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” Dr. Rajkumar said in a statement.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 years. More samples were from men (54.3%) than women (45.7%). Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) odds ratio, 1.06; 95% confidence interval, 1.02-1.11; P = .05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations – individuals with 80% or greater African ancestry and individuals with less than 0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P = .008), compared with the European cohort.
The differences emerged in only the highest and lowest cohorts, they noted. Most patients (60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff for African ancestry was greater than 50%.
The research was supported by the National Cancer Institute and the Mayo Clinic. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
SOURCE: Baughn LB et al. Blood Cancer J. 2018 Oct 10;8(10):96.
FROM BLOOD CANCER JOURNAL
Key clinical point:
Major finding: There was a significantly higher prevalence of t(11;14), t(14;16), and t(14:20) in patients with 80% or greater African ancestry, compared with the European cohort (P = .008).
Study details: The study included 881 samples from patients with an abnormal plasma cell proliferative disorder FISH result and concurrent conventional G-banded chromosome evaluation.
Disclosures: The research was supported by the National Cancer Institute and the Mayo Clinic. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
Source: Baughn LB et al. Blood Cancer J. 2018 Oct 10;8(10):96.
Palliative care guidelines relevant for hematologists, doc says

The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
The latest edition of the national palliative care guidelines provides new clinical strategies relevant to hematology practice in the United States, according to a physician-researcher specializing in hematology.
The Clinical Practice Guidelines for Quality Palliative Care, 4th edition, represents a “blueprint for what it looks like to provide high-quality, comprehensive palliative care to people with serious illness,” said Thomas W. LeBlanc, MD, a physician-researcher at Duke University School of Medicine in Durham, North Carolina.
However, unlike previous editions, this update to the guidelines emphasizes the importance of palliative care provided by both primary care and specialty care clinicians.
“Part of this report is about trying to raise the game of everybody in medicine and provide a higher basic level of primary palliative care to all people with serious illness, but then also to figure out who has higher levels of needs where the specialists should be applied, since they are a scarce resource,” Dr. LeBlanc said.
The latest edition helps establish a foundation for gold standard palliative care for people living with serious illness, regardless of diagnosis, prognosis, setting, or age, according to The National Coalition for Hospice and Palliative Care, which published the clinical practice guidelines.
The update was developed by the National Consensus Project for Quality Palliative Care (NCP), which includes 16 national organizations with palliative care and hospice expertise, and is endorsed by more than 80 national organizations, including the American Society of Hematology.
One key reason for the update, according to NCP, was to acknowledge that today’s healthcare system may not be meeting patients’ palliative care needs.
Specifically, the guidelines call on clinicians who don’t practice palliative care to integrate palliative care principles into their routine assessment of seriously ill patients with conditions such as heart failure, lung disease, and cancer.
That differs from the way palliative care is traditionally practiced, in which specially trained doctors, nurses, and other specialists provide that support.
An issue with that traditional model is a shortage of specialized clinicians to meet palliative care needs, said Dr. LeBlanc, whose clinical practice and research focuses on palliative care needs of patients with hematologic malignancies.
“Palliative care has matured as a field such that we are now actually facing workforce shortage issues and really fundamental questions about who really needs us the most and how we increase our reach to improve the lives of more patients and families facing serious illness,” he said.
That’s a major driver behind the emphasis in the latest guidelines on providing palliative care in the community, coordinating care, and dealing with care transitions, Dr. LeBlanc added.
“I hope that this document will help to demonstrate the value and the need for palliative care specialists and for improvements in primary care in the care of patients with hematologic diseases in general,” he said. “To me, this adds increasing legitimacy to this whole field.”
Three gene types drive MM disparity in African Americans

Researchers say they may have determined why African Americans have a two- to three-fold increased risk of multiple myeloma (MM) compared to European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes—t(11;14), t(14;16), and t(14;20)—that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes compared to individuals with African ancestry less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
The researchers state that previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” said study author Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
He and his colleagues reported their findings in Blood Cancer Journal.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 (range, 26–90), with 35.4% in the 60–69 age category. More samples were from men (n=478, 54.3%) than women (n=403, 45.7%).
Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) (odds ratio=1.06, 95% CI: 1.02–1.11; P=0.05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations—individuals with ≥80.0% African ancestry and individuals with <0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P=0.008) compared to the European cohort.
The researchers said the differences only emerged in the highest (n=120 individuals) and lowest (n=235 individuals) cohorts. Most patients (n=526, 60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff of African ancestry was greater than 50%.
“Our findings provide important information that will help us determine the mechanism by which myeloma is more common in African Americans, as well as help us in our quest to find out what causes myeloma in the first place,” Dr. Rajkumar said.
The research was supported by the National Cancer Institute of the National Institutes of Health and the Mayo Clinic Department of Laboratory Medicine and Pathology and Center for Individualized Medicine. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
Researchers say they may have determined why African Americans have a two- to three-fold increased risk of multiple myeloma (MM) compared to European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes—t(11;14), t(14;16), and t(14;20)—that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes compared to individuals with African ancestry less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
The researchers state that previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” said study author Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
He and his colleagues reported their findings in Blood Cancer Journal.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 (range, 26–90), with 35.4% in the 60–69 age category. More samples were from men (n=478, 54.3%) than women (n=403, 45.7%).
Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) (odds ratio=1.06, 95% CI: 1.02–1.11; P=0.05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations—individuals with ≥80.0% African ancestry and individuals with <0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P=0.008) compared to the European cohort.
The researchers said the differences only emerged in the highest (n=120 individuals) and lowest (n=235 individuals) cohorts. Most patients (n=526, 60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff of African ancestry was greater than 50%.
“Our findings provide important information that will help us determine the mechanism by which myeloma is more common in African Americans, as well as help us in our quest to find out what causes myeloma in the first place,” Dr. Rajkumar said.
The research was supported by the National Cancer Institute of the National Institutes of Health and the Mayo Clinic Department of Laboratory Medicine and Pathology and Center for Individualized Medicine. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.
Researchers say they may have determined why African Americans have a two- to three-fold increased risk of multiple myeloma (MM) compared to European Americans.
The team genotyped 881 MM samples from various racial groups and identified three gene subtypes—t(11;14), t(14;16), and t(14;20)—that explain the racial disparity.
They found that patients with African ancestry of 80% or more had a significantly higher occurrence of these subtypes compared to individuals with African ancestry less than 0.1%.
And these subtypes are driving the disparity in MM diagnoses between the populations.
The researchers state that previous attempts to explain the disparity relied on self-reported race rather than quantitatively measured genetic ancestry, which could result in bias.
“A major new aspect of this study is that we identified the ancestry of each patient through DNA sequencing, which allowed us to determine ancestry more accurately,” said study author Vincent Rajkumar, MD, of the Mayo Clinic in Rochester, Minnesota.
He and his colleagues reported their findings in Blood Cancer Journal.
All 881 samples had abnormal plasma cell FISH, 851 had a normal chromosome study, and 30 had an abnormal study.
Median age for the entire group was 64 (range, 26–90), with 35.4% in the 60–69 age category. More samples were from men (n=478, 54.3%) than women (n=403, 45.7%).
Researchers observed no significant difference between men and women in the proportion of primary cytogenetic abnormalities.
Of the 881 samples, the median African ancestry was 2.3%, the median European ancestry was 64.7%, and Northern European ancestry was 26.6%.
Thirty percent of the entire cohort had less than 0.1% African ancestry, and 13.6% had 80% or greater African ancestry.
Using a logistic regression model, the researchers determined that a 10% increase in the percentage of African ancestry was associated with a 6% increase in the odds of detecting t(11;14), t(14;16), or t(14;20) (odds ratio=1.06, 95% CI: 1.02–1.11; P=0.05).
The researchers plotted the probability of observing these cytogenetic abnormalities with the percentage of African ancestry and found the differences were most striking in the extreme populations—individuals with ≥80.0% African ancestry and individuals with <0.1% African ancestry.
Upon further analysis, the team found a significantly higher prevalence of t(11;14), t(14;16), and t(14;20) in the group of patients with the greatest proportion of African ancestry (P=0.008) compared to the European cohort.
The researchers said the differences only emerged in the highest (n=120 individuals) and lowest (n=235 individuals) cohorts. Most patients (n=526, 60%) were not included in these extreme populations because they had mixed ancestry.
The team observed no significant differences when the cutoff of African ancestry was greater than 50%.
“Our findings provide important information that will help us determine the mechanism by which myeloma is more common in African Americans, as well as help us in our quest to find out what causes myeloma in the first place,” Dr. Rajkumar said.
The research was supported by the National Cancer Institute of the National Institutes of Health and the Mayo Clinic Department of Laboratory Medicine and Pathology and Center for Individualized Medicine. One study author reported relationships with Celgene, Takeda, Prothena, Janssen, Pfizer, Alnylam, and GSK. Two authors reported relationships with the DNA Diagnostics Center.