User login
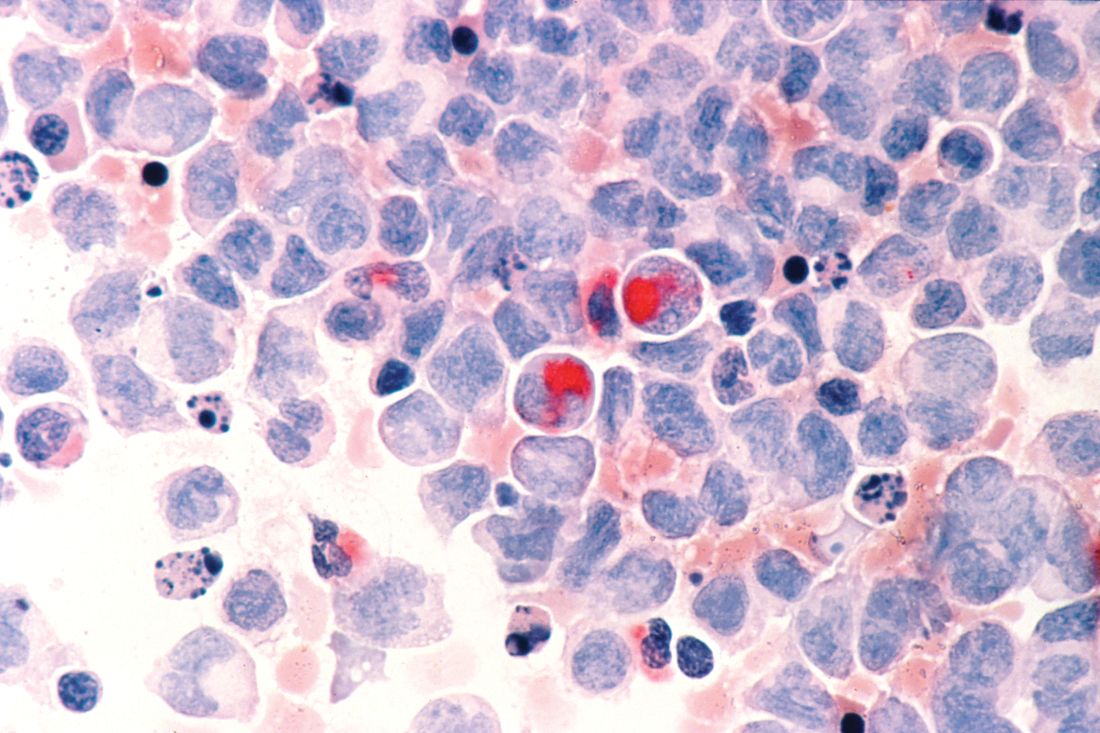
HDACi, HMA combo improves survival for older AML patients
For patients older than 65 years with newly diagnosed acute myeloid leukemia (AML) who were ineligible for standard induction therapy, adding the investigational pan-histone deacetylase (pan-HDAC) inhibitor pracinostat to azacitidine resulted in better complete remission and overall survival rates than azacitidine alone, results of a multicenter phase 2 trial showed.
Among 50 patients treated with the combination, 26 (52%) achieved the primary endpoint of either a complete remission (CR), CR with incomplete recovery of blood counts (CRi), or morphologic leukemia-free state (MLFS).
The median overall survival (OS) was 19.1 months, which compares favorably with historical data on similar patients treated with single-agent azacitidine, reported Guillermo Garcia-Manero, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“[T]his study shows that pracinostat in combination with azacitidine has the potential to be a safe and effective regimen in the frontline treatment of older patients with AML unfit for [induction chemotherapy],” they wrote in Blood Advances.
Pracinostat is an oral pan-HDAC inhibitor that has been shown to have modest activity against AML as a single agent, but synergistic activity when combined with hypomethylating agent azacitidine, a standard of care for older patients with AML in the trial.
The investigators enrolled 50 patients with a median age of 75 years (range, 66-84 years). The cohort included 33 patients with de novo AML, 12 with AML secondary to myelodysplasia syndrome or myleoproliferative neoplasia, and five with therapy-related AML.
The patients were treated with pracinostat 60 mg daily for 3 days each week for 3 consecutive weeks in addition to azacitidine 75 mg/m2 daily for 7 days in a 28-day cycle.
As noted, 26 patients reached the clinical endpoint, including 21 with a CR, 2 with a CRi, and 3 with MLFS. Additionally, two patients had a partial response (PR) and four had a PR with incomplete recovery of blood counts.
The median OS was 19.1 months, and the median progression-free survival (PFS) was 12.6 months. The 1-year OS rate was 62%. The 60-day mortality rate was 10%.
The authors noted that the survival data were superior to those seen in the phase 3 AZA-AML-001 study, which compared azacitidine therapy with conventional regimens in patients older than 65 years with newly diagnosed AML who were not eligible for stem cell transplants. In that trial, median OS was 10.4 months, the CR rate was 19.5% (vs. 49% in the present study), the 1-year OS rate was 46.5%, and the 60-day mortality rate was 16.2%.
They acknowledged, however, that the validity of the comparison is limited by their study’s small sample size, potential differences between the study populations, and lack of a control group in the present study. The investigators also found that clearance rates of baseline somatic mutations correlated with response to treatment.
Grade 3 or greater treatment-emergent adverse events occurred in 43 of the 50 patients, including infections, thrombocytopenias, and febrile neutropenias.
“On the basis of these encouraging results, a phase 3, multicenter, double-blind, randomized study of pracinostat vs. placebo with azacitidine (NCT03151408) is currently ongoing to confirm superiority of the combination in this difficult-to-treat AML population,” the investigators wrote.
The study was supported by research funding from MEI Pharma, which helped develop pracinostat. Dr. Garcia-Manero reported having no disclosures. Multiple coauthors reported financial relationships with MEI and others. One coauthor is an MEI employee.
SOURCE: Garcia-Manero G et al. Blood Adv. 2019 Feb 26;3(4):508-18.
For patients older than 65 years with newly diagnosed acute myeloid leukemia (AML) who were ineligible for standard induction therapy, adding the investigational pan-histone deacetylase (pan-HDAC) inhibitor pracinostat to azacitidine resulted in better complete remission and overall survival rates than azacitidine alone, results of a multicenter phase 2 trial showed.
Among 50 patients treated with the combination, 26 (52%) achieved the primary endpoint of either a complete remission (CR), CR with incomplete recovery of blood counts (CRi), or morphologic leukemia-free state (MLFS).
The median overall survival (OS) was 19.1 months, which compares favorably with historical data on similar patients treated with single-agent azacitidine, reported Guillermo Garcia-Manero, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“[T]his study shows that pracinostat in combination with azacitidine has the potential to be a safe and effective regimen in the frontline treatment of older patients with AML unfit for [induction chemotherapy],” they wrote in Blood Advances.
Pracinostat is an oral pan-HDAC inhibitor that has been shown to have modest activity against AML as a single agent, but synergistic activity when combined with hypomethylating agent azacitidine, a standard of care for older patients with AML in the trial.
The investigators enrolled 50 patients with a median age of 75 years (range, 66-84 years). The cohort included 33 patients with de novo AML, 12 with AML secondary to myelodysplasia syndrome or myleoproliferative neoplasia, and five with therapy-related AML.
The patients were treated with pracinostat 60 mg daily for 3 days each week for 3 consecutive weeks in addition to azacitidine 75 mg/m2 daily for 7 days in a 28-day cycle.
As noted, 26 patients reached the clinical endpoint, including 21 with a CR, 2 with a CRi, and 3 with MLFS. Additionally, two patients had a partial response (PR) and four had a PR with incomplete recovery of blood counts.
The median OS was 19.1 months, and the median progression-free survival (PFS) was 12.6 months. The 1-year OS rate was 62%. The 60-day mortality rate was 10%.
The authors noted that the survival data were superior to those seen in the phase 3 AZA-AML-001 study, which compared azacitidine therapy with conventional regimens in patients older than 65 years with newly diagnosed AML who were not eligible for stem cell transplants. In that trial, median OS was 10.4 months, the CR rate was 19.5% (vs. 49% in the present study), the 1-year OS rate was 46.5%, and the 60-day mortality rate was 16.2%.
They acknowledged, however, that the validity of the comparison is limited by their study’s small sample size, potential differences between the study populations, and lack of a control group in the present study. The investigators also found that clearance rates of baseline somatic mutations correlated with response to treatment.
Grade 3 or greater treatment-emergent adverse events occurred in 43 of the 50 patients, including infections, thrombocytopenias, and febrile neutropenias.
“On the basis of these encouraging results, a phase 3, multicenter, double-blind, randomized study of pracinostat vs. placebo with azacitidine (NCT03151408) is currently ongoing to confirm superiority of the combination in this difficult-to-treat AML population,” the investigators wrote.
The study was supported by research funding from MEI Pharma, which helped develop pracinostat. Dr. Garcia-Manero reported having no disclosures. Multiple coauthors reported financial relationships with MEI and others. One coauthor is an MEI employee.
SOURCE: Garcia-Manero G et al. Blood Adv. 2019 Feb 26;3(4):508-18.
For patients older than 65 years with newly diagnosed acute myeloid leukemia (AML) who were ineligible for standard induction therapy, adding the investigational pan-histone deacetylase (pan-HDAC) inhibitor pracinostat to azacitidine resulted in better complete remission and overall survival rates than azacitidine alone, results of a multicenter phase 2 trial showed.
Among 50 patients treated with the combination, 26 (52%) achieved the primary endpoint of either a complete remission (CR), CR with incomplete recovery of blood counts (CRi), or morphologic leukemia-free state (MLFS).
The median overall survival (OS) was 19.1 months, which compares favorably with historical data on similar patients treated with single-agent azacitidine, reported Guillermo Garcia-Manero, MD, from the University of Texas MD Anderson Cancer Center in Houston and his colleagues.
“[T]his study shows that pracinostat in combination with azacitidine has the potential to be a safe and effective regimen in the frontline treatment of older patients with AML unfit for [induction chemotherapy],” they wrote in Blood Advances.
Pracinostat is an oral pan-HDAC inhibitor that has been shown to have modest activity against AML as a single agent, but synergistic activity when combined with hypomethylating agent azacitidine, a standard of care for older patients with AML in the trial.
The investigators enrolled 50 patients with a median age of 75 years (range, 66-84 years). The cohort included 33 patients with de novo AML, 12 with AML secondary to myelodysplasia syndrome or myleoproliferative neoplasia, and five with therapy-related AML.
The patients were treated with pracinostat 60 mg daily for 3 days each week for 3 consecutive weeks in addition to azacitidine 75 mg/m2 daily for 7 days in a 28-day cycle.
As noted, 26 patients reached the clinical endpoint, including 21 with a CR, 2 with a CRi, and 3 with MLFS. Additionally, two patients had a partial response (PR) and four had a PR with incomplete recovery of blood counts.
The median OS was 19.1 months, and the median progression-free survival (PFS) was 12.6 months. The 1-year OS rate was 62%. The 60-day mortality rate was 10%.
The authors noted that the survival data were superior to those seen in the phase 3 AZA-AML-001 study, which compared azacitidine therapy with conventional regimens in patients older than 65 years with newly diagnosed AML who were not eligible for stem cell transplants. In that trial, median OS was 10.4 months, the CR rate was 19.5% (vs. 49% in the present study), the 1-year OS rate was 46.5%, and the 60-day mortality rate was 16.2%.
They acknowledged, however, that the validity of the comparison is limited by their study’s small sample size, potential differences between the study populations, and lack of a control group in the present study. The investigators also found that clearance rates of baseline somatic mutations correlated with response to treatment.
Grade 3 or greater treatment-emergent adverse events occurred in 43 of the 50 patients, including infections, thrombocytopenias, and febrile neutropenias.
“On the basis of these encouraging results, a phase 3, multicenter, double-blind, randomized study of pracinostat vs. placebo with azacitidine (NCT03151408) is currently ongoing to confirm superiority of the combination in this difficult-to-treat AML population,” the investigators wrote.
The study was supported by research funding from MEI Pharma, which helped develop pracinostat. Dr. Garcia-Manero reported having no disclosures. Multiple coauthors reported financial relationships with MEI and others. One coauthor is an MEI employee.
SOURCE: Garcia-Manero G et al. Blood Adv. 2019 Feb 26;3(4):508-18.
FROM BLOOD ADVANCES
Guadecitabine may be option for certain MDS/AML patients
New research suggests guadecitabine may be an option for select patients with myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) who have failed treatment with azacitidine.
In a phase 2 trial, eight of 56 patients with high-risk MDS or low-blast-count AML responded to guadecitabine after azacitidine failure. Patients were significantly more likely to respond if they had few or no somatic mutations.
Marie Sébert, MD, of Hôpital Saint Louis in Paris and her colleagues conducted this trial and reported the results in Haematologica.
The trial (NCT02197676) included 56 patients with the following disease types:
- Refractory anemia with excess blasts (RAEB) type 2 (n = 31; 55%).
- RAEB type 1 (n = 11; 20%).
- Low-blast-count AML (n = 11; 20%).
- Refractory cytopenias with multilineage dysplasia (RCMD; n = 2; 4%).
- Chronic myelomonocytic leukemia (n = 1; 2%).
The patients had a median age of 75 years (range, 70-79) at baseline, and 37 (66%) were men. Thirty-four patients (61%) had very-high-risk disease according to the revised International Prognostic Scoring System. Forty-nine patients (87.5%) had at least one somatic mutation. The most commonly mutated genes were ASXL1, RUNX1, TP53, U2AF1, and DNMT3A.
Most patients (n = 41, 73%) had relapsed after azacitidine, and 15 (27%) had primary resistance to the drug. Patients had received a median of 13 azacitidine cycles (range, 6-23).
The patients received guadecitabine subcutaneously at 60 mg/m2 on days 1-5 of a 28-day cycle. They were treated until progression, death, unacceptable toxicity, or no response after six to nine cycles. Patients received a median of three cycles (range, 0-27). One patient died of infection before receiving guadecitabine, but the remaining 55 patients received at least one cycle of treatment. Eighteen patients had a dose reduction.
Eight patients (14.3%) responded to guadecitabine. Two patients achieved a complete response (CR) – one who had RAEB-2 and one with AML. Two patients with RAEB-1 had marrow CRs. Two patients – one with RAEB-2 and one with AML – had marrow CRs with hematologic improvement. A patient with RCMD had hematologic improvement, and a patient with RAEB-2 had a partial response.
The researchers said mutation frequency was the only significant predictor of response. The response rate was significantly higher in patients who did not have somatic mutations (P = .036). The median number of mutations was one (range, zero to three) in responders and two (range, zero to six) in nonresponders (P = .035). None of the patients with TP53 mutations achieved a response.
The median duration of response was 11.5 months. The median overall survival was 17.9 months in responders and 7.1 months in the overall population.
In a multivariate analysis, the following factors were significantly associated with longer survival:
- Having low- to high-risk (vs. very-high-risk) disease (P = .03).
- Having experienced primary (vs. secondary) azacitidine failure (P = .01).
- Having a high rate of demethylation in blood during the first treatment cycle (P = .03).
There were 99 serious adverse events (AEs) reported in 44 patients. Most AEs were hematologic events, and the most common of these was myelosuppression (n = 88; 88%). The most common grade 3/4 nonhematologic AE was pulmonary toxicity (n = 7; 12.5%). Thirteen patients were hospitalized for febrile neutropenia for a median of 14 days.
The researchers said patients reported less pain and fewer secondary lesions with guadecitabine than they had with azacitidine.
This trial was sponsored by Groupe Francophone des Myelodysplasies in collaboration with Astex Pharmaceuticals. The researchers reported having no competing interests.
SOURCE: Sébert M et al. Haematologica. 2019 Feb 7. doi: 0.3324/haematol.2018.207118.
New research suggests guadecitabine may be an option for select patients with myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) who have failed treatment with azacitidine.
In a phase 2 trial, eight of 56 patients with high-risk MDS or low-blast-count AML responded to guadecitabine after azacitidine failure. Patients were significantly more likely to respond if they had few or no somatic mutations.
Marie Sébert, MD, of Hôpital Saint Louis in Paris and her colleagues conducted this trial and reported the results in Haematologica.
The trial (NCT02197676) included 56 patients with the following disease types:
- Refractory anemia with excess blasts (RAEB) type 2 (n = 31; 55%).
- RAEB type 1 (n = 11; 20%).
- Low-blast-count AML (n = 11; 20%).
- Refractory cytopenias with multilineage dysplasia (RCMD; n = 2; 4%).
- Chronic myelomonocytic leukemia (n = 1; 2%).
The patients had a median age of 75 years (range, 70-79) at baseline, and 37 (66%) were men. Thirty-four patients (61%) had very-high-risk disease according to the revised International Prognostic Scoring System. Forty-nine patients (87.5%) had at least one somatic mutation. The most commonly mutated genes were ASXL1, RUNX1, TP53, U2AF1, and DNMT3A.
Most patients (n = 41, 73%) had relapsed after azacitidine, and 15 (27%) had primary resistance to the drug. Patients had received a median of 13 azacitidine cycles (range, 6-23).
The patients received guadecitabine subcutaneously at 60 mg/m2 on days 1-5 of a 28-day cycle. They were treated until progression, death, unacceptable toxicity, or no response after six to nine cycles. Patients received a median of three cycles (range, 0-27). One patient died of infection before receiving guadecitabine, but the remaining 55 patients received at least one cycle of treatment. Eighteen patients had a dose reduction.
Eight patients (14.3%) responded to guadecitabine. Two patients achieved a complete response (CR) – one who had RAEB-2 and one with AML. Two patients with RAEB-1 had marrow CRs. Two patients – one with RAEB-2 and one with AML – had marrow CRs with hematologic improvement. A patient with RCMD had hematologic improvement, and a patient with RAEB-2 had a partial response.
The researchers said mutation frequency was the only significant predictor of response. The response rate was significantly higher in patients who did not have somatic mutations (P = .036). The median number of mutations was one (range, zero to three) in responders and two (range, zero to six) in nonresponders (P = .035). None of the patients with TP53 mutations achieved a response.
The median duration of response was 11.5 months. The median overall survival was 17.9 months in responders and 7.1 months in the overall population.
In a multivariate analysis, the following factors were significantly associated with longer survival:
- Having low- to high-risk (vs. very-high-risk) disease (P = .03).
- Having experienced primary (vs. secondary) azacitidine failure (P = .01).
- Having a high rate of demethylation in blood during the first treatment cycle (P = .03).
There were 99 serious adverse events (AEs) reported in 44 patients. Most AEs were hematologic events, and the most common of these was myelosuppression (n = 88; 88%). The most common grade 3/4 nonhematologic AE was pulmonary toxicity (n = 7; 12.5%). Thirteen patients were hospitalized for febrile neutropenia for a median of 14 days.
The researchers said patients reported less pain and fewer secondary lesions with guadecitabine than they had with azacitidine.
This trial was sponsored by Groupe Francophone des Myelodysplasies in collaboration with Astex Pharmaceuticals. The researchers reported having no competing interests.
SOURCE: Sébert M et al. Haematologica. 2019 Feb 7. doi: 0.3324/haematol.2018.207118.
New research suggests guadecitabine may be an option for select patients with myelodysplastic syndromes (MDS) or acute myeloid leukemia (AML) who have failed treatment with azacitidine.
In a phase 2 trial, eight of 56 patients with high-risk MDS or low-blast-count AML responded to guadecitabine after azacitidine failure. Patients were significantly more likely to respond if they had few or no somatic mutations.
Marie Sébert, MD, of Hôpital Saint Louis in Paris and her colleagues conducted this trial and reported the results in Haematologica.
The trial (NCT02197676) included 56 patients with the following disease types:
- Refractory anemia with excess blasts (RAEB) type 2 (n = 31; 55%).
- RAEB type 1 (n = 11; 20%).
- Low-blast-count AML (n = 11; 20%).
- Refractory cytopenias with multilineage dysplasia (RCMD; n = 2; 4%).
- Chronic myelomonocytic leukemia (n = 1; 2%).
The patients had a median age of 75 years (range, 70-79) at baseline, and 37 (66%) were men. Thirty-four patients (61%) had very-high-risk disease according to the revised International Prognostic Scoring System. Forty-nine patients (87.5%) had at least one somatic mutation. The most commonly mutated genes were ASXL1, RUNX1, TP53, U2AF1, and DNMT3A.
Most patients (n = 41, 73%) had relapsed after azacitidine, and 15 (27%) had primary resistance to the drug. Patients had received a median of 13 azacitidine cycles (range, 6-23).
The patients received guadecitabine subcutaneously at 60 mg/m2 on days 1-5 of a 28-day cycle. They were treated until progression, death, unacceptable toxicity, or no response after six to nine cycles. Patients received a median of three cycles (range, 0-27). One patient died of infection before receiving guadecitabine, but the remaining 55 patients received at least one cycle of treatment. Eighteen patients had a dose reduction.
Eight patients (14.3%) responded to guadecitabine. Two patients achieved a complete response (CR) – one who had RAEB-2 and one with AML. Two patients with RAEB-1 had marrow CRs. Two patients – one with RAEB-2 and one with AML – had marrow CRs with hematologic improvement. A patient with RCMD had hematologic improvement, and a patient with RAEB-2 had a partial response.
The researchers said mutation frequency was the only significant predictor of response. The response rate was significantly higher in patients who did not have somatic mutations (P = .036). The median number of mutations was one (range, zero to three) in responders and two (range, zero to six) in nonresponders (P = .035). None of the patients with TP53 mutations achieved a response.
The median duration of response was 11.5 months. The median overall survival was 17.9 months in responders and 7.1 months in the overall population.
In a multivariate analysis, the following factors were significantly associated with longer survival:
- Having low- to high-risk (vs. very-high-risk) disease (P = .03).
- Having experienced primary (vs. secondary) azacitidine failure (P = .01).
- Having a high rate of demethylation in blood during the first treatment cycle (P = .03).
There were 99 serious adverse events (AEs) reported in 44 patients. Most AEs were hematologic events, and the most common of these was myelosuppression (n = 88; 88%). The most common grade 3/4 nonhematologic AE was pulmonary toxicity (n = 7; 12.5%). Thirteen patients were hospitalized for febrile neutropenia for a median of 14 days.
The researchers said patients reported less pain and fewer secondary lesions with guadecitabine than they had with azacitidine.
This trial was sponsored by Groupe Francophone des Myelodysplasies in collaboration with Astex Pharmaceuticals. The researchers reported having no competing interests.
SOURCE: Sébert M et al. Haematologica. 2019 Feb 7. doi: 0.3324/haematol.2018.207118.
REPORTING FROM HAEMATOLOGICA
Understanding AD as immune-driven disease has opened the door to new therapies
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.

“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.
“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
WASHINGTON – The “ of the disease has grown, Emma Guttman-Yassky, MD, PhD, said during a presentation at the annual meeting of the American Academy of Dermatology.
“It’s due to the increased understanding we now have in atopic dermatitis,” Dr. Guttman-Yassky, professor and vice chair for research in the department of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in her presentation.
According to Dr. Guttman-Yassky, therapeutic development was prevented in AD because of the abnormalities present in the disease immune responses and barrier abnormalities. “Frankly, pharma[ceutical] companies didn’t know what they should go after,” she said. “Should they go after the immune abnormalities, or should they go after the barrier? I think that’s why we’re so far behind psoriasis – but don’t worry, we are catching up quite fast because now ... we understand what we need to go after.”
It was when researchers began to look at AD in the same way as psoriasis that they realized the two were “polar” immune diseases, with psoriasis having Th17/interleukin-17 involvement while atopic dermatitis had Th2/IL-13 involvement. The same approach of “bedside-to-bench pathogenic dissection and translational testing of therapeutics” that led to successful advancements in therapies for psoriasis can also be applied to AD, Dr. Guttman-Yassky said.
To create a translational approach to AD, researchers need to have a well-defined molecular phenotype and understanding of inflammatory pathways, good baseline biomarkers of disease activity and treatment responses, and drugs that would selectively target the immune system. Th2-type cytokines such as IL-4 and IL-13 could help link the barrier and immune defects in AD. In addition, all variations of AD subtypes across white, black, Asian, and pediatric populations have “robust Th2 activation” but differ in other areas. “We’ll need to stratify biomarkers specific to different atopic dermatitis phenotypes to really develop a personalized medicine approach in atopic dermatitis,” she said.
High-level systemic immune activation shows that AD is emerging as a systemic disease that leads to atopic comorbidities such as allergy and asthma, as well as cardiovascular and infectious comorbidities. “We need to think about it when we treat our patients, because we really need to give them systemic treatment approaches when they have this moderate to severe disease,” Dr. Guttman-Yassky said. “When adult patients have moderate to severe disease, what is nonlesional today may be lesional tomorrow, and to treat them effectively, you have to offer them some systemic approaches.”
There is evidence that dupilumab, a human monoclonal antibody that targets IL-4 receptor alpha, is “proving the immune hypothesis” of AD, Dr. Guttman-Yassky said. She cited a recent study from her own group that found use of dupilumab to inhibit IL-4/IL-13 signaling improved disease activity for patients with AD, including reducing the expression of genes that caused type 2 inflammation, epidermal hyperplasia, T cells, dendritic cells, and Th17/Th22 activity (J Allergy Clin Immunol. 2019 Jan;143(1):155-72).
“We could postulate it before, but we couldn’t prove it,” she said. “Basically, this opened the door to all the therapy that we now have in atopic dermatitis.”
According to Dr. Guttman-Yassky, the future of AD will be in creating personalized treatments for patients by stratifying biomarkers specific to different AD phenotypes.
“It’s a very hopeful time in atopic dermatitis with this growing knowledge that we have of the biology of [the disease],” she said. “We have many more agents to treat our patients, and I think the future will be about personalized medicine so we really are treating the disease very well.”
Dr. Guttman-Yassky reported relationships with AbbVie, Allergan, Almirall, Anacor Pharmaceuticals, Asana BioSciences, Celgene, Dermira, Eli Lilly, Escalier Biosciences, Galderma Research & Development, Glenmark Generics, Janssen, Kyowa Hakko Kirin, Leo Pharma, Medimmune, Novartis, Pfizer, Regeneron, Sanofi-Aventis, Sanofi/Regeneron, Stiefel, Theravance Biopharma, and Vitae Pharmaceuticals.
EXPERT ANALYSIS FROM AAD 19
Post-HCT azithromycin doesn’t increase relapse risk, study suggests
HOUSTON — When given after transplant, azithromycin does not increase the risk of relapse in patients with moderate to severe chronic graft-versus-host disease (cGVHD) and bronchiolitis obliterans syndrome (BOS), according to a retrospective study.
A prior study, ALLOZITHRO (JAMA. 2017 Aug 8;318[6]:557-66), showed an increased risk of relapse and death in patients who received azithromycin as BOS prophylaxis prior to hematopoietic cell transplant (HCT).
That discovery prompted the Food and Drug Administration to release a safety communication warning prescribers about the risks associated with azithromycin as BOS prophylaxis. However, it wasn’t clear if the same risks exist when azithromycin is given for cGVHD management after HCT.
To gain some insight, Mark Shamoun, MD, of the University of Michigan, Ann Arbor, and his colleagues examined data on patients with moderate to severe cGVHD and BOS who received azithromycin after undergoing HCT to treat a hematologic malignancy.
Dr. Shamoun presented the group’s findings at the Transplantation & Cellular Therapy Meetings.
The researchers reviewed data on 239 patients enrolled in the University of Michigan’s HCT database from 2010 to 2017. The median age at baseline was 55 years (range, 4-72 years).
The patients received transplants to treat acute myeloid leukemia or myelodysplastic syndromes (n = 141), acute lymphoblastic leukemia (n = 40), lymphoma (n = 26), chronic leukemia (n = 24), multiple myeloma (n = 6), and myeloproliferative neoplasms (n = 2).
The patients had matched related donors (43%) or matched unrelated donors (57%). Most patients received peripheral blood transplants (84%), though some received bone marrow (14%) or cord blood (2%). All patients had moderate (38%) or severe (62%) cGVHD.
Patients were split into two cohorts. Patients in cohort A (n = 86) had BOS and received azithromycin for more than 14 days.
Patients in cohort B (n = 153) either did not receive azithromycin or received it for 14 days or less. Fewer than 5% of patients in cohort B had BOS.
Most other baseline characteristics were similar between the cohorts. However, severe cGVHD was more prevalent in cohort A than B — 78% and 51%, respectively.
In cohort A, the median time to the start of azithromycin was 15 months after HCT (range, 3-68 months). The median duration of azithromycin treatment was 26 months (range, 1-77 months).
Results
The 2-year relapse rate was significantly lower in patients who received azithromycin than in those who did not — 4% and 17%, respectively (P = .001).
There was a significant difference in relapse rate both from the time of HCT (P = .001) and from the start of azithromycin or cGVHD (P = .011).
There was no significant difference in overall survival between the cohorts, either from the time of HCT (P = .294) or from the start of azithromycin or cGVHD (P = .428).
Dr. Shamoun said these results suggest azithromycin does not increase the risk of relapse when it is used to manage cGVHD. However, this study is limited by its retrospective nature. In addition, most patients in cohort B did not have BOS, severe cGVHD was more common in cohort A, and the incidence of relapse was not calculated from the time of azithromycin initiation in both cohorts. Therefore, additional investigation is needed.
Dr. Shamoun presented these results at Transplantation & Cellular Therapy Meetings, which is held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At the meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society, the American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Shamoun reported no conflicts of interest.
SOURCE: Shamoun M et al. TCT 2019, Abstract 33.
HOUSTON — When given after transplant, azithromycin does not increase the risk of relapse in patients with moderate to severe chronic graft-versus-host disease (cGVHD) and bronchiolitis obliterans syndrome (BOS), according to a retrospective study.
A prior study, ALLOZITHRO (JAMA. 2017 Aug 8;318[6]:557-66), showed an increased risk of relapse and death in patients who received azithromycin as BOS prophylaxis prior to hematopoietic cell transplant (HCT).
That discovery prompted the Food and Drug Administration to release a safety communication warning prescribers about the risks associated with azithromycin as BOS prophylaxis. However, it wasn’t clear if the same risks exist when azithromycin is given for cGVHD management after HCT.
To gain some insight, Mark Shamoun, MD, of the University of Michigan, Ann Arbor, and his colleagues examined data on patients with moderate to severe cGVHD and BOS who received azithromycin after undergoing HCT to treat a hematologic malignancy.
Dr. Shamoun presented the group’s findings at the Transplantation & Cellular Therapy Meetings.
The researchers reviewed data on 239 patients enrolled in the University of Michigan’s HCT database from 2010 to 2017. The median age at baseline was 55 years (range, 4-72 years).
The patients received transplants to treat acute myeloid leukemia or myelodysplastic syndromes (n = 141), acute lymphoblastic leukemia (n = 40), lymphoma (n = 26), chronic leukemia (n = 24), multiple myeloma (n = 6), and myeloproliferative neoplasms (n = 2).
The patients had matched related donors (43%) or matched unrelated donors (57%). Most patients received peripheral blood transplants (84%), though some received bone marrow (14%) or cord blood (2%). All patients had moderate (38%) or severe (62%) cGVHD.
Patients were split into two cohorts. Patients in cohort A (n = 86) had BOS and received azithromycin for more than 14 days.
Patients in cohort B (n = 153) either did not receive azithromycin or received it for 14 days or less. Fewer than 5% of patients in cohort B had BOS.
Most other baseline characteristics were similar between the cohorts. However, severe cGVHD was more prevalent in cohort A than B — 78% and 51%, respectively.
In cohort A, the median time to the start of azithromycin was 15 months after HCT (range, 3-68 months). The median duration of azithromycin treatment was 26 months (range, 1-77 months).
Results
The 2-year relapse rate was significantly lower in patients who received azithromycin than in those who did not — 4% and 17%, respectively (P = .001).
There was a significant difference in relapse rate both from the time of HCT (P = .001) and from the start of azithromycin or cGVHD (P = .011).
There was no significant difference in overall survival between the cohorts, either from the time of HCT (P = .294) or from the start of azithromycin or cGVHD (P = .428).
Dr. Shamoun said these results suggest azithromycin does not increase the risk of relapse when it is used to manage cGVHD. However, this study is limited by its retrospective nature. In addition, most patients in cohort B did not have BOS, severe cGVHD was more common in cohort A, and the incidence of relapse was not calculated from the time of azithromycin initiation in both cohorts. Therefore, additional investigation is needed.
Dr. Shamoun presented these results at Transplantation & Cellular Therapy Meetings, which is held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At the meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society, the American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Shamoun reported no conflicts of interest.
SOURCE: Shamoun M et al. TCT 2019, Abstract 33.
HOUSTON — When given after transplant, azithromycin does not increase the risk of relapse in patients with moderate to severe chronic graft-versus-host disease (cGVHD) and bronchiolitis obliterans syndrome (BOS), according to a retrospective study.
A prior study, ALLOZITHRO (JAMA. 2017 Aug 8;318[6]:557-66), showed an increased risk of relapse and death in patients who received azithromycin as BOS prophylaxis prior to hematopoietic cell transplant (HCT).
That discovery prompted the Food and Drug Administration to release a safety communication warning prescribers about the risks associated with azithromycin as BOS prophylaxis. However, it wasn’t clear if the same risks exist when azithromycin is given for cGVHD management after HCT.
To gain some insight, Mark Shamoun, MD, of the University of Michigan, Ann Arbor, and his colleagues examined data on patients with moderate to severe cGVHD and BOS who received azithromycin after undergoing HCT to treat a hematologic malignancy.
Dr. Shamoun presented the group’s findings at the Transplantation & Cellular Therapy Meetings.
The researchers reviewed data on 239 patients enrolled in the University of Michigan’s HCT database from 2010 to 2017. The median age at baseline was 55 years (range, 4-72 years).
The patients received transplants to treat acute myeloid leukemia or myelodysplastic syndromes (n = 141), acute lymphoblastic leukemia (n = 40), lymphoma (n = 26), chronic leukemia (n = 24), multiple myeloma (n = 6), and myeloproliferative neoplasms (n = 2).
The patients had matched related donors (43%) or matched unrelated donors (57%). Most patients received peripheral blood transplants (84%), though some received bone marrow (14%) or cord blood (2%). All patients had moderate (38%) or severe (62%) cGVHD.
Patients were split into two cohorts. Patients in cohort A (n = 86) had BOS and received azithromycin for more than 14 days.
Patients in cohort B (n = 153) either did not receive azithromycin or received it for 14 days or less. Fewer than 5% of patients in cohort B had BOS.
Most other baseline characteristics were similar between the cohorts. However, severe cGVHD was more prevalent in cohort A than B — 78% and 51%, respectively.
In cohort A, the median time to the start of azithromycin was 15 months after HCT (range, 3-68 months). The median duration of azithromycin treatment was 26 months (range, 1-77 months).
Results
The 2-year relapse rate was significantly lower in patients who received azithromycin than in those who did not — 4% and 17%, respectively (P = .001).
There was a significant difference in relapse rate both from the time of HCT (P = .001) and from the start of azithromycin or cGVHD (P = .011).
There was no significant difference in overall survival between the cohorts, either from the time of HCT (P = .294) or from the start of azithromycin or cGVHD (P = .428).
Dr. Shamoun said these results suggest azithromycin does not increase the risk of relapse when it is used to manage cGVHD. However, this study is limited by its retrospective nature. In addition, most patients in cohort B did not have BOS, severe cGVHD was more common in cohort A, and the incidence of relapse was not calculated from the time of azithromycin initiation in both cohorts. Therefore, additional investigation is needed.
Dr. Shamoun presented these results at Transplantation & Cellular Therapy Meetings, which is held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At the meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society, the American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Shamoun reported no conflicts of interest.
SOURCE: Shamoun M et al. TCT 2019, Abstract 33.
REPORTING FROM TCT 2019
CLL, GVHD may raise risk for skin cancer after allo-HCT
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Midostaurin maintenance may reduce relapse risk in FLT3-ITD+ AML
HOUSTON – Midostaurin maintenance therapy along with standard-of-care treatment after allogeneic stem cell transplant (alloSCT) in patients with acute myeloid leukemia (AML) appears to reduce the risk of relapse, according to findings from the randomized, phase 2 RADIUS trial.

Notably, the effect of midostaurin in this open-label, exploratory trial was most pronounced in patients with high levels of phosphorylated FLT3 (pFLT3) inhibition as assessed by plasma inhibitor activity assay, Richard T. Maziarz, MD, reported at the Transplantation & Cellular Therapy Meetings.
“The median [pFLT3 reduction] was less than 70% ... those patients who had the deepest level inhibition maintained the highest likelihood of staying free of disease,” Dr. Maziarz, a professor of medicine at Oregon Health & Science University, Portland, said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Midostaurin is a multitargeted tyrosine kinase inhibitor (TKI) that was shown in the pivotal RATIFY trial to significantly improve event-free and overall survival versus placebo when interspersed with induction and consolidation chemotherapy and also when used for maintenance in adults with newly diagnosed FLT3-mutated AML, Dr. Maziarz explained. He noted that patients in the RATIFY study who underwent alloSCT did not receive midostaurin maintenance (N Engl J Med. 2017; 377:454-64).
Although alloSCT provides the greatest likelihood of sustained remission in AML, relapse rates remain high at 30%-59%, he said, adding that, “in the setting of transplantation, FLT3 expression, or FLT3-ITD [internal tandem duplication] ... is a poor risk feature.”
Studies are increasingly suggesting that posttransplant maintenance therapy may improve this outcome. For example, the small, randomized, phase 2 SORMAIN study presented at the 2018 annual meeting of the American Society of Hematology showed a signal for benefit with posttransplant maintenance with the TKI sorafenib. Data regarding midostaurin in this setting are limited, Dr. Maziarz noted.
The RADIUS trial was a small study designed to look for a similar signal with midostaurin and thus was not adequately powered to detect a statistical difference between the arms, he explained.
RADIUS included 60 AML patients aged 18-70 years who underwent myeloablative alloSCT and were in their first complete remission. The primary endpoint was relapse-free survival (RFS) at 18 months after transplant. Results were presented at ASH 2018.
RFS was 89% in 16 of 30 patients who were randomized to receive 50 mg of midostaurin twice daily along with standard-of-care (SOC) treatment and completed 12 4-week cycles. This compared with an RFS rate of 76% in 14 of 30 patients who received SOC only and completed 12 cycles (hazard ratio, 0.46).
The predicted relative reduction in the risk of relapse with the addition of midostaurin was 54%, and at 24 months, both RFS and overall survival were 85% in the midostaurin group and 76% in the SOC-only group, Dr. Maziarz reported.
The median duration of exposure to midostaurin was 10.5 months and the median dose intensity was 93 mg/day, indicating that full-dose therapy was achievable in most patients who stayed on the study.
Treatment was generally well tolerated; there was a comparable number of early discontinuations in the midostaurin and SOC-only arms. The discontinuations were caused mainly by adverse events (typically gastrointestinal toxicities) in the midostaurin arm and by consent withdrawal in the SOC-only arm, he said, adding that there were no significant differences between the groups with respect to serious adverse events or acute or chronic graft-versus-host disease.
Following the presentation of the primary RADIUS results at ASH 2018, an exploratory analysis was conducted to assess midostaurin’s inhibitory effects on FLT3 in plasma.
FLT3 plasma inhibitor activity, assessed by coculturing plasma samples taken on the first day of the treatment cycles with the FLT3-positive AML to look for a reduction in pFLT3, was evaluable in 28 patients in each arm.
“What we see is when you start there are high levels of FLT3, but the pFLT3 drops significantly with exposure to the plasma,” he said, noting that the effect was most prominent during the first two cycles of therapy.
The patients with the highest levels of inhibition had the greatest likelihood of RFS, whereas RFS in those with suboptimal pFLT3 inhibition was similar to that seen in the SOC-only arm, Dr. Maziarz said. Two patients in the midostaurin group who relapsed did so after 12 months – when midostaurin had been discontinued, he noted.
“Our conclusion is that maintenance midostaurin may contribute to a reduction in relapse risk at 18 months post transplant ... and can be safely administered in the posttransplant setting,” Dr. Maziarz said. “pFLT3 inhibition to less than 70% of baseline, at least in this study, was associated with improved relapse-free survival and overall survival, and it was achieved in more than 50% of patients on the midostaurin.”
It is likely that a more definitive answer will be provided by the Blood and Marrow Transplant Clinical Trials Network Protocol 1506, a large, multinational, placebo-controlled trial now recruiting to look at this question of whether maintenance therapy in the posttransplant setting will improve outcomes.
However, it is important to note that no patient in the RADIUS trial received pretransplant midostaurin, as RADIUS was conducted at the same time as the RATIFY trial.
“Patients today who will go to transplant with FLT3-ITD, the vast majority will have been treated during induction ... and we may have a totally different biology going forward,” he said.
Dr. Maziarz reported financial relationships with Incyte, Novartis, Celgene/Juno, Kite/Gilead, Juno Therapeutics, Kite Therapeutics, and Athersys.
HOUSTON – Midostaurin maintenance therapy along with standard-of-care treatment after allogeneic stem cell transplant (alloSCT) in patients with acute myeloid leukemia (AML) appears to reduce the risk of relapse, according to findings from the randomized, phase 2 RADIUS trial.
Notably, the effect of midostaurin in this open-label, exploratory trial was most pronounced in patients with high levels of phosphorylated FLT3 (pFLT3) inhibition as assessed by plasma inhibitor activity assay, Richard T. Maziarz, MD, reported at the Transplantation & Cellular Therapy Meetings.
“The median [pFLT3 reduction] was less than 70% ... those patients who had the deepest level inhibition maintained the highest likelihood of staying free of disease,” Dr. Maziarz, a professor of medicine at Oregon Health & Science University, Portland, said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Midostaurin is a multitargeted tyrosine kinase inhibitor (TKI) that was shown in the pivotal RATIFY trial to significantly improve event-free and overall survival versus placebo when interspersed with induction and consolidation chemotherapy and also when used for maintenance in adults with newly diagnosed FLT3-mutated AML, Dr. Maziarz explained. He noted that patients in the RATIFY study who underwent alloSCT did not receive midostaurin maintenance (N Engl J Med. 2017; 377:454-64).
Although alloSCT provides the greatest likelihood of sustained remission in AML, relapse rates remain high at 30%-59%, he said, adding that, “in the setting of transplantation, FLT3 expression, or FLT3-ITD [internal tandem duplication] ... is a poor risk feature.”
Studies are increasingly suggesting that posttransplant maintenance therapy may improve this outcome. For example, the small, randomized, phase 2 SORMAIN study presented at the 2018 annual meeting of the American Society of Hematology showed a signal for benefit with posttransplant maintenance with the TKI sorafenib. Data regarding midostaurin in this setting are limited, Dr. Maziarz noted.
The RADIUS trial was a small study designed to look for a similar signal with midostaurin and thus was not adequately powered to detect a statistical difference between the arms, he explained.
RADIUS included 60 AML patients aged 18-70 years who underwent myeloablative alloSCT and were in their first complete remission. The primary endpoint was relapse-free survival (RFS) at 18 months after transplant. Results were presented at ASH 2018.
RFS was 89% in 16 of 30 patients who were randomized to receive 50 mg of midostaurin twice daily along with standard-of-care (SOC) treatment and completed 12 4-week cycles. This compared with an RFS rate of 76% in 14 of 30 patients who received SOC only and completed 12 cycles (hazard ratio, 0.46).
The predicted relative reduction in the risk of relapse with the addition of midostaurin was 54%, and at 24 months, both RFS and overall survival were 85% in the midostaurin group and 76% in the SOC-only group, Dr. Maziarz reported.
The median duration of exposure to midostaurin was 10.5 months and the median dose intensity was 93 mg/day, indicating that full-dose therapy was achievable in most patients who stayed on the study.
Treatment was generally well tolerated; there was a comparable number of early discontinuations in the midostaurin and SOC-only arms. The discontinuations were caused mainly by adverse events (typically gastrointestinal toxicities) in the midostaurin arm and by consent withdrawal in the SOC-only arm, he said, adding that there were no significant differences between the groups with respect to serious adverse events or acute or chronic graft-versus-host disease.
Following the presentation of the primary RADIUS results at ASH 2018, an exploratory analysis was conducted to assess midostaurin’s inhibitory effects on FLT3 in plasma.
FLT3 plasma inhibitor activity, assessed by coculturing plasma samples taken on the first day of the treatment cycles with the FLT3-positive AML to look for a reduction in pFLT3, was evaluable in 28 patients in each arm.
“What we see is when you start there are high levels of FLT3, but the pFLT3 drops significantly with exposure to the plasma,” he said, noting that the effect was most prominent during the first two cycles of therapy.
The patients with the highest levels of inhibition had the greatest likelihood of RFS, whereas RFS in those with suboptimal pFLT3 inhibition was similar to that seen in the SOC-only arm, Dr. Maziarz said. Two patients in the midostaurin group who relapsed did so after 12 months – when midostaurin had been discontinued, he noted.
“Our conclusion is that maintenance midostaurin may contribute to a reduction in relapse risk at 18 months post transplant ... and can be safely administered in the posttransplant setting,” Dr. Maziarz said. “pFLT3 inhibition to less than 70% of baseline, at least in this study, was associated with improved relapse-free survival and overall survival, and it was achieved in more than 50% of patients on the midostaurin.”
It is likely that a more definitive answer will be provided by the Blood and Marrow Transplant Clinical Trials Network Protocol 1506, a large, multinational, placebo-controlled trial now recruiting to look at this question of whether maintenance therapy in the posttransplant setting will improve outcomes.
However, it is important to note that no patient in the RADIUS trial received pretransplant midostaurin, as RADIUS was conducted at the same time as the RATIFY trial.
“Patients today who will go to transplant with FLT3-ITD, the vast majority will have been treated during induction ... and we may have a totally different biology going forward,” he said.
Dr. Maziarz reported financial relationships with Incyte, Novartis, Celgene/Juno, Kite/Gilead, Juno Therapeutics, Kite Therapeutics, and Athersys.
HOUSTON – Midostaurin maintenance therapy along with standard-of-care treatment after allogeneic stem cell transplant (alloSCT) in patients with acute myeloid leukemia (AML) appears to reduce the risk of relapse, according to findings from the randomized, phase 2 RADIUS trial.
Notably, the effect of midostaurin in this open-label, exploratory trial was most pronounced in patients with high levels of phosphorylated FLT3 (pFLT3) inhibition as assessed by plasma inhibitor activity assay, Richard T. Maziarz, MD, reported at the Transplantation & Cellular Therapy Meetings.
“The median [pFLT3 reduction] was less than 70% ... those patients who had the deepest level inhibition maintained the highest likelihood of staying free of disease,” Dr. Maziarz, a professor of medicine at Oregon Health & Science University, Portland, said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Midostaurin is a multitargeted tyrosine kinase inhibitor (TKI) that was shown in the pivotal RATIFY trial to significantly improve event-free and overall survival versus placebo when interspersed with induction and consolidation chemotherapy and also when used for maintenance in adults with newly diagnosed FLT3-mutated AML, Dr. Maziarz explained. He noted that patients in the RATIFY study who underwent alloSCT did not receive midostaurin maintenance (N Engl J Med. 2017; 377:454-64).
Although alloSCT provides the greatest likelihood of sustained remission in AML, relapse rates remain high at 30%-59%, he said, adding that, “in the setting of transplantation, FLT3 expression, or FLT3-ITD [internal tandem duplication] ... is a poor risk feature.”
Studies are increasingly suggesting that posttransplant maintenance therapy may improve this outcome. For example, the small, randomized, phase 2 SORMAIN study presented at the 2018 annual meeting of the American Society of Hematology showed a signal for benefit with posttransplant maintenance with the TKI sorafenib. Data regarding midostaurin in this setting are limited, Dr. Maziarz noted.
The RADIUS trial was a small study designed to look for a similar signal with midostaurin and thus was not adequately powered to detect a statistical difference between the arms, he explained.
RADIUS included 60 AML patients aged 18-70 years who underwent myeloablative alloSCT and were in their first complete remission. The primary endpoint was relapse-free survival (RFS) at 18 months after transplant. Results were presented at ASH 2018.
RFS was 89% in 16 of 30 patients who were randomized to receive 50 mg of midostaurin twice daily along with standard-of-care (SOC) treatment and completed 12 4-week cycles. This compared with an RFS rate of 76% in 14 of 30 patients who received SOC only and completed 12 cycles (hazard ratio, 0.46).
The predicted relative reduction in the risk of relapse with the addition of midostaurin was 54%, and at 24 months, both RFS and overall survival were 85% in the midostaurin group and 76% in the SOC-only group, Dr. Maziarz reported.
The median duration of exposure to midostaurin was 10.5 months and the median dose intensity was 93 mg/day, indicating that full-dose therapy was achievable in most patients who stayed on the study.
Treatment was generally well tolerated; there was a comparable number of early discontinuations in the midostaurin and SOC-only arms. The discontinuations were caused mainly by adverse events (typically gastrointestinal toxicities) in the midostaurin arm and by consent withdrawal in the SOC-only arm, he said, adding that there were no significant differences between the groups with respect to serious adverse events or acute or chronic graft-versus-host disease.
Following the presentation of the primary RADIUS results at ASH 2018, an exploratory analysis was conducted to assess midostaurin’s inhibitory effects on FLT3 in plasma.
FLT3 plasma inhibitor activity, assessed by coculturing plasma samples taken on the first day of the treatment cycles with the FLT3-positive AML to look for a reduction in pFLT3, was evaluable in 28 patients in each arm.
“What we see is when you start there are high levels of FLT3, but the pFLT3 drops significantly with exposure to the plasma,” he said, noting that the effect was most prominent during the first two cycles of therapy.
The patients with the highest levels of inhibition had the greatest likelihood of RFS, whereas RFS in those with suboptimal pFLT3 inhibition was similar to that seen in the SOC-only arm, Dr. Maziarz said. Two patients in the midostaurin group who relapsed did so after 12 months – when midostaurin had been discontinued, he noted.
“Our conclusion is that maintenance midostaurin may contribute to a reduction in relapse risk at 18 months post transplant ... and can be safely administered in the posttransplant setting,” Dr. Maziarz said. “pFLT3 inhibition to less than 70% of baseline, at least in this study, was associated with improved relapse-free survival and overall survival, and it was achieved in more than 50% of patients on the midostaurin.”
It is likely that a more definitive answer will be provided by the Blood and Marrow Transplant Clinical Trials Network Protocol 1506, a large, multinational, placebo-controlled trial now recruiting to look at this question of whether maintenance therapy in the posttransplant setting will improve outcomes.
However, it is important to note that no patient in the RADIUS trial received pretransplant midostaurin, as RADIUS was conducted at the same time as the RATIFY trial.
“Patients today who will go to transplant with FLT3-ITD, the vast majority will have been treated during induction ... and we may have a totally different biology going forward,” he said.
Dr. Maziarz reported financial relationships with Incyte, Novartis, Celgene/Juno, Kite/Gilead, Juno Therapeutics, Kite Therapeutics, and Athersys.
REPORTING FROM TCT 2019
Treosulfan may become standard in allo-HCT for AML/MDS
HOUSTON – A treosulfan-based conditioning regimen could become standard prior to allogeneic transplant in elderly or comorbid patients with acute myeloid leukemia or myelodysplastic syndromes, according to the lead investigator in a phase 3 trial.
The treosulfan/fludarabine myeloablative conditioning regimen had noninferior event-free survival, compared with a reduced-intensity busulfan-based regimen in the large, randomized trial that included elderly patients and those with multiple comorbidities, said researcher Dietrich Wilhelm Beelen, MD, PhD.
The experimental regimen was superior to busulfan in overall survival, nonrelapse mortality, and complete donor chimerism in the trial, added Dr. Beelen, who is with the department of bone marrow transplantation at the West German Cancer Center, University Hospital of Essen, Germany.
“The study results point to a potential benefit of the treosulfan/fludarabine regimen, while the early safety profile, engraftment kinetics, acute or chronic graft-versus-host-disease (GvHD), and the relapse risk of both regimens appear comparable,” Dr. Beelen said at the Transplantation & Cellular Therapy Meetings.
Allogeneic hematopoietic cell transplantation (HCT) is challenging in elderly and comorbid patients, who have an increased risk of nonrelapse mortality with standard myeloablative regimens, according to Dr. Beelen, who presented results on behalf of investigators from the international MC-FludT.14/L Study Group.
Their phase 3 randomized trial included patients who were 50-70 years of age, or who had a Hematopoietic Cell Transplantation Comorbidity Index of 2 or greater. The final analysis included 551 patients (352 with AML and 199 with MDS).
The primary endpoint of the study was event-free survival at 2 years. That endpoint comprised relapse/progression of disease, graft failure, or death.
Patient enrollment was terminated early the MC-FludT.14/L study following an interim analysis that investigators said “clearly demonstrated” the noninferiority of the treosulfan/fludarabine regimen versus the reduced intensity busulfan/fludarabine regimen.
In the final analysis, event-free survival at 2 years was about 14.5 percentage points higher in the treosulfan group, at 65.7% versus 51.2% (P = .0000001), Dr. Beelen reported at the meeting.
A number of other secondary endpoints also favored treosulfan/fludarabine over busulfan, including overall survival (P = .0037), nonrelapse mortality (P = .0343), and survival free of chronic GvHD or relapse (P = .0030).
These results help establish the new treosulfan/fludarabine regimen as a “relatively well-tolerable and effective preparative regimen” in elderly or comorbid AML/MDS patients, Dr. Beelen said.
However, treosulfan has not been authorized for use in allogeneic HCT conditioning regimens, and so should be considered experimental in this setting, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Beelen reported honoraria, travel support, and trial documentation support provided by medac GmbH, which sponsored the trial.
SOURCE: Beelen DW et al. TCT 2019, Abstract 4.
HOUSTON – A treosulfan-based conditioning regimen could become standard prior to allogeneic transplant in elderly or comorbid patients with acute myeloid leukemia or myelodysplastic syndromes, according to the lead investigator in a phase 3 trial.
The treosulfan/fludarabine myeloablative conditioning regimen had noninferior event-free survival, compared with a reduced-intensity busulfan-based regimen in the large, randomized trial that included elderly patients and those with multiple comorbidities, said researcher Dietrich Wilhelm Beelen, MD, PhD.
The experimental regimen was superior to busulfan in overall survival, nonrelapse mortality, and complete donor chimerism in the trial, added Dr. Beelen, who is with the department of bone marrow transplantation at the West German Cancer Center, University Hospital of Essen, Germany.
“The study results point to a potential benefit of the treosulfan/fludarabine regimen, while the early safety profile, engraftment kinetics, acute or chronic graft-versus-host-disease (GvHD), and the relapse risk of both regimens appear comparable,” Dr. Beelen said at the Transplantation & Cellular Therapy Meetings.
Allogeneic hematopoietic cell transplantation (HCT) is challenging in elderly and comorbid patients, who have an increased risk of nonrelapse mortality with standard myeloablative regimens, according to Dr. Beelen, who presented results on behalf of investigators from the international MC-FludT.14/L Study Group.
Their phase 3 randomized trial included patients who were 50-70 years of age, or who had a Hematopoietic Cell Transplantation Comorbidity Index of 2 or greater. The final analysis included 551 patients (352 with AML and 199 with MDS).
The primary endpoint of the study was event-free survival at 2 years. That endpoint comprised relapse/progression of disease, graft failure, or death.
Patient enrollment was terminated early the MC-FludT.14/L study following an interim analysis that investigators said “clearly demonstrated” the noninferiority of the treosulfan/fludarabine regimen versus the reduced intensity busulfan/fludarabine regimen.
In the final analysis, event-free survival at 2 years was about 14.5 percentage points higher in the treosulfan group, at 65.7% versus 51.2% (P = .0000001), Dr. Beelen reported at the meeting.
A number of other secondary endpoints also favored treosulfan/fludarabine over busulfan, including overall survival (P = .0037), nonrelapse mortality (P = .0343), and survival free of chronic GvHD or relapse (P = .0030).
These results help establish the new treosulfan/fludarabine regimen as a “relatively well-tolerable and effective preparative regimen” in elderly or comorbid AML/MDS patients, Dr. Beelen said.
However, treosulfan has not been authorized for use in allogeneic HCT conditioning regimens, and so should be considered experimental in this setting, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Beelen reported honoraria, travel support, and trial documentation support provided by medac GmbH, which sponsored the trial.
SOURCE: Beelen DW et al. TCT 2019, Abstract 4.
HOUSTON – A treosulfan-based conditioning regimen could become standard prior to allogeneic transplant in elderly or comorbid patients with acute myeloid leukemia or myelodysplastic syndromes, according to the lead investigator in a phase 3 trial.
The treosulfan/fludarabine myeloablative conditioning regimen had noninferior event-free survival, compared with a reduced-intensity busulfan-based regimen in the large, randomized trial that included elderly patients and those with multiple comorbidities, said researcher Dietrich Wilhelm Beelen, MD, PhD.
The experimental regimen was superior to busulfan in overall survival, nonrelapse mortality, and complete donor chimerism in the trial, added Dr. Beelen, who is with the department of bone marrow transplantation at the West German Cancer Center, University Hospital of Essen, Germany.
“The study results point to a potential benefit of the treosulfan/fludarabine regimen, while the early safety profile, engraftment kinetics, acute or chronic graft-versus-host-disease (GvHD), and the relapse risk of both regimens appear comparable,” Dr. Beelen said at the Transplantation & Cellular Therapy Meetings.
Allogeneic hematopoietic cell transplantation (HCT) is challenging in elderly and comorbid patients, who have an increased risk of nonrelapse mortality with standard myeloablative regimens, according to Dr. Beelen, who presented results on behalf of investigators from the international MC-FludT.14/L Study Group.
Their phase 3 randomized trial included patients who were 50-70 years of age, or who had a Hematopoietic Cell Transplantation Comorbidity Index of 2 or greater. The final analysis included 551 patients (352 with AML and 199 with MDS).
The primary endpoint of the study was event-free survival at 2 years. That endpoint comprised relapse/progression of disease, graft failure, or death.
Patient enrollment was terminated early the MC-FludT.14/L study following an interim analysis that investigators said “clearly demonstrated” the noninferiority of the treosulfan/fludarabine regimen versus the reduced intensity busulfan/fludarabine regimen.
In the final analysis, event-free survival at 2 years was about 14.5 percentage points higher in the treosulfan group, at 65.7% versus 51.2% (P = .0000001), Dr. Beelen reported at the meeting.
A number of other secondary endpoints also favored treosulfan/fludarabine over busulfan, including overall survival (P = .0037), nonrelapse mortality (P = .0343), and survival free of chronic GvHD or relapse (P = .0030).
These results help establish the new treosulfan/fludarabine regimen as a “relatively well-tolerable and effective preparative regimen” in elderly or comorbid AML/MDS patients, Dr. Beelen said.
However, treosulfan has not been authorized for use in allogeneic HCT conditioning regimens, and so should be considered experimental in this setting, he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research. At its meeting, the American Society for Blood and Marrow Transplantation announced a new name for the society: American Society for Transplantation and Cellular Therapy (ASTCT).
Dr. Beelen reported honoraria, travel support, and trial documentation support provided by medac GmbH, which sponsored the trial.
SOURCE: Beelen DW et al. TCT 2019, Abstract 4.
REPORTING FROM TCT 2019
FDA clears test to monitor residual disease in CML
The Food and Drug Administration has cleared Bio-Rad’s digital polymerase chain reaction (PCR) testing solution to monitor patients’ molecular response to treatment for chronic myeloid leukemia.

The QXDx AutoDG ddPCR System combines Bio-Rad’s Droplet Digital PCR technology and the QXDx BCR-ABL %IS Kit, according to the company.
This so-called liquid biopsy test can “precisely and reproducibly” monitor the molecular response to tyrosine kinase inhibitor therapy. The current standard – reverse transcription quantitative PCR – can have variable results, especially at low levels of disease, according to Bio-Rad.
FDA clearance means that the product is “substantially equivalent” to an already-approved product and can be sold in the United States, according to the agency.
The Food and Drug Administration has cleared Bio-Rad’s digital polymerase chain reaction (PCR) testing solution to monitor patients’ molecular response to treatment for chronic myeloid leukemia.
The QXDx AutoDG ddPCR System combines Bio-Rad’s Droplet Digital PCR technology and the QXDx BCR-ABL %IS Kit, according to the company.
This so-called liquid biopsy test can “precisely and reproducibly” monitor the molecular response to tyrosine kinase inhibitor therapy. The current standard – reverse transcription quantitative PCR – can have variable results, especially at low levels of disease, according to Bio-Rad.
FDA clearance means that the product is “substantially equivalent” to an already-approved product and can be sold in the United States, according to the agency.
The Food and Drug Administration has cleared Bio-Rad’s digital polymerase chain reaction (PCR) testing solution to monitor patients’ molecular response to treatment for chronic myeloid leukemia.
The QXDx AutoDG ddPCR System combines Bio-Rad’s Droplet Digital PCR technology and the QXDx BCR-ABL %IS Kit, according to the company.
This so-called liquid biopsy test can “precisely and reproducibly” monitor the molecular response to tyrosine kinase inhibitor therapy. The current standard – reverse transcription quantitative PCR – can have variable results, especially at low levels of disease, according to Bio-Rad.
FDA clearance means that the product is “substantially equivalent” to an already-approved product and can be sold in the United States, according to the agency.
Targeted triplet shows potential for B-cell cancers
A triplet combination of targeted agents ublituximab, umbralisib, and ibrutinib may be a safe and effective regimen for patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and other B-cell malignancies, according to early study results.
The phase 1 trial had an overall response rate of 84% and a favorable safety profile, reported lead author Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center, Houston, and her colleagues. The results suggest that the regimen could eventually serve as a nonchemotherapeutic option for patients with B-cell malignancies.
“Therapeutic targeting of the B-cell receptor signaling pathway has revolutionized the management of B-cell lymphomas,” the investigators wrote in the Lancet Haematology. “Optimum combinations that result in longer periods of remission, possibly allowing for discontinuation of therapy, are needed.”
The present triplet combination included ublituximab, an anti-CD20 monoclonal antibody; ibrutinib, a Bruton tyrosine kinase inhibitor; and umbralisib, a phosphoinositide 3-kinase delta inhibitor.
A total of 46 patients with CLL/SLL or relapsed/refractory B-cell non-Hodgkin lymphoma received at least one dose of the combination in dose-escalation or dose-expansion study sections.
In the dose-escalation group (n = 24), ublituximab was given intravenously at 900 mg, ibrutinib was given orally at 420 mg for CLL and 560 mg for B-cell non-Hodgkin lymphoma, and umbralisib was given orally at three dose levels: 400 mg, 600 mg, and 800 mg.
In the dose-expansion group (n = 22), umbralisib was set at 800 mg while the other agents remained at the previous doses; treatment continued until intolerance or disease progression occurred. The investigators monitored efficacy and safety at defined intervals.
Results showed that 37 out of 44 evaluable patients (84%) had partial or complete responses to therapy.
Among the 22 CLL/SLL patients, there was a 100% overall response rate for both previously treated and untreated patients. Similarly, all three of the patients with marginal zone lymphoma responded, all six of the patients with mantle cell lymphoma responded, and five of seven patients with follicular lymphoma responded. However, only one of the six patients with diffuse large B-cell lymphoma had even a partial response.
The most common adverse events of any kind were diarrhea (59%), fatigue (50%), infusion-related reaction (43%), dizziness (37%), nausea (37%), and cough (35%). The most common grade 3 or higher adverse events were neutropenia (22%) and cellulitis (13%).
Serious adverse events were reported in 24% of patients; pneumonia, rash, sepsis, atrial fibrillation, and syncope occurred in two patients each; abdominal pain, pneumonitis, cellulitis, headache, skin infection, pleural effusion, upper gastrointestinal bleeding, pericardial effusion, weakness, and diarrhea occurred in one patient each. No adverse event–related deaths were reported.
“The findings of this study establish the tolerable safety profile of the ublituximab, umbralisib, and ibrutinib triplet regimen in chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed or refractory B-cell non-Hodgkin lymphoma,” the investigators wrote. “This triplet combination is expected to be investigated further in future clinical trials in different patient populations.”
The study was funded by TG Therapeutics. The authors reported financial relationships with TG Therapeutics and other companies.
SOURCE: Nastoupil LJ et al. Lancet Haematol. 2019 Feb;6(2):e100-9.
A triplet combination of targeted agents ublituximab, umbralisib, and ibrutinib may be a safe and effective regimen for patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and other B-cell malignancies, according to early study results.
The phase 1 trial had an overall response rate of 84% and a favorable safety profile, reported lead author Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center, Houston, and her colleagues. The results suggest that the regimen could eventually serve as a nonchemotherapeutic option for patients with B-cell malignancies.
“Therapeutic targeting of the B-cell receptor signaling pathway has revolutionized the management of B-cell lymphomas,” the investigators wrote in the Lancet Haematology. “Optimum combinations that result in longer periods of remission, possibly allowing for discontinuation of therapy, are needed.”
The present triplet combination included ublituximab, an anti-CD20 monoclonal antibody; ibrutinib, a Bruton tyrosine kinase inhibitor; and umbralisib, a phosphoinositide 3-kinase delta inhibitor.
A total of 46 patients with CLL/SLL or relapsed/refractory B-cell non-Hodgkin lymphoma received at least one dose of the combination in dose-escalation or dose-expansion study sections.
In the dose-escalation group (n = 24), ublituximab was given intravenously at 900 mg, ibrutinib was given orally at 420 mg for CLL and 560 mg for B-cell non-Hodgkin lymphoma, and umbralisib was given orally at three dose levels: 400 mg, 600 mg, and 800 mg.
In the dose-expansion group (n = 22), umbralisib was set at 800 mg while the other agents remained at the previous doses; treatment continued until intolerance or disease progression occurred. The investigators monitored efficacy and safety at defined intervals.
Results showed that 37 out of 44 evaluable patients (84%) had partial or complete responses to therapy.
Among the 22 CLL/SLL patients, there was a 100% overall response rate for both previously treated and untreated patients. Similarly, all three of the patients with marginal zone lymphoma responded, all six of the patients with mantle cell lymphoma responded, and five of seven patients with follicular lymphoma responded. However, only one of the six patients with diffuse large B-cell lymphoma had even a partial response.
The most common adverse events of any kind were diarrhea (59%), fatigue (50%), infusion-related reaction (43%), dizziness (37%), nausea (37%), and cough (35%). The most common grade 3 or higher adverse events were neutropenia (22%) and cellulitis (13%).
Serious adverse events were reported in 24% of patients; pneumonia, rash, sepsis, atrial fibrillation, and syncope occurred in two patients each; abdominal pain, pneumonitis, cellulitis, headache, skin infection, pleural effusion, upper gastrointestinal bleeding, pericardial effusion, weakness, and diarrhea occurred in one patient each. No adverse event–related deaths were reported.
“The findings of this study establish the tolerable safety profile of the ublituximab, umbralisib, and ibrutinib triplet regimen in chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed or refractory B-cell non-Hodgkin lymphoma,” the investigators wrote. “This triplet combination is expected to be investigated further in future clinical trials in different patient populations.”
The study was funded by TG Therapeutics. The authors reported financial relationships with TG Therapeutics and other companies.
SOURCE: Nastoupil LJ et al. Lancet Haematol. 2019 Feb;6(2):e100-9.
A triplet combination of targeted agents ublituximab, umbralisib, and ibrutinib may be a safe and effective regimen for patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and other B-cell malignancies, according to early study results.
The phase 1 trial had an overall response rate of 84% and a favorable safety profile, reported lead author Loretta J. Nastoupil, MD, of the University of Texas MD Anderson Cancer Center, Houston, and her colleagues. The results suggest that the regimen could eventually serve as a nonchemotherapeutic option for patients with B-cell malignancies.
“Therapeutic targeting of the B-cell receptor signaling pathway has revolutionized the management of B-cell lymphomas,” the investigators wrote in the Lancet Haematology. “Optimum combinations that result in longer periods of remission, possibly allowing for discontinuation of therapy, are needed.”
The present triplet combination included ublituximab, an anti-CD20 monoclonal antibody; ibrutinib, a Bruton tyrosine kinase inhibitor; and umbralisib, a phosphoinositide 3-kinase delta inhibitor.
A total of 46 patients with CLL/SLL or relapsed/refractory B-cell non-Hodgkin lymphoma received at least one dose of the combination in dose-escalation or dose-expansion study sections.
In the dose-escalation group (n = 24), ublituximab was given intravenously at 900 mg, ibrutinib was given orally at 420 mg for CLL and 560 mg for B-cell non-Hodgkin lymphoma, and umbralisib was given orally at three dose levels: 400 mg, 600 mg, and 800 mg.
In the dose-expansion group (n = 22), umbralisib was set at 800 mg while the other agents remained at the previous doses; treatment continued until intolerance or disease progression occurred. The investigators monitored efficacy and safety at defined intervals.
Results showed that 37 out of 44 evaluable patients (84%) had partial or complete responses to therapy.
Among the 22 CLL/SLL patients, there was a 100% overall response rate for both previously treated and untreated patients. Similarly, all three of the patients with marginal zone lymphoma responded, all six of the patients with mantle cell lymphoma responded, and five of seven patients with follicular lymphoma responded. However, only one of the six patients with diffuse large B-cell lymphoma had even a partial response.
The most common adverse events of any kind were diarrhea (59%), fatigue (50%), infusion-related reaction (43%), dizziness (37%), nausea (37%), and cough (35%). The most common grade 3 or higher adverse events were neutropenia (22%) and cellulitis (13%).
Serious adverse events were reported in 24% of patients; pneumonia, rash, sepsis, atrial fibrillation, and syncope occurred in two patients each; abdominal pain, pneumonitis, cellulitis, headache, skin infection, pleural effusion, upper gastrointestinal bleeding, pericardial effusion, weakness, and diarrhea occurred in one patient each. No adverse event–related deaths were reported.
“The findings of this study establish the tolerable safety profile of the ublituximab, umbralisib, and ibrutinib triplet regimen in chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed or refractory B-cell non-Hodgkin lymphoma,” the investigators wrote. “This triplet combination is expected to be investigated further in future clinical trials in different patient populations.”
The study was funded by TG Therapeutics. The authors reported financial relationships with TG Therapeutics and other companies.
SOURCE: Nastoupil LJ et al. Lancet Haematol. 2019 Feb;6(2):e100-9.
FROM LANCET HAEMATOLOGY
Key clinical point:
Major finding: Out of 44 patients, 37 (84%) achieved a partial or complete response to therapy.
Study details: A phase 1, multicenter, dose-escalation and dose-expansion trial involving 46 patients with chronic lymphocytic leukemia, small lymphocytic leukemia, or relapsed/refractory non-Hodgkin lymphoma.
Disclosures: The study was funded by TG Therapeutics. The authors reported financial relationships with TG Therapeutics and other companies.
Source: Nastoupil LJ et al. Lancet Haematol. 2019 Feb;6(2):e100-9.
Chronic Myeloid Leukemia: A Review of TKI Therapy
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that arises from a reciprocal translocation between the Abelson (ABL) region on chromosome 9 and the breakpoint cluster region (BCR) of chromosome 22, t(9;22)(q34;q11.2) (the Philadelphia chromosome), resulting in the generation of the BCR-ABL1 fusion gene and its protein product, BCR-ABL tyrosine kinase. BCR-ABL is a constitutively active fusion kinase that confers proliferative and survival advantage to hematopoietic cells through activation of downstream pathways.
CML is divided into 3 phases based on the number of myeloblasts observed in the blood or bone marrow: chronic, accelerated, and blast. Most cases of CML are diagnosed in the chronic phase (CP), which is marked by proliferation of primarily the myeloid element.
The advent of tyrosine kinase inhibitors (TKIs), a class of small molecules targeting the tyrosine kinases, particularly the BCR-ABL tyrosine kinase, led to rapid changes in the management of CML and improved survival for patients. Patients diagnosed with CP-CML now a have life-expectancy that is similar to that of the general population, as long as they receive the appropriate TKI therapy and adhere to treatment. As such, it is crucial to identify patients with CML, ensure they receive a complete, appropriate diagnostic work-up, and select the best therapy for each individual patient. The diagnosis and work-up of CML are reviewed in a separate article; here, the selection of TKI therapy for a patient with newly diagnosed CP-CML is reviewed.
Case Presentation
A 53-year-old woman who recently was diagnosed with CML presents to review her treatment options. The diagnosis was made after she presented to her primary care physician with fatigue, early satiety, left upper quadrant abdominal pain, and an 8-lb unintentional weight loss over the prior month. On physical exam her spleen was palpated 8 cm below the left costal margin. Laboratory evaluation showed a total white blood cell (WBC) count of 124,000/μL with a left-shifted differential including 6% basophils, 3% eosinophils, and 3% blasts; hemoglobin and platelet count were 12.4 g/dL and 801 × 103/µL, respectively. Fluorescent in-situ hybridization for BCR-ABL gene rearrangement using peripheral blood was positive in 87% of cells. Bone marrow biopsy and aspiration showed a 95% cellular bone marrow with granulocytic hyperplasia and 1% blasts. Cytogenetics were 46,XX,t(9;22)(q34;q11.2), and quantitative real-time polymerase chain reaction (RQ-PCR) to measure BCR-ABL1 transcripts in the peripheral blood showed a value of 98% international standard (IS). Her Sokal risk score was 1.42 (high risk). In addition, prior review of her past medical history revealed uncontrolled diabetes, coronary artery disease requiring placement of 3 cardiac stents 2 years prior, and chronic obstructive pulmonary disease (COPD) related to a 30-pack-year history of smoking.
- What factors must be considered when selecting first-line therapy for this patient?
Selection of the most appropriate first-line TKI for newly diagnosed CP-CML patients requires incorporation of many patient-specific factors. These factors include baseline karyotype and confirmation of CP-CML through bone marrow biopsy, Sokal or EURO risk score, and a thorough patient history, including a clear understanding of the patient's comorbidities. In this case, the patient's high Sokal risk score along with her history of diabetes, coronary artery disease, and COPD are all factors that must be accounted for when choosing the most appropriate TKI. The adverse effect profile of all TKIs must be considered in conjunction with the patient's ongoing medical issues in order to decrease the likelihood of worsening her current symptoms or causing a severe complication from TKI therapy.
Imatinib
The management of CML was revolutionized by the development and ultimate regulatory approval of imatinib mesylate in 2001. Imatinib was the first small-molecule cancer therapy developed and approved. It acts by binding to the adenosine triphosphate (ATP) binding site in the catalytic domain of BCR-ABL, thus inhibiting the oncoprotein's tyrosine kinase activity.1
The International Randomized Study of Interferon versus STI571 (IRIS) trial was a randomized phase 3 study that compared imatinib 400 mg daily to interferon α (IFNα) plus cytarabine. More than 1000 CP-CML patients were randomly assigned 1:1 to either imatinib or IFNα plus cytarabine and were assessed for event-free survival, hematologic and cytogenetic responses, freedom from progression to accelerated phase (AP) or blast phase (BP), and toxicity. Imatinib was superior to the prior standard of care for all these outcomes.2 The long-term follow up of the IRIS trial reported an 83% estimated 10-year overall survival (OS) and 79% estimated event-free survival for patients on the imatinib arm of this study.3 The cumulative rate of complete cytogenetic response (CCyR) was 82.8%. Of the 204 imatinib-treated patients who could undergo a molecular response evaluation at 10 years, 93.1% had a major molecular response (MMR) and 63.2% had a molecular response 4.5 (MR4.5), suggesting durable, deep molecular responses for many patients (see Chronic Myeloid Leukemia: Evaluation and Diagnosis for discussion of the hematologic parameters, cytogenetic results, and molecular responses ussed in monitoring response to TKI therapy). The estimated 10-year rate of freedom from progression to AP or BP was 92.1%.
Higher doses of imatinib (600-800 mg daily) have been studied in an attempt to overcome resistance and improve cytogenetic and molecular response rates. The Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) trial was a randomized phase 3 study that compared imatinib 800 mg daily to imatinib 400 mg daily. Although the 6-month assessments found increased rates of CCyR and a MMR in the higher-dose imatinib arm, these differences were no longer present at the 12-month assessment. Furthermore, the higher dose of imatinib led to a significantly higher incidence of grade 3/4 hematologic adverse events, and approximately 50% of patients on imatinib 800 mg daily required a dose reduction to less than 600 mg daily because of toxicity.4
The Therapeutic Intensification in De Novo Leukaemia (TIDEL) -II study used plasma trough levels of imatinib on day 22 of treatment with imatinib 600 mg daily to determine if patients should escalate the imatinib dose to 800 mg daily. In patients who did not meet molecular milestones at 3, 6, or 12 months, cohort 1 was dose escalated to imatinib 800 mg daily and subsequently switched to nilotinib 400 mg twice daily for failing the same target 3 months later, and cohort 2 was switched to nilotinib. At 2 years, 73% of patients achieved MMR and 34% achieved MR4.5, suggesting that initial treatment with higher-dose imatinib subsequently followed by a switch to nilotinib in those failing to achieve desired milestones could be an effective strategy for managing newly diagnosed CP-CML.5
Toxicity
Imatinib 400 mg is considered the standard starting dose in CP-CML patients. The safety profile of imatinib has been very well established. In the IRIS trial, the most common adverse events (all grades in decreasing order of frequency) were peripheral and periorbital edema (60%), nausea (50%), muscle cramps (49%), musculoskeletal pain (47%), diarrhea (45%), rash (40%), fatigue (39%), abdominal pain (37%), headache (37%), and joint pain (31%). Grade 3/4 liver enzyme elevation can occur in 5% of patients.6 In the event of severe liver toxicity or fluid retention, imatinib should be held until the event resolves. At that time, imatinib can be restarted if deemed appropriate, but this is dependent on the severity of the inciting event. Fluid retention can be managed by the use of supportive care, diuretics, imatinib dose reduction, dose interruption, or imatinib discontinuation if the fluid retention is severe. Muscle cramps can be managed by the use of a calcium supplements or tonic water. Management of rash can include topical or systemic steroids, or in some cases imatinib dose reduction, interruption, or discontinuation.7
Grade 3/4 imatinib-induced hematologic toxicity is not uncommon, with 17% of patients experiencing neutropenia, 9% thrombocytopenia, and 4% anemia. These adverse events occurred most commonly during the first year of therapy, and the frequency decreased over time.3,6 Depending on the degree of cytopenias, imatinib dosing should be interrupted until recovery of the absolute neutrophil count or platelet count, and can often be resumed at 400 mg daily. However, if cytopenias recur, imatinib should be held and subsequently restarted at 300 mg daily.7
Dasatinib
Dasatinib is a second-generation TKI that has regulatory approval for treatment of adult patients with newly diagnosed CP-CML or CP-CML in patients with resistance or intolerance to prior TKIs. In addition to dasatinib's ability to inhibit ABL kinases, it is also known to be a potent inhibitor of Src family kinases. Dasatinib has shown efficacy in patients who have developed imatinib-resistant ABL kinase domain mutations.
Dasatinib was initially approved as second-line therapy in patients with resistance or intolerance to imatinib. This indication was based on the results of the phase 3 CA180-034 trial which ultimately identified dasatinib 100 mg daily as the optimal dose. In this trial, 74% of patients enrolled had resistance to imatinib and the remainder were intolerant. The 7-year follow-up of patients randomized to dasatinib 100 mg (n = 167) daily indicated that 46% achieved MMR while on study. Of the 124 imatinib-resistant patients on dasatinib 100 mg daily, the 7-year progression-free survival (PFS) was 39% and OS was 63%. In the 43 imatinib-intolerant patients, the 7-year PFS was 51% and OS was 70%.8
Dasatinib 100 mg daily was compared to imatinib 400 mg daily in newly diagnosed CP-CML patients in the randomized phase 3 DASISION trial. More patients on the dasatinib arm achieved an early molecular response of BCR-ABL1 transcripts ≤10% IS after 3 months on treatment compared to imatinib (84% versus 64%). Furthermore, the 5-year follow-up reports that the cumulative incidence of MMR and MR4.5 in dasatinib-treated patients was 76% and 42%, and was 64% and 33%, with imatinib (P = 0.0022 and P = 0.0251, respectively). Fewer patients treated with dasatinib progressed to AP or BP (4.6%) compared to imatinib (7.3%), but the estimated 5-year OS was similar between the 2 arms (91% for dasatinib versus 90% for imatinib).9 Regulatory approval for dasatinib as first-line therapy in newly diagnosed CML patients was based on results of the DASISION trial.
Toxicity
Most dasatinib-related toxicities are reported as grade 1 or grade 2, but grade 3/4 hematologic adverse events are fairly common. In the DASISION trial, grade 3/4 neutropenia, anemia, and thrombocytopenia occurred in 29%, 13%, and 22% of dasatinib-treated patients, respectively. Cytopenias can generally be managed with temporary dose interruptions or dose reductions.
During the 5-year follow-up of the DASISION trial, pleural effusions were reported in 28% of patients, most of which were grade 1/2. This occurred at a rate of approximately ≤ 8% per year, suggesting a stable incidence over time, and the effusions appear to be dose-dependent.9 Depending on the severity of the effusion, this may be treated with diuretics, dose interruption, and in some instances, steroids or a thoracentesis. Typically, dasatinib can be restarted at 1 dose level lower than the previous dose once the effusion has resolved.7 Other, less common side effects of dasatinib include pulmonary hypertension (5% of patients), as well as abdominal pain, fluid retention, headaches, fatigue, musculoskeletal pain, rash, nausea, and diarrhea. Pulmonary hypertension is typically reversible after cessation of dasatinib, and thus dasatinib should be permanently discontinued once the diagnosis is confirmed. Fluid retention is often treated with diuretics and supportive care. Nausea and diarrhea are generally manageable and occur less frequently when dasatinib is taken with food and a large glass of water. Antiemetics and antidiarrheals can be used as needed. Troublesome rash can be best managed with topical or systemic steroids as well as possible dose reduction or dose interruption.7,9 In the DASISION trial, adverse events led to therapy discontinuation more often in the dasatinib group than in the imatinib group (16% versus 7%).9 Bleeding, particularly in the setting of thrombocytopenia, has been reported in patients being treated with dasatinib as a result of the drug-induced reversible inhibition of platelet aggregation.10
Nilotinib
The structure of nilotinib is similar to that of imatinib; however, it has a markedly increased affinity for the ATP‐binding site on the BCR-ABL1 protein. It was initially given regulatory approval in the setting of imatinib failure. Nilotinib was studied at a dose of 400 mg twice daily in 321 patients who were imatinib-resistant or -intolerant. It proved to be highly effective at inducing cytogenetic remissions in the second-line setting, with 59% of patients achieving a major cytogenetic response (MCyR) and 45% achieving CCyR. With a median follow-up time of 4 years, the OS was 78%.11
Nilotinib gained regulatory approval for use as a first-line TKI after completion of the randomized phase 3 ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients) trial. ENESTnd was a 3-arm study comparing nilotinib 300 mg twice daily versus nilotinib 400 mg twice daily versus imatinib 400 mg daily in newly diagnosed, previously untreated patients diagnosed with CP-CML. The primary endpoint of this clinical trial was rate of MMR at 12 months.12 Nilotinib surpassed imatinib in this regard, with 44% of patients on nilotinib 300 mg twice daily achieving MMR at 12 months versus 43% of nilotinib 400 mg twice daily patients versus 22% of the imatinib-treated patients (P < 0.001 for both comparisons). Furthermore, the rate of CCyR by 12 months was significantly higher for both nilotinib arms compared with imatinib (80% for nilotinib 300 mg, 78% for nilotinib 400 mg, and 65% for imatinib) (P < 0.001).12 Based on this data, nilotinib 300 mg twice daily was chosen as the standard dose of nilotinib in the first-line setting. After 5 years of follow-up on the ENESTnd study, there were fewer progressions to AP/BP CML in nilotinib-treated patients compared with imatinib. MMR was achieved in 77% of nilotinib 300 mg patients compared with 60.4% of patients on the imatinib arm. MR4.5 was also more common in patients treated with nilotinib 300 mg twice daily, with a rate of 53.5% at 5 years versus 31.4% in the imatinib arm.13 In spite of the deeper cytogenetic and molecular responses achieved with nilotinib, this did not translate into a significant improvement in OS. The 5-year OS rate was 93.7% in nilotinib 300 mg patients versus 91.7% in imatinib-treated patients, and this difference lacked statistical significance.13
Toxicity
Although some similarities exist between the toxicity profiles of nilotinib and imatinib, each drug has some distinct adverse events. On the ENESTnd trial, the rate of any grade 3/4 non-hematologic adverse event was fairly low; however, lower-grade toxicities were not uncommon. Patients treated with nilotinib 300 mg twice daily experienced rash (31%), headache (14%), pruritis (15%), and fatigue (11%) most commonly. The most frequently reported laboratory abnormalities included increased total bilirubin (53%), hypophosphatemia (32%), hyperglycemia (36%), elevated lipase (24%), increased alanine aminotransferase (ALT; 66%), and increased aspartate aminotransferase (AST; 40%). Any grade of neutropenia, thrombocytopenia, or anemia occurred at rates of 43%, 48%, and 38%, respectively.12 Although nilotinib has a Black Box Warning from the US Food and Drug Administration for QT interval prolongation, no patients on the ENESTnd trial experienced a QT interval corrected for heart rate greater than 500 msec.12
More recent concerns have emerged regarding the potential for cardiovascular toxicity after long-term use of nilotinib. The 5-year update of ENESTnd reports cardiovascular events, including ischemic heart disease, ischemic cerebrovascular events, or peripheral arterial disease occurring in 7.5% of patients treated with nilotinib 300 mg twice daily compared with a rate of 2.1% in imatinib-treated patients. The frequency of these cardiovascular events increased linearly over time in both arms. Elevations in total cholesterol from baseline occurred in 27.6% of nilotinib patients compared with 3.9% of imatinib patients. Furthermore, clinically meaningful increases in low-density lipoprotein cholesterol and glycated hemoglobin occurred more frequently with nilotinib therapy.12
Nilotinib should be taken on an empty stomach; therefore, patients should be made aware of the need to fast for 2 hours prior to each dose and 1 hour after each dose. Given the potential risk of QT interval prolongation, a baseline electrocardiogram (ECG) is recommended prior to initiating treatment to ensure the QT interval is within a normal range. A repeat ECG should be done approximately 7 days after nilotinib initiation to ensure no prolongation of the QT interval after starting. Close monitoring of potassium and magnesium levels is important to decrease the risk of cardiac arrhythmias, and concomitant use of drugs considered strong CYP3A4 inhibitors should be avoided.7
If the patient experiences any grade 3 or higher laboratory abnormalities, nilotinib should be held until resolution of the toxicity, and then restarted at a lower dose. Similarly, if patients develop significant neutropenia or thrombocytopenia, nilotinib doses should be interrupted until resolution of the cytopenias. At that point, nilotinib can be reinitiated at either the same or a lower dose. Rash can be managed by the use of topical or systemic steroids as well as potential dose reduction, interruption, or discontinuation.
Given the concerns for potential cardiovascular events with long-term use of nilotinib, caution is advised when prescribing it to any patient with a history of cardiovascular disease or peripheral arterial occlusive disease. At the first sign of new occlusive disease, nilotinib should be discontinued.7
Bosutinib
Bosutinib is a second-generation BCR-ABL1 TKI with activity against the Src family of kinases that was initially approved to treat patients with CP-, AP-, or BP-CML after resistance or intolerance to imatinib. Long-term data has been reported from the phase 1/2 trial of bosutinib therapy in patients with CP-CML who developed resistance or intolerance to imatinib plus dasatinib and/or nilotinib. A total of 119 patients were included in the 4-year follow-up; 38 were resistant/intolerant to imatinib and resistant to dasatinib, 50 were resistant/intolerant to imatinib and intolerant to dasatinib, 26 were resistant/intolerant to imatinib and resistant to nilotinib, and 5 were resistant/intolerant to imatinib and intolerant to nilotinib or resistant/intolerant to dasatinib and nilotinib. Bosutinib 400 mg daily was studied in this setting. Of the 38 patients with imatinib resistance/intolerance and dasatinib resistance, 39% achieved MCyR, 22% achieved CCyR, and the OS was 67%. Of the 50 patients with imatinib resistance/intolerance and dasatinib intolerance, 42% achieved MCyR, 40% achieved CCyR, and the OS was 80%. Finally, in the 26 patients with imatinib resistance/intolerance and nilotinib resistance, 38% achieved MCyR, 31% achieved CcyR, and the OS was 87%.14
Five-year follow-up from the phase 1/2 clinical trial which studied bosutinib 500 mg daily in CP-CML patients after imatinib failure reported data on 284 patients. By 5 years on study, 60% of patients had achieved MCyR and 50% achieved CCyR with a 71% and 69% probability, respectively, of maintaining these responses at 5 years. The 5-year OS was 84%.15 These data led to the regulatory approval of bosutinib 500 mg daily as second-line or later therapy.
Bosutinib was initially studied in the first-line setting in the randomized phase 3 BELA (Bosutinib Efficacy and Safety in Newly Diagnosed Chronic Myeloid Leukemia) trial. This trial compared bosutinib 500 mg daily to imatinib 400 mg daily in newly diagnosed, previously untreated CP-CML patients. This trial failed to meet its primary endpoint of increased rate of CCyR at 12 months, with 70% of bosutinib patients achieving this response compared to 68% of imatinib-treated patients (P = 0.601). In spite of this, the rate of MMR at 12 months was significantly higher in the bosutinib arm (41%) compared to the imatinib arm (27%; P = 0.001).16
A second phase 3 trial (BFORE) was designed to study bosutinib 400 mg daily versus imatinib in newly diagnosed, previously untreated CP-CML patients. This study enrolled 536 patients who were randomly assigned 1:1 to bosutinib versus imatinib. The primary endpoint of this trial was rate of MMR at 12 months. A significantly higher number of bosutinib-treated patients achieved this response (47.2%) compared with imatinib-treated patients (36.9%, P = 0.02). Furthermore, by 12 months 77.2% of patients on the bosutinib arm had achieved CCyR compared with 66.4% on the imatinib arm, and this difference did meet statistical significance (P = 0.0075). A lower rate of progression to AP- or BP-CML was noted in bosutinib-treated patients as well (1.6% versus 2.5%). Based on this data, bosutinib gained regulatory approval for first-line therapy in CP-CML at a dose of 400 mg daily.17
Toxicity
On the BFORE trial, the most common treatment-emergent adverse events of any grade reported in the bosutinib-treated patients were diarrhea (70.1%), nausea (35.1%), increased ALT (30.6%), and increased AST (22.8%). Musculoskeletal pain or spasms occurred in 29.5% of patients, rash in 19.8%, fatigue in 19.4%, and headache in 18.7%. Hematologic toxicity was also reported, but most was grade 1/2. Thrombocytopenia was reported in 35.1%, anemia in 18.7%, and neutropenia in 11.2%.17
Cardiovascular events occurred in 5.2% of patients on the bosutinib arm of the BFORE trial, which was similar to the rate observed in imatinib patients. The most common cardiovascular event was QT interval prolongation, which occurred in 1.5% of patients. Pleural effusions were reported in 1.9% of patients treated with bosutinib, and none were grade 3 or higher.17
If liver enzyme elevation occurs at a value greater than 5 times the institutional upper limit of normal, bosutinib should be held until the level recovers to ≤2.5 times the upper limit of normal, at which point bosutinib can be restarted at a lower dose. If recovery takes longer than 4 weeks, bosutinib should be permanently discontinued. Liver enzymes elevated greater than 3 times the institutional upper limit of normal and a concurrent elevation in total bilirubin to 2 times the upper limit of normal is consistent with Hy's law, and bosutinib should be discontinued. Although diarrhea is the most common toxicity associated with bosutinib, it is commonly low grade and transient. Diarrhea occurs most frequently in the first few days after initiating bosutinib. It can often be managed with over-the-counter antidiarrheal medications, but if the diarrhea is grade or higher, bosutinib should be held until recovery to grade 1 or lower. Gastrointestinal side effects may be improved by taking bosutinib with a meal and a large glass of water. Fluid retention can be managed with diuretics and supportive care. Finally, if rash occurs, this can be addressed with topical or systemic steroids as well as bosutinib dose reduction, interruption, or discontinuation.7
Similar to other TKIs, if bosutinib-induced cytopenias occur, treatment should be held and restarted at the same or a lower dose upon blood count recovery.7
Ponatinib
The most common cause of TKI resistance in CP-CML is the development of ABL kinase domain mutations. The majority of imatinib-resistant mutations can be overcome by the use of second-generation TKIs including dasatinib, nilotinib, or bosutinib. However, ponatinib is the only BCR-ABL1 TKI able to overcome a T315I mutation. The phase 2 PACE (Ponatinib Ph-positive ALL and CML Evaluation) trial enrolled patients with CP-, AP-, or BP-CML as well as patients with Ph-positive acute lymphoblastic leukemia who were resistant or intolerant to nilotinib or dasatinib, or who had evidence of a T315I mutation. The starting dose of ponatinib on this trial was 45 mg daily.18 The PACE trial enrolled 267 patients with CP-CML: 203 with resistance or intolerance to nilotinib or dasatinib, and 64 with a T315I mutation. The primary endpoint in the CP cohort was rate of MCyR at any time within 12 months of starting ponatinib. The overall rate of MCyR by 12 months in the CP-CML patients was 56%. In those with a T315I mutation, 70% achieved MCyR, which compared favorably with those with resistance or intolerance to nilotinib or dasatinib, 51% of whom achieved MCyR. CCyR was achieved in 46% of CP-CML patients (40% in the resistant/intolerant cohort and 66% in the T315I cohort). In general, patients with T315I mutations received fewer prior therapies than those in the resistant/intolerant cohort, which likely contributed to the higher response rates in the T315I patients. MR4.5 was achieved in 15% of CP-CML patients by 12 months on the PACE trial.18 The 5-year update to this study reported that 60%, 40%, and 24% of CP-CML patients achieved MCyR, MMR, and MR4.5, respectively. In the patients who achieved MCyR, the probability of maintaining this response for 5 years was 82% and the estimated 5-year OS was 73%.19
Toxicity
In 2013, after the regulatory approval of ponatinib, reports became available that the drug can cause an increase in arterial occlusive events including fatal myocardial infarctions and cerebral vascular accidents. For this reason, dose reductions were implemented in patients who were deriving clinical benefit from ponatinib. In spite of these dose reductions, ≥90% of responders maintained their response for up to 40 months.19 Although the likelihood of developing an arterial occlusive event appears higher in the first year after starting ponatinib than in later years, the cumulative incidence of events continues to increase. The 5-year follow-up to the PACE trial reports 31% of patients experiencing any grade of arterial occlusive event while on ponatinib. Aside from these events, the most common treatment-emergent adverse events in ponatinib-treated patients on the PACE trial included rash (47%), abdominal pain (46%), headache (43%), dry skin (42%), constipation (41%), and hypertension (37%). Hematologic toxicity was also common, with 46% of patients experiencing any grade of thrombocytopenia, 20% experiencing neutropenia, and 20% anemia.19
Patients receiving ponatinib therapy should be monitored closely for any evidence of arterial or venous thrombosis. In the event of an occlusive event, ponatinib should be discontinued. Similarly, in the setting of any new or worsening heart failure symptoms, ponatinib should be promptly discontinued. Management of any underlying cardiovascular risk factors including hypertension, hyperlipidemia, diabetes, or smoking history is recommended, and these patients should be referred to a cardiologist for a full evaluation. In the absence of any contraindications to aspirin, low-dose aspirin should be considered as a means of decreasing cardiovascular risks associated with ponatinib. In patients with known risk factors, a ponatinib starting dose of 30 mg daily rather than the standard 45 mg daily may be a safer option resulting in fewer arterial occlusive events, although the efficacy of this dose is still being studied in comparison to 45 mg daily.7
In the event of ponatinib-induced transaminitis greater than 3 times the upper limit of normal, ponatinib should be held until resolution to less than 3 times the upper limit of normal, at which point it should be resumed at a lower dose. Similarly, in the setting of elevated serum lipase or symptomatic pancreatitis, ponatinib should be held and restarted at a lower dose after resolution of symptoms.7
In the event of neutropenia or thrombocytopenia, ponatinib should be held until blood count recovery and then restarted at the same dose. If cytopenias occur for a second time, the dose of ponatinib should be lowered at the time of treatment reinitiation. If rash occurs, it can be addressed with topical or systemic steroids as well as dose reduction, interruption, or discontinuation.7
Case Conclusion
Given the patient's high-risk Sokal score, ideal first-line treatment is a second-generation TKI in order to increase the likelihood of achieving the desired treatment milestones and improving long-term outcomes. Her history of uncontrolled diabetes and coronary artery disease raises concerns for using nilotinib. Furthermore, her history of COPD makes dasatinib suboptimal because she would have little pulmonary reserve if she were to develop a pleural effusion. For this reason, bosutinib 400 mg daily is chosen as her first-line TKI. Shortly after starting bosutinib, she experiences diarrhea that occurs approximately 3 or 4 times daily during the first week on treatment. She is able to manage this with over-the-counter loperamide and the diarrhea resolves shortly thereafter.
After 3 months of bosutinib therapy, quantitative real-time PCR (RQ-PCR) assay on peripheral blood is done to measure BCR-ABL1 transcripts, and the result is reported at 1.2% IS. This indicates that the patient has achieved an early molecular response, which is defined as a RQ-PCR value of ≤10% IS. She undergoes RQ-PCR monitoring every 3 months, and at 12 months her results indicate a value of 0.07% IS, suggesting she has achieved a MMR.
Conclusion
With the development of imatinib and the subsequent TKIs, dasatinib, nilotinib, bosutinib, and ponatinib, CP-CML has become a chronic disease with a life-expectancy that is similar to the general population. Given the successful treatments available for these patients, it is crucial to identify patients with this diagnosis, ensure they receive a complete, appropriate diagnostic workup including a bone marrow biopsy and aspiration with cytogenetic testing, and select the best therapy for each individual patient. Once on treatment, the importance of frequent monitoring cannot be overstated. This is the only way to be certain patients are achieving the desired treatment milestones that correlate with the favorable long-term outcomes that have been observed with TKI-based treatment of CP-CML.
1. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
2. O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
3. Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917-927.
4. Baccarani M, Druker BJ, Branford S, et al. Long-term response to imatinib is not affected by the initial dose in patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: final update from the Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) study. Int J Hematol. 2014;99:616-624.
5. Yeung DT, Osborn MP, White DL, et al. TIDEL-II: first-line use of imatinib in CML with early switch to nilotinib for failure to achieve time-dependent molecular targets. Blood. 2015;125:915-923.
6. Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
7. Radich JP, Deininger M, Abboud CN, et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:1108-1135.
8. Shah NP, Rousselot P, Schiffer C, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. 2016;91:869-874.
9. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients trial. J Clin Oncol. 2016;34:2333-3340.
10. Quintas-Cardama A, Han X, Kantarjian H, Cortes J. Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood. 2009;114:261-263.
11. Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27:107-112.
12. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251-2259.
13. Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044-1054.
14. Cortes JE, Khoury HJ, Kantarjian HM, et al. Long-term bosutinib for chronic phase chronic myeloid leukemia after failure of imatinib plus dasatinib and/or nilotinib. Am J Hematol. 2016;91:1206-1214.
15. Gambacorti-Passerini C, Cortes JE, Lipton JH, et al. Safety and efficacy of second-line bosutinib for chronic phase chronic myeloid leukemia over a five-year period: final results of a phase I/II study. Haematologica. 2018;103:1298-1307.
16. Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30:3486-3492.
17. Cortes JE, Gambacorti-Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol. 2018;36:231-237.
18. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783-1796.
19. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132:393-404.
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that arises from a reciprocal translocation between the Abelson (ABL) region on chromosome 9 and the breakpoint cluster region (BCR) of chromosome 22, t(9;22)(q34;q11.2) (the Philadelphia chromosome), resulting in the generation of the BCR-ABL1 fusion gene and its protein product, BCR-ABL tyrosine kinase. BCR-ABL is a constitutively active fusion kinase that confers proliferative and survival advantage to hematopoietic cells through activation of downstream pathways.
CML is divided into 3 phases based on the number of myeloblasts observed in the blood or bone marrow: chronic, accelerated, and blast. Most cases of CML are diagnosed in the chronic phase (CP), which is marked by proliferation of primarily the myeloid element.
The advent of tyrosine kinase inhibitors (TKIs), a class of small molecules targeting the tyrosine kinases, particularly the BCR-ABL tyrosine kinase, led to rapid changes in the management of CML and improved survival for patients. Patients diagnosed with CP-CML now a have life-expectancy that is similar to that of the general population, as long as they receive the appropriate TKI therapy and adhere to treatment. As such, it is crucial to identify patients with CML, ensure they receive a complete, appropriate diagnostic work-up, and select the best therapy for each individual patient. The diagnosis and work-up of CML are reviewed in a separate article; here, the selection of TKI therapy for a patient with newly diagnosed CP-CML is reviewed.
Case Presentation
A 53-year-old woman who recently was diagnosed with CML presents to review her treatment options. The diagnosis was made after she presented to her primary care physician with fatigue, early satiety, left upper quadrant abdominal pain, and an 8-lb unintentional weight loss over the prior month. On physical exam her spleen was palpated 8 cm below the left costal margin. Laboratory evaluation showed a total white blood cell (WBC) count of 124,000/μL with a left-shifted differential including 6% basophils, 3% eosinophils, and 3% blasts; hemoglobin and platelet count were 12.4 g/dL and 801 × 103/µL, respectively. Fluorescent in-situ hybridization for BCR-ABL gene rearrangement using peripheral blood was positive in 87% of cells. Bone marrow biopsy and aspiration showed a 95% cellular bone marrow with granulocytic hyperplasia and 1% blasts. Cytogenetics were 46,XX,t(9;22)(q34;q11.2), and quantitative real-time polymerase chain reaction (RQ-PCR) to measure BCR-ABL1 transcripts in the peripheral blood showed a value of 98% international standard (IS). Her Sokal risk score was 1.42 (high risk). In addition, prior review of her past medical history revealed uncontrolled diabetes, coronary artery disease requiring placement of 3 cardiac stents 2 years prior, and chronic obstructive pulmonary disease (COPD) related to a 30-pack-year history of smoking.
- What factors must be considered when selecting first-line therapy for this patient?
Selection of the most appropriate first-line TKI for newly diagnosed CP-CML patients requires incorporation of many patient-specific factors. These factors include baseline karyotype and confirmation of CP-CML through bone marrow biopsy, Sokal or EURO risk score, and a thorough patient history, including a clear understanding of the patient's comorbidities. In this case, the patient's high Sokal risk score along with her history of diabetes, coronary artery disease, and COPD are all factors that must be accounted for when choosing the most appropriate TKI. The adverse effect profile of all TKIs must be considered in conjunction with the patient's ongoing medical issues in order to decrease the likelihood of worsening her current symptoms or causing a severe complication from TKI therapy.
Imatinib
The management of CML was revolutionized by the development and ultimate regulatory approval of imatinib mesylate in 2001. Imatinib was the first small-molecule cancer therapy developed and approved. It acts by binding to the adenosine triphosphate (ATP) binding site in the catalytic domain of BCR-ABL, thus inhibiting the oncoprotein's tyrosine kinase activity.1
The International Randomized Study of Interferon versus STI571 (IRIS) trial was a randomized phase 3 study that compared imatinib 400 mg daily to interferon α (IFNα) plus cytarabine. More than 1000 CP-CML patients were randomly assigned 1:1 to either imatinib or IFNα plus cytarabine and were assessed for event-free survival, hematologic and cytogenetic responses, freedom from progression to accelerated phase (AP) or blast phase (BP), and toxicity. Imatinib was superior to the prior standard of care for all these outcomes.2 The long-term follow up of the IRIS trial reported an 83% estimated 10-year overall survival (OS) and 79% estimated event-free survival for patients on the imatinib arm of this study.3 The cumulative rate of complete cytogenetic response (CCyR) was 82.8%. Of the 204 imatinib-treated patients who could undergo a molecular response evaluation at 10 years, 93.1% had a major molecular response (MMR) and 63.2% had a molecular response 4.5 (MR4.5), suggesting durable, deep molecular responses for many patients (see Chronic Myeloid Leukemia: Evaluation and Diagnosis for discussion of the hematologic parameters, cytogenetic results, and molecular responses ussed in monitoring response to TKI therapy). The estimated 10-year rate of freedom from progression to AP or BP was 92.1%.
Higher doses of imatinib (600-800 mg daily) have been studied in an attempt to overcome resistance and improve cytogenetic and molecular response rates. The Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) trial was a randomized phase 3 study that compared imatinib 800 mg daily to imatinib 400 mg daily. Although the 6-month assessments found increased rates of CCyR and a MMR in the higher-dose imatinib arm, these differences were no longer present at the 12-month assessment. Furthermore, the higher dose of imatinib led to a significantly higher incidence of grade 3/4 hematologic adverse events, and approximately 50% of patients on imatinib 800 mg daily required a dose reduction to less than 600 mg daily because of toxicity.4
The Therapeutic Intensification in De Novo Leukaemia (TIDEL) -II study used plasma trough levels of imatinib on day 22 of treatment with imatinib 600 mg daily to determine if patients should escalate the imatinib dose to 800 mg daily. In patients who did not meet molecular milestones at 3, 6, or 12 months, cohort 1 was dose escalated to imatinib 800 mg daily and subsequently switched to nilotinib 400 mg twice daily for failing the same target 3 months later, and cohort 2 was switched to nilotinib. At 2 years, 73% of patients achieved MMR and 34% achieved MR4.5, suggesting that initial treatment with higher-dose imatinib subsequently followed by a switch to nilotinib in those failing to achieve desired milestones could be an effective strategy for managing newly diagnosed CP-CML.5
Toxicity
Imatinib 400 mg is considered the standard starting dose in CP-CML patients. The safety profile of imatinib has been very well established. In the IRIS trial, the most common adverse events (all grades in decreasing order of frequency) were peripheral and periorbital edema (60%), nausea (50%), muscle cramps (49%), musculoskeletal pain (47%), diarrhea (45%), rash (40%), fatigue (39%), abdominal pain (37%), headache (37%), and joint pain (31%). Grade 3/4 liver enzyme elevation can occur in 5% of patients.6 In the event of severe liver toxicity or fluid retention, imatinib should be held until the event resolves. At that time, imatinib can be restarted if deemed appropriate, but this is dependent on the severity of the inciting event. Fluid retention can be managed by the use of supportive care, diuretics, imatinib dose reduction, dose interruption, or imatinib discontinuation if the fluid retention is severe. Muscle cramps can be managed by the use of a calcium supplements or tonic water. Management of rash can include topical or systemic steroids, or in some cases imatinib dose reduction, interruption, or discontinuation.7
Grade 3/4 imatinib-induced hematologic toxicity is not uncommon, with 17% of patients experiencing neutropenia, 9% thrombocytopenia, and 4% anemia. These adverse events occurred most commonly during the first year of therapy, and the frequency decreased over time.3,6 Depending on the degree of cytopenias, imatinib dosing should be interrupted until recovery of the absolute neutrophil count or platelet count, and can often be resumed at 400 mg daily. However, if cytopenias recur, imatinib should be held and subsequently restarted at 300 mg daily.7
Dasatinib
Dasatinib is a second-generation TKI that has regulatory approval for treatment of adult patients with newly diagnosed CP-CML or CP-CML in patients with resistance or intolerance to prior TKIs. In addition to dasatinib's ability to inhibit ABL kinases, it is also known to be a potent inhibitor of Src family kinases. Dasatinib has shown efficacy in patients who have developed imatinib-resistant ABL kinase domain mutations.
Dasatinib was initially approved as second-line therapy in patients with resistance or intolerance to imatinib. This indication was based on the results of the phase 3 CA180-034 trial which ultimately identified dasatinib 100 mg daily as the optimal dose. In this trial, 74% of patients enrolled had resistance to imatinib and the remainder were intolerant. The 7-year follow-up of patients randomized to dasatinib 100 mg (n = 167) daily indicated that 46% achieved MMR while on study. Of the 124 imatinib-resistant patients on dasatinib 100 mg daily, the 7-year progression-free survival (PFS) was 39% and OS was 63%. In the 43 imatinib-intolerant patients, the 7-year PFS was 51% and OS was 70%.8
Dasatinib 100 mg daily was compared to imatinib 400 mg daily in newly diagnosed CP-CML patients in the randomized phase 3 DASISION trial. More patients on the dasatinib arm achieved an early molecular response of BCR-ABL1 transcripts ≤10% IS after 3 months on treatment compared to imatinib (84% versus 64%). Furthermore, the 5-year follow-up reports that the cumulative incidence of MMR and MR4.5 in dasatinib-treated patients was 76% and 42%, and was 64% and 33%, with imatinib (P = 0.0022 and P = 0.0251, respectively). Fewer patients treated with dasatinib progressed to AP or BP (4.6%) compared to imatinib (7.3%), but the estimated 5-year OS was similar between the 2 arms (91% for dasatinib versus 90% for imatinib).9 Regulatory approval for dasatinib as first-line therapy in newly diagnosed CML patients was based on results of the DASISION trial.
Toxicity
Most dasatinib-related toxicities are reported as grade 1 or grade 2, but grade 3/4 hematologic adverse events are fairly common. In the DASISION trial, grade 3/4 neutropenia, anemia, and thrombocytopenia occurred in 29%, 13%, and 22% of dasatinib-treated patients, respectively. Cytopenias can generally be managed with temporary dose interruptions or dose reductions.
During the 5-year follow-up of the DASISION trial, pleural effusions were reported in 28% of patients, most of which were grade 1/2. This occurred at a rate of approximately ≤ 8% per year, suggesting a stable incidence over time, and the effusions appear to be dose-dependent.9 Depending on the severity of the effusion, this may be treated with diuretics, dose interruption, and in some instances, steroids or a thoracentesis. Typically, dasatinib can be restarted at 1 dose level lower than the previous dose once the effusion has resolved.7 Other, less common side effects of dasatinib include pulmonary hypertension (5% of patients), as well as abdominal pain, fluid retention, headaches, fatigue, musculoskeletal pain, rash, nausea, and diarrhea. Pulmonary hypertension is typically reversible after cessation of dasatinib, and thus dasatinib should be permanently discontinued once the diagnosis is confirmed. Fluid retention is often treated with diuretics and supportive care. Nausea and diarrhea are generally manageable and occur less frequently when dasatinib is taken with food and a large glass of water. Antiemetics and antidiarrheals can be used as needed. Troublesome rash can be best managed with topical or systemic steroids as well as possible dose reduction or dose interruption.7,9 In the DASISION trial, adverse events led to therapy discontinuation more often in the dasatinib group than in the imatinib group (16% versus 7%).9 Bleeding, particularly in the setting of thrombocytopenia, has been reported in patients being treated with dasatinib as a result of the drug-induced reversible inhibition of platelet aggregation.10
Nilotinib
The structure of nilotinib is similar to that of imatinib; however, it has a markedly increased affinity for the ATP‐binding site on the BCR-ABL1 protein. It was initially given regulatory approval in the setting of imatinib failure. Nilotinib was studied at a dose of 400 mg twice daily in 321 patients who were imatinib-resistant or -intolerant. It proved to be highly effective at inducing cytogenetic remissions in the second-line setting, with 59% of patients achieving a major cytogenetic response (MCyR) and 45% achieving CCyR. With a median follow-up time of 4 years, the OS was 78%.11
Nilotinib gained regulatory approval for use as a first-line TKI after completion of the randomized phase 3 ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients) trial. ENESTnd was a 3-arm study comparing nilotinib 300 mg twice daily versus nilotinib 400 mg twice daily versus imatinib 400 mg daily in newly diagnosed, previously untreated patients diagnosed with CP-CML. The primary endpoint of this clinical trial was rate of MMR at 12 months.12 Nilotinib surpassed imatinib in this regard, with 44% of patients on nilotinib 300 mg twice daily achieving MMR at 12 months versus 43% of nilotinib 400 mg twice daily patients versus 22% of the imatinib-treated patients (P < 0.001 for both comparisons). Furthermore, the rate of CCyR by 12 months was significantly higher for both nilotinib arms compared with imatinib (80% for nilotinib 300 mg, 78% for nilotinib 400 mg, and 65% for imatinib) (P < 0.001).12 Based on this data, nilotinib 300 mg twice daily was chosen as the standard dose of nilotinib in the first-line setting. After 5 years of follow-up on the ENESTnd study, there were fewer progressions to AP/BP CML in nilotinib-treated patients compared with imatinib. MMR was achieved in 77% of nilotinib 300 mg patients compared with 60.4% of patients on the imatinib arm. MR4.5 was also more common in patients treated with nilotinib 300 mg twice daily, with a rate of 53.5% at 5 years versus 31.4% in the imatinib arm.13 In spite of the deeper cytogenetic and molecular responses achieved with nilotinib, this did not translate into a significant improvement in OS. The 5-year OS rate was 93.7% in nilotinib 300 mg patients versus 91.7% in imatinib-treated patients, and this difference lacked statistical significance.13
Toxicity
Although some similarities exist between the toxicity profiles of nilotinib and imatinib, each drug has some distinct adverse events. On the ENESTnd trial, the rate of any grade 3/4 non-hematologic adverse event was fairly low; however, lower-grade toxicities were not uncommon. Patients treated with nilotinib 300 mg twice daily experienced rash (31%), headache (14%), pruritis (15%), and fatigue (11%) most commonly. The most frequently reported laboratory abnormalities included increased total bilirubin (53%), hypophosphatemia (32%), hyperglycemia (36%), elevated lipase (24%), increased alanine aminotransferase (ALT; 66%), and increased aspartate aminotransferase (AST; 40%). Any grade of neutropenia, thrombocytopenia, or anemia occurred at rates of 43%, 48%, and 38%, respectively.12 Although nilotinib has a Black Box Warning from the US Food and Drug Administration for QT interval prolongation, no patients on the ENESTnd trial experienced a QT interval corrected for heart rate greater than 500 msec.12
More recent concerns have emerged regarding the potential for cardiovascular toxicity after long-term use of nilotinib. The 5-year update of ENESTnd reports cardiovascular events, including ischemic heart disease, ischemic cerebrovascular events, or peripheral arterial disease occurring in 7.5% of patients treated with nilotinib 300 mg twice daily compared with a rate of 2.1% in imatinib-treated patients. The frequency of these cardiovascular events increased linearly over time in both arms. Elevations in total cholesterol from baseline occurred in 27.6% of nilotinib patients compared with 3.9% of imatinib patients. Furthermore, clinically meaningful increases in low-density lipoprotein cholesterol and glycated hemoglobin occurred more frequently with nilotinib therapy.12
Nilotinib should be taken on an empty stomach; therefore, patients should be made aware of the need to fast for 2 hours prior to each dose and 1 hour after each dose. Given the potential risk of QT interval prolongation, a baseline electrocardiogram (ECG) is recommended prior to initiating treatment to ensure the QT interval is within a normal range. A repeat ECG should be done approximately 7 days after nilotinib initiation to ensure no prolongation of the QT interval after starting. Close monitoring of potassium and magnesium levels is important to decrease the risk of cardiac arrhythmias, and concomitant use of drugs considered strong CYP3A4 inhibitors should be avoided.7
If the patient experiences any grade 3 or higher laboratory abnormalities, nilotinib should be held until resolution of the toxicity, and then restarted at a lower dose. Similarly, if patients develop significant neutropenia or thrombocytopenia, nilotinib doses should be interrupted until resolution of the cytopenias. At that point, nilotinib can be reinitiated at either the same or a lower dose. Rash can be managed by the use of topical or systemic steroids as well as potential dose reduction, interruption, or discontinuation.
Given the concerns for potential cardiovascular events with long-term use of nilotinib, caution is advised when prescribing it to any patient with a history of cardiovascular disease or peripheral arterial occlusive disease. At the first sign of new occlusive disease, nilotinib should be discontinued.7
Bosutinib
Bosutinib is a second-generation BCR-ABL1 TKI with activity against the Src family of kinases that was initially approved to treat patients with CP-, AP-, or BP-CML after resistance or intolerance to imatinib. Long-term data has been reported from the phase 1/2 trial of bosutinib therapy in patients with CP-CML who developed resistance or intolerance to imatinib plus dasatinib and/or nilotinib. A total of 119 patients were included in the 4-year follow-up; 38 were resistant/intolerant to imatinib and resistant to dasatinib, 50 were resistant/intolerant to imatinib and intolerant to dasatinib, 26 were resistant/intolerant to imatinib and resistant to nilotinib, and 5 were resistant/intolerant to imatinib and intolerant to nilotinib or resistant/intolerant to dasatinib and nilotinib. Bosutinib 400 mg daily was studied in this setting. Of the 38 patients with imatinib resistance/intolerance and dasatinib resistance, 39% achieved MCyR, 22% achieved CCyR, and the OS was 67%. Of the 50 patients with imatinib resistance/intolerance and dasatinib intolerance, 42% achieved MCyR, 40% achieved CCyR, and the OS was 80%. Finally, in the 26 patients with imatinib resistance/intolerance and nilotinib resistance, 38% achieved MCyR, 31% achieved CcyR, and the OS was 87%.14
Five-year follow-up from the phase 1/2 clinical trial which studied bosutinib 500 mg daily in CP-CML patients after imatinib failure reported data on 284 patients. By 5 years on study, 60% of patients had achieved MCyR and 50% achieved CCyR with a 71% and 69% probability, respectively, of maintaining these responses at 5 years. The 5-year OS was 84%.15 These data led to the regulatory approval of bosutinib 500 mg daily as second-line or later therapy.
Bosutinib was initially studied in the first-line setting in the randomized phase 3 BELA (Bosutinib Efficacy and Safety in Newly Diagnosed Chronic Myeloid Leukemia) trial. This trial compared bosutinib 500 mg daily to imatinib 400 mg daily in newly diagnosed, previously untreated CP-CML patients. This trial failed to meet its primary endpoint of increased rate of CCyR at 12 months, with 70% of bosutinib patients achieving this response compared to 68% of imatinib-treated patients (P = 0.601). In spite of this, the rate of MMR at 12 months was significantly higher in the bosutinib arm (41%) compared to the imatinib arm (27%; P = 0.001).16
A second phase 3 trial (BFORE) was designed to study bosutinib 400 mg daily versus imatinib in newly diagnosed, previously untreated CP-CML patients. This study enrolled 536 patients who were randomly assigned 1:1 to bosutinib versus imatinib. The primary endpoint of this trial was rate of MMR at 12 months. A significantly higher number of bosutinib-treated patients achieved this response (47.2%) compared with imatinib-treated patients (36.9%, P = 0.02). Furthermore, by 12 months 77.2% of patients on the bosutinib arm had achieved CCyR compared with 66.4% on the imatinib arm, and this difference did meet statistical significance (P = 0.0075). A lower rate of progression to AP- or BP-CML was noted in bosutinib-treated patients as well (1.6% versus 2.5%). Based on this data, bosutinib gained regulatory approval for first-line therapy in CP-CML at a dose of 400 mg daily.17
Toxicity
On the BFORE trial, the most common treatment-emergent adverse events of any grade reported in the bosutinib-treated patients were diarrhea (70.1%), nausea (35.1%), increased ALT (30.6%), and increased AST (22.8%). Musculoskeletal pain or spasms occurred in 29.5% of patients, rash in 19.8%, fatigue in 19.4%, and headache in 18.7%. Hematologic toxicity was also reported, but most was grade 1/2. Thrombocytopenia was reported in 35.1%, anemia in 18.7%, and neutropenia in 11.2%.17
Cardiovascular events occurred in 5.2% of patients on the bosutinib arm of the BFORE trial, which was similar to the rate observed in imatinib patients. The most common cardiovascular event was QT interval prolongation, which occurred in 1.5% of patients. Pleural effusions were reported in 1.9% of patients treated with bosutinib, and none were grade 3 or higher.17
If liver enzyme elevation occurs at a value greater than 5 times the institutional upper limit of normal, bosutinib should be held until the level recovers to ≤2.5 times the upper limit of normal, at which point bosutinib can be restarted at a lower dose. If recovery takes longer than 4 weeks, bosutinib should be permanently discontinued. Liver enzymes elevated greater than 3 times the institutional upper limit of normal and a concurrent elevation in total bilirubin to 2 times the upper limit of normal is consistent with Hy's law, and bosutinib should be discontinued. Although diarrhea is the most common toxicity associated with bosutinib, it is commonly low grade and transient. Diarrhea occurs most frequently in the first few days after initiating bosutinib. It can often be managed with over-the-counter antidiarrheal medications, but if the diarrhea is grade or higher, bosutinib should be held until recovery to grade 1 or lower. Gastrointestinal side effects may be improved by taking bosutinib with a meal and a large glass of water. Fluid retention can be managed with diuretics and supportive care. Finally, if rash occurs, this can be addressed with topical or systemic steroids as well as bosutinib dose reduction, interruption, or discontinuation.7
Similar to other TKIs, if bosutinib-induced cytopenias occur, treatment should be held and restarted at the same or a lower dose upon blood count recovery.7
Ponatinib
The most common cause of TKI resistance in CP-CML is the development of ABL kinase domain mutations. The majority of imatinib-resistant mutations can be overcome by the use of second-generation TKIs including dasatinib, nilotinib, or bosutinib. However, ponatinib is the only BCR-ABL1 TKI able to overcome a T315I mutation. The phase 2 PACE (Ponatinib Ph-positive ALL and CML Evaluation) trial enrolled patients with CP-, AP-, or BP-CML as well as patients with Ph-positive acute lymphoblastic leukemia who were resistant or intolerant to nilotinib or dasatinib, or who had evidence of a T315I mutation. The starting dose of ponatinib on this trial was 45 mg daily.18 The PACE trial enrolled 267 patients with CP-CML: 203 with resistance or intolerance to nilotinib or dasatinib, and 64 with a T315I mutation. The primary endpoint in the CP cohort was rate of MCyR at any time within 12 months of starting ponatinib. The overall rate of MCyR by 12 months in the CP-CML patients was 56%. In those with a T315I mutation, 70% achieved MCyR, which compared favorably with those with resistance or intolerance to nilotinib or dasatinib, 51% of whom achieved MCyR. CCyR was achieved in 46% of CP-CML patients (40% in the resistant/intolerant cohort and 66% in the T315I cohort). In general, patients with T315I mutations received fewer prior therapies than those in the resistant/intolerant cohort, which likely contributed to the higher response rates in the T315I patients. MR4.5 was achieved in 15% of CP-CML patients by 12 months on the PACE trial.18 The 5-year update to this study reported that 60%, 40%, and 24% of CP-CML patients achieved MCyR, MMR, and MR4.5, respectively. In the patients who achieved MCyR, the probability of maintaining this response for 5 years was 82% and the estimated 5-year OS was 73%.19
Toxicity
In 2013, after the regulatory approval of ponatinib, reports became available that the drug can cause an increase in arterial occlusive events including fatal myocardial infarctions and cerebral vascular accidents. For this reason, dose reductions were implemented in patients who were deriving clinical benefit from ponatinib. In spite of these dose reductions, ≥90% of responders maintained their response for up to 40 months.19 Although the likelihood of developing an arterial occlusive event appears higher in the first year after starting ponatinib than in later years, the cumulative incidence of events continues to increase. The 5-year follow-up to the PACE trial reports 31% of patients experiencing any grade of arterial occlusive event while on ponatinib. Aside from these events, the most common treatment-emergent adverse events in ponatinib-treated patients on the PACE trial included rash (47%), abdominal pain (46%), headache (43%), dry skin (42%), constipation (41%), and hypertension (37%). Hematologic toxicity was also common, with 46% of patients experiencing any grade of thrombocytopenia, 20% experiencing neutropenia, and 20% anemia.19
Patients receiving ponatinib therapy should be monitored closely for any evidence of arterial or venous thrombosis. In the event of an occlusive event, ponatinib should be discontinued. Similarly, in the setting of any new or worsening heart failure symptoms, ponatinib should be promptly discontinued. Management of any underlying cardiovascular risk factors including hypertension, hyperlipidemia, diabetes, or smoking history is recommended, and these patients should be referred to a cardiologist for a full evaluation. In the absence of any contraindications to aspirin, low-dose aspirin should be considered as a means of decreasing cardiovascular risks associated with ponatinib. In patients with known risk factors, a ponatinib starting dose of 30 mg daily rather than the standard 45 mg daily may be a safer option resulting in fewer arterial occlusive events, although the efficacy of this dose is still being studied in comparison to 45 mg daily.7
In the event of ponatinib-induced transaminitis greater than 3 times the upper limit of normal, ponatinib should be held until resolution to less than 3 times the upper limit of normal, at which point it should be resumed at a lower dose. Similarly, in the setting of elevated serum lipase or symptomatic pancreatitis, ponatinib should be held and restarted at a lower dose after resolution of symptoms.7
In the event of neutropenia or thrombocytopenia, ponatinib should be held until blood count recovery and then restarted at the same dose. If cytopenias occur for a second time, the dose of ponatinib should be lowered at the time of treatment reinitiation. If rash occurs, it can be addressed with topical or systemic steroids as well as dose reduction, interruption, or discontinuation.7
Case Conclusion
Given the patient's high-risk Sokal score, ideal first-line treatment is a second-generation TKI in order to increase the likelihood of achieving the desired treatment milestones and improving long-term outcomes. Her history of uncontrolled diabetes and coronary artery disease raises concerns for using nilotinib. Furthermore, her history of COPD makes dasatinib suboptimal because she would have little pulmonary reserve if she were to develop a pleural effusion. For this reason, bosutinib 400 mg daily is chosen as her first-line TKI. Shortly after starting bosutinib, she experiences diarrhea that occurs approximately 3 or 4 times daily during the first week on treatment. She is able to manage this with over-the-counter loperamide and the diarrhea resolves shortly thereafter.
After 3 months of bosutinib therapy, quantitative real-time PCR (RQ-PCR) assay on peripheral blood is done to measure BCR-ABL1 transcripts, and the result is reported at 1.2% IS. This indicates that the patient has achieved an early molecular response, which is defined as a RQ-PCR value of ≤10% IS. She undergoes RQ-PCR monitoring every 3 months, and at 12 months her results indicate a value of 0.07% IS, suggesting she has achieved a MMR.
Conclusion
With the development of imatinib and the subsequent TKIs, dasatinib, nilotinib, bosutinib, and ponatinib, CP-CML has become a chronic disease with a life-expectancy that is similar to the general population. Given the successful treatments available for these patients, it is crucial to identify patients with this diagnosis, ensure they receive a complete, appropriate diagnostic workup including a bone marrow biopsy and aspiration with cytogenetic testing, and select the best therapy for each individual patient. Once on treatment, the importance of frequent monitoring cannot be overstated. This is the only way to be certain patients are achieving the desired treatment milestones that correlate with the favorable long-term outcomes that have been observed with TKI-based treatment of CP-CML.
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm that arises from a reciprocal translocation between the Abelson (ABL) region on chromosome 9 and the breakpoint cluster region (BCR) of chromosome 22, t(9;22)(q34;q11.2) (the Philadelphia chromosome), resulting in the generation of the BCR-ABL1 fusion gene and its protein product, BCR-ABL tyrosine kinase. BCR-ABL is a constitutively active fusion kinase that confers proliferative and survival advantage to hematopoietic cells through activation of downstream pathways.
CML is divided into 3 phases based on the number of myeloblasts observed in the blood or bone marrow: chronic, accelerated, and blast. Most cases of CML are diagnosed in the chronic phase (CP), which is marked by proliferation of primarily the myeloid element.
The advent of tyrosine kinase inhibitors (TKIs), a class of small molecules targeting the tyrosine kinases, particularly the BCR-ABL tyrosine kinase, led to rapid changes in the management of CML and improved survival for patients. Patients diagnosed with CP-CML now a have life-expectancy that is similar to that of the general population, as long as they receive the appropriate TKI therapy and adhere to treatment. As such, it is crucial to identify patients with CML, ensure they receive a complete, appropriate diagnostic work-up, and select the best therapy for each individual patient. The diagnosis and work-up of CML are reviewed in a separate article; here, the selection of TKI therapy for a patient with newly diagnosed CP-CML is reviewed.
Case Presentation
A 53-year-old woman who recently was diagnosed with CML presents to review her treatment options. The diagnosis was made after she presented to her primary care physician with fatigue, early satiety, left upper quadrant abdominal pain, and an 8-lb unintentional weight loss over the prior month. On physical exam her spleen was palpated 8 cm below the left costal margin. Laboratory evaluation showed a total white blood cell (WBC) count of 124,000/μL with a left-shifted differential including 6% basophils, 3% eosinophils, and 3% blasts; hemoglobin and platelet count were 12.4 g/dL and 801 × 103/µL, respectively. Fluorescent in-situ hybridization for BCR-ABL gene rearrangement using peripheral blood was positive in 87% of cells. Bone marrow biopsy and aspiration showed a 95% cellular bone marrow with granulocytic hyperplasia and 1% blasts. Cytogenetics were 46,XX,t(9;22)(q34;q11.2), and quantitative real-time polymerase chain reaction (RQ-PCR) to measure BCR-ABL1 transcripts in the peripheral blood showed a value of 98% international standard (IS). Her Sokal risk score was 1.42 (high risk). In addition, prior review of her past medical history revealed uncontrolled diabetes, coronary artery disease requiring placement of 3 cardiac stents 2 years prior, and chronic obstructive pulmonary disease (COPD) related to a 30-pack-year history of smoking.
- What factors must be considered when selecting first-line therapy for this patient?
Selection of the most appropriate first-line TKI for newly diagnosed CP-CML patients requires incorporation of many patient-specific factors. These factors include baseline karyotype and confirmation of CP-CML through bone marrow biopsy, Sokal or EURO risk score, and a thorough patient history, including a clear understanding of the patient's comorbidities. In this case, the patient's high Sokal risk score along with her history of diabetes, coronary artery disease, and COPD are all factors that must be accounted for when choosing the most appropriate TKI. The adverse effect profile of all TKIs must be considered in conjunction with the patient's ongoing medical issues in order to decrease the likelihood of worsening her current symptoms or causing a severe complication from TKI therapy.
Imatinib
The management of CML was revolutionized by the development and ultimate regulatory approval of imatinib mesylate in 2001. Imatinib was the first small-molecule cancer therapy developed and approved. It acts by binding to the adenosine triphosphate (ATP) binding site in the catalytic domain of BCR-ABL, thus inhibiting the oncoprotein's tyrosine kinase activity.1
The International Randomized Study of Interferon versus STI571 (IRIS) trial was a randomized phase 3 study that compared imatinib 400 mg daily to interferon α (IFNα) plus cytarabine. More than 1000 CP-CML patients were randomly assigned 1:1 to either imatinib or IFNα plus cytarabine and were assessed for event-free survival, hematologic and cytogenetic responses, freedom from progression to accelerated phase (AP) or blast phase (BP), and toxicity. Imatinib was superior to the prior standard of care for all these outcomes.2 The long-term follow up of the IRIS trial reported an 83% estimated 10-year overall survival (OS) and 79% estimated event-free survival for patients on the imatinib arm of this study.3 The cumulative rate of complete cytogenetic response (CCyR) was 82.8%. Of the 204 imatinib-treated patients who could undergo a molecular response evaluation at 10 years, 93.1% had a major molecular response (MMR) and 63.2% had a molecular response 4.5 (MR4.5), suggesting durable, deep molecular responses for many patients (see Chronic Myeloid Leukemia: Evaluation and Diagnosis for discussion of the hematologic parameters, cytogenetic results, and molecular responses ussed in monitoring response to TKI therapy). The estimated 10-year rate of freedom from progression to AP or BP was 92.1%.
Higher doses of imatinib (600-800 mg daily) have been studied in an attempt to overcome resistance and improve cytogenetic and molecular response rates. The Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) trial was a randomized phase 3 study that compared imatinib 800 mg daily to imatinib 400 mg daily. Although the 6-month assessments found increased rates of CCyR and a MMR in the higher-dose imatinib arm, these differences were no longer present at the 12-month assessment. Furthermore, the higher dose of imatinib led to a significantly higher incidence of grade 3/4 hematologic adverse events, and approximately 50% of patients on imatinib 800 mg daily required a dose reduction to less than 600 mg daily because of toxicity.4
The Therapeutic Intensification in De Novo Leukaemia (TIDEL) -II study used plasma trough levels of imatinib on day 22 of treatment with imatinib 600 mg daily to determine if patients should escalate the imatinib dose to 800 mg daily. In patients who did not meet molecular milestones at 3, 6, or 12 months, cohort 1 was dose escalated to imatinib 800 mg daily and subsequently switched to nilotinib 400 mg twice daily for failing the same target 3 months later, and cohort 2 was switched to nilotinib. At 2 years, 73% of patients achieved MMR and 34% achieved MR4.5, suggesting that initial treatment with higher-dose imatinib subsequently followed by a switch to nilotinib in those failing to achieve desired milestones could be an effective strategy for managing newly diagnosed CP-CML.5
Toxicity
Imatinib 400 mg is considered the standard starting dose in CP-CML patients. The safety profile of imatinib has been very well established. In the IRIS trial, the most common adverse events (all grades in decreasing order of frequency) were peripheral and periorbital edema (60%), nausea (50%), muscle cramps (49%), musculoskeletal pain (47%), diarrhea (45%), rash (40%), fatigue (39%), abdominal pain (37%), headache (37%), and joint pain (31%). Grade 3/4 liver enzyme elevation can occur in 5% of patients.6 In the event of severe liver toxicity or fluid retention, imatinib should be held until the event resolves. At that time, imatinib can be restarted if deemed appropriate, but this is dependent on the severity of the inciting event. Fluid retention can be managed by the use of supportive care, diuretics, imatinib dose reduction, dose interruption, or imatinib discontinuation if the fluid retention is severe. Muscle cramps can be managed by the use of a calcium supplements or tonic water. Management of rash can include topical or systemic steroids, or in some cases imatinib dose reduction, interruption, or discontinuation.7
Grade 3/4 imatinib-induced hematologic toxicity is not uncommon, with 17% of patients experiencing neutropenia, 9% thrombocytopenia, and 4% anemia. These adverse events occurred most commonly during the first year of therapy, and the frequency decreased over time.3,6 Depending on the degree of cytopenias, imatinib dosing should be interrupted until recovery of the absolute neutrophil count or platelet count, and can often be resumed at 400 mg daily. However, if cytopenias recur, imatinib should be held and subsequently restarted at 300 mg daily.7
Dasatinib
Dasatinib is a second-generation TKI that has regulatory approval for treatment of adult patients with newly diagnosed CP-CML or CP-CML in patients with resistance or intolerance to prior TKIs. In addition to dasatinib's ability to inhibit ABL kinases, it is also known to be a potent inhibitor of Src family kinases. Dasatinib has shown efficacy in patients who have developed imatinib-resistant ABL kinase domain mutations.
Dasatinib was initially approved as second-line therapy in patients with resistance or intolerance to imatinib. This indication was based on the results of the phase 3 CA180-034 trial which ultimately identified dasatinib 100 mg daily as the optimal dose. In this trial, 74% of patients enrolled had resistance to imatinib and the remainder were intolerant. The 7-year follow-up of patients randomized to dasatinib 100 mg (n = 167) daily indicated that 46% achieved MMR while on study. Of the 124 imatinib-resistant patients on dasatinib 100 mg daily, the 7-year progression-free survival (PFS) was 39% and OS was 63%. In the 43 imatinib-intolerant patients, the 7-year PFS was 51% and OS was 70%.8
Dasatinib 100 mg daily was compared to imatinib 400 mg daily in newly diagnosed CP-CML patients in the randomized phase 3 DASISION trial. More patients on the dasatinib arm achieved an early molecular response of BCR-ABL1 transcripts ≤10% IS after 3 months on treatment compared to imatinib (84% versus 64%). Furthermore, the 5-year follow-up reports that the cumulative incidence of MMR and MR4.5 in dasatinib-treated patients was 76% and 42%, and was 64% and 33%, with imatinib (P = 0.0022 and P = 0.0251, respectively). Fewer patients treated with dasatinib progressed to AP or BP (4.6%) compared to imatinib (7.3%), but the estimated 5-year OS was similar between the 2 arms (91% for dasatinib versus 90% for imatinib).9 Regulatory approval for dasatinib as first-line therapy in newly diagnosed CML patients was based on results of the DASISION trial.
Toxicity
Most dasatinib-related toxicities are reported as grade 1 or grade 2, but grade 3/4 hematologic adverse events are fairly common. In the DASISION trial, grade 3/4 neutropenia, anemia, and thrombocytopenia occurred in 29%, 13%, and 22% of dasatinib-treated patients, respectively. Cytopenias can generally be managed with temporary dose interruptions or dose reductions.
During the 5-year follow-up of the DASISION trial, pleural effusions were reported in 28% of patients, most of which were grade 1/2. This occurred at a rate of approximately ≤ 8% per year, suggesting a stable incidence over time, and the effusions appear to be dose-dependent.9 Depending on the severity of the effusion, this may be treated with diuretics, dose interruption, and in some instances, steroids or a thoracentesis. Typically, dasatinib can be restarted at 1 dose level lower than the previous dose once the effusion has resolved.7 Other, less common side effects of dasatinib include pulmonary hypertension (5% of patients), as well as abdominal pain, fluid retention, headaches, fatigue, musculoskeletal pain, rash, nausea, and diarrhea. Pulmonary hypertension is typically reversible after cessation of dasatinib, and thus dasatinib should be permanently discontinued once the diagnosis is confirmed. Fluid retention is often treated with diuretics and supportive care. Nausea and diarrhea are generally manageable and occur less frequently when dasatinib is taken with food and a large glass of water. Antiemetics and antidiarrheals can be used as needed. Troublesome rash can be best managed with topical or systemic steroids as well as possible dose reduction or dose interruption.7,9 In the DASISION trial, adverse events led to therapy discontinuation more often in the dasatinib group than in the imatinib group (16% versus 7%).9 Bleeding, particularly in the setting of thrombocytopenia, has been reported in patients being treated with dasatinib as a result of the drug-induced reversible inhibition of platelet aggregation.10
Nilotinib
The structure of nilotinib is similar to that of imatinib; however, it has a markedly increased affinity for the ATP‐binding site on the BCR-ABL1 protein. It was initially given regulatory approval in the setting of imatinib failure. Nilotinib was studied at a dose of 400 mg twice daily in 321 patients who were imatinib-resistant or -intolerant. It proved to be highly effective at inducing cytogenetic remissions in the second-line setting, with 59% of patients achieving a major cytogenetic response (MCyR) and 45% achieving CCyR. With a median follow-up time of 4 years, the OS was 78%.11
Nilotinib gained regulatory approval for use as a first-line TKI after completion of the randomized phase 3 ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials-Newly Diagnosed Patients) trial. ENESTnd was a 3-arm study comparing nilotinib 300 mg twice daily versus nilotinib 400 mg twice daily versus imatinib 400 mg daily in newly diagnosed, previously untreated patients diagnosed with CP-CML. The primary endpoint of this clinical trial was rate of MMR at 12 months.12 Nilotinib surpassed imatinib in this regard, with 44% of patients on nilotinib 300 mg twice daily achieving MMR at 12 months versus 43% of nilotinib 400 mg twice daily patients versus 22% of the imatinib-treated patients (P < 0.001 for both comparisons). Furthermore, the rate of CCyR by 12 months was significantly higher for both nilotinib arms compared with imatinib (80% for nilotinib 300 mg, 78% for nilotinib 400 mg, and 65% for imatinib) (P < 0.001).12 Based on this data, nilotinib 300 mg twice daily was chosen as the standard dose of nilotinib in the first-line setting. After 5 years of follow-up on the ENESTnd study, there were fewer progressions to AP/BP CML in nilotinib-treated patients compared with imatinib. MMR was achieved in 77% of nilotinib 300 mg patients compared with 60.4% of patients on the imatinib arm. MR4.5 was also more common in patients treated with nilotinib 300 mg twice daily, with a rate of 53.5% at 5 years versus 31.4% in the imatinib arm.13 In spite of the deeper cytogenetic and molecular responses achieved with nilotinib, this did not translate into a significant improvement in OS. The 5-year OS rate was 93.7% in nilotinib 300 mg patients versus 91.7% in imatinib-treated patients, and this difference lacked statistical significance.13
Toxicity
Although some similarities exist between the toxicity profiles of nilotinib and imatinib, each drug has some distinct adverse events. On the ENESTnd trial, the rate of any grade 3/4 non-hematologic adverse event was fairly low; however, lower-grade toxicities were not uncommon. Patients treated with nilotinib 300 mg twice daily experienced rash (31%), headache (14%), pruritis (15%), and fatigue (11%) most commonly. The most frequently reported laboratory abnormalities included increased total bilirubin (53%), hypophosphatemia (32%), hyperglycemia (36%), elevated lipase (24%), increased alanine aminotransferase (ALT; 66%), and increased aspartate aminotransferase (AST; 40%). Any grade of neutropenia, thrombocytopenia, or anemia occurred at rates of 43%, 48%, and 38%, respectively.12 Although nilotinib has a Black Box Warning from the US Food and Drug Administration for QT interval prolongation, no patients on the ENESTnd trial experienced a QT interval corrected for heart rate greater than 500 msec.12
More recent concerns have emerged regarding the potential for cardiovascular toxicity after long-term use of nilotinib. The 5-year update of ENESTnd reports cardiovascular events, including ischemic heart disease, ischemic cerebrovascular events, or peripheral arterial disease occurring in 7.5% of patients treated with nilotinib 300 mg twice daily compared with a rate of 2.1% in imatinib-treated patients. The frequency of these cardiovascular events increased linearly over time in both arms. Elevations in total cholesterol from baseline occurred in 27.6% of nilotinib patients compared with 3.9% of imatinib patients. Furthermore, clinically meaningful increases in low-density lipoprotein cholesterol and glycated hemoglobin occurred more frequently with nilotinib therapy.12
Nilotinib should be taken on an empty stomach; therefore, patients should be made aware of the need to fast for 2 hours prior to each dose and 1 hour after each dose. Given the potential risk of QT interval prolongation, a baseline electrocardiogram (ECG) is recommended prior to initiating treatment to ensure the QT interval is within a normal range. A repeat ECG should be done approximately 7 days after nilotinib initiation to ensure no prolongation of the QT interval after starting. Close monitoring of potassium and magnesium levels is important to decrease the risk of cardiac arrhythmias, and concomitant use of drugs considered strong CYP3A4 inhibitors should be avoided.7
If the patient experiences any grade 3 or higher laboratory abnormalities, nilotinib should be held until resolution of the toxicity, and then restarted at a lower dose. Similarly, if patients develop significant neutropenia or thrombocytopenia, nilotinib doses should be interrupted until resolution of the cytopenias. At that point, nilotinib can be reinitiated at either the same or a lower dose. Rash can be managed by the use of topical or systemic steroids as well as potential dose reduction, interruption, or discontinuation.
Given the concerns for potential cardiovascular events with long-term use of nilotinib, caution is advised when prescribing it to any patient with a history of cardiovascular disease or peripheral arterial occlusive disease. At the first sign of new occlusive disease, nilotinib should be discontinued.7
Bosutinib
Bosutinib is a second-generation BCR-ABL1 TKI with activity against the Src family of kinases that was initially approved to treat patients with CP-, AP-, or BP-CML after resistance or intolerance to imatinib. Long-term data has been reported from the phase 1/2 trial of bosutinib therapy in patients with CP-CML who developed resistance or intolerance to imatinib plus dasatinib and/or nilotinib. A total of 119 patients were included in the 4-year follow-up; 38 were resistant/intolerant to imatinib and resistant to dasatinib, 50 were resistant/intolerant to imatinib and intolerant to dasatinib, 26 were resistant/intolerant to imatinib and resistant to nilotinib, and 5 were resistant/intolerant to imatinib and intolerant to nilotinib or resistant/intolerant to dasatinib and nilotinib. Bosutinib 400 mg daily was studied in this setting. Of the 38 patients with imatinib resistance/intolerance and dasatinib resistance, 39% achieved MCyR, 22% achieved CCyR, and the OS was 67%. Of the 50 patients with imatinib resistance/intolerance and dasatinib intolerance, 42% achieved MCyR, 40% achieved CCyR, and the OS was 80%. Finally, in the 26 patients with imatinib resistance/intolerance and nilotinib resistance, 38% achieved MCyR, 31% achieved CcyR, and the OS was 87%.14
Five-year follow-up from the phase 1/2 clinical trial which studied bosutinib 500 mg daily in CP-CML patients after imatinib failure reported data on 284 patients. By 5 years on study, 60% of patients had achieved MCyR and 50% achieved CCyR with a 71% and 69% probability, respectively, of maintaining these responses at 5 years. The 5-year OS was 84%.15 These data led to the regulatory approval of bosutinib 500 mg daily as second-line or later therapy.
Bosutinib was initially studied in the first-line setting in the randomized phase 3 BELA (Bosutinib Efficacy and Safety in Newly Diagnosed Chronic Myeloid Leukemia) trial. This trial compared bosutinib 500 mg daily to imatinib 400 mg daily in newly diagnosed, previously untreated CP-CML patients. This trial failed to meet its primary endpoint of increased rate of CCyR at 12 months, with 70% of bosutinib patients achieving this response compared to 68% of imatinib-treated patients (P = 0.601). In spite of this, the rate of MMR at 12 months was significantly higher in the bosutinib arm (41%) compared to the imatinib arm (27%; P = 0.001).16
A second phase 3 trial (BFORE) was designed to study bosutinib 400 mg daily versus imatinib in newly diagnosed, previously untreated CP-CML patients. This study enrolled 536 patients who were randomly assigned 1:1 to bosutinib versus imatinib. The primary endpoint of this trial was rate of MMR at 12 months. A significantly higher number of bosutinib-treated patients achieved this response (47.2%) compared with imatinib-treated patients (36.9%, P = 0.02). Furthermore, by 12 months 77.2% of patients on the bosutinib arm had achieved CCyR compared with 66.4% on the imatinib arm, and this difference did meet statistical significance (P = 0.0075). A lower rate of progression to AP- or BP-CML was noted in bosutinib-treated patients as well (1.6% versus 2.5%). Based on this data, bosutinib gained regulatory approval for first-line therapy in CP-CML at a dose of 400 mg daily.17
Toxicity
On the BFORE trial, the most common treatment-emergent adverse events of any grade reported in the bosutinib-treated patients were diarrhea (70.1%), nausea (35.1%), increased ALT (30.6%), and increased AST (22.8%). Musculoskeletal pain or spasms occurred in 29.5% of patients, rash in 19.8%, fatigue in 19.4%, and headache in 18.7%. Hematologic toxicity was also reported, but most was grade 1/2. Thrombocytopenia was reported in 35.1%, anemia in 18.7%, and neutropenia in 11.2%.17
Cardiovascular events occurred in 5.2% of patients on the bosutinib arm of the BFORE trial, which was similar to the rate observed in imatinib patients. The most common cardiovascular event was QT interval prolongation, which occurred in 1.5% of patients. Pleural effusions were reported in 1.9% of patients treated with bosutinib, and none were grade 3 or higher.17
If liver enzyme elevation occurs at a value greater than 5 times the institutional upper limit of normal, bosutinib should be held until the level recovers to ≤2.5 times the upper limit of normal, at which point bosutinib can be restarted at a lower dose. If recovery takes longer than 4 weeks, bosutinib should be permanently discontinued. Liver enzymes elevated greater than 3 times the institutional upper limit of normal and a concurrent elevation in total bilirubin to 2 times the upper limit of normal is consistent with Hy's law, and bosutinib should be discontinued. Although diarrhea is the most common toxicity associated with bosutinib, it is commonly low grade and transient. Diarrhea occurs most frequently in the first few days after initiating bosutinib. It can often be managed with over-the-counter antidiarrheal medications, but if the diarrhea is grade or higher, bosutinib should be held until recovery to grade 1 or lower. Gastrointestinal side effects may be improved by taking bosutinib with a meal and a large glass of water. Fluid retention can be managed with diuretics and supportive care. Finally, if rash occurs, this can be addressed with topical or systemic steroids as well as bosutinib dose reduction, interruption, or discontinuation.7
Similar to other TKIs, if bosutinib-induced cytopenias occur, treatment should be held and restarted at the same or a lower dose upon blood count recovery.7
Ponatinib
The most common cause of TKI resistance in CP-CML is the development of ABL kinase domain mutations. The majority of imatinib-resistant mutations can be overcome by the use of second-generation TKIs including dasatinib, nilotinib, or bosutinib. However, ponatinib is the only BCR-ABL1 TKI able to overcome a T315I mutation. The phase 2 PACE (Ponatinib Ph-positive ALL and CML Evaluation) trial enrolled patients with CP-, AP-, or BP-CML as well as patients with Ph-positive acute lymphoblastic leukemia who were resistant or intolerant to nilotinib or dasatinib, or who had evidence of a T315I mutation. The starting dose of ponatinib on this trial was 45 mg daily.18 The PACE trial enrolled 267 patients with CP-CML: 203 with resistance or intolerance to nilotinib or dasatinib, and 64 with a T315I mutation. The primary endpoint in the CP cohort was rate of MCyR at any time within 12 months of starting ponatinib. The overall rate of MCyR by 12 months in the CP-CML patients was 56%. In those with a T315I mutation, 70% achieved MCyR, which compared favorably with those with resistance or intolerance to nilotinib or dasatinib, 51% of whom achieved MCyR. CCyR was achieved in 46% of CP-CML patients (40% in the resistant/intolerant cohort and 66% in the T315I cohort). In general, patients with T315I mutations received fewer prior therapies than those in the resistant/intolerant cohort, which likely contributed to the higher response rates in the T315I patients. MR4.5 was achieved in 15% of CP-CML patients by 12 months on the PACE trial.18 The 5-year update to this study reported that 60%, 40%, and 24% of CP-CML patients achieved MCyR, MMR, and MR4.5, respectively. In the patients who achieved MCyR, the probability of maintaining this response for 5 years was 82% and the estimated 5-year OS was 73%.19
Toxicity
In 2013, after the regulatory approval of ponatinib, reports became available that the drug can cause an increase in arterial occlusive events including fatal myocardial infarctions and cerebral vascular accidents. For this reason, dose reductions were implemented in patients who were deriving clinical benefit from ponatinib. In spite of these dose reductions, ≥90% of responders maintained their response for up to 40 months.19 Although the likelihood of developing an arterial occlusive event appears higher in the first year after starting ponatinib than in later years, the cumulative incidence of events continues to increase. The 5-year follow-up to the PACE trial reports 31% of patients experiencing any grade of arterial occlusive event while on ponatinib. Aside from these events, the most common treatment-emergent adverse events in ponatinib-treated patients on the PACE trial included rash (47%), abdominal pain (46%), headache (43%), dry skin (42%), constipation (41%), and hypertension (37%). Hematologic toxicity was also common, with 46% of patients experiencing any grade of thrombocytopenia, 20% experiencing neutropenia, and 20% anemia.19
Patients receiving ponatinib therapy should be monitored closely for any evidence of arterial or venous thrombosis. In the event of an occlusive event, ponatinib should be discontinued. Similarly, in the setting of any new or worsening heart failure symptoms, ponatinib should be promptly discontinued. Management of any underlying cardiovascular risk factors including hypertension, hyperlipidemia, diabetes, or smoking history is recommended, and these patients should be referred to a cardiologist for a full evaluation. In the absence of any contraindications to aspirin, low-dose aspirin should be considered as a means of decreasing cardiovascular risks associated with ponatinib. In patients with known risk factors, a ponatinib starting dose of 30 mg daily rather than the standard 45 mg daily may be a safer option resulting in fewer arterial occlusive events, although the efficacy of this dose is still being studied in comparison to 45 mg daily.7
In the event of ponatinib-induced transaminitis greater than 3 times the upper limit of normal, ponatinib should be held until resolution to less than 3 times the upper limit of normal, at which point it should be resumed at a lower dose. Similarly, in the setting of elevated serum lipase or symptomatic pancreatitis, ponatinib should be held and restarted at a lower dose after resolution of symptoms.7
In the event of neutropenia or thrombocytopenia, ponatinib should be held until blood count recovery and then restarted at the same dose. If cytopenias occur for a second time, the dose of ponatinib should be lowered at the time of treatment reinitiation. If rash occurs, it can be addressed with topical or systemic steroids as well as dose reduction, interruption, or discontinuation.7
Case Conclusion
Given the patient's high-risk Sokal score, ideal first-line treatment is a second-generation TKI in order to increase the likelihood of achieving the desired treatment milestones and improving long-term outcomes. Her history of uncontrolled diabetes and coronary artery disease raises concerns for using nilotinib. Furthermore, her history of COPD makes dasatinib suboptimal because she would have little pulmonary reserve if she were to develop a pleural effusion. For this reason, bosutinib 400 mg daily is chosen as her first-line TKI. Shortly after starting bosutinib, she experiences diarrhea that occurs approximately 3 or 4 times daily during the first week on treatment. She is able to manage this with over-the-counter loperamide and the diarrhea resolves shortly thereafter.
After 3 months of bosutinib therapy, quantitative real-time PCR (RQ-PCR) assay on peripheral blood is done to measure BCR-ABL1 transcripts, and the result is reported at 1.2% IS. This indicates that the patient has achieved an early molecular response, which is defined as a RQ-PCR value of ≤10% IS. She undergoes RQ-PCR monitoring every 3 months, and at 12 months her results indicate a value of 0.07% IS, suggesting she has achieved a MMR.
Conclusion
With the development of imatinib and the subsequent TKIs, dasatinib, nilotinib, bosutinib, and ponatinib, CP-CML has become a chronic disease with a life-expectancy that is similar to the general population. Given the successful treatments available for these patients, it is crucial to identify patients with this diagnosis, ensure they receive a complete, appropriate diagnostic workup including a bone marrow biopsy and aspiration with cytogenetic testing, and select the best therapy for each individual patient. Once on treatment, the importance of frequent monitoring cannot be overstated. This is the only way to be certain patients are achieving the desired treatment milestones that correlate with the favorable long-term outcomes that have been observed with TKI-based treatment of CP-CML.
1. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
2. O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
3. Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917-927.
4. Baccarani M, Druker BJ, Branford S, et al. Long-term response to imatinib is not affected by the initial dose in patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: final update from the Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) study. Int J Hematol. 2014;99:616-624.
5. Yeung DT, Osborn MP, White DL, et al. TIDEL-II: first-line use of imatinib in CML with early switch to nilotinib for failure to achieve time-dependent molecular targets. Blood. 2015;125:915-923.
6. Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
7. Radich JP, Deininger M, Abboud CN, et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:1108-1135.
8. Shah NP, Rousselot P, Schiffer C, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. 2016;91:869-874.
9. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients trial. J Clin Oncol. 2016;34:2333-3340.
10. Quintas-Cardama A, Han X, Kantarjian H, Cortes J. Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood. 2009;114:261-263.
11. Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27:107-112.
12. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251-2259.
13. Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044-1054.
14. Cortes JE, Khoury HJ, Kantarjian HM, et al. Long-term bosutinib for chronic phase chronic myeloid leukemia after failure of imatinib plus dasatinib and/or nilotinib. Am J Hematol. 2016;91:1206-1214.
15. Gambacorti-Passerini C, Cortes JE, Lipton JH, et al. Safety and efficacy of second-line bosutinib for chronic phase chronic myeloid leukemia over a five-year period: final results of a phase I/II study. Haematologica. 2018;103:1298-1307.
16. Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30:3486-3492.
17. Cortes JE, Gambacorti-Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol. 2018;36:231-237.
18. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783-1796.
19. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132:393-404.
1. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031-1037.
2. O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994-1004.
3. Hochhaus A, Larson RA, Guilhot F, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917-927.
4. Baccarani M, Druker BJ, Branford S, et al. Long-term response to imatinib is not affected by the initial dose in patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: final update from the Tyrosine Kinase Inhibitor Optimization and Selectivity (TOPS) study. Int J Hematol. 2014;99:616-624.
5. Yeung DT, Osborn MP, White DL, et al. TIDEL-II: first-line use of imatinib in CML with early switch to nilotinib for failure to achieve time-dependent molecular targets. Blood. 2015;125:915-923.
6. Druker BJ, Guilhot F, O'Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408-2417.
7. Radich JP, Deininger M, Abboud CN, et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16:1108-1135.
8. Shah NP, Rousselot P, Schiffer C, et al. Dasatinib in imatinib-resistant or -intolerant chronic-phase, chronic myeloid leukemia patients: 7-year follow-up of study CA180-034. Am J Hematol. 2016;91:869-874.
9. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DASISION: the Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients trial. J Clin Oncol. 2016;34:2333-3340.
10. Quintas-Cardama A, Han X, Kantarjian H, Cortes J. Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood. 2009;114:261-263.
11. Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27:107-112.
12. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251-2259.
13. Hochhaus A, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044-1054.
14. Cortes JE, Khoury HJ, Kantarjian HM, et al. Long-term bosutinib for chronic phase chronic myeloid leukemia after failure of imatinib plus dasatinib and/or nilotinib. Am J Hematol. 2016;91:1206-1214.
15. Gambacorti-Passerini C, Cortes JE, Lipton JH, et al. Safety and efficacy of second-line bosutinib for chronic phase chronic myeloid leukemia over a five-year period: final results of a phase I/II study. Haematologica. 2018;103:1298-1307.
16. Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30:3486-3492.
17. Cortes JE, Gambacorti-Passerini C, Deininger MW, et al. Bosutinib versus imatinib for newly diagnosed chronic myeloid leukemia: results from the randomized BFORE trial. J Clin Oncol. 2018;36:231-237.
18. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783-1796.
19. Cortes JE, Kim DW, Pinilla-Ibarz J, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132:393-404.