User login
Low-calorie diet linked to improved chemo response in leukemia
Children and adolescents with leukemia who were placed on a restrictive diet and exercise regimen concurrent with starting chemotherapy showed responses to treatment that were better than those historically seen in such patients.
This apparently improved response suggests it is possible to boost treatment efficacy without raising the dose – or toxicity – of chemotherapy.
“To our knowledge, this is the first study in any hematologic malignancy to demonstrate potential benefit from caloric restriction via diet and exercise to augment chemotherapy efficacy and improve disease response, the authors reported.
The findings come from the IDEAL pilot trial, conducted in 40 young patients (mean age, 15 years; range, 10-21 years) diagnosed with high-risk B-cell acute lymphoblastic leukemia (B-ALL).
The study was published online April 1 in Blood Advances.
The diet and exercise regimen is a departure from current recommendations for patients with leukemia.
“This was a major paradigm shift – until now, many oncologists encouraged ‘comfort foods’ and increased calories to get through the rigor of chemotherapy,” first author Etan Orgel, MD, of Children’s Hospital Los Angeles and the University of Southern California, also in Los Angeles.
The results from this pilot trial suggest that “the era of encouraging comfort food should be in the past; over-nutrition is likely harmful, and diet and exercise are important tools to harness during chemotherapy,” he said.
Dr. Orgel added that childhood ALL was selected because it is the most common cancer of childhood, but the findings could have potential relevance in other cancer types in children as well as adults.
Commenting on the study, Patrick Brown, MD, director of the pediatric leukemia program at Johns Hopkins University, Baltimore, said the findings are important, albeit preliminary.
“I think the most important contribution of this pilot study is to show that it is possible to change the nutrition and exercise habits of children and adolescents during the initial month of treatment for ALL,” he said in an interview.
“We have to be cautious about the preliminary finding that these changes resulted in deeper remissions – this will need to be confirmed in a larger study,” added Dr. Brown, who was not involved with the research.
Dr. Orgel noted that a prospective, randomized trial, IDEAL-2, is launching later this year to further evaluate the intervention.
Obesity linked to poorer chemotherapy response
Among children and adolescents who start treatment for B-ALL, as many as 40% are overweight or obese, noted the study authors.
Those who are obese have more than a twofold greater risk of having persistent minimal residual disease (MRD) at the end of chemotherapy, considered the strongest patient-level predictor of poor outcome and a common guide for therapy intensification.
The problem is compounded by weight gain that is common during treatment as a result of prolonged chemotherapy and sedentary behavior, they commented.
With studies of obese mice linking calorie and fat restriction to improved survival after chemotherapy, the authors theorized that a calorie- and fat-restrictive diet and exercise could help improve outcomes after chemotherapy in humans.
Participants were enrolled at Children’s Hospital Los Angeles and City of Hope National Medical Center in nearby Duarte. After they were started on chemotherapy, they were placed on a low-carb, low-fat, and low-sugar diet tailored to patient needs and preferences, as well as a moderate daily exercise regimen, and continued on this regimen throughout the 4-week induction phase.
Following the intervention, there were no significant reductions observed in median gain of fat mass at the end of the intervention, compared with baseline (P = .13). However, in the subgroup of patients who were overweight or obese at baseline, the reduction in fat mass was indeed significant versus baseline (+1.5% vs. +9.7% at baseline; P = .02).
Importantly, after adjustment for prognostic factors, adherence to the intervention was associated with a significant reduction in the risk of MRD, compared with recent historical controls who received the same induction therapy at the same institution, but no intervention (odds ratio, 0.30; P = .02).
The intervention was also associated with a lower detectable MRD, compared with the historical controls (OR, 0.16; one-sided P = .002).
“Most importantly, the IDEAL intervention reduced risk of MRD at the end of induction in all patients, irrespective of starting [body mass index] and after accounting for prognostic features,” the authors noted.
Adherence to diet high, exercise low
As many as 82% of study participants achieved the goal of 20% or more caloric deficit throughout the chemotherapy.
“Adherence to the diet was excellent, with caloric deficits and macronutrient goals achieved in nearly all patients, including in the lean group,” the authors reported.
Dr. Orgel added that families embraced the chance to play an active role in the cancer therapy. “In our view, they couldn’t control their disease or their chemotherapy, but this, they could,” he said.
Conversely, adherence to the prescribed exercise was low – just 31.2%, with the inactivity during the first month likely contributed to the similar loss of muscle mass that occurred in both cohorts, Dr. Orgel noted.
“The [low exercise adherence] unfortunately was not a surprise, as it is often difficult to exercise and be active during chemotherapy,” he said.
Key aspects of physical activity will be refined in further studies, Dr. Orgel added.
Insulin sensitivity, adiponectin key factors?
Patients receiving the intervention showed improved insulin sensitivity and reductions in circulating insulin, which are notable in that insulin has been linked to mechanisms that counter chemoresistance, the authors noted.
Furthermore, the decreases in insulin were accompanied by notable elevations in circulating adiponectin, a protein hormone produced and secreted by fat cells.
“Adiponectin was certainly a surprise, as until now it did not appear to play a major role in cancer cell resistance to chemotherapy,” Dr. Orgel said.
“It is too soon to say they are central to the mechanism of the intervention, but the large differences in adiponectin and insulin sensitivity found in children in the trial have definitely highlighted these as important for future study,” he added.
Dr. Orgel, the study coauthors, and Dr. Brown disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Children and adolescents with leukemia who were placed on a restrictive diet and exercise regimen concurrent with starting chemotherapy showed responses to treatment that were better than those historically seen in such patients.
This apparently improved response suggests it is possible to boost treatment efficacy without raising the dose – or toxicity – of chemotherapy.
“To our knowledge, this is the first study in any hematologic malignancy to demonstrate potential benefit from caloric restriction via diet and exercise to augment chemotherapy efficacy and improve disease response, the authors reported.
The findings come from the IDEAL pilot trial, conducted in 40 young patients (mean age, 15 years; range, 10-21 years) diagnosed with high-risk B-cell acute lymphoblastic leukemia (B-ALL).
The study was published online April 1 in Blood Advances.
The diet and exercise regimen is a departure from current recommendations for patients with leukemia.
“This was a major paradigm shift – until now, many oncologists encouraged ‘comfort foods’ and increased calories to get through the rigor of chemotherapy,” first author Etan Orgel, MD, of Children’s Hospital Los Angeles and the University of Southern California, also in Los Angeles.
The results from this pilot trial suggest that “the era of encouraging comfort food should be in the past; over-nutrition is likely harmful, and diet and exercise are important tools to harness during chemotherapy,” he said.
Dr. Orgel added that childhood ALL was selected because it is the most common cancer of childhood, but the findings could have potential relevance in other cancer types in children as well as adults.
Commenting on the study, Patrick Brown, MD, director of the pediatric leukemia program at Johns Hopkins University, Baltimore, said the findings are important, albeit preliminary.
“I think the most important contribution of this pilot study is to show that it is possible to change the nutrition and exercise habits of children and adolescents during the initial month of treatment for ALL,” he said in an interview.
“We have to be cautious about the preliminary finding that these changes resulted in deeper remissions – this will need to be confirmed in a larger study,” added Dr. Brown, who was not involved with the research.
Dr. Orgel noted that a prospective, randomized trial, IDEAL-2, is launching later this year to further evaluate the intervention.
Obesity linked to poorer chemotherapy response
Among children and adolescents who start treatment for B-ALL, as many as 40% are overweight or obese, noted the study authors.
Those who are obese have more than a twofold greater risk of having persistent minimal residual disease (MRD) at the end of chemotherapy, considered the strongest patient-level predictor of poor outcome and a common guide for therapy intensification.
The problem is compounded by weight gain that is common during treatment as a result of prolonged chemotherapy and sedentary behavior, they commented.
With studies of obese mice linking calorie and fat restriction to improved survival after chemotherapy, the authors theorized that a calorie- and fat-restrictive diet and exercise could help improve outcomes after chemotherapy in humans.
Participants were enrolled at Children’s Hospital Los Angeles and City of Hope National Medical Center in nearby Duarte. After they were started on chemotherapy, they were placed on a low-carb, low-fat, and low-sugar diet tailored to patient needs and preferences, as well as a moderate daily exercise regimen, and continued on this regimen throughout the 4-week induction phase.
Following the intervention, there were no significant reductions observed in median gain of fat mass at the end of the intervention, compared with baseline (P = .13). However, in the subgroup of patients who were overweight or obese at baseline, the reduction in fat mass was indeed significant versus baseline (+1.5% vs. +9.7% at baseline; P = .02).
Importantly, after adjustment for prognostic factors, adherence to the intervention was associated with a significant reduction in the risk of MRD, compared with recent historical controls who received the same induction therapy at the same institution, but no intervention (odds ratio, 0.30; P = .02).
The intervention was also associated with a lower detectable MRD, compared with the historical controls (OR, 0.16; one-sided P = .002).
“Most importantly, the IDEAL intervention reduced risk of MRD at the end of induction in all patients, irrespective of starting [body mass index] and after accounting for prognostic features,” the authors noted.
Adherence to diet high, exercise low
As many as 82% of study participants achieved the goal of 20% or more caloric deficit throughout the chemotherapy.
“Adherence to the diet was excellent, with caloric deficits and macronutrient goals achieved in nearly all patients, including in the lean group,” the authors reported.
Dr. Orgel added that families embraced the chance to play an active role in the cancer therapy. “In our view, they couldn’t control their disease or their chemotherapy, but this, they could,” he said.
Conversely, adherence to the prescribed exercise was low – just 31.2%, with the inactivity during the first month likely contributed to the similar loss of muscle mass that occurred in both cohorts, Dr. Orgel noted.
“The [low exercise adherence] unfortunately was not a surprise, as it is often difficult to exercise and be active during chemotherapy,” he said.
Key aspects of physical activity will be refined in further studies, Dr. Orgel added.
Insulin sensitivity, adiponectin key factors?
Patients receiving the intervention showed improved insulin sensitivity and reductions in circulating insulin, which are notable in that insulin has been linked to mechanisms that counter chemoresistance, the authors noted.
Furthermore, the decreases in insulin were accompanied by notable elevations in circulating adiponectin, a protein hormone produced and secreted by fat cells.
“Adiponectin was certainly a surprise, as until now it did not appear to play a major role in cancer cell resistance to chemotherapy,” Dr. Orgel said.
“It is too soon to say they are central to the mechanism of the intervention, but the large differences in adiponectin and insulin sensitivity found in children in the trial have definitely highlighted these as important for future study,” he added.
Dr. Orgel, the study coauthors, and Dr. Brown disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Children and adolescents with leukemia who were placed on a restrictive diet and exercise regimen concurrent with starting chemotherapy showed responses to treatment that were better than those historically seen in such patients.
This apparently improved response suggests it is possible to boost treatment efficacy without raising the dose – or toxicity – of chemotherapy.
“To our knowledge, this is the first study in any hematologic malignancy to demonstrate potential benefit from caloric restriction via diet and exercise to augment chemotherapy efficacy and improve disease response, the authors reported.
The findings come from the IDEAL pilot trial, conducted in 40 young patients (mean age, 15 years; range, 10-21 years) diagnosed with high-risk B-cell acute lymphoblastic leukemia (B-ALL).
The study was published online April 1 in Blood Advances.
The diet and exercise regimen is a departure from current recommendations for patients with leukemia.
“This was a major paradigm shift – until now, many oncologists encouraged ‘comfort foods’ and increased calories to get through the rigor of chemotherapy,” first author Etan Orgel, MD, of Children’s Hospital Los Angeles and the University of Southern California, also in Los Angeles.
The results from this pilot trial suggest that “the era of encouraging comfort food should be in the past; over-nutrition is likely harmful, and diet and exercise are important tools to harness during chemotherapy,” he said.
Dr. Orgel added that childhood ALL was selected because it is the most common cancer of childhood, but the findings could have potential relevance in other cancer types in children as well as adults.
Commenting on the study, Patrick Brown, MD, director of the pediatric leukemia program at Johns Hopkins University, Baltimore, said the findings are important, albeit preliminary.
“I think the most important contribution of this pilot study is to show that it is possible to change the nutrition and exercise habits of children and adolescents during the initial month of treatment for ALL,” he said in an interview.
“We have to be cautious about the preliminary finding that these changes resulted in deeper remissions – this will need to be confirmed in a larger study,” added Dr. Brown, who was not involved with the research.
Dr. Orgel noted that a prospective, randomized trial, IDEAL-2, is launching later this year to further evaluate the intervention.
Obesity linked to poorer chemotherapy response
Among children and adolescents who start treatment for B-ALL, as many as 40% are overweight or obese, noted the study authors.
Those who are obese have more than a twofold greater risk of having persistent minimal residual disease (MRD) at the end of chemotherapy, considered the strongest patient-level predictor of poor outcome and a common guide for therapy intensification.
The problem is compounded by weight gain that is common during treatment as a result of prolonged chemotherapy and sedentary behavior, they commented.
With studies of obese mice linking calorie and fat restriction to improved survival after chemotherapy, the authors theorized that a calorie- and fat-restrictive diet and exercise could help improve outcomes after chemotherapy in humans.
Participants were enrolled at Children’s Hospital Los Angeles and City of Hope National Medical Center in nearby Duarte. After they were started on chemotherapy, they were placed on a low-carb, low-fat, and low-sugar diet tailored to patient needs and preferences, as well as a moderate daily exercise regimen, and continued on this regimen throughout the 4-week induction phase.
Following the intervention, there were no significant reductions observed in median gain of fat mass at the end of the intervention, compared with baseline (P = .13). However, in the subgroup of patients who were overweight or obese at baseline, the reduction in fat mass was indeed significant versus baseline (+1.5% vs. +9.7% at baseline; P = .02).
Importantly, after adjustment for prognostic factors, adherence to the intervention was associated with a significant reduction in the risk of MRD, compared with recent historical controls who received the same induction therapy at the same institution, but no intervention (odds ratio, 0.30; P = .02).
The intervention was also associated with a lower detectable MRD, compared with the historical controls (OR, 0.16; one-sided P = .002).
“Most importantly, the IDEAL intervention reduced risk of MRD at the end of induction in all patients, irrespective of starting [body mass index] and after accounting for prognostic features,” the authors noted.
Adherence to diet high, exercise low
As many as 82% of study participants achieved the goal of 20% or more caloric deficit throughout the chemotherapy.
“Adherence to the diet was excellent, with caloric deficits and macronutrient goals achieved in nearly all patients, including in the lean group,” the authors reported.
Dr. Orgel added that families embraced the chance to play an active role in the cancer therapy. “In our view, they couldn’t control their disease or their chemotherapy, but this, they could,” he said.
Conversely, adherence to the prescribed exercise was low – just 31.2%, with the inactivity during the first month likely contributed to the similar loss of muscle mass that occurred in both cohorts, Dr. Orgel noted.
“The [low exercise adherence] unfortunately was not a surprise, as it is often difficult to exercise and be active during chemotherapy,” he said.
Key aspects of physical activity will be refined in further studies, Dr. Orgel added.
Insulin sensitivity, adiponectin key factors?
Patients receiving the intervention showed improved insulin sensitivity and reductions in circulating insulin, which are notable in that insulin has been linked to mechanisms that counter chemoresistance, the authors noted.
Furthermore, the decreases in insulin were accompanied by notable elevations in circulating adiponectin, a protein hormone produced and secreted by fat cells.
“Adiponectin was certainly a surprise, as until now it did not appear to play a major role in cancer cell resistance to chemotherapy,” Dr. Orgel said.
“It is too soon to say they are central to the mechanism of the intervention, but the large differences in adiponectin and insulin sensitivity found in children in the trial have definitely highlighted these as important for future study,” he added.
Dr. Orgel, the study coauthors, and Dr. Brown disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Cancer screening stopped by pandemic: Repercussions to come?
Last year, cancer screening programs around the world ground to a halt as SARS-CoV-2 infection rates surged globally. The effect of this slowdown is now becoming clear.
Thousands of cancer diagnoses are “missing,” and oncologists worry that this will lead to more advanced cancers and higher mortality for years to come.
“I feel like this is an earthquake that’s rocked our health care system. My guess is that you’ll probably still see repercussions of this over the next couple of years at least,” said Sharon Chang, MD, an attending surgical oncologist in the Permanente Medical Group, Fremont, Calif.
She was senior author of a study that analyzed the effects of the slowdown in mammography screening as a result of California’s “shelter-in-place” order on March 17, 2020. In the 2 months that followed, there were 64% fewer breast cancer diagnoses at 21 Kaiser Permanente medical centers, compared with the same period in 2019 (250 vs. 703).
In effect, approximately 450 breast cancer patients had “disappeared,” said coauthor Annie Tang, MD, a research fellow at the University of California, San Francisco, East Bay surgery program.
“What surprised me most from our data was the sheer number of breast cancer patients that were missing,” Dr. Tang said in an interview.
A similar picture has emerged elsewhere.
In Boston, an estimated 1,438 cancerous and precancerous lesions “went missing” during the first 3 months of pandemic shutdown, according to a study from the Massachusetts General Brigham health care system.
In this study, the investigators assessed screening rates for five cancers – breast cancer (mammography), prostate cancer (prostate-specific antigen testing), colorectal cancer (colonoscopy), cervical cancer (Papanicolaou tests), and lung cancer (low-dose CT).
Screening rates during the first peak of the pandemic (March 2 to June 2, 2020) were compared with those during the preceding and following 3 months and during the same 3 months in 2019.
The results showed a pronounced drop in screening rates during the peak pandemic period, compared with the three control periods. Decreases occurred for all screening tests and ranged from –60% to –82%.
There were also significant decreases in cancer diagnoses resulting from the decreases in screening tests, ranging from –19% to –78%.
“Quantifying the actual problem made us realize how much work needs to be done to get us back to prepandemic numbers,” said senior author Quoc-Dien Trinh, MD, FACS, codirector of the Dana Farber/Brigham and Women’s prostate cancer program.
In the Canadian province of Alberta, a similar decrease in cancer diagnoses occurred during the early days of the pandemic.
By the end of 2020, Alberta was “missing” approximately 2,000 cases of invasive cancers and 1,000 cases of noninvasive cancers, Doug Stewart, MD, senior medical director at the Cancer Strategic Clinical Network (SCN) of Alberta Health Services, told this news organization.
Dr. Stewart is able to track cancer diagnoses in Alberta almost in real time through a mandatory cancer registry. Within a month of shutdown, there was a 30% decrease in diagnoses of invasive cancers and a 50% decrease “in the kind of preinvasive cancers that, for the most part, are picked up by screening programs,” said Dr. Stewart.
After the health care system opened up again in the summer, Stewart said, noninvasive cancer diagnoses continued to be 20% lower than expected. There was a 10% shortfall in invasive cancer diagnoses.
The number of diagnoses had returned to normal by December 2020. However, Dr. Stewart is worried that this fact conceals a terrible truth.
The worry is over the backlog. Although the number of diagnoses is now similar to what it was before the pandemic, “people are presenting later, and maybe the cancer is more advanced,” he speculated.
His team at Alberta Health Services is assessing whether the cancers that are being diagnosed now are more advanced. Initial results are anticipated by late April 2021.
In the United Kingdom, there was a similar halt in cancer screening as a result of the country’s lockdown. Researchers now predict an uptick in cancer diagnoses.
Ajay Aggarwal, MD, PhD, consultant clinical oncologist and associate professor at the London School of Hygiene and Tropical Medicine, and colleagues have estimated that at least 3,500 deaths from breast, colorectal, esophageal, and lung cancer will occur during the next 5 years in England that could have been avoided had it not been for the lockdown measures necessitated by the pandemic.
Speaking to this news organization, Dr. Aggarwal warned that these numbers, which are from a modeling study published in August 2020, are “extremely conservative,” because the investigators considered diagnostic delays over only a 3-month period, the analysis involved only four cancers, and it did not reflect deferral of cancer treatment.
“It felt like it was the tip of the iceberg,” Dr. Aggarwal said. He warns that more recent data suggest that “diagnostic delays are probably worse than we predicted.”
He suspects that there is more at play than screening cancellations.
In another study conducted in the United Kingdom, data show “a falling edge of referrals” from primary care to cancer centers early in the pandemic. In that study, investigators analyzed real-time weekly hospital data from eight large British hospitals and found that urgent cancer referrals fell 70% at their lowest point.
“It really surprised me that the urgent referrals dropped so drastically,” said lead author Alvina Lai, PhD, a lecturer in health data analytics at University College London.
She attributed this in part to patients’ adherence to lockdown rules. “Patients are trying to follow government guidelines to stay home and not go to [general practitioners] unless necessary,” Dr. Lai explained in an interview.
Canada, like the United Kingdom, has a publicly funded health care system. Dr. Stewart came to a similar conclusion. “Some patients who have been diagnosed with cancer ... have told me it took them an extra couple of months to even contact the family doc, because they ... didn’t want to bother the family doctor with something that wasn’t COVID, this kind of guilt. They want to do something good for society. You know, most people are just really nice people, and they don’t want to bother the health care system if they don’t have COVID,” Dr. Stewart said.
Shelley Fuld Nasso, CEO of the National Coalition for Cancer Survivorship, a nonprofit organization based in Silver Spring, Md., agreed that screening shutdowns are not the only danger. “While we agree that screening is really important, we also want to make sure patients are following up with their physicians about symptoms that they have,” she said.
“Some of the speculation or concern about increased mortality for cancer is related to screening, but some of it is related to delayed diagnosis because of not following up on symptoms. ... What concerns me is not everyone has that ability or willingness to advocate for themselves,” she said.
Speaking at a press briefing held by the American Society for Radiation Oncology on March 30, Dr. Nasso related a case involving a patient who experienced severe arm pain. In a teleconsultation with her primary care physician, her condition was diagnosed as arthritis. She was subsequently diagnosed in the ED as having multiple myeloma.
Patients who “feel fine” may postpone their checkups to avoid going to the hospital and risking exposure to COVID-19.
“Some patients are still hesitant about returning for their mammograms or coming in if they feel a breast lump,” Dr. Tang said. “That fear of COVID-19 is still out there, and we don’t know how long patients are going to delay.”
In London, Dr. Aggarwal saw a similar response to the pandemic. “People were overestimating quite significantly what their risk of death was from acquiring COVID-19, and I think that balance was never [redressed] explicitly,” he said.
Public health initiatives to rebalance the messaging are now underway.
Public Health England and National Health Service England launched their Help Us Help You campaign in October 2020. The public information campaign urges people to speak to their doctors if they were “worried about a symptom that could be cancer.”
In Canada, the provincial government in Alberta has launched a public awareness campaign that conveys the message, “cancer has not gone away.”
“Cancer is still the No. 1 cause of potential life-years lost, despite COVID,” Dr. Stewart said. “We need to do what we can to make sure there’s no slippage in survival rates.”
Dr. Tang, Dr. Chang, Dr. Lai, Dr. Stewart, and Dr. Aggarwal have disclosed no relevant financial relationship. Dr. Trinh has received personal fees from Astellas, Bayer, and Janssen and grants from Intuitive Surgical.
A version of this article first appeared on Medscape.com.
Last year, cancer screening programs around the world ground to a halt as SARS-CoV-2 infection rates surged globally. The effect of this slowdown is now becoming clear.
Thousands of cancer diagnoses are “missing,” and oncologists worry that this will lead to more advanced cancers and higher mortality for years to come.
“I feel like this is an earthquake that’s rocked our health care system. My guess is that you’ll probably still see repercussions of this over the next couple of years at least,” said Sharon Chang, MD, an attending surgical oncologist in the Permanente Medical Group, Fremont, Calif.
She was senior author of a study that analyzed the effects of the slowdown in mammography screening as a result of California’s “shelter-in-place” order on March 17, 2020. In the 2 months that followed, there were 64% fewer breast cancer diagnoses at 21 Kaiser Permanente medical centers, compared with the same period in 2019 (250 vs. 703).
In effect, approximately 450 breast cancer patients had “disappeared,” said coauthor Annie Tang, MD, a research fellow at the University of California, San Francisco, East Bay surgery program.
“What surprised me most from our data was the sheer number of breast cancer patients that were missing,” Dr. Tang said in an interview.
A similar picture has emerged elsewhere.
In Boston, an estimated 1,438 cancerous and precancerous lesions “went missing” during the first 3 months of pandemic shutdown, according to a study from the Massachusetts General Brigham health care system.
In this study, the investigators assessed screening rates for five cancers – breast cancer (mammography), prostate cancer (prostate-specific antigen testing), colorectal cancer (colonoscopy), cervical cancer (Papanicolaou tests), and lung cancer (low-dose CT).
Screening rates during the first peak of the pandemic (March 2 to June 2, 2020) were compared with those during the preceding and following 3 months and during the same 3 months in 2019.
The results showed a pronounced drop in screening rates during the peak pandemic period, compared with the three control periods. Decreases occurred for all screening tests and ranged from –60% to –82%.
There were also significant decreases in cancer diagnoses resulting from the decreases in screening tests, ranging from –19% to –78%.
“Quantifying the actual problem made us realize how much work needs to be done to get us back to prepandemic numbers,” said senior author Quoc-Dien Trinh, MD, FACS, codirector of the Dana Farber/Brigham and Women’s prostate cancer program.
In the Canadian province of Alberta, a similar decrease in cancer diagnoses occurred during the early days of the pandemic.
By the end of 2020, Alberta was “missing” approximately 2,000 cases of invasive cancers and 1,000 cases of noninvasive cancers, Doug Stewart, MD, senior medical director at the Cancer Strategic Clinical Network (SCN) of Alberta Health Services, told this news organization.
Dr. Stewart is able to track cancer diagnoses in Alberta almost in real time through a mandatory cancer registry. Within a month of shutdown, there was a 30% decrease in diagnoses of invasive cancers and a 50% decrease “in the kind of preinvasive cancers that, for the most part, are picked up by screening programs,” said Dr. Stewart.
After the health care system opened up again in the summer, Stewart said, noninvasive cancer diagnoses continued to be 20% lower than expected. There was a 10% shortfall in invasive cancer diagnoses.
The number of diagnoses had returned to normal by December 2020. However, Dr. Stewart is worried that this fact conceals a terrible truth.
The worry is over the backlog. Although the number of diagnoses is now similar to what it was before the pandemic, “people are presenting later, and maybe the cancer is more advanced,” he speculated.
His team at Alberta Health Services is assessing whether the cancers that are being diagnosed now are more advanced. Initial results are anticipated by late April 2021.
In the United Kingdom, there was a similar halt in cancer screening as a result of the country’s lockdown. Researchers now predict an uptick in cancer diagnoses.
Ajay Aggarwal, MD, PhD, consultant clinical oncologist and associate professor at the London School of Hygiene and Tropical Medicine, and colleagues have estimated that at least 3,500 deaths from breast, colorectal, esophageal, and lung cancer will occur during the next 5 years in England that could have been avoided had it not been for the lockdown measures necessitated by the pandemic.
Speaking to this news organization, Dr. Aggarwal warned that these numbers, which are from a modeling study published in August 2020, are “extremely conservative,” because the investigators considered diagnostic delays over only a 3-month period, the analysis involved only four cancers, and it did not reflect deferral of cancer treatment.
“It felt like it was the tip of the iceberg,” Dr. Aggarwal said. He warns that more recent data suggest that “diagnostic delays are probably worse than we predicted.”
He suspects that there is more at play than screening cancellations.
In another study conducted in the United Kingdom, data show “a falling edge of referrals” from primary care to cancer centers early in the pandemic. In that study, investigators analyzed real-time weekly hospital data from eight large British hospitals and found that urgent cancer referrals fell 70% at their lowest point.
“It really surprised me that the urgent referrals dropped so drastically,” said lead author Alvina Lai, PhD, a lecturer in health data analytics at University College London.
She attributed this in part to patients’ adherence to lockdown rules. “Patients are trying to follow government guidelines to stay home and not go to [general practitioners] unless necessary,” Dr. Lai explained in an interview.
Canada, like the United Kingdom, has a publicly funded health care system. Dr. Stewart came to a similar conclusion. “Some patients who have been diagnosed with cancer ... have told me it took them an extra couple of months to even contact the family doc, because they ... didn’t want to bother the family doctor with something that wasn’t COVID, this kind of guilt. They want to do something good for society. You know, most people are just really nice people, and they don’t want to bother the health care system if they don’t have COVID,” Dr. Stewart said.
Shelley Fuld Nasso, CEO of the National Coalition for Cancer Survivorship, a nonprofit organization based in Silver Spring, Md., agreed that screening shutdowns are not the only danger. “While we agree that screening is really important, we also want to make sure patients are following up with their physicians about symptoms that they have,” she said.
“Some of the speculation or concern about increased mortality for cancer is related to screening, but some of it is related to delayed diagnosis because of not following up on symptoms. ... What concerns me is not everyone has that ability or willingness to advocate for themselves,” she said.
Speaking at a press briefing held by the American Society for Radiation Oncology on March 30, Dr. Nasso related a case involving a patient who experienced severe arm pain. In a teleconsultation with her primary care physician, her condition was diagnosed as arthritis. She was subsequently diagnosed in the ED as having multiple myeloma.
Patients who “feel fine” may postpone their checkups to avoid going to the hospital and risking exposure to COVID-19.
“Some patients are still hesitant about returning for their mammograms or coming in if they feel a breast lump,” Dr. Tang said. “That fear of COVID-19 is still out there, and we don’t know how long patients are going to delay.”
In London, Dr. Aggarwal saw a similar response to the pandemic. “People were overestimating quite significantly what their risk of death was from acquiring COVID-19, and I think that balance was never [redressed] explicitly,” he said.
Public health initiatives to rebalance the messaging are now underway.
Public Health England and National Health Service England launched their Help Us Help You campaign in October 2020. The public information campaign urges people to speak to their doctors if they were “worried about a symptom that could be cancer.”
In Canada, the provincial government in Alberta has launched a public awareness campaign that conveys the message, “cancer has not gone away.”
“Cancer is still the No. 1 cause of potential life-years lost, despite COVID,” Dr. Stewart said. “We need to do what we can to make sure there’s no slippage in survival rates.”
Dr. Tang, Dr. Chang, Dr. Lai, Dr. Stewart, and Dr. Aggarwal have disclosed no relevant financial relationship. Dr. Trinh has received personal fees from Astellas, Bayer, and Janssen and grants from Intuitive Surgical.
A version of this article first appeared on Medscape.com.
Last year, cancer screening programs around the world ground to a halt as SARS-CoV-2 infection rates surged globally. The effect of this slowdown is now becoming clear.
Thousands of cancer diagnoses are “missing,” and oncologists worry that this will lead to more advanced cancers and higher mortality for years to come.
“I feel like this is an earthquake that’s rocked our health care system. My guess is that you’ll probably still see repercussions of this over the next couple of years at least,” said Sharon Chang, MD, an attending surgical oncologist in the Permanente Medical Group, Fremont, Calif.
She was senior author of a study that analyzed the effects of the slowdown in mammography screening as a result of California’s “shelter-in-place” order on March 17, 2020. In the 2 months that followed, there were 64% fewer breast cancer diagnoses at 21 Kaiser Permanente medical centers, compared with the same period in 2019 (250 vs. 703).
In effect, approximately 450 breast cancer patients had “disappeared,” said coauthor Annie Tang, MD, a research fellow at the University of California, San Francisco, East Bay surgery program.
“What surprised me most from our data was the sheer number of breast cancer patients that were missing,” Dr. Tang said in an interview.
A similar picture has emerged elsewhere.
In Boston, an estimated 1,438 cancerous and precancerous lesions “went missing” during the first 3 months of pandemic shutdown, according to a study from the Massachusetts General Brigham health care system.
In this study, the investigators assessed screening rates for five cancers – breast cancer (mammography), prostate cancer (prostate-specific antigen testing), colorectal cancer (colonoscopy), cervical cancer (Papanicolaou tests), and lung cancer (low-dose CT).
Screening rates during the first peak of the pandemic (March 2 to June 2, 2020) were compared with those during the preceding and following 3 months and during the same 3 months in 2019.
The results showed a pronounced drop in screening rates during the peak pandemic period, compared with the three control periods. Decreases occurred for all screening tests and ranged from –60% to –82%.
There were also significant decreases in cancer diagnoses resulting from the decreases in screening tests, ranging from –19% to –78%.
“Quantifying the actual problem made us realize how much work needs to be done to get us back to prepandemic numbers,” said senior author Quoc-Dien Trinh, MD, FACS, codirector of the Dana Farber/Brigham and Women’s prostate cancer program.
In the Canadian province of Alberta, a similar decrease in cancer diagnoses occurred during the early days of the pandemic.
By the end of 2020, Alberta was “missing” approximately 2,000 cases of invasive cancers and 1,000 cases of noninvasive cancers, Doug Stewart, MD, senior medical director at the Cancer Strategic Clinical Network (SCN) of Alberta Health Services, told this news organization.
Dr. Stewart is able to track cancer diagnoses in Alberta almost in real time through a mandatory cancer registry. Within a month of shutdown, there was a 30% decrease in diagnoses of invasive cancers and a 50% decrease “in the kind of preinvasive cancers that, for the most part, are picked up by screening programs,” said Dr. Stewart.
After the health care system opened up again in the summer, Stewart said, noninvasive cancer diagnoses continued to be 20% lower than expected. There was a 10% shortfall in invasive cancer diagnoses.
The number of diagnoses had returned to normal by December 2020. However, Dr. Stewart is worried that this fact conceals a terrible truth.
The worry is over the backlog. Although the number of diagnoses is now similar to what it was before the pandemic, “people are presenting later, and maybe the cancer is more advanced,” he speculated.
His team at Alberta Health Services is assessing whether the cancers that are being diagnosed now are more advanced. Initial results are anticipated by late April 2021.
In the United Kingdom, there was a similar halt in cancer screening as a result of the country’s lockdown. Researchers now predict an uptick in cancer diagnoses.
Ajay Aggarwal, MD, PhD, consultant clinical oncologist and associate professor at the London School of Hygiene and Tropical Medicine, and colleagues have estimated that at least 3,500 deaths from breast, colorectal, esophageal, and lung cancer will occur during the next 5 years in England that could have been avoided had it not been for the lockdown measures necessitated by the pandemic.
Speaking to this news organization, Dr. Aggarwal warned that these numbers, which are from a modeling study published in August 2020, are “extremely conservative,” because the investigators considered diagnostic delays over only a 3-month period, the analysis involved only four cancers, and it did not reflect deferral of cancer treatment.
“It felt like it was the tip of the iceberg,” Dr. Aggarwal said. He warns that more recent data suggest that “diagnostic delays are probably worse than we predicted.”
He suspects that there is more at play than screening cancellations.
In another study conducted in the United Kingdom, data show “a falling edge of referrals” from primary care to cancer centers early in the pandemic. In that study, investigators analyzed real-time weekly hospital data from eight large British hospitals and found that urgent cancer referrals fell 70% at their lowest point.
“It really surprised me that the urgent referrals dropped so drastically,” said lead author Alvina Lai, PhD, a lecturer in health data analytics at University College London.
She attributed this in part to patients’ adherence to lockdown rules. “Patients are trying to follow government guidelines to stay home and not go to [general practitioners] unless necessary,” Dr. Lai explained in an interview.
Canada, like the United Kingdom, has a publicly funded health care system. Dr. Stewart came to a similar conclusion. “Some patients who have been diagnosed with cancer ... have told me it took them an extra couple of months to even contact the family doc, because they ... didn’t want to bother the family doctor with something that wasn’t COVID, this kind of guilt. They want to do something good for society. You know, most people are just really nice people, and they don’t want to bother the health care system if they don’t have COVID,” Dr. Stewart said.
Shelley Fuld Nasso, CEO of the National Coalition for Cancer Survivorship, a nonprofit organization based in Silver Spring, Md., agreed that screening shutdowns are not the only danger. “While we agree that screening is really important, we also want to make sure patients are following up with their physicians about symptoms that they have,” she said.
“Some of the speculation or concern about increased mortality for cancer is related to screening, but some of it is related to delayed diagnosis because of not following up on symptoms. ... What concerns me is not everyone has that ability or willingness to advocate for themselves,” she said.
Speaking at a press briefing held by the American Society for Radiation Oncology on March 30, Dr. Nasso related a case involving a patient who experienced severe arm pain. In a teleconsultation with her primary care physician, her condition was diagnosed as arthritis. She was subsequently diagnosed in the ED as having multiple myeloma.
Patients who “feel fine” may postpone their checkups to avoid going to the hospital and risking exposure to COVID-19.
“Some patients are still hesitant about returning for their mammograms or coming in if they feel a breast lump,” Dr. Tang said. “That fear of COVID-19 is still out there, and we don’t know how long patients are going to delay.”
In London, Dr. Aggarwal saw a similar response to the pandemic. “People were overestimating quite significantly what their risk of death was from acquiring COVID-19, and I think that balance was never [redressed] explicitly,” he said.
Public health initiatives to rebalance the messaging are now underway.
Public Health England and National Health Service England launched their Help Us Help You campaign in October 2020. The public information campaign urges people to speak to their doctors if they were “worried about a symptom that could be cancer.”
In Canada, the provincial government in Alberta has launched a public awareness campaign that conveys the message, “cancer has not gone away.”
“Cancer is still the No. 1 cause of potential life-years lost, despite COVID,” Dr. Stewart said. “We need to do what we can to make sure there’s no slippage in survival rates.”
Dr. Tang, Dr. Chang, Dr. Lai, Dr. Stewart, and Dr. Aggarwal have disclosed no relevant financial relationship. Dr. Trinh has received personal fees from Astellas, Bayer, and Janssen and grants from Intuitive Surgical.
A version of this article first appeared on Medscape.com.
VEXAS: A novel rheumatologic, hematologic syndrome that’s making waves
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
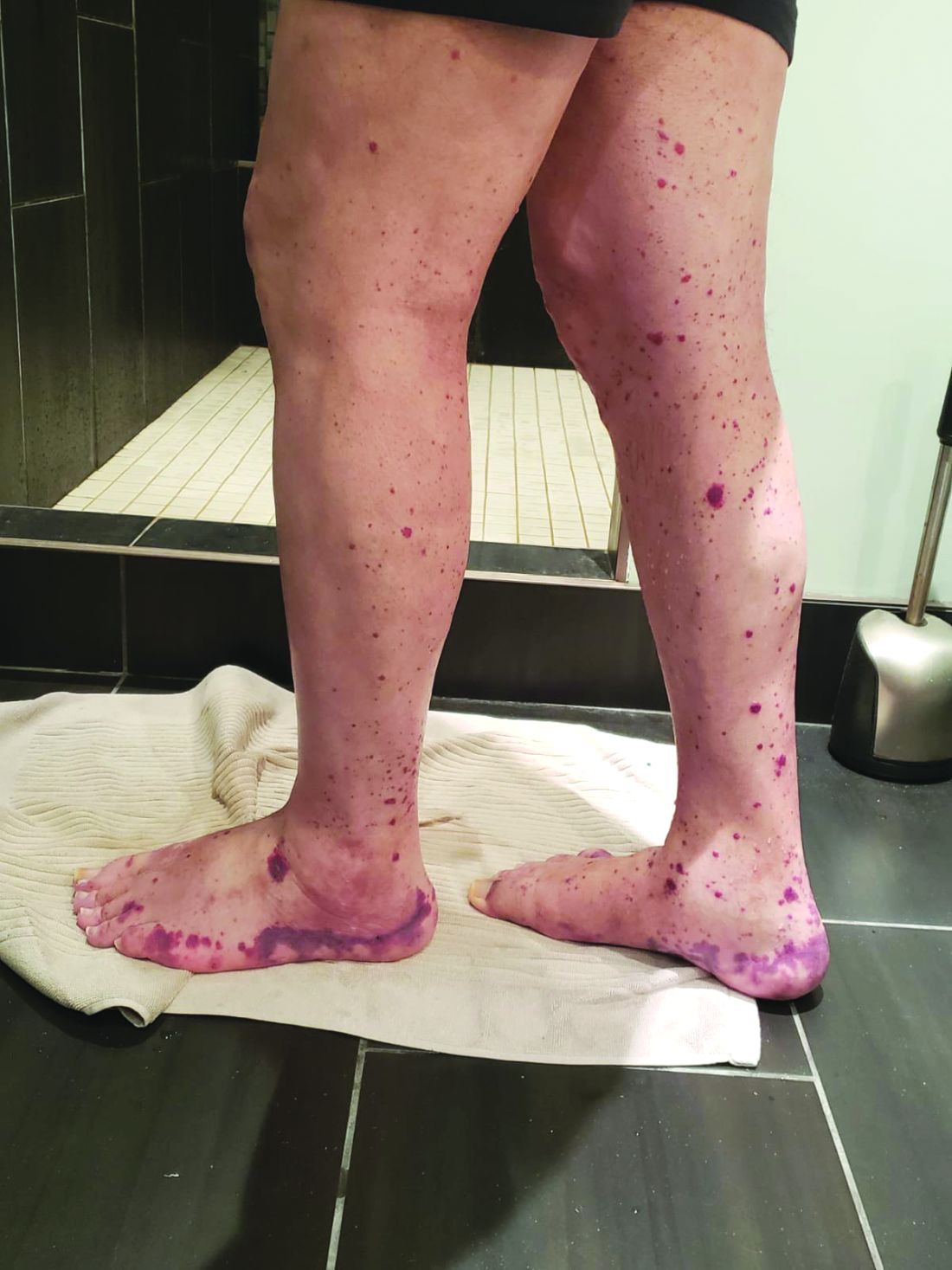
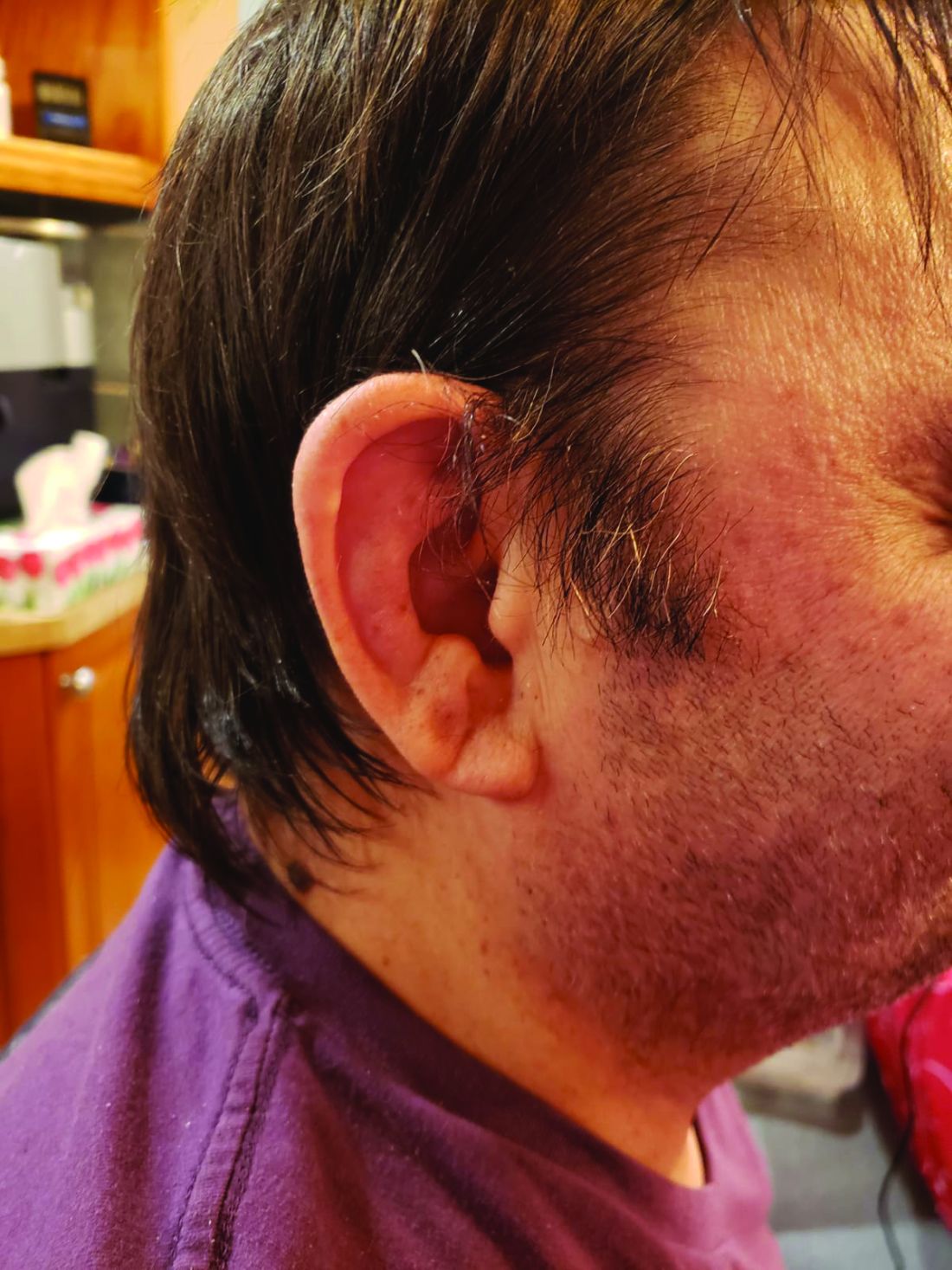
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
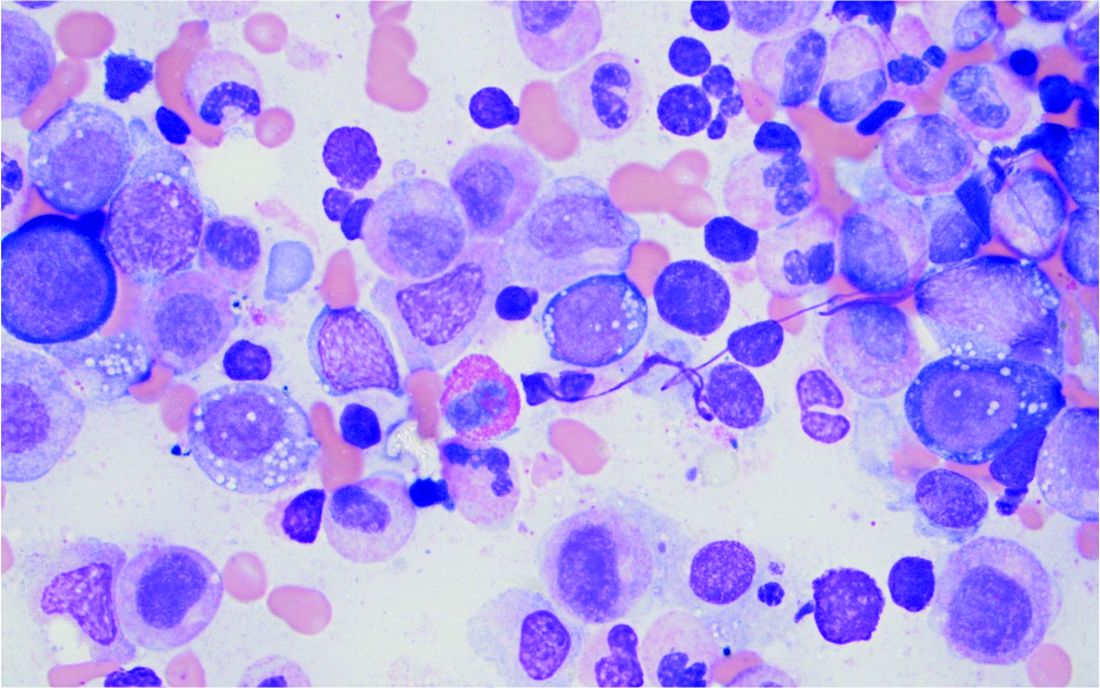
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Older men with a novel adult-onset, severe autoinflammatory syndrome known by the acronym VEXAS are likely hiding in plain sight in many adult rheumatology, hematology, and dermatology practices. New clinical features are being described to fill out the clinical profile of such patients who may be currently misdiagnosed with other conditions, according to researchers who first described the syndrome in the last quarter of 2020.
VEXAS is often misdiagnosed as treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, or giant cell arteritis. These seemingly unrelated disorders are actually tied together by a single thread recently unraveled by David B. Beck, MD, PhD, a clinical fellow at the National Human Genome Research Institute, and colleagues, including rheumatologist Marcela Ferrada, MD, and others at institutes of the National Institutes of Health, Bethesda, Md. The connection between these disparate clinical presentations lies in somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation. VEXAS appears primarily limited to men because the UBA1 gene lies on the X chromosome, although it may be possible for women to have it because of an acquired loss of X chromosome.
VEXAS is an acronym for:
- Vacuoles in bone marrow cells
- E-1 activating enzyme, which is what UBA1 encodes for
- X-linked
- Autoinflammatory
- Somatic mutation featuring hematologic mosaicism
Dr. Beck said that VEXAS is “probably affecting thousands of Americans,” but it is tough to say this early in the understanding of the disease. He estimated that the prevalence of VEXAS could be 1 per 20,000-30,000 individuals.
A new way of looking for disease
VEXAS has caused a major stir among geneticists because of the novel manner in which Dr. Beck and his coinvestigators made their discovery. Instead of starting out in the traditional path to discovery of a new genetic disease – that is, by looking for clinical similarities among patients with undiagnosed diseases and then conducting a search for a gene or genes that might explain the shared patient symptoms – the investigators took a genotype-first approach. They scanned the mapped genomic sequences of patients in the National Institutes of Health Undiagnosed Diseases Network, which led them to zero in on mutations in UBA1 as their top candidate.
“We targeted the ubiquitin-proteasome pathway, because it has been implicated in many autoinflammatory diseases – for example, HA20 [A20 haploinsufficiency] and CANDLE syndrome [Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature]. Many of these recurrent inflammatory diseases are caused by mutations within this pathway,” Dr. Beck said in an interview.
Next, they analyzed the genomes of patients in other NIH databases and patients from other study populations at the University College London and Leeds Teaching Hospitals NHS Trust in the United Kingdom in a search for UBA1 somatic mutations, eventually identifying 25 men with the shared features they called VEXAS. These 25 formed the basis for their initial report on the syndrome in the New England Journal of Medicine.
Most autoinflammatory diseases appear in childhood because they stem from germline mutations. VEXAS syndrome, because of somatic mutations with mosaicism, appears to manifest later in life: The median age of the initial 25-man cohort was 64 years, ranging from 45 to 80 years. It’s a severe disorder. By the time the investigators were preparing their paper for publication, 10 of the 25 patients, or 40%, had died.
“I think that somatic mutations may account for a significant percentage of severe. adult-onset rheumatologic diseases, and it may change the way we think about treating them based on having a genetic diagnosis,” Dr. Beck said.
“This approach could be expanded to look at other pathways we know are important in inflammation, or alternatively, it could be completely unbiased and look for any shared variation that occurs across undiagnosed patients with inflammatory diseases. I think that one thing that’s important about our study is that previously we had been looking for mutations that really in most cases were the same sort of germline mutations present in [pediatric] patients who have disease at early onset, but now we’re thinking about things differently. There may be a different type of genetics that drives adult-onset rheumatologic disease, and this would be somatic mutations which are not present in every cell of the body, just in the blood, and that’s why there’s just this blood-based disease.”
When to suspect VEXAS syndrome
Consider the possibility of VEXAS in middle-aged or older men in a rheumatology clinic with characteristics suggestive of treatment-refractory relapsing polychondritis, giant cell arteritis, polyarteritis nodosa, or Sweet syndrome. In the original series of 25 men, 15 were diagnosed with relapsing polychondritis, 8 with Sweet syndrome, 3 with polyarteritis nodosa, and 1 with giant cell arteritis.
Men with VEXAS often have periodic fevers, pulmonary infiltrates, a history of unprovoked venous thromboembolic events, neutrophilic dermatoses, and/or hematologic abnormalities such as myelodysplastic syndrome, multiple myeloma, or monoclonal gammopathy of unknown origin.
Bone marrow biopsy will show vacuoles in myeloid and erythroid precursor cells. Inflammatory marker levels are very high: In the NIH series, the median C-reactive protein was 73 mg/L and median erythrocyte sedimentation rate was 97 mm/hr. The diagnosis of VEXAS can be confirmed by genetic testing performed by Dr. Beck and his NIH coworkers ([email protected]).
In interviews, Dr. Beck and Dr. Ferrada emphasized that management of VEXAS requires a multidisciplinary team of clinicians including rheumatologists, hematologists, and dermatologists.
Dr. Ferrada said that rheumatologists could suspect VEXAS in patients who have very high inflammatory markers and do not have a clear diagnosis or do not meet all criteria for other rheumatologic diseases, particularly in older men, but it’s possible in younger men as well. Hematologists could also consider VEXAS in patients with macrocytic anemia or macrocytosis without an explanation and inflammatory features, she said.
Dr. Ferrada, Dr. Beck, and colleagues also published a study in Arthritis & Rheumatology that presents a useful clinical algorithm for deciding whether to order genetic screening for VEXAS in patients with relapsing polychondritis.
First off, Dr. Ferrada and colleagues performed whole-exome sequencing and testing for UBA1 variants in an observational cohort of 92 relapsing polychondritis patients to determine the prevalence of VEXAS, which turned out to be 8%. They added an additional 6 patients with relapsing polychondritis and VEXAS from other cohorts, for a total of 13. The investigators determined that patients with VEXAS were older at disease onset, and more likely to have fever, ear chondritis, DVT, pulmonary infiltrates, skin involvement, and periorbital edema. In contrast, the RP cohort had a significantly higher prevalence of airway chondritis, joint involvement, and vestibular symptoms.
Dr. Ferrada’s algorithm for picking out VEXAS in patients who meet diagnostic criteria for relapsing polychondritis is based upon a few simple factors readily apparent in screening patient charts: male sex; age at onset older than 50 years; macrocytic anemia; and thrombocytopenia. Those four variables, when present, identify VEXAS within an RP cohort with 100% sensitivity and 96% specificity. “As we learn more about [VEXAS] and how it presents earlier, I think we are going to be able to find different manifestations or laboratory data that are going to allow us to diagnose these patients earlier,” she said. “The whole role of that algorithm was to guide clinicians who see patients with relapsing polychondritis to test these patients for the mutation, but I think over time that is going to evolve.”
Researchers are taking similar approaches for other clinical diagnoses to see which should be referred for UBA1 testing, Dr. Beck said.
Myelodysplastic syndrome and hematologic abnormalities
While patients with both myelodysplastic syndrome and relapsing polychondritis have been known in the literature for many years, it’s not until now that researchers are seeing a connection between the two, Dr. Ferrada said.
A majority of the VEXAS patients in the NEJM study had a workup for myelodysplastic syndrome, but only 24% met criteria. However, many were within the spectrum of myelodysplastic disease and some did not meet criteria because their anemia was attributed to a rheumatologic diagnosis and they did not have a known genetic driver of myelodysplastic syndrome, Dr. Beck said. It also fits with this new evidence that UBA1 is probably a driver of myelodysplastic syndrome in and of itself, and that anemia and hematologic involvement are not secondary to the rheumatologic disease; they are linked to the same disease process.
Dr. Beck said that there may be a subset of patients who present with primarily hematologic manifestations, noting the NEJM study could have ascertainment bias because the researchers analyzed mainly patients presenting to their clinic with relapsing polychondritis and severe inflammation. NIH researchers also are still looking in their cohort for any association with hematologic malignancies that preceded clinical manifestations, he said.
More cases reported
As of early April, another 27 cases had been reported in the literature as more researchers have begun to look for patients with UBA1 mutations, some with additional presenting clinical features associated with VEXAS, including chronic progressive inflammatory arthritis, Kikuchi-Fujimoto disease, spondyloarthritis, and bacterial pneumonia.
“Many times with rare diseases, we can’t get enough patients to understand the full spectrum of the disease, but this disease seems to be far more common than we would have expected. We’re actually getting many referrals,” Dr. Beck said.
It appears so far that the range of somatic UBA1 mutations that have been discovered in VEXAS patients does make a difference in the severity of clinical presentation and could potentially be useful in prognosis, Dr. Beck said.
Right now, NIH researchers are asking patients about their natural clinical course, assessing disease activity, and determining which treatments get a response, with the ultimate goal of a treatment trial at the NIH.
Treatment
Developing better treatments for VEXAS syndrome is a priority. In the initial report on VEXAS, the researchers found that the only reliably effective therapy is high-dose corticosteroids. Dr. Ferrada said that NIH investigators have begun thinking about agents that target both the hematologic and inflammatory features of VEXAS. “Most patients get exposed to treatments that are targeted to decrease the inflammatory process, and some of these treatments help partially but not completely to decrease the amount of steroids that patients are taking. For example, one of the medications is tocilizumab. [It was used in] patients who had previous diagnosis of relapsing polychondritis, but they still had to take steroids and their hematologic manifestations keep progressing. We’re in the process of figuring out medications that may help in treating both.” Dr. Ferrada added that because the source of the mutation is in the bone marrow, transplantation may be an effective option.
Laboratory work to identify potential treatments for VEXAS in studies of model organisms could identify treatments outside of the classic anti-inflammatory agents, such as targeting certain cell types in the bone marrow or the ubiquitin-proteasome pathway, Dr. Beck said. “We think that however UBA1 works to initiate inflammation may be important not just in VEXAS but in other diseases. Rare diseases may be informing the mechanisms in common diseases.”
The VEXAS NEJM study was sponsored by the NIH Intramural Research Programs and by an EU Horizon 2020 Research and Innovation Program grant. Dr. Beck reported a patent pending on “Diagnosis and Treatment of VEXAS with Mosaic Missense Mutations in UBA1.”
Poor survival with COVID in patients who have had HSCT
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among individuals who have received a hematopoietic stem cell transplant (HSCT), often used in the treatment of blood cancers, rates of survival are poor for those who develop COVID-19.
The probability of survival 30 days after being diagnosed with COVID-19 is only 68% for persons who have received an allogeneic HSCT and 67% for autologous HSCT recipients, according to new data from the Center for International Blood and Marrow Transplant Research.
These findings underscore the need for “stringent surveillance and aggressive treatment measures” in this population, Akshay Sharma, MBBS, of St. Jude Children’s Research Hospital, Memphis, and colleagues wrote.
The findings were published online March 1, 2021, in The Lancet Haematology.
The study is “of importance for physicians caring for HSCT recipients worldwide,” Mathieu Leclerc, MD, and Sébastien Maury, MD, Hôpital Henri Mondor, Créteil, France, commented in an accompanying editorial.
Study details
For their study, Dr. Sharma and colleagues analyzed outcomes for all HSCT recipients who developed COVID-19 and whose cases were reported to the CIBMTR. Of 318 such patients, 184 had undergone allogeneic HSCT, and 134 had undergone autologous HSCT.
Overall, about half of these patients (49%) had mild COVID-19.
Severe COVID-19 that required mechanical ventilation developed in 15% and 13% of the allogeneic and autologous HSCT recipients, respectively.
About one-fifth of patients died: 22% and 19% of allogeneic and autologous HSCT recipients, respectively.
Factors associated with greater mortality risk included age of 50 years or older (hazard ratio, 2.53), male sex (HR, 3.53), and development of COVID-19 within 12 months of undergoing HSCT (HR, 2.67).
Among autologous HSCT recipients, lymphoma was associated with higher mortality risk in comparison with a plasma cell disorder or myeloma (HR, 2.41), the authors noted.
“Two important messages can be drawn from the results reported by Sharma and colleagues,” Dr. Leclerc and Dr. Maury wrote in their editorial. “The first is the confirmation that the prognosis of COVID-19 is particularly poor in HSCT recipients, and that its prevention, in the absence of any specific curative treatment with sufficient efficacy, should be at the forefront of concerns.”
The second relates to the risk factors for death among HSCT recipients who develop COVID-19. In addition to previously known risk factors, such as age and gender, the investigators identified transplant-specific factors potentially associated with prognosis – namely, the nearly threefold increase in death among allogeneic HSCT recipients who develop COVID-19 within 12 months of transplant, they explained.
However, the findings are limited by a substantial amount of missing data, short follow-up, and the possibility of selection bias, they noted.
“Further large and well-designed studies with longer follow-up are needed to confirm and refine the results,” the editorialists wrote.
“[A] better understanding of the distinctive features of COVID-19 infection in HSCT recipients will be a necessary and essential step toward improvement of the remarkably poor prognosis observed in this setting,” they added.
The study was funded by the American Society of Hematology; the Leukemia and Lymphoma Society; the National Cancer Institute; the National Heart, Lung and Blood Institute; the National Institute of Allergy and Infectious Diseases; the National Institutes of Health; the Health Resources and Services Administration; and the Office of Naval Research. Dr. Sharma receives support for the conduct of industry-sponsored trials from Vertex Pharmaceuticals, CRISPR Therapeutics, and Novartis and consulting fees from Spotlight Therapeutics. Dr. Leclerc and Dr. Maury disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Risk factors predict graft failure in pediatric acute leukemia patients
Researchers developed a predictive score for the risk of graft failure in patients with acute leukemia who underwent allogeneic hematopoietic stem cell transplantation (aHSCT) with ex vivo T-cell depletion. T-cell depletion is performed in an effort to prevent subsequent graft-versus-host disease (GVHD) after transplant.
The risk score was based on patient age and the T-lymphocyte population pre-aHSCT with 1 point of risk possible in each category. Patients with 1 point had a graft failure risk of 5% and 13% if they had 2 points, according to the results of the study presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Graft failure is a potentially severe complication in patients treated with aHSCT, but there are few studies analyzing risk factors when ex vivo T-cell depletion is used, Ivan López Torija of the Hospital Infantil Universitario Niño Jesús, Madrid, and colleagues noted in their presentation, which won the Best Young Poster Abstract Award at the meeting.
The researchers assessed 148 pediatric patients (64% boys) with acute leukemia who underwent allogeneic HSCT from haploidentical donors using ex vivo T-cell depletion between 2005 and 2020. About 53% of the patients were diagnosed with acute lymphoblastic leukemia, the rest with acute myeloid leukemia. The donor mean age was 40 years, and all transplant patients received toxicity reduction conditioning based on fludarabine busulfan and thiotepa.
Predictive results
Multivariate analysis showed that T-cell count (CD3+/CD8+ ≥ 350/mL: hazard ratio, 2,6; P = .01) and patient age (less than 9 years: HR; 5.0; P = .04) were associated with graft failure. A risk score was established using these results and based on patient age and T lymphocyte pre-aHSCT with 1 point each for each increased risk category. Patients with 1 point had a graft failure risk of 5% and a risk of 13% if they had 2 points.
However, in this particular population, with a mean follow up of 4 years, the overall survival rate was 60%, with no significant differences seen between patients that presented graft failure and those without graft failure.
“Patient age and pretransplant number of CD3+/CD8+ are associated with [graft failure] in pediatric patients with acute leukemia undergoing ex vivo T-cell–depleted haploidentical transplantation. These findings highlight the importance of preexisting cellular immunity in the transplant recipient and support T-cell population analysis as part of a pretransplant working program,” the researchers concluded.
The authors reported that they had no disclosures.
Researchers developed a predictive score for the risk of graft failure in patients with acute leukemia who underwent allogeneic hematopoietic stem cell transplantation (aHSCT) with ex vivo T-cell depletion. T-cell depletion is performed in an effort to prevent subsequent graft-versus-host disease (GVHD) after transplant.
The risk score was based on patient age and the T-lymphocyte population pre-aHSCT with 1 point of risk possible in each category. Patients with 1 point had a graft failure risk of 5% and 13% if they had 2 points, according to the results of the study presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Graft failure is a potentially severe complication in patients treated with aHSCT, but there are few studies analyzing risk factors when ex vivo T-cell depletion is used, Ivan López Torija of the Hospital Infantil Universitario Niño Jesús, Madrid, and colleagues noted in their presentation, which won the Best Young Poster Abstract Award at the meeting.
The researchers assessed 148 pediatric patients (64% boys) with acute leukemia who underwent allogeneic HSCT from haploidentical donors using ex vivo T-cell depletion between 2005 and 2020. About 53% of the patients were diagnosed with acute lymphoblastic leukemia, the rest with acute myeloid leukemia. The donor mean age was 40 years, and all transplant patients received toxicity reduction conditioning based on fludarabine busulfan and thiotepa.
Predictive results
Multivariate analysis showed that T-cell count (CD3+/CD8+ ≥ 350/mL: hazard ratio, 2,6; P = .01) and patient age (less than 9 years: HR; 5.0; P = .04) were associated with graft failure. A risk score was established using these results and based on patient age and T lymphocyte pre-aHSCT with 1 point each for each increased risk category. Patients with 1 point had a graft failure risk of 5% and a risk of 13% if they had 2 points.
However, in this particular population, with a mean follow up of 4 years, the overall survival rate was 60%, with no significant differences seen between patients that presented graft failure and those without graft failure.
“Patient age and pretransplant number of CD3+/CD8+ are associated with [graft failure] in pediatric patients with acute leukemia undergoing ex vivo T-cell–depleted haploidentical transplantation. These findings highlight the importance of preexisting cellular immunity in the transplant recipient and support T-cell population analysis as part of a pretransplant working program,” the researchers concluded.
The authors reported that they had no disclosures.
Researchers developed a predictive score for the risk of graft failure in patients with acute leukemia who underwent allogeneic hematopoietic stem cell transplantation (aHSCT) with ex vivo T-cell depletion. T-cell depletion is performed in an effort to prevent subsequent graft-versus-host disease (GVHD) after transplant.
The risk score was based on patient age and the T-lymphocyte population pre-aHSCT with 1 point of risk possible in each category. Patients with 1 point had a graft failure risk of 5% and 13% if they had 2 points, according to the results of the study presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Graft failure is a potentially severe complication in patients treated with aHSCT, but there are few studies analyzing risk factors when ex vivo T-cell depletion is used, Ivan López Torija of the Hospital Infantil Universitario Niño Jesús, Madrid, and colleagues noted in their presentation, which won the Best Young Poster Abstract Award at the meeting.
The researchers assessed 148 pediatric patients (64% boys) with acute leukemia who underwent allogeneic HSCT from haploidentical donors using ex vivo T-cell depletion between 2005 and 2020. About 53% of the patients were diagnosed with acute lymphoblastic leukemia, the rest with acute myeloid leukemia. The donor mean age was 40 years, and all transplant patients received toxicity reduction conditioning based on fludarabine busulfan and thiotepa.
Predictive results
Multivariate analysis showed that T-cell count (CD3+/CD8+ ≥ 350/mL: hazard ratio, 2,6; P = .01) and patient age (less than 9 years: HR; 5.0; P = .04) were associated with graft failure. A risk score was established using these results and based on patient age and T lymphocyte pre-aHSCT with 1 point each for each increased risk category. Patients with 1 point had a graft failure risk of 5% and a risk of 13% if they had 2 points.
However, in this particular population, with a mean follow up of 4 years, the overall survival rate was 60%, with no significant differences seen between patients that presented graft failure and those without graft failure.
“Patient age and pretransplant number of CD3+/CD8+ are associated with [graft failure] in pediatric patients with acute leukemia undergoing ex vivo T-cell–depleted haploidentical transplantation. These findings highlight the importance of preexisting cellular immunity in the transplant recipient and support T-cell population analysis as part of a pretransplant working program,” the researchers concluded.
The authors reported that they had no disclosures.
FROM EBMT 2021
Allo-HSCT plus monoclonal antibody treatment can improve survival in patients with r/r B-ALL
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
The use of allogeneic hematopoietic stem cell transplantation (allo-HSCT) can improve survival in minimal residual disease (MRD)–negative remission patients with relapsed/refractory (r/r) B-cell acute lymphoblastic leukemia (B-ALL) after the start of monoclonal antibody treatment, according to the results of a landmark analysis presented at the virtual meeting of the European Society for Blood and Marrow Transplantation.
Previous studies have indicated that allo-HSCT improves the results of treatment in r/r B-ALL patients, compared with chemotherapy alone. In addition, it has been found that the monoclonal antibodies (Mab), anti-CD19-blinatumomab and anti-CD22-inotuzumab ozogamicin, induced remission in a significant proportion of such patients.
To determine if the use of allo-HSCT improves the outcome of patients in MRD-negative remission with or without Mab treatment, researchers performed a landmark analysis of 110 patients who achieved MRD-negative status after Mab treatment. The analysis examined results at 2, 4, and 6 months subsequent to the initiation of Mab treatment, according to poster presentation by Inna V. Markova, MD, and colleagues at Pavlov University, Saint Petersburg, Russian Federation.
Study details
The researchers included 110 patients who achieved MRD-negative status outside of clinical trials at a single institution in the analysis. Forty of the patients (36%) were children and 70 (64%) were adults. The median age for all patients was 23 years and the median follow up was 24 months. Fifty-seven (52%) and 53 (48%) patients received Mab for hematological relapse and persistent measurable residual disease or for molecular relapse, respectively. Therapy with Mab alone without subsequent allo-HSCT was used in 36 (31%) patients (30 received blinatumomab and 6 received inotuzumab ozogamicin). A total of 74 (69%) patients received allo-HSCT from a matched related or unrelated donor (MD-HSCT, n = 38) or haploidentical donor (Haplo-HSCT, n = 36). All patients received posttransplantation cyclophosphamide (PTCY)–based graft-versus host disease (GVHD) prophylaxis. Landmark analysis was performed at 2, 4, and 6 months after Mab therapy initiation to determine the effect of allo-HSCT on the outcome and the optimal timing of HSCT. Overall survival and disease-free survival (DFS) were used as outcomes.
Promising results
No significant differences between the MD-HSCT, Mab alone, and Haplo-HSCT groups were observed in 2-month landmark analysis (P = .4 for OS and P =.65 for DFS). However, the 4-month landmark analysis demonstrated superior overall survival and DFS in patients after MD-HSCT, but not Haplo-HSCT, compared with Mab alone: 2-year OS was 75%, 50%, and 27,7% (P = .032) and DFS was 53.5%, 51.3%, and 16.6% (P = .02) for MD-HSCT, Mab alone and Haplo-HSCT groups, respectively. In addition, 6-month analysis showed that there was no benefit from subsequent transplantation, according to the authors, with regard to overall survival (P = .11).
“Our study demonstrated that at least MD-HSCT with PTCY platform improves survival in MRD-negative remission if performed during the first 4 months after Mab initiation. Haplo-HSCT or MD-HSCT beyond 4 months are not associated with improved outcomes in this groups of patients,” the researchers concluded.
The researchers reported they had no conflicts of interest to declare.
FROM EBMT 2021
Omidubicel improves on umbilical cord blood transplants
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Omidubicel, an investigational enriched umbilical cord blood product being developed by Gamida Cell for transplantation in patients with blood cancers, appears to have some advantages over standard umbilical cord blood.
The results come from a global phase 3 trial (NCT02730299) presented at the annual meeting of the European Society for Blood and Bone Marrow Transplantation.
“Transplantation with omidubicel, compared to standard cord blood transplantation, results in faster hematopoietic recovery, fewer infections, and fewer days in hospital,” said coinvestigator Guillermo F. Sanz, MD, PhD, from the Hospital Universitari i Politècnic la Fe in Valencia, Spain.
“Omidubicel should be considered as the new standard of care for patients eligible for umbilical cord blood transplantation,” Dr. Sanz concluded.
Zachariah DeFilipp, MD, from Mass General Cancer Center in Boston, a hematopoietic stem cell transplantation specialist who was not involved in the study, said in an interview that “omidubicel significantly improves the engraftment after transplant, as compared to standard cord blood transplant. For patients that lack an HLA-matched donor, this approach can help overcome the prolonged cytopenias that occur with standard cord blood transplants in adults.”
Gamida Cell plans to submit these data for approval of omidubicel by the Food and Drug Administration in the fourth quarter of 2021.
Omidubicel is also being evaluated in a phase 1/2 clinical study in patients with severe aplastic anemia (NCT03173937).
Expanding possibilities
Although umbilical cord blood stem cell grafts come from a readily available source and show greater tolerance across HLA barriers than other sources (such as bone marrow), the relatively low dose of stem cells in each unit results in delayed hematopoietic recovery, increased transplant-related morbidity and mortality, and longer hospitalizations, Dr. Sanz said.
Omidubicel consists of two cryopreserved fractions from a single cord blood unit. The product contains both noncultured CD133-negative cells, including T cells, and CD133-positive cells that are then expanded ex vivo for 21 days in the presence of nicotinamide.
“Nicotinamide increases stem and progenitor cells, inhibits differentiation and increases migration, bone marrow homing, and engraftment efficiency while preserving cellular functionality and phenotype,” Dr. Sanz explained during his presentation.
In an earlier phase 1/2 trial in 36 patients with high-risk hematologic malignancies, omidubicel was associated with hematopoietic engraftment lasting at least 10 years.
Details of phase 3 trial results
The global phase 3 trial was conducted in 125 patients (aged 13-65 years) with high-risk malignancies, including acute myeloid and lymphoblastic leukemias, myelodysplastic syndrome, chronic myeloid leukemia, lymphomas, and rare leukemias. These patients were all eligible for allogeneic stem cell transplantation but did not have matched donors.
Patients were randomly assigned to receive hematopoietic reconstitution with either omidubicel (n = 52) or standard cord blood (n = 58).
At 42 days of follow-up, the median time to neutrophil engraftment in the intention-to-treat (ITT) population, the primary endpoint, was 12 days with omidubicel versus 22 days with standard cord blood (P < .001).
In the as-treated population – the 108 patients who actually received omidubicel or standard cord blood – median time to engraftment was 10.0 versus 20.5 days, respectively (P < .001).
Rates of neutrophil engraftment at 42 days were 96% with omidubicel versus 89% with standard cord blood.
The secondary endpoint of time-to-platelet engraftment in the ITT population also favored omidubicel, with a cumulative day 42 incidence rate of 55%, compared with 35% with standard cord blood (P = .028).
In the as-treated population, median times to platelet engraftment were 37 days and 50 days, respectively (P = .023). The cumulative rates of platelet engraftment at 100 days of follow-up were 83% and 73%, respectively.
The incidence of grade 2 or 3 bacterial or invasive fungal infections by day 100 in the ITT population was 37% among patients who received omidubicel, compared with 57% for patients who received standard cord blood (P = .027). Viral infections occurred in 10% versus 26% of patients, respectively.
The incidence of acute graft versus host disease at day 100 was similar between treatment groups, and there was no significant difference at 1 year.
Relapse and nonrelapse mortality rates, as well as disease-free and overall survival rates also did not differ between groups.
In the first 100 days post transplant, patients who received omidubicel were alive and out of the hospital for a median of 60.5 days, compared with 48 days for patients who received standard cord blood (P = .005).
The study was funded by Gamida Cell. Dr. Sanz reported receiving research funding from the company and several others, and consulting fees, honoraria, speakers bureau activity, and travel expenses from other companies. Dr. DeFilipp reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Updated recommendations released on COVID-19 and pediatric ALL
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
The main threat to the vast majority of children with acute lymphoblastic leukemia still remains the ALL itself, according to updated recommendations released by the Leukemia Committee of the French Society for the Fight Against Cancers and Leukemias in Children and Adolescents (SFCE).
“The situation of the current COVID-19 pandemic is continuously evolving. We thus have taken the more recent knowledge into account to update the previous recommendations from the Leukemia Committee,” Jérémie Rouger-Gaudichon, MD, of Pediatric Hemato-Immuno-Oncology Unit, Centre Hospitalier Universitaire, Caen (France), and colleagues wrote on behalf of the SFCE.
The updated recommendations are based on data collected in a real-time prospective survey among the 30 SFCE centers since April 2020. As of December 2020, 127 cases of COVID-19 were reported, most of them being enrolled in the PEDONCOVID study (NCT04433871) according to the report. Of these, eight patients required hospitalization in intensive care unit and one patient with relapsed acute lymphoblastic leukemia (ALL) died from ARDS with multiorgan failure. This confirms earlier reports that SARS-CoV-2 infection can be severe in some children with cancer and/or having hematopoietic stem cell transplant (HSCT), according to the report, which was published online in Bulletin du Cancer.
Recommendations
General recommendations were provided in the report, including the following:
- Test for SARS-CoV-2 (preferably by PCR or at least by immunological tests, on nasopharyngeal swab) before starting intensive induction chemotherapy or other intensive phase of treatment, for ALL patients, with or without symptoms.
- Delay systemic treatment if possible (e.g., absence of major hyperleukocytosis) in positive patients. During later phases, if patients test positive, tests should be repeated over time until negativity, especially before the beginning of an intensive course.
- Isolate any COVID-19–negative child or adolescent to allow treatment to continue (facial mask, social distancing, barrier measures, no contact with individuals suspected of COVID-19 or COVID-19–positive), in particular for patients to be allografted.
- Limit visitation to parents and potentially siblings in patients slated for HSCT and follow all necessary sanitary procedures for those visits.
The report provides a lengthy discussion of more detailed recommendations, including the following for first-line treatment of ALL:
- For patients with high-risk ALL, an individualized decision regarding transplantation and its timing should weigh the risks of transplantation in an epidemic context of COVID-19 against the risk linked to ALL.
- Minimizing hospital visits by the use of home blood tests and partial use of telemedicine may be considered.
- A physical examination should be performed regularly to avoid any delay in the diagnosis of treatment complications or relapse and preventative measures for SARS-CoV-2 should be applied in the home.
Patients with relapsed ALL may be at more risk from the effects of COVID-19 disease, according to the others, so for ALL patients receiving second-line or more treatment the recommendations include the following:
- Testing must be performed before starting a chemotherapy block, and postponing chemotherapy in case of positive test should be discussed in accordance with each specific situation and benefits/risks ratio regarding the leukemia.
- First-relapse patients should follow the INTREALL treatment protocol as much as possible and those who reach appropriate complete remission should be considered promptly for allogeneic transplantation, despite the pandemic.
- Second relapse and refractory relapses require testing and negative results for inclusion in phase I-II trials being conducted by most if not all academic or industrial promoters.
- The indication for treatment with CAR-T cells must be weighed with the center that would perform the procedure to determine the feasibility of performing all necessary procedures including apheresis and manufacturing.
In the case of a SARS-CoV-2 infection diagnosis during the treatment of ALL, discussions should occur with regard to stopping and/or postponing all chemotherapies, according to the severity of the ALL, the stage of treatment and the severity of clinical and/or radiological signs. In addition, any specific anti-COVID-19 treatment must be discussed with the infectious diseases team, according to the report.
“Fortunately, SARS-CoV-2 infection appears nevertheless to be mild in most children with cancer/ALL. Thus, the main threat to the vast majority of children with ALL still remains the ALL itself. Long-term data including well-matched case-control studies will tell if treatment delays/modifications due to COVID-19 have impacted the outcome if children with ALL,” the authors stated. However, “despite extremely rapid advances obtained in less than one year, our knowledge of SARS-CoV-2 and its complications is still incomplete,” they concluded, adding that the recommendations will likely need to be updated within another few months.
The authors reported that they had no conflicts of interest.
FROM BULLETIN DU CANCER
Don’t delay: Cancer patients need both doses of COVID vaccine
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The new findings, which are soon to be published as a preprint, cast doubt on the current U.K. policy of delaying the second dose of the vaccine.
Delaying the second dose can leave most patients with cancer wholly or partially unprotected, according to the researchers. Moreover, such a delay has implications for transmission of SARS-CoV-2 in the cancer patient’s environs as well as for the evolution of virus variants that could be of concern, the researchers concluded.
The data come from a British study that included 151 patients with cancer and 54 healthy control persons. All participants received the COVID-19 mRNA BNT162b2 vaccine (Pfizer-BioNTech).
This vaccine requires two doses. The first few participants in this study were given the second dose 21 days after they had received the first dose, but then national guidelines changed, and the remaining participants had to wait 12 weeks to receive their second dose.
The researchers reported that, among health controls, the immune efficacy of the first dose was very high (97% efficacious). By contrast, among patients with solid tumors, the immune efficacy of a single dose was strikingly low (39%), and it was even lower in patients with hematologic malignancies (13%).
The second dose of vaccine greatly and rapidly increased the immune efficacy in patients with solid tumors (95% within 2 weeks of receiving the second dose), the researchers added.
Too few patients with hematologic cancers had received the second dose before the study ended for clear conclusions to be drawn. Nevertheless, the available data suggest that 50% of patients with hematologic cancers who had received the booster at day 21 were seropositive at 5 weeks vs. only 8% of those who had not received the booster.
“Our data provide the first real-world evidence of immune efficacy following one dose of the Pfizer vaccine in immunocompromised patient populations [and] clearly show that the poor one-dose efficacy in cancer patients can be rescued with an early booster at day 21,” commented senior author Sheeba Irshad, MD, senior clinical lecturer, King’s College London.
“Based on our findings, we would recommend an urgent review of the vaccine strategy for clinically extremely vulnerable groups. Until then, it is important that cancer patients continue to observe all public health measures in place, such as social distancing and shielding when attending hospitals, even after vaccination,” Dr. Irshad added.
The paper, with first author Leticia Monin-Aldama, PhD, is scheduled to appear on the preprint server medRxiv. It has not undergone peer review. The paper was distributed to journalists, with comments from experts not involved in the study, by the UK Science Media Centre.
These data are “of immediate importance” to patients with cancer, commented Shoba Amarnath, PhD, Newcastle University research fellow, Laboratory of T-cell Regulation, Newcastle University Center for Cancer, Newcastle upon Tyne, England.
“These findings are consistent with our understanding. … We know that the immune system within cancer patients is compromised as compared to healthy controls,” Dr. Amarnath said. “The data in the study support the notion that, in solid cancer patients, a considerable delay in second dose will extend the period when cancer patients are at risk of SARS-CoV-2 infection.”
Although more data are required, “this study does raise the issue of whether patients with cancer, other diseases, or those undergoing therapies that affect the body’s immune response should be fast-tracked for their second vaccine dose,” commented Lawrence Young, PhD, professor of molecular oncology and director of the Warwick Cancer Research Center, University of Warwick, Coventry, England.
Stephen Evans, MSc, professor of pharmacoepidemiology, London School of Hygiene and Tropical Medicine, underlined that the study is “essentially” observational and “inevitable limitations must be taken into account.
“Nevertheless, these results do suggest that the vaccines may well not protect those patients with cancer as well as those without cancer,” Mr. Evans said. He added that it is “important that this population continues to observe all COVID-19–associated measures, such as social distancing and shielding when attending hospitals, even after vaccination.”
Study details
Previous studies have shown that some patients with cancer have prolonged responses to SARS-CoV-2 infection, with ongoing immune dysregulation, inefficient seroconversion, and prolonged viral shedding.
There are few data, however, on how these patients respond to COVID-19 vaccination. The authors point out that, among the 18,860 individuals who received the Pfizer vaccine during its development trials, “none with an active oncological diagnosis was included.”
To investigate this issue, they launched the SARS-CoV-2 for Cancer Patients (SOAP-02) study.
The 151 patients with cancer who participated in this study were mostly elderly, the authors noted (75% were older than 65 years; the median age was 73 years). The majority (63%) had solid-tumor malignancies. Of those, 8% had late-stage disease and had been living with their cancer for more than 24 months.
The healthy control persons were vaccine-eligible primary health care workers who were not age matched to the cancer patients.
All participants received the first dose of vaccine; 31 (of 151) patients with cancer and 16 (of 54) healthy control persons received the second dose on day 21.
The remaining participants were scheduled to receive their second dose 12 weeks later (after the study ended), in line with the changes in the national guidelines.
The team reported that, approximately 21 days after receiving the first vaccine dose, the immune efficacy of the vaccine was estimated to be 97% among healthy control persons vs. 39% for patients with solid tumors and only 13% for those with hematologic malignancies (P < .0001 for both).
T-cell responses, as assessed via interferon-gamma and/or interleukin-2 production, were observed in 82% of healthy control persons, 71% of patients with solid tumors, and 50% of those with hematologic cancers.
Vaccine boosting at day 21 resulted in immune efficacy of 100% for healthy control persons and 95% for patients with solid tumors. In contrast, only 43% of those who did not receive the second dose were seropositive 2 weeks later.
Further analysis suggested that participants who did not have a serologic response were “spread evenly” across different cancer types, but the reduced responses were more frequent among patients who had received the vaccine within 15 days of cancer treatment, especially chemotherapy, and had undergone intensive treatments.
The SOAP study is sponsored by King’s College London and Guy’s and St. Thomas Trust Foundation NHS Trust. It is funded from grants from the KCL Charity, Cancer Research UK, and program grants from Breast Cancer Now. The investigators have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
New inhibitor shows promise in previously failed B-cell malignancies
who discontinued prior Bruton’s tyrosine kinase (BTK)–inhibitor treatment due to resistance or intolerance, according to the results of the BRUIN trial, a phase 1/2 study.

Pirtobrutinib (formerly known as LOXO-305) is an oral-dose, highly selective, reversible BTK inhibitor, which might address a growing, unmet need for alternative therapies in BTK-inhibitor treatment failure patients, according to Anthony R. Mato, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues. Their report was published in The Lancet.
The study included 109 women (34%) and 214 men (66%), with a median age of 68 years, who were treated with pirtobrutinib. Of these, 203 patients were assigned to pirtobrutinib (25-300 mg once per day) in the phase 1 portion of the study, and 120 patients were assigned to pirtobrutinib (200 mg once per day) in phase 2.
Promising outcomes
Pirtobrutinib, showed promising efficacy and tolerable safety in patients with CLL or small lymphocytic lymphoma, mantle cell lymphoma, and Waldenström macroglobulinemia who were previously treated with a BTK inhibitor. In 121 efficacy-evaluable patients with CLL or SLL treated with a previous covalent BTK inhibitor, the overall response rate with pirtobrutinib was 62% (95% confidence interval, 53-71). The ORR was similar in CLL patients with previous covalent BTK inhibitor resistance (67%), covalent BTK inhibitor intolerance (52%), BTK C481-mutant (71%), and BTK wild-type (66%) disease.
In 52 efficacy-evaluable patients with mantle cell lymphoma (MCL) previously treated with covalent BTK inhibitors, the ORR was 52% (95% CI, 38-66). Of 117 patients with CLL, SLL, or MCL who responded, all but 8 remain progression free to date, the authors stated.
In 19 efficacy-evaluable patients with Waldenström macroglobulinemia, the ORR was 68%. Among eight patients with follicular lymphoma who were efficacy evaluable, responses were observed in four (50%) patients, and six (75%) of eight efficacy evaluable patients with Richter’s transformation identified before enrollment responded to treatment, the authors stated.
No dose-limiting toxicities were observed and the maximum tolerated dose was not reached, according to the researchers. The recommended phase 2 dose was 200 mg daily. The adverse events, which occurred in at least 10% of 323 patients, were fatigue (20%), diarrhea (17%), and contusion (13%). The most common grade 3 or higher adverse event was neutropenia (10%). Five patients (1%) discontinued treatment because of a treatment-related adverse event.
In this “first-in-human trial of pirtobrutinib, we showed promising efficacy and safety in patients with B-cell malignancies, including CLL or SLL, MCL, Waldenström macroglobulinemia, and follicular lymphoma. Activity was observed in heavily pretreated patients, including patients with resistance and intolerance to previous covalent BTK inhibitor treatment. Global randomized phase 3 studies in CLL or SLL, and MCL are planned,” the researchers concluded.
Birth of a third generation?
“The pirtobrutinib study, by opening the way for a third generation of BTK inhibitors, could improve such a personalized molecular approach in the treatment of B-cell malignancies,” according to accompanying editorial comment by Jean-Marie Michot, MD, and Vincent Ribrag, MD, both of the Institut de Cancérologie Gustave Roussy, Villejuif, France.
They discussed how BTK inhibitors have been a considerable therapeutic advance in the treatment of NHL-B and CLL and how the three currently approved BTK inhibitors, namely ibrutinib, acalabrutinib, and zanubrutinib, are all covalent and irreversible inhibitors at the protein – the C481 binding site. “Ibrutinib was the first approved drug. The second-generation inhibitors, acalabrutinib and zanubrutinib, were designed to be more BTK selective,” they added. However, the covalency and irreversibility of the drugs, considered therapeutic strengths, have resulted in induced resistance mutations occurring at the covalent binding, rendering the drugs inactive. “Two advantages of this new drug class are highlighted. First, the selectivity of the drug on BTK appears to be increased,” they wrote. “Second, this class does not bind BTK to the C481 residue, and the efficacy of the drug is therefore not affected by mutations in the BTK binding site.”
Several of the study authors reported receiving grants and personal fees from Loxo Oncology (a wholly owned subsidiary of Eli Lilly), which sponsored the study, as well as financial relationships with other pharmaceutical and biotechnology companies.
Dr. Michot and Dr. Ribrag reported that they had no disclosures relevant to the discussion.
who discontinued prior Bruton’s tyrosine kinase (BTK)–inhibitor treatment due to resistance or intolerance, according to the results of the BRUIN trial, a phase 1/2 study.
Pirtobrutinib (formerly known as LOXO-305) is an oral-dose, highly selective, reversible BTK inhibitor, which might address a growing, unmet need for alternative therapies in BTK-inhibitor treatment failure patients, according to Anthony R. Mato, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues. Their report was published in The Lancet.
The study included 109 women (34%) and 214 men (66%), with a median age of 68 years, who were treated with pirtobrutinib. Of these, 203 patients were assigned to pirtobrutinib (25-300 mg once per day) in the phase 1 portion of the study, and 120 patients were assigned to pirtobrutinib (200 mg once per day) in phase 2.
Promising outcomes
Pirtobrutinib, showed promising efficacy and tolerable safety in patients with CLL or small lymphocytic lymphoma, mantle cell lymphoma, and Waldenström macroglobulinemia who were previously treated with a BTK inhibitor. In 121 efficacy-evaluable patients with CLL or SLL treated with a previous covalent BTK inhibitor, the overall response rate with pirtobrutinib was 62% (95% confidence interval, 53-71). The ORR was similar in CLL patients with previous covalent BTK inhibitor resistance (67%), covalent BTK inhibitor intolerance (52%), BTK C481-mutant (71%), and BTK wild-type (66%) disease.
In 52 efficacy-evaluable patients with mantle cell lymphoma (MCL) previously treated with covalent BTK inhibitors, the ORR was 52% (95% CI, 38-66). Of 117 patients with CLL, SLL, or MCL who responded, all but 8 remain progression free to date, the authors stated.
In 19 efficacy-evaluable patients with Waldenström macroglobulinemia, the ORR was 68%. Among eight patients with follicular lymphoma who were efficacy evaluable, responses were observed in four (50%) patients, and six (75%) of eight efficacy evaluable patients with Richter’s transformation identified before enrollment responded to treatment, the authors stated.
No dose-limiting toxicities were observed and the maximum tolerated dose was not reached, according to the researchers. The recommended phase 2 dose was 200 mg daily. The adverse events, which occurred in at least 10% of 323 patients, were fatigue (20%), diarrhea (17%), and contusion (13%). The most common grade 3 or higher adverse event was neutropenia (10%). Five patients (1%) discontinued treatment because of a treatment-related adverse event.
In this “first-in-human trial of pirtobrutinib, we showed promising efficacy and safety in patients with B-cell malignancies, including CLL or SLL, MCL, Waldenström macroglobulinemia, and follicular lymphoma. Activity was observed in heavily pretreated patients, including patients with resistance and intolerance to previous covalent BTK inhibitor treatment. Global randomized phase 3 studies in CLL or SLL, and MCL are planned,” the researchers concluded.
Birth of a third generation?
“The pirtobrutinib study, by opening the way for a third generation of BTK inhibitors, could improve such a personalized molecular approach in the treatment of B-cell malignancies,” according to accompanying editorial comment by Jean-Marie Michot, MD, and Vincent Ribrag, MD, both of the Institut de Cancérologie Gustave Roussy, Villejuif, France.
They discussed how BTK inhibitors have been a considerable therapeutic advance in the treatment of NHL-B and CLL and how the three currently approved BTK inhibitors, namely ibrutinib, acalabrutinib, and zanubrutinib, are all covalent and irreversible inhibitors at the protein – the C481 binding site. “Ibrutinib was the first approved drug. The second-generation inhibitors, acalabrutinib and zanubrutinib, were designed to be more BTK selective,” they added. However, the covalency and irreversibility of the drugs, considered therapeutic strengths, have resulted in induced resistance mutations occurring at the covalent binding, rendering the drugs inactive. “Two advantages of this new drug class are highlighted. First, the selectivity of the drug on BTK appears to be increased,” they wrote. “Second, this class does not bind BTK to the C481 residue, and the efficacy of the drug is therefore not affected by mutations in the BTK binding site.”
Several of the study authors reported receiving grants and personal fees from Loxo Oncology (a wholly owned subsidiary of Eli Lilly), which sponsored the study, as well as financial relationships with other pharmaceutical and biotechnology companies.
Dr. Michot and Dr. Ribrag reported that they had no disclosures relevant to the discussion.
who discontinued prior Bruton’s tyrosine kinase (BTK)–inhibitor treatment due to resistance or intolerance, according to the results of the BRUIN trial, a phase 1/2 study.
Pirtobrutinib (formerly known as LOXO-305) is an oral-dose, highly selective, reversible BTK inhibitor, which might address a growing, unmet need for alternative therapies in BTK-inhibitor treatment failure patients, according to Anthony R. Mato, MD, of Memorial Sloan Kettering Cancer Center, New York, and colleagues. Their report was published in The Lancet.
The study included 109 women (34%) and 214 men (66%), with a median age of 68 years, who were treated with pirtobrutinib. Of these, 203 patients were assigned to pirtobrutinib (25-300 mg once per day) in the phase 1 portion of the study, and 120 patients were assigned to pirtobrutinib (200 mg once per day) in phase 2.
Promising outcomes
Pirtobrutinib, showed promising efficacy and tolerable safety in patients with CLL or small lymphocytic lymphoma, mantle cell lymphoma, and Waldenström macroglobulinemia who were previously treated with a BTK inhibitor. In 121 efficacy-evaluable patients with CLL or SLL treated with a previous covalent BTK inhibitor, the overall response rate with pirtobrutinib was 62% (95% confidence interval, 53-71). The ORR was similar in CLL patients with previous covalent BTK inhibitor resistance (67%), covalent BTK inhibitor intolerance (52%), BTK C481-mutant (71%), and BTK wild-type (66%) disease.
In 52 efficacy-evaluable patients with mantle cell lymphoma (MCL) previously treated with covalent BTK inhibitors, the ORR was 52% (95% CI, 38-66). Of 117 patients with CLL, SLL, or MCL who responded, all but 8 remain progression free to date, the authors stated.
In 19 efficacy-evaluable patients with Waldenström macroglobulinemia, the ORR was 68%. Among eight patients with follicular lymphoma who were efficacy evaluable, responses were observed in four (50%) patients, and six (75%) of eight efficacy evaluable patients with Richter’s transformation identified before enrollment responded to treatment, the authors stated.
No dose-limiting toxicities were observed and the maximum tolerated dose was not reached, according to the researchers. The recommended phase 2 dose was 200 mg daily. The adverse events, which occurred in at least 10% of 323 patients, were fatigue (20%), diarrhea (17%), and contusion (13%). The most common grade 3 or higher adverse event was neutropenia (10%). Five patients (1%) discontinued treatment because of a treatment-related adverse event.
In this “first-in-human trial of pirtobrutinib, we showed promising efficacy and safety in patients with B-cell malignancies, including CLL or SLL, MCL, Waldenström macroglobulinemia, and follicular lymphoma. Activity was observed in heavily pretreated patients, including patients with resistance and intolerance to previous covalent BTK inhibitor treatment. Global randomized phase 3 studies in CLL or SLL, and MCL are planned,” the researchers concluded.
Birth of a third generation?
“The pirtobrutinib study, by opening the way for a third generation of BTK inhibitors, could improve such a personalized molecular approach in the treatment of B-cell malignancies,” according to accompanying editorial comment by Jean-Marie Michot, MD, and Vincent Ribrag, MD, both of the Institut de Cancérologie Gustave Roussy, Villejuif, France.
They discussed how BTK inhibitors have been a considerable therapeutic advance in the treatment of NHL-B and CLL and how the three currently approved BTK inhibitors, namely ibrutinib, acalabrutinib, and zanubrutinib, are all covalent and irreversible inhibitors at the protein – the C481 binding site. “Ibrutinib was the first approved drug. The second-generation inhibitors, acalabrutinib and zanubrutinib, were designed to be more BTK selective,” they added. However, the covalency and irreversibility of the drugs, considered therapeutic strengths, have resulted in induced resistance mutations occurring at the covalent binding, rendering the drugs inactive. “Two advantages of this new drug class are highlighted. First, the selectivity of the drug on BTK appears to be increased,” they wrote. “Second, this class does not bind BTK to the C481 residue, and the efficacy of the drug is therefore not affected by mutations in the BTK binding site.”
Several of the study authors reported receiving grants and personal fees from Loxo Oncology (a wholly owned subsidiary of Eli Lilly), which sponsored the study, as well as financial relationships with other pharmaceutical and biotechnology companies.
Dr. Michot and Dr. Ribrag reported that they had no disclosures relevant to the discussion.
FROM THE LANCET