User login
Herpes Zoster Infection
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
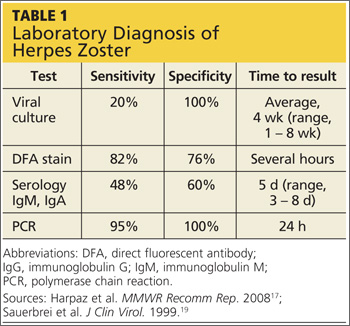
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
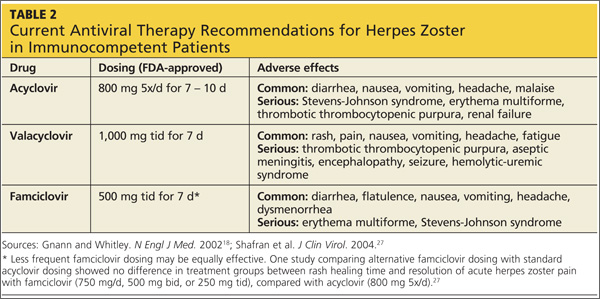
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
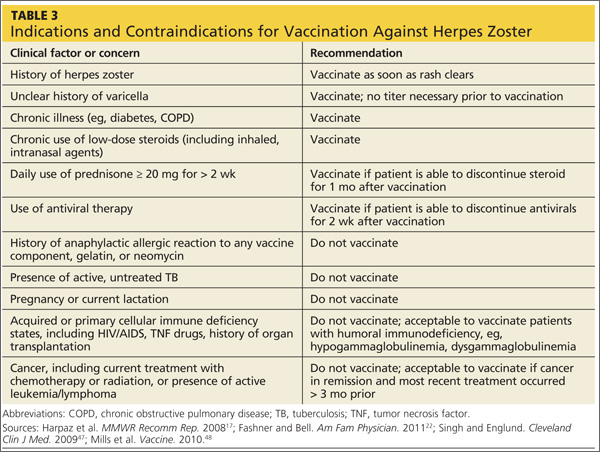
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.

1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.
CE/CME No: CR-1308
PROGRAM OVERVIEW
Earn credit by reading this article and successfully completing the posttest. Successful completion is defined as a cumulative score of at least 70% correct.
EDUCATIONAL OBJECTIVES
• Explain the etiology of herpes zoster infection (HZ), typical and atypical clinical presentation, and diagnostic confirmation, when needed.
• Describe treatment interventions for acute HZ infection, including topical measures, use of antiviral agents, and pain management options.
• Discuss complications of HZ infection, including risk factors and prevention.
• Explain risks, benefits, contraindications, and other considerations for vaccination use to prevent HZ in at-risk adults.
FACULTY
Emily Jacobsen is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at Oregon Health & Science University (OHSU) in Portland, Oregon; she is a practicing Physician Assistant at OHSU Family Medicine at Richmond in Portland. Claire E. Hull is an Assistant Professor in the Department of Family Medicine and in the Division of Physician Assistant Education at OHSU.
The authors have no significant financial relationships to disclose.
ACCREDITATION STATEMENT
![]()
Article begins on next page >>
Herpes zoster (HZ) infection, commonly called shingles, represents a reactivation of the chickenpox virus. Persons older than 50 and those with compromised immune systems are at greatest risk. Most cases resolve spontaneously, but about one-third of patients develop postherpetic neuralgia or other complications, and 1% to 4% require hospitalization. Treatment involves antiviral medications and pain management. Vaccination against HZ, which is recommended for adults 60 and older, incurs benefits and risks that the clinician must be prepared to explain to eligible patients.
Infection with herpes zoster (HZ) affects approximately one million individuals in the United States each year.1-3 The disease is caused by a reactivation of the varicella zoster virus (VZV), which causes chickenpox. Once chickenpox has resolved, VZV remains dormant in the dorsal (spinal) root ganglia, trigeminal nerve, and autonomic ganglia of the nervous system.4 At some later time, VZV may reactivate, causing an extremely painful vesicular rash along the distribution of one or more sensory dermatomes; the rash (as well as the condition in general) is commonly referred to as shingles.
It has been estimated that 90% or more of US adults older than 40 are infected with VZV.1,3 Because the virus is so ubiquitous, virtually anyone may be at risk for the reactivation of VZV in the form of shingles. It is estimated that 10% to 20% of the US population will develop HZ in their lifetime,3 with age and immune status the most significant determinants of persons to be affected.3-6
About half of all cases of shingles in the US occur in persons age 50 or older. Incidence among those older than 75 is approximately 10 cases per 1,000 individuals, compared with about two cases per 1,000 individuals in those younger than 50.3
In addition to age, the integrity of an individual’s immune system plays a key role in the development of shingles. Reactivation of VZV is usually suppressed by the host’s cell-mediated immune response, particularly the T cells.3,5 Thus, if the cell-mediated immune system is compromised, reactivation and widespread dissemination are more likely to occur. Adults with cancer or HIV infection and those taking immunosuppressive drugs have a significantly increased risk for HZ. Psychological or physical stress and trauma have also been shown to play a role in the development of HZ.5 In contrast to chickenpox, HZ has no seasonal predilection.7
Since 1995, with the licensing of Varivax (the vaccination to prevent varicella), the incidence of wild-type varicella infection is now quite low in the US. From 2000 to 2010, varicella wild-type infection declined by 82%.8 Efforts to further quantify the incidence of varicella have been hampered by the absence of reporting requirements for this infection.
Due to the live nature of the Varivax vaccine, patients who have received it remain at risk for HZ infection by way of reactivation of vaccine-type VZV. A population-based surveillance study conducted in California from 2000 to 2006 showed that the incidence of HZ infection decreased by 55% in children 10 years or younger who were vaccinated against varicella.9 This finding, along with similar results in other, older research in immunocompromised hosts, supports the notion that the risk for HZ is substantially reduced among children who have been vaccinated against varicella.10
Incidence of HZ infection seems to be on the rise, both in the US and worldwide1; however, the causes for this are a point of controversy. Fears have been expressed that incidence of HZ infection in adults would increase once varicella vaccination in children became commonplace, based on reasoning that exposure to the virus (which is thought to boost cell-mediated immunity and keep the virus from reactivating) would decline. This concern has put a halt to vaccination against varicella in some European countries.11 At least one US researcher considers the evidence strong for a causal link between the increase in incidence of HZ and the widespread implementation of varicella vaccination.12
Other research has led to different conclusions. Authors of a nationwide, retrospective review of claims data noted an increase in HZ prior to Varivax licensure but did not find any association between vaccination rates and HZ rates geographically.13 Similarly, researchers conducting a case-control study in a Wisconsin clinic found no relationship between HZ and exposure to VZV in the previous 10 years.14
On the next page: Clinical presentation and laboratory diagnosis >>
CLINICAL PRESENTATION
Identifying HZ infection is primarily a clinical diagnosis and not particularly difficult. Approximately 20% of patients will present with prodromal symptoms of fatigue, headache, malaise, and fever. Paresthesias in the involved dermatome often precede the rash by several days and may be manifested as itching, tingling, burning, or severe pain. Physical examination at this stage may reveal tenderness and hyperesthesia of the skin in the involved dermatome.3,5,15,16
Pain and abnormal skin sensations are the most common symptoms of HZ. They often precede and usually accompany the rash. The prodromal pain of HZ can mimic a variety of other conditions, including pleurisy, myocardial infarction, peptic ulcer, appendicitis, or biliary or renal colic, prompting some clinicians to undertake an extensive workup and treatment plan.15,17
Consistent with other herpes infections, the HZ rash initially starts in the form of erythematous papules, which quickly evolve into grouped vesicles or bullae. Within three to four days, these vesicular lesions can become more pustular. In contrast to chickenpox, the rash of shingles is manifested in a dermatomal distribution. The two most commonly affected dermatomes are the first (ophthalmic) division of the trigeminal nerve and the spinal sensory ganglia from T1 to L2.3,5,15,16 The infection is generally limited to one dermatome in previously healthy hosts but can occasionally affect two or three neighboring dermatomes. Some patients have a few scattered vesicles located some distance away from the involved dermatome.15
In immunocompetent hosts, the lesions crust over within seven to 10 days and are no longer considered infectious. The development of new lesions more than a week after presentation should raise concerns regarding possible underlying immunodeficiency.3,5,15,16
LABORATORY DIAGNOSIS
While HZ is generally a clinical diagnosis based on the history and physical exam findings, laboratory testing may be appropriate to confirm the diagnosis when the presentation is atypical, the host is immunocompromised, lesions recur, or serious complications are suspected.17,18
Several laboratory tests are currently available (see Table 117,19). Detection of VZV DNA following amplification of appropriate specimens (most reliably, clear fluid from recently erupted vesicles) by polymerase chain reaction (PCR) is generally recommended if testing is required, because of its high sensitivity and specificity and quick turnaround. However, this test is not available at every laboratory.17-19
When PCR is not available, a suitable alternative is direct fluorescent antibody (DFA) staining of cellular material from fresh vesicles or prevesicular lesions.1 This test uses a modified Tzanck technique to view fluorescein-conjugated monoclonal antibodies. DFA staining can differentiate between herpes simplex and herpes zoster.16,17,20
The original Tzanck smear is inexpensive and may reveal multinucleated giant cells and epithelial cells containing acidophilic intranuclear inclusion bodies.17 However, this test is not often used to confirm a diagnosis of HZ; rather, it is most helpful for distinguishing herpesvirus infections from vesicular lesions of other etiologies (eg, coxsackievirus, echovirus).15,17
Serologic tests measuring immunoglobulin M and A titers (IgM, IgA) may be helpful in cases of zoster without rash (zoster sine herpete), but their sensitivity and specificity are low.15 Positive results may be indicative of primary infection, reinfection, or reactivation.1
On the next page: Treatment >>
TREATMENT
Treatment of HZ infection is focused on limiting the extent, duration, and severity of pain and rash in the primary dermatome, as well as decreasing the risk for complications.
Topical Therapy
Patients should keep the cutaneous lesions clean and dry to reduce the risk for bacterial superinfection. A sterile, nonocclusive, nonadherent dressing placed over the involved dermatome will protect the lesions from contact with clothing. To hasten the drying of vesicular lesions and alleviate pruritus associated with rash, the application of cool compresses, calamine lotion, cornstarch, or baking soda may be helpful.16,21
While antipruritic agents may help prevent infections that can develop when the affected area is scratched, there is no evidence that any of these agents have any real therapeutic effect on the HZ rash or lesions. Topical antiviral agents are not effective.15,18
Antiviral Therapy
Treatment for acute HZ with oral antiviral medication should be considered for any patient who presents within 72 hours of rash onset; antiviral agents initiated within this time frame have been shown to reduce the duration and severity of pain associated with acute HZ.21 Antivirals are recommended and should be given routinely to patients older than 50 and those who have moderate to severe symptoms.15,21
Among patients who present longer than 72 hours after rash onset, antiviral therapy should be considered only for those with new vesicular formation, ophthalmic involvement, or motor or neurologic complications, although evidence is lacking for this recommendation.15,21 A modest reduction in the duration of rash (by 1 to 3 days) has also been reported in patients treated with antivirals,21 most likely because viral replication is slowed within the dorsal root ganglion.16,22
Acyclovir, valacyclovir, and famciclovir—nucleoside analogs that block viral replication—are the only FDA-approved medications for treatment of HZ.18,22 When choosing among these agents, the prescribing clinician should be aware of their differences in bioavailability and pharmacokinetics. Acyclovir, for example, is a second-generation antiviral drug with poor pharmacokinetics, which explains the frequent dosing its use generally requires.22,23 However, the inhibitory dose of acyclovir required for patients with HZ is much lower than that required to treat primary VZV infection.
Valacyclovir and famciclovir, which are third-generation antivirals, feature enhanced absorption from the gastrointestinal tract (77% vs 30% for acyclovir),24 thus improving their bioavailability by three to five times, compared with acyclovir. The superior pharmacokinetics of valacyclovir and famciclovir has been confirmed clinically by researchers who demonstrated median pain duration of 38 days in patients taking valacyclovir, compared with 51 days in those treated with acyclovir.25 In a direct comparison of valacyclovir and famciclovir, resolution of rash and pain times were found comparable.26 Table 218,27 summarizes the current recommendations for antiviral therapy in patients with HZ.
Pain Management
Pain is almost universal once the HZ rash appears. Pain associated with the prodromal period is variable but may be present in 70% to 80% of patients.28,29 The severity of acute pain in HZ is highly variable, ranging from mild to quite severe. Pain can begin weeks or a single day before the rash emerges and persist for several weeks after the rash disappears.
Aggressive pain management is appropriate. A variety of opiate analgesics (eg, hydrocodone, oxycodone, hydromorphone, morphine) and nonopiate analgesics (acetaminophen, NSAIDs) may be effective.17,28 Drug choices, dosage, and scheduling should be tailored to the patient’s level of pain and disability, with any potential contraindications also taken into account. Mild pain can be treated with as-needed dosing, whereas scheduled dosing is preferred for moderate to severe pain.16
If acute pain persists, addition of gabapentin, pregabalin, or nortriptyline is reasonable.17,22,28,30 Although these medications have been studied in the treatment of postherpetic neuralgia (PHN), there is little evidence to support their use for acute zoster pain.22,30
Additional interventions that have been studied for relief of acute HZ pain include topical lidocaine, acupuncture, and interventional pain injections. However, the evidence is either scant or of poor quality. More research is needed before these modalities can be routinely recommended in the clinical setting.17,22
Corticosteroids have been used for acute HZ, but conflicting study results make their routine use controversial.17,31,32 In some studies, corticosteroids reduced acute pain and speeded lesion healing and return to daily activities; others have yielded little evidence to support these findings.22,33 Corticosteroids may offer the greatest benefit when used in combination with effective antiviral therapy.18,22,31,32 In one randomized clinical trial comparing acyclovir with acyclovir plus prednisolone (40 mg/d for three weeks, tapered down) combination therapy was associated with a significant decrease in pain during the initial two weeks.32
Historically, corticosteroids have also been prescribed with the hope that their anti-inflammatory properties might help reduce the risk for PHN. However, a recent Cochrane Review found that these agents do not reliably prevent PHN six months after HZ rash onset.34 Glucocorticoids may improve motor outcomes and acute pain in VZV-induced facial paralysis and cranial polyneuritis, in which compression of affected nerves may contribute to disability.
Before prescribing steroids, clinicians must consider contraindications to their use, including diabetes, osteoporosis, hypertension, glaucoma, and gastritis.16
On the next page: Patient education and complications >>
PATIENT EDUCATION
Patients must be instructed in how to avoid transmitting the HZ virus. The mechanism of transmission was long thought to be restricted to direct contact with lesions; however, molecular studies have shown that the HZ virus can be transmitted via the respiratory route, either through aerosolized virus from skin lesions or from respiratory droplets, as early as 24 to 48 hours before the rash appears.6,17 The risk of transmission by airborne virus is increased in patients with HZ rash that is disseminated beyond the primary and secondary dermatomes. The rash, patients should be informed, generally persists for two to four weeks.1,16,20
HZ continues to be contagious until the lesions crust over.17 Covering the rash greatly reduces patients’ risk for transmitting the virus via airborne or direct contact routes.15,17 A patient with HZ rash can infect a nonimmune person with primary varicella, causing chickenpox.28 The patient with HZ should be advised to avoid exposure to infants younger than 1 year, unvaccinated older children, anyone who is not immune to varicella (either by vaccination or primary infection), susceptible pregnant women, and potentially susceptible immunocompromised persons.16
COMPLICATIONS AND SEQUELAE
The majority of cases of shingles resolve without any complications or long-term sequelae. Complications of HZ that do occur may include superimposed skin infections, such as Streptococcus or Staphylococcus. The virus may be reactivated in the nasociliary branch of the trigeminal nerve and, in 10% to 25% of cases, herpes zoster ophthalmicus (HZO) may develop.17,35 Associated morbidity includes keratitis, corneal ulceration, conjunctivitis, uveitis, episcleritis and scleritis, retinitis, choroiditis, optic neuritis, lid retraction, ptosis, and glaucoma. Patients with HZO should be referred to an ophthalmologist promptly, as this condition can result in permanent loss of vision.35
Other less common complications of HZ include Ramsay Hunt syndrome (facial nerve palsy associated with reactivation in the geniculate ganglion) and zoster paresis (motor weakness in noncranial nerve distributions).17,36,37 Autonomic dysfunction has also been reported in patients with HZ, leading to colonic pseudo-obstruction and urinary retention. Rare but serious neurologic complications include Guillain-Barré syndrome, myelitis, aseptic meningitis, and meningoencephalitis.15,17
Postherpetic Neuralgia
By far, the most common complication of shingles is postherpetic neuralgia, a painfully debilitating and difficult-to-treat condition. Of the one million persons affected by HZ each year, between 9% and 34% will develop PHN.3,7,18 The criteria for diagnosing PHN is variable: While all definitions include the presence of persistent pain after rash resolution, they differ in how long this pain must persist. Some define PHN as pain persisting from 30 days to six months after rash resolution, while others define it as pain continuing three months or longer.3,17,18 While most patients with PHN experience complete resolution, the pain can endure from weeks to months to years.3,18,38
The pathophysiology of PHN is thought to involve replication of the VZV in the basal ganglia, damaging the nerves and thereby causing pain in the affected dermatome.22 Other possible factors include axonal and cell body degeneration, atrophy of the dorsal horn of the spinal cord, scarring of the dorsal root ganglion, and loss of epidermal innervations of the dermatome.17
The risk for PHN increases significantly in patients of advancing age. While PHN is rare in those younger than 50, it complicates HZ in 20% of patients between ages 60 and 65 and in 30% of those 80 and older. Additional risk factors for PHN include female gender, prodromal pain preceding the HZ rash, rash that is moderate to severe, moderate to severe acute pain associated with the rash, and ophthalmic involvement.39
Evidence conflicts regarding the impact of antivirals in patients with HZ on the subsequent development of PHN. Researchers performing a meta-analysis of five randomized clinical trials found no significant difference in the incidence of PHN among patients treated for HZ with oral acyclovir, famciclovir, or placebo.34 In an older, placebo-controlled randomized clinical trial, however, famciclovir-treated patients experienced PHN of reduced duration, compared with controls (63 days vs 119 days, respectively). Six months after development of the HZ rash, 15% of treated patients continued to experience PHN symptoms, compared with 23% of controls.23 More evidence is needed.
Additional Concerns
Recurrence of HZ is uncommon in immunocompetent persons. Despite its ordinarily benign course, 1% to 4% of people with shingles require hospitalization each year, mostly elderly patients.1 In a recent study, it was estimated that 96 US deaths are attributable to HZ each year.40 Almost all HZ-associated deaths occur in elderly patients with compromised or suppressed immune systems.1
On the next page: Prevention >>
PREVENTION
The varicella vaccine was licensed for use in children by the FDA in 1995. In June 2006, the Advisory Committee on Immunization Practices (ACIP) recommended a second dose to boost waning immunity.41
In 2006, the HZ vaccine (Zostavax), a more concentrated formulation of the varicella vaccine, was approved by the FDA for use in adults age 60 or older; in 2011, approval of the vaccine was extended to adults 50 or older.42 As of November 2011, ACIP has continued to recommend routine administration of the HZ vaccine for immunocompetent adults 60 and older, citing lack of evidence for long-term protection in patients vaccinated before age 60, as well as concerns about maintaining sufficient vaccine supplies.
Although ACIP does not recommend routine vaccination against HZ in patients age 50 to 59, health care providers may wish to consider it for patients in this age-group, based on the potential for poor tolerance of HZ or PHN symptoms, anticipated difficulty tolerating the required medications used to treat them, and employment-related considerations.43
Use of the HZ Vaccine
Zostavax is a live attenuated vaccine that increases varicella-specific, cell-mediated immunity in immunocompetent persons.17,42 It should be administered as a single 0.65-mL dose subcutaneously in the deltoid region of the upper arm42; a booster dose is not licensed for the vaccine.17 Adverse effects of the HZ vaccine generally include mild injection-site reactions: pain (54%), erythema (48%), swelling (40%), and pruritus (11%).42 According to researchers for the Shingles Prevention Study38,44 (SPS), these reactions were more common in treated patients than in controls and increasingly common in study participants of advancing age. Less than 2% of patients receiving either the HZ vaccine or placebo experienced serious adverse effects.44
The evidence to support vaccination against HZ comes mainly from the original SPS,38 a randomized, double-blind, placebo-controlled trial in which more than 38,500 adults 60 and older were enrolled. The SPS researchers showed that the vaccine reduced the incidence of HZ by 51.3%, reduced the incidence of PHN by 67%, and reduced the HZ-associated burden of illness (ie, its incidence, severity, and duration of associated pain and discomfort) by 61%2,38,45; they also found vaccination against HZ effective for at least three years.
An ongoing substudy involving 14,270 of the original SPS participants produced data showing that from year 4 to year 5 postvaccination, vaccine efficacy in terms of HZ incidence declined from 51% to 40%, respectively, and its efficacy regarding incidence of PHN, from 67% to 60%.46 Since there is no strong evidence that any treatment intervention started after shingles presents can reduce the risk for PHN, perhaps the vaccine’s most valuable attribute is its potential for preventing this debilitating and common complication of shingles.
Who Should or Should Not Be Vaccinated?
According to the ACIP, there is no upper age limit on vaccination against shingles. This judgment is supported by the fact that the incidence of zoster and PHN both continue to increase among patients of advancing age.17
While vaccination is appropriate for most individuals 60 or older, some contraindications exist (see Table 317,22,47,48). In cases of anticipated immunosuppression (as in patients scheduled to undergo chemotherapy), vaccination is recommended one month before the start of therapy. Additionally, the safety and efficacy of vaccination is unknown in patients receiving immune modulators and recombinant human immune mediators (eg, adalimumab, etanercept, infliximab); thus, these patients too should be vaccinated one month before starting these treatments or one month after their completion.47
On the next page: Conclusion >>
CONCLUSION
Herpes zoster remains a common disease in the US, despite the availability of an effective vaccine. While most cases of shingles resolve spontaneously, life-threatening and permanent complications can occur. Treatment may shorten the length of illness and prevent these complications. Primary care providers should recommend routine vaccination against HZ for their immunocompetent patients 60 or older.
1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
1. CDC. Shingles (herpes zoster). www.cdc.gov/shingles/hcp/clinical-overview.html. Accessed June 26, 2013.
2. Tseng HF, Smith N, Harpaz R, et al. Herpes zoster vaccine in older adults and the risk of subsequent herpes zoster disease. JAMA. 2011;305:160-166.
3. Weinberg JM. Herpes zoster: epidemiology, natural history, and common complications. J Am Acad Dermatol. 2007;57:S130-S135.
4. Kennedy PG, Cohrs RJ. Varicella-zoster virus human ganglionic latency: a current summary. J Neurovirol. 2010;16:411-418.
5. Wilson DD. Herpes zoster: prevention, diagnosis and treatment. Nurse Pract. 2007;32:19-24.
6. Chen TM, George S, Woodruff CA, Hsu S. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
7. Gilden D, Mahalingam R, Nagel MA, et al. Review: the neurobiology of varicella zoster virus infection. Neuropathol Appl Neurobiol. 2011;37:441-463.
8. CDC. Chickenpox (varicella): monitoring the impact of varicella vaccination. www.cdc.gov/chickenpox/hcp/monitoring-varicella.html. Accessed June 26, 2013.
9. Civen R, Chaves SS, Jumaan A, et all. The incidence and clinical characteristics of herpes zoster among children and adolescents after implementation of varicella vaccination. Pediatr Infect Dis J. 2009;28:954-959.
10. Hardy I, Gershon AA, Steinberg SP, LaRussa P; Varicella Vaccine Collaborative Study Group. The incidence of zoster after immunization with live attenuated varicella vaccine: a study in children with leukemia. N Engl J Med. 1991;325:1545-1550.
11. Poletti P, Melegaro A, Ajelli M, et al. Perspectives on the impact of varicella immunization on herpes zoster: a model-based evaluation from three European countries. PLoS One. 2013;8:e60732.
12. Goldman GS, King PG. Review of the United States universal varicella vaccination program: herpes zoster incidence rates, cost-effectiveness, and vaccine efficacy based primarily on the Antelope Valley Varicella Active Surveillance Project data. Vaccine. 2013;31:1680-1694.
13. Leung J, Harpaz R, Molinari NA, et al. Herpes zoster incidence among insured persons in the United States, 1993-2006: evaluation of impact of varicella vaccination. Clin Infect Dis. 2011;52:332-340.
14. Donahue JG, Kieke BA, Gargiullo PM, et al. Herpes zoster and exposure to the varicella zoster virus in an era of varicella vaccination. Am J Public Health. 2010;100:1116-1122.
15. Schmader KE, Oxman MN. Varicella and herpes zoster. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
16. Wilson JF. Herpes zoster. Ann Intern Med. 2011;154:ITC31-ITC15.
17. Harpaz R, Ortega-Sanchez IR, Seward JF. Prevention of herpes zoster: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2008;57(RR-5):1-30.
18. Gnann JW Jr, Whitley RJ. Clinical practice: herpes zoster. N Engl J Med. 2002; 347:340-346.
19. Sauerbrei A, Eichhorn U, Schacke M, Wutzler P. Laboratory diagnosis of herpes zoster. J Clin Virol. 1999;14:31-36.
20. Whitley RJ. A 70-year-old woman with shingles. JAMA. 2009;302:73-80.
21. Galluzzi KE. Managing herpes zoster and postherpetic neuralgia. J Am Osteopath Assoc. 2009;109(6 suppl 2):S7-S12.
22. Fashner J, Bell AL. Herpes zoster and postherpetic neuralgia: prevention and management. Am Fam Physician. 2011;83:1432-1437.
23. Tyring S, Barbarash RA, Nahlik JE, et al; Collaborative Famciclovir Herpes Zoster Study Group. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:89-96.
24. Pavan-Langston D. Herpes zoster: antivirals and pain management. Ophthalmology. 2008;115(2 suppl):S13-S20.
25. Opstelten W, Eekhof J, Neven AK, Verheij T. Treatment of herpes zoster. Can Fam Physician. 2008;54:373-377.
26. Tyring SK, Beutner KR, Tucker BA, et al. Antiviral therapy for herpes zoster: randomized, controlled clinical trial of valacyclovir and famciclovir therapy in immunocompetent patients 50 years and older. Arch Fam Med. 2000;9: 863-869.
27. Shafran SD, Tyring SK, Ashton R, et al. Once, twice, or three times daily famciclovir compared with aciclovir for the oral treatment of herpes zoster in immunocompetent adults: a randomized, multicenter, double-blind clinical trial. J Clin Virol. 2004;29:248-253.
28. Dworkin RH, Johnson RW, Breuer J, et al. Recommendations for the management of herpes zoster. Clin Infect Dis. 2007;44(suppl 1):S1-S26.
29. Benbernou A, Drolet M, Levin MJ, et al. Association between prodromal pain and the severity of acute herpes zoster and utilization of health care resources. Eur J Pain. 2011;15:1100-1106.
30. Gan EY, Tian EA, Tey HL. Management of herpes zoster and post-herpetic neuralgia. Am J Clin Dermatol. 2013;14:77-85.
31. Whitley RJ, Weiss H, Gnann JW Jr, et al; National Institute of Allergy and Infectious Diseases Collaborative Antiviral Study Group. Acyclovir with and without prednisone for the treatment of herpes zoster: a randomized, placebo-controlled trial. Ann Intern Med. 1996;125:376-383.
32. Wood MJ, Johnson RW, McKendrick MW, et al. A randomized trial of acyclovir for 7 days or 21 days with and without prednisolone for treatment of acute herpes zoster. N Engl J Med. 1994;330:896-900.
33. Wareham DW, Breuer J. Herpes zoster. BMJ. 2007;334:1211–1215.
34. Chen N, Yang M, He L, et al. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev. 2010;(12):CD005582.
35. Shaikh S, Ta CN. Evaluation and management of herpes zoster ophthalmicus. Am Fam Physician. 2002;66:1723-1730.
36. Sweeney CJ, Gilden DH. Ramsay Hunt syndrome. J Neurol Neurosurg Psychiatry. 2001;71:149-154.
37. Tilki HE, Mutluer N, Selçuki D, Stålberg E. Zoster paresis. Electromyogr Clin Neurophysiol. 2003;43:231-234.
38. Oxman MN, Levin MJ, Johnson GR, et al; Shingles Prevention Study Group. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271-2284.
39. Opstelten W, Zuithoff NP, van Essen GA, et al. Predicting postherpetic neuralgia in elderly primary care patients with herpes zoster: prospective prognostic study. Pain. 2007;132(suppl 1):S52-S59.
40. Mahamud A, Marin M, Nickell SP, et al. Herpes zoster-related deaths in the United States: validity of death certificates and mortality rates, 1979-2007. Clin Infect Dis. 2012;55:960-6.
41. Marin M, Güris D, Chaves SS, et al. Prevention of varicella: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2007;56(RR-4):1-40.
42. Zostavax® (zoster vaccine live). Highlights of prescribing information (2013). www.merck.com/product/usa/pi_circulars/z/zostavax/zostavax_pi2.pdf. Accessed June 26, 2013.
43. CDC. Update on herpes zoster vaccine: licensure for persons aged 50 through 59 years. MMWR Morb Mortal Wkly Rep. 2011;60:1528.
44. Simberkoff MS, Arbeit RD, Johnson GR, et al; Shingles Prevention Study Group. Safety of herpes zoster vaccine in the shingles prevention study: a randomized trial. Ann Intern Med. 2010;152:545-554.
45. Levin MJ, Oxman MN, Zhang JH, et al; Veterans Affairs Cooperative Studies Program Shingles Prevention Study Investigators. Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis. 2008;197:825-835.
46. Schmader KE, Oxman MN, Levin MJ, et al. Persistence of the efficacy of zoster vaccine in the Shingles Prevention Study and the Short-Term Persistence Substudy. Clin Infect Dis. 2012;55:1320-1328.
47. Singh A, Englund K. Q: Who should receive the shingles vaccine? Cleveland Clin J Med. 2009;76:45-48.
48. Mills R, Tyring SK, Levin MJ, et al. Safety, tolerability, and immunogenicity of zoster vaccine in subjects with a history of herpes zoster. Vaccine. 2010;28:4204-4209.
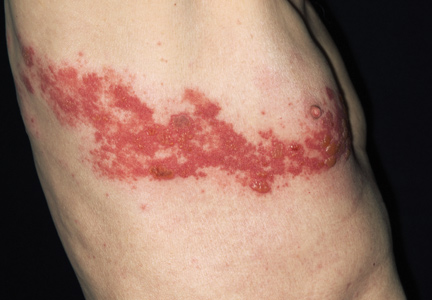
Chest x-rays for asthma doubled in 15 years
Between 1995 and 2009, the use of chest x-rays for children with asthma significantly increased in emergency departments overall, but dropped in pediatric emergency departments – findings that have implications for savings in health care costs and safety, according to Dr. Jane F. Knapp and her associates.
Citing factors that include the average cost of a chest x-ray ($370), the average time that obtaining an x-ray adds to an ED visit (27 minutes), and evidence that many children with respiratory illnesses can be treated safely and effectively without an x-ray, the authors pointed out that "reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure." The results of the retrospective study appear in the August issue of Pediatrics (2013;132:245-52).
Dr. Knapp of Children’s Mercy Hospitals and Clinics, Kansas City, Mo., and her coauthors used national survey data on ED visits from a sample of nonfederal, general, and short-stay hospitals in the United States to compare the rates of x-ray use for three groups of children evaluated between 1995 and 2009: those with moderate to severe asthma, aged 2-18 years; children with bronchiolitis, aged 3 months to 1 year; and children with croup, aged 3 months to 6 years.
During this period, the use of x-rays did not change significantly for the groups with bronchiolitis and croup. But the use of x-rays for children with asthma increased every year, about 7% a year (OR, 1.07). Although this annual increase appears small, it added up to a 2.4-fold increase over the period of time studied, the authors said.
"...Reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure."
In pediatric EDs specifically, the use of x-rays significantly decreased during the period studied for all three respiratory illnesses: asthma (OR, 0.44), bronchiolitis (OR, 0.37), and croup (OR, 0.34). A look at trends according to region showed that the use of x-rays in EDs for all three conditions increased significantly in the Midwest and South compared with the Northeast. In the West, use increased significantly for those with asthma and bronchiolitis only.
The increase in x-ray use among the children with asthma could not be explained by changes in the National Asthma Education and Prevention Program guidelines. The researchers speculated that some factors related to parents, patients, and physicians. They cited evidence that physicians are more likely to order antibiotics for a child or an x-ray for a respiratory illness or low back pain when parents or patients expect such measures. "Pressures on ED physicians to practice more aggressively could also be involved," they wrote.
Although the National Heart, Lung, and Blood Institute guidelines on the use of chest x-rays in children with asthma did not change during the study period, "we also believe that they do not provide the criteria that are sufficiently explicit to affect the discretionary use of x-rays," Dr Knapp and her associates said.
“As we discussed in the article, we need to understand the reasons why more x-rays are being used over time in the ED care of the child with asthma rather than less. The reasons may be multifactorial, but just as we have worked to limit the overuse of antibiotics with guidelines, parental education, and individual and system performance evaluation, we need to find ways to limit the excess radiation and cost associated with the use of x-rays," Dr. Knapp said in an interview.
There are almost 1 million ED visits per year for pediatric asthma, bronchiolitis, and croup, they noted.
The study did not receive external funding and the authors had no disclosures.
This story was updated. 8/5/2013
Between 1995 and 2009, the use of chest x-rays for children with asthma significantly increased in emergency departments overall, but dropped in pediatric emergency departments – findings that have implications for savings in health care costs and safety, according to Dr. Jane F. Knapp and her associates.
Citing factors that include the average cost of a chest x-ray ($370), the average time that obtaining an x-ray adds to an ED visit (27 minutes), and evidence that many children with respiratory illnesses can be treated safely and effectively without an x-ray, the authors pointed out that "reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure." The results of the retrospective study appear in the August issue of Pediatrics (2013;132:245-52).
Dr. Knapp of Children’s Mercy Hospitals and Clinics, Kansas City, Mo., and her coauthors used national survey data on ED visits from a sample of nonfederal, general, and short-stay hospitals in the United States to compare the rates of x-ray use for three groups of children evaluated between 1995 and 2009: those with moderate to severe asthma, aged 2-18 years; children with bronchiolitis, aged 3 months to 1 year; and children with croup, aged 3 months to 6 years.
During this period, the use of x-rays did not change significantly for the groups with bronchiolitis and croup. But the use of x-rays for children with asthma increased every year, about 7% a year (OR, 1.07). Although this annual increase appears small, it added up to a 2.4-fold increase over the period of time studied, the authors said.
"...Reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure."
In pediatric EDs specifically, the use of x-rays significantly decreased during the period studied for all three respiratory illnesses: asthma (OR, 0.44), bronchiolitis (OR, 0.37), and croup (OR, 0.34). A look at trends according to region showed that the use of x-rays in EDs for all three conditions increased significantly in the Midwest and South compared with the Northeast. In the West, use increased significantly for those with asthma and bronchiolitis only.
The increase in x-ray use among the children with asthma could not be explained by changes in the National Asthma Education and Prevention Program guidelines. The researchers speculated that some factors related to parents, patients, and physicians. They cited evidence that physicians are more likely to order antibiotics for a child or an x-ray for a respiratory illness or low back pain when parents or patients expect such measures. "Pressures on ED physicians to practice more aggressively could also be involved," they wrote.
Although the National Heart, Lung, and Blood Institute guidelines on the use of chest x-rays in children with asthma did not change during the study period, "we also believe that they do not provide the criteria that are sufficiently explicit to affect the discretionary use of x-rays," Dr Knapp and her associates said.
“As we discussed in the article, we need to understand the reasons why more x-rays are being used over time in the ED care of the child with asthma rather than less. The reasons may be multifactorial, but just as we have worked to limit the overuse of antibiotics with guidelines, parental education, and individual and system performance evaluation, we need to find ways to limit the excess radiation and cost associated with the use of x-rays," Dr. Knapp said in an interview.
There are almost 1 million ED visits per year for pediatric asthma, bronchiolitis, and croup, they noted.
The study did not receive external funding and the authors had no disclosures.
This story was updated. 8/5/2013
Between 1995 and 2009, the use of chest x-rays for children with asthma significantly increased in emergency departments overall, but dropped in pediatric emergency departments – findings that have implications for savings in health care costs and safety, according to Dr. Jane F. Knapp and her associates.
Citing factors that include the average cost of a chest x-ray ($370), the average time that obtaining an x-ray adds to an ED visit (27 minutes), and evidence that many children with respiratory illnesses can be treated safely and effectively without an x-ray, the authors pointed out that "reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure." The results of the retrospective study appear in the August issue of Pediatrics (2013;132:245-52).
Dr. Knapp of Children’s Mercy Hospitals and Clinics, Kansas City, Mo., and her coauthors used national survey data on ED visits from a sample of nonfederal, general, and short-stay hospitals in the United States to compare the rates of x-ray use for three groups of children evaluated between 1995 and 2009: those with moderate to severe asthma, aged 2-18 years; children with bronchiolitis, aged 3 months to 1 year; and children with croup, aged 3 months to 6 years.
During this period, the use of x-rays did not change significantly for the groups with bronchiolitis and croup. But the use of x-rays for children with asthma increased every year, about 7% a year (OR, 1.07). Although this annual increase appears small, it added up to a 2.4-fold increase over the period of time studied, the authors said.
"...Reversing this trend could improve ED efficiency, decrease costs, and decrease radiation exposure."
In pediatric EDs specifically, the use of x-rays significantly decreased during the period studied for all three respiratory illnesses: asthma (OR, 0.44), bronchiolitis (OR, 0.37), and croup (OR, 0.34). A look at trends according to region showed that the use of x-rays in EDs for all three conditions increased significantly in the Midwest and South compared with the Northeast. In the West, use increased significantly for those with asthma and bronchiolitis only.
The increase in x-ray use among the children with asthma could not be explained by changes in the National Asthma Education and Prevention Program guidelines. The researchers speculated that some factors related to parents, patients, and physicians. They cited evidence that physicians are more likely to order antibiotics for a child or an x-ray for a respiratory illness or low back pain when parents or patients expect such measures. "Pressures on ED physicians to practice more aggressively could also be involved," they wrote.
Although the National Heart, Lung, and Blood Institute guidelines on the use of chest x-rays in children with asthma did not change during the study period, "we also believe that they do not provide the criteria that are sufficiently explicit to affect the discretionary use of x-rays," Dr Knapp and her associates said.
“As we discussed in the article, we need to understand the reasons why more x-rays are being used over time in the ED care of the child with asthma rather than less. The reasons may be multifactorial, but just as we have worked to limit the overuse of antibiotics with guidelines, parental education, and individual and system performance evaluation, we need to find ways to limit the excess radiation and cost associated with the use of x-rays," Dr. Knapp said in an interview.
There are almost 1 million ED visits per year for pediatric asthma, bronchiolitis, and croup, they noted.
The study did not receive external funding and the authors had no disclosures.
This story was updated. 8/5/2013
FROM PEDIATRICS
Asthma medications
One of the most common conditions that complicate pregnancy is maternal asthma. Evidence continues to mount that adequate control of asthma, including appropriate use of medications, is the best approach to optimizing outcomes. Yet questions persist about the effect of asthma itself, as well as specific medications and the risk for major congenital malformations. As with any exposure during pregnancy, answering these questions is challenging because of the rarity of specific birth defects and the various and increasing number of medications that might be used to treat or prevent asthma symptoms.
Previously in this column ("Beta2-agonists for asthma, December 2011), we reviewed two studies that suggested short-acting beta-agonists used for the treatment of asthma were associated with an increased risk of oral clefts and that long-acting beta-agonists might be associated with an increased risk of cardiac anomalies (Hum. Reprod. 2011;26:3147-54; Birth Defects Res. A. Clin. Mol. Teratol. 2011;91:937-47).
What have we learned since then? Two studies published in 2013 add to the body of knowledge. The first, a database analysis using the United Kingdom’s General Practice Research Database, assessed pregnancy outcomes between 1991 and 2002 in 7,911 women exposed to asthma medications in the first trimester of pregnancy and 15,840 women who were not exposed (Pharmacotherapy 2013;33:363-8). Major anomalies were identified up to 1 year of age. Minor anomalies, chromosomal anomalies, and those associated with prematurity were excluded.
The overall risk for any exposure, compared with no exposure, for any congenital anomaly was 1.1 (95% confidence interval [CI], 1.0-1.3). No significant differences were found by class of asthma medication. Specific categories of defects also were evaluated, including musculoskeletal anomalies, oral clefts, cardiovascular defects, and multiple anomalies. Some estimates were elevated for specific medication classes.
For example, the relative risk (RR) of cleft lip or palate associated with exposure to long-acting beta-agonists was 2.4, but the confidence interval included 1 (0.3-21.8) based on 424 exposed pregnancies. The authors concluded that they found no significant increased risk of congenital anomalies associated with exposure to asthma, asthma medications, or any specific asthma medication classes in the first trimester. Limitations of the study included the inability to verify that exposure took place and insufficient data to adjust for confounding by vitamin supplementation, alcohol use, socioeconomic status, or markers of disease severity (other than number of medications prescribed).
The second study addressed the issue of maternal asthma itself and the risk for congenital anomalies (BJOG 2013;120:812-22). Using a meta-analysis approach, 21 cohort studies published between 1975 and 2012 met the criteria for inclusion. Combining major and minor congenital anomalies, the authors found a slight but statistically significant increased risk for any defect (RR, 1.11; 95% CI, 1.01-1.68), but when the analysis was restricted to major defects alone, the summary estimate was elevated but no longer significant (RR, 1.31; 95% CI, 0.57-3.02). When the specific defect grouping of oral clefts was examined, however, there was a significantly elevated overall relative risk of 1.30 (95% CI, 1.01-1.68). Limitations of this study include differing quality of studies and the appropriateness of combining data to derive a summary estimate.
Although both studies provide reassurance about the overall risk of major defects in the offspring of women with asthma, both suggest that more work needs to be done to follow up on the risk for oral clefts and whether this is linked to underlying disease severity and/or use of specific medications. Furthermore, safety of specific long-acting beta-agonist medications in pregnancy should be further examined.
The take-home message, however, continues to be that the risk for major defects in the offspring of pregnant women with asthma appears to be low, which supports the recommendation to follow guidelines for appropriate treatment of women with asthma both during and outside of pregnancy to control symptoms.
Dr. Chambers is professor of pediatrics and family and preventive medicine at the University of California, San Diego. She is director of the California Teratogen Information Service and Clinical Research Program. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said that she had no relevant financial disclosures.
One of the most common conditions that complicate pregnancy is maternal asthma. Evidence continues to mount that adequate control of asthma, including appropriate use of medications, is the best approach to optimizing outcomes. Yet questions persist about the effect of asthma itself, as well as specific medications and the risk for major congenital malformations. As with any exposure during pregnancy, answering these questions is challenging because of the rarity of specific birth defects and the various and increasing number of medications that might be used to treat or prevent asthma symptoms.
Previously in this column ("Beta2-agonists for asthma, December 2011), we reviewed two studies that suggested short-acting beta-agonists used for the treatment of asthma were associated with an increased risk of oral clefts and that long-acting beta-agonists might be associated with an increased risk of cardiac anomalies (Hum. Reprod. 2011;26:3147-54; Birth Defects Res. A. Clin. Mol. Teratol. 2011;91:937-47).
What have we learned since then? Two studies published in 2013 add to the body of knowledge. The first, a database analysis using the United Kingdom’s General Practice Research Database, assessed pregnancy outcomes between 1991 and 2002 in 7,911 women exposed to asthma medications in the first trimester of pregnancy and 15,840 women who were not exposed (Pharmacotherapy 2013;33:363-8). Major anomalies were identified up to 1 year of age. Minor anomalies, chromosomal anomalies, and those associated with prematurity were excluded.
The overall risk for any exposure, compared with no exposure, for any congenital anomaly was 1.1 (95% confidence interval [CI], 1.0-1.3). No significant differences were found by class of asthma medication. Specific categories of defects also were evaluated, including musculoskeletal anomalies, oral clefts, cardiovascular defects, and multiple anomalies. Some estimates were elevated for specific medication classes.
For example, the relative risk (RR) of cleft lip or palate associated with exposure to long-acting beta-agonists was 2.4, but the confidence interval included 1 (0.3-21.8) based on 424 exposed pregnancies. The authors concluded that they found no significant increased risk of congenital anomalies associated with exposure to asthma, asthma medications, or any specific asthma medication classes in the first trimester. Limitations of the study included the inability to verify that exposure took place and insufficient data to adjust for confounding by vitamin supplementation, alcohol use, socioeconomic status, or markers of disease severity (other than number of medications prescribed).
The second study addressed the issue of maternal asthma itself and the risk for congenital anomalies (BJOG 2013;120:812-22). Using a meta-analysis approach, 21 cohort studies published between 1975 and 2012 met the criteria for inclusion. Combining major and minor congenital anomalies, the authors found a slight but statistically significant increased risk for any defect (RR, 1.11; 95% CI, 1.01-1.68), but when the analysis was restricted to major defects alone, the summary estimate was elevated but no longer significant (RR, 1.31; 95% CI, 0.57-3.02). When the specific defect grouping of oral clefts was examined, however, there was a significantly elevated overall relative risk of 1.30 (95% CI, 1.01-1.68). Limitations of this study include differing quality of studies and the appropriateness of combining data to derive a summary estimate.
Although both studies provide reassurance about the overall risk of major defects in the offspring of women with asthma, both suggest that more work needs to be done to follow up on the risk for oral clefts and whether this is linked to underlying disease severity and/or use of specific medications. Furthermore, safety of specific long-acting beta-agonist medications in pregnancy should be further examined.
The take-home message, however, continues to be that the risk for major defects in the offspring of pregnant women with asthma appears to be low, which supports the recommendation to follow guidelines for appropriate treatment of women with asthma both during and outside of pregnancy to control symptoms.
Dr. Chambers is professor of pediatrics and family and preventive medicine at the University of California, San Diego. She is director of the California Teratogen Information Service and Clinical Research Program. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said that she had no relevant financial disclosures.
One of the most common conditions that complicate pregnancy is maternal asthma. Evidence continues to mount that adequate control of asthma, including appropriate use of medications, is the best approach to optimizing outcomes. Yet questions persist about the effect of asthma itself, as well as specific medications and the risk for major congenital malformations. As with any exposure during pregnancy, answering these questions is challenging because of the rarity of specific birth defects and the various and increasing number of medications that might be used to treat or prevent asthma symptoms.
Previously in this column ("Beta2-agonists for asthma, December 2011), we reviewed two studies that suggested short-acting beta-agonists used for the treatment of asthma were associated with an increased risk of oral clefts and that long-acting beta-agonists might be associated with an increased risk of cardiac anomalies (Hum. Reprod. 2011;26:3147-54; Birth Defects Res. A. Clin. Mol. Teratol. 2011;91:937-47).
What have we learned since then? Two studies published in 2013 add to the body of knowledge. The first, a database analysis using the United Kingdom’s General Practice Research Database, assessed pregnancy outcomes between 1991 and 2002 in 7,911 women exposed to asthma medications in the first trimester of pregnancy and 15,840 women who were not exposed (Pharmacotherapy 2013;33:363-8). Major anomalies were identified up to 1 year of age. Minor anomalies, chromosomal anomalies, and those associated with prematurity were excluded.
The overall risk for any exposure, compared with no exposure, for any congenital anomaly was 1.1 (95% confidence interval [CI], 1.0-1.3). No significant differences were found by class of asthma medication. Specific categories of defects also were evaluated, including musculoskeletal anomalies, oral clefts, cardiovascular defects, and multiple anomalies. Some estimates were elevated for specific medication classes.
For example, the relative risk (RR) of cleft lip or palate associated with exposure to long-acting beta-agonists was 2.4, but the confidence interval included 1 (0.3-21.8) based on 424 exposed pregnancies. The authors concluded that they found no significant increased risk of congenital anomalies associated with exposure to asthma, asthma medications, or any specific asthma medication classes in the first trimester. Limitations of the study included the inability to verify that exposure took place and insufficient data to adjust for confounding by vitamin supplementation, alcohol use, socioeconomic status, or markers of disease severity (other than number of medications prescribed).
The second study addressed the issue of maternal asthma itself and the risk for congenital anomalies (BJOG 2013;120:812-22). Using a meta-analysis approach, 21 cohort studies published between 1975 and 2012 met the criteria for inclusion. Combining major and minor congenital anomalies, the authors found a slight but statistically significant increased risk for any defect (RR, 1.11; 95% CI, 1.01-1.68), but when the analysis was restricted to major defects alone, the summary estimate was elevated but no longer significant (RR, 1.31; 95% CI, 0.57-3.02). When the specific defect grouping of oral clefts was examined, however, there was a significantly elevated overall relative risk of 1.30 (95% CI, 1.01-1.68). Limitations of this study include differing quality of studies and the appropriateness of combining data to derive a summary estimate.
Although both studies provide reassurance about the overall risk of major defects in the offspring of women with asthma, both suggest that more work needs to be done to follow up on the risk for oral clefts and whether this is linked to underlying disease severity and/or use of specific medications. Furthermore, safety of specific long-acting beta-agonist medications in pregnancy should be further examined.
The take-home message, however, continues to be that the risk for major defects in the offspring of pregnant women with asthma appears to be low, which supports the recommendation to follow guidelines for appropriate treatment of women with asthma both during and outside of pregnancy to control symptoms.
Dr. Chambers is professor of pediatrics and family and preventive medicine at the University of California, San Diego. She is director of the California Teratogen Information Service and Clinical Research Program. Dr. Chambers is a past president of the Organization of Teratology Information Specialists and past president of the Teratology Society. She said that she had no relevant financial disclosures.

Risk of stopping inhaled corticosteroids
A significant dissatisfier for both clinician and patient is that inhaled corticosteroids, commonly underutilized and potentially lifesaving medications, are almost never (if ever) covered at the lowest tier by insurance companies. We would select a first-tier medication if there were one that we could substitute for an ICS; but frequently there isn’t, so we can’t.
Because of this, patients may be financially motivated to simply stop the medication – especially if they perceive that they are on the lowest doses and believe the medication perhaps is not needed at all. Clinicians, meanwhile, are doing the balancing act of moving patients to the lowest doses in order to avoid side effects while maintaining optimal disease control.
So, what are the risks when patients stop using inhaled corticosteroids?
Dr. Matthew A. Rank of the Mayo Clinic, Rochester, Minn., and his colleagues recently published a systematic review of the literature to answer this question (J. Allergy Clin. Immunol. 2013;131:724-9). In this review, randomized, controlled clinical trials in which the study intervention was continuing or stopping low-dose ICSs were included. Studies had to have 4 or more weeks of a run-in with stable doses of ICSs to ensure a minimum period of asthma stability. Seven studies met inclusion criteria. Two studies were exclusively in children, and one was exclusively in adults.
Asthma exacerbations were more likely among patients who stopped ICSs, compared with those who did not (relative risk, 2.35; 95% CI: 1.88-2.92). The risk for an asthma exacerbation in the next 6 months on low-dose ICSs was 16% if patients continued taking the medications, and 38% if they stopped. For every five patients who stopped ICSs, one patient would be expected to have an asthma exacerbation in the next 6 months – which could have been prevented if steroids had been continued. The mean decrease in forced expiratory volume in 1 second associated with discontinued ICSs use was 130 mL.
Most patients can step down with ICSs if they are on long-acting beta-agonists. Expert panels have suggested that patients should be controlled for 3 months before stepping down therapy. Findings from this study further suggest that patients who discontinue low-dose ICSs are at an increased risk of asthma exacerbation.
We need to help our patients understand the risk of stopping low-dose ICSs and encourage them, as much as they are able, to stay on them.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. He reported having no relevant financial conflicts. The opinions expressed are those of the author.
This column, "What Matters," appears regularly in Internal Medicine News.
A significant dissatisfier for both clinician and patient is that inhaled corticosteroids, commonly underutilized and potentially lifesaving medications, are almost never (if ever) covered at the lowest tier by insurance companies. We would select a first-tier medication if there were one that we could substitute for an ICS; but frequently there isn’t, so we can’t.
Because of this, patients may be financially motivated to simply stop the medication – especially if they perceive that they are on the lowest doses and believe the medication perhaps is not needed at all. Clinicians, meanwhile, are doing the balancing act of moving patients to the lowest doses in order to avoid side effects while maintaining optimal disease control.
So, what are the risks when patients stop using inhaled corticosteroids?
Dr. Matthew A. Rank of the Mayo Clinic, Rochester, Minn., and his colleagues recently published a systematic review of the literature to answer this question (J. Allergy Clin. Immunol. 2013;131:724-9). In this review, randomized, controlled clinical trials in which the study intervention was continuing or stopping low-dose ICSs were included. Studies had to have 4 or more weeks of a run-in with stable doses of ICSs to ensure a minimum period of asthma stability. Seven studies met inclusion criteria. Two studies were exclusively in children, and one was exclusively in adults.
Asthma exacerbations were more likely among patients who stopped ICSs, compared with those who did not (relative risk, 2.35; 95% CI: 1.88-2.92). The risk for an asthma exacerbation in the next 6 months on low-dose ICSs was 16% if patients continued taking the medications, and 38% if they stopped. For every five patients who stopped ICSs, one patient would be expected to have an asthma exacerbation in the next 6 months – which could have been prevented if steroids had been continued. The mean decrease in forced expiratory volume in 1 second associated with discontinued ICSs use was 130 mL.
Most patients can step down with ICSs if they are on long-acting beta-agonists. Expert panels have suggested that patients should be controlled for 3 months before stepping down therapy. Findings from this study further suggest that patients who discontinue low-dose ICSs are at an increased risk of asthma exacerbation.
We need to help our patients understand the risk of stopping low-dose ICSs and encourage them, as much as they are able, to stay on them.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. He reported having no relevant financial conflicts. The opinions expressed are those of the author.
This column, "What Matters," appears regularly in Internal Medicine News.
A significant dissatisfier for both clinician and patient is that inhaled corticosteroids, commonly underutilized and potentially lifesaving medications, are almost never (if ever) covered at the lowest tier by insurance companies. We would select a first-tier medication if there were one that we could substitute for an ICS; but frequently there isn’t, so we can’t.
Because of this, patients may be financially motivated to simply stop the medication – especially if they perceive that they are on the lowest doses and believe the medication perhaps is not needed at all. Clinicians, meanwhile, are doing the balancing act of moving patients to the lowest doses in order to avoid side effects while maintaining optimal disease control.
So, what are the risks when patients stop using inhaled corticosteroids?
Dr. Matthew A. Rank of the Mayo Clinic, Rochester, Minn., and his colleagues recently published a systematic review of the literature to answer this question (J. Allergy Clin. Immunol. 2013;131:724-9). In this review, randomized, controlled clinical trials in which the study intervention was continuing or stopping low-dose ICSs were included. Studies had to have 4 or more weeks of a run-in with stable doses of ICSs to ensure a minimum period of asthma stability. Seven studies met inclusion criteria. Two studies were exclusively in children, and one was exclusively in adults.
Asthma exacerbations were more likely among patients who stopped ICSs, compared with those who did not (relative risk, 2.35; 95% CI: 1.88-2.92). The risk for an asthma exacerbation in the next 6 months on low-dose ICSs was 16% if patients continued taking the medications, and 38% if they stopped. For every five patients who stopped ICSs, one patient would be expected to have an asthma exacerbation in the next 6 months – which could have been prevented if steroids had been continued. The mean decrease in forced expiratory volume in 1 second associated with discontinued ICSs use was 130 mL.
Most patients can step down with ICSs if they are on long-acting beta-agonists. Expert panels have suggested that patients should be controlled for 3 months before stepping down therapy. Findings from this study further suggest that patients who discontinue low-dose ICSs are at an increased risk of asthma exacerbation.
We need to help our patients understand the risk of stopping low-dose ICSs and encourage them, as much as they are able, to stay on them.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. He reported having no relevant financial conflicts. The opinions expressed are those of the author.
This column, "What Matters," appears regularly in Internal Medicine News.
SABAs, not LABAs, for exercise-induced bronchoconstriction
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
Don’t base diagnosis of exercise-induced bronchoconstriction on symptoms alone – but do have patients use inhaled short-acting beta-agonists and do warm-ups before exercise, according to new clinical practice guidelines issued May 1 by the American Thoracic Society.
The guidelines define exercise-induced bronchoconstriction (EIB) as "acute airway narrowing that occurs as a result of exercise."
Considering the high prevalence of EIB, which also affects people without asthma, "evidence-based guidelines for its management are of critical importance," said Dr. Jonathan Parsons, the lead author and chair of the committee that drafted the guidelines, in a statement.
The recommendations "synthesize the latest clinical evidence and will help guide the management of EIB in patients with or without asthma, and in athletes at all levels of competition," added Dr. Parsons, associate professor of internal medicine and associate director of the Ohio State University Asthma Center, Columbus.
The EIB guidelines cover pathogenesis, environmental triggers, diagnosis, treatment, and screening (Am. J. Resp. Crit. Care Med. 2013;187 [doi:10.1164/rccm.201303-0437ST]). Also included is a section on exercise, asthma, and doping – with reminders about which EIB drugs are banned in competitive sports (most beta-agonists) and which are allowed (short-acting inhaled albuterol and inhaled steroids).
Although the guidelines can apply both to adolescents and adults, they cannot be applied reliably to young children, Dr. Parsons noted in an interview.
EIB prevalence among people with asthma is not known, but the estimated prevalence among people who have not been diagnosed with asthma is as high as 20%, according to the ATS. EIB is more prevalent among athletes, affecting 30%-70% of Olympic and elite athletes. Environmental factors likely play a role, such as pollutants emitted from ice surfacing machines in indoor ice rinks, high trichloramine levels in the air of indoor pools, and cold, dry air.
An EIB diagnosis should not be based on symptoms, which are variable, nonspecific, and have poor predictive value. Instead, diagnosis should be made based on changes in lung function provoked by exercise, using serial lung function measurements after a specific exercise or a hyperpnea challenge. Assessing the effects of exercise on forced expiratory volume in 1 second (FEV1) is preferred.
The guidelines grade EIB as mild, moderate, or severe, depending on the percent fall in FEV1 from baseline. They also offer information on alternatives to exercise testing.
The authors rate pharmacologic and nonpharmacologic therapies based on the quality of the supportive evidence. Their first recommendation – administration of an inhaled short-acting beta-agonist (SABA) before exercise – earns a "strong" recommendation based on "high-quality" evidence. Patients typically take SABAs 15 minutes before exercise.
Because of the potential for serious side effects, the authors recommend against daily use of an inhaled long-acting beta-agonist (LABA) for EIB – a strong recommendation based on moderate-quality evidence.
For patients who use an inhaled SABA but continue to have symptoms or need to use the inhaled SABA "daily or more frequently," treatment options before exercise include a daily inhaled corticosteroid (ICS), a daily leukotriene-receptor antagonist, or a mast-cell–stabilizing agent.
"We generally add a daily inhaled ICS or a daily leukotriene-receptor antagonist first, with the choice between these agents made on a case-by-case basis depending upon patient preferences," the guideline authors note. Mast-cell–stabilizing agents and inhaled anticholinergic drugs "play a secondary role," they added. There is also a role for antihistamines in patient with continued symptoms despite treatment, but not for patients without allergies.
Nonpharmacologic measures include interval or combination warm-up exercises before planned exercise, which the guidelines recommend "for all patients" with EIB – a strong recommendation, based on moderate quality evidence. The guidelines cite evidence showing a lower reduction in FEV1 after exercise among people with EIB who engaged in "interval, low-intensity continuous; high-intensity continuous; or combination warm-up" before they exercised.
Another nonpharmacologic recommendation is use of a mask or another device that warms and humidifies the air when patients exercise in a cold climate.
While there is not much evidence supporting dietary modifications, patients interested in this approach can try a low-salt diet, or take fish oil or vitamin C supplements. However, the use of lycopene is not supported, based on the available evidence.
"Our overall recommendations regarding therapy leave a lot of options for the individual patient, which should be discussed with the patient’s physician and tried and evaluated on an ongoing basis," the authors concluded.
The mainstay of treatment "remains maintaining good control of underlying asthma (if present) and preventing or treating symptoms of EIB with SABAs."
The EIB practice guidelines were supported by the ATS and approved by the ATS board of directors. Dr. Parsons’ disclosures include having received lecture fees from AstraZeneca, GlaxoSmithKline, Merck, and Schering Plough. All but one of the other authors disclosed financial relations with a wide range of pharmaceutical companies.
FROM THE AMERICAN JOURNAL OF RESPIRATORY AND CRITICAL CARE MEDICINE
A review of uncommon swelling provides useful reminders

Angioedema associated with insufficient activity of circulating complement component 1 (C1) inhibitor is a rare condition that can be hereditary or acquired. The clinical characteristics of this disorder are nicely reviewed by Drs. Tse and Zuraw in this issue of the Journal. Despite the rarity of this condition, there are good reasons to spend a few minutes with this article.
Angioedema and the urticarias differ in their clinical manifestations and diagnostic and therapeutic implications. Patients who have recurrent urticaria, no matter the severity, need not be evaluated for C1 inhibitor deficiency. Some patients who have recurrent angioedema without urticaria may respond well to antiallergic therapies (eg, antihistamines, corticosteroids), and these patients also are unlikely to have C1 inhibitor deficiency. Other patients who have intermittent localized peripheral swelling (or visceral edema manifest as abdominal pain) without urticaria may not respond, and it is in these patients that a pathophysiologic mechanism other than typical allergy should be considered.
In some patients, bradykinin-mediated angioedema is due to deficient C1 inhibitor activity, in others, to angiotensin-converting enzyme (ACE) inhibition. Although the biochemical site of action is different, both of these conditions result from an imbalance of protease activation and inhibition. If there is not enough C1 inhibition, there is too much kallikrein activity, which leads to excess proteolytic generation of the peptide bradykinin from kininogen. In contrast, ACE inhibitors occasionally cause angioedema by decreasing catabolism of bradykinin. In either case, bradykinin-mediated angioedema is generally clinically resistant to antiallergic therapy.
Thinking about these mechanisms reminds us of the complexity and overlap of many physiologic proteolytic cascades. Although C1 inhibitor is known as an inhibitor of C1 esterase and C4 levels are almost always low when C1 inhibitor activity is depressed, the angioedema in C1-inhibitor-deficiency states is more a result of decreased inhibition of enzymes in the coagulation cascade than in the complement cascade. Similarly, ACE-inhibitor-associated angioedema has little to do with the decreased generation of angiotensin that accounts for these drugs’ antihypertensive effect.
Generalizing these principles should make us vigilant for unpredicted adverse reactions to other newer drugs such as protease and kinase inhibitors, which may variably affect multiple biochemical pathways.
Angioedema associated with insufficient activity of circulating complement component 1 (C1) inhibitor is a rare condition that can be hereditary or acquired. The clinical characteristics of this disorder are nicely reviewed by Drs. Tse and Zuraw in this issue of the Journal. Despite the rarity of this condition, there are good reasons to spend a few minutes with this article.
Angioedema and the urticarias differ in their clinical manifestations and diagnostic and therapeutic implications. Patients who have recurrent urticaria, no matter the severity, need not be evaluated for C1 inhibitor deficiency. Some patients who have recurrent angioedema without urticaria may respond well to antiallergic therapies (eg, antihistamines, corticosteroids), and these patients also are unlikely to have C1 inhibitor deficiency. Other patients who have intermittent localized peripheral swelling (or visceral edema manifest as abdominal pain) without urticaria may not respond, and it is in these patients that a pathophysiologic mechanism other than typical allergy should be considered.
In some patients, bradykinin-mediated angioedema is due to deficient C1 inhibitor activity, in others, to angiotensin-converting enzyme (ACE) inhibition. Although the biochemical site of action is different, both of these conditions result from an imbalance of protease activation and inhibition. If there is not enough C1 inhibition, there is too much kallikrein activity, which leads to excess proteolytic generation of the peptide bradykinin from kininogen. In contrast, ACE inhibitors occasionally cause angioedema by decreasing catabolism of bradykinin. In either case, bradykinin-mediated angioedema is generally clinically resistant to antiallergic therapy.
Thinking about these mechanisms reminds us of the complexity and overlap of many physiologic proteolytic cascades. Although C1 inhibitor is known as an inhibitor of C1 esterase and C4 levels are almost always low when C1 inhibitor activity is depressed, the angioedema in C1-inhibitor-deficiency states is more a result of decreased inhibition of enzymes in the coagulation cascade than in the complement cascade. Similarly, ACE-inhibitor-associated angioedema has little to do with the decreased generation of angiotensin that accounts for these drugs’ antihypertensive effect.
Generalizing these principles should make us vigilant for unpredicted adverse reactions to other newer drugs such as protease and kinase inhibitors, which may variably affect multiple biochemical pathways.
Angioedema associated with insufficient activity of circulating complement component 1 (C1) inhibitor is a rare condition that can be hereditary or acquired. The clinical characteristics of this disorder are nicely reviewed by Drs. Tse and Zuraw in this issue of the Journal. Despite the rarity of this condition, there are good reasons to spend a few minutes with this article.
Angioedema and the urticarias differ in their clinical manifestations and diagnostic and therapeutic implications. Patients who have recurrent urticaria, no matter the severity, need not be evaluated for C1 inhibitor deficiency. Some patients who have recurrent angioedema without urticaria may respond well to antiallergic therapies (eg, antihistamines, corticosteroids), and these patients also are unlikely to have C1 inhibitor deficiency. Other patients who have intermittent localized peripheral swelling (or visceral edema manifest as abdominal pain) without urticaria may not respond, and it is in these patients that a pathophysiologic mechanism other than typical allergy should be considered.
In some patients, bradykinin-mediated angioedema is due to deficient C1 inhibitor activity, in others, to angiotensin-converting enzyme (ACE) inhibition. Although the biochemical site of action is different, both of these conditions result from an imbalance of protease activation and inhibition. If there is not enough C1 inhibition, there is too much kallikrein activity, which leads to excess proteolytic generation of the peptide bradykinin from kininogen. In contrast, ACE inhibitors occasionally cause angioedema by decreasing catabolism of bradykinin. In either case, bradykinin-mediated angioedema is generally clinically resistant to antiallergic therapy.
Thinking about these mechanisms reminds us of the complexity and overlap of many physiologic proteolytic cascades. Although C1 inhibitor is known as an inhibitor of C1 esterase and C4 levels are almost always low when C1 inhibitor activity is depressed, the angioedema in C1-inhibitor-deficiency states is more a result of decreased inhibition of enzymes in the coagulation cascade than in the complement cascade. Similarly, ACE-inhibitor-associated angioedema has little to do with the decreased generation of angiotensin that accounts for these drugs’ antihypertensive effect.
Generalizing these principles should make us vigilant for unpredicted adverse reactions to other newer drugs such as protease and kinase inhibitors, which may variably affect multiple biochemical pathways.
Recognizing and managing hereditary angioedema
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
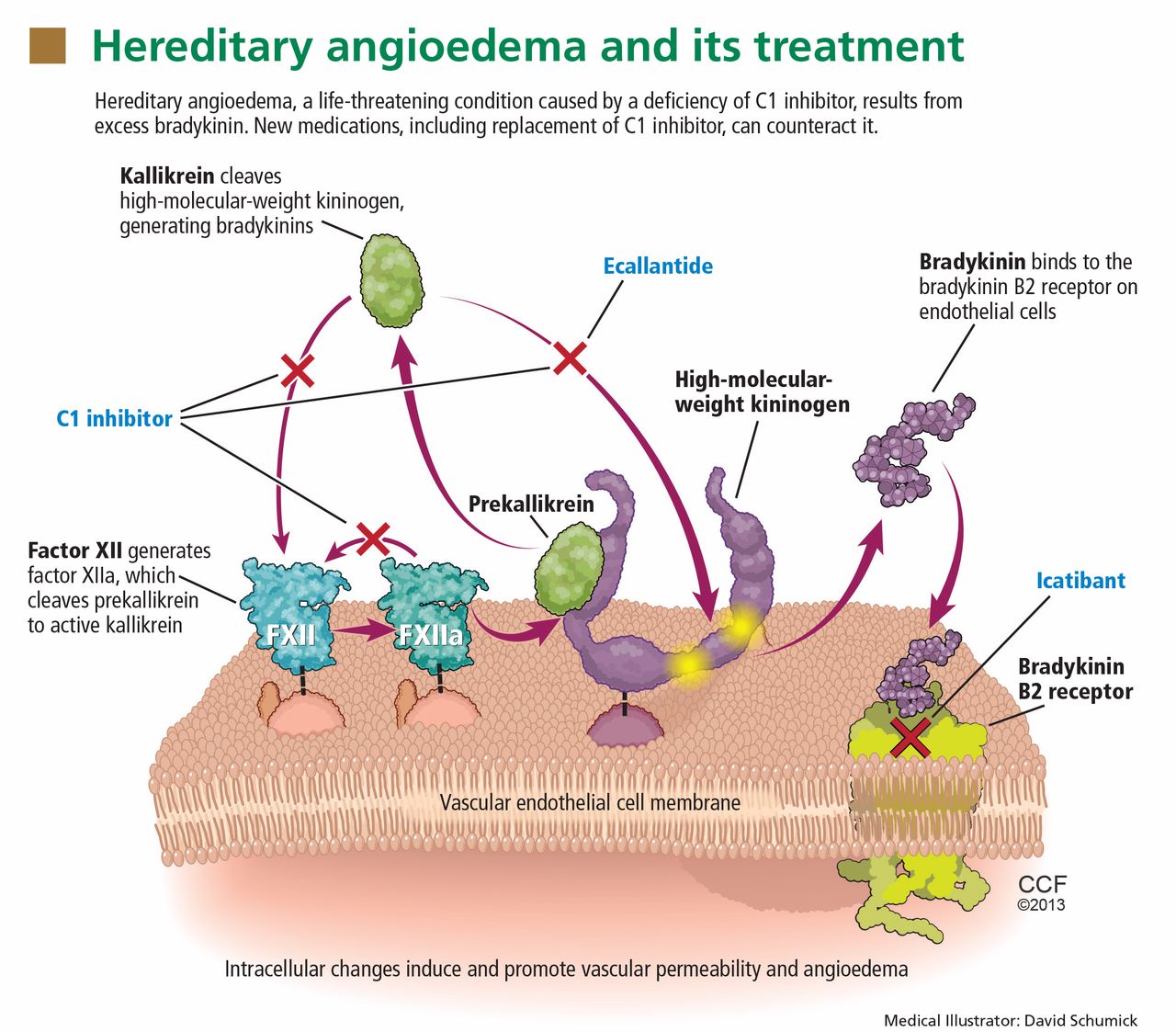
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
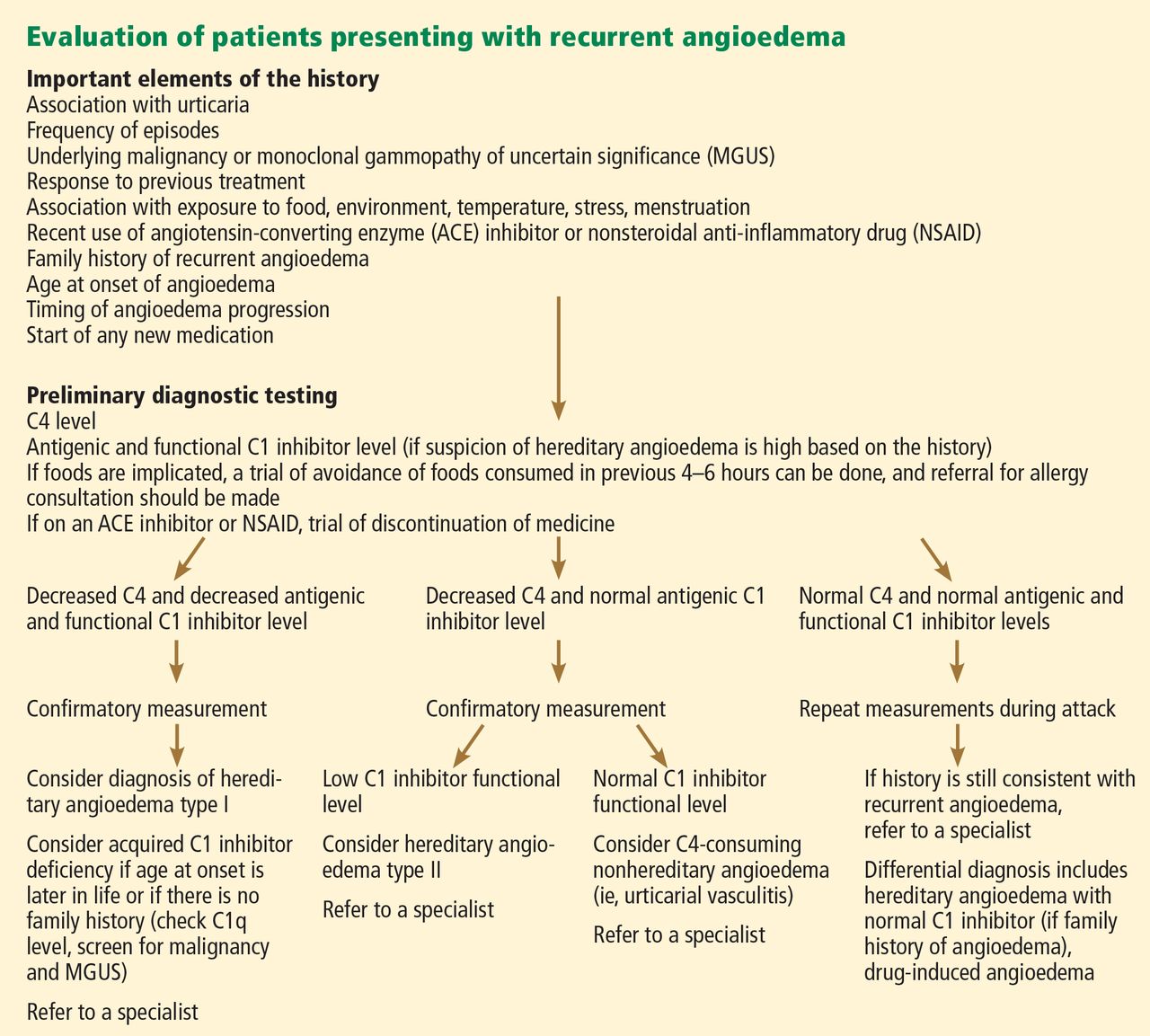
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
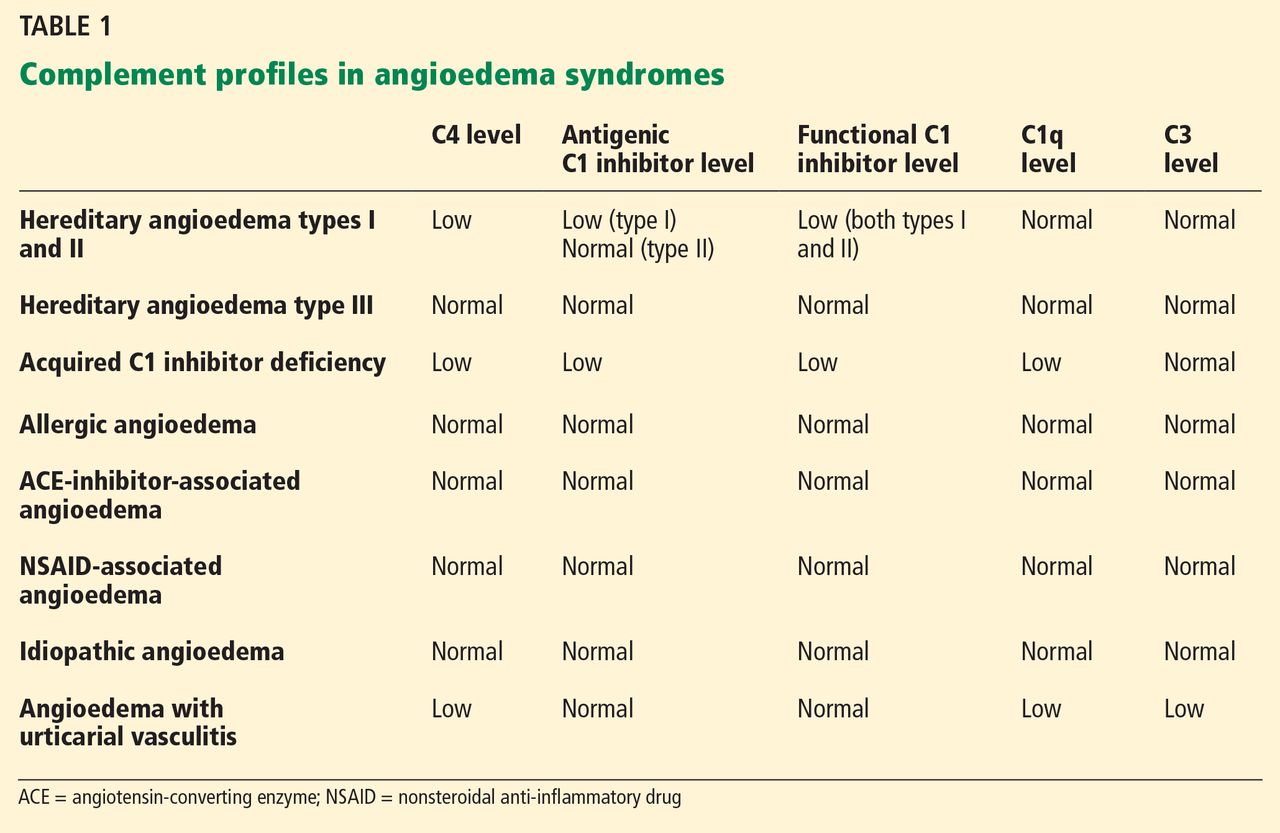
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
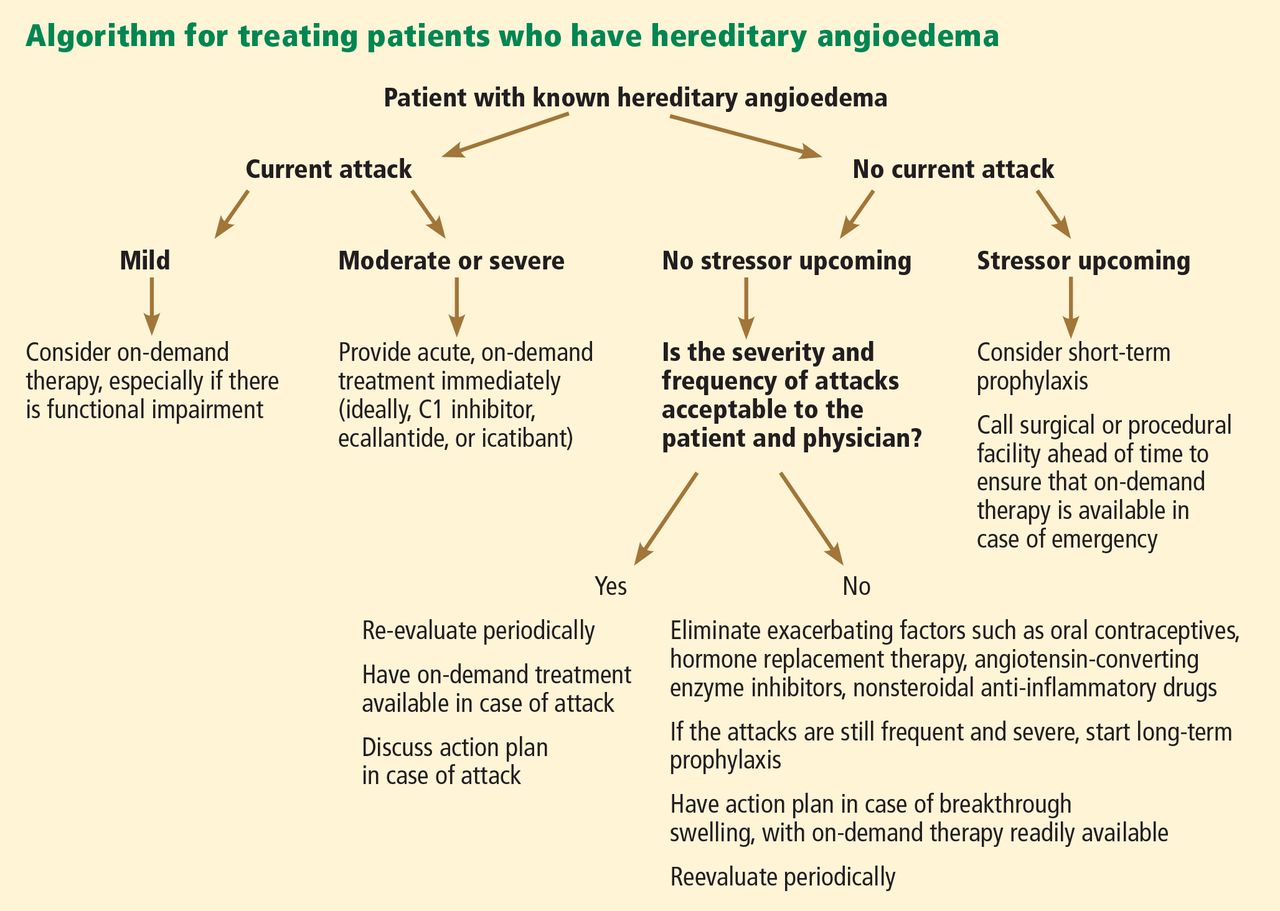
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
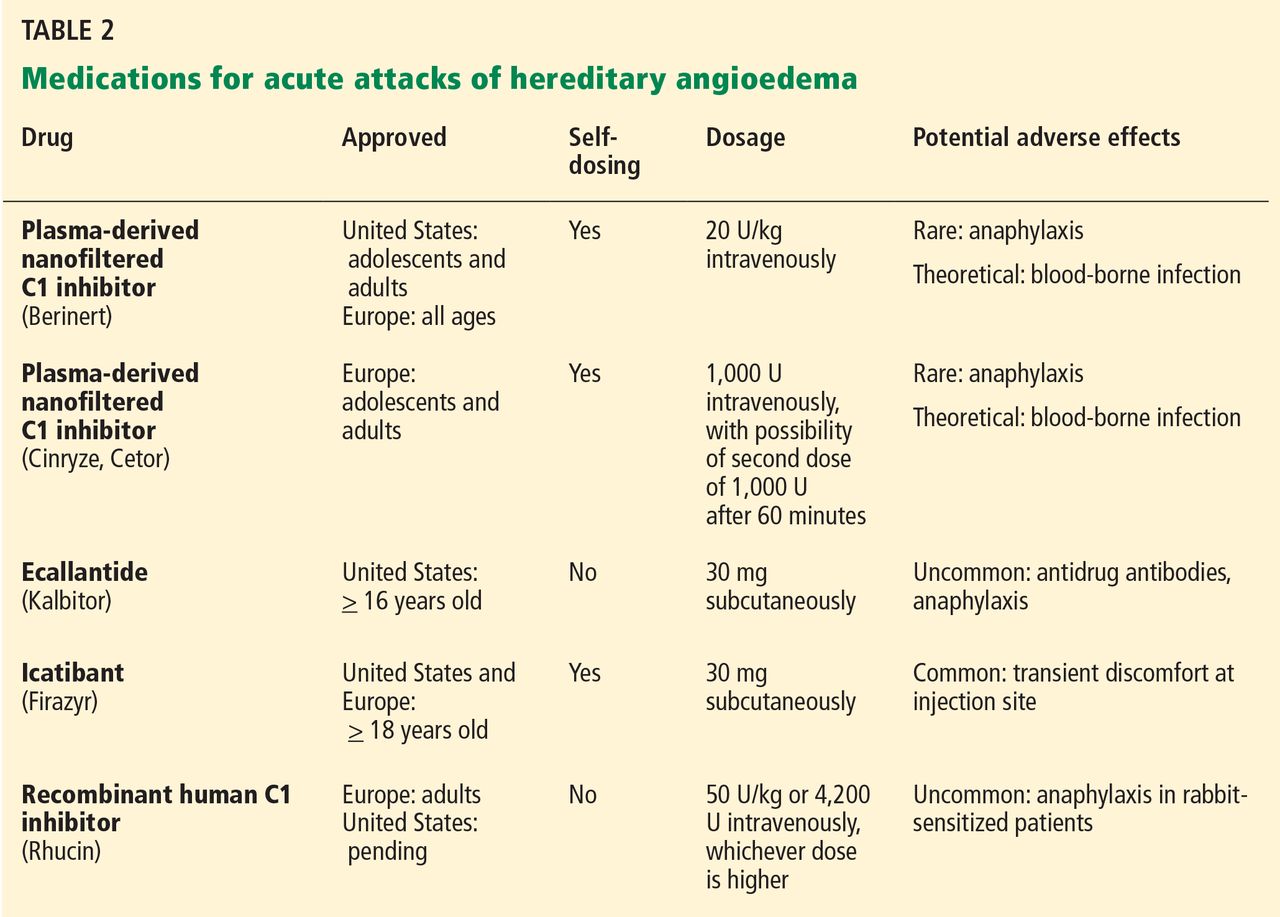
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack

Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack
Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
Hereditary angioedema due to deficiency of C1 inhibitor is a rare autosomal dominant disease that can be life-threatening. It affects about 1 in 50,000 people,1 or about 6,000 people in the United States. There are no known differences in prevalence by ethnicity or sex. A form of hereditary angioedema with normal C1 inhibitor levels has also recently been identified.
Despite a growing awareness of hereditary angioedema in the medical community, repeated surveys have found an average gap of 10 years between the first appearance of symptoms and the correct diagnosis. In view of the risk of morbidity and death, recognizing this disease sooner is critical.
This article will discuss how to recognize hereditary angioedema and how to differentiate it from other forms of recurring angioedema. We will also review its acute and long-term management, with special attention to new therapies and clinical challenges.
EPISODES OF SWELLING WITHOUT HIVES
Hereditary angioedema involves recurrent episodes of nonpruritic, nonpitting, subcutaneous and submucosal edema that can affect the face, tongue, larynx, trunk, extremities, bowels, or genitals. Attacks typically follow a predictable course: swelling that increases slowly and continuously for 24 hours and then gradually subsides over the next 48 to 72 hours. Attacks that involve the oropharynx, larynx, or abdomen carry the highest risk of morbidity and death.1
The frequency and severity of attacks are highly variable and unpredictable. A few patients have no attacks, a few have two attacks per week, and most fall in between.
Hives suggests an allergic or idiopathic rather than hereditary cause and will not be discussed here in detail. A history of angioedema that was rapidly aborted by antihistamines, corticosteroids, or epinephrine also suggests an allergic rather than hereditary cause.
UNCHECKED BRADYKININ PRODUCTION
Substantial evidence indicates that hereditary angioedema results from extravasation of plasma into deeper cutaneous or mucosal compartments as a result of overproduction of the vasoactive mediator bradykinin (Figure 1).
Activated factor XII cleaves plasma prekallikrein to generate active plasma kallikrein (which, in turn, activates more factor XII).2 Once generated, plasma kallikrein cleaves high-molecular-weight kininogen, releasing bradykinin. Bradykinin binds to the B2 bradykinin receptor on endothelial cells, increasing the permeability of the endothelium.
Normally, C1 inhibitor helps control bradykinin production by inhibiting plasma kallikrein and activated factor XII. Without enough C1 inhibitor, the contact system is uninhibited and results in bradykinin being inappropriately generated.
Because the attacks of hereditary angioedema involve excessive bradykinin, they do not respond to the usual treatments for anaphylaxis and allergic angioedema (which involve mast cell degranulation), such as antihistamines, corticosteroids, and epinephrine.
TWO TYPES OF HEREDITARY ANGIOEDEMA
Figure 2 shows the evaluation of patients with suspected hereditary angioedema.
Hereditary angioedema due to C1 inhibitor deficiency
The classic forms of hereditary angioedema (types I and II) involve loss-of-function mutations in SERPING1—the gene that encodes for C1 inhibitor—resulting in low levels of functional C1 inhibitor.3 The mutation is inherited in an autosomal dominant pattern; however, in about 25% of cases, it appears to arise spontaneously,4 so a family history is not required for diagnosis.
Although C1 inhibitor deficiency is present from birth, the clinical disease most commonly presents for the first time when the patient is of school age. Half of patients have their first episode in the first decade of life, and another one-third first develop symptoms over the next 10 years.5
Clinically, types I and II are indistinguishable. Type I, accounting for 85% of cases,1 results from low production of C1 inhibitor. Laboratory studies reveal low antigenic and functional levels of C1 inhibitor.
In type II, the mutant C1 inhibitor protein is present but dysfunctional and unable to inhibit target proteases. On laboratory testing, the functional level of C1 inhibitor is low but its antigenic level is normal (Table 1). Function can be tested by either chromogenic assay or enzyme-linked immunosorbent assay; the former is preferred because it is more sensitive.6
Because C1 inhibitor deficiency results in chronic activation of the complement system, patients with type I or II disease usually have low C4 levels regardless of disease activity, making measuring C4 the most economical screening test. When suspicion for hereditary angioedema is high, based on the presentation and family and clinical history, measuring antigenic and functional C1 inhibitor levels and C4 simultaneously is more efficient.
Hereditary angioedema with normal C1 inhibitor levels
Hereditary angioedema with normal C1 inhibitor levels is also inherited in an autosomal dominant pattern. It is often estrogen-sensitive, making it more severe in women. Symptoms tend to develop slightly later in life than in type I or II disease.7
Angioedema with normal C1 inhibitor levels has been associated with factor XII mutations in a minority of cases, but most patients do not have a specific laboratory abnormality. Because there is no specific laboratory profile, the diagnosis is based on clinical criteria. Hereditary angioedema with normal C1 inhibitor levels should be considered in patients who have recurrent angioedema, normal C4, normal antigenic and functional C1 inhibitor levels, a lack of response to high-dose antihistamines, and either a family history of angioedema without hives or a known factor XII mutation.7 However, other forms of angioedema (allergic, drug-induced, and idiopathic) should also be considered, as C4 and C1 inhibitor levels are normal in these forms as well.
DIFFERENTIAL DIAGNOSIS: OTHER TYPES OF ANGIOEDEMA
Acquired C1 inhibitor deficiency
Symptoms of acquired C1 inhibitor deficiency resemble those of hereditary angioedema but typically do not emerge until the fourth decade of life or later, and patients have no family history of the condition. It is often associated with other diseases, most commonly B-cell lymphoproliferative disorders, which cause uncontrolled complement activation and consumption of C1 inhibitor.
In some patients, autoantibodies to C1 inhibitor develop, greatly reducing its effectiveness and resulting in enhanced consumption. The autoantibody is often associated with a monoclonal gammopathy of unknown significance. The presence of a C1 inhibitor autoantibody does not preclude the presence of an underlying disorder, and vice versa.
Laboratory studies reveal low C4, low C1-inhibitor antigenic and functional levels, and usually a low C1q level owing to consumption of complement. Autoantibodies to C1 inhibitor can be detected by laboratory testing.
Because of the association with autoimmune disease and malignant disorders (especially B-cell malignancy), a patient diagnosed with acquired C1 inhibitor deficiency should be further evaluated for underlying conditions.
Allergic angioedema
Allergic angioedema results from preformed antigen-specific immunoglobulin E (IgE) antibodies that stimulate mast cells to degranulate when patients are exposed to a particular allergen—most commonly food, insect venom, latex, or drugs. IgE-mediated histamine release causes swelling, as histamine is a potent vasodilator.
Symptoms often begin within 2 hours of exposure to the allergen and generally include concurrent urticaria and swelling that last less than 24 hours. Unlike in hereditary angioedema, the swelling responds to antihistamines and corticosteroids. When very severe, these symptoms may also be accompanied by bronchoconstriction and gastrointestinal symptoms, especially if the allergen is ingested.
Histamine-mediated angioedema may also be associated with exercise as part of a syndrome called exercise-induced anaphylaxis or angioedema.
Drug-induced angioedema
Drug-induced angioedema is typically associated with angiotensin-converting enzyme (ACE) inhibitors or nonsteroidal anti-inflammatory drugs (NSAIDs).
Angioedema associated with ACE inhibitors is estimated to affect 0.1% to 6% of patients taking these medications, with African Americans being at significantly higher risk. Although 25% of affected patients develop symptoms of angioedema within the first month of taking the drugs, some tolerate them for as long as 10 years before the first episode.9 The swelling is not allergic or histamine-related. ACE normally degrades bradykinin; therefore, inhibiting ACE leads to accumulation of bradykinin. Because all ACE inhibitors have this effect, this class of drug should be discontinued in any patient who develops isolated angioedema.
NSAID-induced angioedema is often accompanied by other symptoms, including urticaria, rhinitis, cough, hoarseness, or breathlessness.10 The mechanism of NSAID-induced angioedema involves cyclooxygenase (COX) 1 (and to a lesser extent COX-2) inhibition. All NSAIDs (and aspirin) should be avoided in patients with recurrent angioedema. Specific COX-2 inhibitors, while theoretically capable of causing angioedema by the same mechanism, are generally well tolerated in patients who have had COX-1 inhibitor reactions.
Idiopathic angioedema
If no clear cause of recurrent angioedema (at least three episodes in a year) can be found, it is labeled idiopathic.11 Some patients with idiopathic angioedema fail to benefit from high doses of antihistamines, suggesting that the cause is bradykinin-mediated.
CLINICAL MANIFESTATIONS OF HEREDITARY ANGIOEDEMA
Attacks may start at one site and progress to involve additional sites.
Prodromal symptoms may begin up to several days before an attack and include tingling, warmth, burning, or itching at the affected site; increased fatigue or malaise; nausea, abdominal distention, or gassiness; or increased hunger, particularly before an abdominal attack.5 The most characteristic prodromal symptom is erythema marginatum—a raised, serpiginous, nonpruritic rash on the trunk, arms, and legs but often sparing the face.
Abdominal attacks are easily confused with acute abdomen
Almost half of attacks involve the abdomen, and almost all patients with type I or II disease experience at least one such attack.12 Symptoms can include severe abdominal pain, nausea, vomiting, and diarrhea. Abdominal attacks account for many emergency department visits, hospitalizations, and surgical procedures for acute abdomen; about one-third of patients with undiagnosed hereditary angioedema undergo an unnecessary surgery during an abdominal attack. Angioedema of the gastrointestinal tract can result in enough plasma extravasation and vasodilation to cause hypovolemic shock.
Eradicating Helicobacter pylori infection may alleviate abdominal attacks.13
Attacks of the extremities can be painful and disabling
Attacks of the extremities affect 96% of patients12 and can be very disfiguring and disabling. Driving or using the phone is often difficult when the hands are affected. When feet are involved, walking and standing become painful. While these symptoms rarely result in a lengthy hospitalization, they interfere with work and school and require immediate medical attention because they can progress to other parts of the body.
Laryngeal attacks are life-threatening
About half of patients with hereditary angioedema have an attack of laryngeal edema at some point in their lives.12 If not effectively managed, laryngeal angioedema can progress to asphyxiation. A survey of family history in 58 patients with hereditary angioedema suggested a 40% incidence of asphyxiation in untreated laryngeal attacks, and 25% to 30% of patients are estimated to have died of laryngeal edema before effective treatment became available.14
Symptoms of a laryngeal attack include change in voice, hoarseness, trouble swallowing, shortness of breath, and wheezing. Physicians must recognize these symptoms quickly and give effective treatment early in the attack to prevent morbidity and death.
Establishing an airway can be life-saving in the absence of effective therapy, but extensive swelling of the upper airway can make intubation extremely difficult.
Genitourinary attacks also occur
Attacks involving the scrotum and labia have been reported in up to two-thirds of patients with hereditary angioedema at some point in their lives. Attacks involving the bladder and kidneys have also been reported but are less common, affecting about 5% of patients.12 Genitourinary attacks may be triggered by local trauma, such as horseback riding or sexual intercourse, although no trigger may be evident.
MANAGING ACUTE ATTACKS
The goals of treatment are to alleviate acute exacerbations with on-demand treatment and to reduce the number of attacks with prophylaxis. Therapy should be individualized to each patient’s needs. Treatments have advanced greatly in the last several years, and new medications for treating acute attacks and preventing attacks have shown great promise (Figure 3, Table 2).
Patients tend to have recurrent symptoms interspersed with periods of health, suggesting that attacks ought to have identifiable triggers, although in most, no trigger is evident. The most commonly identified are local trauma (including medical and dental procedures), emotional stress, and acute infection. Disease severity may be worsened by menstruation, estrogen-containing oral contraceptives, hormone replacement therapy, ACE inhibitors, and NSAIDs.
It is critical that attacks be treated with an effective medication as soon as possible. Consensus guidelines state that all patients with hereditary angioedema due to C1 inhibitor deficiency, even if they are still asymptomatic, should have access to at least one of the drugs approved for on-demand treatment.15 The guidelines further state that whenever possible, “patients should have the on-demand medicine to treat acute attacks at home and should be trained to self-administer these medicines.”15
Plasma-derived C1 inhibitors
Several plasma-derived C1 inhibitors are available (Cinryze, Berinert, Cetor). They are prepared from fractionated plasma obtained from donors, then pasteurized and nanofiltered.
Berinert and Cinryze were each found to be superior to placebo in double-blind, placebo-controlled trials: attacks usually resolved 30 to 60 minutes after intravenous injection.16,17 Berinert 20 U/kg is associated with the onset of symptom relief as early as half an hour after administration, compared with 1.5 hours with placebo. Early use (at the onset of symptoms) of a plasma-derived C1 inhibitor in a low dose (500 U) can also be effective.18,19 Efficacy appears to be consistent at all sites of attack involvement, including laryngeal edema. Safety and efficacy have been demonstrated during pregnancy and lactation and in young children and babies.20
Plasma-derived C1 inhibitors can be self-administered. The safety and efficacy of self-administration (under physician supervision) were demonstrated in a study of Cinryze and Cetor, in which attack duration, pain medication use, and graded attack severity were significantly less with self-administered therapy than with therapy in the clinic.21
A concern about plasma-derived products is the possibility of blood-borne infection, but this has not been confirmed by experience.22
Recombinant human C1 inhibitor
A recombinant human C1 inhibitor (Rhucin) has been studied in two randomized placebo-controlled trials. Although this product has a shorter half-life than the plasma-derived C1 inhibitors (3 vs more than 24 hours), the two are equipotent: 1 U of recombinant human C1 inhibitor is equivalent to 1 U of plasma-derived C1 inhibitor. Because the supply of recombinant human C1 inhibitor is elastic, dosing has been higher, which may provide more efficacy.23 Similar to plasma-derived C1 inhibitor products, the recombinant human C1 inhibitor resulted in more rapid symptom relief than with saline (66 vs 122 minutes) and in a shorter time to minimal symptoms (247−266 vs 1,210 minutes).24
Allergy is of concern: in one study, a healthy volunteer with undisclosed rabbit allergy experienced an allergic reaction. Patients should be screened by a skin-prick test or serum testing for specific IgE to rabbit epithelium before being prescribed recombinant human C1 inhibitor. No data are available for use during pregnancy or breastfeeding.
Ecallantide
Ecallantide (Kalbitor) is a selective inhibitor of plasma kallikrein that is given in three subcutaneous injections. Ecallantide 30 mg was found superior to placebo during acute attacks.25,26
Ecallantide is well tolerated, with the most common adverse effects being headache, nausea, fatigue, diarrhea, and local injection-site reactions. Antibodies to ecallantide can be found in patients with increasing drug exposure but do not appear to correlate with adverse events. Hypersensitivity reactions have been observed in 2% to 3% of patients receiving repeated doses. Because of anaphylaxis risk, ecallantide must be administered by a health care professional.
Icatibant
Icatibant (Firazyr) is a bradykinin receptor-2 antagonist that is given in a single subcutaneous injection. Icatibant 30 mg significantly shortened time to symptom relief and time to almost complete resolution compared with placebo.27,28 Icatibant’s main adverse effect is transient local pain, swelling, and erythema at the injection site. Icatibant can be self-administered by patients.
Fresh-frozen plasma
Fresh-frozen plasma contains C1 inhibitor and was used before the newer products became available. Several noncontrolled studies reported benefit of its use in acute attacks.29 However, its use is controversial because it also contains contact-system proteins that could provide additional substrate for the generation of bradykinin, which could exacerbate attacks in some patients.1 This may be particularly dangerous in patients presenting with laryngeal edema: in such a situation, the physician should be ready to treat a sudden exacerbation with intubation. The risk of acquiring a blood-borne pathogen is also higher than with plasma-derived C1 inhibitor.
PROPHYLACTIC MANAGEMENT
Short-term and long-term prophylaxis have important roles in preventing attacks (Table 3).
Short-term prophylaxis before an anticipated attack
Short-term prophylaxis is used for patients whose disease is generally well controlled but who anticipate exposure to a potentially exacerbating situation, such as an invasive medical, surgical, or dental procedure. (Routine dental cleanings are generally considered safe and do not require prophylaxis.)
Prophylactic treatments include:
- Plasma-derived C1 inhibitor, 500 to 1,500 U 1 hour before the provoking event
- High-dose 17-alpha alkylated (attenuated) androgens (eg, danazol [Danocrine] 200 mg orally 3 times daily) for 5 to 10 days before the provoking event
- Fresh-frozen plasma, 2 U 1 to 12 hours before the event.1
Yet even with short-term prophylaxis, on-demand treatment should be available.
Long-term prophylaxis
While many patients can be managed with on-demand treatment only, other patients (reflecting the severity of their attacks, as well as their individual needs) may benefit from a combination of on-demand treatment plus long-term prophylaxis. Several options are available (Table 3).
17-alpha alkylated androgens. Patients treated with danazol 600 mg/day were attack-free 90% of the time during a 28-day period compared with only 2.2% of the time in placebo-treated patients.30 Use of anabolic androgens, however, is limited by their adverse effects, including weight gain, virilization, menstrual irregularities, headaches, depression, dyslipidemia, liver enzyme elevation, liver adenomas, and hepatocellular carcinoma. Arterial hypertension occurs in about 25% of treated patients.
Because adverse effects are dose-dependent, treatment should be empirically titrated to find the minimal effective dose, generally recommended to be no more than 200 mg per day of danazol or the equivalent.15
Contraindications include use by women during pregnancy or lactation and by children until growth is complete.
Regular follow-up is recommended every 6 months, with monitoring of liver enzymes, lipids, complete blood counts, alpha fetoprotein, and urinalysis. Abdominal ultrasonography (every 6 months if receiving 100 mg/day or more of danazol, every 12 months if less than 100 mg/day) is advisable for early diagnosis of liver tumors.
Antifibrinolytic drugs. Tranexamic acid (Lysteda) and aminocaproic acid (Amicar) have been found to be effective in reducing the number of attacks of hereditary angioedema compared with placebo but are considered to be less reliable than androgens. These drugs have been used in patients who do not tolerate anabolic androgens, and in children and pregnant women. Tranexamic acid is given at a dose of 20 to 50 mg/kg/day divided into two or three doses per day. The therapeutic dose of aminocaproic acid is 1 g orally three to four times per day.31 Patients with a personal or family history of thromboembolic disease may be at greater risk of venous or arterial thrombosis, but this has not occurred in clinical studies.
Plasma-derived C1 inhibitors. In a 24-week crossover study in 22 patients with hereditary angioedema, Cinryze 1,000 U every 3 to 4 days reduced the rate of attacks by 50% while also reducing their severity and duration.17 An open-label extension study in 146 patients for almost 3 years documented a 90% reduction in attack frequency with no evidence of tachyphylaxis.32
New treatments are costlier
The newer on-demand and prophylactic drugs are substantially costlier than the older alternatives (androgens, antifibrinolytics, and fresh-frozen plasma); however, they have a substantially better benefit-to-risk ratio. Furthermore, the costs of care for an attack requiring emergency treatment are also high. Hereditary angioedema patients are often young, otherwise healthy, and capable of leading normal productive lives. While formal pharmacoeconomic studies of the optimal use of these newer drugs have not yet been done, it is important that the use of these drugs be well justified. Ideally, physicians who prescribe these drugs should be knowledgeable in the management of hereditary angioedema.
SPECIAL CHALLENGES IN WOMEN
Women with hereditary angioedema have more frequent attacks and generally a more severe disease course than men.12 Optimizing care for women is challenging because hormonal changes often cause the disease to flare up in menarche, pregnancy, lactation, and menopause. Women also have a higher rate of discontinuing long-term androgen therapy because of side effects, including virilization and menstrual irregularities. Spironolactone (Aldactone) 100 to 200 mg daily can be used to control hirsutism.33
Contraception
Because estrogen can trigger attacks, progesterone-only formulations, intrauterine devices, or barrier methods are recommended for contraception.33 Progesterone-only pills are preferred and improve symptoms in more than 60% of women. Etonogestrel, another alternative, is available as an implant (Implanon) or vaginal ring (Nuvaring). Intrauterine devices are generally well tolerated, and no prophylaxis is needed during placement. The progesterone-eluting intrauterine device (Mirena) could be beneficial.34
Pregnancy and lactation
Pregnancy and lactation pose particular challenges. Anabolic androgens are contraindicated during pregnancy as well as during breastfeeding because they can be passed on in breast milk. Women receiving androgen prophylaxis should understand that they can still ovulate and need contraception if they are sexually active.34 Patients on attenuated androgens who desire pregnancy should discontinue them 2 months before trying to conceive.
Changes in attack patterns can be unpredictable during pregnancy. Attacks tend to be more severe during the first trimester and more frequent during the third. Due to its safety and efficacy, plasma-derived C1 inhibitor has become the treatment of choice for on-demand or prophylactic treatment during pregnancy and lactation. Antifibrinolytics are considered only when plasma-derived C1 inhibitor is not available.31 Ecallantide and icatibant have not been studied in pregnancy. If neither plasma-derived C1 inhibitor nor antifibrinolytics are available, fresh-frozen plasma or solvent-and-detergent-treated plasma can be used.
Short-term prophylaxis should be considered before amniocentesis, chorionic villous sampling, and dilation and curettage. Delivery should take place in a facility with rapid access to plasma-derived C1 inhibitor as well as consultants in obstetrics, anesthesiology, and perinatology. Although plasma-derived C1 inhibitor should be available at all times during labor and delivery, its prophylactic use is not required unless labor and delivery are particularly traumatic, the underlying hereditary angioedema is very severe, or if forceps, vacuum delivery, or cesarian section is performed. Close monitoring is recommended for at least 72 hours after routine vaginal delivery and for 1 week after cesarian section.
CONCLUSION
The goals of hereditary angioedema treatment are to alleviate morbidity and mortality associated with the disease and to improve the patient’s quality of life. Achieving these goals requires timely diagnosis, patient education, and careful selection of therapeutic modalities that are individualized to the needs of that patient. Treatments have advanced greatly in the last 4 years, and new medications for both the acute and chronic symptoms of hereditary angioedema have shown great promise.
Acknowledgment: K.T. is funded by National Institutes of Health grant T32 AI 07469.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med 2008; 359:1027–1036.
- Kaplan AP. Enzymatic pathways in the pathogenesis of hereditary angioedema: the role of C1 inhibitor therapy. J Allergy Clin Immunol 2010; 126:918–925.
- Davis AE. C1 inhibitor and hereditary angioneurotic edema. Annu Rev Immunol 1988; 6:595–628.
- Pappalardo E, Cicardi M, Duponchel C, et al. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol 2000; 106:1147–1154.
- Frigas E, Park M. Idiopathic recurrent angioedema. Immunol Allergy Clin North Am 2006; 26:739–751.
- Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods 2008; 338:14–20.
- Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol 2010; 6:15.
- Zuraw BL, Bork K, Binkley KE, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc 2012; 33:S145–S156.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am 2006; 26:725–737.
- Busse PJ. Angioedema: differential diagnosis and treatment. Allergy Asthma Proc 2011; 32(suppl 1):S3–S11.
- Prematta MJ, Kemp JG, Gibbs JG, Mende C, Rhoads C, Craig TJ. Frequency, timing, and type of prodromal symptoms associated with hereditary angioedema attacks. Allergy Asthma Proc 2009; 30:506–511.
- Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 2006; 119:267–274.
- Farkas H, Füst G, Fekete B, Karádi I, Varga L. Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 2001; 358:1695–1696.
- Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med 2001; 161:714–718.
- Cicardi M, Bork K, Caballero T, et al; HAWK (Hereditary Angioedema International Working Group). Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 2012; 67:147–157.
- Craig TJ, Levy RJ, Wasserman RL, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 2009; 124:801–808.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 2010; 363:513–522.
- Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion 2005; 45:1774–1784.
- Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion 2009; 49:1987–1995.
- Farkas H, Varga L, Széplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics 2007; 120:e713–e722.
- Tourangeau LM, Castaldo AJ, Davis DK, Koziol J, Christiansen SC, Zuraw BL. Safety and efficacy of physician-supervised self-managed C1 inhibitor replacement therapy. Int Arch Allergy Immunol 2012; 157:417–424.
- De Serres J, Gröner A, Lindner J. Safety and efficacy of pasteurized C1 inhibitor concentrate (Berinert P) in hereditary angioedema: a review. Transfus Apher Sci 2003; 29:247–254.
- Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy 2012; 67:123–130.
- Zuraw B, Cicardi M, Levy RJ, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 2010; 126:821–827.e14.
- Cicardi M, Levy RJ, McNeil DL, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 2010; 363:523–531.
- Levy RJ, Lumry WR, McNeil DL, et al. EDEMA4: a phase 3, double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 2010; 104:523–529.
- Cicardi M, Banerji A, Bracho F, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med 2010; 363:532–541.
- Lumry WR, Li HH, Levy RJ, et al. Randomized placebo-controlled trial of the bradykinin B2 receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol 2011; 107:529–537.
- Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol 2007; 98:383–388.
- Gelfand JA, Sherins RJ, Alling DW, Frank MM. Treatment of hereditary angioedema with danazol. Reversal of clinical and biochemical abnormalities. N Engl J Med 1976; 295:1444–1448.
- Zuraw BL. HAE therapies: past present and future. Allergy Asthma Clin Immunol 2010; 6:23.
- Zuraw BL, Kalfus I. Safety and efficacy of prophylactic nanofiltered C1-inhibitor in hereditary angioedema. Am J Med 2012: Epub ahead of print.
- Caballero T, Farkas H, Bouillet L, et al; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol 2012; 129:308–320.
- Bouillet L, Longhurst H, Boccon-Gibod I, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol 2008; 199:484.e1–e4.
KEY POINTS
- Swelling in the airways is life-threatening and requires rapid treatment.
- Almost half of attacks involve the abdomen, and abdominal attacks account for many emergency department visits, hospitalizations, and unnecessary surgical procedures for acute abdomen.
- Acute attacks can be managed with plasma-derived or recombinant human preparations of C1 inhibitor (which is the deficient factor in this condition), ecallantide (a specific plasma kallikrein inhibitor), or icatibant (a B2 bradykinin receptor antagonist).
- Short-term prophylaxis may be used before events that could provoke attacks (eg, dental work or surgery). Long-term prophylaxis may be used in patients who have frequent or severe attacks or require more stringent control of their disease. Plasma-derived C1 inhibitor is both safe and effective when used as prophylaxis. Attenuated androgens are effective but associated with many adverse effects.
Eczema of the eyelids? Think chemical allergy
LAS VEGAS – If a patient presents with eczema of the eyelids, or swollen eyelids that don’t respond to topical steroids, think about sending them for chemical testing, advised Dr. Janet M. Neigel.
"The eyelids are red and scaly, a little swollen, and it just never goes away," she said in an interview at the annual meeting of the American Academy of Cosmetic Surgery.
Dr. Neigel, a cosmetic surgeon in West Orange, N.J., said that over the past 5 years, she has seen increasing numbers of patients present with eczema localized to the eyelids or eyeball area that recurs like pesky crabgrass.
"I treat them with topical steroids," Dr. Neigel said. "It will get better, but it always comes back. Some of this is seasonal. It may only happen in the winter, when the air is drier and their skin tends to get drier. In others it can be all year long," she said. "It seems to be more common in women, but I see men with this condition, too. In men, it tends to present as a reddish eye and tearing," she noted.
In the majority of cases, the culprit turns out to be an allergy to chemicals including gold, nickel, tin, rubber, preservatives in shampoos and laundry detergent, and formaldehyde resin, which is used in nail polish. "There was one patient who was allergic to the preservative in eyedrops," Dr. Neigel recalled. "She was on several different eyedrops trying to treat the swollen eye area, and it was just making the condition worse."
Another patient’s eczema cleared only after she removed her wedding ring, Dr. Neigel said. "So she couldn’t wear any gold jewelry. In somebody else it was tin and nickel, so she couldn’t wear any cheap jewelry."
Ointments commonly used for cosmetic procedures also can cause trouble. "There is cross-reactivity between neomycin, tobramycin, and Neosporin," Dr. Neigel said. "One patient was applying Neosporin every time she bumped herself on different parts of her body, and her eyelids were the only things flaring up."
Dr. Neigel speculated that the reaction in such cases is localized to the eyelid because "it’s the thinnest skin in the body. It’s the most sensitive, and for some reason, the patients I’m seeing only have reactions there," she noted. So, for patients with allergic conjunctivitis or tearing for a contact dermatitis–type presentation around the eyeball or the eyelids, send them for chemical testing, she advised. "There’s a good chance you might clear things up and figure out what they’re truly reacting to – get to the source instead of just treating the problem symptomatically," she said.
Dr. Neigel said she had no relevant financial disclosures.
LAS VEGAS – If a patient presents with eczema of the eyelids, or swollen eyelids that don’t respond to topical steroids, think about sending them for chemical testing, advised Dr. Janet M. Neigel.
"The eyelids are red and scaly, a little swollen, and it just never goes away," she said in an interview at the annual meeting of the American Academy of Cosmetic Surgery.
Dr. Neigel, a cosmetic surgeon in West Orange, N.J., said that over the past 5 years, she has seen increasing numbers of patients present with eczema localized to the eyelids or eyeball area that recurs like pesky crabgrass.
"I treat them with topical steroids," Dr. Neigel said. "It will get better, but it always comes back. Some of this is seasonal. It may only happen in the winter, when the air is drier and their skin tends to get drier. In others it can be all year long," she said. "It seems to be more common in women, but I see men with this condition, too. In men, it tends to present as a reddish eye and tearing," she noted.
In the majority of cases, the culprit turns out to be an allergy to chemicals including gold, nickel, tin, rubber, preservatives in shampoos and laundry detergent, and formaldehyde resin, which is used in nail polish. "There was one patient who was allergic to the preservative in eyedrops," Dr. Neigel recalled. "She was on several different eyedrops trying to treat the swollen eye area, and it was just making the condition worse."
Another patient’s eczema cleared only after she removed her wedding ring, Dr. Neigel said. "So she couldn’t wear any gold jewelry. In somebody else it was tin and nickel, so she couldn’t wear any cheap jewelry."
Ointments commonly used for cosmetic procedures also can cause trouble. "There is cross-reactivity between neomycin, tobramycin, and Neosporin," Dr. Neigel said. "One patient was applying Neosporin every time she bumped herself on different parts of her body, and her eyelids were the only things flaring up."
Dr. Neigel speculated that the reaction in such cases is localized to the eyelid because "it’s the thinnest skin in the body. It’s the most sensitive, and for some reason, the patients I’m seeing only have reactions there," she noted. So, for patients with allergic conjunctivitis or tearing for a contact dermatitis–type presentation around the eyeball or the eyelids, send them for chemical testing, she advised. "There’s a good chance you might clear things up and figure out what they’re truly reacting to – get to the source instead of just treating the problem symptomatically," she said.
Dr. Neigel said she had no relevant financial disclosures.
LAS VEGAS – If a patient presents with eczema of the eyelids, or swollen eyelids that don’t respond to topical steroids, think about sending them for chemical testing, advised Dr. Janet M. Neigel.
"The eyelids are red and scaly, a little swollen, and it just never goes away," she said in an interview at the annual meeting of the American Academy of Cosmetic Surgery.
Dr. Neigel, a cosmetic surgeon in West Orange, N.J., said that over the past 5 years, she has seen increasing numbers of patients present with eczema localized to the eyelids or eyeball area that recurs like pesky crabgrass.
"I treat them with topical steroids," Dr. Neigel said. "It will get better, but it always comes back. Some of this is seasonal. It may only happen in the winter, when the air is drier and their skin tends to get drier. In others it can be all year long," she said. "It seems to be more common in women, but I see men with this condition, too. In men, it tends to present as a reddish eye and tearing," she noted.
In the majority of cases, the culprit turns out to be an allergy to chemicals including gold, nickel, tin, rubber, preservatives in shampoos and laundry detergent, and formaldehyde resin, which is used in nail polish. "There was one patient who was allergic to the preservative in eyedrops," Dr. Neigel recalled. "She was on several different eyedrops trying to treat the swollen eye area, and it was just making the condition worse."
Another patient’s eczema cleared only after she removed her wedding ring, Dr. Neigel said. "So she couldn’t wear any gold jewelry. In somebody else it was tin and nickel, so she couldn’t wear any cheap jewelry."
Ointments commonly used for cosmetic procedures also can cause trouble. "There is cross-reactivity between neomycin, tobramycin, and Neosporin," Dr. Neigel said. "One patient was applying Neosporin every time she bumped herself on different parts of her body, and her eyelids were the only things flaring up."
Dr. Neigel speculated that the reaction in such cases is localized to the eyelid because "it’s the thinnest skin in the body. It’s the most sensitive, and for some reason, the patients I’m seeing only have reactions there," she noted. So, for patients with allergic conjunctivitis or tearing for a contact dermatitis–type presentation around the eyeball or the eyelids, send them for chemical testing, she advised. "There’s a good chance you might clear things up and figure out what they’re truly reacting to – get to the source instead of just treating the problem symptomatically," she said.
Dr. Neigel said she had no relevant financial disclosures.
AT THE AACS ANNUAL MEETING
Immunotherapy for kids' food allergies is taking baby steps
SAN ANTONIO – Oral and sublingual immunotherapy strategies aren’t yet ready for prime time, but they continue to show promise for inducing tolerance in children with food allergies.
Oral immunotherapy
Preliminary findings from a study of low-dose oral immunotherapy (OIT) for peanut allergy, for example, suggest this approach is an effective early-intervention strategy, Dr. Brian Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In a randomized, controlled trial involving 49 peanut-sensitized children aged 9-36 months, both low- and high-dose immunotherapy resulted in a significant reduction in both peanut-specific IgE (psIgE) and skin prick test values after a median of 19 treatments, said Dr. Vickery, who is a pediatric allergist and immunologist at the University of North Carolina at Chapel Hill.
The degree of change was similar in those treated with low-dose and high-dose oral immunotherapy. The low dose slope coefficient for psIgE was -2.53 for the low dose group, compared with –1.63 for the high-dose group; and the low dose slope coefficient for the skin prick test was –0.007, compared with –0.009 for the high-dose group, he said.
Of eight subjects who met the criteria for tolerance evaluation as of the time of Dr. Vickery’s presentation, seven had successfully achieved tolerance and now eat peanut ad lib, he noted.
Study subjects were enrolled within 6 months of their index reaction or demonstration of psIgE greater than 5 kUA/L. After randomization to the low- or high-dose treatment group, they underwent serial analysis of immune responses. After at least 1 year of maintenance oral immunotherapy, clinical tolerance was assessed using a double-blinded placebo-controlled oral food challenge based on predefined clinical and immunologic benchmarks.
The findings are preliminary but suggest that early-intervention peanut oral immunotherapy is a feasible strategy. In addition, low-dose oral immunotherapy – using a 10-fold lower dose of peanut protein (the equivalent of about 1 vs. 10 peanuts comprised the maintenance doses in the low- and high-dose groups, respectively) may be sufficiently immunomodulatory in young children with newly diagnosed peanut allergy, Dr. Vickery said during a press briefing at the meeting.
Furthermore, the findings suggest that such an approach is technically possible in that young children can be recruited and treated in this manner, he noted.
Dr. Robert A. Wood, who is chief of the division of allergy and immunology at Hopkins Children’s Center at Johns Hopkins University, Baltimore, and who also presented oral immunotherapy data, noted during the press briefing that the approach used in this study "is sort of seizing on the opportunity that maybe kids early in life, when their allergy is less established, may be more amenable to treatment."
Peanut allergy that manifests in early childhood typically intensifies over 5-10 years, he explained.
The findings, however, are very preliminary.
"In order for us to really understand the impact of these two doses, we will need to assess all of the endpoints in all of the subjects who are currently enrolled, and then unblind the study at the end of it, and do an assessment to really understand whether low or high dose therapy was effective," Dr. Vickery said.
Sublingual immunotherapy
Sublingual immunotherapy (SLIT) is another promising intervention for food allergic children, according to findings from a study presented by Dr. A. Wesley Burks, who is chair of pediatrics at the University of North Carolina at Chapel Hill and physician in chief of N.C. Children’s Hospital, also in Chapel Hill.
Interim data from that study of 44 patients showed that after 36 months of dosing, peanut SLIT–induced clinical tolerance with concurrent changes in skin testing and peanut-specific immunoglobulin levels.
Of 11 patients who completed 36 months of dosing, 6 passed a peanut oral food challenge to 5,000 mg of peanut protein. The remaining five patients ingested a median of 3,750 mg of peanut protein. After SLIT discontinuation for 1 month, five of six passed an identical oral food challenge, suggesting clinical tolerance.
Children in this study were aged 2-11 years. All received open-label peanut SLIT with a daily maintenance dose of 2 mg of peanut protein, Dr. Burks said.
The findings do not say anything about long-term efficacy of SLIT, but they do show that tolerance can be induced, at least in the short term, Dr. Burks said.
Predictors of tolerance induction
Another study presented by Dr. Burks shed some light on factors associated with induction of tolerance, namely basophil hyporesponsiveness and a low peanut IgE:IgG4 ratio.
In that study of 12 patients who received SLIT and 27 who received OIT, 5 (41.7%) and 18 (66.7%), respectively, developed tolerance following immunotherapy. In the SLIT subjects, basophil responses were significantly lower among those who developed tolerance than among those who did not. This was true for each of the 4 log-fold dilutions of peanut antigen used in the assay, Dr. Burks said.
The vast majority of tolerant subjects (91.3%) had a peanut-IgE:IgG4 ratio below 0.92, compared with only 20% of nontolerant subjects.
All subjects underwent double-blind placebo-controlled food challenges to assess desensitization while they were on daily immunotherapy, and those passing the challenge ceased daily therapy and avoided all peanut products for 4 weeks. At 4 weeks, a second food challenge was administered.
The findings suggest that basophil suppression and the balance of antigen-specific IgE and IgG4 may be important in the development of tolerance following peanut immunotherapy, Dr. Burks said.
Long-term immunotherapy outcomes
One missing piece of the immunotherapy puzzle are data on long-term outcomes, as most studies report only 1-2 year outcomes. Another study presented by Dr. Wood, however, takes a step toward filling the gap – with underwhelming results.
The small study looked at long-term outcomes following milk oral immunotherapy in children, and showed that at 4.5 year follow-up, only about 25% of 32 patients were tolerating at least one serving of milk daily and 25% were consuming only trace amounts or were on strict avoidance mainly because of reactions. About 40% of patients were having frequent reactions to milk, 20% had required epinephrine for reactions, and 30% had experienced systemic reactions.
The patients were treated in two studies during 2006 and 2007 with a dose escalation to 500 mg of milk protein over about 8 weeks, followed by 3 months of maintenance dosing. Dietary milk was introduced in amounts ranging from 500 to 4,000 mg daily based on the results of a double-blind placebo-controlled food challenge.
Of note, one subject who passed a 16-g challenge without symptoms went on to become reactive again and now consumes only minimal milk, Dr. Wood said
In fact, one of the surprising things in this study was that some of the more dramatic failures long-term were in children who had achieved success in the initial studies.
These were children who "looked like absolute successes" at the end of the study, because there were tolerating large amounts of milk, he said.
"We really thought – and we hesitate to use the word "cure" – that they were about as close to cured as we could really imagine. And now, 3-5 years later, they are having anaphylactic reactions and are back on strict milk avoidance," he said.
As a result, the excitement following the initial studies has waned somewhat.
"We had a very high degree of optimism. I’m not saying we lost that optimism, but it is certainly tempered a bit by looking at where these kids now stand 3-5 years out," he said, concluding that more research with respect to long-term outcomes of food oral immunotherapy is needed, including investigation of whether longer treatment would improve outcomes, and whether certain factors can predict response and failure.
"The most important message is that this sort of adds another caveat about why we need more research before we can bring this out to our patient population. A message that I think all of us who are doing this research have come to agree upon is that we need long-term follow-up and that letting these kids go at the end of the study is not the end of the story."
Taken together, the findings of these studies underscore a need for more research regarding SLIT vs. OIT, higher vs. lower dosing, and treatment duration, as well as long-term follow-up, the investigators agreed.
"The message is that what we are doing so far is very encouraging, but it’s not the final answer. Where we need to be to bring this out to the general public is several steps beyond where we are now," Dr. Wood said.
The investigators reported having no disclosures.
SAN ANTONIO – Oral and sublingual immunotherapy strategies aren’t yet ready for prime time, but they continue to show promise for inducing tolerance in children with food allergies.
Oral immunotherapy
Preliminary findings from a study of low-dose oral immunotherapy (OIT) for peanut allergy, for example, suggest this approach is an effective early-intervention strategy, Dr. Brian Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In a randomized, controlled trial involving 49 peanut-sensitized children aged 9-36 months, both low- and high-dose immunotherapy resulted in a significant reduction in both peanut-specific IgE (psIgE) and skin prick test values after a median of 19 treatments, said Dr. Vickery, who is a pediatric allergist and immunologist at the University of North Carolina at Chapel Hill.
The degree of change was similar in those treated with low-dose and high-dose oral immunotherapy. The low dose slope coefficient for psIgE was -2.53 for the low dose group, compared with –1.63 for the high-dose group; and the low dose slope coefficient for the skin prick test was –0.007, compared with –0.009 for the high-dose group, he said.
Of eight subjects who met the criteria for tolerance evaluation as of the time of Dr. Vickery’s presentation, seven had successfully achieved tolerance and now eat peanut ad lib, he noted.
Study subjects were enrolled within 6 months of their index reaction or demonstration of psIgE greater than 5 kUA/L. After randomization to the low- or high-dose treatment group, they underwent serial analysis of immune responses. After at least 1 year of maintenance oral immunotherapy, clinical tolerance was assessed using a double-blinded placebo-controlled oral food challenge based on predefined clinical and immunologic benchmarks.
The findings are preliminary but suggest that early-intervention peanut oral immunotherapy is a feasible strategy. In addition, low-dose oral immunotherapy – using a 10-fold lower dose of peanut protein (the equivalent of about 1 vs. 10 peanuts comprised the maintenance doses in the low- and high-dose groups, respectively) may be sufficiently immunomodulatory in young children with newly diagnosed peanut allergy, Dr. Vickery said during a press briefing at the meeting.
Furthermore, the findings suggest that such an approach is technically possible in that young children can be recruited and treated in this manner, he noted.
Dr. Robert A. Wood, who is chief of the division of allergy and immunology at Hopkins Children’s Center at Johns Hopkins University, Baltimore, and who also presented oral immunotherapy data, noted during the press briefing that the approach used in this study "is sort of seizing on the opportunity that maybe kids early in life, when their allergy is less established, may be more amenable to treatment."
Peanut allergy that manifests in early childhood typically intensifies over 5-10 years, he explained.
The findings, however, are very preliminary.
"In order for us to really understand the impact of these two doses, we will need to assess all of the endpoints in all of the subjects who are currently enrolled, and then unblind the study at the end of it, and do an assessment to really understand whether low or high dose therapy was effective," Dr. Vickery said.
Sublingual immunotherapy
Sublingual immunotherapy (SLIT) is another promising intervention for food allergic children, according to findings from a study presented by Dr. A. Wesley Burks, who is chair of pediatrics at the University of North Carolina at Chapel Hill and physician in chief of N.C. Children’s Hospital, also in Chapel Hill.
Interim data from that study of 44 patients showed that after 36 months of dosing, peanut SLIT–induced clinical tolerance with concurrent changes in skin testing and peanut-specific immunoglobulin levels.
Of 11 patients who completed 36 months of dosing, 6 passed a peanut oral food challenge to 5,000 mg of peanut protein. The remaining five patients ingested a median of 3,750 mg of peanut protein. After SLIT discontinuation for 1 month, five of six passed an identical oral food challenge, suggesting clinical tolerance.
Children in this study were aged 2-11 years. All received open-label peanut SLIT with a daily maintenance dose of 2 mg of peanut protein, Dr. Burks said.
The findings do not say anything about long-term efficacy of SLIT, but they do show that tolerance can be induced, at least in the short term, Dr. Burks said.
Predictors of tolerance induction
Another study presented by Dr. Burks shed some light on factors associated with induction of tolerance, namely basophil hyporesponsiveness and a low peanut IgE:IgG4 ratio.
In that study of 12 patients who received SLIT and 27 who received OIT, 5 (41.7%) and 18 (66.7%), respectively, developed tolerance following immunotherapy. In the SLIT subjects, basophil responses were significantly lower among those who developed tolerance than among those who did not. This was true for each of the 4 log-fold dilutions of peanut antigen used in the assay, Dr. Burks said.
The vast majority of tolerant subjects (91.3%) had a peanut-IgE:IgG4 ratio below 0.92, compared with only 20% of nontolerant subjects.
All subjects underwent double-blind placebo-controlled food challenges to assess desensitization while they were on daily immunotherapy, and those passing the challenge ceased daily therapy and avoided all peanut products for 4 weeks. At 4 weeks, a second food challenge was administered.
The findings suggest that basophil suppression and the balance of antigen-specific IgE and IgG4 may be important in the development of tolerance following peanut immunotherapy, Dr. Burks said.
Long-term immunotherapy outcomes
One missing piece of the immunotherapy puzzle are data on long-term outcomes, as most studies report only 1-2 year outcomes. Another study presented by Dr. Wood, however, takes a step toward filling the gap – with underwhelming results.
The small study looked at long-term outcomes following milk oral immunotherapy in children, and showed that at 4.5 year follow-up, only about 25% of 32 patients were tolerating at least one serving of milk daily and 25% were consuming only trace amounts or were on strict avoidance mainly because of reactions. About 40% of patients were having frequent reactions to milk, 20% had required epinephrine for reactions, and 30% had experienced systemic reactions.
The patients were treated in two studies during 2006 and 2007 with a dose escalation to 500 mg of milk protein over about 8 weeks, followed by 3 months of maintenance dosing. Dietary milk was introduced in amounts ranging from 500 to 4,000 mg daily based on the results of a double-blind placebo-controlled food challenge.
Of note, one subject who passed a 16-g challenge without symptoms went on to become reactive again and now consumes only minimal milk, Dr. Wood said
In fact, one of the surprising things in this study was that some of the more dramatic failures long-term were in children who had achieved success in the initial studies.
These were children who "looked like absolute successes" at the end of the study, because there were tolerating large amounts of milk, he said.
"We really thought – and we hesitate to use the word "cure" – that they were about as close to cured as we could really imagine. And now, 3-5 years later, they are having anaphylactic reactions and are back on strict milk avoidance," he said.
As a result, the excitement following the initial studies has waned somewhat.
"We had a very high degree of optimism. I’m not saying we lost that optimism, but it is certainly tempered a bit by looking at where these kids now stand 3-5 years out," he said, concluding that more research with respect to long-term outcomes of food oral immunotherapy is needed, including investigation of whether longer treatment would improve outcomes, and whether certain factors can predict response and failure.
"The most important message is that this sort of adds another caveat about why we need more research before we can bring this out to our patient population. A message that I think all of us who are doing this research have come to agree upon is that we need long-term follow-up and that letting these kids go at the end of the study is not the end of the story."
Taken together, the findings of these studies underscore a need for more research regarding SLIT vs. OIT, higher vs. lower dosing, and treatment duration, as well as long-term follow-up, the investigators agreed.
"The message is that what we are doing so far is very encouraging, but it’s not the final answer. Where we need to be to bring this out to the general public is several steps beyond where we are now," Dr. Wood said.
The investigators reported having no disclosures.
SAN ANTONIO – Oral and sublingual immunotherapy strategies aren’t yet ready for prime time, but they continue to show promise for inducing tolerance in children with food allergies.
Oral immunotherapy
Preliminary findings from a study of low-dose oral immunotherapy (OIT) for peanut allergy, for example, suggest this approach is an effective early-intervention strategy, Dr. Brian Vickery reported at the annual meeting of the American Academy of Allergy, Asthma, and Immunology.
In a randomized, controlled trial involving 49 peanut-sensitized children aged 9-36 months, both low- and high-dose immunotherapy resulted in a significant reduction in both peanut-specific IgE (psIgE) and skin prick test values after a median of 19 treatments, said Dr. Vickery, who is a pediatric allergist and immunologist at the University of North Carolina at Chapel Hill.
The degree of change was similar in those treated with low-dose and high-dose oral immunotherapy. The low dose slope coefficient for psIgE was -2.53 for the low dose group, compared with –1.63 for the high-dose group; and the low dose slope coefficient for the skin prick test was –0.007, compared with –0.009 for the high-dose group, he said.
Of eight subjects who met the criteria for tolerance evaluation as of the time of Dr. Vickery’s presentation, seven had successfully achieved tolerance and now eat peanut ad lib, he noted.
Study subjects were enrolled within 6 months of their index reaction or demonstration of psIgE greater than 5 kUA/L. After randomization to the low- or high-dose treatment group, they underwent serial analysis of immune responses. After at least 1 year of maintenance oral immunotherapy, clinical tolerance was assessed using a double-blinded placebo-controlled oral food challenge based on predefined clinical and immunologic benchmarks.
The findings are preliminary but suggest that early-intervention peanut oral immunotherapy is a feasible strategy. In addition, low-dose oral immunotherapy – using a 10-fold lower dose of peanut protein (the equivalent of about 1 vs. 10 peanuts comprised the maintenance doses in the low- and high-dose groups, respectively) may be sufficiently immunomodulatory in young children with newly diagnosed peanut allergy, Dr. Vickery said during a press briefing at the meeting.
Furthermore, the findings suggest that such an approach is technically possible in that young children can be recruited and treated in this manner, he noted.
Dr. Robert A. Wood, who is chief of the division of allergy and immunology at Hopkins Children’s Center at Johns Hopkins University, Baltimore, and who also presented oral immunotherapy data, noted during the press briefing that the approach used in this study "is sort of seizing on the opportunity that maybe kids early in life, when their allergy is less established, may be more amenable to treatment."
Peanut allergy that manifests in early childhood typically intensifies over 5-10 years, he explained.
The findings, however, are very preliminary.
"In order for us to really understand the impact of these two doses, we will need to assess all of the endpoints in all of the subjects who are currently enrolled, and then unblind the study at the end of it, and do an assessment to really understand whether low or high dose therapy was effective," Dr. Vickery said.
Sublingual immunotherapy
Sublingual immunotherapy (SLIT) is another promising intervention for food allergic children, according to findings from a study presented by Dr. A. Wesley Burks, who is chair of pediatrics at the University of North Carolina at Chapel Hill and physician in chief of N.C. Children’s Hospital, also in Chapel Hill.
Interim data from that study of 44 patients showed that after 36 months of dosing, peanut SLIT–induced clinical tolerance with concurrent changes in skin testing and peanut-specific immunoglobulin levels.
Of 11 patients who completed 36 months of dosing, 6 passed a peanut oral food challenge to 5,000 mg of peanut protein. The remaining five patients ingested a median of 3,750 mg of peanut protein. After SLIT discontinuation for 1 month, five of six passed an identical oral food challenge, suggesting clinical tolerance.
Children in this study were aged 2-11 years. All received open-label peanut SLIT with a daily maintenance dose of 2 mg of peanut protein, Dr. Burks said.
The findings do not say anything about long-term efficacy of SLIT, but they do show that tolerance can be induced, at least in the short term, Dr. Burks said.
Predictors of tolerance induction
Another study presented by Dr. Burks shed some light on factors associated with induction of tolerance, namely basophil hyporesponsiveness and a low peanut IgE:IgG4 ratio.
In that study of 12 patients who received SLIT and 27 who received OIT, 5 (41.7%) and 18 (66.7%), respectively, developed tolerance following immunotherapy. In the SLIT subjects, basophil responses were significantly lower among those who developed tolerance than among those who did not. This was true for each of the 4 log-fold dilutions of peanut antigen used in the assay, Dr. Burks said.
The vast majority of tolerant subjects (91.3%) had a peanut-IgE:IgG4 ratio below 0.92, compared with only 20% of nontolerant subjects.
All subjects underwent double-blind placebo-controlled food challenges to assess desensitization while they were on daily immunotherapy, and those passing the challenge ceased daily therapy and avoided all peanut products for 4 weeks. At 4 weeks, a second food challenge was administered.
The findings suggest that basophil suppression and the balance of antigen-specific IgE and IgG4 may be important in the development of tolerance following peanut immunotherapy, Dr. Burks said.
Long-term immunotherapy outcomes
One missing piece of the immunotherapy puzzle are data on long-term outcomes, as most studies report only 1-2 year outcomes. Another study presented by Dr. Wood, however, takes a step toward filling the gap – with underwhelming results.
The small study looked at long-term outcomes following milk oral immunotherapy in children, and showed that at 4.5 year follow-up, only about 25% of 32 patients were tolerating at least one serving of milk daily and 25% were consuming only trace amounts or were on strict avoidance mainly because of reactions. About 40% of patients were having frequent reactions to milk, 20% had required epinephrine for reactions, and 30% had experienced systemic reactions.
The patients were treated in two studies during 2006 and 2007 with a dose escalation to 500 mg of milk protein over about 8 weeks, followed by 3 months of maintenance dosing. Dietary milk was introduced in amounts ranging from 500 to 4,000 mg daily based on the results of a double-blind placebo-controlled food challenge.
Of note, one subject who passed a 16-g challenge without symptoms went on to become reactive again and now consumes only minimal milk, Dr. Wood said
In fact, one of the surprising things in this study was that some of the more dramatic failures long-term were in children who had achieved success in the initial studies.
These were children who "looked like absolute successes" at the end of the study, because there were tolerating large amounts of milk, he said.
"We really thought – and we hesitate to use the word "cure" – that they were about as close to cured as we could really imagine. And now, 3-5 years later, they are having anaphylactic reactions and are back on strict milk avoidance," he said.
As a result, the excitement following the initial studies has waned somewhat.
"We had a very high degree of optimism. I’m not saying we lost that optimism, but it is certainly tempered a bit by looking at where these kids now stand 3-5 years out," he said, concluding that more research with respect to long-term outcomes of food oral immunotherapy is needed, including investigation of whether longer treatment would improve outcomes, and whether certain factors can predict response and failure.
"The most important message is that this sort of adds another caveat about why we need more research before we can bring this out to our patient population. A message that I think all of us who are doing this research have come to agree upon is that we need long-term follow-up and that letting these kids go at the end of the study is not the end of the story."
Taken together, the findings of these studies underscore a need for more research regarding SLIT vs. OIT, higher vs. lower dosing, and treatment duration, as well as long-term follow-up, the investigators agreed.
"The message is that what we are doing so far is very encouraging, but it’s not the final answer. Where we need to be to bring this out to the general public is several steps beyond where we are now," Dr. Wood said.
The investigators reported having no disclosures.
AT THE AAAAI ANNUAL MEETING
Role of food allergy in eczema downplayed
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
MAUI, HAWAII – Current thinking on the role of food allergy in pediatric atopic dermatitis suggests a greatly diminished role for allergy testing compared with times past, according to Dr. Joseph F. Fowler Jr.
Guidelines issued by a National Institute of Allergy and Infectious Diseases expert consensus panel – mostly allergists, with little input from dermatologists – concluded that food allergy is actually fairly uncommon in atopics. It affects less than 10% of children under age 2 who have eczema, and a far smaller percentage of older atopic children.
Moreover, the voluminous 58-page report (J. Allergy Clin. Immunol. 2010;126:S1-58) makes the point that allergy testing is time consuming, costly, and not terribly reliable due to high false-positive rates for both scratch testing and RAST (radioallergosorbent tests).
"The bottom line on all this is you probably don’t need to do food allergy testing very often at all in atopics because in those few who did have an allergy to foods, the big three – eggs, milk, and peanuts – accounted for the vast majority of food allergy. So if you’re not sure, what you can do is eliminate those three from the diet, then add them back in one at a time after a few weeks of elimination. That ought to give you a sense of whether there’s really and truly a food allergy operative in that individual," Dr. Fowler said at the Hawaii Dermatology Seminar sponsored by Global Academy for Medical Education/Skin Disease Education Foundation.
"It’s true that occasionally you see allergy to wheat or fish or chocolate or soy or who knows whatever else, but all those other things are very, very uncommon. So while I wouldn’t say you should never do food allergy testing or send a little atopic with recalcitrant eczema for food allergy testing, I think the yield is really going to be relatively low. Doing the elimination trial first is probably the best thing. A good, motivated, observant caregiver is going to be more informative than what the test will tell you," said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.) and codirector of the SDEF seminar.
SDEF and this news organization are owned by the same parent company.
Dr. Fowler is on the speakers bureaus of Galderma, Ranbaxy, and SmartPractice and has received research grants from numerous pharmaceutical companies.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
MAUI, HAWAII – Current thinking on the role of food allergy in pediatric atopic dermatitis suggests a greatly diminished role for allergy testing compared with times past, according to Dr. Joseph F. Fowler Jr.
Guidelines issued by a National Institute of Allergy and Infectious Diseases expert consensus panel – mostly allergists, with little input from dermatologists – concluded that food allergy is actually fairly uncommon in atopics. It affects less than 10% of children under age 2 who have eczema, and a far smaller percentage of older atopic children.
Moreover, the voluminous 58-page report (J. Allergy Clin. Immunol. 2010;126:S1-58) makes the point that allergy testing is time consuming, costly, and not terribly reliable due to high false-positive rates for both scratch testing and RAST (radioallergosorbent tests).
"The bottom line on all this is you probably don’t need to do food allergy testing very often at all in atopics because in those few who did have an allergy to foods, the big three – eggs, milk, and peanuts – accounted for the vast majority of food allergy. So if you’re not sure, what you can do is eliminate those three from the diet, then add them back in one at a time after a few weeks of elimination. That ought to give you a sense of whether there’s really and truly a food allergy operative in that individual," Dr. Fowler said at the Hawaii Dermatology Seminar sponsored by Global Academy for Medical Education/Skin Disease Education Foundation.
"It’s true that occasionally you see allergy to wheat or fish or chocolate or soy or who knows whatever else, but all those other things are very, very uncommon. So while I wouldn’t say you should never do food allergy testing or send a little atopic with recalcitrant eczema for food allergy testing, I think the yield is really going to be relatively low. Doing the elimination trial first is probably the best thing. A good, motivated, observant caregiver is going to be more informative than what the test will tell you," said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.) and codirector of the SDEF seminar.
SDEF and this news organization are owned by the same parent company.
Dr. Fowler is on the speakers bureaus of Galderma, Ranbaxy, and SmartPractice and has received research grants from numerous pharmaceutical companies.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
Earn 0.25 hours AMA PRA Category 1 credit: Read this article, and click the link at the end to take the post-test.
MAUI, HAWAII – Current thinking on the role of food allergy in pediatric atopic dermatitis suggests a greatly diminished role for allergy testing compared with times past, according to Dr. Joseph F. Fowler Jr.
Guidelines issued by a National Institute of Allergy and Infectious Diseases expert consensus panel – mostly allergists, with little input from dermatologists – concluded that food allergy is actually fairly uncommon in atopics. It affects less than 10% of children under age 2 who have eczema, and a far smaller percentage of older atopic children.
Moreover, the voluminous 58-page report (J. Allergy Clin. Immunol. 2010;126:S1-58) makes the point that allergy testing is time consuming, costly, and not terribly reliable due to high false-positive rates for both scratch testing and RAST (radioallergosorbent tests).
"The bottom line on all this is you probably don’t need to do food allergy testing very often at all in atopics because in those few who did have an allergy to foods, the big three – eggs, milk, and peanuts – accounted for the vast majority of food allergy. So if you’re not sure, what you can do is eliminate those three from the diet, then add them back in one at a time after a few weeks of elimination. That ought to give you a sense of whether there’s really and truly a food allergy operative in that individual," Dr. Fowler said at the Hawaii Dermatology Seminar sponsored by Global Academy for Medical Education/Skin Disease Education Foundation.
"It’s true that occasionally you see allergy to wheat or fish or chocolate or soy or who knows whatever else, but all those other things are very, very uncommon. So while I wouldn’t say you should never do food allergy testing or send a little atopic with recalcitrant eczema for food allergy testing, I think the yield is really going to be relatively low. Doing the elimination trial first is probably the best thing. A good, motivated, observant caregiver is going to be more informative than what the test will tell you," said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.) and codirector of the SDEF seminar.
SDEF and this news organization are owned by the same parent company.
Dr. Fowler is on the speakers bureaus of Galderma, Ranbaxy, and SmartPractice and has received research grants from numerous pharmaceutical companies.
To earn 0.25 hours AMA PRA Category 1 credit after reading this article, take the post-test here.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR
