User login
Bicuspid aortic valve: Basics and beyond
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
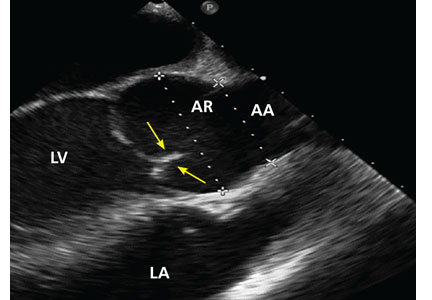
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22


Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22
Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
Bicuspid aortic valve may initially be asymptomatic, but it is associated with progressive valvular and aortic abnormalities that can lead to chronic heart failure and sudden death. Regular monitoring is required with an eye toward surgery when indicated.
This article reviews inheritance patterns and conditions associated with bicuspid aortic valve. We discuss diagnosis, management, and monitoring, and offer surgical recommendations. Special guidance for dental procedures, pregnancy, and athletes is also provided.
A YOUNG MAN WITH PALPITATIONS AND A MURMUR
A 34-year-old man presented to an outpatient clinic with occasional palpitations over the past several months. He reported that he had been diagnosed with a murmur as a child but had received no further testing.
Physical examination at this time revealed a faint systolic crescendo-decrescendo murmur along the right sternal border without radiation to the carotid arteries or to the apex. Transthoracic echocardiography (TTE) showed a bicuspid aortic valve with fusion of the right and left coronary cusps, with no aortic valve stenosis or insufficiency. There was mild dilation of the aortic root, but the mid-ascending aorta could not be evaluated because of limited acoustic windows.
Is further diagnostic testing needed, and if so, what? May he participate in exertional physical activity? Does his newborn son need evaluation?
ABNORMALITIES OCCUR DURING EMBRYOGENESIS
Bicuspid aortic valve develops because of abnormal valvulogenesis. Adjacent cusps fail to separate from each other, resulting in only 2 cusps, with 1 usually larger than the other. Morphology varies according to which commissures are fused.1
Bicuspid aortic valve is associated with abnormalities in the coronary artery anatomy in about 2% of patients, including anomalous origins of the coronary arteries and upwardly displaced coronary ostia.2 Such features need to be considered before surgical intervention.
Bicuspid aortic valve can be found in 1% to 2% of the general population, with a male-to-female predominance of 3:1.1,3,4 It is one of the most common congenital cardiac malformations and is the leading congenital cause of aortic valve stenosis.1,3 However, routine screening of newborns for the condition is not recommended, and most cases are diagnosed incidentally.
GENETIC FACTORS PROMINENT
Bicuspid aortic valve is thought to be primarily inherited in an autosomal-dominant pattern, but there is evidence of genetic heterogeneity, and the pattern may be variable.5,6
No single gene responsible for bicuspid aortic valve has been identified. The condition may occur as a component of different pleiotropic genetic syndromes such as Loeys-Dietz, DiGeorge, and Marfan syndromes,7,8 as well as in patients with Turner syndrome and Williams syndrome.8–11 It also commonly coexists with other congenital heart diseases, including ventricular septal defect, isolated aortic arch obstruction, and patent ductus arteriosus.9
Studies have found a 15% rate of familial clustering.6,12 In a study of 142 patients with bicuspid aortic valve, 20% of first-degree relatives had some cardiac abnormality found by screening, of whom 68% had bicuspid aortic valve. Of these, 71% were newly detected abnormalities.13
CHARACTERISTIC CLICK AND MURMUR
Physical examination findings of a functionally normal bicuspid aortic valve include a systolic ejection click followed by an early peaking systolic murmur at the apex or left lower sternal border. With progression of aortic stenosis, the ejection murmur has a harsher sound, with later peaking, and the S2 sound diminishes or becomes inaudible.14 If aortic regurgitation is present, a diastolic decrescendo murmur is heard best at the left lower sternal border.
DISEASE PROGRESSION
Although bicuspid aortic valve is typically asymptomatic at first, it is commonly associated with progressive valvulopathy and thoracic aortic disease.1,3,4,15 It can lead to chronic heart failure and increase the risk of acute aortic syndromes and sudden cardiac death.15
Michelena et al16 studied 212 cases of asymptomatic bicuspid aortic valve. Although the survival rate 20 years after diagnosis was the same as for an age-matched cohort in the general population, the frequency of adverse cardiovascular events and surgical interventions was higher.
Aortic stenosis progresses rapidly
Aortic stenosis associated with a bicuspid aortic valve tends to affect younger patients and progress more rapidly than when associated with a tricuspid valve.17
In a study of 542 patients with congenital bicuspid aortic valve undergoing aortic valve replacement,3 75% had isolated aortic stenosis, 10% had aortic stenosis with some degree of aortic insufficiency, and 13% had isolated aortic insufficiency. Given the tendency of aortic stenosis to progress rapidly, early surgery is often pursued.17,18
Aneurysmal disease is common
The thoracic aorta is at increased risk of aneurysmal disease, coarctation, and dissection in patients with a bicuspid aortic valve.1,6,15
Michelena et al16 reported that in patients without an aneurysm at the time of bicuspid aortic valve diagnosis, the 25-year risk of aneurysm formation was approximately 26%. In patients with an aneurysm at the time of diagnosis, the 15-year risk of aortic surgery after the diagnosis of aneurysm was about 46% and the risk of aortic dissection after aneurysm diagnosis was 7%.15 Compared with the general population, the age-adjusted relative risk of aortic aneurysm in patients with bicuspid aortic valve was 86.2, and that of aortic dissection was 8.4. Although the absolute incidence of dissection is low in these patients, it is markedly higher than in the general population, particularly in older patients (age > 50) and those with an aneurysm at the time of diagnosis.15
The risk of infective endocarditis
Patients with bicuspid aortic valve are highly prone to infective endocarditis for reasons that remain poorly understood. The pathogens in most cases are staphylococci or viridans streptococci.19 Patients with infective endocarditis typically require emergency surgery. Complications including valvular abscess, myocardial abscess, and overt heart failure are common.19
Lamas and Eykyn20 studied 408 cases of native valve endocarditis; in 12.3%, the patient had a bicuspid aortic valve. In this subset, all were male, the mean age was 39 at diagnosis, 82% needed surgery, and the death rate was 14%.
Patients with bicuspid aortic valve do not routinely need antibiotics before dental and surgical procedures, but if they have had endocarditis in the past, they need antibiotics to prevent a recurrence.21
REGULAR MONITORING NEEDED
Because complications may be life-threatening, early detection of progressive disease by regular screening is critical. Echocardiographic evaluation of valvular function, ventricular dimensions and function, and diameter of the aortic root and ascending aorta should be performed in every patient with bicuspid aortic valve. If initial imaging is normal and there is no aortic dilation, imaging should be repeated every 5 to 10 years. If any abnormality is found, repeat imaging is needed every year.22
Magnetic resonance imaging (MRI) or computed tomographic (CT) angiography may be required to better assess the aorta for patients requiring a surgical intervention, or when aortic dimensions are not clearly visualized on TTE. MRI has 2 advantages over CT angiography: it poses no radiation risk, and it provides more information on left ventricular function and dimensions, in addition to valve assessment.23,24
No published study has compared MRI or CT angiography and transesophageal echocardiography (TEE), but in a study of 174 patients with dilated aortic root, combined TTE and TEE detected aortic valve morphology accurately in 98% of cases. As TEE is more invasive, it is not recommended for regular surveillance (Figures 1 and 2).25
FAMILY SCREENING RECOMMENDED
Close relatives should be evaluated for aortic valve and thoracic aortic disease.12,13,23,26
The American College of Cardiology (ACC) and the American Heart Association (AHA), backed by radiologic and cardiovascular associations, concur in recommending echocardiographic screening and routine screening of the thoracic aorta for aortic root dilation in first-degree relatives (ie, siblings, parents, and children) of patients with bicuspid aortic valve (class I recommendation).22,27,28
A comprehensive physical examination is recommended for family members in addition to TTE, with careful assessment of the aortic valve in short and long axes, and of the aortic root.14 If the aorta cannot be adequately evaluated with TTE, further assessment should be pursued with CT angiography or MRI.
EXERCISE RESTRICTIONS
The 2015 ACC/AHA guidelines for competitive athletes with cardiovascular abnormalities recommend annual screening with TTE or MRI angiography for athletes with bicuspid aortic valve and coexisting dilation of the ascending aorta (aortic diameter 40–42 mm in men and 36–39 mm in women) (class I recommendation, level of evidence C).29
Athletes with a bicuspid aortic valve and a normal aortic root and ascending aorta may participate in all competitive activities.29 However, those with a dilated aorta should avoid strenuous activities because of the increased risk of rupture.30 The ACC/AHA recommendations29 depend on the diameter of the ascending aorta and the nature of the sport:
- For an aortic diameter 40 to 42 mm in men or 36 to 39 mm in women, and no features of connective tissue disease or familial thoracic ascending aortic syndrome, low- and moderate-intensity sports with a low likelihood of significant body contact may be considered; consider avoiding intense weight training (class IIb, level of evidence C)
- For an aortic diameter 43 to 45 mm, low-intensity sports with a low likelihood of body contact may be considered (class IIb, level of evidence C)
- For an aortic diameter greater than 43 mm in men or greater than 40 mm in women, sports involving body collision should be avoided (class III, level of evidence C)
- For an aortic diameter greater than 45 mm, sports activities should be avoided (class III, level of evidence C).
PREGNANCY CONSIDERATIONS
Bicuspid aortic valve is associated with aortic dissection, mainly in the third trimester.31 Patients should ideally undergo echocardiographic screening before conception. The 2010 ACC/AHA guidelines for managing thoracic aortic disease recommend monthly or bimonthly echocardiography until delivery in pregnant women with a dilated thoracic aorta.22
Patients with bicuspid aortic valve and aortic root enlargement of more than 40 mm should have preconception counseling about surgery for aortic root replacement before becoming pregnant. If the diagnosis of enlarged aortic root is made during pregnancy, echocardiographic surveillance at 4- to 6-week intervals is indicated.32
SURGICAL MANAGEMENT
In the past, beta-blockers and angiotensin-converting enzyme inhibitors were recommended to minimize shear stress, with the goal of slowing progression of aortic dilation. However, evidence to support their use is inadequate.33,34
The only definitive treatment is surgery, with various procedures that lower the risk of death or dissection.24,35
The dimensions of the aortic root or ascending aorta should be examined vigilantly, according to the 2014 ACC/AHA guidelines27:
- Repairing the aortic sinuses or replacing the ascending aorta is indicated if the diameter of the aortic sinuses or ascending aorta is greater than 5.5 cm (class I, level of evidence B)
- Repairing the aortic sinuses or replacing the ascending aorta is reasonable if the diameter of the aortic sinuses or ascending aorta is greater than 5.0 cm and the patient has a risk factor for dissection such as a family history of aortic dissection or an increase in diameter of 0.5 cm or greater per year (class IIa, level of evidence C)
- Replacement of the ascending aorta is reasonable if the diameter of the ascending aorta is greater than 4.5 cm and the patient is undergoing aortic valve surgery for severe aortic stenosis or regurgitation.
Valve repair or replacement
Aortic valve repair or replacement is sometimes done separately from aortic root repair.
The value of aortic valve repair is debatable, but a series of 728 patients at Cleveland Clinic showed a very low mortality rate (0.41%) and an annual reoperation rate of 2.6% during up to 15 years of follow-up.36
Aortic valve replacement is usually considered for patients with severe valve dysfunction, abnormal left ventricular dimensions, or symptoms. It is important to determine if the patient is a good surgical candidate and to refer early for surgical evaluation to avoid the higher risk of death associated with emergency surgery.36
Transcatheter aortic valve replacement has been studied in patients deemed to be at too high a risk for surgical replacement. Short- and intermediate-term outcomes have been good in these patients, but long-term data are lacking.37
Surveillance after surgery
The type of operation determines postoperative surveillance.
After isolated aortic valve repair or replacement, patients should continue with surveillance at least annually to monitor for progressive aortopathy, as they remain at increased risk of dissection or rupture after isolated valve surgery, especially if they had aortic insufficiency preoperatively.38
After definitive surgery with replacement or repair of the ascending aorta, no clear recommendations have been established for continued surveillance. However, it is reasonable to image these patients with either MRI or CT angiography 3 to 5 years after their surgery to monitor for anastomotic complications.
CASE QUESTIONS ANSWERED
Our patient should undergo repeat TTE in 1 year. He should also undergo CT angiography of the ascending aorta if it is not seen by TTE. He can participate in low-intensity sports but should avoid intense weight training. His parents, siblings, and children should be screened for bicuspid aortic valve or associated aortopathies.
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
- Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 1970; 26(1):72–83. pmid:5427836
- Michalowska IM, Hryniewiecki T, Kwiatek P, Stoklosa P, Swoboda-Rydz U, Szymanski P. Coronary artery variants and anomalies in patients with bicuspid aortic valve. J Thorac Imaging 2016; 31(3):156–162. doi:10.1097/RTI.0000000000000205
- Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc 1999; 74(1):14–26. doi:10.4065/74.1.14
- Tutar E, Ekici F, Atalay S, Nacar N. The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 2005; 150(3):513–515. doi:10.1016/j.ahj.2004.10.036
- Benson DW. The genetics of congenital heart disease: a point in the revolution. Cardiol Clin 2002; 20(3):385–394. pmid:12371007
- Emanuel R, Withers R, O’Brien K, Ross P, Feizi O. Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families. Br Heart J 1978; 40(12):1402–1407. pmid:737099
- Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high-throughput sequencing approaches on research and diagnosis. Front Physiol 2017; 8:612. doi:10.3389/fphys.2017.00612
- Sachdev V, Matura LA, Sidenko S, et al. Aortic valve disease in Turner syndrome. J Am Coll Cardiol 2008; 51(19):1904–1909. doi:10.1016/j.jacc.2008.02.035
- Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C, Thiene G. Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 1995; 4(6):581–590. pmid:8611973
- De Rubens Figueroa J, Rodríguez LM, Hach JL, Del Castillo Ruíz V, Martínez HO. Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 2008; 35(3):279–285. pmid:18941598
- Yuan SM, Jing H. The bicuspid aortic valve and related disorders. Sao Paulo Med J 2010; 128(5):296–301. pmid:21181071
- Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol 2004; 44(1):138–143. doi:10.1016/j.jacc.2004.03.050
- Kerstjens-Frederikse WS, Sarvaas GJ, Ruiter JS, et al. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart 2011; 97(15):1228–1232. doi:10.1136/hrt.2010.211433
- Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol 2010; 55(25):2789–2800. doi:10.1016/j.jacc.2009.12.068
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112.
- Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 2008; 117(21):2776–2784. doi:10.1161/CIRCULATIONAHA.107.740878
- Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 1993; 71(4):322–327. pmid:8427176
- Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation 2005; 111(7):920–925. doi:10.1161/01.CIR.0000155623.48408.C5
- Yener N, Oktar GL, Erer D, Yardimci MM, Yener A. Bicuspid aortic valve. Ann Thorac Cardiovasc Surg 2002; 8(5):264–267. pmid:12472407
- Lamas CC, Eykyn SJ. Bicuspid aortic valve—a silent danger: analysis of 50 cases of infective endocarditis. Clin Infect Dis 2000; 30(2):336–341. doi:10.1086/313646
- Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation 2007; 116(15):1736–1754. doi:10.1161/CIRCULATIONAHA.106.183095
- Hiratzka L, Bakris G, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. Circulation 2010; 121(13):e266–e369. doi:10.1161/CIR.0b013e3181d4739e
- Chun EJ, Choi SI, Lim C, et al. Aortic stenosis: evaluation with multidetector CT angiography and MR imaging. Korean J Radiol 2008; 9(5):439–448. doi:10.3348/kjr.2008.9.5.439
- Kiefer TL, Wang A, Hughes GC, Bashore TM. Management of patients with bicuspid aortic valve disease. Curr Treat Options Cardiovasc Med 2011; 13(6):489–505. doi:10.1007/s11936-011-0152-7
- Alegret JM, Palazon O, Duran I, Vernis JM. Aortic valve morphology definition with transthoracic combined with transesophageal echocardiography in a population with high prevalence of bicuspid aortic valve. Int J Cardiovasc Imaging 2005; 21(2-3):213–217. doi:10.1007/s10554-004-3901-9
- Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ, Tolstrup K. Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 2009; 53(24):2288–2295. doi:10.1016/j.jacc.2009.03.027
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease. J Thorac Cardiovasc Surg 2014; 148(1):e1-e132. doi:10.1016/j.jtcvs.2014.05.014
- Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. J Am Coll Cardiol 2008; 52(23):e143–e263. doi:10.1016/j.jacc.2008.10.001
- Braverman AC, Harris KM, Kovacs RJ, Maron BJ. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 7: aortic diseases, including Marfan syndrome. Circulation 2015; 132(22):e303–e309. doi:10.1161/CIR.0000000000000243
- De Mozzi P, Longo UG, Galanti G, Maffulli N. Bicuspid aortic valve: a literature review and its impact on sport activity. Br Med Bull 2008; 85:63–85. doi:10.1093/bmb/ldn002
- Thorne SA. Pregnancy in heart disease. Heart 2004; 90(4):450–456. pmid:15020530
- Immer FF, Bansi AG, Immer-Bansi AS, et al. Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 2003; 76(1):309–314. pmid:12842575
- Allen BD, Markl M, Barker AJ, et al. Influence of beta-blocker therapy on aortic blood flow in patients with bicuspid aortic valve. Int J Cardiovasc Imaging 2016; 32(4):621–628. doi:10.1007/s10554-015-0819-3
- Ohnemus D, Oster ME, Gatlin S, Jokhadar M, Mahle WT. The effect of angiotensin-converting enzyme inhibitors on the rate of ascending aorta dilation in patients with bicuspid aortic valve. Congenit Heart Dis 2015; 10(1):E1–E5. doi:10.1111/chd.12184
- Masri A, Kalahasti V, Alkharabsheh S, et al. Characteristics and long-term outcomes of contemporary patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2016; 151(6):1650–1659.e1. doi:10.1016/j.jtcvs.2015.12.019
- Svensson LG, Al Kindi AH, Vivacqua A, et al. Long-term durability of bicuspid aortic valve repair. Ann Thorac Surg 2014; 97(5):1539–1548. doi:10.1016/j.athoracsur.2013.11.036
- Mylotte D, Lefevre T, Sondergaard L, et al. Transcatheter aortic valve replacement in bicuspid aortic valve disease. J Am Coll Cardiol 2014; 64(22):2330–2339. doi:10.1016/j.jacc.2014.09.039
- Girdauskas E, Disha K, Raisin HH, Secknus MA, Borger MA, Kuntze T. Risk of late aortic events after an isolated aortic valve replacement for bicuspid aortic valve stenosis with concomitant ascending aortic dilation. Eur J Cardiothorac Surg 2012; 42(5):832–838. doi:10.1093/ejcts/ezs137
KEY POINTS
- Associated aortopathies such as aortic root dilation, aneurysm, dissection, and coarctation may initially be asymptomatic.
- Regular surveillance with transthoracic echocardiography (TTE) is required.
- Transesophageal echocardiography should be performed if TTE does not clearly show the aorta and aortic root. Magnetic resonance imaging or computed tomographic angiography may also be needed to measure the aortic root and ascending thoracic aorta.
- If initial imaging is normal and there is no aortic dilation, repeat imaging should be done every 5 to 10 years. If any abnormality is found, annual surveillance is needed.
- Women with a bicuspid aortic valve who are contemplating pregnancy should undergo echocardiography first, and some may need to undergo surgery.
When stroke runs in the family
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
.")
.")
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
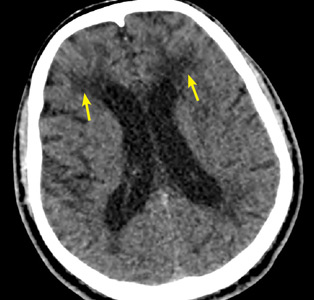
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
A 54-year-old man presented to our hospital with acute-onset left-sided weakness and right facial droop. Three days earlier he had also had migraine-like headaches, which he had never experienced before. He also reported a change in behavior during the past week, which his family had described as inappropriate laughter.
He had no history of hypertension, diabetes, or dyslipidemia. He did not smoke or drink alcohol. However, he had an extensive family history of stroke. His mother had a stroke at age 50, his brother a stroke at age 57, and his sister had been admitted for a stroke 1 month earlier at the age of 52.
On examination, he had weakness of the left arm and leg, right facial droop, and hyperactive reflexes on the left side. He had no sensory or cerebellar deficits. He had episodes of laughter during the examination.
We learned that the patient’s sister had undergone a workup showing mutations in the NOTCH3 gene and a skin biopsy study consistent with CADASIL.
Our patient was started on antiplatelet and high-intensity statin therapy. His symptoms improved, and he was discharged to an acute inpatient rehabilitation facility. He was referred to a CADASIL registry.
STROKE AND HEREDITY
CADASIL is a rare hereditary vascular disorder inherited in an autosomal dominant manner. It is the most common inherited form of small-vessel disease and results from a mutation in the NOTCH3 gene that leads to degeneration of smooth muscle in cerebral blood vessels. It can manifest as migraine with aura, vascular dementia, cognitive impairment, or ischemic stroke.
The diagnosis is based on a clinical picture that typically includes stroke at a young age (age 40 to 50) in the absence of stroke risk factors, or frequent lacunar infarction episodes that can manifest as migraine, lacunar infarct, or dementia.1 Some patients, such as ours, may have subtle nonspecific behavioral changes such as inappropriate laughter, which may herald the development of an infarct.
Characteristic findings on MRI are white matter hyperintensities that tend to be bilateral and symmetrical in the periventricular areas. Symmetrical involvement in the temporal lobes has high sensitivity and specificity for CADASIL.2 Biopsy study of the skin, muscle, or sural nerve shows small-vessel changes that include thickening of the media, granular material positive on periodic acid-Schiff staining, and narrowing of the lumen. However, the gold standard for diagnosis is confirmation of the NOTCH3 mutation on chromosome 19.1,2
There is no known treatment for CADASIL.
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
- Davous P. CADASIL: a review with proposed diagnostic criteria. Eur J Neurol 1998; 5(3):219–233. pmid:10210836
- Stojanov D, Vojinovic S, Aracki-Trenkic A, et al. Imaging characteristics of cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). Bosn J Basic Med Sci 2015; 15(1):1–8. doi:10.17305/bjbms.2015.247
Aortic aneurysm: Fluoroquinolones, genetic counseling
To the Editor: The review of thoracic aortic aneurysm by Cikach et al1 was excellent. However, we noted that referral for clinical genetic counseling and testing is suggested only if 1 or more first-degree relatives have aneurysmal disease.
Absence of a family history does not rule out syndromic aortopathy, which can occur de novo. In addition, a clinical diagnosis of syndromic aortopathy can be made on the basis of physical features that can be very subtle, such as pectus deformities, scoliosis, dolichostenomelia, joint hypermobility or contractures, craniofacial features, or skin fragility.2
Genetic counseling is paramount even if molecular testing is negative or inconclusive, which can occur in more than 50% of patients referred.3 Clinical genetic evaluation would also facilitate testing for other family members who may be affected, and would help to coordinate care for nonvascular conditions that may be associated with the syndrome.
- Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
- McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. OMIM. Online mendelian inheritance in man. https://omim.org. Accessed July 31, 2018.
- Mazine A, Moryousef-Abitbol JH, Faghfoury H, Meza JM, Morel C, Ouzounian M. Yield of genetic testing in patients with thoracic aortic disease. J Am Coll Cardiol 2017; 69(11):2005. doi:10.1016/S0735-1097(17)35394-9
To the Editor: The review of thoracic aortic aneurysm by Cikach et al1 was excellent. However, we noted that referral for clinical genetic counseling and testing is suggested only if 1 or more first-degree relatives have aneurysmal disease.
Absence of a family history does not rule out syndromic aortopathy, which can occur de novo. In addition, a clinical diagnosis of syndromic aortopathy can be made on the basis of physical features that can be very subtle, such as pectus deformities, scoliosis, dolichostenomelia, joint hypermobility or contractures, craniofacial features, or skin fragility.2
Genetic counseling is paramount even if molecular testing is negative or inconclusive, which can occur in more than 50% of patients referred.3 Clinical genetic evaluation would also facilitate testing for other family members who may be affected, and would help to coordinate care for nonvascular conditions that may be associated with the syndrome.
To the Editor: The review of thoracic aortic aneurysm by Cikach et al1 was excellent. However, we noted that referral for clinical genetic counseling and testing is suggested only if 1 or more first-degree relatives have aneurysmal disease.
Absence of a family history does not rule out syndromic aortopathy, which can occur de novo. In addition, a clinical diagnosis of syndromic aortopathy can be made on the basis of physical features that can be very subtle, such as pectus deformities, scoliosis, dolichostenomelia, joint hypermobility or contractures, craniofacial features, or skin fragility.2
Genetic counseling is paramount even if molecular testing is negative or inconclusive, which can occur in more than 50% of patients referred.3 Clinical genetic evaluation would also facilitate testing for other family members who may be affected, and would help to coordinate care for nonvascular conditions that may be associated with the syndrome.
- Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
- McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. OMIM. Online mendelian inheritance in man. https://omim.org. Accessed July 31, 2018.
- Mazine A, Moryousef-Abitbol JH, Faghfoury H, Meza JM, Morel C, Ouzounian M. Yield of genetic testing in patients with thoracic aortic disease. J Am Coll Cardiol 2017; 69(11):2005. doi:10.1016/S0735-1097(17)35394-9
- Cikach F, Desai MY, Roselli EE, Kalahasti V. Thoracic aortic aneurysm: how to counsel, when to refer. Cleve Clin J Med 2018; 85(6):481–492. doi:10.3949/ccjm.85a.17039
- McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. OMIM. Online mendelian inheritance in man. https://omim.org. Accessed July 31, 2018.
- Mazine A, Moryousef-Abitbol JH, Faghfoury H, Meza JM, Morel C, Ouzounian M. Yield of genetic testing in patients with thoracic aortic disease. J Am Coll Cardiol 2017; 69(11):2005. doi:10.1016/S0735-1097(17)35394-9

In reply: Aortic aneurysm: Fluoroquinolones, genetic counseling
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.
In Reply: We thank Drs. Goldstein and Mascitelli for their comments regarding fluoroquinolones and thoracic aortic aneurysms. We acknowledge that fluoroquinolones (particularly ciprofloxacin) have been associated with a risk of aortic aneurysm and dissection based on large observational studies from Taiwan, Canada, and Sweden. Although all of the studies have shown an association between ciprofloxacin and aortic aneurysm, the causative role is not well established. In addition, the numbers of events were very small in these large cohorts of patients. In our large tertiary care practice at Cleveland Clinic, we have very few patients with aortic aneurysm or dissection who have used fluoroquinolones.
We recognize the association; however, our paper was intended to emphasize the more common causes and treatment options that primary care physicians are likely to encounter in routine practice.
We also thank Drs. Ayoubieh and MacCarrick for their comments about genetic counseling. We agree that genetic counseling is important, as is a detailed physical examination for subtle features of genetically mediated aortic aneurysm. In fact, we incorporate the physical examination when patients are seen at our aortic center so as to recognize the physical features. We do routinely recommend screening of first-degree relatives even without significant family history on an individual basis and make appropriate referrals for other conditions that can be seen in these patients. Our article, however, is primarily intended to emphasize the importance of referring these patients for more-focused care at a specialized center, where we incorporate all of the suggestions that were made.

Thoracic aortic aneurysm: How to counsel, when to refer
Thoracic aortic aneurysm (TAA) needs to be detected, monitored, and managed in a timely manner to prevent a serious consequence such as acute dissection or rupture. But only about 5% of patients experience symptoms before an acute event occurs, and for the other 95% the first “symptom” is often death.1 Most cases are detected either incidentally with echocardiography, computed tomography (CT), or magnetic resonance imaging (MRI) during workup for another condition. Patients may also be diagnosed during workup of a murmur or after a family member is found to have an aneurysm. Therefore, its true incidence is difficult to determine.2
With these facts in mind, how would you manage the following 2 cases?
Case 1: Bicuspid aortic valve, ascending aortic aneurysm
A 45-year-old man with stage 1 hypertension presents for evaluation of a bicuspid aortic valve and ascending aortic aneurysm. He has several first-degree relatives with similar conditions, and his brother recently underwent elective aortic repair. At the urging of his primary care physician, he underwent screening echocardiography, which demonstrated a “dilated root and ascending aorta” 4.6 cm in diameter. He presents today to discuss management options and how the aneurysm could affect his everyday life.
Case 2: Marfan syndrome in a young woman
A 24-year-old woman with Marfan syndrome diagnosed in adolescence presents for annual follow-up. She has many family members with the same condition, and several have undergone prophylactic aortic root repair. Her aortic root has been monitored annually for progression of dilation, and today it is 4.6 cm in diameter, a 3-mm increase from the last measurement. She has grade 2+ aortic insufficiency (on a scale of 1+ to 4+) based on echocardiography, but she has no symptoms. She is curious about what size her aortic root will need to reach for surgery to be considered.
LIKELY UNDERDETECTED
TAA is being detected more often than in the past thanks to better detection methods and heightened awareness among physicians and patients. While an incidence rate of 10.4 per 100,000 patient-years is often cited,3 this figure likely underestimates the true incidence of this clinically silent condition. The most robust data come from studies based on in-hospital diagnostic codes coupled with data from autopsies for out-of-hospital deaths.
Olsson et al,4 in a 2016 study in Sweden, found the incidence of TAA and aortic dissection to be 16.3 per 100,000 per year for men and 9.1 per 100,000 per year for women.
Clouse et al5 reported the incidence of thoracic aortic dissection as 3.5 per 100,000 patient-years, and the same figure for thoracic aortic rupture.
Aneurysmal disease accounts for 52,000 deaths per year in the United States, making it the 19th most common cause of death.6 These figures are likely lower than the true mortality rate for this condition, given that aortic dissection is often mistaken for acute myocardial infarction or other acute event if an autopsy is not done to confirm the cause of death.7
RISK FACTORS FOR THORACIC AORTIC ANEURYSM
Risk factors for TAA include genetic conditions that lead to aortic medial weakness or destruction such as Loeys-Dietz syndrome and Marfan syndrome.2 In addition, family history is important even in the absence of known genetic mutations. Other risk factors include conditions that increase aortic wall stress, such as hypertension, cocaine abuse, extreme weightlifting, trauma, and aortic coarctation.2
DIAMETER INCREASES WITH AGE, BODY SURFACE AREA

Normal dimensions for the aortic segments differ depending on age, sex, and body surface area.8,44,45 The size of the aortic root may also vary depending on how it is measured, due to the root’s trefoil shape. Measured sinus to sinus, the root is larger than when measured sinus to commissure on CT angiography or cardiac MRI. It is also larger when measured leading edge to leading edge than inner edge to inner edge on echocardiography.10
TAA is defined as an aortic diameter at least 50% greater than the upper limit of normal.8

Geometric changes in the curvature of the ascending aorta, aortic arch, and descending thoracic aorta can occur as the result of hypertension, atherosclerosis, or connective tissue disease.
HOW IS TAA DIAGNOSED?



Imaging tests
 that tapers to normal dimensions at the sinotubular junction (yellow arrow) and ascending aorta.")


It is particularly important to obtain a gated CTA image in patients with aortic root aneurysm to avoid motion artifact and possible erroneous measurements. Gated CTA is done with electrocardiographic synchronization and allows for image processing to correct for cardiac motion.
HOW IS TAA CLASSIFIED?
TAA can be caused by a variety of inherited and sporadic conditions. These differences in pathogenesis lend themselves to classification of aneurysms into groups. Table 3 highlights the most common conditions associated with TAA.13
Bicuspid aortic valve aortopathy
From 1% to 2% of people have a bicuspid aortic valve, with a 3-to-1 male predominance.14,15 Aortic dilation occurs in 35% to 80% of people who have a bicuspid aortic valve, conferring a risk of dissection 8 times higher than in the general population.16–18
The pathogenic mechanisms that lead to this condition are widely debated, although a combination of genetic defects leading to intrinsic weakening of the aortic wall and hemodynamic effects likely contribute.19 Evidence of hemodynamic contributions to aortic dilation comes from findings that particular patterns of cusp fusion of the bicuspid aortic valve result in changes in transvalvular flow, placing more stress on specific regions of the ascending aorta.20,21 These hemodynamic alterations result in patterns of aortic dilation that depend on cusp fusion and the presence of valvular disease.
Multiple small studies found that replacing bicuspid aortic valves reduced the rate of aortic dilation, suggesting that hemodynamic factors may play a larger role than intrinsic wall properties in genetically susceptible individuals.22,23 However, larger studies are needed before any definitive conclusions can be made.
HOW IS ANEURYSM MANAGED ON AN OUTPATIENT BASIS?
Patients with a new diagnosis of TAA should be referred to a cardiologist with expertise in managing aortic disease or to a cardiac surgeon specializing in aortic surgery, depending on the initial size of the aneurysm.
Control blood pressure with beta-blockers
Medical management for patients with TAA has historically been limited to strict blood pressure control aimed at reducing aortic wall stress, mainly with beta-blockers.
Are angiotensin II receptor blockers (ARBs) beneficial? Studies in a mouse model of Marfan syndrome revealed that the ARB losartan attenuated aortic root growth.24 The results of early, small studies in humans were promising,25–27 but larger randomized trials have shown no advantage of losartan over beta-blockers in slowing aortic root growth.28 These negative results led many to question the effectiveness of losartan, although some point out that no studies have shown even beta-blockers to be beneficial in reducing the clinical end points of death or dissection.29 On the other hand, patients with certain FBN1 mutations respond more readily than others to losartan.30 Additional clinical trials of ARBs in Marfan syndrome are ongoing.
Current guidelines recommend stringent blood pressure control and smoking cessation for patients with a small aneurysm not requiring surgery and for those who are considered unsuitable for surgical or percutaneous intervention (level of evidence C, the lowest).2 For patients with TAA, it is considered reasonable to give beta-blockers. Angiotensin-converting enzyme inhibitors or ARBs may be used in combination with beta-blockers, titrated to the lowest tolerable blood pressure without adverse effects (level of evidence B).2
The recommended target blood pressure is less than 140/90 mm Hg, or 130/80 mm Hg in those with diabetes or chronic kidney disease (level of evidence B).2 However, we recommend more stringent blood pressure control: ie, less than 130/80 mm Hg for all patients with aortic aneurysm and a heart rate goal of 70 beats per minute or less, as tolerated.
Activity restriction
Activity restrictions for patients with TAA are largely based on theory, and certain activities may require more modification than others. For example, heavy lifting should be discouraged, as it may increase blood pressure significantly for short periods of time.2,31 The increased wall stress, in theory, could initiate dissection or rupture. However, moderate-intensity aerobic activity is rarely associated with significant elevations in blood pressure and should be encouraged. Stressful emotional states have been anecdotally associated with aortic dissection; thus, measures to reduce stress may offer some benefit.31
Our recommendations. While there are no published guidelines regarding activity restrictions in patients with TAA, we use a graded approach based on aortic diameter:
- 4.0 to 4.4 cm—lift no more than 75 pounds
- 4.5 to 5 cm—lift no more than 50 pounds
- 5 cm—lift no more than 25 pounds.
We also recommend not lifting anything heavier than half of one’s body weight and to avoid breath-holding or performing the Valsalva maneuver while lifting. Although these recommendations are somewhat arbitrary, based on theory and a large clinical experience at our aortic center, they seem reasonable and practical.
Activity restrictions should be stringent and individualized in patients with Marfan, Loeys-Dietz, or Ehlers-Danlos syndrome due to increased risk of dissection or rupture even if the aorta is normal in size.
We sometimes recommend exercise stress testing to assess the heart rate and blood pressure response to exercise, and we are developing research protocols to help tailor activity recommendations.
WHEN SHOULD A PATIENT BE REFERRED?
To a cardiologist at the time of diagnosis
As soon as TAA is diagnosed, the patient should be referred to a cardiologist who has special interest in aortic disease. This will allow for appropriate and timely decisions about medical management, imaging, follow-up, and referral to surgery. Additional recommendations for screening of family members and referral to clinical geneticists can be discussed at this juncture. Activity restrictions should be reviewed at the initial evaluation.
To a surgeon relatively early
Size thresholds for surgical intervention are discussed below, but one should not wait until these thresholds are reached to send the patient for surgical consultation. It is beneficial to the state of mind of a potential surgical candidate to have early discussions pertaining to the types of operations available, their outcomes, and associated risks and benefits. If a patient’s aortic size remains stable over time, he or she may be followed by the cardiologist until significant size or growth has been documented, at which time the patient and surgeon can reconvene to discuss options for definitive treatment.
To a clinical geneticist
If 1 or more first-degree relatives of a patient with TAA or dissection are found to have aneurysmal disease, referral to a clinical geneticist is very important for genetic testing of multiple genes that have been implicated in thoracic aortic aneurysm and dissection.
WHEN SHOULD TAA BE REPAIRED?
Surgery to prevent rupture or dissection remains the definitive treatment of TAA when size thresholds are reached, and symptomatic aneurysm should be operated on regardless of the size. However, rarely are thoracic aneurysms symptomatic unless they rupture or dissect. The size criteria are based on underlying genetic etiology if known and on the behavior and natural course of TAA.
Size and other factors
Treatment should be tailored to the patient’s clinical scenario, family history, and estimated risk of rupture or dissection, balanced against the individual center’s outcomes of elective aortic replacement.32 For example, young and otherwise healthy patients with TAA and a family history of aortic dissection (who may be more likely to have connective tissue disorders such as Marfan syndrome, Loeys-Dietz syndrome, or vascular Ehler-Danlos syndrome) may elect to undergo repair when the aneurysm reaches or nearly reaches the diameter of that of the family member’s aorta when dissection occurred.2 On the other hand, TAA of degenerative etiology (eg, related to smoking or hypertension) measuring less than 5.5 cm in an older patient with comorbidities poses a lower risk of a catastrophic event such as dissection or rupture than the risk of surgery.11
Thresholds for surgery. Once the diameter of the ascending aorta reaches 6 cm, the likelihood of an acute dissection is 31%.11 A similar threshold is reached for the descending aorta at a size of 7 cm.11 Therefore, to avoid high-risk emergency surgery on an acutely dissected aorta, surgery on an ascending aortic aneurysm of degenerative etiology is usually suggested when the aneurysm reaches 5.5 cm or a documented growth rate greater than 0.5 cm/year.2,33
Additionally, in patients already undergoing surgery for valvular or coronary disease, prophylactic aortic replacement is recommended if the ascending aorta is larger than 4.5 cm. The threshold for intervention is lower in patients with connective tissue disease (> 5.0 cm for Marfan syndrome, 4.4–4.6 cm for Loeys-Dietz syndrome).2,33
Observational studies suggest that the risk of aortic complications in patients with bicuspid aortic valve aortopathy is low overall, though significantly greater than in the general population.18,34,35 These findings led to changes in the 2014 American College of Cardiology/American Heart Association guidelines on valvular heart disease,36 suggesting a surgical threshold of 5.5 cm in the absence of significant valve disease or family history of dissection of an aorta of smaller diameter.
A 2015 study of dissection risk in patients with bicuspid aortic valve aortopathy by our group found a dramatic increase in risk of aortic dissection for ascending aortic diameters greater than 5.3 cm, and a gradual increase in risk for aortic root diameters greater than 5.0 cm.37 In addition, a near-constant 3% to 4% risk of dissection was present for aortic diameters ranging from 4.7 cm to 5.0 cm, revealing that watchful waiting carries its own inherent risks.37 In our surgical experience with this population, the hospital mortality rate and risk of stroke from aortic surgery were 0.25% and 0.75%, respectively.37 Thus, the decision to operate for aortic aneurysm in the setting of a bicuspid aortic valve should take into account patient-specific factors and institutional outcomes.
A statement of clarification in the American College of Cardiology/American Heart Association guidelines was published in 2015, recommending surgery for patients with an aortic diameter of 5.0 cm or greater if the patient is at low risk and the surgery is performed by an experienced surgical team at a center with established surgical expertise in this condition.38 However, current recommendations are for surgery at 5.5 cm if the above conditions are not met.
Ratio of aortic cross-sectional area to height
Although size alone has long been used to guide surgical intervention, a recent review from the International Registry of Aortic Dissection revealed that 59% of patients suffered aortic dissection at diameters less than 5.5 cm, and that patients with certain connective tissue diseases such as Loeys-Dietz syndrome or familial thoracic aneurysm and dissection had a documented propensity for dissection at smaller diameters.39–41
Size indices such as the aortic cross-sectional area indexed to height have been implemented in guidelines for certain patient populations (eg, 10 cm2/m in Marfan syndrome) and provide better risk stratification than size cutoffs alone.2,42
The ratio of aortic cross-sectional area to the patient’s height has also been applied to patients with bicuspid aortic valve-associated aortopathy and to those with a dilated aorta and a tricuspid aortic valve.43,44 Notably, a ratio greater than 10 cm2/m has been associated with aortic dissection in these groups, and this cutoff provides better stratification for prediction of death than traditional size metrics.27,28
HOW SHOULD PATIENTS BE SCREENED? WHAT FOLLOW-UP IS NECESSARY?
Initial screening and follow-up
Follow-up of TAA depends on the initial aortic size or rate of growth, or both. For patients presenting for the first time with TAA, it is reasonable to obtain definitive aortic imaging with CT or magnetic resonance angiography (MRA), then to repeat imaging at 6 months to document stability. If the aortic dimensions remain stable, then annual follow-up with CT or MRA is reasonable.2

Our flow chart of initial screening and follow-up is shown in Figure 5.
Screening of family members
In our center, we routinely recommend screening of all first-degree relatives of patients with TAA. Aortic imaging with echocardiography plus CT or MRI should be considered to detect asymptomatic disease.2 In patients with a strong family history (ie, multiple relatives affected with aortic aneurysm, dissection, or sudden cardiac death), genetic screening and testing for known mutations are recommended for the patient as well as for the family members.
If a mutation is identified in a family, then first-degree relatives should undergo genetic screening for the mutation and aortic imaging.2 Imaging in second-degree relatives may also be considered if one or more first-degree relatives are found to have aortic dilation.2
We recommend similar screening of first-degree family members of patients with bicuspid aortic valve aortopathy. In patients with young children, we recommend obtaining an echocardiogram of the child to look for a bicuspid aortic valve or aortic dilation. If an abnormality is detected or suspected, dedicated imaging with MRA to assess aortic dimensions is warranted.
BACK TO OUR PATIENT WITH A BICUSPID AORTIC VALVE
Our patient with a bicuspid aortic valve had a 4.6-cm root, an ascending aortic aneurysm, and several affected family members.
We would obtain dedicated aortic imaging at this patient’s initial visit with either gated CT with contrast or MRA, and we would obtain a cardioaortic surgery consult. We would repeat these studies at a follow-up visit 6 months later to detect any aortic growth compared with initial studies, and follow up annually thereafter. Echocardiography can also be done at the initial visit to determine if valvular disease is present that may influence clinical decisions.
Surgery would likely be recommended once the root reached a maximum area-to-height ratio greater than 10 cm2/m, or if the valve became severely dysfunctional during follow-up.
BACK TO OUR PATIENT WITH MARFAN SYNDROME
The young woman with Marfan syndrome has a 4.6-cm aortic root aneurysm and 2+ aortic insufficiency. Her question pertains to the threshold at which an operation would be considered. This question is complicated and is influenced by several concurrent clinical features in her presentation.
Starting with size criteria, patients with Marfan syndrome should be considered for elective aortic root repair at a diameter greater than 5 cm. However, an aortic cross-sectional area-to-height ratio greater than 10 cm2/m may provide a more robust metric for clinical decision-making than aortic diameter alone. Additional factors such as degree of aortic insufficiency and deleterious left ventricular remodeling may urge one to consider aortic root repair at a diameter of 4.5 cm.
These factors, including rate of growth and the surgeon’s assessment about his or her ability to preserve the aortic valve during repair, should be considered collectively in this scenario.
- Elefteriades JA, Farkas EA. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J Am Coll Cardiol 2010; 55(9):841–857. doi:10.1016/j.jacc.2009.08.084
- Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary. Anesth Analg 2010; 111(2):279–315. doi:10.1213/ANE.0b013e3181dd869b
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280(22):1926–1929. pmid:9851478
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114(24):2611–2618. doi:10.1161/CIRCULATIONAHA.106.630400
- Clouse WD, Hallett JW Jr, Schaff HV, et al. Acute aortic dissection: population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc 2004; 79(2):176–180. pmid:14959911
- US Centers for Disease Control and Prevention (CDC). National Center for Injury Prevention and Control. WISQARS leading causes of death reports, 1999 – 2007. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10.html. Accessed May 21, 2018.
- Hansen MS, Nogareda GJ, Hutchison SJ. Frequency of and inappropriate treatment of misdiagnosis of acute aortic dissection. Am J Cardiol 2007; 99(6):852–856. doi:10.1016/j.amjcard.2006.10.055
- Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2014; 64(16):1725–1739. doi:10.1016/j.jacc.2014.08.025
- Kumar V, Abbas A, Aster J. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2015.
- Wolak A, Gransar H, Thomson LE, et al. Aortic size assessment by noncontrast cardiac computed tomography: normal limits by age, gender, and body surface area. JACC Cardiovasc Imaging 2008; 1(2):200–209. doi:10.1016/j.jcmg.2007.11.005
- Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg 2002; 74(5):S1877–S1880; discussion S1892–S1898. pmid:12440685
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75(1):7–17. pmid:18236724
- Cury M, Zeidan F, Lobato AC. Aortic disease in the young: genetic aneurysm syndromes, connective tissue disorders, and familial aortic aneurysms and dissections. Int J Vasc Med 2013(2013); 2013:267215. doi:10.1155/2013/267215
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39(12):1890–1900. doi:10.1016/S0735-1097(02)01886-7
- Fedak PW, Verma S, David TE, Leask RL, Weisel RD, Butany J. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation 2002; 106(8):900–904. pmid:12186790
- Della Corte A, Bancone C, Quarto C, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg 2007; 31(3):397–405. doi:10.1016/j.ejcts.2006.12.006
- Jackson V, Petrini J, Caidahl K, et al. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: a study of 300 patients undergoing open-heart surgery. Eur J Cardiothorac Surg 2011; 40(3):e118–e124. doi:10.1016/j.ejcts.2011.04.014
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112. doi:10.1001/jama.2011.1286
- Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 2014; 370(20):1920–1929. doi:10.1056/NEJMra1207059
- Barker AJ, Markl M, Bürk J, et al. Bicuspid aortic valve is associated with altered wall shear stress in the ascending aorta. Circ Cardiovasc Imaging 2012; 5(4):457–466. doi:10.1161/CIRCIMAGING.112.973370
- Hope MD, Hope TA, Meadows AK, et al. Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology 2010; 255(1):53–61. doi:10.1148/radiol.09091437
- Abdulkareem N, Soppa G, Jones S, Valencia O, Smelt J, Jahangiri M. Dilatation of the remaining aorta after aortic valve or aortic root replacement in patients with bicuspid aortic valve: a 5-year follow-up. Ann Thorac Surg 2013; 96(1):43–49. doi:10.1016/j.athoracsur.2013.03.086
- Regeer MV, Versteegh MI, Klautz RJ, et al. Effect of aortic valve replacement on aortic root dilatation rate in patients with bicuspid and tricuspid aortic valves. Ann Thorac Surg 2016; 102(6):1981–1987. doi:10.1016/j.athoracsur.2016.05.038
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312(5770):117–121. doi:10.1126/science.1124287
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358(26):2787–2795. doi:10.1056/NEJMoa0706585
- Chiu HH, Wu MH, Wang JK, et al. Losartan added to ß-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc 2013; 88(3):271–276. doi:10.1016/j.mayocp.2012.11.005
- Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013; 34(45):3491–3500. doi:10.1093/eurheartj/eht334
- Lacro RV, Dietz HC, Sleeper LA, et al; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014; 371(22):2061–2071. doi:10.1056/NEJMoa1404731
- Ziganshin BA, Mukherjee SK, Elefteriades JA, et al. Atenolol versus losartan in Marfan’s syndrome (letters). N Engl J Med 2015; 372(10):977–981. doi:10.1056/NEJMc1500128
- Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet 2015; 8(2):383–388. doi:10.1161/CIRCGENETICS.114.000950
- Elefteriades JA. Thoracic aortic aneurysm: reading the enemy’s playbook. Curr Probl Cardiol 2008; 33(5):203–277. doi:10.1016/j.cpcardiol.2008.01.004
- Idrees JJ, Roselli EE, Lowry AM, et al. Outcomes after elective proximal aortic replacement: a matched comparison of isolated versus multicomponent operations. Ann Thorac Surg 2016; 101(6):2185–2192. doi:10.1016/j.athoracsur.2015.12.026
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Thorac Cardiovasc Surg 2016; 151(4):959–966. doi:10.1016/j.jtcvs.2015.12.001
- Tzemos N, Therrien J, Yip J, et al. Outcomes in adults with bicuspid aortic valves. JAMA 2008; 300(11):1317–1325. doi:10.1001/jama.300.11.1317
- Davies RR, Goldstein LJ, Coady MA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg 2002; 73(1):17–28. pmid:11834007
- Nishimura RA, Otto CM, Bono RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Wojnarski CM, Svensson LG, Roselli EE, et al. Aortic dissection in patients with bicuspid aortic valve–associated aneurysms. Ann Thorac Surg 2015; 100(5):1666–1674. doi:10.1016/j.athoracsur.2015.04.126
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2016; 133(7):680–686. doi:10.1161/CIR.0000000000000331
- Pape LA, Tsai TT, Isselbacher EM, et al; International Registry of Acute Aortic Dissection (IRAD) Investigators. Aortic diameter > or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007; 116(10):1120–1127. doi:10.1161/CIRCULATIONAHA.107.702720
- Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355(8):788–798. doi:10.1056/NEJMoa055695
- Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39(12):1488–1493. doi:10.1038/ng.2007.6
- Svensson LG, Khitin L. Aortic cross-sectional area/height ratio timing of aortic surgery in asymptomatic patients with Marfan syndrome. J Thorac Cardiovasc Surg 2002; 123(2):360–361. pmid:11828302
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection in patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003; 126(3):892–893. pmid:14502185
- Masri A, Kalahasti V, Svensson LG, et al. Aortic cross-sectional area/height ratio and outcomes in patients with a trileaflet aortic valve and a dilated aorta. Circulation 2016; 134(22):1724–1737. doi:10.1161/CIRCULATIONAHA.116.022995
Thoracic aortic aneurysm (TAA) needs to be detected, monitored, and managed in a timely manner to prevent a serious consequence such as acute dissection or rupture. But only about 5% of patients experience symptoms before an acute event occurs, and for the other 95% the first “symptom” is often death.1 Most cases are detected either incidentally with echocardiography, computed tomography (CT), or magnetic resonance imaging (MRI) during workup for another condition. Patients may also be diagnosed during workup of a murmur or after a family member is found to have an aneurysm. Therefore, its true incidence is difficult to determine.2
With these facts in mind, how would you manage the following 2 cases?
Case 1: Bicuspid aortic valve, ascending aortic aneurysm
A 45-year-old man with stage 1 hypertension presents for evaluation of a bicuspid aortic valve and ascending aortic aneurysm. He has several first-degree relatives with similar conditions, and his brother recently underwent elective aortic repair. At the urging of his primary care physician, he underwent screening echocardiography, which demonstrated a “dilated root and ascending aorta” 4.6 cm in diameter. He presents today to discuss management options and how the aneurysm could affect his everyday life.
Case 2: Marfan syndrome in a young woman
A 24-year-old woman with Marfan syndrome diagnosed in adolescence presents for annual follow-up. She has many family members with the same condition, and several have undergone prophylactic aortic root repair. Her aortic root has been monitored annually for progression of dilation, and today it is 4.6 cm in diameter, a 3-mm increase from the last measurement. She has grade 2+ aortic insufficiency (on a scale of 1+ to 4+) based on echocardiography, but she has no symptoms. She is curious about what size her aortic root will need to reach for surgery to be considered.
LIKELY UNDERDETECTED
TAA is being detected more often than in the past thanks to better detection methods and heightened awareness among physicians and patients. While an incidence rate of 10.4 per 100,000 patient-years is often cited,3 this figure likely underestimates the true incidence of this clinically silent condition. The most robust data come from studies based on in-hospital diagnostic codes coupled with data from autopsies for out-of-hospital deaths.
Olsson et al,4 in a 2016 study in Sweden, found the incidence of TAA and aortic dissection to be 16.3 per 100,000 per year for men and 9.1 per 100,000 per year for women.
Clouse et al5 reported the incidence of thoracic aortic dissection as 3.5 per 100,000 patient-years, and the same figure for thoracic aortic rupture.
Aneurysmal disease accounts for 52,000 deaths per year in the United States, making it the 19th most common cause of death.6 These figures are likely lower than the true mortality rate for this condition, given that aortic dissection is often mistaken for acute myocardial infarction or other acute event if an autopsy is not done to confirm the cause of death.7
RISK FACTORS FOR THORACIC AORTIC ANEURYSM
Risk factors for TAA include genetic conditions that lead to aortic medial weakness or destruction such as Loeys-Dietz syndrome and Marfan syndrome.2 In addition, family history is important even in the absence of known genetic mutations. Other risk factors include conditions that increase aortic wall stress, such as hypertension, cocaine abuse, extreme weightlifting, trauma, and aortic coarctation.2
DIAMETER INCREASES WITH AGE, BODY SURFACE AREA
Normal dimensions for the aortic segments differ depending on age, sex, and body surface area.8,44,45 The size of the aortic root may also vary depending on how it is measured, due to the root’s trefoil shape. Measured sinus to sinus, the root is larger than when measured sinus to commissure on CT angiography or cardiac MRI. It is also larger when measured leading edge to leading edge than inner edge to inner edge on echocardiography.10
TAA is defined as an aortic diameter at least 50% greater than the upper limit of normal.8
Geometric changes in the curvature of the ascending aorta, aortic arch, and descending thoracic aorta can occur as the result of hypertension, atherosclerosis, or connective tissue disease.
HOW IS TAA DIAGNOSED?
Imaging tests
It is particularly important to obtain a gated CTA image in patients with aortic root aneurysm to avoid motion artifact and possible erroneous measurements. Gated CTA is done with electrocardiographic synchronization and allows for image processing to correct for cardiac motion.
HOW IS TAA CLASSIFIED?
TAA can be caused by a variety of inherited and sporadic conditions. These differences in pathogenesis lend themselves to classification of aneurysms into groups. Table 3 highlights the most common conditions associated with TAA.13
Bicuspid aortic valve aortopathy
From 1% to 2% of people have a bicuspid aortic valve, with a 3-to-1 male predominance.14,15 Aortic dilation occurs in 35% to 80% of people who have a bicuspid aortic valve, conferring a risk of dissection 8 times higher than in the general population.16–18
The pathogenic mechanisms that lead to this condition are widely debated, although a combination of genetic defects leading to intrinsic weakening of the aortic wall and hemodynamic effects likely contribute.19 Evidence of hemodynamic contributions to aortic dilation comes from findings that particular patterns of cusp fusion of the bicuspid aortic valve result in changes in transvalvular flow, placing more stress on specific regions of the ascending aorta.20,21 These hemodynamic alterations result in patterns of aortic dilation that depend on cusp fusion and the presence of valvular disease.
Multiple small studies found that replacing bicuspid aortic valves reduced the rate of aortic dilation, suggesting that hemodynamic factors may play a larger role than intrinsic wall properties in genetically susceptible individuals.22,23 However, larger studies are needed before any definitive conclusions can be made.
HOW IS ANEURYSM MANAGED ON AN OUTPATIENT BASIS?
Patients with a new diagnosis of TAA should be referred to a cardiologist with expertise in managing aortic disease or to a cardiac surgeon specializing in aortic surgery, depending on the initial size of the aneurysm.
Control blood pressure with beta-blockers
Medical management for patients with TAA has historically been limited to strict blood pressure control aimed at reducing aortic wall stress, mainly with beta-blockers.
Are angiotensin II receptor blockers (ARBs) beneficial? Studies in a mouse model of Marfan syndrome revealed that the ARB losartan attenuated aortic root growth.24 The results of early, small studies in humans were promising,25–27 but larger randomized trials have shown no advantage of losartan over beta-blockers in slowing aortic root growth.28 These negative results led many to question the effectiveness of losartan, although some point out that no studies have shown even beta-blockers to be beneficial in reducing the clinical end points of death or dissection.29 On the other hand, patients with certain FBN1 mutations respond more readily than others to losartan.30 Additional clinical trials of ARBs in Marfan syndrome are ongoing.
Current guidelines recommend stringent blood pressure control and smoking cessation for patients with a small aneurysm not requiring surgery and for those who are considered unsuitable for surgical or percutaneous intervention (level of evidence C, the lowest).2 For patients with TAA, it is considered reasonable to give beta-blockers. Angiotensin-converting enzyme inhibitors or ARBs may be used in combination with beta-blockers, titrated to the lowest tolerable blood pressure without adverse effects (level of evidence B).2
The recommended target blood pressure is less than 140/90 mm Hg, or 130/80 mm Hg in those with diabetes or chronic kidney disease (level of evidence B).2 However, we recommend more stringent blood pressure control: ie, less than 130/80 mm Hg for all patients with aortic aneurysm and a heart rate goal of 70 beats per minute or less, as tolerated.
Activity restriction
Activity restrictions for patients with TAA are largely based on theory, and certain activities may require more modification than others. For example, heavy lifting should be discouraged, as it may increase blood pressure significantly for short periods of time.2,31 The increased wall stress, in theory, could initiate dissection or rupture. However, moderate-intensity aerobic activity is rarely associated with significant elevations in blood pressure and should be encouraged. Stressful emotional states have been anecdotally associated with aortic dissection; thus, measures to reduce stress may offer some benefit.31
Our recommendations. While there are no published guidelines regarding activity restrictions in patients with TAA, we use a graded approach based on aortic diameter:
- 4.0 to 4.4 cm—lift no more than 75 pounds
- 4.5 to 5 cm—lift no more than 50 pounds
- 5 cm—lift no more than 25 pounds.
We also recommend not lifting anything heavier than half of one’s body weight and to avoid breath-holding or performing the Valsalva maneuver while lifting. Although these recommendations are somewhat arbitrary, based on theory and a large clinical experience at our aortic center, they seem reasonable and practical.
Activity restrictions should be stringent and individualized in patients with Marfan, Loeys-Dietz, or Ehlers-Danlos syndrome due to increased risk of dissection or rupture even if the aorta is normal in size.
We sometimes recommend exercise stress testing to assess the heart rate and blood pressure response to exercise, and we are developing research protocols to help tailor activity recommendations.
WHEN SHOULD A PATIENT BE REFERRED?
To a cardiologist at the time of diagnosis
As soon as TAA is diagnosed, the patient should be referred to a cardiologist who has special interest in aortic disease. This will allow for appropriate and timely decisions about medical management, imaging, follow-up, and referral to surgery. Additional recommendations for screening of family members and referral to clinical geneticists can be discussed at this juncture. Activity restrictions should be reviewed at the initial evaluation.
To a surgeon relatively early
Size thresholds for surgical intervention are discussed below, but one should not wait until these thresholds are reached to send the patient for surgical consultation. It is beneficial to the state of mind of a potential surgical candidate to have early discussions pertaining to the types of operations available, their outcomes, and associated risks and benefits. If a patient’s aortic size remains stable over time, he or she may be followed by the cardiologist until significant size or growth has been documented, at which time the patient and surgeon can reconvene to discuss options for definitive treatment.
To a clinical geneticist
If 1 or more first-degree relatives of a patient with TAA or dissection are found to have aneurysmal disease, referral to a clinical geneticist is very important for genetic testing of multiple genes that have been implicated in thoracic aortic aneurysm and dissection.
WHEN SHOULD TAA BE REPAIRED?
Surgery to prevent rupture or dissection remains the definitive treatment of TAA when size thresholds are reached, and symptomatic aneurysm should be operated on regardless of the size. However, rarely are thoracic aneurysms symptomatic unless they rupture or dissect. The size criteria are based on underlying genetic etiology if known and on the behavior and natural course of TAA.
Size and other factors
Treatment should be tailored to the patient’s clinical scenario, family history, and estimated risk of rupture or dissection, balanced against the individual center’s outcomes of elective aortic replacement.32 For example, young and otherwise healthy patients with TAA and a family history of aortic dissection (who may be more likely to have connective tissue disorders such as Marfan syndrome, Loeys-Dietz syndrome, or vascular Ehler-Danlos syndrome) may elect to undergo repair when the aneurysm reaches or nearly reaches the diameter of that of the family member’s aorta when dissection occurred.2 On the other hand, TAA of degenerative etiology (eg, related to smoking or hypertension) measuring less than 5.5 cm in an older patient with comorbidities poses a lower risk of a catastrophic event such as dissection or rupture than the risk of surgery.11
Thresholds for surgery. Once the diameter of the ascending aorta reaches 6 cm, the likelihood of an acute dissection is 31%.11 A similar threshold is reached for the descending aorta at a size of 7 cm.11 Therefore, to avoid high-risk emergency surgery on an acutely dissected aorta, surgery on an ascending aortic aneurysm of degenerative etiology is usually suggested when the aneurysm reaches 5.5 cm or a documented growth rate greater than 0.5 cm/year.2,33
Additionally, in patients already undergoing surgery for valvular or coronary disease, prophylactic aortic replacement is recommended if the ascending aorta is larger than 4.5 cm. The threshold for intervention is lower in patients with connective tissue disease (> 5.0 cm for Marfan syndrome, 4.4–4.6 cm for Loeys-Dietz syndrome).2,33
Observational studies suggest that the risk of aortic complications in patients with bicuspid aortic valve aortopathy is low overall, though significantly greater than in the general population.18,34,35 These findings led to changes in the 2014 American College of Cardiology/American Heart Association guidelines on valvular heart disease,36 suggesting a surgical threshold of 5.5 cm in the absence of significant valve disease or family history of dissection of an aorta of smaller diameter.
A 2015 study of dissection risk in patients with bicuspid aortic valve aortopathy by our group found a dramatic increase in risk of aortic dissection for ascending aortic diameters greater than 5.3 cm, and a gradual increase in risk for aortic root diameters greater than 5.0 cm.37 In addition, a near-constant 3% to 4% risk of dissection was present for aortic diameters ranging from 4.7 cm to 5.0 cm, revealing that watchful waiting carries its own inherent risks.37 In our surgical experience with this population, the hospital mortality rate and risk of stroke from aortic surgery were 0.25% and 0.75%, respectively.37 Thus, the decision to operate for aortic aneurysm in the setting of a bicuspid aortic valve should take into account patient-specific factors and institutional outcomes.
A statement of clarification in the American College of Cardiology/American Heart Association guidelines was published in 2015, recommending surgery for patients with an aortic diameter of 5.0 cm or greater if the patient is at low risk and the surgery is performed by an experienced surgical team at a center with established surgical expertise in this condition.38 However, current recommendations are for surgery at 5.5 cm if the above conditions are not met.
Ratio of aortic cross-sectional area to height
Although size alone has long been used to guide surgical intervention, a recent review from the International Registry of Aortic Dissection revealed that 59% of patients suffered aortic dissection at diameters less than 5.5 cm, and that patients with certain connective tissue diseases such as Loeys-Dietz syndrome or familial thoracic aneurysm and dissection had a documented propensity for dissection at smaller diameters.39–41
Size indices such as the aortic cross-sectional area indexed to height have been implemented in guidelines for certain patient populations (eg, 10 cm2/m in Marfan syndrome) and provide better risk stratification than size cutoffs alone.2,42
The ratio of aortic cross-sectional area to the patient’s height has also been applied to patients with bicuspid aortic valve-associated aortopathy and to those with a dilated aorta and a tricuspid aortic valve.43,44 Notably, a ratio greater than 10 cm2/m has been associated with aortic dissection in these groups, and this cutoff provides better stratification for prediction of death than traditional size metrics.27,28
HOW SHOULD PATIENTS BE SCREENED? WHAT FOLLOW-UP IS NECESSARY?
Initial screening and follow-up
Follow-up of TAA depends on the initial aortic size or rate of growth, or both. For patients presenting for the first time with TAA, it is reasonable to obtain definitive aortic imaging with CT or magnetic resonance angiography (MRA), then to repeat imaging at 6 months to document stability. If the aortic dimensions remain stable, then annual follow-up with CT or MRA is reasonable.2
Our flow chart of initial screening and follow-up is shown in Figure 5.
Screening of family members
In our center, we routinely recommend screening of all first-degree relatives of patients with TAA. Aortic imaging with echocardiography plus CT or MRI should be considered to detect asymptomatic disease.2 In patients with a strong family history (ie, multiple relatives affected with aortic aneurysm, dissection, or sudden cardiac death), genetic screening and testing for known mutations are recommended for the patient as well as for the family members.
If a mutation is identified in a family, then first-degree relatives should undergo genetic screening for the mutation and aortic imaging.2 Imaging in second-degree relatives may also be considered if one or more first-degree relatives are found to have aortic dilation.2
We recommend similar screening of first-degree family members of patients with bicuspid aortic valve aortopathy. In patients with young children, we recommend obtaining an echocardiogram of the child to look for a bicuspid aortic valve or aortic dilation. If an abnormality is detected or suspected, dedicated imaging with MRA to assess aortic dimensions is warranted.
BACK TO OUR PATIENT WITH A BICUSPID AORTIC VALVE
Our patient with a bicuspid aortic valve had a 4.6-cm root, an ascending aortic aneurysm, and several affected family members.
We would obtain dedicated aortic imaging at this patient’s initial visit with either gated CT with contrast or MRA, and we would obtain a cardioaortic surgery consult. We would repeat these studies at a follow-up visit 6 months later to detect any aortic growth compared with initial studies, and follow up annually thereafter. Echocardiography can also be done at the initial visit to determine if valvular disease is present that may influence clinical decisions.
Surgery would likely be recommended once the root reached a maximum area-to-height ratio greater than 10 cm2/m, or if the valve became severely dysfunctional during follow-up.
BACK TO OUR PATIENT WITH MARFAN SYNDROME
The young woman with Marfan syndrome has a 4.6-cm aortic root aneurysm and 2+ aortic insufficiency. Her question pertains to the threshold at which an operation would be considered. This question is complicated and is influenced by several concurrent clinical features in her presentation.
Starting with size criteria, patients with Marfan syndrome should be considered for elective aortic root repair at a diameter greater than 5 cm. However, an aortic cross-sectional area-to-height ratio greater than 10 cm2/m may provide a more robust metric for clinical decision-making than aortic diameter alone. Additional factors such as degree of aortic insufficiency and deleterious left ventricular remodeling may urge one to consider aortic root repair at a diameter of 4.5 cm.
These factors, including rate of growth and the surgeon’s assessment about his or her ability to preserve the aortic valve during repair, should be considered collectively in this scenario.
Thoracic aortic aneurysm (TAA) needs to be detected, monitored, and managed in a timely manner to prevent a serious consequence such as acute dissection or rupture. But only about 5% of patients experience symptoms before an acute event occurs, and for the other 95% the first “symptom” is often death.1 Most cases are detected either incidentally with echocardiography, computed tomography (CT), or magnetic resonance imaging (MRI) during workup for another condition. Patients may also be diagnosed during workup of a murmur or after a family member is found to have an aneurysm. Therefore, its true incidence is difficult to determine.2
With these facts in mind, how would you manage the following 2 cases?
Case 1: Bicuspid aortic valve, ascending aortic aneurysm
A 45-year-old man with stage 1 hypertension presents for evaluation of a bicuspid aortic valve and ascending aortic aneurysm. He has several first-degree relatives with similar conditions, and his brother recently underwent elective aortic repair. At the urging of his primary care physician, he underwent screening echocardiography, which demonstrated a “dilated root and ascending aorta” 4.6 cm in diameter. He presents today to discuss management options and how the aneurysm could affect his everyday life.
Case 2: Marfan syndrome in a young woman
A 24-year-old woman with Marfan syndrome diagnosed in adolescence presents for annual follow-up. She has many family members with the same condition, and several have undergone prophylactic aortic root repair. Her aortic root has been monitored annually for progression of dilation, and today it is 4.6 cm in diameter, a 3-mm increase from the last measurement. She has grade 2+ aortic insufficiency (on a scale of 1+ to 4+) based on echocardiography, but she has no symptoms. She is curious about what size her aortic root will need to reach for surgery to be considered.
LIKELY UNDERDETECTED
TAA is being detected more often than in the past thanks to better detection methods and heightened awareness among physicians and patients. While an incidence rate of 10.4 per 100,000 patient-years is often cited,3 this figure likely underestimates the true incidence of this clinically silent condition. The most robust data come from studies based on in-hospital diagnostic codes coupled with data from autopsies for out-of-hospital deaths.
Olsson et al,4 in a 2016 study in Sweden, found the incidence of TAA and aortic dissection to be 16.3 per 100,000 per year for men and 9.1 per 100,000 per year for women.
Clouse et al5 reported the incidence of thoracic aortic dissection as 3.5 per 100,000 patient-years, and the same figure for thoracic aortic rupture.
Aneurysmal disease accounts for 52,000 deaths per year in the United States, making it the 19th most common cause of death.6 These figures are likely lower than the true mortality rate for this condition, given that aortic dissection is often mistaken for acute myocardial infarction or other acute event if an autopsy is not done to confirm the cause of death.7
RISK FACTORS FOR THORACIC AORTIC ANEURYSM
Risk factors for TAA include genetic conditions that lead to aortic medial weakness or destruction such as Loeys-Dietz syndrome and Marfan syndrome.2 In addition, family history is important even in the absence of known genetic mutations. Other risk factors include conditions that increase aortic wall stress, such as hypertension, cocaine abuse, extreme weightlifting, trauma, and aortic coarctation.2
DIAMETER INCREASES WITH AGE, BODY SURFACE AREA
Normal dimensions for the aortic segments differ depending on age, sex, and body surface area.8,44,45 The size of the aortic root may also vary depending on how it is measured, due to the root’s trefoil shape. Measured sinus to sinus, the root is larger than when measured sinus to commissure on CT angiography or cardiac MRI. It is also larger when measured leading edge to leading edge than inner edge to inner edge on echocardiography.10
TAA is defined as an aortic diameter at least 50% greater than the upper limit of normal.8
Geometric changes in the curvature of the ascending aorta, aortic arch, and descending thoracic aorta can occur as the result of hypertension, atherosclerosis, or connective tissue disease.
HOW IS TAA DIAGNOSED?
Imaging tests
It is particularly important to obtain a gated CTA image in patients with aortic root aneurysm to avoid motion artifact and possible erroneous measurements. Gated CTA is done with electrocardiographic synchronization and allows for image processing to correct for cardiac motion.
HOW IS TAA CLASSIFIED?
TAA can be caused by a variety of inherited and sporadic conditions. These differences in pathogenesis lend themselves to classification of aneurysms into groups. Table 3 highlights the most common conditions associated with TAA.13
Bicuspid aortic valve aortopathy
From 1% to 2% of people have a bicuspid aortic valve, with a 3-to-1 male predominance.14,15 Aortic dilation occurs in 35% to 80% of people who have a bicuspid aortic valve, conferring a risk of dissection 8 times higher than in the general population.16–18
The pathogenic mechanisms that lead to this condition are widely debated, although a combination of genetic defects leading to intrinsic weakening of the aortic wall and hemodynamic effects likely contribute.19 Evidence of hemodynamic contributions to aortic dilation comes from findings that particular patterns of cusp fusion of the bicuspid aortic valve result in changes in transvalvular flow, placing more stress on specific regions of the ascending aorta.20,21 These hemodynamic alterations result in patterns of aortic dilation that depend on cusp fusion and the presence of valvular disease.
Multiple small studies found that replacing bicuspid aortic valves reduced the rate of aortic dilation, suggesting that hemodynamic factors may play a larger role than intrinsic wall properties in genetically susceptible individuals.22,23 However, larger studies are needed before any definitive conclusions can be made.
HOW IS ANEURYSM MANAGED ON AN OUTPATIENT BASIS?
Patients with a new diagnosis of TAA should be referred to a cardiologist with expertise in managing aortic disease or to a cardiac surgeon specializing in aortic surgery, depending on the initial size of the aneurysm.
Control blood pressure with beta-blockers
Medical management for patients with TAA has historically been limited to strict blood pressure control aimed at reducing aortic wall stress, mainly with beta-blockers.
Are angiotensin II receptor blockers (ARBs) beneficial? Studies in a mouse model of Marfan syndrome revealed that the ARB losartan attenuated aortic root growth.24 The results of early, small studies in humans were promising,25–27 but larger randomized trials have shown no advantage of losartan over beta-blockers in slowing aortic root growth.28 These negative results led many to question the effectiveness of losartan, although some point out that no studies have shown even beta-blockers to be beneficial in reducing the clinical end points of death or dissection.29 On the other hand, patients with certain FBN1 mutations respond more readily than others to losartan.30 Additional clinical trials of ARBs in Marfan syndrome are ongoing.
Current guidelines recommend stringent blood pressure control and smoking cessation for patients with a small aneurysm not requiring surgery and for those who are considered unsuitable for surgical or percutaneous intervention (level of evidence C, the lowest).2 For patients with TAA, it is considered reasonable to give beta-blockers. Angiotensin-converting enzyme inhibitors or ARBs may be used in combination with beta-blockers, titrated to the lowest tolerable blood pressure without adverse effects (level of evidence B).2
The recommended target blood pressure is less than 140/90 mm Hg, or 130/80 mm Hg in those with diabetes or chronic kidney disease (level of evidence B).2 However, we recommend more stringent blood pressure control: ie, less than 130/80 mm Hg for all patients with aortic aneurysm and a heart rate goal of 70 beats per minute or less, as tolerated.
Activity restriction
Activity restrictions for patients with TAA are largely based on theory, and certain activities may require more modification than others. For example, heavy lifting should be discouraged, as it may increase blood pressure significantly for short periods of time.2,31 The increased wall stress, in theory, could initiate dissection or rupture. However, moderate-intensity aerobic activity is rarely associated with significant elevations in blood pressure and should be encouraged. Stressful emotional states have been anecdotally associated with aortic dissection; thus, measures to reduce stress may offer some benefit.31
Our recommendations. While there are no published guidelines regarding activity restrictions in patients with TAA, we use a graded approach based on aortic diameter:
- 4.0 to 4.4 cm—lift no more than 75 pounds
- 4.5 to 5 cm—lift no more than 50 pounds
- 5 cm—lift no more than 25 pounds.
We also recommend not lifting anything heavier than half of one’s body weight and to avoid breath-holding or performing the Valsalva maneuver while lifting. Although these recommendations are somewhat arbitrary, based on theory and a large clinical experience at our aortic center, they seem reasonable and practical.
Activity restrictions should be stringent and individualized in patients with Marfan, Loeys-Dietz, or Ehlers-Danlos syndrome due to increased risk of dissection or rupture even if the aorta is normal in size.
We sometimes recommend exercise stress testing to assess the heart rate and blood pressure response to exercise, and we are developing research protocols to help tailor activity recommendations.
WHEN SHOULD A PATIENT BE REFERRED?
To a cardiologist at the time of diagnosis
As soon as TAA is diagnosed, the patient should be referred to a cardiologist who has special interest in aortic disease. This will allow for appropriate and timely decisions about medical management, imaging, follow-up, and referral to surgery. Additional recommendations for screening of family members and referral to clinical geneticists can be discussed at this juncture. Activity restrictions should be reviewed at the initial evaluation.
To a surgeon relatively early
Size thresholds for surgical intervention are discussed below, but one should not wait until these thresholds are reached to send the patient for surgical consultation. It is beneficial to the state of mind of a potential surgical candidate to have early discussions pertaining to the types of operations available, their outcomes, and associated risks and benefits. If a patient’s aortic size remains stable over time, he or she may be followed by the cardiologist until significant size or growth has been documented, at which time the patient and surgeon can reconvene to discuss options for definitive treatment.
To a clinical geneticist
If 1 or more first-degree relatives of a patient with TAA or dissection are found to have aneurysmal disease, referral to a clinical geneticist is very important for genetic testing of multiple genes that have been implicated in thoracic aortic aneurysm and dissection.
WHEN SHOULD TAA BE REPAIRED?
Surgery to prevent rupture or dissection remains the definitive treatment of TAA when size thresholds are reached, and symptomatic aneurysm should be operated on regardless of the size. However, rarely are thoracic aneurysms symptomatic unless they rupture or dissect. The size criteria are based on underlying genetic etiology if known and on the behavior and natural course of TAA.
Size and other factors
Treatment should be tailored to the patient’s clinical scenario, family history, and estimated risk of rupture or dissection, balanced against the individual center’s outcomes of elective aortic replacement.32 For example, young and otherwise healthy patients with TAA and a family history of aortic dissection (who may be more likely to have connective tissue disorders such as Marfan syndrome, Loeys-Dietz syndrome, or vascular Ehler-Danlos syndrome) may elect to undergo repair when the aneurysm reaches or nearly reaches the diameter of that of the family member’s aorta when dissection occurred.2 On the other hand, TAA of degenerative etiology (eg, related to smoking or hypertension) measuring less than 5.5 cm in an older patient with comorbidities poses a lower risk of a catastrophic event such as dissection or rupture than the risk of surgery.11
Thresholds for surgery. Once the diameter of the ascending aorta reaches 6 cm, the likelihood of an acute dissection is 31%.11 A similar threshold is reached for the descending aorta at a size of 7 cm.11 Therefore, to avoid high-risk emergency surgery on an acutely dissected aorta, surgery on an ascending aortic aneurysm of degenerative etiology is usually suggested when the aneurysm reaches 5.5 cm or a documented growth rate greater than 0.5 cm/year.2,33
Additionally, in patients already undergoing surgery for valvular or coronary disease, prophylactic aortic replacement is recommended if the ascending aorta is larger than 4.5 cm. The threshold for intervention is lower in patients with connective tissue disease (> 5.0 cm for Marfan syndrome, 4.4–4.6 cm for Loeys-Dietz syndrome).2,33
Observational studies suggest that the risk of aortic complications in patients with bicuspid aortic valve aortopathy is low overall, though significantly greater than in the general population.18,34,35 These findings led to changes in the 2014 American College of Cardiology/American Heart Association guidelines on valvular heart disease,36 suggesting a surgical threshold of 5.5 cm in the absence of significant valve disease or family history of dissection of an aorta of smaller diameter.
A 2015 study of dissection risk in patients with bicuspid aortic valve aortopathy by our group found a dramatic increase in risk of aortic dissection for ascending aortic diameters greater than 5.3 cm, and a gradual increase in risk for aortic root diameters greater than 5.0 cm.37 In addition, a near-constant 3% to 4% risk of dissection was present for aortic diameters ranging from 4.7 cm to 5.0 cm, revealing that watchful waiting carries its own inherent risks.37 In our surgical experience with this population, the hospital mortality rate and risk of stroke from aortic surgery were 0.25% and 0.75%, respectively.37 Thus, the decision to operate for aortic aneurysm in the setting of a bicuspid aortic valve should take into account patient-specific factors and institutional outcomes.
A statement of clarification in the American College of Cardiology/American Heart Association guidelines was published in 2015, recommending surgery for patients with an aortic diameter of 5.0 cm or greater if the patient is at low risk and the surgery is performed by an experienced surgical team at a center with established surgical expertise in this condition.38 However, current recommendations are for surgery at 5.5 cm if the above conditions are not met.
Ratio of aortic cross-sectional area to height
Although size alone has long been used to guide surgical intervention, a recent review from the International Registry of Aortic Dissection revealed that 59% of patients suffered aortic dissection at diameters less than 5.5 cm, and that patients with certain connective tissue diseases such as Loeys-Dietz syndrome or familial thoracic aneurysm and dissection had a documented propensity for dissection at smaller diameters.39–41
Size indices such as the aortic cross-sectional area indexed to height have been implemented in guidelines for certain patient populations (eg, 10 cm2/m in Marfan syndrome) and provide better risk stratification than size cutoffs alone.2,42
The ratio of aortic cross-sectional area to the patient’s height has also been applied to patients with bicuspid aortic valve-associated aortopathy and to those with a dilated aorta and a tricuspid aortic valve.43,44 Notably, a ratio greater than 10 cm2/m has been associated with aortic dissection in these groups, and this cutoff provides better stratification for prediction of death than traditional size metrics.27,28
HOW SHOULD PATIENTS BE SCREENED? WHAT FOLLOW-UP IS NECESSARY?
Initial screening and follow-up
Follow-up of TAA depends on the initial aortic size or rate of growth, or both. For patients presenting for the first time with TAA, it is reasonable to obtain definitive aortic imaging with CT or magnetic resonance angiography (MRA), then to repeat imaging at 6 months to document stability. If the aortic dimensions remain stable, then annual follow-up with CT or MRA is reasonable.2
Our flow chart of initial screening and follow-up is shown in Figure 5.
Screening of family members
In our center, we routinely recommend screening of all first-degree relatives of patients with TAA. Aortic imaging with echocardiography plus CT or MRI should be considered to detect asymptomatic disease.2 In patients with a strong family history (ie, multiple relatives affected with aortic aneurysm, dissection, or sudden cardiac death), genetic screening and testing for known mutations are recommended for the patient as well as for the family members.
If a mutation is identified in a family, then first-degree relatives should undergo genetic screening for the mutation and aortic imaging.2 Imaging in second-degree relatives may also be considered if one or more first-degree relatives are found to have aortic dilation.2
We recommend similar screening of first-degree family members of patients with bicuspid aortic valve aortopathy. In patients with young children, we recommend obtaining an echocardiogram of the child to look for a bicuspid aortic valve or aortic dilation. If an abnormality is detected or suspected, dedicated imaging with MRA to assess aortic dimensions is warranted.
BACK TO OUR PATIENT WITH A BICUSPID AORTIC VALVE
Our patient with a bicuspid aortic valve had a 4.6-cm root, an ascending aortic aneurysm, and several affected family members.
We would obtain dedicated aortic imaging at this patient’s initial visit with either gated CT with contrast or MRA, and we would obtain a cardioaortic surgery consult. We would repeat these studies at a follow-up visit 6 months later to detect any aortic growth compared with initial studies, and follow up annually thereafter. Echocardiography can also be done at the initial visit to determine if valvular disease is present that may influence clinical decisions.
Surgery would likely be recommended once the root reached a maximum area-to-height ratio greater than 10 cm2/m, or if the valve became severely dysfunctional during follow-up.
BACK TO OUR PATIENT WITH MARFAN SYNDROME
The young woman with Marfan syndrome has a 4.6-cm aortic root aneurysm and 2+ aortic insufficiency. Her question pertains to the threshold at which an operation would be considered. This question is complicated and is influenced by several concurrent clinical features in her presentation.
Starting with size criteria, patients with Marfan syndrome should be considered for elective aortic root repair at a diameter greater than 5 cm. However, an aortic cross-sectional area-to-height ratio greater than 10 cm2/m may provide a more robust metric for clinical decision-making than aortic diameter alone. Additional factors such as degree of aortic insufficiency and deleterious left ventricular remodeling may urge one to consider aortic root repair at a diameter of 4.5 cm.
These factors, including rate of growth and the surgeon’s assessment about his or her ability to preserve the aortic valve during repair, should be considered collectively in this scenario.
- Elefteriades JA, Farkas EA. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J Am Coll Cardiol 2010; 55(9):841–857. doi:10.1016/j.jacc.2009.08.084
- Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary. Anesth Analg 2010; 111(2):279–315. doi:10.1213/ANE.0b013e3181dd869b
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280(22):1926–1929. pmid:9851478
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114(24):2611–2618. doi:10.1161/CIRCULATIONAHA.106.630400
- Clouse WD, Hallett JW Jr, Schaff HV, et al. Acute aortic dissection: population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc 2004; 79(2):176–180. pmid:14959911
- US Centers for Disease Control and Prevention (CDC). National Center for Injury Prevention and Control. WISQARS leading causes of death reports, 1999 – 2007. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10.html. Accessed May 21, 2018.
- Hansen MS, Nogareda GJ, Hutchison SJ. Frequency of and inappropriate treatment of misdiagnosis of acute aortic dissection. Am J Cardiol 2007; 99(6):852–856. doi:10.1016/j.amjcard.2006.10.055
- Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2014; 64(16):1725–1739. doi:10.1016/j.jacc.2014.08.025
- Kumar V, Abbas A, Aster J. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2015.
- Wolak A, Gransar H, Thomson LE, et al. Aortic size assessment by noncontrast cardiac computed tomography: normal limits by age, gender, and body surface area. JACC Cardiovasc Imaging 2008; 1(2):200–209. doi:10.1016/j.jcmg.2007.11.005
- Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg 2002; 74(5):S1877–S1880; discussion S1892–S1898. pmid:12440685
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75(1):7–17. pmid:18236724
- Cury M, Zeidan F, Lobato AC. Aortic disease in the young: genetic aneurysm syndromes, connective tissue disorders, and familial aortic aneurysms and dissections. Int J Vasc Med 2013(2013); 2013:267215. doi:10.1155/2013/267215
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39(12):1890–1900. doi:10.1016/S0735-1097(02)01886-7
- Fedak PW, Verma S, David TE, Leask RL, Weisel RD, Butany J. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation 2002; 106(8):900–904. pmid:12186790
- Della Corte A, Bancone C, Quarto C, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg 2007; 31(3):397–405. doi:10.1016/j.ejcts.2006.12.006
- Jackson V, Petrini J, Caidahl K, et al. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: a study of 300 patients undergoing open-heart surgery. Eur J Cardiothorac Surg 2011; 40(3):e118–e124. doi:10.1016/j.ejcts.2011.04.014
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112. doi:10.1001/jama.2011.1286
- Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 2014; 370(20):1920–1929. doi:10.1056/NEJMra1207059
- Barker AJ, Markl M, Bürk J, et al. Bicuspid aortic valve is associated with altered wall shear stress in the ascending aorta. Circ Cardiovasc Imaging 2012; 5(4):457–466. doi:10.1161/CIRCIMAGING.112.973370
- Hope MD, Hope TA, Meadows AK, et al. Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology 2010; 255(1):53–61. doi:10.1148/radiol.09091437
- Abdulkareem N, Soppa G, Jones S, Valencia O, Smelt J, Jahangiri M. Dilatation of the remaining aorta after aortic valve or aortic root replacement in patients with bicuspid aortic valve: a 5-year follow-up. Ann Thorac Surg 2013; 96(1):43–49. doi:10.1016/j.athoracsur.2013.03.086
- Regeer MV, Versteegh MI, Klautz RJ, et al. Effect of aortic valve replacement on aortic root dilatation rate in patients with bicuspid and tricuspid aortic valves. Ann Thorac Surg 2016; 102(6):1981–1987. doi:10.1016/j.athoracsur.2016.05.038
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312(5770):117–121. doi:10.1126/science.1124287
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358(26):2787–2795. doi:10.1056/NEJMoa0706585
- Chiu HH, Wu MH, Wang JK, et al. Losartan added to ß-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc 2013; 88(3):271–276. doi:10.1016/j.mayocp.2012.11.005
- Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013; 34(45):3491–3500. doi:10.1093/eurheartj/eht334
- Lacro RV, Dietz HC, Sleeper LA, et al; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014; 371(22):2061–2071. doi:10.1056/NEJMoa1404731
- Ziganshin BA, Mukherjee SK, Elefteriades JA, et al. Atenolol versus losartan in Marfan’s syndrome (letters). N Engl J Med 2015; 372(10):977–981. doi:10.1056/NEJMc1500128
- Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet 2015; 8(2):383–388. doi:10.1161/CIRCGENETICS.114.000950
- Elefteriades JA. Thoracic aortic aneurysm: reading the enemy’s playbook. Curr Probl Cardiol 2008; 33(5):203–277. doi:10.1016/j.cpcardiol.2008.01.004
- Idrees JJ, Roselli EE, Lowry AM, et al. Outcomes after elective proximal aortic replacement: a matched comparison of isolated versus multicomponent operations. Ann Thorac Surg 2016; 101(6):2185–2192. doi:10.1016/j.athoracsur.2015.12.026
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Thorac Cardiovasc Surg 2016; 151(4):959–966. doi:10.1016/j.jtcvs.2015.12.001
- Tzemos N, Therrien J, Yip J, et al. Outcomes in adults with bicuspid aortic valves. JAMA 2008; 300(11):1317–1325. doi:10.1001/jama.300.11.1317
- Davies RR, Goldstein LJ, Coady MA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg 2002; 73(1):17–28. pmid:11834007
- Nishimura RA, Otto CM, Bono RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Wojnarski CM, Svensson LG, Roselli EE, et al. Aortic dissection in patients with bicuspid aortic valve–associated aneurysms. Ann Thorac Surg 2015; 100(5):1666–1674. doi:10.1016/j.athoracsur.2015.04.126
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2016; 133(7):680–686. doi:10.1161/CIR.0000000000000331
- Pape LA, Tsai TT, Isselbacher EM, et al; International Registry of Acute Aortic Dissection (IRAD) Investigators. Aortic diameter > or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007; 116(10):1120–1127. doi:10.1161/CIRCULATIONAHA.107.702720
- Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355(8):788–798. doi:10.1056/NEJMoa055695
- Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39(12):1488–1493. doi:10.1038/ng.2007.6
- Svensson LG, Khitin L. Aortic cross-sectional area/height ratio timing of aortic surgery in asymptomatic patients with Marfan syndrome. J Thorac Cardiovasc Surg 2002; 123(2):360–361. pmid:11828302
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection in patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003; 126(3):892–893. pmid:14502185
- Masri A, Kalahasti V, Svensson LG, et al. Aortic cross-sectional area/height ratio and outcomes in patients with a trileaflet aortic valve and a dilated aorta. Circulation 2016; 134(22):1724–1737. doi:10.1161/CIRCULATIONAHA.116.022995
- Elefteriades JA, Farkas EA. Thoracic aortic aneurysm clinically pertinent controversies and uncertainties. J Am Coll Cardiol 2010; 55(9):841–857. doi:10.1016/j.jacc.2009.08.084
- Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary. Anesth Analg 2010; 111(2):279–315. doi:10.1213/ANE.0b013e3181dd869b
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280(22):1926–1929. pmid:9851478
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114(24):2611–2618. doi:10.1161/CIRCULATIONAHA.106.630400
- Clouse WD, Hallett JW Jr, Schaff HV, et al. Acute aortic dissection: population-based incidence compared with degenerative aortic aneurysm rupture. Mayo Clin Proc 2004; 79(2):176–180. pmid:14959911
- US Centers for Disease Control and Prevention (CDC). National Center for Injury Prevention and Control. WISQARS leading causes of death reports, 1999 – 2007. https://webappa.cdc.gov/sasweb/ncipc/leadcaus10.html. Accessed May 21, 2018.
- Hansen MS, Nogareda GJ, Hutchison SJ. Frequency of and inappropriate treatment of misdiagnosis of acute aortic dissection. Am J Cardiol 2007; 99(6):852–856. doi:10.1016/j.amjcard.2006.10.055
- Goldfinger JZ, Halperin JL, Marin ML, Stewart AS, Eagle KA, Fuster V. Thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2014; 64(16):1725–1739. doi:10.1016/j.jacc.2014.08.025
- Kumar V, Abbas A, Aster J. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2015.
- Wolak A, Gransar H, Thomson LE, et al. Aortic size assessment by noncontrast cardiac computed tomography: normal limits by age, gender, and body surface area. JACC Cardiovasc Imaging 2008; 1(2):200–209. doi:10.1016/j.jcmg.2007.11.005
- Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg 2002; 74(5):S1877–S1880; discussion S1892–S1898. pmid:12440685
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75(1):7–17. pmid:18236724
- Cury M, Zeidan F, Lobato AC. Aortic disease in the young: genetic aneurysm syndromes, connective tissue disorders, and familial aortic aneurysms and dissections. Int J Vasc Med 2013(2013); 2013:267215. doi:10.1155/2013/267215
- Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002; 39(12):1890–1900. doi:10.1016/S0735-1097(02)01886-7
- Fedak PW, Verma S, David TE, Leask RL, Weisel RD, Butany J. Clinical and pathophysiological implications of a bicuspid aortic valve. Circulation 2002; 106(8):900–904. pmid:12186790
- Della Corte A, Bancone C, Quarto C, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg 2007; 31(3):397–405. doi:10.1016/j.ejcts.2006.12.006
- Jackson V, Petrini J, Caidahl K, et al. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: a study of 300 patients undergoing open-heart surgery. Eur J Cardiothorac Surg 2011; 40(3):e118–e124. doi:10.1016/j.ejcts.2011.04.014
- Michelena HI, Khanna AD, Mahoney D, et al. Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 2011; 306(10):1104–1112. doi:10.1001/jama.2011.1286
- Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med 2014; 370(20):1920–1929. doi:10.1056/NEJMra1207059
- Barker AJ, Markl M, Bürk J, et al. Bicuspid aortic valve is associated with altered wall shear stress in the ascending aorta. Circ Cardiovasc Imaging 2012; 5(4):457–466. doi:10.1161/CIRCIMAGING.112.973370
- Hope MD, Hope TA, Meadows AK, et al. Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology 2010; 255(1):53–61. doi:10.1148/radiol.09091437
- Abdulkareem N, Soppa G, Jones S, Valencia O, Smelt J, Jahangiri M. Dilatation of the remaining aorta after aortic valve or aortic root replacement in patients with bicuspid aortic valve: a 5-year follow-up. Ann Thorac Surg 2013; 96(1):43–49. doi:10.1016/j.athoracsur.2013.03.086
- Regeer MV, Versteegh MI, Klautz RJ, et al. Effect of aortic valve replacement on aortic root dilatation rate in patients with bicuspid and tricuspid aortic valves. Ann Thorac Surg 2016; 102(6):1981–1987. doi:10.1016/j.athoracsur.2016.05.038
- Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312(5770):117–121. doi:10.1126/science.1124287
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358(26):2787–2795. doi:10.1056/NEJMoa0706585
- Chiu HH, Wu MH, Wang JK, et al. Losartan added to ß-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc 2013; 88(3):271–276. doi:10.1016/j.mayocp.2012.11.005
- Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013; 34(45):3491–3500. doi:10.1093/eurheartj/eht334
- Lacro RV, Dietz HC, Sleeper LA, et al; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014; 371(22):2061–2071. doi:10.1056/NEJMoa1404731
- Ziganshin BA, Mukherjee SK, Elefteriades JA, et al. Atenolol versus losartan in Marfan’s syndrome (letters). N Engl J Med 2015; 372(10):977–981. doi:10.1056/NEJMc1500128
- Franken R, den Hartog AW, Radonic T, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet 2015; 8(2):383–388. doi:10.1161/CIRCGENETICS.114.000950
- Elefteriades JA. Thoracic aortic aneurysm: reading the enemy’s playbook. Curr Probl Cardiol 2008; 33(5):203–277. doi:10.1016/j.cpcardiol.2008.01.004
- Idrees JJ, Roselli EE, Lowry AM, et al. Outcomes after elective proximal aortic replacement: a matched comparison of isolated versus multicomponent operations. Ann Thorac Surg 2016; 101(6):2185–2192. doi:10.1016/j.athoracsur.2015.12.026
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Thorac Cardiovasc Surg 2016; 151(4):959–966. doi:10.1016/j.jtcvs.2015.12.001
- Tzemos N, Therrien J, Yip J, et al. Outcomes in adults with bicuspid aortic valves. JAMA 2008; 300(11):1317–1325. doi:10.1001/jama.300.11.1317
- Davies RR, Goldstein LJ, Coady MA, et al. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg 2002; 73(1):17–28. pmid:11834007
- Nishimura RA, Otto CM, Bono RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: executive summary: a report of the American College of Cardiology/American heart Association Task Force on Practice Guidelines. Circulation 2014; 129(23):2440–2492. doi:10.1161/CIR.0000000000000029
- Wojnarski CM, Svensson LG, Roselli EE, et al. Aortic dissection in patients with bicuspid aortic valve–associated aneurysms. Ann Thorac Surg 2015; 100(5):1666–1674. doi:10.1016/j.athoracsur.2015.04.126
- Hiratzka LF, Creager MA, Isselbacher EM, et al. Surgery for aortic dilatation in patients with bicuspid aortic valves: a statement of clarification from the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2016; 133(7):680–686. doi:10.1161/CIR.0000000000000331
- Pape LA, Tsai TT, Isselbacher EM, et al; International Registry of Acute Aortic Dissection (IRAD) Investigators. Aortic diameter > or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007; 116(10):1120–1127. doi:10.1161/CIRCULATIONAHA.107.702720
- Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355(8):788–798. doi:10.1056/NEJMoa055695
- Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007; 39(12):1488–1493. doi:10.1038/ng.2007.6
- Svensson LG, Khitin L. Aortic cross-sectional area/height ratio timing of aortic surgery in asymptomatic patients with Marfan syndrome. J Thorac Cardiovasc Surg 2002; 123(2):360–361. pmid:11828302
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection in patients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003; 126(3):892–893. pmid:14502185
- Masri A, Kalahasti V, Svensson LG, et al. Aortic cross-sectional area/height ratio and outcomes in patients with a trileaflet aortic valve and a dilated aorta. Circulation 2016; 134(22):1724–1737. doi:10.1161/CIRCULATIONAHA.116.022995
KEY POINTS
- Screening and referral depend on clinical context. A size-based model to determine screening, referral, follow-up, and management serves most cases but should be modified in the context of connective tissue disease or family history of aneurysm and dissection.
- Medical management involves strict blood pressure and heart rate control with beta-blockers and angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers. Activity modifications should be tailored to the individual, although extreme isometric exercises and heavy lifting should be discouraged.
- Patients with TAA should be followed up annually, unless the patient is presenting for initial evaluation or significant changes are seen with dedicated imaging.

Hypertrophic cardiomyopathy: A complex disease
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
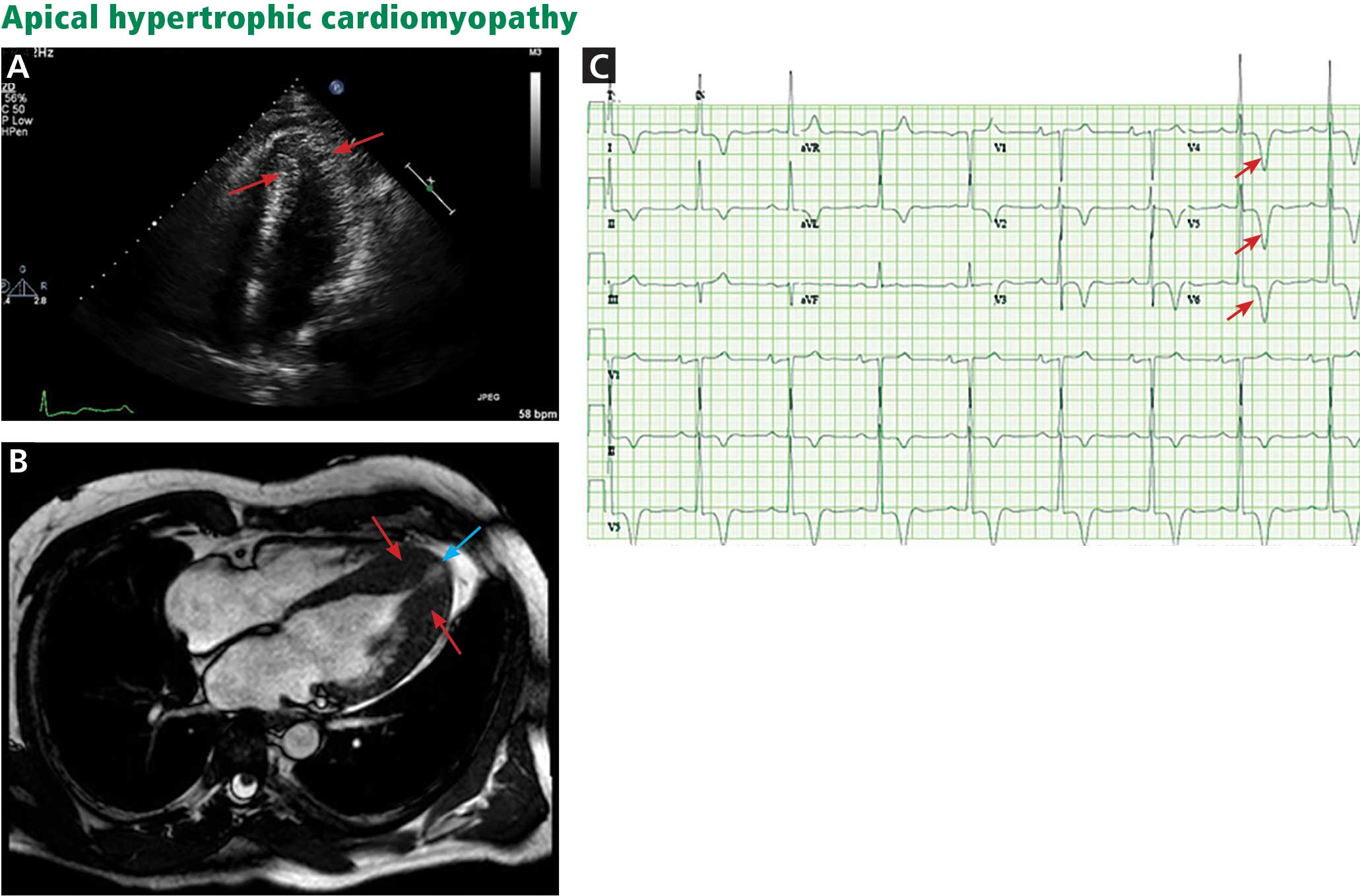
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
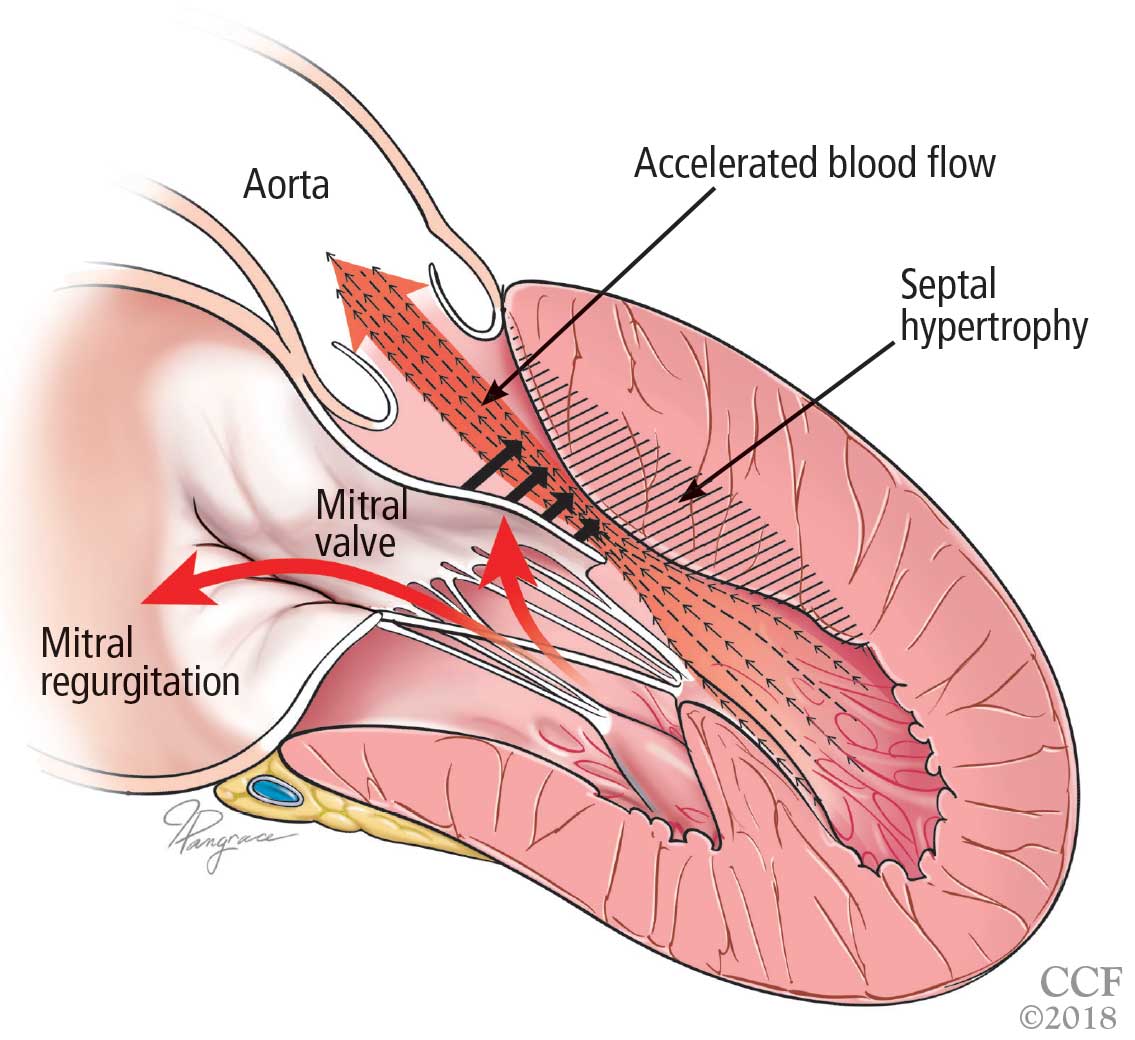
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
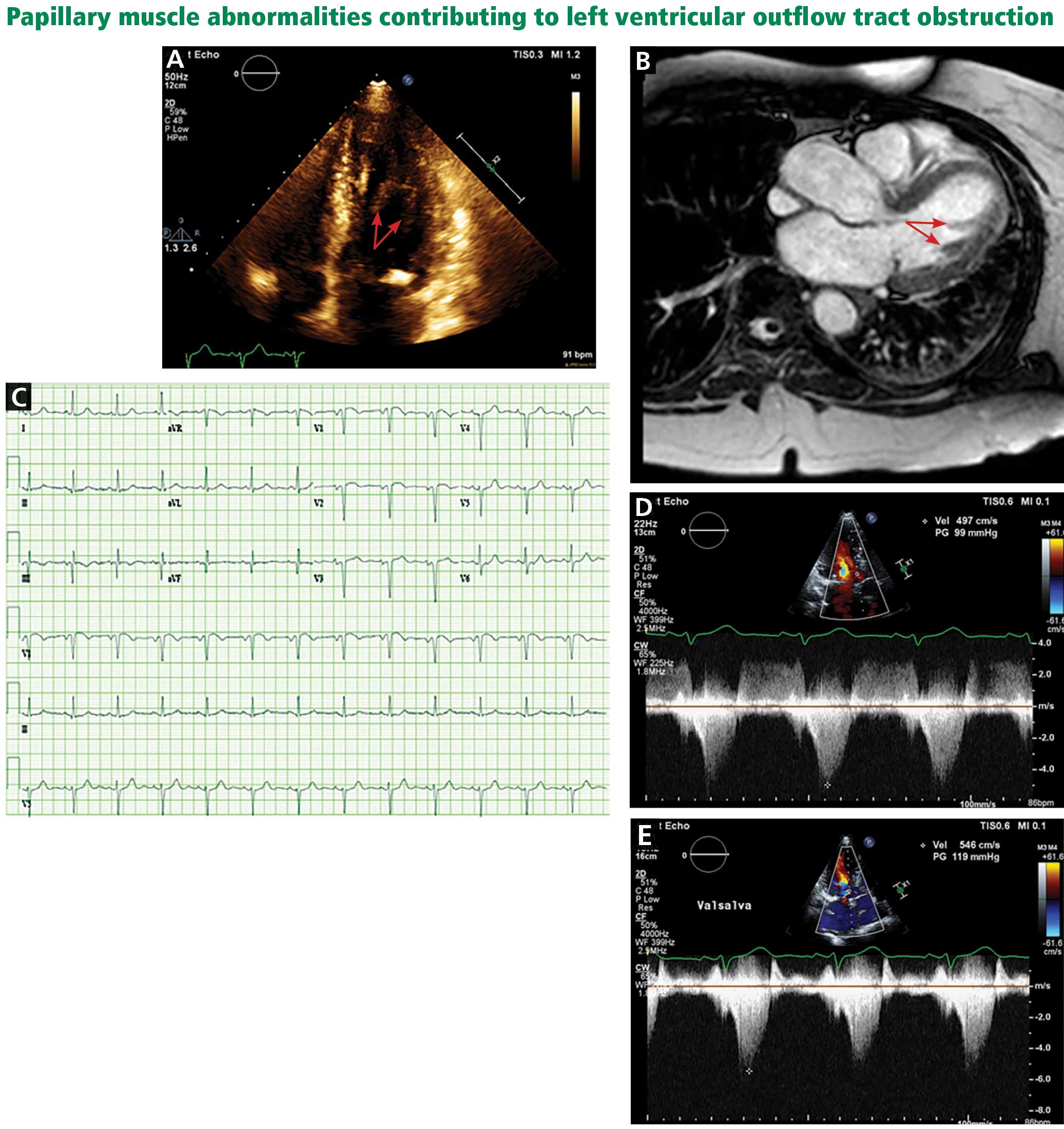
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
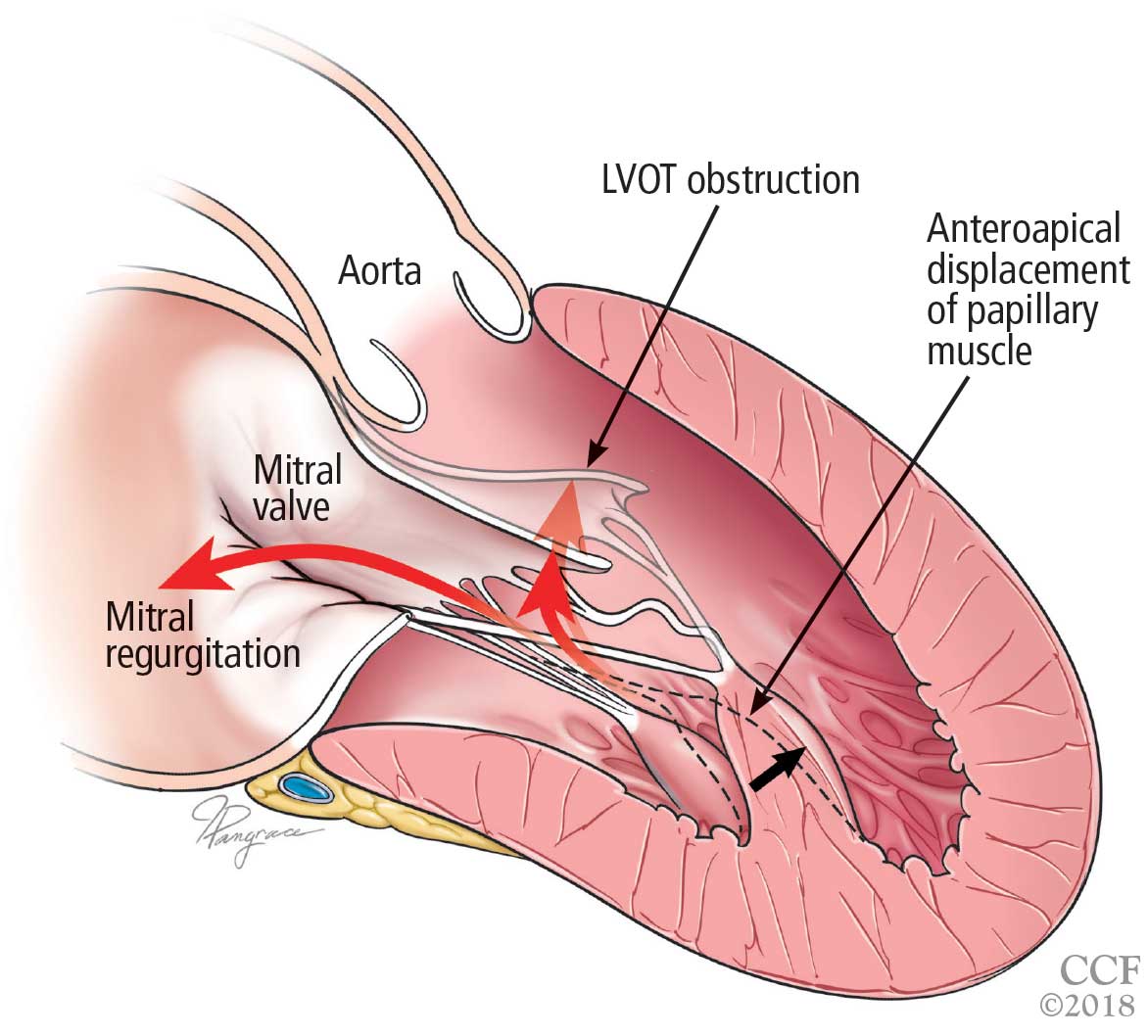
Physical findings are nonspecific

It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM

A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models

The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.

Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
Hypertrophic cardiomyopathy (HCM) is a complex disease. Most people who carry the mutations that cause it are never affected at any point in their life, but some are affected at a young age. And in rare but tragic cases, some die suddenly while competing in sports. With such a wide range of phenotypic expressions, a single therapy does not fit all.
HCM is more common than once thought. Since the discovery of its genetic predisposition in 1960, it has come to be recognized as the most common heritable cardiovascular disease.1 Although earlier epidemiologic studies had estimated a prevalence of 1 in 500 (0.2%) of the general population, genetic testing and cardiac magnetic resonance imaging (MRI) now show that up to 1 in 200 (0.5%) of all people may be affected.1,2 Its prevalence is significant in all ethnic groups.
This review outlines our expanding knowledge of the pathophysiology, diagnosis, and clinical management of HCM.
A PLETHORA OF MUTATIONS IN CARDIAC SARCOMERIC GENES
The genetic differences within HCM result in varying degrees and locations of left ventricular hypertrophy. Any segment of the ventricle can be involved, although HCM is classically asymmetric and mainly involves the septum (Figure 1). A variant form of HCM involves the apex of the heart (Figure 2).
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
Only in the last decade has the significance of left ventricular outflow tract obstruction in HCM been truly appreciated. The degree of obstruction in HCM is dynamic, as opposed to the fixed obstruction in patients with aortic stenosis or congenital subvalvular membranes. Therefore, in HCM, exercise or drugs (eg, dobutamine) that increase cardiac contractility increase the obstruction, as do maneuvers or drugs (the Valsalva maneuver, nitrates) that reduce filling of the left ventricle.
A less common source of dynamic obstruction is the papillary muscles (Figure 4). Hypertrophy of the papillary muscles can result in obstruction by these muscles themselves, which is visible on echocardiography. Anatomic variations include anteroapical displacement or bifid papillary muscles, and these variants can be associated with dynamic left ventricular outflow tract obstruction, even with no evidence of septal thickening (Figure 5).7,8 Recognizing this patient subset has important implications for management, as discussed below.
DIAGNOSTIC EVALUATION
The clinical presentation varies
Even if patients harbor the same genetic variant, the clinical presentation can differ widely. Although the most feared presentation is sudden cardiac death, particularly in young athletes, most patients have no symptoms and can anticipate a normal life expectancy. The annual incidence of sudden cardiac death in all HCM patients is estimated at less than 1%.10 Sudden cardiac death in HCM patients is most often due to ventricular tachyarrhythmias and most often occurs in asymptomatic patients under age 35.
Physical findings are nonspecific
It can be difficult to distinguish the murmur of left ventricular outflow tract obstruction in HCM from a murmur related to aortic stenosis by auscultation alone. The simplest clinical method for telling them apart involves the Valsalva maneuver: bearing down creates a positive intrathoracic pressure and limits venous return, thus decreasing intracardiac filling pressure. This in turn results in less separation between the mitral valve and the ventricular septum in HCM, which increases obstruction and therefore makes the murmur louder. In contrast, in patients with fixed obstruction due to aortic stenosis, the murmur will decrease in intensity owing to the reduced flow associated with reduced preload.
Laboratory testing for phenocopies of HCM
A metabolic panel will show derangements in liver function and glucose levels in patients with glycogen storage disorders such as Pompe disease.
Serum creatinine. Renal dysfunction will be seen in patients with Fabry disease or amyloidosis.
Creatine kinase may be elevated in patients with Danon disease.
Electrocardiographic findings are common
More than 90% of HCM patients have electrocardiographic abnormalities. Although the findings can vary widely, common manifestations include:
- Left ventricular hypertrophy
- A pseudoinfarct pattern with Q waves in the anterolateral leads
- Repolarization changes such as T-wave inversions and horizontal or down-sloping ST segments.
Apical HCM, seen mainly in Asian populations, often presents with giant T-wave inversion (> 10 mm) in the anterolateral leads, most prominent in V4, V5, and V6.
Notably, the degree of electrocardiographic abnormalities does not correlate with the severity or pattern of hypertrophy.9 Electrocardiography lacks specificity for definitive diagnosis, and further diagnostic testing should therefore be pursued.
Echocardiography: Initial imaging test
Transthoracic echocardiography is the initial imaging test in patients with suspected HCM, allowing for cost-effective quantitative and qualitative assessment of left ventricular morphology and function. Left ventricular hypertrophy is considered pathologic if wall thickness is 15 mm or greater without a known cause. Transthoracic echocardiography also allows for evaluation of left atrial volume and mitral valve anatomy and function.
Speckle tracking imaging is an advanced echocardiographic technique that measures strain. Its major advantage is in identifying early abnormalities in genotype-positive, phenotype-negative HCM patients, ie, people who harbor mutations but who have no clinical symptoms or signs of HCM, potentially allowing for modification of the natural history of HCM.12 Strain imaging can also differentiate between physiologic hypertrophy (“athlete’s heart”) and hypertension and HCM.13,14
The utility of echocardiography in HCM is heavily influenced by the sonographer’s experience in obtaining adequate acoustic windows. This may be more difficult in obese patients, patients with advanced obstructive lung disease or pleural effusions, and women with breast implants.
Magnetic resonance imaging
MRI has an emerging role in both diagnosing and predicting risk in HCM, and is routinely done as an adjunct to transthoracic echocardiography on initial diagnosis in our tertiary referral center. It is particularly useful in patients suspected of having apical hypertrophy (Figure 2), in whom the diagnosis may be missed in up to 10% on transthoracic echocardiography alone.15 MRI can also enhance the assessment of left ventricular hypertrophy and has been shown to improve the diagnostic classification of HCM.16 It is the best way to assess myocardial tissue abnormalities, and late gadolinium enhancement to detect interstitial fibrosis can be used for further prognostication. While historically the primary role of MRI in HCM has been in phenotype classification, there is currently much interest in its role in risk stratification of HCM patients for ICD implantation.
MRI with late gadolinium enhancement provides insight into the location, pattern, and extent of myocardial fibrosis; the extent of fibrosis has been shown to be a strong independent predictor of poor outcomes, including sudden cardiac death.17–20 However, late gadolinium enhancement can be technically challenging, as variations in the timing of postcontrast imaging, sequences for measuring late gadolinium enhancement, or detection thresholds can result in widely variable image quality. Cardiac MRI should therefore be performed at an experienced center with standardized imaging protocols in place.
Current guidelines recommend considering cardiac MRI if a patient’s risk of sudden cardiac death remains inconclusive after conventional risk stratification, as discussed below.9,21
Stress testing for risk stratification
Exercise stress electrocardiography. Treadmill exercise stress testing with electrocardiography and hemodynamic monitoring was one of the first tools used for risk stratification in HCM.
Although systolic blood pressure normally increases by at least 20 mm Hg with exercise, one-quarter of HCM patients have either a blunted response (failure of systolic blood pressure to increase by at least 20 mm Hg) or a hypotensive response (a drop in systolic blood pressure of 20 mm Hg or more, either continuously or after an initial increase). Studies have shown that HCM patients who have abnormal blood pressure responses during exercise have a higher risk of sudden cardiac death.22–24
Exercise stress echocardiography can be useful to evaluate for provoked increases in the left ventricular outflow tract gradient, which may contribute to a patient’s symptoms even if the resting left ventricular outflow tract gradient is normal. Exercise testing is preferred over pharmacologic stimulation because it can provide functional assessment of whether a patient’s clinical symptoms are truly related to hemodynamic changes due to the hypertrophied ventricle, or whether alternative mechanisms should be explored.
Cardiopulmonary stress testing can readily add prognostic value with additional measurements of functional capacity. HCM patients who cannot achieve their predicted maximal exercise value such as peak rate of oxygen consumption, ventilation efficiency, or anaerobic threshold have higher rates of morbidity and mortality.25,26 Stress testing can also be useful for risk stratification in asymptomatic patients, with one study showing that those who achieve more than 100% of their age- and sex-predicted metabolic equivalents have a low event rate.27
Ambulatory electrocardiographic monitoring in all patients at diagnosis
Ambulatory electrocardiographic monitoring for 24 to 48 hours is recommended for all HCM patients at the time of diagnosis, even if they have no symptoms. Any evidence of nonsustained ventricular tachycardia suggests a substantially higher risk of sudden cardiac death.28,29
In patients with no symptoms or history of arrhythmia, current guidelines suggest ambulatory electrocardiographic monitoring every 1 to 2 years.9,21
Two risk-stratification models
The North American model was the first risk-stratification tool and considers 5 risk factors.9 However, if this algorithm were strictly followed, up to 60% of HCM patients would be candidates for cardioverter-defibrillator implantation.
The European model. This concern led to the development of the HCM Risk-SCD (sudden cardiac death), a risk-stratification tool introduced in the 2014 European Society of Cardiology HCM guidelines.30 This web-based calculator estimates a patient’s 5-year risk of sudden cardiac death using a complex calculation based on 7 clinical risk factors. If a patient’s calculated 5-year risk of sudden cardiac death is 6% or higher, cardioverter-defibrillator implantation is recommended for primary prevention.
The HCM Risk-SCD calculator was validated and compared with classic risk factors alone in a retrospective cohort study in 48 HCM patients.30 Compared with the North American model, the European model results in a lower rate of cardioverter-defibrillator implantation (20% to 26%).31,32
Despite the better specificity of the European model, a large retrospective cohort analysis showed that a significant number of patients stratified as being at low risk for sudden cardiac death were ultimately found to be at high risk in clinical practice.31 Further research is needed to find the optimal risk-stratification approach in HCM patients at low to intermediate risk.
GENETIC TESTING, COUNSELING, AND FAMILY SCREENING
Genetic testing is becoming more widely available and has rapidly expanded in clinical practice. Genetic counseling must be performed alongside genetic testing and requires professionals trained to handle the clinical and social implications of genetic testing. With this in mind, genetic testing can provide a definitive means of identifying family members at risk of HCM.
Given the autosomal dominant nature of HCM, screening for HCM is recommended in all first-degree relatives of an affected patient. Genetic testing may be a means to achieve this if a pathogenic mutation has been identified in the affected patient. However, serial electrocardiographic and transthoracic echocardiographic monitoring is an acceptable alternative in those without a clear genetic mutation association or in those who do not want to undergo genetic testing. If these first-degree relatives who do not undergo genetic testing are adult athletes or adolescents, they should undergo surveillance monitoring, with echocardiography and electrocardiography, whereas adults not participating in athletics should be monitored every 5 years.9,21
As genetic counseling and testing become more widely available, more patients are being found who harbor a mutation but have no phenotypic manifestations of HCM on initial presentation. Clinical expression varies, so continued monitoring of these patients is important. Expert guidelines again recommend serial electrocardiography, transthoracic echocardiography, and clinical assessment every 5 years for adults.9
Recent data suggest that up to 40% of HCM cases are nonfamilial, ie, their inheritance is sporadic with no known family history and no sarcomeric gene mutation evident on screening.33,34 The clinical course in this subgroup seems to be more benign, with later clinical presentations (age > 40) and lower risk of major adverse cardiovascular events.
MANAGEMENT
Conservative management
Asymptomatic HCM can usually be managed with lifestyle modifications.
Avoiding high-risk physical activities is the most important modification. All HCM patients should be counseled on the risk of sudden cardiac death and advised against participating in competitive sports or intense physical activity.35 Aerobic exercise is preferable to isometric exercises such as weightlifting, which may prompt the Valsalva maneuver with worsening of left ventricular outflow tract obstruction leading to syncope. A recent study showed that moderate-intensity aerobic exercise can safely improve exercise capacity, which may ultimately improve functional status and quality of life.36
Avoiding dehydration and excessive alcohol intake are also important in maintaining adequate preload to prevent an increasing left ventricular outflow tract gradient, given the dynamic nature of the left ventricular outflow tract obstruction in HCM.
Medical management: Beta-blockers, then calcium channel blockers
Beta-blockers are the first-line therapy for symptomatic HCM related to left ventricular outflow tract obstruction. Their negative inotropic effect reduces the contractile force of the ventricle, effectively reducing the pressure gradient across the outflow tract. Reduced contractility also means that the overall myocardial workload is less, which ultimately translates to a reduced oxygen demand. With their negative chronotropic effect, beta-blockers lower the heart rate and thereby lengthen the diastolic filling phase, allowing for optimization of preload conditions to help prevent increasing the left ventricular outflow tract gradient.37,38
Beta-blockers can be titrated according to the patient’s symptoms and tolerance. Fatigue and loss of libido are among the most common side effects.
Nondihydropyridine calcium channel blockers can be a second-line therapy in patients who cannot tolerate beta-blockers. Several studies have shown improvement in surrogate outcomes such as estimated left ventricular mass and QRS amplitude on electrocardiography, but currently no available data show that these drugs improve symptoms.28,39,40 They should be avoided in those with severe left ventricular outflow tract obstruction (gradient ≥ 100 mm Hg), as they can lead to critical outflow tract obstruction owing to their peripheral vasodilatory effect.
Dihydropyridine calcium channel blockers should be avoided altogether, as they produce even more peripheral vasodilation and afterload reduction than nondihydropyridine calcium channel blockers.
Disopyramide, a class IA antiarrhythmic, has been shown to effectively reduce outflow gradients and relieve symptoms. However, in view of its adverse effects, it is a third-line therapy, given to those for whom beta-blockers and calcium channel blockers have failed. Its most worrisome adverse effect is QT prolongation, and the QT interval should therefore be closely monitored at the start of treatment. Anticholinergic effects are common and include dry eyes and mouth, urinary retention, and drowsiness.
Disopyramide is usually used in combination with beta-blockers for symptom control as a bridge to a planned invasive intervention.41
Use with caution
Any medication that causes afterload reduction, peripheral vasodilation, intravascular volume depletion, or positive inotropy can worsen the dynamic left ventricular outflow tract obstruction in a patient with HCM and should be avoided.
Angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and nitrates must be used with extreme caution in these patients.
Diuretics. Even restrained use of diuretics can cause significant hemodynamic compromise in patients with obstructive physiology. Therefore, diuretics should be used sparingly in these patients.
Digoxin should not be used for managing atrial fibrillation in these patients, as its positive inotropic effect increases contractility and increases the left ventricular outflow tract gradient.
Norepinephrine and inotropic agents such as dobutamine and dopamine should be avoided for the same reason as digoxin. In patients with circulatory shock requiring vasopressor support, pure alpha-agonists such as phenylephrine are preferred, as they increase peripheral resistance without an inotropic effect.
Anticoagulation for atrial tachyarrhythmias
The risk of systemic thromboembolic events is significantly increased in HCM patients with atrial fibrillation or flutter, regardless of their estimated risk using conventional risk-stratification tools such as the CHADS2 score.42–44 In accordance with current American Heart Association and American College of Cardiology guidelines, we recommend anticoagulation therapy for all HCM patients with a history of atrial fibrillation or flutter. Warfarin is the preferred anticoagulant; direct oral anticoagulants can be considered, but there are currently no data on their use in HCM.9
Standard heart failure treatments
End-stage systolic heart failure is a consequence of HCM but affects only 3% to 4% of patients.45 While most randomized controlled trials of heart failure treatment have excluded HCM patients, current guidelines recommend the same evidence-based medical therapies used in other patients who have heart failure with reduced ejection fraction. This includes ACE inhibitors, ARBs, beta-blockers, and aldosterone antagonists if indicated.9,21
Heart transplant should be considered in patients with class III or IV New York Heart Association functional status despite optimization of their HCM treatment regimen. Heart transplant outcomes for HCM patients are comparable to outcomes for patients who receive a transplant for non-HCM cardiovascular disease.45,46
Septal reduction therapy
If medical therapy fails or is not tolerated in patients with severe symptoms, surgery can be considered for obstructive HCM.
Ventricular septal myectomy has been the long-standing gold standard of invasive therapy. Multiple studies have demonstrated long-term survival after myectomy to be equivalent to that in the general population and better than that of HCM patients who do not undergo this surgery.47–50 Factors that may be associated with better surgical outcomes include age younger than 50, left atrial size less than 46 mm, and resolution of atrial fibrillation during follow-up.51
Septal reduction therapy may also be considered in patients at high risk of sudden cardiac death based on a history of recurrent ventricular tachycardia or risk-stratification models as described above. Retrospective analyses have shown that surgical myectomy can markedly reduce the incidence of appropriate implantable cardioverter-defibrillator discharges and the risk of sudden cardiac death.52
Alcohol septal ablation is an alternative. This percutaneous procedure, first described in the mid-1990s, consists of injecting a small amount of alcohol into the artery supplying the septum to induce myocardial necrosis, ultimately leading to scarring and widening of the left ventricular outflow tract.53
Up to 50% of patients develop right bundle branch block after alcohol septal ablation, and the risk of complete heart block is highest in those with preexisting left bundle branch block. Nevertheless, studies have shown significant symptomatic improvement after alcohol septal ablation, with long-term survival comparable to that in the general population.53–56
Several meta-analyses compared alcohol septal ablation and septal myectomy and found that the rates of functional improvement and long-term mortality were similar.57–59 However, the less-invasive approach with alcohol septal ablation comes at the cost of a higher incidence of conduction abnormalities and higher left ventricular outflow tract gradients afterward. One meta-analysis found that alcohol septal ablation patients may have 5 times the risk of needing additional septal reduction therapy compared with their myectomy counterparts.
Current US guidelines recommend septal myectomy, performed at an experienced center, as the first-line interventional treatment, leaving alcohol septal ablation to be considered in those who have contraindications to myectomy.9 The treatment strategy should ultimately be individualized based on a patient’s comorbidities and personal preferences following informed consent.
A nationwide database study recently suggested that postmyectomy mortality rates may be as high as 5.9%,60 although earlier studies at high-volume centers showed much lower mortality rates (< 1%).50–52,61 This discrepancy highlights the critical role of expert centers in optimizing surgical management of these patients. Regardless of the approach, interventional therapies for HCM should be performed by a multidisciplinary team at a medical center able to handle the complexity of these cases.
Additional surgical procedures
A handful of other procedures may benefit specific patient subgroups.
Apical myectomy has been shown to improve functional status in patients with isolated apical hypertrophy by reducing left ventricular end-diastolic pressure and thereby allowing for improved diastolic filling.63
Mitral valve surgery may need to be considered at the time of myectomy in patients with degenerative valve disease. As in the general population, mitral valve repair is preferred to replacement if possible.
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
- Maron BJ. Hypertrophic cardiomyopathy: an important global disease. Am J Med 2004; 116(1):63–65. pmid:14706671
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60(8):705–715. doi:10.1016/j.jacc.2012.02.068
- Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol 2000; 35(1):36–44. pmid:10636256
- Sherrid MV, Chu CK, Delia E, Mogtader A, Dwyer EM Jr. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol 1993; 22(3):816–825. pmid:8354817
- Ro R, Halpern D, Sahn DJ, et al. Vector flow mapping in obstructive hypertrophic cardiomyopathy to assess the relationship of early systolic left ventricular flow and the mitral valve. J Am Coll Cardiol 2014; 64(19):1984–1995. doi:10.1016/j.jacc.2014.04.090
- Kwon DH, Setser RM, Thamilarasan M, et al. Abnormal papillary muscle morphology is independently associated with increased left ventricular outflow tract obstruction in hypertrophic cardiomyopathy. Heart 2008; 94(10):1295–1301. doi:10.1136/hrt.2007.118018
- Patel P, Dhillon A, Popovic ZB, et al. Left ventricular outflow tract obstruction in hypertrophic cardiomyopathy patients without severe septal hypertrophy: implications of mitral valve and papillary muscle abnormalities assessed using cardiac magnetic resonance and echocardiography. Circ Cardiovasc Imaging 2015; 8(7):e003132. doi:10.1161/CIRCIMAGING.115.003132
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2011; 142(6):e153-e203. doi:10.1016/j.jtcvs.2011.10.020
- Elliott PM, Gimeno JR, Thaman R, et al. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006; 92(6):785–791. doi:10.1136/hrt.2005.068577
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006; 114(21):2232–2239. doi:10.1161/CIRCULATIONAHA.106.644682
- Ho CY, Carlsen C, Thune JJ, et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2009; 2(4):314–321. doi:10.1161/CIRCGENETICS.109.862128
- Wasfy MM, Weiner RB. Differentiating the athlete’s heart from hypertrophic cardiomyopathy. Curr Opin Cardiol 2015; 30(5):500–505. doi:10.1097/HCO.0000000000000203
- Palka P, Lange A, Fleming AD, et al. Differences in myocardial velocity gradient measured throughout the cardiac cycle in patients with hypertrophic cardiomyopathy, athletes and patients with left ventricular hypertrophy due to hypertension. J Am Coll Cardiol 1997; 30(3):760–768. pmid:9283537
- Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2002; 39(4):638–645. pmid:11849863
- Rickers C, Wilke NM, Jerosch-Herold M, et al. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005; 112(6):855–861. doi:10.1161/CIRCULATIONAHA.104.507723
- Kwon DH, Setser RM, Popovic ZB, et al. Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging 2008; 24(6):617–625. doi:10.1007/s10554-008-9292-6
- Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(1):51–58. doi:10.1161/CIRCHEARTFAILURE.109.854026
- O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):867–874. doi:10.1016/j.jacc.2010.05.010
- Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 56(11):875–887. doi:10.1016/j.jacc.2010.05.007
- Authors/Task Force members, Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014; 35(39):2733–2779. doi:10.1093/eurheartj/ehu284
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol 1999; 33(7):2044–2051. pmid:10362212
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation 1997; 96(9):2987–2991. pmid:9386166
- Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol 2000; 36(7):2212–2218. pmid:11127463
- Masri A, Pierson LM, Smedira NG, et al. Predictors of long-term outcomes in patients with hypertrophic cardiomyopathy undergoing cardiopulmonary stress testing and echocardiography. Am Heart J 2015; 169(5):684–692.e1. doi:10.1016/j.ahj.2015.02.006
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary exercise testing and prognosis in hypertrophic cardiomyopathy. Circ Heart Fail 2015; 8(6):1022–1031. doi:10.1161/CIRCHEARTFAILURE.114.002248
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging 2014; 7(1):26–36. doi:10.1016/j.jcmg.2013.08.010
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med 1997; 336(11):775–785. doi:10.1056/NEJM199703133361107
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic implications of nonsustained ventricular tachycardia in high-risk patients with hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 2017; 10(3)e004604. doi:10.1161/CIRCEP.116.004604
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J 2014; 35(30):2010–2020. doi:10.1093/eurheartj/eht439
- Maron BJ, Casey SA, Chan RH, Garberich RF, Rowin EJ, Maron MS. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol 2015; 116(5):757–764. doi:10.1016/j.amjcard.2015.05.047
- Jahnlová D, Tomašov P, Zemánek D, Veselka J. Transatlantic differences in assessment of risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Int J Cardiol 2015; 186:3–4. doi:10.1016/j.ijcard.2015.03.207
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017; 10(2)e001620. doi:10.1161/CIRCGENETICS.116.001620
- Ko C, Arscott P, Concannon M, et al. Genetic testing impacts the utility of prospective familial screening in hypertrophic cardiomyopathy through identification of a nonfamilial subgroup. Genet Med 2017; 20(1):69–75. doi:10.1038/gim.2017.79
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004; 109(22):2807–2816. doi:10.1161/01.CIR.0000128363.85581.E1
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. JAMA 2017; 317(13):1349–1357. doi:10.1001/jama.2017.2503
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: an echographic study. Am Heart J 1985; 109(6):1311–1316. pmid:4039882
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. Eur Heart J 2012; 33(14):1724–1733. doi:10.1093/eurheartj/ehs150
- Choudhury L, Elliott P, Rimoldi O, et al. Transmural myocardial blood flow distribution in hypertrophic cardiomyopathy and effect of treatment. Basic Res Cardiol 1999; 94(1):49–59. pmid:10097830
- Kaltenbach M, Hopf R, Kober G, Bussmann WD, Keller M, Petersen Y. Treatment of hypertrophic obstructive cardiomyopathy with verapamil. Br Heart J 1979; 42(1):35–42. doi:10.1136/hrt.42.1.35
- Sherrid MV, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ Heart Fail 2013; 6(4):694–702. doi:10.1161/CIRCHEARTFAILURE.112.000122
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart 2014; 100(6):465–472. doi:10.1136/heartjnl-2013-304276
- Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation 2001; 104(21):2517–2524. pmid:11714644
- Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population. Circulation 2000; 102(8):858–864. pmid:10952953
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006; 114(3):216-225. doi:10.1161/CIRCULATIONAHA.105.583500
- Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail 2010; 3(5):574–579. doi:10.1161/CIRCHEARTFAILURE.109.922872
- Smedira NG, Lytle BW, Lever HM, et al. Current effectiveness and risks of isolated septal myectomy for hypertrophic obstructive cardiomyopathy. Ann Thorac Surg 2008; 85(1):127–133. doi:10.1016/j.athoracsur.2007.07.063
- Robbins RC, Stinson EB. Long-term results of left ventricular myotomy and myectomy for obstructive hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 1996; 111(3):586–594. pmid:8601973
- Heric B, Lytle BW, Miller DP, Rosenkranz ER, Lever HM, Cosgrove DM. Surgical management of hypertrophic obstructive cardiomyopathy. Early and late results. J Thorac Cardiovasc Surg 1995; 110(1):195–208. pmid:7609544
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol 2005; 46(3):470–476. doi:10.1016/j.jacc.2005.02.090
- Desai MY, Bhonsale A, Smedira NG, et al. Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction. Circulation 2013; 128(3):209–216. doi:10.1161/CIRCULATIONAHA.112.000849
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J 2007; 28(21):2583–2588. doi:10.1093/eurheartj/ehm117
- Veselka J, Tomasov P, Zemanek D. Long-term effects of varying alcohol dosing in percutaneous septal ablation for obstructive hypertrophic cardiomyopathy: a randomized study with a follow-up up to 11 years. Can J Cardiol 2011; 27(6):763–767. doi:10.1016/j.cjca.2011.09.001
- Veselka J, Jensen MK, Liebregts M, et al. Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol 2016; 209:194–195. doi:10.1016/j.ijcard.2016.02.077
- Veselka J, Krejci J, Tomašov P, Zemánek D. Long-term survival after alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a comparison with general population. Eur Heart J 2014; 35(30):2040–2045. doi:10.1093/eurheartj/eht495
- Sorajja P, Ommen SR, Holmes DR Jr, et al. Survival after alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation 2012; 126(20):2374–2380. doi:10.1161/CIRCULATIONAHA.111.076257
- Agarwal S, Tuzcu EM, Desai MY, et al. Updated meta-analysis of septal alcohol ablation versus myectomy for hypertrophic cardiomyopathy. J Am Coll Cardiol 2010; 55(8):823–834. doi:10.1016/j.jacc.2009.09.047
- Leonardi RA, Kransdorf EP, Simel DL, Wang A. Meta-analyses of septal reduction therapies for obstructive hypertrophic cardiomyopathy: comparative rates of overall mortality and sudden cardiac death after treatment. Circ Cardiovasc Interv 2010; 3(2):97–104. doi:10.1161/CIRCINTERVENTIONS.109.916676
- Liebregts M, Vriesendorp PA, Mahmoodi BK, Schinkel AF, Michels M, ten Berg JM. A systematic review and meta-analysis of long-term outcomes after septal reduction therapy in patients with hypertrophic cardiomyopathy. JACC Heart Fail 2015; 3(11):896–905. doi:10.1016/j.jchf.2015.06.011
- Panaich SS, Badheka AO, Chothani A, et al. Results of ventricular septal myectomy and hypertrophic cardiomyopathy (from Nationwide Inpatient Sample [1998-2010]). Am J Cardiol 2014; 114(9):1390–1395. doi:10.1016/j.amjcard.2014.07.075
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol 2015; 66(11):1307–1308. doi:10.1016/j.jacc.2015.06.1333
- Kwon DH, Smedira NG, Thamilarasan M, Lytle BW, Lever H, Desai MY. Characteristics and surgical outcomes of symptomatic patients with hypertrophic cardiomyopathy with abnormal papillary muscle morphology undergoing papillary muscle reorientation. J Thorac Cardiovasc Surg 2010; 140(2):317–324. doi:10.1016/j.jtcvs.2009.10.045
- Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg 2010; 139(3):634–640. doi:10.1016/j.jtcvs.2009.07.079
KEY POINTS
- Obstruction of the left ventricular outflow tract is a key pathophysiologic mechanism in HCM.
- Because most of the genetic variants that contribute to HCM are autosomal dominant, genetic counseling and testing are suggested for patients and their first-degree relatives.
- Transthoracic echocardiography is the first-line imaging test, followed by magnetic resonance imaging.
- Beta-blockers are the first-line drugs for treating symptoms of HCM.
- An implantable cardioverter-defibrillator can be considered for patients at risk of sudden cardiac death.
- When medical therapy fails or is not tolerated in patients with severe symptoms of obstructive HCM, surgery to reduce the size of the ventricular septum can be considered. Alcohol septal ablation is an alternative.

Improving Strength and Balance for Long-Term Care Residents At Risk for Falling: Suggestions for Practice
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search. 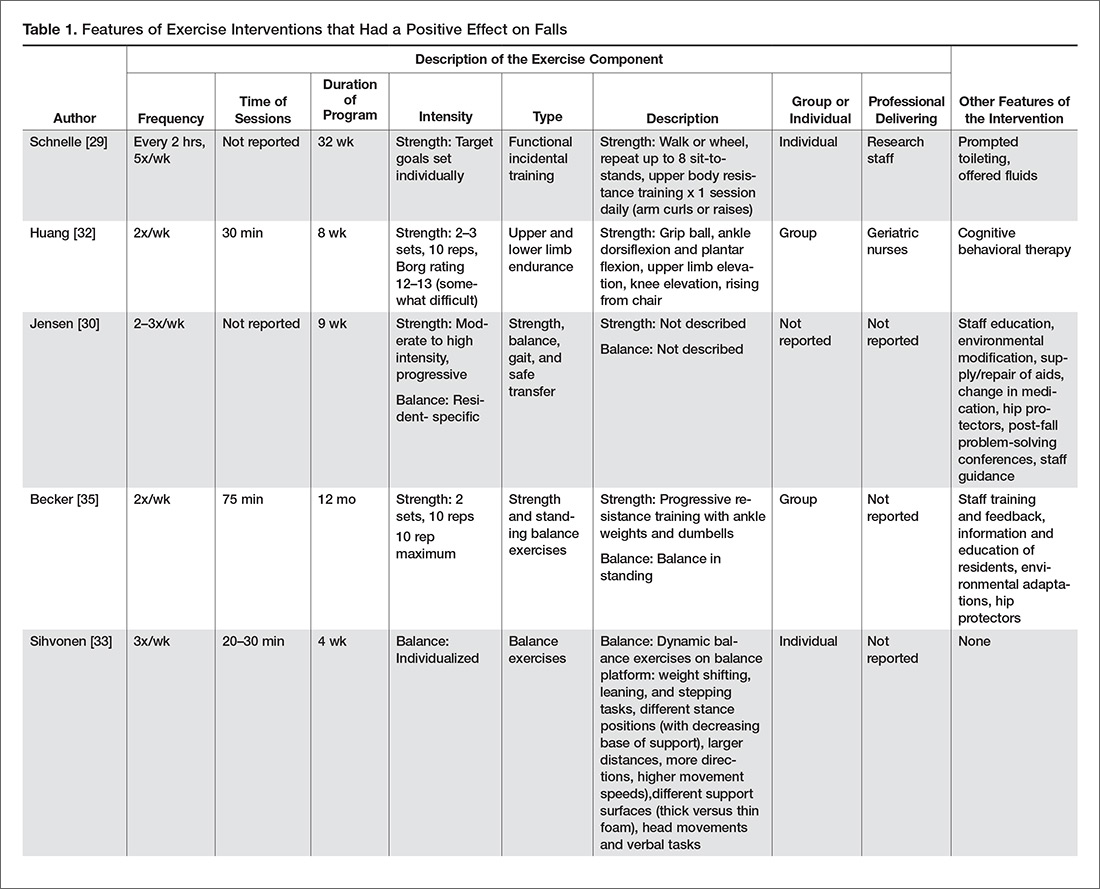
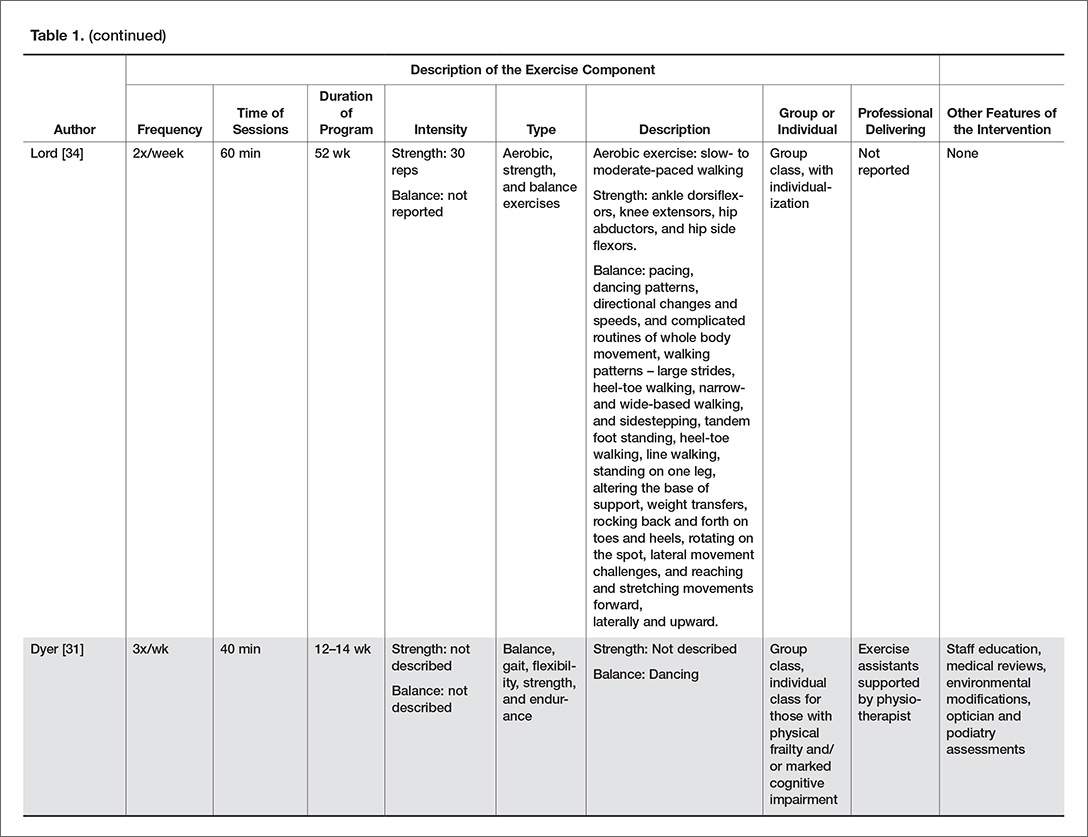
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure) 
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48]. 
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search.
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure)
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48].
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
From the Geriatric Education and Research in Aging Sciences Centre, McMaster University Hamilton, ON (Dr. McArthur) and the University of Waterloo and Research Institute for Aging, Waterloo, ON (Dr. Giangregorio), Canada
Abstract
- Objective: To synthesize the available literature on exercise and falls reduction interventions in long-term care (LTC) and provide practical information for clinicians and other decision makers.
- Methods: Review of positive trials included in systematic reviews.
- Results: Falls are a major concern for residents, families, clinicians, and decision-makers in LTC. Exercise is recommended as part of a multifactorial falls prevention program for residents in LTC. Strength and balance exercises should be incorporated into the multifactorial falls prevention program. They should be challenging and progressed as the residents’ abilities improve. Evidence suggests that exercises should be completed 2 to 3 times per week for a period longer than 6 months. Exercise programs in LTC should be resident-centered and should consider residents’ potential physical and cognitive impairments. Exercises in standing should be prioritized where appropriate.
- Conclusion: Appropriately challenging and progressive strength and balance exercises should be included in a multifactorial falls prevention program for residents in LTC.
Key words: long-term care; nursing homes; falls reduction; exercise.
Falls are common in long-term care (LTC) homes: the estimated falls rate is 1.5 falls per bed per year, which is 3 times greater than that for older adults living in the community [1]. Falls can have significant consequences for residents in LTC, including functional disability, fractures, pain, reduced quality of life, and death [1–6]. Indeed, 25% of residents who are hospitalized after a fall die within 1 year [3]. Consequently, falls prevention programs are important to help in reducing falls and averting the associated negative consequences.
Exercise may address the circumstances and physical deconditioning that often contribute to falls in LTC residents. Weight shifting [7], walking, and transferring [8–10], are common activities that precede falls, suggesting that balance, gait, and functional mobility training may be possible targets for prevention. Additionally, it is estimated that LTC residents spend three quarters of their waking time in sedentary activities [11,12] and have a high prevalence of sarcopenia [13–16]. Challenging balance training and resistance exercise are well-known intervention for reducing falls [17] and improving muscle strength for community-dwelling older adults [18]. However, evidence around balance and strength training for preventing falls in LTC is mixed [17,19,20], and careful planning and modification of exercises is necessary to meet the needs of LTC residents.
Residents in LTC are often medically complex, with multiple comorbidities [21] that can affect their ability to meaningfully participate in exercise. In Canada, 56.3% of residents have a diagnosis of Alzheimer’s or other dementias, 25.0% have diabetes, 14.4% have chronic obstructive pulmonary disease, and 21.2% have experienced a stroke [21]. Residents also often have significant functional impairments. For example, 97% of residents require assistance with basic activities of daily living [21]. Therefore, the lack of effect of exercise as a single falls prevention strategy observed in previous studies may be because the often complex, multimorbid LTC population likely requires a multifactorial approach to fall prevention [17]. Additionally, organizational aspects of LTC homes (eg, specific funds dedicated to employing exercise professionals and to support exercise programming) can affect residents’ engagement in exercise [22,23]. Subsequently, prescribing exercises in the LTC context must consider both resident characteristics and organizational features of the LTC home (eg, professionals available to support exercise programming).
A comprehensive exercise prescription describes the elements of an appropriate exercise program to facilitate implementation of that program. The exercise prescription should include a description of the type (eg, balance, strength) and intensity of exercises (eg, subjective or objective measurement of how hard the resident is working) included in the program [24]. The prescription should also include a description of the dose of exercise: frequency of exercise participation (eg, 2 days per week), duration of individual exercise sessions (eg, 30-minute sessions), and duration of exercise program (eg, 12-week program) [24]. Lastly, the prescription should describe the setting of the exercise program (eg, group or individual basis) and the professional delivering the program (eg, physiotherapist, fitness instructor) [24].
Therefore, the objectives of this article are to (1) synthesize studies demonstrating a positive effect of exercise on reducing falls for residents in LTC; (2) provide an overview of the principles of balance and strength training to guide clinicians in designing appropriate exercise prescription; and (3) make suggestions for clinical practice regarding an appropriate strength and balance exercise protocol by considering the influence of the LTC context.
Methods
To provide clinicians and other policy-makers with a description of which balance and strength exercises may be effective for preventing falls, we synthesized trials that demonstrated a positive effect on reducing falls or falls risk for residents in LTC. Studies were identified through a database search for systematic reviews in PubMed, Ovid, and Google Scholar using the keywords falls, long-term care, nursing homes, exercise, strength, balance, and systematic reviews. Our purpose was to provide practical information on what works to prevent falls through balance and strength training for residents in LTC rather than to evaluate the available evidence. Therefore, only positive trials from systematic reviews were discussed, as we wanted to present exercises that seem to have a positive effect on decreasing falls. Positive trials were defined as those included in identified systematic reviews with a risk or rate ratio and confidence intervals below 1.0.
We first provide an overview of the conclusions of the systematic reviews found in our search. Next, for each positive trial we describe the following elements of the exercise component of the intervention: frequency, time of sessions, length of program, intensity, type of exercise including a description of the specific exercises performed, whether the intervention was delivered in a group or on an individual basis, the professional delivering the intervention, and any other features of the intervention aside from the exercise component. We used the ProFaNE taxonomy definitions [25] to identify and describe each element of the exercise interventions. Frequency is the number of times per week that residents engage in sessions, time of sessions is the amount allocated to each exercise session, duration of program is how long the resident participates in the exercise program, and intensity is the subjective or objective report of how hard the resident is working [25]. The types of exercises described were those targeting balance defined as “...the efficient transfer of bodyweight from one part of the body to another or challenges specific aspects of the balance systems (eg, vestibular system)” [25], and strength defined as “...contracting the muscles against a resistance to ‘overload’ and bring about a training effect in the muscular system” [25]. Strength could be either an external resistance (eg, dumbbell) or using body weight against gravity (eg, squat) [25].
Results
We found 3 systematic reviews that include exercise programs to reduce falls in LTC homes [17,19,20]. Overall, evidence suggests that exercise should be included as part of a multifactorial falls prevention program for residents in LTC. There is limited evidence that exercise as a single intervention prevents falls, and some trials, albeit underpowered, even demonstrate an increased risk of falling in the exercise group compared to control [19]. With regards to specific exercise programs, the Cochrane review found that gait, balance, and functional training decrease the rate of falls but not the risk of falling [26–28], and the 2013 review by Silva et al [20] concluded that combined exercise programs (ie, multiple types of exercise) that include balance tasks, are completed frequently (2–3 times per week), and over a long term (greater than 6 months) were most effective at preventing falls [20].
A more recent systematic review and meta-analysis [17] also concluded that there was no evidence that exercise as a single intervention can prevent falls for residents in LTC. Table 1 provides a description of the exercise component of the seven positive trials [29–35] that were included in the 3 systematic reviews we identified in our search.
Type of Exercise
Balance Exercises
There were 4 positive trials that included balance exercises in their intervention [31,33–35]. Trials that had a positive effect on reducing falls and included balance training employed mostly dynamic balance exercises in standing (Table 1). However, only 2 of the 7 trials provided a detailed description of their balance exercises (Table 1) [26,34]. Jensen et al [30] and Dyer et al [31] did not include a description of the balance training performed but stated that balance was part of the multicomponent exercise program. Becker et al [36] stated that participants performed standing balance exercises, while Schnelle et al [39] and Huang et al [32] did not include balance training in their trial.
Strength Exercises
Of the 7 positive trials included in this review, 6 included strength exercises [29–32,34,35]. The strength activities used in trials where exercise had a positive effect on decreasing falls included functional activities [29,31] and progressive resistance training [31,36] (Table 1). Functional activities are those that replicate what a resident might be required to do in their everyday life, such as performing sit-to-stands out of a chair (Figure)
Frequency, Time of Sessions, Duration of Program
In our description of positive trials, exercise was performed on 2 to 3 days per week for 20 to 75 minutes per session, for periods ranging from 4 to 52 weeks (Table 1).
Intensity
For the trials including balance exercises, one trial described the intensity as resident-specific [37] and another as individualized [33]. Two studies did not describe the intensity of their balance exercises [31,34]. The intensity of strength exercises included in the positive trials was individualized for one of the trial [29]. Two trials had participants complete 2 to 3 sets of 10 repetitions [32,35], with one indicating an intensity of 12–13 or “somewhat difficult” on the Borg Rating of Perceived Exertion Scale [32] and the other using a 10-rep max [35]. Two studies described their strength exercises as progressive [31,37], and one at a moderate to high intensity [30]. Lord et al prescribed 30 repetitions of each strength exercise [34].
Delivery of Intervention
Exercise was delivered in a group setting for 4 of the trials [31,32,34,36], individually for 2 of the trials [26,29], and the setting was not described for one of the trials (Table 1) [30]. Finally, only 3 of the 7 articles reported the professional delivering the intervention: one was research staff [29], one was geriatric nurses [32], and one was exercise assistants supported by a physiotherapist [31].
Discussion
There is limited evidence to support the use of strength and balance exercise as a single intervention to prevent falls in LTC. However, exercise should be included as part of a multifactorial falls prevention program. Trials that had a positive effect on decreasing falls training used dynamic balance exercises in standing, functional training, and progressive resistance training on 2 to 3 days per week, for 20 to 75 minutes per session, over 4 to 52 weeks. The intensity of balance exercises was individualized, and strength exercises were described as somewhat difficult or performed at a moderate to high intensity. Exercise was performed in a group or individually, and was delivered by research staff, geriatric nurses, exercise assistants supervised by physiotherapists, or more frequently, it was not reported who delivered the intervention.
Balance Training
Our work suggests that standing, dynamic balance exercises may be best to decrease falls. Example balance exercises include reducing the base of support (eg, standing with feet together instead of apart, or tandem with one foot in front), moving the center of gravity and control body position while standing (eg, reaching, weight shifting, stepping up or down), and standing without using arms for support or reducing reliance on the upper limbs for support (eg, use one hand on a handrail instead of two, or two fingers instead of the whole hand) [17]. It is well established that balance training programs, especially those including challenging exercises, can prevent falls in community-dwelling older adults [17]. However, the relationship is not as clear in LTC.
Strength Training
Reduced muscle strength has been identified as an important risk factor for falls [38]. There are also many psychological and metabolic benefits to strength training [39]. To induce change in muscular strength, resistance exercises need to be challenging and progressive. Our work suggests that strength training that is effective at decreasing falls is functional and progressive, and is completed at a moderate to high intensity. A resident should be able to do a strength exercise for one to two sets of 6 to 8 repetitions before being fatigued [40]. Once the resident can complete two sets of 13 to 15 repetitions easily the exercise should be progressed. Residents who are particularly deconditioned may need to begin with lower intensity strength exercises (eg, only do one set, with a lower resistance and progress to a higher resistance) [40]. Residents should perform resistance exercises for all major muscle groups [40]. Progression could include increasing the number of sets (eg, increase from one to two sets), the resistance (eg, holding dumbbells while squatting), or the intensity of the exercise (eg, squat lower or faster) [41].
Implementing Exercise Programs in LTC
Implementation of exercise programs into LTC homes should consider the dose of exercise (eg, time and frequency of sessions, duration of program), if they are delivered in a group or individual setting, and who is delivering them. First, trials included in this paper suggest that strength and balance exercises to prevent falls were delivered 2 to 3 times per week, for 20 to 75 minutes per session, over 4 to 52 weeks. Second, previous work has established that exercise programs delivered on 2 to 3 days per week over a period of more than 6 months are most effective at reducing falls in LTC [20]. Finally, a recent task force report from an international group of clinician researchers in LTC recommends twice weekly exercise sessions lasting 35 to 45 minutes each [40]. Therefore, strength and balance exercises to prevent falls in LTC should be delivered at least twice per week, for at least 20 minutes, for greater than 6 weeks’ duration.
Whether exercise should be performed in a group or individual setting remains unclear. Two of the 6 positive trials in this paper were completed individually, while 3 were in a group. The aforementioned task force also recommended that every resident who does not have contraindications to exercise must have an individualized exercise program as part of their health care plan [40]. However, whether the exercise program is provided on an individual basis or in a group setting was not delineated. Indeed, there are currently no recommendations concerning prioritizing group or individual exercise programs. Therefore, exercise programs being implemented into LTC homes should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings.
Finally, which professionals should deliver the exercise program is also uncertain. Only 3 of the positive trials in this paper described the professional delivering the intervention, with one being research staff, one geriatric nurses, and one exercise assistants supported by a physiotherapist. We suggest that professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC.
Modifications for Physical Impairments
Residents in LTC often have complex health needs, with multiple comorbidities (eg, stroke, Parkinson’s disease, multiple sclerosis) [21]. Modifications of strength and balance exercises may be required to accommodate for physical impairments (eg, hemiplegia, drop foot, freezing gait). For example, if a resident has hemiplegia and cannot fully activate the muscles of one arm, one can do resistance exercises with a dumbbell on the functioning side and active assisted range of motion (ie, the exercise provider assists the resident to achieve full range of motion against gravity) on the hemiparetic side. A resident with Parkinson’s disease who has freezing gait may need visual or rhythmical verbal cues to be able to accomplish standing balance tasks such as altered walking patterns (eg, wide or narrow stepping) [42].
Modifications for Cognitive Impairments
More than 80% of residents in LTC have some degree of cognitive impairment [21]. Cognitive impairment may be the result of stroke, depression, traumatic injuries, medications, and degenerative diseases such as Parkinson’s and Alzheimer’s disease [43]. A common misconception is that residents with cognitive impairment cannot benefit from exercise because they cannot learn new skills and have difficulty following directions. On the contrary, evidence suggests that exercise can improve functional mobility for residents with cognitive impairment [44,45].
Residents with cognitive impairment may require a different approach to facilitate participation in the desired exercises because of difficulty following multi-step directions, responsive behaviors, or increased distractibility [46]. Clear communication is key in improving the quality of interaction for residents with cognitive impairment. The Alzheimer Society of Ontario suggests 10 strategies for communicating with people with dementia [47], and we have provided suggestions of how to apply these communication strategies to the exercise context in LTC (Table 2). Other suggestions for engaging residents with cognitive impairment in strength and balance training include making the exercises functional (eg, ask them to pick something up of the floor to perform a squat, or reach a point on the wall to do calf raises) and playful (eg, toss a ball back and forth or sing a song about rowing to promote weight shifting) [48].
Standing versus Seated Exercises
Residents may not be able to participate in standing exercises for several reasons: perhaps the resident cannot stand or has severe balance impairments and a high falls risk; the resident may have poor insight into which exercises are safe to perform in standing versus sitting; or there may be limited supervision of a large group exercise class where the risk of falls is a concern. If balance impairments are a concern, where the risk of injury or falling while completing exercises in standing outweighs the benefit of doing the exercises, then seated exercises are appropriate. However, when residents are able, we recommend encouraging some or all exercises in standing, to facilitate carry over of strength gains into functional tasks such as being able to rise from a chair and walking. A recent study, comparing standing versus seated exercises for community dwelling older adults, saw greater functional gains for those who completed the standing exercises [49]. Therefore, strength and balance exercises should be performed in standing, where appropriate.
Resident-Centered Exercise for Falls Prevention
Putting the resident at the center of falls prevention is important. Previous work has found that older adults have expressed a strong preference for care that transcends traditional biomedical care and that values efficiency, consistency, and hierarchical decision making [50]. On the contrary, resident-centered care emphasizes well-being and quality of life as defined by the resident, values giving residents greater control over the nature of services they receive, and respects their rights to be involved in every day decision making [51,52]. Indeed, residents may choose to engage in risky behaviors that increase their risk of falls but also increases their quality of life. Previous work has found disconnects between residents’ perceived frailty and the potential ability of protective devices to prevent adverse events, such as falls and fractures [53]. Additionally, one study identified that older residents feared being labelled, so instead hid impairments and chose to refuse assistance and assistive devices [54]. For example, a resident with impaired balance and gait may choose to walk independently when they have been deemed as requiring a gait aid (eg, rollator walker). However, they may value walking without a gait aid and accept the increased risk of falling. Therefore, it is essential to find the delicate balance between respecting a resident’s right to make their own decisions and preventing adverse events, such as falls [52]. An example of this would be respecting a resident’s right to refuse to attend exercise programming even though the team may think they can benefit from strength and balance training.
There is limited evidence around falls prevention and resident-centered care. A recent systematic review [55] revealed that resident-centered care may increase falls rates [56,57]. However, the authors of the review attributed the increase in falls to differences in frailty between the control and intervention group [56], and to environmental factors (eg, slippery flooring material, lack of handrails) [57]. Additionally, these trials did not include an exercise program as part of the resident-centered care program. On the other hand, resident-centered care has been associated with reduction of boredom, helplessness, and depression [58,59]. Most studies included in the review were quasi-experimental, which significantly limits the evidence quality [55]. At this point in time, the evidence suggests that resident-centered care is important for mood and quality of life but may have a negative or no effect on reducing falls.
Multifactorial Falls Prevention Programs
While there are mixed results about the effect of exercise as a single intervention for reducing falls for residents in LTC, the literature clearly supports exercise as part of a multifactorial falls prevention program [17,20,60–62]. A 2015 umbrella review [62] of meta-analyses of randomized controlled trials of falls prevention interventions in LTC concluded that multifactorial interventions were the most effective at preventing falls in LTC. Additionally, recently developed recommendations for fracture prevention in LTC [61] suggest that balance, strength, and functional training should be included for residents who are not at high risk of fracture, while for those at high risk, exercise should be provided as part of a multifactorial falls prevention intervention. Clinicians must therefore incorporate elements aside from exercise into their falls prevention strategies. Interventions that have shown positive effects on reducing falls when delivered as part of multifactorial interventions include: staff and resident education [31,35,37], environmental modifications [31,35], supply/repair/provision of assistive devices [30], falls problem-solving conferences [30], urinary incontinence management [29], medication review [30], optician review [31], and cognitive behavioral therapy [32].
Conclusion and Suggestions for Clinical Practice
We suggest incorporating strength and balance exercises as part of a multifactorial falls prevention program for residents in LTC. Balance exercises should be challenging and dynamic (eg, weight shifting). Strength exercises should be of a moderate to high intensity (eg, can complete one to sets of 6 to 8 repetitions) and need to be progressed as the residents’ abilities improve. Residents should participate in strength and balance training on 2 to 3 days per week, for 30- to 45-minute sessions, for at least 6 months. Exercises in standing should be prioritized where appropriate. Exercise could be delivered in a group or individual format, but should consider the residents’ preferences, the social benefits of group exercise, and the feasibility of individualizing exercises for the complex needs of residents in LTC in large group settings. Professionals delivering an exercise program should be trained in exercise planning, delivery, and progression, be familiar with the principles of balance and strength training, and have training in working with older adults in LTC. Exercise programs in LTC should be resident-centered and consider residents’ potential physical and cognitive impairments.
Funding/support: Dr. Giangregorio was supported by grants from the Canadian Frailty Network and Canadian Institutes of Health Research.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
1. Harris IA, Yong S, McEvoy L, Thorn L. A prospective study of the effect of nursing home residency on mortality following hip fracture. ANZ J Surg 2010;80:447–50.
2. Ooms ME, Vlasman P, Lips P, et al. The incidence of hip fractures in independent and institutionalized elderly people. Osteoporos Int 1994;4:6–10.
3. Ayoung-Chee P, McIntyre L, Ebel BE, et al. Long-term outcomes of ground-level falls in the elderly. J Trauma Acute Care Surg 2014;76:498–503.
4. Heinrich S, Rapp K, Rissmann U, et al. Cost of falls in old age: a systematic review. Osteoporos Int 2010;21: 891–902.
5. Rubenstein LZ, Josephson KR, Robbins AS. Falls in the nursing home. Ann Intern Med 1994;121:442–51.
6. Hartholt KA, van Beeck EF, Polinder S, et al. Societal consequences of falls in the older population: injuries, healthcare costs, and long-term reduced quality of life. J Trauma
2011;71:748–53.
7. Robinovitch SN, Feldman F, Yang Y, et al. Video capture of the circumstances of falls in elderly people residing in long-term care: an observational study. Lancet 2013;381:
47–54.
8. Rapp K, Becker C, Cameron ID, et al. Epidemiology of falls in residential aged care: analysis of more than 70,000 falls from residents of bavarian nursing homes. J Am Med Dir Assoc 2012;13:187.
9. Büchele G, Becker C, Cameron ID, et al. Predictors of serious consequences of falls in residential aged care: analysis of more than 70,000 falls from residents of Bavarian nursing homes. J Am Med Dir Assoc 2014;15:559–63.
10. McArthur C, Gonzalez DA, Roy E, Giangregorio L. What are the circumstances of falls and fractures in long-term care? Can J Aging / La Rev Can du Vieil 2016;35:491–8.
11. Chin A Paw MJM, van Poppel MNM, van Mechelen W. Effects of resistance and functional-skills training on habitual activity and constipation among older adults living in long-term care facilities: a randomized controlled trial. BMC Geriatr 2006;6:9.
12. Ikezoe T, Asakawa Y, Shima H, et al. Daytime physical activity patterns and physical fitness in institutionalized elderly women: an exploratory study. Arch Gerontol Geriatr 2013;57:221–5.
13. Senior HE, Henwood TR, Beller EM, et al. Prevalence and risk factors of sarcopenia among adults living in nursing homes. Maturitas 2015;82:418–23.
14. Smoliner C, Sieber CC, Wirth R. Prevalence of sarcopenia in geriatric hospitalized patients. J Am Med Dir Assoc 2014;15:267–72.
15. Landi F, Liperoti R, Fusco D, et al. Sarcopenia and mortality among older nursing home residents. J Am Med Dir Assoc 2012;13:121–6.
16. Yalcin A, Aras S, Atmis V, et al. Sarcopenia prevalence and factors associated with sarcopenia in older people living in a nursing home in Ankara Turkey. Geriatr Gerontol Int
2016;16:903–10.
17. Sherrington C, Michaleff ZA, Fairhall N, et al. Exercise to prevent falls in older adults: an updated systematic review and meta-analysis. Br J Sports Med October 2016.
18. Liu C, Latham NK. Progressive resistance strength training for improving physical function in older adults. In: Liu C, ed. Cochrane Database Syst Rev;2009:CD002759.
19. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev;2012:CD005465.
20. Silva RB, Eslick GD, Duque G. Exercise for falls and fracture prevention in long term care facilities: a systematic review and meta-analysis. J Am Med Dir Assoc 2013;14:685–9.
21. Hirdes JP, Mitchell L, Maxwell CJ, White N. Beyond the “iron lungs of gerontology”: Using evidence to shape the future of nursing homes in Canada. Can J Aging 2011;30: 371–90.
22. Benjamin K, Edwards N, Guitard P, et al. Factors that influence physical activity in long-term care: Perspectives of residents, staff, and significant others. Can J Aging 2011;30:247–58.
23. Benjamin K, Edwards N, Ploeg J, Legault F. Barriers to physical activity and restorative care for residents in long-term care: A review of the literature. J Aging Phys Act 2014;22:154–65.
24. American College of Sports Medicine. ACSM’s guidelines for exercise testing and prescription. 9th ed. American College of Sports Medicine; 2013.
25. Prevention of Falls Network Europe. Prevention of Falls Network Europe. Accessed 27 Nov 2017 at www.profane.eu.org/.
26. Sihvonen SE, Sipilä S, Era PA. Changes in postural balance in frail elderly women during a 4-week visual feedback training: a randomized controlled trial. Gerontology 2004;50:87–95.
27. Sakamoto K, Nakamura T, Hagino H, et al. Effects of unipedal standing balance exercise on the prevention of falls and hip fracture among clinically defined high-risk elderly individuals: a randomized controlled trial. J Orthop Sci 2006;11:467–72.
28. Shimada H, Obuchi S, Furuna T, Suzuki T. New intervention program for preventing falls among frail elderly people: the effects of perturbed walking exercise using a bilateral separated treadmill. Am J Phys Med Rehabil 2004;83:493–9.
29. Schnelle JF, Kapur K, Alessi C, et al. Does an exercise and incontinence intervention save healthcare costs in a nursing home population? J Am Geriatr Soc 2003;51:161–8.
30. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities: A cluster randomized trial. Ann Intern Med 2002;136:733–41.
31. Dyer CAE. Falls prevention in residential care homes: a randomised controlled trial. Age Ageing 2004;33:596–602.
32. Huang T-T, Chung M-L, Chen F-R, Chin Y-F, Wang B-H. Evaluation of a combined cognitive-behavioural and exercise intervention to manage fear of falling among elderly residents in nursing homes. Aging Ment Health 2016;20:2–12.
33. Sihvonen S, Sipilä S, Taskinen S, Era P. Fall incidence in frail older women after individualized visual feedback-based balance training. Gerontology 2004;50:411–6.
34. Lord SR, Castell S, Corcoran J, et al. The effect of group exercise on physical functioning and falls in frail older people living in retirement villages: a randomized, controlled trial. J Am Geriatr Soc 2003;51:1685–92.
35. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
36. Becker C, Kron M, Lindemann U, et al. Effectiveness of a multifaceted intervention on falls in nursing home residents. J Am Geriatr Soc 2003;51:306–13.
37. Jensen J, Lundin-Olsson L, Nyberg L, Gustafson Y. Fall and injury prevention in older people living in residential care facilities. A cluster randomized trial. Ann Intern Med 2002;136:733–41.
38. Moreland JD, Richardson JA, Goldsmith CH, Clase CM. Muscle weakness and falls in older adults: a systematic review and meta-analysis. J Am Geriatr Soc 2004;52: 1121–9.
39. Chodzko-Zajko WJ, Proctor DN, Fiatarone Singh MA, et al. Exercise and physical activity for older adults. Med Sci Sport Exerc 2009;41:1510–30.
40. de Souto Barreto P, Morley JE, Chodzko-Zajko W, et al. Recommendations on physical activity and exercise for older adults living in long-term care facilities: a taskforce report. J Am Med Dir Assoc 2016;17:381–92.
41. American College of Sports Medicine. Progression models in resistance training for healthy adults. Med Sci Sport Exerc 2009;41:687–708.
42. Fietzek UM, Schroeteler FE, Ziegler K, et al. Randomized cross-over trial to investigate the efficacy of a two-week physiotherapy programme with repetitive exercises of cueing to reduce the severity of freezing of gait in patients with Parkinson’s disease. Clin Rehabil 2014;28:902–11.
43. Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: A systematic evidence review. Alzheimer Dement 2007;3:341–7.
44. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
45. Christofoletti G, Oliani MM, Gobbi S, et al. A controlled clinical trial on the effects of motor intervention on balance and cognition in institutionalized elderly patients with dementia. Clin Rehabil 2008;22:618–26.
46. van Alphen HJM, Hortobágyi T, van Heuvelen MJG. Barriers, motivators, and facilitators of physical activity in dementia patients: A systematic review. Arch Gerontol Geriatr 2016;66:109–18.
47. Alzheimer Society of Ontario. Rethink Dementia. Accessed 18 Sep 2017 at http://rethinkdementia.ca/.
48. Roach KE, Tappen RM, Kirk-Sanchez N, et al. A randomized controlled trial of an activity specific exercise program for individuals with Alzheimer disease in long-term care settings. J Geriatr Phys Ther 2011;34:50–6.
49. Brach JS, Perera S, Gilmore S, et al. Effectiveness of a timing and coordination group exercise program to improve mobility in community-dwelling older adults. JAMA Intern Med August 2017.
50. Rosher RB, Robinson S. Impact of the Eden alternative on family satisfaction. J Am Med Dir Assoc 2005;6:189–93.
51. Crandall LG, White DL, Schuldheis S, Talerico KA. Initiating person-centered care practices in long-term care facilities. J Gerontol Nurs 2007;33:47–56.
52. Sims-Gould J, McKay HA, Feldman F, et al. Autonomy, choice, patient-centered care, and hip protectors: the experience of residents and staff in long-term care. J Appl Gerontol 2014;33:690–709.
53. Robinovitch SN, Cronin T. Perception of postural limits in elderly nursing home and day care participants. J Gerontol A Biol Sci Med Sci 1999;54:B124-30.
54. Perkins MM, Ball MM, Whittington FJ, Hollingsworth C. Relational autonomy in assisted living: a focus on diverse care settings for older adults. J Aging Stud 2012;26:214–25.
55. Brownie S, Nancarrow S. Effects of person-centered care on residents and staff in aged-care facilities: a systematic review. Clin Interv Aging 2013;8:1–10.
56. Coleman MT, Looney S, O’Brien J, et al. The Eden Alternative: findings after 1 year of implementation. J Gerontol A Biol Sci Med Sci 2002;57:M422–7.
57. Chenoweth L, King MT, Jeon Y-H, et al. Caring for Aged Dementia Care Resident Study (CADRES) of personcentred care, dementia-care mapping, and usual care in dementia: a cluster-randomised trial. Lancet Neurol 2009;8: 317–25.
58. Bergman-Evans B. Beyond the basics. Effects of the Eden Alternative model on quality of life issues. J Gerontol Nurs 2004;30:27–34.
59. Robinson SB, Rosher RB. Tangling with the barriers to culture change: creating a resident-centered nursing home environment. J Gerontol Nurs 2006;32:19–25.
60. Cameron ID, Gillespie LD, Robertson MC, et al. Interventions for preventing falls in older people in care facilities and hospitals. Cochrane Database Syst Rev 2012;12.
61. Papaioannou A, Santesso N, Morin SN, et al. Recommendations for preventing fracture in long-term care. Can Med Assoc J 2015;187:1135–44.
62. Stubbs B, Denkinger MD, Brefka S, Dallmeier D. What works to prevent falls in older adults dwelling in long term care facilities and hospitals? An umbrella review of meta-analyses of randomised controlled trials. Maturitas 2015;81:335–42.
Genomic Testing in the Management of Early-Stage Breast Cancer
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
From the University of Arizona Cancer Center, Tucson, AZ (Dr. Ehsani), and University of Wisconsin Carbone Cancer Center and School of Medicine and Public Health, Madison, WI (Dr. Wisinski).
Abstract
- Objectives: To describe common genomic tests being used clinically to assess prognosis and guide adjuvant chemotherapy and endocrine therapy decisions for early-stage breast cancer.
- Methods: Case presentation and review of the literature.
- Results: Hormone receptor–positive (HR-positive) breast cancers, which express the estrogen and/or progesterone receptor, account for the majority of breast cancers. Endocrine therapy can be highly effective for patients with these HR-positive tumors, and identification of HR-positive breast cancers that do not require the addition of chemotherapy is critical. Clinicopathological features of the breast cancer, including tumor size, nodal involvement, grading, and HR status, are insufficient in predicting the risk for recurrence or the need for chemotherapy. Furthermore, a portion of HR-positive breast cancers have an ongoing risk for late recurrence, and longer durations of endocrine therapy are being used to reduce this risk.
- Conclusion: There is sufficient evidence for use of genomic testing in early-stage HR-positive breast cancer to aid in chemotherapy recommendations. Further confirmation of genomic assays for prediction of benefit from prolonged endocrine therapy is needed.
Key words: molecular testing; decision aids; HR-positive cancer; recurrence risk; adjuvant chemotherapy; endocrine therapy.
Despite the increase in incidence of breast cancer, breast cancer mortality has decreased over the past several decades. This is likely due to both early detection and advances in systemic therapy. However, with more widespread use of screening mammography, there are increasing concerns regarding potential overdiagnosis of cancer [1]. One key challenge is that breast cancer is a heterogeneous disease. Thus, improved tools for determining breast cancer biology can help physicians individualize treatments, with low-risk cancers approached with less aggressive treatments, thus preventing unnecessary toxicities, and higher-risk cancers treated appropriately.
Traditionally, adjuvant chemotherapy was recommended based on tumor features such as stage (tumor size, regional nodal involvement), grade, expression of hormone receptors (estrogen receptor [ER] and progesterone receptor [PR]) and human epidermal growth factor receptor-2 (HER2), and patient features (age, menopausal status). However, this approach is not accurate enough to guide individualized treatment recommendations, which are based on the risk for recurrence and the reduction in this risk that can be achieved with various systemic treatments. In particular, there are individuals with low-risk HR-positive, HER2-negative breast cancers who could be spared the toxicities of cytotoxic chemotherapies without compromising the prognosis.
Beyond chemotherapy, endocrine therapies also have risks, especially when given for extended durations. Recently, extended endocrine therapy has been shown to prevent late recurrences of HR-positive breast cancers. In the MA.17R study, extended endocrine therapy with letrozole for a total of 10 years (beyond 5 years of an aromatase inhibitor [AI]) decreased the risk for breast cancer recurrence or the occurrence of contralateral breast cancer by 34% [2]. However, the overall survival was similar between the 2 groups and the results were not confirmed in other studies [3–5]. Identifying the subgroup of patients who benefit from this extended AI therapy is important in the era of personalized medicine. Several tumor genomic assays have been developed to provide additional prognostic and predictive information with the goal of individualizing adjuvant therapies for breast cancer. Although assays are also being evaluated in HER2-positive and triple negative breast cancer, this review will focus on HR-positive, HER2-negative breast cancer.
Case Study
Initial Presentation
A 54-year-old postmenopausal woman with no significant past medical history presents with an abnormal screening mammogram, which shows a focal asymmetry in the 10 o’clock position at middle depth of the left breast. Further work-up with a diagnostic mammogram and ultrasound of the left breast shows a suspicious hypoechoic solid mass with irregular margins measuring 17 mm. The patient undergoes an ultrasound-guided core needle biopsy of the suspicious mass, the results of which are consistent with an invasive ductal carcinoma, Nottingham grade 2, ER strongly positive (95%), PR weakly positive (5%), HER2 negative, and Ki-67 of 15%. She undergoes a left partial mastectomy and sentinel lymph node biopsy, with final pathology demonstrating a single focus of invasive ductal carcinoma, measuring 2.2 cm in greatest dimension with no evidence of lymphovascular invasion. Margins are clear and 2 sentinel lymph nodes are negative for metastatic disease (final pathologic stage IIA, pT2 pN0 cM0). She is referred to medical oncology to discuss adjuvant systemic therapy.
Can additional testing be used to determine prognosis and guide systemic therapy rec-ommendations for early-stage HR-positive/HER2-negative breast cancer?
After a diagnosis of early-stage breast cancer, the key clinical question faced by the patient and medical oncologist is: what is the individual’s risk for a metastatic breast cancer recurrence and thus the risk for death due to breast cancer? Once the risk for recurrence is established, systemic adjuvant chemotherapy, endocrine therapy, and/or HER2-directed therapy are considered based on the receptor status (ER/PR and HER2) to reduce this risk. Hormone receptor (HR)–positive, HER2-negative breast cancer is the most common type of breast cancer. Although adjuvant endocrine therapy has significantly reduced the risk for recurrence and improved survival for HR-positive breast cancer [6], the role of adjuvant chemotherapy for this subset of breast cancer remains unclear. Prior to genomic testing, the recommendation for adjuvant chemotherapy for HR-positive/HER2-negative tumors was primarily based on patient age and tumor stage and grade. However, chemotherapy overtreatment remained a concern given the potential short- and long-term risks of chemotherapy. Further studies into HR-positive/HER2-negative tumors have shown that these tumors can be divided into 2 main subtypes, luminal A and luminal B [7]. These subtypes represent unique biology and differ in terms of prognosis and response to endocrine therapy and chemotherapy. Luminal A tumors are strongly endocrine responsive and have a good prognosis, while luminal B tumors are less endocrine responsive and are associated with a poorer prognosis; the addition of adjuvant chemotherapy is often considered for luminal B tumors [8]. Several tests, including tumor genomic assays, are now available to help with delineating the tumor subtype and aid in decision-making regarding adjuvant chemotherapy for HR-positive/HER2-negative breast cancers.
Tests for Guiding Adjuvant Chemotherapy Decisions
Ki-67 Assays, Including IHC4 and PEPI
Chronic proliferation is a hallmark of cancer cells [9]. Ki-67, a nuclear nonhistone protein whose expression varies in intensity throughout the cell cycle, has been used as a measurement of tumor cell proliferation [10]. Two large meta-analyses have demonstrated that high Ki-67 expression in breast tumors is independently associated with worse disease-free and overall survival rates [11,12]. Ki-67 expression has also been used to classify HR-positive tumors as luminal A or B. After classifying tumor subtypes based on intrinsic gene expression profiling, Cheang et al determined that a Ki-67 cut point of 13.25% differentiated luminal A and B tumors [13]. However, the ideal cut point for Ki-67 remains unclear, as the sensitivity and specificity in this study was 77% and 78%, respectively. Others have combined Ki-67 with standard ER, PR, and HER2 testing. This IHC4 score, which weighs each of these variables, was validated in postmenopausal patients from the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial who had ER-positive tumors and did not receive chemotherapy [14]. The prognostic information from the IHC4 was similar to that seen with the 21-gene recurrence score (Oncotype DX), which is discussed later in this article. The key challenge with Ki-67 testing currently is the lack of a validated test methodology, and intraobserver variability in interpreting the Ki-67 results [15]. Recent series have suggested that Ki-67 be considered as a continuous marker rather than a set cut point [16]. These issues continue to impact the clinical utility of Ki-67 for decision making for adjuvant chemotherapy.
Ki-67 and the preoperative endocrine prognostic index (PEPI) score have been explored in the neoadjuvant setting to separate postmenopausal women with endocrine-sensitive versus intrinsically resistant disease and identify patients at risk for recurrent disease [17]. The on-treatment levels of Ki-67 in response to endocrine therapy have been shown to be more prognostic than baseline values, and a decrease in Ki-67 as early as 2 weeks after initiation of neoadjuvant endocrine therapy is associated with endocrine-sensitive tumors and improved outcome. The PEPI score was developed through retrospective analysis of the P024 trial [18] to evaluate the relationship between post-neoadjuvant endocrine therapy tumor characteristics and risk for early relapse. This was subsequently validated in an independent data set from the IMPACT trial [19]. Patients with low pathological stage (0 or 1) and a favorable biomarker profile (PEPI score 0) at surgery had the best prognosis in the absence of chemotherapy. On the other hand, higher pathological stage at surgery and a poor biomarker profile with loss of ER positivity or persistently elevated Ki-67 (PEPI score of 3) identified de novo endocrine-resistant tumors which are at higher risk for early relapse [20]. The ongoing Alliance A011106 ALTERNATE trial (ALTernate approaches for clinical stage II or III Estrogen Receptor positive breast cancer NeoAdjuvant TrEatment in postmenopausal women, NCT01953588) is a phase 3 study to prospectively test this hypothesis.
21-Gene Recurrence Score (Oncotype DX Assay)
The 21-gene Oncotype DX assay is conducted on paraffin-embedded tumor tissue and measures the expression of 16 cancer-related genes and 5 reference genes using quantitative polymerase chain reaction. The genes included in this assay are mainly related to proliferation (including Ki-67), invasion, and HER2 or estrogen signaling [21]. Originally, the 21-gene recurrence score assay was analyzed as a prognostic biomarker tool in a prospective-retrospective biomarker substudy of the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14 clinical trial in which patients with node-negative, ER-positive tumors were randomly assigned to receive tamoxifen or placebo without chemotherapy [22]. Using the standard reported values of low risk (< 18), intermediate risk (18–30), or high risk (≥ 31) for recurrence, among the tamoxifen-treated patients, cancers with a high-risk recurrence score had a significantly worse rate of distant recurrence and overall survival [21]. Inferior breast cancer survival with a high recurrence score was also confirmed in other series of endocrine-treated patients with node-negative and node-positive disease [23–25].
The predictive utility of the 21-gene recurrence score for endocrine therapy has also been evaluated. A comparison of the placebo- and tamoxifen-treated patients from the NSABP B-14 trial demonstrated that the 21-gene recurrence score predicted benefit from tamoxifen in cancers with low- or intermediate-risk recurrence scores [26]. However, there was no benefit from the use of tamoxifen over placebo in cancers with high-risk recurrence scores. To date, this intriguing data has not been prospectively confirmed, and thus the 21-gene recurrence score is not used to avoid endocrine therapy.
The 21-gene recurrence score is primarily used by oncologists to aid in decision-making regarding adjuvant chemotherapy in patients with node-negative and node-positive (with up to 3 positive lymph nodes), HR-positive/HER2-negative breast cancers. The predictive utility of the 21-gene recurrence score for adjuvant chemotherapy was initially tested using tumor samples from the NSABP B-20 study. This study initially compared adjuvant tamoxifen alone with tamoxifen plus chemotherapy in patients with node-negative, HR-positive tumors. The prospective-retrospective biomarker analysis showed that the patients with high-risk 21-gene recurrence scores benefited from the addition of chemotherapy, whereas those with low- or intermediate-risk did not have an improved freedom from distant recurrence with chemotherapy [27]. Similarly, an analysis from the prospective phase 3 Southwest Oncology Group (SWOG) 8814 trial comparing tamoxifen to tamoxifen with chemotherapy showed that for node-positive tumors, chemotherapy benefit was only seen in those with high 21-gene recurrence scores [24].
Prospective studies are now starting to report results regarding the predictive role of the 21-gene recurrence score. The TAILORx (Trial Assigning Individualized Options for Treatment) trial includes women with node-negative, HR-positive and HER2-negative tumors measuring 0.6 to 5 cm. All patients were treated with standard of care endocrine therapy for at least 5 years. Chemotherapy was determined based on the 21-gene recurrence score results on the primary tumor. The 21-gene recurrence score cutoffs were changed to low (0–10), intermediate (11–25), and high (≥ 26). Patients with scores of 26 or higher were treated with chemotherapy, and those with intermediate scores were randomly assigned to hemotherapy or no chemotherapy; results from this cohort are still pending. However, excellent breast cancer outcomes with endocrine therapy alone were reported from the 1626 (15.9% of total cohort) prospectively followed patients with low-recurrence score tumors. The 5-year invasive disease-free survival was 93.8%, with overall survival of 98% [28]. Given that 5 years is appropriate follow-up to see any chemotherapy benefit, this data supports the recommendation for no chemotherapy in this cohort of patients with very low 21-gene recurrence scores.
The RxPONDER (Rx for Positive Node, Endocrine Responsive Breast Cancer) trial is evaluating women with 1 to 3 node-positive, HR-positive, HER2-negative tumors. In this trial, patients with 21-gene recurrence scores of 0 to 25 were assigned to adjuvant chemotherapy or none. Those with scores of 26 or higher were assigned to chemotherapy. All patients received standard adjuvant endocrine therapy. This study has completed accrual and results are pending. Of note, TAILORx and RxPONDER did not investigate the potential lack of benefit of endocrine therapy in cancers with high recurrence scores. Furthermore, despite data suggesting that chemotherapy may not even benefit women with 4 or more nodes involved but who have a low recurrence score [24], due to the lack of prospective data in this cohort and the quite high risk for distant recurrence, chemotherapy continues to be the standard of care for these patients.
PAM50 (Breast Cancer Prognostic Gene Signature)
Using microarray and quantitative reverse transcriptase PCR (RT-PCR) on formalin-fixed paraffin-embedded (FFPE) tissues, the Breast Cancer Prognostic Gene Signature (PAM50) assay was initially developed to identify intrinsic breast cancer subtypes, including luminal A, luminal B, HER2-enriched, and basal-like [7,29]. Based on the prediction analysis of microarray (PAM) method, the assay measures the expression levels of 50 genes, provides a risk category (low, intermediate, and high), and generates a numerical risk of recurrence score (ROR). The intrinsic subtype and ROR have been shown to add significant prognostic value to the clinicopathological characteristics of tumors. Clinical validity of PAM50 was evaluated in postmenopausal women with HR-positive, early-stage breast cancer treated in the prospective ATAC and ABCSG-8 (Austrian Breast and Colorectal Cancer Study Group 8) trials [30,31]. In 1017 patients with ER-positive breast cancer treated with anastrozole or tamoxifen in the ATAC trial, ROR added significant prognostic information beyond the clinical treatment score (integrated prognostic information from nodal status, tumor size, histopathologic grade, age, and anastrozole or tamoxifen treatment) in all patients. Also, compared with the 21-gene recurrence score, ROR provided more prognostic information in ER-positive, node-negative disease and better differentiation of intermediate- and higher-risk groups. Fewer patients were categorized as intermediate risk by ROR and more as high risk, which could reduce the uncertainty in the estimate of clinical benefit from chemotherapy [30]. The clinical utility of PAM50 as a prognostic model was also validated in 1478 postmenopausal women with ER-positive early-stage breast cancer enrolled in the ABCSG-8 trial. In this study, ROR assigned 47% of patients with node-negative disease to the low-risk category. In this low-risk group, the 10-year metastasis risk was less than 3.5 %, indicating lack of benefit from additional chemotherapy [31]. A key limitation of the PAM50 is the lack of any prospective studies with this assay.
PAM50 has been designed to be carried out in any qualified pathology laboratory. Moreover, the ROR score provides additional prognostic information about risk of late recurrence, which will be discussed in the next section.
70-Gene Breast Cancer Recurrence Assay (MammaPrint)
MammaPrint is a 70-gene assay that was initially developed using an unsupervised, hierarchical clustering algorithm on whole-genome expression arrays with early-stage breast cancer. Among 295 consecutive patients who had MammaPrint testing, those classified with a good-prognosis tumor signature (n = 115) had an excellent 10-year survival rate (94.5%) compared to those with a poor-prognosis signature (54.5%), and the signature remained prognostic upon multivariate analysis [32]. Subsequently, a pooled analysis comparing outcomes by MammaPrint score in patients with node-negative or 1 to 3 node-positive breast cancers treated as per discretion of their medical team with either adjuvant chemotherapy plus endocrine therapy or endocrine therapy alone reported that only those patients with a high-risk score benefited from chemotherapy [33]. Recently, a prospective phase 3 study (MINDACT [Microarray In Node negative Disease may Avoid ChemoTherapy]) evaluating the utility of MammaPrint for adjuvant chemotherapy decision-making reported results [34]. In this study, 6693 women with early-stage breast cancer were assessed by clinical risk and genomic risk using MammaPrint. Those with low clinical and genomic risk did not receive chemotherapy, while those with high clinical and genomic risk all received chemotherapy. The primary goal of the study was to assess whether forgoing chemotherapy would be associated with a low rate of recurrence in those patients with a low-risk prognostic MammaPrint signature but high clinical risk. A total of 1550 patients (23.2%) were in the discordant group, and the majority of these patients had HR-positive disease (98.1%). Without chemotherapy, the rate of survival without distant metastasis at 5 years in this group was 94.7% (95% confidence interval [CI] 92.5% to 96.2%), which met the primary endpoint. Of note, initially, MammaPrint was only available for fresh tissue analysis, but recent advances in RNA processing now allow for this analysis on FFPE tissue [35].
Summary
Case Continued
The patient undergoes 21-gene recurrence score testing, which shows a low recurrence score of 10, estimating the 10-year risk of distant recurrence to be approximately 7% with 5 years of tamoxifen. Chemo-therapy is not recommended. The patient completes adjuvant whole breast radiation therapy, and then, based on data supporting AIs over tamoxifen in postmenopausal women, she is started on anastrozole [36]. She initially experiences mild side effects from treatment, including fatigue, arthralgia, and vaginal dryness, but her symptoms are able to be managed. As she approaches 5 years of adjuvant endocrine therapy with anastrozole, she is struggling with rotator cuff injury and is anxious about recurrence, but has no evidence of recurrent cancer. Her bone density scan in the beginning of her fourth year of therapy shows a decrease in bone mineral density, with the lowest T score of –1.5 at the left femoral neck, consistent with osteopenia. She has been treated with calcium and vitamin D supplements.
How long should this patient continue treatment with anastrozole?
The risk for recurrence is highest during the first 5 years after diagnosis for all patients with early breast cancer [37]. Although HR-positive breast cancers have a better prognosis than HR-negative disease, the pattern of recurrence is different between the 2 groups, and it is estimated that approximately half of the recurrences among patients with HR-positive early breast cancer occur after the first 5 years from diagnosis. Annualized hazard of recurrence in HR-positive breast cancer has been shown to remain elevated and fairly stable beyond 10 years, even for those with low tumor burden and node-negative disease [38]. Prospective trials showed that for women with HR-positive early breast cancer, 5 years of adjuvant tamoxifen could substantially reduce recurrence rates and improve survival, and this became the standard of care [39]. AIs are considered the standard of care for adjuvant endocrine therapy in most postmenopausal women, as they result in a significantly lower recurrence rate compared with tamoxifen, either as initial adjuvant therapy or sequentially following 2 to 3 years of tamoxifen [40].
However, extending AI therapy from 5 years to 10 years is not clearly beneficial. In the MA.17R trial, although longer AI therapy resulted in significantly better disease-free survival (95% versus 91%, hazard ratio 0.66; P = 0.01), this was primarily due to a lower incidence of contralateral breast cancer in those taking the AI compared with placebo. The distant recurrence risks were similar and low (4.4% versus 5.5%), and there was no overall survival difference [2]. Also, the NSABP B-42 study, which was presented at the 2016 San Antonio Breast Cancer Symposium, did not meet its predefined endpoint for benefit from extending adjuvant AI therapy with letrozole beyond 5 years [3]. Thus, the absolute benefit from extended endocrine therapy has been modest across these studies. Although endocrine therapy is considered relatively safe and well tolerated, side effects can be significant and even associated with morbidity. Ideally, extended endocrine therapy should be offered to the subset of patients who would benefit the most. Several genomic diagnostic assays, including the EndoPredict test, PAM50, and the Breast Cancer Index (BCI) tests, specifically assess the risk for late recurrence in HR-positive cancers.
Tests for Assessing Risk for Late Recurrence
PAM50
Studies suggest that the ROR score also has value in predicting late recurrences. Analysis of data in patients enrolled in the ABCSG-8 trial showed that ROR could identify patients with endocrine-sensitive disease who are at low risk for late relapse and could be spared from unwanted toxicities of extended endocrine therapies. In 1246 ABCSG-8 patients between years 5 and 15, the PAM50 ROR demonstrated an absolute risk of distant recurrence of 2.4% in the low-risk group, as compared with 17.5% in the high-risk group [44]. Also, a combined analysis of patients from both the ATAC and ABCSG-8 trials demonstrated the utility of ROR in identifying this subgroup of patients with low risk for late relapse [45].
EndoPredict
EndoPredict (EP) is another quantitative RT-PCR–based assay which uses FFPE tissues to calculate a risk score based on 8 cancer-related and 3 reference genes. The score is combined with clinicopathological factors including tumor size and nodal status to make a comprehensive risk score (EPclin). EPclin is used to dichotomize patients into EP low- and EP high-risk groups. EP has been validated in 2 cohorts of patients enrolled in separate randomized studies, ABCSG-6 and ABCSG-8. EP provided prognostic information beyond clinicopathological variables to predict distant recurrence in patients with HR-positive, HER2-negative early breast cancer [46]. More important, EP has been shown to predict early (years 0–5) versus late (> 5 years after diagnosis) recurrences and identify a low-risk subset of patients who would not be expected to benefit from further treatment beyond 5 years of endocrine therapy [47]. Recently, EP and EPclin were compared with the 21-gene (Oncotype DX) recurrence score in a patient population from the TransATAC study. Both EP and EPclin provided more prognostic information compared to the 21-gene recurrence score and identified early and late relapse events [48]. EndoPredict is the first multigene expression assay that could be routinely performed in decentral molecular pathological laboratories with a short turnaround time [49].
Breast Cancer Index
The BCI is a RT-PCR–based gene expression assay that consists of 2 gene expression biomarkers: molecular grade index (MGI) and HOXB13/IL17BR (H/I). The BCI was developed as a prognostic test to assess risk for breast cancer recurrence using a cohort of ER-positive patients (n = 588) treated with adjuvant tamoxifen versus observation from the prospective randomized Stockholm trial [50]. In this blinded retrospective study, H/I and MGI were measured and a continuous risk model (BCI) was developed in the tamoxifen-treated group. More than 50% of the patients in this group were classified as having a low risk of recurrence. The rate of distant recurrence or death in this low-risk group at 10 years was less than 3%. The performance of the BCI model was then tested in the untreated arm of the Stockholm trial. In the untreated arm, BCI classified 53%, 27%, and 20% of patients as low, intermediate, and high risk, respectively. The rate of distant metastasis at 10 years in these risk groups was 8.3% (95% CI 4.7% to 14.4%), 22.9% (95% CI 14.5% to 35.2%), and 28.5% (95% CI 17.9% to 43.6%), respectively, and the rate of breast cancer–specific mortality was 5.1% (95% CI 1.3% to 8.7%), 19.8% (95% CI 10.0% to 28.6%), and 28.8% (95% CI 15.3% to 40.2%) [50].
The prognostic and predictive values of the BCI have been validated in other large, randomized studies and in patients with both node-negative and node-positive disease [51,52]. The predictive value of the endocrine-response biomarker, the H/I ratio, has been demonstrated in randomized studies. In the MA.17 trial, a high H/I ratio was associated with increased risk for late recurrence in the absence of letrozole. However, extended endocrine therapy with letrozole in patients with high H/I ratios predicted benefit from therapy and decreased the probability of late disease recurrence [53]. BCI was also compared to IHC4 and the 21-gene recurrence score in the TransATAC study and was the only test to show prognostic significance for both early (0–5 years) and late (5–10 year) recurrence [54].
The impact of the BCI results on physicians’ recommendations for extended endocrine therapy was assessed by a prospective study. This study showed that the test result had a significant effect on both physician treatment recommendation and patient satisfaction. BCI testing resulted in a change in physician recommendations for extended endocrine therapy, with an overall decrease in recommendations for extended endocrine therapy from 74% to 54%. Knowledge of the test result also led to improved patient satisfaction and decreased anxiety [55].
Summary
Due to the risk for late recurrence, extended endocrine therapy is being recommended for many patients with HR-positive breast cancers. Multiple genomic assays are being developed to better understand an individual’s risk for late recurrence and the potential for benefit from extended endocrine therapies. However, none of the assays have been validated in prospective randomized studies. Further validation is needed prior to routine use of these assays.
Case Continued
A BCI test is done and the result shows 4.3% BCI low-risk category in years 5–10; low likelihood of benefit from extended endocrine therapy. After discussing the results of the BCI test in the context of no survival benefit from extending AIs beyond 5 years, both the patient and her oncologist feel comfortable with discontinuing endocrine therapy at the end of 5 years.
Conclusion
Reduction in breast cancer mortality is mainly the result of improved systemic treatments. With advances in breast cancer screening tools in recent years, the rate of cancer detection has increased. This has raised concerns regarding overdiagnosis. To prevent unwanted toxicities associated with overtreatment, better treatment decision tools are needed. Several genomic assays are currently available and widely used to provide prognostic and predictive information and aid in decisions regarding appropriate use of adjuvant chemotherapy in HR-positive/HER2-negative early-stage breast cancer. Ongoing studies are refining the cutoffs for these assays and expanding the applicability to node-positive breast cancers. Furthermore, with several studies now showing benefit from the use of extended endocrine therapy, some of these assays may be able to identify the subset of patients who are at increased risk for late recurrence and who might benefit from extended endocrine therapy. Advances in molecular testing has enabled clinicians to offer more personalized treatments to their patients, improve patient’s compliance, and decrease anxiety and conflict associated with management decisions. Although small numbers of patients with HER2-positive and triple negative breast cancers were also included in some of these studies, use of genomic assays in this subset of patients is very limited and currently not recommended.
Corresponding author: Kari Braun Wisinski, MD, 1111 Highland Avenue, 6033 Wisconsin Institute for Medical Research, Madison, WI 53705-2275, [email protected].
Financial disclosures: This work was supported by the NCI Cancer Center Support Grant P30 CA014520.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
1. Welch HG, Prorok PC, O'Malley AJ, Kramer BS. Breast-cancer tumor size, overdiagnosis, and mammography screening effectiveness. N Engl J Med 2016;375:1438–47.
2. Goss PE, Ingle JN, Pritchard KI, et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N Engl J Med 2016;375:209–19.
3. Mamounas E, Bandos H, Lembersky B. A randomized, double-blinded, placebo-controlled clinical trial of extended adjuvant endocrine therapy with letrozole in postmenopausal women with hormone-receptor-positive breast cancer who have completed previous adjuvant treatment with an aromatase inhibitor. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-05.
4. Tjan-Heijnen VC, Van Hellemond IE, Peer PG, et al: First results from the multicenter phase III DATA study comparing 3 versus 6 years of anastrozole after 2-3 years of tamoxifen in postmenopausal women with hormone receptor-positive early breast cancer. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-03.
5. Blok EJ, Van de Velde CJH, Meershoek-Klein Kranenbarg EM, et al: Optimal duration of extended letrozole treatment after 5 years of adjuvant endocrine therapy. In: Proceedings from the San Antonio Breast Cancer Symposium; December 6–10, 2016; San Antonio, TX. Abstract S1-04.
6. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet 2005;365:1687–717.
7. Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–52.
8. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol 2015;26:1533–46.
9. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
10. Urruticoechea A, Smith IE, Dowsett M. Proliferation marker Ki-67 in early breast cancer. J Clin Oncol 2005;23:7212–20.
11. de Azambuja E, Cardoso F, de Castro G Jr, et al. Ki-67 as prognostic marker in early breast cancer: a meta-analysis of published studies involving 12,155 patients. Br J Cancer 2007;96:1504–13.
12. Petrelli F, Viale G, Cabiddu M, Barni S. Prognostic value of different cut-off levels of Ki-67 in breast cancer: a systematic review and meta-analysis of 64,196 patients. Breast Cancer Res Treat 2015;153:477–91.
13. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009;101:736–50.
14. Cuzick J, Dowsett M, Pineda S, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and com-parison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol 2011;29:4273–8.
15. Pathmanathan N, Balleine RL. Ki67 and proliferation in breast cancer. J Clin Pathol 2013;66:512–6.
16. Denkert C, Budczies J, von Minckwitz G, et al. Strategies for developing Ki67 as a useful biomarker in breast cancer. Breast 2015; 24 Suppl 2:S67–72.
17. Ma CX, Bose R, Ellis MJ. Prognostic and predictive biomarkers of endocrine responsiveness for estrogen receptor positive breast cancer. Adv Exp Med Biol 2016;882:125–54.
18. Eiermann W, Paepke S, Appfelstaedt J, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: a randomized double-blind multicenter study. Ann Oncol 2001;12:1527–32.
19. Smith IE, Dowsett M, Ebbs SR, et al. Neoadjuvant treatment of postmenopausal breast cancer with anastrozole, tamoxifen, or both in combination: the Immediate Preoperative Anas-trozole, Tamoxifen, or Combined with Tamoxifen (IMPACT) multicenter double-blind randomized trial. J Clin Oncol 2005;23:5108–16.
20. Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008;100:1380–8.
21. Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26.
22. Fisher B, Jeong JH, Bryant J, et al. Treatment of lymph-node-negative, oestrogen-receptor-positive breast cancer: long-term findings from National Surgical Adjuvant Breast and Bowel Project randomised clinical trials. Lancet 2004;364:858–68.
23. Habel LA, Shak S, Jacobs MK, et al. A population-based study of tumor gene expression and risk of breast cancer death among lymph node-negative patients. Breast Cancer Res 2006;8:R25.
24. Albain KS, Barlow WE, Shak S, et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol 2010;11:55–65.
25. Dowsett M, Cuzick J, Wale C, et al. Prediction of risk of distant recurrence using the 21-gene recurrence score in node-negative and node-positive postmenopausal patients with breast cancer treated with anastrozole or tamoxifen: a TransATAC study. J Clin Oncol 2010;28:1829–34.
26. Paik S, Shak S, Tang G, et al. Expression of the 21 genes in the recurrence score assay and tamoxifen clinical benefit in the NSABP study B-14 of node negative, estrogen receptor positive breast cancer. J Clin Oncol 2005;23: suppl:510.
27. Paik S, Tang G, Shak S, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol2006;24:3726–34.
28. Sparano JA, Gray RJ, Makower DF, et al. Prospective validation of a 21-gene expression assay in breast cancer. N Engl J Med 2015;373:2005–14.
29. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7.
30. Dowsett M, Sestak I, Lopez-Knowles E, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90.
31. Gnant M, Filipits M, Greil R, et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: using the PAM50 Risk of Recurrence score in 1478 post-menopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann Oncol 2014;25:339–45.
32. van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
33. Knauer M, Mook S, Rutgers EJ, et al. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res Treat 2010;120:655–61.
34. Cardoso F, van't Veer LJ, Bogaerts J, et al. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. N Engl J Med 2016;375:717–29.
35. Sapino A, Roepman P, Linn SC, et al. MammaPrint molecular diagnostics on formalin-fixed, paraffin-embedded tissue. J Mol Diagn 2014;16:190–7.
36. Burstein HJ, Griggs JJ, Prestrud AA, Temin S. American society of clinical oncology clinical practice guideline update on adjuvant endocrine therapy for women with hormone receptor-positive breast cancer. J Oncol Pract 2010;6:243–6.
37. Saphner T, Tormey DC, Gray R. Annual hazard rates of recurrence for breast cancer after primary therapy. J Clin Oncol 1996;14:2738–46.
38. Colleoni M, Sun Z, Price KN, et al. Annual hazard rates of recurrence for breast cancer during 24 years of follow-up: results from the International Breast Cancer Study Group Trials I to V. J Clin Oncol 2016;34:927–35.
39. Davies C, Godwin J, Gray R, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771–84.
40. Dowsett M, Forbes JF, Bradley R, et al. Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015;386:1341–52.
41. Davies C, Pan H, Godwin J, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013;381:805–16.
42. Gray R, Rea D, Handley K, et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6,953 women with early breast cancer. J Clin Oncol 2013;31 (suppl):5.
43. Goss PE, Ingle JN, Martino S, et al. Randomized trial of letrozole following tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: updated findings from NCIC CTG MA.17. J Natl Can-cer Inst 2005;97:1262–71.
44. Filipits M, Nielsen TO, Rudas M, et al. The PAM50 risk-of-recurrence score predicts risk for late distant recurrence after endocrine therapy in postmenopausal women with endocrine-responsive early breast cancer. Clin Cancer Res 2014;20:1298–305.
45. Sestak I, Cuzick J, Dowsett M, et al. Prediction of late distant recurrence after 5 years of endocrine treatment: a combined analysis of patients from the Austrian breast and colorectal cancer study group 8 and arimidex, tamoxifen alone or in combination randomized trials using the PAM50 risk of recurrence score. J Clin Oncol 2015;33:916–22.
46. Filipits M, Rudas M, Jakesz R, et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res 2011;17:6012–20.
47. Dubsky P, Brase JC, Jakesz R, et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br J Cancer 2013;109:2959–64.
48. Buus R, Sestak I, Kronenwett R, et al. Comparison of EndoPredict and EPclin with Oncotype DX Recurrence Score for prediction of risk of distant recurrence after endocrine therapy. J Natl Cancer Inst 2016;108:djw149.
49. Muller BM, Keil E, Lehmann A, et al. The EndoPredict gene-expression assay in clinical practice - performance and impact on clinical decisions. PLoS One 2013;8:e68252.
50. Jerevall PL, Ma XJ, Li H, et al. Prognostic utility of HOXB13:IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br J Cancer 2011;104:1762–9.
51. Sgroi DC, Chapman JA, Badovinac-Crnjevic T, et al. Assessment of the prognostic and predictive utility of the Breast Cancer Index (BCI): an NCIC CTG MA.14 study. Breast Cancer Res 2016;18:1.
52. Zhang Y, Schnabel CA, Schroeder BE, et al. Breast cancer index identifies early-stage estrogen receptor-positive breast cancer patients at risk for early- and late-distant recurrence. Clin Cancer Res 2013;19:4196–205.
53. Sgroi DC, Carney E, Zarrella E, et al. Prediction of late disease recurrence and extended adjuvant letrozole benefit by the HOXB13/IL17BR biomarker. J Natl Cancer Inst 2013;105:1036–42.
54. Sgroi DC, Sestak I, Cuzick J, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol 2013;14:1067–76.
55. Sanft T, Aktas B, Schroeder B, et al. Prospective assessment of the decision-making impact of the Breast Cancer Index in recommending extended adjuvant endocrine therapy for patients with early-stage ER-positive breast cancer. Breast Cancer Res Treat 2015;154:533–41.
56. Nielsen TO, Parker JS, Leung S, et al. A comparison of PAM50 Insrinsic Subtyping with Immunohistochemistry and Clinical Prognostic Factors in Tamoxifen-Treated Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res 2010;16:5222–32.
57. Mamounas EP, Jeong JH, Wickerham DL, et al. Benefit from exemestane as extended adjuvant therapy after 5 years of adjuvant tamoxifen: intention-to-treat analysis of the National Surgical Adjuvant Breast And Bowel Project B-33 trial. J Clin Oncol 2008;26:1965–71.
Eruptive xanthoma: Warning sign of systemic disease
A 30-year-old man presented with multiple asymptomatic skin-colored and yellowish papules and nodules over the elbows, wrists, feet, buttocks, hands, and forearms (Figure 1) that had appeared suddenly 2 weeks before.
His medical history included diabetes mellitus, Hansen disease diagnosed 6 years earlier and treated with a multibacillary regimen for 1 year, and pancreatitis diagnosed 6 months earlier.
Laboratory testing. When a serum sample was centrifuged at 1,500 rpm for 15 minutes, a large lipid layer formed at the top (Figure 2). Other results:
- Triglycerides 5,742 mg/dL (reference range < 160)
- Total cholesterol 293 mg/dL (< 240 mg/dL)
- Fasting blood glucose 473 mg/dL
- Low-density and high-density lipoprotein cholesterol levels normal
- Complete blood cell count within normal limits.
Other testing. The electrocardiogram was normal. Retinal examination showed evidence of lipemia retinalis. Histologic study of a biopsy specimen from one of the lesions showed foamy macrophages in the superficial dermis with lymphocytic infiltrate, confirming the diagnosis of eruptive xanthoma.
An urgent medical referral was sought. The patient was started on statins and fibrates, and the treatment resulted in remarkable improvement.
ERUPTIVE XANTHOMA
Cutaneous manifestations can be warning signs of systemic disease, and physicians should be aware of the dermatologic presentations of common medical conditions.
Eruptive xanthoma is a sign of severe hypertriglyceridemia and is almost always due to an acquired cause such as hyperchylomicronemia or an inherited lipoprotein disorder (eg, lipoprotein lipase deficiency in children, common familial hypertriglyceridemia type 5 in adults).1
Chylomicronemia syndrome is characterized by triglyceride levels greater than 1,000 mg/dL in a patient with eruptive xanthoma, lipemia retinalis, or abdominal pain or pancreatitis. The syndrome has a prevalence of 1.7 out of 10,000 patients.2 Treatment is a strict low-fat diet, with a minimal role for fibrates and nicotinic acid.
Eruptive xanthoma is seen at the time of presentation in 8.5% of patients with severe hypertriglyceridemia (serum level > 1,772 mg/dL).3 In patients with serum triglyceride levels greater than 1,000 mg/dL, the risk of acute pancreatitis is 5%, and at levels above 2,000 mg/dL, the risk is 10% to 20%.4
In our patient, the diagnosis of chylomicronemia was based on the appearance of the skin lesions, his history of pancreatitis and diabetes mellitus, and the results of the initial workup, which revealed severe hypertriglyceridemia and lipemia retinalis. The presence of eruptive xanthomas should raise suspicion for a grossly elevated triglyceride level. Therefore, in view of the increased risk of acute pancreatitis and pancreatic necrosis associated with chylomicronemic syndrome,2 recognizing the cutaneous signs is mandatory.
- Chalès G, Coiffier G, Guggenbuhl P. Miscellaneous non-inflammatory musculoskeletal conditions. Rare thesaurismosis and xanthomatosis. Best Pract Res Clin Rheumatol 2011; 25:683–701.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Sandhu S, Al-Sarraf A, Taraboanta C, Frohlich J, Francis GA. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis 2011; 10:157.
- Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol 2014; 48:195–203.
A 30-year-old man presented with multiple asymptomatic skin-colored and yellowish papules and nodules over the elbows, wrists, feet, buttocks, hands, and forearms (Figure 1) that had appeared suddenly 2 weeks before.
His medical history included diabetes mellitus, Hansen disease diagnosed 6 years earlier and treated with a multibacillary regimen for 1 year, and pancreatitis diagnosed 6 months earlier.
Laboratory testing. When a serum sample was centrifuged at 1,500 rpm for 15 minutes, a large lipid layer formed at the top (Figure 2). Other results:
- Triglycerides 5,742 mg/dL (reference range < 160)
- Total cholesterol 293 mg/dL (< 240 mg/dL)
- Fasting blood glucose 473 mg/dL
- Low-density and high-density lipoprotein cholesterol levels normal
- Complete blood cell count within normal limits.
Other testing. The electrocardiogram was normal. Retinal examination showed evidence of lipemia retinalis. Histologic study of a biopsy specimen from one of the lesions showed foamy macrophages in the superficial dermis with lymphocytic infiltrate, confirming the diagnosis of eruptive xanthoma.
An urgent medical referral was sought. The patient was started on statins and fibrates, and the treatment resulted in remarkable improvement.
ERUPTIVE XANTHOMA
Cutaneous manifestations can be warning signs of systemic disease, and physicians should be aware of the dermatologic presentations of common medical conditions.
Eruptive xanthoma is a sign of severe hypertriglyceridemia and is almost always due to an acquired cause such as hyperchylomicronemia or an inherited lipoprotein disorder (eg, lipoprotein lipase deficiency in children, common familial hypertriglyceridemia type 5 in adults).1
Chylomicronemia syndrome is characterized by triglyceride levels greater than 1,000 mg/dL in a patient with eruptive xanthoma, lipemia retinalis, or abdominal pain or pancreatitis. The syndrome has a prevalence of 1.7 out of 10,000 patients.2 Treatment is a strict low-fat diet, with a minimal role for fibrates and nicotinic acid.
Eruptive xanthoma is seen at the time of presentation in 8.5% of patients with severe hypertriglyceridemia (serum level > 1,772 mg/dL).3 In patients with serum triglyceride levels greater than 1,000 mg/dL, the risk of acute pancreatitis is 5%, and at levels above 2,000 mg/dL, the risk is 10% to 20%.4
In our patient, the diagnosis of chylomicronemia was based on the appearance of the skin lesions, his history of pancreatitis and diabetes mellitus, and the results of the initial workup, which revealed severe hypertriglyceridemia and lipemia retinalis. The presence of eruptive xanthomas should raise suspicion for a grossly elevated triglyceride level. Therefore, in view of the increased risk of acute pancreatitis and pancreatic necrosis associated with chylomicronemic syndrome,2 recognizing the cutaneous signs is mandatory.
A 30-year-old man presented with multiple asymptomatic skin-colored and yellowish papules and nodules over the elbows, wrists, feet, buttocks, hands, and forearms (Figure 1) that had appeared suddenly 2 weeks before.
His medical history included diabetes mellitus, Hansen disease diagnosed 6 years earlier and treated with a multibacillary regimen for 1 year, and pancreatitis diagnosed 6 months earlier.
Laboratory testing. When a serum sample was centrifuged at 1,500 rpm for 15 minutes, a large lipid layer formed at the top (Figure 2). Other results:
- Triglycerides 5,742 mg/dL (reference range < 160)
- Total cholesterol 293 mg/dL (< 240 mg/dL)
- Fasting blood glucose 473 mg/dL
- Low-density and high-density lipoprotein cholesterol levels normal
- Complete blood cell count within normal limits.
Other testing. The electrocardiogram was normal. Retinal examination showed evidence of lipemia retinalis. Histologic study of a biopsy specimen from one of the lesions showed foamy macrophages in the superficial dermis with lymphocytic infiltrate, confirming the diagnosis of eruptive xanthoma.
An urgent medical referral was sought. The patient was started on statins and fibrates, and the treatment resulted in remarkable improvement.
ERUPTIVE XANTHOMA
Cutaneous manifestations can be warning signs of systemic disease, and physicians should be aware of the dermatologic presentations of common medical conditions.
Eruptive xanthoma is a sign of severe hypertriglyceridemia and is almost always due to an acquired cause such as hyperchylomicronemia or an inherited lipoprotein disorder (eg, lipoprotein lipase deficiency in children, common familial hypertriglyceridemia type 5 in adults).1
Chylomicronemia syndrome is characterized by triglyceride levels greater than 1,000 mg/dL in a patient with eruptive xanthoma, lipemia retinalis, or abdominal pain or pancreatitis. The syndrome has a prevalence of 1.7 out of 10,000 patients.2 Treatment is a strict low-fat diet, with a minimal role for fibrates and nicotinic acid.
Eruptive xanthoma is seen at the time of presentation in 8.5% of patients with severe hypertriglyceridemia (serum level > 1,772 mg/dL).3 In patients with serum triglyceride levels greater than 1,000 mg/dL, the risk of acute pancreatitis is 5%, and at levels above 2,000 mg/dL, the risk is 10% to 20%.4
In our patient, the diagnosis of chylomicronemia was based on the appearance of the skin lesions, his history of pancreatitis and diabetes mellitus, and the results of the initial workup, which revealed severe hypertriglyceridemia and lipemia retinalis. The presence of eruptive xanthomas should raise suspicion for a grossly elevated triglyceride level. Therefore, in view of the increased risk of acute pancreatitis and pancreatic necrosis associated with chylomicronemic syndrome,2 recognizing the cutaneous signs is mandatory.
- Chalès G, Coiffier G, Guggenbuhl P. Miscellaneous non-inflammatory musculoskeletal conditions. Rare thesaurismosis and xanthomatosis. Best Pract Res Clin Rheumatol 2011; 25:683–701.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Sandhu S, Al-Sarraf A, Taraboanta C, Frohlich J, Francis GA. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis 2011; 10:157.
- Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol 2014; 48:195–203.
- Chalès G, Coiffier G, Guggenbuhl P. Miscellaneous non-inflammatory musculoskeletal conditions. Rare thesaurismosis and xanthomatosis. Best Pract Res Clin Rheumatol 2011; 25:683–701.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Sandhu S, Al-Sarraf A, Taraboanta C, Frohlich J, Francis GA. Incidence of pancreatitis, secondary causes, and treatment of patients referred to a specialty lipid clinic with severe hypertriglyceridemia: a retrospective cohort study. Lipids Health Dis 2011; 10:157.
- Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol 2014; 48:195–203.
Alpha-1 antitrypsin deficiency: An underrecognized, treatable cause of COPD
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
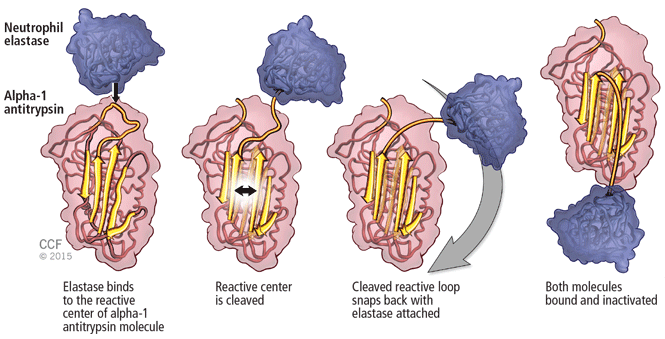
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.

In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina ([email protected]) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina ([email protected]) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
Alpha-1 antitrypsin deficiency is a common but underrecognized genetic condition that increases the risk of chronic obstructive pulmonary disease (COPD) and liver disease. Primary care providers can play a critical role in detecting it and managing patients who have it.
RECOGNIZED CASES ARE THE TIP OF THE ICEBERG
First described in 1963,1 alpha-1 antitrypsin deficiency is estimated to affect 100,000 Americans, fewer than 15,000 of whom have received a clinical diagnosis. As further evidence of its underrecognition,2–7 many patients experience long delays between their first symptoms and the diagnosis. Early studies indicated that the average diagnostic delay was 7.2 years,4 and the latest studies, as recent as 2013, indicate a similar diagnostic delay.7
Furthermore, many patients see multiple healthcare providers before receiving the correct diagnosis. A 1994 survey by this author4 found that 43.7% of patients who had severe deficiency of alpha-1 antitrypsin saw at least three physicians before the correct diagnosis was made.
Why is the disease underrecognized?
Several reasons may account for underrecognition of this disease. Many clinicians—including, unfortunately, many pulmonologists—do not know much about it,7,8 do not adhere to clinical guidelines,9,10 or harbor the misperception that there is no therapy available and, therefore, no compelling reason to make a diagnosis.7
Regarding inadequate knowledge, in a study by Taliercio, Chatburn, and this author,8 internal medicine residents scored only 63% correct on a 10-question quiz on diagnostic features of alpha-1 antitrypsin deficiency. There was no evidence of a training effect—senior residents scored no higher than interns.
Similarly, when Greulich et al7 surveyed German and Italian internists, general practitioners, and pulmonologists, one-fourth to one-half of them (depending on specialty and country) stated that they knew either very little or nothing at all about alpha-1 antitrypsin deficiency. In addition, 7% to 8% agreed with the statement, “There is no treatment available for this disease.”7
Nonadoption of clinical guidelines has been widely recognized in medicine and is evident in the failure to implement various recommended practices,9,10 such as low-stretch ventilation for acute respiratory distress syndrome and prophylaxis against deep vein thrombosis.
Finding the rest of the iceberg
Efforts to enhance compliance with guidelines on testing for alpha-1 antitrypsin deficiency have included using the electronic medical record to prompt physicians to test appropriate candidates.11–13
Jain et al13 examined the effect of installing such a prompting system to remind physicians to test for alpha-1 antitrypsin deficiency in patients with airflow obstruction that does not reverse with a bronchodilator—a recognized indication for testing for this disease according to standards endorsed by the American Thoracic Society and European Respiratory Society.14 At baseline, only 4.7% of appropriate candidates were being tested; after a prompt was installed in the electronic medical record, the rate rose to 15.1%, still a minority of candidates.
Another strategy is to empower respiratory therapists who perform pulmonary function tests to invite patients to be tested if their pulmonary function tests show postbronchodilator airflow obstruction. Rahaghi et al15 showed that using this strategy, 20 (0.63%) of 3,152 patients who were found to have fixed airflow obstruction when they underwent pulmonary function testing were newly diagnosed with severe deficiency of alpha-1 antitrypsin. Other targeted detection studies in patients with COPD estimated the prevalence of alpha-1 antitrypsin deficiency at up to 12%.3
PHYSIOLOGY AND PATHOPHYSIOLOGY OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Alpha-1 antitrypsin is a single-chain, 394-amino acid glycoprotein with three carbohydrate side chains found at asparagine residues along the primary structure.16
A major physiologic function of this molecule is to bind neutrophil elastase, which it does avidly. In a “mousetrap-like” mechanism,16 an active site on the alpha-1 antitrypsin molecule captures the neutrophil elastase and is cleaved, releasing steric energy in the molecule, catapulting the neutrophil elastase to the opposite side of the alpha-1 antitrypsin molecule, and inactivating it (Figure 1).
MM is normal, ZZ is not
Alpha-1 antitrypsin deficiency is inherited as an autosomal-codominant condition.17
The SERPINA1 gene, which codes for alpha-1 antitrypsin, is located on the long arm of the 14th chromosome, and more than 150 alleles of this gene have been identified to date. The normal allele is denoted M, and the allele most commonly associated with severe deficiency is denoted Z. People who are homozygous for the M allele (ie, normal) are called PI*MM (PI stands for “protease inhibitor”), and those who are homozygous for the Z allele are PI*ZZ. More than 90% of patients with severe alpha-1 antitrypsin deficiency are PI*ZZ.18
The Z allele has a single amino acid substitution (glutamic acid-to-lysine at position 342), which results in abnormal folding and formation of polymers of the Z molecule within hepatocytes.19,20 These polymers are recognized on liver biopsy as periodic acid-Schiff diastase-resistant eosinophilic inclusion bodies on histologic staining (Figure 2).
With alpha-1 antitrypsin trapped as Z-molecule polymers in the liver, the amount in the bloodstream falls, and there is a consequent decrease in the amount available in the lung to oppose the proteolytic burden of neutrophil elastase, especially in people who smoke or work in dusty environments.21
Tan et al22 have shown that some of the polymerized Z protein can escape the liver and circulate in the blood and that alveolar macrophages may also produce Z polymers. These Z polymers are chemotactic for neutrophils,23 so that their presence in the lung fuels the inflammatory cascade by recruiting more neutrophils to the lung, thereby increasing the proteolytic burden to the lung and increasing the risk of emphysema. Z monomers that do circulate can bind neutrophil elastase, but their binding avidity to neutrophil elastase is substantially lower than that of M-type alpha-1 antitrypsin.
CLINICAL MANIFESTATIONS
Alpha-1 antitrypsin deficiency of the PI*ZZ type is associated with two major clinical manifestations:
- Emphysema, resulting from the loss of proteolytic protection of the lung by alpha-1 antitrypsin (a toxic loss of function), and
- Liver diseases such as cirrhosis and chronic hepatitis, which result from abnormal accumulation of alpha-1 antitrypsin within hepatocytes (a toxic gain of function), and hepatoma.17
Other clinical manifestations of PI*ZZ alpha-1 antitrypsin deficiency include panniculitis and an association with cytoplasmic antineutrophil cytoplasmic antibody-positive vasculitis.17
Some uncertainty exists regarding the risk associated with the PI*MZ heterozygous state because there has been no systematic longitudinal study of people with this genotype. However, the weight of available experience suggests that PI*MZ individuals who have never smoked are not at increased risk of developing emphysema.24
Findings from a national registry: PI*ZZ COPD resembles ‘usual’ COPD
Distinguishing patients with alpha-1 antitrypsin deficiency from those with “usual” COPD (ie, without alpha-1 antitrypsin deficiency) can be difficult, as shown in data from the National Heart, Lung, and Blood Institute’s Alpha-1 Antitrypsin Deficiency Registry study.18 This multicenter, longitudinal, observational study contains the largest well-characterized cohort with severe deficiency of alpha-1 antitrypsin (PI*ZZ, PI*ZNull, etc), with 1,129 patients.
Pulmonary function test results were consistent with emphysema in most of the patients in the registry. Mean postbronchodilator pulmonary function values (± standard error of the mean) were:
- Forced expiratory volume in 1 second (FEV1) 46.7% of predicted (± 30%)
- Ratio of FEV1 to forced vital capacity 42.9% (± 20.4% )
- Mean diffusing capacity for carbon monoxide 50.3% of predicted (± 22.5%).
Like many patients with usual COPD, 60% of the registry patients demonstrated a component of airway reactivity, with significant reversal of airflow obstruction over three spirometries after receiving a dose of an inhaled bronchodilator (characterized by a 12% and 200-mL postbronchodilator rise in FEV1). Moreover, 78 patients had normal lung function.
Symptoms also resembled those in patients with usual emphysema, chronic bronchitis, or both. On enrollment in the registry, 83.9% of the patients had shortness of breath on exertion, 75.5% had wheezing with upper respiratory infections, 65.3% had wheezing without upper respiratory infection, 67.6% had recent debilitating chest illness, 42.4% had “usual” cough, and 49.6% had annual cough and phlegm episodes.
Imaging findings. Although the classic teaching is that emphysema due to alpha-1 antitrypsin deficiency produces lower-lobe hyperlucency on plain films, relying on this sign would lead to underrecognition, as 36% of PI*ZZ patients have apical-predominant emphysema on chest computed tomography,24 which resembles the usual centriacinar emphysema pattern. Figure 3 shows axial computed tomographic scans through the apices and the bases of the lungs of a patient with alpha-1 antitrypsin deficiency.
In view of these difficulties, guidelines from the American Thoracic Society and European Respiratory Society14 endorse testing for alpha-1 antitrypsin deficiency in all adults who have symptoms and fixed airflow obstruction (Table 1).
CONSEQUENCES OF ALPHA-1 ANTITRYPSIN DEFICIENCY
Two large screening studies2,3,25,26 followed people who were identified at birth as having alpha-1 antitrypsin deficiency to examine the natural course of the disease.
The larger of the two studies27 tested 200,000 Swedish newborns. Follow-up of this cohort to age 35 indicated that 35-year-old never-smoking PI*ZZ individuals have normal lung function and no excess emphysema on computed tomography compared with normal peers matched for age and sex.27 In contrast, the few PI*ZZ ever-smokers demonstrated a lower level of transfer factor and significantly more emphysema on computed tomography than normal (PI*MM) never-smokers.
Faster decline in lung function
Data from the National Heart, Lung, and Blood Institute registry indicate that, on average, people with severe alpha-1 antitrypsin deficiency lose lung function faster than people without the disease.28 Specifically, in never-smokers in the registry, the average rate of FEV1 decline was 67 mL/year, and among ex-smokers, it was 54 mL/year. Both of these values exceed the general age-related rate of FEV1 decline of approximately 20 to 25 mL/year in never-smoking, normal adults. Among current smokers in the registry with severe alpha-1 antitrypsin deficiency, the rate of FEV1 decline was 109 mL/year.
Rates of FEV1 decline over time vary among groups with differing degrees of airflow obstruction. For example, PI*ZZ patients with moderate COPD (stage II of the four-stage Global Initiative for Chronic Obstructive Lung Disease classification system) lose lung function faster than patients with either milder or more severe degrees of airflow obstruction.29
As with COPD in general, exacerbations of COPD in people with severe deficiency of alpha-1 antitrypsin are associated with worsened clinical status. In one series,30 54% of 265 PI*ZZ patients experienced an exacerbation in the first year of follow-up, and 18% experienced at least three. Such exacerbations occurred in December and January in 32% of these individuals, likely due to a viral precipitant.
Increased mortality
Severe deficiency of alpha-1 antitrypsin is associated not only with severe morbidity but also death. In the national registry, the overall rate of death was 18.6% at 5 years of follow-up, or approximately 3% per year.28
A low FEV1 at entry was a bad sign. Patients entering the registry with FEV1 values below 15% of predicted had a 36% mortality rate at 3 years, compared with 2.6% in those whose baseline FEV1 exceeded 50% of predicted.
Underlying causes of death in registry participants included emphysema (accounting for 72% of deaths) and cirrhosis (10%),31 which were the only causes of death more frequent than in age- and sex-matched controls. In a series of never-smokers who had PI*ZZ alpha-1 antitrypsin deficiency,32 death was less frequently attributed to emphysema than in the national registry (46%) and more often attributed to cirrhosis (28%), indicating that never-smokers may more frequently escape the ravages of emphysema but experience a higher rate of developing cirrhosis later in life.33
DIAGNOSING ALPHA-1 ANTITRYPSIN DEFICIENCY
Available blood tests for alpha-1 antitrypsin deficiency include:
The serum alpha-1 antitrypsin level, most often done by nephelometry. Normal serum levels generally range from 100 to 220 mg/dL.
Phenotyping, usually performed by isoelectric focusing, which can identify different band patterns associated with different alleles.
Genotyping involves determining which alpha-1 antitrypsin alleles are present, most often using polymerase chain reaction testing targeting the S and Z alleles and occasionally set up to detect less common alleles such as F and I.17
Gene sequencing is occasionally necessary to achieve an accurate, definitive diagnosis.
Free, confidential testing is available
Clinical testing most often involves checking both a serum level and a phenotype or genotype. Such tests are often available in hospital laboratories and commercial laboratories, with testing also facilitated by the availability of free testing kits from several manufacturers of drugs for alpha-1 antitrypsin deficiency.
The Alpha-1 Foundation (www.alpha1.org)34 also offers a free, home-based confidential testing kit through a research protocol at the Medical University of South Carolina ([email protected]) called the Alpha-1 Coded Testing (ACT) study. Patients can receive a kit and lancet at home, submit the dried blood-spot specimen, and receive in the mail a confidential serum level and genotype.
The availability of such home-based confidential testing allows patients to seek testing without a physician’s order and makes it easier for facilitated allied health providers, such as respiratory therapists, to recommend testing in appropriate clinical circumstances.15
TREATMENT OF ALPHA-1 ANTITRYPSIN DEFICIENCY
The treatment of patients with severe deficiency of alpha-1 antitrypsin and emphysema generally resembles that of patients with usual COPD. Specifically, smoking cessation, bronchodilators, occasionally inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation are indicated as per usual clinical assessment.
Lung volume reduction surgery, which is beneficial in appropriate subsets of COPD patients, is generally less effective in those with severe alpha-1 antitrypsin deficiency,35 specifically because the magnitude of FEV1 increase and the duration of such a rise are lower than in usual COPD patients.
Augmentation therapy
Specific therapy for alpha-1 antitrypsin deficiency currently involves weekly intravenous infusions of purified, pooled human-plasma-derived alpha-1 antitrypsin, so-called augmentation therapy. Four drugs have been approved for use in the United States:
- Prolastin-C (Grifols, Barcelona, Spain)
- Aralast NP (Baxalta, Bonneckborn, IL)
- Zemaira (CSL Behring, King of Prussia, PA)
- Glassia (Baxalta, Bonneckborn, IL, and Kamada, Ness Ziona, Israel).
All of these were approved for use in the United States on the basis of biochemical efficacy. Specifically, infusion of these drugs has been shown to raise serum levels above a protective threshold value (generally considered 57 mg/dL, the value below which the risk of developing emphysema increases beyond normal).
Randomized controlled trials36,37 have addressed the efficacy of intravenous augmentation therapy, and although no single trial has been definitive, the weight of evidence shows that augmentation therapy can slow the progression of emphysema. For example, in a study by Dirksen et al,37 augmentation therapy was associated with a slower progression of emphysema as assessed by the rate of loss of lung density on computed tomography.
On the basis of the available evidence, the American Thoracic Society and European Respiratory Society14 have recommended augmentation therapy in individuals with “established airflow obstruction from alpha-1 antitrypsin deficiency.”14 Their guidelines go on to say that the evidence that augmentation therapy is beneficial “is stronger for individuals with moderate airflow obstruction (eg, FEV1 35%–60% of predicted) than for those with severe airflow obstruction. Augmentation therapy is not currently recommended for individuals without emphysema.”
The guidelines recognize that although augmentation therapy does not satisfy the usual criteria for cost-effectiveness (< $50,000 per quality-adjusted life year) due to its high cost (approximately $100,000 per year if paid for out of pocket),38 it is recommended for appropriate candidates because it is the only available specific therapy for severe deficiency of alpha-1 antitrypsin.
Novel therapies
In addition to current treatment approaches of augmentation therapy, a number of novel treatment strategies are being investigated, several of which hold much promise.
Gene therapy, using adeno-associated virus to transfect the normal human gene into individuals with severe deficiency of alpha-1 antitrypsin, has been undertaken and is currently under study. In addition, a variety of approaches to interdict production of abnormal Z protein from the liver are being examined, as well as inhaled hyaluronic acid to protect the lung.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
- Laurell C, Eriksson A. The electrophoretic alpha-1 globulin pattern of serum in alpha-1 antitrypsin deficiency. Scand J Clin Lab Invest 1963; 15:132–140.
- Aboussouan LS, Stoller JK. Detection of alpha-1 antitrypsin deficiency: a review. Respir Med 2009; 103:335–341.
- Stoller JK, Brantly M. The challenge of detecting alpha-1 antitrypsin deficiency. COPD 2013; 10(suppl 1):26–34.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61:461–467.
- Stoller JK, Sandhaus RA, Turino G, Dickson R, Rodgers K, Strange C. Delay in diagnosis of alpha1-antitrypsin deficiency: a continuing problem. Chest 2005; 128:1989–1994.
- Campos MA, Wanner A, Zhang G, Sandhaus RA. Trends in the diagnosis of symptomatic patients with alpha1-antitrypsin deficiency between 1968 and 2003. Chest 2005; 128:1179–1186.
- Greulich T, Ottaviani S, Bals R, et al. Alpha1-antitrypsin deficiency—diagnostic testing and disease awareness in Germany and Italy. Respir Med 2013; 107:1400–1408.
- Taliercio RM, Chatburn RL, Stoller JK. Knowledge of alpha-1 antitrypsin deficiency among internal medicine house officers and respiratory therapists: results of a survey. Respir Care 2010; 55:322–327.
- Rubenfeld GD, Cooper C, Carter G, Thompson BT, Hudson LD. Barriers to providing lung-protective ventilation to patients with acute lung injury. Crit Care Med 2004; 32:1289–1293.
- Cabana MD, Rand CS, Powe NR, et al. Why don’t physicians follow clinical practice guidelines? A framework for improvement. JAMA 1999; 282:1458–1465.
- Rahaghi F, Ortega I, Rahaghi N, et al. Physician alert suggesting alpha-1 antitrypsin deficiency testing in pulmonary function test (PFT) results. COPD 2009; 6:26–30.
- Campos M, Hagenlocker B, Martinez N, et al. Impact of an electronic medical record clinical reminder to improve detection of COPD and alpha-1 antitrypsin deficiency in the Veterans Administration (VA) system (abstract). Am J Respir Crit Care Med 2011;183:A5356. www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2011.183.1_MeetingAbstracts.A5356. Accessed May 24, 2016.
- Jain A, McCarthy K, Xu M, Stoller JK. Impact of a clinical decision support system in an electronic health record to enhance detection of alpha(1)-antitrypsin deficiency. Chest 2011;140:198–204.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168:818–900.
- Rahaghi FF, Sandhaus RA, Brantly ML, et al. The prevalence of alpha-1 antitrypsin deficiency among patients found to have airflow obstruction. COPD 2012; 9:352–358.
- Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency—a model for conformational diseases. N Engl J Med 2002; 346:45–53.
- Stoller JK, Aboussouan LS. A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2012; 185:246–259.
- McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of Alpha 1-Antitrypsin Deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest 1997; 111:394–403.
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357:605–607.
- Lomas DA, Finch JT, Seyama K, Nukiwa T, Carrell RW. Alpha 1-antitrypsin Siiyama (Ser53-->Phe). Further evidence for intracellular loop-sheet polymerization. J Biol Chem 1993; 268:15333–15335.
- Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med 2000; 162:553–558.
- Tan L, Dickens JA, Demeo DL, et al. Circulating polymers in alpha1-antitrypsin deficiency. Eur Respir J 2014; 43:1501–1504.
- Parmar JS, Mahadeva R, Reed BJ, et al. Polymers of alpha(1)-antitrypsin are chemotactic for human neutrophils: a new paradigm for the pathogenesis of emphysema. Am J Respir Cell Mol Biol 2002; 26:723–730.
- Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in alpha1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med 2014; 189:419–427.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med 2004; 170:1172–1178.
- Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294:1316–1321.
- O’Brien ML, Buist NR, Murphey WH. Neonatal screening for alpha1-antitrypsin deficiency. J Pediatr 1978; 92:1006–1010.
- Piitulainen E, Montero LC, Nystedt-Duzakin M, et al. Lung function and CT densitometry in subjects with alpha-1-antitrypsin deficiency and healthy controls at 35 years of age. COPD 2015; 12:162–167.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158:49–59.
- Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1338–1344.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: clinical manifestations and natural history. Thorax 2004; 59:441–445.
- Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the National Registry for Individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol 2004; 35:1452–1461.
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63:1091–1095.
- Walsh JW, Snider GL, Stoller JK. A review of the Alpha-1 Foundation: its formation, impact, and critical success factors. Respir Care 2006; 51:526–531.
- Rokadia HK, Stoller JK. Surgical and bronchoscopic lung volume reduction treatment for a-1 antitrypsin deficiency. Clin Pulm Med 2015; 22:279–285.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33:1345–1353.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160:1468–1472.
- Gildea TR, Shermock KM, Singer ME, Stoller JK. Cost-effectiveness analysis of augmentation therapy for severe alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 2003; 167:1387–1392.
KEY POINTS
- Only about 15% of people who have alpha-1 antitrypsin disease have received a diagnosis of it.
- The disease is genetic. People who are homozygous for the Z allele of the gene that codes for alpha-1 antitrypsin are at increased risk of lung and liver disease.
- Chronic obstructive pulmonary disease (COPD) due to alpha-1 antitrypsin deficiency is difficult to distinguish from “usual” COPD on a clinical basis, but blood tests are available.
- The basic care of a patient with COPD due to alpha-1 antitrypsin disease is the same as for any patient with COPD, ie, with bronchodilators, inhaled steroids, supplemental oxygen, preventive vaccinations, and pulmonary rehabilitation as indicated. Specific treatment consists of weekly infusions of alpha-1 antitrypsin (augmentation therapy).