User login
Grand Rounds: Man, 61, With Painful Oral Ulcerations
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
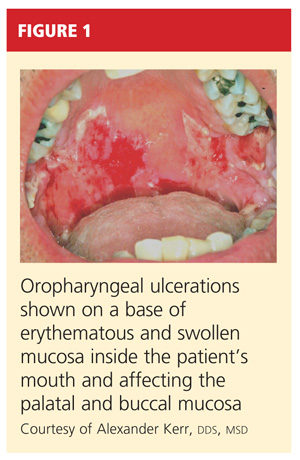
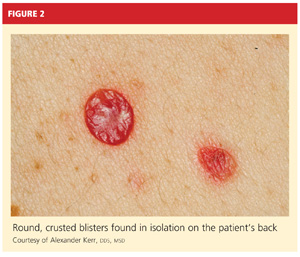
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
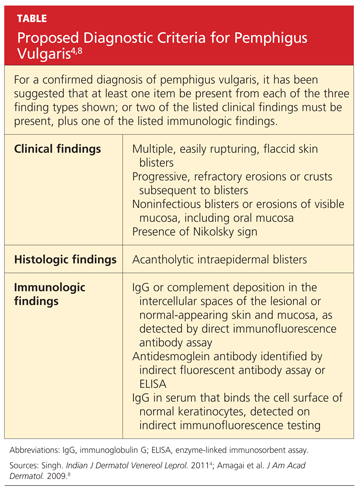
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
A 61-year-old man, who had recently emigrated from the Ukraine, presented to his primary care provider with a chief complaint of painful oral lesions and weight loss. The patient described the gradual onset of a severe sore throat and mouth pain three months earlier. Originally, he attributed his symptoms to an upper respiratory infection but became concerned when his symptoms did not resolve.
He reported that the pain had worsened over time and that he was now barely able to swallow solid food or tolerate acidic beverages due to considerable discomfort. His son, who accompanied him to the appointment, had also noted weight loss.
The patient denied any concomitant symptoms, including fever, cough, night sweats, fatigue, lymphadenopathy, abdominal pain, diarrhea, melena, or concomitant rash. His medical history was remarkable only for stage 1 hypertension, which had been well controlled on hydrochlorothiazide 12.5 mg/d for the previous three years. However, the patient had received only minimal preventive health care while living in the Ukraine. His family history was unknown.
One week earlier, the patient had seen a dentist complaining of mouth pain, and was referred to an oral medicine specialist; this specialist, in turn, referred the patient to a primary care nurse practitioner for lab work to confirm the suspected diagnosis of pemphigus vulgaris.
On physical examination, the patient appeared older than his stated age. He was a thin, mildly ill–appearing man, afebrile and normotensive, with heart rate and respirations within normal limits. However, intraoral examination revealed multiple oropharyngeal ulcerations of varying size on a base of erythematous and swollen mucosa on the inside of the man’s cheek and palatal and buccal mucosa (see Figure 1). On his upper back, two round, crusted blisters were noted in isolation (Figure 2). The remaining findings in the physical examination were unremarkable.
Based on the patient’s physical exam findings and clinical guideline recommendations regarding chronic oral ulcerations of unknown etiology,1,2 the patient was scheduled for a cytologic smear to be performed by oral medicine, followed by a gingival biopsy for a direct immunofluorescence test and routine histopathology.3 Unfortunately, due to extensive involvement and concern for possible mucosal shredding, an oral biopsy was not deemed possible.
However, the oral medicine specialist, because he strongly suspected pemphigus vulgaris, recommended testing for circulating autoantibodies against the antigens desmogleins 1 and/or 3 in the epidermis, which are responsible for cellular adhesion. (A positive test result supports, but does not confirm, a diagnosis of pemphigus vulgaris.4)
Additionally, baseline labs were performed for signs of systemic illness, including infection, anemia, and liver and kidney disease. Frequent monitoring was conducted for steroid-induced symptoms of elevated blood sugars; the primary care provider was responsible for monitoring the patient for weight gain and steroid-induced psychosis. The patient was referred to gastroenterology for a colonoscopy to ensure that his weight loss and anorexia were not the result of gastrointestinal malignancy. However, the patient declined this test.
DISCUSSION
Painful oral lesions can have numerous etiologies of varying severity and complexity, including herpes simplex virus infection, aphthae, lichen planus, erythema multiforme, squamous cell and other oral carcinomas, primary HIV infection, lupus, and pemphigus. Differentiating among these conditions requires a careful medical history and complete physical exam.5
Pemphigus vulgaris (PV) is the most common variant of pemphigus, a group of chronic autoimmune diseases that cause blistering and ulceration of the mucous membranes and the skin.6 From the Greek pemphix (bubble), PV is more common in people of Ashkenazi Jewish or Mediterranean descent,6,7 usually occurs in middle-aged and older persons, and occurs about 1.5 times more commonly in women than men.5,7 Until the introduction of systemic steroids, pemphigus was often a fatal disease. Significant mortality still exists, mainly as a result of infection or adverse reactions to medication therapy.5
In patients with PV, flaccid bullae are formed on the skin in a process called acantholysis, in which epidermal cells lose their ability to adhere to one another. This results in rapidly expanding, thin-walled blisters on the oral mucosa, scalp, face, axillae, and groin. The blisters burst easily, leaving irregularly shaped, painful ulcerations.4 Painful oral mucosal membrane erosions are the first presenting sign of PV and often the only sign for an average of five months before other skin lesions develop.3 These lesions are noninfectious.
To make a definitive diagnosis of PV, clinical lesions must be present, with a confirmation of histologic findings, acantholysis on biopsy, and a confirmation of autoantibodies present in tissue and/or serum.4 (For proposed detailed diagnostic criteria, see table4,8.)
Initial misdiagnoses, which often lead to delayed or incorrect treatment, usually include aphthous stomatitis, gingivostomatitis, erythema multiforme, erosive lichen planus, herpes simplex virus, and/or oral candidiasis.3
Common Differentials
Herpes simplex virus. Affecting between 15% and 45% of the population, herpes simplex virus (HSV) infection, also known as cold sores, is the most common cause of recurrent oral ulcers.9 HSV is transmitted through direct contact with lesions or via viral shedding. Primary infection, which may occur with flu-like symptoms, causes the sudden onset of multiple clustered vesicles on an erythematous base that quickly ulcerate and crust. Recurrent infections tend to be less severe and are accompanied by minimal systemic symptoms.10
Diagnosis is usually made through history and physical exam. However, diagnostic tests, including Tzanck smears, biopsy, polymerase chain reaction (PCR) assay, and/or viral isolation in culture, are sometimes used to confirm a suspected case.10
Oral lichen planus (OLP). This is a common, chronic, mucocutaneous inflammatory disease of unknown etiology that affects skin and mucous membranes of the mouth, including the buccal mucosa, tongue, and/or gums. These lesions are noninfectious and are an immunologically mediated disease. Stress, anxiety, genetic predisposition, NSAID use, antihypertensive medications (eg, captopril, enalapril, propranolol; considered an oral lichenoid drug reaction), and altered cell-mediated immune response have been considered possible causative factors.11,12 Recent reports suggest an association between hepatitis C virus and OLP.13
Affecting about 4% of the general population, and more predominate in perimenopausal women, OLP lesions appear as white, lacey patches; red, swollen tissues; or open sores, most commonly on the inside of the mouth bilaterally. Patients will present with complaints of burning, roughness, or pain in the mouth, dry mouth, sensitivity to hot or spicy foods, and difficulty swallowing if the throat is involved. Diagnosis is based on history and physical examination and often a confirmatory biopsy. Topical high-potency corticosteroids are generally first-line therapy, with systemic medications such as oral prednisone used to treat severe cases.14,15
Oral candidiasis. Up to 80% of healthy individuals carry Candida albicans in their mouths16; this pathogen accounts for about half of all cases of oral candidiasis (oral thrush). Oral infections occur only with an underlying predisposing condition in the host. Oral thrush presents as creamy white lesions on the oral mucosa; a diagnostic feature is that the plaques can be removed to reveal an erythematous base.16,17
In the chronic form of candidiasis, the mucosal surface is bright red and smooth. When the tongue is involved, it may appear dry, fissured, or cracked. Patients may report a dry mouth, burning pain, and difficulty eating. Infection can be confirmed with periodic acid-Schiff staining of a smear to detect candidal hyphae.9
Use of antifungal creams and lozenges, as well as improved oral hygiene, will often lead to resolution of symptoms.9 Management of any associated underlying conditions, such as diabetes, asthma requiring long-term use of steroid inhalers, or infection with HIV/AIDS, is essential.18
Oral aphthae. Recurrent aphthous ulcers (commonly called canker sores; also referred to as recurrent aphthous stomatitis [RAS]) are a common oral condition. Etiology is unknown and most likely multifactorial, with a strong genetic tendency and multiple predisposing factors, including trauma, stress, food allergies, hormones, and smoking.19 Certain chronic illnesses, including celiac disease, inflammatory bowel disease (IBD), HIV, and neutropenia may also predispose patients to RAS or RAS-like syndromes.
Aphthous ulcers are classified as minor or major. Minor aphthae, which account for 90% of RAS cases, present as single or multiple, small, oval or round ulcers with an erythematous halo on the buccal or labial mucosa or tongue.19 The ulcers last 7 to 10 days and heal spontaneously without scarring.
Diagnosis, based on history and clinical presentation, may include evaluation for systemic causes of oral ulcers. Treatment for both minor and major apthae is palliative, with mainstays including topical corticosteroids, mouth rinses, and, in severe cases, thalidomide, although randomized controlled trials have not shown this agent to be of benefit.9
Treatment for Pemphigus Vulgaris
The outcome goal for management of pemphigus is to achieve and maintain remission. This includes the epithelialization of all skin and mucosal lesions, prevention of relapse, minimization of adverse treatment effects, and successful withdrawal of therapeutic medications.20
The response to treatment varies greatly among patients, as the optimal therapeutic regimen for pemphigus is unknown.20 Systemic glucocorticoids are considered the gold standard of treatment and management, but their use has been associated with several adverse effects, including weight gain and elevated blood sugar levels. Recently, the combination of IV immune globulin and biological therapies (eg, rituximab) that target specific molecules in the inflammatory process have been demonstrated as effective in cases of refractory pemphigus.21,22
PATIENT MANAGEMENT AND OUTCOME
Several referrals were made, including dermatology, for its familiarity with autoimmune diseases of the skin. There, the patient was fully examined and found to have a small truncal lesion compatible with PV. He was referred to an otolaryngologist for a nasal endoscopy to determine the extent of the lesions. They were found to extend far beyond his oral cavity into his esophagus.
Based on a positive enzyme-linked immunosorbent assay (ELISA) for PV antibodies, a cytologic smear with acantholytic cells, and a classic clinical presentation of PV, the patient was started on prednisone 80 mg/d with azathioprine 50 mg/d for the first 14 days.23,24 He responded quickly to these oral medications and underwent a confirmatory oral biopsy within a few weeks.
After several months, the patient was slowly titrated down to lower maintenance doses of prednisone and azathioprine. Now in remission, he continues to receive collaborative management from oral medicine, dermatology, and a nurse practitioner–managed primary care practice. Health care maintenance has included appropriate vaccination and discussion regarding prostate cancer screening, per 2010 guidelines from the US Preventive Services Task Force.25
CONCLUSION
Since the differential diagnosis for pemphigus vulgaris is extensive and the diagnostic criteria are exacting, many affected patients are undiagnosed or misdiagnosed, with a resulting delay in effective treatment. It is important for the primary care clinician to undertake a frequent review of common oral infections, particularly those with similar presentations.
The authors extend their thanks to Alexander Kerr, DDS, MSD, Clinical Associate Professor, Department of Oral and Maxillary Pathology, Radiology and Medicine, New York University College of Dentistry, for the images included in this article and for Dr. Kerr’s clinical expertise and partnership.
REFERENCES
1. Sciubba JJ. Oral mucosal diseases in the office setting. Part II. Oral lichen planus, pemphigus vulgaris, and mucosal pemphigoid. Gen Dent. 2007;55(5):464-476.
2. Muñoz-Corcuera M, Esparza-Gómez G, González-Moles MA, Bascones-Martínez A. Oral ulcers: clinical aspects. A tool for dermatologists. Part II. Chronic ulcers. Clin Exp Dermatol. 2009; 34(4):456-461.
3. Dagistan S, Goregen M, Miloglu O, Cakur B. Oral pemphigus vulgaris: a case report with review of the literature. J Oral Sci. 2008;50(3):359-362.
4. Singh S. Evidence-based treatments for pemphigus vulgaris, pemphigus foliaceus and bullous pemphigoid: a systematic review. Indian J Dermatol Venereol Leprol. 2011;77(4):456-469.
5. Ohta M, Osawa S, Endo H, et al. Pemphigus vulgaris confined to the gingiva: a case report. Int J Dent. 2011;2011:207153. Epub 2011 May 11.
6. Mignona MD, Fortuna G, Leuci S. Oral pemphigus. Minerva Stomatol. 2009;58(10):501-518.
7. Mimouni D, Bar H, Gdalevich M, et al. Pemphigus: analysis of epidemiological factors in 155 patients. J Eur Acad Dermatol Venereol. 2008; 22(10):1232-1235.
8. Amagai M, Ikeda S, Shimizu H, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60(4):595-603.
9. Gonsalves WC, Chi AC, Neville BW. Common oral lesions: Part I. Superficial mucosal lesions. Am Fam Physician. 2007;75(4):501-507.
10. Fatahzadeh M, Schwartz R. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57(5):737-763.
11. Sugerman PB, Savage NW. Oral lichen planus: causes, diagnosis and management. Aust Dent J. 2002;47(4):290-297.
12. Kaomongkolgit R. Oral lichenoid drug reaction associated with antihypertensive and hypoglycemic drugs. J Drugs Dermatol. 2010;9(1):73-75.
13. Petti S, Rabiei M, De Luca M, Scully C. The magnitude of the association between hepatitis C virus infection and oral lichen planus: meta-analysis and case control study. Odontology. 2011;99(2):168-178.
14. Usatine RP, Tinitigan M. Diagnosis and treatment of lichen planus. Am Fam Physician. 2011;84(1): 53-60.
15. Thongprasom K, Carrozzo M, Furness S, Lodi G. Interventions for treating oral lichen planus. Cochrane Database Syst Rev. 2011 Jul 6; (7):CD001168.
16. Giannini PJ, Shetty KV. Diagnosis and management of oral candidiasis. Otolaryngol Clin North Am. 2011;44(1):231-240, vii.
17. Lynch DP. Oral candidiasis. History, classification, and clinical presentation. Oral Surg Oral Med Oral Pathol. 1994;78(2):189-193.
18. Williams D, Lewis M. Pathogenesis and treatment of oral candidosis. J Oral Microbiol. 2011 Jan 28;3. doi: 10.3402/jom.v3i0.5771.
19. Scully C, Challacombe SJ. Pemphigus vulgaris: update on etiopathogenesis, oral manifestations, and management. Crit Rev Oral Biol Med. 2002;13(5):397-408.
20. Martin LK, Werth V, Villanueva E, Murrell DF. A systematic review of randomized controlled trials for pemphigus vulgaris and pemphigus foliaceus. J Am Acad Dermatol. 2011;64(5):903-908.
21. Joly P, Mouquet H, Roujeau JC, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545-552.
22. Diaz LA. Rituximab and pemphigus: a therapeutic advance. N Engl J Med. 2007;357(6):605-607.
23. Anstey AV, Wakelin S, Reynolds NJ. Guidelines for prescribing azathioprine in dermatology. Br J Dermatol. 2004;151(6):1123-1132.
24. Chams-Davatchi C, Daneshpazhooh M. Prednisolone dosage in pemphigus vulgaris. J Am Acad Dermatol. 2005;53(3):547.
25. Agency for Healthcare Research and Quality. Guide to Clinical Preventive Services, 2010-2011: recommendations of the US Preventive Services Task Force. AHRQ Publication No. 10-05145, September 2010. www.ahrq.gov/clinic/pocketgd1011/pocketgd1011.pdf. Accessed January 23, 2012.
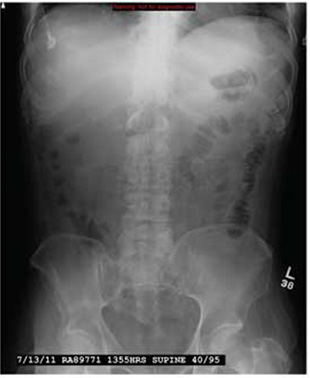
Woman with Abdominal Pain Following Severe Car Crash
ANSWER
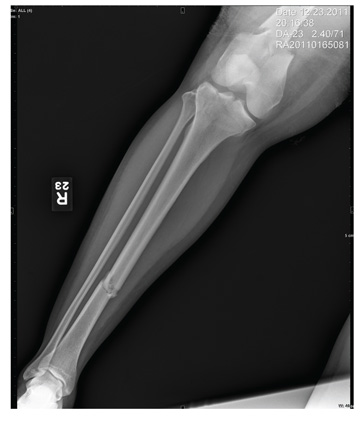
The radiograph shows a comminuted fracture at the midshaft of the tibia. In addition, there is a comminuted fracture of the proximal tibial metaphysis extending to the tibia plateau. Also noted is a comminuted fracture of the distal femur metaphysis extending to the intercondylar notch
ANSWER
The radiograph shows a comminuted fracture at the midshaft of the tibia. In addition, there is a comminuted fracture of the proximal tibial metaphysis extending to the tibia plateau. Also noted is a comminuted fracture of the distal femur metaphysis extending to the intercondylar notch
ANSWER
The radiograph shows a comminuted fracture at the midshaft of the tibia. In addition, there is a comminuted fracture of the proximal tibial metaphysis extending to the tibia plateau. Also noted is a comminuted fracture of the distal femur metaphysis extending to the intercondylar notch
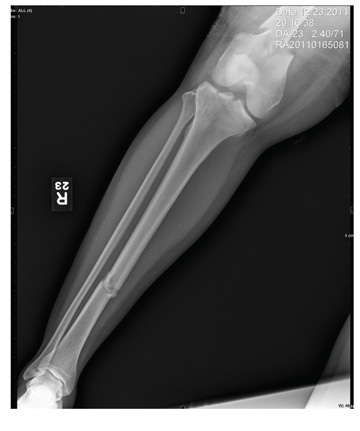
A 43-year-old woman is airlifted to your facility from an outlying area following a severe motor vehicle collision. Details are unclear, but there were known fatalities at the scene. Her primary complaints are abdominal pain and noted deformities of the lower extremities, according to the transporting medical personnel. On arrival, she is noted to be semi-arousable and is moving distal portions of all four extremities. Her heart rate is 150 beats/min, with a blood pressure of 80/40 mm Hg. She responds to initial fluid and volume resuscitation. She has no pertinent medical history. Her response to the fluid resuscitation is sufficient to stabilize her for transport to the CT scanner for additional imaging. Prior to the transfer, though, a portable radiograph of her right tibia is obtained. What is your impression?
Grand Rounds: Woman, 29, With Persistent Migraine
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
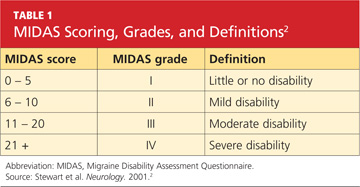
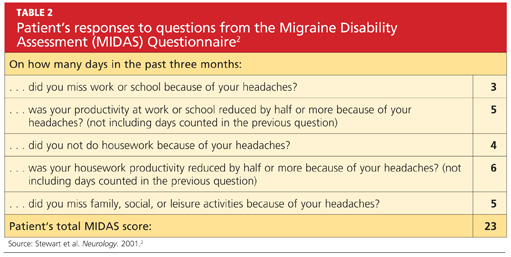
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
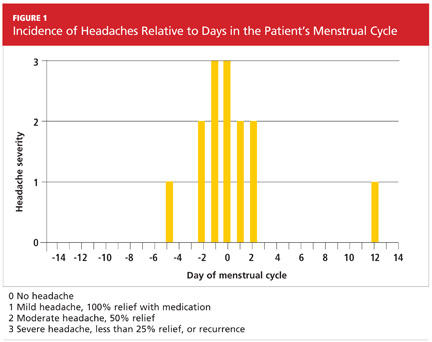
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
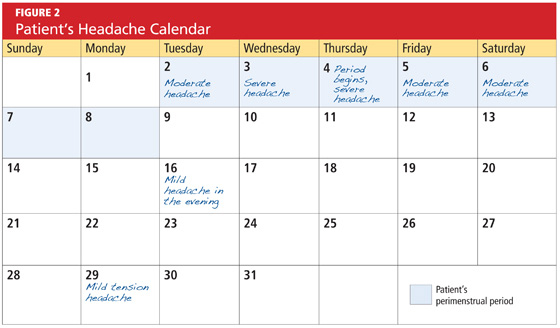
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
A 29-year-old woman with a history of frequent migraines presented to her primary care provider for a refill of medication. For the past two years she had been taking rizatriptan 10 mg, but with little relief. She stated that she had continued to experience discrete migraines several days per month, often clustered around menses. The severity of the headaches had negatively affected her work attendance, productivity, and social interactions. She wondered if she should be taking a different kind of medication.
The patient had been diagnosed with migraines at age 12, just prior to menarche. She described her headache as a unilateral, sharp throbbing pain associated with increased sensitivity to light and sound as well as nausea. She denied any history of head trauma. She had no allergies, and the only other medications she was taking at the time were an oral contraceptive (ethinyl estradiol/norgestimate 0.035 mg/0.18 mg with an oral triphasic 21/7 treatment cycle) and fluoxetine 20 mg for depression.
The patient worked daytime hours as a sales representative. She considered herself active, exercised regularly, ate a balanced diet, and slept well. She consumed no more than two to four alcoholic drinks per month and denied the use of herbals, dietary supplements, tobacco, or illegal drugs.
The patient stated that her mother had frequent headaches but had never sought a medical explanation or treatment. She was unaware of any other family history of headaches, and there was no family history of cardiovascular disease. Her sister had been diagnosed with a prolactinoma at age 25. At age 26, the patient had undergone a pituitary protocol MRI of the head with and without contrast, with negative results.
On examination, the patient was alert and oriented with normal vital signs. Her pupils were equal and reactive to light, and no papilledema was evident on fundoscopic examination. The cranial nerves were grossly intact and no other neurologic deficits were appreciated. No carotid bruits were present on cardiovascular exam.
Based on the patient’s history and physical exam, she met the International Classification of Headache Disorders (ICHD-II)1 diagnostic criteria for migraine without aura (1.1). When asked to recall the onset and frequency of attacks she had had in the previous four weeks, she noted that they regularly occurred during her menstrual cycle.
She was subsequently asked to begin a diary to record her headache characteristics, severity, and duration, with days of menstruation noted. The Migraine Disability Assessment (MIDAS) questionnaire2 (see Tables 1 and 22) was performed to measure the migraine attacks’ impact on the patient’s life; her score indicated that the headaches were causing her severe disability.
The patient’s abortive migraine medication was changed from rizatriptan 10 mg to the combination sumatriptan/naproxen sodium 85 mg/500 mg. She was instructed to take the initial dose as soon as she noticed signs of an impending migraine and to repeat the dose in two hours if symptoms persisted. The possibility of starting a preventive medication was discussed, but the patient wanted to evaluate her response to the combination triptan/NSAID before considering migraine prophylaxis.
Three months later, the patient returned for follow-up, including a review of her headache diary. She stated that the frequency and intensity of attacks had not decreased; acute treatment with sumatriptan/naproxen sodium made her headaches more bearable but did not ameliorate symptoms. The patient had recorded a detailed account of each migraine which, based on the ICHD-II criteria,1 demonstrated a pattern of headache occurrences consistent with menstrually related migraine. She reported a total of 18 headaches in the previous three months, 12 of which had occurred within the five-day perimenstrual period (see Figure 1).
Based on this information and the fact that the patient’s headaches were resistant to previous treatments, it was decided to alter the approach to her migraine management once more. In an effort to limit estrogen fluctuations during her menstrual cycle, her oral contraceptive was changed from ethinyl estradiol/norgestimate to a 12-week placebo-free monophasic regimen of ethinyl estradiol/levonorgestrel 20 mg/90 mcg. For intermittent prophylaxis, she was instructed to take frovatriptan 2.5 mg twice daily, beginning two days prior to the start of menses and continuing through the last day of her cycle. For acute treatment of breakthrough migraines, she was prescribed sumatriptan 20-mg nasal spray to take at the first sign of migraine symptoms and instructed to repeat the dose if the pain persisted or returned.
The patient continued to track her headaches in the diary and was seen in the office after three months of following the revised menstrual migraine management plan. She reported fewer migraines associated with her menstrual cycle and noted that they were less severe and shorter in duration. When she repeated the MIDAS test, her score was reduced from 23 to 10. In the subsequent nine months she has reported a consistent decrease in migraine prevalence and now rarely needs the abortive therapy.
DISCUSSION
Migraine, though commonly encountered in clinical practice, is a complex disorder. For women, migraine headaches have been recognized by the World Health Organization as the 12th leading cause of “life lived with a disabling condition.”3 Pure menstrual migraine and menstrually related migraine will be the focus of discussion here.
Etiology
Menstrually related migraine (comparable to pure menstrual migraine, although the latter is distinguished by occurring only during the perimenstrual period1) is recognized as a distinct type of migraine associated with perimenstrual hormone fluctuations.4 Of women who experience migraine, 42% to 61% can associate their attacks with the perimenstrual period5; this is defined as two days before to three days after the start of menstruation.
It has also been determined that women are more likely to have migraine attacks during the late luteal and early follicular phases (when there is a natural drop in estrogen levels) than in other phases (when estrogen levels are higher).6 Despite clinical evidence to support this estrogen withdrawal theory, the pathophysiology is not completely understood. It is possible that affected women are more sensitive than other women to the decrease in estrogen levels that occurs with menstruation.7
History and Physical Findings of Menstrual Migraines
Almost every woman with perimenstrual migraines reports an absence of aura.7 In the evaluation of headache, the same criteria for migraine without aura pertain to the classifications of pure menstrual migraine (PMM) or menstrually related migraine (MRM).1 Correlation of migraine attacks to the onset of menses is the key finding in the patient history to differentiate menstrual migraine from migraine without aura in women.8 Furthermore, perimenstrual migraines are often of longer duration and more difficult to treat than migraines not associated with hormone fluctuations.9
In order to distinguish between PMM and MRM, it is important to understand that pure menstrual migraine attacks take place exclusively in the five-day perimenstrual window and at no other times of the cycle. The criteria for MRM allow for attacks at other times of the cycle.1
In addition to causing physical pain, menstrual migraines can impact work performance, household activities, and personal relationships. The MIDAS questionnaire is a disability assessment tool that can reveal to the practitioner how migraines have affected the patient’s life over the previous three months.10 This is a useful method to identify patients with disabling migraines, determine their need for treatment, and monitor treatment efficacy.
Diagnosis
Menstrual migraine is a clinical diagnosis made by findings from the patient’s history. The International Headache Society has established specific diagnostic criteria in the ICHD-II for both PMM and MRM.1 An accurate and detailed migraine history is invaluable for the diagnosis of menstrual migraine. Although a formal questionnaire can serve as a good screening tool, it relies on the patient’s ability to recall specific times and dates with accuracy.11 Recall bias can be misleading in any attempt to confirm a diagnosis. The patient’s conscientious use of a daily headache diary or calendar (see Figure 2, for example) can lead to a precise record of the characteristics and timing of migraines, overcoming these obstacles.
Brain imaging is necessary if the patient’s symptoms suggest a critical etiology that requires immediate diagnosis and management. Red flags include sudden onset of a severe headache, a headache characterized as “the worst headache of the patient’s life,” a change in headache pattern, altered mental status, an abnormal neurologic examination, or fever with neck stiffness.12
Treatment Options for Menstrual Migraine
There is no FDA-approved treatment specific for menstrual migraines; however, medications used for management of nonmenstrual migraines are also those most commonly prescribed for women with menstrual migraine headaches.13 Because these headaches are frequently more severe and of longer duration than nonmenstrual migraine headaches, a combination of intermittent preventive therapy, hormone manipulation, and acute treatment strategies is often necessary.4
Acute therapy is aimed to treat migraine pain quickly and effectively with minimal adverse effects or need for additional medication. Triptans have been the mainstay of menstrual migraine treatment and have been proven effective for both acute attacks and prevention.4 Sumatriptan has a rapid onset of action and may be given orally as a 50- or 100-mg tablet, as a 6-mg subcutaneous injection, or as a 20-mg nasal spray.14
Abortive therapies are most effective when taken at the first sign of an attack. Patients can repeat the dose in two hours if the headache persists or recurs, to a maximum of two doses in 24 hours.15 Rizatriptan is another triptan used for acute treatment of menstrual migraine headaches. Its initial 10-mg dose can be repeated every two hours, to a maximum of 30 mg per 24 hours. NSAIDs, such as naproxen sodium, have also been recommended in acute migraine attacks. They seem to work synergistically with triptans, inhibiting prostaglandin synthesis and blocking neurogenic inflammation.15
Clinical study results have demonstrated superior pain relief and decreased migraine recurrence when a triptan and NSAID are used in combination, compared with use of either medication alone.4 A single-tablet formulation of sumatriptan 85 mg and naproxen sodium 500 mg may be considered for initial therapy in hard-to-treat patients.14
Preventive therapy should be considered when responsiveness to acute treatment is inadequate.4 Nonhormonal intermittent prophylactic treatment is recommended two days prior to the beginning of menses, continuing for five days.16 Longer-acting triptans, such as frovatriptan 2.5 mg and naratriptan 1.0 mg, dosed twice daily, have been demonstrated as effective in clinical trials when used during the perimenstrual period.17,18
The advantage of short-term therapy over daily prophylaxis is the potential to avoid adverse effects seen with continuous exposure to the drug.3 However, successful therapy relies on consistency in menstruation, and therefore may not be ideal for women with irregular cycles or those with coexisting nonmenstrual migraines.16 Estrogen-based therapy is an option for these women and for those who have failed nonhormonal methods.19
The goal of hormone prophylaxis is to prevent or reduce the physiologic decline in estradiol that occurs in the late luteal phase.4 Clinical studies have been conducted using various hormonal strategies to maintain steady estradiol levels, all of which decreased migraine prevalence.19 Estrogen fluctuations can be minimized by eliminating the placebo week in traditional estrogen/progestin oral contraceptives to achieve an extended-cycle regimen, resembling that of the 12-week ethinyl estradiol/levonorgestrel formulation.19
Continuous use of combined oral contraceptives is also an option for relief of menstrual migraine. When cyclic or extended-cycle regimens allow for menses, supplemental estrogen (10- to 20-mg ethinyl estradiol) is recommended during the hormone-free week.14
CONCLUSION
Proper diagnosis of menstrual migraines, using screening tools and the MIDAS questionnaire, can help practitioners provide the most effective migraine management for their patients. The most important step toward a good prognosis is acknowledging menstrual migraine as a unique headache disorder and formulating a precise diagnosis in order to identify individually tailored treatment options. With proper identification and integrated acute and prophylactic treatment, women with menstrual migraines are able to lead a healthier, more satisfying life.
REFERENCES
1. International Headache Society. The International Classification of Headache Disorders. 2nd ed. Cephalalgia. 2004;24(suppl 1):1-160.
2. Stewart WF, Lipton RB, Dowson AJ, Sawyer J. Development and testing of the Migraine Disability Assessment (MIDAS) Questionnaire to assess headache-related disability. Neurology. 2001;56(6 suppl 1):S20-S28.
3. MacGregor EA. Perimenstrual headaches: unmet needs. Curr Pain Headache Rep. 2008;12(6):468-474.
4. Mannix LK. Menstrual-related pain conditions: dysmenorrhea and migraine. J Womens Health (Larchmt). 2008;17(5):879-891.
5. Martin VT. New theories in the pathogenesis of menstrual migraine. Curr Pain Headache Rep. 2008;12(6):453-462.
6. MacGregor EA. Migraine headache in perimenopausal and menopausal women. Curr Pain Headache Rep. 2009;13(5):399-403.
7. Martin VT, Wernke S, Mandell K, et al. Symptoms of premenstrual syndrome and their association with migraine headache. Headache. 2006; 46(1):125-137.
8. Martin VT, Behbehani M. Ovarian hormones and migraine headache: understanding mechanisms and pathogenesis—part 2. Headache. 2006;46(3):365-386.
9. Granella F, Sances G, Allais G, et al. Characteristics of menstrual and nonmenstrual attacks in women with menstrually related migraine referred to headache centres. Cephalalgia. 2004;24(9):707-716.
10. Hutchinson SL, Silberstein SD. Menstrual migraine: case studies of women with estrogen-related headaches. Headache. 2008;48 suppl 3:S131-S141.
11. Tepper SJ, Zatochill M, Szeto M, et al. Development of a simple menstrual migraine screening tool for obstetric and gynecology clinics: the Menstrual Migraine Assessment Tool. Headache. 2008; 48(10):1419-1425.
12. Marcus DA. Focus on primary care diagnosis and management of headache in women. Obstet Gynecol Surv. 1999;54(6):395-402.
13. Tepper SJ. Tailoring management strategies for the patient with menstrual migraine: focus on prevention and treatment. Headache. 2006;46(suppl 2):S61-S68.
14. Lay CL, Payne R. Recognition and treatment of menstrual migraine. Neurologist. 2007;13(4):197-204.
15. Henry KA, Cohen CI. Perimenstrual headache: treatment options. Curr Pain Headache Rep. 2009;13(1):82-88.
16. Calhoun AH. Estrogen-associated migraine. www.uptodate.com/contents/estrogen-associated-migraine. Accessed May 4, 2011.
17. Silberstein SD, Elkind AH, Schreiber C, et al. A randomized trial of frovatriptan for the intermittent prevention of menstrual migraine. Neurology. 2004;63:261-269.
18. Mannix LK, Savani N, Landy S, et al. Efficacy and tolerability of naratriptan for short-term prevention of menstrually related migraine: data from two randomized, double-blind, placebo-controlled studies. Headache. 2007;47(7):1037-1049.
19. Calhoun AH, Hutchinson S. Hormonal therapies for menstrual migraine. Curr Pain Headache Rep. 2009;13(5):381-385.
Man Run Over By Vehicle
ANSWER
The radiograph shows some deformity within the mid-to-distal diaphysis of both the radius and ulna; however, no acute fracture is seen. This is most likely related to remote trauma.
Of note, closer to the elbow, there are some small avulsion fractures near the area of the lateral epicondyle. The patient was treated with a posterior splint and referred to orthopedics.
ANSWER
The radiograph shows some deformity within the mid-to-distal diaphysis of both the radius and ulna; however, no acute fracture is seen. This is most likely related to remote trauma.
Of note, closer to the elbow, there are some small avulsion fractures near the area of the lateral epicondyle. The patient was treated with a posterior splint and referred to orthopedics.
ANSWER
The radiograph shows some deformity within the mid-to-distal diaphysis of both the radius and ulna; however, no acute fracture is seen. This is most likely related to remote trauma.
Of note, closer to the elbow, there are some small avulsion fractures near the area of the lateral epicondyle. The patient was treated with a posterior splint and referred to orthopedics.
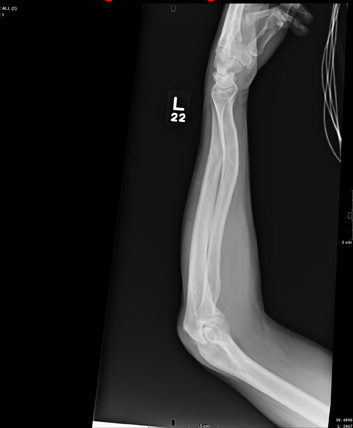
After accidentally being run over by a vehicle, a 54-year-old man presents to the emergency department for evaluation of pain in his elbow and left arm. He was leaning down behind the vehicle and was not seen when the driver backed up. The patient states that one of the tires went over his left shoulder and arm. Primary complaint is pain and decreased range of motion. He denies any significant medical history, except for medication-controlled hypertension and gallbladder surgery. His vital signs are stable. Exammination of the left arm demonstrates some abrasions and contusions over the shoulder and forearm, as well as some swelling over the elbow. The patient has good color, distal pulses, and sensation. There is localized tenderness over the elbow and midforearm. Flexion of the elbow is somewhat limited secondary to pain. Radiograph of the forearm is obtained and shown. What is your impression?
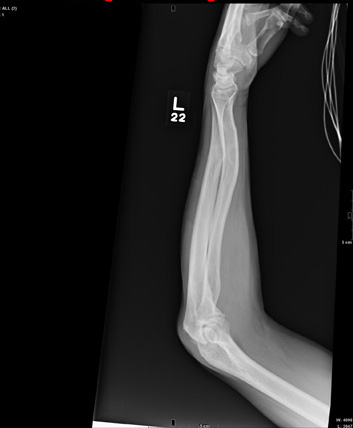
Man with Debilitating Back Pain
ANSWER
The radiograph demonstrates generalized degenerative changes. Of note are fairly significant destructive changes in the endplate at the L3-4 level. Such changes are generally consistent with osteomyelitis and diskitis.
On further questioning, the patient reported that when he was admitted previously, he was told that he had an “infection in his back” and was treated with IV antibiotics. This patient was again admitted for additional workup and treatment.
ANSWER
The radiograph demonstrates generalized degenerative changes. Of note are fairly significant destructive changes in the endplate at the L3-4 level. Such changes are generally consistent with osteomyelitis and diskitis.
On further questioning, the patient reported that when he was admitted previously, he was told that he had an “infection in his back” and was treated with IV antibiotics. This patient was again admitted for additional workup and treatment.
ANSWER
The radiograph demonstrates generalized degenerative changes. Of note are fairly significant destructive changes in the endplate at the L3-4 level. Such changes are generally consistent with osteomyelitis and diskitis.
On further questioning, the patient reported that when he was admitted previously, he was told that he had an “infection in his back” and was treated with IV antibiotics. This patient was again admitted for additional workup and treatment.
A 54-year-old man presents with a complaint of a two-week history of severe low back pain. He denies any injury or trauma. The pain is so severe that it limits his ability to walk. He states he had similar episodes earlier this year, some of which required him to be admitted to the hospital. His medical history is significant for hypertension, diabetes, and coronary artery disease. He admits to recently having subjective fever and chills, as well as some nausea. Physical exam shows a deconditioned male who is uncomfortable but in no obvious distress. He is afebrile, with a blood pressure of 92/57 mm Hg, a heart rate of 97 beats/min, and a respiratory rate of 20 breaths/min. He has mild tenderness to his lumbosacral area; no deformity, step-off, or crepitus is appreciated. He does have decreased range of motion in his lower extremities, although this may be a consequence of his back pain. While trying to pull up his previous medical records, you order some basic labwork and lumbar spine radiographs. Lateral lumbar spine radiograph is shown; what is your impression?
A 54-year-old woman with pancytopenia
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
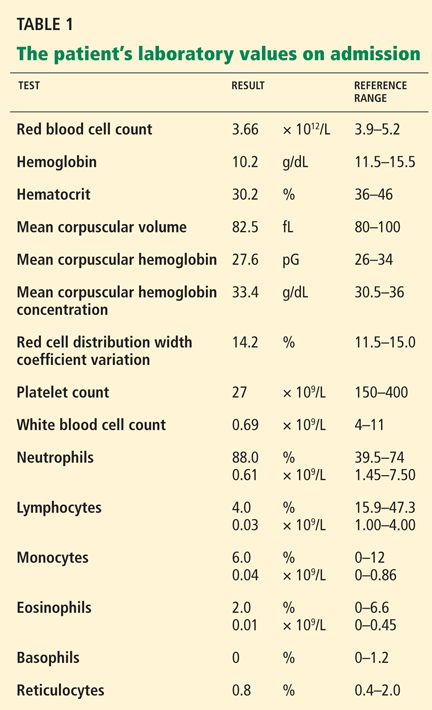
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
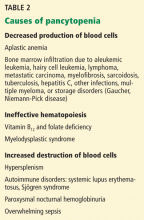
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other

Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
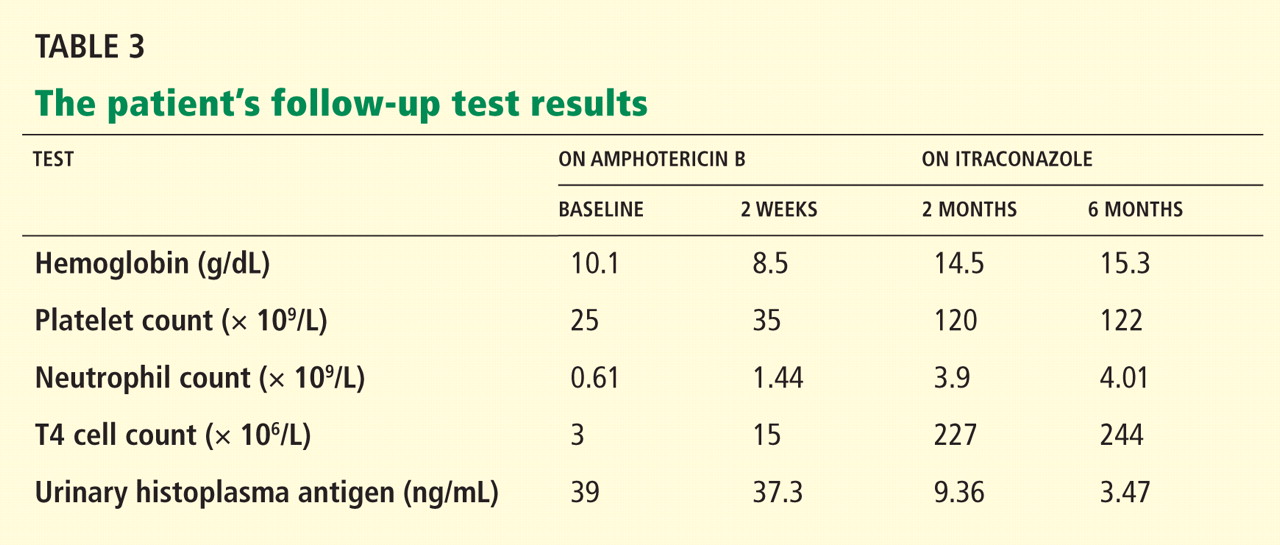
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
A 54-year-old woman with a 1-month history of progressive weakness was transported to the emergency department of a local hospital when a family member found her unresponsive. Before this event, the patient had said she had been feeling tired and cold and looking pale for several weeks.
In the emergency department, her temperature was low. Cableomputed tomography (CT) of the head showed a 1.4-cm hyperdense extraaxial mass. Imaging of the chest showed focal consolidations within the anterior segment of the right upper lobe and the left and right lower lobes.
A urine toxicology screen was positive for acetaminophen (Tylenol), opiates, and benzodiazepines. She was given three doses of naloxone (Narcan), which raised her level of arousal; however, she later became obtunded again and was intubated and transferred to Cleveland Clinic.
A new CT scan of the head confirmed a small left temporal, extradural, calcified lesion with no mass effect or overt bleeding; it appeared most compatible with a solitary calcified meningioma—a likely benign finding.
Her medical history includes hypertension, type 2 diabetes (controlled with diet), and osteoarthritis of the spine. In 1999, she had undergone a hysterectomy that necessitated a blood transfusion. She has never smoked tobacco and does not consume alcohol or use illicit drugs. In the past she worked as a nurse’s aid in a nursing home. However, for the past several years she has stayed at home. Her only avocation of note is gardening.
Initial physical examination
The patient is intubated and sedated. Her temperature is 35.3°C (95.5°F), blood pressure 122/81 mm Hg, heart rate 83 beats per minute, and respiratory rate 14 on assist-controlled ventilator settings with an Fio2 of 100% and a positive end-expiratory pressure of 5 cm H2O.
Her pupils are round, equal, and reactive to light. Her face is symmetric and notable for hirsutism over the chin. Her neck is supple and without lymphadenopathy or thyromegaly.
Rhonchi can be heard at both lung bases. She has normal bowel sounds, and her abdomen is soft and nondistended, with no masses or palpable hepatosplenomegaly. She has no pedal edema on either side, and no clubbing or cyanosis. Her skin is intact, without rashes, lesions, or tattoos. She is able to withdraw from painful stimuli in all four extremities.
INITIAL TESTS PROVIDE A CLUE
1. Which of the following is the likely cause of this patient’s pancytopenia?
- Folate deficiency
- Gastrointestinal bleeding secondary to colon cancer
- Paroxysmal nocturnal hemoglobinuria
- Myelophthisis
- Other
Causes of pancytopenia are listed in Table 2.
Folate deficiency
Folate is necessary for thymidylate synthesis, a rate-limiting step in DNA synthesis. The minimum daily requirement for dietary folate intake is 50 μg.
Severe deficiency of folate has been reported to cause pancytopenia in alcoholics.1 Abuse of alcohol leads to an abrupt decrease in serum folate (within 2 to 4 days of ceasing intake of proper amounts of folate, as in an alcoholic binge) by inhibiting its absorption in the proximal jejunum as well as its metabolism in the liver.2 The resulting folate deficiency, if sustained, can develop into megaloblastosis in 5 to 10 weeks.
The duration of weakness and pallor reported by this patient would raise suspicion of folate deficiency if she had a history of malnutrition or of alcohol abuse, but she has neither. Further, her mean corpuscular volume is 82.5 fL, red blood cell folate 391 ng/mL (reference range 257–800 ng/mL), and serum vitamin B12 1,886 pg/mL (22–700 pg/mL), and she has no macro-ovalocytes or hypersegmented neutrophils on a peripheral blood smear. This makes folate or vitamin B12 deficiency less likely.
Gastrointestinal bleeding due to colon cancer
Iron-deficiency anemia, hematochezia, melena, a change in bowel habits, and abdominal pain may be manifestations of colon cancer. Cancers of the colon originate from adenomatous polyps arising from the colonic mucosa.
The quantity of occult blood loss depends on the site of the tumor. Patients with tumors in the cecum or ascending colon lose an average of 9 mL/day, whereas those with tumors in the transverse, descending, or sigmoid colon or rectum lose less than 2 mL/day.3
Pertinent laboratory findings in iron-deficiency anemia are a low iron concentration, a low transferrin saturation, a depleted serum ferritin, and a normal to high total iron-binding capacity. An initial microcytic normochromic anemia eventually progresses to a microcytic hypochromic anemia that has a tendency to increasingly demonstrate anisocytosis and poikilocytosis.
Our patient’s symptoms, signs, and laboratory values (with normocytic normochromic anemia) are inconsistent with symptomatic colon cancer leading to iron-deficiency anemia.
Acute myeloid leukemia
Acute myeloid leukemia generally manifests with symptoms related to pancytopenia, with weakness and fatigability being the most common.4
In this condition, genetic alterations in hematopoietic precursor cells result in reduced differentiation capacity and accumulation of leukemic blasts in the bone marrow, peripheral blood, and other tissues.
Peripheral blood analysis usually reveals normocytic normochromic anemia with blasts. To establish a diagnosis of acute myeloid leukemia, one must observe at least 20% myeloblasts in the blood, the bone marrow, or both.
No blasts are seen on our patient’s peripheral blood smear, making acute myeloid leukemia less likely.
Paroxysmal nocturnal hemoglobinuria
Paroxysmal nocturnal hemoglobinuria is a possibility in the setting of intravascular hemolytic anemia, bone marrow failure, and thrombosis.
These processes are due to a defect in the glycosyl phosphatidyl inositol (GPI) anchor caused by an abnormality in the PIG-A gene. Partial or complete absence of the GPI anchor allows for activation of complement-mediated hemolysis. A diminished rate of hematopoiesis is presumably responsible for reticulocytopenia, granulocytopenia, or thrombocytopenia, though reticulocytosis can also be seen.5,6 The highly thrombogenic state is believed to occur because of microparticles rich in phosphatidylserine.7
Our patient’s peripheral smear has rare fragmented red blood cells and lacks teardrop red cells. Although paroxysmal nocturnal hemoglobinuria does not have characteristic morphologic features in the peripheral blood, there are no signs of thrombosis in our patient. Her lactate dehydrogenase level is 395 U/L (reference range 100–220 U/L), and her haptoglobin level is less than 20 mg/dL (33–246). These findings could indicate a low level of intravascular hemolysis.
Myelophthisis
Myelophthisis refers to any disorder in which an abnormal cell process invades the bone marrow, damaging hematopoietic tissue. These processes include neoplastic diseases, storage disorders, and a variety of infections. A decrease in all three cell types may result, depending on the severity of invasion. Documented infectious causes include hepatitis viruses, Epstein-Barr virus, human immunodeficiency virus (HIV), mycobacteria, and fungi.
Our patient’s condition is likely due to a marrow-based process of uncertain etiology. In myelophthisic processes, one may see teardrop red cells, which are not seen in this patient’s smear. However, on her chest imaging, the finding of focal consolidations within the anterior segment of the right upper lobe and both lower lobes raises suspicion of an infectious cause.
CASE CONTINUED: SHE UNDERGOES DIAGNOSTIC TESTING
Let us recap some of the laboratory studies that document the extent of our patient’s pancytopenia and the pattern of her anemia:
- Hemoglobin 10.2 g/dL (reference range 11.5–15.5 g/dL)
- Platelet count 27 × 109/L (150–400)
- Leukopenia with profound T-cell lymphopenia
- Iron 59 μg/dL (30–140)
- Total iron-binding capacity 110 μg/dL (210–415)
- Ferritin 3,004 ng/mL (18–300)
- Transferrin saturation 54% (11%–46%).
2. Which of the following would be the best test to obtain next?
- Bone marrow examination
- Blood cultures
- Tuberculin skin test
- Liver biopsy
- Positron emission tomography and CT
Our patient has unexplained pancytopenia. While all the tests listed above might shed light on her condition, a bone marrow examination would be the best test to obtain next.
Urine histoplasma antigen studies are positive at greater than 39 ng/mL (normal 0, low positive < 0.6–3.9, moderate positive 4.0–19.9, high positive 20–39 ng/mL). A culture of the marrow subsequently grows this organism.
3. Which of the following tests would establish a definitive diagnosis in this patient?
- Methenamine silver stain of the marrow
- Serum antibody testing
- Fungal culture
- Peripheral blood smear
- Carbolfuchsin stain of marrow
- Urine histoplasma antigen
A prompt diagnosis is critical in patients with acute pulmonary histoplasmosis or progressive disseminated histoplasmosis because early treatment may shorten the clinical course and length of treatment and, in cases of disseminated histoplasmosis, prevent death.8–10
Histopathologic examination of the bone marrow gives the most rapid results, although biopsy to obtain the tissue is invasive. It can give a definitive diagnosis if it reveals the typical 2- to 4-μm yeast structures of H capsulatum. These are observed on an aspirate smear of the patient’s bone marrow biopsy (Figure 1) and can be confirmed by methenamine silver or periodic acid-Schiff staining of the tissue.
Antibody detection is less practical because the antibodies take 2 to 6 weeks after infection to form.11 Also, it is less useful in cases of disseminated infection because many of these patients are immunosuppressed.
Fungal culture remains the gold standard diagnostic test for histoplasmosis. However, results may take up to 1 month and may be falsely negative in less severe cases.
Histoplasma antigen testing is of greater utility in patients with severe disease, including cases of disseminated histoplasmosis. Rates of antigen detection approach 90% in urine specimens from non-AIDS patients with disseminated infection.12 The urine assay has a greater sensitivity and specificity than the serum assay. The rate of detection is lower (ie, around 82%) in patients with acute pulmonary histoplasmosis when both the serum and urine specimens are tested.13
The immunoassay for histoplasma antigen is particularly useful for monitoring the response to therapy. Antigen levels should be measured before treatment is started and at 2 weeks, 1 month, and then approximately every 3 months during therapy.14 If the treatment is effective, antigens should decline by at least 20% in the first month of treatment and by another 20% in each of the following 3-month intervals. Antigen testing should be done every 3 months until a negative antigen level is achieved. The antigen level should also be followed for at least 6 months after treatment has stopped.14
HISTOPLASMA IS INHALED
H capsulatum is the cause of one of the most common pulmonary and systemic mycotic infections in the world, with hundreds of thousands of new cases annually. In areas where the soil is contaminated by bird or bat guano, the fungus is inhaled, resulting in an asymptomatic or a self-limiting influenza-like syndrome in an immunocompetent individual.15
An antigen-specific CD4+ T lymphocytemediated immunity occurs. The immune response of the host is thought to be fungistatic rather than fungicidal, resulting in a persistent inactive infection capable of reactivation in the presence of a host-pathogen imbalance.16
Most infections are asymptomatic or self-limited. For every 2,000 acute infections there is one that results in severe and progressive dissemination, usually in an immunocompromised host.17,18
TREATMENT OF HISTOPLASMOSIS
4. What is the appropriate initial choice of treatment for a severe case of disseminated histoplasmosis?
- Amphotericin B in a lipid complex formulation (Abelcet)
- Itraconazole (Sporanox)
- Fluconazole (Diflucan)
- Ketoconazole (Nizoral)
Untreated, acute disseminated histoplasmosis can progress over a period of 2 to 12 weeks, ultimately killing the patient.17,19
The leading therapies include amphotericin B in a lipid formulation and azole drugs, in particular itraconazole. Fluconazole and ketoconazole are not first-line options in severe cases because they are less predictably effective, and ketoconazole has a higher rate of side effects.20–23 The current recommendation is to treat severely ill hospitalized patients with one of the liposomal formulations or the lipid complex formulation of amphotericin B. Itraconazole is used for patients who have mild to moderate symptoms and as a step-down therapy in patients who improve after initial use of amphotericin B.
CASE CONCLUDED: THE PATIENT RECOVERS
At the time of the initial patient encounter, there was no history of or obvious cause of immunosuppression in this patient. She was found to be HIV-negative and was subsequently diagnosed with “profound immunosuppression of unknown etiology” resulting in a low CD4 count.
The patient receives trimethoprim-sulfamethoxazole (Bactrim, Septra) and azithromycin (Zithromax) for prophylaxis against Pneumocystis carinii pneumonia and Mycobacterium avium intracellulare infection. Two months after the hospitalization, she recalls being at a corn maze 1 month before becoming ill.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
- Clarke V, Weston-Smith S. Severe folate-deficiency pancytopenia. BMJ Case Reports 2010; published online.
- Anthony AC. Megaloblastic anemias. In:Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohebn HJ, Silberstein LE, editors. Hematology: Basic Principles and Practice, 2nd ed. New York, NY: Churchill Livingston, 1995:552–586.
- Macrae FA, St John DJ. Relationship between patterns of bleeding and Hemoccult sensitivity in patients with colorectal cancers or adenomas. Gastroenterology 1982; 82:891–898.
- Meyers CA, Albitar M, Estey E. Cognitive impairment, fatigue, and cytokine levels in patients with acute myelogenous leukemia or myelodysplastic syndrome. Cancer 2005; 104:788–793.
- Parker CJ. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria. Hematol Oncol Clin North Am 2009; 23:333–346.
- Young NS, Maciejewski JP, Sloand E, et al. The relationship of aplastic anemia and PNH. Int J Hematol 2002; 76(suppl 2):168–172.
- Rosse W. A new way to prevent thrombosis? Blood 2007; 110:3821.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Meals LT, McKinney WP. Acute pulmonary histoplasmosis: progressive pneumonia resulting from high inoculum exposure. J Ky Med Assoc 1998; 96:258–260.
- Salomon J, Flament Saillour M, De Truchis P, et al. An outbreak of acute pulmonary histoplasmosis in members of a trekking trip in Martinique, French West Indies. J Travel Med 2003; 10:87–93.
- Joseph Wheat L. Current diagnosis of histoplasmosis. Trends Microbiol 2003; 11:488–494.
- Wheat LJ, Kauffman CA. Histoplasmosis. Infect Dis Clin North Am 2003; 17:1–19.
- Swartzentruber S, Rhodes L, Kurkjian K, et al. Diagnosis of acute pulmonary histoplasmosis by antigen detection. Clin Infect Dis 2009; 49:1878–1882.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007; 45:807–825.
- Retallack DM, Woods JP. Molecular epidemiology, pathogenesis, and genetics of the dimorphic fungus Histoplasma capsulatum. Microbes Infect 1999; 1:817–825.
- Deepe GS. The immune response to Histoplasma capsulatum: unearthing its secrets. J Lab Clin Med 1994; 123:201–205.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Wheat LJ, Connolly-Stringfield PA, Baker RL, et al. Disseminated histoplasmosis in the acquired immune deficiency syndrome: clinical findings, diagnosis and treatment, and review of the literature. Medicine (Baltimore) 1990; 69:361–374.
- Rubin H, Furcolow ML, Yates JL, Brasher CA. The course and prognosis of histoplasmosis. Am J Med 1959; 27:278–288.
- Wheat J, MaWhinney S, Hafner R, et al. Treatment of histoplasmosis with fluconazole in patients with acquired immunodeficiency syndrome. National Institute of Allergy and Infectious Diseases Acquired Immunodeficiency Syndrome Clinical Trials Group and Mycoses Study Group. Am J Med 1997; 103:223–232.
- McKinsey DS, Kauffman CA, Pappas PG, et al. Fluconazole therapy for histoplasmosis. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Clin Infect Dis 1996; 23:996–1001.
- Slama TG. Treatment of disseminated and progressive cavitary histoplasmosis with ketoconazole. Am J Med 1983; 74:70–73.
- Treatment of blastomycosis and histoplasmosis with ketoconazole. Results of a prospective randomized clinical trial. National Institute of Allergy and Infectious Diseases Mycoses Study Group. Ann Intern Med 1985; 103:861–872.
Grand Rounds: Pregnant Woman, 33, With Leg Pain and Numbness
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
A 33-year-old woman in her 32nd week of pregnancy (gravida 3, para 2) presented to the emergency department (ED) with a five-day history of weakness and ascending numbness below the right knee. She related a two-week history of right-sided low back pain that radiated to the right buttock and was associated with severe right lower extremity (RLE) pain, most prominent in the posterolateral aspect of the right calf. She denied perianal numbness, incontinence, or other changes in bowel or bladder function. She also denied left lower extremity involvement or trauma.
The patient had had one uneventful pregnancy to date. Her medical history included hypothyroidism, treated with levothyroxine; and anxiety, for which she was taking sertraline. She denied any history of allergies, alcohol consumption, smoking, or illicit drug use. She had been evaluated twice and received reassurance in the two weeks before her presentation to the ED. She was admitted to the obstetric service secondary to pain, and a stat MRI rather than x-ray was ordered by obstetrics. An orthopedic consult was ordered. A spine surgeon happened to be on call.
Examination revealed that the patient walked plantigrade, with her right foot slightly externally rotated. She was unable to dorsiflex or plantarflex her right foot. She was unable to heel- or toe-walk on the right side, possessed 0 out of 5 strength at the right extensor hallucis longus and 2 to 3 out of 5 at the right tibialis anterior and gastroc soleus complex. She complained of pain with right leg elevation exceeding 30° and had very limited sensation to light touch in the right L5 and S1 dermatomes. Deep tendon reflex was absent at the right ankle. The patient refused a rectal exam or post-void evaluation.
The initial diagnosis considered by the ED clinician was sciatica, with a differential diagnosis that included pelvic pain of pregnancy, lumbar sprain strain, sciatica, lumbar disk, herniated nucleus pulposus with radiculopathy, and cauda equina syndrome. Trauma was considered and ruled out, as were malignancies; inflammatory, infectious, or degenerative conditions; or other compressive processes.1
Lumbar MRI demonstrated a very large, right-sided disk herniation at L5-S1 with an extruded fragment that was severely compressing the thecal sac and the right S1 nerve root, causing severe right foraminal stenosis at the level of L5-S1. Degenerative changes were noted at L4-5 with disk dessication and no lesions seen.
The patient was diagnosed with cauda equina syndrome, which was felt to be causing severe RLE weakness and ascending numbness. The options of observation, analgesia, physical therapy, and epidural injections were discussed with the patient; however, surgery was strongly recommended due to her profound weakness and the severity of pain she was experiencing, in addition to the size of the disk herniation. She opted for surgery.
The patient was given epidural anesthesia at the L3-4 level, with a catheter left in place during the procedure. A test dose of lidocaine (1.5 cc) with epinephrine was injected to ensure proper placement, and bupivacaine 0.5% was given in increments of 5.0 cc three times during the case. Propofol was administered for sedation, and a 2.0-mg dose of a long-acting morphine was given to the patient before removal of the epidural catheter. Fetal monitoring was performed by obstetrics throughout the procedure.
A laminotomy, partial facetectomy, and diskectomy were performed at L5-S1 with excision of a free fragment. Surgical pathology described the disk as fibrocartilaginous tissue measuring 3.5 cm x 1.4 cm x 0.6 cm.
DISCUSSION
Although nearly half of pregnant women experience low back pain, cauda equina syndrome (CES), a complication of lumbar disk herniation, is extremely rare in the gravid patient.2 In a decade-long review of 48,760 consecutive deliveries, LaBan et al3 identified symptomatic lumbar herniated nucleus pulposus in only five patients (approximately one in 10,000 pregnancies). In pregnant women who do experience CES, symptoms most commonly develop between the fifth and seventh month of pregnancy.4 According to Small et al,5 “The major pitfall in diagnosis is not including CES in the back pain differential.”
True CES presents as a triad of symptoms: lower extremity weakness, altered sensation in the skin of the buttocks and upper posterior thighs (saddle anesthesia), and dysfunction or paralysis of the bowel and bladder. However, few patients present with all of the classic symptoms,6 and patients with CES are often dismissed by several clinicians in their search for relief before presenting to a subspecialist. Kostuik et al7 consider “unilateral sciatica with motor and sensory disturbance” a more common presentation of CES; also indicative of this condition, they report, is “urinary dysfunction combined with motor and sensory loss in the presence of a disc lesion.”
The polypeptide relaxin, which is secreted by the corpus luteum to promote joint laxity in late pregnancy, has been associated with low back pain and pelvic pain of pregnancy; it has also been suggested as a possible contributing cause of CES during pregnancy.8,9 Additionally, increased lumbar lordosis with positional and postural stress may cause direct pressure by the gravid uterus on nerve roots. The great vessels may also be compressed by the uterus, resulting in ischemia of the neural element and back pain that radiates to the legs.10 Many cases of lumbar disk prolapse occur during the first and second trimesters. The most clinically incapacitated patients have been found to have the highest levels of relaxin.9
The Diagnosis
Early diagnosis of CES, through proper physical examination and radiologic studies, is paramount. A rectal examination should be performed to assess for sphincter tone (which may be diminished in 80% of patients) and to assess for perineal sensation.5 Catheterization yielding a postvoid residual urine greater than 100/200 cc is reported to have a specificity and sensitivity of 90% or greater for CES. Small et al5 recommend a straight leg raise maneuver to assess for radiculopathy.
Various studies in the literature support the use of MRI in the gravid patient to confirm the diagnosis of CES and to identify the degree and level of disk protrusion.2-4,11
Treatment
CES requires urgent surgical decompression.11 Early recognition of CES attributable to lumbar disk prolapse, report O’Laoire et al,12 is essential to prevent irreversible sphincter paralysis. They liken the condition’s urgency to that of extradural hematoma in a head injury.
Disk surgery during pregnancy—preferably a team effort, with obstetrics performing perioperative fetal monitoring—has been deemed a safe management method.2,4 Spinal or general anesthesia during nonobstetric surgery is generally considered safe for both mother and fetus.13,14 Adequate oxygenation without risk for hyperventilation is considered essential.15
PATIENT OUTCOME
In the immediate postoperative period, the patient continued to complain of RLE pain, which abated significantly by the time she was discharged. When she was seen in follow-up four days later, she was able to heel- and toe-walk on the right side, and her strength had improved to 3 or 4 out of 5 at the RLE. She continued to experience diminished sensation to the plantar aspect of the right foot, which persisted at the one-month follow up. At that visit, the patient also reported occasional pain in the right buttock. Physical therapy was started to strengthen the RLE.
By three months postsurgery, the patient had undergone uneventful vaginal delivery. She had an entirely benign exam with 5 out of 5 strength at the RLE and no neurologic deficits. She was cleared to return to light weightlifting with good technique and lumbar support but was told to refrain from running until the sixth month postsurgery.
CONCLUSION
Although the case patient did not have a “true” (ie, typical) presentation of CES, her symptoms warranted a full workup and treatment to prevent possible long-term sequelae. Medical practitioners should be familiar with the triad presentation of CES. They must differentiate lower back pain of muscular origin from lumbar disk herniation and be able to appreciate the degree of symptom severity reported by the gravid patient. A thorough history and physical assessment must be performed in every such case. When in doubt, the clinician must err on the side of caution, referring the patient for MRI and consulting with a specialist.
REFERENCES
1. Johnston RA. The management of acute spinal cord compression. J Neurol Neurosurg Psychiatr. 1993;56(10):1046-1054.
2. Brown MD, Levi AD. Surgery for lumbar disc herniation during pregnancy. Spine (Phila PA 1976). 2001;26(5):440-443.
3. LaBan MM, Perrin JCS, Latimer FR. Pregnancy and the herniated lumbar disc. Arch Phys Med Rehabil. 1983;64(7):319-321.
4. LaBan MM, Rapp NS, Van Oeyen P, Meerschaert JR. The lumbar herniated disk of pregnancy: a report of six cases identified by magnetic resonance imaging. Arch Phys Med Rehabil. 1995;76(5):476-479.
5. Small SA, Perron AD, Brady WJ. Orthopedic pitfalls: cauda equina syndrome. Am J Emerg Med. 2005;23(2):159-163.
6. Tay EC, Chacha PB. Midline prolapse of a lumbar intervertebral disc with compression of the cauda equina. J Bone Joint Surg. 1979;61(1):43-46.
7. Kostuik JP, Harrington I, Alexander D, et al. Cauda equina syndrome and lumbar disc herniation. J Bone Joint Surg Am. 1986;68(3):386-391.
8. Russell R, Reynolds F. Back pain, pregnancy, and childbirth. BMJ. 1997;314(7087):1062-1063.
9. MacLennan AH, Nicholson R, Green RC, Bath M. Serum relaxin and pelvic pain of pregnancy. Lancet. 1986;2(8501):243-245.
10. Ashkan K, Casey AT, Powell M, Crockard HA. Back pain during pregnancy and after childbirth: an unusual cause not to miss. J R Soc Med. 1998;91(2):88-90.
11. Busse JW, Bhandari M, Schnittker JB, et al. Delayed presentation of cauda equina syndrome secondary to lumbar disc herniation: functional outcomes and health-related quality of life. CJEM. 2001;3(4):285-291.
12. O’Laoire SA, Crockard HA, Thomas DG. Prognosis for sphincter recovery after operation for cauda equina compression owing to lumbar disc prolapse. Br Med J (Clin Res Ed). 1981;282(6279):1852-1854.
13. Kuczkowski KM. The safety of anaesthetics in pregnant women. Expert Opin Drug Saf. 2006; 5(2):251-264.
14. Kuczkowski KM. Nonobstetric surgery during pregnancy: what are the risks of anesthesia? Obstet Gynecol Surv. 2004;59(1):52-56.
15. Birnbach DJ, Browne IM. Anesthesia for obstetrics. In: Miller RD, Eriksson LI, Fleisher LA, et al. Miller’s Anesthesia. Philadelphia, PA: Churchill Livingston, Elsevier Health Science; 2010: 2203-2240.
Woman with Foot Pain After Neurosurgery Service
ANSWER
The radiograph demonstrates mild soft-tissue swelling and an oblique, mildly displaced fracture of the distal fibula. In addition, there is a small bone density adjacent to the medial malleolus, which could represent either an unfused accessory ossification center or a sequela of prior trauma.
On closer questioning, the patient acknowledged that a few months prior, she had stepped in a hole and been treated for a “broken bone.” She had been in a cast for an undisclosed period of time; at a follow-up appointment, the cast had been removed and the bone had been declared “healed.”
A new orthopedic consultation was obtained prior to the patient’s discharge from the hospital, which resulted in placement of a short leg cast.
ANSWER
The radiograph demonstrates mild soft-tissue swelling and an oblique, mildly displaced fracture of the distal fibula. In addition, there is a small bone density adjacent to the medial malleolus, which could represent either an unfused accessory ossification center or a sequela of prior trauma.
On closer questioning, the patient acknowledged that a few months prior, she had stepped in a hole and been treated for a “broken bone.” She had been in a cast for an undisclosed period of time; at a follow-up appointment, the cast had been removed and the bone had been declared “healed.”
A new orthopedic consultation was obtained prior to the patient’s discharge from the hospital, which resulted in placement of a short leg cast.
ANSWER
The radiograph demonstrates mild soft-tissue swelling and an oblique, mildly displaced fracture of the distal fibula. In addition, there is a small bone density adjacent to the medial malleolus, which could represent either an unfused accessory ossification center or a sequela of prior trauma.
On closer questioning, the patient acknowledged that a few months prior, she had stepped in a hole and been treated for a “broken bone.” She had been in a cast for an undisclosed period of time; at a follow-up appointment, the cast had been removed and the bone had been declared “healed.”
A new orthopedic consultation was obtained prior to the patient’s discharge from the hospital, which resulted in placement of a short leg cast.
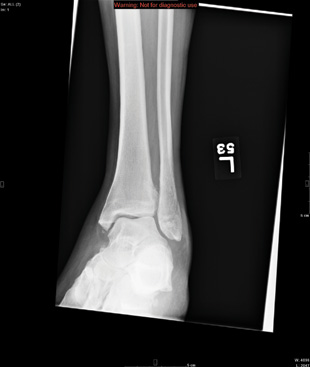
A 60-year-old woman is admitted electively to the neurosurgery service for a multilevel posterior cervical decompression and fusion. The procedure is completed uneventfully and without complication. The patient is admitted to the neurosurgical floor. In the middle of the night, you receive a call from the nurse stating that the patient is complaining of moderate pain and swelling in her left ankle and foot, which occurred after she got up to use the bathroom. The patient denies injuring herself. She complains of pain with weight-bearing. Since the pain is tolerable and pain medication has already been given, it is decided to assess the complaint later in the morning. On evaluation, the left foot and ankle are mildly swollen, with tenderness on the lateral aspect. There is minimal tenderness on the foot. Distal pulses and color are good. The calf is nontender. The patient’s medical history is significant only for mild hypertension. Radiographs of the left ankle are obtained. What is your impression?
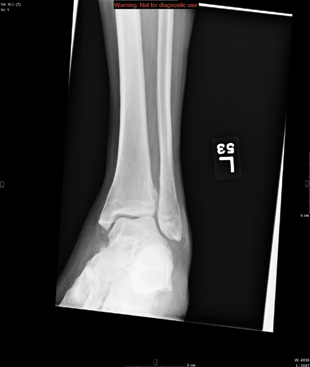
Osborn waves: An inverse correlation with core body temperature
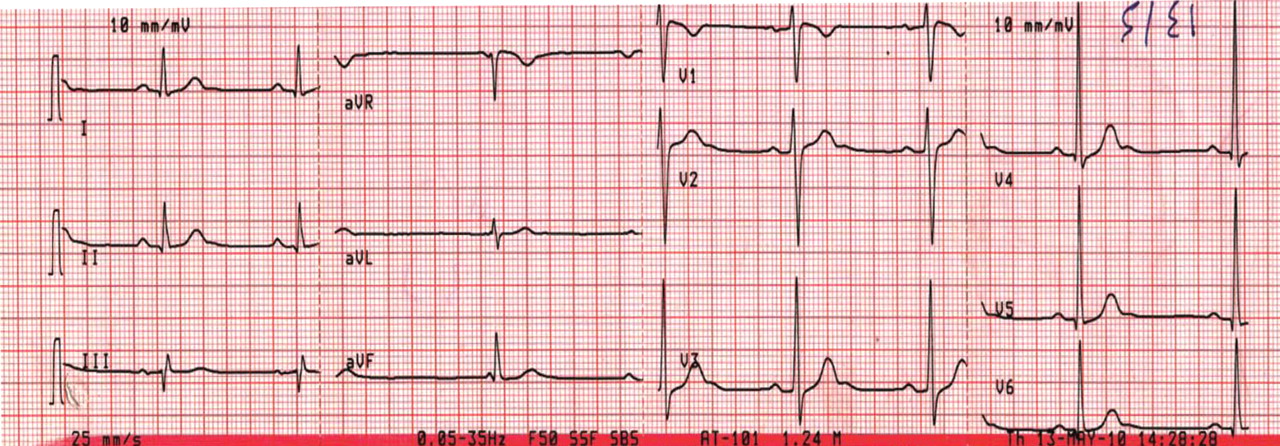
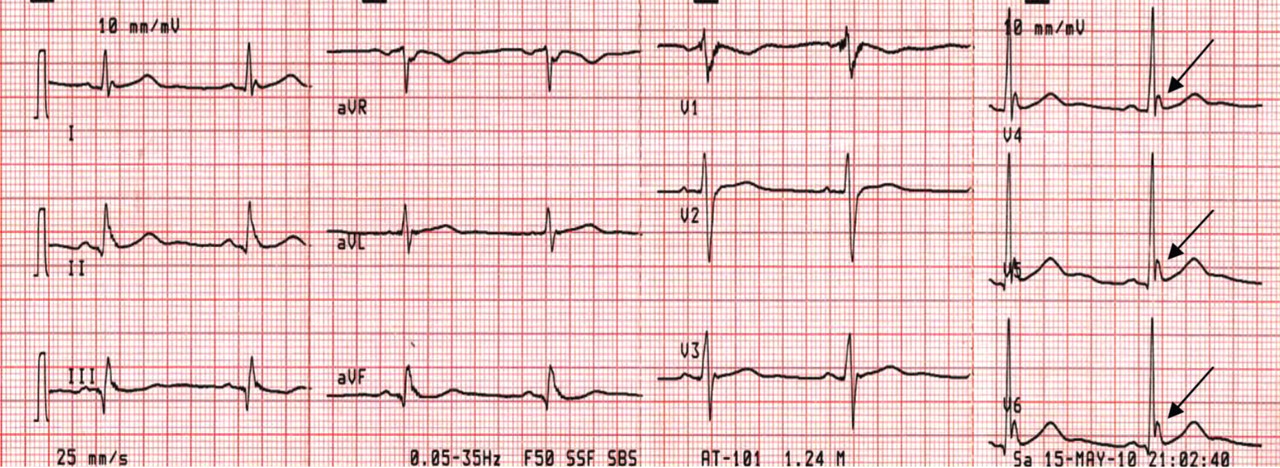
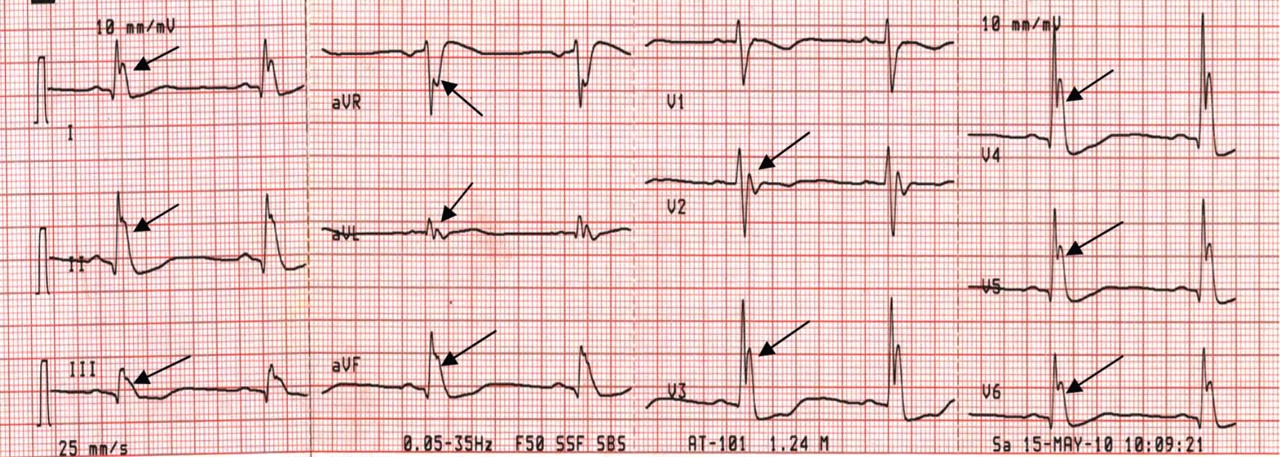
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
- Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
- The lower the core body temperature, the higher the amplitude of the J wave.
- The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
- As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
- Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol 1953; 175:389–398.
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
- Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
- The lower the core body temperature, the higher the amplitude of the J wave.
- The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
- As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
Although Osborn waves are a marker of hypothermia, they also occur in nonhypothermic conditions. Brainstem death is a precursor of the J wave, and this is explained by impaired thermoregulatory ability resulting from hypothalamic dysfunction and subsequent hypothermia.
The three electrocardiograms presented here illustrate several points:
- Classic findings in hypothermia include J waves, sinus bradycardia, prolongation of the PR interval, widening of the QRS complex, and prolongation of the QT interval.
- The lower the core body temperature, the higher the amplitude of the J wave.
- The J wave in brain death (unlike hypothermic causes of the J wave) is not associated with the characteristic signs of shivering in the surface electrocardiogram.
- As hypothermia becomes more profound, the J wave becomes evident in all leads, not only the inferolateral leads.
- Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol 1953; 175:389–398.
- Osborn JJ. Experimental hypothermia; respiratory and blood pH changes in relation to cardiac function. Am J Physiol 1953; 175:389–398.
Dehydration Leads to Incidental Finding
ANSWER
The radiograph demonstrates that the nasogastric tube is within a decompressed stomach. Normal gas pattern is noted within the bowel and colon.
Of note, however, are tiny calcifications within the right upper quadrant. This finding is suggestive of cholelithiasis—in this case, likely an incidental finding.
ANSWER
The radiograph demonstrates that the nasogastric tube is within a decompressed stomach. Normal gas pattern is noted within the bowel and colon.
Of note, however, are tiny calcifications within the right upper quadrant. This finding is suggestive of cholelithiasis—in this case, likely an incidental finding.
ANSWER
The radiograph demonstrates that the nasogastric tube is within a decompressed stomach. Normal gas pattern is noted within the bowel and colon.
Of note, however, are tiny calcifications within the right upper quadrant. This finding is suggestive of cholelithiasis—in this case, likely an incidental finding.
A 57-year-old man is admitted to your facility for weakness and dehydration. His medical history is significant for rectal carcinoma, diabetes, and hypertension. His family states that his oral intake has been progressively less in the past several weeks. The patient’s vital signs are stable. Overall examination demonstrates a weak, frail-appearing man with dry mucous membranes and no other acute abnormalities. Lab work reveals that his serum sodium level and BUN/creatinine ratio are slightly elevated. To facilitate hydration, administration of medications, and nourishment, placement of a nasogastric tube is ordered; this is done just prior to your arrival at the patient’s room during rounds. As per protocol, an abdominal radiograph is obtained, and the nurses ask you to check it to confirm placement. The radiograph is shown. What is your impression?