User login
Flu or strep? Rapid tests can mislead
A 62-year-old woman presented to our emergency department with fever, chills, hoarseness, pain on swallowing, and a painful neck. Her symptoms had begun 1 day earlier. Because acetaminophen brought no improvement, she went to an urgent care facility, where a nasal swab polymerase chain reaction test was positive for influenza A, and a throat swab rapid test was positive for group A streptococci. She was then referred to our emergency department.
She reported no pre-existing conditions predisposing her to infection. Her temperature was 99.9°F (37.7°C), pulse 112 beats per minute, and respiratory rate 24 breaths per minute. The physical examination was unremarkable except for bilateral anterior cervical adenopathy and bilateral anterior neck tenderness. Her pharynx was not injected, and no exudate, palatal edema, or petechiae were noted.
Results of initial laboratory testing were as follows:
- White blood cell count 20.5 × 109/L (reference range 3.9–11)
- Neutrophils 76% (42%–75%)
- Bands 15% (0%–5%)
- Lymphocytes 3% (21%–51%)
- Erythrocyte sedimentation rate 75 mm/h (< 20 mm/h)
- C-reactive protein 247.14 mg/L (≤ 3 mg/L)
- Serum aminotransferase levels were normal.
- Polymerase chain reaction testing of a nasal swab was negative for viral infection.
Throat swabs and blood samples were sent for culture.
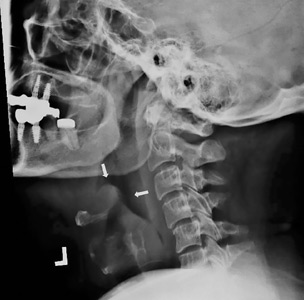
 visible on lateral neck radiography.")
She was started on ceftriaxone 1 g intravenously every 24 hours, with close observation in the medical intensive care unit, where she was admitted because of epiglottitis. On hospital day 3, the throat culture was reported as negative, but the blood culture was reported as positive for Haemophilus influenzae. Thus, the clinical diagnosis was acute epiglottitis due to H influenzae, not group A streptococci.
The patient completed 10 days of ceftriaxone therapy; her recovery was uneventful, and she was discharged on hospital day 10.
INFLUENZA: CHALLENGES TO PROMPT, ACCURATE DIAGNOSIS
During influenza season, emergency departments are inundated with adults with influenza A and other viral respiratory infections. This makes prompt, accurate diagnosis a challenge,1 given the broad differential diagnosis.2,3 Adults with influenza and its complications as well as unrelated conditions can present a special challenge.4
Our patient presented with acute-onset influenza A and was then found to have acute epiglottitis, an unexpected complication of influenza A.5 A positive rapid test for group A streptococci done at an urgent care facility led emergency department physicians to assume that the acute epiglottitis was due to group A streptococci. Unless correlated with clinical findings, results of rapid diagnostic tests may mislead the unwary practitioner. Accurate diagnosis should be based mainly on the history and physical findings. Results of rapid diagnostic tests can be helpful if interpreted in the clinical context.6–8
The rapid test for streptococci is appropriate for the diagnosis of pharyngitis due to group A streptococci in people under age 30 with acute-onset sore throat, fever, and bilateral acute cervical adenopathy, without fatigue or myalgias. However, the rapid test does not differentiate colonization from infection. Group A streptococci are common colonizers with viral pharyngitis. In 30% of cases of Epstein-Barr virus pharyngitis, there is colonization with group A streptococci. A positive rapid test in such cases can result in the wrong diagnosis, ie, pharyngitis due to group A streptococci rather than Epstein-Barr virus.
- Cunha BA. The clinical diagnosis of severe viral influenza A. Infection 2008; 36(1):92–93. doi:10.1007/s15010-007-7255-9
- Cunha BA, Klein NC, Strollo S, Syed U, Mickail N, Laguerre M. Legionnaires’ disease mimicking swine influenza (H1N1) pneumonia during the “herald wave” of the pandemic. Heart Lung 2010; 39(3):242–248. doi:10.1016/j.hrtlng.2009.10.009
- Cunha BA, Raza M. During influenza season: all influenza-like illnesses are not due to influenza: dengue mimicking influenza. J Emerg Med 2015; 48(5):e117–e120. doi:10.1016/j.jemermed.2014.12.051
- Cunha CB. Infectious disease differential diagnosis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:493–526.
- Cunha BA. Pharyngitis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:42–47.
- Cohen JF, Chalumeau M, Levy C, et al. Effect of clinical spectrum, inoculum size and physician characteristics on sensitivity of rapid antigen detection test for group A streptococcal pharyngitis. Eur J Clin Microbiol Infect Dis 2013; 32(6):787–793. doi:10.1007/s10096-012-1809-1
- Dimatteo LA, Lowenstein SR, Brimhall B, Reiquam W, Gonzales R. The relationship between the clinical features of pharyngitis and the sensitivity of a rapid antigen test: evidence of spectrum bias. Ann Emerg Med 2001; 38(6):648–652. doi:10.1067/mem.2001.119850
- Cunha BA. A positive rapid strep test in a young adult with acute pharyngitis: be careful what you wish for! IDCases 2017; 10:58–59. doi:10.1016/j.idcr.2017.08.012
A 62-year-old woman presented to our emergency department with fever, chills, hoarseness, pain on swallowing, and a painful neck. Her symptoms had begun 1 day earlier. Because acetaminophen brought no improvement, she went to an urgent care facility, where a nasal swab polymerase chain reaction test was positive for influenza A, and a throat swab rapid test was positive for group A streptococci. She was then referred to our emergency department.
She reported no pre-existing conditions predisposing her to infection. Her temperature was 99.9°F (37.7°C), pulse 112 beats per minute, and respiratory rate 24 breaths per minute. The physical examination was unremarkable except for bilateral anterior cervical adenopathy and bilateral anterior neck tenderness. Her pharynx was not injected, and no exudate, palatal edema, or petechiae were noted.
Results of initial laboratory testing were as follows:
- White blood cell count 20.5 × 109/L (reference range 3.9–11)
- Neutrophils 76% (42%–75%)
- Bands 15% (0%–5%)
- Lymphocytes 3% (21%–51%)
- Erythrocyte sedimentation rate 75 mm/h (< 20 mm/h)
- C-reactive protein 247.14 mg/L (≤ 3 mg/L)
- Serum aminotransferase levels were normal.
- Polymerase chain reaction testing of a nasal swab was negative for viral infection.
Throat swabs and blood samples were sent for culture.
She was started on ceftriaxone 1 g intravenously every 24 hours, with close observation in the medical intensive care unit, where she was admitted because of epiglottitis. On hospital day 3, the throat culture was reported as negative, but the blood culture was reported as positive for Haemophilus influenzae. Thus, the clinical diagnosis was acute epiglottitis due to H influenzae, not group A streptococci.
The patient completed 10 days of ceftriaxone therapy; her recovery was uneventful, and she was discharged on hospital day 10.
INFLUENZA: CHALLENGES TO PROMPT, ACCURATE DIAGNOSIS
During influenza season, emergency departments are inundated with adults with influenza A and other viral respiratory infections. This makes prompt, accurate diagnosis a challenge,1 given the broad differential diagnosis.2,3 Adults with influenza and its complications as well as unrelated conditions can present a special challenge.4
Our patient presented with acute-onset influenza A and was then found to have acute epiglottitis, an unexpected complication of influenza A.5 A positive rapid test for group A streptococci done at an urgent care facility led emergency department physicians to assume that the acute epiglottitis was due to group A streptococci. Unless correlated with clinical findings, results of rapid diagnostic tests may mislead the unwary practitioner. Accurate diagnosis should be based mainly on the history and physical findings. Results of rapid diagnostic tests can be helpful if interpreted in the clinical context.6–8
The rapid test for streptococci is appropriate for the diagnosis of pharyngitis due to group A streptococci in people under age 30 with acute-onset sore throat, fever, and bilateral acute cervical adenopathy, without fatigue or myalgias. However, the rapid test does not differentiate colonization from infection. Group A streptococci are common colonizers with viral pharyngitis. In 30% of cases of Epstein-Barr virus pharyngitis, there is colonization with group A streptococci. A positive rapid test in such cases can result in the wrong diagnosis, ie, pharyngitis due to group A streptococci rather than Epstein-Barr virus.
A 62-year-old woman presented to our emergency department with fever, chills, hoarseness, pain on swallowing, and a painful neck. Her symptoms had begun 1 day earlier. Because acetaminophen brought no improvement, she went to an urgent care facility, where a nasal swab polymerase chain reaction test was positive for influenza A, and a throat swab rapid test was positive for group A streptococci. She was then referred to our emergency department.
She reported no pre-existing conditions predisposing her to infection. Her temperature was 99.9°F (37.7°C), pulse 112 beats per minute, and respiratory rate 24 breaths per minute. The physical examination was unremarkable except for bilateral anterior cervical adenopathy and bilateral anterior neck tenderness. Her pharynx was not injected, and no exudate, palatal edema, or petechiae were noted.
Results of initial laboratory testing were as follows:
- White blood cell count 20.5 × 109/L (reference range 3.9–11)
- Neutrophils 76% (42%–75%)
- Bands 15% (0%–5%)
- Lymphocytes 3% (21%–51%)
- Erythrocyte sedimentation rate 75 mm/h (< 20 mm/h)
- C-reactive protein 247.14 mg/L (≤ 3 mg/L)
- Serum aminotransferase levels were normal.
- Polymerase chain reaction testing of a nasal swab was negative for viral infection.
Throat swabs and blood samples were sent for culture.
She was started on ceftriaxone 1 g intravenously every 24 hours, with close observation in the medical intensive care unit, where she was admitted because of epiglottitis. On hospital day 3, the throat culture was reported as negative, but the blood culture was reported as positive for Haemophilus influenzae. Thus, the clinical diagnosis was acute epiglottitis due to H influenzae, not group A streptococci.
The patient completed 10 days of ceftriaxone therapy; her recovery was uneventful, and she was discharged on hospital day 10.
INFLUENZA: CHALLENGES TO PROMPT, ACCURATE DIAGNOSIS
During influenza season, emergency departments are inundated with adults with influenza A and other viral respiratory infections. This makes prompt, accurate diagnosis a challenge,1 given the broad differential diagnosis.2,3 Adults with influenza and its complications as well as unrelated conditions can present a special challenge.4
Our patient presented with acute-onset influenza A and was then found to have acute epiglottitis, an unexpected complication of influenza A.5 A positive rapid test for group A streptococci done at an urgent care facility led emergency department physicians to assume that the acute epiglottitis was due to group A streptococci. Unless correlated with clinical findings, results of rapid diagnostic tests may mislead the unwary practitioner. Accurate diagnosis should be based mainly on the history and physical findings. Results of rapid diagnostic tests can be helpful if interpreted in the clinical context.6–8
The rapid test for streptococci is appropriate for the diagnosis of pharyngitis due to group A streptococci in people under age 30 with acute-onset sore throat, fever, and bilateral acute cervical adenopathy, without fatigue or myalgias. However, the rapid test does not differentiate colonization from infection. Group A streptococci are common colonizers with viral pharyngitis. In 30% of cases of Epstein-Barr virus pharyngitis, there is colonization with group A streptococci. A positive rapid test in such cases can result in the wrong diagnosis, ie, pharyngitis due to group A streptococci rather than Epstein-Barr virus.
- Cunha BA. The clinical diagnosis of severe viral influenza A. Infection 2008; 36(1):92–93. doi:10.1007/s15010-007-7255-9
- Cunha BA, Klein NC, Strollo S, Syed U, Mickail N, Laguerre M. Legionnaires’ disease mimicking swine influenza (H1N1) pneumonia during the “herald wave” of the pandemic. Heart Lung 2010; 39(3):242–248. doi:10.1016/j.hrtlng.2009.10.009
- Cunha BA, Raza M. During influenza season: all influenza-like illnesses are not due to influenza: dengue mimicking influenza. J Emerg Med 2015; 48(5):e117–e120. doi:10.1016/j.jemermed.2014.12.051
- Cunha CB. Infectious disease differential diagnosis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:493–526.
- Cunha BA. Pharyngitis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:42–47.
- Cohen JF, Chalumeau M, Levy C, et al. Effect of clinical spectrum, inoculum size and physician characteristics on sensitivity of rapid antigen detection test for group A streptococcal pharyngitis. Eur J Clin Microbiol Infect Dis 2013; 32(6):787–793. doi:10.1007/s10096-012-1809-1
- Dimatteo LA, Lowenstein SR, Brimhall B, Reiquam W, Gonzales R. The relationship between the clinical features of pharyngitis and the sensitivity of a rapid antigen test: evidence of spectrum bias. Ann Emerg Med 2001; 38(6):648–652. doi:10.1067/mem.2001.119850
- Cunha BA. A positive rapid strep test in a young adult with acute pharyngitis: be careful what you wish for! IDCases 2017; 10:58–59. doi:10.1016/j.idcr.2017.08.012
- Cunha BA. The clinical diagnosis of severe viral influenza A. Infection 2008; 36(1):92–93. doi:10.1007/s15010-007-7255-9
- Cunha BA, Klein NC, Strollo S, Syed U, Mickail N, Laguerre M. Legionnaires’ disease mimicking swine influenza (H1N1) pneumonia during the “herald wave” of the pandemic. Heart Lung 2010; 39(3):242–248. doi:10.1016/j.hrtlng.2009.10.009
- Cunha BA, Raza M. During influenza season: all influenza-like illnesses are not due to influenza: dengue mimicking influenza. J Emerg Med 2015; 48(5):e117–e120. doi:10.1016/j.jemermed.2014.12.051
- Cunha CB. Infectious disease differential diagnosis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:493–526.
- Cunha BA. Pharyngitis. In: Cunha CB, Cunha BA, eds. Antibiotic Essentials. Jaypee Brothers Medical Pub: New Delhi, India; 2017:42–47.
- Cohen JF, Chalumeau M, Levy C, et al. Effect of clinical spectrum, inoculum size and physician characteristics on sensitivity of rapid antigen detection test for group A streptococcal pharyngitis. Eur J Clin Microbiol Infect Dis 2013; 32(6):787–793. doi:10.1007/s10096-012-1809-1
- Dimatteo LA, Lowenstein SR, Brimhall B, Reiquam W, Gonzales R. The relationship between the clinical features of pharyngitis and the sensitivity of a rapid antigen test: evidence of spectrum bias. Ann Emerg Med 2001; 38(6):648–652. doi:10.1067/mem.2001.119850
- Cunha BA. A positive rapid strep test in a young adult with acute pharyngitis: be careful what you wish for! IDCases 2017; 10:58–59. doi:10.1016/j.idcr.2017.08.012
Delaying antibiotics in elderly with UTI linked to higher sepsis, death rates
results of a large, population-based study suggest.
The risk of bloodstream infection was more than seven times greater in patients who did not receive antibiotics immediately after seeing a general practitioner for a UTI versus those who did, according to results of the study based on primary care records and other data for nearly 160,000 U.K. patients aged 65 years or older. Death rates and hospital admissions were significantly higher for these patients, according to the study published in The BMJ by Myriam Gharbi, PharmD, Phd, Imperial College London, and her colleagues.
The publication of these findings coincides with an increase in Escherichia coli bloodstream infections in England.
“Our study suggests the early initiation of antibiotics for UTI in older high risk adult populations (especially men aged [older than] 85 years) should be recommended to prevent serious complications,” Dr. Gharbi and her coauthors said in their report.
The population-based cohort study comprised 157,264 adult primary care patients at least 65 years of age who had one or more suspected or confirmed lower UTIs from November 2007 to May 2015. The researchers found that health care providers had diagnosed a total of 312,896 UTI episodes in these patients during the period they studied. In 7.2% (22,534) of the UTI episodes, the researchers were unable to find records of the patients having been prescribed antibiotics by a general practitioner within 7 days of the UTI diagnosis. These 22,534 episodes included those that occurred in patients who had a complication before an antibiotic was prescribed. An additional 6.2% (19,292) of the episodes occurred in patients who were prescribed antibiotics, but not during their first UTI-related visit to a general practitioner or on the same day of such a visit. The researchers classified this group of patients as having been prescribed antibiotics on a deferred or delayed basis, as they were not prescribed such drugs within 7 days of their visit.
Overall, there were 1,539 cases (0.5% of the total number of UTIs) of bloodstream infection within 60 days of the initial urinary tract infection diagnosis, the researchers reported.
The bloodstream infection rate was 2.9% for patients who were not prescribed antibiotics ever or prior to an infection occurring, 2.2% in those who were prescribed antibiotics on a deferred basis, and 0.2% in those who were prescribed antibiotics immediately, meaning during their first visit to a general practitioner for a UTI or on the same day of such a visit (P less than .001). After adjustment for potential confounding variables such as age, sex, and region, the patients classified as having not been prescribed antibiotics or having been prescribed antibiotics on a deferred basis were significantly more likely to have a bloodstream infection within 60 days of their visit to a health care provider, compared with those who received antibiotics immediately, with odds ratios of 8.08 (95% confidence interval, 7.12-9.16) and 7.12 (95% CI, 6.22-8.14), respectively.
Hospital admissions after a UTI episode were nearly twice as high in the no- or deferred-antibiotics groups (27.0% and 26.8%, respectively), compared with the group that received antibiotics right away (14.8%), the investigators reported. The lengths of hospital stays were 12.1 days for the group classified as having not been prescribed antibiotics, 7.7 days for the group subject to delayed antibiotic prescribing, and 6.3 days for the group who received antibiotics immediately.
Deaths within 60 days of experiencing a urinary tract infection occurred in 5.4% of patients in the no-antibiotics group, 2.8% of the deferred-antibiotics group, and 1.6% of the immediate-antibiotics group. After adjustment for covariates, a regression analysis showed the risks for all-cause mortality were 1.16 and 2.18 times higher in the deferred-antibiotics group and the no-antibiotics group, respectively, according to the paper.
In the immediate-antibiotics group, those patients who received nitrofurantoin had a “small but significant increase” in 60-day survival versus those who received trimethoprim, the investigators noted in the discussion section of their report.
“This increase could reflect either higher levels of resistance to trimethoprim or a healthier population treated with nitrofurantoin, the latest being not recommended for patients with poor kidney function,” the researchers wrote.
This study was supported by the National Institute for Health Research and other U.K. sources. One study coauthor reported working as an epidemiologist with GSK in areas not related to the study.
SOURCE: Gharbi M et al. BMJ. 2019 Feb 27. doi: 10.1136/bmj.l525.
This study linking primary care prescribing to serious infections in elderly patients with urinary tract infections is timely, as rates of bloodstream infection and mortality are increasing in this age group, according to Alastair D. Hay, MB.ChB, a professor at University of Bristol, England.
“Prompt treatment should be offered to older patients, men (who are at higher risk than women), and those living in areas of greater socioeconomic deprivation who are at the highest risk of bloodstream infections,” Dr. Hay said in an editorial accompanying the report by Gharbi et al.
That said, the link between prescribing and infection in this particular study may not be causal: “The implications are likely to be more nuanced than primary care doctors risking the health of older adults to meet targets for antimicrobial stewardship,” Dr. Hay noted.
Doctors are cautious when managing infections in vulnerable groups, evidence shows, and the deferred prescribing reported in this study is likely not the same as the delayed prescribing seen in primary care, he explained.
“Most clinicians issue a prescription on the day of presentation, with verbal advice to delay treatment, rather than waiting for a patient to return or issuing a postdated prescription,” he said. “The group given immediate antibiotics in the study by Gharbi and colleagues likely contained some patients managed in this way.”
Patients who apparently had no prescription in this retrospective analysis may have had a same-day admission with a bloodstream infection; moreover, a number of bloodstream infections in older people are due to urinary tract bacteria, and so would not be prevented by treatment for urinary tract infection, Dr. Hay said.
“Further research is needed to establish whether treatment should be initiated with a broad or a narrow spectrum antibiotic and to identify those in whom delaying treatment (while awaiting investigation) is safe,” he concluded.
Dr. Hay is a professor in the Centre for Academic Primary Care, University of Bristol, England. His editorial appears in The BMJ (2019 Feb 27. doi: 10.1136/bmj.l780). Dr. Hay declared that he is a member of the managing common infections guideline committee for the National Institute for Health and Care Excellence (NICE).
This study linking primary care prescribing to serious infections in elderly patients with urinary tract infections is timely, as rates of bloodstream infection and mortality are increasing in this age group, according to Alastair D. Hay, MB.ChB, a professor at University of Bristol, England.
“Prompt treatment should be offered to older patients, men (who are at higher risk than women), and those living in areas of greater socioeconomic deprivation who are at the highest risk of bloodstream infections,” Dr. Hay said in an editorial accompanying the report by Gharbi et al.
That said, the link between prescribing and infection in this particular study may not be causal: “The implications are likely to be more nuanced than primary care doctors risking the health of older adults to meet targets for antimicrobial stewardship,” Dr. Hay noted.
Doctors are cautious when managing infections in vulnerable groups, evidence shows, and the deferred prescribing reported in this study is likely not the same as the delayed prescribing seen in primary care, he explained.
“Most clinicians issue a prescription on the day of presentation, with verbal advice to delay treatment, rather than waiting for a patient to return or issuing a postdated prescription,” he said. “The group given immediate antibiotics in the study by Gharbi and colleagues likely contained some patients managed in this way.”
Patients who apparently had no prescription in this retrospective analysis may have had a same-day admission with a bloodstream infection; moreover, a number of bloodstream infections in older people are due to urinary tract bacteria, and so would not be prevented by treatment for urinary tract infection, Dr. Hay said.
“Further research is needed to establish whether treatment should be initiated with a broad or a narrow spectrum antibiotic and to identify those in whom delaying treatment (while awaiting investigation) is safe,” he concluded.
Dr. Hay is a professor in the Centre for Academic Primary Care, University of Bristol, England. His editorial appears in The BMJ (2019 Feb 27. doi: 10.1136/bmj.l780). Dr. Hay declared that he is a member of the managing common infections guideline committee for the National Institute for Health and Care Excellence (NICE).
This study linking primary care prescribing to serious infections in elderly patients with urinary tract infections is timely, as rates of bloodstream infection and mortality are increasing in this age group, according to Alastair D. Hay, MB.ChB, a professor at University of Bristol, England.
“Prompt treatment should be offered to older patients, men (who are at higher risk than women), and those living in areas of greater socioeconomic deprivation who are at the highest risk of bloodstream infections,” Dr. Hay said in an editorial accompanying the report by Gharbi et al.
That said, the link between prescribing and infection in this particular study may not be causal: “The implications are likely to be more nuanced than primary care doctors risking the health of older adults to meet targets for antimicrobial stewardship,” Dr. Hay noted.
Doctors are cautious when managing infections in vulnerable groups, evidence shows, and the deferred prescribing reported in this study is likely not the same as the delayed prescribing seen in primary care, he explained.
“Most clinicians issue a prescription on the day of presentation, with verbal advice to delay treatment, rather than waiting for a patient to return or issuing a postdated prescription,” he said. “The group given immediate antibiotics in the study by Gharbi and colleagues likely contained some patients managed in this way.”
Patients who apparently had no prescription in this retrospective analysis may have had a same-day admission with a bloodstream infection; moreover, a number of bloodstream infections in older people are due to urinary tract bacteria, and so would not be prevented by treatment for urinary tract infection, Dr. Hay said.
“Further research is needed to establish whether treatment should be initiated with a broad or a narrow spectrum antibiotic and to identify those in whom delaying treatment (while awaiting investigation) is safe,” he concluded.
Dr. Hay is a professor in the Centre for Academic Primary Care, University of Bristol, England. His editorial appears in The BMJ (2019 Feb 27. doi: 10.1136/bmj.l780). Dr. Hay declared that he is a member of the managing common infections guideline committee for the National Institute for Health and Care Excellence (NICE).
results of a large, population-based study suggest.
The risk of bloodstream infection was more than seven times greater in patients who did not receive antibiotics immediately after seeing a general practitioner for a UTI versus those who did, according to results of the study based on primary care records and other data for nearly 160,000 U.K. patients aged 65 years or older. Death rates and hospital admissions were significantly higher for these patients, according to the study published in The BMJ by Myriam Gharbi, PharmD, Phd, Imperial College London, and her colleagues.
The publication of these findings coincides with an increase in Escherichia coli bloodstream infections in England.
“Our study suggests the early initiation of antibiotics for UTI in older high risk adult populations (especially men aged [older than] 85 years) should be recommended to prevent serious complications,” Dr. Gharbi and her coauthors said in their report.
The population-based cohort study comprised 157,264 adult primary care patients at least 65 years of age who had one or more suspected or confirmed lower UTIs from November 2007 to May 2015. The researchers found that health care providers had diagnosed a total of 312,896 UTI episodes in these patients during the period they studied. In 7.2% (22,534) of the UTI episodes, the researchers were unable to find records of the patients having been prescribed antibiotics by a general practitioner within 7 days of the UTI diagnosis. These 22,534 episodes included those that occurred in patients who had a complication before an antibiotic was prescribed. An additional 6.2% (19,292) of the episodes occurred in patients who were prescribed antibiotics, but not during their first UTI-related visit to a general practitioner or on the same day of such a visit. The researchers classified this group of patients as having been prescribed antibiotics on a deferred or delayed basis, as they were not prescribed such drugs within 7 days of their visit.
Overall, there were 1,539 cases (0.5% of the total number of UTIs) of bloodstream infection within 60 days of the initial urinary tract infection diagnosis, the researchers reported.
The bloodstream infection rate was 2.9% for patients who were not prescribed antibiotics ever or prior to an infection occurring, 2.2% in those who were prescribed antibiotics on a deferred basis, and 0.2% in those who were prescribed antibiotics immediately, meaning during their first visit to a general practitioner for a UTI or on the same day of such a visit (P less than .001). After adjustment for potential confounding variables such as age, sex, and region, the patients classified as having not been prescribed antibiotics or having been prescribed antibiotics on a deferred basis were significantly more likely to have a bloodstream infection within 60 days of their visit to a health care provider, compared with those who received antibiotics immediately, with odds ratios of 8.08 (95% confidence interval, 7.12-9.16) and 7.12 (95% CI, 6.22-8.14), respectively.
Hospital admissions after a UTI episode were nearly twice as high in the no- or deferred-antibiotics groups (27.0% and 26.8%, respectively), compared with the group that received antibiotics right away (14.8%), the investigators reported. The lengths of hospital stays were 12.1 days for the group classified as having not been prescribed antibiotics, 7.7 days for the group subject to delayed antibiotic prescribing, and 6.3 days for the group who received antibiotics immediately.
Deaths within 60 days of experiencing a urinary tract infection occurred in 5.4% of patients in the no-antibiotics group, 2.8% of the deferred-antibiotics group, and 1.6% of the immediate-antibiotics group. After adjustment for covariates, a regression analysis showed the risks for all-cause mortality were 1.16 and 2.18 times higher in the deferred-antibiotics group and the no-antibiotics group, respectively, according to the paper.
In the immediate-antibiotics group, those patients who received nitrofurantoin had a “small but significant increase” in 60-day survival versus those who received trimethoprim, the investigators noted in the discussion section of their report.
“This increase could reflect either higher levels of resistance to trimethoprim or a healthier population treated with nitrofurantoin, the latest being not recommended for patients with poor kidney function,” the researchers wrote.
This study was supported by the National Institute for Health Research and other U.K. sources. One study coauthor reported working as an epidemiologist with GSK in areas not related to the study.
SOURCE: Gharbi M et al. BMJ. 2019 Feb 27. doi: 10.1136/bmj.l525.
results of a large, population-based study suggest.
The risk of bloodstream infection was more than seven times greater in patients who did not receive antibiotics immediately after seeing a general practitioner for a UTI versus those who did, according to results of the study based on primary care records and other data for nearly 160,000 U.K. patients aged 65 years or older. Death rates and hospital admissions were significantly higher for these patients, according to the study published in The BMJ by Myriam Gharbi, PharmD, Phd, Imperial College London, and her colleagues.
The publication of these findings coincides with an increase in Escherichia coli bloodstream infections in England.
“Our study suggests the early initiation of antibiotics for UTI in older high risk adult populations (especially men aged [older than] 85 years) should be recommended to prevent serious complications,” Dr. Gharbi and her coauthors said in their report.
The population-based cohort study comprised 157,264 adult primary care patients at least 65 years of age who had one or more suspected or confirmed lower UTIs from November 2007 to May 2015. The researchers found that health care providers had diagnosed a total of 312,896 UTI episodes in these patients during the period they studied. In 7.2% (22,534) of the UTI episodes, the researchers were unable to find records of the patients having been prescribed antibiotics by a general practitioner within 7 days of the UTI diagnosis. These 22,534 episodes included those that occurred in patients who had a complication before an antibiotic was prescribed. An additional 6.2% (19,292) of the episodes occurred in patients who were prescribed antibiotics, but not during their first UTI-related visit to a general practitioner or on the same day of such a visit. The researchers classified this group of patients as having been prescribed antibiotics on a deferred or delayed basis, as they were not prescribed such drugs within 7 days of their visit.
Overall, there were 1,539 cases (0.5% of the total number of UTIs) of bloodstream infection within 60 days of the initial urinary tract infection diagnosis, the researchers reported.
The bloodstream infection rate was 2.9% for patients who were not prescribed antibiotics ever or prior to an infection occurring, 2.2% in those who were prescribed antibiotics on a deferred basis, and 0.2% in those who were prescribed antibiotics immediately, meaning during their first visit to a general practitioner for a UTI or on the same day of such a visit (P less than .001). After adjustment for potential confounding variables such as age, sex, and region, the patients classified as having not been prescribed antibiotics or having been prescribed antibiotics on a deferred basis were significantly more likely to have a bloodstream infection within 60 days of their visit to a health care provider, compared with those who received antibiotics immediately, with odds ratios of 8.08 (95% confidence interval, 7.12-9.16) and 7.12 (95% CI, 6.22-8.14), respectively.
Hospital admissions after a UTI episode were nearly twice as high in the no- or deferred-antibiotics groups (27.0% and 26.8%, respectively), compared with the group that received antibiotics right away (14.8%), the investigators reported. The lengths of hospital stays were 12.1 days for the group classified as having not been prescribed antibiotics, 7.7 days for the group subject to delayed antibiotic prescribing, and 6.3 days for the group who received antibiotics immediately.
Deaths within 60 days of experiencing a urinary tract infection occurred in 5.4% of patients in the no-antibiotics group, 2.8% of the deferred-antibiotics group, and 1.6% of the immediate-antibiotics group. After adjustment for covariates, a regression analysis showed the risks for all-cause mortality were 1.16 and 2.18 times higher in the deferred-antibiotics group and the no-antibiotics group, respectively, according to the paper.
In the immediate-antibiotics group, those patients who received nitrofurantoin had a “small but significant increase” in 60-day survival versus those who received trimethoprim, the investigators noted in the discussion section of their report.
“This increase could reflect either higher levels of resistance to trimethoprim or a healthier population treated with nitrofurantoin, the latest being not recommended for patients with poor kidney function,” the researchers wrote.
This study was supported by the National Institute for Health Research and other U.K. sources. One study coauthor reported working as an epidemiologist with GSK in areas not related to the study.
SOURCE: Gharbi M et al. BMJ. 2019 Feb 27. doi: 10.1136/bmj.l525.
FROM THE BMJ
Big pharma says it can’t drop drug list prices alone
Top pharmaceutical executives expressed willingness to lower the list prices of their drugs, but only if there were cooperation among all sectors to reform how drugs get from manufacturer to patient.

That theme was common in the testimony of seven pharmaceutical executives before the Senate Finance Committee during a Feb. 26 hearing.
“We are in a system that used to be fit for purpose and really drove enormous savings over the last few years but it is no longer fit for purpose,” Pascal Soriot, executive director and CEO of AstraZeneca, testified before the committee. “It’s one of those situations where nobody in the system can do anything, can fix it by themselves.”
The problem, the executives agreed, is the financial structure of drug delivery that ties list prices and their associated rebates to formulary placement.
“If you went back a few years ago, when we negotiated to get our drugs on formulary, our goal was to have the lowest copay by patients,” Kenneth Frazier, chairman and CEO of Merck, testified before the committee. “Today, the goal is to pay into the supply chain the biggest rebate. That actually puts the patient at a disadvantage since they are the only ones that are paying a portion of the list price. The list price is actually working against the patient.”
When asked why the list prices of prescription drugs are so high, Olivier Brandicourt, MD, CEO of Sanofi, said, “We are trying to get formulary position with those high list price-high rebate. It’s a preferred position. Unfortunately that preferred position doesn’t automatically ensure affordability.”
Mr. Frazier added that if a manufacturer brings a product “with a low list price in this system, you get punished financially and you get no uptake because everyone in the supply chain makes money as a result of a higher list price.”
Executives noted that when accounting for financial incentives such as rebates, discounts, and coupons, net prices for pharmaceuticals have actually come down even as list prices are on the rise to accommodate competition on formulary placement.
But that is obscured at the pharmacy counter, where patients are paying higher and higher out-of-pocket costs because more often than not, payment is tied to the list price of the drug, not the net price after all rebates and other discounts have been taken into consideration.
This is a particular problem in Medicare Part D, said AbbVie Chairman and CEO Richard Gonzalez.
“Due to the structure of the Part D benefit design, patients are charged out-of-pocket costs on a medicine’s list price which does not reflect the market-based rebates that Medicare receives,” he testified.
Despite acknowledging that this is a problem, the executives gathered were hesitant to commit to simply lowering the list prices, or anything for that matter.
The closest the panel came to a commitment to lowering the list prices of their drugs was to do so if all rebates went away in both the public and private sector.
But beyond that, the pharma executives continued to assign responsibility for high out-of-pocket drug costs to other players in the health care system, adding that the only way to change the situation would be to have everyone come to the table simultaneously.
“I understand the dissatisfaction with our industry,” Mr. Frazier said. “I understand why patients are frustrated because they need these medicines and they can’t afford them. I would pledge to do everything that we could, but I would urge you to recognize that the system itself is complex and it is interdependent and no one company can unilaterally lower list prices without running into financial and operating disadvantages that make it impossible to do that. But if we all bring the parties together around the table with the goal of doing what’s best for the patient, I think we can some up with a system that works for all Americans.”
Ultimately, the panel suggested, legislation is going to be required to change the system.
Top pharmaceutical executives expressed willingness to lower the list prices of their drugs, but only if there were cooperation among all sectors to reform how drugs get from manufacturer to patient.
That theme was common in the testimony of seven pharmaceutical executives before the Senate Finance Committee during a Feb. 26 hearing.
“We are in a system that used to be fit for purpose and really drove enormous savings over the last few years but it is no longer fit for purpose,” Pascal Soriot, executive director and CEO of AstraZeneca, testified before the committee. “It’s one of those situations where nobody in the system can do anything, can fix it by themselves.”
The problem, the executives agreed, is the financial structure of drug delivery that ties list prices and their associated rebates to formulary placement.
“If you went back a few years ago, when we negotiated to get our drugs on formulary, our goal was to have the lowest copay by patients,” Kenneth Frazier, chairman and CEO of Merck, testified before the committee. “Today, the goal is to pay into the supply chain the biggest rebate. That actually puts the patient at a disadvantage since they are the only ones that are paying a portion of the list price. The list price is actually working against the patient.”
When asked why the list prices of prescription drugs are so high, Olivier Brandicourt, MD, CEO of Sanofi, said, “We are trying to get formulary position with those high list price-high rebate. It’s a preferred position. Unfortunately that preferred position doesn’t automatically ensure affordability.”
Mr. Frazier added that if a manufacturer brings a product “with a low list price in this system, you get punished financially and you get no uptake because everyone in the supply chain makes money as a result of a higher list price.”
Executives noted that when accounting for financial incentives such as rebates, discounts, and coupons, net prices for pharmaceuticals have actually come down even as list prices are on the rise to accommodate competition on formulary placement.
But that is obscured at the pharmacy counter, where patients are paying higher and higher out-of-pocket costs because more often than not, payment is tied to the list price of the drug, not the net price after all rebates and other discounts have been taken into consideration.
This is a particular problem in Medicare Part D, said AbbVie Chairman and CEO Richard Gonzalez.
“Due to the structure of the Part D benefit design, patients are charged out-of-pocket costs on a medicine’s list price which does not reflect the market-based rebates that Medicare receives,” he testified.
Despite acknowledging that this is a problem, the executives gathered were hesitant to commit to simply lowering the list prices, or anything for that matter.
The closest the panel came to a commitment to lowering the list prices of their drugs was to do so if all rebates went away in both the public and private sector.
But beyond that, the pharma executives continued to assign responsibility for high out-of-pocket drug costs to other players in the health care system, adding that the only way to change the situation would be to have everyone come to the table simultaneously.
“I understand the dissatisfaction with our industry,” Mr. Frazier said. “I understand why patients are frustrated because they need these medicines and they can’t afford them. I would pledge to do everything that we could, but I would urge you to recognize that the system itself is complex and it is interdependent and no one company can unilaterally lower list prices without running into financial and operating disadvantages that make it impossible to do that. But if we all bring the parties together around the table with the goal of doing what’s best for the patient, I think we can some up with a system that works for all Americans.”
Ultimately, the panel suggested, legislation is going to be required to change the system.
Top pharmaceutical executives expressed willingness to lower the list prices of their drugs, but only if there were cooperation among all sectors to reform how drugs get from manufacturer to patient.
That theme was common in the testimony of seven pharmaceutical executives before the Senate Finance Committee during a Feb. 26 hearing.
“We are in a system that used to be fit for purpose and really drove enormous savings over the last few years but it is no longer fit for purpose,” Pascal Soriot, executive director and CEO of AstraZeneca, testified before the committee. “It’s one of those situations where nobody in the system can do anything, can fix it by themselves.”
The problem, the executives agreed, is the financial structure of drug delivery that ties list prices and their associated rebates to formulary placement.
“If you went back a few years ago, when we negotiated to get our drugs on formulary, our goal was to have the lowest copay by patients,” Kenneth Frazier, chairman and CEO of Merck, testified before the committee. “Today, the goal is to pay into the supply chain the biggest rebate. That actually puts the patient at a disadvantage since they are the only ones that are paying a portion of the list price. The list price is actually working against the patient.”
When asked why the list prices of prescription drugs are so high, Olivier Brandicourt, MD, CEO of Sanofi, said, “We are trying to get formulary position with those high list price-high rebate. It’s a preferred position. Unfortunately that preferred position doesn’t automatically ensure affordability.”
Mr. Frazier added that if a manufacturer brings a product “with a low list price in this system, you get punished financially and you get no uptake because everyone in the supply chain makes money as a result of a higher list price.”
Executives noted that when accounting for financial incentives such as rebates, discounts, and coupons, net prices for pharmaceuticals have actually come down even as list prices are on the rise to accommodate competition on formulary placement.
But that is obscured at the pharmacy counter, where patients are paying higher and higher out-of-pocket costs because more often than not, payment is tied to the list price of the drug, not the net price after all rebates and other discounts have been taken into consideration.
This is a particular problem in Medicare Part D, said AbbVie Chairman and CEO Richard Gonzalez.
“Due to the structure of the Part D benefit design, patients are charged out-of-pocket costs on a medicine’s list price which does not reflect the market-based rebates that Medicare receives,” he testified.
Despite acknowledging that this is a problem, the executives gathered were hesitant to commit to simply lowering the list prices, or anything for that matter.
The closest the panel came to a commitment to lowering the list prices of their drugs was to do so if all rebates went away in both the public and private sector.
But beyond that, the pharma executives continued to assign responsibility for high out-of-pocket drug costs to other players in the health care system, adding that the only way to change the situation would be to have everyone come to the table simultaneously.
“I understand the dissatisfaction with our industry,” Mr. Frazier said. “I understand why patients are frustrated because they need these medicines and they can’t afford them. I would pledge to do everything that we could, but I would urge you to recognize that the system itself is complex and it is interdependent and no one company can unilaterally lower list prices without running into financial and operating disadvantages that make it impossible to do that. But if we all bring the parties together around the table with the goal of doing what’s best for the patient, I think we can some up with a system that works for all Americans.”
Ultimately, the panel suggested, legislation is going to be required to change the system.
REPORTING FROM SENATE FINANCE COMMITTEE HEARING
Antidepressants may be best add-on to antipsychotics in schizophrenia
Antidepressants could be the best adjunctive treatment for adult outpatients with schizophrenia who are taking a second-generation antipsychotic and need a change in medication, results of an observational study suggest. Patients who added antidepressants to their treatment had a lower risk of psychiatric hospitalization and emergency room visits than did those who tried an alternative antipsychotic, and those who took mood stabilizers and benzodiazepines were significantly more likely to die over 365 days.
Specifically, “the possibility that adjunctive use of gabapentin is associated with increased risk of death raises a serious concern,” wrote T. Scott Stroup, MD, MPH, of the department of psychiatry at Columbia University, New York, and his associates in JAMA Psychiatry.
Often, Dr. Stroup and his associates noted, second-generation antipsychotics often are insufficient to alleviate symptoms and leave patients with functional limitations. said Dr. Stroup, who is also affiliated with the New York State Psychiatric Institute, and his associates.
Using a Medicaid database, the researchers retrospectively tracked 81,921 outpatients with schizophrenia (aged 18-64 years; mean age, 41 years; 46% women) who were treated with a single antipsychotic from 2001-2010. Each patient added an antidepressant (31,117), a benzodiazepine (11,941), a mood stabilizer (12,849), or another second-generation antipsychotic (26,014).
The researchers examined treatment outcomes over a yearlong period after patients began their new treatment and compared the various groups to the reference group (those who began taking an additional antipsychotic medication).
Compared with the reference group, patients who took an antidepressant had a lower risk of psychiatric hospitalization (hazard ratio, 0.84; 95% confidence interval, 0.80-0.88), while the benzodiazepine group had a higher risk (HR, 1.08; 95% CI, 1.02-1.15), and the mood stabilizer group saw no major difference (HR, 0.98; 95% CI, 0.94-1.03). Similar results were found for the risk of psychiatric emergency department visits, compared with the reference group: The HR with the addition of an antidepressant was 0.92 (95% CI, 0.88-0.96), 1.12 with a benzodiazepine (95% CI, 1.07-1.19), and 0.99 with a mood stabilizer (95% CI, 0.94-1.04).
In regard to mortality, the researchers found that mood stabilizers and benzodiazepines stood apart on the risk front with HRs of 1.31 (95% CI, 1.04-1.66) and 1.22 (95% CI, 0.98-1.52), respectively. Among mood stabilizer use, Gabapentin accounted for 1,755 initiations (13.7%) and was associated with 45 deaths (28.0%), the researchers reported. “No other mood stabilizer appeared to be associated with a higher rate of death than the others.”
Dr. Stroup and his associates cited several limitations. One is that the results might not be generalizable because the investigators looked only at patients who were enrolled in the Medicaid program. Nevertheless, “improved pharmacologic treatment of schizophrenia and consequent reduced need for hospitalization and ED visits associated with more antidepressant and less benzodiazepine use would represent a significant benefit for individuals and for public health,” they wrote.
The study authors reported various relationships with drugmakers, including Auspex, Intra-Cellular Therapies, Eli Lilly, Bristol-Myers Squibb, and Merck. The study was funded by a Patient-Centered Outcomes Research Institute award.
SOURCE: Stroup TS et al. JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4489.
Much of the research into adjunctive therapy in schizophrenia is of poor quality, and other hurdles make it difficult to understand the best treatment approach. The new study links the addition of an antidepressant to a substantial lowering of psychiatric hospitalization risk, compared with initiating another antipsychotic, wrote Donald C. Goff, MD. Previous randomized controlled trials (RCTs) have suggested that adding on antidepressants can moderately reduce symptoms – mainly negative ones – in schizophrenia. The study findings are preliminary and suggest that an RCT is in order.
Dr. Goff disclosed grants from Avanir.
These statements are based on an accompanying editorial by Dr. Goff of New York University (JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4318).
Much of the research into adjunctive therapy in schizophrenia is of poor quality, and other hurdles make it difficult to understand the best treatment approach. The new study links the addition of an antidepressant to a substantial lowering of psychiatric hospitalization risk, compared with initiating another antipsychotic, wrote Donald C. Goff, MD. Previous randomized controlled trials (RCTs) have suggested that adding on antidepressants can moderately reduce symptoms – mainly negative ones – in schizophrenia. The study findings are preliminary and suggest that an RCT is in order.
Dr. Goff disclosed grants from Avanir.
These statements are based on an accompanying editorial by Dr. Goff of New York University (JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4318).
Much of the research into adjunctive therapy in schizophrenia is of poor quality, and other hurdles make it difficult to understand the best treatment approach. The new study links the addition of an antidepressant to a substantial lowering of psychiatric hospitalization risk, compared with initiating another antipsychotic, wrote Donald C. Goff, MD. Previous randomized controlled trials (RCTs) have suggested that adding on antidepressants can moderately reduce symptoms – mainly negative ones – in schizophrenia. The study findings are preliminary and suggest that an RCT is in order.
Dr. Goff disclosed grants from Avanir.
These statements are based on an accompanying editorial by Dr. Goff of New York University (JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4318).
Antidepressants could be the best adjunctive treatment for adult outpatients with schizophrenia who are taking a second-generation antipsychotic and need a change in medication, results of an observational study suggest. Patients who added antidepressants to their treatment had a lower risk of psychiatric hospitalization and emergency room visits than did those who tried an alternative antipsychotic, and those who took mood stabilizers and benzodiazepines were significantly more likely to die over 365 days.
Specifically, “the possibility that adjunctive use of gabapentin is associated with increased risk of death raises a serious concern,” wrote T. Scott Stroup, MD, MPH, of the department of psychiatry at Columbia University, New York, and his associates in JAMA Psychiatry.
Often, Dr. Stroup and his associates noted, second-generation antipsychotics often are insufficient to alleviate symptoms and leave patients with functional limitations. said Dr. Stroup, who is also affiliated with the New York State Psychiatric Institute, and his associates.
Using a Medicaid database, the researchers retrospectively tracked 81,921 outpatients with schizophrenia (aged 18-64 years; mean age, 41 years; 46% women) who were treated with a single antipsychotic from 2001-2010. Each patient added an antidepressant (31,117), a benzodiazepine (11,941), a mood stabilizer (12,849), or another second-generation antipsychotic (26,014).
The researchers examined treatment outcomes over a yearlong period after patients began their new treatment and compared the various groups to the reference group (those who began taking an additional antipsychotic medication).
Compared with the reference group, patients who took an antidepressant had a lower risk of psychiatric hospitalization (hazard ratio, 0.84; 95% confidence interval, 0.80-0.88), while the benzodiazepine group had a higher risk (HR, 1.08; 95% CI, 1.02-1.15), and the mood stabilizer group saw no major difference (HR, 0.98; 95% CI, 0.94-1.03). Similar results were found for the risk of psychiatric emergency department visits, compared with the reference group: The HR with the addition of an antidepressant was 0.92 (95% CI, 0.88-0.96), 1.12 with a benzodiazepine (95% CI, 1.07-1.19), and 0.99 with a mood stabilizer (95% CI, 0.94-1.04).
In regard to mortality, the researchers found that mood stabilizers and benzodiazepines stood apart on the risk front with HRs of 1.31 (95% CI, 1.04-1.66) and 1.22 (95% CI, 0.98-1.52), respectively. Among mood stabilizer use, Gabapentin accounted for 1,755 initiations (13.7%) and was associated with 45 deaths (28.0%), the researchers reported. “No other mood stabilizer appeared to be associated with a higher rate of death than the others.”
Dr. Stroup and his associates cited several limitations. One is that the results might not be generalizable because the investigators looked only at patients who were enrolled in the Medicaid program. Nevertheless, “improved pharmacologic treatment of schizophrenia and consequent reduced need for hospitalization and ED visits associated with more antidepressant and less benzodiazepine use would represent a significant benefit for individuals and for public health,” they wrote.
The study authors reported various relationships with drugmakers, including Auspex, Intra-Cellular Therapies, Eli Lilly, Bristol-Myers Squibb, and Merck. The study was funded by a Patient-Centered Outcomes Research Institute award.
SOURCE: Stroup TS et al. JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4489.
Antidepressants could be the best adjunctive treatment for adult outpatients with schizophrenia who are taking a second-generation antipsychotic and need a change in medication, results of an observational study suggest. Patients who added antidepressants to their treatment had a lower risk of psychiatric hospitalization and emergency room visits than did those who tried an alternative antipsychotic, and those who took mood stabilizers and benzodiazepines were significantly more likely to die over 365 days.
Specifically, “the possibility that adjunctive use of gabapentin is associated with increased risk of death raises a serious concern,” wrote T. Scott Stroup, MD, MPH, of the department of psychiatry at Columbia University, New York, and his associates in JAMA Psychiatry.
Often, Dr. Stroup and his associates noted, second-generation antipsychotics often are insufficient to alleviate symptoms and leave patients with functional limitations. said Dr. Stroup, who is also affiliated with the New York State Psychiatric Institute, and his associates.
Using a Medicaid database, the researchers retrospectively tracked 81,921 outpatients with schizophrenia (aged 18-64 years; mean age, 41 years; 46% women) who were treated with a single antipsychotic from 2001-2010. Each patient added an antidepressant (31,117), a benzodiazepine (11,941), a mood stabilizer (12,849), or another second-generation antipsychotic (26,014).
The researchers examined treatment outcomes over a yearlong period after patients began their new treatment and compared the various groups to the reference group (those who began taking an additional antipsychotic medication).
Compared with the reference group, patients who took an antidepressant had a lower risk of psychiatric hospitalization (hazard ratio, 0.84; 95% confidence interval, 0.80-0.88), while the benzodiazepine group had a higher risk (HR, 1.08; 95% CI, 1.02-1.15), and the mood stabilizer group saw no major difference (HR, 0.98; 95% CI, 0.94-1.03). Similar results were found for the risk of psychiatric emergency department visits, compared with the reference group: The HR with the addition of an antidepressant was 0.92 (95% CI, 0.88-0.96), 1.12 with a benzodiazepine (95% CI, 1.07-1.19), and 0.99 with a mood stabilizer (95% CI, 0.94-1.04).
In regard to mortality, the researchers found that mood stabilizers and benzodiazepines stood apart on the risk front with HRs of 1.31 (95% CI, 1.04-1.66) and 1.22 (95% CI, 0.98-1.52), respectively. Among mood stabilizer use, Gabapentin accounted for 1,755 initiations (13.7%) and was associated with 45 deaths (28.0%), the researchers reported. “No other mood stabilizer appeared to be associated with a higher rate of death than the others.”
Dr. Stroup and his associates cited several limitations. One is that the results might not be generalizable because the investigators looked only at patients who were enrolled in the Medicaid program. Nevertheless, “improved pharmacologic treatment of schizophrenia and consequent reduced need for hospitalization and ED visits associated with more antidepressant and less benzodiazepine use would represent a significant benefit for individuals and for public health,” they wrote.
The study authors reported various relationships with drugmakers, including Auspex, Intra-Cellular Therapies, Eli Lilly, Bristol-Myers Squibb, and Merck. The study was funded by a Patient-Centered Outcomes Research Institute award.
SOURCE: Stroup TS et al. JAMA Psychiatry. 2019 Feb 20. doi: 10.1001/jamapsychiatry.2018.4489.
FROM JAMA PSYCHIATRY
McAneny: Transparency needed for meaningful talk on drug pricing
WASHINGTON – As the rising cost of prescription drugs continues to garner heightened scrutiny from the federal government, one thing is missing from the conversation that would make any solution more effective, according to American Medical Association President Barbara L. McAneny, MD.

“We would like to see transparency from end to end in this pipeline because that is the way we will have the ability to look for savings,” she said in an interview at a national advocacy conference sponsored by the American Medical Association.
“I think we don’t have the information we need to make rational decisions,” she said. “I think the first thing we need to do is to understand the entire pipeline from the basic research that results in a drug’s clinical trials that results in a new drug to the pharmacy benefit managers and all of the ways that they have increased the cost of the drugs all the way to when the patient actually gets it.”
In particular, Dr. McAneny targeted the need for transparency in the role and financial impact pharmacy benefit managers have on the cost of prescription drugs.
“We were told at our state advocacy conference, which we held in January, by an expert who studied pharmacy benefit managers, that 42% of the cost of any drug is attributable to the profits of pharmacy benefit managers,” she said. “To me, that makes me wonder what value do they add that is worth 42% of these exorbitant costs and if they are not adding value, why do we have them in this process?”
She also called on the pharmaceutical manufacturers to be more forthcoming with their financial information regarding marketing and advertising, but she stressed that it needs to be done in a way that does not hinder future development of life-saving therapies.
“We do not want to stifle innovation.” Dr. McAneny said. “As a cancer doctor, I have seen diseases that I used to treat with morphine and sympathy now be diseases that I can treat, where I can restore people to good quality of life and buy them additional years of life because of these drugs. They are amazing and I don’t want to do without them and I want more of them. ... We want to support that research. That is probably worth a lot of the price tag. But how much of that goes to direct-to-consumer advertising? How much of that is the advertising budget? Where can we cut some of this out of the system so that we get the innovation, but we get innovation that all of our patients can afford?”
Another issue that is looming for physicians is a payment rate freeze from 2020-2025 under the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program, which was created under the Medicare Access and CHIP Reauthorization Act (MACRA).
“A 5-year freeze when my expenses do not freeze is a terrifying thing and could be a practice-ending expense for a lot of practices,” Dr. McAneny warned. “I will point out that independent practices, if they sell to a hospital, the cost of care immediately doubles. The amount that is paid for it, not the cost of delivering it. So that is not a great solution. But we also have this increasing practice expense and a flat rate is not going to help practices survive. So I am very concerned.”
Related to the QPP, Dr. McAneny also wants to see the Centers for Medicare & Medicaid Services do more with physician-developed alternative payment models. The Physician-Focused Payment Model Technical Advisory Committee (PTAC) continues to review and evaluate submissions, but to date, the CMS has yet to implement any of the committee’s recommendations.
“I read all of the PTAC submissions that went to CMS. Some of them I thought were a little on the weak side, but there are a lot of them that I thought were really good ideas. I do not know why CMS did not approve them and fund them and let them be tried,” she said, although she did offer an opinion on why the agency has yet to act on any of them.
“I read their paper saying why they didn’t approve [the recommended physician-developed alternative payment models]. It seemed to me they are waiting for one silver bullet that will fix all of health care. I don’t think a silver bullet is going to fix health care. It’s not an issue. It’s a complicated set of issues. You will not have a quick fix.”
WASHINGTON – As the rising cost of prescription drugs continues to garner heightened scrutiny from the federal government, one thing is missing from the conversation that would make any solution more effective, according to American Medical Association President Barbara L. McAneny, MD.
“We would like to see transparency from end to end in this pipeline because that is the way we will have the ability to look for savings,” she said in an interview at a national advocacy conference sponsored by the American Medical Association.
“I think we don’t have the information we need to make rational decisions,” she said. “I think the first thing we need to do is to understand the entire pipeline from the basic research that results in a drug’s clinical trials that results in a new drug to the pharmacy benefit managers and all of the ways that they have increased the cost of the drugs all the way to when the patient actually gets it.”
In particular, Dr. McAneny targeted the need for transparency in the role and financial impact pharmacy benefit managers have on the cost of prescription drugs.
“We were told at our state advocacy conference, which we held in January, by an expert who studied pharmacy benefit managers, that 42% of the cost of any drug is attributable to the profits of pharmacy benefit managers,” she said. “To me, that makes me wonder what value do they add that is worth 42% of these exorbitant costs and if they are not adding value, why do we have them in this process?”
She also called on the pharmaceutical manufacturers to be more forthcoming with their financial information regarding marketing and advertising, but she stressed that it needs to be done in a way that does not hinder future development of life-saving therapies.
“We do not want to stifle innovation.” Dr. McAneny said. “As a cancer doctor, I have seen diseases that I used to treat with morphine and sympathy now be diseases that I can treat, where I can restore people to good quality of life and buy them additional years of life because of these drugs. They are amazing and I don’t want to do without them and I want more of them. ... We want to support that research. That is probably worth a lot of the price tag. But how much of that goes to direct-to-consumer advertising? How much of that is the advertising budget? Where can we cut some of this out of the system so that we get the innovation, but we get innovation that all of our patients can afford?”
Another issue that is looming for physicians is a payment rate freeze from 2020-2025 under the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program, which was created under the Medicare Access and CHIP Reauthorization Act (MACRA).
“A 5-year freeze when my expenses do not freeze is a terrifying thing and could be a practice-ending expense for a lot of practices,” Dr. McAneny warned. “I will point out that independent practices, if they sell to a hospital, the cost of care immediately doubles. The amount that is paid for it, not the cost of delivering it. So that is not a great solution. But we also have this increasing practice expense and a flat rate is not going to help practices survive. So I am very concerned.”
Related to the QPP, Dr. McAneny also wants to see the Centers for Medicare & Medicaid Services do more with physician-developed alternative payment models. The Physician-Focused Payment Model Technical Advisory Committee (PTAC) continues to review and evaluate submissions, but to date, the CMS has yet to implement any of the committee’s recommendations.
“I read all of the PTAC submissions that went to CMS. Some of them I thought were a little on the weak side, but there are a lot of them that I thought were really good ideas. I do not know why CMS did not approve them and fund them and let them be tried,” she said, although she did offer an opinion on why the agency has yet to act on any of them.
“I read their paper saying why they didn’t approve [the recommended physician-developed alternative payment models]. It seemed to me they are waiting for one silver bullet that will fix all of health care. I don’t think a silver bullet is going to fix health care. It’s not an issue. It’s a complicated set of issues. You will not have a quick fix.”
WASHINGTON – As the rising cost of prescription drugs continues to garner heightened scrutiny from the federal government, one thing is missing from the conversation that would make any solution more effective, according to American Medical Association President Barbara L. McAneny, MD.
“We would like to see transparency from end to end in this pipeline because that is the way we will have the ability to look for savings,” she said in an interview at a national advocacy conference sponsored by the American Medical Association.
“I think we don’t have the information we need to make rational decisions,” she said. “I think the first thing we need to do is to understand the entire pipeline from the basic research that results in a drug’s clinical trials that results in a new drug to the pharmacy benefit managers and all of the ways that they have increased the cost of the drugs all the way to when the patient actually gets it.”
In particular, Dr. McAneny targeted the need for transparency in the role and financial impact pharmacy benefit managers have on the cost of prescription drugs.
“We were told at our state advocacy conference, which we held in January, by an expert who studied pharmacy benefit managers, that 42% of the cost of any drug is attributable to the profits of pharmacy benefit managers,” she said. “To me, that makes me wonder what value do they add that is worth 42% of these exorbitant costs and if they are not adding value, why do we have them in this process?”
She also called on the pharmaceutical manufacturers to be more forthcoming with their financial information regarding marketing and advertising, but she stressed that it needs to be done in a way that does not hinder future development of life-saving therapies.
“We do not want to stifle innovation.” Dr. McAneny said. “As a cancer doctor, I have seen diseases that I used to treat with morphine and sympathy now be diseases that I can treat, where I can restore people to good quality of life and buy them additional years of life because of these drugs. They are amazing and I don’t want to do without them and I want more of them. ... We want to support that research. That is probably worth a lot of the price tag. But how much of that goes to direct-to-consumer advertising? How much of that is the advertising budget? Where can we cut some of this out of the system so that we get the innovation, but we get innovation that all of our patients can afford?”
Another issue that is looming for physicians is a payment rate freeze from 2020-2025 under the Merit-based Incentive Payment System (MIPS) track of the Quality Payment Program, which was created under the Medicare Access and CHIP Reauthorization Act (MACRA).
“A 5-year freeze when my expenses do not freeze is a terrifying thing and could be a practice-ending expense for a lot of practices,” Dr. McAneny warned. “I will point out that independent practices, if they sell to a hospital, the cost of care immediately doubles. The amount that is paid for it, not the cost of delivering it. So that is not a great solution. But we also have this increasing practice expense and a flat rate is not going to help practices survive. So I am very concerned.”
Related to the QPP, Dr. McAneny also wants to see the Centers for Medicare & Medicaid Services do more with physician-developed alternative payment models. The Physician-Focused Payment Model Technical Advisory Committee (PTAC) continues to review and evaluate submissions, but to date, the CMS has yet to implement any of the committee’s recommendations.
“I read all of the PTAC submissions that went to CMS. Some of them I thought were a little on the weak side, but there are a lot of them that I thought were really good ideas. I do not know why CMS did not approve them and fund them and let them be tried,” she said, although she did offer an opinion on why the agency has yet to act on any of them.
“I read their paper saying why they didn’t approve [the recommended physician-developed alternative payment models]. It seemed to me they are waiting for one silver bullet that will fix all of health care. I don’t think a silver bullet is going to fix health care. It’s not an issue. It’s a complicated set of issues. You will not have a quick fix.”
REPORTING FROM THE AMA NATIONAL ADVOCACY CONFERENCE
CMS proposes coverage of CAR T-cell therapy in trials
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
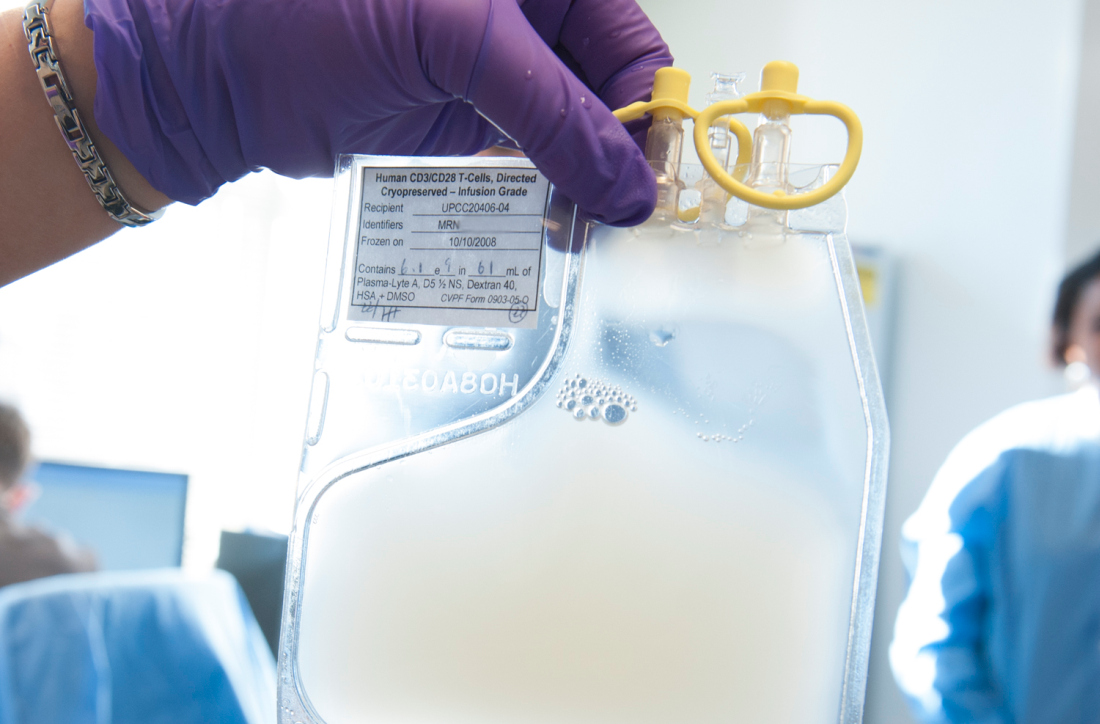
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
The Centers for Medicare & Medicaid Services has proposed to cover chimeric antigen receptor (CAR) T-cell therapy for cancer patients participating in clinical trials that study the treatment’s effectiveness, according to a Feb. 15 announcement.
The proposed national coverage determination would require CMS to cover CAR T-cell therapies nationwide when the treatment is offered in CMS-approved registries or clinical studies in which patients are monitored for 2 or more years following treatment.
Results from the studies would help CMS identify which patients benefit most from CAR T-cell therapies and inform future coverage decisions, CMS Administrator Seema Verma said.
“CAR T-cell therapy was the first FDA-approved gene therapy, marking the beginning of an entirely new approach to treating serious and even life-threatening diseases,” Ms. Verma said in a statement. “Today’s proposed coverage decision would improve access to this therapy while deepening CMS’s understanding of how patients in Medicare respond to it, so the agency can ensure that it is paying for CAR T-cell therapy for cases in which the benefits outweigh the risks.”
As part of the proposal, CMS would cover autologous treatment with T cells expressing at least one chimeric antigen receptor (CAR) through coverage with evidence development when prescribed by a treating oncologist and performed in a hospital, according to a summary of the proposal.
The patient and hospital must meet specific criteria to be eligible for coverage, including that patients have relapsed or refractory cancer and do not have a comorbidity that would otherwise preclude patient benefit.
Hospitals, meanwhile, must have a cellular therapy program consisting of an integrated medical team that includes a clinical program director, a quality manager, and at least one physician experienced in cellular therapy, among other requirements.
CMS also would require that treatment is an FDA-approved biologic, providing targeted therapy for a known antigen expressed in the patient’s cancer according to an FDA indication. Repeat treatment would be covered only when a new primary cancer diagnosis is made by the treating oncologist and certain patient conditions are met.
Both inpatient and outpatient settings for the CAR T-cell therapy treatment are acceptable under the proposal. In either case, the patient and the hospital must be participating in a prospective, national, audited registry that consecutively enrolls patients, accepts all manufactured products, follows the patient for at least 2 years, and addresses a set of approved evidence-development questions. Additionally, all registries must be reviewed and approved by CMS.
The proposed national coverage determination was the result of an Aug. 22, 2018 meeting of the Medicare Evidence Development & Coverage Advisory Committee. The committee provides CMS with an external assessment of the appropriateness of therapies under review.
Public comments about the CAR T-cell therapy proposal will be accepted online here until March 15. A final decision on the proposal is expected by May 2019.
The agency’s proposal follows an Aug. 17 final rule by CMS that sets a new payment scheme for inpatient administration of two CAR T-cell therapies. The rule categorizes CAR T-cell therapies under the umbrella of the renamed Medicare Severity–Diagnosis Related Groups 016 – Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy – and assigns ICD-10 PCS procedure codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019, which began in October 2018. CMS also approved a temporary New Technology Add-On Payment for use of the therapies with a maximum threshold of $186,500.
In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
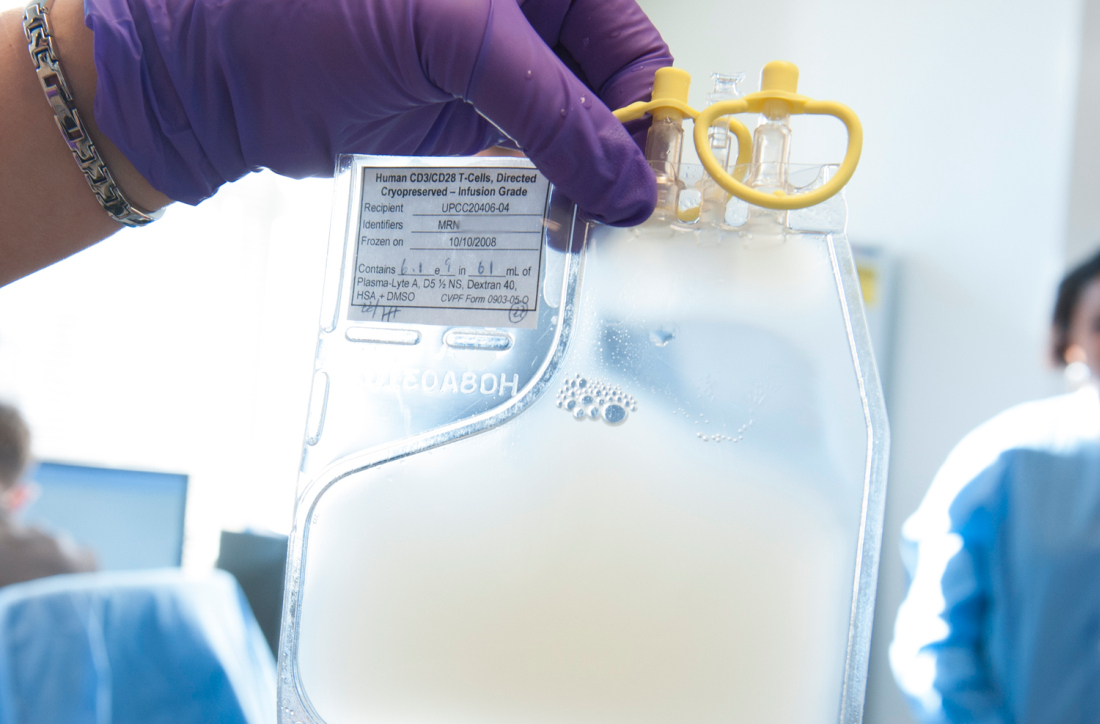
HHS to target step therapy, Stark Law in 2019
WASHINGTON –

Speaking Feb. 12 at the American Medical Association’s National Advocacy Conference, Secretary Azar said the agency will be looking into ensuring that patients on medical plans who have found a working drug after going through a step-therapy protocol will not have to restart on a drug that has already failed for them if they switch insurance providers.
“I was very disturbed to hear that stable patients switching among insurance plans, like switching among Medicare Advantage plans, can often be required to start over again on a step therapy regimen,” he said.
“This is not just potentially injurious to their health, it’s also penny-wise and pound-foolish,” Secretary Azar continued. “We know that getting a patient on the right drug, at the right time, is one of the best investments we can make in their health, and we do not want to impede physicians from making that happen. We’re looking at how we can address that issue now.”
The other area Secretary Azar highlighted that the agency is working on is making changes to the Stark Law.
“The Stark Law was written with noble purposes in mind, but it was designed for a fee-for-service system, not the kind of system we are moving toward today,” he said. “We’ve heard from many, many stakeholders, including the AMA, about the need to update the enumerated exceptions in the Stark Law to include value-based approaches to care.”
He added that how care coordination interacts with the antikickback statutes and HIPAA are also going to be examined.
Secretary Azar did not offer any timelines or other more specific details about how the agency plans to tackle these issues.
He used most of his speech to discuss recent regulatory actions around drug pricing and pushed for support for the Part B drug pricing model that the agency is preparing for a formal proposed rule, despite having received a critical reception from medical societies.
“If you have a small practice that uses infusions, and you don’t want to bear the risk of buy and bill, now you’re off the hook,” he said. “We’ll allow you to work with private vendors who can take the risk for buying the drugs in a way that isn’t possible today. But if you’re part of a much larger practice that’s able to drive a better deal than you could on your own, or want to band together with other practices to do the purchasing, then you can do that, too.”
He continued: “Next is the launch of the actual proposed rule, followed by the rule itself, which, I’ll remind you, is just a model.”
However, despite it being a model under test from the Center for Medicare & Medicaid Innovation, the advanced notice of proposed rule making that was issued in October 2018 suggested that participation in the so-called International Pricing Index model would be mandatory.
Secretary Azar did not acknowledge any mandatory participation in his pitch for support, noting that CMMI models “are carefully assessed. We will closely monitor how the model will affect clinical outcomes, including patients’ adherence to their drugs. We believe that the lower costs will, of course, mean better patient access to drugs, better adherence, and better outcomes for the care you provide. That is the goal.”
WASHINGTON –
Speaking Feb. 12 at the American Medical Association’s National Advocacy Conference, Secretary Azar said the agency will be looking into ensuring that patients on medical plans who have found a working drug after going through a step-therapy protocol will not have to restart on a drug that has already failed for them if they switch insurance providers.
“I was very disturbed to hear that stable patients switching among insurance plans, like switching among Medicare Advantage plans, can often be required to start over again on a step therapy regimen,” he said.
“This is not just potentially injurious to their health, it’s also penny-wise and pound-foolish,” Secretary Azar continued. “We know that getting a patient on the right drug, at the right time, is one of the best investments we can make in their health, and we do not want to impede physicians from making that happen. We’re looking at how we can address that issue now.”
The other area Secretary Azar highlighted that the agency is working on is making changes to the Stark Law.
“The Stark Law was written with noble purposes in mind, but it was designed for a fee-for-service system, not the kind of system we are moving toward today,” he said. “We’ve heard from many, many stakeholders, including the AMA, about the need to update the enumerated exceptions in the Stark Law to include value-based approaches to care.”
He added that how care coordination interacts with the antikickback statutes and HIPAA are also going to be examined.
Secretary Azar did not offer any timelines or other more specific details about how the agency plans to tackle these issues.
He used most of his speech to discuss recent regulatory actions around drug pricing and pushed for support for the Part B drug pricing model that the agency is preparing for a formal proposed rule, despite having received a critical reception from medical societies.
“If you have a small practice that uses infusions, and you don’t want to bear the risk of buy and bill, now you’re off the hook,” he said. “We’ll allow you to work with private vendors who can take the risk for buying the drugs in a way that isn’t possible today. But if you’re part of a much larger practice that’s able to drive a better deal than you could on your own, or want to band together with other practices to do the purchasing, then you can do that, too.”
He continued: “Next is the launch of the actual proposed rule, followed by the rule itself, which, I’ll remind you, is just a model.”
However, despite it being a model under test from the Center for Medicare & Medicaid Innovation, the advanced notice of proposed rule making that was issued in October 2018 suggested that participation in the so-called International Pricing Index model would be mandatory.
Secretary Azar did not acknowledge any mandatory participation in his pitch for support, noting that CMMI models “are carefully assessed. We will closely monitor how the model will affect clinical outcomes, including patients’ adherence to their drugs. We believe that the lower costs will, of course, mean better patient access to drugs, better adherence, and better outcomes for the care you provide. That is the goal.”
WASHINGTON –
Speaking Feb. 12 at the American Medical Association’s National Advocacy Conference, Secretary Azar said the agency will be looking into ensuring that patients on medical plans who have found a working drug after going through a step-therapy protocol will not have to restart on a drug that has already failed for them if they switch insurance providers.
“I was very disturbed to hear that stable patients switching among insurance plans, like switching among Medicare Advantage plans, can often be required to start over again on a step therapy regimen,” he said.
“This is not just potentially injurious to their health, it’s also penny-wise and pound-foolish,” Secretary Azar continued. “We know that getting a patient on the right drug, at the right time, is one of the best investments we can make in their health, and we do not want to impede physicians from making that happen. We’re looking at how we can address that issue now.”
The other area Secretary Azar highlighted that the agency is working on is making changes to the Stark Law.
“The Stark Law was written with noble purposes in mind, but it was designed for a fee-for-service system, not the kind of system we are moving toward today,” he said. “We’ve heard from many, many stakeholders, including the AMA, about the need to update the enumerated exceptions in the Stark Law to include value-based approaches to care.”
He added that how care coordination interacts with the antikickback statutes and HIPAA are also going to be examined.
Secretary Azar did not offer any timelines or other more specific details about how the agency plans to tackle these issues.
He used most of his speech to discuss recent regulatory actions around drug pricing and pushed for support for the Part B drug pricing model that the agency is preparing for a formal proposed rule, despite having received a critical reception from medical societies.
“If you have a small practice that uses infusions, and you don’t want to bear the risk of buy and bill, now you’re off the hook,” he said. “We’ll allow you to work with private vendors who can take the risk for buying the drugs in a way that isn’t possible today. But if you’re part of a much larger practice that’s able to drive a better deal than you could on your own, or want to band together with other practices to do the purchasing, then you can do that, too.”
He continued: “Next is the launch of the actual proposed rule, followed by the rule itself, which, I’ll remind you, is just a model.”
However, despite it being a model under test from the Center for Medicare & Medicaid Innovation, the advanced notice of proposed rule making that was issued in October 2018 suggested that participation in the so-called International Pricing Index model would be mandatory.
Secretary Azar did not acknowledge any mandatory participation in his pitch for support, noting that CMMI models “are carefully assessed. We will closely monitor how the model will affect clinical outcomes, including patients’ adherence to their drugs. We believe that the lower costs will, of course, mean better patient access to drugs, better adherence, and better outcomes for the care you provide. That is the goal.”
REPORTING FROM AMA NATIONAL ADVOCACY CONFERENCE
Cloud of inconsistency hangs over cannabis data
More people are using medical cannabis as it becomes legal in more states, but the lack of standardization in states’ data collection hindered investigators’ efforts to track that use.
Legalized medical cannabis is now available in 33 states and the District of Columbia, and the number of users has risen from just over 72,000 in 2009 to almost 814,000 in 2017. That 814,000, however, covers only 16 states and D.C., since 1 state (Connecticut) does not publish reports on medical cannabis use, 12 did not have statistics available, 2 (New York and Vermont) didn’t report data for 2017, and 2 (California and Maine) have voluntary registries that are unlikely to be accurate, according to Kevin F. Boehnke, PhD, of the University of Michigan, Ann Arbor, and his associates.
Michigan had the largest reported number of patients enrolled in its medical cannabis program in 2017, almost 270,000. California – the state with the oldest medical cannabis legislation (passed in 1996) and the largest overall population but a voluntary cannabis registry – reported its highest number of enrollees, 12,659, in 2009-2010, the investigators said. Colorado had more than 116,000 patients in its medical cannabis program in 2010 (Health Aff. 2019;38[2]:295-302).
The “many inconsistencies in data quality across states [suggest] the need for further standardization of data collection. Such standardization would add transparency to understanding how medical cannabis programs are used, which would help guide both research and policy needs,” Dr. Boehnke and his associates wrote.
More consistency was seen in the reasons for using medical cannabis. Chronic pain made up 62.2% of all qualifying conditions reported by patients during 1999-2016, with the annual average varying between 33.3% and 73%. Multiple sclerosis spasticity symptoms had the second-highest number of reports over the study period, followed by chemotherapy-induced nausea and vomiting, posttraumatic stress disorder, and cancer, they reported.
The investigators also looked at the appropriateness of cannabis and determined that its use in 85.5% of patient-reported conditions was “supported by conclusive or substantial evidence of therapeutic effectiveness, according to the 2017 National Academies report” on the health effects of cannabis.
“We believe not only that it is inappropriate for cannabis to remain a Schedule I substance, but also that state and federal policy makers should begin evaluating evidence-based ways for safely integrating cannabis research and products into the health care system,” they concluded.
SOURCE: Boehnke KF et al. Health Aff. 2019;38(2):295-302.
More people are using medical cannabis as it becomes legal in more states, but the lack of standardization in states’ data collection hindered investigators’ efforts to track that use.
Legalized medical cannabis is now available in 33 states and the District of Columbia, and the number of users has risen from just over 72,000 in 2009 to almost 814,000 in 2017. That 814,000, however, covers only 16 states and D.C., since 1 state (Connecticut) does not publish reports on medical cannabis use, 12 did not have statistics available, 2 (New York and Vermont) didn’t report data for 2017, and 2 (California and Maine) have voluntary registries that are unlikely to be accurate, according to Kevin F. Boehnke, PhD, of the University of Michigan, Ann Arbor, and his associates.
Michigan had the largest reported number of patients enrolled in its medical cannabis program in 2017, almost 270,000. California – the state with the oldest medical cannabis legislation (passed in 1996) and the largest overall population but a voluntary cannabis registry – reported its highest number of enrollees, 12,659, in 2009-2010, the investigators said. Colorado had more than 116,000 patients in its medical cannabis program in 2010 (Health Aff. 2019;38[2]:295-302).
The “many inconsistencies in data quality across states [suggest] the need for further standardization of data collection. Such standardization would add transparency to understanding how medical cannabis programs are used, which would help guide both research and policy needs,” Dr. Boehnke and his associates wrote.
More consistency was seen in the reasons for using medical cannabis. Chronic pain made up 62.2% of all qualifying conditions reported by patients during 1999-2016, with the annual average varying between 33.3% and 73%. Multiple sclerosis spasticity symptoms had the second-highest number of reports over the study period, followed by chemotherapy-induced nausea and vomiting, posttraumatic stress disorder, and cancer, they reported.
The investigators also looked at the appropriateness of cannabis and determined that its use in 85.5% of patient-reported conditions was “supported by conclusive or substantial evidence of therapeutic effectiveness, according to the 2017 National Academies report” on the health effects of cannabis.
“We believe not only that it is inappropriate for cannabis to remain a Schedule I substance, but also that state and federal policy makers should begin evaluating evidence-based ways for safely integrating cannabis research and products into the health care system,” they concluded.
SOURCE: Boehnke KF et al. Health Aff. 2019;38(2):295-302.
More people are using medical cannabis as it becomes legal in more states, but the lack of standardization in states’ data collection hindered investigators’ efforts to track that use.
Legalized medical cannabis is now available in 33 states and the District of Columbia, and the number of users has risen from just over 72,000 in 2009 to almost 814,000 in 2017. That 814,000, however, covers only 16 states and D.C., since 1 state (Connecticut) does not publish reports on medical cannabis use, 12 did not have statistics available, 2 (New York and Vermont) didn’t report data for 2017, and 2 (California and Maine) have voluntary registries that are unlikely to be accurate, according to Kevin F. Boehnke, PhD, of the University of Michigan, Ann Arbor, and his associates.
Michigan had the largest reported number of patients enrolled in its medical cannabis program in 2017, almost 270,000. California – the state with the oldest medical cannabis legislation (passed in 1996) and the largest overall population but a voluntary cannabis registry – reported its highest number of enrollees, 12,659, in 2009-2010, the investigators said. Colorado had more than 116,000 patients in its medical cannabis program in 2010 (Health Aff. 2019;38[2]:295-302).
The “many inconsistencies in data quality across states [suggest] the need for further standardization of data collection. Such standardization would add transparency to understanding how medical cannabis programs are used, which would help guide both research and policy needs,” Dr. Boehnke and his associates wrote.
More consistency was seen in the reasons for using medical cannabis. Chronic pain made up 62.2% of all qualifying conditions reported by patients during 1999-2016, with the annual average varying between 33.3% and 73%. Multiple sclerosis spasticity symptoms had the second-highest number of reports over the study period, followed by chemotherapy-induced nausea and vomiting, posttraumatic stress disorder, and cancer, they reported.
The investigators also looked at the appropriateness of cannabis and determined that its use in 85.5% of patient-reported conditions was “supported by conclusive or substantial evidence of therapeutic effectiveness, according to the 2017 National Academies report” on the health effects of cannabis.
“We believe not only that it is inappropriate for cannabis to remain a Schedule I substance, but also that state and federal policy makers should begin evaluating evidence-based ways for safely integrating cannabis research and products into the health care system,” they concluded.
SOURCE: Boehnke KF et al. Health Aff. 2019;38(2):295-302.
FROM HEALTH AFFAIRS
Heart failure guidelines: What you need to know about the 2017 focused update
In 2017, the American College of Cardiology (ACC), American Heart Association (AHA), and Heart Failure Society of America (HFSA) jointly released a focused update1 of the 2013 ACC/AHA guideline for managing heart failure.2 This is the second focused update of the 2013 guidelines; the first update,3 in 2016, covered 2 new drugs (sacubitril-valsartan and ivabradine) for chronic stage C heart failure with reduced ejection fraction (HFrEF).
Rather than focus on new medication classes, this second update provides recommendations regarding:
- Preventing the progression to left ventricular dysfunction or heart failure in patients at high risk (stage A) through screening with B-type natriuretic peptide (BNP) and aiming for more aggressive blood pressure control
- Inpatient biomarker use
- Medications in heart failure with preserved ejection fraction (HFpEF, or diastolic heart failure)
- Blood pressure targets in stage C heart failure
- Managing important comorbidities such as iron deficiency and sleep-disordered breathing to decrease morbidity, improve functional capacity, and enhance quality of life.
These guidelines and the data that underlie them are explored below. We also discuss potential applications to the management of hospitalization for acute decompensated heart failure (ADHF).
COMMON, COSTLY, AND DEBILITATING
Heart failure—defined by the ACC/AHA as the complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood—remains one of the most common, costly, and debilitating diseases in the United States.2 Based on National Health and Nutrition Examination Survey data from 2011 to 2014, an estimated 6.5 million US adults have it, with projections of more than 8 million by 2030.4,5 More than 960,000 new cases are thought to occur annually, with a lifetime risk of developing it of roughly 20% to 45%.6
Despite ever-growing familiarity and some significant strides in management, the death rate in this syndrome is substantial. After admissions for heart failure (which number 1 million per year), the mortality rate is roughly 10% at 1 year and 40% at 5 years.6 Also staggering are the associated costs, with $30.7 billion attributed to heart failure in 2012 and a projected $69.7 billion annually by 2030.5 Thus, we must direct efforts not only to treatment, but also to prevention.

Preventive efforts would target patients with ACC/AHA stage A heart failure—those at high risk for developing but currently without evidence of structural heart disease or heart failure symptoms (Table 1).7 This group may represent up to one-third of the US adult population, or 75 million people, when including the well-recognized risk factors of coronary artery disease, hypertension, diabetes mellitus, and chronic kidney disease in those without left ventricular dysfunction or heart failure.8
BIOMARKERS FOR PREVENTION
Past ACC/AHA heart failure guidelines2 have included recommendations on the use of biomarkers to aid in diagnosis and prognosis and, to a lesser degree, to guide treatment of heart failure. Largely based on 2 trials (see below), the 2017 guidelines go further, issuing a recommendation on the use of natriuretic peptide biomarkers in a screening strategy to prompt early intervention and prevent the progression to clinical heart failure in high-risk patients (stage A heart failure).
The PONTIAC trial
The NT-proBNP Selected Prevention of Cardiac Events in a Population of Diabetic Patients Without a History of Cardiac Disease (PONTIAC) trial9 randomized 300 outpatients with type 2 diabetes mellitus and an elevated N-terminal proBNP (NT-proBNP) level (> 125 pg/mL) to standard medical care vs standard care plus intensive up-titration of renin-angiotensin system antagonists and beta-blockers in a cardiac clinic over 2 years.
Earlier studies10 had shown NT-proBNP levels to have predictive value for cardiac events in diabetic patients, while the neurohormonal treatments were thought to have an established record of preventing primary and secondary cardiovascular events. In PONTIAC, a significant reduction was seen in the primary end point of hospitalization or death due to cardiac disease (hazard ratio [HR] 0.351, P = .044), as well as in the secondary end point of hospitalization due to heart failure (P < .05), in the aggressive-intervention group. These results laid the foundation for the larger St. Vincent’s Screening to Prevent Heart Failure (STOP-HF) trial.11
The STOP-HF trial
The STOP-HF trial randomized 1,235 outpatients who were at high risk but without left ventricular dysfunction or heart failure symptoms (stage A) to annual screening alone vs annual screening plus BNP testing, in which a BNP level higher than 50 pg/mL triggered echocardiography and evaluation by a cardiologist who would then assist with medications.11
Eligible patients were over age 40 and had 1 or more of the following risk factors:
- Diabetes mellitus
- Hypertension
- Hypercholesterolemia
- Obesity (body mass index > 30 kg/m2)
- Vascular disease (coronary, cerebral, or peripheral arterial disease)
- Arrhythmia requiring treatment
- Moderate to severe valvular disease.
After a mean follow-up of 4.3 years, the primary end point, ie, asymptomatic left ventricular dysfunction with or without newly diagnosed heart failure, was found in 9.7% of the control group and in only 5.9% of the intervention group with BNP screening, a 42% relative risk reduction (P = .013).
Similarly, the incidence of secondary end points of emergency hospitalization for a cardiovascular event (arrhythmia, transient ischemic attack, stroke, myocardial infarction, peripheral or pulmonary thrombosis or embolization, or heart failure) was also lower at 45.2 vs 24.4 per 1,000 patient-years, a 46% relative risk reduction.
An important difference in medications between the 2 groups was an increase in subsequently prescribed renin-angiotensin-aldosterone system therapy, mainly consisting of angiotensin II receptor blockers (ARBs), in those with elevated BNP in the intervention group. Notably, blood pressure was about the same in the 2 groups.11
Although these findings are encouraging, larger studies are needed, as the lack of blinding, low event rates, and small absolute risk reduction make the results difficult to generalize.
New or modified recommendations for screening

Employing this novel prevention strategy in the extremely large number of patients with stage A heart failure, thought to be up to one-third of the US adult population, may serve as a way to best direct and utilize limited medical resources.8
BIOMARKERS FOR PROGNOSIS OR ADDED RISK STRATIFICATION
The 2013 guidelines2 recognized that a significant body of work had accumulated showing that natriuretic peptide levels can predict outcomes in both chronic and acute heart failure. Thus, in both conditions, the guidelines contained separate class Ia recommendations to obtain a natriuretic peptide level, troponin level, or both to establish prognosis or disease severity.
The 2017 update1 underscores the importance of timing in measuring natriuretic peptide levels during admission for ADHF, with emphasis on obtaining them at admission and at discharge for acute and postdischarge prognosis. The completely new class IIa recommendation to obtain a predischarge natriuretic peptide level for postdischarge prognosis was based on a number of observational studies, some of which we explore below.
The ELAN-HF meta-analysis
The European Collaboration on Acute Decompensated Heart Failure (ELAN-HF)12 performed a meta-analysis to develop a discharge prognostication score for ADHF that included both absolute level and percent change in natriuretic peptide levels at the time of discharge.
Using data from 7 prospective cohorts totaling 1,301 patients, the authors found that incorporation of these values into a subsequently validated risk model led to significant improvements in the ability to predict the end points of all-cause mortality and the combined end point of all-cause mortality or first readmission for a cardiovascular reason within 180 days.
The OPTIMIZE-HF retrospective analysis
Data from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure (OPTIMIZE-HF) were retrospectively analyzed13 to determine whether postdischarge outcomes were best predicted by natriuretic peptide levels at admission or discharge or by the relative change in natriuretic peptide level. More than 7,000 patients age 65 or older, in 220 hospitals, were included, and Cox prediction models were compared using clinical variables alone or in combination with the natriuretic peptide levels.
The model that included the discharge natriuretic peptide level was found to be the most predictive, with a c-index of 0.693 for predicting mortality and a c-index of 0.606 for mortality or rehospitalization at 1 year.
New or modified recommendations on biomarkers for prognosis
The 2017 update1 modified the earlier recommendation to obtain a natriuretic peptide or troponin level or both at admission for ADHF to establish prognosis. This now has a class Ia recommendation, emphasizing that such levels be obtained on admission. In addition, a new class IIa recommendation is made to obtain a predischarge natriuretic peptide level for postdischarge prognosis. The former class Ia recommendation to obtain a natriuretic peptide level in chronic heart failure to establish prognosis or disease severity remains unchanged.
Also worth noting is what the 2017 update does not recommend in regard to obtaining biomarker levels. It emphasizes that many patients, particularly those with advanced (stage D) heart failure, have a poor prognosis that is well established with or without biomarker levels. Additionally, there are many cardiac and noncardiac causes of natriuretic peptide elevation; thus, clinical judgment remains paramount.
The 2017 update1 also cautions against setting targets of percent change in or absolute levels of natriuretic peptide at discharge despite observational and retrospective studies demonstrating better outcomes when levels are reduced, as treating for any specific target has never been studied in a large prospective study. Thus, doing so may result in unintended harm. Rather, clinical judgment and optimization of guideline-directed management and therapy are encouraged (Table 2).
PHARMACOLOGIC TREATMENT FOR STAGE C HFpEF
Although the 2013 guidelines2 contain many class I recommendations for various medications in chronic HFrEF, not a single such recommendation is found for chronic HFpEF. A review by Okwuosa et al7 covered HFrEF, including the most recent additions on which the 2016 update was based, sacubitril-valsartan and ivabradine. The 2016 update was similarly devoid of recommendations regarding specific medications in HFpEF, leaving only the 2013 class IIb recommendation to consider using an ARB to decrease hospitalizations in HFpEF.
Evidence behind this recommendation came from the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity program’s randomized controlled trial in 3,025 patients with New York Heart Association (NYHA) class II to IV heart failure and left ventricular ejection fraction over 40%, who were treated with candesartan or placebo.14 Over a median follow-up of 36.6 months, there was no significant difference in the primary composite outcome of cardiovascular death or admission for heart failure, but significantly fewer patients in the candesartan arm were admitted (230 vs 270, P = .017). Thus the recommendation.
Although this finding was encouraging, it was clear that no blockbuster drug for HFpEF had been identified. Considering that roughly half of all heart failure patients have preserved ejection fraction, the discovery of such a drug for HFpEF would be met with much excitement.15 Subsequently, other medication classes have been evaluated in the hope of benefit, allowing the 2017 update to provide specific recommendations for aldosterone antagonists, nitrates, and phosphodiesterase-5 inhibitors in HFpEF.
ALDOSTERONE ANTAGONISTS FOR HFpEF
Mineralocorticoid receptor antagonists had previously been shown to significantly reduce morbidity and mortality rates in patients with HFrEF.16 In addition to aldosterone’s effects on sodium retention and many other pathophysiologic mechanisms relating to heart failure, this hormone is also known to play a role in promoting myocardial fibrosis.17 Accordingly, some have wondered whether aldosterone antagonists could improve diastolic dysfunction, and perhaps outcomes, in HFpEF.
The Aldo-DHF trial
The Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial investigated whether the aldosterone antagonist spironolactone would improve diastolic function or maximal exercise capacity in chronic HFpEF.18 It randomized 422 ambulatory patients with NYHA stage II or III heart failure, preserved left ventricular ejection fraction (≥ 50%), and echocardiographic evidence of diastolic dysfunction to receive spironolactone 25 mg daily or placebo.
Although no significant difference was seen in maximal exercise capacity, follow-up over 1 year nevertheless showed significant improvement in echocardiographic diastolic dysfunction (E/e') and perhaps reverse remodeling (decreased left ventricular mass index). These improvements spurred larger trials powered to detect whether clinical outcomes could also be improved.
The TOPCAT trial
The Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial19 was a large, multicenter, international, double-blind, placebo-controlled trial that investigated whether spironolactone could improve clinical outcomes in HFpEF. It randomized 3,445 patients with symptomatic heart failure and left ventricular ejection fraction of 45% or more to spironolactone 15 to 45 mg daily or placebo.
The effect on a composite primary outcome of death from cardiovascular cause, aborted cardiac arrest, or hospitalization for heart failure was evaluated over a mean follow-up of 3.3 years, with only a small (HR 0.89), nonclinically significant reduction evident. Those in the spironolactone group did have a significantly lower incidence of hospitalization for heart failure (12.0% vs 14.2%, P = .04).
Although the results were disappointing in this essentially negative trial, significant regional variations evident on post hoc analysis prompted further investigation and much controversy since the trial’s publication in 2014.
Participants came in roughly equal proportions from the Americas (United States, Canada, Brazil, and Argentina—51%) and from Russia and Georgia (49%), but outcomes between the two groups were markedly different. Concern was first raised when immediate review discovered a 4-fold lower rate of the primary outcome in the placebo groups from Russia and Georgia (8.4%), a rate in fact similar to that in patients without heart failure.19 This led to further exploration that identified other red flags that called into question the data integrity from the non-American sites.20
Not only did patients receiving spironolactone in Russia and Georgia not experience the reduction in clinical outcomes seen in their American counterparts, they also did not manifest the expected elevations in potassium and creatinine, and spironolactone metabolites were undetectable in almost one-third of patients.21
These findings prompted a post hoc analysis that included only the 51% (1,767 patients) of the study population coming from the Americas; in this subgroup, treatment with spironolactone was associated with a statistically significant 18% relative risk reduction in the primary composite outcome, a 26% reduction in cardiovascular mortality, and an 18% reduction in hospitalization for heart failure.20
New or modified recommendations on aldosterone receptor antagonists

Nitrates and phosphodiesterase-5 inhibitors
Earlier studies indicated that long-acting nitrates are prescribed in 15% to 50% of patients with HFpEF, perhaps based on extrapolation from studies in HFrEF suggesting that they might improve exercise intolerance.22 Some have speculated that the hemodynamic effects of nitrates, such as decreasing pulmonary congestion, might improve exercise intolerance in those with the stiff ventricles of HFpEF as well, prompting further study.
The NEAT-HFpEF trial
The Nitrate’s Effect on Activity Tolerance in Heart Failure With Preserved Ejection Fraction (NEAT-HFpEF) trial22 investigated whether extended-release isosorbide mononitrate would increase daily activity levels in patients with HFpEF. This double-blind, crossover study randomized 110 patients with HFpEF (ejection fraction ≥ 50%) and persistent dyspnea to escalating doses of isosorbide mononitrate or placebo over 6 weeks, then to the other arm for another 6 weeks. Daily activity levels during the 120-mg phase were measured with a continuously worn accelerometer.
No beneficial effect of nitrates was evident, with a nonsignificant trend towards decreased activity levels, a significant decrease in hours of activity per day (–0.30 hours, P = .02), and no change in the other secondary end points such as quality-of-life score, 6-minute walk distance, or natriuretic peptide level.
Suggested explanations for these negative findings include the possibility of rapid dose escalation leading to increased subtle side effects (headache, dizziness, fatigue) that, in turn, decreased activity. Additionally, given the imprecise diagnostic criteria for HFpEF, difficulties with patient selection may have led to inclusion of a large number of patients without elevated left-sided filling pressures.23
The RELAX trial
The Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure With Preserved Ejection Fraction (RELAX) trial24 investigated whether the phosphodiesterase-5 inhibitor sildenafil would improve exercise capacity in HFpEF. Improvements in both exercise capacity and clinical outcomes had already been seen in earlier trials in patients with pulmonary hypertension, as well as in those with HFrEF.25 A smaller study in HFpEF patients with pulmonary hypertension was also encouraging.26
Thus, it was disappointing that, after randomizing 216 outpatients with HFpEF to sildenafil or placebo for 24 weeks, no benefit was seen in the primary end point of change in peak oxygen consumption or in secondary end points of change in 6-minute walk distance or composite clinical score. Unlike in NEAT-HFpEF, patients here were required to have elevated natriuretic peptide levels or elevated invasively measured filling pressures.
The study authors speculated that pulmonary arterial hypertension and right ventricular systolic failure might need to be significant for patients with HFpEF to benefit from phosphodiesterase-5 inhibitors, with their known effects of dilation of pulmonary vasculature and increasing contractility of the right ventricle.24
New or modified recommendations on nitrates or phosphodiesterase-5 drugs
Given these disappointing results, the 2017 update provides a class III (no benefit) recommendation against the routine use of nitrates or phosphodiesterase-5 inhibitors to improve exercise tolerance or quality of life in HFpEF, citing them as ineffective (Table 3).1
IRON DEFICIENCY IN HEART FAILURE
Not only is iron deficiency present in roughly 50% of patients with symptomatic heart failure (stage C and D HFrEF),27 it is also associated with increased heart failure symptoms such as fatigue and exercise intolerance,28 reduced functional capacity, decreased quality of life, and increased mortality.
Notably, this association exists regardless of the hemoglobin level.29 In fact, even in those without heart failure or anemia, iron deficiency alone results in worsened aerobic performance, exercise intolerance, and increased fatigue.30 Conversely, improvement in symptoms, exercise tolerance, and cognition have been shown with repletion of iron stores in such patients.31
At the time of the 2013 guidelines, only a single large trial of intravenous iron in HFrEF and iron deficiency had been carried out (see below), and although the results were promising, it was felt that the evidence base on which to make recommendations was inadequate. Thus, recommendations were deferred until more data could be obtained.
Of note, in all the trials discussed below, iron deficiency was diagnosed in the setting of heart failure as ferritin less than 100 mg/mL (absolute iron deficiency) or as ferritin 100 to 300 mg/mL with transferrin saturation less than 20% (relative deficiency).32
The CONFIRM-HF trial
As in the Ferinject Assessment in Patients With Iron Deficiency and Chronic Heart Failure (FAIR-HF) trial,33 the subsequent Ferric Carboxymaltose Evaluation on Performance in Patients With Iron Deficiency in Combination With Chronic Heart Failure (CONFIRM-HF) trial34 involved the intravenous infusion of iron (ferric carboxymaltose) in outpatients with symptomatic HFrEF and iron deficiency. It showed that benefits remained evident with a more objective primary end point (change in 6-minute walk test distance at 24 weeks), and that such benefits were sustained, as seen in numerous secondary end points related to functional capacity at 52 weeks. Benefits in CONFIRM-HF were evident independently from anemia, specifically whether hemoglobin was under or over 12 g/dL.
Although these results were promising, it remained unclear whether such improvements could be obtained with a much easier to administer, more readily available, and less expensive oral iron formulation.
The IRONOUT-HF trial
The Iron Repletion Effects on Oxygen Uptake in Heart Failure (IRONOUT-HF) trial35 investigated whether oral, rather than intravenous, iron supplementation could improve peak exercise capacity in patients with HFrEF and iron deficiency. This double-blind, placebo-controlled trial randomized 225 patients with NYHA class II to IV HFrEF and iron deficiency to treatment with oral iron polysaccharide (150 mg twice daily) or placebo for 16 weeks.
Contrary to the supportive findings above, no significant change was seen in the primary end point of change in peak oxygen uptake or in any of the secondary end points (change in 6-minute walk, quality of life). Also, despite a 15-fold increase in the amount of iron administered in oral form compared with intravenously, little change was evident in the indices of iron stores over the course of the study, with only a 3% increase in transferrin saturation and an 11 ng/mL increase in ferritin. The intravenous trials resulted in a 4-fold greater increase in transferrin saturation and a 20-fold greater increase in ferritin.36
What keeps heart failure patients from absorbing oral iron? It is unclear why oral iron administration in HFrEF, such as in IRONOUT-HF, seems to be so ineffective, but hepcidin—a protein hormone made by the liver that shuts down intestinal iron absorption and iron release from macrophages—may play a central role.37 When iron stores are adequate, hepcidin is upregulated to prevent iron overload. However, hepcidin is also increased in inflammatory states, and chronic heart failure is often associated with inflammation.
With this in mind, the IRONOUT-HF investigators measured baseline hepcidin levels at the beginning and at the end of the 16 weeks and found that high baseline hepcidin levels predicted poorer response to oral iron. Other inflammatory mediators, such as interleukin 6, may also play a role.38,39 Unlike oral iron formulations such as iron polysaccharide, intravenous iron (ferric carboxymaltose) bypasses these regulatory mechanisms, which may partly explain its much more significant effect on the indices of iron stores and outcomes.
New or modified recommendations on iron
The 2017 update1 makes recommendations regarding iron deficiency and anemia in heart failure for the first time.
A class IIb recommendation states that it might be reasonable to treat NYHA class II and III heart failure patients with iron deficiency with intravenous iron to improve functional status and quality of life. A strong recommendation has been deferred until more is known about morbidity and mortality effects from adequately powered trials, some of which are under way and explored further below.
The 2017 update also withholds any recommendations regarding oral iron supplementation in heart failure, citing an uncertain evidence base. Certainly, the subsequent IRONOUT-HF trial does not lend enthusiasm for this approach.
Lastly, given the lack of benefit coupled with the increased risk of thromboembolic events evident in a trial of darbepoetin alfa vs placebo in non-iron deficiency-related anemia in HFrEF,40,41 the 2017 update provides a class III (no benefit) recommendation against using erythropoietin-stimulating agents in heart failure and anemia.
HYPERTENSION IN HEART FAILURE
The 2013 guidelines for the management of heart failure simply provided a class I recommendation to control hypertension and lipid disorders in accordance with contemporary guidelines to lower the risk of heart failure.1
SPRINT
The Systolic Blood Pressure Intervention Trial (SPRINT)42 sought to determine whether a lower systolic blood pressure target (120 vs 140 mm Hg) would reduce clinical events in patients at high risk for cardiovascular events but without diabetes mellitus. Patients at high risk were defined as over age 75, or with known vascular disease, chronic kidney disease, or a Framingham Risk Score higher than 15%. This multicenter, open-label controlled trial randomized 9,361 patients to intensive treatment (goal systolic blood pressure < 120 mm Hg) or standard treatment (goal systolic blood pressure < 140 mm Hg).
SPRINT was stopped early at a median follow-up of 3.26 years when a 25% relative risk reduction in the primary composite outcome of myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes became evident in the intensive-treatment group (1.65% vs 2.19% per year, HR 0.75, P < .0001).
All-cause mortality was also lower in the intensive-treatment group (HR 0.73, P = .003), while the incidence of serious adverse events (hypotension, syncope, electrolyte abnormalities, acute kidney injury, and noninjurious falls) was only slightly higher (38.3% vs 37.1%, P = .25). Most pertinent, a significant 38% relative risk reduction in heart failure and a 43% relative risk reduction in cardiovascular events were also evident.
Of note, blood pressure measurements were taken as the average of 3 measurements obtained by an automated cuff taken after the patient had been sitting quietly alone in a room for 5 minutes.
New or modified recommendations on hypertension in heart failure
Given the impressive 25% relative risk reduction in myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes in SPRINT,42 the 2017 update1 incorporated the intensive targets of SPRINT into its recommendations. However, to compensate for what are expected to be higher blood pressures obtained in real-world clinical practice as opposed to the near-perfect conditions used in SPRINT, a slightly higher blood pressure goal of less than 130/80 mm Hg was set.

Although not specifically included in SPRINT, given the lack of trial data on specific blood pressure targets in HFrEF and the decreased cardiovascular events noted above, a class I (level of evidence C, expert opinion) recommendation to target a goal systolic blood pressure less than 130 mm Hg in stage C HFrEF with hypertension is also given. Standard guideline-directed medications in the treatment of HFrEF are to be used (Table 4).
Similarly, a new class I (level of evidence C, expert opinion) recommendation is given for hypertension in HFpEF to target a systolic blood pressure of less than 130 mm Hg, with special mention to first manage any element of volume overload with diuretics. Other than avoiding nitrates (unless used for angina) and phosphodiesterase inhibitors, it is noted that few data exist to guide the choice of antihypertensive further, although perhaps renin-angiotensin-aldosterone system inhibition, especially aldosterone antagonists, may be considered. These recommendations are fully in line with the 2017 ACC/AHA high blood pressure clinical practice guidelines,43 ie, that renin-angiotensin-aldosterone system inhibition with an angiotensin-converting enzyme (ACE) inhibitor or ARB and especially mineralocorticoid receptor antagonists would be the preferred choice (Table 4).
SLEEP-DISORDERED BREATHING IN HEART FAILURE
Sleep-disordered breathing, either obstructive sleep apnea (OSA) or central sleep apnea, is quite commonly associated with symptomatic HFrEF.44 Whereas OSA is found in roughly 18% and central sleep apnea in 1% of the general population, sleep-disordered breathing is found in nearly 60% of patients with HFrEF, with some studies showing a nearly equal proportion of OSA and central sleep apnea.45 A similar prevalence is seen in HFpEF, although with a much higher proportion of OSA.46 Central sleep apnea tends to be a marker of more severe heart failure, as it is strongly associated with severe cardiac systolic dysfunction and worse functional capacity.47
Not surprisingly, the underlying mechanism of central sleep apnea is quite different from that of OSA. Whereas OSA predominantly occurs because of repeated obstruction of the pharynx due to nocturnal pharyngeal muscle relaxation, no such airway patency issues or strained breathing patterns exist in central sleep apnea. Central sleep apnea, which can manifest as Cheyne-Stokes respirations, is thought to occur due to an abnormal ventilatory control system with complex pathophysiology such as altered sensitivity of central chemoreceptors to carbon dioxide, interplay of pulmonary congestion, subsequent hyperventilation, and prolonged circulation times due to reduced cardiac output.48
What the two types of sleep-disordered breathing have in common is an association with negative health outcomes. Both appear to induce inflammation and sympathetic nervous system activity via oxidative stress from intermittent nocturnal hypoxemia and hypercapnea.49 OSA was already known to be associated with significant morbidity and mortality rates in the general population,50 and central sleep apnea had been identified as an independent predictor of mortality in HFrEF.51

At the time of the 2013 guidelines, only small or observational studies with limited results had been done evaluating treatment effects of continuous positive airway pressure therapy (CPAP) on OSA and central sleep apnea. Given the relative paucity of data, only a single class IIa recommendation stating that CPAP could be beneficial to increase left ventricular ejection fraction and functional status in concomitant sleep apnea and heart failure was given in 2013. However, many larger trials were under way,52–59 some with surprising results such as a significant increase in cardiovascular and all-cause mortality (Table 5).54
New or modified recommendations on sleep-disordered breathing

Given the common association with heart failure (60%)45 and the marked variation in response to treatment, including potential for harm with adaptive servo-ventilation and central sleep apnea, a class IIa recommendation is made stating that it is reasonable to obtain a formal sleep study in any patient with symptomatic (NYHA class II–IV) heart failure.1
Due to the potential for harm with adaptive servo-ventilation in patients with central sleep apnea and NYHA class II to IV HFrEF, a class III (harm) recommendation is made against its use.
Largely based on the results of the Sleep Apnea Cardiovascular Endpoints (SAVE) trial,56 a class IIb, level of evidence B-R (moderate, based on randomized trials) recommendation is given, stating that the use of CPAP in those with OSA and known cardiovascular disease may be reasonable to improve sleep quality and reduce daytime sleepiness.
POTENTIAL APPLICATIONS IN ACUTE DECOMPENSATED HEART FAILURE
Although the 2017 update1 is directed mostly toward managing chronic heart failure, it is worth considering how it might apply to the management of ADHF.
SHOULD WE USE BIOMARFER TARGETS TO GUIDE THERAPY IN ADHF?
The 2017 update1 does offer direct recommendations regarding the use of biomarker levels during admissions for ADHF. Mainly, they emphasize that the admission biomarker levels provide valuable information regarding acute prognosis and risk stratification (class I recommendation), while natriuretic peptide levels just before discharge provide the same for the postdischarge timeframe (class IIa recommendation).
The update also explicitly cautions against using a natriuretic peptide level-guided treatment strategy, such as setting targets for predischarge absolute level or percent change in level of natriuretic peptides during admissions for ADHF. Although observational and retrospective studies have shown better outcomes when levels are reduced at discharge, treating for any specific inpatient target has never been tested in any large, prospective study; thus, doing so could result in unintended harm.
So what do we know?
McQuade et al systematic review
McQuade et al57 performed a systematic review of more than 40 ADHF trials, which showed that, indeed, patients who achieved a target absolute natriuretic peptide level (BNP ≤ 250 pg/mL) or percent reduction (≥ 30%) at time of discharge had significantly improved outcomes such as reduced postdischarge all-cause mortality and rehospitalization rates. However, these were mostly prospective cohort studies that did not use any type of natriuretic peptide level-guided treatment protocol, leaving it unclear whether such a strategy could positively influence outcomes.
For this reason, both McQuade et al57 and, in an accompanying editorial, Felker et al58 called for properly designed, randomized controlled trials to investigate such a strategy. Felker noted that only 2 such phase II trials in ADHF have been completed,59,60 with unconvincing results.
PRIMA II
The Multicenter, Randomized Clinical Trial to Study the Impact of In-hospital Guidance for Acute Decompensated Heart Failure Treatment by a Predefined NT-ProBNP Target on the Reduction of Readmission and Mortality Rates (PRIMA II)60 randomized patients to natriuretic peptide level-guided treatment or standard care during admission for ADHF.
Many participants (60%) reached the predetermined target of 30% reduction in natriuretic peptide levels at the time of clinical stabilization and randomization; 405 patients were randomized. Patients in the natriuretic peptide level-guided treatment group underwent a prespecified treatment algorithm, with repeat natriuretic peptide levels measured again after the protocol.
Natriuretic peptide-guided therapy failed to show any significant benefit in any clinical outcomes, including the primary composite end point of mortality or heart failure readmissions at 180 days (36% vs 38%, HR 0.99, 95% confidence interval 0.72–1.36). Consistent with the review by McQuade et al,57 achieving the 30% reduction in natriuretic peptide at discharge, in either arm, was associated with a better prognosis, with significantly lower mortality and readmission rates at 180 days (HR 0.39 for rehospitalization or death, 95% confidence interval 0.27–0.55).
As in the observational studies, those who achieved the target natriuretic peptide level at the time of discharge had a better prognosis than those who did not, but neither study showed an improvement in clinical outcomes using a natriuretic peptide level-targeting treatment strategy.
No larger randomized controlled trial results are available for guided therapy in ADHF. However, additional insight may be gained from a subsequent trial61 that evaluated biomarker-guided titration of guideline-directed medical therapy in outpatients with chronic HFrEF.
The GUIDE-IT trial
That trial, the Guiding Evidence Based Therapy Using Biomarker Intensified Treatment in Heart Failure (GUIDE-IT)61 trial, was a large multicenter attempt to determine whether a natriuretic peptide-guided treatment strategy was more effective than standard care in the management of 894 high-risk outpatients with chronic HFrEF. Earlier, promising results had been obtained in a meta-analysis62 of more than 11 similar trials in 2,000 outpatients, with a decreased mortality rate (HR 0.62) seen in the biomarker-guided arm. However, the results had not been definitive due to being underpowered.62
Unfortunately, the results of GUIDE-IT were disappointing, with no significant difference in either the combined primary end point of mortality or hospitalization for heart failure, or the secondary end points evident at 15 months, prompting early termination for futility.61 Among other factors, the study authors postulated that this may have partly resulted from a patient population with more severe heart failure and resultant azotemia, limiting the ability to titrate neurohormonal medications to the desired dosage.
The question of whether patients who cannot achieve such biomarker targets need more intensive therapy or whether their heart failure is too severe to respond adequately echoes the question often raised in discussions of inpatient biomarker-guided therapy.58 Thus, only limited insight is gained, and it remains unclear whether a natriuretic peptide-guided treatment strategy can improve outpatient or inpatient outcomes. Until this is clarified, clinical judgment and optimization of guideline-directed management and therapy should remain the bedrock of treatment.
SHOULD ALDOSTERONE ANTAGONISTS BE USED IN ACUTE HFpEF?
Given the encouraging results in chronic HFpEF from post hoc analyses of TOPCAT, are there any additional recent data suggesting a role for aldosterone antagonists such as spironolactone in acute HFpEF?
The ATHENA-HF trial
The Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial63 compared treatment with high-dose spironolactone (100 mg) for 96 hours vs usual care in 360 patients with ADHF. The patient population included those with HFrEF and HFpEF, and usual care included low-dose spironolactone (12.5–25 mg) in roughly 15% of patients. High-dose mineralocorticoid receptor antagonists have been shown to overcome diuretic resistance, improve pulmonary vascular congestion, and partially combat the adverse neurohormonal activation seen in ADHF.
Unfortunately, the trial was completely neutral in regard to the primary end point of reduction in natriuretic peptide levels as well as to the secondary end points of 30-day mortality rate, heart failure readmission, clinical congestion scores, urine output, and change in weight. No suggestion of additional benefit was seen in subgroup analysis of patients with acute HFpEF (ejection fraction > 45%), which yielded similar results.63
Given these lackluster findings, routine use of high-dose spironolactone in ADHF is not recommended.64 However, the treatment was well tolerated, without significant adverse effects of hyperkalemia or kidney injury, leaving the door open as to whether it may have utility in selected patients with diuretic resistance.
Should ARNIs and ivabradine be started during ADHF admissions?
The first half of the focused update3 of the 2013 guidelines,2 reviewed by Okwuosa et al,7 provided recommendations for the use of sacubitril-valsartan, an angiotensin-neprilysin inhibitor (ARNI), and ivabradine, a selective sinoatrial node If channel inhibitor, in chronic HFrEF.
Sacubitril-valsartan was given a class I recommendation for use in patients with NYHA class II or III chronic HFrEF who tolerate an ACE inhibitor or an ARB. This recommendation was given largely based on the benefits in mortality and heart failure hospitalizations seen in PARADIGM-HF (the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure)65 compared with enalapril (HR 0.80, 95% CI 0.73–0.87, P < .001).
There is currently no recommendation on initiation or use of ARNIs during admissions for ADHF, but a recent trial may lend some insight.66
THE PIONEER-HF trial
The Comparison of Sacubitril/Valsartan vs Enalapril on Effect on NT-proBNP in Patients Stabilized From an Acute Heart Failure Episode (PIONEER-HF) trial66 randomized patients admitted for acute HFrEF, once stabilized, to sacubitril-valsartan or enalapril. Encouragingly, the percentage change of natriuretic peptide levels from the time of inpatient initiation to 4 and 8 weeks thereafter, the primary efficacy end point, was 46.7% with sacubitril-valsartan versus 25.3% with enalapril alone (ratio of change 0.71, 95% CI 0.63–0.81, P < .001). Although not powered for such, a prespecified analysis of a composite of clinical outcomes was also favorable for sacubitril-valsartan, largely driven by a 44% decreased rate of rehospitalization. More definitive, and quite reassuring, was that no significant difference was seen in the key safety outcomes of worsening renal function, hyperkalemia, symptomatic hypotension, and angioedema. These results were also applicable to the one-third of study participants who had no former diagnosis of heart failure, the one-third identifying as African American, and the one-third who had not been taking an ACE inhibitor or ARB. These results, taken together with the notion that at study completion the patients become similar to those included in PARADIGM-HF, have led some to assert that PIONEER-HF has the potential to change clinical practice.
Ivabradine was given a class IIa recommendation for use in patients with NYHA class II or III chronic HFrEF with a resting heart rate of at least 70 bpm, in sinus rhythm, despite being on optimal medical therapy including a beta-blocker at a maximum tolerated dose.
This recommendation was largely based on SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which randomized patients to ivabradine or placebo to evaluate the effects of isolated lowering of the heart rate on the composite primary outcome of cardiovascular death or hospitalization. A significant reduction was seen in the ivabradine arm (HR 0.82, 95% CI 0.75–0.90, P < .0001), mainly driven by decreased hospitalizations.67
Subsequently, a small unblinded single-center study was undertaken to evaluate the efficacy and safety of initiating ivabradine during admissions for ADHF.68
THE ETHIC-AHF trial
The Effect of Early Treatment With Ivabradine Combined With Beta-Blockers vs Beta-Blockers Alone in Patients Hospitalized With Heart Failure and Reduced Left Ventricular Ejection Fraction (ETHIC-AHF) trial68 sought to determine the safety and effectiveness of early coadministration of ivabradine with beta-blockers in patients with acute HFrEF.
This single-center, unblinded study randomized 71 patients to ivabradine and beta-blockade or beta-blockade alone upon clinical stabilization (24–48 hours) after admission for acute decompensated HFrEF.
The primary end point was heart rate at 28 days, with the ivabradine group showing a statistically significant decrease (64 vs 70 bpm, P = .01), which persisted at 4 months. There was no significant difference in the secondary end points of adverse drug effects or the composite of clinical event outcomes (all-cause mortality, admission for heart failure or cardiovascular cause), but a number of surrogate end points including left ventricular ejection fraction, BNP level, and NYHA functional class at 4 months showed mild improvement.
Although this study provided evidence that the coadministration of ivabradine and a beta-blocker is safe and was positive in regard to clinical outcomes, the significant limitations due to its size and study design (single-center, unblinded, 4-month follow-up) simply serve to support the pursuit of larger studies with more stringent design and longer follow-up in order to determine the clinical efficacy.
The PRIME-HF trial
The Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF) trial69 is a randomized, open-label, multicenter trial comparing standard care vs the initiation of ivabradine before discharge, but after clinical stabilization, during admissions for ADHF in patients with chronic HFrEF (left ventricular ejection fraction ≤ 35%). At subsequent outpatient visits, the dosage can be modified in the ivabradine group, or ivabradine can be initiated at the provider’s discretion in the usual-care group.
PRIME-HF is attempting to determine whether initiating ivabradine before discharge will result in more patients taking ivabradine at 180 days, its primary end point, as well as in changes in secondary end points including heart rate and patient-centered outcomes. The study is active, with reporting expected in 2019.
As these trials all come to completion, it will not be long before we have further guidance regarding the inpatient initiation of these new and exciting therapeutic agents.
SHOULD INTRAVENOUS IRON BE GIVEN DURING ADHF ADMISSIONS?
Given the high prevalence of iron deficiency in symptomatic HFrEF, its independent association with mortality, improvements in quality of life and functional capacity suggested by repleting with intravenous iron (in FAIR-HF and CONFIRM-HF), the seeming inefficacy of oral iron in IRONOUT, and the logistical challenges of intravenous administration during standard clinic visits, could giving intravenous iron soon be incorporated into admissions for ADHF?
Caution has been advised for several reasons. As discussed above, larger randomized controlled trials powered to detect more definitive clinical end points such as death and the rate of hospitalization are still needed before a stronger recommendation can be made for intravenous iron in HFrEF. Also, without such data, it seems unwise to add the considerable economic burden of routinely assessing for iron deficiency and providing intravenous iron during ADHF admissions to the already staggering costs of heart failure.

The effects seen on morbidity and mortality that become evident in these trials over the next 5 years will help determine future guidelines and whether intravenous iron is routinely administered in bridge clinics, during inpatient admissions for ADHF, or not at all in patients with HFrEF and iron deficiency.
INTERNISTS ARE KEY
Heart failure remains one of the most common, morbid, complex, and costly diseases in the United States, and its prevalence is expected only to increase.4,5 The 2017 update1 of the 2013 guideline2 for the management of heart failure provides recommendations aimed not only at management of heart failure, but also at its comorbidities and, for the first time ever, at its prevention.
Internists provide care for the majority of heart failure patients, as well as for their comorbidities, and are most often the first to come into contact with patients at high risk of developing heart failure. Thus, a thorough understanding of these guidelines and how to apply them to the management of acute decompensated heart failure is of critical importance.
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2017; 70(6):776–803. doi:10.1016/j.jacc.2017.04.025
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 128(16):e240–e327. doi:10.1161/CIR.0b013e31829e8776
- Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2016; 134(13):e282–e293. doi:10.1161/CIR.0000000000000435
- Benjamin EJ, Blaha MJ, Chiuve SE, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 2017; 135(10):e146–e603. doi:10.1161/CIR.0000000000000485
- Heidenreich PA, Albert NM, Allen LA, et al; American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 2013; 6(3):606–619. doi:10.1161/HHF.0b013e318291329a
- Huffman MD, Berry JD, Ning H, et al. Lifetime risk for heart failure among white and black Americans: cardiovascular lifetime risk pooling project. J Am Coll Cardiol 2013; 61(14):1510–1517. doi:10.1016/j.jacc.2013.01.022
- Okwuosa IS, Princewill O, Nwabueze C, et al. The ABCs of managing systolic heart failure: past, present, and future. Cleve Clin J Med 2016; 83(10):753–765. doi:10.3949/ccjm.83a.16006
- Kovell LC, Juraschek SP, Russell SD. Stage A heart failure is not adequately recognized in US adults: analysis of the National Health and Nutrition Examination Surveys, 2007–2010. PLoS One 2015; 10(7):e0132228. doi:10.1371/journal.pone.0132228
- Huelsmann M, Neuhold S, Resl M, et al. PONTIAC (NT-proBNP selected prevention of cardiac events in a population of diabetic patients without a history of cardiac disease): a prospective randomized controlled trial. J Am Coll Cardiol 2013; 62(15):1365–1372. doi:10.1016/j.jacc.2013.05.069
- Clodi M, Resl M, Neuhold S, et al. A comparison of NT-proBNP and albuminuria for predicting cardiac events in patients with diabetes mellitus. Eur J Prev Cardiol 2012; 19(5):944–951. doi:10.1177/1741826711420015
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310(1):66–74. doi:10.1001/jama.2013.7588
- Salah K, Kok WE, Eurlings LW, et al. A novel discharge risk model for patients hospitalised for acute decompensated heart failure incorporating N-terminal pro-B-type natriuretic peptide levels: a European coLlaboration on Acute decompeNsated Heart Failure: ELAN-HF Score. Heart 2014; 100(2):115–125. doi:10.1136/heartjnl-2013-303632
- Kociol RD, Horton JR, Fonarow GC, et al. Admission, discharge, or change in B-type natriuretic peptide and long-term outcomes: data from Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) linked to Medicare claims. Circ Heart Fail 2011; 4(5):628–636. doi:10.1161/CIRCHEARTFAILURE.111.962290
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362(9386):777–781. doi:10.1016/S0140-6736(03)14285-7
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355(3):251–259. doi:10.1056/NEJMoa052256
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341(10):709–717. doi:10.1056/NEJM199909023411001
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35(1):30–34. pmid:9302344
- Edelmann F, Wachter R, Schmidt AG, et al; Aldo-DHF Investigators. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA 2013; 309(8):781–791. doi:10.1001/jama.2013.905
- Pitt B, Pfeffer MA, Assmann SF, et al; TOPCAT Investigators. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 2014; 370(15):1383–1392. doi:10.1056/NEJMoa1313731
- Pfeffer MA, Claggett B, Assmann SF, et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015; 31(1):34–42. doi:10.1161/CIRCULATIONAHA.114.013255
- de Denus S, O’Meara E, Desai AS, et al. Spironolactone metabolites in TOPCAT—new insights into regional variation. N Engl J Med 2017; 376(17):1690–1692. doi:10.1056/NEJMc1612601
- Redfield MM, Anstrom KJ, Levine JA, et al; NHLBI Heart Failure Clinical Research Network. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 2015; 373(24):2314–2324. doi:10.1056/NEJMoa1510774
- Walton-Shirley M. Succinct thoughts on NEAT-HFpEF: true, true, and unrelated? Medscape 2015. https://www.medscape.com/viewarticle/854116. Accessed January 17, 2019.
- Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309(12):1268–1277. doi:10.1001/jama.2013.2024
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo controlled study. Circ Heart Fail 2011; 4(1):8–17. doi:10.1161/CIRCHEARTFAILURE.110.944694
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011; 124(2):164–174. doi:10.1161/CIRCULATIONAHA.110.983866
- Klip IT, Comin-Colet J, Voors AA, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013; 165(4):575–582.e3. doi:10.1016/j.ahj.2013.01.017
- Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 2013; 34(11):816–829. doi:10.1093/eurheartj/ehs224
- Jankowska EA, Rozentryt P, Witkowska A, et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J Card Fail 2011; 17(11):899–906. doi:10.1016/j.cardfail.2011.08.003
- Haas JD, Brownlie T 4th. Iron deficiency and reduced work capacity: a critical review of the research to determine a causal relationship. J Nutr 2001; 131(2S–2):676S-690S. doi:10.1093/jn/131.2.676S
- Davies KJ, Maguire JJ, Brooks GA, Dallman PR, Packer L. Muscle mitochondrial bioenergetics, oxygen supply, and work capacity during dietary iron deficiency and repletion. Am J Physiol 1982; 242(6):E418–E427. doi:10.1152/ajpendo.1982.242.6.E418
- Drozd M, Jankowska EA, Banasiak W, Ponikowski P. Iron therapy in patients with heart failure and iron deficiency: review of iron preparations for practitioners. Am J Cardiovasc Drugs 2017; 17(3):183–201. doi:10.1007/s40256-016-0211-2
- Anker SD, Comin Colet J, Filippatos G, et al; FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009; 361(25):2436–2448. doi:10.1056/NEJMoa0908355
- Ponikowski P, van Veldhuisen DJ, Comin-Colet J, et al; CONFIRM-HF Investigators. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J 2015; 36(11):657–668. doi:10.1093/eurheartj/ehu385
- Lewis GD, Malhotra R, Hernandez AF, et al; NHLBI Heart Failure Clinical Research Network. Effect of Oral Iron Repletion on Exercise Capacity in Patients With Heart Failure With Reduced Ejection Fraction and Iron Deficiency: The IRONOUT HF randomized clinical trial. JAMA 2017; 317(19):1958–1966. doi:10.1001/jama.2017.5427
- Wendling P. Iron supplementation in HF: trials support IV but not oral. Medscape 2016. https://www.medscape.com/viewarticle/872088. Accessed January 17, 2019.
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood 2011; 117(17):4425–4433. doi:10.1182/blood-2011-01-258467
- Jankowska EA, Kasztura M, Sokolski M, et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur Heart J 2014; 35(36):2468–2476. doi:10.1093/eurheartj/ehu235
- Jankowska EA, Malyszko J, Ardehali H, et al. Iron status in patients with chronic heart failure. Eur Heart J 2013; 34(11):827–834. doi:10.1093/eurheartj/ehs377
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368(13):1210–1219. doi:10.1056/NEJMoa1214865
- Ghali JK, Anand IS, Abraham WT, et al; Study of Anemia in Heart Failure Trial (STAMINA-HeFT) Group. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation 2008; 117(4):526–535. doi:10.1161/CIRCULATIONAHA.107.698514
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Whelton PK, Carey RM, Arnow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162(8):893–900. pmid:11966340
- MacDonald M, Fang J, Pittman SD, White DP, Malhotra A.The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med 2008; 4(1):38-42. pmid:18350960
- Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail 2009; 11(6):602–608. doi:10.1093/eurjhf/hfp057
- Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 1999; 160(4):1101–1106. doi:10.1164/ajrccm.160.4.9903020
- Ng AC, Freedman SB. Sleep disordered breathing in chronic heart failure. Heart Fail Rev 2009; 14(2):89–99. doi:10.1007/s10741-008-9096-8
- Kasai T, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. J Am Coll Cardiol 2011; 57(2):119–127. doi:10.1016/j.jacc.2010.08.627
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365(9464):1046–1053. doi:10.1016/S0140-6736(05)71141-7
- Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol 2007; 49(20):2028–2034. doi:10.1016/j.jacc.2007.01.084
- Bradley TD, Logan AG, Kimoff RJ, et al; CANPAP Investigators. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 2005; 353(19):2025–2033. doi:10.1056/NEJMoa051001
- Arzt M, Floras JS, Logan AG, et al; CANPAP Investigators. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: a post hoc analysis of the Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure Trial (CANPAP). Circulation 2007; 115(25):3173–3180. doi:10.1161/CIRCULATIONAHA.106.683482
- Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med 2015; 373(12):1095–1105. doi:10.1056/NEJMoa1506459
- O’Connor CM, Whellan DJ, Fiuzat M, et al. Cardiovascular outcomes with minute ventilation-targeted adaptive servo-ventilation therapy in heart failure: the CAT-HF Trial. J Am Coll Cardiol 2017; 69(12):1577–1587. doi:10.1016/j.jacc.2017.01.041
- McEvoy RD, Antic NA, Heeley E, et al; SAVE Investigators and Coordinators. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med 2016; 375(10):919–931. doi:10.1056/NEJMoa1606599
- McQuade CN, Mizus M, Wald JW, Goldberg L, Jessup M, Umscheid CA. Brain-type natriuretic peptide and amino-terminal pro-brain-type natriuretic peptide discharge thresholds for acute decompensated heart failure: a systematic review. Ann Intern Med 2017; 166(3):180–190. doi:10.7326/M16-1468
- Felker GM, Whellan DJ. Inpatient management of heart failure: are we shooting at the right target? Ann Intern Med 2017; 166(3):223–224. doi:10.7326/M16-2667
- Carubelli V, Lombardi C, Lazzarini V, et al. N-terminal pro-B-type natriuretic peptide-guided therapy in patients hospitalized for acute heart failure. J Cardiovasc Med (Hagerstown) 2016; 17(11):828–839. doi:10.2459/JCM.0000000000000419
- Stienen S, Salah K, Moons AH, et al. Rationale and design of PRIMA II: a multicenter, randomized clinical trial to study the impact of in-hospital guidance for acute decompensated heart failure treatment by a predefined NT-PRoBNP target on the reduction of readmIssion and mortality rates. Am Heart J 2014; 168(1):30–36. doi:10.1016/j.ahj.2014.04.008
- Felker GM, Anstrom KJ, Adams KF, et al. Effect of natriuretic peptide-guided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 2017; 318(8):713–720. doi:10.1001/jama.2017.10565
- Troughton RW, Frampton CM, Brunner-La Rocca HP, et al. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: an individual patient meta-analysis. Eur Heart J 2014; 35(23):1559–1567. doi:10.1093/eurheartj/ehu090
- van Vliet AA, Donker AJ, Nauta JJ, Verheugt FW. Spironolactone in congestive heart failure refractory to high-dose loop diuretic and low-dose angiotensin-converting enzyme inhibitor. Am J Cardiol 1993; 71(3):21A–28A. pmid:8422000
- Butler J, Anstrom KJ, Felker GM, et al; National Heart Lung and Blood Institute Heart Failure Clinical Research Network. Efficacy and safety of spironolactone in acute heart failure. The ATHENA-HF randomized clinical trial. JAMA Cardiol 2017; 2(9):950–958. doi:10.1001/jamacardio.2017.2198
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371(11):993–1004. doi:10.1056/NEJMoa1409077
- ClinicalTrials.gov. ComParIson Of Sacubitril/valsartaN Versus Enalapril on Effect on NTpRo-BNP in patients stabilized from an acute Heart Failure episode (PIONEER-HF). https://clinicaltrials.gov/ct2/show/NCT02554890. Accessed January 17, 2019.
- Swedberg K, Komajda M, Böhm M, et al; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 2010; 376(9744):875–885. doi:10.1016/S0140-6736(10)61198-1
- Hidalgo FJ, Anguita M, Castillo JC, et al. Effect of early treatment with ivabradine combined with beta-blockers versus beta-blockers alone in patients hospitalised with heart failure and reduced left ventricular ejection fraction (ETHIC-AHF): a randomised study. Int J Cardiol 2016; 217:7–11. doi:10.1016/j.ijcard.2016.04.136
- ClinicalTrials.gov. Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF). https://clinicaltrials.gov/ct2/show/NCT02827500. Accessed January 17, 2019.
- Anker SD, Kirwan BA, van Veldhuisen DJ, et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: an individual patient data meta-analysis. Eur J Heart Fail 2018; 20(1):125–133. doi:10.1002/ejhf.823
- ClinicalTrials.gov. Intravenous Iron in Patients With Systolic Heart Failure and Iron Deficiency to Improve Morbidity and Mortality (FAIR-HF2). https://clinicaltrials.gov/ct2/show/NCT03036462. Accessed January 17, 2019.
- ClinicalTrials.gov. Study to Compare Ferric Carboxymaltose With Placebo in Patients With Acute Heart Failure and Iron Deficiency (AFFIRM-AHF). https://clinicaltrials.gov/ct2/show/record/NCT02937454. Accessed January 17, 2019.
- ClinicalTrials.gov. Randomized Placebo-controlled Trial of Ferric Carboxymaltose as Treatment for Heart Failure With Iron Deficiency (HEART-FID). https://clinicaltrials.gov/ct2/show/NCT03037931. Accessed January 17, 2019.
- ClinicalTrials.gov. Intravenous Iron Treatment in Patients With Heart Failure and Iron Deficiency (IRONMAN). https://clinicaltrials.gov/ct2/show/NCT02642562. Accessed January 17, 2019.
In 2017, the American College of Cardiology (ACC), American Heart Association (AHA), and Heart Failure Society of America (HFSA) jointly released a focused update1 of the 2013 ACC/AHA guideline for managing heart failure.2 This is the second focused update of the 2013 guidelines; the first update,3 in 2016, covered 2 new drugs (sacubitril-valsartan and ivabradine) for chronic stage C heart failure with reduced ejection fraction (HFrEF).
Rather than focus on new medication classes, this second update provides recommendations regarding:
- Preventing the progression to left ventricular dysfunction or heart failure in patients at high risk (stage A) through screening with B-type natriuretic peptide (BNP) and aiming for more aggressive blood pressure control
- Inpatient biomarker use
- Medications in heart failure with preserved ejection fraction (HFpEF, or diastolic heart failure)
- Blood pressure targets in stage C heart failure
- Managing important comorbidities such as iron deficiency and sleep-disordered breathing to decrease morbidity, improve functional capacity, and enhance quality of life.
These guidelines and the data that underlie them are explored below. We also discuss potential applications to the management of hospitalization for acute decompensated heart failure (ADHF).
COMMON, COSTLY, AND DEBILITATING
Heart failure—defined by the ACC/AHA as the complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood—remains one of the most common, costly, and debilitating diseases in the United States.2 Based on National Health and Nutrition Examination Survey data from 2011 to 2014, an estimated 6.5 million US adults have it, with projections of more than 8 million by 2030.4,5 More than 960,000 new cases are thought to occur annually, with a lifetime risk of developing it of roughly 20% to 45%.6
Despite ever-growing familiarity and some significant strides in management, the death rate in this syndrome is substantial. After admissions for heart failure (which number 1 million per year), the mortality rate is roughly 10% at 1 year and 40% at 5 years.6 Also staggering are the associated costs, with $30.7 billion attributed to heart failure in 2012 and a projected $69.7 billion annually by 2030.5 Thus, we must direct efforts not only to treatment, but also to prevention.
Preventive efforts would target patients with ACC/AHA stage A heart failure—those at high risk for developing but currently without evidence of structural heart disease or heart failure symptoms (Table 1).7 This group may represent up to one-third of the US adult population, or 75 million people, when including the well-recognized risk factors of coronary artery disease, hypertension, diabetes mellitus, and chronic kidney disease in those without left ventricular dysfunction or heart failure.8
BIOMARKERS FOR PREVENTION
Past ACC/AHA heart failure guidelines2 have included recommendations on the use of biomarkers to aid in diagnosis and prognosis and, to a lesser degree, to guide treatment of heart failure. Largely based on 2 trials (see below), the 2017 guidelines go further, issuing a recommendation on the use of natriuretic peptide biomarkers in a screening strategy to prompt early intervention and prevent the progression to clinical heart failure in high-risk patients (stage A heart failure).
The PONTIAC trial
The NT-proBNP Selected Prevention of Cardiac Events in a Population of Diabetic Patients Without a History of Cardiac Disease (PONTIAC) trial9 randomized 300 outpatients with type 2 diabetes mellitus and an elevated N-terminal proBNP (NT-proBNP) level (> 125 pg/mL) to standard medical care vs standard care plus intensive up-titration of renin-angiotensin system antagonists and beta-blockers in a cardiac clinic over 2 years.
Earlier studies10 had shown NT-proBNP levels to have predictive value for cardiac events in diabetic patients, while the neurohormonal treatments were thought to have an established record of preventing primary and secondary cardiovascular events. In PONTIAC, a significant reduction was seen in the primary end point of hospitalization or death due to cardiac disease (hazard ratio [HR] 0.351, P = .044), as well as in the secondary end point of hospitalization due to heart failure (P < .05), in the aggressive-intervention group. These results laid the foundation for the larger St. Vincent’s Screening to Prevent Heart Failure (STOP-HF) trial.11
The STOP-HF trial
The STOP-HF trial randomized 1,235 outpatients who were at high risk but without left ventricular dysfunction or heart failure symptoms (stage A) to annual screening alone vs annual screening plus BNP testing, in which a BNP level higher than 50 pg/mL triggered echocardiography and evaluation by a cardiologist who would then assist with medications.11
Eligible patients were over age 40 and had 1 or more of the following risk factors:
- Diabetes mellitus
- Hypertension
- Hypercholesterolemia
- Obesity (body mass index > 30 kg/m2)
- Vascular disease (coronary, cerebral, or peripheral arterial disease)
- Arrhythmia requiring treatment
- Moderate to severe valvular disease.
After a mean follow-up of 4.3 years, the primary end point, ie, asymptomatic left ventricular dysfunction with or without newly diagnosed heart failure, was found in 9.7% of the control group and in only 5.9% of the intervention group with BNP screening, a 42% relative risk reduction (P = .013).
Similarly, the incidence of secondary end points of emergency hospitalization for a cardiovascular event (arrhythmia, transient ischemic attack, stroke, myocardial infarction, peripheral or pulmonary thrombosis or embolization, or heart failure) was also lower at 45.2 vs 24.4 per 1,000 patient-years, a 46% relative risk reduction.
An important difference in medications between the 2 groups was an increase in subsequently prescribed renin-angiotensin-aldosterone system therapy, mainly consisting of angiotensin II receptor blockers (ARBs), in those with elevated BNP in the intervention group. Notably, blood pressure was about the same in the 2 groups.11
Although these findings are encouraging, larger studies are needed, as the lack of blinding, low event rates, and small absolute risk reduction make the results difficult to generalize.
New or modified recommendations for screening
Employing this novel prevention strategy in the extremely large number of patients with stage A heart failure, thought to be up to one-third of the US adult population, may serve as a way to best direct and utilize limited medical resources.8
BIOMARKERS FOR PROGNOSIS OR ADDED RISK STRATIFICATION
The 2013 guidelines2 recognized that a significant body of work had accumulated showing that natriuretic peptide levels can predict outcomes in both chronic and acute heart failure. Thus, in both conditions, the guidelines contained separate class Ia recommendations to obtain a natriuretic peptide level, troponin level, or both to establish prognosis or disease severity.
The 2017 update1 underscores the importance of timing in measuring natriuretic peptide levels during admission for ADHF, with emphasis on obtaining them at admission and at discharge for acute and postdischarge prognosis. The completely new class IIa recommendation to obtain a predischarge natriuretic peptide level for postdischarge prognosis was based on a number of observational studies, some of which we explore below.
The ELAN-HF meta-analysis
The European Collaboration on Acute Decompensated Heart Failure (ELAN-HF)12 performed a meta-analysis to develop a discharge prognostication score for ADHF that included both absolute level and percent change in natriuretic peptide levels at the time of discharge.
Using data from 7 prospective cohorts totaling 1,301 patients, the authors found that incorporation of these values into a subsequently validated risk model led to significant improvements in the ability to predict the end points of all-cause mortality and the combined end point of all-cause mortality or first readmission for a cardiovascular reason within 180 days.
The OPTIMIZE-HF retrospective analysis
Data from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure (OPTIMIZE-HF) were retrospectively analyzed13 to determine whether postdischarge outcomes were best predicted by natriuretic peptide levels at admission or discharge or by the relative change in natriuretic peptide level. More than 7,000 patients age 65 or older, in 220 hospitals, were included, and Cox prediction models were compared using clinical variables alone or in combination with the natriuretic peptide levels.
The model that included the discharge natriuretic peptide level was found to be the most predictive, with a c-index of 0.693 for predicting mortality and a c-index of 0.606 for mortality or rehospitalization at 1 year.
New or modified recommendations on biomarkers for prognosis
The 2017 update1 modified the earlier recommendation to obtain a natriuretic peptide or troponin level or both at admission for ADHF to establish prognosis. This now has a class Ia recommendation, emphasizing that such levels be obtained on admission. In addition, a new class IIa recommendation is made to obtain a predischarge natriuretic peptide level for postdischarge prognosis. The former class Ia recommendation to obtain a natriuretic peptide level in chronic heart failure to establish prognosis or disease severity remains unchanged.
Also worth noting is what the 2017 update does not recommend in regard to obtaining biomarker levels. It emphasizes that many patients, particularly those with advanced (stage D) heart failure, have a poor prognosis that is well established with or without biomarker levels. Additionally, there are many cardiac and noncardiac causes of natriuretic peptide elevation; thus, clinical judgment remains paramount.
The 2017 update1 also cautions against setting targets of percent change in or absolute levels of natriuretic peptide at discharge despite observational and retrospective studies demonstrating better outcomes when levels are reduced, as treating for any specific target has never been studied in a large prospective study. Thus, doing so may result in unintended harm. Rather, clinical judgment and optimization of guideline-directed management and therapy are encouraged (Table 2).
PHARMACOLOGIC TREATMENT FOR STAGE C HFpEF
Although the 2013 guidelines2 contain many class I recommendations for various medications in chronic HFrEF, not a single such recommendation is found for chronic HFpEF. A review by Okwuosa et al7 covered HFrEF, including the most recent additions on which the 2016 update was based, sacubitril-valsartan and ivabradine. The 2016 update was similarly devoid of recommendations regarding specific medications in HFpEF, leaving only the 2013 class IIb recommendation to consider using an ARB to decrease hospitalizations in HFpEF.
Evidence behind this recommendation came from the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity program’s randomized controlled trial in 3,025 patients with New York Heart Association (NYHA) class II to IV heart failure and left ventricular ejection fraction over 40%, who were treated with candesartan or placebo.14 Over a median follow-up of 36.6 months, there was no significant difference in the primary composite outcome of cardiovascular death or admission for heart failure, but significantly fewer patients in the candesartan arm were admitted (230 vs 270, P = .017). Thus the recommendation.
Although this finding was encouraging, it was clear that no blockbuster drug for HFpEF had been identified. Considering that roughly half of all heart failure patients have preserved ejection fraction, the discovery of such a drug for HFpEF would be met with much excitement.15 Subsequently, other medication classes have been evaluated in the hope of benefit, allowing the 2017 update to provide specific recommendations for aldosterone antagonists, nitrates, and phosphodiesterase-5 inhibitors in HFpEF.
ALDOSTERONE ANTAGONISTS FOR HFpEF
Mineralocorticoid receptor antagonists had previously been shown to significantly reduce morbidity and mortality rates in patients with HFrEF.16 In addition to aldosterone’s effects on sodium retention and many other pathophysiologic mechanisms relating to heart failure, this hormone is also known to play a role in promoting myocardial fibrosis.17 Accordingly, some have wondered whether aldosterone antagonists could improve diastolic dysfunction, and perhaps outcomes, in HFpEF.
The Aldo-DHF trial
The Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial investigated whether the aldosterone antagonist spironolactone would improve diastolic function or maximal exercise capacity in chronic HFpEF.18 It randomized 422 ambulatory patients with NYHA stage II or III heart failure, preserved left ventricular ejection fraction (≥ 50%), and echocardiographic evidence of diastolic dysfunction to receive spironolactone 25 mg daily or placebo.
Although no significant difference was seen in maximal exercise capacity, follow-up over 1 year nevertheless showed significant improvement in echocardiographic diastolic dysfunction (E/e') and perhaps reverse remodeling (decreased left ventricular mass index). These improvements spurred larger trials powered to detect whether clinical outcomes could also be improved.
The TOPCAT trial
The Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial19 was a large, multicenter, international, double-blind, placebo-controlled trial that investigated whether spironolactone could improve clinical outcomes in HFpEF. It randomized 3,445 patients with symptomatic heart failure and left ventricular ejection fraction of 45% or more to spironolactone 15 to 45 mg daily or placebo.
The effect on a composite primary outcome of death from cardiovascular cause, aborted cardiac arrest, or hospitalization for heart failure was evaluated over a mean follow-up of 3.3 years, with only a small (HR 0.89), nonclinically significant reduction evident. Those in the spironolactone group did have a significantly lower incidence of hospitalization for heart failure (12.0% vs 14.2%, P = .04).
Although the results were disappointing in this essentially negative trial, significant regional variations evident on post hoc analysis prompted further investigation and much controversy since the trial’s publication in 2014.
Participants came in roughly equal proportions from the Americas (United States, Canada, Brazil, and Argentina—51%) and from Russia and Georgia (49%), but outcomes between the two groups were markedly different. Concern was first raised when immediate review discovered a 4-fold lower rate of the primary outcome in the placebo groups from Russia and Georgia (8.4%), a rate in fact similar to that in patients without heart failure.19 This led to further exploration that identified other red flags that called into question the data integrity from the non-American sites.20
Not only did patients receiving spironolactone in Russia and Georgia not experience the reduction in clinical outcomes seen in their American counterparts, they also did not manifest the expected elevations in potassium and creatinine, and spironolactone metabolites were undetectable in almost one-third of patients.21
These findings prompted a post hoc analysis that included only the 51% (1,767 patients) of the study population coming from the Americas; in this subgroup, treatment with spironolactone was associated with a statistically significant 18% relative risk reduction in the primary composite outcome, a 26% reduction in cardiovascular mortality, and an 18% reduction in hospitalization for heart failure.20
New or modified recommendations on aldosterone receptor antagonists
Nitrates and phosphodiesterase-5 inhibitors
Earlier studies indicated that long-acting nitrates are prescribed in 15% to 50% of patients with HFpEF, perhaps based on extrapolation from studies in HFrEF suggesting that they might improve exercise intolerance.22 Some have speculated that the hemodynamic effects of nitrates, such as decreasing pulmonary congestion, might improve exercise intolerance in those with the stiff ventricles of HFpEF as well, prompting further study.
The NEAT-HFpEF trial
The Nitrate’s Effect on Activity Tolerance in Heart Failure With Preserved Ejection Fraction (NEAT-HFpEF) trial22 investigated whether extended-release isosorbide mononitrate would increase daily activity levels in patients with HFpEF. This double-blind, crossover study randomized 110 patients with HFpEF (ejection fraction ≥ 50%) and persistent dyspnea to escalating doses of isosorbide mononitrate or placebo over 6 weeks, then to the other arm for another 6 weeks. Daily activity levels during the 120-mg phase were measured with a continuously worn accelerometer.
No beneficial effect of nitrates was evident, with a nonsignificant trend towards decreased activity levels, a significant decrease in hours of activity per day (–0.30 hours, P = .02), and no change in the other secondary end points such as quality-of-life score, 6-minute walk distance, or natriuretic peptide level.
Suggested explanations for these negative findings include the possibility of rapid dose escalation leading to increased subtle side effects (headache, dizziness, fatigue) that, in turn, decreased activity. Additionally, given the imprecise diagnostic criteria for HFpEF, difficulties with patient selection may have led to inclusion of a large number of patients without elevated left-sided filling pressures.23
The RELAX trial
The Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure With Preserved Ejection Fraction (RELAX) trial24 investigated whether the phosphodiesterase-5 inhibitor sildenafil would improve exercise capacity in HFpEF. Improvements in both exercise capacity and clinical outcomes had already been seen in earlier trials in patients with pulmonary hypertension, as well as in those with HFrEF.25 A smaller study in HFpEF patients with pulmonary hypertension was also encouraging.26
Thus, it was disappointing that, after randomizing 216 outpatients with HFpEF to sildenafil or placebo for 24 weeks, no benefit was seen in the primary end point of change in peak oxygen consumption or in secondary end points of change in 6-minute walk distance or composite clinical score. Unlike in NEAT-HFpEF, patients here were required to have elevated natriuretic peptide levels or elevated invasively measured filling pressures.
The study authors speculated that pulmonary arterial hypertension and right ventricular systolic failure might need to be significant for patients with HFpEF to benefit from phosphodiesterase-5 inhibitors, with their known effects of dilation of pulmonary vasculature and increasing contractility of the right ventricle.24
New or modified recommendations on nitrates or phosphodiesterase-5 drugs
Given these disappointing results, the 2017 update provides a class III (no benefit) recommendation against the routine use of nitrates or phosphodiesterase-5 inhibitors to improve exercise tolerance or quality of life in HFpEF, citing them as ineffective (Table 3).1
IRON DEFICIENCY IN HEART FAILURE
Not only is iron deficiency present in roughly 50% of patients with symptomatic heart failure (stage C and D HFrEF),27 it is also associated with increased heart failure symptoms such as fatigue and exercise intolerance,28 reduced functional capacity, decreased quality of life, and increased mortality.
Notably, this association exists regardless of the hemoglobin level.29 In fact, even in those without heart failure or anemia, iron deficiency alone results in worsened aerobic performance, exercise intolerance, and increased fatigue.30 Conversely, improvement in symptoms, exercise tolerance, and cognition have been shown with repletion of iron stores in such patients.31
At the time of the 2013 guidelines, only a single large trial of intravenous iron in HFrEF and iron deficiency had been carried out (see below), and although the results were promising, it was felt that the evidence base on which to make recommendations was inadequate. Thus, recommendations were deferred until more data could be obtained.
Of note, in all the trials discussed below, iron deficiency was diagnosed in the setting of heart failure as ferritin less than 100 mg/mL (absolute iron deficiency) or as ferritin 100 to 300 mg/mL with transferrin saturation less than 20% (relative deficiency).32
The CONFIRM-HF trial
As in the Ferinject Assessment in Patients With Iron Deficiency and Chronic Heart Failure (FAIR-HF) trial,33 the subsequent Ferric Carboxymaltose Evaluation on Performance in Patients With Iron Deficiency in Combination With Chronic Heart Failure (CONFIRM-HF) trial34 involved the intravenous infusion of iron (ferric carboxymaltose) in outpatients with symptomatic HFrEF and iron deficiency. It showed that benefits remained evident with a more objective primary end point (change in 6-minute walk test distance at 24 weeks), and that such benefits were sustained, as seen in numerous secondary end points related to functional capacity at 52 weeks. Benefits in CONFIRM-HF were evident independently from anemia, specifically whether hemoglobin was under or over 12 g/dL.
Although these results were promising, it remained unclear whether such improvements could be obtained with a much easier to administer, more readily available, and less expensive oral iron formulation.
The IRONOUT-HF trial
The Iron Repletion Effects on Oxygen Uptake in Heart Failure (IRONOUT-HF) trial35 investigated whether oral, rather than intravenous, iron supplementation could improve peak exercise capacity in patients with HFrEF and iron deficiency. This double-blind, placebo-controlled trial randomized 225 patients with NYHA class II to IV HFrEF and iron deficiency to treatment with oral iron polysaccharide (150 mg twice daily) or placebo for 16 weeks.
Contrary to the supportive findings above, no significant change was seen in the primary end point of change in peak oxygen uptake or in any of the secondary end points (change in 6-minute walk, quality of life). Also, despite a 15-fold increase in the amount of iron administered in oral form compared with intravenously, little change was evident in the indices of iron stores over the course of the study, with only a 3% increase in transferrin saturation and an 11 ng/mL increase in ferritin. The intravenous trials resulted in a 4-fold greater increase in transferrin saturation and a 20-fold greater increase in ferritin.36
What keeps heart failure patients from absorbing oral iron? It is unclear why oral iron administration in HFrEF, such as in IRONOUT-HF, seems to be so ineffective, but hepcidin—a protein hormone made by the liver that shuts down intestinal iron absorption and iron release from macrophages—may play a central role.37 When iron stores are adequate, hepcidin is upregulated to prevent iron overload. However, hepcidin is also increased in inflammatory states, and chronic heart failure is often associated with inflammation.
With this in mind, the IRONOUT-HF investigators measured baseline hepcidin levels at the beginning and at the end of the 16 weeks and found that high baseline hepcidin levels predicted poorer response to oral iron. Other inflammatory mediators, such as interleukin 6, may also play a role.38,39 Unlike oral iron formulations such as iron polysaccharide, intravenous iron (ferric carboxymaltose) bypasses these regulatory mechanisms, which may partly explain its much more significant effect on the indices of iron stores and outcomes.
New or modified recommendations on iron
The 2017 update1 makes recommendations regarding iron deficiency and anemia in heart failure for the first time.
A class IIb recommendation states that it might be reasonable to treat NYHA class II and III heart failure patients with iron deficiency with intravenous iron to improve functional status and quality of life. A strong recommendation has been deferred until more is known about morbidity and mortality effects from adequately powered trials, some of which are under way and explored further below.
The 2017 update also withholds any recommendations regarding oral iron supplementation in heart failure, citing an uncertain evidence base. Certainly, the subsequent IRONOUT-HF trial does not lend enthusiasm for this approach.
Lastly, given the lack of benefit coupled with the increased risk of thromboembolic events evident in a trial of darbepoetin alfa vs placebo in non-iron deficiency-related anemia in HFrEF,40,41 the 2017 update provides a class III (no benefit) recommendation against using erythropoietin-stimulating agents in heart failure and anemia.
HYPERTENSION IN HEART FAILURE
The 2013 guidelines for the management of heart failure simply provided a class I recommendation to control hypertension and lipid disorders in accordance with contemporary guidelines to lower the risk of heart failure.1
SPRINT
The Systolic Blood Pressure Intervention Trial (SPRINT)42 sought to determine whether a lower systolic blood pressure target (120 vs 140 mm Hg) would reduce clinical events in patients at high risk for cardiovascular events but without diabetes mellitus. Patients at high risk were defined as over age 75, or with known vascular disease, chronic kidney disease, or a Framingham Risk Score higher than 15%. This multicenter, open-label controlled trial randomized 9,361 patients to intensive treatment (goal systolic blood pressure < 120 mm Hg) or standard treatment (goal systolic blood pressure < 140 mm Hg).
SPRINT was stopped early at a median follow-up of 3.26 years when a 25% relative risk reduction in the primary composite outcome of myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes became evident in the intensive-treatment group (1.65% vs 2.19% per year, HR 0.75, P < .0001).
All-cause mortality was also lower in the intensive-treatment group (HR 0.73, P = .003), while the incidence of serious adverse events (hypotension, syncope, electrolyte abnormalities, acute kidney injury, and noninjurious falls) was only slightly higher (38.3% vs 37.1%, P = .25). Most pertinent, a significant 38% relative risk reduction in heart failure and a 43% relative risk reduction in cardiovascular events were also evident.
Of note, blood pressure measurements were taken as the average of 3 measurements obtained by an automated cuff taken after the patient had been sitting quietly alone in a room for 5 minutes.
New or modified recommendations on hypertension in heart failure
Given the impressive 25% relative risk reduction in myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes in SPRINT,42 the 2017 update1 incorporated the intensive targets of SPRINT into its recommendations. However, to compensate for what are expected to be higher blood pressures obtained in real-world clinical practice as opposed to the near-perfect conditions used in SPRINT, a slightly higher blood pressure goal of less than 130/80 mm Hg was set.
Although not specifically included in SPRINT, given the lack of trial data on specific blood pressure targets in HFrEF and the decreased cardiovascular events noted above, a class I (level of evidence C, expert opinion) recommendation to target a goal systolic blood pressure less than 130 mm Hg in stage C HFrEF with hypertension is also given. Standard guideline-directed medications in the treatment of HFrEF are to be used (Table 4).
Similarly, a new class I (level of evidence C, expert opinion) recommendation is given for hypertension in HFpEF to target a systolic blood pressure of less than 130 mm Hg, with special mention to first manage any element of volume overload with diuretics. Other than avoiding nitrates (unless used for angina) and phosphodiesterase inhibitors, it is noted that few data exist to guide the choice of antihypertensive further, although perhaps renin-angiotensin-aldosterone system inhibition, especially aldosterone antagonists, may be considered. These recommendations are fully in line with the 2017 ACC/AHA high blood pressure clinical practice guidelines,43 ie, that renin-angiotensin-aldosterone system inhibition with an angiotensin-converting enzyme (ACE) inhibitor or ARB and especially mineralocorticoid receptor antagonists would be the preferred choice (Table 4).
SLEEP-DISORDERED BREATHING IN HEART FAILURE
Sleep-disordered breathing, either obstructive sleep apnea (OSA) or central sleep apnea, is quite commonly associated with symptomatic HFrEF.44 Whereas OSA is found in roughly 18% and central sleep apnea in 1% of the general population, sleep-disordered breathing is found in nearly 60% of patients with HFrEF, with some studies showing a nearly equal proportion of OSA and central sleep apnea.45 A similar prevalence is seen in HFpEF, although with a much higher proportion of OSA.46 Central sleep apnea tends to be a marker of more severe heart failure, as it is strongly associated with severe cardiac systolic dysfunction and worse functional capacity.47
Not surprisingly, the underlying mechanism of central sleep apnea is quite different from that of OSA. Whereas OSA predominantly occurs because of repeated obstruction of the pharynx due to nocturnal pharyngeal muscle relaxation, no such airway patency issues or strained breathing patterns exist in central sleep apnea. Central sleep apnea, which can manifest as Cheyne-Stokes respirations, is thought to occur due to an abnormal ventilatory control system with complex pathophysiology such as altered sensitivity of central chemoreceptors to carbon dioxide, interplay of pulmonary congestion, subsequent hyperventilation, and prolonged circulation times due to reduced cardiac output.48
What the two types of sleep-disordered breathing have in common is an association with negative health outcomes. Both appear to induce inflammation and sympathetic nervous system activity via oxidative stress from intermittent nocturnal hypoxemia and hypercapnea.49 OSA was already known to be associated with significant morbidity and mortality rates in the general population,50 and central sleep apnea had been identified as an independent predictor of mortality in HFrEF.51
At the time of the 2013 guidelines, only small or observational studies with limited results had been done evaluating treatment effects of continuous positive airway pressure therapy (CPAP) on OSA and central sleep apnea. Given the relative paucity of data, only a single class IIa recommendation stating that CPAP could be beneficial to increase left ventricular ejection fraction and functional status in concomitant sleep apnea and heart failure was given in 2013. However, many larger trials were under way,52–59 some with surprising results such as a significant increase in cardiovascular and all-cause mortality (Table 5).54
New or modified recommendations on sleep-disordered breathing
Given the common association with heart failure (60%)45 and the marked variation in response to treatment, including potential for harm with adaptive servo-ventilation and central sleep apnea, a class IIa recommendation is made stating that it is reasonable to obtain a formal sleep study in any patient with symptomatic (NYHA class II–IV) heart failure.1
Due to the potential for harm with adaptive servo-ventilation in patients with central sleep apnea and NYHA class II to IV HFrEF, a class III (harm) recommendation is made against its use.
Largely based on the results of the Sleep Apnea Cardiovascular Endpoints (SAVE) trial,56 a class IIb, level of evidence B-R (moderate, based on randomized trials) recommendation is given, stating that the use of CPAP in those with OSA and known cardiovascular disease may be reasonable to improve sleep quality and reduce daytime sleepiness.
POTENTIAL APPLICATIONS IN ACUTE DECOMPENSATED HEART FAILURE
Although the 2017 update1 is directed mostly toward managing chronic heart failure, it is worth considering how it might apply to the management of ADHF.
SHOULD WE USE BIOMARFER TARGETS TO GUIDE THERAPY IN ADHF?
The 2017 update1 does offer direct recommendations regarding the use of biomarker levels during admissions for ADHF. Mainly, they emphasize that the admission biomarker levels provide valuable information regarding acute prognosis and risk stratification (class I recommendation), while natriuretic peptide levels just before discharge provide the same for the postdischarge timeframe (class IIa recommendation).
The update also explicitly cautions against using a natriuretic peptide level-guided treatment strategy, such as setting targets for predischarge absolute level or percent change in level of natriuretic peptides during admissions for ADHF. Although observational and retrospective studies have shown better outcomes when levels are reduced at discharge, treating for any specific inpatient target has never been tested in any large, prospective study; thus, doing so could result in unintended harm.
So what do we know?
McQuade et al systematic review
McQuade et al57 performed a systematic review of more than 40 ADHF trials, which showed that, indeed, patients who achieved a target absolute natriuretic peptide level (BNP ≤ 250 pg/mL) or percent reduction (≥ 30%) at time of discharge had significantly improved outcomes such as reduced postdischarge all-cause mortality and rehospitalization rates. However, these were mostly prospective cohort studies that did not use any type of natriuretic peptide level-guided treatment protocol, leaving it unclear whether such a strategy could positively influence outcomes.
For this reason, both McQuade et al57 and, in an accompanying editorial, Felker et al58 called for properly designed, randomized controlled trials to investigate such a strategy. Felker noted that only 2 such phase II trials in ADHF have been completed,59,60 with unconvincing results.
PRIMA II
The Multicenter, Randomized Clinical Trial to Study the Impact of In-hospital Guidance for Acute Decompensated Heart Failure Treatment by a Predefined NT-ProBNP Target on the Reduction of Readmission and Mortality Rates (PRIMA II)60 randomized patients to natriuretic peptide level-guided treatment or standard care during admission for ADHF.
Many participants (60%) reached the predetermined target of 30% reduction in natriuretic peptide levels at the time of clinical stabilization and randomization; 405 patients were randomized. Patients in the natriuretic peptide level-guided treatment group underwent a prespecified treatment algorithm, with repeat natriuretic peptide levels measured again after the protocol.
Natriuretic peptide-guided therapy failed to show any significant benefit in any clinical outcomes, including the primary composite end point of mortality or heart failure readmissions at 180 days (36% vs 38%, HR 0.99, 95% confidence interval 0.72–1.36). Consistent with the review by McQuade et al,57 achieving the 30% reduction in natriuretic peptide at discharge, in either arm, was associated with a better prognosis, with significantly lower mortality and readmission rates at 180 days (HR 0.39 for rehospitalization or death, 95% confidence interval 0.27–0.55).
As in the observational studies, those who achieved the target natriuretic peptide level at the time of discharge had a better prognosis than those who did not, but neither study showed an improvement in clinical outcomes using a natriuretic peptide level-targeting treatment strategy.
No larger randomized controlled trial results are available for guided therapy in ADHF. However, additional insight may be gained from a subsequent trial61 that evaluated biomarker-guided titration of guideline-directed medical therapy in outpatients with chronic HFrEF.
The GUIDE-IT trial
That trial, the Guiding Evidence Based Therapy Using Biomarker Intensified Treatment in Heart Failure (GUIDE-IT)61 trial, was a large multicenter attempt to determine whether a natriuretic peptide-guided treatment strategy was more effective than standard care in the management of 894 high-risk outpatients with chronic HFrEF. Earlier, promising results had been obtained in a meta-analysis62 of more than 11 similar trials in 2,000 outpatients, with a decreased mortality rate (HR 0.62) seen in the biomarker-guided arm. However, the results had not been definitive due to being underpowered.62
Unfortunately, the results of GUIDE-IT were disappointing, with no significant difference in either the combined primary end point of mortality or hospitalization for heart failure, or the secondary end points evident at 15 months, prompting early termination for futility.61 Among other factors, the study authors postulated that this may have partly resulted from a patient population with more severe heart failure and resultant azotemia, limiting the ability to titrate neurohormonal medications to the desired dosage.
The question of whether patients who cannot achieve such biomarker targets need more intensive therapy or whether their heart failure is too severe to respond adequately echoes the question often raised in discussions of inpatient biomarker-guided therapy.58 Thus, only limited insight is gained, and it remains unclear whether a natriuretic peptide-guided treatment strategy can improve outpatient or inpatient outcomes. Until this is clarified, clinical judgment and optimization of guideline-directed management and therapy should remain the bedrock of treatment.
SHOULD ALDOSTERONE ANTAGONISTS BE USED IN ACUTE HFpEF?
Given the encouraging results in chronic HFpEF from post hoc analyses of TOPCAT, are there any additional recent data suggesting a role for aldosterone antagonists such as spironolactone in acute HFpEF?
The ATHENA-HF trial
The Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial63 compared treatment with high-dose spironolactone (100 mg) for 96 hours vs usual care in 360 patients with ADHF. The patient population included those with HFrEF and HFpEF, and usual care included low-dose spironolactone (12.5–25 mg) in roughly 15% of patients. High-dose mineralocorticoid receptor antagonists have been shown to overcome diuretic resistance, improve pulmonary vascular congestion, and partially combat the adverse neurohormonal activation seen in ADHF.
Unfortunately, the trial was completely neutral in regard to the primary end point of reduction in natriuretic peptide levels as well as to the secondary end points of 30-day mortality rate, heart failure readmission, clinical congestion scores, urine output, and change in weight. No suggestion of additional benefit was seen in subgroup analysis of patients with acute HFpEF (ejection fraction > 45%), which yielded similar results.63
Given these lackluster findings, routine use of high-dose spironolactone in ADHF is not recommended.64 However, the treatment was well tolerated, without significant adverse effects of hyperkalemia or kidney injury, leaving the door open as to whether it may have utility in selected patients with diuretic resistance.
Should ARNIs and ivabradine be started during ADHF admissions?
The first half of the focused update3 of the 2013 guidelines,2 reviewed by Okwuosa et al,7 provided recommendations for the use of sacubitril-valsartan, an angiotensin-neprilysin inhibitor (ARNI), and ivabradine, a selective sinoatrial node If channel inhibitor, in chronic HFrEF.
Sacubitril-valsartan was given a class I recommendation for use in patients with NYHA class II or III chronic HFrEF who tolerate an ACE inhibitor or an ARB. This recommendation was given largely based on the benefits in mortality and heart failure hospitalizations seen in PARADIGM-HF (the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure)65 compared with enalapril (HR 0.80, 95% CI 0.73–0.87, P < .001).
There is currently no recommendation on initiation or use of ARNIs during admissions for ADHF, but a recent trial may lend some insight.66
THE PIONEER-HF trial
The Comparison of Sacubitril/Valsartan vs Enalapril on Effect on NT-proBNP in Patients Stabilized From an Acute Heart Failure Episode (PIONEER-HF) trial66 randomized patients admitted for acute HFrEF, once stabilized, to sacubitril-valsartan or enalapril. Encouragingly, the percentage change of natriuretic peptide levels from the time of inpatient initiation to 4 and 8 weeks thereafter, the primary efficacy end point, was 46.7% with sacubitril-valsartan versus 25.3% with enalapril alone (ratio of change 0.71, 95% CI 0.63–0.81, P < .001). Although not powered for such, a prespecified analysis of a composite of clinical outcomes was also favorable for sacubitril-valsartan, largely driven by a 44% decreased rate of rehospitalization. More definitive, and quite reassuring, was that no significant difference was seen in the key safety outcomes of worsening renal function, hyperkalemia, symptomatic hypotension, and angioedema. These results were also applicable to the one-third of study participants who had no former diagnosis of heart failure, the one-third identifying as African American, and the one-third who had not been taking an ACE inhibitor or ARB. These results, taken together with the notion that at study completion the patients become similar to those included in PARADIGM-HF, have led some to assert that PIONEER-HF has the potential to change clinical practice.
Ivabradine was given a class IIa recommendation for use in patients with NYHA class II or III chronic HFrEF with a resting heart rate of at least 70 bpm, in sinus rhythm, despite being on optimal medical therapy including a beta-blocker at a maximum tolerated dose.
This recommendation was largely based on SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which randomized patients to ivabradine or placebo to evaluate the effects of isolated lowering of the heart rate on the composite primary outcome of cardiovascular death or hospitalization. A significant reduction was seen in the ivabradine arm (HR 0.82, 95% CI 0.75–0.90, P < .0001), mainly driven by decreased hospitalizations.67
Subsequently, a small unblinded single-center study was undertaken to evaluate the efficacy and safety of initiating ivabradine during admissions for ADHF.68
THE ETHIC-AHF trial
The Effect of Early Treatment With Ivabradine Combined With Beta-Blockers vs Beta-Blockers Alone in Patients Hospitalized With Heart Failure and Reduced Left Ventricular Ejection Fraction (ETHIC-AHF) trial68 sought to determine the safety and effectiveness of early coadministration of ivabradine with beta-blockers in patients with acute HFrEF.
This single-center, unblinded study randomized 71 patients to ivabradine and beta-blockade or beta-blockade alone upon clinical stabilization (24–48 hours) after admission for acute decompensated HFrEF.
The primary end point was heart rate at 28 days, with the ivabradine group showing a statistically significant decrease (64 vs 70 bpm, P = .01), which persisted at 4 months. There was no significant difference in the secondary end points of adverse drug effects or the composite of clinical event outcomes (all-cause mortality, admission for heart failure or cardiovascular cause), but a number of surrogate end points including left ventricular ejection fraction, BNP level, and NYHA functional class at 4 months showed mild improvement.
Although this study provided evidence that the coadministration of ivabradine and a beta-blocker is safe and was positive in regard to clinical outcomes, the significant limitations due to its size and study design (single-center, unblinded, 4-month follow-up) simply serve to support the pursuit of larger studies with more stringent design and longer follow-up in order to determine the clinical efficacy.
The PRIME-HF trial
The Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF) trial69 is a randomized, open-label, multicenter trial comparing standard care vs the initiation of ivabradine before discharge, but after clinical stabilization, during admissions for ADHF in patients with chronic HFrEF (left ventricular ejection fraction ≤ 35%). At subsequent outpatient visits, the dosage can be modified in the ivabradine group, or ivabradine can be initiated at the provider’s discretion in the usual-care group.
PRIME-HF is attempting to determine whether initiating ivabradine before discharge will result in more patients taking ivabradine at 180 days, its primary end point, as well as in changes in secondary end points including heart rate and patient-centered outcomes. The study is active, with reporting expected in 2019.
As these trials all come to completion, it will not be long before we have further guidance regarding the inpatient initiation of these new and exciting therapeutic agents.
SHOULD INTRAVENOUS IRON BE GIVEN DURING ADHF ADMISSIONS?
Given the high prevalence of iron deficiency in symptomatic HFrEF, its independent association with mortality, improvements in quality of life and functional capacity suggested by repleting with intravenous iron (in FAIR-HF and CONFIRM-HF), the seeming inefficacy of oral iron in IRONOUT, and the logistical challenges of intravenous administration during standard clinic visits, could giving intravenous iron soon be incorporated into admissions for ADHF?
Caution has been advised for several reasons. As discussed above, larger randomized controlled trials powered to detect more definitive clinical end points such as death and the rate of hospitalization are still needed before a stronger recommendation can be made for intravenous iron in HFrEF. Also, without such data, it seems unwise to add the considerable economic burden of routinely assessing for iron deficiency and providing intravenous iron during ADHF admissions to the already staggering costs of heart failure.
The effects seen on morbidity and mortality that become evident in these trials over the next 5 years will help determine future guidelines and whether intravenous iron is routinely administered in bridge clinics, during inpatient admissions for ADHF, or not at all in patients with HFrEF and iron deficiency.
INTERNISTS ARE KEY
Heart failure remains one of the most common, morbid, complex, and costly diseases in the United States, and its prevalence is expected only to increase.4,5 The 2017 update1 of the 2013 guideline2 for the management of heart failure provides recommendations aimed not only at management of heart failure, but also at its comorbidities and, for the first time ever, at its prevention.
Internists provide care for the majority of heart failure patients, as well as for their comorbidities, and are most often the first to come into contact with patients at high risk of developing heart failure. Thus, a thorough understanding of these guidelines and how to apply them to the management of acute decompensated heart failure is of critical importance.
In 2017, the American College of Cardiology (ACC), American Heart Association (AHA), and Heart Failure Society of America (HFSA) jointly released a focused update1 of the 2013 ACC/AHA guideline for managing heart failure.2 This is the second focused update of the 2013 guidelines; the first update,3 in 2016, covered 2 new drugs (sacubitril-valsartan and ivabradine) for chronic stage C heart failure with reduced ejection fraction (HFrEF).
Rather than focus on new medication classes, this second update provides recommendations regarding:
- Preventing the progression to left ventricular dysfunction or heart failure in patients at high risk (stage A) through screening with B-type natriuretic peptide (BNP) and aiming for more aggressive blood pressure control
- Inpatient biomarker use
- Medications in heart failure with preserved ejection fraction (HFpEF, or diastolic heart failure)
- Blood pressure targets in stage C heart failure
- Managing important comorbidities such as iron deficiency and sleep-disordered breathing to decrease morbidity, improve functional capacity, and enhance quality of life.
These guidelines and the data that underlie them are explored below. We also discuss potential applications to the management of hospitalization for acute decompensated heart failure (ADHF).
COMMON, COSTLY, AND DEBILITATING
Heart failure—defined by the ACC/AHA as the complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood—remains one of the most common, costly, and debilitating diseases in the United States.2 Based on National Health and Nutrition Examination Survey data from 2011 to 2014, an estimated 6.5 million US adults have it, with projections of more than 8 million by 2030.4,5 More than 960,000 new cases are thought to occur annually, with a lifetime risk of developing it of roughly 20% to 45%.6
Despite ever-growing familiarity and some significant strides in management, the death rate in this syndrome is substantial. After admissions for heart failure (which number 1 million per year), the mortality rate is roughly 10% at 1 year and 40% at 5 years.6 Also staggering are the associated costs, with $30.7 billion attributed to heart failure in 2012 and a projected $69.7 billion annually by 2030.5 Thus, we must direct efforts not only to treatment, but also to prevention.
Preventive efforts would target patients with ACC/AHA stage A heart failure—those at high risk for developing but currently without evidence of structural heart disease or heart failure symptoms (Table 1).7 This group may represent up to one-third of the US adult population, or 75 million people, when including the well-recognized risk factors of coronary artery disease, hypertension, diabetes mellitus, and chronic kidney disease in those without left ventricular dysfunction or heart failure.8
BIOMARKERS FOR PREVENTION
Past ACC/AHA heart failure guidelines2 have included recommendations on the use of biomarkers to aid in diagnosis and prognosis and, to a lesser degree, to guide treatment of heart failure. Largely based on 2 trials (see below), the 2017 guidelines go further, issuing a recommendation on the use of natriuretic peptide biomarkers in a screening strategy to prompt early intervention and prevent the progression to clinical heart failure in high-risk patients (stage A heart failure).
The PONTIAC trial
The NT-proBNP Selected Prevention of Cardiac Events in a Population of Diabetic Patients Without a History of Cardiac Disease (PONTIAC) trial9 randomized 300 outpatients with type 2 diabetes mellitus and an elevated N-terminal proBNP (NT-proBNP) level (> 125 pg/mL) to standard medical care vs standard care plus intensive up-titration of renin-angiotensin system antagonists and beta-blockers in a cardiac clinic over 2 years.
Earlier studies10 had shown NT-proBNP levels to have predictive value for cardiac events in diabetic patients, while the neurohormonal treatments were thought to have an established record of preventing primary and secondary cardiovascular events. In PONTIAC, a significant reduction was seen in the primary end point of hospitalization or death due to cardiac disease (hazard ratio [HR] 0.351, P = .044), as well as in the secondary end point of hospitalization due to heart failure (P < .05), in the aggressive-intervention group. These results laid the foundation for the larger St. Vincent’s Screening to Prevent Heart Failure (STOP-HF) trial.11
The STOP-HF trial
The STOP-HF trial randomized 1,235 outpatients who were at high risk but without left ventricular dysfunction or heart failure symptoms (stage A) to annual screening alone vs annual screening plus BNP testing, in which a BNP level higher than 50 pg/mL triggered echocardiography and evaluation by a cardiologist who would then assist with medications.11
Eligible patients were over age 40 and had 1 or more of the following risk factors:
- Diabetes mellitus
- Hypertension
- Hypercholesterolemia
- Obesity (body mass index > 30 kg/m2)
- Vascular disease (coronary, cerebral, or peripheral arterial disease)
- Arrhythmia requiring treatment
- Moderate to severe valvular disease.
After a mean follow-up of 4.3 years, the primary end point, ie, asymptomatic left ventricular dysfunction with or without newly diagnosed heart failure, was found in 9.7% of the control group and in only 5.9% of the intervention group with BNP screening, a 42% relative risk reduction (P = .013).
Similarly, the incidence of secondary end points of emergency hospitalization for a cardiovascular event (arrhythmia, transient ischemic attack, stroke, myocardial infarction, peripheral or pulmonary thrombosis or embolization, or heart failure) was also lower at 45.2 vs 24.4 per 1,000 patient-years, a 46% relative risk reduction.
An important difference in medications between the 2 groups was an increase in subsequently prescribed renin-angiotensin-aldosterone system therapy, mainly consisting of angiotensin II receptor blockers (ARBs), in those with elevated BNP in the intervention group. Notably, blood pressure was about the same in the 2 groups.11
Although these findings are encouraging, larger studies are needed, as the lack of blinding, low event rates, and small absolute risk reduction make the results difficult to generalize.
New or modified recommendations for screening
Employing this novel prevention strategy in the extremely large number of patients with stage A heart failure, thought to be up to one-third of the US adult population, may serve as a way to best direct and utilize limited medical resources.8
BIOMARKERS FOR PROGNOSIS OR ADDED RISK STRATIFICATION
The 2013 guidelines2 recognized that a significant body of work had accumulated showing that natriuretic peptide levels can predict outcomes in both chronic and acute heart failure. Thus, in both conditions, the guidelines contained separate class Ia recommendations to obtain a natriuretic peptide level, troponin level, or both to establish prognosis or disease severity.
The 2017 update1 underscores the importance of timing in measuring natriuretic peptide levels during admission for ADHF, with emphasis on obtaining them at admission and at discharge for acute and postdischarge prognosis. The completely new class IIa recommendation to obtain a predischarge natriuretic peptide level for postdischarge prognosis was based on a number of observational studies, some of which we explore below.
The ELAN-HF meta-analysis
The European Collaboration on Acute Decompensated Heart Failure (ELAN-HF)12 performed a meta-analysis to develop a discharge prognostication score for ADHF that included both absolute level and percent change in natriuretic peptide levels at the time of discharge.
Using data from 7 prospective cohorts totaling 1,301 patients, the authors found that incorporation of these values into a subsequently validated risk model led to significant improvements in the ability to predict the end points of all-cause mortality and the combined end point of all-cause mortality or first readmission for a cardiovascular reason within 180 days.
The OPTIMIZE-HF retrospective analysis
Data from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients With Heart Failure (OPTIMIZE-HF) were retrospectively analyzed13 to determine whether postdischarge outcomes were best predicted by natriuretic peptide levels at admission or discharge or by the relative change in natriuretic peptide level. More than 7,000 patients age 65 or older, in 220 hospitals, were included, and Cox prediction models were compared using clinical variables alone or in combination with the natriuretic peptide levels.
The model that included the discharge natriuretic peptide level was found to be the most predictive, with a c-index of 0.693 for predicting mortality and a c-index of 0.606 for mortality or rehospitalization at 1 year.
New or modified recommendations on biomarkers for prognosis
The 2017 update1 modified the earlier recommendation to obtain a natriuretic peptide or troponin level or both at admission for ADHF to establish prognosis. This now has a class Ia recommendation, emphasizing that such levels be obtained on admission. In addition, a new class IIa recommendation is made to obtain a predischarge natriuretic peptide level for postdischarge prognosis. The former class Ia recommendation to obtain a natriuretic peptide level in chronic heart failure to establish prognosis or disease severity remains unchanged.
Also worth noting is what the 2017 update does not recommend in regard to obtaining biomarker levels. It emphasizes that many patients, particularly those with advanced (stage D) heart failure, have a poor prognosis that is well established with or without biomarker levels. Additionally, there are many cardiac and noncardiac causes of natriuretic peptide elevation; thus, clinical judgment remains paramount.
The 2017 update1 also cautions against setting targets of percent change in or absolute levels of natriuretic peptide at discharge despite observational and retrospective studies demonstrating better outcomes when levels are reduced, as treating for any specific target has never been studied in a large prospective study. Thus, doing so may result in unintended harm. Rather, clinical judgment and optimization of guideline-directed management and therapy are encouraged (Table 2).
PHARMACOLOGIC TREATMENT FOR STAGE C HFpEF
Although the 2013 guidelines2 contain many class I recommendations for various medications in chronic HFrEF, not a single such recommendation is found for chronic HFpEF. A review by Okwuosa et al7 covered HFrEF, including the most recent additions on which the 2016 update was based, sacubitril-valsartan and ivabradine. The 2016 update was similarly devoid of recommendations regarding specific medications in HFpEF, leaving only the 2013 class IIb recommendation to consider using an ARB to decrease hospitalizations in HFpEF.
Evidence behind this recommendation came from the Candesartan in Heart Failure Assessment of Reduction in Mortality and Morbidity program’s randomized controlled trial in 3,025 patients with New York Heart Association (NYHA) class II to IV heart failure and left ventricular ejection fraction over 40%, who were treated with candesartan or placebo.14 Over a median follow-up of 36.6 months, there was no significant difference in the primary composite outcome of cardiovascular death or admission for heart failure, but significantly fewer patients in the candesartan arm were admitted (230 vs 270, P = .017). Thus the recommendation.
Although this finding was encouraging, it was clear that no blockbuster drug for HFpEF had been identified. Considering that roughly half of all heart failure patients have preserved ejection fraction, the discovery of such a drug for HFpEF would be met with much excitement.15 Subsequently, other medication classes have been evaluated in the hope of benefit, allowing the 2017 update to provide specific recommendations for aldosterone antagonists, nitrates, and phosphodiesterase-5 inhibitors in HFpEF.
ALDOSTERONE ANTAGONISTS FOR HFpEF
Mineralocorticoid receptor antagonists had previously been shown to significantly reduce morbidity and mortality rates in patients with HFrEF.16 In addition to aldosterone’s effects on sodium retention and many other pathophysiologic mechanisms relating to heart failure, this hormone is also known to play a role in promoting myocardial fibrosis.17 Accordingly, some have wondered whether aldosterone antagonists could improve diastolic dysfunction, and perhaps outcomes, in HFpEF.
The Aldo-DHF trial
The Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF) trial investigated whether the aldosterone antagonist spironolactone would improve diastolic function or maximal exercise capacity in chronic HFpEF.18 It randomized 422 ambulatory patients with NYHA stage II or III heart failure, preserved left ventricular ejection fraction (≥ 50%), and echocardiographic evidence of diastolic dysfunction to receive spironolactone 25 mg daily or placebo.
Although no significant difference was seen in maximal exercise capacity, follow-up over 1 year nevertheless showed significant improvement in echocardiographic diastolic dysfunction (E/e') and perhaps reverse remodeling (decreased left ventricular mass index). These improvements spurred larger trials powered to detect whether clinical outcomes could also be improved.
The TOPCAT trial
The Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial19 was a large, multicenter, international, double-blind, placebo-controlled trial that investigated whether spironolactone could improve clinical outcomes in HFpEF. It randomized 3,445 patients with symptomatic heart failure and left ventricular ejection fraction of 45% or more to spironolactone 15 to 45 mg daily or placebo.
The effect on a composite primary outcome of death from cardiovascular cause, aborted cardiac arrest, or hospitalization for heart failure was evaluated over a mean follow-up of 3.3 years, with only a small (HR 0.89), nonclinically significant reduction evident. Those in the spironolactone group did have a significantly lower incidence of hospitalization for heart failure (12.0% vs 14.2%, P = .04).
Although the results were disappointing in this essentially negative trial, significant regional variations evident on post hoc analysis prompted further investigation and much controversy since the trial’s publication in 2014.
Participants came in roughly equal proportions from the Americas (United States, Canada, Brazil, and Argentina—51%) and from Russia and Georgia (49%), but outcomes between the two groups were markedly different. Concern was first raised when immediate review discovered a 4-fold lower rate of the primary outcome in the placebo groups from Russia and Georgia (8.4%), a rate in fact similar to that in patients without heart failure.19 This led to further exploration that identified other red flags that called into question the data integrity from the non-American sites.20
Not only did patients receiving spironolactone in Russia and Georgia not experience the reduction in clinical outcomes seen in their American counterparts, they also did not manifest the expected elevations in potassium and creatinine, and spironolactone metabolites were undetectable in almost one-third of patients.21
These findings prompted a post hoc analysis that included only the 51% (1,767 patients) of the study population coming from the Americas; in this subgroup, treatment with spironolactone was associated with a statistically significant 18% relative risk reduction in the primary composite outcome, a 26% reduction in cardiovascular mortality, and an 18% reduction in hospitalization for heart failure.20
New or modified recommendations on aldosterone receptor antagonists
Nitrates and phosphodiesterase-5 inhibitors
Earlier studies indicated that long-acting nitrates are prescribed in 15% to 50% of patients with HFpEF, perhaps based on extrapolation from studies in HFrEF suggesting that they might improve exercise intolerance.22 Some have speculated that the hemodynamic effects of nitrates, such as decreasing pulmonary congestion, might improve exercise intolerance in those with the stiff ventricles of HFpEF as well, prompting further study.
The NEAT-HFpEF trial
The Nitrate’s Effect on Activity Tolerance in Heart Failure With Preserved Ejection Fraction (NEAT-HFpEF) trial22 investigated whether extended-release isosorbide mononitrate would increase daily activity levels in patients with HFpEF. This double-blind, crossover study randomized 110 patients with HFpEF (ejection fraction ≥ 50%) and persistent dyspnea to escalating doses of isosorbide mononitrate or placebo over 6 weeks, then to the other arm for another 6 weeks. Daily activity levels during the 120-mg phase were measured with a continuously worn accelerometer.
No beneficial effect of nitrates was evident, with a nonsignificant trend towards decreased activity levels, a significant decrease in hours of activity per day (–0.30 hours, P = .02), and no change in the other secondary end points such as quality-of-life score, 6-minute walk distance, or natriuretic peptide level.
Suggested explanations for these negative findings include the possibility of rapid dose escalation leading to increased subtle side effects (headache, dizziness, fatigue) that, in turn, decreased activity. Additionally, given the imprecise diagnostic criteria for HFpEF, difficulties with patient selection may have led to inclusion of a large number of patients without elevated left-sided filling pressures.23
The RELAX trial
The Phosphodiesterase-5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure With Preserved Ejection Fraction (RELAX) trial24 investigated whether the phosphodiesterase-5 inhibitor sildenafil would improve exercise capacity in HFpEF. Improvements in both exercise capacity and clinical outcomes had already been seen in earlier trials in patients with pulmonary hypertension, as well as in those with HFrEF.25 A smaller study in HFpEF patients with pulmonary hypertension was also encouraging.26
Thus, it was disappointing that, after randomizing 216 outpatients with HFpEF to sildenafil or placebo for 24 weeks, no benefit was seen in the primary end point of change in peak oxygen consumption or in secondary end points of change in 6-minute walk distance or composite clinical score. Unlike in NEAT-HFpEF, patients here were required to have elevated natriuretic peptide levels or elevated invasively measured filling pressures.
The study authors speculated that pulmonary arterial hypertension and right ventricular systolic failure might need to be significant for patients with HFpEF to benefit from phosphodiesterase-5 inhibitors, with their known effects of dilation of pulmonary vasculature and increasing contractility of the right ventricle.24
New or modified recommendations on nitrates or phosphodiesterase-5 drugs
Given these disappointing results, the 2017 update provides a class III (no benefit) recommendation against the routine use of nitrates or phosphodiesterase-5 inhibitors to improve exercise tolerance or quality of life in HFpEF, citing them as ineffective (Table 3).1
IRON DEFICIENCY IN HEART FAILURE
Not only is iron deficiency present in roughly 50% of patients with symptomatic heart failure (stage C and D HFrEF),27 it is also associated with increased heart failure symptoms such as fatigue and exercise intolerance,28 reduced functional capacity, decreased quality of life, and increased mortality.
Notably, this association exists regardless of the hemoglobin level.29 In fact, even in those without heart failure or anemia, iron deficiency alone results in worsened aerobic performance, exercise intolerance, and increased fatigue.30 Conversely, improvement in symptoms, exercise tolerance, and cognition have been shown with repletion of iron stores in such patients.31
At the time of the 2013 guidelines, only a single large trial of intravenous iron in HFrEF and iron deficiency had been carried out (see below), and although the results were promising, it was felt that the evidence base on which to make recommendations was inadequate. Thus, recommendations were deferred until more data could be obtained.
Of note, in all the trials discussed below, iron deficiency was diagnosed in the setting of heart failure as ferritin less than 100 mg/mL (absolute iron deficiency) or as ferritin 100 to 300 mg/mL with transferrin saturation less than 20% (relative deficiency).32
The CONFIRM-HF trial
As in the Ferinject Assessment in Patients With Iron Deficiency and Chronic Heart Failure (FAIR-HF) trial,33 the subsequent Ferric Carboxymaltose Evaluation on Performance in Patients With Iron Deficiency in Combination With Chronic Heart Failure (CONFIRM-HF) trial34 involved the intravenous infusion of iron (ferric carboxymaltose) in outpatients with symptomatic HFrEF and iron deficiency. It showed that benefits remained evident with a more objective primary end point (change in 6-minute walk test distance at 24 weeks), and that such benefits were sustained, as seen in numerous secondary end points related to functional capacity at 52 weeks. Benefits in CONFIRM-HF were evident independently from anemia, specifically whether hemoglobin was under or over 12 g/dL.
Although these results were promising, it remained unclear whether such improvements could be obtained with a much easier to administer, more readily available, and less expensive oral iron formulation.
The IRONOUT-HF trial
The Iron Repletion Effects on Oxygen Uptake in Heart Failure (IRONOUT-HF) trial35 investigated whether oral, rather than intravenous, iron supplementation could improve peak exercise capacity in patients with HFrEF and iron deficiency. This double-blind, placebo-controlled trial randomized 225 patients with NYHA class II to IV HFrEF and iron deficiency to treatment with oral iron polysaccharide (150 mg twice daily) or placebo for 16 weeks.
Contrary to the supportive findings above, no significant change was seen in the primary end point of change in peak oxygen uptake or in any of the secondary end points (change in 6-minute walk, quality of life). Also, despite a 15-fold increase in the amount of iron administered in oral form compared with intravenously, little change was evident in the indices of iron stores over the course of the study, with only a 3% increase in transferrin saturation and an 11 ng/mL increase in ferritin. The intravenous trials resulted in a 4-fold greater increase in transferrin saturation and a 20-fold greater increase in ferritin.36
What keeps heart failure patients from absorbing oral iron? It is unclear why oral iron administration in HFrEF, such as in IRONOUT-HF, seems to be so ineffective, but hepcidin—a protein hormone made by the liver that shuts down intestinal iron absorption and iron release from macrophages—may play a central role.37 When iron stores are adequate, hepcidin is upregulated to prevent iron overload. However, hepcidin is also increased in inflammatory states, and chronic heart failure is often associated with inflammation.
With this in mind, the IRONOUT-HF investigators measured baseline hepcidin levels at the beginning and at the end of the 16 weeks and found that high baseline hepcidin levels predicted poorer response to oral iron. Other inflammatory mediators, such as interleukin 6, may also play a role.38,39 Unlike oral iron formulations such as iron polysaccharide, intravenous iron (ferric carboxymaltose) bypasses these regulatory mechanisms, which may partly explain its much more significant effect on the indices of iron stores and outcomes.
New or modified recommendations on iron
The 2017 update1 makes recommendations regarding iron deficiency and anemia in heart failure for the first time.
A class IIb recommendation states that it might be reasonable to treat NYHA class II and III heart failure patients with iron deficiency with intravenous iron to improve functional status and quality of life. A strong recommendation has been deferred until more is known about morbidity and mortality effects from adequately powered trials, some of which are under way and explored further below.
The 2017 update also withholds any recommendations regarding oral iron supplementation in heart failure, citing an uncertain evidence base. Certainly, the subsequent IRONOUT-HF trial does not lend enthusiasm for this approach.
Lastly, given the lack of benefit coupled with the increased risk of thromboembolic events evident in a trial of darbepoetin alfa vs placebo in non-iron deficiency-related anemia in HFrEF,40,41 the 2017 update provides a class III (no benefit) recommendation against using erythropoietin-stimulating agents in heart failure and anemia.
HYPERTENSION IN HEART FAILURE
The 2013 guidelines for the management of heart failure simply provided a class I recommendation to control hypertension and lipid disorders in accordance with contemporary guidelines to lower the risk of heart failure.1
SPRINT
The Systolic Blood Pressure Intervention Trial (SPRINT)42 sought to determine whether a lower systolic blood pressure target (120 vs 140 mm Hg) would reduce clinical events in patients at high risk for cardiovascular events but without diabetes mellitus. Patients at high risk were defined as over age 75, or with known vascular disease, chronic kidney disease, or a Framingham Risk Score higher than 15%. This multicenter, open-label controlled trial randomized 9,361 patients to intensive treatment (goal systolic blood pressure < 120 mm Hg) or standard treatment (goal systolic blood pressure < 140 mm Hg).
SPRINT was stopped early at a median follow-up of 3.26 years when a 25% relative risk reduction in the primary composite outcome of myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes became evident in the intensive-treatment group (1.65% vs 2.19% per year, HR 0.75, P < .0001).
All-cause mortality was also lower in the intensive-treatment group (HR 0.73, P = .003), while the incidence of serious adverse events (hypotension, syncope, electrolyte abnormalities, acute kidney injury, and noninjurious falls) was only slightly higher (38.3% vs 37.1%, P = .25). Most pertinent, a significant 38% relative risk reduction in heart failure and a 43% relative risk reduction in cardiovascular events were also evident.
Of note, blood pressure measurements were taken as the average of 3 measurements obtained by an automated cuff taken after the patient had been sitting quietly alone in a room for 5 minutes.
New or modified recommendations on hypertension in heart failure
Given the impressive 25% relative risk reduction in myocardial infarction, other acute coronary syndromes, stroke, heart failure, or death from cardiovascular causes in SPRINT,42 the 2017 update1 incorporated the intensive targets of SPRINT into its recommendations. However, to compensate for what are expected to be higher blood pressures obtained in real-world clinical practice as opposed to the near-perfect conditions used in SPRINT, a slightly higher blood pressure goal of less than 130/80 mm Hg was set.
Although not specifically included in SPRINT, given the lack of trial data on specific blood pressure targets in HFrEF and the decreased cardiovascular events noted above, a class I (level of evidence C, expert opinion) recommendation to target a goal systolic blood pressure less than 130 mm Hg in stage C HFrEF with hypertension is also given. Standard guideline-directed medications in the treatment of HFrEF are to be used (Table 4).
Similarly, a new class I (level of evidence C, expert opinion) recommendation is given for hypertension in HFpEF to target a systolic blood pressure of less than 130 mm Hg, with special mention to first manage any element of volume overload with diuretics. Other than avoiding nitrates (unless used for angina) and phosphodiesterase inhibitors, it is noted that few data exist to guide the choice of antihypertensive further, although perhaps renin-angiotensin-aldosterone system inhibition, especially aldosterone antagonists, may be considered. These recommendations are fully in line with the 2017 ACC/AHA high blood pressure clinical practice guidelines,43 ie, that renin-angiotensin-aldosterone system inhibition with an angiotensin-converting enzyme (ACE) inhibitor or ARB and especially mineralocorticoid receptor antagonists would be the preferred choice (Table 4).
SLEEP-DISORDERED BREATHING IN HEART FAILURE
Sleep-disordered breathing, either obstructive sleep apnea (OSA) or central sleep apnea, is quite commonly associated with symptomatic HFrEF.44 Whereas OSA is found in roughly 18% and central sleep apnea in 1% of the general population, sleep-disordered breathing is found in nearly 60% of patients with HFrEF, with some studies showing a nearly equal proportion of OSA and central sleep apnea.45 A similar prevalence is seen in HFpEF, although with a much higher proportion of OSA.46 Central sleep apnea tends to be a marker of more severe heart failure, as it is strongly associated with severe cardiac systolic dysfunction and worse functional capacity.47
Not surprisingly, the underlying mechanism of central sleep apnea is quite different from that of OSA. Whereas OSA predominantly occurs because of repeated obstruction of the pharynx due to nocturnal pharyngeal muscle relaxation, no such airway patency issues or strained breathing patterns exist in central sleep apnea. Central sleep apnea, which can manifest as Cheyne-Stokes respirations, is thought to occur due to an abnormal ventilatory control system with complex pathophysiology such as altered sensitivity of central chemoreceptors to carbon dioxide, interplay of pulmonary congestion, subsequent hyperventilation, and prolonged circulation times due to reduced cardiac output.48
What the two types of sleep-disordered breathing have in common is an association with negative health outcomes. Both appear to induce inflammation and sympathetic nervous system activity via oxidative stress from intermittent nocturnal hypoxemia and hypercapnea.49 OSA was already known to be associated with significant morbidity and mortality rates in the general population,50 and central sleep apnea had been identified as an independent predictor of mortality in HFrEF.51
At the time of the 2013 guidelines, only small or observational studies with limited results had been done evaluating treatment effects of continuous positive airway pressure therapy (CPAP) on OSA and central sleep apnea. Given the relative paucity of data, only a single class IIa recommendation stating that CPAP could be beneficial to increase left ventricular ejection fraction and functional status in concomitant sleep apnea and heart failure was given in 2013. However, many larger trials were under way,52–59 some with surprising results such as a significant increase in cardiovascular and all-cause mortality (Table 5).54
New or modified recommendations on sleep-disordered breathing
Given the common association with heart failure (60%)45 and the marked variation in response to treatment, including potential for harm with adaptive servo-ventilation and central sleep apnea, a class IIa recommendation is made stating that it is reasonable to obtain a formal sleep study in any patient with symptomatic (NYHA class II–IV) heart failure.1
Due to the potential for harm with adaptive servo-ventilation in patients with central sleep apnea and NYHA class II to IV HFrEF, a class III (harm) recommendation is made against its use.
Largely based on the results of the Sleep Apnea Cardiovascular Endpoints (SAVE) trial,56 a class IIb, level of evidence B-R (moderate, based on randomized trials) recommendation is given, stating that the use of CPAP in those with OSA and known cardiovascular disease may be reasonable to improve sleep quality and reduce daytime sleepiness.
POTENTIAL APPLICATIONS IN ACUTE DECOMPENSATED HEART FAILURE
Although the 2017 update1 is directed mostly toward managing chronic heart failure, it is worth considering how it might apply to the management of ADHF.
SHOULD WE USE BIOMARFER TARGETS TO GUIDE THERAPY IN ADHF?
The 2017 update1 does offer direct recommendations regarding the use of biomarker levels during admissions for ADHF. Mainly, they emphasize that the admission biomarker levels provide valuable information regarding acute prognosis and risk stratification (class I recommendation), while natriuretic peptide levels just before discharge provide the same for the postdischarge timeframe (class IIa recommendation).
The update also explicitly cautions against using a natriuretic peptide level-guided treatment strategy, such as setting targets for predischarge absolute level or percent change in level of natriuretic peptides during admissions for ADHF. Although observational and retrospective studies have shown better outcomes when levels are reduced at discharge, treating for any specific inpatient target has never been tested in any large, prospective study; thus, doing so could result in unintended harm.
So what do we know?
McQuade et al systematic review
McQuade et al57 performed a systematic review of more than 40 ADHF trials, which showed that, indeed, patients who achieved a target absolute natriuretic peptide level (BNP ≤ 250 pg/mL) or percent reduction (≥ 30%) at time of discharge had significantly improved outcomes such as reduced postdischarge all-cause mortality and rehospitalization rates. However, these were mostly prospective cohort studies that did not use any type of natriuretic peptide level-guided treatment protocol, leaving it unclear whether such a strategy could positively influence outcomes.
For this reason, both McQuade et al57 and, in an accompanying editorial, Felker et al58 called for properly designed, randomized controlled trials to investigate such a strategy. Felker noted that only 2 such phase II trials in ADHF have been completed,59,60 with unconvincing results.
PRIMA II
The Multicenter, Randomized Clinical Trial to Study the Impact of In-hospital Guidance for Acute Decompensated Heart Failure Treatment by a Predefined NT-ProBNP Target on the Reduction of Readmission and Mortality Rates (PRIMA II)60 randomized patients to natriuretic peptide level-guided treatment or standard care during admission for ADHF.
Many participants (60%) reached the predetermined target of 30% reduction in natriuretic peptide levels at the time of clinical stabilization and randomization; 405 patients were randomized. Patients in the natriuretic peptide level-guided treatment group underwent a prespecified treatment algorithm, with repeat natriuretic peptide levels measured again after the protocol.
Natriuretic peptide-guided therapy failed to show any significant benefit in any clinical outcomes, including the primary composite end point of mortality or heart failure readmissions at 180 days (36% vs 38%, HR 0.99, 95% confidence interval 0.72–1.36). Consistent with the review by McQuade et al,57 achieving the 30% reduction in natriuretic peptide at discharge, in either arm, was associated with a better prognosis, with significantly lower mortality and readmission rates at 180 days (HR 0.39 for rehospitalization or death, 95% confidence interval 0.27–0.55).
As in the observational studies, those who achieved the target natriuretic peptide level at the time of discharge had a better prognosis than those who did not, but neither study showed an improvement in clinical outcomes using a natriuretic peptide level-targeting treatment strategy.
No larger randomized controlled trial results are available for guided therapy in ADHF. However, additional insight may be gained from a subsequent trial61 that evaluated biomarker-guided titration of guideline-directed medical therapy in outpatients with chronic HFrEF.
The GUIDE-IT trial
That trial, the Guiding Evidence Based Therapy Using Biomarker Intensified Treatment in Heart Failure (GUIDE-IT)61 trial, was a large multicenter attempt to determine whether a natriuretic peptide-guided treatment strategy was more effective than standard care in the management of 894 high-risk outpatients with chronic HFrEF. Earlier, promising results had been obtained in a meta-analysis62 of more than 11 similar trials in 2,000 outpatients, with a decreased mortality rate (HR 0.62) seen in the biomarker-guided arm. However, the results had not been definitive due to being underpowered.62
Unfortunately, the results of GUIDE-IT were disappointing, with no significant difference in either the combined primary end point of mortality or hospitalization for heart failure, or the secondary end points evident at 15 months, prompting early termination for futility.61 Among other factors, the study authors postulated that this may have partly resulted from a patient population with more severe heart failure and resultant azotemia, limiting the ability to titrate neurohormonal medications to the desired dosage.
The question of whether patients who cannot achieve such biomarker targets need more intensive therapy or whether their heart failure is too severe to respond adequately echoes the question often raised in discussions of inpatient biomarker-guided therapy.58 Thus, only limited insight is gained, and it remains unclear whether a natriuretic peptide-guided treatment strategy can improve outpatient or inpatient outcomes. Until this is clarified, clinical judgment and optimization of guideline-directed management and therapy should remain the bedrock of treatment.
SHOULD ALDOSTERONE ANTAGONISTS BE USED IN ACUTE HFpEF?
Given the encouraging results in chronic HFpEF from post hoc analyses of TOPCAT, are there any additional recent data suggesting a role for aldosterone antagonists such as spironolactone in acute HFpEF?
The ATHENA-HF trial
The Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy in Heart Failure (ATHENA-HF) trial63 compared treatment with high-dose spironolactone (100 mg) for 96 hours vs usual care in 360 patients with ADHF. The patient population included those with HFrEF and HFpEF, and usual care included low-dose spironolactone (12.5–25 mg) in roughly 15% of patients. High-dose mineralocorticoid receptor antagonists have been shown to overcome diuretic resistance, improve pulmonary vascular congestion, and partially combat the adverse neurohormonal activation seen in ADHF.
Unfortunately, the trial was completely neutral in regard to the primary end point of reduction in natriuretic peptide levels as well as to the secondary end points of 30-day mortality rate, heart failure readmission, clinical congestion scores, urine output, and change in weight. No suggestion of additional benefit was seen in subgroup analysis of patients with acute HFpEF (ejection fraction > 45%), which yielded similar results.63
Given these lackluster findings, routine use of high-dose spironolactone in ADHF is not recommended.64 However, the treatment was well tolerated, without significant adverse effects of hyperkalemia or kidney injury, leaving the door open as to whether it may have utility in selected patients with diuretic resistance.
Should ARNIs and ivabradine be started during ADHF admissions?
The first half of the focused update3 of the 2013 guidelines,2 reviewed by Okwuosa et al,7 provided recommendations for the use of sacubitril-valsartan, an angiotensin-neprilysin inhibitor (ARNI), and ivabradine, a selective sinoatrial node If channel inhibitor, in chronic HFrEF.
Sacubitril-valsartan was given a class I recommendation for use in patients with NYHA class II or III chronic HFrEF who tolerate an ACE inhibitor or an ARB. This recommendation was given largely based on the benefits in mortality and heart failure hospitalizations seen in PARADIGM-HF (the Prospective Comparison of ARNI With ACEI to Determine Impact on Global Mortality and Morbidity in Heart Failure)65 compared with enalapril (HR 0.80, 95% CI 0.73–0.87, P < .001).
There is currently no recommendation on initiation or use of ARNIs during admissions for ADHF, but a recent trial may lend some insight.66
THE PIONEER-HF trial
The Comparison of Sacubitril/Valsartan vs Enalapril on Effect on NT-proBNP in Patients Stabilized From an Acute Heart Failure Episode (PIONEER-HF) trial66 randomized patients admitted for acute HFrEF, once stabilized, to sacubitril-valsartan or enalapril. Encouragingly, the percentage change of natriuretic peptide levels from the time of inpatient initiation to 4 and 8 weeks thereafter, the primary efficacy end point, was 46.7% with sacubitril-valsartan versus 25.3% with enalapril alone (ratio of change 0.71, 95% CI 0.63–0.81, P < .001). Although not powered for such, a prespecified analysis of a composite of clinical outcomes was also favorable for sacubitril-valsartan, largely driven by a 44% decreased rate of rehospitalization. More definitive, and quite reassuring, was that no significant difference was seen in the key safety outcomes of worsening renal function, hyperkalemia, symptomatic hypotension, and angioedema. These results were also applicable to the one-third of study participants who had no former diagnosis of heart failure, the one-third identifying as African American, and the one-third who had not been taking an ACE inhibitor or ARB. These results, taken together with the notion that at study completion the patients become similar to those included in PARADIGM-HF, have led some to assert that PIONEER-HF has the potential to change clinical practice.
Ivabradine was given a class IIa recommendation for use in patients with NYHA class II or III chronic HFrEF with a resting heart rate of at least 70 bpm, in sinus rhythm, despite being on optimal medical therapy including a beta-blocker at a maximum tolerated dose.
This recommendation was largely based on SHIFT (Systolic Heart Failure Treatment With the If Inhibitor Ivabradine Trial), which randomized patients to ivabradine or placebo to evaluate the effects of isolated lowering of the heart rate on the composite primary outcome of cardiovascular death or hospitalization. A significant reduction was seen in the ivabradine arm (HR 0.82, 95% CI 0.75–0.90, P < .0001), mainly driven by decreased hospitalizations.67
Subsequently, a small unblinded single-center study was undertaken to evaluate the efficacy and safety of initiating ivabradine during admissions for ADHF.68
THE ETHIC-AHF trial
The Effect of Early Treatment With Ivabradine Combined With Beta-Blockers vs Beta-Blockers Alone in Patients Hospitalized With Heart Failure and Reduced Left Ventricular Ejection Fraction (ETHIC-AHF) trial68 sought to determine the safety and effectiveness of early coadministration of ivabradine with beta-blockers in patients with acute HFrEF.
This single-center, unblinded study randomized 71 patients to ivabradine and beta-blockade or beta-blockade alone upon clinical stabilization (24–48 hours) after admission for acute decompensated HFrEF.
The primary end point was heart rate at 28 days, with the ivabradine group showing a statistically significant decrease (64 vs 70 bpm, P = .01), which persisted at 4 months. There was no significant difference in the secondary end points of adverse drug effects or the composite of clinical event outcomes (all-cause mortality, admission for heart failure or cardiovascular cause), but a number of surrogate end points including left ventricular ejection fraction, BNP level, and NYHA functional class at 4 months showed mild improvement.
Although this study provided evidence that the coadministration of ivabradine and a beta-blocker is safe and was positive in regard to clinical outcomes, the significant limitations due to its size and study design (single-center, unblinded, 4-month follow-up) simply serve to support the pursuit of larger studies with more stringent design and longer follow-up in order to determine the clinical efficacy.
The PRIME-HF trial
The Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF) trial69 is a randomized, open-label, multicenter trial comparing standard care vs the initiation of ivabradine before discharge, but after clinical stabilization, during admissions for ADHF in patients with chronic HFrEF (left ventricular ejection fraction ≤ 35%). At subsequent outpatient visits, the dosage can be modified in the ivabradine group, or ivabradine can be initiated at the provider’s discretion in the usual-care group.
PRIME-HF is attempting to determine whether initiating ivabradine before discharge will result in more patients taking ivabradine at 180 days, its primary end point, as well as in changes in secondary end points including heart rate and patient-centered outcomes. The study is active, with reporting expected in 2019.
As these trials all come to completion, it will not be long before we have further guidance regarding the inpatient initiation of these new and exciting therapeutic agents.
SHOULD INTRAVENOUS IRON BE GIVEN DURING ADHF ADMISSIONS?
Given the high prevalence of iron deficiency in symptomatic HFrEF, its independent association with mortality, improvements in quality of life and functional capacity suggested by repleting with intravenous iron (in FAIR-HF and CONFIRM-HF), the seeming inefficacy of oral iron in IRONOUT, and the logistical challenges of intravenous administration during standard clinic visits, could giving intravenous iron soon be incorporated into admissions for ADHF?
Caution has been advised for several reasons. As discussed above, larger randomized controlled trials powered to detect more definitive clinical end points such as death and the rate of hospitalization are still needed before a stronger recommendation can be made for intravenous iron in HFrEF. Also, without such data, it seems unwise to add the considerable economic burden of routinely assessing for iron deficiency and providing intravenous iron during ADHF admissions to the already staggering costs of heart failure.
The effects seen on morbidity and mortality that become evident in these trials over the next 5 years will help determine future guidelines and whether intravenous iron is routinely administered in bridge clinics, during inpatient admissions for ADHF, or not at all in patients with HFrEF and iron deficiency.
INTERNISTS ARE KEY
Heart failure remains one of the most common, morbid, complex, and costly diseases in the United States, and its prevalence is expected only to increase.4,5 The 2017 update1 of the 2013 guideline2 for the management of heart failure provides recommendations aimed not only at management of heart failure, but also at its comorbidities and, for the first time ever, at its prevention.
Internists provide care for the majority of heart failure patients, as well as for their comorbidities, and are most often the first to come into contact with patients at high risk of developing heart failure. Thus, a thorough understanding of these guidelines and how to apply them to the management of acute decompensated heart failure is of critical importance.
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2017; 70(6):776–803. doi:10.1016/j.jacc.2017.04.025
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 128(16):e240–e327. doi:10.1161/CIR.0b013e31829e8776
- Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2016; 134(13):e282–e293. doi:10.1161/CIR.0000000000000435
- Benjamin EJ, Blaha MJ, Chiuve SE, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 2017; 135(10):e146–e603. doi:10.1161/CIR.0000000000000485
- Heidenreich PA, Albert NM, Allen LA, et al; American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 2013; 6(3):606–619. doi:10.1161/HHF.0b013e318291329a
- Huffman MD, Berry JD, Ning H, et al. Lifetime risk for heart failure among white and black Americans: cardiovascular lifetime risk pooling project. J Am Coll Cardiol 2013; 61(14):1510–1517. doi:10.1016/j.jacc.2013.01.022
- Okwuosa IS, Princewill O, Nwabueze C, et al. The ABCs of managing systolic heart failure: past, present, and future. Cleve Clin J Med 2016; 83(10):753–765. doi:10.3949/ccjm.83a.16006
- Kovell LC, Juraschek SP, Russell SD. Stage A heart failure is not adequately recognized in US adults: analysis of the National Health and Nutrition Examination Surveys, 2007–2010. PLoS One 2015; 10(7):e0132228. doi:10.1371/journal.pone.0132228
- Huelsmann M, Neuhold S, Resl M, et al. PONTIAC (NT-proBNP selected prevention of cardiac events in a population of diabetic patients without a history of cardiac disease): a prospective randomized controlled trial. J Am Coll Cardiol 2013; 62(15):1365–1372. doi:10.1016/j.jacc.2013.05.069
- Clodi M, Resl M, Neuhold S, et al. A comparison of NT-proBNP and albuminuria for predicting cardiac events in patients with diabetes mellitus. Eur J Prev Cardiol 2012; 19(5):944–951. doi:10.1177/1741826711420015
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310(1):66–74. doi:10.1001/jama.2013.7588
- Salah K, Kok WE, Eurlings LW, et al. A novel discharge risk model for patients hospitalised for acute decompensated heart failure incorporating N-terminal pro-B-type natriuretic peptide levels: a European coLlaboration on Acute decompeNsated Heart Failure: ELAN-HF Score. Heart 2014; 100(2):115–125. doi:10.1136/heartjnl-2013-303632
- Kociol RD, Horton JR, Fonarow GC, et al. Admission, discharge, or change in B-type natriuretic peptide and long-term outcomes: data from Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) linked to Medicare claims. Circ Heart Fail 2011; 4(5):628–636. doi:10.1161/CIRCHEARTFAILURE.111.962290
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362(9386):777–781. doi:10.1016/S0140-6736(03)14285-7
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355(3):251–259. doi:10.1056/NEJMoa052256
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341(10):709–717. doi:10.1056/NEJM199909023411001
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35(1):30–34. pmid:9302344
- Edelmann F, Wachter R, Schmidt AG, et al; Aldo-DHF Investigators. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA 2013; 309(8):781–791. doi:10.1001/jama.2013.905
- Pitt B, Pfeffer MA, Assmann SF, et al; TOPCAT Investigators. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 2014; 370(15):1383–1392. doi:10.1056/NEJMoa1313731
- Pfeffer MA, Claggett B, Assmann SF, et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015; 31(1):34–42. doi:10.1161/CIRCULATIONAHA.114.013255
- de Denus S, O’Meara E, Desai AS, et al. Spironolactone metabolites in TOPCAT—new insights into regional variation. N Engl J Med 2017; 376(17):1690–1692. doi:10.1056/NEJMc1612601
- Redfield MM, Anstrom KJ, Levine JA, et al; NHLBI Heart Failure Clinical Research Network. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 2015; 373(24):2314–2324. doi:10.1056/NEJMoa1510774
- Walton-Shirley M. Succinct thoughts on NEAT-HFpEF: true, true, and unrelated? Medscape 2015. https://www.medscape.com/viewarticle/854116. Accessed January 17, 2019.
- Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309(12):1268–1277. doi:10.1001/jama.2013.2024
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo controlled study. Circ Heart Fail 2011; 4(1):8–17. doi:10.1161/CIRCHEARTFAILURE.110.944694
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011; 124(2):164–174. doi:10.1161/CIRCULATIONAHA.110.983866
- Klip IT, Comin-Colet J, Voors AA, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013; 165(4):575–582.e3. doi:10.1016/j.ahj.2013.01.017
- Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 2013; 34(11):816–829. doi:10.1093/eurheartj/ehs224
- Jankowska EA, Rozentryt P, Witkowska A, et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J Card Fail 2011; 17(11):899–906. doi:10.1016/j.cardfail.2011.08.003
- Haas JD, Brownlie T 4th. Iron deficiency and reduced work capacity: a critical review of the research to determine a causal relationship. J Nutr 2001; 131(2S–2):676S-690S. doi:10.1093/jn/131.2.676S
- Davies KJ, Maguire JJ, Brooks GA, Dallman PR, Packer L. Muscle mitochondrial bioenergetics, oxygen supply, and work capacity during dietary iron deficiency and repletion. Am J Physiol 1982; 242(6):E418–E427. doi:10.1152/ajpendo.1982.242.6.E418
- Drozd M, Jankowska EA, Banasiak W, Ponikowski P. Iron therapy in patients with heart failure and iron deficiency: review of iron preparations for practitioners. Am J Cardiovasc Drugs 2017; 17(3):183–201. doi:10.1007/s40256-016-0211-2
- Anker SD, Comin Colet J, Filippatos G, et al; FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009; 361(25):2436–2448. doi:10.1056/NEJMoa0908355
- Ponikowski P, van Veldhuisen DJ, Comin-Colet J, et al; CONFIRM-HF Investigators. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J 2015; 36(11):657–668. doi:10.1093/eurheartj/ehu385
- Lewis GD, Malhotra R, Hernandez AF, et al; NHLBI Heart Failure Clinical Research Network. Effect of Oral Iron Repletion on Exercise Capacity in Patients With Heart Failure With Reduced Ejection Fraction and Iron Deficiency: The IRONOUT HF randomized clinical trial. JAMA 2017; 317(19):1958–1966. doi:10.1001/jama.2017.5427
- Wendling P. Iron supplementation in HF: trials support IV but not oral. Medscape 2016. https://www.medscape.com/viewarticle/872088. Accessed January 17, 2019.
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood 2011; 117(17):4425–4433. doi:10.1182/blood-2011-01-258467
- Jankowska EA, Kasztura M, Sokolski M, et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur Heart J 2014; 35(36):2468–2476. doi:10.1093/eurheartj/ehu235
- Jankowska EA, Malyszko J, Ardehali H, et al. Iron status in patients with chronic heart failure. Eur Heart J 2013; 34(11):827–834. doi:10.1093/eurheartj/ehs377
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368(13):1210–1219. doi:10.1056/NEJMoa1214865
- Ghali JK, Anand IS, Abraham WT, et al; Study of Anemia in Heart Failure Trial (STAMINA-HeFT) Group. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation 2008; 117(4):526–535. doi:10.1161/CIRCULATIONAHA.107.698514
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Whelton PK, Carey RM, Arnow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162(8):893–900. pmid:11966340
- MacDonald M, Fang J, Pittman SD, White DP, Malhotra A.The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med 2008; 4(1):38-42. pmid:18350960
- Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail 2009; 11(6):602–608. doi:10.1093/eurjhf/hfp057
- Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 1999; 160(4):1101–1106. doi:10.1164/ajrccm.160.4.9903020
- Ng AC, Freedman SB. Sleep disordered breathing in chronic heart failure. Heart Fail Rev 2009; 14(2):89–99. doi:10.1007/s10741-008-9096-8
- Kasai T, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. J Am Coll Cardiol 2011; 57(2):119–127. doi:10.1016/j.jacc.2010.08.627
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365(9464):1046–1053. doi:10.1016/S0140-6736(05)71141-7
- Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol 2007; 49(20):2028–2034. doi:10.1016/j.jacc.2007.01.084
- Bradley TD, Logan AG, Kimoff RJ, et al; CANPAP Investigators. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 2005; 353(19):2025–2033. doi:10.1056/NEJMoa051001
- Arzt M, Floras JS, Logan AG, et al; CANPAP Investigators. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: a post hoc analysis of the Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure Trial (CANPAP). Circulation 2007; 115(25):3173–3180. doi:10.1161/CIRCULATIONAHA.106.683482
- Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med 2015; 373(12):1095–1105. doi:10.1056/NEJMoa1506459
- O’Connor CM, Whellan DJ, Fiuzat M, et al. Cardiovascular outcomes with minute ventilation-targeted adaptive servo-ventilation therapy in heart failure: the CAT-HF Trial. J Am Coll Cardiol 2017; 69(12):1577–1587. doi:10.1016/j.jacc.2017.01.041
- McEvoy RD, Antic NA, Heeley E, et al; SAVE Investigators and Coordinators. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med 2016; 375(10):919–931. doi:10.1056/NEJMoa1606599
- McQuade CN, Mizus M, Wald JW, Goldberg L, Jessup M, Umscheid CA. Brain-type natriuretic peptide and amino-terminal pro-brain-type natriuretic peptide discharge thresholds for acute decompensated heart failure: a systematic review. Ann Intern Med 2017; 166(3):180–190. doi:10.7326/M16-1468
- Felker GM, Whellan DJ. Inpatient management of heart failure: are we shooting at the right target? Ann Intern Med 2017; 166(3):223–224. doi:10.7326/M16-2667
- Carubelli V, Lombardi C, Lazzarini V, et al. N-terminal pro-B-type natriuretic peptide-guided therapy in patients hospitalized for acute heart failure. J Cardiovasc Med (Hagerstown) 2016; 17(11):828–839. doi:10.2459/JCM.0000000000000419
- Stienen S, Salah K, Moons AH, et al. Rationale and design of PRIMA II: a multicenter, randomized clinical trial to study the impact of in-hospital guidance for acute decompensated heart failure treatment by a predefined NT-PRoBNP target on the reduction of readmIssion and mortality rates. Am Heart J 2014; 168(1):30–36. doi:10.1016/j.ahj.2014.04.008
- Felker GM, Anstrom KJ, Adams KF, et al. Effect of natriuretic peptide-guided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 2017; 318(8):713–720. doi:10.1001/jama.2017.10565
- Troughton RW, Frampton CM, Brunner-La Rocca HP, et al. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: an individual patient meta-analysis. Eur Heart J 2014; 35(23):1559–1567. doi:10.1093/eurheartj/ehu090
- van Vliet AA, Donker AJ, Nauta JJ, Verheugt FW. Spironolactone in congestive heart failure refractory to high-dose loop diuretic and low-dose angiotensin-converting enzyme inhibitor. Am J Cardiol 1993; 71(3):21A–28A. pmid:8422000
- Butler J, Anstrom KJ, Felker GM, et al; National Heart Lung and Blood Institute Heart Failure Clinical Research Network. Efficacy and safety of spironolactone in acute heart failure. The ATHENA-HF randomized clinical trial. JAMA Cardiol 2017; 2(9):950–958. doi:10.1001/jamacardio.2017.2198
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371(11):993–1004. doi:10.1056/NEJMoa1409077
- ClinicalTrials.gov. ComParIson Of Sacubitril/valsartaN Versus Enalapril on Effect on NTpRo-BNP in patients stabilized from an acute Heart Failure episode (PIONEER-HF). https://clinicaltrials.gov/ct2/show/NCT02554890. Accessed January 17, 2019.
- Swedberg K, Komajda M, Böhm M, et al; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 2010; 376(9744):875–885. doi:10.1016/S0140-6736(10)61198-1
- Hidalgo FJ, Anguita M, Castillo JC, et al. Effect of early treatment with ivabradine combined with beta-blockers versus beta-blockers alone in patients hospitalised with heart failure and reduced left ventricular ejection fraction (ETHIC-AHF): a randomised study. Int J Cardiol 2016; 217:7–11. doi:10.1016/j.ijcard.2016.04.136
- ClinicalTrials.gov. Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF). https://clinicaltrials.gov/ct2/show/NCT02827500. Accessed January 17, 2019.
- Anker SD, Kirwan BA, van Veldhuisen DJ, et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: an individual patient data meta-analysis. Eur J Heart Fail 2018; 20(1):125–133. doi:10.1002/ejhf.823
- ClinicalTrials.gov. Intravenous Iron in Patients With Systolic Heart Failure and Iron Deficiency to Improve Morbidity and Mortality (FAIR-HF2). https://clinicaltrials.gov/ct2/show/NCT03036462. Accessed January 17, 2019.
- ClinicalTrials.gov. Study to Compare Ferric Carboxymaltose With Placebo in Patients With Acute Heart Failure and Iron Deficiency (AFFIRM-AHF). https://clinicaltrials.gov/ct2/show/record/NCT02937454. Accessed January 17, 2019.
- ClinicalTrials.gov. Randomized Placebo-controlled Trial of Ferric Carboxymaltose as Treatment for Heart Failure With Iron Deficiency (HEART-FID). https://clinicaltrials.gov/ct2/show/NCT03037931. Accessed January 17, 2019.
- ClinicalTrials.gov. Intravenous Iron Treatment in Patients With Heart Failure and Iron Deficiency (IRONMAN). https://clinicaltrials.gov/ct2/show/NCT02642562. Accessed January 17, 2019.
- Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2017; 70(6):776–803. doi:10.1016/j.jacc.2017.04.025
- Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2013; 128(16):e240–e327. doi:10.1161/CIR.0b013e31829e8776
- Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation 2016; 134(13):e282–e293. doi:10.1161/CIR.0000000000000435
- Benjamin EJ, Blaha MJ, Chiuve SE, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation 2017; 135(10):e146–e603. doi:10.1161/CIR.0000000000000485
- Heidenreich PA, Albert NM, Allen LA, et al; American Heart Association Advocacy Coordinating Committee; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Stroke Council. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 2013; 6(3):606–619. doi:10.1161/HHF.0b013e318291329a
- Huffman MD, Berry JD, Ning H, et al. Lifetime risk for heart failure among white and black Americans: cardiovascular lifetime risk pooling project. J Am Coll Cardiol 2013; 61(14):1510–1517. doi:10.1016/j.jacc.2013.01.022
- Okwuosa IS, Princewill O, Nwabueze C, et al. The ABCs of managing systolic heart failure: past, present, and future. Cleve Clin J Med 2016; 83(10):753–765. doi:10.3949/ccjm.83a.16006
- Kovell LC, Juraschek SP, Russell SD. Stage A heart failure is not adequately recognized in US adults: analysis of the National Health and Nutrition Examination Surveys, 2007–2010. PLoS One 2015; 10(7):e0132228. doi:10.1371/journal.pone.0132228
- Huelsmann M, Neuhold S, Resl M, et al. PONTIAC (NT-proBNP selected prevention of cardiac events in a population of diabetic patients without a history of cardiac disease): a prospective randomized controlled trial. J Am Coll Cardiol 2013; 62(15):1365–1372. doi:10.1016/j.jacc.2013.05.069
- Clodi M, Resl M, Neuhold S, et al. A comparison of NT-proBNP and albuminuria for predicting cardiac events in patients with diabetes mellitus. Eur J Prev Cardiol 2012; 19(5):944–951. doi:10.1177/1741826711420015
- Ledwidge M, Gallagher J, Conlon C, et al. Natriuretic peptide-based screening and collaborative care for heart failure: the STOP-HF randomized trial. JAMA 2013; 310(1):66–74. doi:10.1001/jama.2013.7588
- Salah K, Kok WE, Eurlings LW, et al. A novel discharge risk model for patients hospitalised for acute decompensated heart failure incorporating N-terminal pro-B-type natriuretic peptide levels: a European coLlaboration on Acute decompeNsated Heart Failure: ELAN-HF Score. Heart 2014; 100(2):115–125. doi:10.1136/heartjnl-2013-303632
- Kociol RD, Horton JR, Fonarow GC, et al. Admission, discharge, or change in B-type natriuretic peptide and long-term outcomes: data from Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) linked to Medicare claims. Circ Heart Fail 2011; 4(5):628–636. doi:10.1161/CIRCHEARTFAILURE.111.962290
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362(9386):777–781. doi:10.1016/S0140-6736(03)14285-7
- Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355(3):251–259. doi:10.1056/NEJMoa052256
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341(10):709–717. doi:10.1056/NEJM199909023411001
- MacFadyen RJ, Barr CS, Struthers AD. Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 1997; 35(1):30–34. pmid:9302344
- Edelmann F, Wachter R, Schmidt AG, et al; Aldo-DHF Investigators. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo-DHF randomized controlled trial. JAMA 2013; 309(8):781–791. doi:10.1001/jama.2013.905
- Pitt B, Pfeffer MA, Assmann SF, et al; TOPCAT Investigators. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 2014; 370(15):1383–1392. doi:10.1056/NEJMoa1313731
- Pfeffer MA, Claggett B, Assmann SF, et al. Regional variation in patients and outcomes in the Treatment of Preserved Cardiac Function Heart Failure With an Aldosterone Antagonist (TOPCAT) trial. Circulation 2015; 31(1):34–42. doi:10.1161/CIRCULATIONAHA.114.013255
- de Denus S, O’Meara E, Desai AS, et al. Spironolactone metabolites in TOPCAT—new insights into regional variation. N Engl J Med 2017; 376(17):1690–1692. doi:10.1056/NEJMc1612601
- Redfield MM, Anstrom KJ, Levine JA, et al; NHLBI Heart Failure Clinical Research Network. Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 2015; 373(24):2314–2324. doi:10.1056/NEJMoa1510774
- Walton-Shirley M. Succinct thoughts on NEAT-HFpEF: true, true, and unrelated? Medscape 2015. https://www.medscape.com/viewarticle/854116. Accessed January 17, 2019.
- Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309(12):1268–1277. doi:10.1001/jama.2013.2024
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo controlled study. Circ Heart Fail 2011; 4(1):8–17. doi:10.1161/CIRCHEARTFAILURE.110.944694
- Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation 2011; 124(2):164–174. doi:10.1161/CIRCULATIONAHA.110.983866
- Klip IT, Comin-Colet J, Voors AA, et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013; 165(4):575–582.e3. doi:10.1016/j.ahj.2013.01.017
- Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 2013; 34(11):816–829. doi:10.1093/eurheartj/ehs224
- Jankowska EA, Rozentryt P, Witkowska A, et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J Card Fail 2011; 17(11):899–906. doi:10.1016/j.cardfail.2011.08.003
- Haas JD, Brownlie T 4th. Iron deficiency and reduced work capacity: a critical review of the research to determine a causal relationship. J Nutr 2001; 131(2S–2):676S-690S. doi:10.1093/jn/131.2.676S
- Davies KJ, Maguire JJ, Brooks GA, Dallman PR, Packer L. Muscle mitochondrial bioenergetics, oxygen supply, and work capacity during dietary iron deficiency and repletion. Am J Physiol 1982; 242(6):E418–E427. doi:10.1152/ajpendo.1982.242.6.E418
- Drozd M, Jankowska EA, Banasiak W, Ponikowski P. Iron therapy in patients with heart failure and iron deficiency: review of iron preparations for practitioners. Am J Cardiovasc Drugs 2017; 17(3):183–201. doi:10.1007/s40256-016-0211-2
- Anker SD, Comin Colet J, Filippatos G, et al; FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009; 361(25):2436–2448. doi:10.1056/NEJMoa0908355
- Ponikowski P, van Veldhuisen DJ, Comin-Colet J, et al; CONFIRM-HF Investigators. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J 2015; 36(11):657–668. doi:10.1093/eurheartj/ehu385
- Lewis GD, Malhotra R, Hernandez AF, et al; NHLBI Heart Failure Clinical Research Network. Effect of Oral Iron Repletion on Exercise Capacity in Patients With Heart Failure With Reduced Ejection Fraction and Iron Deficiency: The IRONOUT HF randomized clinical trial. JAMA 2017; 317(19):1958–1966. doi:10.1001/jama.2017.5427
- Wendling P. Iron supplementation in HF: trials support IV but not oral. Medscape 2016. https://www.medscape.com/viewarticle/872088. Accessed January 17, 2019.
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood 2011; 117(17):4425–4433. doi:10.1182/blood-2011-01-258467
- Jankowska EA, Kasztura M, Sokolski M, et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur Heart J 2014; 35(36):2468–2476. doi:10.1093/eurheartj/ehu235
- Jankowska EA, Malyszko J, Ardehali H, et al. Iron status in patients with chronic heart failure. Eur Heart J 2013; 34(11):827–834. doi:10.1093/eurheartj/ehs377
- Swedberg K, Young JB, Anand IS, et al. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013; 368(13):1210–1219. doi:10.1056/NEJMoa1214865
- Ghali JK, Anand IS, Abraham WT, et al; Study of Anemia in Heart Failure Trial (STAMINA-HeFT) Group. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation 2008; 117(4):526–535. doi:10.1161/CIRCULATIONAHA.107.698514
- SPRINT Research Group; Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood pressure control. N Engl J Med 2015; 373(22):2103–2116. doi:10.1056/NEJMoa1511939
- Whelton PK, Carey RM, Arnow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018; 71(19):e127–e248. doi:10.1016/j.jacc.2017.11.006
- Young T, Shahar E, Nieto FJ, et al; Sleep Heart Health Study Research Group. Predictors of sleep-disordered breathing in community dwelling adults: the Sleep Heart Health Study. Arch Intern Med 2002; 162(8):893–900. pmid:11966340
- MacDonald M, Fang J, Pittman SD, White DP, Malhotra A.The current prevalence of sleep disordered breathing in congestive heart failure patients treated with beta-blockers. J Clin Sleep Med 2008; 4(1):38-42. pmid:18350960
- Bitter T, Faber L, Hering D, Langer C, Horstkotte D, Oldenburg O. Sleep-disordered breathing in heart failure with normal left ventricular ejection fraction. Eur J Heart Fail 2009; 11(6):602–608. doi:10.1093/eurjhf/hfp057
- Sin DD, Fitzgerald F, Parker JD, Newton G, Floras JS, Bradley TD. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med 1999; 160(4):1101–1106. doi:10.1164/ajrccm.160.4.9903020
- Ng AC, Freedman SB. Sleep disordered breathing in chronic heart failure. Heart Fail Rev 2009; 14(2):89–99. doi:10.1007/s10741-008-9096-8
- Kasai T, Bradley TD. Obstructive sleep apnea and heart failure: pathophysiologic and therapeutic implications. J Am Coll Cardiol 2011; 57(2):119–127. doi:10.1016/j.jacc.2010.08.627
- Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005; 365(9464):1046–1053. doi:10.1016/S0140-6736(05)71141-7
- Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol 2007; 49(20):2028–2034. doi:10.1016/j.jacc.2007.01.084
- Bradley TD, Logan AG, Kimoff RJ, et al; CANPAP Investigators. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med 2005; 353(19):2025–2033. doi:10.1056/NEJMoa051001
- Arzt M, Floras JS, Logan AG, et al; CANPAP Investigators. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: a post hoc analysis of the Canadian Continuous Positive Airway Pressure for Patients with Central Sleep Apnea and Heart Failure Trial (CANPAP). Circulation 2007; 115(25):3173–3180. doi:10.1161/CIRCULATIONAHA.106.683482
- Cowie MR, Woehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med 2015; 373(12):1095–1105. doi:10.1056/NEJMoa1506459
- O’Connor CM, Whellan DJ, Fiuzat M, et al. Cardiovascular outcomes with minute ventilation-targeted adaptive servo-ventilation therapy in heart failure: the CAT-HF Trial. J Am Coll Cardiol 2017; 69(12):1577–1587. doi:10.1016/j.jacc.2017.01.041
- McEvoy RD, Antic NA, Heeley E, et al; SAVE Investigators and Coordinators. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med 2016; 375(10):919–931. doi:10.1056/NEJMoa1606599
- McQuade CN, Mizus M, Wald JW, Goldberg L, Jessup M, Umscheid CA. Brain-type natriuretic peptide and amino-terminal pro-brain-type natriuretic peptide discharge thresholds for acute decompensated heart failure: a systematic review. Ann Intern Med 2017; 166(3):180–190. doi:10.7326/M16-1468
- Felker GM, Whellan DJ. Inpatient management of heart failure: are we shooting at the right target? Ann Intern Med 2017; 166(3):223–224. doi:10.7326/M16-2667
- Carubelli V, Lombardi C, Lazzarini V, et al. N-terminal pro-B-type natriuretic peptide-guided therapy in patients hospitalized for acute heart failure. J Cardiovasc Med (Hagerstown) 2016; 17(11):828–839. doi:10.2459/JCM.0000000000000419
- Stienen S, Salah K, Moons AH, et al. Rationale and design of PRIMA II: a multicenter, randomized clinical trial to study the impact of in-hospital guidance for acute decompensated heart failure treatment by a predefined NT-PRoBNP target on the reduction of readmIssion and mortality rates. Am Heart J 2014; 168(1):30–36. doi:10.1016/j.ahj.2014.04.008
- Felker GM, Anstrom KJ, Adams KF, et al. Effect of natriuretic peptide-guided therapy on hospitalization or cardiovascular mortality in high-risk patients with heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 2017; 318(8):713–720. doi:10.1001/jama.2017.10565
- Troughton RW, Frampton CM, Brunner-La Rocca HP, et al. Effect of B-type natriuretic peptide-guided treatment of chronic heart failure on total mortality and hospitalization: an individual patient meta-analysis. Eur Heart J 2014; 35(23):1559–1567. doi:10.1093/eurheartj/ehu090
- van Vliet AA, Donker AJ, Nauta JJ, Verheugt FW. Spironolactone in congestive heart failure refractory to high-dose loop diuretic and low-dose angiotensin-converting enzyme inhibitor. Am J Cardiol 1993; 71(3):21A–28A. pmid:8422000
- Butler J, Anstrom KJ, Felker GM, et al; National Heart Lung and Blood Institute Heart Failure Clinical Research Network. Efficacy and safety of spironolactone in acute heart failure. The ATHENA-HF randomized clinical trial. JAMA Cardiol 2017; 2(9):950–958. doi:10.1001/jamacardio.2017.2198
- McMurray JJ, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371(11):993–1004. doi:10.1056/NEJMoa1409077
- ClinicalTrials.gov. ComParIson Of Sacubitril/valsartaN Versus Enalapril on Effect on NTpRo-BNP in patients stabilized from an acute Heart Failure episode (PIONEER-HF). https://clinicaltrials.gov/ct2/show/NCT02554890. Accessed January 17, 2019.
- Swedberg K, Komajda M, Böhm M, et al; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 2010; 376(9744):875–885. doi:10.1016/S0140-6736(10)61198-1
- Hidalgo FJ, Anguita M, Castillo JC, et al. Effect of early treatment with ivabradine combined with beta-blockers versus beta-blockers alone in patients hospitalised with heart failure and reduced left ventricular ejection fraction (ETHIC-AHF): a randomised study. Int J Cardiol 2016; 217:7–11. doi:10.1016/j.ijcard.2016.04.136
- ClinicalTrials.gov. Predischarge Initiation of Ivabradine in the Management of Heart Failure (PRIME-HF). https://clinicaltrials.gov/ct2/show/NCT02827500. Accessed January 17, 2019.
- Anker SD, Kirwan BA, van Veldhuisen DJ, et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: an individual patient data meta-analysis. Eur J Heart Fail 2018; 20(1):125–133. doi:10.1002/ejhf.823
- ClinicalTrials.gov. Intravenous Iron in Patients With Systolic Heart Failure and Iron Deficiency to Improve Morbidity and Mortality (FAIR-HF2). https://clinicaltrials.gov/ct2/show/NCT03036462. Accessed January 17, 2019.
- ClinicalTrials.gov. Study to Compare Ferric Carboxymaltose With Placebo in Patients With Acute Heart Failure and Iron Deficiency (AFFIRM-AHF). https://clinicaltrials.gov/ct2/show/record/NCT02937454. Accessed January 17, 2019.
- ClinicalTrials.gov. Randomized Placebo-controlled Trial of Ferric Carboxymaltose as Treatment for Heart Failure With Iron Deficiency (HEART-FID). https://clinicaltrials.gov/ct2/show/NCT03037931. Accessed January 17, 2019.
- ClinicalTrials.gov. Intravenous Iron Treatment in Patients With Heart Failure and Iron Deficiency (IRONMAN). https://clinicaltrials.gov/ct2/show/NCT02642562. Accessed January 17, 2019.
KEY POINTS
- Despite advances in treatment, heart failure remains highly morbid, common, and costly. Prevention is key.
- Strategies to prevent progression to clinical heart failure in high-risk patients include new blood pressure targets (< 130/80 mm Hg) and B-type natriuretic peptide screening to prompt referral to a cardiovascular specialist.
- An aldosterone receptor antagonist might be considered to decrease hospitalizations in appropriately selected stage C HFpEF patients. Routine use of nitrates or phosphodiesterase-5 inhibitors in such patients is not recommended.
- Outpatient intravenous iron infusions are reasonable in persistently symptomatic New York Heart Association stage II to III heart failure with reduced ejection fraction (HFrEF) to improve functional capacity and quality of life.
- The new systolic blood pressure target is less than 130 mm Hg for stage A heart failure, stage C HFrEF, and stage C HFpEF.

Assessment of Cardiovascular Disease Risk in Rheumatoid Arthritis
From the Division of Rheumatology & Immunology, University of Nebraska Medical Center, and Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE.
Abstract
- Objective: To review cardiovascular disease (CVD) risk assessment in patients with rheumatoid arthritis (RA).
- Methods: Literature review of the assessment of CVD risk in RA.
- Results: CVD is the leading cause of death among RA patients.
Because of the increased risk of CVD events and CVD mortality in patients with RA, regular assessment of CVD risk and aggressive management of CVD risk in these patients is crucial. CVD risk estimation typically centers on the use of well-established CVD risk calculators. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing, while novel RA-derived CVD risk scores that incorporate RA-related factors have had limited external validity testing. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. - Conclusion: Primary care and rheumatology providers must be aware of the increased risk of CVD in RA, a risk that approaches that of diabetic patients.
Routine assessment of CVD risk is an essential first step in minimizing CVD risk in this population. Until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Keywords: rheumatoid arthritis; cardiovascular disease; cardiovascular risk assessment.
Editor’s note: This article is part 1 of a 2-part article. “Management of Cardiovascular Disease Risk in Rheumatoid Arthritis” was published in the March/April 2019 issue.
Rheumatoid arthritis (RA) is a chronic, autoimmune inflammatory arthritis affecting up to 1% of the US population that can lead to joint damage, functional disability, and reduced quality of life.1 In addition to articular involvement, systemic inflammation accompanying RA may lead to extra-articular manifestations and increase the risk of premature death.2 Cardiovascular disease (CVD), accounting for nearly half of all deaths among RA patients, is now recognized as a critical extra-articular manifestation of RA.2,3 As such, assessment and management of CVD risk is essential to the comprehensive care of the RA patient. This article reviews the approach to assessing CVD risk in patients with RA; the management of both traditional and RA-specific risk factors is discussed in a separate article.
Scope of the Problem
In a large meta-analysis of observational studies that included more than 111,000 patients with RA, CVD-related mortality rates were 1.5 times higher among RA patients than among general population controls.4 The risk of overall CVD, including nonfatal events, is similar; a separate meta-analysis of observational studies that included more than 41,000 patients with RA calculated a pooled relative risk for incident CVD of 1.48.5 Individual analyses identified heightened risk of acute coronary syndrome (ACS), cerebrovascular accident, and congestive heart failure (CHF).5 Perhaps more illustrative of the magnitude of the problem, the risk of CVD in RA approaches that observed among individuals with diabetes mellitus.6,7
Coronary artery disease (CAD) accounts for a significant portion of the CVD risk in RA, but its presentation may be atypical in RA patients. RA patients are at higher risk of suffering unrecognized myocardial infarction (MI) and sudden cardiac death.8 The reasons for silent ischemia in RA are not fully known, but have been hypothesized to include imbalances of inflammatory cytokines, alterations in pain sensitization, or the female predominance of RA (with women more often presenting with atypical symptoms of myocardial ischemia).9 Alarmingly, a retrospective chart review study reported that RA patients admitted for an acute MI were less likely to receive appropriate reperfusion therapy as well as secondary prevention with beta-blockers and lipid-lowering agents.10 Even with appropriate therapy, long-term outcomes such as mortality and recurrent ischemic events are more likely to occur in RA patients after acute MI.11-13
Independent of ischemic heart disease, RA patients are at increased risk of CHF.14-16 RA patients are at particular risk for CHF with preserved ejection fraction,17 which may be a result of systemic inflammation causing left ventricular stiffening.18,19 Similar to CAD, patients with RA are less likely to present with typical CHF symptoms, are less likely to receive guideline-concordant care, and have higher mortality rates following presentation with CHF.17
Although accounting for a lower proportion of the excess CVD morbidity and mortality in RA, the risk of noncardiac vascular disease is also increased in RA patients. Large meta-analyses have identified positive associations between RA with both ischemic (odds ratio [OR], 1.64 [95% confidence interval {CI}, 1.32-2.05]) and hemorrhagic (OR, 1.68 [95% CI, 1.11-2.53]) stroke.20 Similarly, RA patients appear to have an approximately twofold higher risk of venous thromboembolic events.21 Less frequently studied than other forms of CVD, peripheral arterial disease may be increased in RA patients independent of other CVD and CVD risk factors.22,23
Assessing CVD Risk in RA
CVD Risk Scores
In order to identify patients who may benefit from primary prevention interventions, such as lipid-lowering therapy, CVD risk estimation typically centers on the use of well-established CVD risk calculators (Table). CVD risk scores such as the Framingham Risk Score (FRS), Systematic Coronary Risk Evaluation (SCORE), and American College of Cardiology/ American Heart Association (ACC/AHA) Pooled Cohort Equation incorporate traditional CVD risk factors, including age, sex, smoking status, blood pressure, lipid levels, and presence of diabetes mellitus.24,25 However, CVD risk in RA patients appears to be inadequately explained by traditional CVD risk factors,26 with disease activity and inflammation being associated with higher CVD risk. Recognizing that inflammation may contribute to CVD risk even among non-RA patients, the Reynolds Risk Score includes high-sensitivity C-reactive protein (hsCRP) in its calculation.27 In contrast to more robust performance in the general population, these well-established CVD risk scores have had variable predictive potential of incident CVD in RA patients.28-30

Several models, or adaptations to existing models, have been proposed to improve CVD risk assessment in RA populations (Table). In 2009, the European League Against Rheumatism (EULAR) task force suggested using a correction factor of 1.5 with traditional CVD risk models in RA patients with 2 of the following criteria: disease duration exceeding 10 years, rheumatoid factor or anti-cyclic citrullinated peptide (CCP) antibody positivity, or extra-articular manifestations of RA.31 An update to these recommendations in 2015 continued to propose the use of a 1.5 correction factor, but suggested applying this to all RA patients.32 QRISK2, a modification to QRISK1 which was developed to predict CVD in the UK general population, includes the diagnosis of RA as a risk factor, and in early validation efforts more accurately discriminated patients in the general population at increased risk of CVD compared to the FRS.33 Additional disease-specific risk factors such as systemic lupus, steroid use, severe mental illness, and steroid and atypical antipsychotic use were incorporated in the QRISK3 algorithm, with model performance similar to the QRISK2.34 The Expanded Cardiovascular Risk Prediction Score for RA (ERS-RA) was specifically developed to assess CVD risk in RA patients by including RA disease activity, level of physical disability, RA disease duration, and prednisone use.35 Despite efforts to develop “RA-specific” risk scores, these have not consistently outperformed traditional CVD risk calculators.36-38 In one study involving more than 1700 RA patients, the ERS-RA performed similarly to the FRS and Reynolds Risk Score, with a net reclassification index of just 2.3% versus the FRS.36
Imaging Modalities
Imaging modalities may assist in characterizing the increased risk of CVD in RA and the subclinical CVD manifestations that occur. For example, RA patients were shown to have more prevalent and unstable coronary plaque, higher carotid intima media thickness, and impaired myocardial function with computed tomography (CT) angiography and carotid ultrasound.39,40 However, studies harnessing noninvasive imaging to augment CVD risk assessment in RA patients are limited.
Carotid ultrasound has been the most extensively studied imaging modality for CVD risk assessment in RA. In a cohort of 599 RA patients with no history of ACS, rates of ACS were nearly 4 times higher in RA patients with bilateral carotid plaque on carotid ultrasound, and the association with ACS was independent of other traditional and RA-related risk factors.41 Presence of bilateral carotid plaques was similarly associated with an increased risk of overall CVD events (hazard ratio [HR], 3.34 [95% CI, 1.21-9.22]), ACS alone (HR, 6.31 [95% CI, 1.27-31.40]), and a lower mean CVD event-free survival (13.9 versus 15.2 years, P = 0.01) in a separate inception cohort of 105 RA patients with no prior history of CVD.42 The most useful application of carotid ultrasound may be in conjunction with clinical CVD risk models. Use of carotid ultrasound improved CVD risk stratification among RA patients who were considered at moderate risk by the EULAR-modified SCORE calculator.43 Beyond carotid ultrasound, measurement of arterial stiffness through ultrasound could also aid in CVD risk stratification. Aortic pulse wave velocity and augmentation index, measures of arterial stiffness, are predictive of CVD in the general population as well as RA patients and improve with reduction in RA disease activity.44,45 Peripheral arterial stiffness (brachial-ankle elasticity index) is impaired in RA patients and predictive of CVD morbidity and mortality in the general population.46,47
CT coronary angiography and coronary artery calcium (CAC) scores are reliable measures of coronary artery atherosclerosis and have been validated for CVD risk assessment in the general population.48-52 While the association between RA and CT-related findings of atherosclerosis is well established, assessment of CT-mediated evaluation as a prognostic tool for CVD in RA is limited. In one cohort study, CAC predicted higher rates of CVD events in Chinese patients with RA and systemic lupus erythematosus in a pooled analysis, although results were limited by low event rates and the absence of RA-only subanalyses.53
While the aforementioned imaging modalities have focused on enhancing the identification of atherosclerosis, echocardiography or cardiac magnetic resonance imaging (MRI) may be useful for detecting subclinical structural and/or functional abnormalities that predispose to CHF. Structural abnormalities including increased left ventricular mass and hypertrophy are more prevalent in RA patients and predict incident CHF in the general population.54-56 MRI measures of myocardial inflammation, including T1 mapping and extracellular volume, are associated with higher mortality rates and also appear to be elevated in RA patients.57,58 Whether identification of these imaging findings influences the cost-effective clinical management of RA patients needs further study.
Biomarkers
Serum biomarkers, such as the anti-CCP antibody, have become crucial to the evaluation of patients suspected to have RA. With the growing understanding of the role pro-inflammatory mediators play in CVD pathogenesis and the relative ease with which they can be measured, serum biomarkers have potential to inform CVD risk assessment. In the general population, hsCRP concentrations are predictive of CVD and are included in the Reynolds Risk Score.27 In RA, CRP concentrations are typically much higher than those observed among individuals in the general population solely at increased CVD risk, yet elevated levels remain predictive of CVD death independent of RA disease activity and traditional CVD risk factors.59 Several additional cytokines, chemokines, and adhesion molecules have been associated with surrogate markers of CVD in RA patients, although further study is needed to elucidate thresholds that signify increased CVD risk in a population characterized by the presence of systemic inflammation.60
Cardiac biomarkers used frequently in the general population may be useful to assess CVD risk in RA patients. N-terminal-pro brain natriuretic peptide (NT-pro BNP) is a biomarker typically used to evaluate CHF severity, but it may also predict long-term mortality in patients with coronary heart disease.61,62 Circulating NT-pro BNP concentrations are elevated in RA independent of prevalent CHF and may serve as a useful tool to identify subclinical cardiac disease in RA patients.63 High-sensitivity cardiac troponin I (HS-cTnI) assays are capable of detecting levels of cardiac troponin below the threshold typically used to diagnose ACS. HS-cTnI levels are increased in RA patients independent of additional CVD risk factors, and elevated levels (> 1.5 pg/mL) were associated with more severe CT angiography findings of coronary plaque as well as increased risk of CVD events.64,65
Clinical Application
A fully validated algorithm for CVD risk assessment in RA is lacking. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing. In contrast, novel RA-derived CVD risk scores incorporate RA-related factors, but have had limited external validity testing. Additionally, RA-derived risk scores are less likely to be utilized and adopted by primary care providers and cardiologists involved in RA patients’ care. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. Therefore, until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Conclusion
RA patients are at increased risk of CVD and CVD-related mortality relative to the general population. The disproportionate CVD burden seen in RA appears to be multifactorial, owing to the complex effects of systemic inflammation, endothelial dysfunction, and pro-atherogenic lipoprotein modifications. Additionally, many traditional CVD risk factors are more prevalent and suboptimally managed in RA patients. To mitigate the increased risk of CVD in RA, primary care and subspecialty providers alike must be aware of this heightened risk in RA, perform frequent assessment of CVD risk, and
Corresponding author: Bryant R. England, MD; 986270 Nebraska Medical Center, Omaha, NE 68198-6270; [email protected].
Financial disclosures: Dr. England is supported by UNMC Internal Medicine Scientist Development Award, UNMC Physician-Scientist Training Program, the UNMC Mentored Scholars Program, and the Rheumatology Research Foundation Scientist Development Award. Dr. Mikuls is supported by a VA Merit Award (CX000896) and grants from the National Institutes of Health: National Institute of General Medical Sciences (U54GM115458), National Institute on Alcohol Abuse and Alcoholism (R25AA020818), and National Institute of Arthritis and Musculoskeletal and Skin Diseases (2P50AR60772).
1. Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the united states. part I. Arthritis Rheum. 2008;58:15-25.
2. England BR, Sayles H, Michaud K, et al. Cause-specific mortality in male US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2016;68:36-45.
3. Sokka T, Abelson B, Pincus T. Mortality in rheumatoid arthritis: 2008 update. Clin Exp Rheumatol. 2008;26:S35-61.
4. Avina-Zubieta JA, Choi HK, Sadatsafavi M, et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690-1697.
5. Avina-Zubieta JA, Thomas J, Sadatsafavi M, et al. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524-1529.
6. van Halm VP, Peters MJ, Voskuyl AE, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE investigation. Ann Rheum Dis. 2009;68:1395-1400.
7. Peters MJ, van Halm VP, Voskuyl AE, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571-1579.
8. Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: A population-based cohort study. Arthritis Rheum. 2005;52:402-411.
9. Cardiovascular disease in women--often silent and fatal. Lancet. 2011;378:200,6736(11)61108-61112.
10. Van Doornum S, Brand C, Sundararajan V, et al. Rheumatoid arthritis patients receive less frequent acute reperfusion and secondary prevention therapy after myocardial infarction compared with the general population. Arthritis Res Ther. 2010;12:R183.
11. Sodergren A, Stegmayr B, Lundberg V, et al. Increased incidence of and impaired prognosis after acute myocardial infarction among patients with seropositive rheumatoid arthritis. Ann Rheum Dis. 2007;66:263-266.
12. Douglas KM, Pace AV, Treharne GJ, et al. Excess recurrent cardiac events in rheumatoid arthritis patients with acute coronary syndrome. Ann Rheum Dis. 2006;65:348-353.
13. McCoy SS, Crowson CS, Maradit-Kremers H, et al. Long-term outcomes and treatment after myocardial infarction in patients with rheumatoid arthritis. J Rheumatol. 2013;40:605-610.
14. Mantel A, Holmqvist M, Andersson DC, et al. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J Am Coll Cardiol. 2017;69:1275-1285.
15. Crowson CS, Nicola PJ, Kremers HM, et al. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52:3039-3044.
16. Nicola PJ, Maradit-Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: A population-based study over 46 years. Arthritis Rheum. 2005;52:412-420.
17. Davis JM,3rd, Roger VL, Crowson CS, et al. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58:2603-2611.
18. Arslan S, Bozkurt E, Sari RA, Erol MK. Diastolic function abnormalities in active rheumatoid arthritis evaluation by conventional doppler and tissue doppler: Relation with duration of disease. Clin Rheumatol. 2006;25:294-299.
19. Liang KP, Myasoedova E, Crowson CS, et al. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1665-1670.
20. Wiseman SJ, Ralston SH, Wardlaw JM. Cerebrovascular disease in rheumatic diseases: A systematic review and meta-analysis. Stroke. 2016;47:943-950.
21. Ungprasert P, Srivali N, Spanuchart I, et al. Risk of venous thromboembolism in patients with rheumatoid arthritis: A systematic review and meta-analysis. Clin Rheumatol. 2014;33:297-304.
22. Stamatelopoulos KS, Kitas GD, Papamichael CM, et al. Subclinical peripheral arterial disease in rheumatoid arthritis. Atherosclerosis. 2010;212:305-309.
23. Chuang YW, Yu MC, Lin CL, et al. Risk of peripheral arterial occlusive disease in patients with rheumatoid arthritis. A nationwide population-based cohort study. Thromb Haemost. 2016;115:439-445.
24. Conroy RM, Pyorala K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in europe: The SCORE project. Eur Heart J. 2003;24:987-1003.
25. D’Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: The Framingham heart study. Circulation. 2008;117:743-753.
26. del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737-2745.
27. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: The Reynolds Risk Score. JAMA. 2007;297:611-619.
28. Arts EE, Popa C, Den Broeder AA, et al. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis. 2015;74:668-674.
29. Crowson CS, Matteson EL, Roger VL, et al. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol. 2012;110:420-424.
30. Kawai VK, Chung CP, Solus JF, et al. The ability of the 2013 American College of Cardiology/American Heart Association cardiovascular risk score to identify rheumatoid arthritis patients with high coronary artery calcification scores. Arthritis Rheumatol. 2015;67:381-385.
31. Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325-331.
32. Agca R, Heslinga SC, Rollefstad S, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17-28.
33. Hippisley-Cox J, Coupland C, Vinogradova Y, et al. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ. 2008;336:1475-1482.
34. Hippisley-Cox J, Coupland C, Brindle P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: Prospective cohort study. BMJ. 2017;357:j2099.
35. Solomon DH, Greenberg J, Curtis JR, et al. Derivation and internal validation of an expanded cardiovascular risk prediction score for rheumatoid arthritis: A consortium of rheumatology researchers of north america registry study. Arthritis Rheumatol. 2015;67:1995-2003.
36. Crowson CS, Gabriel SE, Semb AG, et al. Rheumatoid arthritis-specific cardiovascular risk scores are not superior to general risk scores: A validation analysis of patients from seven countries. Rheumatology (Oxford). 2017;56:1102-1110.
37. Alemao E, Cawston H, Bourhis F, et al. Comparison of cardiovascular risk algorithms in patients with vs without rheumatoid arthritis and the role of C-reactive protein in predicting cardiovascular outcomes in rheumatoid arthritis. Rheumatology (Oxford). 2017;56:777-786.
38. Crowson CS, Rollefstad S, Kitas GD, et al. Challenges of developing a cardiovascular risk calculator for patients with rheumatoid arthritis. PLoS One. 2017;12: e0174656.
39. Karpouzas GA, Malpeso J, Choi TY, et al. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis. 2014;73:1797-1804.
40. van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: A meta-analysis. Semin Arthritis Rheum. 2011;40:3893-97.
41. Evans MR, Escalante A, Battafarano DF, et al. Carotid atherosclerosis predicts incident acute coronary syndromes in rheumatoid arthritis. Arthritis Rheum. 2011;63:1211-1220.
42. Ajeganova S, de Faire U, Jogestrand T, et al. Carotid atherosclerosis, disease measures, oxidized low-density lipoproteins, and atheroprotective natural antibodies for cardiovascular disease in early rheumatoid arthritis--an inception cohort study. J Rheumatol. 2012;39:1146-1154.
43. Corrales A, Gonzalez-Juanatey C, Peiro ME, et al. Carotid ultrasound is useful for the cardiovascular risk stratification of patients with rheumatoid arthritis: Results of a population-based study. Ann Rheum Dis. 2014;73:722-727.
44. Ikdahl E, Rollefstad S, Wibetoe G, et al. Predictive value of arterial stiffness and subclinical carotid atherosclerosis for cardiovascular disease in patients with rheumatoid arthritis. J Rheumatol. 2016;43:1622-1630.
45. Provan SA, Semb AG, Hisdal J, et al. Remission is the goal for cardiovascular risk management in patients with rheumatoid arthritis: A cross-sectional comparative study. Ann Rheum Dis. 2011;70:812-817.
46. Vlachopoulos C, Aznaouridis K, Terentes-Printzios D, et al. Prediction of cardiovascular events and all-cause mortality with brachial-ankle elasticity index: A systematic review and meta-analysis. Hypertension. 2012;60:556-562.
47. Ambrosino P, Tasso M, Lupoli R, et al. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: A systematic review and meta-analysis of literature studies. Ann Med. 2015;47:457-467.
48. Rumberger JA, Simons DB, Fitzpatrick LA, et al. Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. A histopathologic correlative study. Circulation. 1995;92:2157-2162.
49. Detrano R, Guerci AD, Carr JJ, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336-1345.
50. Task Force Members, Montalescot G, Sechtem U, et al. 2013 ESC guidelines on the management of stable coronary artery disease: The task force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34:2949-3003.
51. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2935-2959.
52. Hou ZH, Lu B, Gao Y, et al. Prognostic value of coronary CT angiography and calcium score for major adverse cardiac events in outpatients. JACC Cardiovasc Imaging. 2012;5:990-999.
53. Yiu KH, Mok MY, Wang S, et al. Prognostic role of coronary calcification in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Rheumatol. 2012;30:345-350.
54. Wright K, Crowson CS, Gabriel SE. Cardiovascular comorbidity in rheumatic diseases: A focus on heart failure. Heart Fail Clin. 2014;10:339-352.
55. Rudominer RL, Roman MJ, Devereux RB, et al. Independent association of rheumatoid arthritis with increased left ventricular mass but not with reduced ejection fraction. Arthritis Rheum. 2009;60:22-29.
56. Bluemke DA, Kronmal RA, Lima JA, et al. The relationship of left ventricular mass and geometry to incident cardiovascular events: The MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;52:2148-2155.
57. Ntusi NAB, Piechnik SK, Francis JM, et al. Diffuse myocardial fibrosis and inflammation in rheumatoid arthritis: Insights from CMR T1 mapping. JACC Cardiovasc Imaging. 2015;8:526-536.
58. Wong TC, Piehler K, Meier CG, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012;126:1206-1216.
59. Goodson NJ, Symmons DP, Scott DG, et al. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: A ten-year followup study of a primary care-based inception cohort. Arthritis Rheum. 2005;52:2293-2299.
60. Kozera L, Andrews J, Morgan AW. Cardiovascular risk and rheumatoid arthritis--the next step: Differentiating true soluble biomarkers of cardiovascular risk from surrogate measures of inflammation. Rheumatology (Oxford). 2011;50:1944-1954.
61. Cardarelli R, Lumicao TG Jr. B-type natriuretic peptide: A review of its diagnostic, prognostic, and therapeutic monitoring value in heart failure for primary care physicians. J Am Board Fam Pract. 2003;16:327-333.
62. Kragelund C, Gronning B, Kober L, et al. N-terminal pro-B-type natriuretic peptide and long-term mortality in stable coronary heart disease. N Engl J Med. 2005;352:666-675.
63. Harney SM, Timperley J, Daly C, et al. Brain natriuretic peptide is a potentially useful screening tool for the detection of cardiovascular disease in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:136.
64. Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One. 2012;7:e38930.
65. Karpouzas GA, Estis J, Rezaeian P, et al. High-sensitivity cardiac troponin I is a biomarker for occult coronary plaque burden and cardiovascular events in patients with rheumatoid arthritis. Rheumatology (Oxford). 2018;57:1080-1088.
66. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
67. Assmann G, Cullen P, Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular munster (PROCAM) study. Circulation. 2002;105:310-315.
From the Division of Rheumatology & Immunology, University of Nebraska Medical Center, and Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE.
Abstract
- Objective: To review cardiovascular disease (CVD) risk assessment in patients with rheumatoid arthritis (RA).
- Methods: Literature review of the assessment of CVD risk in RA.
- Results: CVD is the leading cause of death among RA patients.
Because of the increased risk of CVD events and CVD mortality in patients with RA, regular assessment of CVD risk and aggressive management of CVD risk in these patients is crucial. CVD risk estimation typically centers on the use of well-established CVD risk calculators. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing, while novel RA-derived CVD risk scores that incorporate RA-related factors have had limited external validity testing. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. - Conclusion: Primary care and rheumatology providers must be aware of the increased risk of CVD in RA, a risk that approaches that of diabetic patients.
Routine assessment of CVD risk is an essential first step in minimizing CVD risk in this population. Until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Keywords: rheumatoid arthritis; cardiovascular disease; cardiovascular risk assessment.
Editor’s note: This article is part 1 of a 2-part article. “Management of Cardiovascular Disease Risk in Rheumatoid Arthritis” was published in the March/April 2019 issue.
Rheumatoid arthritis (RA) is a chronic, autoimmune inflammatory arthritis affecting up to 1% of the US population that can lead to joint damage, functional disability, and reduced quality of life.1 In addition to articular involvement, systemic inflammation accompanying RA may lead to extra-articular manifestations and increase the risk of premature death.2 Cardiovascular disease (CVD), accounting for nearly half of all deaths among RA patients, is now recognized as a critical extra-articular manifestation of RA.2,3 As such, assessment and management of CVD risk is essential to the comprehensive care of the RA patient. This article reviews the approach to assessing CVD risk in patients with RA; the management of both traditional and RA-specific risk factors is discussed in a separate article.
Scope of the Problem
In a large meta-analysis of observational studies that included more than 111,000 patients with RA, CVD-related mortality rates were 1.5 times higher among RA patients than among general population controls.4 The risk of overall CVD, including nonfatal events, is similar; a separate meta-analysis of observational studies that included more than 41,000 patients with RA calculated a pooled relative risk for incident CVD of 1.48.5 Individual analyses identified heightened risk of acute coronary syndrome (ACS), cerebrovascular accident, and congestive heart failure (CHF).5 Perhaps more illustrative of the magnitude of the problem, the risk of CVD in RA approaches that observed among individuals with diabetes mellitus.6,7
Coronary artery disease (CAD) accounts for a significant portion of the CVD risk in RA, but its presentation may be atypical in RA patients. RA patients are at higher risk of suffering unrecognized myocardial infarction (MI) and sudden cardiac death.8 The reasons for silent ischemia in RA are not fully known, but have been hypothesized to include imbalances of inflammatory cytokines, alterations in pain sensitization, or the female predominance of RA (with women more often presenting with atypical symptoms of myocardial ischemia).9 Alarmingly, a retrospective chart review study reported that RA patients admitted for an acute MI were less likely to receive appropriate reperfusion therapy as well as secondary prevention with beta-blockers and lipid-lowering agents.10 Even with appropriate therapy, long-term outcomes such as mortality and recurrent ischemic events are more likely to occur in RA patients after acute MI.11-13
Independent of ischemic heart disease, RA patients are at increased risk of CHF.14-16 RA patients are at particular risk for CHF with preserved ejection fraction,17 which may be a result of systemic inflammation causing left ventricular stiffening.18,19 Similar to CAD, patients with RA are less likely to present with typical CHF symptoms, are less likely to receive guideline-concordant care, and have higher mortality rates following presentation with CHF.17
Although accounting for a lower proportion of the excess CVD morbidity and mortality in RA, the risk of noncardiac vascular disease is also increased in RA patients. Large meta-analyses have identified positive associations between RA with both ischemic (odds ratio [OR], 1.64 [95% confidence interval {CI}, 1.32-2.05]) and hemorrhagic (OR, 1.68 [95% CI, 1.11-2.53]) stroke.20 Similarly, RA patients appear to have an approximately twofold higher risk of venous thromboembolic events.21 Less frequently studied than other forms of CVD, peripheral arterial disease may be increased in RA patients independent of other CVD and CVD risk factors.22,23
Assessing CVD Risk in RA
CVD Risk Scores
In order to identify patients who may benefit from primary prevention interventions, such as lipid-lowering therapy, CVD risk estimation typically centers on the use of well-established CVD risk calculators (Table). CVD risk scores such as the Framingham Risk Score (FRS), Systematic Coronary Risk Evaluation (SCORE), and American College of Cardiology/ American Heart Association (ACC/AHA) Pooled Cohort Equation incorporate traditional CVD risk factors, including age, sex, smoking status, blood pressure, lipid levels, and presence of diabetes mellitus.24,25 However, CVD risk in RA patients appears to be inadequately explained by traditional CVD risk factors,26 with disease activity and inflammation being associated with higher CVD risk. Recognizing that inflammation may contribute to CVD risk even among non-RA patients, the Reynolds Risk Score includes high-sensitivity C-reactive protein (hsCRP) in its calculation.27 In contrast to more robust performance in the general population, these well-established CVD risk scores have had variable predictive potential of incident CVD in RA patients.28-30
Several models, or adaptations to existing models, have been proposed to improve CVD risk assessment in RA populations (Table). In 2009, the European League Against Rheumatism (EULAR) task force suggested using a correction factor of 1.5 with traditional CVD risk models in RA patients with 2 of the following criteria: disease duration exceeding 10 years, rheumatoid factor or anti-cyclic citrullinated peptide (CCP) antibody positivity, or extra-articular manifestations of RA.31 An update to these recommendations in 2015 continued to propose the use of a 1.5 correction factor, but suggested applying this to all RA patients.32 QRISK2, a modification to QRISK1 which was developed to predict CVD in the UK general population, includes the diagnosis of RA as a risk factor, and in early validation efforts more accurately discriminated patients in the general population at increased risk of CVD compared to the FRS.33 Additional disease-specific risk factors such as systemic lupus, steroid use, severe mental illness, and steroid and atypical antipsychotic use were incorporated in the QRISK3 algorithm, with model performance similar to the QRISK2.34 The Expanded Cardiovascular Risk Prediction Score for RA (ERS-RA) was specifically developed to assess CVD risk in RA patients by including RA disease activity, level of physical disability, RA disease duration, and prednisone use.35 Despite efforts to develop “RA-specific” risk scores, these have not consistently outperformed traditional CVD risk calculators.36-38 In one study involving more than 1700 RA patients, the ERS-RA performed similarly to the FRS and Reynolds Risk Score, with a net reclassification index of just 2.3% versus the FRS.36
Imaging Modalities
Imaging modalities may assist in characterizing the increased risk of CVD in RA and the subclinical CVD manifestations that occur. For example, RA patients were shown to have more prevalent and unstable coronary plaque, higher carotid intima media thickness, and impaired myocardial function with computed tomography (CT) angiography and carotid ultrasound.39,40 However, studies harnessing noninvasive imaging to augment CVD risk assessment in RA patients are limited.
Carotid ultrasound has been the most extensively studied imaging modality for CVD risk assessment in RA. In a cohort of 599 RA patients with no history of ACS, rates of ACS were nearly 4 times higher in RA patients with bilateral carotid plaque on carotid ultrasound, and the association with ACS was independent of other traditional and RA-related risk factors.41 Presence of bilateral carotid plaques was similarly associated with an increased risk of overall CVD events (hazard ratio [HR], 3.34 [95% CI, 1.21-9.22]), ACS alone (HR, 6.31 [95% CI, 1.27-31.40]), and a lower mean CVD event-free survival (13.9 versus 15.2 years, P = 0.01) in a separate inception cohort of 105 RA patients with no prior history of CVD.42 The most useful application of carotid ultrasound may be in conjunction with clinical CVD risk models. Use of carotid ultrasound improved CVD risk stratification among RA patients who were considered at moderate risk by the EULAR-modified SCORE calculator.43 Beyond carotid ultrasound, measurement of arterial stiffness through ultrasound could also aid in CVD risk stratification. Aortic pulse wave velocity and augmentation index, measures of arterial stiffness, are predictive of CVD in the general population as well as RA patients and improve with reduction in RA disease activity.44,45 Peripheral arterial stiffness (brachial-ankle elasticity index) is impaired in RA patients and predictive of CVD morbidity and mortality in the general population.46,47
CT coronary angiography and coronary artery calcium (CAC) scores are reliable measures of coronary artery atherosclerosis and have been validated for CVD risk assessment in the general population.48-52 While the association between RA and CT-related findings of atherosclerosis is well established, assessment of CT-mediated evaluation as a prognostic tool for CVD in RA is limited. In one cohort study, CAC predicted higher rates of CVD events in Chinese patients with RA and systemic lupus erythematosus in a pooled analysis, although results were limited by low event rates and the absence of RA-only subanalyses.53
While the aforementioned imaging modalities have focused on enhancing the identification of atherosclerosis, echocardiography or cardiac magnetic resonance imaging (MRI) may be useful for detecting subclinical structural and/or functional abnormalities that predispose to CHF. Structural abnormalities including increased left ventricular mass and hypertrophy are more prevalent in RA patients and predict incident CHF in the general population.54-56 MRI measures of myocardial inflammation, including T1 mapping and extracellular volume, are associated with higher mortality rates and also appear to be elevated in RA patients.57,58 Whether identification of these imaging findings influences the cost-effective clinical management of RA patients needs further study.
Biomarkers
Serum biomarkers, such as the anti-CCP antibody, have become crucial to the evaluation of patients suspected to have RA. With the growing understanding of the role pro-inflammatory mediators play in CVD pathogenesis and the relative ease with which they can be measured, serum biomarkers have potential to inform CVD risk assessment. In the general population, hsCRP concentrations are predictive of CVD and are included in the Reynolds Risk Score.27 In RA, CRP concentrations are typically much higher than those observed among individuals in the general population solely at increased CVD risk, yet elevated levels remain predictive of CVD death independent of RA disease activity and traditional CVD risk factors.59 Several additional cytokines, chemokines, and adhesion molecules have been associated with surrogate markers of CVD in RA patients, although further study is needed to elucidate thresholds that signify increased CVD risk in a population characterized by the presence of systemic inflammation.60
Cardiac biomarkers used frequently in the general population may be useful to assess CVD risk in RA patients. N-terminal-pro brain natriuretic peptide (NT-pro BNP) is a biomarker typically used to evaluate CHF severity, but it may also predict long-term mortality in patients with coronary heart disease.61,62 Circulating NT-pro BNP concentrations are elevated in RA independent of prevalent CHF and may serve as a useful tool to identify subclinical cardiac disease in RA patients.63 High-sensitivity cardiac troponin I (HS-cTnI) assays are capable of detecting levels of cardiac troponin below the threshold typically used to diagnose ACS. HS-cTnI levels are increased in RA patients independent of additional CVD risk factors, and elevated levels (> 1.5 pg/mL) were associated with more severe CT angiography findings of coronary plaque as well as increased risk of CVD events.64,65
Clinical Application
A fully validated algorithm for CVD risk assessment in RA is lacking. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing. In contrast, novel RA-derived CVD risk scores incorporate RA-related factors, but have had limited external validity testing. Additionally, RA-derived risk scores are less likely to be utilized and adopted by primary care providers and cardiologists involved in RA patients’ care. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. Therefore, until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Conclusion
RA patients are at increased risk of CVD and CVD-related mortality relative to the general population. The disproportionate CVD burden seen in RA appears to be multifactorial, owing to the complex effects of systemic inflammation, endothelial dysfunction, and pro-atherogenic lipoprotein modifications. Additionally, many traditional CVD risk factors are more prevalent and suboptimally managed in RA patients. To mitigate the increased risk of CVD in RA, primary care and subspecialty providers alike must be aware of this heightened risk in RA, perform frequent assessment of CVD risk, and
Corresponding author: Bryant R. England, MD; 986270 Nebraska Medical Center, Omaha, NE 68198-6270; [email protected].
Financial disclosures: Dr. England is supported by UNMC Internal Medicine Scientist Development Award, UNMC Physician-Scientist Training Program, the UNMC Mentored Scholars Program, and the Rheumatology Research Foundation Scientist Development Award. Dr. Mikuls is supported by a VA Merit Award (CX000896) and grants from the National Institutes of Health: National Institute of General Medical Sciences (U54GM115458), National Institute on Alcohol Abuse and Alcoholism (R25AA020818), and National Institute of Arthritis and Musculoskeletal and Skin Diseases (2P50AR60772).
From the Division of Rheumatology & Immunology, University of Nebraska Medical Center, and Veterans Affairs Nebraska-Western Iowa Health Care System, Omaha, NE.
Abstract
- Objective: To review cardiovascular disease (CVD) risk assessment in patients with rheumatoid arthritis (RA).
- Methods: Literature review of the assessment of CVD risk in RA.
- Results: CVD is the leading cause of death among RA patients.
Because of the increased risk of CVD events and CVD mortality in patients with RA, regular assessment of CVD risk and aggressive management of CVD risk in these patients is crucial. CVD risk estimation typically centers on the use of well-established CVD risk calculators. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing, while novel RA-derived CVD risk scores that incorporate RA-related factors have had limited external validity testing. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. - Conclusion: Primary care and rheumatology providers must be aware of the increased risk of CVD in RA, a risk that approaches that of diabetic patients.
Routine assessment of CVD risk is an essential first step in minimizing CVD risk in this population. Until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Keywords: rheumatoid arthritis; cardiovascular disease; cardiovascular risk assessment.
Editor’s note: This article is part 1 of a 2-part article. “Management of Cardiovascular Disease Risk in Rheumatoid Arthritis” was published in the March/April 2019 issue.
Rheumatoid arthritis (RA) is a chronic, autoimmune inflammatory arthritis affecting up to 1% of the US population that can lead to joint damage, functional disability, and reduced quality of life.1 In addition to articular involvement, systemic inflammation accompanying RA may lead to extra-articular manifestations and increase the risk of premature death.2 Cardiovascular disease (CVD), accounting for nearly half of all deaths among RA patients, is now recognized as a critical extra-articular manifestation of RA.2,3 As such, assessment and management of CVD risk is essential to the comprehensive care of the RA patient. This article reviews the approach to assessing CVD risk in patients with RA; the management of both traditional and RA-specific risk factors is discussed in a separate article.
Scope of the Problem
In a large meta-analysis of observational studies that included more than 111,000 patients with RA, CVD-related mortality rates were 1.5 times higher among RA patients than among general population controls.4 The risk of overall CVD, including nonfatal events, is similar; a separate meta-analysis of observational studies that included more than 41,000 patients with RA calculated a pooled relative risk for incident CVD of 1.48.5 Individual analyses identified heightened risk of acute coronary syndrome (ACS), cerebrovascular accident, and congestive heart failure (CHF).5 Perhaps more illustrative of the magnitude of the problem, the risk of CVD in RA approaches that observed among individuals with diabetes mellitus.6,7
Coronary artery disease (CAD) accounts for a significant portion of the CVD risk in RA, but its presentation may be atypical in RA patients. RA patients are at higher risk of suffering unrecognized myocardial infarction (MI) and sudden cardiac death.8 The reasons for silent ischemia in RA are not fully known, but have been hypothesized to include imbalances of inflammatory cytokines, alterations in pain sensitization, or the female predominance of RA (with women more often presenting with atypical symptoms of myocardial ischemia).9 Alarmingly, a retrospective chart review study reported that RA patients admitted for an acute MI were less likely to receive appropriate reperfusion therapy as well as secondary prevention with beta-blockers and lipid-lowering agents.10 Even with appropriate therapy, long-term outcomes such as mortality and recurrent ischemic events are more likely to occur in RA patients after acute MI.11-13
Independent of ischemic heart disease, RA patients are at increased risk of CHF.14-16 RA patients are at particular risk for CHF with preserved ejection fraction,17 which may be a result of systemic inflammation causing left ventricular stiffening.18,19 Similar to CAD, patients with RA are less likely to present with typical CHF symptoms, are less likely to receive guideline-concordant care, and have higher mortality rates following presentation with CHF.17
Although accounting for a lower proportion of the excess CVD morbidity and mortality in RA, the risk of noncardiac vascular disease is also increased in RA patients. Large meta-analyses have identified positive associations between RA with both ischemic (odds ratio [OR], 1.64 [95% confidence interval {CI}, 1.32-2.05]) and hemorrhagic (OR, 1.68 [95% CI, 1.11-2.53]) stroke.20 Similarly, RA patients appear to have an approximately twofold higher risk of venous thromboembolic events.21 Less frequently studied than other forms of CVD, peripheral arterial disease may be increased in RA patients independent of other CVD and CVD risk factors.22,23
Assessing CVD Risk in RA
CVD Risk Scores
In order to identify patients who may benefit from primary prevention interventions, such as lipid-lowering therapy, CVD risk estimation typically centers on the use of well-established CVD risk calculators (Table). CVD risk scores such as the Framingham Risk Score (FRS), Systematic Coronary Risk Evaluation (SCORE), and American College of Cardiology/ American Heart Association (ACC/AHA) Pooled Cohort Equation incorporate traditional CVD risk factors, including age, sex, smoking status, blood pressure, lipid levels, and presence of diabetes mellitus.24,25 However, CVD risk in RA patients appears to be inadequately explained by traditional CVD risk factors,26 with disease activity and inflammation being associated with higher CVD risk. Recognizing that inflammation may contribute to CVD risk even among non-RA patients, the Reynolds Risk Score includes high-sensitivity C-reactive protein (hsCRP) in its calculation.27 In contrast to more robust performance in the general population, these well-established CVD risk scores have had variable predictive potential of incident CVD in RA patients.28-30
Several models, or adaptations to existing models, have been proposed to improve CVD risk assessment in RA populations (Table). In 2009, the European League Against Rheumatism (EULAR) task force suggested using a correction factor of 1.5 with traditional CVD risk models in RA patients with 2 of the following criteria: disease duration exceeding 10 years, rheumatoid factor or anti-cyclic citrullinated peptide (CCP) antibody positivity, or extra-articular manifestations of RA.31 An update to these recommendations in 2015 continued to propose the use of a 1.5 correction factor, but suggested applying this to all RA patients.32 QRISK2, a modification to QRISK1 which was developed to predict CVD in the UK general population, includes the diagnosis of RA as a risk factor, and in early validation efforts more accurately discriminated patients in the general population at increased risk of CVD compared to the FRS.33 Additional disease-specific risk factors such as systemic lupus, steroid use, severe mental illness, and steroid and atypical antipsychotic use were incorporated in the QRISK3 algorithm, with model performance similar to the QRISK2.34 The Expanded Cardiovascular Risk Prediction Score for RA (ERS-RA) was specifically developed to assess CVD risk in RA patients by including RA disease activity, level of physical disability, RA disease duration, and prednisone use.35 Despite efforts to develop “RA-specific” risk scores, these have not consistently outperformed traditional CVD risk calculators.36-38 In one study involving more than 1700 RA patients, the ERS-RA performed similarly to the FRS and Reynolds Risk Score, with a net reclassification index of just 2.3% versus the FRS.36
Imaging Modalities
Imaging modalities may assist in characterizing the increased risk of CVD in RA and the subclinical CVD manifestations that occur. For example, RA patients were shown to have more prevalent and unstable coronary plaque, higher carotid intima media thickness, and impaired myocardial function with computed tomography (CT) angiography and carotid ultrasound.39,40 However, studies harnessing noninvasive imaging to augment CVD risk assessment in RA patients are limited.
Carotid ultrasound has been the most extensively studied imaging modality for CVD risk assessment in RA. In a cohort of 599 RA patients with no history of ACS, rates of ACS were nearly 4 times higher in RA patients with bilateral carotid plaque on carotid ultrasound, and the association with ACS was independent of other traditional and RA-related risk factors.41 Presence of bilateral carotid plaques was similarly associated with an increased risk of overall CVD events (hazard ratio [HR], 3.34 [95% CI, 1.21-9.22]), ACS alone (HR, 6.31 [95% CI, 1.27-31.40]), and a lower mean CVD event-free survival (13.9 versus 15.2 years, P = 0.01) in a separate inception cohort of 105 RA patients with no prior history of CVD.42 The most useful application of carotid ultrasound may be in conjunction with clinical CVD risk models. Use of carotid ultrasound improved CVD risk stratification among RA patients who were considered at moderate risk by the EULAR-modified SCORE calculator.43 Beyond carotid ultrasound, measurement of arterial stiffness through ultrasound could also aid in CVD risk stratification. Aortic pulse wave velocity and augmentation index, measures of arterial stiffness, are predictive of CVD in the general population as well as RA patients and improve with reduction in RA disease activity.44,45 Peripheral arterial stiffness (brachial-ankle elasticity index) is impaired in RA patients and predictive of CVD morbidity and mortality in the general population.46,47
CT coronary angiography and coronary artery calcium (CAC) scores are reliable measures of coronary artery atherosclerosis and have been validated for CVD risk assessment in the general population.48-52 While the association between RA and CT-related findings of atherosclerosis is well established, assessment of CT-mediated evaluation as a prognostic tool for CVD in RA is limited. In one cohort study, CAC predicted higher rates of CVD events in Chinese patients with RA and systemic lupus erythematosus in a pooled analysis, although results were limited by low event rates and the absence of RA-only subanalyses.53
While the aforementioned imaging modalities have focused on enhancing the identification of atherosclerosis, echocardiography or cardiac magnetic resonance imaging (MRI) may be useful for detecting subclinical structural and/or functional abnormalities that predispose to CHF. Structural abnormalities including increased left ventricular mass and hypertrophy are more prevalent in RA patients and predict incident CHF in the general population.54-56 MRI measures of myocardial inflammation, including T1 mapping and extracellular volume, are associated with higher mortality rates and also appear to be elevated in RA patients.57,58 Whether identification of these imaging findings influences the cost-effective clinical management of RA patients needs further study.
Biomarkers
Serum biomarkers, such as the anti-CCP antibody, have become crucial to the evaluation of patients suspected to have RA. With the growing understanding of the role pro-inflammatory mediators play in CVD pathogenesis and the relative ease with which they can be measured, serum biomarkers have potential to inform CVD risk assessment. In the general population, hsCRP concentrations are predictive of CVD and are included in the Reynolds Risk Score.27 In RA, CRP concentrations are typically much higher than those observed among individuals in the general population solely at increased CVD risk, yet elevated levels remain predictive of CVD death independent of RA disease activity and traditional CVD risk factors.59 Several additional cytokines, chemokines, and adhesion molecules have been associated with surrogate markers of CVD in RA patients, although further study is needed to elucidate thresholds that signify increased CVD risk in a population characterized by the presence of systemic inflammation.60
Cardiac biomarkers used frequently in the general population may be useful to assess CVD risk in RA patients. N-terminal-pro brain natriuretic peptide (NT-pro BNP) is a biomarker typically used to evaluate CHF severity, but it may also predict long-term mortality in patients with coronary heart disease.61,62 Circulating NT-pro BNP concentrations are elevated in RA independent of prevalent CHF and may serve as a useful tool to identify subclinical cardiac disease in RA patients.63 High-sensitivity cardiac troponin I (HS-cTnI) assays are capable of detecting levels of cardiac troponin below the threshold typically used to diagnose ACS. HS-cTnI levels are increased in RA patients independent of additional CVD risk factors, and elevated levels (> 1.5 pg/mL) were associated with more severe CT angiography findings of coronary plaque as well as increased risk of CVD events.64,65
Clinical Application
A fully validated algorithm for CVD risk assessment in RA is lacking. Most CVD risk scores from the general population do not contain RA-related factors predictive of CVD but have had more extensive performance testing. In contrast, novel RA-derived CVD risk scores incorporate RA-related factors, but have had limited external validity testing. Additionally, RA-derived risk scores are less likely to be utilized and adopted by primary care providers and cardiologists involved in RA patients’ care. Neither set of risk scores incorporates novel imaging modalities or serum biomarkers, which are most likely to be helpful among individuals at intermediate risk. Therefore, until the performance of RA-specific CVD risk scores can be better established, we recommend the use of nationally endorsed CVD risk scores, with the frequency of reassessment based on CVD risk.
Conclusion
RA patients are at increased risk of CVD and CVD-related mortality relative to the general population. The disproportionate CVD burden seen in RA appears to be multifactorial, owing to the complex effects of systemic inflammation, endothelial dysfunction, and pro-atherogenic lipoprotein modifications. Additionally, many traditional CVD risk factors are more prevalent and suboptimally managed in RA patients. To mitigate the increased risk of CVD in RA, primary care and subspecialty providers alike must be aware of this heightened risk in RA, perform frequent assessment of CVD risk, and
Corresponding author: Bryant R. England, MD; 986270 Nebraska Medical Center, Omaha, NE 68198-6270; [email protected].
Financial disclosures: Dr. England is supported by UNMC Internal Medicine Scientist Development Award, UNMC Physician-Scientist Training Program, the UNMC Mentored Scholars Program, and the Rheumatology Research Foundation Scientist Development Award. Dr. Mikuls is supported by a VA Merit Award (CX000896) and grants from the National Institutes of Health: National Institute of General Medical Sciences (U54GM115458), National Institute on Alcohol Abuse and Alcoholism (R25AA020818), and National Institute of Arthritis and Musculoskeletal and Skin Diseases (2P50AR60772).
1. Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the united states. part I. Arthritis Rheum. 2008;58:15-25.
2. England BR, Sayles H, Michaud K, et al. Cause-specific mortality in male US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2016;68:36-45.
3. Sokka T, Abelson B, Pincus T. Mortality in rheumatoid arthritis: 2008 update. Clin Exp Rheumatol. 2008;26:S35-61.
4. Avina-Zubieta JA, Choi HK, Sadatsafavi M, et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690-1697.
5. Avina-Zubieta JA, Thomas J, Sadatsafavi M, et al. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524-1529.
6. van Halm VP, Peters MJ, Voskuyl AE, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE investigation. Ann Rheum Dis. 2009;68:1395-1400.
7. Peters MJ, van Halm VP, Voskuyl AE, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571-1579.
8. Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: A population-based cohort study. Arthritis Rheum. 2005;52:402-411.
9. Cardiovascular disease in women--often silent and fatal. Lancet. 2011;378:200,6736(11)61108-61112.
10. Van Doornum S, Brand C, Sundararajan V, et al. Rheumatoid arthritis patients receive less frequent acute reperfusion and secondary prevention therapy after myocardial infarction compared with the general population. Arthritis Res Ther. 2010;12:R183.
11. Sodergren A, Stegmayr B, Lundberg V, et al. Increased incidence of and impaired prognosis after acute myocardial infarction among patients with seropositive rheumatoid arthritis. Ann Rheum Dis. 2007;66:263-266.
12. Douglas KM, Pace AV, Treharne GJ, et al. Excess recurrent cardiac events in rheumatoid arthritis patients with acute coronary syndrome. Ann Rheum Dis. 2006;65:348-353.
13. McCoy SS, Crowson CS, Maradit-Kremers H, et al. Long-term outcomes and treatment after myocardial infarction in patients with rheumatoid arthritis. J Rheumatol. 2013;40:605-610.
14. Mantel A, Holmqvist M, Andersson DC, et al. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J Am Coll Cardiol. 2017;69:1275-1285.
15. Crowson CS, Nicola PJ, Kremers HM, et al. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52:3039-3044.
16. Nicola PJ, Maradit-Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: A population-based study over 46 years. Arthritis Rheum. 2005;52:412-420.
17. Davis JM,3rd, Roger VL, Crowson CS, et al. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58:2603-2611.
18. Arslan S, Bozkurt E, Sari RA, Erol MK. Diastolic function abnormalities in active rheumatoid arthritis evaluation by conventional doppler and tissue doppler: Relation with duration of disease. Clin Rheumatol. 2006;25:294-299.
19. Liang KP, Myasoedova E, Crowson CS, et al. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1665-1670.
20. Wiseman SJ, Ralston SH, Wardlaw JM. Cerebrovascular disease in rheumatic diseases: A systematic review and meta-analysis. Stroke. 2016;47:943-950.
21. Ungprasert P, Srivali N, Spanuchart I, et al. Risk of venous thromboembolism in patients with rheumatoid arthritis: A systematic review and meta-analysis. Clin Rheumatol. 2014;33:297-304.
22. Stamatelopoulos KS, Kitas GD, Papamichael CM, et al. Subclinical peripheral arterial disease in rheumatoid arthritis. Atherosclerosis. 2010;212:305-309.
23. Chuang YW, Yu MC, Lin CL, et al. Risk of peripheral arterial occlusive disease in patients with rheumatoid arthritis. A nationwide population-based cohort study. Thromb Haemost. 2016;115:439-445.
24. Conroy RM, Pyorala K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in europe: The SCORE project. Eur Heart J. 2003;24:987-1003.
25. D’Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: The Framingham heart study. Circulation. 2008;117:743-753.
26. del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737-2745.
27. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: The Reynolds Risk Score. JAMA. 2007;297:611-619.
28. Arts EE, Popa C, Den Broeder AA, et al. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis. 2015;74:668-674.
29. Crowson CS, Matteson EL, Roger VL, et al. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol. 2012;110:420-424.
30. Kawai VK, Chung CP, Solus JF, et al. The ability of the 2013 American College of Cardiology/American Heart Association cardiovascular risk score to identify rheumatoid arthritis patients with high coronary artery calcification scores. Arthritis Rheumatol. 2015;67:381-385.
31. Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325-331.
32. Agca R, Heslinga SC, Rollefstad S, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17-28.
33. Hippisley-Cox J, Coupland C, Vinogradova Y, et al. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ. 2008;336:1475-1482.
34. Hippisley-Cox J, Coupland C, Brindle P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: Prospective cohort study. BMJ. 2017;357:j2099.
35. Solomon DH, Greenberg J, Curtis JR, et al. Derivation and internal validation of an expanded cardiovascular risk prediction score for rheumatoid arthritis: A consortium of rheumatology researchers of north america registry study. Arthritis Rheumatol. 2015;67:1995-2003.
36. Crowson CS, Gabriel SE, Semb AG, et al. Rheumatoid arthritis-specific cardiovascular risk scores are not superior to general risk scores: A validation analysis of patients from seven countries. Rheumatology (Oxford). 2017;56:1102-1110.
37. Alemao E, Cawston H, Bourhis F, et al. Comparison of cardiovascular risk algorithms in patients with vs without rheumatoid arthritis and the role of C-reactive protein in predicting cardiovascular outcomes in rheumatoid arthritis. Rheumatology (Oxford). 2017;56:777-786.
38. Crowson CS, Rollefstad S, Kitas GD, et al. Challenges of developing a cardiovascular risk calculator for patients with rheumatoid arthritis. PLoS One. 2017;12: e0174656.
39. Karpouzas GA, Malpeso J, Choi TY, et al. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis. 2014;73:1797-1804.
40. van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: A meta-analysis. Semin Arthritis Rheum. 2011;40:3893-97.
41. Evans MR, Escalante A, Battafarano DF, et al. Carotid atherosclerosis predicts incident acute coronary syndromes in rheumatoid arthritis. Arthritis Rheum. 2011;63:1211-1220.
42. Ajeganova S, de Faire U, Jogestrand T, et al. Carotid atherosclerosis, disease measures, oxidized low-density lipoproteins, and atheroprotective natural antibodies for cardiovascular disease in early rheumatoid arthritis--an inception cohort study. J Rheumatol. 2012;39:1146-1154.
43. Corrales A, Gonzalez-Juanatey C, Peiro ME, et al. Carotid ultrasound is useful for the cardiovascular risk stratification of patients with rheumatoid arthritis: Results of a population-based study. Ann Rheum Dis. 2014;73:722-727.
44. Ikdahl E, Rollefstad S, Wibetoe G, et al. Predictive value of arterial stiffness and subclinical carotid atherosclerosis for cardiovascular disease in patients with rheumatoid arthritis. J Rheumatol. 2016;43:1622-1630.
45. Provan SA, Semb AG, Hisdal J, et al. Remission is the goal for cardiovascular risk management in patients with rheumatoid arthritis: A cross-sectional comparative study. Ann Rheum Dis. 2011;70:812-817.
46. Vlachopoulos C, Aznaouridis K, Terentes-Printzios D, et al. Prediction of cardiovascular events and all-cause mortality with brachial-ankle elasticity index: A systematic review and meta-analysis. Hypertension. 2012;60:556-562.
47. Ambrosino P, Tasso M, Lupoli R, et al. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: A systematic review and meta-analysis of literature studies. Ann Med. 2015;47:457-467.
48. Rumberger JA, Simons DB, Fitzpatrick LA, et al. Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. A histopathologic correlative study. Circulation. 1995;92:2157-2162.
49. Detrano R, Guerci AD, Carr JJ, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336-1345.
50. Task Force Members, Montalescot G, Sechtem U, et al. 2013 ESC guidelines on the management of stable coronary artery disease: The task force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34:2949-3003.
51. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2935-2959.
52. Hou ZH, Lu B, Gao Y, et al. Prognostic value of coronary CT angiography and calcium score for major adverse cardiac events in outpatients. JACC Cardiovasc Imaging. 2012;5:990-999.
53. Yiu KH, Mok MY, Wang S, et al. Prognostic role of coronary calcification in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Rheumatol. 2012;30:345-350.
54. Wright K, Crowson CS, Gabriel SE. Cardiovascular comorbidity in rheumatic diseases: A focus on heart failure. Heart Fail Clin. 2014;10:339-352.
55. Rudominer RL, Roman MJ, Devereux RB, et al. Independent association of rheumatoid arthritis with increased left ventricular mass but not with reduced ejection fraction. Arthritis Rheum. 2009;60:22-29.
56. Bluemke DA, Kronmal RA, Lima JA, et al. The relationship of left ventricular mass and geometry to incident cardiovascular events: The MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;52:2148-2155.
57. Ntusi NAB, Piechnik SK, Francis JM, et al. Diffuse myocardial fibrosis and inflammation in rheumatoid arthritis: Insights from CMR T1 mapping. JACC Cardiovasc Imaging. 2015;8:526-536.
58. Wong TC, Piehler K, Meier CG, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012;126:1206-1216.
59. Goodson NJ, Symmons DP, Scott DG, et al. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: A ten-year followup study of a primary care-based inception cohort. Arthritis Rheum. 2005;52:2293-2299.
60. Kozera L, Andrews J, Morgan AW. Cardiovascular risk and rheumatoid arthritis--the next step: Differentiating true soluble biomarkers of cardiovascular risk from surrogate measures of inflammation. Rheumatology (Oxford). 2011;50:1944-1954.
61. Cardarelli R, Lumicao TG Jr. B-type natriuretic peptide: A review of its diagnostic, prognostic, and therapeutic monitoring value in heart failure for primary care physicians. J Am Board Fam Pract. 2003;16:327-333.
62. Kragelund C, Gronning B, Kober L, et al. N-terminal pro-B-type natriuretic peptide and long-term mortality in stable coronary heart disease. N Engl J Med. 2005;352:666-675.
63. Harney SM, Timperley J, Daly C, et al. Brain natriuretic peptide is a potentially useful screening tool for the detection of cardiovascular disease in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:136.
64. Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One. 2012;7:e38930.
65. Karpouzas GA, Estis J, Rezaeian P, et al. High-sensitivity cardiac troponin I is a biomarker for occult coronary plaque burden and cardiovascular events in patients with rheumatoid arthritis. Rheumatology (Oxford). 2018;57:1080-1088.
66. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
67. Assmann G, Cullen P, Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular munster (PROCAM) study. Circulation. 2002;105:310-315.
1. Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the united states. part I. Arthritis Rheum. 2008;58:15-25.
2. England BR, Sayles H, Michaud K, et al. Cause-specific mortality in male US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2016;68:36-45.
3. Sokka T, Abelson B, Pincus T. Mortality in rheumatoid arthritis: 2008 update. Clin Exp Rheumatol. 2008;26:S35-61.
4. Avina-Zubieta JA, Choi HK, Sadatsafavi M, et al. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690-1697.
5. Avina-Zubieta JA, Thomas J, Sadatsafavi M, et al. Risk of incident cardiovascular events in patients with rheumatoid arthritis: A meta-analysis of observational studies. Ann Rheum Dis. 2012;71:1524-1529.
6. van Halm VP, Peters MJ, Voskuyl AE, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: A cross-sectional study, the CARRE investigation. Ann Rheum Dis. 2009;68:1395-1400.
7. Peters MJ, van Halm VP, Voskuyl AE, et al. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571-1579.
8. Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: A population-based cohort study. Arthritis Rheum. 2005;52:402-411.
9. Cardiovascular disease in women--often silent and fatal. Lancet. 2011;378:200,6736(11)61108-61112.
10. Van Doornum S, Brand C, Sundararajan V, et al. Rheumatoid arthritis patients receive less frequent acute reperfusion and secondary prevention therapy after myocardial infarction compared with the general population. Arthritis Res Ther. 2010;12:R183.
11. Sodergren A, Stegmayr B, Lundberg V, et al. Increased incidence of and impaired prognosis after acute myocardial infarction among patients with seropositive rheumatoid arthritis. Ann Rheum Dis. 2007;66:263-266.
12. Douglas KM, Pace AV, Treharne GJ, et al. Excess recurrent cardiac events in rheumatoid arthritis patients with acute coronary syndrome. Ann Rheum Dis. 2006;65:348-353.
13. McCoy SS, Crowson CS, Maradit-Kremers H, et al. Long-term outcomes and treatment after myocardial infarction in patients with rheumatoid arthritis. J Rheumatol. 2013;40:605-610.
14. Mantel A, Holmqvist M, Andersson DC, et al. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J Am Coll Cardiol. 2017;69:1275-1285.
15. Crowson CS, Nicola PJ, Kremers HM, et al. How much of the increased incidence of heart failure in rheumatoid arthritis is attributable to traditional cardiovascular risk factors and ischemic heart disease? Arthritis Rheum. 2005;52:3039-3044.
16. Nicola PJ, Maradit-Kremers H, Roger VL, et al. The risk of congestive heart failure in rheumatoid arthritis: A population-based study over 46 years. Arthritis Rheum. 2005;52:412-420.
17. Davis JM,3rd, Roger VL, Crowson CS, et al. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008;58:2603-2611.
18. Arslan S, Bozkurt E, Sari RA, Erol MK. Diastolic function abnormalities in active rheumatoid arthritis evaluation by conventional doppler and tissue doppler: Relation with duration of disease. Clin Rheumatol. 2006;25:294-299.
19. Liang KP, Myasoedova E, Crowson CS, et al. Increased prevalence of diastolic dysfunction in rheumatoid arthritis. Ann Rheum Dis. 2010;69:1665-1670.
20. Wiseman SJ, Ralston SH, Wardlaw JM. Cerebrovascular disease in rheumatic diseases: A systematic review and meta-analysis. Stroke. 2016;47:943-950.
21. Ungprasert P, Srivali N, Spanuchart I, et al. Risk of venous thromboembolism in patients with rheumatoid arthritis: A systematic review and meta-analysis. Clin Rheumatol. 2014;33:297-304.
22. Stamatelopoulos KS, Kitas GD, Papamichael CM, et al. Subclinical peripheral arterial disease in rheumatoid arthritis. Atherosclerosis. 2010;212:305-309.
23. Chuang YW, Yu MC, Lin CL, et al. Risk of peripheral arterial occlusive disease in patients with rheumatoid arthritis. A nationwide population-based cohort study. Thromb Haemost. 2016;115:439-445.
24. Conroy RM, Pyorala K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in europe: The SCORE project. Eur Heart J. 2003;24:987-1003.
25. D’Agostino RB, Vasan RS, Pencina MJ, et al. General cardiovascular risk profile for use in primary care: The Framingham heart study. Circulation. 2008;117:743-753.
26. del Rincon ID, Williams K, Stern MP, et al. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum. 2001;44:2737-2745.
27. Ridker PM, Buring JE, Rifai N, Cook NR. Development and validation of improved algorithms for the assessment of global cardiovascular risk in women: The Reynolds Risk Score. JAMA. 2007;297:611-619.
28. Arts EE, Popa C, Den Broeder AA, et al. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis. 2015;74:668-674.
29. Crowson CS, Matteson EL, Roger VL, et al. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol. 2012;110:420-424.
30. Kawai VK, Chung CP, Solus JF, et al. The ability of the 2013 American College of Cardiology/American Heart Association cardiovascular risk score to identify rheumatoid arthritis patients with high coronary artery calcification scores. Arthritis Rheumatol. 2015;67:381-385.
31. Peters MJ, Symmons DP, McCarey D, et al. EULAR evidence-based recommendations for cardiovascular risk management in patients with rheumatoid arthritis and other forms of inflammatory arthritis. Ann Rheum Dis. 2010;69:325-331.
32. Agca R, Heslinga SC, Rollefstad S, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17-28.
33. Hippisley-Cox J, Coupland C, Vinogradova Y, et al. Predicting cardiovascular risk in England and Wales: Prospective derivation and validation of QRISK2. BMJ. 2008;336:1475-1482.
34. Hippisley-Cox J, Coupland C, Brindle P. Development and validation of QRISK3 risk prediction algorithms to estimate future risk of cardiovascular disease: Prospective cohort study. BMJ. 2017;357:j2099.
35. Solomon DH, Greenberg J, Curtis JR, et al. Derivation and internal validation of an expanded cardiovascular risk prediction score for rheumatoid arthritis: A consortium of rheumatology researchers of north america registry study. Arthritis Rheumatol. 2015;67:1995-2003.
36. Crowson CS, Gabriel SE, Semb AG, et al. Rheumatoid arthritis-specific cardiovascular risk scores are not superior to general risk scores: A validation analysis of patients from seven countries. Rheumatology (Oxford). 2017;56:1102-1110.
37. Alemao E, Cawston H, Bourhis F, et al. Comparison of cardiovascular risk algorithms in patients with vs without rheumatoid arthritis and the role of C-reactive protein in predicting cardiovascular outcomes in rheumatoid arthritis. Rheumatology (Oxford). 2017;56:777-786.
38. Crowson CS, Rollefstad S, Kitas GD, et al. Challenges of developing a cardiovascular risk calculator for patients with rheumatoid arthritis. PLoS One. 2017;12: e0174656.
39. Karpouzas GA, Malpeso J, Choi TY, et al. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis. 2014;73:1797-1804.
40. van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: A meta-analysis. Semin Arthritis Rheum. 2011;40:3893-97.
41. Evans MR, Escalante A, Battafarano DF, et al. Carotid atherosclerosis predicts incident acute coronary syndromes in rheumatoid arthritis. Arthritis Rheum. 2011;63:1211-1220.
42. Ajeganova S, de Faire U, Jogestrand T, et al. Carotid atherosclerosis, disease measures, oxidized low-density lipoproteins, and atheroprotective natural antibodies for cardiovascular disease in early rheumatoid arthritis--an inception cohort study. J Rheumatol. 2012;39:1146-1154.
43. Corrales A, Gonzalez-Juanatey C, Peiro ME, et al. Carotid ultrasound is useful for the cardiovascular risk stratification of patients with rheumatoid arthritis: Results of a population-based study. Ann Rheum Dis. 2014;73:722-727.
44. Ikdahl E, Rollefstad S, Wibetoe G, et al. Predictive value of arterial stiffness and subclinical carotid atherosclerosis for cardiovascular disease in patients with rheumatoid arthritis. J Rheumatol. 2016;43:1622-1630.
45. Provan SA, Semb AG, Hisdal J, et al. Remission is the goal for cardiovascular risk management in patients with rheumatoid arthritis: A cross-sectional comparative study. Ann Rheum Dis. 2011;70:812-817.
46. Vlachopoulos C, Aznaouridis K, Terentes-Printzios D, et al. Prediction of cardiovascular events and all-cause mortality with brachial-ankle elasticity index: A systematic review and meta-analysis. Hypertension. 2012;60:556-562.
47. Ambrosino P, Tasso M, Lupoli R, et al. Non-invasive assessment of arterial stiffness in patients with rheumatoid arthritis: A systematic review and meta-analysis of literature studies. Ann Med. 2015;47:457-467.
48. Rumberger JA, Simons DB, Fitzpatrick LA, et al. Coronary artery calcium area by electron-beam computed tomography and coronary atherosclerotic plaque area. A histopathologic correlative study. Circulation. 1995;92:2157-2162.
49. Detrano R, Guerci AD, Carr JJ, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336-1345.
50. Task Force Members, Montalescot G, Sechtem U, et al. 2013 ESC guidelines on the management of stable coronary artery disease: The task force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34:2949-3003.
51. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2935-2959.
52. Hou ZH, Lu B, Gao Y, et al. Prognostic value of coronary CT angiography and calcium score for major adverse cardiac events in outpatients. JACC Cardiovasc Imaging. 2012;5:990-999.
53. Yiu KH, Mok MY, Wang S, et al. Prognostic role of coronary calcification in patients with rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Rheumatol. 2012;30:345-350.
54. Wright K, Crowson CS, Gabriel SE. Cardiovascular comorbidity in rheumatic diseases: A focus on heart failure. Heart Fail Clin. 2014;10:339-352.
55. Rudominer RL, Roman MJ, Devereux RB, et al. Independent association of rheumatoid arthritis with increased left ventricular mass but not with reduced ejection fraction. Arthritis Rheum. 2009;60:22-29.
56. Bluemke DA, Kronmal RA, Lima JA, et al. The relationship of left ventricular mass and geometry to incident cardiovascular events: The MESA (Multi-Ethnic Study of Atherosclerosis) study. J Am Coll Cardiol. 2008;52:2148-2155.
57. Ntusi NAB, Piechnik SK, Francis JM, et al. Diffuse myocardial fibrosis and inflammation in rheumatoid arthritis: Insights from CMR T1 mapping. JACC Cardiovasc Imaging. 2015;8:526-536.
58. Wong TC, Piehler K, Meier CG, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012;126:1206-1216.
59. Goodson NJ, Symmons DP, Scott DG, et al. Baseline levels of C-reactive protein and prediction of death from cardiovascular disease in patients with inflammatory polyarthritis: A ten-year followup study of a primary care-based inception cohort. Arthritis Rheum. 2005;52:2293-2299.
60. Kozera L, Andrews J, Morgan AW. Cardiovascular risk and rheumatoid arthritis--the next step: Differentiating true soluble biomarkers of cardiovascular risk from surrogate measures of inflammation. Rheumatology (Oxford). 2011;50:1944-1954.
61. Cardarelli R, Lumicao TG Jr. B-type natriuretic peptide: A review of its diagnostic, prognostic, and therapeutic monitoring value in heart failure for primary care physicians. J Am Board Fam Pract. 2003;16:327-333.
62. Kragelund C, Gronning B, Kober L, et al. N-terminal pro-B-type natriuretic peptide and long-term mortality in stable coronary heart disease. N Engl J Med. 2005;352:666-675.
63. Harney SM, Timperley J, Daly C, et al. Brain natriuretic peptide is a potentially useful screening tool for the detection of cardiovascular disease in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:136.
64. Bradham WS, Bian A, Oeser A, et al. High-sensitivity cardiac troponin-I is elevated in patients with rheumatoid arthritis, independent of cardiovascular risk factors and inflammation. PLoS One. 2012;7:e38930.
65. Karpouzas GA, Estis J, Rezaeian P, et al. High-sensitivity cardiac troponin I is a biomarker for occult coronary plaque burden and cardiovascular events in patients with rheumatoid arthritis. Rheumatology (Oxford). 2018;57:1080-1088.
66. Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2014;63:2889-2934.
67. Assmann G, Cullen P, Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular munster (PROCAM) study. Circulation. 2002;105:310-315.