User login
Anatomy dictates endovascular repair of popliteal artery aneurysms
CHICAGO – Endovascular repair of popliteal artery aneurysms is a relatively safe and viable off-label alternative to open repair, but appropriate anatomy is essential. This includes a landing zone of at least 2 cm above and below the aneurysm, minimal discrepancy in size between the proximal and distal landing zones, and lack of extensive vessel tortuosity due to potential kinking of the endograft, Dr. Neal S. Cayne said at a symposium on vascular surgery sponsored by Northwestern University.
The risk of kinking and graft thrombosis also excludes patients who frequently flex their knee more than 90 degrees, such as carpenters and gardeners.
Patients with a contraindication to antiplatelet medication are also off limits, as clopidogrel (Plavix) has been shown to be a predictor of success, said Dr. Cayne, director of endovascular surgery at New York University Langone Medical Center.
In 2012, his team reported technical success in 25 of 26 endovascular popliteal artery aneurysm (PAA) repairs performed in 21 consecutive patients between January 2004 and January 2011, with the one technical failure due to stent graft infolding (J. Vasc. Surg. 2012;55:1647-53).
Primary and secondary patency rates were both 91.2% at 1 year, and were 85.5% and 91.2%, respectively, at 2 years. All patients were maintained on aspirin or clopidogrel.
No limb loss was reported, but three occlusions occurred during follow-up at 4, 14, and 26 months. One patient required a tibial artery bypass for a nonhealing wound, and two were successfully repaired with open thrombectomy. All three occlusion patients had single-vessel runoff.
Based on our data, in general, I will not stent someone "with single-vessel outflow, and I also will not stent someone who, for some reason, can’t take antiplatelet agents," Dr. Cayne said.
Even when patients present urgently and the PAA has thrombosed, endovascular repair is not an option if there is only one outflow vessel. "I would do a bypass; there’s nothing wrong with open surgery," he said.
A recent unpublished review of 79 PAAs treated at Langone from 1998 to 2012 with both approaches found 5-year primary patencies of 67% for open repair and 80% for endovascular repair (P less than .05). Secondary patency was 90% in both groups.
One amputation occurred in the open group, but occlusion rates were higher using endovascular repair with one-vessel runoff (P = .003), Dr. Cayne said.
As expected, length of stay was shorter with endovascular repair (1.9 vs. 6.4 days; P less than .001).
Follow-up was longer for the 36 open PAAs than the 43 endovascular PAAs (75 vs. 34 months), but patients in both groups were similar with respect to age, comorbidities, PAA size, runoff, and symptoms, he said.
During a discussion following the presentation, some attendees said they still prefer to use bypass for all patients with PAA, and asked how candidates are selected for stent placement.
"Number one and most important is the anatomy," Dr. Cayne said. "The one advantage of endovascular repair is that if you have a patient too sick to get general or even regional anesthesia, you can do it under local [anesthesia] almost all the time with a small cutdown or puncture. But you do have that long discussion with the patient that this is a non–FDA-approved, off-label use. We provide them with the data, but some patients in New York will come in with pages and pages of literature and say, ‘Nope, I want a stent, I want this particular stent, and this is the way I want you to do it.’ "
The stent of choice at Langone has been Gore’s Viabahn covered stent graft, which is FDA approved for treating occlusive disease rather than PAA. The device is usually oversized by 10%-15%, but no more than that, because of the risk of graft infolding, Dr. Cayne said.
If more than one graft is needed, a maximum of no more than 1-mm size differential between grafts is suggested. A minimum overlap of 2-3 cm between grafts is also preferred.
Last year, surgeons reported a dismal 50% occlusion rate within just 6 weeks in the endovascular treatment of six PAAs using a novel, multilayer stent (Cardiatis’s Multilayer Aneurysm Repair System) (J. Endovasc. Ther. 2013;20:381-8), he observed.
Dr. Cayne reported receiving honoraria as a consultant from Cook Medical.
CHICAGO – Endovascular repair of popliteal artery aneurysms is a relatively safe and viable off-label alternative to open repair, but appropriate anatomy is essential. This includes a landing zone of at least 2 cm above and below the aneurysm, minimal discrepancy in size between the proximal and distal landing zones, and lack of extensive vessel tortuosity due to potential kinking of the endograft, Dr. Neal S. Cayne said at a symposium on vascular surgery sponsored by Northwestern University.
The risk of kinking and graft thrombosis also excludes patients who frequently flex their knee more than 90 degrees, such as carpenters and gardeners.
Patients with a contraindication to antiplatelet medication are also off limits, as clopidogrel (Plavix) has been shown to be a predictor of success, said Dr. Cayne, director of endovascular surgery at New York University Langone Medical Center.
In 2012, his team reported technical success in 25 of 26 endovascular popliteal artery aneurysm (PAA) repairs performed in 21 consecutive patients between January 2004 and January 2011, with the one technical failure due to stent graft infolding (J. Vasc. Surg. 2012;55:1647-53).
Primary and secondary patency rates were both 91.2% at 1 year, and were 85.5% and 91.2%, respectively, at 2 years. All patients were maintained on aspirin or clopidogrel.
No limb loss was reported, but three occlusions occurred during follow-up at 4, 14, and 26 months. One patient required a tibial artery bypass for a nonhealing wound, and two were successfully repaired with open thrombectomy. All three occlusion patients had single-vessel runoff.
Based on our data, in general, I will not stent someone "with single-vessel outflow, and I also will not stent someone who, for some reason, can’t take antiplatelet agents," Dr. Cayne said.
Even when patients present urgently and the PAA has thrombosed, endovascular repair is not an option if there is only one outflow vessel. "I would do a bypass; there’s nothing wrong with open surgery," he said.
A recent unpublished review of 79 PAAs treated at Langone from 1998 to 2012 with both approaches found 5-year primary patencies of 67% for open repair and 80% for endovascular repair (P less than .05). Secondary patency was 90% in both groups.
One amputation occurred in the open group, but occlusion rates were higher using endovascular repair with one-vessel runoff (P = .003), Dr. Cayne said.
As expected, length of stay was shorter with endovascular repair (1.9 vs. 6.4 days; P less than .001).
Follow-up was longer for the 36 open PAAs than the 43 endovascular PAAs (75 vs. 34 months), but patients in both groups were similar with respect to age, comorbidities, PAA size, runoff, and symptoms, he said.
During a discussion following the presentation, some attendees said they still prefer to use bypass for all patients with PAA, and asked how candidates are selected for stent placement.
"Number one and most important is the anatomy," Dr. Cayne said. "The one advantage of endovascular repair is that if you have a patient too sick to get general or even regional anesthesia, you can do it under local [anesthesia] almost all the time with a small cutdown or puncture. But you do have that long discussion with the patient that this is a non–FDA-approved, off-label use. We provide them with the data, but some patients in New York will come in with pages and pages of literature and say, ‘Nope, I want a stent, I want this particular stent, and this is the way I want you to do it.’ "
The stent of choice at Langone has been Gore’s Viabahn covered stent graft, which is FDA approved for treating occlusive disease rather than PAA. The device is usually oversized by 10%-15%, but no more than that, because of the risk of graft infolding, Dr. Cayne said.
If more than one graft is needed, a maximum of no more than 1-mm size differential between grafts is suggested. A minimum overlap of 2-3 cm between grafts is also preferred.
Last year, surgeons reported a dismal 50% occlusion rate within just 6 weeks in the endovascular treatment of six PAAs using a novel, multilayer stent (Cardiatis’s Multilayer Aneurysm Repair System) (J. Endovasc. Ther. 2013;20:381-8), he observed.
Dr. Cayne reported receiving honoraria as a consultant from Cook Medical.
CHICAGO – Endovascular repair of popliteal artery aneurysms is a relatively safe and viable off-label alternative to open repair, but appropriate anatomy is essential. This includes a landing zone of at least 2 cm above and below the aneurysm, minimal discrepancy in size between the proximal and distal landing zones, and lack of extensive vessel tortuosity due to potential kinking of the endograft, Dr. Neal S. Cayne said at a symposium on vascular surgery sponsored by Northwestern University.
The risk of kinking and graft thrombosis also excludes patients who frequently flex their knee more than 90 degrees, such as carpenters and gardeners.
Patients with a contraindication to antiplatelet medication are also off limits, as clopidogrel (Plavix) has been shown to be a predictor of success, said Dr. Cayne, director of endovascular surgery at New York University Langone Medical Center.
In 2012, his team reported technical success in 25 of 26 endovascular popliteal artery aneurysm (PAA) repairs performed in 21 consecutive patients between January 2004 and January 2011, with the one technical failure due to stent graft infolding (J. Vasc. Surg. 2012;55:1647-53).
Primary and secondary patency rates were both 91.2% at 1 year, and were 85.5% and 91.2%, respectively, at 2 years. All patients were maintained on aspirin or clopidogrel.
No limb loss was reported, but three occlusions occurred during follow-up at 4, 14, and 26 months. One patient required a tibial artery bypass for a nonhealing wound, and two were successfully repaired with open thrombectomy. All three occlusion patients had single-vessel runoff.
Based on our data, in general, I will not stent someone "with single-vessel outflow, and I also will not stent someone who, for some reason, can’t take antiplatelet agents," Dr. Cayne said.
Even when patients present urgently and the PAA has thrombosed, endovascular repair is not an option if there is only one outflow vessel. "I would do a bypass; there’s nothing wrong with open surgery," he said.
A recent unpublished review of 79 PAAs treated at Langone from 1998 to 2012 with both approaches found 5-year primary patencies of 67% for open repair and 80% for endovascular repair (P less than .05). Secondary patency was 90% in both groups.
One amputation occurred in the open group, but occlusion rates were higher using endovascular repair with one-vessel runoff (P = .003), Dr. Cayne said.
As expected, length of stay was shorter with endovascular repair (1.9 vs. 6.4 days; P less than .001).
Follow-up was longer for the 36 open PAAs than the 43 endovascular PAAs (75 vs. 34 months), but patients in both groups were similar with respect to age, comorbidities, PAA size, runoff, and symptoms, he said.
During a discussion following the presentation, some attendees said they still prefer to use bypass for all patients with PAA, and asked how candidates are selected for stent placement.
"Number one and most important is the anatomy," Dr. Cayne said. "The one advantage of endovascular repair is that if you have a patient too sick to get general or even regional anesthesia, you can do it under local [anesthesia] almost all the time with a small cutdown or puncture. But you do have that long discussion with the patient that this is a non–FDA-approved, off-label use. We provide them with the data, but some patients in New York will come in with pages and pages of literature and say, ‘Nope, I want a stent, I want this particular stent, and this is the way I want you to do it.’ "
The stent of choice at Langone has been Gore’s Viabahn covered stent graft, which is FDA approved for treating occlusive disease rather than PAA. The device is usually oversized by 10%-15%, but no more than that, because of the risk of graft infolding, Dr. Cayne said.
If more than one graft is needed, a maximum of no more than 1-mm size differential between grafts is suggested. A minimum overlap of 2-3 cm between grafts is also preferred.
Last year, surgeons reported a dismal 50% occlusion rate within just 6 weeks in the endovascular treatment of six PAAs using a novel, multilayer stent (Cardiatis’s Multilayer Aneurysm Repair System) (J. Endovasc. Ther. 2013;20:381-8), he observed.
Dr. Cayne reported receiving honoraria as a consultant from Cook Medical.
AT THE NORTHWESTERN VASCULAR SYMPOSIUM
Major finding: Five-year primary patency was 67% for open repair and 80% for endovascular repair (P less than .05).
Data source: Expert opinion and retrospective study of 79 popliteal artery aneurysm repairs.
Disclosures: Dr. Cayne reported receiving honoraria as a consultant from Cook Medical.
DRIL particularly effective for access-related hand ischemia
CHICAGO – The best strategy for access-related hand ischemia is to have a plan in place before it occurs.
"The whole approach for patients at high risk of developing hand ischemia is to think about it preoperatively" and devise a remedial plan, Dr. Thomas S. Huber said at the symposium. "I always tell our trainees and fellows: ‘What are you going to do in 20 minutes when the recovery room nurse says the patient has motor compromise and unbelievable hand pain?’ You have to have a remedial plan."
Access-related hand ischemia (ARHI), or steal syndrome, occurs in just 2% of radial artery–based access procedures, but up to 20% of brachial artery–based access procedures. Roughly half of these events are severe enough to merit remedial treatment. With more than 8 million arterial catheters placed perioperatively in the United States each year and 350,000 patients on dialysis, the numbers add up.
"There are multiple complementary treatment options that you ought to have in your armamentarium," said Dr. Huber, chief of vascular surgery and endovascular therapy, University of Florida Health Science Center, Gainesville, Fla.
"The DRIL has worked remarkably well for us."
The distal revascularization and interval ligation (DRIL) procedure involves a brachial-brachial bypass with ligation of the brachial artery immediately distal to the fistula anastomosis.

In their hands, DRIL significantly increased wrist-brachial and digital-brachial indices (0.31 and 0.25) in 126 patients undergoing 134 procedures from 2002 to 2011 (J. Vasc. Surg. 2013;57:451-8). Ischemic symptoms were relieved in 82% of patients, and the index access was salvaged in 85%.
"Really, the DRIL procedure has almost no impact on the success of the access," Dr. Huber said. "For those patients [in which] we perform the DRIL [procedure] before their access is mature, our maturation rate is comparable whether we do the DRIL or not."
Primary patency was 95% at 1 year and 78% at 5 years. The overall complication rate, driven mostly by wound complications, reached 27%, and 30-day mortality was 2%.
"I sheepishly stand before you to say that our mortality rate for all our access procedures is about 3%," he said. "It’s a pretty minimal operation so, my gosh, how could that possibly be? But it’s a very morbid population. In our country, the 1-year mortality rate for people starting dialysis is right at 23%. So they’re older, sicker patients, with a limited life expectancy."
All-cause mortality in the series was 28% and 79% at 1 and 5 years, respectively. Age greater than 40 years, grade 3 ischemia, any DRIL complication, and smoking history were all significant multivariable predictors of mortality.
An extensive list of clinical ARHI predictors has been identified, such as female sex, advanced age, and large conduits, but none reliably predicts when access should not be attempted, Dr. Huber said. The use of preoperative noninvasive imaging also is helpful to identify patients at risk for ARHI, but the predictive values have not been sufficient to avoid attempting the access procedure.
In their experience, presentation of hand ischemia has been trimodal, with a third of patients presenting within 7 days of their index procedure, a third within 7-30 days, and a third thereafter. A weak ulnar or radial pulse on physical exam after creating a dialysis access has misled some to dismiss hand ischemia, while a prior episode of ARHI is a red flag for high risk of recurrence.
"I would contend that the incidence of developing hand ischemia on the right arm if you had it on the left arm is about 100%," dispelling the concept of simply ligating the access that led to hand ischemia and then repositioning it on the contralateral extremity, Dr. Huber said.
Their overall treatment approach for ARHI in poor operative-risk patients, particularly older patients with a prosthetic access, is to proceed to ligation. Good-risk patients undergo some type of inflow assessment, and if a significant inflow lesion is present, it is corrected, usually at the same time as the DRIL, he said. Proximalization of the arterial inflow would be considered for patients with persistent symptoms and no available conduit, while DRIL is the go-to procedure for those with available vein conduits more than 3 cm in diameter, preferably the greater saphenous vein.
Noticeably absent in their algorithm is some type of flow-limiting strategy, such as banding, and the revision using distal inflow procedure. These approaches may have a role, but the published literature is somewhat inconclusive, Dr. Huber said at the symposium, sponsored by Northwestern University.
Despite the overwhelming number of Americans on dialysis, less than 300 DRIL cases have been reported in the literature since its introduction in 1988. The University of Florida experience has been corroborated by a recent report of complete symptom resolution in 82% of 81 DRIL procedures, with a 17% complication rate, and five-year access and bypass survival rates of 56% and 97% (J. Vasc. Surg. 2013;54:1073-8).
Surgeons in Texas reported full symptom resolution in 77% of 33 patients, but cautioned against potential complications such as bypass failure that led to gangrene and transmetacarpal amputation in one patient (J. Vasc. Access 2012;13:299-304).
Dr. Huber reported having no financial disclosures.
CHICAGO – The best strategy for access-related hand ischemia is to have a plan in place before it occurs.
"The whole approach for patients at high risk of developing hand ischemia is to think about it preoperatively" and devise a remedial plan, Dr. Thomas S. Huber said at the symposium. "I always tell our trainees and fellows: ‘What are you going to do in 20 minutes when the recovery room nurse says the patient has motor compromise and unbelievable hand pain?’ You have to have a remedial plan."
Access-related hand ischemia (ARHI), or steal syndrome, occurs in just 2% of radial artery–based access procedures, but up to 20% of brachial artery–based access procedures. Roughly half of these events are severe enough to merit remedial treatment. With more than 8 million arterial catheters placed perioperatively in the United States each year and 350,000 patients on dialysis, the numbers add up.
"There are multiple complementary treatment options that you ought to have in your armamentarium," said Dr. Huber, chief of vascular surgery and endovascular therapy, University of Florida Health Science Center, Gainesville, Fla.
"The DRIL has worked remarkably well for us."
The distal revascularization and interval ligation (DRIL) procedure involves a brachial-brachial bypass with ligation of the brachial artery immediately distal to the fistula anastomosis.
In their hands, DRIL significantly increased wrist-brachial and digital-brachial indices (0.31 and 0.25) in 126 patients undergoing 134 procedures from 2002 to 2011 (J. Vasc. Surg. 2013;57:451-8). Ischemic symptoms were relieved in 82% of patients, and the index access was salvaged in 85%.
"Really, the DRIL procedure has almost no impact on the success of the access," Dr. Huber said. "For those patients [in which] we perform the DRIL [procedure] before their access is mature, our maturation rate is comparable whether we do the DRIL or not."
Primary patency was 95% at 1 year and 78% at 5 years. The overall complication rate, driven mostly by wound complications, reached 27%, and 30-day mortality was 2%.
"I sheepishly stand before you to say that our mortality rate for all our access procedures is about 3%," he said. "It’s a pretty minimal operation so, my gosh, how could that possibly be? But it’s a very morbid population. In our country, the 1-year mortality rate for people starting dialysis is right at 23%. So they’re older, sicker patients, with a limited life expectancy."
All-cause mortality in the series was 28% and 79% at 1 and 5 years, respectively. Age greater than 40 years, grade 3 ischemia, any DRIL complication, and smoking history were all significant multivariable predictors of mortality.
An extensive list of clinical ARHI predictors has been identified, such as female sex, advanced age, and large conduits, but none reliably predicts when access should not be attempted, Dr. Huber said. The use of preoperative noninvasive imaging also is helpful to identify patients at risk for ARHI, but the predictive values have not been sufficient to avoid attempting the access procedure.
In their experience, presentation of hand ischemia has been trimodal, with a third of patients presenting within 7 days of their index procedure, a third within 7-30 days, and a third thereafter. A weak ulnar or radial pulse on physical exam after creating a dialysis access has misled some to dismiss hand ischemia, while a prior episode of ARHI is a red flag for high risk of recurrence.
"I would contend that the incidence of developing hand ischemia on the right arm if you had it on the left arm is about 100%," dispelling the concept of simply ligating the access that led to hand ischemia and then repositioning it on the contralateral extremity, Dr. Huber said.
Their overall treatment approach for ARHI in poor operative-risk patients, particularly older patients with a prosthetic access, is to proceed to ligation. Good-risk patients undergo some type of inflow assessment, and if a significant inflow lesion is present, it is corrected, usually at the same time as the DRIL, he said. Proximalization of the arterial inflow would be considered for patients with persistent symptoms and no available conduit, while DRIL is the go-to procedure for those with available vein conduits more than 3 cm in diameter, preferably the greater saphenous vein.
Noticeably absent in their algorithm is some type of flow-limiting strategy, such as banding, and the revision using distal inflow procedure. These approaches may have a role, but the published literature is somewhat inconclusive, Dr. Huber said at the symposium, sponsored by Northwestern University.
Despite the overwhelming number of Americans on dialysis, less than 300 DRIL cases have been reported in the literature since its introduction in 1988. The University of Florida experience has been corroborated by a recent report of complete symptom resolution in 82% of 81 DRIL procedures, with a 17% complication rate, and five-year access and bypass survival rates of 56% and 97% (J. Vasc. Surg. 2013;54:1073-8).
Surgeons in Texas reported full symptom resolution in 77% of 33 patients, but cautioned against potential complications such as bypass failure that led to gangrene and transmetacarpal amputation in one patient (J. Vasc. Access 2012;13:299-304).
Dr. Huber reported having no financial disclosures.
CHICAGO – The best strategy for access-related hand ischemia is to have a plan in place before it occurs.
"The whole approach for patients at high risk of developing hand ischemia is to think about it preoperatively" and devise a remedial plan, Dr. Thomas S. Huber said at the symposium. "I always tell our trainees and fellows: ‘What are you going to do in 20 minutes when the recovery room nurse says the patient has motor compromise and unbelievable hand pain?’ You have to have a remedial plan."
Access-related hand ischemia (ARHI), or steal syndrome, occurs in just 2% of radial artery–based access procedures, but up to 20% of brachial artery–based access procedures. Roughly half of these events are severe enough to merit remedial treatment. With more than 8 million arterial catheters placed perioperatively in the United States each year and 350,000 patients on dialysis, the numbers add up.
"There are multiple complementary treatment options that you ought to have in your armamentarium," said Dr. Huber, chief of vascular surgery and endovascular therapy, University of Florida Health Science Center, Gainesville, Fla.
"The DRIL has worked remarkably well for us."
The distal revascularization and interval ligation (DRIL) procedure involves a brachial-brachial bypass with ligation of the brachial artery immediately distal to the fistula anastomosis.
In their hands, DRIL significantly increased wrist-brachial and digital-brachial indices (0.31 and 0.25) in 126 patients undergoing 134 procedures from 2002 to 2011 (J. Vasc. Surg. 2013;57:451-8). Ischemic symptoms were relieved in 82% of patients, and the index access was salvaged in 85%.
"Really, the DRIL procedure has almost no impact on the success of the access," Dr. Huber said. "For those patients [in which] we perform the DRIL [procedure] before their access is mature, our maturation rate is comparable whether we do the DRIL or not."
Primary patency was 95% at 1 year and 78% at 5 years. The overall complication rate, driven mostly by wound complications, reached 27%, and 30-day mortality was 2%.
"I sheepishly stand before you to say that our mortality rate for all our access procedures is about 3%," he said. "It’s a pretty minimal operation so, my gosh, how could that possibly be? But it’s a very morbid population. In our country, the 1-year mortality rate for people starting dialysis is right at 23%. So they’re older, sicker patients, with a limited life expectancy."
All-cause mortality in the series was 28% and 79% at 1 and 5 years, respectively. Age greater than 40 years, grade 3 ischemia, any DRIL complication, and smoking history were all significant multivariable predictors of mortality.
An extensive list of clinical ARHI predictors has been identified, such as female sex, advanced age, and large conduits, but none reliably predicts when access should not be attempted, Dr. Huber said. The use of preoperative noninvasive imaging also is helpful to identify patients at risk for ARHI, but the predictive values have not been sufficient to avoid attempting the access procedure.
In their experience, presentation of hand ischemia has been trimodal, with a third of patients presenting within 7 days of their index procedure, a third within 7-30 days, and a third thereafter. A weak ulnar or radial pulse on physical exam after creating a dialysis access has misled some to dismiss hand ischemia, while a prior episode of ARHI is a red flag for high risk of recurrence.
"I would contend that the incidence of developing hand ischemia on the right arm if you had it on the left arm is about 100%," dispelling the concept of simply ligating the access that led to hand ischemia and then repositioning it on the contralateral extremity, Dr. Huber said.
Their overall treatment approach for ARHI in poor operative-risk patients, particularly older patients with a prosthetic access, is to proceed to ligation. Good-risk patients undergo some type of inflow assessment, and if a significant inflow lesion is present, it is corrected, usually at the same time as the DRIL, he said. Proximalization of the arterial inflow would be considered for patients with persistent symptoms and no available conduit, while DRIL is the go-to procedure for those with available vein conduits more than 3 cm in diameter, preferably the greater saphenous vein.
Noticeably absent in their algorithm is some type of flow-limiting strategy, such as banding, and the revision using distal inflow procedure. These approaches may have a role, but the published literature is somewhat inconclusive, Dr. Huber said at the symposium, sponsored by Northwestern University.
Despite the overwhelming number of Americans on dialysis, less than 300 DRIL cases have been reported in the literature since its introduction in 1988. The University of Florida experience has been corroborated by a recent report of complete symptom resolution in 82% of 81 DRIL procedures, with a 17% complication rate, and five-year access and bypass survival rates of 56% and 97% (J. Vasc. Surg. 2013;54:1073-8).
Surgeons in Texas reported full symptom resolution in 77% of 33 patients, but cautioned against potential complications such as bypass failure that led to gangrene and transmetacarpal amputation in one patient (J. Vasc. Access 2012;13:299-304).
Dr. Huber reported having no financial disclosures.
AT THE NORTHWESTERN VASCULAR SYMPOSIUM

Partial flail chest stabilization may suffice
NAPLES, FLA. – Partial repair of flail segment rib fractures may suffice in patients whose anatomy precludes full stabilization, a small study suggests.
After a median follow-up of 189 days, only predicted total lung capacity at 3 months (90% vs. 72%; P = .02) and 6 months (94% vs. 75%; P = .038) significantly differed between patients undergoing complete flail stabilization (CFS) vs. partial flail stabilization (PFS).
No other differences in pulmonary and clinical outcomes were observed between the 43 patients, Dr. Terry P. Nickerson reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
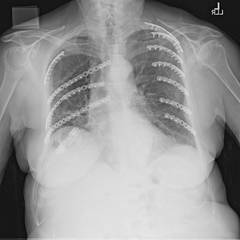
Flail chest is defined by two or more consecutive ribs fractured in two places and a segment of chest wall unable to contribute to respiratory mechanics.
Benefits of surgical stabilization of flail chest reported in the heterogeneous literature include decreased ICU stay, lower rates of pneumonia, increased return to work, improved quality of life, and low surgical morbidity, Dr. Nickerson noted.
The retrospective analysis, included all patients (aged 30-85 years) who underwent surgical rib fracture stabilization for flail chest from August 2009 through February 2013 at the Mayo Clinic Hospital, St. Marys Campus, Rochester, Minn. In all, 23 patients had CFS, defined as all fractures involved in a flail segment undergoing full fixation, and 20 had PFS or at least one flail segment not completely repaired.
The CFS and PFS groups were similar with respect to median age (63 years vs. 58 years), sex (52% male vs. 65% male), operating time (186 minutes vs. 183 minutes), Injury Severity Score (20 vs. 17), hospital length of stay (10 days for both), ICU stay (1 day vs. 2 days), and narcotic use at 1-month follow-up (50% vs. 47%), according to the poster presentation.
At 6 months, there were no significant differences between the CFS and PFS groups in predicted vital capacity (86% vs. 76%), forced vital capacity (85% vs. 77%), forced expiratory volume in 1 second (71% vs. 78%), and FVC/FEV1 ratio (68% vs. 74%).
Rates of pneumonia were also similar in the two groups (21% vs. 20%; P = .39), reported Dr. Nickerson, a general surgeon in Rochester.
No patient or provider observed a significant chest wall deformity in either group during follow-up and no reoperations were required for incomplete repair in the PFS.
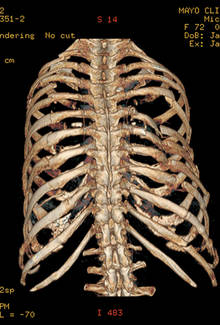
"Despite advances in surgical technique, not all flail segment rib fractures are accessible for repair via standard thoracotomy," the authors wrote. "Our data suggest that PFS is acceptable and that extending or creating additional incisions for exposure for CFS is unwarranted."
Additional research assessing late complications, performance/functional outcomes, and perceived benefits of the operation is needed to determine whether partial repair is truly comparable with full stabilization, coauthor Dr. Brian Kim, associate medical director Mayo Clinic Trauma Center, Rochester, said in an interview.
In their experience, 6 months’ postoperative follow-up has been sufficient with respect to bony healing, convalescence from pain, and monitoring of chest wall integrity.
He noted that some surgeons may be dissuaded from pursuing or recommending operative stabilization of a flail segment with fractures deemed outside the boundaries of repair, particularly if the injury is in the elderly or obese patient.
"The results of our study lend support to the practice of repairing the most anatomically disrupted and/or symptomatic fractures within the flail segment," he said. "Age and/or BMI [body mass index] are not absolute contraindications for flail segment stabilization in our practice."
Poster session moderator Dr. Alexander Eastman of University of Texas Southwestern Medical Center in Dallas, said the low patient numbers and short follow-up in the study make it difficult to draw big conclusions, other than the need for further research.
His group uses surgical fixation of flail chest for patients with instability and pain management for those patients who are difficult to wean from the ventilator.
"I think many surgeons still are skeptical of the data supporting fixation of flail chest," he noted in an interview.
Dr. Nickerson and his coauthors reported having no financial disclosures.
NAPLES, FLA. – Partial repair of flail segment rib fractures may suffice in patients whose anatomy precludes full stabilization, a small study suggests.
After a median follow-up of 189 days, only predicted total lung capacity at 3 months (90% vs. 72%; P = .02) and 6 months (94% vs. 75%; P = .038) significantly differed between patients undergoing complete flail stabilization (CFS) vs. partial flail stabilization (PFS).
No other differences in pulmonary and clinical outcomes were observed between the 43 patients, Dr. Terry P. Nickerson reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Flail chest is defined by two or more consecutive ribs fractured in two places and a segment of chest wall unable to contribute to respiratory mechanics.
Benefits of surgical stabilization of flail chest reported in the heterogeneous literature include decreased ICU stay, lower rates of pneumonia, increased return to work, improved quality of life, and low surgical morbidity, Dr. Nickerson noted.
The retrospective analysis, included all patients (aged 30-85 years) who underwent surgical rib fracture stabilization for flail chest from August 2009 through February 2013 at the Mayo Clinic Hospital, St. Marys Campus, Rochester, Minn. In all, 23 patients had CFS, defined as all fractures involved in a flail segment undergoing full fixation, and 20 had PFS or at least one flail segment not completely repaired.
The CFS and PFS groups were similar with respect to median age (63 years vs. 58 years), sex (52% male vs. 65% male), operating time (186 minutes vs. 183 minutes), Injury Severity Score (20 vs. 17), hospital length of stay (10 days for both), ICU stay (1 day vs. 2 days), and narcotic use at 1-month follow-up (50% vs. 47%), according to the poster presentation.
At 6 months, there were no significant differences between the CFS and PFS groups in predicted vital capacity (86% vs. 76%), forced vital capacity (85% vs. 77%), forced expiratory volume in 1 second (71% vs. 78%), and FVC/FEV1 ratio (68% vs. 74%).
Rates of pneumonia were also similar in the two groups (21% vs. 20%; P = .39), reported Dr. Nickerson, a general surgeon in Rochester.
No patient or provider observed a significant chest wall deformity in either group during follow-up and no reoperations were required for incomplete repair in the PFS.
"Despite advances in surgical technique, not all flail segment rib fractures are accessible for repair via standard thoracotomy," the authors wrote. "Our data suggest that PFS is acceptable and that extending or creating additional incisions for exposure for CFS is unwarranted."
Additional research assessing late complications, performance/functional outcomes, and perceived benefits of the operation is needed to determine whether partial repair is truly comparable with full stabilization, coauthor Dr. Brian Kim, associate medical director Mayo Clinic Trauma Center, Rochester, said in an interview.
In their experience, 6 months’ postoperative follow-up has been sufficient with respect to bony healing, convalescence from pain, and monitoring of chest wall integrity.
He noted that some surgeons may be dissuaded from pursuing or recommending operative stabilization of a flail segment with fractures deemed outside the boundaries of repair, particularly if the injury is in the elderly or obese patient.
"The results of our study lend support to the practice of repairing the most anatomically disrupted and/or symptomatic fractures within the flail segment," he said. "Age and/or BMI [body mass index] are not absolute contraindications for flail segment stabilization in our practice."
Poster session moderator Dr. Alexander Eastman of University of Texas Southwestern Medical Center in Dallas, said the low patient numbers and short follow-up in the study make it difficult to draw big conclusions, other than the need for further research.
His group uses surgical fixation of flail chest for patients with instability and pain management for those patients who are difficult to wean from the ventilator.
"I think many surgeons still are skeptical of the data supporting fixation of flail chest," he noted in an interview.
Dr. Nickerson and his coauthors reported having no financial disclosures.
NAPLES, FLA. – Partial repair of flail segment rib fractures may suffice in patients whose anatomy precludes full stabilization, a small study suggests.
After a median follow-up of 189 days, only predicted total lung capacity at 3 months (90% vs. 72%; P = .02) and 6 months (94% vs. 75%; P = .038) significantly differed between patients undergoing complete flail stabilization (CFS) vs. partial flail stabilization (PFS).
No other differences in pulmonary and clinical outcomes were observed between the 43 patients, Dr. Terry P. Nickerson reported at the annual scientific assembly of the Eastern Association for the Surgery of Trauma.
Flail chest is defined by two or more consecutive ribs fractured in two places and a segment of chest wall unable to contribute to respiratory mechanics.
Benefits of surgical stabilization of flail chest reported in the heterogeneous literature include decreased ICU stay, lower rates of pneumonia, increased return to work, improved quality of life, and low surgical morbidity, Dr. Nickerson noted.
The retrospective analysis, included all patients (aged 30-85 years) who underwent surgical rib fracture stabilization for flail chest from August 2009 through February 2013 at the Mayo Clinic Hospital, St. Marys Campus, Rochester, Minn. In all, 23 patients had CFS, defined as all fractures involved in a flail segment undergoing full fixation, and 20 had PFS or at least one flail segment not completely repaired.
The CFS and PFS groups were similar with respect to median age (63 years vs. 58 years), sex (52% male vs. 65% male), operating time (186 minutes vs. 183 minutes), Injury Severity Score (20 vs. 17), hospital length of stay (10 days for both), ICU stay (1 day vs. 2 days), and narcotic use at 1-month follow-up (50% vs. 47%), according to the poster presentation.
At 6 months, there were no significant differences between the CFS and PFS groups in predicted vital capacity (86% vs. 76%), forced vital capacity (85% vs. 77%), forced expiratory volume in 1 second (71% vs. 78%), and FVC/FEV1 ratio (68% vs. 74%).
Rates of pneumonia were also similar in the two groups (21% vs. 20%; P = .39), reported Dr. Nickerson, a general surgeon in Rochester.
No patient or provider observed a significant chest wall deformity in either group during follow-up and no reoperations were required for incomplete repair in the PFS.
"Despite advances in surgical technique, not all flail segment rib fractures are accessible for repair via standard thoracotomy," the authors wrote. "Our data suggest that PFS is acceptable and that extending or creating additional incisions for exposure for CFS is unwarranted."
Additional research assessing late complications, performance/functional outcomes, and perceived benefits of the operation is needed to determine whether partial repair is truly comparable with full stabilization, coauthor Dr. Brian Kim, associate medical director Mayo Clinic Trauma Center, Rochester, said in an interview.
In their experience, 6 months’ postoperative follow-up has been sufficient with respect to bony healing, convalescence from pain, and monitoring of chest wall integrity.
He noted that some surgeons may be dissuaded from pursuing or recommending operative stabilization of a flail segment with fractures deemed outside the boundaries of repair, particularly if the injury is in the elderly or obese patient.
"The results of our study lend support to the practice of repairing the most anatomically disrupted and/or symptomatic fractures within the flail segment," he said. "Age and/or BMI [body mass index] are not absolute contraindications for flail segment stabilization in our practice."
Poster session moderator Dr. Alexander Eastman of University of Texas Southwestern Medical Center in Dallas, said the low patient numbers and short follow-up in the study make it difficult to draw big conclusions, other than the need for further research.
His group uses surgical fixation of flail chest for patients with instability and pain management for those patients who are difficult to wean from the ventilator.
"I think many surgeons still are skeptical of the data supporting fixation of flail chest," he noted in an interview.
Dr. Nickerson and his coauthors reported having no financial disclosures.
AT THE EAST SCIENTIFIC ASSEMBLY
Major finding: Only predicted total lung capacity at 3 months (90% vs. 72%; P = .02) and 6 months (94% vs. 75%; P = .038) significantly differed between CFS and PFS patients.
Data source: A retrospective study of 43 patients undergoing surgical rib fracture stabilization for flail chest.
Disclosures: Dr. Nickerson and his coauthors reported having no financial disclosures.
FDA panel backs approval of novel antiplatelet drug vorapaxar
SILVER SPRING, MD. – Vorapaxar has won the backing of a Food and Drug Administration advisory panel for secondary prevention of atherothrombotic events.
At a meeting on Jan. 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization; it is contraindicated in patients with a history of stroke or transient ischemic attack and a history of intracranial hemorrhage (ICH).
Vorapaxar is an antagonist of protease-activated receptor-1, which inhibits the action of thrombin on the platelet, according to the company. If approved, it will be the first marketed drug of that class.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The proposed indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo. The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient. "Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
If approved, Merck plans to market vorapaxar as Zontivity. The FDA usually follows the recommendations of its advisory panels. Members of advisory panels have been cleared of potential conflicts of interest, although occasionally they are given a waiver, but not at this meeting.
The FDA is due to make a decision on approval during the first half of this year, Merck said.
SILVER SPRING, MD. – Vorapaxar has won the backing of a Food and Drug Administration advisory panel for secondary prevention of atherothrombotic events.
At a meeting on Jan. 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization; it is contraindicated in patients with a history of stroke or transient ischemic attack and a history of intracranial hemorrhage (ICH).
Vorapaxar is an antagonist of protease-activated receptor-1, which inhibits the action of thrombin on the platelet, according to the company. If approved, it will be the first marketed drug of that class.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The proposed indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo. The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient. "Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
If approved, Merck plans to market vorapaxar as Zontivity. The FDA usually follows the recommendations of its advisory panels. Members of advisory panels have been cleared of potential conflicts of interest, although occasionally they are given a waiver, but not at this meeting.
The FDA is due to make a decision on approval during the first half of this year, Merck said.
SILVER SPRING, MD. – Vorapaxar has won the backing of a Food and Drug Administration advisory panel for secondary prevention of atherothrombotic events.
At a meeting on Jan. 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization; it is contraindicated in patients with a history of stroke or transient ischemic attack and a history of intracranial hemorrhage (ICH).
Vorapaxar is an antagonist of protease-activated receptor-1, which inhibits the action of thrombin on the platelet, according to the company. If approved, it will be the first marketed drug of that class.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The proposed indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo. The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient. "Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
If approved, Merck plans to market vorapaxar as Zontivity. The FDA usually follows the recommendations of its advisory panels. Members of advisory panels have been cleared of potential conflicts of interest, although occasionally they are given a waiver, but not at this meeting.
The FDA is due to make a decision on approval during the first half of this year, Merck said.
FROM AN FDA PANEL MEETING
Platelet inhibition test helps predict surgical bleeding
SAN FRANCISCO – Preoperative light transmission aggregometry assessments of platelet aggregation may help identify which patients on dual antiplatelet therapy are at greater risk of sustained bleeding from noncardiac surgery, a prospective study of 147 consecutive patients suggests.
The light transmission aggregometry (LTA) assessments of blood drawn immediately before noncardiac surgery were significantly lower in the 32% of patients with sustained bleeding than in the other patients.
All patients were on dual antiplatelet therapy, 95% of them on maintenance therapy with aspirin plus clopidogrel. Timing of the surgery was at the discretion of the surgeons. Treating physicians were blinded to LTA results. The mean preoperative washout period for dual antiplatelet therapy was 1.5 days. Patients had vascular (76%), orthopedic (10%), abdominal (7%), or other (7%) surgery.
The ongoing study might help define a "bleeding cutoff" measure by LTA to better individualize the timing of surgery, Dr. Wolfgang Toller and his colleagues said in a poster session at the annual meeting of the American Society of Anesthesiologists.
In general, approximately 5% of patients in their first year of dual antiplatelet therapy undergo noncardiac surgery, which creates a conundrum for management. Discontinuing dual antiplatelet therapy before noncardiac surgery has been associated with a 20% risk of major adverse cardiac events, but there’s a 20%-40% risk of moderate to severe bleeding if dual antiplatelet therapy is continued during noncardiac surgery, said Dr. Toller of the Medical University of Graz, Austria.
The 147 patients in the study underwent vascular surgery (76%), orthopedic surgery (10%), abdominal surgery (7%), or other surgical procedures (7%). All had been on P2Y12 receptor inhibitors within 7 days before surgery.
Investigators used the Chronolog 700 Lumi-Aggregometer to assess platelet aggregation in preoperative blood, using 5 mcm of adenosine diphosphate as the specific inductor for platelet aggregation.
Overall, they found an average 40% maximum change in light transmission from baseline after adding the adenosine diphosphate to blood samples. In patients with increased bleeding, however, the mean maximum change in light transmission was approximately 30% (suggesting less platelet aggregation), compared with a more than 40% change in patients who bled less from the surgery.
Dr. Toller reported having no financial disclosures.
Surgery adds risk of perioperative bleeding
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
SAN FRANCISCO – Preoperative light transmission aggregometry assessments of platelet aggregation may help identify which patients on dual antiplatelet therapy are at greater risk of sustained bleeding from noncardiac surgery, a prospective study of 147 consecutive patients suggests.
The light transmission aggregometry (LTA) assessments of blood drawn immediately before noncardiac surgery were significantly lower in the 32% of patients with sustained bleeding than in the other patients.
All patients were on dual antiplatelet therapy, 95% of them on maintenance therapy with aspirin plus clopidogrel. Timing of the surgery was at the discretion of the surgeons. Treating physicians were blinded to LTA results. The mean preoperative washout period for dual antiplatelet therapy was 1.5 days. Patients had vascular (76%), orthopedic (10%), abdominal (7%), or other (7%) surgery.
The ongoing study might help define a "bleeding cutoff" measure by LTA to better individualize the timing of surgery, Dr. Wolfgang Toller and his colleagues said in a poster session at the annual meeting of the American Society of Anesthesiologists.
In general, approximately 5% of patients in their first year of dual antiplatelet therapy undergo noncardiac surgery, which creates a conundrum for management. Discontinuing dual antiplatelet therapy before noncardiac surgery has been associated with a 20% risk of major adverse cardiac events, but there’s a 20%-40% risk of moderate to severe bleeding if dual antiplatelet therapy is continued during noncardiac surgery, said Dr. Toller of the Medical University of Graz, Austria.
The 147 patients in the study underwent vascular surgery (76%), orthopedic surgery (10%), abdominal surgery (7%), or other surgical procedures (7%). All had been on P2Y12 receptor inhibitors within 7 days before surgery.
Investigators used the Chronolog 700 Lumi-Aggregometer to assess platelet aggregation in preoperative blood, using 5 mcm of adenosine diphosphate as the specific inductor for platelet aggregation.
Overall, they found an average 40% maximum change in light transmission from baseline after adding the adenosine diphosphate to blood samples. In patients with increased bleeding, however, the mean maximum change in light transmission was approximately 30% (suggesting less platelet aggregation), compared with a more than 40% change in patients who bled less from the surgery.
Dr. Toller reported having no financial disclosures.
Surgery adds risk of perioperative bleeding
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
SAN FRANCISCO – Preoperative light transmission aggregometry assessments of platelet aggregation may help identify which patients on dual antiplatelet therapy are at greater risk of sustained bleeding from noncardiac surgery, a prospective study of 147 consecutive patients suggests.
The light transmission aggregometry (LTA) assessments of blood drawn immediately before noncardiac surgery were significantly lower in the 32% of patients with sustained bleeding than in the other patients.
All patients were on dual antiplatelet therapy, 95% of them on maintenance therapy with aspirin plus clopidogrel. Timing of the surgery was at the discretion of the surgeons. Treating physicians were blinded to LTA results. The mean preoperative washout period for dual antiplatelet therapy was 1.5 days. Patients had vascular (76%), orthopedic (10%), abdominal (7%), or other (7%) surgery.
The ongoing study might help define a "bleeding cutoff" measure by LTA to better individualize the timing of surgery, Dr. Wolfgang Toller and his colleagues said in a poster session at the annual meeting of the American Society of Anesthesiologists.
In general, approximately 5% of patients in their first year of dual antiplatelet therapy undergo noncardiac surgery, which creates a conundrum for management. Discontinuing dual antiplatelet therapy before noncardiac surgery has been associated with a 20% risk of major adverse cardiac events, but there’s a 20%-40% risk of moderate to severe bleeding if dual antiplatelet therapy is continued during noncardiac surgery, said Dr. Toller of the Medical University of Graz, Austria.
The 147 patients in the study underwent vascular surgery (76%), orthopedic surgery (10%), abdominal surgery (7%), or other surgical procedures (7%). All had been on P2Y12 receptor inhibitors within 7 days before surgery.
Investigators used the Chronolog 700 Lumi-Aggregometer to assess platelet aggregation in preoperative blood, using 5 mcm of adenosine diphosphate as the specific inductor for platelet aggregation.
Overall, they found an average 40% maximum change in light transmission from baseline after adding the adenosine diphosphate to blood samples. In patients with increased bleeding, however, the mean maximum change in light transmission was approximately 30% (suggesting less platelet aggregation), compared with a more than 40% change in patients who bled less from the surgery.
Dr. Toller reported having no financial disclosures.
Surgery adds risk of perioperative bleeding
Dr. Lary Robinson, FCCP, comments: The vast majority of patients on dual platelet therapy (aspirin plus another agent such as clopidogrel) have had implantation of a drug-eluting coronary stent, who need to remain on this regimen for 1 year, after which it may be safely reduced to aspirin alone in most cases. In this first year, urgent surgery, such as that needed for cancer, as well as vascular surgery for ischemia or trauma, comes with the added risk of significant perioperative bleeding.
Anesthesiologists Wolfgang Toller and associates describe using the in vitro platelet function test, called light transmission aggregometry, in 147 patients. When used just prior to surgery, the test was somewhat predictive of which patients were at elevated bleeding risk. They propose that this test may better define and individualize the timing of surgery. However, the differences in test results are small and recommendations are not yet definite. Nevertheless, refinements in testing may lead to more specific recommendations about which patients should have surgery postponed due to a much greater bleeding risk.
Major finding: The mean maximum change in light transmission on LTA assessment of preoperative blood was approximately 30% in patients with increased bleeding from surgery, significantly lower than the more than 40% change in those who bled less.
Data source: Prospective study of 147 patients on dual antiplatelet therapy who underwent noncardiac surgery at one institution.
Disclosures: Dr. Toller reported having no financial disclosures.
Radiation exposure: Unwanted baggage in the endo suite
CHICAGO – The growing number of endovascular interventions over the past 2 decades has changed the face of surgery, but also has brought with it increased radiation exposure for patients, surgeons, and medical personnel involved in patient care.
"It’s unwanted baggage. The downside of more endovascular procedures is more radiation exposure" said Dr. Raghu L. Motaganahalli, with Indiana University School of Medicine, in Indianapolis.
The most recent 2009 report from the National Council on Radiation Protection and Measurements says the average effective dose received by Americans from medical uses, excluding radiotherapy, is 3.0 millisieverts or essentially the same as that received from natural background radiation.

Still, the use of computed tomography imaging has doubled every 2 years since the early 1980s to nearly 70 million scans in 2007 and at least 2% of all cancers are estimated to caused by CT radiation scan exposure, he said.
While patient exposure garners the lion’s share of attention, Dr. Motaganahalli urged surgeons at the annual meeting of the Midwestern Vascular Surgical Society to play a more active role in reducing radiation exposure. Personal monitoring is the first step, with the nonprofit International Commission on Radiological Protection recommending the use of two dosimeters – one at the neck and the second under the lead shielding at the waist – for the greatest measurement accuracy.
Lead aprons should be worn by all medical staff within about six feet of the patient, while thyroid collars and lead glasses also should be part of protective gear to reduce the risk of cancers or cataracts.
Distance from the radiation source in the interventional suite is also critical, he said. A recent study demonstrated that radiation doses vary widely around the perimeter of the angiography table and change according to the imaging angle (J. Vasc. Surg. 2013 [doi:10.1016/j.jvs.2013.01.025]).
The highest radiation doses were seen on the emitter side of the table, and thus, special emphasis should be paid to moving staff away from the scatter source, that is the patient, when standing on this side of the table, Dr. Motaganahalli said.
Other efforts to reduce radiation scatter to staff include minimizing the use of fluoroscopy and digital acquisition ("cine") times. The dose rate during a cine run is 10-20 times that during fluoroscopy. Using the "last image hold" capabilities may reduce the need for additional fluoroscopic time.
Pulsed fluoroscopy is also a better option than continuous fluoroscopy because the short bursts of x-ray decrease the exposure time and radiation exposure to the patient, he said. If the pulse rate is increased to approximately 30 pulses per second, however, the dose rate is virtually equivalent to that of continuous fluoroscopy.
High-dose fluoroscopy should be reserved for procedures requiring very high levels of detail because it allows exposure rates of up to 20 roentgens (R) per minute, whereas the normal operating mode is limited to 10 R/min., the Food and Drug Administration limit, Dr. Motaganahalli said.
Similarly, the use of magnification modes should be limited to the extent practicable because higher magnification modes result in higher radiation doses to smaller areas of the skin.
The "good news" is that vascular surgeons are exposed to about 70% of the ICRP occupational exposure limits over a 1-year-period (J. Vasc. Surg. 2000;32:704-10), he said. A more recent study reports even lower effective eye and hand doses, even after adjustments for longer fluoroscopy time, possibly because of extra protective devices such as a table-side lead shield and mobile lead shield reducing scatter radiation (J. Vasc. Surg. 2007;46:455-9).
A number of protection devices have been created including portable shields, under-table shields, lead caps, and zero gravity lead suits, although audience members commented that the suit has proven too unwieldy for practical use.
Dr. Motaganahalli reported having no conflicts of interest.
CHICAGO – The growing number of endovascular interventions over the past 2 decades has changed the face of surgery, but also has brought with it increased radiation exposure for patients, surgeons, and medical personnel involved in patient care.
"It’s unwanted baggage. The downside of more endovascular procedures is more radiation exposure" said Dr. Raghu L. Motaganahalli, with Indiana University School of Medicine, in Indianapolis.
The most recent 2009 report from the National Council on Radiation Protection and Measurements says the average effective dose received by Americans from medical uses, excluding radiotherapy, is 3.0 millisieverts or essentially the same as that received from natural background radiation.
Still, the use of computed tomography imaging has doubled every 2 years since the early 1980s to nearly 70 million scans in 2007 and at least 2% of all cancers are estimated to caused by CT radiation scan exposure, he said.
While patient exposure garners the lion’s share of attention, Dr. Motaganahalli urged surgeons at the annual meeting of the Midwestern Vascular Surgical Society to play a more active role in reducing radiation exposure. Personal monitoring is the first step, with the nonprofit International Commission on Radiological Protection recommending the use of two dosimeters – one at the neck and the second under the lead shielding at the waist – for the greatest measurement accuracy.
Lead aprons should be worn by all medical staff within about six feet of the patient, while thyroid collars and lead glasses also should be part of protective gear to reduce the risk of cancers or cataracts.
Distance from the radiation source in the interventional suite is also critical, he said. A recent study demonstrated that radiation doses vary widely around the perimeter of the angiography table and change according to the imaging angle (J. Vasc. Surg. 2013 [doi:10.1016/j.jvs.2013.01.025]).
The highest radiation doses were seen on the emitter side of the table, and thus, special emphasis should be paid to moving staff away from the scatter source, that is the patient, when standing on this side of the table, Dr. Motaganahalli said.
Other efforts to reduce radiation scatter to staff include minimizing the use of fluoroscopy and digital acquisition ("cine") times. The dose rate during a cine run is 10-20 times that during fluoroscopy. Using the "last image hold" capabilities may reduce the need for additional fluoroscopic time.
Pulsed fluoroscopy is also a better option than continuous fluoroscopy because the short bursts of x-ray decrease the exposure time and radiation exposure to the patient, he said. If the pulse rate is increased to approximately 30 pulses per second, however, the dose rate is virtually equivalent to that of continuous fluoroscopy.
High-dose fluoroscopy should be reserved for procedures requiring very high levels of detail because it allows exposure rates of up to 20 roentgens (R) per minute, whereas the normal operating mode is limited to 10 R/min., the Food and Drug Administration limit, Dr. Motaganahalli said.
Similarly, the use of magnification modes should be limited to the extent practicable because higher magnification modes result in higher radiation doses to smaller areas of the skin.
The "good news" is that vascular surgeons are exposed to about 70% of the ICRP occupational exposure limits over a 1-year-period (J. Vasc. Surg. 2000;32:704-10), he said. A more recent study reports even lower effective eye and hand doses, even after adjustments for longer fluoroscopy time, possibly because of extra protective devices such as a table-side lead shield and mobile lead shield reducing scatter radiation (J. Vasc. Surg. 2007;46:455-9).
A number of protection devices have been created including portable shields, under-table shields, lead caps, and zero gravity lead suits, although audience members commented that the suit has proven too unwieldy for practical use.
Dr. Motaganahalli reported having no conflicts of interest.
CHICAGO – The growing number of endovascular interventions over the past 2 decades has changed the face of surgery, but also has brought with it increased radiation exposure for patients, surgeons, and medical personnel involved in patient care.
"It’s unwanted baggage. The downside of more endovascular procedures is more radiation exposure" said Dr. Raghu L. Motaganahalli, with Indiana University School of Medicine, in Indianapolis.
The most recent 2009 report from the National Council on Radiation Protection and Measurements says the average effective dose received by Americans from medical uses, excluding radiotherapy, is 3.0 millisieverts or essentially the same as that received from natural background radiation.
Still, the use of computed tomography imaging has doubled every 2 years since the early 1980s to nearly 70 million scans in 2007 and at least 2% of all cancers are estimated to caused by CT radiation scan exposure, he said.
While patient exposure garners the lion’s share of attention, Dr. Motaganahalli urged surgeons at the annual meeting of the Midwestern Vascular Surgical Society to play a more active role in reducing radiation exposure. Personal monitoring is the first step, with the nonprofit International Commission on Radiological Protection recommending the use of two dosimeters – one at the neck and the second under the lead shielding at the waist – for the greatest measurement accuracy.
Lead aprons should be worn by all medical staff within about six feet of the patient, while thyroid collars and lead glasses also should be part of protective gear to reduce the risk of cancers or cataracts.
Distance from the radiation source in the interventional suite is also critical, he said. A recent study demonstrated that radiation doses vary widely around the perimeter of the angiography table and change according to the imaging angle (J. Vasc. Surg. 2013 [doi:10.1016/j.jvs.2013.01.025]).
The highest radiation doses were seen on the emitter side of the table, and thus, special emphasis should be paid to moving staff away from the scatter source, that is the patient, when standing on this side of the table, Dr. Motaganahalli said.
Other efforts to reduce radiation scatter to staff include minimizing the use of fluoroscopy and digital acquisition ("cine") times. The dose rate during a cine run is 10-20 times that during fluoroscopy. Using the "last image hold" capabilities may reduce the need for additional fluoroscopic time.
Pulsed fluoroscopy is also a better option than continuous fluoroscopy because the short bursts of x-ray decrease the exposure time and radiation exposure to the patient, he said. If the pulse rate is increased to approximately 30 pulses per second, however, the dose rate is virtually equivalent to that of continuous fluoroscopy.
High-dose fluoroscopy should be reserved for procedures requiring very high levels of detail because it allows exposure rates of up to 20 roentgens (R) per minute, whereas the normal operating mode is limited to 10 R/min., the Food and Drug Administration limit, Dr. Motaganahalli said.
Similarly, the use of magnification modes should be limited to the extent practicable because higher magnification modes result in higher radiation doses to smaller areas of the skin.
The "good news" is that vascular surgeons are exposed to about 70% of the ICRP occupational exposure limits over a 1-year-period (J. Vasc. Surg. 2000;32:704-10), he said. A more recent study reports even lower effective eye and hand doses, even after adjustments for longer fluoroscopy time, possibly because of extra protective devices such as a table-side lead shield and mobile lead shield reducing scatter radiation (J. Vasc. Surg. 2007;46:455-9).
A number of protection devices have been created including portable shields, under-table shields, lead caps, and zero gravity lead suits, although audience members commented that the suit has proven too unwieldy for practical use.
Dr. Motaganahalli reported having no conflicts of interest.

Abrupt increase noted in LVAD thrombosis
An abrupt and unexpected increase in pump thrombosis has occurred since March 2011 in patients with advanced heart failure who received the HeartMate II left ventricular assist device, according to a report published online Nov. 27 in the New England Journal of Medicine.
Those who develop LVAD thrombosis are at substantial risk of death and morbidity unless the device is replaced or cardiac transplantation is performed, said Dr. Randall C. Starling of the Cleveland Clinic and his associates.
The reason for the sudden rise in pump thrombosis is not yet known, but it may be related to excess "deposition of material (fibrin and denatured protein) in proximity to the inflow bearing" of the device, which has been observed by clinicians in at least three cardiac centers.
"Our data suggest that the HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation," Dr. Starling and his colleagues noted.
The HeartMate II, made by Thoratec, is a small axial-flow LVAD that "rapidly became integral in the treatment of patients with advanced heart failure" after it was introduced. Early trials and postmarketing approval studies reported a 2%-4% incidence of thrombosis. But a quality review at the Cleveland Clinic showed a sudden, marked increase in that rate beginning in March 2011.
Researchers at the Cleveland Clinic then pooled their data on the HeartMate II (from October 2004 to February 2013) with data on the device from two other high-volume cardiology centers: Washington University Barnes-Jewish Hospital, St. Louis (January 2004 to April 2013) and Duke University Medical Center, Durham, N.C. (May 2005 to May 2013). They now report on their findings from 895 HeartMate II LVADs implanted in 837 patients during that time.
"The HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation..."
The mean patient age was 55 years, and 21% of patients were women.
The primary endpoint of the study was confirmed LVAD thrombosis, defined as a finding at pump replacement, urgent transplantation, or autopsy of a thrombus on the blood-contacting surface of the device, its inflow cannula, or its outflow conduit. A total of 72 such thromboses were found in 66 patients; in addition, 36 cases of suspected thromboses occurred.
The rate of confirmed pump thrombosis was initially stable at roughly 2.2% for several years, then rose steeply after March 2011 to 8.4%. This pattern was seen at all three medical centers and across all the cardiac surgeons at those centers.
The median time interval from implantation to pump thrombosis had been 18.6 months before March 2011, but that rate plummeted to 2.7 months during and after March 2011, Dr. Starling and his associates reported (N. Engl. J. Med. 2013 Nov. 27 [doi: 10.1056/NEJMoa1313385]).
The investigators also reviewed serial measurements of lactate dehydrogenase (LDH) that had been taken in recipients of 568 of the devices. Elevated LDH indicates hemolysis that may herald impending thrombosis.
"The graph of the occurrence of LDH levels above 1000 IU per liter ... was nearly superimposable on the graph of the occurrence of confirmed pump thrombosis. The LDH data showed a parallel tendency for elevations to occur early after implantation beginning in approximately March 2011," they said.
The LDH level typically rose from an average of 540 IU/L to an average of 1,490 IU/L during the weeks leading up to LVAD thrombosis at all three medical centers. This confirms that LDH level is a useful clinical biomarker of hemolysis associated with LVAD thrombosis, the researchers said.
Of the 72 pump thromboses, 11 were managed by heart transplantation and 21 by pump replacement. Of the remaining 40 thromboses, 2 were managed by LVAD removal because left ventricular function had improved, and 38 were managed medically, with augmentation of anticoagulation therapy and thrombolytic agents. This stabilized the clinical course in some patients, but others elected withdrawal of care, citing "futility."
Mortality was 48% among the patients who could not be managed by heart transplantation or pump replacement, and this elevated rate was consistent across the three medical centers.
The reasons for this abrupt, dramatic rise in thromboses associated with the HeartMate II are not known. There were no changes in LVAD implantation techniques at any of the three medical centers during the course of this study, "and we could find no association between pump thrombosis and the surgeon performing the implantation." The design of the HeartMate II was changed during this interval – "modification of the outflow graft and bend relief, inflow conduit, and software" was introduced – but these changes have not been linked to pump thrombosis as yet.
Dr. Starling and his associates did note the deposition of fibrin and denatured protein around the inflow bearing, which could narrow the inflow pathway, "increasing shear stress on the red cells and, if the deposition is large enough, decreasing the ability of the pump to unload the left ventricle." But it remains to be seen whether this or other factors played a role.
"Further investigation of predisposing patient and device factors and preventive and therapeutic strategies are urgently needed to resolve this important safety issue," the investigators said.
They added that a fourth cardiology center, the University of Pennsylvania, Philadelphia, found a similar pattern in a preliminary analysis that was reported as the article went to press. In that report on 150 HeartMate II LVADs implanted in 148 patients between November 2005 and September 2013, there were 15 thrombotic events. "Similar to the findings in our study, the event rate has increased abruptly and unexpectedly, with a rate of confirmed thrombosis that continues to rise," Dr. Starling and his associates said.
Dr. Starling reported ties to Thoratec and HeartWare, and his associates reported ties to Thoratec, HeartWare, Abiomed, and Syncardia.
An abrupt and unexpected increase in pump thrombosis has occurred since March 2011 in patients with advanced heart failure who received the HeartMate II left ventricular assist device, according to a report published online Nov. 27 in the New England Journal of Medicine.
Those who develop LVAD thrombosis are at substantial risk of death and morbidity unless the device is replaced or cardiac transplantation is performed, said Dr. Randall C. Starling of the Cleveland Clinic and his associates.
The reason for the sudden rise in pump thrombosis is not yet known, but it may be related to excess "deposition of material (fibrin and denatured protein) in proximity to the inflow bearing" of the device, which has been observed by clinicians in at least three cardiac centers.
"Our data suggest that the HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation," Dr. Starling and his colleagues noted.
The HeartMate II, made by Thoratec, is a small axial-flow LVAD that "rapidly became integral in the treatment of patients with advanced heart failure" after it was introduced. Early trials and postmarketing approval studies reported a 2%-4% incidence of thrombosis. But a quality review at the Cleveland Clinic showed a sudden, marked increase in that rate beginning in March 2011.
Researchers at the Cleveland Clinic then pooled their data on the HeartMate II (from October 2004 to February 2013) with data on the device from two other high-volume cardiology centers: Washington University Barnes-Jewish Hospital, St. Louis (January 2004 to April 2013) and Duke University Medical Center, Durham, N.C. (May 2005 to May 2013). They now report on their findings from 895 HeartMate II LVADs implanted in 837 patients during that time.
"The HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation..."
The mean patient age was 55 years, and 21% of patients were women.
The primary endpoint of the study was confirmed LVAD thrombosis, defined as a finding at pump replacement, urgent transplantation, or autopsy of a thrombus on the blood-contacting surface of the device, its inflow cannula, or its outflow conduit. A total of 72 such thromboses were found in 66 patients; in addition, 36 cases of suspected thromboses occurred.
The rate of confirmed pump thrombosis was initially stable at roughly 2.2% for several years, then rose steeply after March 2011 to 8.4%. This pattern was seen at all three medical centers and across all the cardiac surgeons at those centers.
The median time interval from implantation to pump thrombosis had been 18.6 months before March 2011, but that rate plummeted to 2.7 months during and after March 2011, Dr. Starling and his associates reported (N. Engl. J. Med. 2013 Nov. 27 [doi: 10.1056/NEJMoa1313385]).
The investigators also reviewed serial measurements of lactate dehydrogenase (LDH) that had been taken in recipients of 568 of the devices. Elevated LDH indicates hemolysis that may herald impending thrombosis.
"The graph of the occurrence of LDH levels above 1000 IU per liter ... was nearly superimposable on the graph of the occurrence of confirmed pump thrombosis. The LDH data showed a parallel tendency for elevations to occur early after implantation beginning in approximately March 2011," they said.
The LDH level typically rose from an average of 540 IU/L to an average of 1,490 IU/L during the weeks leading up to LVAD thrombosis at all three medical centers. This confirms that LDH level is a useful clinical biomarker of hemolysis associated with LVAD thrombosis, the researchers said.
Of the 72 pump thromboses, 11 were managed by heart transplantation and 21 by pump replacement. Of the remaining 40 thromboses, 2 were managed by LVAD removal because left ventricular function had improved, and 38 were managed medically, with augmentation of anticoagulation therapy and thrombolytic agents. This stabilized the clinical course in some patients, but others elected withdrawal of care, citing "futility."
Mortality was 48% among the patients who could not be managed by heart transplantation or pump replacement, and this elevated rate was consistent across the three medical centers.
The reasons for this abrupt, dramatic rise in thromboses associated with the HeartMate II are not known. There were no changes in LVAD implantation techniques at any of the three medical centers during the course of this study, "and we could find no association between pump thrombosis and the surgeon performing the implantation." The design of the HeartMate II was changed during this interval – "modification of the outflow graft and bend relief, inflow conduit, and software" was introduced – but these changes have not been linked to pump thrombosis as yet.
Dr. Starling and his associates did note the deposition of fibrin and denatured protein around the inflow bearing, which could narrow the inflow pathway, "increasing shear stress on the red cells and, if the deposition is large enough, decreasing the ability of the pump to unload the left ventricle." But it remains to be seen whether this or other factors played a role.
"Further investigation of predisposing patient and device factors and preventive and therapeutic strategies are urgently needed to resolve this important safety issue," the investigators said.
They added that a fourth cardiology center, the University of Pennsylvania, Philadelphia, found a similar pattern in a preliminary analysis that was reported as the article went to press. In that report on 150 HeartMate II LVADs implanted in 148 patients between November 2005 and September 2013, there were 15 thrombotic events. "Similar to the findings in our study, the event rate has increased abruptly and unexpectedly, with a rate of confirmed thrombosis that continues to rise," Dr. Starling and his associates said.
Dr. Starling reported ties to Thoratec and HeartWare, and his associates reported ties to Thoratec, HeartWare, Abiomed, and Syncardia.
An abrupt and unexpected increase in pump thrombosis has occurred since March 2011 in patients with advanced heart failure who received the HeartMate II left ventricular assist device, according to a report published online Nov. 27 in the New England Journal of Medicine.
Those who develop LVAD thrombosis are at substantial risk of death and morbidity unless the device is replaced or cardiac transplantation is performed, said Dr. Randall C. Starling of the Cleveland Clinic and his associates.
The reason for the sudden rise in pump thrombosis is not yet known, but it may be related to excess "deposition of material (fibrin and denatured protein) in proximity to the inflow bearing" of the device, which has been observed by clinicians in at least three cardiac centers.
"Our data suggest that the HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation," Dr. Starling and his colleagues noted.
The HeartMate II, made by Thoratec, is a small axial-flow LVAD that "rapidly became integral in the treatment of patients with advanced heart failure" after it was introduced. Early trials and postmarketing approval studies reported a 2%-4% incidence of thrombosis. But a quality review at the Cleveland Clinic showed a sudden, marked increase in that rate beginning in March 2011.
Researchers at the Cleveland Clinic then pooled their data on the HeartMate II (from October 2004 to February 2013) with data on the device from two other high-volume cardiology centers: Washington University Barnes-Jewish Hospital, St. Louis (January 2004 to April 2013) and Duke University Medical Center, Durham, N.C. (May 2005 to May 2013). They now report on their findings from 895 HeartMate II LVADs implanted in 837 patients during that time.
"The HeartMate II may have a more narrow tolerance with respect to thrombus formation than originally understood, and may be vulnerable to the timing and intensity of anticoagulation..."
The mean patient age was 55 years, and 21% of patients were women.
The primary endpoint of the study was confirmed LVAD thrombosis, defined as a finding at pump replacement, urgent transplantation, or autopsy of a thrombus on the blood-contacting surface of the device, its inflow cannula, or its outflow conduit. A total of 72 such thromboses were found in 66 patients; in addition, 36 cases of suspected thromboses occurred.
The rate of confirmed pump thrombosis was initially stable at roughly 2.2% for several years, then rose steeply after March 2011 to 8.4%. This pattern was seen at all three medical centers and across all the cardiac surgeons at those centers.
The median time interval from implantation to pump thrombosis had been 18.6 months before March 2011, but that rate plummeted to 2.7 months during and after March 2011, Dr. Starling and his associates reported (N. Engl. J. Med. 2013 Nov. 27 [doi: 10.1056/NEJMoa1313385]).
The investigators also reviewed serial measurements of lactate dehydrogenase (LDH) that had been taken in recipients of 568 of the devices. Elevated LDH indicates hemolysis that may herald impending thrombosis.
"The graph of the occurrence of LDH levels above 1000 IU per liter ... was nearly superimposable on the graph of the occurrence of confirmed pump thrombosis. The LDH data showed a parallel tendency for elevations to occur early after implantation beginning in approximately March 2011," they said.
The LDH level typically rose from an average of 540 IU/L to an average of 1,490 IU/L during the weeks leading up to LVAD thrombosis at all three medical centers. This confirms that LDH level is a useful clinical biomarker of hemolysis associated with LVAD thrombosis, the researchers said.
Of the 72 pump thromboses, 11 were managed by heart transplantation and 21 by pump replacement. Of the remaining 40 thromboses, 2 were managed by LVAD removal because left ventricular function had improved, and 38 were managed medically, with augmentation of anticoagulation therapy and thrombolytic agents. This stabilized the clinical course in some patients, but others elected withdrawal of care, citing "futility."
Mortality was 48% among the patients who could not be managed by heart transplantation or pump replacement, and this elevated rate was consistent across the three medical centers.
The reasons for this abrupt, dramatic rise in thromboses associated with the HeartMate II are not known. There were no changes in LVAD implantation techniques at any of the three medical centers during the course of this study, "and we could find no association between pump thrombosis and the surgeon performing the implantation." The design of the HeartMate II was changed during this interval – "modification of the outflow graft and bend relief, inflow conduit, and software" was introduced – but these changes have not been linked to pump thrombosis as yet.
Dr. Starling and his associates did note the deposition of fibrin and denatured protein around the inflow bearing, which could narrow the inflow pathway, "increasing shear stress on the red cells and, if the deposition is large enough, decreasing the ability of the pump to unload the left ventricle." But it remains to be seen whether this or other factors played a role.
"Further investigation of predisposing patient and device factors and preventive and therapeutic strategies are urgently needed to resolve this important safety issue," the investigators said.
They added that a fourth cardiology center, the University of Pennsylvania, Philadelphia, found a similar pattern in a preliminary analysis that was reported as the article went to press. In that report on 150 HeartMate II LVADs implanted in 148 patients between November 2005 and September 2013, there were 15 thrombotic events. "Similar to the findings in our study, the event rate has increased abruptly and unexpectedly, with a rate of confirmed thrombosis that continues to rise," Dr. Starling and his associates said.
Dr. Starling reported ties to Thoratec and HeartWare, and his associates reported ties to Thoratec, HeartWare, Abiomed, and Syncardia.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major finding: The rate of pump thrombosis was stable at approximately 2.2% for several years, then rose steeply after March 2011 to 8.4%; the median interval from implantation to pump thrombosis, which had been stable at 18.6 months for several years, plummeted to 2.7 months after March 2011.
Data source: A retrospective study involving 837 recipients of 895 HeartMate II LVADs at three U.S. medical centers during 2004-2013, in which 72 LVAD thromboses were confirmed in 66 patients and an additional 36 thromboses were suspected.
Disclosures: Dr. Starling reported ties to Thoratec and HeartWare, and his associates reported ties to Thoratec, HeartWare, Abiomed, and Syncardia.
Electrocautery incision of lymph nodes improved biopsy yield
Endobronchial ultrasound–guided biopsies made after an electrocautery incision to the lymph node improved biopsy yields from 39% to 71% in 38 nodes, according to a small study presented at the annual meeting of the American College of Chest Physicians meeting.
"Because it is not always possible to pass biopsy forceps through defects in the lymph node – the literature indicates a failure rate of between 10% and 29% – we developed a novel technique," said presenter Dr. Kyle Bramley of Yale University, New Haven, Conn.
The technique employs EBUS, and involves passing an electrocautery knife activated at 40 W through the working channel of the scope in order to make an incision in the bronchial wall and enlarge the defect in the lymph node. This facilitates passage of the forceps into the node so that a larger biopsy sample can be obtained.
To test their technique, Dr. Bramley and his colleagues designed a prospective observational cohort study at a single tertiary academic medical center. Twenty patients (mean age, 68 years), including 11 women, who were undergoing EBUS were enrolled. An associated lung mass was present in 14 (70%) of the participants; 6 (30%) had isolated lymphadenopathy. One patient had prior lymphoma, and two others had prior lung cancer.
The researchers evaluated 68 nodes in all; 19 patients had nodes greater than 9 mm. Cautery was only used when initial attempts failed to biopsy nodes 9 mm or larger using EBUS-guided miniforceps of 1.2 mm.
The average node size biopsied using EBUS-transbronchial needle aspiration (EBUS-TBNA) was 5.7 mm. The average forceps-biopsied node was 15.8 mm.
In all, 23 nodes were biopsied successfully on the first pass using EBUS-TBNA only. The biopsies yielded diagnostic material such as lymphocytes, malignancy, or granulomas in 15 of these nodes.
Of the 15 nodes that required cautery, 12 yielded diagnostic material, and 3 had no diagnostic material.
The overall yield increased from 39% (15 out of 38) without cautery to 71% (27 out of 38) when cautery was used.
Notably, four patients had clinically relevant discrepancies between their cytologies and histopathologies. "In all four, TBNA provided a definitive diagnosis," said Dr. Bramley. "The forceps provided fibroconnective tissue or necrotic debris."
These results did not negate the efficacy of the cautery technique, according to Dr. Bramley. "We think we had a forceps issue ... the 1.2 mm are flexible, but they were unable to push all the way through a tough lymph node capsule."
Dr. Bramley also said that other factors, including the operator learning curve, the smaller size of the nodes the investigators attempted to biopsy, and the "nonideal" population they were studying, contributed to these results.
He and his colleagues have since adjusted the procedure to make cauterization routine and to include a 1.9-mm transbronchial biopsy forceps needle, "which, incidentally, is a lot less expensive than the larger forceps we’d been using," he said.
Although more study is needed, Dr. Bramley said he and his team believed that this technique would be appropriate for future use in isolated mediastinal lymphadenopathy, especially with a low suspicion of non–small cell lung carcinoma; evaluation of lymphoma; and clinical trials requiring core biopsy.
Dr. Bramley had no relevant disclosures.
Dr. Frank Podbielski, FCCP, comments: The authors have again proven that a larger pathology specimen obtained at the time of biopsy significantly improves diagnostic accuracy, especially in the setting of mediastinal nodes that are difficult to access and thus require an electrocautery incision through the airway in concert with EBUS guidance.
Dr. Francis J. Podbielski leads the Lung Cancer Program at Jordan Hospital in Plymouth, Mass.
Dr. Frank Podbielski, FCCP, comments: The authors have again proven that a larger pathology specimen obtained at the time of biopsy significantly improves diagnostic accuracy, especially in the setting of mediastinal nodes that are difficult to access and thus require an electrocautery incision through the airway in concert with EBUS guidance.
Dr. Francis J. Podbielski leads the Lung Cancer Program at Jordan Hospital in Plymouth, Mass.
Dr. Frank Podbielski, FCCP, comments: The authors have again proven that a larger pathology specimen obtained at the time of biopsy significantly improves diagnostic accuracy, especially in the setting of mediastinal nodes that are difficult to access and thus require an electrocautery incision through the airway in concert with EBUS guidance.
Dr. Francis J. Podbielski leads the Lung Cancer Program at Jordan Hospital in Plymouth, Mass.
Endobronchial ultrasound–guided biopsies made after an electrocautery incision to the lymph node improved biopsy yields from 39% to 71% in 38 nodes, according to a small study presented at the annual meeting of the American College of Chest Physicians meeting.
"Because it is not always possible to pass biopsy forceps through defects in the lymph node – the literature indicates a failure rate of between 10% and 29% – we developed a novel technique," said presenter Dr. Kyle Bramley of Yale University, New Haven, Conn.
The technique employs EBUS, and involves passing an electrocautery knife activated at 40 W through the working channel of the scope in order to make an incision in the bronchial wall and enlarge the defect in the lymph node. This facilitates passage of the forceps into the node so that a larger biopsy sample can be obtained.
To test their technique, Dr. Bramley and his colleagues designed a prospective observational cohort study at a single tertiary academic medical center. Twenty patients (mean age, 68 years), including 11 women, who were undergoing EBUS were enrolled. An associated lung mass was present in 14 (70%) of the participants; 6 (30%) had isolated lymphadenopathy. One patient had prior lymphoma, and two others had prior lung cancer.
The researchers evaluated 68 nodes in all; 19 patients had nodes greater than 9 mm. Cautery was only used when initial attempts failed to biopsy nodes 9 mm or larger using EBUS-guided miniforceps of 1.2 mm.
The average node size biopsied using EBUS-transbronchial needle aspiration (EBUS-TBNA) was 5.7 mm. The average forceps-biopsied node was 15.8 mm.
In all, 23 nodes were biopsied successfully on the first pass using EBUS-TBNA only. The biopsies yielded diagnostic material such as lymphocytes, malignancy, or granulomas in 15 of these nodes.
Of the 15 nodes that required cautery, 12 yielded diagnostic material, and 3 had no diagnostic material.
The overall yield increased from 39% (15 out of 38) without cautery to 71% (27 out of 38) when cautery was used.
Notably, four patients had clinically relevant discrepancies between their cytologies and histopathologies. "In all four, TBNA provided a definitive diagnosis," said Dr. Bramley. "The forceps provided fibroconnective tissue or necrotic debris."
These results did not negate the efficacy of the cautery technique, according to Dr. Bramley. "We think we had a forceps issue ... the 1.2 mm are flexible, but they were unable to push all the way through a tough lymph node capsule."
Dr. Bramley also said that other factors, including the operator learning curve, the smaller size of the nodes the investigators attempted to biopsy, and the "nonideal" population they were studying, contributed to these results.
He and his colleagues have since adjusted the procedure to make cauterization routine and to include a 1.9-mm transbronchial biopsy forceps needle, "which, incidentally, is a lot less expensive than the larger forceps we’d been using," he said.
Although more study is needed, Dr. Bramley said he and his team believed that this technique would be appropriate for future use in isolated mediastinal lymphadenopathy, especially with a low suspicion of non–small cell lung carcinoma; evaluation of lymphoma; and clinical trials requiring core biopsy.
Dr. Bramley had no relevant disclosures.
Endobronchial ultrasound–guided biopsies made after an electrocautery incision to the lymph node improved biopsy yields from 39% to 71% in 38 nodes, according to a small study presented at the annual meeting of the American College of Chest Physicians meeting.
"Because it is not always possible to pass biopsy forceps through defects in the lymph node – the literature indicates a failure rate of between 10% and 29% – we developed a novel technique," said presenter Dr. Kyle Bramley of Yale University, New Haven, Conn.
The technique employs EBUS, and involves passing an electrocautery knife activated at 40 W through the working channel of the scope in order to make an incision in the bronchial wall and enlarge the defect in the lymph node. This facilitates passage of the forceps into the node so that a larger biopsy sample can be obtained.
To test their technique, Dr. Bramley and his colleagues designed a prospective observational cohort study at a single tertiary academic medical center. Twenty patients (mean age, 68 years), including 11 women, who were undergoing EBUS were enrolled. An associated lung mass was present in 14 (70%) of the participants; 6 (30%) had isolated lymphadenopathy. One patient had prior lymphoma, and two others had prior lung cancer.
The researchers evaluated 68 nodes in all; 19 patients had nodes greater than 9 mm. Cautery was only used when initial attempts failed to biopsy nodes 9 mm or larger using EBUS-guided miniforceps of 1.2 mm.
The average node size biopsied using EBUS-transbronchial needle aspiration (EBUS-TBNA) was 5.7 mm. The average forceps-biopsied node was 15.8 mm.
In all, 23 nodes were biopsied successfully on the first pass using EBUS-TBNA only. The biopsies yielded diagnostic material such as lymphocytes, malignancy, or granulomas in 15 of these nodes.
Of the 15 nodes that required cautery, 12 yielded diagnostic material, and 3 had no diagnostic material.
The overall yield increased from 39% (15 out of 38) without cautery to 71% (27 out of 38) when cautery was used.
Notably, four patients had clinically relevant discrepancies between their cytologies and histopathologies. "In all four, TBNA provided a definitive diagnosis," said Dr. Bramley. "The forceps provided fibroconnective tissue or necrotic debris."
These results did not negate the efficacy of the cautery technique, according to Dr. Bramley. "We think we had a forceps issue ... the 1.2 mm are flexible, but they were unable to push all the way through a tough lymph node capsule."
Dr. Bramley also said that other factors, including the operator learning curve, the smaller size of the nodes the investigators attempted to biopsy, and the "nonideal" population they were studying, contributed to these results.
He and his colleagues have since adjusted the procedure to make cauterization routine and to include a 1.9-mm transbronchial biopsy forceps needle, "which, incidentally, is a lot less expensive than the larger forceps we’d been using," he said.
Although more study is needed, Dr. Bramley said he and his team believed that this technique would be appropriate for future use in isolated mediastinal lymphadenopathy, especially with a low suspicion of non–small cell lung carcinoma; evaluation of lymphoma; and clinical trials requiring core biopsy.
Dr. Bramley had no relevant disclosures.
Major finding: EBUS-guided lymph node biopsies made after electrocautery incision improved biopsy yields from 39% to 71% in 38 lymph nodes.
Data source: Prospective observational cohort study of 20 patients at a single tertiary academic medical center.
Disclosures: Dr. Bramley had no relevant disclosures.
First U.S. TAVR experience fares well post-market
Postmarket outcomes with the first commercially available U.S. transcatheter aortic valve replacement device are comparable with randomized trial and international registry experience, national registry results suggest.
The Edwards LifeSciences Sapien XT valve was successfully implanted in 92% of 7,710 TAVR patients, two-thirds with a transfemoral approach.
The primary outcomes of in-hospital all-cause mortality and stroke were 5.5% and 2%, respectively, Dr. Michael J. Mack reported at the American Heart Association scientific sessions.
Conversion to open-heart surgery was needed in only 1% of patients, but associated with a 49% incidence of in-hospital mortality.
Major bleeding and vascular complications were relatively low, at 3.5% and 6.4%, respectively, despite the overall lack of experience and use of larger sheath delivery systems. Acute renal insufficiency occurred in 5.5% of patients.
The 30-day death and stroke rates were 7.6% and 2.8% among 3,113 patients with sufficient follow-up. Most deaths were due to noncardiovascular causes, he noted.
This is the first public report from the national Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy (TVT) Registry, and comes amid calls for increased postmarket surveillance of high-risk medical devices.
The results also help address whether outcomes after a controlled commercial rollout of the first-generation valve hold up to those from clinical trials and the global TAVR experience, now based on second- and third-generation devices, according to Dr. Mack, past STS president and director of cardiovascular research and medicine at the Heart Hospital Baylor Plano in Plano, Texas.
The Food and Drug Administration initially approved the Edwards valve for severe, symptomatic aortic stenosis in inoperable patients in 2011 and expanded its label in September 2012 to include operable, high-risk patients using either a transfemoral or transapical approach.
The TVT analysis comprised 1,559 inoperable and 6,151 high-risk, operable patients who underwent TAVR at 224 participating registry hospitals from November 2011 to May 2013. Their median STS predicted risk of operative mortality (PROM) was 7%, but varied widely from 1.2% to 17.4%. Their median age was 84 years.
The risk profile of patients was generally lower in the registry than in randomized trials and European reports, and may have contributed to the good outcomes, Dr. Mack acknowledged. The mean PROM was 11.2% among inoperable patients and 11.8% among high-risk, operable patients in the pivotal PARTNER trial, which was used for the device’s approval.
Although incomplete data may have been entered into the risk calculator, the other plausible explanation is "risk creep," or that patients with lower surgical risk are being treated with the less-invasive procedure, leading to better outcomes, he noted.
Other possible contributors to the registry outcomes include a shorter U.S. learning curve due to international collaboration, inclusion of at least 35 highly experienced PARTNER study centers in the registry, and mandated training by the manufacturer including a company employed clinical specialist during all procedures.
Still, the results stack up well. Germany, which has led the TAVR revolution, had an in-hospital mortality of 5.1% with a transfemoral approach and 7.7% with a transapical approach in 2011 in the German Aortic Valve Registry.
In the PARTNER trial, 30-day mortality was 5% among inoperable patients, 3.7% among high-risk, operable patients by a transfemoral approach, and 8.7% among high-risk, operable patients by a transapical approach.
This compares with 30-day mortality rates of 6.1%, 4.6%, and 9.8%, respectively, in the TVT registry, Dr. Mack reported.
The TVT registry captured an estimated 88% of TAVR procedures performed between May 2012, when the Centers for Medicare and Medicaid Services issued its national coverage determination, and the end of the study. Notably, 30-day outcomes were reported from only 114 hospitals or 51% of the 244 hospitals in the registry. The results were published simultaneously with Dr. Mack’s presentation (JAMA 2013;310:2069-77. doi:10.1001/jama/2013.282043).
More complete long-term outcomes at 30 days and 1 year are needed to have a more comprehensive assessment of TAVR outcomes in the United States, Dr. Mack cautioned. Patient health status and quality of life outcomes will also be addressed in subsequent reports.
So far, it’s known that most (63%) patients were discharged home, 6% experienced new-onset atrial fibrillation, and 6.6% needed a new permanent pacemaker or implantable cardioverter-defibrillator.
The study was funded by the American College of Cardiology Foundation and the Society of Thoracic Surgeons. Dr. Mack reported no other disclosures; a co-author reported travel support from the STS and a second reported consulting for Janssen, Boehringer Ingelheim, and Sanofi.
Postmarket outcomes with the first commercially available U.S. transcatheter aortic valve replacement device are comparable with randomized trial and international registry experience, national registry results suggest.
The Edwards LifeSciences Sapien XT valve was successfully implanted in 92% of 7,710 TAVR patients, two-thirds with a transfemoral approach.
The primary outcomes of in-hospital all-cause mortality and stroke were 5.5% and 2%, respectively, Dr. Michael J. Mack reported at the American Heart Association scientific sessions.
Conversion to open-heart surgery was needed in only 1% of patients, but associated with a 49% incidence of in-hospital mortality.
Major bleeding and vascular complications were relatively low, at 3.5% and 6.4%, respectively, despite the overall lack of experience and use of larger sheath delivery systems. Acute renal insufficiency occurred in 5.5% of patients.
The 30-day death and stroke rates were 7.6% and 2.8% among 3,113 patients with sufficient follow-up. Most deaths were due to noncardiovascular causes, he noted.
This is the first public report from the national Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy (TVT) Registry, and comes amid calls for increased postmarket surveillance of high-risk medical devices.
The results also help address whether outcomes after a controlled commercial rollout of the first-generation valve hold up to those from clinical trials and the global TAVR experience, now based on second- and third-generation devices, according to Dr. Mack, past STS president and director of cardiovascular research and medicine at the Heart Hospital Baylor Plano in Plano, Texas.
The Food and Drug Administration initially approved the Edwards valve for severe, symptomatic aortic stenosis in inoperable patients in 2011 and expanded its label in September 2012 to include operable, high-risk patients using either a transfemoral or transapical approach.
The TVT analysis comprised 1,559 inoperable and 6,151 high-risk, operable patients who underwent TAVR at 224 participating registry hospitals from November 2011 to May 2013. Their median STS predicted risk of operative mortality (PROM) was 7%, but varied widely from 1.2% to 17.4%. Their median age was 84 years.
The risk profile of patients was generally lower in the registry than in randomized trials and European reports, and may have contributed to the good outcomes, Dr. Mack acknowledged. The mean PROM was 11.2% among inoperable patients and 11.8% among high-risk, operable patients in the pivotal PARTNER trial, which was used for the device’s approval.
Although incomplete data may have been entered into the risk calculator, the other plausible explanation is "risk creep," or that patients with lower surgical risk are being treated with the less-invasive procedure, leading to better outcomes, he noted.
Other possible contributors to the registry outcomes include a shorter U.S. learning curve due to international collaboration, inclusion of at least 35 highly experienced PARTNER study centers in the registry, and mandated training by the manufacturer including a company employed clinical specialist during all procedures.
Still, the results stack up well. Germany, which has led the TAVR revolution, had an in-hospital mortality of 5.1% with a transfemoral approach and 7.7% with a transapical approach in 2011 in the German Aortic Valve Registry.
In the PARTNER trial, 30-day mortality was 5% among inoperable patients, 3.7% among high-risk, operable patients by a transfemoral approach, and 8.7% among high-risk, operable patients by a transapical approach.
This compares with 30-day mortality rates of 6.1%, 4.6%, and 9.8%, respectively, in the TVT registry, Dr. Mack reported.
The TVT registry captured an estimated 88% of TAVR procedures performed between May 2012, when the Centers for Medicare and Medicaid Services issued its national coverage determination, and the end of the study. Notably, 30-day outcomes were reported from only 114 hospitals or 51% of the 244 hospitals in the registry. The results were published simultaneously with Dr. Mack’s presentation (JAMA 2013;310:2069-77. doi:10.1001/jama/2013.282043).
More complete long-term outcomes at 30 days and 1 year are needed to have a more comprehensive assessment of TAVR outcomes in the United States, Dr. Mack cautioned. Patient health status and quality of life outcomes will also be addressed in subsequent reports.
So far, it’s known that most (63%) patients were discharged home, 6% experienced new-onset atrial fibrillation, and 6.6% needed a new permanent pacemaker or implantable cardioverter-defibrillator.
The study was funded by the American College of Cardiology Foundation and the Society of Thoracic Surgeons. Dr. Mack reported no other disclosures; a co-author reported travel support from the STS and a second reported consulting for Janssen, Boehringer Ingelheim, and Sanofi.
Postmarket outcomes with the first commercially available U.S. transcatheter aortic valve replacement device are comparable with randomized trial and international registry experience, national registry results suggest.
The Edwards LifeSciences Sapien XT valve was successfully implanted in 92% of 7,710 TAVR patients, two-thirds with a transfemoral approach.
The primary outcomes of in-hospital all-cause mortality and stroke were 5.5% and 2%, respectively, Dr. Michael J. Mack reported at the American Heart Association scientific sessions.
Conversion to open-heart surgery was needed in only 1% of patients, but associated with a 49% incidence of in-hospital mortality.
Major bleeding and vascular complications were relatively low, at 3.5% and 6.4%, respectively, despite the overall lack of experience and use of larger sheath delivery systems. Acute renal insufficiency occurred in 5.5% of patients.
The 30-day death and stroke rates were 7.6% and 2.8% among 3,113 patients with sufficient follow-up. Most deaths were due to noncardiovascular causes, he noted.
This is the first public report from the national Society of Thoracic Surgeons/American College of Cardiology Transcatheter Valve Therapy (TVT) Registry, and comes amid calls for increased postmarket surveillance of high-risk medical devices.
The results also help address whether outcomes after a controlled commercial rollout of the first-generation valve hold up to those from clinical trials and the global TAVR experience, now based on second- and third-generation devices, according to Dr. Mack, past STS president and director of cardiovascular research and medicine at the Heart Hospital Baylor Plano in Plano, Texas.
The Food and Drug Administration initially approved the Edwards valve for severe, symptomatic aortic stenosis in inoperable patients in 2011 and expanded its label in September 2012 to include operable, high-risk patients using either a transfemoral or transapical approach.
The TVT analysis comprised 1,559 inoperable and 6,151 high-risk, operable patients who underwent TAVR at 224 participating registry hospitals from November 2011 to May 2013. Their median STS predicted risk of operative mortality (PROM) was 7%, but varied widely from 1.2% to 17.4%. Their median age was 84 years.
The risk profile of patients was generally lower in the registry than in randomized trials and European reports, and may have contributed to the good outcomes, Dr. Mack acknowledged. The mean PROM was 11.2% among inoperable patients and 11.8% among high-risk, operable patients in the pivotal PARTNER trial, which was used for the device’s approval.
Although incomplete data may have been entered into the risk calculator, the other plausible explanation is "risk creep," or that patients with lower surgical risk are being treated with the less-invasive procedure, leading to better outcomes, he noted.
Other possible contributors to the registry outcomes include a shorter U.S. learning curve due to international collaboration, inclusion of at least 35 highly experienced PARTNER study centers in the registry, and mandated training by the manufacturer including a company employed clinical specialist during all procedures.
Still, the results stack up well. Germany, which has led the TAVR revolution, had an in-hospital mortality of 5.1% with a transfemoral approach and 7.7% with a transapical approach in 2011 in the German Aortic Valve Registry.
In the PARTNER trial, 30-day mortality was 5% among inoperable patients, 3.7% among high-risk, operable patients by a transfemoral approach, and 8.7% among high-risk, operable patients by a transapical approach.
This compares with 30-day mortality rates of 6.1%, 4.6%, and 9.8%, respectively, in the TVT registry, Dr. Mack reported.
The TVT registry captured an estimated 88% of TAVR procedures performed between May 2012, when the Centers for Medicare and Medicaid Services issued its national coverage determination, and the end of the study. Notably, 30-day outcomes were reported from only 114 hospitals or 51% of the 244 hospitals in the registry. The results were published simultaneously with Dr. Mack’s presentation (JAMA 2013;310:2069-77. doi:10.1001/jama/2013.282043).
More complete long-term outcomes at 30 days and 1 year are needed to have a more comprehensive assessment of TAVR outcomes in the United States, Dr. Mack cautioned. Patient health status and quality of life outcomes will also be addressed in subsequent reports.
So far, it’s known that most (63%) patients were discharged home, 6% experienced new-onset atrial fibrillation, and 6.6% needed a new permanent pacemaker or implantable cardioverter-defibrillator.
The study was funded by the American College of Cardiology Foundation and the Society of Thoracic Surgeons. Dr. Mack reported no other disclosures; a co-author reported travel support from the STS and a second reported consulting for Janssen, Boehringer Ingelheim, and Sanofi.
FROM THE AHA SCIENTIFIC SESSIONS
Major finding: The 30-day death rate was 7.6% and stroke rate 2.8%.
Data source: A retrospective analysis of 7,710 patients undergoing post-approval TAVR with the Edwards Sapien XT device from November 2011 to May 2013.
Disclosures: The study was funded by the American College of Cardiology Foundation and the Society of Thoracic Surgeons. Dr. Mack reported no other disclosures; a co-author reported travel support from the STS and a second reported consulting for Janssen, Boehringer Ingelheim, and Sanofi.
For dyspnea, details should drive choice of lung volume reduction therapy
CHICAGO – Taking a personalized approach to treating dyspnea will result in better outcomes, and will make choosing between surgical and the increasing number of nonsurgical techniques an easier process, according to Dr. Frank Sciurba, a presenter at the annual meeting of the American College of Chest Physicians.
In a talk that reviewed current and trial surgical and bronchoscopic treatments of dyspnea in chronic obstructive pulmonary disease, Dr. Sciurba said, "Just treating diseases that are now naively classified as COPD or [interstitial lung disease] is not enough. We can instead look at variations within those diseases that may or may not be responsive to different therapies."
For example, because the Impact of Heterogeneity on Outcome Following Endobronchial Valves (VENT) trial data showed that fissure integrity (collateral tracts) significantly influenced target and adjacent lobe volume changes, Dr. Sciurba said that medical device manufacturers have begun to develop technologies that are more specific to the patient.
Straight nitinol coils (PneumRx), which are placed bronchoscopically, are implanted in stages, and according to collateral tracts. "The concept is to target the most affected areas of the lung, allowing regional expansion of the least affected lung. It’s not dependent on just lobar re-expansion," said Dr. Sciurba, director of the emphysema research center at the University of Pittsburgh Medical Center.
Pilot trial data for this technique published in CHEST earlier this year showed that patients (n = 56) had a 17.5% improvement in forced expiratory volume in 1 second (FEV1) and a greater than 10% drop in residual volume, and clinical meaningful improvements in 6-minute walk distances at more than a 28% improvement from baseline: 73% had a greater than 25 meter improvement at 6 months post treatment.
The hydro-gel foam, AeriSeal (Aeris) is another bronchoscopic technique currently undergoing a small (n = 20) pilot trial. After fibrinogen was eliminated from the sealant, this polymeric lung volume reduction technology was cleared by the Food and Drug Administration for testing in humans.
The sealant is administered into specific subsegments of the lungs, where the foam adheres to surrounding tissues; air and water in the foam are reabsorbed when collapse occurs, with durable absorption in atelectasis.
The results will soon be published, although Dr. Sciurba said that at this point, "the mechanical benefits seem to exceed the symptomatic benefits," but that a trial in a larger population would produce more definitive results.
Other factors to consider include "understanding the pulmonary physiologic interaction in lung volume reduction, and how that translates downstream, and the importance of linking the mechanical intervention with pulmonary rehab."
Expanding the ‘tool chest’
In determining whether bronchoscopic solutions can achieve the same benefits of surgical ones, while also minimizing adverse effects, Dr. Sciurba said, the FDA is beginning to take a more personalized view when approving trials, which he hopes will increase the "tool chest" available to physicians.
Clinical trials going forward may need to consider selection criteria such as interlobar collaterals, regional emphysema heterogeneity, and the degree of hyperinflation, as well as the most relevant outcomes when determining adverse events, Dr. Sciurba said.
Whether therapies are reversible also will be relevant, and will have an impact on future criteria for lung volume reduction surgery and transplant candidacy.
"If we actually look in a little more detail and start to classify these patients both on physiologic and clinical patterns, and as we evolve, on genetic patterns and molecular patterns, we will isolate groups of patients who are home run responders from those in whom certain therapies may not be cost effective."
Dr. Sciurba disclosed that he has received support from AstraZeneca, GlaxoSmithKline, Pfizer, and other companies, as well as grant monies from the National Institutes of Health and the University of Pittsburgh.
CHICAGO – Taking a personalized approach to treating dyspnea will result in better outcomes, and will make choosing between surgical and the increasing number of nonsurgical techniques an easier process, according to Dr. Frank Sciurba, a presenter at the annual meeting of the American College of Chest Physicians.
In a talk that reviewed current and trial surgical and bronchoscopic treatments of dyspnea in chronic obstructive pulmonary disease, Dr. Sciurba said, "Just treating diseases that are now naively classified as COPD or [interstitial lung disease] is not enough. We can instead look at variations within those diseases that may or may not be responsive to different therapies."
For example, because the Impact of Heterogeneity on Outcome Following Endobronchial Valves (VENT) trial data showed that fissure integrity (collateral tracts) significantly influenced target and adjacent lobe volume changes, Dr. Sciurba said that medical device manufacturers have begun to develop technologies that are more specific to the patient.
Straight nitinol coils (PneumRx), which are placed bronchoscopically, are implanted in stages, and according to collateral tracts. "The concept is to target the most affected areas of the lung, allowing regional expansion of the least affected lung. It’s not dependent on just lobar re-expansion," said Dr. Sciurba, director of the emphysema research center at the University of Pittsburgh Medical Center.
Pilot trial data for this technique published in CHEST earlier this year showed that patients (n = 56) had a 17.5% improvement in forced expiratory volume in 1 second (FEV1) and a greater than 10% drop in residual volume, and clinical meaningful improvements in 6-minute walk distances at more than a 28% improvement from baseline: 73% had a greater than 25 meter improvement at 6 months post treatment.
The hydro-gel foam, AeriSeal (Aeris) is another bronchoscopic technique currently undergoing a small (n = 20) pilot trial. After fibrinogen was eliminated from the sealant, this polymeric lung volume reduction technology was cleared by the Food and Drug Administration for testing in humans.
The sealant is administered into specific subsegments of the lungs, where the foam adheres to surrounding tissues; air and water in the foam are reabsorbed when collapse occurs, with durable absorption in atelectasis.
The results will soon be published, although Dr. Sciurba said that at this point, "the mechanical benefits seem to exceed the symptomatic benefits," but that a trial in a larger population would produce more definitive results.
Other factors to consider include "understanding the pulmonary physiologic interaction in lung volume reduction, and how that translates downstream, and the importance of linking the mechanical intervention with pulmonary rehab."
Expanding the ‘tool chest’
In determining whether bronchoscopic solutions can achieve the same benefits of surgical ones, while also minimizing adverse effects, Dr. Sciurba said, the FDA is beginning to take a more personalized view when approving trials, which he hopes will increase the "tool chest" available to physicians.
Clinical trials going forward may need to consider selection criteria such as interlobar collaterals, regional emphysema heterogeneity, and the degree of hyperinflation, as well as the most relevant outcomes when determining adverse events, Dr. Sciurba said.
Whether therapies are reversible also will be relevant, and will have an impact on future criteria for lung volume reduction surgery and transplant candidacy.
"If we actually look in a little more detail and start to classify these patients both on physiologic and clinical patterns, and as we evolve, on genetic patterns and molecular patterns, we will isolate groups of patients who are home run responders from those in whom certain therapies may not be cost effective."
Dr. Sciurba disclosed that he has received support from AstraZeneca, GlaxoSmithKline, Pfizer, and other companies, as well as grant monies from the National Institutes of Health and the University of Pittsburgh.
CHICAGO – Taking a personalized approach to treating dyspnea will result in better outcomes, and will make choosing between surgical and the increasing number of nonsurgical techniques an easier process, according to Dr. Frank Sciurba, a presenter at the annual meeting of the American College of Chest Physicians.
In a talk that reviewed current and trial surgical and bronchoscopic treatments of dyspnea in chronic obstructive pulmonary disease, Dr. Sciurba said, "Just treating diseases that are now naively classified as COPD or [interstitial lung disease] is not enough. We can instead look at variations within those diseases that may or may not be responsive to different therapies."
For example, because the Impact of Heterogeneity on Outcome Following Endobronchial Valves (VENT) trial data showed that fissure integrity (collateral tracts) significantly influenced target and adjacent lobe volume changes, Dr. Sciurba said that medical device manufacturers have begun to develop technologies that are more specific to the patient.
Straight nitinol coils (PneumRx), which are placed bronchoscopically, are implanted in stages, and according to collateral tracts. "The concept is to target the most affected areas of the lung, allowing regional expansion of the least affected lung. It’s not dependent on just lobar re-expansion," said Dr. Sciurba, director of the emphysema research center at the University of Pittsburgh Medical Center.
Pilot trial data for this technique published in CHEST earlier this year showed that patients (n = 56) had a 17.5% improvement in forced expiratory volume in 1 second (FEV1) and a greater than 10% drop in residual volume, and clinical meaningful improvements in 6-minute walk distances at more than a 28% improvement from baseline: 73% had a greater than 25 meter improvement at 6 months post treatment.
The hydro-gel foam, AeriSeal (Aeris) is another bronchoscopic technique currently undergoing a small (n = 20) pilot trial. After fibrinogen was eliminated from the sealant, this polymeric lung volume reduction technology was cleared by the Food and Drug Administration for testing in humans.
The sealant is administered into specific subsegments of the lungs, where the foam adheres to surrounding tissues; air and water in the foam are reabsorbed when collapse occurs, with durable absorption in atelectasis.
The results will soon be published, although Dr. Sciurba said that at this point, "the mechanical benefits seem to exceed the symptomatic benefits," but that a trial in a larger population would produce more definitive results.
Other factors to consider include "understanding the pulmonary physiologic interaction in lung volume reduction, and how that translates downstream, and the importance of linking the mechanical intervention with pulmonary rehab."
Expanding the ‘tool chest’
In determining whether bronchoscopic solutions can achieve the same benefits of surgical ones, while also minimizing adverse effects, Dr. Sciurba said, the FDA is beginning to take a more personalized view when approving trials, which he hopes will increase the "tool chest" available to physicians.
Clinical trials going forward may need to consider selection criteria such as interlobar collaterals, regional emphysema heterogeneity, and the degree of hyperinflation, as well as the most relevant outcomes when determining adverse events, Dr. Sciurba said.
Whether therapies are reversible also will be relevant, and will have an impact on future criteria for lung volume reduction surgery and transplant candidacy.
"If we actually look in a little more detail and start to classify these patients both on physiologic and clinical patterns, and as we evolve, on genetic patterns and molecular patterns, we will isolate groups of patients who are home run responders from those in whom certain therapies may not be cost effective."
Dr. Sciurba disclosed that he has received support from AstraZeneca, GlaxoSmithKline, Pfizer, and other companies, as well as grant monies from the National Institutes of Health and the University of Pittsburgh.
EXPERT ANALYSIS FROM CHEST 2013