User login
Radiation-induced heart disease: A practical guide to diagnosis and management
Advances in radiotherapy over the past 50 years have dramatically improved outcomes in patients with malignancy. Five-year overall survival rates for Hodgkin lymphoma and non-Hodgkin lymphoma now stand at 80%, and breast cancer survival is 90%.1
Increased longevity, however, has come at the cost of late side effects such as radiation-induced heart disease (RIHD). Cardiac dysfunction due to radiation involves a spectrum of disease processes in patients who have undergone mediastinal, thoracic, or breast radiotherapy and may involve any cardiac structure, including the pericardium, myocardium, valves, conduction system, and coronary arteries.
Overall, compared with nonirradiated patients, patients who have undergone chest radiotherapy have a 2% higher absolute risk of cardiac morbidity and death at 5 years and a 23% increased absolute risk after 20 years.2
This article will review the pathophysiology and epidemiology of RIHD and will offer a practical approach to its diagnosis and management.
MOST DAMAGE IS ENDOTHELIAL
Cardiac myocytes are relatively resistant to radiation damage because of their postmitotic state. But endothelial cells remain sensitive to radiation, and the pathophysiology of most forms of RIHD appears to be associated with damage to endothelial cells. Conventional cardiac risk factors such as hyperlipidemia and smoking have been shown to compound and accelerate radiation-induced endothelial damage in animal models.3
Radiation is believed to result in transient increases in oxidative stress, resulting in formation of reactive oxygen species and a subsequent inflammatory response that includes activation of nuclear factor-kappa B. Upregulation of proinflammatory pathways results in increased expression of matrix metalloproteinases, adhesion molecules, and proinflammatory cytokines and downregulation of vasculoprotective nitric oxide.4 Indirect evidence for radiation-induced vascular inflammation comes from numerous studies that demonstrated increased levels of the proinflammatory cytokines interleukin 6, tumor necrosis factor alpha, and interferon gamma in Japanese atomic bomb survivors.5
RISK FACTORS

Risk factors for RIHD are summarized in Table 1.
The volume of heart irradiated is a major determinant of the development of RIHD.6 A retrospective study of 960 breast cancer patients in Stockholm between 1971 and 1976 found that those who had received the highest doses and volumes of cardiac radiation had a threefold higher risk of cardiac death. By comparison, those with lesser volumes of the heart exposed to radiation had no increase in risk of cardiac death compared with the general population.7
Younger age at the time of radiotherapy is associated with an increased risk of RIHD in breast cancer and lymphoma patients. A retrospective analysis of 635 patients under age 21 with Hodgkin lymphoma treated with radiotherapy showed a relative risk of fatal myocardial infarction of 41.5 compared with a general population matched for age, sex, and race.8
Conventional cardiac risk factors such as smoking, hypertension, diabetes, and hyperlipidemia further increase the risk of RIHD, and radiation increases the cardiotoxicity of chemotherapeutic agents such as anthracyclines.9
In general, high-risk patients are defined as those with at least one risk factor for RIHD who underwent anterior or left-sided chest irradiation (Table 1).10
CORONARY ARTERY DISEASE
Ischemic heart disease is the most common cause of cardiac death in patients who have undergone radiation therapy. Atherosclerotic lesions in RIHD are morphologically identical to those in nonirradiated vessels and are characterized by intimal proliferation, accumulation of lipid-rich macrophages, and plaque formation.11
A retrospective single-institution study of 415 patients with Hodgkin lymphoma who had undergone radiation therapy found the incidence of coronary artery disease 20 years later to be 10%. The mean time to development of coronary artery disease was 9 years, and all patients who developed it had at least one conventional cardiac risk factor.12
A meta-analysis of more than 20,000 breast cancer patients who received radiotherapy in 40 randomized controlled trials found an increase in the rate of non-breast-cancer deaths, primarily from vascular causes (annual event ratio 1.27, P < .0001).13
A randomized controlled trial comparing breast cancer patients who underwent preoperative or postoperative radiotherapy vs those who had surgery alone revealed a significantly higher death rate from coronary artery disease in the postradiotherapy group.7
The risk of radiation-induced coronary artery disease is proportional to both the dose and the duration of radiation therapy. A retrospective study of more than 2,000 women undergoing radiotherapy for breast cancer found that the relative risk of coronary artery disease increased linearly by about 7.4% per Gy of radiation to the heart, with no apparent ceiling.14
The distribution of atherosclerotic coronary arteries correlates well with the areas exposed to the highest doses of radiation. For instance, in left-sided breast cancer, the apex and anterior wall of the heart typically receive the highest doses of radiation; consequently, the left anterior descending and distal diagonal branches are most prominently involved.15 In patients with lymphoma who undergo radiotherapy to mediastinal nodes and in breast cancer patients receiving radiotherapy to the internal mammary chain, basal structures may be exposed as well. Ostial lesions can also be seen in these patients.16
The clinical presentation of coronary artery disease in radiotherapy recipients does not differ significantly from that in the general population. Ischemia may be silent, may lead to classic anginal symptoms, or may cause sudden cardiac death. The incidence of silent myocardial infarction has been reported to be higher after mediastinal radiotherapy than it is in the general population, possibly from damage to nerve endings within the radiation field.17
Management of radiation-associated coronary artery disease
Managing patients with radiation-associated coronary artery disease is challenging, but the therapeutic options remain the same as those in nonirradiated patients and include medical therapy, percutaneous coronary intervention, and coronary artery bypass grafting, depending on the site and extent of disease.18 Although results are conflicting, there does not seem to be a significant difference in the rates of stent restenosis between patients with a history of radiation therapy and the general population.
Percutaneous coronary intervention is generally preferred to coronary artery bypass grafting in these patients for several reasons. Radiation-induced fibrosis of surrounding structures generally makes surgical procedures more difficult,19 and inclusion of the internal mammary artery or internal thoracic artery in the radiation field may result in stenosis of these vessels, rendering them unsuitable for harvesting.20 Moreover, many patients with RIHD have concurrent radiation-induced lung damage, which increases the risk of perioperative pulmonary complications.21
If the coronary lesions are not amenable to percutaneous intervention, a careful valvular evaluation should be performed preoperatively in view of the frequency of radiation-associated valvular disease. In a study of 72 patients with RIHD undergoing coronary artery bypass grafting, 40% required valvular surgery at the time of surgery or shortly thereafter.22
Results of studies of coronary artery bypass graft outcomes in patients with a history of thoracic radiation therapy have been conflicting, but success seems to depend on the status of the internal mammary and internal thoracic arteries.23 Therefore, the patency of these vessels should be elucidated preoperatively by angiography and intraoperatively by visual inspection of the vessels for fibrosis.
A large single-institution study by Wu et al24 revealed higher short-term and long-term mortality rates in patients with RIHD undergoing cardiac surgery than in control patients without RIHD undergoing similar procedures.
VALVULAR DISEASE
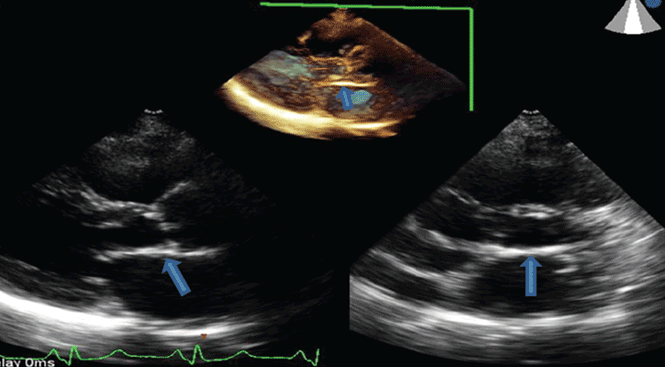
Radiation therapy may directly affect heart valves, and both stenotic (Figure 1) and regurgitant lesions have been described. Pathologic findings include leaflet retraction, fibrotic thickening, and late calcification.25
The precise mechanism of radiation-induced valvular disease is unknown but is thought to be a change in the phenotype of valvular interstitial cells from a myofibroblast to an osteoblast-like cell. Radiation results in significant expression of osteogenic factors such as bone morphogenic protein 2, osteopontin, alkaline phosphatase, and runt-related transcription factor 2 by valvular interstitial cells.26
Valvular heart disease is evident in as many as 81% of patients with RIHD, with the aortic and mitral valves affected more commonly than the tricuspid and pulmonic valves.27 Why there are more left-sided valve lesions than pulmonic valve lesions, despite the pulmonic valve’s anterior position in the heart, is unknown but may be due to higher pressures across the left-sided heart valves.
Although valvular disease is common in patients with RIHD, clinically significant disease is not; more than 70% of patients with radiation-induced valvular disease have no symptoms. A study of 38 cases of radiation-induced valvular disease reported a mean time to development of asymptomatic valvular lesions of 11.5 years and an average time to symptomatic valvular dysfunction of 16.5 years, indicating that 5 years seems to be the interval required for progression from asymptomatic to symptomatic valvular RIHD.28
The thickness of the aortomitral curtain (the junction between the base of the anterior mitral leaflet and the aortic root) is an independent predictor of the long-term risk of death in patients with valvular RIHD.29
Management of radiation-induced valvular disease
Management of patients with valvular RIHD poses a major clinical conundrum because of the high rates of perioperative morbidity and death in patients with a history of chest radiotherapy. In one study,23 the long-term mortality rate was 45% in postradiotherapy patients undergoing single-valve surgery and 61% in those undergoing surgery on two or more valves, compared with 13% and 17% in patients with no history of chest radiotherapy.23
Furthermore, valve repair is an unattractive option in these patients because of high failure rates of mitral valve and tricuspid valve repair attributed to ongoing radiotherapy-induced valvular changes after repair.30
As a result, valve replacement is generally preferred in this group. Patients should be advised of the higher risk of perioperative and long-term morbidity and death associated with open heart surgery than in the general population, and that the risks are even higher with repeat open heart surgery.
This risk has implications for the choice of replacement valves in younger patients. Bioprosthetic valves, which deteriorate over time, may not be advisable. Transcatheter aortic valve replacement has been successful in radiation-induced valvular disease and may become the preferred method of aortic valve replacement.31
PERICARDIAL DISEASE
Pericardial disease is a frequent manifestation of RIHD and covers a spectrum of manifestations from acute pericarditis, pericardial effusion, and tamponade to constrictive pericarditis. In a necropsy study, 70% of patients with RIHD were found to have pericardial involvement.32
The mechanism is believed to be radiation-induced microvascular injury resulting in increased capillary permeability and the sometimes rapid development of a protein-rich exudate. Associated inflammation may cause acute pericarditis, which may eventually be complicated by chronic pericarditis. The parietal surface tends to be affected more severely than the epicardium.33
Perhaps as a result of recent advances such as lower radiation doses, equal weighting of the anterior and posterior fields, and subcarinal blocking, incidence rates of pericarditis as low as 2.5% have been reported.34
Pericardial RIHD may be divided into early acute pericarditis, delayed chronic pericardial effusion, and constrictive pericarditis.
Early acute pericarditis is rare and is thought to represent a reaction to tumor necrosis. It is defined as occurring during radiotherapy and occurs almost exclusively with high-dose radiotherapy for lymphoma. Due to the relatively benign course of acute pericarditis and fear of tumor recurrence, it is not an indication to withhold radiotherapy.35
Delayed chronic pericardial effusion occurs months to years after radiotherapy, is typically asymptomatic, and presents as an enlarged cardiac silhouette on chest imaging.35 Delayed pericardial effusion is followed with imaging. While in many cases it resolves within 2 years, it may also be long-standing. Pericardiocentesis or a pericardial window may be performed to treat symptomatic effusion or delayed effusion causing hemodynamic compromise.35–37 Hypothyroidism should be ruled out, as it can complicate mantle irradiation and result in chronic pericardial effusion.38
Constrictive pericarditis may occur as a late complication of radiotherapy and typically causes symptoms of congestive heart failure. Pericardial stripping in these patients is complicated by the possibility of coexisting RIHD of the valves, myocardium, or coronary arteries, as well as mediastinal fibrosis. A study of 163 patients who underwent pericardial stripping for chronic pericarditis found a 7-year overall survival rate of only 27%, far lower than the rate for those who had no history of radiation exposure.39 Therefore, these patients are often treated for symptom control with diuretics and a low-salt diet rather than with surgery.
MYOCARDIAL DISEASE
Microvascular injury in the myocardium results in chronic ischemia, which may lead to myocardial fibrosis, typically manifesting as diastolic dysfunction. Chest radiotherapy may result in both systolic and diastolic dysfunction, and dilated and restrictive cardiomyopathy are well-recognized complications.40
Historically, high radiation doses resulted in systolic dysfunction in more than half of patients who underwent thoracic radiotherapy.41 Now, however, fewer than 5% of patients develop reductions in left ventricular ejection fraction, and most cases of radiotherapy-induced cardiomyopathy have a restrictive pattern.42
In a single-institution study, diastolic dysfunction was reported in as many as 14% of patients who underwent thoracic radiotherapy for Hodgkin lymphoma.40 Systolic dysfunction is now seen almost exclusively in patients treated concurrently with cardiotoxic chemotherapeutic agents such as anthracyclines in addition to radiotherapy.43
In a childhood cancer survival series, the hazard ratio of congestive heart failure in patients who had undergone radiotherapy for Wilms tumor was 6.6—almost identical to the occurrence in sibling controls. By contrast, the hazard ratio increased to 18.3 in those who received doxorubicin in addition to radiotherapy.44
Treatment of radiation-induced cardiomyopathy
Treatment of radiation-induced cardiomyopathy is similar to that for other forms of cardiomyopathy, with an emphasis on symptom management.
Heart transplant may be an option for highly selected patients with end-stage heart failure secondary to RIHD. In one report, a series of four RIHD patients received a heart transplant, and all four survived past 48 months.45 However, data from the United Network of Organ Sharing revealed an increase in the all-cause mortality rate in patients undergoing heart transplant for RIHD compared with those undergoing transplant for cardiomyopathy due to other causes.46 This trend may be confounded by a higher prevalence of prior cardiac surgery in the RIHD group—itself an established risk factor for poor posttransplant outcomes.
CONDUCTION SYSTEM DISEASE
Life-threatening arrhythmias have been reported that are distinct from the common, asymptomatic repolarization abnormalities that occur during radiotherapy. Atrioventricular nodal bradycardia, all degrees of heart block, and sick sinus syndrome have all been reported after chest radiotherapy. As conduction abnormalities do not typically manifest until years after radiotherapy, it is difficult to establish causation and, consequently, to define incidence.
Right bundle branch block is the most common conduction abnormality because of the proximity of the right bundle to the endocardium on the right side.47
Chest radiotherapy is also associated with prolongation of the corrected QT interval (QTc). A study in patients with a history of thoracic radiotherapy found that the QTc characteristically increased with exercise, a poor prognostic indicator.48 In a study of 134 survivors of childhood cancer, 12.5% of those who had undergone radiotherapy had a resting QTc of 0.44 msec or more.49
Furthermore, a study of 69 breast cancer survivors found a higher incidence of conduction abnormalities at 6 months and 10 years after radiotherapy compared with baseline. The characteristic electrocardiographic changes at 6 months were T-wave changes. At 10 years, the T-wave abnormalities had resolved and were replaced by ST depression.50
As mentioned above, establishing radiotherapy as a cause for these conduction abnormalities is challenging, given the lag between radiation therapy and electrocardiographic changes. The following criteria have been proposed for establishing a link between atrioventricular blockade and prior radiation51:
- Total radiation dose to the heart > 40 Gy
- Delay of 10 years or more since therapy
- Abnormal interval electrocardiographic changes such as bundle branch block
- Prior pericardial involvement
- Associated cardiac or mediastinal lesions.
SCREENING GUIDELINES
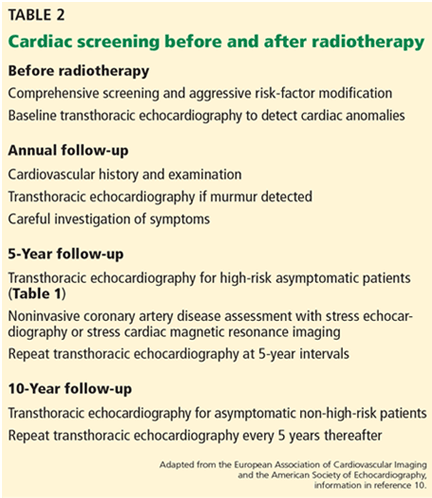
Consensus guidelines for identifying and monitoring RIHD have been published by the European Association of Cardiovascular Imaging and the American Society of Echocardiography (Table 2).10 The European Society of Medical Oncology has also issued guidelines for the prevention, diagnosis, and management of cardiovascular disease associated with cancer therapy.
Briefly, the guidelines call for aggressive cardiac risk-factor modification through weight loss, exercise, blood pressure control, and smoking cessation, in addition to early detection of RIHD. Cardiovascular screening for risk factors and a careful clinical examination should be performed in all patients. Baseline comprehensive transthoracic echocardiography is advocated in all patients before starting radiotherapy to detect cardiac anomalies. Beyond this, an annual history and physical examination, paying close attention to the signs and symptoms of cardiopulmonary disease, is essential. The development of new cardiopulmonary symptoms or a new physical finding such as a murmur should prompt evaluation with transthoracic echocardiography.
In patients without symptoms, screening transthoracic echocardiography at 10 years after the start of radiotherapy is recommended in light of the high probability of diagnosing cardiac disease at this juncture. In patients with no preexisting cardiac disease, surveillance transthoracic echocardiography should be at 5-year intervals thereafter.
In high-risk patients without symptoms (those who have undergone anterior or left-sided radiotherapy and have at least one risk factor for RIHD), initial screening transthoracic echocardiography is recommended 5 years after radiotherapy. These patients have a heightened risk of coronary events as described above and, consequently, are recommended to undergo noninvasive imaging 5 to 10 years after radiation exposure. If this initial examination is negative, stress testing should be repeated at 5-year intervals. Stress echocardiography and stress cardiac magnetic resonance imaging have higher specificity than stress electrocardiography and therefore are generally preferred. Stress scintigraphy should be used with caution, as it adds to the cumulative radiation exposure.
The role of magnetic resonance imaging and computed tomography depends on the results of initial transthoracic echocardiography and the clinical indication, in addition to the center’s expertise and facilities. However, there are currently no data advocating their use as screening tools, except for early detection of porcelain aorta in high-risk patients.10
MODERN RADIOTHERAPY TECHNIQUES
In recent years, there has been emphasis on exposing the patient to as little radiation as possible without compromising cure.52 The three major strategies employed to decrease cardiac exposure include reducing the radiation dose, reducing the radiation field and volume, and using newer planning and delivery techniques.
Reducing the radiation dose. It is well recognized that the mean dose of radiation to the heart is a significant predictor of cardiovascular disease, with one study demonstrating a linear increase in the risk of coronary artery disease with increasing mean heart radiation dose (excess relative risk per Gy 7.4%, 95% confidence interval 3.3%–14.8%).53
Reducing the radiation field and volume. Modern strategies and computed tomography-based radiotherapy planning have enabled a transition from older techniques such as extended-field radiation therapy, mantle-field radiation therapy, and involved-field radiation therapy to new techniques such as involved-node and involved-site radiation therapy.54 These have shown promise. For instance, a study in patients with early Hodgkin lymphoma found a mean heart dose of 27.5 Gy with mantle-field therapy compared with 7.7 Gy with involved-node therapy. This decrease in mean heart dose was associated with a reduction in the 25-year absolute excess cardiac risk from 9.1% to 1.4% and a reduction in cardiac mortality from 2.1% to 1%.55
Employing newer planning and delivery systems has also demonstrated some promise in reducing rates of cardiac morbidity and mortality. Extended-field radiation therapy, mantle-field radiotherapy, and involved-field radiation therapy were traditionally based on two-dimensional planning and often resulted in large volumes of myocardium being unnecessarily exposed to large doses of radiation because of the uncertainty in targeting. Involved-site and involved-node radiotherapy are based on computed tomography, resulting in more accurate targeting and sparing of normal tissue.
In addition, newer techniques such as intensity-modulated radiotherapy and proton beam therapy have resulted in further improvements in conformality compared with three-dimensional conformal radiotherapy.56,57 Respiratory motion management, including deep inspiration breath-holding and end-inspiration breath-holding, have decreased the radiation dose to the heart in patients undergoing mediastinal radiotherapy.58,59
TOWARD THE GOALS OF PREVENTION AND EARLIER DETECTION
As survival from breast cancer and lymphoma has increased, we continue to see legacy or latent effects of therapy, such as RIHD. Radiation therapy can affect any cardiac structure and is a major cause of morbidity and death in cancer survivors.
Modern radiation techniques use a variety of mechanisms to decrease the radiation dose to the heart. A large body of evidence emanating from an era of higher radiation doses and a lack of knowledge of the cardiac effects of radiation highlight the perilous cardiac consequences of chest radiation. With advances in radiotherapy and the development and widespread implementation of consensus guidelines, we envision earlier detection and less frequent occurrence of RIHD, although the latter trend could be blunted by increased cardiovascular risk factors within the population. Given the lag between irradiation and the cardiac consequences, it may be a number of years before any comparisons can be drawn.
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225–249.
- Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood 2011; 117:412–418.
- Amromin GD, Gildenhorn HL, Solomon RD, Nadkarni BB. The synergism of x-irradiation and cholesterol-fat feeding on the development of coronary artery lesions. J Atheroscler Res 1964; 4:325–334.
- Tribble DL, Barcellos-Hoff MH, Chu BM, Gong EL. Ionizing radiation accelerates aortic lesion formation in fat-fed mice via SOD-inhibitable processes. Arterioscler Thromb Vasc Biol 1999; 19:1387–1392.
- Hayashi T, Morishita Y, Kubo Y, et al. Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am J Med 2005; 118:83–86.
- Gagliardi G, Constine LS, Moiseenko V, et al. Radiation dose-volume effects in the heart. Int J Radiat Oncol Biol Phys 2010; 76(suppl 3):S77–S85.
- Rutqvist LE, Lax I, Fornander T, Johansson H. Cardiovascular mortality in a randomized trial of adjuvant radiation therapy versus surgery alone in primary breast cancer. Int J Radiat Oncol Biol Phys 1992; 22:887–896.
- Hancock SL, Donaldson SS, Hoppe RT. Cardiac disease following treatment of Hodgkin’s disease in children and adolescents. J Clin Oncol 1993; 11:1208–1215.
- Meyer RM, Gospodarowicz MK, Connors JM, et al; NCIC Clinical Trials Group; Eastern Cooperative Oncology Group. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012; 366:399–408.
- Lancellotti P, Nkomo VT, Badano LP, et al; European Society of Cardiology Working Groups on Nuclear Cardiology and Cardiac Computed Tomography and Cardiovascular Magnetic Resonance; American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J Am Soc Echocardiogr 2013; 26:1013–1032.
- Cheng RK, Lee MS, Seki A, et al. Radiation coronary arteritis refractory to surgical and percutaneous revascularization culminating in orthotopic heart transplantation. Cardiovasc Pathol 2013; 22:303–308.
- Hull MC, Morris CG, Pepine CJ, Mendenhall NP. Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of Hodgkin lymphoma treated with radiation therapy. JAMA 2003; 290:2831–2837.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 2013; 368:987–998.
- Lind PA, Pagnanelli R, Marks LB, et al. Myocardial perfusion changes in patients irradiated for left-sided breast cancer and correlation with coronary artery distribution. Int J Radiat Oncol Biol Phys 2003; 55:914–920.
- Rademaker J, Schöder H, Ariaratnam NS, et al. Coronary artery disease after radiation therapy for Hodgkin’s lymphoma: coronary CT angiography findings and calcium scores in nine asymptomatic patients. AJR Am J Roentgenol 2008; 191:32–37.
- Orzan F, Brusca A, Conte MR, Presbitero P, Figliomeni MC. Severe coronary artery disease after radiation therapy of the chest and mediastinum: clinical presentation and treatment. Br Heart J 1993; 69:496–500.
- Mousavi N, Nohria A. Radiation-induced cardiovascular disease. Curr Treat Options Cardiovasc Med 2013; 15:507–517.
- McEniery PT, Dorosti K, Schiavone WA, Pedrick TJ, Sheldon WC. Clinical and angiographic features of coronary artery disease after chest irradiation. Am J Cardiol 1987; 60:1020–1024.
- Renner SM, Massel D, Moon BC. Mediastinal irradiation: a risk factor for atherosclerosis of the internal thoracic arteries. Can J Cardiol 1999; 15:597–600.
- Chang AS, Smedira NG, Chang CL, et al. Cardiac surgery after mediastinal radiation: extent of exposure influences outcome. J Thorac Cardiovasc Surg 2007; 133:404–413.
- Handa N, McGregor CG, Danielson GK, et al. Coronary artery bypass grafting in patients with previous mediastinal radiation therapy. J Thorac Cardiovasc Surg 1999; 117:1136–1142.
- Gharagozloo F, Clements IP, Mullany CJ. Use of the internal mammary artery for myocardial revascularization in a patient with radiation-induced coronary artery disease. Mayo Clin Proc 1992; 67:1081–1084.
- Wu W, Masri A, Popovic ZB, et al. Long-term survival of patients with radiation heart disease undergoing cardiac surgery: a cohort study. Circulation 2013; 127:1476–1485.
- Brand MD, Abadi CA, Aurigemma GP, Dauerman HL, Meyer TE. Radiation-associated valvular heart disease in Hodgkin’s disease is associated with characteristic thickening and fibrosis of the aortic-mitral curtain. J Heart Valve Dis 2001; 10:681–685.
- Nadlonek NA, Weyant MJ, Yu JA, et al. Radiation induces osteogenesis in human aortic valve interstitial cells. J Thorac Cardiovasc Surg 2012; 144:1466–1470.
- Tamura A, Takahara Y, Mogi K, Katsumata M. Radiation-induced valvular disease is the logical consequence of irradiation. Gen Thorac Cardiovasc Surg 2007; 55:53–56.
- Carlson RG, Mayfield WR, Normann S, Alexander JA. Radiation-associated valvular disease. Chest 1991; 99:538–545.
- Desai MY, Wu W, Masri A, et al. Increased aorto-mitral curtain thickness independently predicts mortality in patients with radiation-associated cardiac disease undergoing cardiac surgery. Ann Thorac Surg 2014; 97:1348–1355.
- Crestanello JA, McGregor CG, Danielson GK, et al. Mitral and tricuspid valve repair in patients with previous mediastinal radiation therapy. Ann Thorac Surg 2004; 78:826–831.
- Latib A, Montorfano M, Figini F, et al. Percutaneous valve replacement in a young adult for radiation-induced aortic stenosis. J Cardiovasc Med (Hagerstown) 2012; 13:397–398.
- Veinot JP, Edwards WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol 1996; 27:766–773.
- Carver JR, Shapiro CL, Ng A, et al; ASCO Cancer Survivorship Expert Panel. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 2007; 25:3991–4008.
- Carmel RJ, Kaplan HS. Mantle irradiation in Hodgkin’s disease. An analysis of technique, tumor eradication, and complications. Cancer 1976; 37:2813–2825.
- Morton DL, Glancy DL, Joseph WL, Adkins PC. Management of patients with radiation-induced pericarditis with effusion: a note on the development of aortic regurgitation in two of them. Chest 1973; 64:291–297.
- Arsenian MA. Cardiovascular sequelae of therapeutic thoracic radiation. Prog Cardiovasc Dis 1991; 33:299–311.
- Imazio M, Brucato A, Mayosi BM, et al. Medical therapy of pericardial diseases: part II: Noninfectious pericarditis, pericardial effusion and constrictive pericarditis. J Cardiovasc Med (Hagerstown). 2010; 11:785–794.
- Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and the heart. Circulation 1993; 87:1435–1441.
- Bertog SC, Thambidorai SK, Parakh K, et al. Constrictive pericarditis: etiology and cause-specific survival after pericardiectomy. J Am Coll Cardiol 2004; 43:1445–1452.
- Heidenreich PA, Hancock SL, Vagelos RH, Lee BK, Schnittger I. Diastolic dysfunction after mediastinal irradiation. Am Heart J 2005; 150:977–982.
- Burns RJ, Bar-Shlomo BZ, Druck MN, et al. Detection of radiation cardiomyopathy by gated radionuclide angiography. Am J Med 1983; 74:297–302.
- Constine LS, Schwartz RG, Savage DE, King V, Muhs A. Cardiac function, perfusion, and morbidity in irradiated long-term survivors of Hodgkin’s disease. Int J Radiat Oncol Biol Phys 1997; 39:897–906.
- Tolba KA, Deliargyris EN. Cardiotoxicity of cancer therapy. Cancer Invest 1999; 17:408–422.
- Termuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 2011; 57:1210–1216.
- Handa N, McGregor CG, Daly RC, et al. Heart transplantation for radiation-associated end-stage heart failure. Transpl Int 2000; 13:162–165.
- DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant 2012; 31:1269–1275.
- Adams MJ, Lipshultz SE, Schwartz C, Fajardo LF, Coen V, Constine LS. Radiation-associated cardiovascular disease: manifestations and management. Semin Radiat Oncol 2003; 13:346–356.
- Schwartz CL, Hobbie WL, Truesdell S, Constine LC, Clark EB. Corrected QT interval prolongation in anthracycline-treated survivors of childhood cancer. J Clin Oncol 1993; 11:1906–1910.
- Orzan F, Brusca A, Gaita F, Giustetto C, Figliomeni MC, Libero L. Associated cardiac lesions in patients with radiation-induced complete heart block. Int J Cardiol 1993; 39:151–156.
- Larsen RL, Jakacki RI, Vetter VL, Meadows AT, Silber JH, Barber G. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am J Cardiol 1992; 70:73–77.
- Shapiro CL, Hardenbergh PH, Gelman R, et al. Cardiac effects of adjuvant doxorubicin and radiation therapy in breast cancer patients. J Clin Oncol 1998; 16:3493–3501.
- Armstrong GT, Chen Y, Yasui Y, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med 2016; 374:833–842.
- van Nimwegen FA, Schaapveld M, Cutter DJ, et al. Radiation dose-response relationship for risk of coronary heart disease in survivors of Hodgkin lymphoma. J Clin Oncol 2016; 34:235–243.
- Maraldo MV, Ng AK. Minimizing cardiac risks with contemporary radiation therapy for Hodgkin lymphoma. J Clin Oncol 2016; 34:208–210.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012; 83:1232–1237.
- Maraldo MV, Specht L. A decade of comparative dose planning studies for early-stage Hodgkin lymphoma: what can we learn? Int J Radiat Oncol Biol Phys 2014; 90:1126–1135.
- Hoppe BS, Flampouri S, Su Z, et al. Consolidative involved-node proton therapy for Stage IA-IIIB mediastinal Hodgkin lymphoma: preliminary dosimetric outcomes from a Phase II study. Int J Radiat Oncol Biol Phys 2012; 83:260–267.
- Petersen PM, Aznar MC, Berthelsen AK, et al. Prospective phase II trial of image-guided radiotherapy in Hodgkin lymphoma: benefit of deep inspiration breath-hold. Acta Oncol 2015; 54:60–66.
- Aznar MC, Maraldo MV, Schut DA, et al. Minimizing late effects for patients with mediastinal Hodgkin lymphoma: deep inspiration breath-hold, IMRT, or both? Int J Radiat Oncol Biol Phys 2015; 92:169–174.
Advances in radiotherapy over the past 50 years have dramatically improved outcomes in patients with malignancy. Five-year overall survival rates for Hodgkin lymphoma and non-Hodgkin lymphoma now stand at 80%, and breast cancer survival is 90%.1
Increased longevity, however, has come at the cost of late side effects such as radiation-induced heart disease (RIHD). Cardiac dysfunction due to radiation involves a spectrum of disease processes in patients who have undergone mediastinal, thoracic, or breast radiotherapy and may involve any cardiac structure, including the pericardium, myocardium, valves, conduction system, and coronary arteries.
Overall, compared with nonirradiated patients, patients who have undergone chest radiotherapy have a 2% higher absolute risk of cardiac morbidity and death at 5 years and a 23% increased absolute risk after 20 years.2
This article will review the pathophysiology and epidemiology of RIHD and will offer a practical approach to its diagnosis and management.
MOST DAMAGE IS ENDOTHELIAL
Cardiac myocytes are relatively resistant to radiation damage because of their postmitotic state. But endothelial cells remain sensitive to radiation, and the pathophysiology of most forms of RIHD appears to be associated with damage to endothelial cells. Conventional cardiac risk factors such as hyperlipidemia and smoking have been shown to compound and accelerate radiation-induced endothelial damage in animal models.3
Radiation is believed to result in transient increases in oxidative stress, resulting in formation of reactive oxygen species and a subsequent inflammatory response that includes activation of nuclear factor-kappa B. Upregulation of proinflammatory pathways results in increased expression of matrix metalloproteinases, adhesion molecules, and proinflammatory cytokines and downregulation of vasculoprotective nitric oxide.4 Indirect evidence for radiation-induced vascular inflammation comes from numerous studies that demonstrated increased levels of the proinflammatory cytokines interleukin 6, tumor necrosis factor alpha, and interferon gamma in Japanese atomic bomb survivors.5
RISK FACTORS
Risk factors for RIHD are summarized in Table 1.
The volume of heart irradiated is a major determinant of the development of RIHD.6 A retrospective study of 960 breast cancer patients in Stockholm between 1971 and 1976 found that those who had received the highest doses and volumes of cardiac radiation had a threefold higher risk of cardiac death. By comparison, those with lesser volumes of the heart exposed to radiation had no increase in risk of cardiac death compared with the general population.7
Younger age at the time of radiotherapy is associated with an increased risk of RIHD in breast cancer and lymphoma patients. A retrospective analysis of 635 patients under age 21 with Hodgkin lymphoma treated with radiotherapy showed a relative risk of fatal myocardial infarction of 41.5 compared with a general population matched for age, sex, and race.8
Conventional cardiac risk factors such as smoking, hypertension, diabetes, and hyperlipidemia further increase the risk of RIHD, and radiation increases the cardiotoxicity of chemotherapeutic agents such as anthracyclines.9
In general, high-risk patients are defined as those with at least one risk factor for RIHD who underwent anterior or left-sided chest irradiation (Table 1).10
CORONARY ARTERY DISEASE
Ischemic heart disease is the most common cause of cardiac death in patients who have undergone radiation therapy. Atherosclerotic lesions in RIHD are morphologically identical to those in nonirradiated vessels and are characterized by intimal proliferation, accumulation of lipid-rich macrophages, and plaque formation.11
A retrospective single-institution study of 415 patients with Hodgkin lymphoma who had undergone radiation therapy found the incidence of coronary artery disease 20 years later to be 10%. The mean time to development of coronary artery disease was 9 years, and all patients who developed it had at least one conventional cardiac risk factor.12
A meta-analysis of more than 20,000 breast cancer patients who received radiotherapy in 40 randomized controlled trials found an increase in the rate of non-breast-cancer deaths, primarily from vascular causes (annual event ratio 1.27, P < .0001).13
A randomized controlled trial comparing breast cancer patients who underwent preoperative or postoperative radiotherapy vs those who had surgery alone revealed a significantly higher death rate from coronary artery disease in the postradiotherapy group.7
The risk of radiation-induced coronary artery disease is proportional to both the dose and the duration of radiation therapy. A retrospective study of more than 2,000 women undergoing radiotherapy for breast cancer found that the relative risk of coronary artery disease increased linearly by about 7.4% per Gy of radiation to the heart, with no apparent ceiling.14
The distribution of atherosclerotic coronary arteries correlates well with the areas exposed to the highest doses of radiation. For instance, in left-sided breast cancer, the apex and anterior wall of the heart typically receive the highest doses of radiation; consequently, the left anterior descending and distal diagonal branches are most prominently involved.15 In patients with lymphoma who undergo radiotherapy to mediastinal nodes and in breast cancer patients receiving radiotherapy to the internal mammary chain, basal structures may be exposed as well. Ostial lesions can also be seen in these patients.16
The clinical presentation of coronary artery disease in radiotherapy recipients does not differ significantly from that in the general population. Ischemia may be silent, may lead to classic anginal symptoms, or may cause sudden cardiac death. The incidence of silent myocardial infarction has been reported to be higher after mediastinal radiotherapy than it is in the general population, possibly from damage to nerve endings within the radiation field.17
Management of radiation-associated coronary artery disease
Managing patients with radiation-associated coronary artery disease is challenging, but the therapeutic options remain the same as those in nonirradiated patients and include medical therapy, percutaneous coronary intervention, and coronary artery bypass grafting, depending on the site and extent of disease.18 Although results are conflicting, there does not seem to be a significant difference in the rates of stent restenosis between patients with a history of radiation therapy and the general population.
Percutaneous coronary intervention is generally preferred to coronary artery bypass grafting in these patients for several reasons. Radiation-induced fibrosis of surrounding structures generally makes surgical procedures more difficult,19 and inclusion of the internal mammary artery or internal thoracic artery in the radiation field may result in stenosis of these vessels, rendering them unsuitable for harvesting.20 Moreover, many patients with RIHD have concurrent radiation-induced lung damage, which increases the risk of perioperative pulmonary complications.21
If the coronary lesions are not amenable to percutaneous intervention, a careful valvular evaluation should be performed preoperatively in view of the frequency of radiation-associated valvular disease. In a study of 72 patients with RIHD undergoing coronary artery bypass grafting, 40% required valvular surgery at the time of surgery or shortly thereafter.22
Results of studies of coronary artery bypass graft outcomes in patients with a history of thoracic radiation therapy have been conflicting, but success seems to depend on the status of the internal mammary and internal thoracic arteries.23 Therefore, the patency of these vessels should be elucidated preoperatively by angiography and intraoperatively by visual inspection of the vessels for fibrosis.
A large single-institution study by Wu et al24 revealed higher short-term and long-term mortality rates in patients with RIHD undergoing cardiac surgery than in control patients without RIHD undergoing similar procedures.
VALVULAR DISEASE
Radiation therapy may directly affect heart valves, and both stenotic (Figure 1) and regurgitant lesions have been described. Pathologic findings include leaflet retraction, fibrotic thickening, and late calcification.25
The precise mechanism of radiation-induced valvular disease is unknown but is thought to be a change in the phenotype of valvular interstitial cells from a myofibroblast to an osteoblast-like cell. Radiation results in significant expression of osteogenic factors such as bone morphogenic protein 2, osteopontin, alkaline phosphatase, and runt-related transcription factor 2 by valvular interstitial cells.26
Valvular heart disease is evident in as many as 81% of patients with RIHD, with the aortic and mitral valves affected more commonly than the tricuspid and pulmonic valves.27 Why there are more left-sided valve lesions than pulmonic valve lesions, despite the pulmonic valve’s anterior position in the heart, is unknown but may be due to higher pressures across the left-sided heart valves.
Although valvular disease is common in patients with RIHD, clinically significant disease is not; more than 70% of patients with radiation-induced valvular disease have no symptoms. A study of 38 cases of radiation-induced valvular disease reported a mean time to development of asymptomatic valvular lesions of 11.5 years and an average time to symptomatic valvular dysfunction of 16.5 years, indicating that 5 years seems to be the interval required for progression from asymptomatic to symptomatic valvular RIHD.28
The thickness of the aortomitral curtain (the junction between the base of the anterior mitral leaflet and the aortic root) is an independent predictor of the long-term risk of death in patients with valvular RIHD.29
Management of radiation-induced valvular disease
Management of patients with valvular RIHD poses a major clinical conundrum because of the high rates of perioperative morbidity and death in patients with a history of chest radiotherapy. In one study,23 the long-term mortality rate was 45% in postradiotherapy patients undergoing single-valve surgery and 61% in those undergoing surgery on two or more valves, compared with 13% and 17% in patients with no history of chest radiotherapy.23
Furthermore, valve repair is an unattractive option in these patients because of high failure rates of mitral valve and tricuspid valve repair attributed to ongoing radiotherapy-induced valvular changes after repair.30
As a result, valve replacement is generally preferred in this group. Patients should be advised of the higher risk of perioperative and long-term morbidity and death associated with open heart surgery than in the general population, and that the risks are even higher with repeat open heart surgery.
This risk has implications for the choice of replacement valves in younger patients. Bioprosthetic valves, which deteriorate over time, may not be advisable. Transcatheter aortic valve replacement has been successful in radiation-induced valvular disease and may become the preferred method of aortic valve replacement.31
PERICARDIAL DISEASE
Pericardial disease is a frequent manifestation of RIHD and covers a spectrum of manifestations from acute pericarditis, pericardial effusion, and tamponade to constrictive pericarditis. In a necropsy study, 70% of patients with RIHD were found to have pericardial involvement.32
The mechanism is believed to be radiation-induced microvascular injury resulting in increased capillary permeability and the sometimes rapid development of a protein-rich exudate. Associated inflammation may cause acute pericarditis, which may eventually be complicated by chronic pericarditis. The parietal surface tends to be affected more severely than the epicardium.33
Perhaps as a result of recent advances such as lower radiation doses, equal weighting of the anterior and posterior fields, and subcarinal blocking, incidence rates of pericarditis as low as 2.5% have been reported.34
Pericardial RIHD may be divided into early acute pericarditis, delayed chronic pericardial effusion, and constrictive pericarditis.
Early acute pericarditis is rare and is thought to represent a reaction to tumor necrosis. It is defined as occurring during radiotherapy and occurs almost exclusively with high-dose radiotherapy for lymphoma. Due to the relatively benign course of acute pericarditis and fear of tumor recurrence, it is not an indication to withhold radiotherapy.35
Delayed chronic pericardial effusion occurs months to years after radiotherapy, is typically asymptomatic, and presents as an enlarged cardiac silhouette on chest imaging.35 Delayed pericardial effusion is followed with imaging. While in many cases it resolves within 2 years, it may also be long-standing. Pericardiocentesis or a pericardial window may be performed to treat symptomatic effusion or delayed effusion causing hemodynamic compromise.35–37 Hypothyroidism should be ruled out, as it can complicate mantle irradiation and result in chronic pericardial effusion.38
Constrictive pericarditis may occur as a late complication of radiotherapy and typically causes symptoms of congestive heart failure. Pericardial stripping in these patients is complicated by the possibility of coexisting RIHD of the valves, myocardium, or coronary arteries, as well as mediastinal fibrosis. A study of 163 patients who underwent pericardial stripping for chronic pericarditis found a 7-year overall survival rate of only 27%, far lower than the rate for those who had no history of radiation exposure.39 Therefore, these patients are often treated for symptom control with diuretics and a low-salt diet rather than with surgery.
MYOCARDIAL DISEASE
Microvascular injury in the myocardium results in chronic ischemia, which may lead to myocardial fibrosis, typically manifesting as diastolic dysfunction. Chest radiotherapy may result in both systolic and diastolic dysfunction, and dilated and restrictive cardiomyopathy are well-recognized complications.40
Historically, high radiation doses resulted in systolic dysfunction in more than half of patients who underwent thoracic radiotherapy.41 Now, however, fewer than 5% of patients develop reductions in left ventricular ejection fraction, and most cases of radiotherapy-induced cardiomyopathy have a restrictive pattern.42
In a single-institution study, diastolic dysfunction was reported in as many as 14% of patients who underwent thoracic radiotherapy for Hodgkin lymphoma.40 Systolic dysfunction is now seen almost exclusively in patients treated concurrently with cardiotoxic chemotherapeutic agents such as anthracyclines in addition to radiotherapy.43
In a childhood cancer survival series, the hazard ratio of congestive heart failure in patients who had undergone radiotherapy for Wilms tumor was 6.6—almost identical to the occurrence in sibling controls. By contrast, the hazard ratio increased to 18.3 in those who received doxorubicin in addition to radiotherapy.44
Treatment of radiation-induced cardiomyopathy
Treatment of radiation-induced cardiomyopathy is similar to that for other forms of cardiomyopathy, with an emphasis on symptom management.
Heart transplant may be an option for highly selected patients with end-stage heart failure secondary to RIHD. In one report, a series of four RIHD patients received a heart transplant, and all four survived past 48 months.45 However, data from the United Network of Organ Sharing revealed an increase in the all-cause mortality rate in patients undergoing heart transplant for RIHD compared with those undergoing transplant for cardiomyopathy due to other causes.46 This trend may be confounded by a higher prevalence of prior cardiac surgery in the RIHD group—itself an established risk factor for poor posttransplant outcomes.
CONDUCTION SYSTEM DISEASE
Life-threatening arrhythmias have been reported that are distinct from the common, asymptomatic repolarization abnormalities that occur during radiotherapy. Atrioventricular nodal bradycardia, all degrees of heart block, and sick sinus syndrome have all been reported after chest radiotherapy. As conduction abnormalities do not typically manifest until years after radiotherapy, it is difficult to establish causation and, consequently, to define incidence.
Right bundle branch block is the most common conduction abnormality because of the proximity of the right bundle to the endocardium on the right side.47
Chest radiotherapy is also associated with prolongation of the corrected QT interval (QTc). A study in patients with a history of thoracic radiotherapy found that the QTc characteristically increased with exercise, a poor prognostic indicator.48 In a study of 134 survivors of childhood cancer, 12.5% of those who had undergone radiotherapy had a resting QTc of 0.44 msec or more.49
Furthermore, a study of 69 breast cancer survivors found a higher incidence of conduction abnormalities at 6 months and 10 years after radiotherapy compared with baseline. The characteristic electrocardiographic changes at 6 months were T-wave changes. At 10 years, the T-wave abnormalities had resolved and were replaced by ST depression.50
As mentioned above, establishing radiotherapy as a cause for these conduction abnormalities is challenging, given the lag between radiation therapy and electrocardiographic changes. The following criteria have been proposed for establishing a link between atrioventricular blockade and prior radiation51:
- Total radiation dose to the heart > 40 Gy
- Delay of 10 years or more since therapy
- Abnormal interval electrocardiographic changes such as bundle branch block
- Prior pericardial involvement
- Associated cardiac or mediastinal lesions.
SCREENING GUIDELINES
Consensus guidelines for identifying and monitoring RIHD have been published by the European Association of Cardiovascular Imaging and the American Society of Echocardiography (Table 2).10 The European Society of Medical Oncology has also issued guidelines for the prevention, diagnosis, and management of cardiovascular disease associated with cancer therapy.
Briefly, the guidelines call for aggressive cardiac risk-factor modification through weight loss, exercise, blood pressure control, and smoking cessation, in addition to early detection of RIHD. Cardiovascular screening for risk factors and a careful clinical examination should be performed in all patients. Baseline comprehensive transthoracic echocardiography is advocated in all patients before starting radiotherapy to detect cardiac anomalies. Beyond this, an annual history and physical examination, paying close attention to the signs and symptoms of cardiopulmonary disease, is essential. The development of new cardiopulmonary symptoms or a new physical finding such as a murmur should prompt evaluation with transthoracic echocardiography.
In patients without symptoms, screening transthoracic echocardiography at 10 years after the start of radiotherapy is recommended in light of the high probability of diagnosing cardiac disease at this juncture. In patients with no preexisting cardiac disease, surveillance transthoracic echocardiography should be at 5-year intervals thereafter.
In high-risk patients without symptoms (those who have undergone anterior or left-sided radiotherapy and have at least one risk factor for RIHD), initial screening transthoracic echocardiography is recommended 5 years after radiotherapy. These patients have a heightened risk of coronary events as described above and, consequently, are recommended to undergo noninvasive imaging 5 to 10 years after radiation exposure. If this initial examination is negative, stress testing should be repeated at 5-year intervals. Stress echocardiography and stress cardiac magnetic resonance imaging have higher specificity than stress electrocardiography and therefore are generally preferred. Stress scintigraphy should be used with caution, as it adds to the cumulative radiation exposure.
The role of magnetic resonance imaging and computed tomography depends on the results of initial transthoracic echocardiography and the clinical indication, in addition to the center’s expertise and facilities. However, there are currently no data advocating their use as screening tools, except for early detection of porcelain aorta in high-risk patients.10
MODERN RADIOTHERAPY TECHNIQUES
In recent years, there has been emphasis on exposing the patient to as little radiation as possible without compromising cure.52 The three major strategies employed to decrease cardiac exposure include reducing the radiation dose, reducing the radiation field and volume, and using newer planning and delivery techniques.
Reducing the radiation dose. It is well recognized that the mean dose of radiation to the heart is a significant predictor of cardiovascular disease, with one study demonstrating a linear increase in the risk of coronary artery disease with increasing mean heart radiation dose (excess relative risk per Gy 7.4%, 95% confidence interval 3.3%–14.8%).53
Reducing the radiation field and volume. Modern strategies and computed tomography-based radiotherapy planning have enabled a transition from older techniques such as extended-field radiation therapy, mantle-field radiation therapy, and involved-field radiation therapy to new techniques such as involved-node and involved-site radiation therapy.54 These have shown promise. For instance, a study in patients with early Hodgkin lymphoma found a mean heart dose of 27.5 Gy with mantle-field therapy compared with 7.7 Gy with involved-node therapy. This decrease in mean heart dose was associated with a reduction in the 25-year absolute excess cardiac risk from 9.1% to 1.4% and a reduction in cardiac mortality from 2.1% to 1%.55
Employing newer planning and delivery systems has also demonstrated some promise in reducing rates of cardiac morbidity and mortality. Extended-field radiation therapy, mantle-field radiotherapy, and involved-field radiation therapy were traditionally based on two-dimensional planning and often resulted in large volumes of myocardium being unnecessarily exposed to large doses of radiation because of the uncertainty in targeting. Involved-site and involved-node radiotherapy are based on computed tomography, resulting in more accurate targeting and sparing of normal tissue.
In addition, newer techniques such as intensity-modulated radiotherapy and proton beam therapy have resulted in further improvements in conformality compared with three-dimensional conformal radiotherapy.56,57 Respiratory motion management, including deep inspiration breath-holding and end-inspiration breath-holding, have decreased the radiation dose to the heart in patients undergoing mediastinal radiotherapy.58,59
TOWARD THE GOALS OF PREVENTION AND EARLIER DETECTION
As survival from breast cancer and lymphoma has increased, we continue to see legacy or latent effects of therapy, such as RIHD. Radiation therapy can affect any cardiac structure and is a major cause of morbidity and death in cancer survivors.
Modern radiation techniques use a variety of mechanisms to decrease the radiation dose to the heart. A large body of evidence emanating from an era of higher radiation doses and a lack of knowledge of the cardiac effects of radiation highlight the perilous cardiac consequences of chest radiation. With advances in radiotherapy and the development and widespread implementation of consensus guidelines, we envision earlier detection and less frequent occurrence of RIHD, although the latter trend could be blunted by increased cardiovascular risk factors within the population. Given the lag between irradiation and the cardiac consequences, it may be a number of years before any comparisons can be drawn.
Advances in radiotherapy over the past 50 years have dramatically improved outcomes in patients with malignancy. Five-year overall survival rates for Hodgkin lymphoma and non-Hodgkin lymphoma now stand at 80%, and breast cancer survival is 90%.1
Increased longevity, however, has come at the cost of late side effects such as radiation-induced heart disease (RIHD). Cardiac dysfunction due to radiation involves a spectrum of disease processes in patients who have undergone mediastinal, thoracic, or breast radiotherapy and may involve any cardiac structure, including the pericardium, myocardium, valves, conduction system, and coronary arteries.
Overall, compared with nonirradiated patients, patients who have undergone chest radiotherapy have a 2% higher absolute risk of cardiac morbidity and death at 5 years and a 23% increased absolute risk after 20 years.2
This article will review the pathophysiology and epidemiology of RIHD and will offer a practical approach to its diagnosis and management.
MOST DAMAGE IS ENDOTHELIAL
Cardiac myocytes are relatively resistant to radiation damage because of their postmitotic state. But endothelial cells remain sensitive to radiation, and the pathophysiology of most forms of RIHD appears to be associated with damage to endothelial cells. Conventional cardiac risk factors such as hyperlipidemia and smoking have been shown to compound and accelerate radiation-induced endothelial damage in animal models.3
Radiation is believed to result in transient increases in oxidative stress, resulting in formation of reactive oxygen species and a subsequent inflammatory response that includes activation of nuclear factor-kappa B. Upregulation of proinflammatory pathways results in increased expression of matrix metalloproteinases, adhesion molecules, and proinflammatory cytokines and downregulation of vasculoprotective nitric oxide.4 Indirect evidence for radiation-induced vascular inflammation comes from numerous studies that demonstrated increased levels of the proinflammatory cytokines interleukin 6, tumor necrosis factor alpha, and interferon gamma in Japanese atomic bomb survivors.5
RISK FACTORS
Risk factors for RIHD are summarized in Table 1.
The volume of heart irradiated is a major determinant of the development of RIHD.6 A retrospective study of 960 breast cancer patients in Stockholm between 1971 and 1976 found that those who had received the highest doses and volumes of cardiac radiation had a threefold higher risk of cardiac death. By comparison, those with lesser volumes of the heart exposed to radiation had no increase in risk of cardiac death compared with the general population.7
Younger age at the time of radiotherapy is associated with an increased risk of RIHD in breast cancer and lymphoma patients. A retrospective analysis of 635 patients under age 21 with Hodgkin lymphoma treated with radiotherapy showed a relative risk of fatal myocardial infarction of 41.5 compared with a general population matched for age, sex, and race.8
Conventional cardiac risk factors such as smoking, hypertension, diabetes, and hyperlipidemia further increase the risk of RIHD, and radiation increases the cardiotoxicity of chemotherapeutic agents such as anthracyclines.9
In general, high-risk patients are defined as those with at least one risk factor for RIHD who underwent anterior or left-sided chest irradiation (Table 1).10
CORONARY ARTERY DISEASE
Ischemic heart disease is the most common cause of cardiac death in patients who have undergone radiation therapy. Atherosclerotic lesions in RIHD are morphologically identical to those in nonirradiated vessels and are characterized by intimal proliferation, accumulation of lipid-rich macrophages, and plaque formation.11
A retrospective single-institution study of 415 patients with Hodgkin lymphoma who had undergone radiation therapy found the incidence of coronary artery disease 20 years later to be 10%. The mean time to development of coronary artery disease was 9 years, and all patients who developed it had at least one conventional cardiac risk factor.12
A meta-analysis of more than 20,000 breast cancer patients who received radiotherapy in 40 randomized controlled trials found an increase in the rate of non-breast-cancer deaths, primarily from vascular causes (annual event ratio 1.27, P < .0001).13
A randomized controlled trial comparing breast cancer patients who underwent preoperative or postoperative radiotherapy vs those who had surgery alone revealed a significantly higher death rate from coronary artery disease in the postradiotherapy group.7
The risk of radiation-induced coronary artery disease is proportional to both the dose and the duration of radiation therapy. A retrospective study of more than 2,000 women undergoing radiotherapy for breast cancer found that the relative risk of coronary artery disease increased linearly by about 7.4% per Gy of radiation to the heart, with no apparent ceiling.14
The distribution of atherosclerotic coronary arteries correlates well with the areas exposed to the highest doses of radiation. For instance, in left-sided breast cancer, the apex and anterior wall of the heart typically receive the highest doses of radiation; consequently, the left anterior descending and distal diagonal branches are most prominently involved.15 In patients with lymphoma who undergo radiotherapy to mediastinal nodes and in breast cancer patients receiving radiotherapy to the internal mammary chain, basal structures may be exposed as well. Ostial lesions can also be seen in these patients.16
The clinical presentation of coronary artery disease in radiotherapy recipients does not differ significantly from that in the general population. Ischemia may be silent, may lead to classic anginal symptoms, or may cause sudden cardiac death. The incidence of silent myocardial infarction has been reported to be higher after mediastinal radiotherapy than it is in the general population, possibly from damage to nerve endings within the radiation field.17
Management of radiation-associated coronary artery disease
Managing patients with radiation-associated coronary artery disease is challenging, but the therapeutic options remain the same as those in nonirradiated patients and include medical therapy, percutaneous coronary intervention, and coronary artery bypass grafting, depending on the site and extent of disease.18 Although results are conflicting, there does not seem to be a significant difference in the rates of stent restenosis between patients with a history of radiation therapy and the general population.
Percutaneous coronary intervention is generally preferred to coronary artery bypass grafting in these patients for several reasons. Radiation-induced fibrosis of surrounding structures generally makes surgical procedures more difficult,19 and inclusion of the internal mammary artery or internal thoracic artery in the radiation field may result in stenosis of these vessels, rendering them unsuitable for harvesting.20 Moreover, many patients with RIHD have concurrent radiation-induced lung damage, which increases the risk of perioperative pulmonary complications.21
If the coronary lesions are not amenable to percutaneous intervention, a careful valvular evaluation should be performed preoperatively in view of the frequency of radiation-associated valvular disease. In a study of 72 patients with RIHD undergoing coronary artery bypass grafting, 40% required valvular surgery at the time of surgery or shortly thereafter.22
Results of studies of coronary artery bypass graft outcomes in patients with a history of thoracic radiation therapy have been conflicting, but success seems to depend on the status of the internal mammary and internal thoracic arteries.23 Therefore, the patency of these vessels should be elucidated preoperatively by angiography and intraoperatively by visual inspection of the vessels for fibrosis.
A large single-institution study by Wu et al24 revealed higher short-term and long-term mortality rates in patients with RIHD undergoing cardiac surgery than in control patients without RIHD undergoing similar procedures.
VALVULAR DISEASE
Radiation therapy may directly affect heart valves, and both stenotic (Figure 1) and regurgitant lesions have been described. Pathologic findings include leaflet retraction, fibrotic thickening, and late calcification.25
The precise mechanism of radiation-induced valvular disease is unknown but is thought to be a change in the phenotype of valvular interstitial cells from a myofibroblast to an osteoblast-like cell. Radiation results in significant expression of osteogenic factors such as bone morphogenic protein 2, osteopontin, alkaline phosphatase, and runt-related transcription factor 2 by valvular interstitial cells.26
Valvular heart disease is evident in as many as 81% of patients with RIHD, with the aortic and mitral valves affected more commonly than the tricuspid and pulmonic valves.27 Why there are more left-sided valve lesions than pulmonic valve lesions, despite the pulmonic valve’s anterior position in the heart, is unknown but may be due to higher pressures across the left-sided heart valves.
Although valvular disease is common in patients with RIHD, clinically significant disease is not; more than 70% of patients with radiation-induced valvular disease have no symptoms. A study of 38 cases of radiation-induced valvular disease reported a mean time to development of asymptomatic valvular lesions of 11.5 years and an average time to symptomatic valvular dysfunction of 16.5 years, indicating that 5 years seems to be the interval required for progression from asymptomatic to symptomatic valvular RIHD.28
The thickness of the aortomitral curtain (the junction between the base of the anterior mitral leaflet and the aortic root) is an independent predictor of the long-term risk of death in patients with valvular RIHD.29
Management of radiation-induced valvular disease
Management of patients with valvular RIHD poses a major clinical conundrum because of the high rates of perioperative morbidity and death in patients with a history of chest radiotherapy. In one study,23 the long-term mortality rate was 45% in postradiotherapy patients undergoing single-valve surgery and 61% in those undergoing surgery on two or more valves, compared with 13% and 17% in patients with no history of chest radiotherapy.23
Furthermore, valve repair is an unattractive option in these patients because of high failure rates of mitral valve and tricuspid valve repair attributed to ongoing radiotherapy-induced valvular changes after repair.30
As a result, valve replacement is generally preferred in this group. Patients should be advised of the higher risk of perioperative and long-term morbidity and death associated with open heart surgery than in the general population, and that the risks are even higher with repeat open heart surgery.
This risk has implications for the choice of replacement valves in younger patients. Bioprosthetic valves, which deteriorate over time, may not be advisable. Transcatheter aortic valve replacement has been successful in radiation-induced valvular disease and may become the preferred method of aortic valve replacement.31
PERICARDIAL DISEASE
Pericardial disease is a frequent manifestation of RIHD and covers a spectrum of manifestations from acute pericarditis, pericardial effusion, and tamponade to constrictive pericarditis. In a necropsy study, 70% of patients with RIHD were found to have pericardial involvement.32
The mechanism is believed to be radiation-induced microvascular injury resulting in increased capillary permeability and the sometimes rapid development of a protein-rich exudate. Associated inflammation may cause acute pericarditis, which may eventually be complicated by chronic pericarditis. The parietal surface tends to be affected more severely than the epicardium.33
Perhaps as a result of recent advances such as lower radiation doses, equal weighting of the anterior and posterior fields, and subcarinal blocking, incidence rates of pericarditis as low as 2.5% have been reported.34
Pericardial RIHD may be divided into early acute pericarditis, delayed chronic pericardial effusion, and constrictive pericarditis.
Early acute pericarditis is rare and is thought to represent a reaction to tumor necrosis. It is defined as occurring during radiotherapy and occurs almost exclusively with high-dose radiotherapy for lymphoma. Due to the relatively benign course of acute pericarditis and fear of tumor recurrence, it is not an indication to withhold radiotherapy.35
Delayed chronic pericardial effusion occurs months to years after radiotherapy, is typically asymptomatic, and presents as an enlarged cardiac silhouette on chest imaging.35 Delayed pericardial effusion is followed with imaging. While in many cases it resolves within 2 years, it may also be long-standing. Pericardiocentesis or a pericardial window may be performed to treat symptomatic effusion or delayed effusion causing hemodynamic compromise.35–37 Hypothyroidism should be ruled out, as it can complicate mantle irradiation and result in chronic pericardial effusion.38
Constrictive pericarditis may occur as a late complication of radiotherapy and typically causes symptoms of congestive heart failure. Pericardial stripping in these patients is complicated by the possibility of coexisting RIHD of the valves, myocardium, or coronary arteries, as well as mediastinal fibrosis. A study of 163 patients who underwent pericardial stripping for chronic pericarditis found a 7-year overall survival rate of only 27%, far lower than the rate for those who had no history of radiation exposure.39 Therefore, these patients are often treated for symptom control with diuretics and a low-salt diet rather than with surgery.
MYOCARDIAL DISEASE
Microvascular injury in the myocardium results in chronic ischemia, which may lead to myocardial fibrosis, typically manifesting as diastolic dysfunction. Chest radiotherapy may result in both systolic and diastolic dysfunction, and dilated and restrictive cardiomyopathy are well-recognized complications.40
Historically, high radiation doses resulted in systolic dysfunction in more than half of patients who underwent thoracic radiotherapy.41 Now, however, fewer than 5% of patients develop reductions in left ventricular ejection fraction, and most cases of radiotherapy-induced cardiomyopathy have a restrictive pattern.42
In a single-institution study, diastolic dysfunction was reported in as many as 14% of patients who underwent thoracic radiotherapy for Hodgkin lymphoma.40 Systolic dysfunction is now seen almost exclusively in patients treated concurrently with cardiotoxic chemotherapeutic agents such as anthracyclines in addition to radiotherapy.43
In a childhood cancer survival series, the hazard ratio of congestive heart failure in patients who had undergone radiotherapy for Wilms tumor was 6.6—almost identical to the occurrence in sibling controls. By contrast, the hazard ratio increased to 18.3 in those who received doxorubicin in addition to radiotherapy.44
Treatment of radiation-induced cardiomyopathy
Treatment of radiation-induced cardiomyopathy is similar to that for other forms of cardiomyopathy, with an emphasis on symptom management.
Heart transplant may be an option for highly selected patients with end-stage heart failure secondary to RIHD. In one report, a series of four RIHD patients received a heart transplant, and all four survived past 48 months.45 However, data from the United Network of Organ Sharing revealed an increase in the all-cause mortality rate in patients undergoing heart transplant for RIHD compared with those undergoing transplant for cardiomyopathy due to other causes.46 This trend may be confounded by a higher prevalence of prior cardiac surgery in the RIHD group—itself an established risk factor for poor posttransplant outcomes.
CONDUCTION SYSTEM DISEASE
Life-threatening arrhythmias have been reported that are distinct from the common, asymptomatic repolarization abnormalities that occur during radiotherapy. Atrioventricular nodal bradycardia, all degrees of heart block, and sick sinus syndrome have all been reported after chest radiotherapy. As conduction abnormalities do not typically manifest until years after radiotherapy, it is difficult to establish causation and, consequently, to define incidence.
Right bundle branch block is the most common conduction abnormality because of the proximity of the right bundle to the endocardium on the right side.47
Chest radiotherapy is also associated with prolongation of the corrected QT interval (QTc). A study in patients with a history of thoracic radiotherapy found that the QTc characteristically increased with exercise, a poor prognostic indicator.48 In a study of 134 survivors of childhood cancer, 12.5% of those who had undergone radiotherapy had a resting QTc of 0.44 msec or more.49
Furthermore, a study of 69 breast cancer survivors found a higher incidence of conduction abnormalities at 6 months and 10 years after radiotherapy compared with baseline. The characteristic electrocardiographic changes at 6 months were T-wave changes. At 10 years, the T-wave abnormalities had resolved and were replaced by ST depression.50
As mentioned above, establishing radiotherapy as a cause for these conduction abnormalities is challenging, given the lag between radiation therapy and electrocardiographic changes. The following criteria have been proposed for establishing a link between atrioventricular blockade and prior radiation51:
- Total radiation dose to the heart > 40 Gy
- Delay of 10 years or more since therapy
- Abnormal interval electrocardiographic changes such as bundle branch block
- Prior pericardial involvement
- Associated cardiac or mediastinal lesions.
SCREENING GUIDELINES
Consensus guidelines for identifying and monitoring RIHD have been published by the European Association of Cardiovascular Imaging and the American Society of Echocardiography (Table 2).10 The European Society of Medical Oncology has also issued guidelines for the prevention, diagnosis, and management of cardiovascular disease associated with cancer therapy.
Briefly, the guidelines call for aggressive cardiac risk-factor modification through weight loss, exercise, blood pressure control, and smoking cessation, in addition to early detection of RIHD. Cardiovascular screening for risk factors and a careful clinical examination should be performed in all patients. Baseline comprehensive transthoracic echocardiography is advocated in all patients before starting radiotherapy to detect cardiac anomalies. Beyond this, an annual history and physical examination, paying close attention to the signs and symptoms of cardiopulmonary disease, is essential. The development of new cardiopulmonary symptoms or a new physical finding such as a murmur should prompt evaluation with transthoracic echocardiography.
In patients without symptoms, screening transthoracic echocardiography at 10 years after the start of radiotherapy is recommended in light of the high probability of diagnosing cardiac disease at this juncture. In patients with no preexisting cardiac disease, surveillance transthoracic echocardiography should be at 5-year intervals thereafter.
In high-risk patients without symptoms (those who have undergone anterior or left-sided radiotherapy and have at least one risk factor for RIHD), initial screening transthoracic echocardiography is recommended 5 years after radiotherapy. These patients have a heightened risk of coronary events as described above and, consequently, are recommended to undergo noninvasive imaging 5 to 10 years after radiation exposure. If this initial examination is negative, stress testing should be repeated at 5-year intervals. Stress echocardiography and stress cardiac magnetic resonance imaging have higher specificity than stress electrocardiography and therefore are generally preferred. Stress scintigraphy should be used with caution, as it adds to the cumulative radiation exposure.
The role of magnetic resonance imaging and computed tomography depends on the results of initial transthoracic echocardiography and the clinical indication, in addition to the center’s expertise and facilities. However, there are currently no data advocating their use as screening tools, except for early detection of porcelain aorta in high-risk patients.10
MODERN RADIOTHERAPY TECHNIQUES
In recent years, there has been emphasis on exposing the patient to as little radiation as possible without compromising cure.52 The three major strategies employed to decrease cardiac exposure include reducing the radiation dose, reducing the radiation field and volume, and using newer planning and delivery techniques.
Reducing the radiation dose. It is well recognized that the mean dose of radiation to the heart is a significant predictor of cardiovascular disease, with one study demonstrating a linear increase in the risk of coronary artery disease with increasing mean heart radiation dose (excess relative risk per Gy 7.4%, 95% confidence interval 3.3%–14.8%).53
Reducing the radiation field and volume. Modern strategies and computed tomography-based radiotherapy planning have enabled a transition from older techniques such as extended-field radiation therapy, mantle-field radiation therapy, and involved-field radiation therapy to new techniques such as involved-node and involved-site radiation therapy.54 These have shown promise. For instance, a study in patients with early Hodgkin lymphoma found a mean heart dose of 27.5 Gy with mantle-field therapy compared with 7.7 Gy with involved-node therapy. This decrease in mean heart dose was associated with a reduction in the 25-year absolute excess cardiac risk from 9.1% to 1.4% and a reduction in cardiac mortality from 2.1% to 1%.55
Employing newer planning and delivery systems has also demonstrated some promise in reducing rates of cardiac morbidity and mortality. Extended-field radiation therapy, mantle-field radiotherapy, and involved-field radiation therapy were traditionally based on two-dimensional planning and often resulted in large volumes of myocardium being unnecessarily exposed to large doses of radiation because of the uncertainty in targeting. Involved-site and involved-node radiotherapy are based on computed tomography, resulting in more accurate targeting and sparing of normal tissue.
In addition, newer techniques such as intensity-modulated radiotherapy and proton beam therapy have resulted in further improvements in conformality compared with three-dimensional conformal radiotherapy.56,57 Respiratory motion management, including deep inspiration breath-holding and end-inspiration breath-holding, have decreased the radiation dose to the heart in patients undergoing mediastinal radiotherapy.58,59
TOWARD THE GOALS OF PREVENTION AND EARLIER DETECTION
As survival from breast cancer and lymphoma has increased, we continue to see legacy or latent effects of therapy, such as RIHD. Radiation therapy can affect any cardiac structure and is a major cause of morbidity and death in cancer survivors.
Modern radiation techniques use a variety of mechanisms to decrease the radiation dose to the heart. A large body of evidence emanating from an era of higher radiation doses and a lack of knowledge of the cardiac effects of radiation highlight the perilous cardiac consequences of chest radiation. With advances in radiotherapy and the development and widespread implementation of consensus guidelines, we envision earlier detection and less frequent occurrence of RIHD, although the latter trend could be blunted by increased cardiovascular risk factors within the population. Given the lag between irradiation and the cardiac consequences, it may be a number of years before any comparisons can be drawn.
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225–249.
- Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood 2011; 117:412–418.
- Amromin GD, Gildenhorn HL, Solomon RD, Nadkarni BB. The synergism of x-irradiation and cholesterol-fat feeding on the development of coronary artery lesions. J Atheroscler Res 1964; 4:325–334.
- Tribble DL, Barcellos-Hoff MH, Chu BM, Gong EL. Ionizing radiation accelerates aortic lesion formation in fat-fed mice via SOD-inhibitable processes. Arterioscler Thromb Vasc Biol 1999; 19:1387–1392.
- Hayashi T, Morishita Y, Kubo Y, et al. Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am J Med 2005; 118:83–86.
- Gagliardi G, Constine LS, Moiseenko V, et al. Radiation dose-volume effects in the heart. Int J Radiat Oncol Biol Phys 2010; 76(suppl 3):S77–S85.
- Rutqvist LE, Lax I, Fornander T, Johansson H. Cardiovascular mortality in a randomized trial of adjuvant radiation therapy versus surgery alone in primary breast cancer. Int J Radiat Oncol Biol Phys 1992; 22:887–896.
- Hancock SL, Donaldson SS, Hoppe RT. Cardiac disease following treatment of Hodgkin’s disease in children and adolescents. J Clin Oncol 1993; 11:1208–1215.
- Meyer RM, Gospodarowicz MK, Connors JM, et al; NCIC Clinical Trials Group; Eastern Cooperative Oncology Group. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012; 366:399–408.
- Lancellotti P, Nkomo VT, Badano LP, et al; European Society of Cardiology Working Groups on Nuclear Cardiology and Cardiac Computed Tomography and Cardiovascular Magnetic Resonance; American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J Am Soc Echocardiogr 2013; 26:1013–1032.
- Cheng RK, Lee MS, Seki A, et al. Radiation coronary arteritis refractory to surgical and percutaneous revascularization culminating in orthotopic heart transplantation. Cardiovasc Pathol 2013; 22:303–308.
- Hull MC, Morris CG, Pepine CJ, Mendenhall NP. Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of Hodgkin lymphoma treated with radiation therapy. JAMA 2003; 290:2831–2837.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 2013; 368:987–998.
- Lind PA, Pagnanelli R, Marks LB, et al. Myocardial perfusion changes in patients irradiated for left-sided breast cancer and correlation with coronary artery distribution. Int J Radiat Oncol Biol Phys 2003; 55:914–920.
- Rademaker J, Schöder H, Ariaratnam NS, et al. Coronary artery disease after radiation therapy for Hodgkin’s lymphoma: coronary CT angiography findings and calcium scores in nine asymptomatic patients. AJR Am J Roentgenol 2008; 191:32–37.
- Orzan F, Brusca A, Conte MR, Presbitero P, Figliomeni MC. Severe coronary artery disease after radiation therapy of the chest and mediastinum: clinical presentation and treatment. Br Heart J 1993; 69:496–500.
- Mousavi N, Nohria A. Radiation-induced cardiovascular disease. Curr Treat Options Cardiovasc Med 2013; 15:507–517.
- McEniery PT, Dorosti K, Schiavone WA, Pedrick TJ, Sheldon WC. Clinical and angiographic features of coronary artery disease after chest irradiation. Am J Cardiol 1987; 60:1020–1024.
- Renner SM, Massel D, Moon BC. Mediastinal irradiation: a risk factor for atherosclerosis of the internal thoracic arteries. Can J Cardiol 1999; 15:597–600.
- Chang AS, Smedira NG, Chang CL, et al. Cardiac surgery after mediastinal radiation: extent of exposure influences outcome. J Thorac Cardiovasc Surg 2007; 133:404–413.
- Handa N, McGregor CG, Danielson GK, et al. Coronary artery bypass grafting in patients with previous mediastinal radiation therapy. J Thorac Cardiovasc Surg 1999; 117:1136–1142.
- Gharagozloo F, Clements IP, Mullany CJ. Use of the internal mammary artery for myocardial revascularization in a patient with radiation-induced coronary artery disease. Mayo Clin Proc 1992; 67:1081–1084.
- Wu W, Masri A, Popovic ZB, et al. Long-term survival of patients with radiation heart disease undergoing cardiac surgery: a cohort study. Circulation 2013; 127:1476–1485.
- Brand MD, Abadi CA, Aurigemma GP, Dauerman HL, Meyer TE. Radiation-associated valvular heart disease in Hodgkin’s disease is associated with characteristic thickening and fibrosis of the aortic-mitral curtain. J Heart Valve Dis 2001; 10:681–685.
- Nadlonek NA, Weyant MJ, Yu JA, et al. Radiation induces osteogenesis in human aortic valve interstitial cells. J Thorac Cardiovasc Surg 2012; 144:1466–1470.
- Tamura A, Takahara Y, Mogi K, Katsumata M. Radiation-induced valvular disease is the logical consequence of irradiation. Gen Thorac Cardiovasc Surg 2007; 55:53–56.
- Carlson RG, Mayfield WR, Normann S, Alexander JA. Radiation-associated valvular disease. Chest 1991; 99:538–545.
- Desai MY, Wu W, Masri A, et al. Increased aorto-mitral curtain thickness independently predicts mortality in patients with radiation-associated cardiac disease undergoing cardiac surgery. Ann Thorac Surg 2014; 97:1348–1355.
- Crestanello JA, McGregor CG, Danielson GK, et al. Mitral and tricuspid valve repair in patients with previous mediastinal radiation therapy. Ann Thorac Surg 2004; 78:826–831.
- Latib A, Montorfano M, Figini F, et al. Percutaneous valve replacement in a young adult for radiation-induced aortic stenosis. J Cardiovasc Med (Hagerstown) 2012; 13:397–398.
- Veinot JP, Edwards WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol 1996; 27:766–773.
- Carver JR, Shapiro CL, Ng A, et al; ASCO Cancer Survivorship Expert Panel. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 2007; 25:3991–4008.
- Carmel RJ, Kaplan HS. Mantle irradiation in Hodgkin’s disease. An analysis of technique, tumor eradication, and complications. Cancer 1976; 37:2813–2825.
- Morton DL, Glancy DL, Joseph WL, Adkins PC. Management of patients with radiation-induced pericarditis with effusion: a note on the development of aortic regurgitation in two of them. Chest 1973; 64:291–297.
- Arsenian MA. Cardiovascular sequelae of therapeutic thoracic radiation. Prog Cardiovasc Dis 1991; 33:299–311.
- Imazio M, Brucato A, Mayosi BM, et al. Medical therapy of pericardial diseases: part II: Noninfectious pericarditis, pericardial effusion and constrictive pericarditis. J Cardiovasc Med (Hagerstown). 2010; 11:785–794.
- Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and the heart. Circulation 1993; 87:1435–1441.
- Bertog SC, Thambidorai SK, Parakh K, et al. Constrictive pericarditis: etiology and cause-specific survival after pericardiectomy. J Am Coll Cardiol 2004; 43:1445–1452.
- Heidenreich PA, Hancock SL, Vagelos RH, Lee BK, Schnittger I. Diastolic dysfunction after mediastinal irradiation. Am Heart J 2005; 150:977–982.
- Burns RJ, Bar-Shlomo BZ, Druck MN, et al. Detection of radiation cardiomyopathy by gated radionuclide angiography. Am J Med 1983; 74:297–302.
- Constine LS, Schwartz RG, Savage DE, King V, Muhs A. Cardiac function, perfusion, and morbidity in irradiated long-term survivors of Hodgkin’s disease. Int J Radiat Oncol Biol Phys 1997; 39:897–906.
- Tolba KA, Deliargyris EN. Cardiotoxicity of cancer therapy. Cancer Invest 1999; 17:408–422.
- Termuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 2011; 57:1210–1216.
- Handa N, McGregor CG, Daly RC, et al. Heart transplantation for radiation-associated end-stage heart failure. Transpl Int 2000; 13:162–165.
- DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant 2012; 31:1269–1275.
- Adams MJ, Lipshultz SE, Schwartz C, Fajardo LF, Coen V, Constine LS. Radiation-associated cardiovascular disease: manifestations and management. Semin Radiat Oncol 2003; 13:346–356.
- Schwartz CL, Hobbie WL, Truesdell S, Constine LC, Clark EB. Corrected QT interval prolongation in anthracycline-treated survivors of childhood cancer. J Clin Oncol 1993; 11:1906–1910.
- Orzan F, Brusca A, Gaita F, Giustetto C, Figliomeni MC, Libero L. Associated cardiac lesions in patients with radiation-induced complete heart block. Int J Cardiol 1993; 39:151–156.
- Larsen RL, Jakacki RI, Vetter VL, Meadows AT, Silber JH, Barber G. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am J Cardiol 1992; 70:73–77.
- Shapiro CL, Hardenbergh PH, Gelman R, et al. Cardiac effects of adjuvant doxorubicin and radiation therapy in breast cancer patients. J Clin Oncol 1998; 16:3493–3501.
- Armstrong GT, Chen Y, Yasui Y, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med 2016; 374:833–842.
- van Nimwegen FA, Schaapveld M, Cutter DJ, et al. Radiation dose-response relationship for risk of coronary heart disease in survivors of Hodgkin lymphoma. J Clin Oncol 2016; 34:235–243.
- Maraldo MV, Ng AK. Minimizing cardiac risks with contemporary radiation therapy for Hodgkin lymphoma. J Clin Oncol 2016; 34:208–210.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012; 83:1232–1237.
- Maraldo MV, Specht L. A decade of comparative dose planning studies for early-stage Hodgkin lymphoma: what can we learn? Int J Radiat Oncol Biol Phys 2014; 90:1126–1135.
- Hoppe BS, Flampouri S, Su Z, et al. Consolidative involved-node proton therapy for Stage IA-IIIB mediastinal Hodgkin lymphoma: preliminary dosimetric outcomes from a Phase II study. Int J Radiat Oncol Biol Phys 2012; 83:260–267.
- Petersen PM, Aznar MC, Berthelsen AK, et al. Prospective phase II trial of image-guided radiotherapy in Hodgkin lymphoma: benefit of deep inspiration breath-hold. Acta Oncol 2015; 54:60–66.
- Aznar MC, Maraldo MV, Schut DA, et al. Minimizing late effects for patients with mediastinal Hodgkin lymphoma: deep inspiration breath-hold, IMRT, or both? Int J Radiat Oncol Biol Phys 2015; 92:169–174.
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225–249.
- Galper SL, Yu JB, Mauch PM, et al. Clinically significant cardiac disease in patients with Hodgkin lymphoma treated with mediastinal irradiation. Blood 2011; 117:412–418.
- Amromin GD, Gildenhorn HL, Solomon RD, Nadkarni BB. The synergism of x-irradiation and cholesterol-fat feeding on the development of coronary artery lesions. J Atheroscler Res 1964; 4:325–334.
- Tribble DL, Barcellos-Hoff MH, Chu BM, Gong EL. Ionizing radiation accelerates aortic lesion formation in fat-fed mice via SOD-inhibitable processes. Arterioscler Thromb Vasc Biol 1999; 19:1387–1392.
- Hayashi T, Morishita Y, Kubo Y, et al. Long-term effects of radiation dose on inflammatory markers in atomic bomb survivors. Am J Med 2005; 118:83–86.
- Gagliardi G, Constine LS, Moiseenko V, et al. Radiation dose-volume effects in the heart. Int J Radiat Oncol Biol Phys 2010; 76(suppl 3):S77–S85.
- Rutqvist LE, Lax I, Fornander T, Johansson H. Cardiovascular mortality in a randomized trial of adjuvant radiation therapy versus surgery alone in primary breast cancer. Int J Radiat Oncol Biol Phys 1992; 22:887–896.
- Hancock SL, Donaldson SS, Hoppe RT. Cardiac disease following treatment of Hodgkin’s disease in children and adolescents. J Clin Oncol 1993; 11:1208–1215.
- Meyer RM, Gospodarowicz MK, Connors JM, et al; NCIC Clinical Trials Group; Eastern Cooperative Oncology Group. ABVD alone versus radiation-based therapy in limited-stage Hodgkin’s lymphoma. N Engl J Med 2012; 366:399–408.
- Lancellotti P, Nkomo VT, Badano LP, et al; European Society of Cardiology Working Groups on Nuclear Cardiology and Cardiac Computed Tomography and Cardiovascular Magnetic Resonance; American Society of Nuclear Cardiology, Society for Cardiovascular Magnetic Resonance, and Society of Cardiovascular Computed Tomography. Expert consensus for multi-modality imaging evaluation of cardiovascular complications of radiotherapy in adults: a report from the European Association of Cardiovascular Imaging and the American Society of Echocardiography. J Am Soc Echocardiogr 2013; 26:1013–1032.
- Cheng RK, Lee MS, Seki A, et al. Radiation coronary arteritis refractory to surgical and percutaneous revascularization culminating in orthotopic heart transplantation. Cardiovasc Pathol 2013; 22:303–308.
- Hull MC, Morris CG, Pepine CJ, Mendenhall NP. Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of Hodgkin lymphoma treated with radiation therapy. JAMA 2003; 290:2831–2837.
- Clarke M, Collins R, Darby S, et al; Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 366:2087–2106.
- Darby SC, Ewertz M, McGale P, et al. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 2013; 368:987–998.
- Lind PA, Pagnanelli R, Marks LB, et al. Myocardial perfusion changes in patients irradiated for left-sided breast cancer and correlation with coronary artery distribution. Int J Radiat Oncol Biol Phys 2003; 55:914–920.
- Rademaker J, Schöder H, Ariaratnam NS, et al. Coronary artery disease after radiation therapy for Hodgkin’s lymphoma: coronary CT angiography findings and calcium scores in nine asymptomatic patients. AJR Am J Roentgenol 2008; 191:32–37.
- Orzan F, Brusca A, Conte MR, Presbitero P, Figliomeni MC. Severe coronary artery disease after radiation therapy of the chest and mediastinum: clinical presentation and treatment. Br Heart J 1993; 69:496–500.
- Mousavi N, Nohria A. Radiation-induced cardiovascular disease. Curr Treat Options Cardiovasc Med 2013; 15:507–517.
- McEniery PT, Dorosti K, Schiavone WA, Pedrick TJ, Sheldon WC. Clinical and angiographic features of coronary artery disease after chest irradiation. Am J Cardiol 1987; 60:1020–1024.
- Renner SM, Massel D, Moon BC. Mediastinal irradiation: a risk factor for atherosclerosis of the internal thoracic arteries. Can J Cardiol 1999; 15:597–600.
- Chang AS, Smedira NG, Chang CL, et al. Cardiac surgery after mediastinal radiation: extent of exposure influences outcome. J Thorac Cardiovasc Surg 2007; 133:404–413.
- Handa N, McGregor CG, Danielson GK, et al. Coronary artery bypass grafting in patients with previous mediastinal radiation therapy. J Thorac Cardiovasc Surg 1999; 117:1136–1142.
- Gharagozloo F, Clements IP, Mullany CJ. Use of the internal mammary artery for myocardial revascularization in a patient with radiation-induced coronary artery disease. Mayo Clin Proc 1992; 67:1081–1084.
- Wu W, Masri A, Popovic ZB, et al. Long-term survival of patients with radiation heart disease undergoing cardiac surgery: a cohort study. Circulation 2013; 127:1476–1485.
- Brand MD, Abadi CA, Aurigemma GP, Dauerman HL, Meyer TE. Radiation-associated valvular heart disease in Hodgkin’s disease is associated with characteristic thickening and fibrosis of the aortic-mitral curtain. J Heart Valve Dis 2001; 10:681–685.
- Nadlonek NA, Weyant MJ, Yu JA, et al. Radiation induces osteogenesis in human aortic valve interstitial cells. J Thorac Cardiovasc Surg 2012; 144:1466–1470.
- Tamura A, Takahara Y, Mogi K, Katsumata M. Radiation-induced valvular disease is the logical consequence of irradiation. Gen Thorac Cardiovasc Surg 2007; 55:53–56.
- Carlson RG, Mayfield WR, Normann S, Alexander JA. Radiation-associated valvular disease. Chest 1991; 99:538–545.
- Desai MY, Wu W, Masri A, et al. Increased aorto-mitral curtain thickness independently predicts mortality in patients with radiation-associated cardiac disease undergoing cardiac surgery. Ann Thorac Surg 2014; 97:1348–1355.
- Crestanello JA, McGregor CG, Danielson GK, et al. Mitral and tricuspid valve repair in patients with previous mediastinal radiation therapy. Ann Thorac Surg 2004; 78:826–831.
- Latib A, Montorfano M, Figini F, et al. Percutaneous valve replacement in a young adult for radiation-induced aortic stenosis. J Cardiovasc Med (Hagerstown) 2012; 13:397–398.
- Veinot JP, Edwards WD. Pathology of radiation-induced heart disease: a surgical and autopsy study of 27 cases. Hum Pathol 1996; 27:766–773.
- Carver JR, Shapiro CL, Ng A, et al; ASCO Cancer Survivorship Expert Panel. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 2007; 25:3991–4008.
- Carmel RJ, Kaplan HS. Mantle irradiation in Hodgkin’s disease. An analysis of technique, tumor eradication, and complications. Cancer 1976; 37:2813–2825.
- Morton DL, Glancy DL, Joseph WL, Adkins PC. Management of patients with radiation-induced pericarditis with effusion: a note on the development of aortic regurgitation in two of them. Chest 1973; 64:291–297.
- Arsenian MA. Cardiovascular sequelae of therapeutic thoracic radiation. Prog Cardiovasc Dis 1991; 33:299–311.
- Imazio M, Brucato A, Mayosi BM, et al. Medical therapy of pericardial diseases: part II: Noninfectious pericarditis, pericardial effusion and constrictive pericarditis. J Cardiovasc Med (Hagerstown). 2010; 11:785–794.
- Polikar R, Burger AG, Scherrer U, Nicod P. The thyroid and the heart. Circulation 1993; 87:1435–1441.
- Bertog SC, Thambidorai SK, Parakh K, et al. Constrictive pericarditis: etiology and cause-specific survival after pericardiectomy. J Am Coll Cardiol 2004; 43:1445–1452.
- Heidenreich PA, Hancock SL, Vagelos RH, Lee BK, Schnittger I. Diastolic dysfunction after mediastinal irradiation. Am Heart J 2005; 150:977–982.
- Burns RJ, Bar-Shlomo BZ, Druck MN, et al. Detection of radiation cardiomyopathy by gated radionuclide angiography. Am J Med 1983; 74:297–302.
- Constine LS, Schwartz RG, Savage DE, King V, Muhs A. Cardiac function, perfusion, and morbidity in irradiated long-term survivors of Hodgkin’s disease. Int J Radiat Oncol Biol Phys 1997; 39:897–906.
- Tolba KA, Deliargyris EN. Cardiotoxicity of cancer therapy. Cancer Invest 1999; 17:408–422.
- Termuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 2011; 57:1210–1216.
- Handa N, McGregor CG, Daly RC, et al. Heart transplantation for radiation-associated end-stage heart failure. Transpl Int 2000; 13:162–165.
- DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant 2012; 31:1269–1275.
- Adams MJ, Lipshultz SE, Schwartz C, Fajardo LF, Coen V, Constine LS. Radiation-associated cardiovascular disease: manifestations and management. Semin Radiat Oncol 2003; 13:346–356.
- Schwartz CL, Hobbie WL, Truesdell S, Constine LC, Clark EB. Corrected QT interval prolongation in anthracycline-treated survivors of childhood cancer. J Clin Oncol 1993; 11:1906–1910.
- Orzan F, Brusca A, Gaita F, Giustetto C, Figliomeni MC, Libero L. Associated cardiac lesions in patients with radiation-induced complete heart block. Int J Cardiol 1993; 39:151–156.
- Larsen RL, Jakacki RI, Vetter VL, Meadows AT, Silber JH, Barber G. Electrocardiographic changes and arrhythmias after cancer therapy in children and young adults. Am J Cardiol 1992; 70:73–77.
- Shapiro CL, Hardenbergh PH, Gelman R, et al. Cardiac effects of adjuvant doxorubicin and radiation therapy in breast cancer patients. J Clin Oncol 1998; 16:3493–3501.
- Armstrong GT, Chen Y, Yasui Y, et al. Reduction in late mortality among 5-year survivors of childhood cancer. N Engl J Med 2016; 374:833–842.
- van Nimwegen FA, Schaapveld M, Cutter DJ, et al. Radiation dose-response relationship for risk of coronary heart disease in survivors of Hodgkin lymphoma. J Clin Oncol 2016; 34:235–243.
- Maraldo MV, Ng AK. Minimizing cardiac risks with contemporary radiation therapy for Hodgkin lymphoma. J Clin Oncol 2016; 34:208–210.
- Maraldo MV, Brodin NP, Vogelius IR, et al. Risk of developing cardiovascular disease after involved node radiotherapy versus mantle field for Hodgkin lymphoma. Int J Radiat Oncol Biol Phys 2012; 83:1232–1237.
- Maraldo MV, Specht L. A decade of comparative dose planning studies for early-stage Hodgkin lymphoma: what can we learn? Int J Radiat Oncol Biol Phys 2014; 90:1126–1135.
- Hoppe BS, Flampouri S, Su Z, et al. Consolidative involved-node proton therapy for Stage IA-IIIB mediastinal Hodgkin lymphoma: preliminary dosimetric outcomes from a Phase II study. Int J Radiat Oncol Biol Phys 2012; 83:260–267.
- Petersen PM, Aznar MC, Berthelsen AK, et al. Prospective phase II trial of image-guided radiotherapy in Hodgkin lymphoma: benefit of deep inspiration breath-hold. Acta Oncol 2015; 54:60–66.
- Aznar MC, Maraldo MV, Schut DA, et al. Minimizing late effects for patients with mediastinal Hodgkin lymphoma: deep inspiration breath-hold, IMRT, or both? Int J Radiat Oncol Biol Phys 2015; 92:169–174.
KEY POINTS
- Ischemic heart disease is the most common cause of cardiac death after radiotherapy. Valvular, pericardial, myocardial, and conduction system disease are also common.
- Surgery may not be an attractive option because of radiation-induced fibrosis of surrounding structures. Consequently, conservative interventions are preferred.
- The incidence of RIHD is expected to decline, as lower doses of radiation are being used in radiotherapy than in the past.
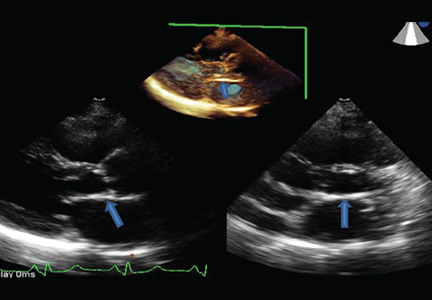
Women’s health 2016: An update for internists
Women's health encompasses a variety of topics relevant to the daily practice of internists. Staying up to date with the evidence in this wide field is a challenge.
This article reviews important studies published in 2015 and early 2016 on treatment of urinary tract infections, the optimal duration of bisphosphonate use, ovarian cancer screening, the impact of oral contraceptives and lactation on mortality rates, and the risks and benefits of intrauterine contraception. We critically appraised the studies and judged that their methodology was strong and appropriate for inclusion in this review.
IBUPROFEN FOR URINARY TRACT INFECTIONS
A 36-year-old woman reports 4 days of mild to moderate dysuria, frequency, and urgency. She denies fever, nausea, or back pain. Her last urinary tract infection was 2 years ago. Office urinalysis reveals leukocyte esterase and nitrites. She has read an article about antibiotic resistance and Clostridium difficile infection and asks you if antibiotics are truly necessary. What do you recommend?
Urinary tract infections are often self-limited
Uncomplicated urinary tract infections account for 25% of antibiotic prescriptions in primary care.1
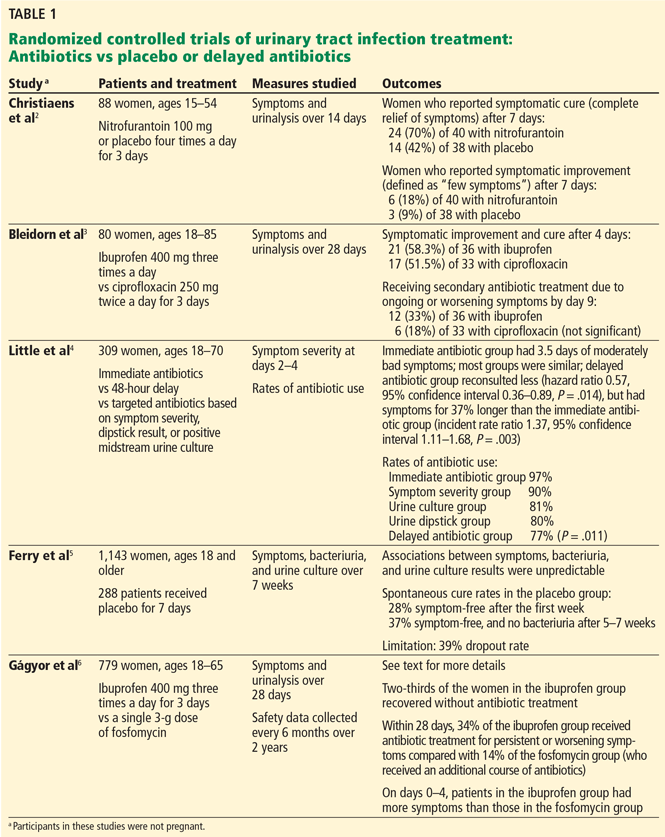
Several small studies have suggested that many of these infections are self-limited, resolving within 3 to 14 days without antibiotics (Table 1).2–6 A potential disadvantage of withholding treatment is slower bacterial clearance and resolution of symptoms, but reducing the number of antibiotic prescriptions may help slow antibiotic resistance.7,8 Surveys and qualitative studies have suggested that women are concerned about the harms of antibiotic treatment and so may be willing to avoid or postpone antibiotic use.9–11
Ibuprofen vs fosfomycin
Gágyor et al6 conducted a double-blind, randomized multicenter trial in 42 general practices in Germany to assess whether treating the symptoms of uncomplicated urinary tract infection with ibuprofen would reduce antibiotic use without worsening outcomes.
Of the 779 eligible women with suspected urinary tract infection, 281 declined to participate in the study, 4 did not participate for reasons not specified, 246 received a single dose of fosfomycin 3 g, and 248 were treated with ibuprofen 400 mg three times a day for 3 days. Participants scored their daily symptoms and activity impairment, and safety data were collected for adverse events and relapses up to day 28 and within 6 and 12 months. In both groups, if symptoms worsened or persisted, antibiotic therapy was initiated at the discretion of the treating physician.
Exclusion criteria included fever, “loin” (back) tenderness, pregnancy, renal disease, a previous urinary tract infection within 2 weeks, urinary catheterization, and a contraindication to nonsteroidal anti-inflammatory medications.
Results. Within 28 days of symptom onset, women in the ibuprofen group had received 81 courses of antibiotics for symptoms of urinary tract infection (plus another 13 courses for other reasons), compared with 277 courses for urinary tract infection in the fosfomycin group (plus 6 courses for other reasons), for a relative rate reduction in antibiotic use of 66.5% (95% confidence interval [CI] 58.8%–74.4%, P < .001). The women who received ibuprofen were more likely to need antibiotics after initial treatment because of refractory symptoms but were still less likely to receive antibiotics overall (Table 1).
The mean duration of symptoms was slightly shorter in the fosfomycin group (4.6 vs 5.6 days, P < .001). However, the percentage of patients who had a recurrent urinary tract infection within 2 to 4 weeks was higher in the fosfomycin-treated patients (11% vs 6% P = .049).
Although the study was not powered to show significant differences in pyelonephritis, five patients in the ibuprofen group developed pyelonephritis compared with one in the antibiotic-treated group (P = .12).
An important limitation of the study was that nonparticipants had higher symptom scores, which may mean that the results are not generalizable to women who have recurrent urinary tract infections, longer duration of symptoms, or symptoms that are more severe. The strengths of the study were that more than half of all potentially eligible women were enrolled, and baseline data were collected from nonparticipants.
Can our patient avoid antibiotics?
Given the mild nature of her symptoms, the clinician should discuss with her the risks vs benefits of delaying antibiotics, once it has been determined that she has no risk factors for severe urinary tract infection. Her symptoms are likely to resolve within 1 week even if she declines antibiotic treatment, though they may last a day longer with ibuprofen alone than if she had received antibiotics. She should watch for symptoms of pyelonephritis (eg, flank pain, fever, chills, vomiting) and should seek prompt medical care if such symptoms occur.
DISCONTINUING BISPHOSPHONATES
A 64-year-old woman has taken alendronate for her osteoporosis for 5 years. She has no history of fractures. Her original bone density scans showed a T-score of –2.6 at the spine and –1.5 at the hip. Since she started to take alendronate, there has been no further loss in bone mineral density. She is tolerating the drug well and does not take any other medications. Should she continue the bisphosphonate?
Optimal duration of therapy unknown
The risks and benefits of long-term bisphosphonate use are debated.
In the Fracture Intervention Trial (FIT),12 women with low bone mineral density of the femoral neck were randomized to receive alendronate or placebo and were followed for 36 months. The alendronate group had significantly fewer vertebral fractures and clinical fractures overall. Then, in the FIT Long-term Extension (FLEX) study,13 1,009 alendronate-treated women in the FIT study were rerandomized to receive 5 years of additional treatment or to stop treatment. Bone density in the untreated women decreased, although not to the level it was before treatment. At the end of the study, there was no difference in hip fracture rate between the two groups (3% of each group had had a hip fracture), although women in the treated group had a lower rate of clinical vertebral fracture (2% vs 5%, relative risk 0.5, 95% CI 0.2–0.8).
In addition, rare but serious risks have been associated with bisphosphonate use, specifically atypical femoral fracture and osteonecrosis of the jaw. A US Food and Drug Administration (FDA) evaluation of long-term bisphosphonate use concluded that there was evidence of an increased risk of osteonecrosis of the jaw with longer duration of use, but causality was not established. The evaluation also noted conflicting results about the association with atypical femoral fracture.14
Based on this report and focusing on the absence of nonspine benefit after 5 years, the FDA suggested that bisphosphonates may be safely discontinued in some patients without compromising therapeutic gains, but no adequate clinical trial has yet delineated how long the benefits of treatment are maintained after cessation. A periodic reevaluation of continued need was recommended.14
New recommendations from the American Society for Bone and Mineral Research
Age is the greatest risk factor for fracture.15 Therefore, deciding whether to discontinue a bisphosphonate when a woman is older, and hence at higher risk, is a challenge.
A task force of the American Society for Bone and Mineral Research (ASBMR) has developed an evidence-based guideline on managing osteoporosis in patients on long-term bisphosphonate treatment.16 The goal was to provide guidance on the duration of bisphosphonate therapy from the perspective of risk vs benefit. The authors conducted a systematic review focusing on two randomized controlled trials (FLEX13 and the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Pivotal Fracture Trial17) that provided data on long-term bisphosphonate use.
The task force recommended16 that after 5 years of oral bisphosphonates or 3 years of intravenous bisphosphonates, risk should be reassessed. In women at high fracture risk, they recommended continuing the oral bisphosphonate for 10 years or the intravenous bisphosphonate for 6 years. Factors that favored continuation of bisphosphonate therapy were as follows:
- An osteoporotic fracture before or during therapy
- A hip bone mineral density T-score ≤ –2.5
- High risk of fracture, defined as age older than 70 or 75, other strong risk factors for fracture, or a FRAX fracture risk score18 above a country-specific threshold.
(The FRAX score is based on age, sex, weight, height, previous fracture, hip fracture in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, alcohol use, and bone mineral density in the femoral neck. It gives an estimate of the 10-year risk of major osteoporotic fracture and hip fracture. High risk would be a 10-year risk of major osteoporotic fracture greater than 20% or a 10-year risk of hip fracture greater than 3%.)
For women at high risk, the risks of atypical femoral fracture and osteonecrosis of the jaw are outweighed by the benefit of a reduction in vertebral fracture risk. For women not at high risk of fracture, a drug holiday of 2 to 3 years can be considered after 3 to 5 years of treatment.
Although the task force recommended reassessment after 2 to 3 years of drug holiday, how best to do this is not clear. The task force did not recommend a specific approach to reassessment, so decisions about when to restart therapy after a drug holiday could potentially be informed by subsequent bone mineral density testing if it were to show persistent bone loss. Another option could be to restart bisphosphonates after a defined amount of time (eg, 3–5 years) for women who have previously experienced benefit.
The task force recommendations are in line with those of other societies, the FDA, and expert opinion.19–23
The American Association of Clinical Endocrinologists recommends considering a drug holiday in low-risk patients after 4 to 5 years of treatment. For high-risk patients, they recommend 1 to 2 years of drug holiday after 10 years of treatment. They encourage restarting treatment if bone mineral density decreases, bone turnover markers rise, or fracture occurs.19 This is a grade C recommendation, meaning the advice is based on descriptive studies and expert opinion.
Although some clinicians restart bisphosphonates when markers of bone turnover such as NTX (N-telopeptide of type 1 collagen) rise to premenopausal levels, there is no evidence to support this strategy.24
The task force recommendations are based on limited evidence that primarily comes from white postmenopausal women. Another important limitation is that the outcomes are primarily vertebral fractures. However, until additional evidence is available, these guidelines can be useful in guiding decision-making.
Should our patient continue therapy?
Our patient is relatively young and does not have any of the high-risk features noted within the task force recommendations. She has responded well to bisphosphonate treatment and so can consider a drug holiday at this time.
OVARIAN CANCER SCREENING
A 50-year-old woman requests screening for ovarian cancer. She is postmenopausal and has no personal or family history of cancer. She is concerned because a friend forwarded an e-mail stating, “Please tell all your female friends and relatives to insist on a cancer antigen (CA) 125 blood test every year as part of their annual exam. This is an inexpensive and simple blood test. Don’t take no for an answer. If I had known then what I know now, we would have caught my cancer much earlier, before it was stage III!” What should you tell the patient?
Ovarian cancer is the most deadly of female reproductive cancers, largely because in most patients the cancer has already spread beyond the ovary by the time of clinical detection. Death rates from ovarian cancer have decreased only slightly in the past 30 years.
Little benefit and considerable harm of screening
In 2011, the Prostate Lung Colorectal Ovarian (PLCO) Cancer Screening trial25 randomized more than 68,000 women ages 55 to 74 from the general US population to annual screening with CA 125 testing and transvaginal ultrasonography compared with usual care. They were followed for a median of 12.4 years.
Screening did not affect stage at diagnosis (77%–78% were in stage III or IV in both the screening and usual care groups), nor did it reduce the rate of death from ovarian cancer. In addition, false-positive findings led to some harm: nearly one in three women who had a positive screening test underwent surgery. Of 3,285 women with false-positive results, 1,080 underwent surgery, and 15% of these had at least one serious complication. The trial was stopped early due to evidence of futility.
A new UK study also found no benefit from screening
In the PLCO study, a CA 125 result of 35 U/mL or greater was classified as abnormal. However, researchers in the United Kingdom postulated that instead of using a single cutoff for a normal or abnormal CA 125 level, it would be better to interpret the CA 125 result according to a somewhat complicated (and proprietary) algorithm called the Risk of Ovarian Cancer Algorithm (ROCA).26,27 The ROCA takes into account a woman’s age, menopausal status, known genetic mutations (BRCA 1 or 2 or Lynch syndrome), Ashkenazi Jewish descent, and family history of ovarian or breast cancer, as well as any change in CA 125 level over time.
In a 2016 UK study,26 202,638 postmenopausal women ages 50 to 74 were randomized to no screening, annual screening with transvaginal ultrasonography, or multimodal screening with an annual CA 125 blood test interpreted with the ROCA algorithm, adding transvaginal ultrasonography as a second-line test when needed if the CA 125 level was abnormal based on the ROCA. Women with abnormal findings on multimodal screening or ultrasonography had repeat tests, and women with persistent abnormalities underwent clinical evaluation and, when appropriate, surgery.
Participants were at average risk of ovarian cancer; those with suspected familial ovarian cancer syndrome were excluded, as were those with a personal history of ovarian cancer or other active cancer.
Results. At a median follow-up of 11.1 years, the percentage of women who were diagnosed with ovarian cancer was 0.7% in the multimodal screening group, 0.6% in the screening ultrasonography group, and 0.6% in the no-screening group. Comparing either multimodal or screening ultrasonography with no screening, there was no statistically significant reduction in mortality rate over 14 years of follow-up.
Screening had significant costs and potential harms. For every ovarian or peritoneal cancer detected by screening, an additional 2 women in the multimodal screening group and 10 women in the ultrasonography group underwent needless surgery.
Strengths of this trial included its large size, allowing adequate power to detect differences in outcomes, its multicenter setting, its high compliance rate, and the low crossover rate in the no-screening group. However, the design of the study makes it difficult to anticipate the late effects of screening. Also, the patient must purchase ROCA testing online and must also pay a consultation fee. Insurance providers do not cover this test.
Should our patient proceed with ovarian cancer screening?
No. Current evidence shows no clear benefit to ovarian cancer screening for average-risk women, and we should not recommend yearly ultrasonography and CA 125 level testing, as they are likely to cause harm without providing benefit. The US Preventive Services Task Force recommends against screening for ovarian cancer.28 For premenopausal women, pregnancy, hormonal contraception, and breastfeeding all significantly decrease ovarian cancer risk by suppressing ovulation.29–31
REPRODUCTIVE FACTORS AND THE RISK OF DEATH
A 26-year-old woman comes in to discuss her contraceptive options. She has been breastfeeding since the birth of her first baby 6 months ago, and wonders how lactation and contraception may affect her long-term health.
Questions about the safety of contraceptive options are common, especially in breastfeeding mothers.
In 2010, the long-term Royal College of General Practitioners’ Oral Contraceptive Study reported that the all-cause mortality rate was actually lower in women who used oral contraceptives.32 Similarly, in 2013, an Oxford study that followed 17,032 women for over 30 years reported no association between oral contraceptives and breast cancer.33
However, in 2014, results from the Nurses’ Health Study indicated that breast cancer rates were higher in oral contraceptive users, although reassuringly, the study found no difference in all-cause mortality rates in women who had used oral contraception.34
The European Prospective Investigation Into Cancer and Nutrition
To further characterize relationships between reproductive characteristics and mortality rates, investigators analyzed data from the European Prospective Investigation Into Cancer and Nutrition,35 which recruited 322,972 women from 10 countries between 1992 and 2000. Analyses were stratified by study center and participant age and were adjusted for body mass index, physical activity, education level, smoking, and menopausal status; alcohol intake was examined as a potential confounder but was excluded from final models.
Findings. Over an average 13 years of follow-up, the rate of all-cause mortality was 20% lower in parous than in nulliparous women. In parous women, the all-cause mortality rate was additionally 18% lower in those who had breastfed vs those who had never breastfed, although breastfeeding duration was not associated with mortality. Use of oral contraceptives lowered all-cause mortality by 10% among nonsmokers; in smokers, no association with all-cause mortality was seen for oral contraceptive use, as smoking is such a powerful risk factor for mortality. The primary contributor to all-cause mortality appeared to be ischemic heart disease, the incidence of which was significantly lower in parous women (by 14%) and those who breastfed (by 20%) and was not related to oral contraceptive use.35
Strengths of this study included the large sample size recruited from countries across Europe, with varying rates of breastfeeding and contraceptive use. However, as with all observational studies, it remains subject to the possibility of residual confounding.
What should we tell this patient?
After congratulating her for breastfeeding, we can reassure her about the safety of all available contraceptives. According to the US Centers for Disease Control and Prevention (CDC),36 after 42 days postpartum most women can use combined hormonal contraception. All other methods can be used immediately postpartum, including progestin-only pills.
As lactational amenorrhea is only effective while mothers are exclusively breastfeeding, and short interpregnancy intervals have been associated with higher rates of adverse pregnancy outcomes,37 this patient will likely benefit from promptly starting a prescription contraceptive.
HIGHLY EFFECTIVE REVERSIBLE CONTRACEPTION
This same 26-year-old patient is concerned that she will not remember to take an oral contraceptive every day, and expresses interest in a more convenient method of contraception. However, she is concerned about the potential risks.
Although intrauterine contraceptives (IUCs) are typically 20 times more effective than oral contraceptives38 and have been used by millions of women worldwide, rates of use in the United States have been lower than in many other countries.39
A study of intrauterine contraception
To clarify the safety of IUCs, researchers followed 61,448 women who underwent IUC placement in six European countries between 2006 and 2013.40 Most participants received an IUC containing levonorgestrel, while 30% received a copper IUC.
Findings. Overall, rates of uterine perforation were low (approximately 1 per 1,000 insertions). The most significant risk factors for perforation were breastfeeding at the time of insertion and insertion less than 36 weeks after the last delivery. None of the perforations in the study led to serious illness or injury of intra-abdominal or pelvic structures. Interestingly, women using a levonorgestrel IUC were considerably less likely to experience a contraceptive failure than those using a copper IUC.41
Strengths of this study included the prospective data collection and power to examine rare clinical outcomes. However, it was industry-funded.
The risk of pelvic infection with an IUC is so low that the CDC does not recommend prophylactic antibiotics with the insertion procedure. If women have other indications for testing for sexually transmitted disease, an IUC can be placed the same day as testing, and before results are available.42 If a woman is found to have a sexually transmitted disease while she has an IUC in place, she should be treated with antibiotics, and there is no need to remove the IUC.43
Subdermal implants
Another highly effective contraceptive option for this patient is the progestin-only subdermal contraceptive implant (marketed in the United States as Nexplanon). Implants have been well-studied and found to have no adverse effect on lactation.44
Learning to place a subdermal contraceptive is far easier than learning to place an IUC, but it requires a few hours of FDA-mandated in-person training. Unfortunately, relatively few clinicians have obtained this training.45 As placing a subdermal contraceptive is like placing an intravenous line without needing to hit the vein, this procedure can easily be incorporated into a primary care practice. Training from the manufacturer is available to providers who request it.
What should we tell this patient?
An IUC is a great option for many women. When pregnancy is desired, the device is easily removed. Of the three IUCs now available in the United States, those containing 52 mg of levonorgestrel (marketed in the United States as Mirena and Liletta) are the most effective.
The only option more effective than these IUCs is subdermal contraception.46 These reversible contraceptives are typically more effective than permanent contraceptives (ie, tubal ligation)47 and can be removed at any time if a patient wishes to switch to another method or to become pregnant.
Pregnancy rates following attempts at “sterilization” are higher than many realize. There are a variety of approaches to “tying tubes,” some of which may not result in complete tubal occlusion. The failure rate of the laparoscopic approach, according to the US Collaborative Review of Sterilization, ranges from 7.5 per 1,000 procedures for unipolar coagulation to a high of 36.5 per 1,000 for the spring clip.48 The relatively commonly used Filshie clip was not included in this study, but its failure rate is reported to be between 1% and 2%.
- Hooton TM. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med 2012; 366:1028–1037.
- Christiaens TC, De Meyere M, Verschraegen G, et al. Randomised controlled trial of nitrofurantoin versus placebo in the treatment of uncomplicated urinary tract infection in adult women. Br J Gen Pract 2002; 52:729–734.
- Bleidorn J, Gágyor I, Kochen MM, Wegscheider K, Hummers-Pradier E. Symptomatic treatment (ibuprofen) or antibiotics (ciprofloxacin) for uncomplicated urinary tract infection?—results of a randomized controlled pilot trial. BMC Med 2010; 8:30. doi: 10.1186/1741-7015-8-30.
- Little P, Moore MV, Turner S, et al. Effectiveness of five different approaches in management of urinary tract infection: randomised controlled trial. BMJ 2010; 340:c199.
- Ferry SA, Holm SE, Stenlund H, Lundholm R, Monsen TJ. The natural course of uncomplicated lower urinary tract infection in women illustrated by a randomized placebo controlled study. Scand J Infect Dis 2004; 36:296–301.
- Gágyor I, Bleidorn J, Kochen MM, Schmiemann G, Wegscheider K, Hummers-Pradier E. Ibuprofen versus fosfomycin for uncomplicated urinary tract infection in women: randomised controlled trial. BMJ 2015; 351:h6544. doi: 10.1136/bmj.h6544.
- Butler CC, Dunstan F, Heginbothom M, et al. Containing antibiotic resistance: decreased antibiotic-resistant coliform urinary tract infections with reduction in antibiotic prescribing by general practices. Br J Gen Pract 2007; 57:785–792.
- Gottesman BS, Carmeli Y, Shitrit P, Chowers M. Impact of quinolone restriction on resistance patterns of Escherichia coli isolated from urine by culture in a community setting. Clin Infect Dis 2009; 49:869–875.
- Knottnerus BJ, Geerlings SE, Moll van Charante EP, ter Riet G. Women with symptoms of uncomplicated urinary tract infection are often willing to delay antibiotic treatment: a prospective cohort study. BMC Fam Pract 2013; 14:71. doi: 10.1186/1471-2296-14-71.
- Leydon GM, Turner S, Smith H, Little P; UTIS team. Women’s views about management and cause of urinary tract infection: qualitative interview study. BMJ 2010; 340:c279. doi: 10.1136/bmj.c279.
- Willems CS, van den Broek D’Obrenan J, Numans ME, Verheij TJ, van der Velden AW. Cystitis: antibiotic prescribing, consultation, attitudes and opinions. Fam Pract 2014; 31:149–155.
- Black DM, Cummings SR, Karpf DB et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- Black DM, Schwartz AV, Ensrud KE, et al; FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 2006; 296:2927–2938.
- US Food and Drug Administration. Background document for meeting of Advisory Committee for Reproductive Health Drugs and Drug Safety and Risk Management Advisory Committee. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM270958.pdf. Accessed November 3, 2016.
- Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int 2005; 16:581–589.
- Adler RA, El-Hajj Fuleihan G, Bauer DC, et al. Managing osteoporosis in patients on long-term bisphosphonate treatment: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2016; 31:16–35.
- Black DM, Reid IR, Boonen S, et al. The effect of 3 versus 6 years of zoledronic acid treatment of osteoporosis: a randomized extension to the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 2012; 27:243–254.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed October 7, 2016.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010; 16(suppl 3):1–37.
- Whitaker M, Guo J, Kehoe T, Benson G. Bisphosphonates for osteoporosis—where do we go from here? N Engl J Med 2012; 366:2048–2051.
- Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med 2012; 366:2051–2053.
- Brown JP, Morin S, Leslie W, et al. Bisphosphonates for treatment of osteoporosis: expected benefits, potential harms, and drug holidays. Can Fam Physician 2014; 60:324–333.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab 2010; 95:1555–1565.
- Bauer DC, Schwartz A, Palermo L, et al. Fracture prediction after discontinuation of 4 to 5 years of alendronate therapy: the FLEX study. JAMA Intern Med 2014; 174:1126–1134.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011; 305:2295–2303.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet 2016; 387:945–956.
- Abcodia Inc. The ROCA test. www.therocatest.co.uk/for-clinicians/about-roca. Accessed November 3, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med 2012; 157:900–904.
- Titus-Ernstoff L, Perez K, Cramer DW, Harlow BL, Baron JA, Greenberg ER. Menstrual and reproductive factors in relation to ovarian cancer risk. Br J Cancer 2001; 84:714–721.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer, Beral V, Doll R, Hermon C, Peto R, Reeves G. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008; 371:303–314.
- Chowdhury R, Sinha B, Sankar MJ, et al. Breastfeeding and maternal health outcomes: a systematic review and meta-analysis. Acta Paediatr 2015; 104:96–113.
- Hannaford PC, Iversen L, Macfarlane TV, Elliott AM, Angus V, Lee AJ. Mortality among contraceptive pill users: cohort evidence from Royal College of General Practitioners’ Oral Contraception Study. BMJ 2010; 340:c927. doi: 10.1136/bmj.c927.
- Vessey M, Yeates D. Oral contraceptive use and cancer: final report from the Oxford-Family Planning Association contraceptive study. Contraception 2013; 88:678–683.
- Charlton BM, Rich-Edwards JW, Colditz GA, et al. Oral contraceptive use and mortality after 36 years of follow-up in the Nurses’ Health Study: prospective cohort study. BMJ 2014; 349:g6356. doi: 10.1136/bmj.g6356.
- Merritt MA, Riboli E, Murphy N, et al. Reproductive factors and risk of mortality in the European Prospective Investigation into Cancer and Nutrition; a cohort study. BMC Med 2015; 13:252. doi: 10.1186/s12916-015-0484-3.
- Centers for Disease Control and Prevention (CDC). Update to CDC’s U.S. Medical Eligibility Criteria for Contraceptive Use, 2010: revised recommendations for the use of contraceptive methods during the postpartum period. MMWR Morb Mortal Wkly Rep 2011; 60:878–883.
- Bigelow CA, Bryant AS. Short interpregnancy intervals: an evidence-based guide for clinicians. Obstet Gynecol Surv 2015; 70:458–464.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Buhling KJ, Zite NB, Lotke P, Black K; INTRA Writing Group. Worldwide use of intrauterine contraception: a review. Contraception 2014; 89:162–173.
- Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception 2015; 91:274–279.
- Heinemann K, Reed S, Moehner S, Minh TD. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception 2015; 91:280–283.
- Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement. Am J Obstet Gynecol 2016 May 12. pii: S0002-9378(16)30212-5. doi: 10.1016/j.ajog.2016.05.017. [Epub ahead of print]
- Division of Reproductive health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). U.S. Selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62(RR-05):1–60.
- Gurtcheff SE, Turok DK, Stoddard G, Murphy PA, Gibson M, Jones KP. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol 2011; 117:1114–1121.
- Nisen MB, Peterson LE, Cochrane A, Rubin SE. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception 2016; 93:432–437.
- Polis CB, Bradley SE, Bankole A, Onda T, Croft T, Singh S. Typical-use contraceptive failure rates in 43 countries with Demographic and Health Survey data: summary of a detailed report. Contraception 2016; 94:11–17.
- Gariepy AM, Creinin MD, Smith KJ, Xu X. Probability of pregnancy after sterilization: a comparison of hysteroscopic versus laparoscopic sterilization. Contraception 2014; 90:174–181.
- Peterson HB, Xia Z, Hughes JM, Wilcox LS, Tylor LR, Trussel J. The risk of pregnancy after tubal sterilization: findings from the U.S. Collaborative Rerview of Sterilization. Am J Obstet Gynecol 1996; 174:1161–1168.
Women's health encompasses a variety of topics relevant to the daily practice of internists. Staying up to date with the evidence in this wide field is a challenge.
This article reviews important studies published in 2015 and early 2016 on treatment of urinary tract infections, the optimal duration of bisphosphonate use, ovarian cancer screening, the impact of oral contraceptives and lactation on mortality rates, and the risks and benefits of intrauterine contraception. We critically appraised the studies and judged that their methodology was strong and appropriate for inclusion in this review.
IBUPROFEN FOR URINARY TRACT INFECTIONS
A 36-year-old woman reports 4 days of mild to moderate dysuria, frequency, and urgency. She denies fever, nausea, or back pain. Her last urinary tract infection was 2 years ago. Office urinalysis reveals leukocyte esterase and nitrites. She has read an article about antibiotic resistance and Clostridium difficile infection and asks you if antibiotics are truly necessary. What do you recommend?
Urinary tract infections are often self-limited
Uncomplicated urinary tract infections account for 25% of antibiotic prescriptions in primary care.1
Several small studies have suggested that many of these infections are self-limited, resolving within 3 to 14 days without antibiotics (Table 1).2–6 A potential disadvantage of withholding treatment is slower bacterial clearance and resolution of symptoms, but reducing the number of antibiotic prescriptions may help slow antibiotic resistance.7,8 Surveys and qualitative studies have suggested that women are concerned about the harms of antibiotic treatment and so may be willing to avoid or postpone antibiotic use.9–11
Ibuprofen vs fosfomycin
Gágyor et al6 conducted a double-blind, randomized multicenter trial in 42 general practices in Germany to assess whether treating the symptoms of uncomplicated urinary tract infection with ibuprofen would reduce antibiotic use without worsening outcomes.
Of the 779 eligible women with suspected urinary tract infection, 281 declined to participate in the study, 4 did not participate for reasons not specified, 246 received a single dose of fosfomycin 3 g, and 248 were treated with ibuprofen 400 mg three times a day for 3 days. Participants scored their daily symptoms and activity impairment, and safety data were collected for adverse events and relapses up to day 28 and within 6 and 12 months. In both groups, if symptoms worsened or persisted, antibiotic therapy was initiated at the discretion of the treating physician.
Exclusion criteria included fever, “loin” (back) tenderness, pregnancy, renal disease, a previous urinary tract infection within 2 weeks, urinary catheterization, and a contraindication to nonsteroidal anti-inflammatory medications.
Results. Within 28 days of symptom onset, women in the ibuprofen group had received 81 courses of antibiotics for symptoms of urinary tract infection (plus another 13 courses for other reasons), compared with 277 courses for urinary tract infection in the fosfomycin group (plus 6 courses for other reasons), for a relative rate reduction in antibiotic use of 66.5% (95% confidence interval [CI] 58.8%–74.4%, P < .001). The women who received ibuprofen were more likely to need antibiotics after initial treatment because of refractory symptoms but were still less likely to receive antibiotics overall (Table 1).
The mean duration of symptoms was slightly shorter in the fosfomycin group (4.6 vs 5.6 days, P < .001). However, the percentage of patients who had a recurrent urinary tract infection within 2 to 4 weeks was higher in the fosfomycin-treated patients (11% vs 6% P = .049).
Although the study was not powered to show significant differences in pyelonephritis, five patients in the ibuprofen group developed pyelonephritis compared with one in the antibiotic-treated group (P = .12).
An important limitation of the study was that nonparticipants had higher symptom scores, which may mean that the results are not generalizable to women who have recurrent urinary tract infections, longer duration of symptoms, or symptoms that are more severe. The strengths of the study were that more than half of all potentially eligible women were enrolled, and baseline data were collected from nonparticipants.
Can our patient avoid antibiotics?
Given the mild nature of her symptoms, the clinician should discuss with her the risks vs benefits of delaying antibiotics, once it has been determined that she has no risk factors for severe urinary tract infection. Her symptoms are likely to resolve within 1 week even if she declines antibiotic treatment, though they may last a day longer with ibuprofen alone than if she had received antibiotics. She should watch for symptoms of pyelonephritis (eg, flank pain, fever, chills, vomiting) and should seek prompt medical care if such symptoms occur.
DISCONTINUING BISPHOSPHONATES
A 64-year-old woman has taken alendronate for her osteoporosis for 5 years. She has no history of fractures. Her original bone density scans showed a T-score of –2.6 at the spine and –1.5 at the hip. Since she started to take alendronate, there has been no further loss in bone mineral density. She is tolerating the drug well and does not take any other medications. Should she continue the bisphosphonate?
Optimal duration of therapy unknown
The risks and benefits of long-term bisphosphonate use are debated.
In the Fracture Intervention Trial (FIT),12 women with low bone mineral density of the femoral neck were randomized to receive alendronate or placebo and were followed for 36 months. The alendronate group had significantly fewer vertebral fractures and clinical fractures overall. Then, in the FIT Long-term Extension (FLEX) study,13 1,009 alendronate-treated women in the FIT study were rerandomized to receive 5 years of additional treatment or to stop treatment. Bone density in the untreated women decreased, although not to the level it was before treatment. At the end of the study, there was no difference in hip fracture rate between the two groups (3% of each group had had a hip fracture), although women in the treated group had a lower rate of clinical vertebral fracture (2% vs 5%, relative risk 0.5, 95% CI 0.2–0.8).
In addition, rare but serious risks have been associated with bisphosphonate use, specifically atypical femoral fracture and osteonecrosis of the jaw. A US Food and Drug Administration (FDA) evaluation of long-term bisphosphonate use concluded that there was evidence of an increased risk of osteonecrosis of the jaw with longer duration of use, but causality was not established. The evaluation also noted conflicting results about the association with atypical femoral fracture.14
Based on this report and focusing on the absence of nonspine benefit after 5 years, the FDA suggested that bisphosphonates may be safely discontinued in some patients without compromising therapeutic gains, but no adequate clinical trial has yet delineated how long the benefits of treatment are maintained after cessation. A periodic reevaluation of continued need was recommended.14
New recommendations from the American Society for Bone and Mineral Research
Age is the greatest risk factor for fracture.15 Therefore, deciding whether to discontinue a bisphosphonate when a woman is older, and hence at higher risk, is a challenge.
A task force of the American Society for Bone and Mineral Research (ASBMR) has developed an evidence-based guideline on managing osteoporosis in patients on long-term bisphosphonate treatment.16 The goal was to provide guidance on the duration of bisphosphonate therapy from the perspective of risk vs benefit. The authors conducted a systematic review focusing on two randomized controlled trials (FLEX13 and the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Pivotal Fracture Trial17) that provided data on long-term bisphosphonate use.
The task force recommended16 that after 5 years of oral bisphosphonates or 3 years of intravenous bisphosphonates, risk should be reassessed. In women at high fracture risk, they recommended continuing the oral bisphosphonate for 10 years or the intravenous bisphosphonate for 6 years. Factors that favored continuation of bisphosphonate therapy were as follows:
- An osteoporotic fracture before or during therapy
- A hip bone mineral density T-score ≤ –2.5
- High risk of fracture, defined as age older than 70 or 75, other strong risk factors for fracture, or a FRAX fracture risk score18 above a country-specific threshold.
(The FRAX score is based on age, sex, weight, height, previous fracture, hip fracture in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, alcohol use, and bone mineral density in the femoral neck. It gives an estimate of the 10-year risk of major osteoporotic fracture and hip fracture. High risk would be a 10-year risk of major osteoporotic fracture greater than 20% or a 10-year risk of hip fracture greater than 3%.)
For women at high risk, the risks of atypical femoral fracture and osteonecrosis of the jaw are outweighed by the benefit of a reduction in vertebral fracture risk. For women not at high risk of fracture, a drug holiday of 2 to 3 years can be considered after 3 to 5 years of treatment.
Although the task force recommended reassessment after 2 to 3 years of drug holiday, how best to do this is not clear. The task force did not recommend a specific approach to reassessment, so decisions about when to restart therapy after a drug holiday could potentially be informed by subsequent bone mineral density testing if it were to show persistent bone loss. Another option could be to restart bisphosphonates after a defined amount of time (eg, 3–5 years) for women who have previously experienced benefit.
The task force recommendations are in line with those of other societies, the FDA, and expert opinion.19–23
The American Association of Clinical Endocrinologists recommends considering a drug holiday in low-risk patients after 4 to 5 years of treatment. For high-risk patients, they recommend 1 to 2 years of drug holiday after 10 years of treatment. They encourage restarting treatment if bone mineral density decreases, bone turnover markers rise, or fracture occurs.19 This is a grade C recommendation, meaning the advice is based on descriptive studies and expert opinion.
Although some clinicians restart bisphosphonates when markers of bone turnover such as NTX (N-telopeptide of type 1 collagen) rise to premenopausal levels, there is no evidence to support this strategy.24
The task force recommendations are based on limited evidence that primarily comes from white postmenopausal women. Another important limitation is that the outcomes are primarily vertebral fractures. However, until additional evidence is available, these guidelines can be useful in guiding decision-making.
Should our patient continue therapy?
Our patient is relatively young and does not have any of the high-risk features noted within the task force recommendations. She has responded well to bisphosphonate treatment and so can consider a drug holiday at this time.
OVARIAN CANCER SCREENING
A 50-year-old woman requests screening for ovarian cancer. She is postmenopausal and has no personal or family history of cancer. She is concerned because a friend forwarded an e-mail stating, “Please tell all your female friends and relatives to insist on a cancer antigen (CA) 125 blood test every year as part of their annual exam. This is an inexpensive and simple blood test. Don’t take no for an answer. If I had known then what I know now, we would have caught my cancer much earlier, before it was stage III!” What should you tell the patient?
Ovarian cancer is the most deadly of female reproductive cancers, largely because in most patients the cancer has already spread beyond the ovary by the time of clinical detection. Death rates from ovarian cancer have decreased only slightly in the past 30 years.
Little benefit and considerable harm of screening
In 2011, the Prostate Lung Colorectal Ovarian (PLCO) Cancer Screening trial25 randomized more than 68,000 women ages 55 to 74 from the general US population to annual screening with CA 125 testing and transvaginal ultrasonography compared with usual care. They were followed for a median of 12.4 years.
Screening did not affect stage at diagnosis (77%–78% were in stage III or IV in both the screening and usual care groups), nor did it reduce the rate of death from ovarian cancer. In addition, false-positive findings led to some harm: nearly one in three women who had a positive screening test underwent surgery. Of 3,285 women with false-positive results, 1,080 underwent surgery, and 15% of these had at least one serious complication. The trial was stopped early due to evidence of futility.
A new UK study also found no benefit from screening
In the PLCO study, a CA 125 result of 35 U/mL or greater was classified as abnormal. However, researchers in the United Kingdom postulated that instead of using a single cutoff for a normal or abnormal CA 125 level, it would be better to interpret the CA 125 result according to a somewhat complicated (and proprietary) algorithm called the Risk of Ovarian Cancer Algorithm (ROCA).26,27 The ROCA takes into account a woman’s age, menopausal status, known genetic mutations (BRCA 1 or 2 or Lynch syndrome), Ashkenazi Jewish descent, and family history of ovarian or breast cancer, as well as any change in CA 125 level over time.
In a 2016 UK study,26 202,638 postmenopausal women ages 50 to 74 were randomized to no screening, annual screening with transvaginal ultrasonography, or multimodal screening with an annual CA 125 blood test interpreted with the ROCA algorithm, adding transvaginal ultrasonography as a second-line test when needed if the CA 125 level was abnormal based on the ROCA. Women with abnormal findings on multimodal screening or ultrasonography had repeat tests, and women with persistent abnormalities underwent clinical evaluation and, when appropriate, surgery.
Participants were at average risk of ovarian cancer; those with suspected familial ovarian cancer syndrome were excluded, as were those with a personal history of ovarian cancer or other active cancer.
Results. At a median follow-up of 11.1 years, the percentage of women who were diagnosed with ovarian cancer was 0.7% in the multimodal screening group, 0.6% in the screening ultrasonography group, and 0.6% in the no-screening group. Comparing either multimodal or screening ultrasonography with no screening, there was no statistically significant reduction in mortality rate over 14 years of follow-up.
Screening had significant costs and potential harms. For every ovarian or peritoneal cancer detected by screening, an additional 2 women in the multimodal screening group and 10 women in the ultrasonography group underwent needless surgery.
Strengths of this trial included its large size, allowing adequate power to detect differences in outcomes, its multicenter setting, its high compliance rate, and the low crossover rate in the no-screening group. However, the design of the study makes it difficult to anticipate the late effects of screening. Also, the patient must purchase ROCA testing online and must also pay a consultation fee. Insurance providers do not cover this test.
Should our patient proceed with ovarian cancer screening?
No. Current evidence shows no clear benefit to ovarian cancer screening for average-risk women, and we should not recommend yearly ultrasonography and CA 125 level testing, as they are likely to cause harm without providing benefit. The US Preventive Services Task Force recommends against screening for ovarian cancer.28 For premenopausal women, pregnancy, hormonal contraception, and breastfeeding all significantly decrease ovarian cancer risk by suppressing ovulation.29–31
REPRODUCTIVE FACTORS AND THE RISK OF DEATH
A 26-year-old woman comes in to discuss her contraceptive options. She has been breastfeeding since the birth of her first baby 6 months ago, and wonders how lactation and contraception may affect her long-term health.
Questions about the safety of contraceptive options are common, especially in breastfeeding mothers.
In 2010, the long-term Royal College of General Practitioners’ Oral Contraceptive Study reported that the all-cause mortality rate was actually lower in women who used oral contraceptives.32 Similarly, in 2013, an Oxford study that followed 17,032 women for over 30 years reported no association between oral contraceptives and breast cancer.33
However, in 2014, results from the Nurses’ Health Study indicated that breast cancer rates were higher in oral contraceptive users, although reassuringly, the study found no difference in all-cause mortality rates in women who had used oral contraception.34
The European Prospective Investigation Into Cancer and Nutrition
To further characterize relationships between reproductive characteristics and mortality rates, investigators analyzed data from the European Prospective Investigation Into Cancer and Nutrition,35 which recruited 322,972 women from 10 countries between 1992 and 2000. Analyses were stratified by study center and participant age and were adjusted for body mass index, physical activity, education level, smoking, and menopausal status; alcohol intake was examined as a potential confounder but was excluded from final models.
Findings. Over an average 13 years of follow-up, the rate of all-cause mortality was 20% lower in parous than in nulliparous women. In parous women, the all-cause mortality rate was additionally 18% lower in those who had breastfed vs those who had never breastfed, although breastfeeding duration was not associated with mortality. Use of oral contraceptives lowered all-cause mortality by 10% among nonsmokers; in smokers, no association with all-cause mortality was seen for oral contraceptive use, as smoking is such a powerful risk factor for mortality. The primary contributor to all-cause mortality appeared to be ischemic heart disease, the incidence of which was significantly lower in parous women (by 14%) and those who breastfed (by 20%) and was not related to oral contraceptive use.35
Strengths of this study included the large sample size recruited from countries across Europe, with varying rates of breastfeeding and contraceptive use. However, as with all observational studies, it remains subject to the possibility of residual confounding.
What should we tell this patient?
After congratulating her for breastfeeding, we can reassure her about the safety of all available contraceptives. According to the US Centers for Disease Control and Prevention (CDC),36 after 42 days postpartum most women can use combined hormonal contraception. All other methods can be used immediately postpartum, including progestin-only pills.
As lactational amenorrhea is only effective while mothers are exclusively breastfeeding, and short interpregnancy intervals have been associated with higher rates of adverse pregnancy outcomes,37 this patient will likely benefit from promptly starting a prescription contraceptive.
HIGHLY EFFECTIVE REVERSIBLE CONTRACEPTION
This same 26-year-old patient is concerned that she will not remember to take an oral contraceptive every day, and expresses interest in a more convenient method of contraception. However, she is concerned about the potential risks.
Although intrauterine contraceptives (IUCs) are typically 20 times more effective than oral contraceptives38 and have been used by millions of women worldwide, rates of use in the United States have been lower than in many other countries.39
A study of intrauterine contraception
To clarify the safety of IUCs, researchers followed 61,448 women who underwent IUC placement in six European countries between 2006 and 2013.40 Most participants received an IUC containing levonorgestrel, while 30% received a copper IUC.
Findings. Overall, rates of uterine perforation were low (approximately 1 per 1,000 insertions). The most significant risk factors for perforation were breastfeeding at the time of insertion and insertion less than 36 weeks after the last delivery. None of the perforations in the study led to serious illness or injury of intra-abdominal or pelvic structures. Interestingly, women using a levonorgestrel IUC were considerably less likely to experience a contraceptive failure than those using a copper IUC.41
Strengths of this study included the prospective data collection and power to examine rare clinical outcomes. However, it was industry-funded.
The risk of pelvic infection with an IUC is so low that the CDC does not recommend prophylactic antibiotics with the insertion procedure. If women have other indications for testing for sexually transmitted disease, an IUC can be placed the same day as testing, and before results are available.42 If a woman is found to have a sexually transmitted disease while she has an IUC in place, she should be treated with antibiotics, and there is no need to remove the IUC.43
Subdermal implants
Another highly effective contraceptive option for this patient is the progestin-only subdermal contraceptive implant (marketed in the United States as Nexplanon). Implants have been well-studied and found to have no adverse effect on lactation.44
Learning to place a subdermal contraceptive is far easier than learning to place an IUC, but it requires a few hours of FDA-mandated in-person training. Unfortunately, relatively few clinicians have obtained this training.45 As placing a subdermal contraceptive is like placing an intravenous line without needing to hit the vein, this procedure can easily be incorporated into a primary care practice. Training from the manufacturer is available to providers who request it.
What should we tell this patient?
An IUC is a great option for many women. When pregnancy is desired, the device is easily removed. Of the three IUCs now available in the United States, those containing 52 mg of levonorgestrel (marketed in the United States as Mirena and Liletta) are the most effective.
The only option more effective than these IUCs is subdermal contraception.46 These reversible contraceptives are typically more effective than permanent contraceptives (ie, tubal ligation)47 and can be removed at any time if a patient wishes to switch to another method or to become pregnant.
Pregnancy rates following attempts at “sterilization” are higher than many realize. There are a variety of approaches to “tying tubes,” some of which may not result in complete tubal occlusion. The failure rate of the laparoscopic approach, according to the US Collaborative Review of Sterilization, ranges from 7.5 per 1,000 procedures for unipolar coagulation to a high of 36.5 per 1,000 for the spring clip.48 The relatively commonly used Filshie clip was not included in this study, but its failure rate is reported to be between 1% and 2%.
Women's health encompasses a variety of topics relevant to the daily practice of internists. Staying up to date with the evidence in this wide field is a challenge.
This article reviews important studies published in 2015 and early 2016 on treatment of urinary tract infections, the optimal duration of bisphosphonate use, ovarian cancer screening, the impact of oral contraceptives and lactation on mortality rates, and the risks and benefits of intrauterine contraception. We critically appraised the studies and judged that their methodology was strong and appropriate for inclusion in this review.
IBUPROFEN FOR URINARY TRACT INFECTIONS
A 36-year-old woman reports 4 days of mild to moderate dysuria, frequency, and urgency. She denies fever, nausea, or back pain. Her last urinary tract infection was 2 years ago. Office urinalysis reveals leukocyte esterase and nitrites. She has read an article about antibiotic resistance and Clostridium difficile infection and asks you if antibiotics are truly necessary. What do you recommend?
Urinary tract infections are often self-limited
Uncomplicated urinary tract infections account for 25% of antibiotic prescriptions in primary care.1
Several small studies have suggested that many of these infections are self-limited, resolving within 3 to 14 days without antibiotics (Table 1).2–6 A potential disadvantage of withholding treatment is slower bacterial clearance and resolution of symptoms, but reducing the number of antibiotic prescriptions may help slow antibiotic resistance.7,8 Surveys and qualitative studies have suggested that women are concerned about the harms of antibiotic treatment and so may be willing to avoid or postpone antibiotic use.9–11
Ibuprofen vs fosfomycin
Gágyor et al6 conducted a double-blind, randomized multicenter trial in 42 general practices in Germany to assess whether treating the symptoms of uncomplicated urinary tract infection with ibuprofen would reduce antibiotic use without worsening outcomes.
Of the 779 eligible women with suspected urinary tract infection, 281 declined to participate in the study, 4 did not participate for reasons not specified, 246 received a single dose of fosfomycin 3 g, and 248 were treated with ibuprofen 400 mg three times a day for 3 days. Participants scored their daily symptoms and activity impairment, and safety data were collected for adverse events and relapses up to day 28 and within 6 and 12 months. In both groups, if symptoms worsened or persisted, antibiotic therapy was initiated at the discretion of the treating physician.
Exclusion criteria included fever, “loin” (back) tenderness, pregnancy, renal disease, a previous urinary tract infection within 2 weeks, urinary catheterization, and a contraindication to nonsteroidal anti-inflammatory medications.
Results. Within 28 days of symptom onset, women in the ibuprofen group had received 81 courses of antibiotics for symptoms of urinary tract infection (plus another 13 courses for other reasons), compared with 277 courses for urinary tract infection in the fosfomycin group (plus 6 courses for other reasons), for a relative rate reduction in antibiotic use of 66.5% (95% confidence interval [CI] 58.8%–74.4%, P < .001). The women who received ibuprofen were more likely to need antibiotics after initial treatment because of refractory symptoms but were still less likely to receive antibiotics overall (Table 1).
The mean duration of symptoms was slightly shorter in the fosfomycin group (4.6 vs 5.6 days, P < .001). However, the percentage of patients who had a recurrent urinary tract infection within 2 to 4 weeks was higher in the fosfomycin-treated patients (11% vs 6% P = .049).
Although the study was not powered to show significant differences in pyelonephritis, five patients in the ibuprofen group developed pyelonephritis compared with one in the antibiotic-treated group (P = .12).
An important limitation of the study was that nonparticipants had higher symptom scores, which may mean that the results are not generalizable to women who have recurrent urinary tract infections, longer duration of symptoms, or symptoms that are more severe. The strengths of the study were that more than half of all potentially eligible women were enrolled, and baseline data were collected from nonparticipants.
Can our patient avoid antibiotics?
Given the mild nature of her symptoms, the clinician should discuss with her the risks vs benefits of delaying antibiotics, once it has been determined that she has no risk factors for severe urinary tract infection. Her symptoms are likely to resolve within 1 week even if she declines antibiotic treatment, though they may last a day longer with ibuprofen alone than if she had received antibiotics. She should watch for symptoms of pyelonephritis (eg, flank pain, fever, chills, vomiting) and should seek prompt medical care if such symptoms occur.
DISCONTINUING BISPHOSPHONATES
A 64-year-old woman has taken alendronate for her osteoporosis for 5 years. She has no history of fractures. Her original bone density scans showed a T-score of –2.6 at the spine and –1.5 at the hip. Since she started to take alendronate, there has been no further loss in bone mineral density. She is tolerating the drug well and does not take any other medications. Should she continue the bisphosphonate?
Optimal duration of therapy unknown
The risks and benefits of long-term bisphosphonate use are debated.
In the Fracture Intervention Trial (FIT),12 women with low bone mineral density of the femoral neck were randomized to receive alendronate or placebo and were followed for 36 months. The alendronate group had significantly fewer vertebral fractures and clinical fractures overall. Then, in the FIT Long-term Extension (FLEX) study,13 1,009 alendronate-treated women in the FIT study were rerandomized to receive 5 years of additional treatment or to stop treatment. Bone density in the untreated women decreased, although not to the level it was before treatment. At the end of the study, there was no difference in hip fracture rate between the two groups (3% of each group had had a hip fracture), although women in the treated group had a lower rate of clinical vertebral fracture (2% vs 5%, relative risk 0.5, 95% CI 0.2–0.8).
In addition, rare but serious risks have been associated with bisphosphonate use, specifically atypical femoral fracture and osteonecrosis of the jaw. A US Food and Drug Administration (FDA) evaluation of long-term bisphosphonate use concluded that there was evidence of an increased risk of osteonecrosis of the jaw with longer duration of use, but causality was not established. The evaluation also noted conflicting results about the association with atypical femoral fracture.14
Based on this report and focusing on the absence of nonspine benefit after 5 years, the FDA suggested that bisphosphonates may be safely discontinued in some patients without compromising therapeutic gains, but no adequate clinical trial has yet delineated how long the benefits of treatment are maintained after cessation. A periodic reevaluation of continued need was recommended.14
New recommendations from the American Society for Bone and Mineral Research
Age is the greatest risk factor for fracture.15 Therefore, deciding whether to discontinue a bisphosphonate when a woman is older, and hence at higher risk, is a challenge.
A task force of the American Society for Bone and Mineral Research (ASBMR) has developed an evidence-based guideline on managing osteoporosis in patients on long-term bisphosphonate treatment.16 The goal was to provide guidance on the duration of bisphosphonate therapy from the perspective of risk vs benefit. The authors conducted a systematic review focusing on two randomized controlled trials (FLEX13 and the Health Outcomes and Reduced Incidence With Zoledronic Acid Once Yearly Pivotal Fracture Trial17) that provided data on long-term bisphosphonate use.
The task force recommended16 that after 5 years of oral bisphosphonates or 3 years of intravenous bisphosphonates, risk should be reassessed. In women at high fracture risk, they recommended continuing the oral bisphosphonate for 10 years or the intravenous bisphosphonate for 6 years. Factors that favored continuation of bisphosphonate therapy were as follows:
- An osteoporotic fracture before or during therapy
- A hip bone mineral density T-score ≤ –2.5
- High risk of fracture, defined as age older than 70 or 75, other strong risk factors for fracture, or a FRAX fracture risk score18 above a country-specific threshold.
(The FRAX score is based on age, sex, weight, height, previous fracture, hip fracture in a parent, current smoking, use of glucocorticoids, rheumatoid arthritis, secondary osteoporosis, alcohol use, and bone mineral density in the femoral neck. It gives an estimate of the 10-year risk of major osteoporotic fracture and hip fracture. High risk would be a 10-year risk of major osteoporotic fracture greater than 20% or a 10-year risk of hip fracture greater than 3%.)
For women at high risk, the risks of atypical femoral fracture and osteonecrosis of the jaw are outweighed by the benefit of a reduction in vertebral fracture risk. For women not at high risk of fracture, a drug holiday of 2 to 3 years can be considered after 3 to 5 years of treatment.
Although the task force recommended reassessment after 2 to 3 years of drug holiday, how best to do this is not clear. The task force did not recommend a specific approach to reassessment, so decisions about when to restart therapy after a drug holiday could potentially be informed by subsequent bone mineral density testing if it were to show persistent bone loss. Another option could be to restart bisphosphonates after a defined amount of time (eg, 3–5 years) for women who have previously experienced benefit.
The task force recommendations are in line with those of other societies, the FDA, and expert opinion.19–23
The American Association of Clinical Endocrinologists recommends considering a drug holiday in low-risk patients after 4 to 5 years of treatment. For high-risk patients, they recommend 1 to 2 years of drug holiday after 10 years of treatment. They encourage restarting treatment if bone mineral density decreases, bone turnover markers rise, or fracture occurs.19 This is a grade C recommendation, meaning the advice is based on descriptive studies and expert opinion.
Although some clinicians restart bisphosphonates when markers of bone turnover such as NTX (N-telopeptide of type 1 collagen) rise to premenopausal levels, there is no evidence to support this strategy.24
The task force recommendations are based on limited evidence that primarily comes from white postmenopausal women. Another important limitation is that the outcomes are primarily vertebral fractures. However, until additional evidence is available, these guidelines can be useful in guiding decision-making.
Should our patient continue therapy?
Our patient is relatively young and does not have any of the high-risk features noted within the task force recommendations. She has responded well to bisphosphonate treatment and so can consider a drug holiday at this time.
OVARIAN CANCER SCREENING
A 50-year-old woman requests screening for ovarian cancer. She is postmenopausal and has no personal or family history of cancer. She is concerned because a friend forwarded an e-mail stating, “Please tell all your female friends and relatives to insist on a cancer antigen (CA) 125 blood test every year as part of their annual exam. This is an inexpensive and simple blood test. Don’t take no for an answer. If I had known then what I know now, we would have caught my cancer much earlier, before it was stage III!” What should you tell the patient?
Ovarian cancer is the most deadly of female reproductive cancers, largely because in most patients the cancer has already spread beyond the ovary by the time of clinical detection. Death rates from ovarian cancer have decreased only slightly in the past 30 years.
Little benefit and considerable harm of screening
In 2011, the Prostate Lung Colorectal Ovarian (PLCO) Cancer Screening trial25 randomized more than 68,000 women ages 55 to 74 from the general US population to annual screening with CA 125 testing and transvaginal ultrasonography compared with usual care. They were followed for a median of 12.4 years.
Screening did not affect stage at diagnosis (77%–78% were in stage III or IV in both the screening and usual care groups), nor did it reduce the rate of death from ovarian cancer. In addition, false-positive findings led to some harm: nearly one in three women who had a positive screening test underwent surgery. Of 3,285 women with false-positive results, 1,080 underwent surgery, and 15% of these had at least one serious complication. The trial was stopped early due to evidence of futility.
A new UK study also found no benefit from screening
In the PLCO study, a CA 125 result of 35 U/mL or greater was classified as abnormal. However, researchers in the United Kingdom postulated that instead of using a single cutoff for a normal or abnormal CA 125 level, it would be better to interpret the CA 125 result according to a somewhat complicated (and proprietary) algorithm called the Risk of Ovarian Cancer Algorithm (ROCA).26,27 The ROCA takes into account a woman’s age, menopausal status, known genetic mutations (BRCA 1 or 2 or Lynch syndrome), Ashkenazi Jewish descent, and family history of ovarian or breast cancer, as well as any change in CA 125 level over time.
In a 2016 UK study,26 202,638 postmenopausal women ages 50 to 74 were randomized to no screening, annual screening with transvaginal ultrasonography, or multimodal screening with an annual CA 125 blood test interpreted with the ROCA algorithm, adding transvaginal ultrasonography as a second-line test when needed if the CA 125 level was abnormal based on the ROCA. Women with abnormal findings on multimodal screening or ultrasonography had repeat tests, and women with persistent abnormalities underwent clinical evaluation and, when appropriate, surgery.
Participants were at average risk of ovarian cancer; those with suspected familial ovarian cancer syndrome were excluded, as were those with a personal history of ovarian cancer or other active cancer.
Results. At a median follow-up of 11.1 years, the percentage of women who were diagnosed with ovarian cancer was 0.7% in the multimodal screening group, 0.6% in the screening ultrasonography group, and 0.6% in the no-screening group. Comparing either multimodal or screening ultrasonography with no screening, there was no statistically significant reduction in mortality rate over 14 years of follow-up.
Screening had significant costs and potential harms. For every ovarian or peritoneal cancer detected by screening, an additional 2 women in the multimodal screening group and 10 women in the ultrasonography group underwent needless surgery.
Strengths of this trial included its large size, allowing adequate power to detect differences in outcomes, its multicenter setting, its high compliance rate, and the low crossover rate in the no-screening group. However, the design of the study makes it difficult to anticipate the late effects of screening. Also, the patient must purchase ROCA testing online and must also pay a consultation fee. Insurance providers do not cover this test.
Should our patient proceed with ovarian cancer screening?
No. Current evidence shows no clear benefit to ovarian cancer screening for average-risk women, and we should not recommend yearly ultrasonography and CA 125 level testing, as they are likely to cause harm without providing benefit. The US Preventive Services Task Force recommends against screening for ovarian cancer.28 For premenopausal women, pregnancy, hormonal contraception, and breastfeeding all significantly decrease ovarian cancer risk by suppressing ovulation.29–31
REPRODUCTIVE FACTORS AND THE RISK OF DEATH
A 26-year-old woman comes in to discuss her contraceptive options. She has been breastfeeding since the birth of her first baby 6 months ago, and wonders how lactation and contraception may affect her long-term health.
Questions about the safety of contraceptive options are common, especially in breastfeeding mothers.
In 2010, the long-term Royal College of General Practitioners’ Oral Contraceptive Study reported that the all-cause mortality rate was actually lower in women who used oral contraceptives.32 Similarly, in 2013, an Oxford study that followed 17,032 women for over 30 years reported no association between oral contraceptives and breast cancer.33
However, in 2014, results from the Nurses’ Health Study indicated that breast cancer rates were higher in oral contraceptive users, although reassuringly, the study found no difference in all-cause mortality rates in women who had used oral contraception.34
The European Prospective Investigation Into Cancer and Nutrition
To further characterize relationships between reproductive characteristics and mortality rates, investigators analyzed data from the European Prospective Investigation Into Cancer and Nutrition,35 which recruited 322,972 women from 10 countries between 1992 and 2000. Analyses were stratified by study center and participant age and were adjusted for body mass index, physical activity, education level, smoking, and menopausal status; alcohol intake was examined as a potential confounder but was excluded from final models.
Findings. Over an average 13 years of follow-up, the rate of all-cause mortality was 20% lower in parous than in nulliparous women. In parous women, the all-cause mortality rate was additionally 18% lower in those who had breastfed vs those who had never breastfed, although breastfeeding duration was not associated with mortality. Use of oral contraceptives lowered all-cause mortality by 10% among nonsmokers; in smokers, no association with all-cause mortality was seen for oral contraceptive use, as smoking is such a powerful risk factor for mortality. The primary contributor to all-cause mortality appeared to be ischemic heart disease, the incidence of which was significantly lower in parous women (by 14%) and those who breastfed (by 20%) and was not related to oral contraceptive use.35
Strengths of this study included the large sample size recruited from countries across Europe, with varying rates of breastfeeding and contraceptive use. However, as with all observational studies, it remains subject to the possibility of residual confounding.
What should we tell this patient?
After congratulating her for breastfeeding, we can reassure her about the safety of all available contraceptives. According to the US Centers for Disease Control and Prevention (CDC),36 after 42 days postpartum most women can use combined hormonal contraception. All other methods can be used immediately postpartum, including progestin-only pills.
As lactational amenorrhea is only effective while mothers are exclusively breastfeeding, and short interpregnancy intervals have been associated with higher rates of adverse pregnancy outcomes,37 this patient will likely benefit from promptly starting a prescription contraceptive.
HIGHLY EFFECTIVE REVERSIBLE CONTRACEPTION
This same 26-year-old patient is concerned that she will not remember to take an oral contraceptive every day, and expresses interest in a more convenient method of contraception. However, she is concerned about the potential risks.
Although intrauterine contraceptives (IUCs) are typically 20 times more effective than oral contraceptives38 and have been used by millions of women worldwide, rates of use in the United States have been lower than in many other countries.39
A study of intrauterine contraception
To clarify the safety of IUCs, researchers followed 61,448 women who underwent IUC placement in six European countries between 2006 and 2013.40 Most participants received an IUC containing levonorgestrel, while 30% received a copper IUC.
Findings. Overall, rates of uterine perforation were low (approximately 1 per 1,000 insertions). The most significant risk factors for perforation were breastfeeding at the time of insertion and insertion less than 36 weeks after the last delivery. None of the perforations in the study led to serious illness or injury of intra-abdominal or pelvic structures. Interestingly, women using a levonorgestrel IUC were considerably less likely to experience a contraceptive failure than those using a copper IUC.41
Strengths of this study included the prospective data collection and power to examine rare clinical outcomes. However, it was industry-funded.
The risk of pelvic infection with an IUC is so low that the CDC does not recommend prophylactic antibiotics with the insertion procedure. If women have other indications for testing for sexually transmitted disease, an IUC can be placed the same day as testing, and before results are available.42 If a woman is found to have a sexually transmitted disease while she has an IUC in place, she should be treated with antibiotics, and there is no need to remove the IUC.43
Subdermal implants
Another highly effective contraceptive option for this patient is the progestin-only subdermal contraceptive implant (marketed in the United States as Nexplanon). Implants have been well-studied and found to have no adverse effect on lactation.44
Learning to place a subdermal contraceptive is far easier than learning to place an IUC, but it requires a few hours of FDA-mandated in-person training. Unfortunately, relatively few clinicians have obtained this training.45 As placing a subdermal contraceptive is like placing an intravenous line without needing to hit the vein, this procedure can easily be incorporated into a primary care practice. Training from the manufacturer is available to providers who request it.
What should we tell this patient?
An IUC is a great option for many women. When pregnancy is desired, the device is easily removed. Of the three IUCs now available in the United States, those containing 52 mg of levonorgestrel (marketed in the United States as Mirena and Liletta) are the most effective.
The only option more effective than these IUCs is subdermal contraception.46 These reversible contraceptives are typically more effective than permanent contraceptives (ie, tubal ligation)47 and can be removed at any time if a patient wishes to switch to another method or to become pregnant.
Pregnancy rates following attempts at “sterilization” are higher than many realize. There are a variety of approaches to “tying tubes,” some of which may not result in complete tubal occlusion. The failure rate of the laparoscopic approach, according to the US Collaborative Review of Sterilization, ranges from 7.5 per 1,000 procedures for unipolar coagulation to a high of 36.5 per 1,000 for the spring clip.48 The relatively commonly used Filshie clip was not included in this study, but its failure rate is reported to be between 1% and 2%.
- Hooton TM. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med 2012; 366:1028–1037.
- Christiaens TC, De Meyere M, Verschraegen G, et al. Randomised controlled trial of nitrofurantoin versus placebo in the treatment of uncomplicated urinary tract infection in adult women. Br J Gen Pract 2002; 52:729–734.
- Bleidorn J, Gágyor I, Kochen MM, Wegscheider K, Hummers-Pradier E. Symptomatic treatment (ibuprofen) or antibiotics (ciprofloxacin) for uncomplicated urinary tract infection?—results of a randomized controlled pilot trial. BMC Med 2010; 8:30. doi: 10.1186/1741-7015-8-30.
- Little P, Moore MV, Turner S, et al. Effectiveness of five different approaches in management of urinary tract infection: randomised controlled trial. BMJ 2010; 340:c199.
- Ferry SA, Holm SE, Stenlund H, Lundholm R, Monsen TJ. The natural course of uncomplicated lower urinary tract infection in women illustrated by a randomized placebo controlled study. Scand J Infect Dis 2004; 36:296–301.
- Gágyor I, Bleidorn J, Kochen MM, Schmiemann G, Wegscheider K, Hummers-Pradier E. Ibuprofen versus fosfomycin for uncomplicated urinary tract infection in women: randomised controlled trial. BMJ 2015; 351:h6544. doi: 10.1136/bmj.h6544.
- Butler CC, Dunstan F, Heginbothom M, et al. Containing antibiotic resistance: decreased antibiotic-resistant coliform urinary tract infections with reduction in antibiotic prescribing by general practices. Br J Gen Pract 2007; 57:785–792.
- Gottesman BS, Carmeli Y, Shitrit P, Chowers M. Impact of quinolone restriction on resistance patterns of Escherichia coli isolated from urine by culture in a community setting. Clin Infect Dis 2009; 49:869–875.
- Knottnerus BJ, Geerlings SE, Moll van Charante EP, ter Riet G. Women with symptoms of uncomplicated urinary tract infection are often willing to delay antibiotic treatment: a prospective cohort study. BMC Fam Pract 2013; 14:71. doi: 10.1186/1471-2296-14-71.
- Leydon GM, Turner S, Smith H, Little P; UTIS team. Women’s views about management and cause of urinary tract infection: qualitative interview study. BMJ 2010; 340:c279. doi: 10.1136/bmj.c279.
- Willems CS, van den Broek D’Obrenan J, Numans ME, Verheij TJ, van der Velden AW. Cystitis: antibiotic prescribing, consultation, attitudes and opinions. Fam Pract 2014; 31:149–155.
- Black DM, Cummings SR, Karpf DB et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- Black DM, Schwartz AV, Ensrud KE, et al; FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 2006; 296:2927–2938.
- US Food and Drug Administration. Background document for meeting of Advisory Committee for Reproductive Health Drugs and Drug Safety and Risk Management Advisory Committee. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM270958.pdf. Accessed November 3, 2016.
- Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int 2005; 16:581–589.
- Adler RA, El-Hajj Fuleihan G, Bauer DC, et al. Managing osteoporosis in patients on long-term bisphosphonate treatment: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2016; 31:16–35.
- Black DM, Reid IR, Boonen S, et al. The effect of 3 versus 6 years of zoledronic acid treatment of osteoporosis: a randomized extension to the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 2012; 27:243–254.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed October 7, 2016.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010; 16(suppl 3):1–37.
- Whitaker M, Guo J, Kehoe T, Benson G. Bisphosphonates for osteoporosis—where do we go from here? N Engl J Med 2012; 366:2048–2051.
- Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med 2012; 366:2051–2053.
- Brown JP, Morin S, Leslie W, et al. Bisphosphonates for treatment of osteoporosis: expected benefits, potential harms, and drug holidays. Can Fam Physician 2014; 60:324–333.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab 2010; 95:1555–1565.
- Bauer DC, Schwartz A, Palermo L, et al. Fracture prediction after discontinuation of 4 to 5 years of alendronate therapy: the FLEX study. JAMA Intern Med 2014; 174:1126–1134.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011; 305:2295–2303.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet 2016; 387:945–956.
- Abcodia Inc. The ROCA test. www.therocatest.co.uk/for-clinicians/about-roca. Accessed November 3, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med 2012; 157:900–904.
- Titus-Ernstoff L, Perez K, Cramer DW, Harlow BL, Baron JA, Greenberg ER. Menstrual and reproductive factors in relation to ovarian cancer risk. Br J Cancer 2001; 84:714–721.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer, Beral V, Doll R, Hermon C, Peto R, Reeves G. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008; 371:303–314.
- Chowdhury R, Sinha B, Sankar MJ, et al. Breastfeeding and maternal health outcomes: a systematic review and meta-analysis. Acta Paediatr 2015; 104:96–113.
- Hannaford PC, Iversen L, Macfarlane TV, Elliott AM, Angus V, Lee AJ. Mortality among contraceptive pill users: cohort evidence from Royal College of General Practitioners’ Oral Contraception Study. BMJ 2010; 340:c927. doi: 10.1136/bmj.c927.
- Vessey M, Yeates D. Oral contraceptive use and cancer: final report from the Oxford-Family Planning Association contraceptive study. Contraception 2013; 88:678–683.
- Charlton BM, Rich-Edwards JW, Colditz GA, et al. Oral contraceptive use and mortality after 36 years of follow-up in the Nurses’ Health Study: prospective cohort study. BMJ 2014; 349:g6356. doi: 10.1136/bmj.g6356.
- Merritt MA, Riboli E, Murphy N, et al. Reproductive factors and risk of mortality in the European Prospective Investigation into Cancer and Nutrition; a cohort study. BMC Med 2015; 13:252. doi: 10.1186/s12916-015-0484-3.
- Centers for Disease Control and Prevention (CDC). Update to CDC’s U.S. Medical Eligibility Criteria for Contraceptive Use, 2010: revised recommendations for the use of contraceptive methods during the postpartum period. MMWR Morb Mortal Wkly Rep 2011; 60:878–883.
- Bigelow CA, Bryant AS. Short interpregnancy intervals: an evidence-based guide for clinicians. Obstet Gynecol Surv 2015; 70:458–464.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Buhling KJ, Zite NB, Lotke P, Black K; INTRA Writing Group. Worldwide use of intrauterine contraception: a review. Contraception 2014; 89:162–173.
- Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception 2015; 91:274–279.
- Heinemann K, Reed S, Moehner S, Minh TD. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception 2015; 91:280–283.
- Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement. Am J Obstet Gynecol 2016 May 12. pii: S0002-9378(16)30212-5. doi: 10.1016/j.ajog.2016.05.017. [Epub ahead of print]
- Division of Reproductive health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). U.S. Selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62(RR-05):1–60.
- Gurtcheff SE, Turok DK, Stoddard G, Murphy PA, Gibson M, Jones KP. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol 2011; 117:1114–1121.
- Nisen MB, Peterson LE, Cochrane A, Rubin SE. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception 2016; 93:432–437.
- Polis CB, Bradley SE, Bankole A, Onda T, Croft T, Singh S. Typical-use contraceptive failure rates in 43 countries with Demographic and Health Survey data: summary of a detailed report. Contraception 2016; 94:11–17.
- Gariepy AM, Creinin MD, Smith KJ, Xu X. Probability of pregnancy after sterilization: a comparison of hysteroscopic versus laparoscopic sterilization. Contraception 2014; 90:174–181.
- Peterson HB, Xia Z, Hughes JM, Wilcox LS, Tylor LR, Trussel J. The risk of pregnancy after tubal sterilization: findings from the U.S. Collaborative Rerview of Sterilization. Am J Obstet Gynecol 1996; 174:1161–1168.
- Hooton TM. Clinical practice. Uncomplicated urinary tract infection. N Engl J Med 2012; 366:1028–1037.
- Christiaens TC, De Meyere M, Verschraegen G, et al. Randomised controlled trial of nitrofurantoin versus placebo in the treatment of uncomplicated urinary tract infection in adult women. Br J Gen Pract 2002; 52:729–734.
- Bleidorn J, Gágyor I, Kochen MM, Wegscheider K, Hummers-Pradier E. Symptomatic treatment (ibuprofen) or antibiotics (ciprofloxacin) for uncomplicated urinary tract infection?—results of a randomized controlled pilot trial. BMC Med 2010; 8:30. doi: 10.1186/1741-7015-8-30.
- Little P, Moore MV, Turner S, et al. Effectiveness of five different approaches in management of urinary tract infection: randomised controlled trial. BMJ 2010; 340:c199.
- Ferry SA, Holm SE, Stenlund H, Lundholm R, Monsen TJ. The natural course of uncomplicated lower urinary tract infection in women illustrated by a randomized placebo controlled study. Scand J Infect Dis 2004; 36:296–301.
- Gágyor I, Bleidorn J, Kochen MM, Schmiemann G, Wegscheider K, Hummers-Pradier E. Ibuprofen versus fosfomycin for uncomplicated urinary tract infection in women: randomised controlled trial. BMJ 2015; 351:h6544. doi: 10.1136/bmj.h6544.
- Butler CC, Dunstan F, Heginbothom M, et al. Containing antibiotic resistance: decreased antibiotic-resistant coliform urinary tract infections with reduction in antibiotic prescribing by general practices. Br J Gen Pract 2007; 57:785–792.
- Gottesman BS, Carmeli Y, Shitrit P, Chowers M. Impact of quinolone restriction on resistance patterns of Escherichia coli isolated from urine by culture in a community setting. Clin Infect Dis 2009; 49:869–875.
- Knottnerus BJ, Geerlings SE, Moll van Charante EP, ter Riet G. Women with symptoms of uncomplicated urinary tract infection are often willing to delay antibiotic treatment: a prospective cohort study. BMC Fam Pract 2013; 14:71. doi: 10.1186/1471-2296-14-71.
- Leydon GM, Turner S, Smith H, Little P; UTIS team. Women’s views about management and cause of urinary tract infection: qualitative interview study. BMJ 2010; 340:c279. doi: 10.1136/bmj.c279.
- Willems CS, van den Broek D’Obrenan J, Numans ME, Verheij TJ, van der Velden AW. Cystitis: antibiotic prescribing, consultation, attitudes and opinions. Fam Pract 2014; 31:149–155.
- Black DM, Cummings SR, Karpf DB et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 1996; 348:1535–1541.
- Black DM, Schwartz AV, Ensrud KE, et al; FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 2006; 296:2927–2938.
- US Food and Drug Administration. Background document for meeting of Advisory Committee for Reproductive Health Drugs and Drug Safety and Risk Management Advisory Committee. www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/DrugSafetyandRiskManagementAdvisoryCommittee/UCM270958.pdf. Accessed November 3, 2016.
- Kanis JA, Borgstrom F, De Laet C, et al. Assessment of fracture risk. Osteoporos Int 2005; 16:581–589.
- Adler RA, El-Hajj Fuleihan G, Bauer DC, et al. Managing osteoporosis in patients on long-term bisphosphonate treatment: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2016; 31:16–35.
- Black DM, Reid IR, Boonen S, et al. The effect of 3 versus 6 years of zoledronic acid treatment of osteoporosis: a randomized extension to the HORIZON-Pivotal Fracture Trial (PFT). J Bone Miner Res 2012; 27:243–254.
- World Health Organization Collaborating Centre for Metabolic Bone Diseases. FRAX WHO fracture risk assessment tool. www.shef.ac.uk/FRAX/. Accessed October 7, 2016.
- Watts NB, Bilezikian JP, Camacho PM, et al; AACE Osteoporosis Task Force. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of postmenopausal osteoporosis. Endocr Pract 2010; 16(suppl 3):1–37.
- Whitaker M, Guo J, Kehoe T, Benson G. Bisphosphonates for osteoporosis—where do we go from here? N Engl J Med 2012; 366:2048–2051.
- Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ. Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med 2012; 366:2051–2053.
- Brown JP, Morin S, Leslie W, et al. Bisphosphonates for treatment of osteoporosis: expected benefits, potential harms, and drug holidays. Can Fam Physician 2014; 60:324–333.
- Watts NB, Diab DL. Long-term use of bisphosphonates in osteoporosis. J Clin Endocrinol Metab 2010; 95:1555–1565.
- Bauer DC, Schwartz A, Palermo L, et al. Fracture prediction after discontinuation of 4 to 5 years of alendronate therapy: the FLEX study. JAMA Intern Med 2014; 174:1126–1134.
- Buys SS, Partridge E, Black A, et al; PLCO Project Team. Effect of screening on ovarian cancer mortality: the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011; 305:2295–2303.
- Jacobs IJ, Menon U, Ryan A, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet 2016; 387:945–956.
- Abcodia Inc. The ROCA test. www.therocatest.co.uk/for-clinicians/about-roca. Accessed November 3, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for ovarian cancer: US Preventive Services Task Force reaffirmation recommendation statement. Ann Intern Med 2012; 157:900–904.
- Titus-Ernstoff L, Perez K, Cramer DW, Harlow BL, Baron JA, Greenberg ER. Menstrual and reproductive factors in relation to ovarian cancer risk. Br J Cancer 2001; 84:714–721.
- Collaborative Group on Epidemiological Studies of Ovarian Cancer, Beral V, Doll R, Hermon C, Peto R, Reeves G. Ovarian cancer and oral contraceptives: collaborative reanalysis of data from 45 epidemiological studies including 23,257 women with ovarian cancer and 87,303 controls. Lancet 2008; 371:303–314.
- Chowdhury R, Sinha B, Sankar MJ, et al. Breastfeeding and maternal health outcomes: a systematic review and meta-analysis. Acta Paediatr 2015; 104:96–113.
- Hannaford PC, Iversen L, Macfarlane TV, Elliott AM, Angus V, Lee AJ. Mortality among contraceptive pill users: cohort evidence from Royal College of General Practitioners’ Oral Contraception Study. BMJ 2010; 340:c927. doi: 10.1136/bmj.c927.
- Vessey M, Yeates D. Oral contraceptive use and cancer: final report from the Oxford-Family Planning Association contraceptive study. Contraception 2013; 88:678–683.
- Charlton BM, Rich-Edwards JW, Colditz GA, et al. Oral contraceptive use and mortality after 36 years of follow-up in the Nurses’ Health Study: prospective cohort study. BMJ 2014; 349:g6356. doi: 10.1136/bmj.g6356.
- Merritt MA, Riboli E, Murphy N, et al. Reproductive factors and risk of mortality in the European Prospective Investigation into Cancer and Nutrition; a cohort study. BMC Med 2015; 13:252. doi: 10.1186/s12916-015-0484-3.
- Centers for Disease Control and Prevention (CDC). Update to CDC’s U.S. Medical Eligibility Criteria for Contraceptive Use, 2010: revised recommendations for the use of contraceptive methods during the postpartum period. MMWR Morb Mortal Wkly Rep 2011; 60:878–883.
- Bigelow CA, Bryant AS. Short interpregnancy intervals: an evidence-based guide for clinicians. Obstet Gynecol Surv 2015; 70:458–464.
- Winner B, Peipert JF, Zhao Q, et al. Effectiveness of long-acting reversible contraception. N Engl J Med 2012; 366:1998–2007.
- Buhling KJ, Zite NB, Lotke P, Black K; INTRA Writing Group. Worldwide use of intrauterine contraception: a review. Contraception 2014; 89:162–173.
- Heinemann K, Reed S, Moehner S, Minh TD. Risk of uterine perforation with levonorgestrel-releasing and copper intrauterine devices in the European Active Surveillance Study on Intrauterine Devices. Contraception 2015; 91:274–279.
- Heinemann K, Reed S, Moehner S, Minh TD. Comparative contraceptive effectiveness of levonorgestrel-releasing and copper intrauterine devices: the European Active Surveillance Study for Intrauterine Devices. Contraception 2015; 91:280–283.
- Turok DK, Eisenberg DL, Teal SB, Keder LM, Creinin MD. A prospective assessment of pelvic infection risk following same-day sexually transmitted infection testing and levonorgestrel intrauterine system placement. Am J Obstet Gynecol 2016 May 12. pii: S0002-9378(16)30212-5. doi: 10.1016/j.ajog.2016.05.017. [Epub ahead of print]
- Division of Reproductive health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention (CDC). U.S. Selected practice recommendations for contraceptive use, 2013: adapted from the World Health Organization selected practice recommendations for contraceptive use, 2nd edition. MMWR Recomm Rep 2013; 62(RR-05):1–60.
- Gurtcheff SE, Turok DK, Stoddard G, Murphy PA, Gibson M, Jones KP. Lactogenesis after early postpartum use of the contraceptive implant: a randomized controlled trial. Obstet Gynecol 2011; 117:1114–1121.
- Nisen MB, Peterson LE, Cochrane A, Rubin SE. US family physicians’ intrauterine and implantable contraception provision: results from a national survey. Contraception 2016; 93:432–437.
- Polis CB, Bradley SE, Bankole A, Onda T, Croft T, Singh S. Typical-use contraceptive failure rates in 43 countries with Demographic and Health Survey data: summary of a detailed report. Contraception 2016; 94:11–17.
- Gariepy AM, Creinin MD, Smith KJ, Xu X. Probability of pregnancy after sterilization: a comparison of hysteroscopic versus laparoscopic sterilization. Contraception 2014; 90:174–181.
- Peterson HB, Xia Z, Hughes JM, Wilcox LS, Tylor LR, Trussel J. The risk of pregnancy after tubal sterilization: findings from the U.S. Collaborative Rerview of Sterilization. Am J Obstet Gynecol 1996; 174:1161–1168.
KEY POINTS
- Many women with mild uncomplicated urinary tract infections can avoid taking antibiotics and instead receive treatment for symptoms alone.
- The American Society for Bone and Mineral Research now recommends reassessing the risk of osteoporotic fracture after 3 to 5 years of bisphosphonate therapy. Women at high risk may benefit from extending bisphosphonate therapy to 10 years.
- Current evidence shows no clear benefit of ovarian cancer screening for women at average risk, and we should not recommend yearly ultrasonography or cancer antigen 125 level testing, either of which is likely to cause harm without providing benefit.
- A large observational study found death rates were lower in parous than in nulliparous women, in women who had breastfed than in those who had never breastfed, and in nonsmokers who had used oral contraceptives.
- Intrauterine contraception and subdermal implants are safe and are the most effective contraceptive options.

Taurine, energy drinks, and neuroendocrine effects
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
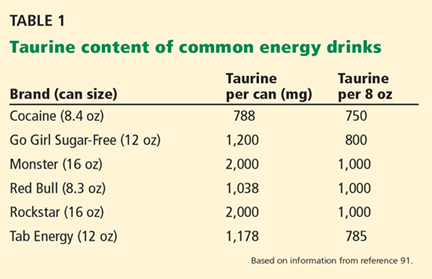
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
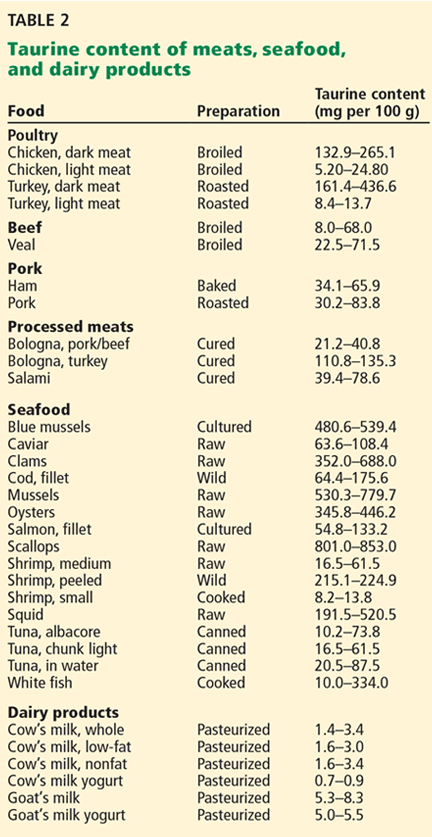
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
- US Food and Drug Administration (FDA). Caffeine intake by the US population. www.fda.gov/downloads/AboutFDA/ CentersOffices/OfficeofFoods/CFSAN/CFSANFOIAElectronicReadingRoom/UCM333191.pdf. Accessed October 4, 2016.
- McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
- Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
- Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
- Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
- Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
- Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
- Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
- Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
- Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
- Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
- Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
- Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
- Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
- Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
- Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
- Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
- Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
- Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
- Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
- Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
- Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
- Beverage Industry. 2013 state of the industry: energy drinks & shots. www.bevindustry.com/articles/86552-state-of-the-industry-energy-drinks-shots. Accessed October 4, 2016.
- Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
- Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
- Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
- Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
- Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
- Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
- Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
- Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
- Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
- Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
- Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
- Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
- Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
- Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
- Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
- Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
- Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
- Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
- Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
- Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
- Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
- Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
- Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
- Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
- Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
- Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
- Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
- Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
- Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
- Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
- Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
- Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
- Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
- Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
- Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
- Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
- Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
- Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
- Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
- Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
- Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
- Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
- Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
- Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
- Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
- Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
- Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
- Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
- Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
- Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
- Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
- Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
- Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
- Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
- Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
- Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
- Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
- Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
- Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
- Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
- Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
- Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
- Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
- Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
- Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
- Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
- Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
- Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
- Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
- Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
- Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
- Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
- Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
- Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
- Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
- Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
- Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
- Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
- Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
- Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
- Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
- Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
- Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
- Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
- Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
- Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
- Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
- Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
- Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
- Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
- Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
- Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
- Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
- Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
- Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
- Lerdweeraphon W, Wyss JM, Boonmars T, Roysommuti S. Perinatal taurine exposure affects adult oxidative stress. Am J Physiol Regul Integr Comp Physiol 2013; 305:R95–R97.
- Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
- Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
- Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
- Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
- Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
- Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
- D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
- Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
- Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
- Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
- Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
- Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
- vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
- Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
- Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
- Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
- Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
- Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
- Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
- European Commission; Scientific Committee on Food. Opinion on additional information on energy drinks. http://ec.europa.eu/food/fs/sc/scf/out169_en.pdf. Accessed October 4, 2016.
- Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
- Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
- Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
- El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
- Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
- Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
- El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
- Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
- Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
- Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
- Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
- El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
- Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
- Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
- Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
- Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
Taurine—an amino acid found in abundance in the human brain, retina, heart, and reproductive organs, as well as in meat and seafood—is also a major ingredient in “energy drinks” (Table 1).1,2 Given the tremendous popularity of these drinks in the United States, it would seem important to know and to recognize taurine’s neuroendocrine effects. Unfortunately, little is known about the effects of taurine supplementation in humans.
This paper reviews the sparse data to provide clinicians some background on the structure, synthesis, distribution, metabolism, mechanisms, effects, safety, and proposed therapeutic targets of taurine.
TAURINE’S THERAPEUTIC POTENTIAL
Taurine has been reported to have widespread anti-inflammatory actions.3,4 Taurine supplementation has been proposed to have beneficial effects in the treatment of epilepsy,5 heart failure,6,7 cystic fibrosis,8 and diabetes9 and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.10–16
In addition, taurine analogues such as homotaurine and N-acetyl-homotaurine (acamprosate) have been probed for possible therapeutic applications. Homotaurine has been shown to have antiamyloid activity that could in theory protect against the progression of Alzheimer disease,17 and acamprosate is approved by the US Food and Drug Administration (FDA) for the treatment of alcohol use disorders.18
TAURINE CONSUMPTION
Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.1 In 2012, US sales of energy drinks exceeded $12 billion,19 with young men, particularly those in the military deployed in war zones, being the biggest consumers.20–22 Analyses have found that of 49 nonalcoholic energy drinks tested, the average concentration of taurine was 3,180 mg/L, or approximately 750 mg per 8-oz serving.23,24 Popular brands include Red Bull, Monster, Rockstar (Table 1), NOS, Amp, and Full Throttle.
Taurine is plentiful in the human body, which contains up to 1 g of taurine per kg.25 Foods such as poultry, beef, pork, seafood, and processed meats have a high taurine content (Table 2).26–29 People who eat meat and seafood have plentiful taurine intake, whereas vegetarians and vegans consume much less and have significantly lower circulating levels30 because plants do not contain taurine in appreciable amounts.26,29
The typical American diet provides between 123 and 178 mg of taurine daily.26 Consumption of one 8-oz energy drink can increase the average intake 6 to 16 times. A lacto-ovo vegetarian diet provides only about 17 mg of taurine daily, and an 8-oz energy drink can increase the average intake by 44 to 117 mg.26 And since a vegan diet provides essentially no taurine,30 energy drink intake in any amount would constitute a major relative increase in taurine consumption.
ATTEMPTS TO STUDY TAURINE'S EFFECTS
Since most clinical trials to date have looked at the effects of taurine in combination with other ingredients such as caffeine, creatine, and glucose31–35 in drinks such as Red Bull, these studies cannot be used to determine the effects of taurine alone. In the few clinical trials that have tested isolated taurine consumption, data are not sufficient to make a conclusion on direct effects on energy metabolism.
Rutherford et al36 tested the effect of oral taurine supplementation (1,660 mg) on endurance in trained male cyclists 1 hour before exercise, but observed no effect on fluid intake, heart rate, subjective exertion, or time-trial performance. A small increase (16%) in total fat oxidation was observed during the 90-minute exercise period. Since mitochondria are the main location of fatty acid degradation, this effect may be attributed to taurine supplementation, with subsequent improvement in mitochondrial function.
Zhang et al37 found a 30-second increase in cycling energy capacity after 7 days of 6 g oral taurine supplementation, but the study was neither blinded nor placebo-controlled.
Kammerer et al38 tested the effect of 1 g of taurine supplementation on physical and mental performance in young adult soldiers 45 minutes before physical fitness and cognitive testing. This double-blind, placebo-controlled randomized trial found no effect of taurine on cardiorespiratory fitness indices, concentration, or immediate memory, nor did it find any effect of an 80-mg dose of caffeine.
In sum, the available data are far from sufficient to determine the direct effect of taurine consumption on energy metabolism in healthy people.
PHARMACOLOGY OF TAURINE
Chemical structure
Taurine, or 2-aminoethane sulfonic acid, is a conditionally essential amino acid, ie, we can usually make enough in our own bodies. It was first prepared on a large scale for physiologic investigation almost 90 years ago, through the purification of ox bile.39 It can be obtained either exogenously through dietary sources or endogenously through biosynthesis from methionine and cysteine precursors, both essential sulfur-containing alpha-amino acids.40 Both sources are important to maintain physiologic levels of taurine, and either can help compensate for the other in cases of deficiency.41
The structure of taurine has two main differences from the essential amino acids. First, taurine’s amino group is attached to the beta-carbon rather than the alpha-carbon, making it a beta-amino acid instead of an alpha-amino acid.42 Second, the acid group in taurine is sulfonic acid, whereas the essential amino acids have a carboxylic acid.43 Because of its distinctive structure, taurine is not used as a structural unit in proteins,43 existing mostly as a free amino acid within cells, readily positioned to perform several unique functions.
Synthesis
De novo synthesis of taurine involves several enzymes and at least five pathways,44 mostly differing by the order in which sulfur is oxidized and decarboxylated.45
The rate-limiting enzyme of the predominant pathway is thought to be cysteine sulfinate decarboxylase (CSD), and its presence within an organ indicates involvement in taurine production.44 CSD has been found in the liver,46 the primary site of taurine biosynthesis, as well as in the retina,47 brain,48 kidney,49 mammary glands,50,51 and reproductive organs.52
Distribution
Taurine levels are highest in electrically excitable tissues such as the central nervous system, retina, and heart; in secretory structures such as the pineal gland and the pituitary gland (including the posterior lobe or neurohypophysis); and in platelets25 and neutrophils.53
In the fetal brain, the taurine concentration is higher than that of any other amino acid,54 but the concentration in the brain decreases with advancing age, whereas glutamate levels increase over time to make it the predominant amino acid in the adult brain.54 Regardless, taurine is still the second most prevalent amino acid in the adult brain, its levels comparable to those of gamma-aminobutyric acid (GABA).55
Taurine has also been found in variable amounts in the liver, muscle, kidney, pancreas, spleen, small intestine, and lungs,56 as well as in several other locations.45,57
Taurine is also present in the male and female reproductive organs. In male rats, taurine and taurine biosynthesis have been localized to Leydig cells of the testes, the cellular source of testosterone in males, as well as the cremaster muscle, efferent ducts, and peritubular myoid cells surrounding seminiferous tubules.58 More recently, taurine has been detected in the testes of humans59 and is also found in sperm and seminal fluid.60 Levels of taurine in spermatozoa are correlated with sperm quality, presumably by protecting against lipid peroxidation through taurine’s antioxidant effects,61,62 as well as through contribution to the spermatozoa maturation process by facilitating the capacitation, motility, and acrosomal reaction of sperm.63
In female rats, taurine has been found in uterine tissue,64 oviducts,65 uterine fluid (where it is the predominant amino acid),66 and thecal cells of developing follicles of ovaries, cells responsible for the synthesis of androgens such as testosterone and androstenedione.65 Taurine is also a major component of human breast milk67 and is important for proper neonatal nutrition.68
Metabolism and excretion
Ninety-five percent of taurine is excreted in urine, about 70% as taurine itself, and the rest as sulfate. Most of the sulfate derived from taurine is produced by bacterial metabolism in the gut and then absorbed.69 However, taurine can also be conjugated with bile acids to act as a detergent in lipid emulsification.70 In this form, it may be subjected to the enterohepatic circulation, which gives bacteria another chance to convert it into inorganic sulfate for excretion in urine.69
MECHANISMS AND NEUROENDOCRINE EFFECTS
As a free amino acid, taurine has widespread distribution and unique biochemical and physiologic properties and exhibits several organ-specific functions; however, indisputable evidence of a taurine-specific receptor is lacking, and its putative existence71 is controversial.72 Nonetheless, taurine is a neuromodulator with a variety of actions.
Neurotransmission
Taurine is known to be an inhibitory neurotransmitter and neuromodulator.73 It is structurally analogous to GABA, the main inhibitory neurotransmitter in the brain.45 Accordingly, it binds to GABA receptors to serve as an agonist,74,75 causing neuronal hyperpolarization and inhibition. Taurine has an even higher affinity for glycine receptors75 where it has long been known to act as an agonist.76 GABA and glycine receptors both belong to the Cys-loop receptor superfamily,77 with conservation of subunits that allows taurine to bind each receptor, albeit at different affinities. The binding effects of taurine on GABA and glycine receptors have not been well documented quantitatively; however, it is known that taurine has a substantially lower affinity than GABA and glycine for their respective receptors.76
Catecholamines and the sympathetic nervous system
Surprisingly little is known about the effects of taurine on norepinephrine, dopamine, and the human sympathetic nervous system.78 Humans with borderline hypertension given 6 g of taurine orally for 7 days79 experienced decreases in epinephrine secretion and blood pressure, but normotensive study participants did not experience similar results, possibly because of a better ability to regulate sympathetic tone. Mizushima et al80 showed that a longer period of taurine intake (6 g orally for 3 weeks) could elicit a decrease in norepinephrine in healthy men with normal blood pressure. Other similar studies81–83 also suggested interplay between taurine and catecholamines, but the extent is still undetermined.
Growth hormone, prolactin, sex hormones, and cortisol
Taurine appears to have a complex relationship with several hormones, although its direct effects on hormone secretion remain obscure. Clinical studies of the acute and chronic neuroendocrine effects of taurine loading in humans are needed.
In female rats, secretion of prolactin is increased by the intraventricular injection of 5 μL of 2.0 μmol taurine over a 10-minute period.84 Ikuyama et al85 found an increase in prolactin and growth hormone secretion in adult male rats given 10 μL of 0.25 μmol and 1.0 μmol taurine intraventricularly, yet a higher dose of 4.0 μmol had no effect on either hormone. Furthermore, prolactin receptor deficiency is seen in CSD knockout mice, but the receptor is restored with taurine supplementation.86
Mantovani and DeVivo87 reported that 375 to 8,000 mg/day of taurine given orally for 4 to 6 months to epileptic patients stimulated the secretion of growth hormone. However, in another study, a single 75-mg/kg dose of oral taurine did not trigger an acute increase in levels of growth hormone or prolactin in humans.88 Energy drinks may contain up to 1,000 mg of taurine per 8-oz serving, but the effects of larger doses on growth hormone, which is banned as a supplement by major athletic organizations because of its anabolic and possible performance-enhancing effects, remain to be determined.
Taurine may have effects on human sex hormones, based on the limited observations in rodents.89–94
Although human salivary cortisol concentrations were purportedly assessed in response to 2,000 mg of oral taurine,95 the methods and reported data are not adequate to draw any conclusions.
Energy metabolism
Mammals are unable to directly use taurine in energy production because they cannot directly reduce it.25 Instead, bacteria in the gut use it as a source of energy, carbon, nitrogen, and sulfur.96 However, taurine deficiency appears to impair the cellular respiratory chain, resulting in diminished production of adenosine triphosphate and diminished uptake of long-chain fatty acids by mitochondria, at least in the heart.97
Taurine is present in human mitochondria and regulates mitochondrial function. For example, taurine in mitochondria assists in conjugation of transfer RNA for leucine, lysine, glutamate, and glutamine.98 In TauT knockout mice, deficiency of taurine causes mitochondrial dysfunction, triggering a greater than 80% decrease in exercise capacity.99 Several studies in rodents have shown increased exercise capacity after taurine supplementation.100–102 In addition, taurine is critical for the growth of blastocytes, skeletal muscle, and myocardium; it is necessary for mitochondrial development and is also important for muscular endurance.103,104
Antioxidation, anti-inflammation, and other functions
Taurine is a major antioxidant, scavenging reactive oxygen and protecting against oxidative stress to organs including the brain,97,105,106 where it increasingly appears to have neuroprotective effects.107,108
Cellular taurine also has anti-inflammatory actions.3 One of the proposed mechanisms is taurine inhibition of NF-kappa B, an important transcription factor for the synthesis of pro-inflammatory cytokines.4 This function may be important in protecting polyunsaturated fatty acids from oxidative stress—helping to maintain and stabilize the structure and function of plasma membranes within the lungs,109 heart,110 brain,111 liver,112 and spermatozoa.61,62
Taurine is also conjugated to bile acids synthesized in the liver, forming bile salts70 that act as detergents to help emulsify and digest lipids in the body. In addition, taurine facilitates xenobiotic detoxification in the liver by conjugating with several drugs to aid in their excretion.25 Taurine is also implicated in calcium modulation113 and homeostasis.114 Through inhibition of several types of calcium channels, taurine has been shown to decrease calcium influx into cells, effectively serving a cytoprotective role against calcium overload.115,116
TAURINE DEFICIENCY
Fetal and neonatal deficiency
Though taurine is considered nonessential in adults because it can be readily synthesized endogenously, it is thought to be conditionally essential in neonatal nutrition.68 It is the second most abundant free amino acid in human breast milk117 and the most abundant free amino acid in fetal brain.118 In cases of long-term parenteral nutrition, neonates can become drastically taurine deficient119 due to suboptimal CSD activity,118 leading to retinal dysfunction.41 Taurine deficiencies can lead to functional and structural brain damage.118 Moreover, maternal taurine deficiency results in neurologic abnormalities in offspring120 and may lead to oxidative stress throughout life.121
In 1984, the FDA approved the inclusion of taurine in infant formulas based on research showing that taurine-deficient infants had impaired fat absorption, bile acid secretion, retinal function, and hepatic function.122 But still under debate are the amount and duration of taurine supplementation required by preterm and low-birth-weight infants, as several randomized controlled trials failed to show statistically significant effects on growth.123 Nonetheless, given the alleged detrimental ramifications of a lack of taurine supplementation, as well as the ethical dilemma of performing additional research trials on infants, it is presumed that infant formulas and parenteral nutrition for preterm and low-birth-weight infants will continue to contain taurine.
Age- and disease-related deficiency
Although taurine deficiency is rare in neonates, it is perhaps inevitable with advancing age. Healthy elderly patients ages 61 to 81 have up to a 49% decrease in plasma taurine concentration compared with healthy individuals ages 27 to 57.124 While reduced renal retention125 and taurine intake126 can account for depressed taurine levels, Eppler and Dawson127 found that tissue and circulating taurine concentrations decrease over the human life span primarily due to an age-dependent depletion of CSD activity in the liver. This effectively impairs the biosynthesis of endogenous taurine from cysteine or methionine or both, forcing a greater reliance on exogenous sources.
While specific mechanisms have not been fully elucidated, taurine deficiency has also been identified in patients suffering from diseases including but not limited to disorders of bone (osteogenesis imperfecta, osteoporosis),128 blood (acute myelogenous leukemia),129 central nervous system (schizophrenia, Friedreich ataxia-spinocerebellar degeneration),130,131 retina (retinitis pigmentosa),132 circulatory system and heart (essential hypertension, atherosclerosis),133 digestion (Gaucher disease),134 absorption (short-bowel syndrome),135 cellular proliferation (cancer),136 and membrane channels (cystic fibrosis),137 as well as in patients restricted to long-term parenteral nutrition.138 However, the apparent correlation between taurine deficiency and these conditions does not necessarily mean causation; more study is needed to elucidate a direct connection.
SAFETY AND TOXICITY OF TAURINE SUPPLEMENTATION
An upper safe level of intake for taurine has not been established. To date, several studies have involved heavy taurine supplementation without serious adverse effects. While the largest dosage of taurine tested in humans appears to be 10 g/day for 6 months,139 a number of studies have used 1 to 6 g/day for periods of 1 week to 1 year.140 However, the assessment of potential acute, subacute, and chronic adverse effects has not been comprehensive. The Scientific Committee on Food of the European Commission141 reviewed several toxicologic studies on taurine through 2003 and were unable to expose any carcinogenic or teratogenic potential. Nevertheless, based on the available data from trials in humans and lower animals, Shao and Hathcock140 suggested an observed safe level of taurine of 3 g/day, a conservatively smaller dose that carries a higher level of confidence. Because there is no “observed adverse effect level” for daily taurine intake,141 more research must be done to ensure safety of higher amounts of taurine administration and to define a tolerable upper limit of intake.
Interactions with medications
To date, the literature is scarce regarding potential interactions between taurine and commonly used medications.
Although no evidence specifically links taurine with adverse effects when used concurrently with other medications, there may be a link between taurine supplementation and various cytochrome P450 systems responsible for hepatic drug metabolism. Specifically, taurine inhibits cytochrome P450 2E1, a highly conserved xenobiotic-metabolizing P450 responsible for the breakdown of more than 70 substrates, including several commonly used anesthetics, analgesics, antidepressants, antibacterials, and antiepileptics.142 Of note, taurine may contribute to the attenuation of oxidative stress in the liver in the presence of alcohol143 and acetaminophen,144 two substances frequently used and abused. Since the P450 2E1 system catalyzes comparable reactions in rodents and humans,142 rodents should plausibly serve as a model for further testing of the effects of taurine on various substrates.
POTENTIAL THERAPEUTIC APPLICATIONS
More analysis is needed to fully unlock and understand taurine’s potential value in healthcare.
Correction of late-life taurine decline in humans could be beneficial for cognitive performance, energy metabolism, sexual function, and vision, but clinical studies remain to be performed. While a decline in taurine with age may intensify the stress caused by reactive oxygen species, taurine supplementation has been shown to decrease the presence of oxidative markers127 and to serve a neuroprotective role in rodents.145,146 Taurine levels increase in the hippocampus after experimentally induced gliosis147 and are neuroprotective against glutamate excitotoxicity.148,149 Furthermore, data in Alzheimer disease, Huntington disease, and brain ischemia experimental models show that taurine inhibits neuronal death (apoptosis).13,150,151 Taurine has even been proposed as a potential preventive treatment for Alzheimer dementia, as it stabilizes protein conformations to prevent their aggregation and subsequent dysfunction.152 Although improvement in memory and cognitive performance has been linked to taurine supplementation in old mice,145,153 similar results have not been found in adult mice whose taurine levels are within normal limits. Taurine also has transient anticonvulsant effects in some epileptic humans.154
Within the male reproductive organs, the age-related decline in taurine may or may not have implications regarding sexuality, as only very limited rat data are available.89–91
In cats, taurine supplementation has been found to prevent the progressive degeneration of retinal photoreceptors seen in retinitis pigmentosa, a genetic disease that causes the loss of vision.155
While several energy drink companies have advertised that taurine plays a role in improving cognitive and physical performance, there are few human studies that examine this contention in the absence of confounding factors such as caffeine or glucose.36,37,95 Taurine supplementation in patients with heart failure has been shown to increase exercise capacity vs placebo.156 This supports the idea that in cases of taurine deficiency, such as those seen in cardiomyopathy,157 taurine supplementation could have restorative effects. However, we are not aware of any double-blind, placebo-controlled clinical trial of taurine alone in healthy patients that measured energy parameters as clinical outcomes.
Although it remains possible that acute supraphysiologic taurine levels achieved by supplementation could transiently trigger various psychoneuroendocrine responses in healthy people, clinical research is needed in which taurine is the sole intervention. At present, the most compelling clinical reason to prescribe or recommend taurine supplementation is taurine deficiency.
- US Food and Drug Administration (FDA). Caffeine intake by the US population. www.fda.gov/downloads/AboutFDA/ CentersOffices/OfficeofFoods/CFSAN/CFSANFOIAElectronicReadingRoom/UCM333191.pdf. Accessed October 4, 2016.
- McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
- Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
- Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
- Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
- Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
- Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
- Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
- Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
- Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
- Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
- Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
- Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
- Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
- Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
- Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
- Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
- Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
- Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
- Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
- Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
- Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
- Beverage Industry. 2013 state of the industry: energy drinks & shots. www.bevindustry.com/articles/86552-state-of-the-industry-energy-drinks-shots. Accessed October 4, 2016.
- Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
- Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
- Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
- Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
- Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
- Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
- Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
- Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
- Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
- Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
- Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
- Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
- Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
- Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
- Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
- Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
- Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
- Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
- Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
- Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
- Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
- Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
- Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
- Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
- Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
- Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
- Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
- Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
- Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
- Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
- Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
- Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
- Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
- Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
- Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
- Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
- Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
- Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
- Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
- Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
- Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
- Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
- Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
- Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
- Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
- Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
- Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
- Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
- Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
- Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
- Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
- Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
- Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
- Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
- Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
- Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
- Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
- Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
- Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
- Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
- Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
- Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
- Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
- Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
- Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
- Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
- Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
- Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
- Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
- Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
- Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
- Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
- Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
- Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
- Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
- Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
- Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
- Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
- Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
- Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
- Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
- Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
- Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
- Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
- Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
- Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
- Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
- Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
- Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
- Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
- Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
- Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
- Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
- Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
- Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
- Lerdweeraphon W, Wyss JM, Boonmars T, Roysommuti S. Perinatal taurine exposure affects adult oxidative stress. Am J Physiol Regul Integr Comp Physiol 2013; 305:R95–R97.
- Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
- Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
- Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
- Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
- Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
- Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
- D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
- Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
- Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
- Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
- Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
- Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
- vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
- Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
- Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
- Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
- Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
- Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
- Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
- European Commission; Scientific Committee on Food. Opinion on additional information on energy drinks. http://ec.europa.eu/food/fs/sc/scf/out169_en.pdf. Accessed October 4, 2016.
- Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
- Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
- Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
- El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
- Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
- Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
- El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
- Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
- Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
- Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
- Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
- El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
- Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
- Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
- Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
- Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
- US Food and Drug Administration (FDA). Caffeine intake by the US population. www.fda.gov/downloads/AboutFDA/ CentersOffices/OfficeofFoods/CFSAN/CFSANFOIAElectronicReadingRoom/UCM333191.pdf. Accessed October 4, 2016.
- McLellan TM, Lieberman HR. Do energy drinks contain active components other than caffeine? Nutr Rev 2012; 70:730–744.
- Park E, Quinn MR, Wright CE, Schuller-Levis G. Taurine chloramine inhibits the synthesis of nitric oxide and the release of tumor necrosis factor in activated RAW 264.7 cells. J Leukoc Biol 1993; 54:119–124.
- Kontny E, Szczepanska K, Kowalczewski J, et al. The mechanism of taurine chloramine inhibition of cytokine (interleukin-6, interleukin-8) production by rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum 2000; 43:2169–2177.
- Barbeau A, Inoue N, Tsukada Y, Butterworth RF. The neuropharmacology of taurine. Life Sci 1975; 17:669–677.
- Kramer JH, Chovan JP, Schaffer SW. Effect of taurine on calcium paradox and ischemic heart failure. Am J Physiol 1981; 240:H238–H246.
- Azuma J, Sawamura A, Awata N, et al. Therapeutic effect of taurine in congestive heart failure: a double-blind crossover trial. Clin Cardiol 1985; 8:276–282.
- Darling PB, Lepage G, Leroy C, Masson P, Roy CC. Effect of taurine supplements on fat absorption in cystic fibrosis. Pediatr Res 1985; 19:578–582.
- Franconi F, Di Leo MA, Bennardini F, Ghirlanda G. Is taurine beneficial in reducing risk factors for diabetes mellitus? Neurochem Res 2004; 29:143–150.
- Malcangio M, Bartolini A, Ghelardini C, et al. Effect of ICV taurine on the impairment of learning, convulsions and death caused by hypoxia. Psychopharmacology (Berl) 1989; 98:316–320.
- Rivas-Arancibia S, Dorado-Martínez C, Borgonio-Pérez G, et al. Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ Res 2000; 82:7–17.
- Vohra BP, Hui X. Improvement of impaired memory in mice by taurine. Neural Plast 2000; 7:245–259.
- Tadros MG, Khalifa AE, Abdel-Naim AB, Arafa HM. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav 2005; 82:574–582.
- Zhu DM, Wang M, She JQ, Yu K, Ruan DY. Protection by a taurine supplemented diet from lead-induced deficits of long-term potentiation/depotentiation in dentate gyrus of rats in vivo. Neuroscience 2005; 134:215–224.
- Chepkova AN, Sergeeva OA, Haas HL. Taurine rescues hippocampal long-term potentiation from ammonia-induced impairment. Neurobiol Dis 2006; 23:512–521.
- Wang GH, Jiang ZL, Fan XJ, Zhang L, Li X, Ke KF. Neuroprotective effect of taurine against focal cerebral ischemia in rats possibly mediated by activation of both GABAA and glycine receptors. Neuropharmacology 2007; 52:1199–1209.
- Caltagirone C, Ferrannini L, Marchionni N, Nappi G, Scapagnini G, Trabucchi M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: a review. Aging Clin Exp Res 2012; 24:580–587.
- Jonas DE, Amick HR, Feltner C, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889–1900.
- Sorkin BC, Camp KM, Haggans CJ, et al. Executive summary of NIH workshop on the use and biology of energy drinks: current knowledge and critical gaps. Nutr Rev 2014; 72(suppl 1):1–8.
- Centers for Disease Control and Prevention (CDC). Energy drink consumption and its association with sleep problems among US service members on a combat deployment—Afghanistan, 2010. MMWR Morb Mortal Wkly Rep 2012; 61:895–898.
- Bailey RL, Saldanha LG, Dwyer JT. Estimating caffeine intake from energy drinks and dietary supplements in the United States. Nutr Rev 2014; 72(suppl 1):9–13.
- Stephens MB, Attipoe S, Jones D, Ledford CJ, Deuster PA. Energy drink and energy shot use in the military. Nutr Rev 2014; 72(suppl 1):72–77.
- Triebel S, Sproll C, Reusch H, Godelmann R, Lachenmeier DW. Rapid analysis of taurine in energy drinks using amino acid analyzer and Fourier transform infrared (FTIR) spectroscopy as basis for toxicological evaluation. Amino Acids 2007; 33:451–457.
- Beverage Industry. 2013 state of the industry: energy drinks & shots. www.bevindustry.com/articles/86552-state-of-the-industry-energy-drinks-shots. Accessed October 4, 2016.
- Huxtable RJ. Physiological actions of taurine. Physiol Rev 1992; 72:101–163.
- Laidlaw SA, Grosvenor M, Kopple JD. The taurine content of common foodstuffs. JPEN J Parenter Enteral Nutr 1990; 14:183–188.
- Manzi P, Pizzoferrato L. Taurine in milk and yoghurt marketed in Italy. Int J Food Sci Nutr 2013; 64:112–116.
- Dragnes BT, Larsen R, Ernstsen MH, Mæhre H, Elvevoll EO. Impact of processing on the taurine content in processed seafood and their corresponding unprocessed raw materials. Int J Food Sci Nutr 2009; 60:143–152.
- Laidlaw SA, Shultz TD, Cecchino JT, Kopple JD. Plasma and urine taurine levels in vegans. Am J Clin Nutr 1988; 47:660–663.
- Rana SK, Sanders TA. Taurine concentrations in the diet, plasma, urine and breast milk of vegans compared with omnivores. Br J Nutr 1986; 56:17–27.
- Alford C, Cox H, Wescott R. The effects of Red Bull energy drink on human performance and mood. Amino Acids 2001; 21:139–150.
- Hoffman JR, Ratamess NA, Ross R, Shanklin M, Kang J, Faigenbaum AD. Effect of a pre-exercise energy supplement on the acute hormonal response to resistance exercise. J Strength Cond Res 2008; 22:874–882.
- Ivy JL, Kammer L, Ding Z, et al. Improved cycling time-trial performance after ingestion of a caffeine energy drink. Int J Sport Nutr Exerc Metab 2009; 19:61–78.
- Gonzalez AM, Walsh AL, Ratamess NA, Kang J, Hoffman JR. Effect of a pre-workout energy supplement on acute multi-joint resistance exercise. J Sports Sci Med 2011; 10:261–266.
- Yeh TS, Chan KH, Hsu MC, Liu JF. Supplementation with soybean peptides, taurine, pueraria isoflavone, and ginseng saponin complex improves endurance exercise capacity in humans. J Med Food 2011; 14:219–225.
- Rutherford JA, Spriet LL, Stellingwerff T. The effect of acute taurine ingestion on endurance performance and metabolism in well-trained cyclists. Int J Sport Nutr Exerc Metab 2010; 20:322–329.
- Zhang M, Izumi I, Kagamimori S, et al. Role of taurine supplementation to prevent exercise-induced oxidative stress in healthy young men. Amino Acids 2004; 26:203–207.
- Kammerer M, Jaramillo JA, García A, Calderón JC, Valbuena LH. Effects of energy drink major bioactive compounds on the performance of young adults in fitness and cognitive tests: a randomized controlled trial. J Int Soc Sports Nutr 2014; 11:44.
- Kermack WO, Slater RH. The preparation of taurine in considerable quantity. Biochem J 1927; 21:1065–1067.
- Huxtable RJ, Lippincott SE. Diet and biosynthesis as sources of taurine in the mouse. J Nutr 1982; 112:1003–1010.
- Ohkuma S, Tamura J, Kuriyama K. Roles of endogenous and exogenous taurine and glycine in the formation of conjugated bile acids: analyses using freshly isolated and primary cultured rat hepatocytes. Jpn J Pharmacol 1984; 35:347–358.
- Chapman GE, Greenwood CE. Taurine in nutrition and brain development. Nutrition Research 1988; 8:955–968.
- Andrews S, Schmidt CLA. Titration curves of taurine and cysteic acid. J Biol Chem 1927; 73:651–654.
- Huxtable RJ. Taurine in the central nervous system and the mammalian actions of taurine. Prog Neurobiol 1989; 32:471–533.
- Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424–511.
- Hope DB. Pyridoxal phosphate as the coenzyme of the mammalian decarboxylase for L-cysteine sulphinic and L-cysteic acids. Biochem J 1955; 59:497–500.
- Pasantes-Morales H, Lopez-Colome AM, Salceda R, Mandel P. Cysteine sulphinate decarboxylase in chick and rat retina during development. J Neurochem 1976; 27:1103–1106.
- Oertel WH, Schmechel DE, Weise VK, et al. Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 1981; 6:2701–2714.
- Reymond I, Bitoun M, Levillain O, Tappaz M. Regional expression and histological localization of cysteine sulfinate decarboxylase mRNA in the rat kidney. J Histochem Cytochem 2000; 48:1461–1468.
- Hu JM, Ikemura R, Chang KT, Suzuki M, Nishihara M, Takahashi M. Expression of cysteine sulfinate decarboxylase mRNA in rat mammary gland. J Vet Med Sci 2000; 62:829–834.
- Ueki I, Stipanuk MH. Enzymes of the taurine biosynthetic pathway are expressed in rat mammary gland. J Nutr 2007; 137:1887–1894.
- Li JH, Ling YQ, Fan JJ, Zhang XP, Cui S. Expression of cysteine sulfinate decarboxylase (CSD) in male reproductive organs of mice. Histochem Cell Biol 2006; 125:607–613.
- Green TR, Fellman JH, Eicher AL, Pratt KL. Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1991; 1073:91–97.
- Lefauconnier JM, Portemer C, Chatagner F. Free amino acids and related substances in human glial tumours and in fetal brain: comparison with normal adult brain. Brain Res 1976; 117:105–113.
- Sturman JA, Rassin DK, Gaull GE. Taurine in development. Life Sci 1977; 21:1–22.
- Sturman JA. Taurine pool sizes in the rat: effects of vitamin B-6 deficiency and high taurine diet. J Nutr 1973; 103:1566–1580.
- Terauchi A, Nakazaw A, Johkura K, Yan L, Usuda N. Immunohistochemical localization of taurine in various tissues of the mouse. Amino Acids 1998; 15:151–160.
- Lobo MV, Alonso FJ, del Río RM. Immunohistochemical localization of taurine in the male reproductive organs of the rat. J Histochem Cytochem 2000; 48:313–320.
- Aaronson DS, Iman R, Walsh TJ, Kurhanewicz J, Turek PJ. A novel application of 1H magnetic resonance spectroscopy: non-invasive identification of spermatogenesis in men with non-obstructive azoospermia. Hum Reprod 2010; 25:847–852.
- Holmes RP, Goodman HO, Shihabi ZK, Jarow JP. The taurine and hypotaurine content of human semen. J Androl 1992; 13:289–292.
- Alvarez JG, Storey BT. Taurine, hypotaurine, epinephrine and albumin inhibit lipid peroxidation in rabbit spermatozoa and protect against loss of motility. Biol Reprod 1983; 29:548–555.
- Das J, Ghosh J, Manna P, Sinha M, Sil PC. Taurine protects rat testes against NaAsO(2)-induced oxidative stress and apoptosis via mitochondrial dependent and independent pathways. Toxicol Lett 2009; 187:201–210.
- Mrsny RJ, Meizel S. Inhibition of hamster sperm Na+, K+-ATPase activity by taurine and hypotaurine. Life Sci 1985; 36:271–275.
- Phoenix J, Wray S. Changes in human and rat uterine phosphoethanolamine and taurine with pregnancy and parturition. Exp Physiol 1994; 79:601–604.
- Lobo MV, Alonso FJ, Latorre A, del Río RM. Immunohistochemical localization of taurine in the rat ovary, oviduct, and uterus. J Histochem Cytochem 2001; 49:1133–1142.
- Casslén BG. Free amino acids in human uterine fluid. Possible role of high taurine concentration. J Reprod Med 1987; 32:181–184.
- Pamblanco M, Portolés M, Paredes C, Ten A, Comín J. Free amino acids in preterm and term milk from mothers delivering appropriate- or small-for-gestational-age infants. Am J Clin Nutr 1989; 50:778–781.
- Rigo J, Senterre J. Is taurine essential for the neonates? Biol Neonate 1977; 32:73–76.
- Sturman JA, Hepner GW, Hofmann AF, Thomas PJ. Metabolism of [35S]taurine in man. J Nutr 1975; 105:1206–1214.
- Bremer J. Species differences in the conjugation of free bile acids with taurine and glycine. Biochem J 1956; 63:507–513.
- Wu JY, Tang XW, Tsai WH. Taurine receptor: kinetic analysis and pharmacological studies. Adv Exp Med Biol 1992; 315:263–268.
- Ripps H, Shen W. Review: taurine: a “very essential” amino acid. Mol Vis 2012; 18:2673–2686.
- Haas HL, Hösli L. The depression of brain stem neurones by taurine and its interaction with strychnine and bicuculline. Brain Res 1973; 52:399–402.
- Bureau MH, Olsen RW. Taurine acts on a subclass of GABAA receptors in mammalian brain in vitro. Eur J Pharmacol 1991; 207:9–16.
- Bhattarai JP, Park SJ, Chun SW, Cho DH, Han SK. Activation of synaptic and extrasynaptic glycine receptors by taurine in preoptic hypothalamic neurons. Neurosci Lett 2015; 608:51–56.
- Horikoshi T, Asanuma A, Yanagisawa K, Anzai K, Goto S. Taurine and beta-alanine act on both GABA and glycine receptors in Xenopus oocyte injected with mouse brain messenger RNA. Brain Res 1988; 464:97–105.
- Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci 2010; 31:161–174.
- Fujita T, Sato Y. Hypotensive effect of taurine. Possible involvement of the sympathetic nervous system and endogenous opiates. J Clin Invest 1988; 82:993–997.
- Fujita T, Ando K, Noda H, Ito Y, Sato Y. Effects of increased adrenomedullary activity and taurine in young patients with borderline hypertension. Circulation 1987; 75:525–532.
- Mizushima S, Nara Y, Sawamura M, Yamori Y. Effects of oral taurine supplementation on lipids and sympathetic nerve tone. Adv Exp Med Biol 1996; 403:615–622.
- Fujita T, Sato Y. The antihypertensive effect of taurine in DOCA-salt rats. J Hypertens Suppl 1984; 2:S563–S565.
- Fujita T, Sato Y, Ando K. Changes in cardiac and hypothalamic noradrenergic activity with taurine in DOCA-salt rats. Am J Physiol 1986; 251:H926–H933.
- Sato Y, Ando K, Fujita T. Role of sympathetic nervous system in hypotensive action of taurine in DOCA-salt rats. Hypertension 1987; 9:81–87.
- Scheibel J, Elsasser T, Ondo JG. A neuromodulatory role for taurine in controlling prolactin secretion in female rats. Psychoneuroendocrinology 1981; 6:139–144.
- Ikuyama S, Okajima T, Kato K, Ibayashi H. Effect of taurine on growth hormone and prolactin secretion in rats: possible interaction with opioid peptidergic system. Life Sci 1988; 43:807–812.
- Park E, Park SY, Dobkin C, Schuller-Levis G. Development of a novel cysteine sulfinic acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014; 2014:346809.
- Mantovani J, DeVivo DC. Effects of taurine on seizures and growth hormone release in epileptic patients. Arch Neurol 1979; 36:672–674.
- Carlson HE, Miglietta JT, Roginsky MS, Stegink LD. Stimulation of pituitary hormone secretion by neurotransmitter amino acids in humans. Metabolism 1989; 38:1179–1182.
- Yang J, Wu G, Feng Y, Lv Q, Lin S, Hu J. Effects of taurine on male reproduction in rats of different ages. J Biomed Sci 2010; 17(suppl 1):S9.
- Yang J, Lin S, Feng Y, Wu G, Hu J. Taurine enhances the sexual response and mating ability in aged male rats. Adv Exp Med Biol 2013; 776:347–355.
- Yang J, Zong X, Wu G, Lin S, Feng Y, Hu J. Taurine increases testicular function in aged rats by inhibiting oxidative stress and apoptosis. Amino Acids 2015; 47:1549–1558.
- Scheibel J, Elsasser T, Ondo JG. Stimulation of LH and FSH secretion following intraventricular injection of cysteic acid but not taurine. Brain Res 1980; 201:99–106.
- Price MT, Olney JW, Mitchell MV, Fuller T, Cicero TJ. Luteinizing hormone releasing action of N-methyl aspartate is blocked by GABA or taurine but not by dopamine antagonists. Brain Res 1978; 158:461–465.
- Ma Q, Zhao J, Cao W, Liu J, Cui S. Estradiol decreases taurine level by reducing cysteine sulfinic acid decarboxylase via the estrogen receptor-a in female mice liver. Am J Physiol Gastrointest Liver Physiol 2015; 308:G277–G286.
- Giles GE, Mahoney CR, Brunyé TT, Gardony AL, Taylor HA, Kanarek RB. Differential cognitive effects of energy drink ingredients: caffeine, taurine, and glucose. Pharmacol Biochem Behav 2012; 102:569–577.
- Giehl TJ, Qoronfleh MW, Wilkinson BJ. Transport, nutritional and metabolic studies of taurine in staphylococci. J Gen Microbiol 1987; 133:849–856.
- Schaffer SW, Shimada-Takaura K, Jong CJ, Ito T, Takahashi K. Impaired energy metabolism of the taurine-deficient heart. Amino Acids 2015 Oct 16. Epub ahead of print.
- Schaffer SW, Jong CJ, Ito T, Azuma J. Role of taurine in the pathologies of MELAS and MERRF. Amino Acids 2014; 46:47–56.
- Warskulat U, Heller-Stilb B, Oermann E, et al. Phenotype of the taurine transporter knockout mouse. Methods Enzymol 2007; 428:439–458.
- Yatabe Y, Miyakawa S, Miyazaki T, Matsuzaki Y, Ochiai N. Effects of taurine administration in rat skeletal muscles on exercise. J Orthop Sci 2003; 8:415–419.
- Miyazaki T, Matsuzaki Y, Ikegami T, et al. Optimal and effective oral dose of taurine to prolong exercise performance in rat. Amino Acids 2004; 27:291–298.
- Goodman CA, Horvath D, Stathis C, et al. Taurine supplementation increases skeletal muscle force production and protects muscle function during and after high-frequency in vitro stimulation. J Appl Physiol (1985) 2009; 107:144–154.
- Van Winkle LJ, Patel M, Wasserlauf HG, Dickinson HR, Campione AL. Osmotic regulation of taurine transport via system beta and novel processes in mouse preimplantation conceptuses. Biochim Biophys Acta 1994; 1191:244–255.
- Ito T, Kimura Y, Uozumi Y, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol 2008; 44:927–937.
- Aydın AF, Çoban J, Dogan-Ekici I, Betül-Kalaz E, Dogru-Abbasoglu S, Uysal M. Carnosine and taurine treatments diminished brain oxidative stress and apoptosis in D-galactose aging model. Metab Brain Dis 2016; 31:337–345.
- Nagai K, Fukuno S, Oda A, Konishi H. Protective effects of taurine on doxorubicin-induced acute hepatotoxicity through suppression of oxidative stress and apoptotic responses. Anticancer Drugs 2016; 27:17–23.
- Caletti G, Almeida FB, Agnes G, Nin MS, Barros HM, Gomez R. Antidepressant dose of taurine increases mRNA expression of GABAA receptor a2 subunit and BDNF in the hippocampus of diabetic rats. Behav Brain Res 2015; 283:11–55.
- Gu Y, Zhao Y, Qian K, Sun M. Taurine attenuates hippocampal and corpus callosum damage, and enhances neurological recovery after closed head injury in rats. Neuroscience 2015; 291:331–340.
- Banks MA, Porter DW, Pailes WH, Schwegler-Berry D, Martin WG, Castranova V. Taurine content of isolated rat alveolar type I cells. Comp Biochem Physiol B 1991; 100:795–799.
- Raschke P, Massoudy P, Becker BF. Taurine protects the heart from neutrophil-induced reperfusion injury. Free Radic Biol Med 1995; 19:461–471.
- Pushpakiran G, Mahalakshmi K, Anuradha CV. Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 2004; 27:91–96.
- Refik Mas M, Comert B, Oncu K, et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatol Res 2004; 28:207–215.
- Sebring LA, Huxtable RJ. Taurine modulation of calcium binding to cardiac sarcolemma. J Pharmacol Exp Ther 1985; 232:445–451.
- Schaffer SW, Kramer J, Chovan JP. Regulation of calcium homeostasis in the heart by taurine. Fed Proc 1980; 39:2691–2694.
- Foos TM, Wu JY. The role of taurine in the central nervous system and the modulation of intracellular calcium homeostasis. Neurochem Res 2002; 27:21–26.
- Ito T, Schaffer S, Azuma J. The effect of taurine on chronic heart failure: actions of taurine against catecholamine and angiotensin II. Amino Acids 2014; 46:111–119.
- Gaull GE. Taurine in pediatric nutrition: review and update. Pediatrics 1989; 83:433–442.
- Sturman JA, Rassin DK, Gaull GE. Taurine in developing rat brain: transfer of [35S] taurine to pups via the milk. Pediatr Res 1977; 11:28–33.
- Geggel HS, Ament ME, Heckenlively JR, Martin DA, Kopple JD. Nutritional requirement for taurine in patients receiving long-term parenteral nutrition. N Engl J Med 1985; 312:142–146.
- Sturman JA, Gargano AD, Messing JM, Imaki H. Feline maternal taurine deficiency: effect on mother and offspring. J Nutr 1986; 116:655–667.
- Lerdweeraphon W, Wyss JM, Boonmars T, Roysommuti S. Perinatal taurine exposure affects adult oxidative stress. Am J Physiol Regul Integr Comp Physiol 2013; 305:R95–R97.
- Chesney RW, Helms RA, Christensen M, Budreau AM, Han X, Sturman JA. The role of taurine in infant nutrition. Adv Exp Med Biol 1998; 442:463–476.
- Verner A, Craig S, McGuire W. Effect of taurine supplementation on growth and development in preterm or low birth weight infants. Cochrane Database Syst Rev 2007; 4:CD006072.
- Jeevanandam M, Young DH, Ramias L, Schiller WR. Effect of major trauma on plasma free amino acid concentrations in geriatric patients. Am J Clin Nutr 1990; 51:1040–1045.
- Dawson R Jr, Eppler B, Patterson TA, Shih D, Liu S. The effects of taurine in a rodent model of aging. Adv Exp Med Biol 1996; 403:37–50.
- Han X, Budreau AM, Chesney RW. Molecular cloning and functional expression of an LLC-PK1 cell taurine transporter that is adaptively regulated by taurine. Adv Exp Med Biol 1998; 442:261–268.
- Eppler B, Dawson R Jr. Dietary taurine manipulations in aged male Fischer 344 rat tissue: taurine concentration, taurine biosynthesis, and oxidative markers. Biochem Pharmacol 2001; 62:29–39.
- D’Eufemia P, Finocchiaro R, Celli M, et al. Taurine deficiency in thalassemia major-induced osteoporosis treated with neridronate. Biomed Pharmacother 2010; 64:271–274.
- Muscaritoli M, Conversano L, Petti MC, et al. Plasma amino acid concentrations in patients with acute myelogenous leukemia. Nutrition 1999; 15:195–199.
- Do KQ, Lauer CJ, Schreiber W, et al. Gamma-glutamylglutamine and taurine concentrations are decreased in the cerebrospinal fluid of drug-naive patients with schizophrenic disorders. J Neurochem 1995; 65:2652–2662.
- Lemieux B, Giguère R, Shapcott D. Studies on the role of taurine in Friedreich’s ataxia. Can J Neurol Sci 1984; 11(suppl 4):610–615.
- Airaksinen EM, Oja SS, Marnela KM, Sihvola P. Taurine and other amino acids of platelets and plasma in retinitis pigmentosa. Ann Clin Res 1980; 12:52–54.
- Kohashi N, Katori R. Decrease of urinary taurine in essential hypertension. Jpn Heart J 1983; 24:91–102.
- vom Dahl S, Mönnighoff I, Häussinger D. Decrease of plasma taurine in Gaucher disease and its sustained correction during enzyme replacement therapy. Amino Acids 2000; 19:585–592.
- Schneider SM, Joly F, Gehrardt MF, et al. Taurine status and response to intravenous taurine supplementation in adults with short-bowel syndrome undergoing long-term parenteral nutrition: a pilot study. Br J Nutr 2006; 96:365–370.
- Gray GE, Landel AM, Meguid MM. Taurine-supplemented total parenteral nutrition and taurine status of malnourished cancer patients. Nutrition 1994; 10:11–15.
- Thompson GN. Assessment of taurine deficiency in cystic fibrosis. Clin Chim Acta 1988; 171:233–237.
- Cho KH, Kim ES, Chen JD. Taurine intake and excretion in patients undergoing long term enteral nutrition. Adv Exp Med Biol 2000; 483:605–612.
- Durelli L, Mutani R, Fassio F. The treatment of myotonia: evaluation of chronic oral taurine therapy. Neurology 1983; 33:599–603.
- Shao A, Hathcock JN. Risk assessment for the amino acids taurine, L-glutamine and L-arginine. Regul Toxicol Pharmacol 2008; 50:376–399.
- European Commission; Scientific Committee on Food. Opinion on additional information on energy drinks. http://ec.europa.eu/food/fs/sc/scf/out169_en.pdf. Accessed October 4, 2016.
- Tanaka E, Terada M, Misawa S. Cytochrome P450 2E1: its clinical and toxicological role. J Clin Pharm Ther 2000; 25:165–175.
- Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA. Reversal of ethanol-induced hepatic steatosis and lipid peroxidation by taurine: a study in rats. Alcohol Alcohol 1999; 34:529–541.
- Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010; 44:340–355.
- El Idrissi A, Shen CH, L’amoreaux WJ. Neuroprotective role of taurine during aging. Amino Acids 2013; 45:735–750.
- Gharibani P, Modi J, Menzie J, et al. Comparison between single and combined post-treatment with S-methyl-N,N-diethylthiolcarbamate sulfoxide and taurine following transient focal cerebral ischemia in rat brain. Neuroscience 2015; 300:460–473.
- Junyent F, De Lemos L, Utrera J, et al. Content and traffic of taurine in hippocampal reactive astrocytes. Hippocampus 2011; 21:185–197.
- El Idrissi A, Trenkner E. Growth factors and taurine protect against excitotoxicity by stabilizing calcium homeostasis and energy metabolism. J Neurosci 1999; 19:9459–9468.
- Wu H, Jin Y, Wei J, Jin H, Sha D, Wu JY. Mode of action of taurine as a neuroprotector. Brain Res 2005; 1038:123–131.
- Paula-Lima AC, De Felice FG, Brito-Moreira J, Ferreira ST. Activation of GABA(A) receptors by taurine and muscimol blocks the neurotoxicity of beta-amyloid in rat hippocampal and cortical neurons. Neuropharmacology 2005; 49:1140–1148.
- Takatani T, Takahashi K, Uozumi Y, et al. Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 2004; 287:C949–C953.
- Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis 2010; 20(suppl 2):S439–S452.
- El Idrissi A. Taurine improves learning and retention in aged mice. Neurosci Lett 2008; 436:19–22.
- Oja SS, Saransaari P. Taurine and epilepsy. Epilepsy Res 2013; 104:187–194.
- Berson EL, Hayes KC, Rabin AR, Schmidt SY, Watson G. Retinal degeneration in cats fed casein. II. Supplementation with methionine, cysteine, or taurine. Invest Ophthalmol 1976; 15:52–58.
- Beyranvand MR, Khalafi MK, Roshan VD, Choobineh S, Parsa SA, Piranfar MA. Effect of taurine supplementation on exercise capacity of patients with heart failure. J Cardiol 2011; 57:333–337.
- Eby G, Halcomb WW. Elimination of cardiac arrhythmias using oral taurine with l-arginine with case histories: hypothesis for nitric oxide stabilization of the sinus node. Med Hypotheses 2006; 67:1200–1204.
KEY POINTS
- Energy drinks are widely consumed in the United States, with an estimated 354 million gallons sold in 2009, or approximately 5.25 L/year per person over age 10.
- Taurine has been reported to have anti-inflammatory action. Supplementation has been proposed to have beneficial effects in epilepsy, heart failure, cystic fibrosis, and diabetes, and has been shown in animal studies to protect against neurotoxic insults from alcohol, ammonia, lead, and other substances.
- Taurine is an inhibitory neurotransmitter and neuromodulator. It is structurally analogous to gamma-aminobutyric acid, the main inhibitory neurotransmitter in the brain.

Self-criticism and self-compassion: Risk and resilience
Once thought to only be associated with depression, self-criticism is a transdiagnostic risk factor for diverse forms of psychopathology.1,2 However, research has shown that self-compassion is a robust resilience factor when faced with feelings of personal inadequacy.3,4
Self-critical individuals experience feelings of unworthiness, inferiority, failure, and guilt. They engage in constant and harsh self-scrutiny and evaluation, and fear being disapproved and criticized and losing the approval and acceptance of others.5 Self-compassion involves treating oneself with care and concern when confronted with personal inadequacies, mistakes, failures, and painful life situations.6,7
Although self-criticism is destructive across clinical disorders and interpersonal relationships, self-compassion is associated with healthy relationships, emotional well-being, and better treatment outcomes.
Recent research shows how clinicians can teach their patients how to be less self-critical and more self-compassionate. Neff6,7 proposes that self-compassion involves treating yourself with care and concern when being confronted with personal inadequacies, mistakes, failures, and painful life situations. It consists of 3 interacting components, each of which has a positive and negative pole:
- self-kindness vs self-judgment
- a sense of common humanity vs isolation
- mindfulness vs over-identification.
Self-kindness refers to being caring and understanding with oneself rather than harshly judgmental. Instead of attacking and berating oneself for personal shortcomings, the self is offered warmth and unconditional acceptance.
Humanity involves recognizing that humans are imperfect, that all people fail, make mistakes, and have serious life challenges. By remembering that imperfection is part of life, we feel less isolated when we are in pain.
Mindfulness in the context of self-compassion involves being aware of one’s painful experiences in a balanced way that neither ignores and avoids nor exaggerates painful thoughts and emotions.
Self-compassion is more than the absence of self-judgment, although a defining feature of self-compassion is the lack of self-judgment, and self-judgment overlaps with self-criticism. Rather, self-compassion provides several access points for reducing self-criticism. For example, being kind and understanding when confronting personal inadequacies (eg, “it’s okay not to be perfect”) can counter harsh self-talk (eg, “I’m not defective”). Mindfulness of emotional pain (eg, “this is hard”) can facilitate a kind and warm response (eg, “what can I do to take care of myself right now?”) and therefore lessen self-blame (eg, “blaming myself is just causing me more suffering”). Similarly, remembering that failure is part of the human experience (eg, “it’s normal to mess up sometimes”) can lessen egocentric feelings of isolation (eg, “it’s not just me”) and over-identification (eg, “it’s not the end of the world”), resulting in lessened self-criticism (eg, “maybe it’s not just because I’m a bad person”).
Depression
Several studies have found that self-criticism predicts depression. In 3 epidemiological studies, “feeling worthless” was among the top 2 symptoms predicting a depression diagnosis and later depressive episodes.10 Self-criticism in fourth-year medical students predicted depression 2 years later, and—in males—10 years later in their medical careers better than a history of depression.11 Self-critical perfectionism also is associated with suicidal ideation and lethality of suicide attempts.12
Self-criticism has been shown to predict depressive relapse and residual self-devaluative symptoms in recovered depressed patients.13 In one study, currently depressed and remitted depressed patients had higher self-criticism and lower self-compassion compared with healthy controls. Both factors were more strongly associated with depression status than higher perfectionistic beliefs and cognitions, rumination, and maladaptive emotional regulation.14
Self-criticism and response to treatment. In the National Institute of Mental Health Treatment of Depression Collaborative Research Program,15 self-critical perfectionism predicted a poorer outcome across all 4 treatments (cognitive-behavioral therapy [CBT], interpersonal psychotherapy [IPT], pharmacotherapy plus clinical management, and placebo plus clinical management). Subsequent studies found that self-criticism predicted poorer response to CBT16 and IPT.17 The authors suggest that self-criticism could interfere with treatment because self-critical patients might have difficulty developing a strong therapeutic alliance.18,19
Anxiety disorders
Self-criticism is common across psychiatric disorders. In a study of 5,877 respondents in the National Comorbidity Survey (NCS), self-criticism was associated with social phobia, findings that were significant after controlling for current emotional distress, neuroticism, and lifetime history of mood, anxiety, and substance use disorders.20 Further, in a CBT treatment study, baseline self-criticism was associated with severity of social phobia and changes in self-criticism predicted treatment outcome.21 Self-criticism might be an important core psychological process in the development, maintenance, and course of social phobia. Patients with social anxiety disorder have less self-compassion than healthy controls and greater fear of negative evaluation.
In the NCS, self-criticism was associated with posttraumatic stress disorder (PTSD) even after controlling for lifetime history of affective and anxiety disorders.20 Self-criticism predicted greater severity of combat-related PTSD in hospitalized male veterans,22 and those with PTSD had higher scores on self-criticism scales than those with major depressive disorder.23 In a study of Holocaust survivors, those with PTSD scored higher on self-criticism than survivors without PTSD.24 Self-criticism also distinguished between female victims of domestic violence with and without PTSD.25
Self-compassion could be a protective factor for posttraumatic stress.26 Combat veterans with higher levels of self-compassion showed lower levels of psychopathology, better functioning in daily life, and fewer symptoms of posttraumatic stress.27 In fact, self-compassion has been found to be a stronger predictor of PTSD than level of combat exposure.28
In an early study, self-criticism scores were higher in patients with panic disorder than in healthy controls, but lower than in patients with depression.29 In a study of a mixed sample of anxiety disorder patients, symptoms of generalized anxiety disorder were associated with shame proneness.30 Consistent with these results, Hoge et al31 found that self-compassion was lower in generalized anxiety disorder patients compared with healthy controls with elevated stress. Low self-compassion has been associated with severity of obsessive-compulsive disorder.32
Eating disorders
Self-criticism is correlated with eating disorder severity.33 In a study of patients with binge eating disorder, Dunkley and Grilo34 found that self-criticism was associated with the over-evaluation of shape and weight independently of self-esteem and depression. Self-criticism also is associated with body dissatisfaction, independent of self-esteem and depression. Dunkley et al35 found that self-criticism, but not global self-esteem, in patients with binge eating disorder mediated the relationship between childhood abuse and body dissatisfaction and depression. Numerous studies have shown that shame is associated with more severe eating disorder pathology.33
Self-compassion seems to buffer against body image concerns. It is associated with less body dissatisfaction, body preoccupation, and weight worries,36 greater body appreciation37 and less disordered eating.37-39 Early decreases in shame during eating disorder treatment was associated with more rapid reduction in eating disorder symptoms.40
Interpersonal relationships
Several studies have shown that self-criticism has negative effects on interpersonal relationships throughout life.5,41,42
- Self-criticism at age 12 predicted less involvement in high school activities and, at age 31, personal and social maladjustment.43
- High school students with high self-criticism reported more interpersonal problems.44
- Self-criticism was associated with loneliness, depression, and lack of intimacy with opposite sex friends or partners during the transition to college.45
- In a study of college roommates,46 self-criticism was associated with increased likelihood of rejection.
- Whiffen and Aube47 found that self-criticism was associated with marital dissatisfaction and depression.
- Self-critical mothers with postpartum depression were less satisfied with social support and were more vulnerable to depression.48
Self-compassion appears to enhance interpersonal relationships. In a study of heterosexual couples,49 self-compassionate individuals were described by their partners as being more emotionally connected, as well as accepting and supporting autonomy, while being less detached, controlling, and verbally or physically aggressive than those lacking self-compassion. Because self-compassionate people give themselves care and support, they seem to have more emotional resources available to give to others.
See the Box examining the evidence on the role of self-compassion in borderline personality disorder and non-suicidal self-injury.

Achieving goals
Powers et al50 suggest that self-critics approach goals based on motivation to avoid failure and disapproval, rather than on intrinsic interest and personal meaning. In studies of college students pursuing academic, social, or weight loss goals, self-criticism was associated with less progress to that goal. Self-criticism was associated with rumination and procrastination, which the authors suggest might have focused the self-critic on potential failure, negative evaluation from others, and loss of self-esteem. Additional studies showed the deleterious effects of self-criticism on college students’ progress on obtaining academic or music performance goals and on community residents’ weight loss goals.51
Not surprisingly, self-compassion is associated with successful goal pursuit and resilience when goals are not met52 and less procrastination and academic worry.53 Self-compassion also is associated with intrinsic motivation, goals based on mastery rather than performance, and less fear of academic failure.54
How self-criticism and self-compassion develop
Studies have explored the impact of early relationships with parents and development of self-criticism. Parental overcontrol and restrictiveness and lack of warmth consistently have been identified as parenting styles associated with development of self-criticism in children.55 One study found that self-criticism fully mediated the relationship between childhood verbal abuse from parents and depression and anxiety in adulthood.56 Reports from parents on their current parenting styles are consistent with these studies.57 Amitay et al57 states that “[s]elf-critics’ negative childhood experiences thus seem to contribute to a pattern of entering, creating, or manipulating subsequent interpersonal environments in ways that perpetuate their negative self-image and increase vulnerability to depression.” Not surprisingly, self-criticism is associated with a fearful avoidant attachment style.58 Review of the developmental origins of self-criticism confirms these factors and presents findings that peer relationships also are important factors in the development of self-criticism.59,60
Early positive relationships with caregivers are associated with self-compassion. Recollections of maternal support are correlated with self-compassion and secure attachment styles in adolescents and adults.61 Pepping et al62 found that retrospective reports of parental rejection, overprotection, and low parental warmth was associated with low self-compassion.
Benefits of self-compassion
A growing body of research suggests that self-compassion is strongly linked to mental health. Greater self-compassion consistently has been associated with lower levels of depression and anxiety,3 with a large effect size.4 Of course, central to self-compassion is the lack of self-criticism, but self-compassion still protects against anxiety and depression when controlling for self-criticism and negative affect.6,63 Self-compassion is a strong predictor of symptom severity and quality of life among individuals with anxious distress.64
The benefits of self-compassion stem partly from a greater ability to cope with negative emotions.6,63,65 Self-compassionate people are less likely to ruminate on their negative thoughts and emotions or suppress them,6,66 which helps to explain why self-compassion is a negative predictor of depression.67
Self-compassion also enhances positive mind states. A number of studies have found links between self-compassion and positive psychological qualities, such as happiness, optimism, wisdom, curiosity, and exploration, and personal initiative.63,65,68,69 By embracing one’s suffering with compassion, negative states are ameliorated when positive emotions of kindness, connectedness, and mindful presence are generated.
Misconceptions about self-compassion
A common misconception is that abandoning self-criticism in favor of self-compassion will undermine motivation70; however, research indicates the opposite. Although self-compassion is negatively associated with maladaptive perfectionism, it is not correlated with self-adopted performance standards.6 Self-compassionate people have less fear of failure54 and, when they do fail, they are more likely to try again.71 Breines and Chen72 found in a series of experimental studies that engendering feelings of self-compassion for personal weaknesses, failures, and past transgressions resulted in more motivation to change, to try harder to learn, and to avoid repeating past mistakes.
Another common misunderstanding is that self-compassion is a weakness. In fact, research suggests that self-compassion is a powerful way to cope with life challenges.73
Although some fear that self-compassion leads to self-indulgence, there is evidence that self-compassion promotes health-related behaviors. Self-compassionate individuals are more likely to seek medical treatment when needed,74 exercise for intrinsic reasons,75 and drink less alcohol.76 Inducing self-compassion has been found to help people stick to their diets77 and quit smoking.78
Self-compassion interventions
Individuals can develop self-compassion. Shapira and Mongrain79 found that adults who wrote a compassionate letter to themselves once a day for a week about the distressing events they were experiencing showed significant reductions in depression up to 3 months and significant increases in happiness up to 6 months compared with a control group who wrote about early memories. Albertson et al80 found that, compared with a wait-list control group, 3 weeks of self-compassion meditation training improved body dissatisfaction, body shame, and body appreciation among women with body image concerns. Similarly, Smeets et al81 found that 3 weeks of self-compassion training for female college students led to significantly greater increases in mindfulness, optimism, and self-efficacy, as well as greater decreases in rumination compared with a time management control group.
The Box6,70,82-86 describes rating scales that can measure self-compassion and self-criticism.

Mindful self-compassion (MSC), developed by Neff and Germer,87 is an 8-week group intervention designed to teach people how to be more self-compassionate through meditation and informal practices in daily life. Results of a randomized controlled trial found that, compared with a wait-list control group, participants using MSC reported significantly greater increases in self-compassion, compassion for others, mindfulness, and life satisfaction, and greater decreases in depression, anxiety, stress, and emotional avoidance, with large effect sizes indicated. These results were maintained up to 1 year.
Compassion-focused therapy (CFT) is designed to enhance self-compassion in clinical populations.88 The approach uses a number of imagery and experiential exercises to enhance patients’ abilities to extend feelings of reassurance, safeness, and understanding toward themselves. CFT has shown promise in treating a diverse group of clinical disorders such as depression and shame,8,89 social anxiety and shame,90 eating disorders,91 psychosis,92 and patients with acquired brain injury.93 A group-based CFT intervention with a heterogeneous group of community mental health patients led to significant reductions in depression, anxiety, stress, and self-criticism.94 See Leaviss and Utley95 for a review of the benefits of CFT.
Fears of developing self-compassion
It is important to note that some people can access self-compassion more easily than others. Highly self-critical patients could feel anxious when learning to be compassionate to themselves, a phenomenon known as “fear of compassion”96 or “backdraft.”97 Backdraft occurs when a firefighter opens a door with a hot fire behind it. Oxygen rushes in, causing a burst of flame. Similarly, when the door of the heart is opened with compassion, intense pain could be released. Unconditional love reveals the conditions under which we were unloved in the past. Some individuals, especially those with a history of childhood abuse or neglect, are fearful of compassion because it activates grief associated with feelings of wanting, but not receiving, affection and care from significant others in childhood.
Clinicians should be aware that anxiety could arise and should help patients learn how to go slowly and stabilize themselves if overwhelming emotions occur as a part of self-compassion practice. Both CFT and MSC have processes to deal with fear of compassion in their protocols,98,99 with the focus on explaining to individuals that although such fears may occur, they are a normal and necessary part of the healing process. Individuals also are taught to focus on the breath, feeling the sensations in the soles of their feet, or other mindfulness practices to ground and stabilize attention when overwhelming feelings arise.
Clinical interventions
Self-compassion interventions that I (R.W.) find most helpful, in the order I administer them, are:
- exploring perceived advantages and disadvantages of self-criticism
- presenting self-compassion as a way to get the perceived advantages of self-criticism without the disadvantages
- discussing what it means to be compassionate for someone else who is suffering, and then asking what it would be like if they treated themselves with the same compassion
- exploring patients’ misconceptions and fears of self-compassion
- directing patients to the self-compassion Web site to get an understanding of what self-compassion is and how it differs from self-esteem
- taking an example of a recent situation in which the patient was self-critical and exploring how a self-compassionate response would differ.
Asking what they would say to a friend often is an effective way to get at this. In a later therapy session, self-compassionate imagery is a useful way to get the patient to experience self-compassion on an emotional level. See Neff100 and Gilbert98 for other techniques to enhance self-compassion.
1. Shahar B, Doron G, Ohad S. Childhood maltreatment, shame-proneness and self-criticism in social anxiety disorder: a sequential mediational model. Clin Psychol Psychother. 2015:22(6):570-579.
2. Kannan D, Levitt HM. A review of client self-criticism in psychotherapy. J Psychother Integr. 2013;23(2):166-178.
3. Barnard LK, Curry JF. Self-compassion: conceptualizations, correlates, and interventions. Rev Gen Psychol. 2011;15(4):289-303.
4. MacBeth A, Gumley A. Exploring compassion: a meta-analysis of the association between self-compassion and psychopathology. Clin Psychol Rev. 2012;32(6):545-552
5. Blatt SJ, Zuroff DC. Interpersonal relatedness and self-definition: two prototypes for depression. Clin Psychol Rev. 1992;12(5):527-562.
6. Neff KD. The development and validation of a scale to measure self-compassion. Self Identity. 2003;2(2):223-250.
7. Neff KD. Self-compassion: an alternative conceptualization of a healthy attitude toward oneself. Self Identity. 2003;(2)2:85-101.
8. Gilbert P, Procter S. Compassionate mind training for people with high shame and self-criticism: overview and pilot study of a group therapy approach. Clin Psychol Psychother. 2006;13(6):353-379.
9. Dunkley DM, Zuroff DC, Blankstein KR. Specific perfectionism components versus self-criticism in predicting maladjustment. Pers Individ Dif. 2006;40(4):665-676.
10. Murphy JM, Nierenberg AA, Monson RR, et al. Self-disparagement as feature and forerunner of depression: Mindfindings from the Stirling County Study. Compr Psychiatry. 2002;43(1):13-21.
11. Brewin CR, Firth-Cozens J. Dependency and self-criticism as predictors of depression in young doctors. J Occup Health Psychol. 1997;2(3):242-246.
12. Fazaa N, Page S. Dependency and self-criticism as predictors of suicidal behavior. Suicide Life Threat Behav. 2003;33(2):172-185.
13. Teasdale JD, Cox SG. Dysphoria: self-devaluative and affective components in recovered depressed patients and never depressed controls. Psychol Med. 2001;31(7):1311-1316.
14. Ehret AM, Joormann J, Berking M. Examining risk and resilience factors for depression: the role of self-criticism and self-compassion. Cogn Emot. 2015;29(8):1496-1504.
15. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health Treatment of Depression Collaborative Research Program. General effectiveness of treatments. Arch Gen Psychiatry. 1989;46(11):971-982; discussion 983.
16. Rector NA, Bagby RM, Segal ZV, et al. Self-criticism and dependency in depressed patients treated with cognitive therapy or pharmacotherapy. Cognit Ther Res. 2000;24(5):571-584.
17. Marshall MB, Zuroff DC, McBride C, et al. Self-criticism predicts differential response to treatment for major depression. J Clin Psychol. 2008;64(3):231-244.
18. Zuroff DC, Blatt SJ, Sotsky SM, et al. Relation of therapeutic alliance and perfectionism to outcome in brief outpatient treatment of depression. J Consult Clin Psychol. 2000;68(1):114-124.
19. Whelton WJ, Greenberg LS. Emotion in self-criticism. Pers Individ Dif. 2005;38(7):1583-1595.
20. Cox BJ, Fleet C, Stein MB. Self-criticism and social phobia in the US national comorbidity survey. J Affect Disord. 2004;82(2):227-234.
21. Cox BJ, Walker JR, Enns MW, et al. Self-criticism in generalized social phobia and response to cognitive-behavioral treatment. Behav Ther. 2002;33(4):479-491.
22. McCranie EW, Hyer LA. Self-critical depressive experience in posttraumatic stress disorder. Psychol Rep. 1995;77(3 pt 1):880-882.
23. Southwick SM, Yehuda R, Giller EL Jr. Characterization of depression in war-related posttraumatic stress disorder. Am J Psychiatry. 1991;148(2):179-183.
24. Yehuda R, Kahana B, Southwick SM, et al. Depressive features in Holocaust survivors with post-traumatic stress disorder. J Traumatic Stress. 1994;7(4):699-704.
25. Sharhabani-Arzy R, Amir M, Swisa A. Self-criticism, dependency and posttraumatic stress disorder among a female group of help-seeking victims of domestic violence in Israel. Pers Individ Dif. 2005;38(5):1231-1240.
26. Beaumont E, Galpin A, Jenkins P. ‘Being kinder to myself’: a prospective comparative study, exploring post-trauma therapy outcome measures, for two groups of clients, receiving either cognitive behaviour therapy or cognitive behaviour therapy and compassionate mind training. Counselling Psychol Rev. 2012;27(1):31-43.
27. Dahm KA. Mindfulness and self-compassion as predictors of functional outcomes and psychopathology in OEF/OIF veterans exposed to trauma. https://repositories.lib.utexas.edu/handle/2152/21635. Published August 2013. Accessed November 8, 2016.
28. Hiraoka R, Meyer EC, Kimbrel NA, et al. Self-compassion as a prospective predictor of PTSD symptom severity among trauma-exposed US Iraq and Afghanistan war veterans. J Trauma Stress. 2015;28(2):127-133.
29. Bagby RM, Cox BJ, Schuller DR, et al. Diagnostic specificity of the dependent and self-critical personality dimensions in major depression. J Affect Disord. 1992;26(1):59-63.
30. Hedman E, Ström P, Stünkel A, et al. Shame and guilt in social anxiety disorder: effects of cognitive behavior therapy and association with social anxiety and depressive symptoms. PLoS One. 2013;8(4):e61713. doi: 10.1371/journal.pone.0061713.
31. Hoge EA, Hölzel BK, Marques L, et al. Mindfulness and self-compassion in generalized anxiety disorder: examining predictors of disability. Evid Based Complement Alternat Med. 2013;2013:576258. doi: 10.1155/2013/576258.
32. Wetterneck CT, Lee EB, Smith AH, et al. Courage, self-compassion, and values in obsessive-compulsive disorder. J Contextual Behav Sci. 2013;2(3-4):68-73.
33. Kelly AC, Carter JC. Why self-critical patients present with more severe eating disorder pathology: The mediating role of shame. Br J Clin Psychol. 2013;52(2):148-161.
34. Dunkley DM, Grilo CM. Self-criticism, low self-esteem, depressive symptoms, and over-evaluation of shape and weight in binge eating disorder patients. Behav Res Ther. 2007;45(1):139-149.
35. Dunkley DM, Masheb RM, Grilo CM. Childhood maltreatment, depressive symptoms, and body dissatisfaction in patients with binge eating disorder: the mediating role of self-criticism. Int J Eat Disord. 2010;43(3):274-281.
36. Wasylkiw L, MacKinnon AL, MacLellan AM. Exploring the link between self-compassion and body image in university women. Body Image. 2012;9(2):236-245.
37. Ferreira C, Pinto-Gouveia J, Duarte C. Self-compassion in the face of shame and body image dissatisfaction: implications for eating disorders. Eat Behavs. 2013;14(2):207-210.
38. Kelly AC, Carter JC, Zuroff DC, et al. Self-compassion and fear of self-compassion interact to predict response to eating disorders treatment: a preliminary investigation. Psychother Res. 2013;23(3):252-264.
39. Webb JB, Forman MJ. Evaluating the indirect effect of self-compassion on binge eating severity through cognitive-affective self-regulatory pathways. Eat Behavs. 2013;14(2):224-228.
40. Kelly AC, Carter JC, Borairi S. Are improvements in shame and self-compassion early in eating disorders treatment associated with better patient outcomes? Int J Eat Disord. 2014;47(1):54-64.
41. Wiseman H, Raz A, Sharabany R. Depressive personality styles and interpersonal problems in young adults with difficulties in establishing long-term romantic relationships. Isr J Psychiatry Rel Sci. 2007;44(4):280-291.
42. Besser A, Priel B. A multisource approach to self-critical vulnerability to depression: the moderating role of attachment. J Pers. 2003;71(4):515-555.
43. Zuroff DC, Koestner R, Powers TA. Self-criticism at age 12: a longitudinal-study of adjustment. Cognit Ther Res. 1994;18(4):367-385.
44. Fichman L, Koestner R, Zuroff DC. Depressive styles in adolescence: Assessment, relation to social functioning, and developmental trends. J Youth Adolesc. 1994;23(3):315-330.
45. Wiseman H. Interpersonal relatedness and self-definition in the experience of loneliness during the transition to university. Personal Relationships. 1997;4(3):285-299.
46. Mongrain M, Lubbers R, Struthers W. The power of love: mediation of rejection in roommate relationships of dependents and self-critics. Pers Soc Psychol Bull. 2004;30(1):94-105.
47. Whiffen VE, Aube JA. Personality, interpersonal context and depression in couples. J Soc Pers Relat. 1999;16(3):369-383.
48. Priel B, Besser A. Dependency and self-criticism among first-time mothers: the roles of global and specific support. J Soc Clin Psychol. 2000;19(4):437-450.
49. Neff KD, Beretvas SN. The role of self-compassion in romantic relationships. Self Identity. 2013;12(1):78-98.
50. Powers TA, Koestner R, Zuroff DC. Self-criticism, goal motivation, and goal progress. J Soc Clin Psychol. 2007;26(7):826-840.
51. Powers TA, Koestner R, Zuroff DC, et al. The effects of self-criticism and self-oriented perfectionism on goal pursuit. Pers Soc Psychol Bull. 2011;37(7):964-975.
52. Hope N, Koestner R, Milyavskaya M. The role of self-compassion in goal pursuit and well-being among university freshmen. Self Identity. 2014;13(5):579-593.
53. Williams JG, Stark SK, Foster EE. Start today or the very last day? The relationships among self-compassion, motivation, and procrastination. Am J Psychol Res. 2008;4(1):37-44.
54. Neff KD, Hseih Y, Dejittherat K. Self-compassion, achievement goals, and coping with academic failure. Self Identity. 2005;4(3):263-287.
55. Campos RC, Besser A, Blatt SJ. The mediating role of self-criticism and dependency in the association between perceptions of maternal caring and depressive symptoms. Depress Anxiety. 2010;27(12):1149-1157.
56. Sachs-Ericsson N, Verona E, Joiner T, et al. Parental verbal abuse and the mediating role of self-criticism in adult internalizing disorders. J Affect Disord. 2006;93(1-3):71-78.
57. Amitay OA, Mongrain M, Fazaa N. Love and control: self-criticism in parents and daughters and perceptions of relationship partners. Pers Individ Dif. 2008;44(1):75-85.
58. Zuroff DC, Fitzpatrick DK. Depressive personality styles: implications for adult attachment. Pers Individ Dif. 1995;18(2):253-265.
59. Kopala-Sibley DC, Zuroff DC. The developmental origins of personality factors from the self-definitional and relatedness domains: a review of theory and research. Rev Gen Psychol. 2014;18(3):137-155.
60. Kopala-Sibley DC, Zuroff DC, Leybman MJ, et al. Recalled peer relationship experiences and current levels of self-criticism and self-reassurance. Psychol Psychother. 2013;86(1):33-51.
61. Neff KD, McGehee P. Self-compassion and psychological resilience among adolescents and young adults. Self Identity. 2010;9(3):225-240.
62. Pepping CA, Davis PJ, O’Donovan A, et al. Individual differences in self-compassion: the role of attachment and experiences of parenting in childhood. Self Identity. 2015;14(1):104-117.
63. Neff KD, Rude SS, Kirkpatrick KL. An examination of self-compassion in relation to positive psychological functioning and personality traits. J Res Pers. 2007;41(4):908-916.
64. Van Dam NT, Sheppard SC, Forsyth JP, et al. Self-compassion is a better predictor than mindfulness of symptom severity and quality of life in mixed anxiety and depression. J Anxiety Disord. 2011;25(1):123-130.
65. Heffernan M, Quinn MT, McNulty SR, et al. Self-compassion and emotional intelligence in nurses. Int J Nursing Practice. 2010;16(4):366-373.
66. Neff KD, Kirkpatrick KL, Rude SS. Self-compassion and adaptive psychological functioning. J Res Pers. 2007;41(1):139-154.
67. Krieger T, Altenstein D, Baettig I, et al. Self-compassion in depression: associations with depressive symptoms, rumination, and avoidance in depressed outpatients. Behav Ther. 2013;44(3):501-513.
68. Breen WE, Kashdan TB, Lenser ML, et al. Gratitude and forgiveness: convergence and divergence on self-report and informant ratings. Pers Individ Dif. 2010;49(8):932-937.
69. Hollis-Walker L, Colosimo K. Mindfulness, self-compassion, and happiness in non-meditators: A theoretical and empirical examination. Pers Individ Dif. 2011;50(2):222-227.
70. Gilbert P, McEwan K, Matos M, et al. Fears of compassion: development of three self-report measures. Psychol Psychother. 2011;84(3):239-255.
71. Neely ME, Schallert DL, Mohammed SS, et al. Self-kindness when facing stress: the role of self-compassion, goal regulation, and support in college students’ well-being. Motiv Emot. 2009;33(1):88-97.
72. Breines JG, Chen S. Self-compassion increases self-improvement motivation. Pers Soc Psychol Bull. 2012;38(9):1133-1143.
73. Allen AB, Leary MR. Self-compassion, stress, and coping. Soc Pers Psychol Compass. 2010;4(2):107-118.
74. Terry ML, Leary MR. Self-compassion, self-regulation, and health. Self Identity. 2011;10(3):352-362.
75. Magnus CMR, Kowalski KC, McHugh TF. The role of self-compassion in women’s self-determined motives to exercise and exercise-related outcomes. Self Identity. 2010;9(4):363-382.
76. Brooks M, Kay-Lambkin F, Bowman J, et al. Self-compassion amongst clients with problematic alcohol use. Mindfulness. 2012;3(4):308-317.
77. Adams CE, Leary MR. Promoting self-compassionate attitudes toward eating among restrictive and guilty eaters. J Soc Clin Psychol. 2007;26(10):1120-1144.
78. Kelly AC, Zuroff DC, Foa CL, et al. Who benefits from training in self-compassionate self-regulation? A study of smoking reduction. J Soc Clin Psychol. 2010;29(7):727-755.
79. Shapira LB, Mongrain M. The benefits of self-compassion and optimism exercises for individuals vulnerable to depression. J Posit Psychol. 2010;5(5):377-389.
80. Albertson ER, Neff KD, Dill-Shackleford KE. Self-compassion and body dissatisfaction in women: a randomized controlled trial of a brief meditation intervention. Mindfulness. 2015;6(3):444-454.
81. Smeets E, Neff K, Alberts H, et al. Meeting suffering with kindness: effects of a brief self-compassion intervention for female college students. J Clinical Psychol. 2014;70(9):794-807.
82. Blatt SJ, D’Afflitti JP, Quinlan DM. Depressive experiences questionnaire. New Haven, CT: Yale University Press; 1976.
83. Weissman AN, Beck AT. Development and validation of the dysfunctional attitude scale: a preliminary investigation. Paper presented at: 62nd Annual Meeting of the Association for Advanced Behavior Therapy; March 27-31, 1978; Toronto, Ontario, Canada.
84. Gilbert P, Clarke M, Hempel S, et al. Criticizing and reassuring oneself: an exploration of forms, styles and reasons in female students. Br J Clin Psychol. 2004;43(pt 1):31-50.
85. Baião R, Gilbert P, McEwan K, et al. Forms of self-criticising/attacking & self-reassuring scale: psychometric properties and normative study. Psychol Psychother. 2015;88(4):438-452.
86. Neff KD. The self-compassion scale is a valid and theoretically coherent measure of self-compassion. Mindfulness. 2016;7(1):264-274.
87. Neff KD, Germer CK. A pilot study and randomized controlled trial of the mindful self-compassion program. J Clinical Psychol. 2013;69(1):28-44.
88. Gilbert P. Introducing compassion-focused therapy. Adv Psychiatr Treat. 2009;15(3):199-208.
89. Kelly AC, Zuroff DC, Shapira LB. Soothing oneself and resisting self-attacks: the treatment of two intrapersonal deficits in depression vulnerability. Cognit Ther Res. 2009;33(3):301-313.
90. Boersma K, Hakanson A, Salomonsson E, et al. Compassion focused therapy to counteract shame, self-criticism and isolation. A replicated single case experimental study of individuals with social anxiety. J Contemp Psychother. 2015;45(2):89-98.
91. Gale C, Gilbert P, Read N, et al. An evaluation of the impact of introducing compassion focused therapy to a standard treatment programme for people with eating disorders. Clin Psychol Psychother. 2014;21(1):1-12.
92. Braehler C, Gumley A, Harper J, et al. Exploring change processes in compassion focused therapy in psychosis: results of a feasibility randomized controlled trial. Br J Clin Psychol. 2013;52(2):199-214.
93. Ashworth F, Clarke A, Jones L, et al. An exploration of compassion focused therapy following acquired brain injury. Psychol Psychother. 2014;88(2):143-162.
94. Judge L, Cleghorn A, McEwan K, et al. An exploration of group-based compassion focused therapy for a heterogeneous range of clients presenting to a community mental health team. Int J Cogn Ther. 2012;5(4):420-429.
95. Leaviss J, Utley L. Psychotherapeutic benefits of compassion-focused therapy: an early systematic review. Psychol Med. 2015;45(5):927-945.
96. Gilbert P, McEwan K, Gibbons L, et al. Fears of compassion and happiness in relation to alexithymia, mindfulness, and self‐criticism. Psychol Psychother. 2012;85(4):374-390.
97. Germer CK, Neff KD. Cultivating self-compassion in trauma survivors. In: Follette VM, Briere J, Rozelle D, et al, eds. Mindfulness-oriented interventions for trauma: integrating contemplative practices. New York, NY: Guilford Press; 2015:43-58.
98. Gilbert P. Compassion focused therapy: the CBT distinctive features series. London, United Kingdom: Routledge; 2010.
99. Germer C, Neff K. The mindful self-compassion training program. In: Singer T, Bolz M, eds. Compassion: bridging theory and practice: a multimedia book. Leipzig, Germany: Max-Planck Institute; 2013:365-396.
100. Neff K. Self-compassion: the proven power of being kind to yourself. New York, NY: HarperCollins; 2015.
Once thought to only be associated with depression, self-criticism is a transdiagnostic risk factor for diverse forms of psychopathology.1,2 However, research has shown that self-compassion is a robust resilience factor when faced with feelings of personal inadequacy.3,4
Self-critical individuals experience feelings of unworthiness, inferiority, failure, and guilt. They engage in constant and harsh self-scrutiny and evaluation, and fear being disapproved and criticized and losing the approval and acceptance of others.5 Self-compassion involves treating oneself with care and concern when confronted with personal inadequacies, mistakes, failures, and painful life situations.6,7
Although self-criticism is destructive across clinical disorders and interpersonal relationships, self-compassion is associated with healthy relationships, emotional well-being, and better treatment outcomes.
Recent research shows how clinicians can teach their patients how to be less self-critical and more self-compassionate. Neff6,7 proposes that self-compassion involves treating yourself with care and concern when being confronted with personal inadequacies, mistakes, failures, and painful life situations. It consists of 3 interacting components, each of which has a positive and negative pole:
- self-kindness vs self-judgment
- a sense of common humanity vs isolation
- mindfulness vs over-identification.
Self-kindness refers to being caring and understanding with oneself rather than harshly judgmental. Instead of attacking and berating oneself for personal shortcomings, the self is offered warmth and unconditional acceptance.
Humanity involves recognizing that humans are imperfect, that all people fail, make mistakes, and have serious life challenges. By remembering that imperfection is part of life, we feel less isolated when we are in pain.
Mindfulness in the context of self-compassion involves being aware of one’s painful experiences in a balanced way that neither ignores and avoids nor exaggerates painful thoughts and emotions.
Self-compassion is more than the absence of self-judgment, although a defining feature of self-compassion is the lack of self-judgment, and self-judgment overlaps with self-criticism. Rather, self-compassion provides several access points for reducing self-criticism. For example, being kind and understanding when confronting personal inadequacies (eg, “it’s okay not to be perfect”) can counter harsh self-talk (eg, “I’m not defective”). Mindfulness of emotional pain (eg, “this is hard”) can facilitate a kind and warm response (eg, “what can I do to take care of myself right now?”) and therefore lessen self-blame (eg, “blaming myself is just causing me more suffering”). Similarly, remembering that failure is part of the human experience (eg, “it’s normal to mess up sometimes”) can lessen egocentric feelings of isolation (eg, “it’s not just me”) and over-identification (eg, “it’s not the end of the world”), resulting in lessened self-criticism (eg, “maybe it’s not just because I’m a bad person”).
Depression
Several studies have found that self-criticism predicts depression. In 3 epidemiological studies, “feeling worthless” was among the top 2 symptoms predicting a depression diagnosis and later depressive episodes.10 Self-criticism in fourth-year medical students predicted depression 2 years later, and—in males—10 years later in their medical careers better than a history of depression.11 Self-critical perfectionism also is associated with suicidal ideation and lethality of suicide attempts.12
Self-criticism has been shown to predict depressive relapse and residual self-devaluative symptoms in recovered depressed patients.13 In one study, currently depressed and remitted depressed patients had higher self-criticism and lower self-compassion compared with healthy controls. Both factors were more strongly associated with depression status than higher perfectionistic beliefs and cognitions, rumination, and maladaptive emotional regulation.14
Self-criticism and response to treatment. In the National Institute of Mental Health Treatment of Depression Collaborative Research Program,15 self-critical perfectionism predicted a poorer outcome across all 4 treatments (cognitive-behavioral therapy [CBT], interpersonal psychotherapy [IPT], pharmacotherapy plus clinical management, and placebo plus clinical management). Subsequent studies found that self-criticism predicted poorer response to CBT16 and IPT.17 The authors suggest that self-criticism could interfere with treatment because self-critical patients might have difficulty developing a strong therapeutic alliance.18,19
Anxiety disorders
Self-criticism is common across psychiatric disorders. In a study of 5,877 respondents in the National Comorbidity Survey (NCS), self-criticism was associated with social phobia, findings that were significant after controlling for current emotional distress, neuroticism, and lifetime history of mood, anxiety, and substance use disorders.20 Further, in a CBT treatment study, baseline self-criticism was associated with severity of social phobia and changes in self-criticism predicted treatment outcome.21 Self-criticism might be an important core psychological process in the development, maintenance, and course of social phobia. Patients with social anxiety disorder have less self-compassion than healthy controls and greater fear of negative evaluation.
In the NCS, self-criticism was associated with posttraumatic stress disorder (PTSD) even after controlling for lifetime history of affective and anxiety disorders.20 Self-criticism predicted greater severity of combat-related PTSD in hospitalized male veterans,22 and those with PTSD had higher scores on self-criticism scales than those with major depressive disorder.23 In a study of Holocaust survivors, those with PTSD scored higher on self-criticism than survivors without PTSD.24 Self-criticism also distinguished between female victims of domestic violence with and without PTSD.25
Self-compassion could be a protective factor for posttraumatic stress.26 Combat veterans with higher levels of self-compassion showed lower levels of psychopathology, better functioning in daily life, and fewer symptoms of posttraumatic stress.27 In fact, self-compassion has been found to be a stronger predictor of PTSD than level of combat exposure.28
In an early study, self-criticism scores were higher in patients with panic disorder than in healthy controls, but lower than in patients with depression.29 In a study of a mixed sample of anxiety disorder patients, symptoms of generalized anxiety disorder were associated with shame proneness.30 Consistent with these results, Hoge et al31 found that self-compassion was lower in generalized anxiety disorder patients compared with healthy controls with elevated stress. Low self-compassion has been associated with severity of obsessive-compulsive disorder.32
Eating disorders
Self-criticism is correlated with eating disorder severity.33 In a study of patients with binge eating disorder, Dunkley and Grilo34 found that self-criticism was associated with the over-evaluation of shape and weight independently of self-esteem and depression. Self-criticism also is associated with body dissatisfaction, independent of self-esteem and depression. Dunkley et al35 found that self-criticism, but not global self-esteem, in patients with binge eating disorder mediated the relationship between childhood abuse and body dissatisfaction and depression. Numerous studies have shown that shame is associated with more severe eating disorder pathology.33
Self-compassion seems to buffer against body image concerns. It is associated with less body dissatisfaction, body preoccupation, and weight worries,36 greater body appreciation37 and less disordered eating.37-39 Early decreases in shame during eating disorder treatment was associated with more rapid reduction in eating disorder symptoms.40
Interpersonal relationships
Several studies have shown that self-criticism has negative effects on interpersonal relationships throughout life.5,41,42
- Self-criticism at age 12 predicted less involvement in high school activities and, at age 31, personal and social maladjustment.43
- High school students with high self-criticism reported more interpersonal problems.44
- Self-criticism was associated with loneliness, depression, and lack of intimacy with opposite sex friends or partners during the transition to college.45
- In a study of college roommates,46 self-criticism was associated with increased likelihood of rejection.
- Whiffen and Aube47 found that self-criticism was associated with marital dissatisfaction and depression.
- Self-critical mothers with postpartum depression were less satisfied with social support and were more vulnerable to depression.48
Self-compassion appears to enhance interpersonal relationships. In a study of heterosexual couples,49 self-compassionate individuals were described by their partners as being more emotionally connected, as well as accepting and supporting autonomy, while being less detached, controlling, and verbally or physically aggressive than those lacking self-compassion. Because self-compassionate people give themselves care and support, they seem to have more emotional resources available to give to others.
See the Box examining the evidence on the role of self-compassion in borderline personality disorder and non-suicidal self-injury.
Achieving goals
Powers et al50 suggest that self-critics approach goals based on motivation to avoid failure and disapproval, rather than on intrinsic interest and personal meaning. In studies of college students pursuing academic, social, or weight loss goals, self-criticism was associated with less progress to that goal. Self-criticism was associated with rumination and procrastination, which the authors suggest might have focused the self-critic on potential failure, negative evaluation from others, and loss of self-esteem. Additional studies showed the deleterious effects of self-criticism on college students’ progress on obtaining academic or music performance goals and on community residents’ weight loss goals.51
Not surprisingly, self-compassion is associated with successful goal pursuit and resilience when goals are not met52 and less procrastination and academic worry.53 Self-compassion also is associated with intrinsic motivation, goals based on mastery rather than performance, and less fear of academic failure.54
How self-criticism and self-compassion develop
Studies have explored the impact of early relationships with parents and development of self-criticism. Parental overcontrol and restrictiveness and lack of warmth consistently have been identified as parenting styles associated with development of self-criticism in children.55 One study found that self-criticism fully mediated the relationship between childhood verbal abuse from parents and depression and anxiety in adulthood.56 Reports from parents on their current parenting styles are consistent with these studies.57 Amitay et al57 states that “[s]elf-critics’ negative childhood experiences thus seem to contribute to a pattern of entering, creating, or manipulating subsequent interpersonal environments in ways that perpetuate their negative self-image and increase vulnerability to depression.” Not surprisingly, self-criticism is associated with a fearful avoidant attachment style.58 Review of the developmental origins of self-criticism confirms these factors and presents findings that peer relationships also are important factors in the development of self-criticism.59,60
Early positive relationships with caregivers are associated with self-compassion. Recollections of maternal support are correlated with self-compassion and secure attachment styles in adolescents and adults.61 Pepping et al62 found that retrospective reports of parental rejection, overprotection, and low parental warmth was associated with low self-compassion.
Benefits of self-compassion
A growing body of research suggests that self-compassion is strongly linked to mental health. Greater self-compassion consistently has been associated with lower levels of depression and anxiety,3 with a large effect size.4 Of course, central to self-compassion is the lack of self-criticism, but self-compassion still protects against anxiety and depression when controlling for self-criticism and negative affect.6,63 Self-compassion is a strong predictor of symptom severity and quality of life among individuals with anxious distress.64
The benefits of self-compassion stem partly from a greater ability to cope with negative emotions.6,63,65 Self-compassionate people are less likely to ruminate on their negative thoughts and emotions or suppress them,6,66 which helps to explain why self-compassion is a negative predictor of depression.67
Self-compassion also enhances positive mind states. A number of studies have found links between self-compassion and positive psychological qualities, such as happiness, optimism, wisdom, curiosity, and exploration, and personal initiative.63,65,68,69 By embracing one’s suffering with compassion, negative states are ameliorated when positive emotions of kindness, connectedness, and mindful presence are generated.
Misconceptions about self-compassion
A common misconception is that abandoning self-criticism in favor of self-compassion will undermine motivation70; however, research indicates the opposite. Although self-compassion is negatively associated with maladaptive perfectionism, it is not correlated with self-adopted performance standards.6 Self-compassionate people have less fear of failure54 and, when they do fail, they are more likely to try again.71 Breines and Chen72 found in a series of experimental studies that engendering feelings of self-compassion for personal weaknesses, failures, and past transgressions resulted in more motivation to change, to try harder to learn, and to avoid repeating past mistakes.
Another common misunderstanding is that self-compassion is a weakness. In fact, research suggests that self-compassion is a powerful way to cope with life challenges.73
Although some fear that self-compassion leads to self-indulgence, there is evidence that self-compassion promotes health-related behaviors. Self-compassionate individuals are more likely to seek medical treatment when needed,74 exercise for intrinsic reasons,75 and drink less alcohol.76 Inducing self-compassion has been found to help people stick to their diets77 and quit smoking.78
Self-compassion interventions
Individuals can develop self-compassion. Shapira and Mongrain79 found that adults who wrote a compassionate letter to themselves once a day for a week about the distressing events they were experiencing showed significant reductions in depression up to 3 months and significant increases in happiness up to 6 months compared with a control group who wrote about early memories. Albertson et al80 found that, compared with a wait-list control group, 3 weeks of self-compassion meditation training improved body dissatisfaction, body shame, and body appreciation among women with body image concerns. Similarly, Smeets et al81 found that 3 weeks of self-compassion training for female college students led to significantly greater increases in mindfulness, optimism, and self-efficacy, as well as greater decreases in rumination compared with a time management control group.
The Box6,70,82-86 describes rating scales that can measure self-compassion and self-criticism.
Mindful self-compassion (MSC), developed by Neff and Germer,87 is an 8-week group intervention designed to teach people how to be more self-compassionate through meditation and informal practices in daily life. Results of a randomized controlled trial found that, compared with a wait-list control group, participants using MSC reported significantly greater increases in self-compassion, compassion for others, mindfulness, and life satisfaction, and greater decreases in depression, anxiety, stress, and emotional avoidance, with large effect sizes indicated. These results were maintained up to 1 year.
Compassion-focused therapy (CFT) is designed to enhance self-compassion in clinical populations.88 The approach uses a number of imagery and experiential exercises to enhance patients’ abilities to extend feelings of reassurance, safeness, and understanding toward themselves. CFT has shown promise in treating a diverse group of clinical disorders such as depression and shame,8,89 social anxiety and shame,90 eating disorders,91 psychosis,92 and patients with acquired brain injury.93 A group-based CFT intervention with a heterogeneous group of community mental health patients led to significant reductions in depression, anxiety, stress, and self-criticism.94 See Leaviss and Utley95 for a review of the benefits of CFT.
Fears of developing self-compassion
It is important to note that some people can access self-compassion more easily than others. Highly self-critical patients could feel anxious when learning to be compassionate to themselves, a phenomenon known as “fear of compassion”96 or “backdraft.”97 Backdraft occurs when a firefighter opens a door with a hot fire behind it. Oxygen rushes in, causing a burst of flame. Similarly, when the door of the heart is opened with compassion, intense pain could be released. Unconditional love reveals the conditions under which we were unloved in the past. Some individuals, especially those with a history of childhood abuse or neglect, are fearful of compassion because it activates grief associated with feelings of wanting, but not receiving, affection and care from significant others in childhood.
Clinicians should be aware that anxiety could arise and should help patients learn how to go slowly and stabilize themselves if overwhelming emotions occur as a part of self-compassion practice. Both CFT and MSC have processes to deal with fear of compassion in their protocols,98,99 with the focus on explaining to individuals that although such fears may occur, they are a normal and necessary part of the healing process. Individuals also are taught to focus on the breath, feeling the sensations in the soles of their feet, or other mindfulness practices to ground and stabilize attention when overwhelming feelings arise.
Clinical interventions
Self-compassion interventions that I (R.W.) find most helpful, in the order I administer them, are:
- exploring perceived advantages and disadvantages of self-criticism
- presenting self-compassion as a way to get the perceived advantages of self-criticism without the disadvantages
- discussing what it means to be compassionate for someone else who is suffering, and then asking what it would be like if they treated themselves with the same compassion
- exploring patients’ misconceptions and fears of self-compassion
- directing patients to the self-compassion Web site to get an understanding of what self-compassion is and how it differs from self-esteem
- taking an example of a recent situation in which the patient was self-critical and exploring how a self-compassionate response would differ.
Asking what they would say to a friend often is an effective way to get at this. In a later therapy session, self-compassionate imagery is a useful way to get the patient to experience self-compassion on an emotional level. See Neff100 and Gilbert98 for other techniques to enhance self-compassion.
Once thought to only be associated with depression, self-criticism is a transdiagnostic risk factor for diverse forms of psychopathology.1,2 However, research has shown that self-compassion is a robust resilience factor when faced with feelings of personal inadequacy.3,4
Self-critical individuals experience feelings of unworthiness, inferiority, failure, and guilt. They engage in constant and harsh self-scrutiny and evaluation, and fear being disapproved and criticized and losing the approval and acceptance of others.5 Self-compassion involves treating oneself with care and concern when confronted with personal inadequacies, mistakes, failures, and painful life situations.6,7
Although self-criticism is destructive across clinical disorders and interpersonal relationships, self-compassion is associated with healthy relationships, emotional well-being, and better treatment outcomes.
Recent research shows how clinicians can teach their patients how to be less self-critical and more self-compassionate. Neff6,7 proposes that self-compassion involves treating yourself with care and concern when being confronted with personal inadequacies, mistakes, failures, and painful life situations. It consists of 3 interacting components, each of which has a positive and negative pole:
- self-kindness vs self-judgment
- a sense of common humanity vs isolation
- mindfulness vs over-identification.
Self-kindness refers to being caring and understanding with oneself rather than harshly judgmental. Instead of attacking and berating oneself for personal shortcomings, the self is offered warmth and unconditional acceptance.
Humanity involves recognizing that humans are imperfect, that all people fail, make mistakes, and have serious life challenges. By remembering that imperfection is part of life, we feel less isolated when we are in pain.
Mindfulness in the context of self-compassion involves being aware of one’s painful experiences in a balanced way that neither ignores and avoids nor exaggerates painful thoughts and emotions.
Self-compassion is more than the absence of self-judgment, although a defining feature of self-compassion is the lack of self-judgment, and self-judgment overlaps with self-criticism. Rather, self-compassion provides several access points for reducing self-criticism. For example, being kind and understanding when confronting personal inadequacies (eg, “it’s okay not to be perfect”) can counter harsh self-talk (eg, “I’m not defective”). Mindfulness of emotional pain (eg, “this is hard”) can facilitate a kind and warm response (eg, “what can I do to take care of myself right now?”) and therefore lessen self-blame (eg, “blaming myself is just causing me more suffering”). Similarly, remembering that failure is part of the human experience (eg, “it’s normal to mess up sometimes”) can lessen egocentric feelings of isolation (eg, “it’s not just me”) and over-identification (eg, “it’s not the end of the world”), resulting in lessened self-criticism (eg, “maybe it’s not just because I’m a bad person”).
Depression
Several studies have found that self-criticism predicts depression. In 3 epidemiological studies, “feeling worthless” was among the top 2 symptoms predicting a depression diagnosis and later depressive episodes.10 Self-criticism in fourth-year medical students predicted depression 2 years later, and—in males—10 years later in their medical careers better than a history of depression.11 Self-critical perfectionism also is associated with suicidal ideation and lethality of suicide attempts.12
Self-criticism has been shown to predict depressive relapse and residual self-devaluative symptoms in recovered depressed patients.13 In one study, currently depressed and remitted depressed patients had higher self-criticism and lower self-compassion compared with healthy controls. Both factors were more strongly associated with depression status than higher perfectionistic beliefs and cognitions, rumination, and maladaptive emotional regulation.14
Self-criticism and response to treatment. In the National Institute of Mental Health Treatment of Depression Collaborative Research Program,15 self-critical perfectionism predicted a poorer outcome across all 4 treatments (cognitive-behavioral therapy [CBT], interpersonal psychotherapy [IPT], pharmacotherapy plus clinical management, and placebo plus clinical management). Subsequent studies found that self-criticism predicted poorer response to CBT16 and IPT.17 The authors suggest that self-criticism could interfere with treatment because self-critical patients might have difficulty developing a strong therapeutic alliance.18,19
Anxiety disorders
Self-criticism is common across psychiatric disorders. In a study of 5,877 respondents in the National Comorbidity Survey (NCS), self-criticism was associated with social phobia, findings that were significant after controlling for current emotional distress, neuroticism, and lifetime history of mood, anxiety, and substance use disorders.20 Further, in a CBT treatment study, baseline self-criticism was associated with severity of social phobia and changes in self-criticism predicted treatment outcome.21 Self-criticism might be an important core psychological process in the development, maintenance, and course of social phobia. Patients with social anxiety disorder have less self-compassion than healthy controls and greater fear of negative evaluation.
In the NCS, self-criticism was associated with posttraumatic stress disorder (PTSD) even after controlling for lifetime history of affective and anxiety disorders.20 Self-criticism predicted greater severity of combat-related PTSD in hospitalized male veterans,22 and those with PTSD had higher scores on self-criticism scales than those with major depressive disorder.23 In a study of Holocaust survivors, those with PTSD scored higher on self-criticism than survivors without PTSD.24 Self-criticism also distinguished between female victims of domestic violence with and without PTSD.25
Self-compassion could be a protective factor for posttraumatic stress.26 Combat veterans with higher levels of self-compassion showed lower levels of psychopathology, better functioning in daily life, and fewer symptoms of posttraumatic stress.27 In fact, self-compassion has been found to be a stronger predictor of PTSD than level of combat exposure.28
In an early study, self-criticism scores were higher in patients with panic disorder than in healthy controls, but lower than in patients with depression.29 In a study of a mixed sample of anxiety disorder patients, symptoms of generalized anxiety disorder were associated with shame proneness.30 Consistent with these results, Hoge et al31 found that self-compassion was lower in generalized anxiety disorder patients compared with healthy controls with elevated stress. Low self-compassion has been associated with severity of obsessive-compulsive disorder.32
Eating disorders
Self-criticism is correlated with eating disorder severity.33 In a study of patients with binge eating disorder, Dunkley and Grilo34 found that self-criticism was associated with the over-evaluation of shape and weight independently of self-esteem and depression. Self-criticism also is associated with body dissatisfaction, independent of self-esteem and depression. Dunkley et al35 found that self-criticism, but not global self-esteem, in patients with binge eating disorder mediated the relationship between childhood abuse and body dissatisfaction and depression. Numerous studies have shown that shame is associated with more severe eating disorder pathology.33
Self-compassion seems to buffer against body image concerns. It is associated with less body dissatisfaction, body preoccupation, and weight worries,36 greater body appreciation37 and less disordered eating.37-39 Early decreases in shame during eating disorder treatment was associated with more rapid reduction in eating disorder symptoms.40
Interpersonal relationships
Several studies have shown that self-criticism has negative effects on interpersonal relationships throughout life.5,41,42
- Self-criticism at age 12 predicted less involvement in high school activities and, at age 31, personal and social maladjustment.43
- High school students with high self-criticism reported more interpersonal problems.44
- Self-criticism was associated with loneliness, depression, and lack of intimacy with opposite sex friends or partners during the transition to college.45
- In a study of college roommates,46 self-criticism was associated with increased likelihood of rejection.
- Whiffen and Aube47 found that self-criticism was associated with marital dissatisfaction and depression.
- Self-critical mothers with postpartum depression were less satisfied with social support and were more vulnerable to depression.48
Self-compassion appears to enhance interpersonal relationships. In a study of heterosexual couples,49 self-compassionate individuals were described by their partners as being more emotionally connected, as well as accepting and supporting autonomy, while being less detached, controlling, and verbally or physically aggressive than those lacking self-compassion. Because self-compassionate people give themselves care and support, they seem to have more emotional resources available to give to others.
See the Box examining the evidence on the role of self-compassion in borderline personality disorder and non-suicidal self-injury.
Achieving goals
Powers et al50 suggest that self-critics approach goals based on motivation to avoid failure and disapproval, rather than on intrinsic interest and personal meaning. In studies of college students pursuing academic, social, or weight loss goals, self-criticism was associated with less progress to that goal. Self-criticism was associated with rumination and procrastination, which the authors suggest might have focused the self-critic on potential failure, negative evaluation from others, and loss of self-esteem. Additional studies showed the deleterious effects of self-criticism on college students’ progress on obtaining academic or music performance goals and on community residents’ weight loss goals.51
Not surprisingly, self-compassion is associated with successful goal pursuit and resilience when goals are not met52 and less procrastination and academic worry.53 Self-compassion also is associated with intrinsic motivation, goals based on mastery rather than performance, and less fear of academic failure.54
How self-criticism and self-compassion develop
Studies have explored the impact of early relationships with parents and development of self-criticism. Parental overcontrol and restrictiveness and lack of warmth consistently have been identified as parenting styles associated with development of self-criticism in children.55 One study found that self-criticism fully mediated the relationship between childhood verbal abuse from parents and depression and anxiety in adulthood.56 Reports from parents on their current parenting styles are consistent with these studies.57 Amitay et al57 states that “[s]elf-critics’ negative childhood experiences thus seem to contribute to a pattern of entering, creating, or manipulating subsequent interpersonal environments in ways that perpetuate their negative self-image and increase vulnerability to depression.” Not surprisingly, self-criticism is associated with a fearful avoidant attachment style.58 Review of the developmental origins of self-criticism confirms these factors and presents findings that peer relationships also are important factors in the development of self-criticism.59,60
Early positive relationships with caregivers are associated with self-compassion. Recollections of maternal support are correlated with self-compassion and secure attachment styles in adolescents and adults.61 Pepping et al62 found that retrospective reports of parental rejection, overprotection, and low parental warmth was associated with low self-compassion.
Benefits of self-compassion
A growing body of research suggests that self-compassion is strongly linked to mental health. Greater self-compassion consistently has been associated with lower levels of depression and anxiety,3 with a large effect size.4 Of course, central to self-compassion is the lack of self-criticism, but self-compassion still protects against anxiety and depression when controlling for self-criticism and negative affect.6,63 Self-compassion is a strong predictor of symptom severity and quality of life among individuals with anxious distress.64
The benefits of self-compassion stem partly from a greater ability to cope with negative emotions.6,63,65 Self-compassionate people are less likely to ruminate on their negative thoughts and emotions or suppress them,6,66 which helps to explain why self-compassion is a negative predictor of depression.67
Self-compassion also enhances positive mind states. A number of studies have found links between self-compassion and positive psychological qualities, such as happiness, optimism, wisdom, curiosity, and exploration, and personal initiative.63,65,68,69 By embracing one’s suffering with compassion, negative states are ameliorated when positive emotions of kindness, connectedness, and mindful presence are generated.
Misconceptions about self-compassion
A common misconception is that abandoning self-criticism in favor of self-compassion will undermine motivation70; however, research indicates the opposite. Although self-compassion is negatively associated with maladaptive perfectionism, it is not correlated with self-adopted performance standards.6 Self-compassionate people have less fear of failure54 and, when they do fail, they are more likely to try again.71 Breines and Chen72 found in a series of experimental studies that engendering feelings of self-compassion for personal weaknesses, failures, and past transgressions resulted in more motivation to change, to try harder to learn, and to avoid repeating past mistakes.
Another common misunderstanding is that self-compassion is a weakness. In fact, research suggests that self-compassion is a powerful way to cope with life challenges.73
Although some fear that self-compassion leads to self-indulgence, there is evidence that self-compassion promotes health-related behaviors. Self-compassionate individuals are more likely to seek medical treatment when needed,74 exercise for intrinsic reasons,75 and drink less alcohol.76 Inducing self-compassion has been found to help people stick to their diets77 and quit smoking.78
Self-compassion interventions
Individuals can develop self-compassion. Shapira and Mongrain79 found that adults who wrote a compassionate letter to themselves once a day for a week about the distressing events they were experiencing showed significant reductions in depression up to 3 months and significant increases in happiness up to 6 months compared with a control group who wrote about early memories. Albertson et al80 found that, compared with a wait-list control group, 3 weeks of self-compassion meditation training improved body dissatisfaction, body shame, and body appreciation among women with body image concerns. Similarly, Smeets et al81 found that 3 weeks of self-compassion training for female college students led to significantly greater increases in mindfulness, optimism, and self-efficacy, as well as greater decreases in rumination compared with a time management control group.
The Box6,70,82-86 describes rating scales that can measure self-compassion and self-criticism.
Mindful self-compassion (MSC), developed by Neff and Germer,87 is an 8-week group intervention designed to teach people how to be more self-compassionate through meditation and informal practices in daily life. Results of a randomized controlled trial found that, compared with a wait-list control group, participants using MSC reported significantly greater increases in self-compassion, compassion for others, mindfulness, and life satisfaction, and greater decreases in depression, anxiety, stress, and emotional avoidance, with large effect sizes indicated. These results were maintained up to 1 year.
Compassion-focused therapy (CFT) is designed to enhance self-compassion in clinical populations.88 The approach uses a number of imagery and experiential exercises to enhance patients’ abilities to extend feelings of reassurance, safeness, and understanding toward themselves. CFT has shown promise in treating a diverse group of clinical disorders such as depression and shame,8,89 social anxiety and shame,90 eating disorders,91 psychosis,92 and patients with acquired brain injury.93 A group-based CFT intervention with a heterogeneous group of community mental health patients led to significant reductions in depression, anxiety, stress, and self-criticism.94 See Leaviss and Utley95 for a review of the benefits of CFT.
Fears of developing self-compassion
It is important to note that some people can access self-compassion more easily than others. Highly self-critical patients could feel anxious when learning to be compassionate to themselves, a phenomenon known as “fear of compassion”96 or “backdraft.”97 Backdraft occurs when a firefighter opens a door with a hot fire behind it. Oxygen rushes in, causing a burst of flame. Similarly, when the door of the heart is opened with compassion, intense pain could be released. Unconditional love reveals the conditions under which we were unloved in the past. Some individuals, especially those with a history of childhood abuse or neglect, are fearful of compassion because it activates grief associated with feelings of wanting, but not receiving, affection and care from significant others in childhood.
Clinicians should be aware that anxiety could arise and should help patients learn how to go slowly and stabilize themselves if overwhelming emotions occur as a part of self-compassion practice. Both CFT and MSC have processes to deal with fear of compassion in their protocols,98,99 with the focus on explaining to individuals that although such fears may occur, they are a normal and necessary part of the healing process. Individuals also are taught to focus on the breath, feeling the sensations in the soles of their feet, or other mindfulness practices to ground and stabilize attention when overwhelming feelings arise.
Clinical interventions
Self-compassion interventions that I (R.W.) find most helpful, in the order I administer them, are:
- exploring perceived advantages and disadvantages of self-criticism
- presenting self-compassion as a way to get the perceived advantages of self-criticism without the disadvantages
- discussing what it means to be compassionate for someone else who is suffering, and then asking what it would be like if they treated themselves with the same compassion
- exploring patients’ misconceptions and fears of self-compassion
- directing patients to the self-compassion Web site to get an understanding of what self-compassion is and how it differs from self-esteem
- taking an example of a recent situation in which the patient was self-critical and exploring how a self-compassionate response would differ.
Asking what they would say to a friend often is an effective way to get at this. In a later therapy session, self-compassionate imagery is a useful way to get the patient to experience self-compassion on an emotional level. See Neff100 and Gilbert98 for other techniques to enhance self-compassion.
1. Shahar B, Doron G, Ohad S. Childhood maltreatment, shame-proneness and self-criticism in social anxiety disorder: a sequential mediational model. Clin Psychol Psychother. 2015:22(6):570-579.
2. Kannan D, Levitt HM. A review of client self-criticism in psychotherapy. J Psychother Integr. 2013;23(2):166-178.
3. Barnard LK, Curry JF. Self-compassion: conceptualizations, correlates, and interventions. Rev Gen Psychol. 2011;15(4):289-303.
4. MacBeth A, Gumley A. Exploring compassion: a meta-analysis of the association between self-compassion and psychopathology. Clin Psychol Rev. 2012;32(6):545-552
5. Blatt SJ, Zuroff DC. Interpersonal relatedness and self-definition: two prototypes for depression. Clin Psychol Rev. 1992;12(5):527-562.
6. Neff KD. The development and validation of a scale to measure self-compassion. Self Identity. 2003;2(2):223-250.
7. Neff KD. Self-compassion: an alternative conceptualization of a healthy attitude toward oneself. Self Identity. 2003;(2)2:85-101.
8. Gilbert P, Procter S. Compassionate mind training for people with high shame and self-criticism: overview and pilot study of a group therapy approach. Clin Psychol Psychother. 2006;13(6):353-379.
9. Dunkley DM, Zuroff DC, Blankstein KR. Specific perfectionism components versus self-criticism in predicting maladjustment. Pers Individ Dif. 2006;40(4):665-676.
10. Murphy JM, Nierenberg AA, Monson RR, et al. Self-disparagement as feature and forerunner of depression: Mindfindings from the Stirling County Study. Compr Psychiatry. 2002;43(1):13-21.
11. Brewin CR, Firth-Cozens J. Dependency and self-criticism as predictors of depression in young doctors. J Occup Health Psychol. 1997;2(3):242-246.
12. Fazaa N, Page S. Dependency and self-criticism as predictors of suicidal behavior. Suicide Life Threat Behav. 2003;33(2):172-185.
13. Teasdale JD, Cox SG. Dysphoria: self-devaluative and affective components in recovered depressed patients and never depressed controls. Psychol Med. 2001;31(7):1311-1316.
14. Ehret AM, Joormann J, Berking M. Examining risk and resilience factors for depression: the role of self-criticism and self-compassion. Cogn Emot. 2015;29(8):1496-1504.
15. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health Treatment of Depression Collaborative Research Program. General effectiveness of treatments. Arch Gen Psychiatry. 1989;46(11):971-982; discussion 983.
16. Rector NA, Bagby RM, Segal ZV, et al. Self-criticism and dependency in depressed patients treated with cognitive therapy or pharmacotherapy. Cognit Ther Res. 2000;24(5):571-584.
17. Marshall MB, Zuroff DC, McBride C, et al. Self-criticism predicts differential response to treatment for major depression. J Clin Psychol. 2008;64(3):231-244.
18. Zuroff DC, Blatt SJ, Sotsky SM, et al. Relation of therapeutic alliance and perfectionism to outcome in brief outpatient treatment of depression. J Consult Clin Psychol. 2000;68(1):114-124.
19. Whelton WJ, Greenberg LS. Emotion in self-criticism. Pers Individ Dif. 2005;38(7):1583-1595.
20. Cox BJ, Fleet C, Stein MB. Self-criticism and social phobia in the US national comorbidity survey. J Affect Disord. 2004;82(2):227-234.
21. Cox BJ, Walker JR, Enns MW, et al. Self-criticism in generalized social phobia and response to cognitive-behavioral treatment. Behav Ther. 2002;33(4):479-491.
22. McCranie EW, Hyer LA. Self-critical depressive experience in posttraumatic stress disorder. Psychol Rep. 1995;77(3 pt 1):880-882.
23. Southwick SM, Yehuda R, Giller EL Jr. Characterization of depression in war-related posttraumatic stress disorder. Am J Psychiatry. 1991;148(2):179-183.
24. Yehuda R, Kahana B, Southwick SM, et al. Depressive features in Holocaust survivors with post-traumatic stress disorder. J Traumatic Stress. 1994;7(4):699-704.
25. Sharhabani-Arzy R, Amir M, Swisa A. Self-criticism, dependency and posttraumatic stress disorder among a female group of help-seeking victims of domestic violence in Israel. Pers Individ Dif. 2005;38(5):1231-1240.
26. Beaumont E, Galpin A, Jenkins P. ‘Being kinder to myself’: a prospective comparative study, exploring post-trauma therapy outcome measures, for two groups of clients, receiving either cognitive behaviour therapy or cognitive behaviour therapy and compassionate mind training. Counselling Psychol Rev. 2012;27(1):31-43.
27. Dahm KA. Mindfulness and self-compassion as predictors of functional outcomes and psychopathology in OEF/OIF veterans exposed to trauma. https://repositories.lib.utexas.edu/handle/2152/21635. Published August 2013. Accessed November 8, 2016.
28. Hiraoka R, Meyer EC, Kimbrel NA, et al. Self-compassion as a prospective predictor of PTSD symptom severity among trauma-exposed US Iraq and Afghanistan war veterans. J Trauma Stress. 2015;28(2):127-133.
29. Bagby RM, Cox BJ, Schuller DR, et al. Diagnostic specificity of the dependent and self-critical personality dimensions in major depression. J Affect Disord. 1992;26(1):59-63.
30. Hedman E, Ström P, Stünkel A, et al. Shame and guilt in social anxiety disorder: effects of cognitive behavior therapy and association with social anxiety and depressive symptoms. PLoS One. 2013;8(4):e61713. doi: 10.1371/journal.pone.0061713.
31. Hoge EA, Hölzel BK, Marques L, et al. Mindfulness and self-compassion in generalized anxiety disorder: examining predictors of disability. Evid Based Complement Alternat Med. 2013;2013:576258. doi: 10.1155/2013/576258.
32. Wetterneck CT, Lee EB, Smith AH, et al. Courage, self-compassion, and values in obsessive-compulsive disorder. J Contextual Behav Sci. 2013;2(3-4):68-73.
33. Kelly AC, Carter JC. Why self-critical patients present with more severe eating disorder pathology: The mediating role of shame. Br J Clin Psychol. 2013;52(2):148-161.
34. Dunkley DM, Grilo CM. Self-criticism, low self-esteem, depressive symptoms, and over-evaluation of shape and weight in binge eating disorder patients. Behav Res Ther. 2007;45(1):139-149.
35. Dunkley DM, Masheb RM, Grilo CM. Childhood maltreatment, depressive symptoms, and body dissatisfaction in patients with binge eating disorder: the mediating role of self-criticism. Int J Eat Disord. 2010;43(3):274-281.
36. Wasylkiw L, MacKinnon AL, MacLellan AM. Exploring the link between self-compassion and body image in university women. Body Image. 2012;9(2):236-245.
37. Ferreira C, Pinto-Gouveia J, Duarte C. Self-compassion in the face of shame and body image dissatisfaction: implications for eating disorders. Eat Behavs. 2013;14(2):207-210.
38. Kelly AC, Carter JC, Zuroff DC, et al. Self-compassion and fear of self-compassion interact to predict response to eating disorders treatment: a preliminary investigation. Psychother Res. 2013;23(3):252-264.
39. Webb JB, Forman MJ. Evaluating the indirect effect of self-compassion on binge eating severity through cognitive-affective self-regulatory pathways. Eat Behavs. 2013;14(2):224-228.
40. Kelly AC, Carter JC, Borairi S. Are improvements in shame and self-compassion early in eating disorders treatment associated with better patient outcomes? Int J Eat Disord. 2014;47(1):54-64.
41. Wiseman H, Raz A, Sharabany R. Depressive personality styles and interpersonal problems in young adults with difficulties in establishing long-term romantic relationships. Isr J Psychiatry Rel Sci. 2007;44(4):280-291.
42. Besser A, Priel B. A multisource approach to self-critical vulnerability to depression: the moderating role of attachment. J Pers. 2003;71(4):515-555.
43. Zuroff DC, Koestner R, Powers TA. Self-criticism at age 12: a longitudinal-study of adjustment. Cognit Ther Res. 1994;18(4):367-385.
44. Fichman L, Koestner R, Zuroff DC. Depressive styles in adolescence: Assessment, relation to social functioning, and developmental trends. J Youth Adolesc. 1994;23(3):315-330.
45. Wiseman H. Interpersonal relatedness and self-definition in the experience of loneliness during the transition to university. Personal Relationships. 1997;4(3):285-299.
46. Mongrain M, Lubbers R, Struthers W. The power of love: mediation of rejection in roommate relationships of dependents and self-critics. Pers Soc Psychol Bull. 2004;30(1):94-105.
47. Whiffen VE, Aube JA. Personality, interpersonal context and depression in couples. J Soc Pers Relat. 1999;16(3):369-383.
48. Priel B, Besser A. Dependency and self-criticism among first-time mothers: the roles of global and specific support. J Soc Clin Psychol. 2000;19(4):437-450.
49. Neff KD, Beretvas SN. The role of self-compassion in romantic relationships. Self Identity. 2013;12(1):78-98.
50. Powers TA, Koestner R, Zuroff DC. Self-criticism, goal motivation, and goal progress. J Soc Clin Psychol. 2007;26(7):826-840.
51. Powers TA, Koestner R, Zuroff DC, et al. The effects of self-criticism and self-oriented perfectionism on goal pursuit. Pers Soc Psychol Bull. 2011;37(7):964-975.
52. Hope N, Koestner R, Milyavskaya M. The role of self-compassion in goal pursuit and well-being among university freshmen. Self Identity. 2014;13(5):579-593.
53. Williams JG, Stark SK, Foster EE. Start today or the very last day? The relationships among self-compassion, motivation, and procrastination. Am J Psychol Res. 2008;4(1):37-44.
54. Neff KD, Hseih Y, Dejittherat K. Self-compassion, achievement goals, and coping with academic failure. Self Identity. 2005;4(3):263-287.
55. Campos RC, Besser A, Blatt SJ. The mediating role of self-criticism and dependency in the association between perceptions of maternal caring and depressive symptoms. Depress Anxiety. 2010;27(12):1149-1157.
56. Sachs-Ericsson N, Verona E, Joiner T, et al. Parental verbal abuse and the mediating role of self-criticism in adult internalizing disorders. J Affect Disord. 2006;93(1-3):71-78.
57. Amitay OA, Mongrain M, Fazaa N. Love and control: self-criticism in parents and daughters and perceptions of relationship partners. Pers Individ Dif. 2008;44(1):75-85.
58. Zuroff DC, Fitzpatrick DK. Depressive personality styles: implications for adult attachment. Pers Individ Dif. 1995;18(2):253-265.
59. Kopala-Sibley DC, Zuroff DC. The developmental origins of personality factors from the self-definitional and relatedness domains: a review of theory and research. Rev Gen Psychol. 2014;18(3):137-155.
60. Kopala-Sibley DC, Zuroff DC, Leybman MJ, et al. Recalled peer relationship experiences and current levels of self-criticism and self-reassurance. Psychol Psychother. 2013;86(1):33-51.
61. Neff KD, McGehee P. Self-compassion and psychological resilience among adolescents and young adults. Self Identity. 2010;9(3):225-240.
62. Pepping CA, Davis PJ, O’Donovan A, et al. Individual differences in self-compassion: the role of attachment and experiences of parenting in childhood. Self Identity. 2015;14(1):104-117.
63. Neff KD, Rude SS, Kirkpatrick KL. An examination of self-compassion in relation to positive psychological functioning and personality traits. J Res Pers. 2007;41(4):908-916.
64. Van Dam NT, Sheppard SC, Forsyth JP, et al. Self-compassion is a better predictor than mindfulness of symptom severity and quality of life in mixed anxiety and depression. J Anxiety Disord. 2011;25(1):123-130.
65. Heffernan M, Quinn MT, McNulty SR, et al. Self-compassion and emotional intelligence in nurses. Int J Nursing Practice. 2010;16(4):366-373.
66. Neff KD, Kirkpatrick KL, Rude SS. Self-compassion and adaptive psychological functioning. J Res Pers. 2007;41(1):139-154.
67. Krieger T, Altenstein D, Baettig I, et al. Self-compassion in depression: associations with depressive symptoms, rumination, and avoidance in depressed outpatients. Behav Ther. 2013;44(3):501-513.
68. Breen WE, Kashdan TB, Lenser ML, et al. Gratitude and forgiveness: convergence and divergence on self-report and informant ratings. Pers Individ Dif. 2010;49(8):932-937.
69. Hollis-Walker L, Colosimo K. Mindfulness, self-compassion, and happiness in non-meditators: A theoretical and empirical examination. Pers Individ Dif. 2011;50(2):222-227.
70. Gilbert P, McEwan K, Matos M, et al. Fears of compassion: development of three self-report measures. Psychol Psychother. 2011;84(3):239-255.
71. Neely ME, Schallert DL, Mohammed SS, et al. Self-kindness when facing stress: the role of self-compassion, goal regulation, and support in college students’ well-being. Motiv Emot. 2009;33(1):88-97.
72. Breines JG, Chen S. Self-compassion increases self-improvement motivation. Pers Soc Psychol Bull. 2012;38(9):1133-1143.
73. Allen AB, Leary MR. Self-compassion, stress, and coping. Soc Pers Psychol Compass. 2010;4(2):107-118.
74. Terry ML, Leary MR. Self-compassion, self-regulation, and health. Self Identity. 2011;10(3):352-362.
75. Magnus CMR, Kowalski KC, McHugh TF. The role of self-compassion in women’s self-determined motives to exercise and exercise-related outcomes. Self Identity. 2010;9(4):363-382.
76. Brooks M, Kay-Lambkin F, Bowman J, et al. Self-compassion amongst clients with problematic alcohol use. Mindfulness. 2012;3(4):308-317.
77. Adams CE, Leary MR. Promoting self-compassionate attitudes toward eating among restrictive and guilty eaters. J Soc Clin Psychol. 2007;26(10):1120-1144.
78. Kelly AC, Zuroff DC, Foa CL, et al. Who benefits from training in self-compassionate self-regulation? A study of smoking reduction. J Soc Clin Psychol. 2010;29(7):727-755.
79. Shapira LB, Mongrain M. The benefits of self-compassion and optimism exercises for individuals vulnerable to depression. J Posit Psychol. 2010;5(5):377-389.
80. Albertson ER, Neff KD, Dill-Shackleford KE. Self-compassion and body dissatisfaction in women: a randomized controlled trial of a brief meditation intervention. Mindfulness. 2015;6(3):444-454.
81. Smeets E, Neff K, Alberts H, et al. Meeting suffering with kindness: effects of a brief self-compassion intervention for female college students. J Clinical Psychol. 2014;70(9):794-807.
82. Blatt SJ, D’Afflitti JP, Quinlan DM. Depressive experiences questionnaire. New Haven, CT: Yale University Press; 1976.
83. Weissman AN, Beck AT. Development and validation of the dysfunctional attitude scale: a preliminary investigation. Paper presented at: 62nd Annual Meeting of the Association for Advanced Behavior Therapy; March 27-31, 1978; Toronto, Ontario, Canada.
84. Gilbert P, Clarke M, Hempel S, et al. Criticizing and reassuring oneself: an exploration of forms, styles and reasons in female students. Br J Clin Psychol. 2004;43(pt 1):31-50.
85. Baião R, Gilbert P, McEwan K, et al. Forms of self-criticising/attacking & self-reassuring scale: psychometric properties and normative study. Psychol Psychother. 2015;88(4):438-452.
86. Neff KD. The self-compassion scale is a valid and theoretically coherent measure of self-compassion. Mindfulness. 2016;7(1):264-274.
87. Neff KD, Germer CK. A pilot study and randomized controlled trial of the mindful self-compassion program. J Clinical Psychol. 2013;69(1):28-44.
88. Gilbert P. Introducing compassion-focused therapy. Adv Psychiatr Treat. 2009;15(3):199-208.
89. Kelly AC, Zuroff DC, Shapira LB. Soothing oneself and resisting self-attacks: the treatment of two intrapersonal deficits in depression vulnerability. Cognit Ther Res. 2009;33(3):301-313.
90. Boersma K, Hakanson A, Salomonsson E, et al. Compassion focused therapy to counteract shame, self-criticism and isolation. A replicated single case experimental study of individuals with social anxiety. J Contemp Psychother. 2015;45(2):89-98.
91. Gale C, Gilbert P, Read N, et al. An evaluation of the impact of introducing compassion focused therapy to a standard treatment programme for people with eating disorders. Clin Psychol Psychother. 2014;21(1):1-12.
92. Braehler C, Gumley A, Harper J, et al. Exploring change processes in compassion focused therapy in psychosis: results of a feasibility randomized controlled trial. Br J Clin Psychol. 2013;52(2):199-214.
93. Ashworth F, Clarke A, Jones L, et al. An exploration of compassion focused therapy following acquired brain injury. Psychol Psychother. 2014;88(2):143-162.
94. Judge L, Cleghorn A, McEwan K, et al. An exploration of group-based compassion focused therapy for a heterogeneous range of clients presenting to a community mental health team. Int J Cogn Ther. 2012;5(4):420-429.
95. Leaviss J, Utley L. Psychotherapeutic benefits of compassion-focused therapy: an early systematic review. Psychol Med. 2015;45(5):927-945.
96. Gilbert P, McEwan K, Gibbons L, et al. Fears of compassion and happiness in relation to alexithymia, mindfulness, and self‐criticism. Psychol Psychother. 2012;85(4):374-390.
97. Germer CK, Neff KD. Cultivating self-compassion in trauma survivors. In: Follette VM, Briere J, Rozelle D, et al, eds. Mindfulness-oriented interventions for trauma: integrating contemplative practices. New York, NY: Guilford Press; 2015:43-58.
98. Gilbert P. Compassion focused therapy: the CBT distinctive features series. London, United Kingdom: Routledge; 2010.
99. Germer C, Neff K. The mindful self-compassion training program. In: Singer T, Bolz M, eds. Compassion: bridging theory and practice: a multimedia book. Leipzig, Germany: Max-Planck Institute; 2013:365-396.
100. Neff K. Self-compassion: the proven power of being kind to yourself. New York, NY: HarperCollins; 2015.
1. Shahar B, Doron G, Ohad S. Childhood maltreatment, shame-proneness and self-criticism in social anxiety disorder: a sequential mediational model. Clin Psychol Psychother. 2015:22(6):570-579.
2. Kannan D, Levitt HM. A review of client self-criticism in psychotherapy. J Psychother Integr. 2013;23(2):166-178.
3. Barnard LK, Curry JF. Self-compassion: conceptualizations, correlates, and interventions. Rev Gen Psychol. 2011;15(4):289-303.
4. MacBeth A, Gumley A. Exploring compassion: a meta-analysis of the association between self-compassion and psychopathology. Clin Psychol Rev. 2012;32(6):545-552
5. Blatt SJ, Zuroff DC. Interpersonal relatedness and self-definition: two prototypes for depression. Clin Psychol Rev. 1992;12(5):527-562.
6. Neff KD. The development and validation of a scale to measure self-compassion. Self Identity. 2003;2(2):223-250.
7. Neff KD. Self-compassion: an alternative conceptualization of a healthy attitude toward oneself. Self Identity. 2003;(2)2:85-101.
8. Gilbert P, Procter S. Compassionate mind training for people with high shame and self-criticism: overview and pilot study of a group therapy approach. Clin Psychol Psychother. 2006;13(6):353-379.
9. Dunkley DM, Zuroff DC, Blankstein KR. Specific perfectionism components versus self-criticism in predicting maladjustment. Pers Individ Dif. 2006;40(4):665-676.
10. Murphy JM, Nierenberg AA, Monson RR, et al. Self-disparagement as feature and forerunner of depression: Mindfindings from the Stirling County Study. Compr Psychiatry. 2002;43(1):13-21.
11. Brewin CR, Firth-Cozens J. Dependency and self-criticism as predictors of depression in young doctors. J Occup Health Psychol. 1997;2(3):242-246.
12. Fazaa N, Page S. Dependency and self-criticism as predictors of suicidal behavior. Suicide Life Threat Behav. 2003;33(2):172-185.
13. Teasdale JD, Cox SG. Dysphoria: self-devaluative and affective components in recovered depressed patients and never depressed controls. Psychol Med. 2001;31(7):1311-1316.
14. Ehret AM, Joormann J, Berking M. Examining risk and resilience factors for depression: the role of self-criticism and self-compassion. Cogn Emot. 2015;29(8):1496-1504.
15. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health Treatment of Depression Collaborative Research Program. General effectiveness of treatments. Arch Gen Psychiatry. 1989;46(11):971-982; discussion 983.
16. Rector NA, Bagby RM, Segal ZV, et al. Self-criticism and dependency in depressed patients treated with cognitive therapy or pharmacotherapy. Cognit Ther Res. 2000;24(5):571-584.
17. Marshall MB, Zuroff DC, McBride C, et al. Self-criticism predicts differential response to treatment for major depression. J Clin Psychol. 2008;64(3):231-244.
18. Zuroff DC, Blatt SJ, Sotsky SM, et al. Relation of therapeutic alliance and perfectionism to outcome in brief outpatient treatment of depression. J Consult Clin Psychol. 2000;68(1):114-124.
19. Whelton WJ, Greenberg LS. Emotion in self-criticism. Pers Individ Dif. 2005;38(7):1583-1595.
20. Cox BJ, Fleet C, Stein MB. Self-criticism and social phobia in the US national comorbidity survey. J Affect Disord. 2004;82(2):227-234.
21. Cox BJ, Walker JR, Enns MW, et al. Self-criticism in generalized social phobia and response to cognitive-behavioral treatment. Behav Ther. 2002;33(4):479-491.
22. McCranie EW, Hyer LA. Self-critical depressive experience in posttraumatic stress disorder. Psychol Rep. 1995;77(3 pt 1):880-882.
23. Southwick SM, Yehuda R, Giller EL Jr. Characterization of depression in war-related posttraumatic stress disorder. Am J Psychiatry. 1991;148(2):179-183.
24. Yehuda R, Kahana B, Southwick SM, et al. Depressive features in Holocaust survivors with post-traumatic stress disorder. J Traumatic Stress. 1994;7(4):699-704.
25. Sharhabani-Arzy R, Amir M, Swisa A. Self-criticism, dependency and posttraumatic stress disorder among a female group of help-seeking victims of domestic violence in Israel. Pers Individ Dif. 2005;38(5):1231-1240.
26. Beaumont E, Galpin A, Jenkins P. ‘Being kinder to myself’: a prospective comparative study, exploring post-trauma therapy outcome measures, for two groups of clients, receiving either cognitive behaviour therapy or cognitive behaviour therapy and compassionate mind training. Counselling Psychol Rev. 2012;27(1):31-43.
27. Dahm KA. Mindfulness and self-compassion as predictors of functional outcomes and psychopathology in OEF/OIF veterans exposed to trauma. https://repositories.lib.utexas.edu/handle/2152/21635. Published August 2013. Accessed November 8, 2016.
28. Hiraoka R, Meyer EC, Kimbrel NA, et al. Self-compassion as a prospective predictor of PTSD symptom severity among trauma-exposed US Iraq and Afghanistan war veterans. J Trauma Stress. 2015;28(2):127-133.
29. Bagby RM, Cox BJ, Schuller DR, et al. Diagnostic specificity of the dependent and self-critical personality dimensions in major depression. J Affect Disord. 1992;26(1):59-63.
30. Hedman E, Ström P, Stünkel A, et al. Shame and guilt in social anxiety disorder: effects of cognitive behavior therapy and association with social anxiety and depressive symptoms. PLoS One. 2013;8(4):e61713. doi: 10.1371/journal.pone.0061713.
31. Hoge EA, Hölzel BK, Marques L, et al. Mindfulness and self-compassion in generalized anxiety disorder: examining predictors of disability. Evid Based Complement Alternat Med. 2013;2013:576258. doi: 10.1155/2013/576258.
32. Wetterneck CT, Lee EB, Smith AH, et al. Courage, self-compassion, and values in obsessive-compulsive disorder. J Contextual Behav Sci. 2013;2(3-4):68-73.
33. Kelly AC, Carter JC. Why self-critical patients present with more severe eating disorder pathology: The mediating role of shame. Br J Clin Psychol. 2013;52(2):148-161.
34. Dunkley DM, Grilo CM. Self-criticism, low self-esteem, depressive symptoms, and over-evaluation of shape and weight in binge eating disorder patients. Behav Res Ther. 2007;45(1):139-149.
35. Dunkley DM, Masheb RM, Grilo CM. Childhood maltreatment, depressive symptoms, and body dissatisfaction in patients with binge eating disorder: the mediating role of self-criticism. Int J Eat Disord. 2010;43(3):274-281.
36. Wasylkiw L, MacKinnon AL, MacLellan AM. Exploring the link between self-compassion and body image in university women. Body Image. 2012;9(2):236-245.
37. Ferreira C, Pinto-Gouveia J, Duarte C. Self-compassion in the face of shame and body image dissatisfaction: implications for eating disorders. Eat Behavs. 2013;14(2):207-210.
38. Kelly AC, Carter JC, Zuroff DC, et al. Self-compassion and fear of self-compassion interact to predict response to eating disorders treatment: a preliminary investigation. Psychother Res. 2013;23(3):252-264.
39. Webb JB, Forman MJ. Evaluating the indirect effect of self-compassion on binge eating severity through cognitive-affective self-regulatory pathways. Eat Behavs. 2013;14(2):224-228.
40. Kelly AC, Carter JC, Borairi S. Are improvements in shame and self-compassion early in eating disorders treatment associated with better patient outcomes? Int J Eat Disord. 2014;47(1):54-64.
41. Wiseman H, Raz A, Sharabany R. Depressive personality styles and interpersonal problems in young adults with difficulties in establishing long-term romantic relationships. Isr J Psychiatry Rel Sci. 2007;44(4):280-291.
42. Besser A, Priel B. A multisource approach to self-critical vulnerability to depression: the moderating role of attachment. J Pers. 2003;71(4):515-555.
43. Zuroff DC, Koestner R, Powers TA. Self-criticism at age 12: a longitudinal-study of adjustment. Cognit Ther Res. 1994;18(4):367-385.
44. Fichman L, Koestner R, Zuroff DC. Depressive styles in adolescence: Assessment, relation to social functioning, and developmental trends. J Youth Adolesc. 1994;23(3):315-330.
45. Wiseman H. Interpersonal relatedness and self-definition in the experience of loneliness during the transition to university. Personal Relationships. 1997;4(3):285-299.
46. Mongrain M, Lubbers R, Struthers W. The power of love: mediation of rejection in roommate relationships of dependents and self-critics. Pers Soc Psychol Bull. 2004;30(1):94-105.
47. Whiffen VE, Aube JA. Personality, interpersonal context and depression in couples. J Soc Pers Relat. 1999;16(3):369-383.
48. Priel B, Besser A. Dependency and self-criticism among first-time mothers: the roles of global and specific support. J Soc Clin Psychol. 2000;19(4):437-450.
49. Neff KD, Beretvas SN. The role of self-compassion in romantic relationships. Self Identity. 2013;12(1):78-98.
50. Powers TA, Koestner R, Zuroff DC. Self-criticism, goal motivation, and goal progress. J Soc Clin Psychol. 2007;26(7):826-840.
51. Powers TA, Koestner R, Zuroff DC, et al. The effects of self-criticism and self-oriented perfectionism on goal pursuit. Pers Soc Psychol Bull. 2011;37(7):964-975.
52. Hope N, Koestner R, Milyavskaya M. The role of self-compassion in goal pursuit and well-being among university freshmen. Self Identity. 2014;13(5):579-593.
53. Williams JG, Stark SK, Foster EE. Start today or the very last day? The relationships among self-compassion, motivation, and procrastination. Am J Psychol Res. 2008;4(1):37-44.
54. Neff KD, Hseih Y, Dejittherat K. Self-compassion, achievement goals, and coping with academic failure. Self Identity. 2005;4(3):263-287.
55. Campos RC, Besser A, Blatt SJ. The mediating role of self-criticism and dependency in the association between perceptions of maternal caring and depressive symptoms. Depress Anxiety. 2010;27(12):1149-1157.
56. Sachs-Ericsson N, Verona E, Joiner T, et al. Parental verbal abuse and the mediating role of self-criticism in adult internalizing disorders. J Affect Disord. 2006;93(1-3):71-78.
57. Amitay OA, Mongrain M, Fazaa N. Love and control: self-criticism in parents and daughters and perceptions of relationship partners. Pers Individ Dif. 2008;44(1):75-85.
58. Zuroff DC, Fitzpatrick DK. Depressive personality styles: implications for adult attachment. Pers Individ Dif. 1995;18(2):253-265.
59. Kopala-Sibley DC, Zuroff DC. The developmental origins of personality factors from the self-definitional and relatedness domains: a review of theory and research. Rev Gen Psychol. 2014;18(3):137-155.
60. Kopala-Sibley DC, Zuroff DC, Leybman MJ, et al. Recalled peer relationship experiences and current levels of self-criticism and self-reassurance. Psychol Psychother. 2013;86(1):33-51.
61. Neff KD, McGehee P. Self-compassion and psychological resilience among adolescents and young adults. Self Identity. 2010;9(3):225-240.
62. Pepping CA, Davis PJ, O’Donovan A, et al. Individual differences in self-compassion: the role of attachment and experiences of parenting in childhood. Self Identity. 2015;14(1):104-117.
63. Neff KD, Rude SS, Kirkpatrick KL. An examination of self-compassion in relation to positive psychological functioning and personality traits. J Res Pers. 2007;41(4):908-916.
64. Van Dam NT, Sheppard SC, Forsyth JP, et al. Self-compassion is a better predictor than mindfulness of symptom severity and quality of life in mixed anxiety and depression. J Anxiety Disord. 2011;25(1):123-130.
65. Heffernan M, Quinn MT, McNulty SR, et al. Self-compassion and emotional intelligence in nurses. Int J Nursing Practice. 2010;16(4):366-373.
66. Neff KD, Kirkpatrick KL, Rude SS. Self-compassion and adaptive psychological functioning. J Res Pers. 2007;41(1):139-154.
67. Krieger T, Altenstein D, Baettig I, et al. Self-compassion in depression: associations with depressive symptoms, rumination, and avoidance in depressed outpatients. Behav Ther. 2013;44(3):501-513.
68. Breen WE, Kashdan TB, Lenser ML, et al. Gratitude and forgiveness: convergence and divergence on self-report and informant ratings. Pers Individ Dif. 2010;49(8):932-937.
69. Hollis-Walker L, Colosimo K. Mindfulness, self-compassion, and happiness in non-meditators: A theoretical and empirical examination. Pers Individ Dif. 2011;50(2):222-227.
70. Gilbert P, McEwan K, Matos M, et al. Fears of compassion: development of three self-report measures. Psychol Psychother. 2011;84(3):239-255.
71. Neely ME, Schallert DL, Mohammed SS, et al. Self-kindness when facing stress: the role of self-compassion, goal regulation, and support in college students’ well-being. Motiv Emot. 2009;33(1):88-97.
72. Breines JG, Chen S. Self-compassion increases self-improvement motivation. Pers Soc Psychol Bull. 2012;38(9):1133-1143.
73. Allen AB, Leary MR. Self-compassion, stress, and coping. Soc Pers Psychol Compass. 2010;4(2):107-118.
74. Terry ML, Leary MR. Self-compassion, self-regulation, and health. Self Identity. 2011;10(3):352-362.
75. Magnus CMR, Kowalski KC, McHugh TF. The role of self-compassion in women’s self-determined motives to exercise and exercise-related outcomes. Self Identity. 2010;9(4):363-382.
76. Brooks M, Kay-Lambkin F, Bowman J, et al. Self-compassion amongst clients with problematic alcohol use. Mindfulness. 2012;3(4):308-317.
77. Adams CE, Leary MR. Promoting self-compassionate attitudes toward eating among restrictive and guilty eaters. J Soc Clin Psychol. 2007;26(10):1120-1144.
78. Kelly AC, Zuroff DC, Foa CL, et al. Who benefits from training in self-compassionate self-regulation? A study of smoking reduction. J Soc Clin Psychol. 2010;29(7):727-755.
79. Shapira LB, Mongrain M. The benefits of self-compassion and optimism exercises for individuals vulnerable to depression. J Posit Psychol. 2010;5(5):377-389.
80. Albertson ER, Neff KD, Dill-Shackleford KE. Self-compassion and body dissatisfaction in women: a randomized controlled trial of a brief meditation intervention. Mindfulness. 2015;6(3):444-454.
81. Smeets E, Neff K, Alberts H, et al. Meeting suffering with kindness: effects of a brief self-compassion intervention for female college students. J Clinical Psychol. 2014;70(9):794-807.
82. Blatt SJ, D’Afflitti JP, Quinlan DM. Depressive experiences questionnaire. New Haven, CT: Yale University Press; 1976.
83. Weissman AN, Beck AT. Development and validation of the dysfunctional attitude scale: a preliminary investigation. Paper presented at: 62nd Annual Meeting of the Association for Advanced Behavior Therapy; March 27-31, 1978; Toronto, Ontario, Canada.
84. Gilbert P, Clarke M, Hempel S, et al. Criticizing and reassuring oneself: an exploration of forms, styles and reasons in female students. Br J Clin Psychol. 2004;43(pt 1):31-50.
85. Baião R, Gilbert P, McEwan K, et al. Forms of self-criticising/attacking & self-reassuring scale: psychometric properties and normative study. Psychol Psychother. 2015;88(4):438-452.
86. Neff KD. The self-compassion scale is a valid and theoretically coherent measure of self-compassion. Mindfulness. 2016;7(1):264-274.
87. Neff KD, Germer CK. A pilot study and randomized controlled trial of the mindful self-compassion program. J Clinical Psychol. 2013;69(1):28-44.
88. Gilbert P. Introducing compassion-focused therapy. Adv Psychiatr Treat. 2009;15(3):199-208.
89. Kelly AC, Zuroff DC, Shapira LB. Soothing oneself and resisting self-attacks: the treatment of two intrapersonal deficits in depression vulnerability. Cognit Ther Res. 2009;33(3):301-313.
90. Boersma K, Hakanson A, Salomonsson E, et al. Compassion focused therapy to counteract shame, self-criticism and isolation. A replicated single case experimental study of individuals with social anxiety. J Contemp Psychother. 2015;45(2):89-98.
91. Gale C, Gilbert P, Read N, et al. An evaluation of the impact of introducing compassion focused therapy to a standard treatment programme for people with eating disorders. Clin Psychol Psychother. 2014;21(1):1-12.
92. Braehler C, Gumley A, Harper J, et al. Exploring change processes in compassion focused therapy in psychosis: results of a feasibility randomized controlled trial. Br J Clin Psychol. 2013;52(2):199-214.
93. Ashworth F, Clarke A, Jones L, et al. An exploration of compassion focused therapy following acquired brain injury. Psychol Psychother. 2014;88(2):143-162.
94. Judge L, Cleghorn A, McEwan K, et al. An exploration of group-based compassion focused therapy for a heterogeneous range of clients presenting to a community mental health team. Int J Cogn Ther. 2012;5(4):420-429.
95. Leaviss J, Utley L. Psychotherapeutic benefits of compassion-focused therapy: an early systematic review. Psychol Med. 2015;45(5):927-945.
96. Gilbert P, McEwan K, Gibbons L, et al. Fears of compassion and happiness in relation to alexithymia, mindfulness, and self‐criticism. Psychol Psychother. 2012;85(4):374-390.
97. Germer CK, Neff KD. Cultivating self-compassion in trauma survivors. In: Follette VM, Briere J, Rozelle D, et al, eds. Mindfulness-oriented interventions for trauma: integrating contemplative practices. New York, NY: Guilford Press; 2015:43-58.
98. Gilbert P. Compassion focused therapy: the CBT distinctive features series. London, United Kingdom: Routledge; 2010.
99. Germer C, Neff K. The mindful self-compassion training program. In: Singer T, Bolz M, eds. Compassion: bridging theory and practice: a multimedia book. Leipzig, Germany: Max-Planck Institute; 2013:365-396.
100. Neff K. Self-compassion: the proven power of being kind to yourself. New York, NY: HarperCollins; 2015.

When to use an anticonvulsant to treat alcohol withdrawal
Alcohol withdrawal is an uncomfortable and potentially life-threatening condition that must be treated before patients can achieve sobriety. Benzodiazepines remain the first-line treatment for alcohol withdrawal; however, these agents could:
- exacerbate agitation
- interact adversely with other medications, particularly opioids
- be unsafe for outpatients at risk of drinking again.


Off-label use of anticonvulsants could reduce these risks. In our emergency department, we routinely use these agents as monotherapy for patients discharging to outpatient detoxification or as adjunctive treatment for patients who require admission for severe withdrawal (Table1,2).
Gabapentin is safe for patients with liver disease and has few drug–drug interactions.1 Dosages of at least 1,200 mg/d seems to be comparable to lorazepam for alcohol withdrawal and could help prevent relapse after the withdrawal period.1 Many patients report that gabapentin helps them sleep. Gabapentin could cause gastrointestinal upset or slight dizziness; patients with severe renal disease might require dosage adjustments.
Carbamazepine. In a randomized double-blind trial, carbamazepine was superior to lorazepam in preventing rebound withdrawal symptoms and reducing post-treatment drinking, although both agents were effective in decreasing withdrawal symptoms.2 Avoid this agent in patients with serum liver enzymes 3 times higher than normal values
Divalproex with as-needed benzodiazepines reduces the duration of withdrawal and risk of medical complications.3 Avoid using divalproex in patients with thrombocytopenia, leukopenia, or severe liver disease.
1. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.
2. Malcom R, Myrick H, Roberts J, et al. The effects of carbamazepine and lorazepam on a single versus multiple previous alcohol withdrawals in an outpatient randomized trial. J Gen Int Med. 2002;17(5):349-355.
3. Eyer F, Schreckenberg M, Adorjan K, et al. Carbamazepine and valproate as adjuncts in the treatment of alcohol withdrawal syndrome: a retrospective cohort study. Alcohol Alcohol. 2011;46(2):177-184.
Alcohol withdrawal is an uncomfortable and potentially life-threatening condition that must be treated before patients can achieve sobriety. Benzodiazepines remain the first-line treatment for alcohol withdrawal; however, these agents could:
- exacerbate agitation
- interact adversely with other medications, particularly opioids
- be unsafe for outpatients at risk of drinking again.
Off-label use of anticonvulsants could reduce these risks. In our emergency department, we routinely use these agents as monotherapy for patients discharging to outpatient detoxification or as adjunctive treatment for patients who require admission for severe withdrawal (Table1,2).
Gabapentin is safe for patients with liver disease and has few drug–drug interactions.1 Dosages of at least 1,200 mg/d seems to be comparable to lorazepam for alcohol withdrawal and could help prevent relapse after the withdrawal period.1 Many patients report that gabapentin helps them sleep. Gabapentin could cause gastrointestinal upset or slight dizziness; patients with severe renal disease might require dosage adjustments.
Carbamazepine. In a randomized double-blind trial, carbamazepine was superior to lorazepam in preventing rebound withdrawal symptoms and reducing post-treatment drinking, although both agents were effective in decreasing withdrawal symptoms.2 Avoid this agent in patients with serum liver enzymes 3 times higher than normal values
Divalproex with as-needed benzodiazepines reduces the duration of withdrawal and risk of medical complications.3 Avoid using divalproex in patients with thrombocytopenia, leukopenia, or severe liver disease.
Alcohol withdrawal is an uncomfortable and potentially life-threatening condition that must be treated before patients can achieve sobriety. Benzodiazepines remain the first-line treatment for alcohol withdrawal; however, these agents could:
- exacerbate agitation
- interact adversely with other medications, particularly opioids
- be unsafe for outpatients at risk of drinking again.
Off-label use of anticonvulsants could reduce these risks. In our emergency department, we routinely use these agents as monotherapy for patients discharging to outpatient detoxification or as adjunctive treatment for patients who require admission for severe withdrawal (Table1,2).
Gabapentin is safe for patients with liver disease and has few drug–drug interactions.1 Dosages of at least 1,200 mg/d seems to be comparable to lorazepam for alcohol withdrawal and could help prevent relapse after the withdrawal period.1 Many patients report that gabapentin helps them sleep. Gabapentin could cause gastrointestinal upset or slight dizziness; patients with severe renal disease might require dosage adjustments.
Carbamazepine. In a randomized double-blind trial, carbamazepine was superior to lorazepam in preventing rebound withdrawal symptoms and reducing post-treatment drinking, although both agents were effective in decreasing withdrawal symptoms.2 Avoid this agent in patients with serum liver enzymes 3 times higher than normal values
Divalproex with as-needed benzodiazepines reduces the duration of withdrawal and risk of medical complications.3 Avoid using divalproex in patients with thrombocytopenia, leukopenia, or severe liver disease.
1. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.
2. Malcom R, Myrick H, Roberts J, et al. The effects of carbamazepine and lorazepam on a single versus multiple previous alcohol withdrawals in an outpatient randomized trial. J Gen Int Med. 2002;17(5):349-355.
3. Eyer F, Schreckenberg M, Adorjan K, et al. Carbamazepine and valproate as adjuncts in the treatment of alcohol withdrawal syndrome: a retrospective cohort study. Alcohol Alcohol. 2011;46(2):177-184.
1. Myrick H, Malcolm R, Randall PK, et al. A double-blind trial of gabapentin versus lorazepam in the treatment of alcohol withdrawal. Alcohol Clin Exp Res. 2009;33(9):1582-1588.
2. Malcom R, Myrick H, Roberts J, et al. The effects of carbamazepine and lorazepam on a single versus multiple previous alcohol withdrawals in an outpatient randomized trial. J Gen Int Med. 2002;17(5):349-355.
3. Eyer F, Schreckenberg M, Adorjan K, et al. Carbamazepine and valproate as adjuncts in the treatment of alcohol withdrawal syndrome: a retrospective cohort study. Alcohol Alcohol. 2011;46(2):177-184.
Opioids for chronic pain: The CDC’s 12 recommendations
Earlier this year, the Centers for Disease Control and Prevention (CDC) published a clinical practice guideline aimed at decreasing opioid use in the treatment of chronic pain.1 It developed this guideline in response to the increasing problem of opioid abuse and opioid-related mortality in the United States.
The CDC notes that an estimated 1.9 million people abused or were dependent on prescription opioid pain medication in 2013.1 Between 1999 and 2014, more than 165,000 people in the United States died from an overdose of opioid pain medication, with that rate increasing markedly in the past decade.1 In 2011, an estimated 420,000 emergency department visits were related to the abuse of narcotic pain relievers.2
While the problem of increasing opioid-related abuse and deaths has been apparent for some time, effective interventions have been elusive. Evidence remains sparse on the benefits and harms of long-term opioid therapy for chronic pain, except for those at the end of life. Evidence has been insufficient to determine long-term benefits of opioid therapy vs no opioid therapy, although the potential for harms from high doses of opioids are documented. There is not much evidence comparing nonpharmacologic and non-opioid pharmacologic treatments with long-term opioid therapy.
This lack of an evidence base is reflected in the CDC guideline. Of the guideline’s 12 recommendations, not one has high-level supporting evidence and only one has even moderate-level evidence behind it. Four recommendations are supported by low-level evidence, and 7 by very-low-level evidence. Yet 11 of the 12 are given an A recommendation, meaning that the guideline panel feels that most patients should receive this course of action.
Methodology used to create the guideline
The guideline committee used a modified GRADE approach (Grading of Recommendations Assessment, Development, and Evaluation) to develop the guideline. It is the same system the Advisory Committee on Immunization Practices adopted to assess and make recommendations on vaccines.3 The system’s classification of levels of evidence and recommendation categories are described in FIGURE 1.1

The committee started by assessing evidence with a report on the long-term effectiveness of opioids for chronic pain, produced by the Agency for Health Care Research and Quality in 2014;4 it then augmented that report by performing an updated search for new evidence published since the report came out.5 The committee then conducted a “contextual evidence review”6 on the following 4 areas:
- the effectiveness of nonpharmacologic (cognitive behavioral therapy, exercise therapy, interventional treatments, multimodal pain treatment) and non-opioid pharmacologic treatments (acetaminophen, nonsteroidal anti-inflammatory drugs, antidepressants, anticonvulsants)
- the benefits and harms of opioid therapy
- clinician and patient values and preferences related to opioids and medication risks, benefits, and use
- resource allocation, including costs and economic analyses.
The guideline wording indicates that, for this contextual analysis, the committee used a rapid systematic review methodology, in part because of time constraints given the imperative to produce a guideline to address a pressing problem, and because of a recognition that evidence on the questions would be scant and not of high quality.1 The 12 recommendations are categorized under 3 main headings.
Determining when to initiate or continue opioids for chronic pain
1. Nonpharmacologic therapy and non-opioid pharmacologic therapy are preferred for chronic pain. Consider opioid therapy only if you anticipate that benefits for both pain and function will outweigh risks to the patient. If opioids are used, combine them as appropriate with nonpharmacologic therapy and non-opioid pharmacologic therapy. (Recommendation category: A; evidence type: 3)
2. Before starting opioid therapy for chronic pain, establish treatment goals with the patient, including realistic goals for pain and function, and consider how therapy will be discontinued if the benefits do not outweigh the risks. Continue opioid therapy only if there is clinically meaningful improvement in pain and function that outweighs risks to patient safety. (Recommendation category: A; evidence type: 4)
3. Before starting opioid therapy, and periodically during its course, discuss with patients known risks and realistic benefits of opioid therapy and patient and clinician responsibilities for managing therapy. (Recommendation category: A; evidence type: 3)
Opioid selection, dosage, duration, follow-up, and discontinuation
4. When starting opioid therapy for chronic pain, prescribe immediate-release opioids instead of extended-release/long-acting (ER/LA) agents. (Recommendation category: A; evidence type: 4)
5. When starting opioids, prescribe the lowest effective dosage. Use caution when prescribing opioids at any dosage; carefully reassess the evidence for individual benefits and risks when increasing the dosage to ≥50 morphine milligram equivalents (MME)/d; and avoid increasing the dosage to ≥90 MME/d (or carefully justify such a decision, if made). (Recommendation category: A; evidence type: 3)
6. Long-term opioid use often begins with treatment of acute pain. When opioids are used for acute pain, prescribe the lowest effective dose of immediate-release opioids at a quantity no greater than is needed for the expected duration of pain severe enough to require opioids. Three days or less will often be sufficient; more than 7 days will rarely be needed. (Recommendation category: A; evidence type: 4)
7. In monitoring opioid therapy for chronic pain, reevaluate benefits and harms with patients within one to 4 weeks of starting opioid therapy or escalating the dose. Also, evaluate the benefits and harms of continued therapy with patients every 3 months or more frequently. If the benefits of continued opioid therapy do not outweigh the harms, optimize other therapies and work with patients to taper opioids to lower dosages or to taper and discontinue them. (Recommendation category: A; evidence type: 4)
Assessing risk and addressing harms of opioid use
8. Before starting opioid therapy, and periodically during its continuation, evaluate risk factors for opioid-related harms. Incorporate strategies into the management plan to mitigate risk; consider offering naloxone when factors are present that increase the risk for opioid overdose—eg, a history of overdose, history of substance use disorder, higher opioid dosages (≥50 MME/d), or concurrent benzodiazepine use. (Recommendation category: A; evidence type: 4)
9. Review the patient’s history of controlled substance prescriptions. Use data from the state prescription drug monitoring program (PDMP) to determine whether the patient is receiving opioid dosages or dangerous combinations that put him or her at high risk for overdose. (State Web sites are available at http://www.pdmpassist.org/content/state-pdmp-websites.) Review PDMP data when starting opioid therapy for chronic pain and periodically during its continuation, at least every 3 months and with each new prescription. (Recommendation category: A; evidence type: 4)
10. Before prescribing opioids for chronic pain, use urine drug testing to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs, and consider urine drug testing at least annually. (Recommendation category: B; evidence type: 4)
11. Avoid prescribing opioid pain medication and benzodiazepines concurrently whenever possible. (Recommendation category: A; evidence type: 3)
12. For patients with opioid use disorder, offer or arrange for evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies). (Recommendation category: A; evidence type: 2)
Aids for guideline implementation
The CDC has produced materials to assist physicians in implementing this guideline, including checklists for prescribing or continuing opioids. The checklist for initiation of opioids is reproduced in FIGURE 2.7
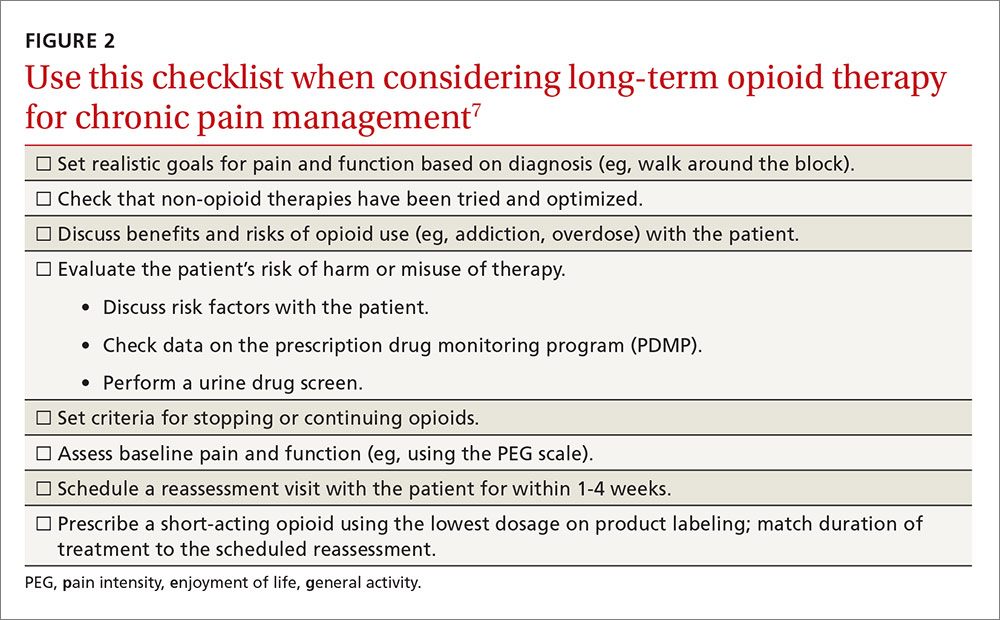
The CDC is addressing a severe public health problem and doing so by using contemporary evidence-based methodology and guideline development processes. The lack of high-quality evidence on the topic and the use of a less-than-optimal evidence review process for some key questions may hamper this effort. However, given the prominence of the CDC, this clinical guideline will likely be considered the standard of care for family physicians.
1. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1–49. Available at: https://www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed October 17, 20
2. Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. The DAWN Report: Highlights of the 2011 Drug Abuse Warning Network (DAWN) Findings on Drug-Related Emergency Department Visits. 20
3. Ahmed F, Temte JL, Campos-Outcalt D, et al; for the ACIP Evidence Based Recommendations Work Group (EBRWG). Methods for developing evidence-based recommendations by the Advisory Committee on Immunization Practices (ACIP) of the U.S. Centers for Disease Control and Prevention (CDC). Vaccine. 2011;29:9171-9176.
4. Chou R, Deyo R, Devine B, et al. The effectiveness and risks of long-term opioid treatment of chronic pain. Evidence Report/Technology Assessment No. 218. AHRQ Publication No. 14-E005-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2014. Available at: http://www.effectivehealthcare.ahrq.gov/ehc/products/557/1971/chronic-pain-opioid-treatment-report-141205.pdf. Accessed October 17, 2016.
5. Centers for Disease Control and Prevention. Clinical evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain-United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38026. Accessed October 17, 2016.
6. Centers for Disease Control and Prevention. Contextual evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain – United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38027. Accessed October 17, 2016.
7. Centers for Disease Control and Prevention. Checklist for prescribing opioids for chronic pain. Available at: https://stacks.cdc.gov/view/cdc/38025. Accessed October 17, 2016.
Earlier this year, the Centers for Disease Control and Prevention (CDC) published a clinical practice guideline aimed at decreasing opioid use in the treatment of chronic pain.1 It developed this guideline in response to the increasing problem of opioid abuse and opioid-related mortality in the United States.
The CDC notes that an estimated 1.9 million people abused or were dependent on prescription opioid pain medication in 2013.1 Between 1999 and 2014, more than 165,000 people in the United States died from an overdose of opioid pain medication, with that rate increasing markedly in the past decade.1 In 2011, an estimated 420,000 emergency department visits were related to the abuse of narcotic pain relievers.2
While the problem of increasing opioid-related abuse and deaths has been apparent for some time, effective interventions have been elusive. Evidence remains sparse on the benefits and harms of long-term opioid therapy for chronic pain, except for those at the end of life. Evidence has been insufficient to determine long-term benefits of opioid therapy vs no opioid therapy, although the potential for harms from high doses of opioids are documented. There is not much evidence comparing nonpharmacologic and non-opioid pharmacologic treatments with long-term opioid therapy.
This lack of an evidence base is reflected in the CDC guideline. Of the guideline’s 12 recommendations, not one has high-level supporting evidence and only one has even moderate-level evidence behind it. Four recommendations are supported by low-level evidence, and 7 by very-low-level evidence. Yet 11 of the 12 are given an A recommendation, meaning that the guideline panel feels that most patients should receive this course of action.
Methodology used to create the guideline
The guideline committee used a modified GRADE approach (Grading of Recommendations Assessment, Development, and Evaluation) to develop the guideline. It is the same system the Advisory Committee on Immunization Practices adopted to assess and make recommendations on vaccines.3 The system’s classification of levels of evidence and recommendation categories are described in FIGURE 1.1
The committee started by assessing evidence with a report on the long-term effectiveness of opioids for chronic pain, produced by the Agency for Health Care Research and Quality in 2014;4 it then augmented that report by performing an updated search for new evidence published since the report came out.5 The committee then conducted a “contextual evidence review”6 on the following 4 areas:
- the effectiveness of nonpharmacologic (cognitive behavioral therapy, exercise therapy, interventional treatments, multimodal pain treatment) and non-opioid pharmacologic treatments (acetaminophen, nonsteroidal anti-inflammatory drugs, antidepressants, anticonvulsants)
- the benefits and harms of opioid therapy
- clinician and patient values and preferences related to opioids and medication risks, benefits, and use
- resource allocation, including costs and economic analyses.
The guideline wording indicates that, for this contextual analysis, the committee used a rapid systematic review methodology, in part because of time constraints given the imperative to produce a guideline to address a pressing problem, and because of a recognition that evidence on the questions would be scant and not of high quality.1 The 12 recommendations are categorized under 3 main headings.
Determining when to initiate or continue opioids for chronic pain
1. Nonpharmacologic therapy and non-opioid pharmacologic therapy are preferred for chronic pain. Consider opioid therapy only if you anticipate that benefits for both pain and function will outweigh risks to the patient. If opioids are used, combine them as appropriate with nonpharmacologic therapy and non-opioid pharmacologic therapy. (Recommendation category: A; evidence type: 3)
2. Before starting opioid therapy for chronic pain, establish treatment goals with the patient, including realistic goals for pain and function, and consider how therapy will be discontinued if the benefits do not outweigh the risks. Continue opioid therapy only if there is clinically meaningful improvement in pain and function that outweighs risks to patient safety. (Recommendation category: A; evidence type: 4)
3. Before starting opioid therapy, and periodically during its course, discuss with patients known risks and realistic benefits of opioid therapy and patient and clinician responsibilities for managing therapy. (Recommendation category: A; evidence type: 3)
Opioid selection, dosage, duration, follow-up, and discontinuation
4. When starting opioid therapy for chronic pain, prescribe immediate-release opioids instead of extended-release/long-acting (ER/LA) agents. (Recommendation category: A; evidence type: 4)
5. When starting opioids, prescribe the lowest effective dosage. Use caution when prescribing opioids at any dosage; carefully reassess the evidence for individual benefits and risks when increasing the dosage to ≥50 morphine milligram equivalents (MME)/d; and avoid increasing the dosage to ≥90 MME/d (or carefully justify such a decision, if made). (Recommendation category: A; evidence type: 3)
6. Long-term opioid use often begins with treatment of acute pain. When opioids are used for acute pain, prescribe the lowest effective dose of immediate-release opioids at a quantity no greater than is needed for the expected duration of pain severe enough to require opioids. Three days or less will often be sufficient; more than 7 days will rarely be needed. (Recommendation category: A; evidence type: 4)
7. In monitoring opioid therapy for chronic pain, reevaluate benefits and harms with patients within one to 4 weeks of starting opioid therapy or escalating the dose. Also, evaluate the benefits and harms of continued therapy with patients every 3 months or more frequently. If the benefits of continued opioid therapy do not outweigh the harms, optimize other therapies and work with patients to taper opioids to lower dosages or to taper and discontinue them. (Recommendation category: A; evidence type: 4)
Assessing risk and addressing harms of opioid use
8. Before starting opioid therapy, and periodically during its continuation, evaluate risk factors for opioid-related harms. Incorporate strategies into the management plan to mitigate risk; consider offering naloxone when factors are present that increase the risk for opioid overdose—eg, a history of overdose, history of substance use disorder, higher opioid dosages (≥50 MME/d), or concurrent benzodiazepine use. (Recommendation category: A; evidence type: 4)
9. Review the patient’s history of controlled substance prescriptions. Use data from the state prescription drug monitoring program (PDMP) to determine whether the patient is receiving opioid dosages or dangerous combinations that put him or her at high risk for overdose. (State Web sites are available at http://www.pdmpassist.org/content/state-pdmp-websites.) Review PDMP data when starting opioid therapy for chronic pain and periodically during its continuation, at least every 3 months and with each new prescription. (Recommendation category: A; evidence type: 4)
10. Before prescribing opioids for chronic pain, use urine drug testing to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs, and consider urine drug testing at least annually. (Recommendation category: B; evidence type: 4)
11. Avoid prescribing opioid pain medication and benzodiazepines concurrently whenever possible. (Recommendation category: A; evidence type: 3)
12. For patients with opioid use disorder, offer or arrange for evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies). (Recommendation category: A; evidence type: 2)
Aids for guideline implementation
The CDC has produced materials to assist physicians in implementing this guideline, including checklists for prescribing or continuing opioids. The checklist for initiation of opioids is reproduced in FIGURE 2.7
The CDC is addressing a severe public health problem and doing so by using contemporary evidence-based methodology and guideline development processes. The lack of high-quality evidence on the topic and the use of a less-than-optimal evidence review process for some key questions may hamper this effort. However, given the prominence of the CDC, this clinical guideline will likely be considered the standard of care for family physicians.
Earlier this year, the Centers for Disease Control and Prevention (CDC) published a clinical practice guideline aimed at decreasing opioid use in the treatment of chronic pain.1 It developed this guideline in response to the increasing problem of opioid abuse and opioid-related mortality in the United States.
The CDC notes that an estimated 1.9 million people abused or were dependent on prescription opioid pain medication in 2013.1 Between 1999 and 2014, more than 165,000 people in the United States died from an overdose of opioid pain medication, with that rate increasing markedly in the past decade.1 In 2011, an estimated 420,000 emergency department visits were related to the abuse of narcotic pain relievers.2
While the problem of increasing opioid-related abuse and deaths has been apparent for some time, effective interventions have been elusive. Evidence remains sparse on the benefits and harms of long-term opioid therapy for chronic pain, except for those at the end of life. Evidence has been insufficient to determine long-term benefits of opioid therapy vs no opioid therapy, although the potential for harms from high doses of opioids are documented. There is not much evidence comparing nonpharmacologic and non-opioid pharmacologic treatments with long-term opioid therapy.
This lack of an evidence base is reflected in the CDC guideline. Of the guideline’s 12 recommendations, not one has high-level supporting evidence and only one has even moderate-level evidence behind it. Four recommendations are supported by low-level evidence, and 7 by very-low-level evidence. Yet 11 of the 12 are given an A recommendation, meaning that the guideline panel feels that most patients should receive this course of action.
Methodology used to create the guideline
The guideline committee used a modified GRADE approach (Grading of Recommendations Assessment, Development, and Evaluation) to develop the guideline. It is the same system the Advisory Committee on Immunization Practices adopted to assess and make recommendations on vaccines.3 The system’s classification of levels of evidence and recommendation categories are described in FIGURE 1.1
The committee started by assessing evidence with a report on the long-term effectiveness of opioids for chronic pain, produced by the Agency for Health Care Research and Quality in 2014;4 it then augmented that report by performing an updated search for new evidence published since the report came out.5 The committee then conducted a “contextual evidence review”6 on the following 4 areas:
- the effectiveness of nonpharmacologic (cognitive behavioral therapy, exercise therapy, interventional treatments, multimodal pain treatment) and non-opioid pharmacologic treatments (acetaminophen, nonsteroidal anti-inflammatory drugs, antidepressants, anticonvulsants)
- the benefits and harms of opioid therapy
- clinician and patient values and preferences related to opioids and medication risks, benefits, and use
- resource allocation, including costs and economic analyses.
The guideline wording indicates that, for this contextual analysis, the committee used a rapid systematic review methodology, in part because of time constraints given the imperative to produce a guideline to address a pressing problem, and because of a recognition that evidence on the questions would be scant and not of high quality.1 The 12 recommendations are categorized under 3 main headings.
Determining when to initiate or continue opioids for chronic pain
1. Nonpharmacologic therapy and non-opioid pharmacologic therapy are preferred for chronic pain. Consider opioid therapy only if you anticipate that benefits for both pain and function will outweigh risks to the patient. If opioids are used, combine them as appropriate with nonpharmacologic therapy and non-opioid pharmacologic therapy. (Recommendation category: A; evidence type: 3)
2. Before starting opioid therapy for chronic pain, establish treatment goals with the patient, including realistic goals for pain and function, and consider how therapy will be discontinued if the benefits do not outweigh the risks. Continue opioid therapy only if there is clinically meaningful improvement in pain and function that outweighs risks to patient safety. (Recommendation category: A; evidence type: 4)
3. Before starting opioid therapy, and periodically during its course, discuss with patients known risks and realistic benefits of opioid therapy and patient and clinician responsibilities for managing therapy. (Recommendation category: A; evidence type: 3)
Opioid selection, dosage, duration, follow-up, and discontinuation
4. When starting opioid therapy for chronic pain, prescribe immediate-release opioids instead of extended-release/long-acting (ER/LA) agents. (Recommendation category: A; evidence type: 4)
5. When starting opioids, prescribe the lowest effective dosage. Use caution when prescribing opioids at any dosage; carefully reassess the evidence for individual benefits and risks when increasing the dosage to ≥50 morphine milligram equivalents (MME)/d; and avoid increasing the dosage to ≥90 MME/d (or carefully justify such a decision, if made). (Recommendation category: A; evidence type: 3)
6. Long-term opioid use often begins with treatment of acute pain. When opioids are used for acute pain, prescribe the lowest effective dose of immediate-release opioids at a quantity no greater than is needed for the expected duration of pain severe enough to require opioids. Three days or less will often be sufficient; more than 7 days will rarely be needed. (Recommendation category: A; evidence type: 4)
7. In monitoring opioid therapy for chronic pain, reevaluate benefits and harms with patients within one to 4 weeks of starting opioid therapy or escalating the dose. Also, evaluate the benefits and harms of continued therapy with patients every 3 months or more frequently. If the benefits of continued opioid therapy do not outweigh the harms, optimize other therapies and work with patients to taper opioids to lower dosages or to taper and discontinue them. (Recommendation category: A; evidence type: 4)
Assessing risk and addressing harms of opioid use
8. Before starting opioid therapy, and periodically during its continuation, evaluate risk factors for opioid-related harms. Incorporate strategies into the management plan to mitigate risk; consider offering naloxone when factors are present that increase the risk for opioid overdose—eg, a history of overdose, history of substance use disorder, higher opioid dosages (≥50 MME/d), or concurrent benzodiazepine use. (Recommendation category: A; evidence type: 4)
9. Review the patient’s history of controlled substance prescriptions. Use data from the state prescription drug monitoring program (PDMP) to determine whether the patient is receiving opioid dosages or dangerous combinations that put him or her at high risk for overdose. (State Web sites are available at http://www.pdmpassist.org/content/state-pdmp-websites.) Review PDMP data when starting opioid therapy for chronic pain and periodically during its continuation, at least every 3 months and with each new prescription. (Recommendation category: A; evidence type: 4)
10. Before prescribing opioids for chronic pain, use urine drug testing to assess for prescribed medications, as well as other controlled prescription drugs and illicit drugs, and consider urine drug testing at least annually. (Recommendation category: B; evidence type: 4)
11. Avoid prescribing opioid pain medication and benzodiazepines concurrently whenever possible. (Recommendation category: A; evidence type: 3)
12. For patients with opioid use disorder, offer or arrange for evidence-based treatment (usually medication-assisted treatment with buprenorphine or methadone in combination with behavioral therapies). (Recommendation category: A; evidence type: 2)
Aids for guideline implementation
The CDC has produced materials to assist physicians in implementing this guideline, including checklists for prescribing or continuing opioids. The checklist for initiation of opioids is reproduced in FIGURE 2.7
The CDC is addressing a severe public health problem and doing so by using contemporary evidence-based methodology and guideline development processes. The lack of high-quality evidence on the topic and the use of a less-than-optimal evidence review process for some key questions may hamper this effort. However, given the prominence of the CDC, this clinical guideline will likely be considered the standard of care for family physicians.
1. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1–49. Available at: https://www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed October 17, 20
2. Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. The DAWN Report: Highlights of the 2011 Drug Abuse Warning Network (DAWN) Findings on Drug-Related Emergency Department Visits. 20
3. Ahmed F, Temte JL, Campos-Outcalt D, et al; for the ACIP Evidence Based Recommendations Work Group (EBRWG). Methods for developing evidence-based recommendations by the Advisory Committee on Immunization Practices (ACIP) of the U.S. Centers for Disease Control and Prevention (CDC). Vaccine. 2011;29:9171-9176.
4. Chou R, Deyo R, Devine B, et al. The effectiveness and risks of long-term opioid treatment of chronic pain. Evidence Report/Technology Assessment No. 218. AHRQ Publication No. 14-E005-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2014. Available at: http://www.effectivehealthcare.ahrq.gov/ehc/products/557/1971/chronic-pain-opioid-treatment-report-141205.pdf. Accessed October 17, 2016.
5. Centers for Disease Control and Prevention. Clinical evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain-United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38026. Accessed October 17, 2016.
6. Centers for Disease Control and Prevention. Contextual evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain – United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38027. Accessed October 17, 2016.
7. Centers for Disease Control and Prevention. Checklist for prescribing opioids for chronic pain. Available at: https://stacks.cdc.gov/view/cdc/38025. Accessed October 17, 2016.
1. Dowell D, Haegerich TM, Chou R. CDC guideline for prescribing opioids for chronic pain — United States, 2016. MMWR Recomm Rep. 2016;65:1–49. Available at: https://www.cdc.gov/mmwr/volumes/65/rr/rr6501e1.htm. Accessed October 17, 20
2. Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. The DAWN Report: Highlights of the 2011 Drug Abuse Warning Network (DAWN) Findings on Drug-Related Emergency Department Visits. 20
3. Ahmed F, Temte JL, Campos-Outcalt D, et al; for the ACIP Evidence Based Recommendations Work Group (EBRWG). Methods for developing evidence-based recommendations by the Advisory Committee on Immunization Practices (ACIP) of the U.S. Centers for Disease Control and Prevention (CDC). Vaccine. 2011;29:9171-9176.
4. Chou R, Deyo R, Devine B, et al. The effectiveness and risks of long-term opioid treatment of chronic pain. Evidence Report/Technology Assessment No. 218. AHRQ Publication No. 14-E005-EF. Rockville, MD: Agency for Healthcare Research and Quality; 2014. Available at: http://www.effectivehealthcare.ahrq.gov/ehc/products/557/1971/chronic-pain-opioid-treatment-report-141205.pdf. Accessed October 17, 2016.
5. Centers for Disease Control and Prevention. Clinical evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain-United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38026. Accessed October 17, 2016.
6. Centers for Disease Control and Prevention. Contextual evidence review for the CDC Guideline for Prescribing Opioids for Chronic Pain – United States, 2016. Available at: https://stacks.cdc.gov/view/cdc/38027. Accessed October 17, 2016.
7. Centers for Disease Control and Prevention. Checklist for prescribing opioids for chronic pain. Available at: https://stacks.cdc.gov/view/cdc/38025. Accessed October 17, 2016.

Never gonna give you up: Intrusive musical imagery as compulsions
Intrusive musical imagery (IMI) is characterized by recalling pieces of music,1 usually repetitions of 15 to 30 seconds,2 without pathology of the ear or nervous system.1 Also known as earworm—ohrwurm in German—or involuntary musical imagery, bits of music can become a constant cause of distress.1
IMI is prevalent in the general population; in an internet survey >85% of respondents reported experiencing IMI at least weekly.2 IMI can be generated by:
- hearing music
- reading song lyrics
- being in contact with an environment or people who are linked to specific song, such as department stores that play holiday music.2,3
IMI also is associated with stressful situations or neurological insult.1
Any song or segment of music can be the basis of IMI. The content of IMI change over time (ie, a new song can become a source of IMI).3 The frequency of experiencing IMI is correlated to how much music a person is exposed to and the importance a person places on music.2 Most episodes are intermittent; however, continuous musical episodes are known to occur.3 Episodes of IMI with obsessive-compulsive features can be classified as musical obsessions (MO).1 MO may be part of obsessive-compulsive symptoms, including washing, checking, aggression, sexual obsessions, and religious obsessions or other obsessions.1
Diagnosing musical obsessions
No current measures are adequate to diagnose MO. The Yale-Brown Obsessive Compulsive Scale does not distinguish MO from other intrusive auditory imagery.1
It is important to differentiate MO from:
- Musical preoccupations or recollections in which an individual repeatedly listens or recalls a particular song or part of a song, but does not have the urge to listen or recall music in an obsessive-compulsive pattern.1 These individuals do not display fear and avoidant behaviors that could be seen in patients with MO.1
- Musical hallucinations lack an input stimulus and the patient believes the music comes from an outside source and interprets it as reality. Misdiagnosing MO as a psychotic symptom is common and can result in improper treatment.1
Management
Pharmacotherapy. MO responds to the same medications used to treat obsessive-compulsive disorder, such as selective serotonin reuptake inhibitors and clomipramine.1 Cognitive-behavioral interventions could help patients address dysfunctional beliefs, without trying to suppress them.1
Distraction. Encourage patients to sing a different song that does not have obsessive quality1 or engage in a task that uses working memory.3
Exposure and response prevention therapy. Some case reports have reported efficacy in treating MO.1
1. Taylor S, McKay D, Miguel EC, et al. Musical obsessions: a comprehensive review of neglected clinical phenomena. J Anxiety Disord. 2014;28(6):580-589.
Intrusive musical imagery (IMI) is characterized by recalling pieces of music,1 usually repetitions of 15 to 30 seconds,2 without pathology of the ear or nervous system.1 Also known as earworm—ohrwurm in German—or involuntary musical imagery, bits of music can become a constant cause of distress.1
IMI is prevalent in the general population; in an internet survey >85% of respondents reported experiencing IMI at least weekly.2 IMI can be generated by:
- hearing music
- reading song lyrics
- being in contact with an environment or people who are linked to specific song, such as department stores that play holiday music.2,3
IMI also is associated with stressful situations or neurological insult.1
Any song or segment of music can be the basis of IMI. The content of IMI change over time (ie, a new song can become a source of IMI).3 The frequency of experiencing IMI is correlated to how much music a person is exposed to and the importance a person places on music.2 Most episodes are intermittent; however, continuous musical episodes are known to occur.3 Episodes of IMI with obsessive-compulsive features can be classified as musical obsessions (MO).1 MO may be part of obsessive-compulsive symptoms, including washing, checking, aggression, sexual obsessions, and religious obsessions or other obsessions.1
Diagnosing musical obsessions
No current measures are adequate to diagnose MO. The Yale-Brown Obsessive Compulsive Scale does not distinguish MO from other intrusive auditory imagery.1
It is important to differentiate MO from:
- Musical preoccupations or recollections in which an individual repeatedly listens or recalls a particular song or part of a song, but does not have the urge to listen or recall music in an obsessive-compulsive pattern.1 These individuals do not display fear and avoidant behaviors that could be seen in patients with MO.1
- Musical hallucinations lack an input stimulus and the patient believes the music comes from an outside source and interprets it as reality. Misdiagnosing MO as a psychotic symptom is common and can result in improper treatment.1
Management
Pharmacotherapy. MO responds to the same medications used to treat obsessive-compulsive disorder, such as selective serotonin reuptake inhibitors and clomipramine.1 Cognitive-behavioral interventions could help patients address dysfunctional beliefs, without trying to suppress them.1
Distraction. Encourage patients to sing a different song that does not have obsessive quality1 or engage in a task that uses working memory.3
Exposure and response prevention therapy. Some case reports have reported efficacy in treating MO.1
Intrusive musical imagery (IMI) is characterized by recalling pieces of music,1 usually repetitions of 15 to 30 seconds,2 without pathology of the ear or nervous system.1 Also known as earworm—ohrwurm in German—or involuntary musical imagery, bits of music can become a constant cause of distress.1
IMI is prevalent in the general population; in an internet survey >85% of respondents reported experiencing IMI at least weekly.2 IMI can be generated by:
- hearing music
- reading song lyrics
- being in contact with an environment or people who are linked to specific song, such as department stores that play holiday music.2,3
IMI also is associated with stressful situations or neurological insult.1
Any song or segment of music can be the basis of IMI. The content of IMI change over time (ie, a new song can become a source of IMI).3 The frequency of experiencing IMI is correlated to how much music a person is exposed to and the importance a person places on music.2 Most episodes are intermittent; however, continuous musical episodes are known to occur.3 Episodes of IMI with obsessive-compulsive features can be classified as musical obsessions (MO).1 MO may be part of obsessive-compulsive symptoms, including washing, checking, aggression, sexual obsessions, and religious obsessions or other obsessions.1
Diagnosing musical obsessions
No current measures are adequate to diagnose MO. The Yale-Brown Obsessive Compulsive Scale does not distinguish MO from other intrusive auditory imagery.1
It is important to differentiate MO from:
- Musical preoccupations or recollections in which an individual repeatedly listens or recalls a particular song or part of a song, but does not have the urge to listen or recall music in an obsessive-compulsive pattern.1 These individuals do not display fear and avoidant behaviors that could be seen in patients with MO.1
- Musical hallucinations lack an input stimulus and the patient believes the music comes from an outside source and interprets it as reality. Misdiagnosing MO as a psychotic symptom is common and can result in improper treatment.1
Management
Pharmacotherapy. MO responds to the same medications used to treat obsessive-compulsive disorder, such as selective serotonin reuptake inhibitors and clomipramine.1 Cognitive-behavioral interventions could help patients address dysfunctional beliefs, without trying to suppress them.1
Distraction. Encourage patients to sing a different song that does not have obsessive quality1 or engage in a task that uses working memory.3
Exposure and response prevention therapy. Some case reports have reported efficacy in treating MO.1
1. Taylor S, McKay D, Miguel EC, et al. Musical obsessions: a comprehensive review of neglected clinical phenomena. J Anxiety Disord. 2014;28(6):580-589.
1. Taylor S, McKay D, Miguel EC, et al. Musical obsessions: a comprehensive review of neglected clinical phenomena. J Anxiety Disord. 2014;28(6):580-589.
Benefits and challenges of caring for international patients
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
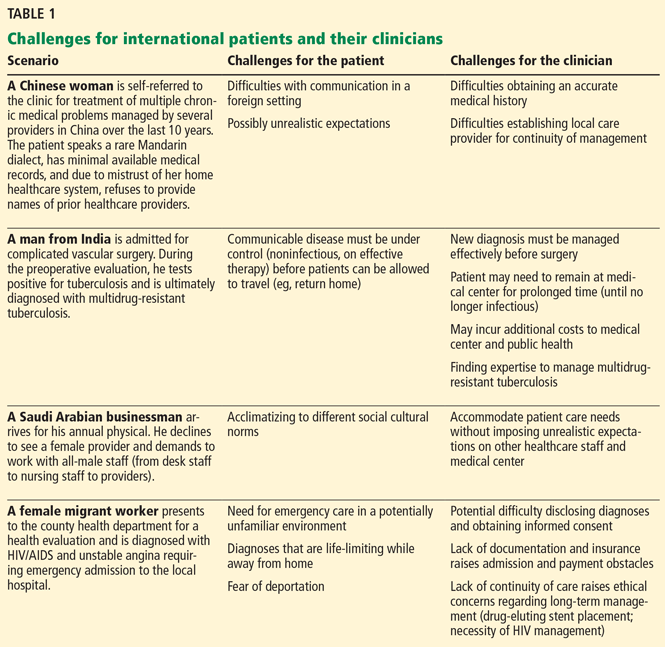
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.

Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement

Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
It is much more important to know what sort of a patient has a disease than what sort of a disease a patient has.
—Attributed to Sir William Osler1
Recent years have seen an increase in people traveling away from their home region for healthcare, often for care that is less expensive or unavailable where they live.2–4 Many Americans seek care abroad (engaging in “medical tourism”); conversely, the United States annually receives thousands of foreign travelers for medical evaluations, a trend projected to increase.2,3,5 Additionally, US healthcare providers often see foreign travelers for unexpected ailments that develop during their time here.
Traveling for healthcare can be stressful for patients, and caring for international patients may pose challenges for providers and medical centers. On the other hand, such encounters also provide many mutual benefits. Unfortunately, there is little published guidance addressing these issues.2 In this article, we therefore discuss many of the benefits and challenges, with the hope of improving the quality of care delivered and the clinical experience for both providers and patients.
CHALLENGES FOR INTERNATIONAL PATIENTS AND THEIR PROVIDERS
Some scenarios that illustrate challenges faced by international patients and their healthcare providers are presented in Table 1.
For patients, heightened anxiety
Many international patients feel anxious, isolated, and vulnerable, particularly if they have never been away from home before. These feelings arise from multiple factors, including the stress of traveling, lack of family or social support, an unfamiliar environment, contrasting cultural practices, and high expectations.3,4 Language barriers, especially for patients who speak uncommon dialects, and lack of continuously available interpretive services often augment the unsettled emotions of international patients.
Cultural differences
International patients may quickly notice significant differences from their home country in how healthcare is practiced and culturally applied.4,6 Such differences may include dress codes and the comparatively equal role of women vis-à-vis men in the Western medical profession.
For cultural, personal, or religious reasons, some patients feel uncomfortable with healthcare providers of the opposite sex. This discomfort can be heightened if the patient needs a potentially uncomfortable and humiliating procedure such as a gynecologic or rectal examination.
The multidisciplinary team approach to healthcare, which can include trainees, nurses, and pharmacists, may leave patients confused about who their primary health provider is.
Decision-making also has cultural implications. In Western medicine, we respect individual autonomy and expect patients to participate in decisions about their care. However, in many areas of the world, medical decision-making is deferred to extended family members or cultural leaders.2 Additional and often repeated conversations may be needed with both the patient and family members to ensure appropriate understanding and ethical consent for care.
Some international patients may have expectations that are quite different from those of the healthcare provider and that are sometimes unrealistic.2,6
Institutional challenges
Many medical conditions require prolonged treatment and longitudinal care, a notable challenge when that care is delivered outside of one’s home country. Practice models within a clinic may not allow for prolonged subsequent visits, which may be needed to accommodate language-translation services. Complex multidisciplinary plans of care must somehow effectively utilize available appointment slots and be time-efficient.
Criteria for hospitalization differ widely among different countries, often based on resources, and may necessitate additional dialogue between the patient and healthcare provider.
Obtaining, interpreting the patient’s record
Medical records from foreign institutions are often unavailable, incomplete, or illegible. Further, depending on the country, it may be difficult to contact local providers for supplemental information. Differences in time zones, limited access to technology, language barriers, and handwritten notes all pose problems when trying to obtain additional information.
Many under-resourced foreign medical centers cannot duplicate medical records and radiographic films for the patient to bring to the United States. Medical records from foreign laboratories often raise questions about the quality, accuracy, and methodology of the testing platform used.2 Thus, the provider may need to start over and repeat the entire clinical, radiologic, and laboratory evaluation.
Communicating with the patient
Difficulties in communication between patients and providers can hinder the development of a positive and productive relationship, reducing patient autonomy and complicating informed consent.2 Obtaining a medical history from non–English-speaking patients can be arduous and time-consuming. Colloquial language may further alter interpretation and understanding, even for formally trained interpreters. Language differences may make it more difficult to explain differential diagnoses, diagnostic approaches, and management plans.
Many US medical centers provide interpreters for many languages, but the great number of languages spoken around the world ensures that barriers in communication persist. Telephone language lines and other commercial language services are available but may feel less personal to patients or evoke concerns about medical confidentiality. For less commonly spoken languages and dialects, appropriate translation services may not even be available.6
Filling in information gaps
Medical conditions, medications, and treatments may have different names in different countries. The quality of pharmaceuticals in some regions may be questionable, and herbal supplements may be unique to a particular location. Many medications available abroad are not available in the United States, potentially confusing US providers as to medication appropriateness, efficacy, and potential toxicities.
Lacking adequate medical records and trying to obtain a new medical history from patients and their family members, providers may struggle with continued gaps of information, hindering a timely diagnosis and composition of an appropriate management plan.
A culturally sensitive but complete physical examination
Every effort should be made to complete a thorough and comprehensive physical examination, even if the patient’s culture differs on this point. This may require a “chaperone” to be present or, if available, a clinician of the same sex as the patient to perform the examination. A compromised examination will impede making the correct diagnosis.
Religious, cultural, and other patient-specific attitudes and beliefs that may affect a medical evaluation should ideally be addressed before scheduling the appointment. A preexamination discussion with the patient and family can help avert unintentional actions and behavior misperceived as offensive, while strengthening the level of trust between patient and provider.2
Money matters
Foreign patients typically have limited or no medical insurance coverage and thus may be paying out of pocket or through limited governmental subsidies. Many refugees and asylum-seekers have no insurance or money to pay for care. (A full discussion of refugee care is beyond the scope of this article). Thus, it is necessary to ascertain in advance who will pay for the care.
Clinicians must be sensitive to the exorbitant costs of medical care and medications in the United States, particularly from the perspective of foreign patients. We strive to provide the best cost-effective care, but what is considered cost-effective and standard care for a patient with US health insurance may be viewed differently by international patients. For some foreign patients, some tests and treatments may be just too expensive, raising personal and institutional ethical concerns regarding how best to evaluate and manage these patients. Ideally, these issues should also be addressed before the patient’s appointment is scheduled.
Clinicians must optimize diagnostic and medical management while minimizing unnecessary testing. This principle further underscores the importance of obtaining a complete medical history and physical examination within a time-sensitive and well-coordinated plan of care.2,4
Continuity of care after the patient leaves
As the medical evaluation and care plan approach completion, ensuring some form of continued medical care can become challenging. Some foreign patients may have the financial or legal means (eg, through an extended medical visa) to remain for further care and follow-up, but most do not.
Finding an available, willing health provider in the patient’s native country for continued management may be difficult and time-consuming. Most US medical centers have no established system to identify available foreign health providers, and usually the patient and family are responsible for arranging continued healthcare back in their home country.
Opportunities for possible improvement of care are noted in Table 2.
ADVANTAGES OF CARING FOR INTERNATIONAL PATIENTS
Despite the possible challenges, there are many benefits of caring for international patients.
Gaining medical knowledge
In US medical centers caring for both regional and referred patients, providers are often exposed to medical conditions that range from common ailments to the rare conditions (or “zebras”) taught during residency training. From the medical education standpoint, international patients provide US health providers heightened opportunities to encounter diseases not commonly seen in the United States (eg, infections such as malaria, schistosomiasis, drug-resistant tuberculosis, and advanced or end-stage forms of noncommunicable diseases). Although not limited to international patients, chronically neglected diseases often give providers first-hand experience in the natural history of select disease progression.
Gaining cultural knowledge
Caring for international patients also enables health providers to learn about different cultures, societal norms, and regional beliefs affecting healthcare. In essence, international patients enable US providers to become more diversified and enlightened with communication skills and assorted managerial strategies on a global scale.
These patients remind us of the stark differences regarding access and quality of medical care globally, particularly in lesser-resourced locations. In a busy domestic medical practice with its own daily challenges, many of us forget these international healthcare disparities, and often take for granted the comparative abundance of healthcare resources available in the United States. Provider frustrations about domestic policies and concerns for a “broken” healthcare system often blind us to the available resources we are fortunate to have at our disposal.
Further, as members of the global community, we have the opportunity to learn from international patients while broadening our view of humanity, thereby enhancing our awareness and empathy toward patients and communities struggling with under-resourced healthcare systems. Healthcare providers are often touched by the gratitude of patients for the opportunity to receive treatments that may otherwise be unavailable. Such experiences may motivate many US health providers to become more engaged in coordinated strategies for global health improvement.
Reimbursement is possible
Caring for international patients should not financially deter US health care centers. Complex, multidisciplinary care evaluations may incur notable expenses; however, alternative and more lucrative payer systems, including government subsidies, can be involved to maintain revenue, reimbursements, and even possibly lead to increased donations.3–5 Given the potential for high costs to be incurred, US providers and institutions need to continually ensure appropriate evidence-based use of resources and cost-effective care without compromising the quality of care provided. The price of certain drugs has been rising astonishingly in the United States, and some patients may therefore prefer to obtain them for long-term use upon return to their home country.
High-quality cost-effective care is satisfying to the patient, provider, and institution, and also may save money that can be reallocated.4 Providers also may find personal fulfillment in striving for and achieving such goals, despite the potential challenges throughout the course of care.
Opportunities for improvement
Regardless of the challenges presented by international patients, participating medical centers often enjoy the prestige and credibility of becoming an “international healthcare center.”4,7 From the standpoint of medical education, these centers have the potential to train providers with increased clinical and cultural competencies along with expanding healthcare services to include clinical, educational and research opportunities abroad.
Research is needed to provide evidence-based guidance on best strategies for patients, clinicians, and healthcare systems to effectively care for international patients.
Suggested opportunities for maximizing advantages are noted in Table 3.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
- William Osler. BrainyQuote.com, Xplore Inc, 2016. www.brainyquote.com/quotes/quotes/w/williamosl391388.html. Accessed September 21, 2016.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006; 81:189–192.
- Bower LC, Johnson TJ, Hohmann SF, Garman AN, Allen M, Meurer SJ. An evaluation of international patient length of stay. Int J Healthc Manag 2014; 7:200–205.
- Satjapot SP, Johnson TJ, Garman AN. International medical travelers, length of stay, and the continuum of care: inquiry and comparison. Qual Manag Health Care 2011; 20:76–83.
- Donohoe M. Luxury primary care, academic medical centers, and the erosion of science and professional ethics. J Gen Intern Med 2004; 19:90–94.
- Dogan H, Tschudin V, Hot I, Özkan I. Patients’ transcultural needs and carers’ ethical responses. Nurs Ethics 2009; 16:683–696.
- Bauer AM, Alegria M. Impact of patient language proficiency and interpreter service use on the quality of psychiatric care: a systematic review. Psychiatr Serv 2010; 61:765–773.
KEY POINTS
- Challenges in caring for international patients include cultural differences, institutional barriers, communication difficulties, sparse medical records, and financial considerations.
- Understanding should be reached beforehand on potentially sensitive issues such as physical examinations, payment, tests, and treatment.
- Benefits to the provider and institution include enhanced medical skills, cultural competency, personal satisfaction, and institutional prestige.

Serotonin syndrome: Preventing, recognizing, and treating it
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16

(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16
(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
With a substantial increase in antidepressant use in the United States over the last 2 decades, serotonin syndrome has become an increasingly common and significant clinical concern. In 1999, 6.5% of adults age 18 and older were taking antidepressants; by 2010, the percentage had increased to 10.4%.1 Though the true incidence of serotonin syndrome is difficult to determine, the number of ingestions of selective serotonin reuptake inhibitors (SSRIs) associated with moderate to major effects reported to US poison control centers increased from 7,349 in 20022 to 8,585 in 2005.3
Though the clinical manifestations are often mild to moderate, patients with serotonin syndrome can deteriorate rapidly and require intensive care. Unlike neuroleptic malignant syndrome, serotonin syndrome should not be considered an extremely rare idiosyncratic reaction to medication, but rather a progression of serotonergic toxicity based on increasing concentration levels that can occur in any patient regardless of age.4
Because it has a nonspecific prodrome and protean manifestations, serotonin syndrome can easily be overlooked, misdiagnosed, or exacerbated if not carefully assessed. Diagnosis requires a low threshold for suspicion and a meticulous history and physical examination. In the syndrome’s mildest stage, symptoms are often misattributed to other causes, and in its most severe form, it can easily be mistaken for neuroleptic malignant syndrome.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome classically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms are a result of increased serotonin levels affecting the central and peripheral nervous systems. Serotonin affects a family of receptors that has seven members, of which 5-HT1A and 5-HT2A are most often responsible for serotonin syndrome.5
Conditions that can alter the regulation of serotonin include therapeutic doses, drug interactions, intentional or unintentional overdoses, and overlapping transitions between medications. As a result, drugs that have been associated with serotonin syndrome can be classified into the following five categories as shown below and in Table 1:
Drugs that decrease serotonin breakdown include monoamine oxidase inhibitors (MAOIs), linezolid,6 methylene blue, procarbazine, and Syrian rue.
Drugs that decrease serotonin reuptake include SSRIs, serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, opioids (meperidine, buprenorphine, tramadol, tapentadol, dextromethorphan), antiepileptics (carbamazepine, valproate), and antiemetics (ondansetron, granisetron, metoclopramide), and the herbal preparation St. John’s wort.
Drugs that increase serotonin precursors or agonists include tryptophan, lithium, fentanyl, and lysergic acid diethylamide (LSD).
Drugs that increase serotonin release include fenfluramine, amphetamines, and methylenedioxymethamphetamine (ecstasy).
Drugs that prevent breakdown of the agents listed above are CYP2D6 and CYP3A4 inhibitors, eg, erythromycin,7 ciprofloxacin, fluconazole, ritonavir, and grapefruit juice.
However, the only drugs that have been reliably confirmed to precipitate serotonin syndrome are MAOIs, SSRIs, SNRIs, and serotonin releasers. Other listed drug interactions are based on case reports and have not been thoroughly evaluated.6–9
Currently, SSRIs are the most commonly prescribed antidepressant medications and, consequently, they are the ones most often implicated in serotonergic toxicity.1,10 An estimated 15% of SSRI overdoses lead to mild or moderate serotonin toxicity.11 Serotonergic agents used in conjunction can increase the risk for severe serotonin syndrome; an SSRI and an MAOI in combination poses the greatest risk.5
Ultimately, the incidence of serotonin syndrome is difficult to assess, but it is believed to be underreported because it is easy to misdiagnose and mild symptoms may be dismissed.
WHO IS AT RISK OF SEROTONIN SYNDROME?
Long-term antidepressant use has disproportionately increased in middle-aged and older adults and non-Hispanic whites.1,12,13 Intuitively, as the risk for depression increases dramatically in patients with chronic medical conditions, serotonin syndrome should be more prevalent among the elderly. In addition, patients with multiple comorbidities take more medications, increasing the risk of polypharmacy and adverse drug reactions.14
Although the epidemiology of serotonin syndrome has yet to be extensively studied, the combination of age and comorbidities may increase the risk for this condition.
HOW DOES IT PRESENT?
Serotonin syndrome characteristically presents as the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. However, these symptoms may not occur simultaneously: autonomic dysfunction is present in 40% of patients, neuromuscular excitation in 50%, and altered mental status in 40%.15 The symptoms can range from mild to life-threatening (Table 2).16
Autonomic dysfunction. Diaphoresis is present in 48.8% of cases, tachycardia in 44%, nausea and vomiting in 26.8%, and mydriasis in 19.5%. Other signs are hyperactive bowel sounds, diarrhea, and flushing.16
Neuromuscular excitation. Myoclonus is present in 48.8%, hyperreflexia in 41%, hyperthermia in 26.8%, and hypertonicity and rigidity in 19.5%. Other signs are spontaneous or inducible clonus, ocular clonus (continuous rhythmic oscillations of gaze), and tremor.
Altered mental status. Confusion is present in 41.2% and agitation in 36.5%. Other signs are anxiety, lethargy, and coma.
Symptoms of serotonin toxicity arise within an hour of a precipitating event (eg, ingestion) in approximately 28% of patients, and within 6 hours in 61%.16 Highly diagnostic features include hyperreflexia and induced or spontaneous clonus that are generally more pronounced in the lower limbs.11 Clonus can be elicited with ankle dorsiflexion.
In mild toxicity, patients may present with tremor or twitching and anxiety, as well as with hyperreflexia, tachycardia, diaphoresis, and mydriasis. Further investigation may uncover a recently initiated antidepressant or a cold-and-cough medication that contains dextromethorphan.15,17
In moderate toxicity, patients present in significant distress, with agitation and restlessness. Features may include hyperreflexia and clonus of the lower extremities, opsoclonus, hyperactive bowel sounds, diarrhea, nausea, vomiting, tachycardia, hypertension, diaphoresis, mydriasis, and hyperthermia (< 40°C, 104°F). The patient’s history may reveal use of ecstasy or combined treatment with serotonin-potentiating agents such as an antidepressant with a proserotonergic opioid, antiepileptic, or CYP2D6 or CYP3A4 inhibitor.15
Severe serotonin toxicity is a life-threatening condition that can lead to multiorgan failure within hours. It can be characterized by muscle rigidity, which can cause the body temperature to elevate rapidly to over 40°C. This hypertonicity can mask the classic and diagnostic signs of hyperreflexia and clonus. Patients may have unstable and dynamic vital signs with confusion or delirium and can experience tonic-clonic seizures.
If the muscle rigidity and resulting hyperthermia are not managed properly, patients can develop cellular damage and enzyme dysfunction leading to rhabdomyolysis, myoglobinuria, renal failure, metabolic acidosis, acute respiratory distress syndrome, and disseminated intravascular coagulation.16,18
Serotonin crisis is usually caused by the co-ingestion of multiple serotonergic agents, such as an antidepressant with an aforementioned opioid and antiemetic19; combining an SSRI and an MAOI poses the greatest risk. Alternatively, patients may have recently switched antidepressants without observing a safe washout period, leading to an overlap of serotonin levels.16
HOW DO WE DIAGNOSE SEROTONIN SYNDROME?
Serotonin syndrome is a clinical diagnosis and therefore requires a thorough review of medications and physical examination. Serum serotonin levels are an unreliable indicator of toxicity and do not correlate well with the clinical presentation.16
(based on information in reference 9).
Currently, there are two clinical tools for diagnosing serotonin syndrome: the Hunter serotonin toxicity criteria (Figure 1) and the Sternbach criteria.
The Hunter criteria are based more heavily on physical findings. The patient must have taken a serotonergic agent and have one of the following:
- Spontaneous clonus
- Inducible clonus plus agitation or diaphoresis
- Ocular clonus plus agitation or diaphoresis
- Inducible clonus or ocular clonus, plus hypertonia and hyperthermia
- Tremor plus hyperreflexia.
The Sternbach criteria. The patient must be using a serotonergic agent, must have no other causes of symptoms, must not have recently used a neuroleptic agent, and must have three of the following:
- Mental status changes
- Agitation
- Hyperreflexia
- Myoclonus
- Diaphoresis
- Shivering
- Tremor
- Diarrhea
- Incoordination
- Fever
The Hunter criteria are recommended and are more specific (97% vs 96%) and more sensitive (84% vs 75%) than the Sternbach criteria when compared with the gold standard of diagnosis by a clinical toxicologist.1 The Hunter criteria are also less likely to yield false-positive results.11
Differential diagnosis
The differential diagnosis for serotonin syndrome includes neuroleptic malignant syndrome, anticholinergic poisoning (Table 3), metastatic carcinoma, central nervous system infection, gastroenteritis, and sepsis.
Neuroleptic malignant syndrome, the disorder most often misdiagnosed as serotonin syndrome, is an idiosyncratic reaction to a dopamine antagonist (eg, haloperidol, fluphenazine) that develops over days to weeks.20 In 70% of patients, agitated delirium with confusion appears first, followed by lead pipe rigidity and cogwheel tremor, then hyperthermia with body temperature greater than 40°C, and finally, profuse diaphoresis, tachycardia, hypertension, and tachypnea.21
Key elements that distinguish neuroleptic malignant syndrome are the timeline of the clinical course, bradyreflexia, and the absence of clonus. Prodromal symptoms of nausea, vomiting, and diarrhea are also rare in neuroleptic malignant syndrome. Neuroleptic malignant syndrome typically requires an average of 9 days to resolve.
Anticholinergic poisoning usually develops within 1 to 2 hours of oral ingestion. Symptoms include flushing, anhidrosis, anhidrotic hyperthermia, mydriasis, urinary retention, decreased bowel sounds, agitated delirium, and visual hallucinations. In contrast to serotonin syndrome, reflexes and muscle tone are normal with anticholinergic poisoning.
HOW CAN WE TREAT SEROTONIN SYNDROME?
The two mainstays of serotonin syndrome management are to discontinue the serotonergic agent and to give supportive care. Most patients improve within 24 hours of stopping the precipitating drug and starting therapy.16
For mild serotonin syndrome, treatment involves discontinuing the offending agent and supportive therapy with intravenous fluids, correction of vital signs, and symptomatic treatment with a benzodiazepine. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For moderate serotonin syndrome, treatment also involves stopping the serotonergic agent and giving supportive care. Symptomatic treatment with a benzodiazepine and nonserotonergic antiemetics is recommended, and standard cooling measures should be implemented for hyperthermia. Patients should be admitted and observed for 12 to 24 hours to prevent exacerbation.
For severe serotonin toxicity, treatment should focus on management of airway, breathing, and circulation—ie, the “ABCs.” The two primary life-threatening concerns are hyperthermia (temperature > 40°C or 104°F) and rigidity, which can lead to hypoventilation.1,22 Controlling hyperthermia and rigidity can prevent other grave complications. Patients with severe serotonin toxicity should be sedated, paralyzed, and intubated.21 This will reverse ventilatory hypertonia and allow for mechanical ventilation. Paralysis will also prevent the exacerbation of hyperthermia, which is caused by muscle rigidity. Antipyretics have no role in the treatment of serotonin syndrome since the hyperthermia is not caused by a change in the hypothalamic temperature set point.21 Standard cooling measures should be used to manage hyperthermia.
Serotonin antagonists
Serotonin antagonists have had some success in case reports, but further studies are needed to confirm this.4,23,24
Cyproheptadine is a potent 5-HT2A antagonist; patients usually respond within 1 to 2 hours of administration. Signs and symptoms have resolved completely within times ranging from 20 minutes to 48 hours, depending on the severity of toxicity.
The recommended initial dose of cyproheptadine is 12 mg, followed by 2 mg every 2 hours if symptoms continue.16 Maintenance dosing with 8 mg every 6 hours should be prescribed once stabilization is achieved. The total daily dose for adults should not exceed 0.5 mg/kg/day. Cyproheptadine is available only in oral form but can be crushed and administered via a nasogastric tube.21
Chlorpromazine is a 5-HT1A and 5-HT2A antagonist and can be given intramuscularly. Despite case reports citing its effectiveness, the risk of hypotension, dystonic reactions, and neuroleptic malignant syndrome may make it a less desirable option.4,25
Cyproheptadine, chlorpromazine, and other serotonin receptor antagonists require further investigation beyond individual case reports to determine their effectiveness and reliability in treating serotonin syndrome.
Other agents
Benzodiazepines are considered a mainstay for symptomatic relief because of their anxiolytic and muscle relaxant effects.26 However, animal studies showed that treatment with benzodiazepines attenuated hyperthermia but had no effect on time to recovery or outcome.27
Neuromuscular blocking agents. The suggested neuromuscular blocking agent for severe toxicity is a nondepolarizing agent such as vecuronium. Succinylcholine should be avoided, as it can exacerbate rhabdomyolysis and hyperkalemia.21
Dantrolene has also been suggested for its muscle-relaxing effects and use in malignant hyperthermia. However, this treatment has not been successful in isolated case reports and has been ineffective in animal models.4,28
Physical restraints are ill-advised, since isometric muscle contractions can exacerbate hyperthermia and lactic acidosis in agitated patients.21 If physical restraints are necessary to deliver medications, they should be removed as soon as possible.
HOW CAN WE PREVENT SEROTONIN SYNDROME?
Prevention of serotonin syndrome begins with improving education and awareness in patients and healthcare providers. Patients should be primarily concerned with taking their medications carefully as prescribed and recognizing early signs and symptoms of serotonin toxicity.
As use of antidepressants among an aging population continues to increase, and as physicians in multiple disciplines prescribe them for evolving indications (eg, duloxetine to treat osteoarthritis, diabetic neuropathy, fibromyalgia, and chemotherapy-induced peripheral neuropathy), healthcare providers need to be prepared to see more cases of serotonin syndrome and its deleterious effects.29–31 Physicians should be vigilant in minimizing unnecessary use of serotonergic agents and reviewing drug regimens regularly to limit polypharmacy.
Electronic ordering systems should be designed to detect and alert the prescriber to possible interactions that can potentiate serotonin syndrome, and to not place the order until the prescriber overrides the alert. Combinations of SSRIs and MAOIs have the highest risk for inducing severe serotonin syndrome and should always be avoided.
If a patient is transitioning between serotonergic agents, physicians should observe a safe washout period to prevent overlap.16,32 Washout periods may differ among medications depending on their half-lives. For example, sertraline has a washout period of 2 weeks, while fluoxetine requires a washout period of 5 to 6 weeks.33 Consulting a pharmacist may be helpful when considering half-lives and washout periods.
We believe that educating both patients and physicians regarding prevention will help minimize the risk for serotonergic syndrome and will improve efficiency in assessment and management should toxicity develop.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
- Mojtabai R, Olfson M. National trends in long-term use of antidepressant medications: results from the US National Health and Nutrition Examination Survey. J Clin Psychiatry 2014; 75:169–177.
- Watson WA, Litovitz TL, Rodgers GC Jr, et al. 2002 Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System. Am J Emerg Med. 2003;21:353–421.
- Lai MW, Klein-Schwartz W, Rodgers GC, et al. 2005 Annual Report of the American Association of Poison Control Centers’ national poisoning and exopsure database. Clin Toxicol (Phila) 2006; 44:803–932.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999; 13:100–109.
- Isbister GK, Buckley NA. The pathophysiology of serotonin toxicity in animals and humans: implications for diagnosis and treatment. Clin Neuropharmacol 2005; 28:205–214.
- Woytowish MR, Maynor LM. Clinical relevance of linezolid-associated serotonin toxicity. Ann Pharmacother 2013; 47:388–397.
- Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999; 19:894–896.
- Gillman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): a review. Headache 2010; 50:264–272.
- Isbister GK, Buckley NA, Whyte IM. Serotonin toxicity: a practical approach to diagnosis and treatment. Med J Aust 2007; 187:361–365.
- Mowry JB, Spyker DA, Cantilena LR Jr, McMillan N, Ford M. 2013 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 31st Annual Report. Clin Toxicol (Phila) 2014; 52:1032–1283.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Karkare SU, Bhattacharjee S, Kamble P, Aparasu R. Prevalence and predictors of antidepressant prescribing in nursing home residents in the United States. Am J Geriatr Pharmacother 2011; 9:109–119.
- Weissman J, Meyers BS, Ghosh S, Bruce ML. Demographic, clinical, and functional factors associated with antidepressant use in the home healthcare elderly. Am J Geriatr Psychiatry 2011; 19:1042–1045.
- Caughey GE, Roughead EE, Shakib S, Vitry AI, Gilbert AL. Co-morbidity and potential treatment conflicts in elderly heart failure patients: a retrospective, cross-sectional study of administrative claims data. Drugs Aging 2011; 28:575–581.
- Iqbal MM, Basil MJ, Kaplan J, Iqbal MT. Overview of serotonin syndrome. Ann Clin Psychiatry 2012; 24:310–318.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- Prakash S, Patel V, Kakked S, Patel I, Yadav R. Mild serotonin syndrome: a report of 12 cases. Ann Indian Acad Neurol 2015; 18:226–230.
- Davies O, Batajoo-Shrestha B, Sosa-Popoteur J, Olibrice M. Full recovery after severe serotonin syndrome, severe rhabdomyolysis, multi-organ failure and disseminated intravascular coagulopathy from MDMA. Heart Lung 2014; 43:117–119.
- Pedavally S, Fugate JE, Rabinstein AA. Serotonin syndrome in the intensive care unit: clinical presentations and precipitating medications. Neurocrit Care 2014; 21:108–113.
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Velamoor VR, Norman RM, Caroff SN, Mann SC, Sullivan KA, Antelo RE. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994; 182:168–173.
- Isbister GK, Hackett LP, Dawson AH, Whyte IM, Smith AJ. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol 2003; 56:441–450.
- Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med 1998; 16:615–619.
- Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med 1994; 331:1021–1022.
- Gillman PK. Successful treatment of serotonin syndrome with chlorpromazine. Med J Aust 1996; 165:345–346.
- Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ 2014; 348:g1626.
- Nisijima K, Shioda K, Yoshino T, Takano K, Kato S. Diazepam and chlormethiazole attenuate the development of hyperthermia in an animal model of the serotonin syndrome. Neurochem Int 2003; 43:155–164.
- Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001; 890:23–31.
- Micca JL, Ruff D, Ahl J, Wohlreich MM. Safety and efficacy of duloxetine treatment in older and younger patients with osteoarthritis knee pain: a post hoc, subgroup analysis of two randomized, placebo-controlled trials. BMC Musculoskelet Disord 2013; 14:137.
- Smith EM, Pang H, Cirrincione C, et al; Alliance for Clinical Trials in Oncology. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA 2013; 309:1359–1367.
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database Syst Rev 2014; 1:CD007115.
- Caughey GE, Roughead EE, Shakib S, McDermott RA, Vitry AI, Gilbert AL. Comorbidity of chronic disease and potential treatment conflicts in older people dispensed antidepressants. Age Ageing 2010; 39:488–494.
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half-life and clinical applicability. Encephale 1999; 25:470–476. French.
KEY POINTS
- Serotonin syndrome is caused by elevated serotonin levels in the central and peripheral nervous systems.
- The classic presentation is the triad of autonomic dysfunction, neuromuscular excitation, and altered mental status. These symptoms vary based on the severity of serotonergic toxicity and often do not present concomitantly.
- Early recognition is critical to ensure appropriate resuscitative measures and to limit further use of drugs that can exacerbate symptoms.

Update on the management of intestinal failure
Intestinal failure, the inability of the gut to maintain nutritional homeostasis,1 is a complication of vascular thrombosis, inflammatory bowel disease, radiation enteritis, obstruction, and other conditions, and of removing segments of the small and large intestines in response to these diseases.1,2 Imbalances of fluids and electrolytes, dehydration, malabsorption, vitamin and mineral deficiencies, chronic diarrhea, and increased ostomy output contribute to a decline in the quality of life and in the survival rate in these patients.2,3
Referral to an intestinal rehabilitation program that combines gastroenterology, nutrition, pharmacy, nursing, and social work can improve nutritional status and quality of life.4 Whenever possible, the goal of rehabilitation is nutritional autonomy, helping the patient make the transition to an independent oral diet.4 In selected patients in whom rehabilitation is not effective, intestinal transplant may be an option.
In this article, we review the intestinal adaptations that follow surgical resection and provide an update on intestinal rehabilitation techniques such as dietary modification, drug therapy, and parenteral nutrition. We also review experience with intestinal transplant in patients with intestinal failure.
INTESTINAL FAILURE
Intestinal failure results from reduction in enterocyte cell mass, obstruction, dysmotility, surgical resection, congenital defects, or disease-associated loss of absorption with suboptimal nutritional autonomy.5 Patients often suffer from extensive nutrient, electrolyte, and fluid abnormalities proportional to the remnant length and part of the intestine removed.5

Epidemiologic studies have demonstrated that short-bowel syndrome is the most common cause of intestinal failure in adults and children.6,7 Short-bowel syndrome is defined as a small-bowel length less than 200 cm, most commonly from extensive resections for inflammatory bowel disease.6 In children, the syndrome is also defined by a residual small-bowel length of less than 25% expected for gestational age.7
Table 1 lists the frequencies of the underlying disorders leading to intestinal failure or short-bowel syndrome in one series.8
INTESTINAL ADAPTATION
The gastrointestinal tract is the only organ for nutrient, fluid, and electrolyte absorption.9 Every day, 8 to 9 L of fluids and secretions reach the small intestine, comprising about 2 to 3 L of oral fluids, 1 L of saliva, 2 L of gastric juices, 1 L of bile, and 2 L of pancreatic juices.9 Approximately 7 to 8 L are reabsorbed by the small intestine and 1 to 2 L by the colon.9
Although carbohydrates, lipids, and proteins are absorbed through the entire small intestine and colon, site-specific digestion and absorption of different nutrients occur in different parts of the gastrointestinal tract.10 Also, certain nutrients may need site-specific receptors or transporters for their absorption,10 for example:
- Iron in the duodenum and proximal jejunum1
- Lactose in the brush border membrane of the jejunum and proximal ileum, where most of the enzyme lactase is present
- Vitamin B12 and bile salts in the distal ileum.
Hence, resection of a specific part of the intestine may predict deficiencies the patient may encounter after surgery.
The diarrhea that occurs in short-bowel syndrome may be due partly to loss of neurohumoral mediators that govern gastrointestinal transit time, most importantly cholecystokinin, peptide YY, and glucagon-like peptide 1.11 After contact with lipid- or protein-rich nutrients, cholecystokinin is released from the proximal small intestine, which decreases the gastric emptying to maximize nutrient digestion.12 Additionally, release of peptide YY and glucagon-like peptide 1 from the ileal L cells decreases gastric and intestinal motility. These mediators prolong gastrointestinal transit, increase nutrient processing time, and enhance absorption.12
After massive intestinal resection, the remnant bowel undergoes physiologic and functional adaptation to maintain nutritional homeostasis.13 Enterocytes express membrane-bound transporters and undergo accelerated cell division to enhance the absorptive surface area.13 Intestinal hypertrophy, which includes an increase in villous diameter and crypt height, continues for 2 years or more after intestinal resection, leading to greater absorptive surface area.14 It is estimated that villous height may increase by as much as 80%, illustrating a dynamic process in response to intestinal stress.15
Luminal nutrients are essential to the stimulation of enterocyte cells through paracrine mechanisms as well as through the up-regulation of colonic peptide transporter PepT1.15 Furthermore, gut motility is initially decreased in order to increase the concentration of local luminal growth factors.16
Other factors that may affect intestinal adaptation are the length of the residual colon and small intestine, enteral growth, and enterotropic factors.16 And especially in patients with short-bowel syndrome, complications such as malabsorption secondary to pancreatic insufficiency or rapid transit, excessive gastric acid secretion, bile acid wasting due to terminal ileum resection, and bacterial overgrowth in the small intestine result in worsened nutritional status and poor quality of life.16
Key factors that affect the degree of nutritional deficiencies
The degree of nutritional deficiencies and fluid and electrolyte imbalances depends on the length and location of resection and whether the colon is still continuous with the small intestine.17 Normal small-bowel length in adults is highly variable and can be up to 600 cm. Malnutrition after surgical resection usually occurs when more than three-fourths of intestinal tissue is removed.17 However, because of intestinal adaptation, patients with 50% of remnant small bowel may be able to achieve nutritional autonomy.18 Furthermore, because absorption of nutrients occurs primarily in the first 150 cm of the small intestine, resections of this anatomic region have the highest probability of resulting in malnutrition.18
After extensive intestinal resection, absorption of water and electrolytes is better and intestinal transit time is longer if the colon is still continuous with the rest of the gastrointestinal system.19 Approximately 100 cm of remnant intestinal tissue without colonic continuity or 60 cm with colonic continuity is needed to ensure the possibility of nutritional autonomy and independence from parenteral nutrition.19 Severe malnutrition and fluid and electrolyte imbalances can be prevented by appropriate and timely multidisciplinary care and early referral for intestinal rehabilitation.
INTESTINAL REHABILITATION AND NUTRITIONAL AUTONOMY

The aim of intestinal rehabilitation is to improve quality of life by reversing malnutrition and promoting nutritional autonomy, ie, independence from parenteral nutrition (Table 2).20 The complex nature of intestinal failure necessitates collaboration of multiple specialists—gastroenterologists, surgeons, dietitians, nurses, psychiatrists or psychologists, pharmacists, and social workers.20
Although most patients with intestinal failure initially require parenteral nutrition to maintain nutritional homeostasis, progressive adaptation of the remnant intestine enables a transition to enteral nutrition.21 Stimulation of the remnant intestine by enteral feeding reduces the complications of parenteral nutrition and encourages intestinal adaptation.21
Outpatient participation in an intestinal rehabilitation program can facilitate weaning from parenteral nutrition. Patients are monitored and supported during dietary modification, pharmacologic interventions, and reconstructive surgeries.21 A study of 61 patients with short-bowel syndrome undergoing a 3-week program of intestinal rehabilitation (recombinant human growth hormone, glutamine, enteral nutrition, and parenteral nutrition) reported an overall survival rate of 95% with an 85% success rate in weaning from parenteral nutrition during a mean follow-up of 50 (± 24) months.22 Permanent dependence on parenteral nutrition despite rehabilitation was predicted by length of the small bowel less than 100 cm and by the absence of terminal ileum and colon.22
Permanent intestinal failure, defined by the inability to wean from parenteral nutrition and restore nutrition autonomy, may require early referral for evaluation for intestinal and multivisceral transplant. Early referral improves survival rates, possibly because of fewer complications from parenteral nutrition.4
DIETARY MODIFICATION

Dietary modification is the single most effective means of weaning patients safely from parenteral nutrition (Table 3).23,24 Small, frequent feedings help reduce symptoms associated with rapid intestinal transit and increase the activity of luminal growth factors.23 Likewise, limits on intake of simple sugars, stimulants such as caffeine or insoluble fiber, and hypo- or hypertonic fluids decrease intestinal losses and the risk of dehydration.23 Low sugar loads also aim to reduce the occurrence of d-lactic acidosis and bacterial overgrowth in the small intestine.23 Patients who cannot maintain positive fluid balance may require standardized oral rehydration (Table 4) to improve absorption by way of the sodium-glucose coupled transporters at the brush border membrane, or they may require intravenous fluid supplementation.25
Colonic continuity

Other dietary recommendations depend on colonic continuity. In 1994, Nordgaard et al26 compared the effects of high-carbohydrate and high-fat diets in eight patients with colonic continuity and six patients with jejunostomies. The authors noted that a high-carbohydrate diet (60% carbohydrate, 20% fat) reduced fecal loss of energy and increased energy absorption in patients with colonic continuity. However, patients with an end-jejunostomy experienced equal fecal losses of carbohydrates and fat proportional to the amount consumed. The authors concluded that the presence of colonic bacteria promoted carbohydrate salvage, ie, the fermentation of malabsorbed carbohydrates to easily absorbed short-chain fatty acids.26
The colon can salvage as much as 1,000 kcal/day in patients with less than 200 cm of small bowel, and the presence of at least 50% of colon in continuity has been shown to reduce parenteral nutrition requirements by half in patients with less than 100 cm of small bowel.27 As a result, a diet high in complex carbohydrates and soluble fiber supplements is recommended in cases of preserved colon to promote adaptation and nutritional autonomy.27
Another aim of a high-carbohydrate, low- fat diet is to prevent calcium oxalate-related nephrolithiasis and choleretic diarrhea.26
In summary, patients with short-bowel syndrome with or without colonic continuity need different dietary regimens to attain nutritional autonomy.
DRUG THERAPY
In addition to diet therapy, most patients with intestinal failure require pharmacologic therapy.28 High stool or stoma effluent is most commonly treated with an antidiarrheal to increase transit time; diphenoxylate-atropine, loperamide, codeine sulfate, paregoric, and opium tincture are commonly prescribed (Table 5).27 In severe high-output states, a somatostatin analogue (eg, octreotide) may be added.29
Postoperative increases in gastric secretion may be countered by histamine 2 receptor antagonists and proton pump inhibitors, but long-term use of these drugs may lead to nutritional deficiencies and bacterial overgrowth in the small intestine.29 Bile acid sequestrants (in cases of distal ileal resection) and pancreatic enzymes target fat malabsorption, resultant cases of choleretic diarrhea, deficiency of essential fatty acids, kidney stones, and deficiency of fat-soluble vitamins.29 Probiotics and antibiotics can also be given for prevention and treatment of small-intestinal bacterial overgrowth.29
When traditional dietary modification and medical therapy fail to achieve nutritional homeostasis, another option to consider is a glucagon-like peptide-2 analogue to enhance intestinal adaptation.30 Produced in the native distal ileum and colon, glucagon-like peptide 2 moderates the rate of gastric emptying and small-bowel transit and enhances epithelial cell proliferation, thereby promoting intestinal adaptation.30 Further, a randomized controlled trial of 83 patients reported efficacy of these agents in reducing parenteral nutrition requirements in patients with intestinal failure.31
Hence, in patients with intestinal failure who have increased stoma effluent, drug therapy may play an important role in maintaining fluid and nutritional homeostasis.
THE ROLE OF PARENTERAL NUTRITION IN INTESTINAL FAILURE
Despite the best efforts of an intestinal rehabilitation program, not all patients gain nutritional autonomy.32 Physiologic, psychological, social, and economic factors may contribute to dependence on parenteral nutrition.32 Currently, more than 40,000 US patients depend on it for survival.33
The need for short-term or long-term parenteral nutrition is determined by the patient’s medical needs.33 Patients requiring short-term parenteral nutrition (2–6 weeks) include those whose bowel function has not returned to normal postoperatively, and those who were severely malnourished preoperatively.34 Patients needing it long-term (from months to years to lifelong) are those with gastrointestinal dysmotility and short-bowel syndrome due to extensive bowel resections.33
Complications of parenteral nutrition
Catheter-related bloodstream infection is the most common complication and cause of hospitalization. Infection can be localized to the exit site or tunnel or can be systemic (eg, line sepsis).35Staphylococcus aureus and coagulase-negative staphylococci are most often implicated in catheter infection.35 When possible, catheter salvage is desirable, but the central venous catheter must be removed in cases of tunnel infection, port abscess, septic shock, paired blood cultures positive for fungi or highly virulent bacteria, endocarditis, septic thrombosis, and other conditions.35,36
Liver disease is a serious complication of long-term parenteral nutrition and may occur in up to 55% of patients on therapy for more than 2 years; it carries a mortality rate of 15%.37
Risk factors include younger age and use of excessive carbohydrate and fat compositions, mainly soybean-oil–based lipid emulsions.37 However, fish-oil–based lipid emulsions have recently shown promise in preventing and reversing parenteral nutrition-associated liver failure and cholestasis, especially in a pediatric population.38
Catheter thrombosis may occur in up to 30% of patients on long-term parenteral nutrition.39 However, this risk is reduced with appropriate positioning of the catheter tip in the mid or lower superior vena cava.37 Treatment of thrombosis of the central access includes either anticoagulation or thrombolysis.37
Hence, appropriate and timely follow-up of patients on parenteral nutrition is essential in reducing associated complications. Monitoring weight, fluid status, serum glucose, and patency of central access are critical to ensure that the patient maintains nutritional status effectively.40 To prevent complications, a specialized nutritional support team should monitor the patient’s parenteral nutrition both in the hospital and at home.
RECONSTRUCTIVE SURGERY
Patients with intestinal failure due to short- bowel syndrome should be considered for reconstructive surgery during different phases of the adaptation process. Options include reversed-segment procedures, stricturoplasty, bowel-lengthening procedures (eg, the Bianchi procedure), and serial transverse enteroplasty.41,42 If reconstructive surgery is ineffective, referral to an intestinal transplant program should be considered.
INTESTINAL AND MULTIVISCERAL TRANSPLANT
For patients who develop permanent intestinal failure and require lifelong parenteral nutrition, and for patients who experience significant complications of parenteral nutrition, such as infections and liver disease,43 intestinal transplant has emerged as a way to restore clinical nutritional autonomy.44 In one study, the 1-year survival rate after intestinal transplant was approximately 90%.44
There are currently three transplant procedures: isolated intestine transplant, combined liver-intestine transplant, and multivisceral transplant with or without a liver, depending on the presence of parenteral nutrition-associated liver disease.42,45 Close postoperative care is required to help the patient transition from parenteral to enteral nutrition.42 An intestinal rehabilitation team is equipped to provide this level of postoperative care.42
Intestinal and multivisceral transplant gained momentum in the early 1960s in preclinical and clinical studies.46,47 Since that time, the field has experienced remarkable advances due to standardization of surgical techniques, novel immunosuppressive therapies and induction protocols, and improved postoperative care.48 With the advent of tacrolimus in 1989, the rates of allograft rejection improved significantly, and the field of transplant emerged as a potentially lifesaving therapy for patients with permanent intestinal failure.48
Since 1990, more than 2,300 intestinal transplant procedures have been performed for various etiologies of intestinal failure, with short-bowel syndrome being the most common.49

The indications for intestinal transplant approved by the US Centers for Medicare and Medicaid services are detailed in Table 6.50 Despite ongoing challenges of graft rejection and maintenance immunosuppression, posttransplant quality-of-life questionnaires have indicated a significant improvement in functional status and a decrease in depressive symptoms.51 As such, it is evident that intestinal and multivisceral transplant offers substantial promise in restoring a patient’s quality of life and nutritional status.
- Parekh NR, Steiger E. Short bowel syndrome. Curr Treat Options Gastroenterol 2007; 10:10–23.
- Williamson RC. Intestinal adaptation (first of two parts). Structural, functional and cytokinetic changes. N Engl J Med 1978; 298:1393–1402.
- Vantini I, Benini L, Bonfante F, et al. Survival rate and prognostic factors in patients with intestinal failure. Dig Liver Dis 2004; 36:46–55.
- Abu-Elmagd KM, Bond GJ, Matarese L, et al. Gut rehabilitation and intestinal transplantation. Therapy 2005; 2:853–864.
- Nightingale JMD, Lennard-Jones JE. The short bowel syndrome: what’s new and old? Dig Dis 1993; 11:12–31.
- Parekh N, Seidner D, Steiger E. Managing short bowel syndrome: making the most of what the patient still has. Cleve Clin J Med 2005; 72:833–838.
- Wales PW. Surgical therapy for short bowel syndrome. Pediatr Surg Int 2004; 20:647–657.
- Parekh NR, Steiger E, Seidner DL. Determination of residual bowel length via surgical, radiological or historical data in patients with short bowel syndrome and intestinal failure (abstract). Gastroenterology 2006; 130:A605.
- Shatnawei A, Parekh NR, Rhoda KM, et al. Intestinal failure management at the Cleveland Clinic. Arch Surg 2010; 145:521–527.
- Kelly DG, Tappenden KA, Winkler MF. Short bowel syndrome: highlights of patient management, quality of life, and survival. JPEN J Parenter Enteral Nutr 2014; 38:427–437.
- Efsen E, Jeppesen PB. Modern treatment of adult short bowel syndrome patients. Minerva Gastroenterol Dietol 2011; 57:405–417.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Dowling RH, Booth DB. Functional compensation after small bowel resection in man. Lancet 1996; 2:146–147.
- Tappenden KA. Intestinal adaptation following resection. JPEN J Parenter Enteral Nutr 2014; 38(suppl 1):23S–31S.
- Friedman HI, Chandler JG, Peck CC, Nemeth TJ, Odum SK. Alterations in intestinal structure, fat absorption and body weight after intestinal bypass for morbid obesity. Surg Gynecol Obstet 1978; 146:757–767.
- O’Keefe SJ, Buchman AL, Fishbein TM, Jeejeebhoy KN, Jeppesen PB, Shaffer J. Short bowel syndrome and intestinal failure: consensus definitions and overview. Clin Gastroenterol Hepatol 2006; 4:6–10.
- Lennard-Jones JE. Review article: practical management of the short bowel. Aliment Pharmacol Ther 1994; 8:563–577.
- Goulet O, Colomb-Jung V, Joly F. Role of the colon in short bowel syndrome and intestinal transplantation. J Pediatr Gastroenterol Nutr 2009; 48(suppl 2):S66–S71.
- Jeppesen PB, Mortensen PB. Colonic digestion and absorption of energy from carbohydrates and medium-chain fat in small bowel failure. JPEN J Parenter Enteral Nutr 1999; 23(suppl 5):S101–S105.
- Buchman AL. Etiology and initial management of short bowel syndrome. Gastroenterology 2006; 130(suppl 1):S5–S15.
- Donohoe CL, Reynolds JV. Short bowel syndrome. Surgeon 2010; 8:270–279.
- Gong JF, Zhu WM, Yu WK, Li N, Li JS. Role of enteral nutrition in adult short bowel syndrome undergoing intestinal rehabilitation: the long-term outcome. Asia Pac J Clin Nutr 2009; 18:155–163.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol 2002; 34:207–220.
- Byrne TA, Wilmore DW, Iyer K, et al. Growth hormone, glutamine, and an optimal diet reduces parenteral nutrition in patients with short bowel syndrome: a prospective, randomized, placebo-controlled, double-blind clinical trial. Ann Surg 2005; 242:655–661.
- Matarese LE, Steiger E. Dietary and medical management of short bowel syndrome in adult patients. J Clin Gastroenterol 2006; 40(suppl 2):S85–S93.
- Nordgaard I, Hansen BS, Mortensen PB. Colon as a digestive organ in patients with short bowel. Lancet 1994; 343:373–376.
- Ukleja A, Scolapio JS, Buchman AL. Nutritional management of short bowel syndrome. Semin Gastrointest Dis 2002; 13:161–168.
- Jeejeebhoy KN. Short bowel syndrome: a nutritional and medical approach. CMAJ 2002; 166:1297–1302.
- Seetharam P, Rodrigues G. Short bowel syndrome: a review of management options. Saudi J Gastroenterol 2011; 17:229–235.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 2011; 60:902–914.
- Pironi L, Joly F, Forbes A, et al; Home Artificial Nutrition & Chronic Intestinal Failure Working Group of the European Society for Clinical Nutrition and Metabolism (ESPEN). Long-term follow-up of patients on home parenteral nutrition in Europe: implications for intestinal transplantation. Gut 2011; 60:17–25.
- Ekema G, Milianti S, Boroni G. Total parenteral nutrition in patients with short bowel syndrome. Minerva Pediatr 2009; 61:283–291.
- Messing B, Crenn P, Beau P, Boutron-Ruault MC, Rambaud JC, Matuchansky C. Long-term survival and parenteral nutrition dependence in adult patients with the short bowel syndrome. Gastroenterology 1999; 117:1043–1050.
- Opilla M. Epidemiology of bloodstream infection associated with parenteral nutrition. Am J Infect Control 2008; 36:S173.e5–e8.
- Ukleja A, Romano MM. Complications of parenteral nutrition. Gastroenterol Clin North Am 2007; 36:23–46.
- Buchman AI, Iyer K, Fryer J. Parenteral nutrition-associated liver disease and the role for isolated intestine and intestine/liver transplantation. Hepatology 2006; 43:9–19.
- Fürst P, Kuhn KS. Fish oil emulsions: what benefits can they bring? Clin Nutr 2000; 19:7–14.
- Verso M, Agnelli G. Venous thromboembolism associated with long-term use of central venous catheters in cancer patients. J Clin Oncol 2003; 21:3665–3675.
- McMahon MM, Nystrom E, Braunschweig C, Miles J, Compher C; American Society for Parenteral and Enteral Nutrition (ASPEN) Board of Directors; American Society for Parenteral and Enteral Nutrition. American Society of Parenteral and Enteral Nutrition (ASPEN) Board of Directors. A.S.P.E.N. clinical guidelines: nutrition support of adult patients with hyperglycemia. JPEN J Parenter Enteral Nutr 2013; 37:23–36.
- Kim HB, Fauza D, Garza J, Oh JT, Nurko S, Jaksic T. Serial transverse enteroplasty (STEP): a novel bowel lengthening procedure. J Pediatr Surg 2003; 38:425–429.
- King B, Carlson G, Khalil BA, Morabito A. Intestinal bowel lengthening in children with short bowel syndrome: systematic review of the Bianchi and STEP procedures. World J Surg 2013; 37:694–704.
- Matarese LE, O’Keefe SJ, Kandil HM, Costa G, Abu-Elmagd KM. Short bowel syndrome: clinical guidelines for nutrition management. Nutr Clin Pract 2005; 20:493–502.
- Abu-Elmagd KM, Costa G, Bond GJ, et al. Five hundred intestinal and multivisceral transplantations at a single center: major advances with new challenges. Ann Surg 2009; 250:567–581.
- Abu-Elmagd K. The concept of gut rehabilitation and the future of visceral transplantation. Nat Rev Gastroenterol Hepatol 2015; 12:108–120.
- Lillehei RC, Goott B, Miller FA. The physiological response of the small bowel of the dog to ischemia including prolonged in vitro preservation of the bowel with successful replacement and survival. Ann Surg 1959; 150:543–559.
- Starzl TE, Kaupp HA. Mass homotransplantation of abdominal organs in dogs. Surg Forum 1960; 11:28–30.
- O’Keefe SJ, Matarese L. Small bowel transplantation. Curr Gastroenterol Rep 2006; 8:360–366.
- Horslen SP. Optimal management of the post-intestinal transplant patient. Gastroenterology 2006; 130(suppl 1):S163–S169.
- Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003; 124:1111–1134.
- DiMartini A, Rovera GM, Graham TO, et al. Quality of life after small intestinal transplantation and among home parenteral nutrition patients. JPEN J Parenter Enteral Nutr 1998; 22:357–362.
Intestinal failure, the inability of the gut to maintain nutritional homeostasis,1 is a complication of vascular thrombosis, inflammatory bowel disease, radiation enteritis, obstruction, and other conditions, and of removing segments of the small and large intestines in response to these diseases.1,2 Imbalances of fluids and electrolytes, dehydration, malabsorption, vitamin and mineral deficiencies, chronic diarrhea, and increased ostomy output contribute to a decline in the quality of life and in the survival rate in these patients.2,3
Referral to an intestinal rehabilitation program that combines gastroenterology, nutrition, pharmacy, nursing, and social work can improve nutritional status and quality of life.4 Whenever possible, the goal of rehabilitation is nutritional autonomy, helping the patient make the transition to an independent oral diet.4 In selected patients in whom rehabilitation is not effective, intestinal transplant may be an option.
In this article, we review the intestinal adaptations that follow surgical resection and provide an update on intestinal rehabilitation techniques such as dietary modification, drug therapy, and parenteral nutrition. We also review experience with intestinal transplant in patients with intestinal failure.
INTESTINAL FAILURE
Intestinal failure results from reduction in enterocyte cell mass, obstruction, dysmotility, surgical resection, congenital defects, or disease-associated loss of absorption with suboptimal nutritional autonomy.5 Patients often suffer from extensive nutrient, electrolyte, and fluid abnormalities proportional to the remnant length and part of the intestine removed.5
Epidemiologic studies have demonstrated that short-bowel syndrome is the most common cause of intestinal failure in adults and children.6,7 Short-bowel syndrome is defined as a small-bowel length less than 200 cm, most commonly from extensive resections for inflammatory bowel disease.6 In children, the syndrome is also defined by a residual small-bowel length of less than 25% expected for gestational age.7
Table 1 lists the frequencies of the underlying disorders leading to intestinal failure or short-bowel syndrome in one series.8
INTESTINAL ADAPTATION
The gastrointestinal tract is the only organ for nutrient, fluid, and electrolyte absorption.9 Every day, 8 to 9 L of fluids and secretions reach the small intestine, comprising about 2 to 3 L of oral fluids, 1 L of saliva, 2 L of gastric juices, 1 L of bile, and 2 L of pancreatic juices.9 Approximately 7 to 8 L are reabsorbed by the small intestine and 1 to 2 L by the colon.9
Although carbohydrates, lipids, and proteins are absorbed through the entire small intestine and colon, site-specific digestion and absorption of different nutrients occur in different parts of the gastrointestinal tract.10 Also, certain nutrients may need site-specific receptors or transporters for their absorption,10 for example:
- Iron in the duodenum and proximal jejunum1
- Lactose in the brush border membrane of the jejunum and proximal ileum, where most of the enzyme lactase is present
- Vitamin B12 and bile salts in the distal ileum.
Hence, resection of a specific part of the intestine may predict deficiencies the patient may encounter after surgery.
The diarrhea that occurs in short-bowel syndrome may be due partly to loss of neurohumoral mediators that govern gastrointestinal transit time, most importantly cholecystokinin, peptide YY, and glucagon-like peptide 1.11 After contact with lipid- or protein-rich nutrients, cholecystokinin is released from the proximal small intestine, which decreases the gastric emptying to maximize nutrient digestion.12 Additionally, release of peptide YY and glucagon-like peptide 1 from the ileal L cells decreases gastric and intestinal motility. These mediators prolong gastrointestinal transit, increase nutrient processing time, and enhance absorption.12
After massive intestinal resection, the remnant bowel undergoes physiologic and functional adaptation to maintain nutritional homeostasis.13 Enterocytes express membrane-bound transporters and undergo accelerated cell division to enhance the absorptive surface area.13 Intestinal hypertrophy, which includes an increase in villous diameter and crypt height, continues for 2 years or more after intestinal resection, leading to greater absorptive surface area.14 It is estimated that villous height may increase by as much as 80%, illustrating a dynamic process in response to intestinal stress.15
Luminal nutrients are essential to the stimulation of enterocyte cells through paracrine mechanisms as well as through the up-regulation of colonic peptide transporter PepT1.15 Furthermore, gut motility is initially decreased in order to increase the concentration of local luminal growth factors.16
Other factors that may affect intestinal adaptation are the length of the residual colon and small intestine, enteral growth, and enterotropic factors.16 And especially in patients with short-bowel syndrome, complications such as malabsorption secondary to pancreatic insufficiency or rapid transit, excessive gastric acid secretion, bile acid wasting due to terminal ileum resection, and bacterial overgrowth in the small intestine result in worsened nutritional status and poor quality of life.16
Key factors that affect the degree of nutritional deficiencies
The degree of nutritional deficiencies and fluid and electrolyte imbalances depends on the length and location of resection and whether the colon is still continuous with the small intestine.17 Normal small-bowel length in adults is highly variable and can be up to 600 cm. Malnutrition after surgical resection usually occurs when more than three-fourths of intestinal tissue is removed.17 However, because of intestinal adaptation, patients with 50% of remnant small bowel may be able to achieve nutritional autonomy.18 Furthermore, because absorption of nutrients occurs primarily in the first 150 cm of the small intestine, resections of this anatomic region have the highest probability of resulting in malnutrition.18
After extensive intestinal resection, absorption of water and electrolytes is better and intestinal transit time is longer if the colon is still continuous with the rest of the gastrointestinal system.19 Approximately 100 cm of remnant intestinal tissue without colonic continuity or 60 cm with colonic continuity is needed to ensure the possibility of nutritional autonomy and independence from parenteral nutrition.19 Severe malnutrition and fluid and electrolyte imbalances can be prevented by appropriate and timely multidisciplinary care and early referral for intestinal rehabilitation.
INTESTINAL REHABILITATION AND NUTRITIONAL AUTONOMY
The aim of intestinal rehabilitation is to improve quality of life by reversing malnutrition and promoting nutritional autonomy, ie, independence from parenteral nutrition (Table 2).20 The complex nature of intestinal failure necessitates collaboration of multiple specialists—gastroenterologists, surgeons, dietitians, nurses, psychiatrists or psychologists, pharmacists, and social workers.20
Although most patients with intestinal failure initially require parenteral nutrition to maintain nutritional homeostasis, progressive adaptation of the remnant intestine enables a transition to enteral nutrition.21 Stimulation of the remnant intestine by enteral feeding reduces the complications of parenteral nutrition and encourages intestinal adaptation.21
Outpatient participation in an intestinal rehabilitation program can facilitate weaning from parenteral nutrition. Patients are monitored and supported during dietary modification, pharmacologic interventions, and reconstructive surgeries.21 A study of 61 patients with short-bowel syndrome undergoing a 3-week program of intestinal rehabilitation (recombinant human growth hormone, glutamine, enteral nutrition, and parenteral nutrition) reported an overall survival rate of 95% with an 85% success rate in weaning from parenteral nutrition during a mean follow-up of 50 (± 24) months.22 Permanent dependence on parenteral nutrition despite rehabilitation was predicted by length of the small bowel less than 100 cm and by the absence of terminal ileum and colon.22
Permanent intestinal failure, defined by the inability to wean from parenteral nutrition and restore nutrition autonomy, may require early referral for evaluation for intestinal and multivisceral transplant. Early referral improves survival rates, possibly because of fewer complications from parenteral nutrition.4
DIETARY MODIFICATION
Dietary modification is the single most effective means of weaning patients safely from parenteral nutrition (Table 3).23,24 Small, frequent feedings help reduce symptoms associated with rapid intestinal transit and increase the activity of luminal growth factors.23 Likewise, limits on intake of simple sugars, stimulants such as caffeine or insoluble fiber, and hypo- or hypertonic fluids decrease intestinal losses and the risk of dehydration.23 Low sugar loads also aim to reduce the occurrence of d-lactic acidosis and bacterial overgrowth in the small intestine.23 Patients who cannot maintain positive fluid balance may require standardized oral rehydration (Table 4) to improve absorption by way of the sodium-glucose coupled transporters at the brush border membrane, or they may require intravenous fluid supplementation.25
Colonic continuity
Other dietary recommendations depend on colonic continuity. In 1994, Nordgaard et al26 compared the effects of high-carbohydrate and high-fat diets in eight patients with colonic continuity and six patients with jejunostomies. The authors noted that a high-carbohydrate diet (60% carbohydrate, 20% fat) reduced fecal loss of energy and increased energy absorption in patients with colonic continuity. However, patients with an end-jejunostomy experienced equal fecal losses of carbohydrates and fat proportional to the amount consumed. The authors concluded that the presence of colonic bacteria promoted carbohydrate salvage, ie, the fermentation of malabsorbed carbohydrates to easily absorbed short-chain fatty acids.26
The colon can salvage as much as 1,000 kcal/day in patients with less than 200 cm of small bowel, and the presence of at least 50% of colon in continuity has been shown to reduce parenteral nutrition requirements by half in patients with less than 100 cm of small bowel.27 As a result, a diet high in complex carbohydrates and soluble fiber supplements is recommended in cases of preserved colon to promote adaptation and nutritional autonomy.27
Another aim of a high-carbohydrate, low- fat diet is to prevent calcium oxalate-related nephrolithiasis and choleretic diarrhea.26
In summary, patients with short-bowel syndrome with or without colonic continuity need different dietary regimens to attain nutritional autonomy.
DRUG THERAPY
In addition to diet therapy, most patients with intestinal failure require pharmacologic therapy.28 High stool or stoma effluent is most commonly treated with an antidiarrheal to increase transit time; diphenoxylate-atropine, loperamide, codeine sulfate, paregoric, and opium tincture are commonly prescribed (Table 5).27 In severe high-output states, a somatostatin analogue (eg, octreotide) may be added.29
Postoperative increases in gastric secretion may be countered by histamine 2 receptor antagonists and proton pump inhibitors, but long-term use of these drugs may lead to nutritional deficiencies and bacterial overgrowth in the small intestine.29 Bile acid sequestrants (in cases of distal ileal resection) and pancreatic enzymes target fat malabsorption, resultant cases of choleretic diarrhea, deficiency of essential fatty acids, kidney stones, and deficiency of fat-soluble vitamins.29 Probiotics and antibiotics can also be given for prevention and treatment of small-intestinal bacterial overgrowth.29
When traditional dietary modification and medical therapy fail to achieve nutritional homeostasis, another option to consider is a glucagon-like peptide-2 analogue to enhance intestinal adaptation.30 Produced in the native distal ileum and colon, glucagon-like peptide 2 moderates the rate of gastric emptying and small-bowel transit and enhances epithelial cell proliferation, thereby promoting intestinal adaptation.30 Further, a randomized controlled trial of 83 patients reported efficacy of these agents in reducing parenteral nutrition requirements in patients with intestinal failure.31
Hence, in patients with intestinal failure who have increased stoma effluent, drug therapy may play an important role in maintaining fluid and nutritional homeostasis.
THE ROLE OF PARENTERAL NUTRITION IN INTESTINAL FAILURE
Despite the best efforts of an intestinal rehabilitation program, not all patients gain nutritional autonomy.32 Physiologic, psychological, social, and economic factors may contribute to dependence on parenteral nutrition.32 Currently, more than 40,000 US patients depend on it for survival.33
The need for short-term or long-term parenteral nutrition is determined by the patient’s medical needs.33 Patients requiring short-term parenteral nutrition (2–6 weeks) include those whose bowel function has not returned to normal postoperatively, and those who were severely malnourished preoperatively.34 Patients needing it long-term (from months to years to lifelong) are those with gastrointestinal dysmotility and short-bowel syndrome due to extensive bowel resections.33
Complications of parenteral nutrition
Catheter-related bloodstream infection is the most common complication and cause of hospitalization. Infection can be localized to the exit site or tunnel or can be systemic (eg, line sepsis).35Staphylococcus aureus and coagulase-negative staphylococci are most often implicated in catheter infection.35 When possible, catheter salvage is desirable, but the central venous catheter must be removed in cases of tunnel infection, port abscess, septic shock, paired blood cultures positive for fungi or highly virulent bacteria, endocarditis, septic thrombosis, and other conditions.35,36
Liver disease is a serious complication of long-term parenteral nutrition and may occur in up to 55% of patients on therapy for more than 2 years; it carries a mortality rate of 15%.37
Risk factors include younger age and use of excessive carbohydrate and fat compositions, mainly soybean-oil–based lipid emulsions.37 However, fish-oil–based lipid emulsions have recently shown promise in preventing and reversing parenteral nutrition-associated liver failure and cholestasis, especially in a pediatric population.38
Catheter thrombosis may occur in up to 30% of patients on long-term parenteral nutrition.39 However, this risk is reduced with appropriate positioning of the catheter tip in the mid or lower superior vena cava.37 Treatment of thrombosis of the central access includes either anticoagulation or thrombolysis.37
Hence, appropriate and timely follow-up of patients on parenteral nutrition is essential in reducing associated complications. Monitoring weight, fluid status, serum glucose, and patency of central access are critical to ensure that the patient maintains nutritional status effectively.40 To prevent complications, a specialized nutritional support team should monitor the patient’s parenteral nutrition both in the hospital and at home.
RECONSTRUCTIVE SURGERY
Patients with intestinal failure due to short- bowel syndrome should be considered for reconstructive surgery during different phases of the adaptation process. Options include reversed-segment procedures, stricturoplasty, bowel-lengthening procedures (eg, the Bianchi procedure), and serial transverse enteroplasty.41,42 If reconstructive surgery is ineffective, referral to an intestinal transplant program should be considered.
INTESTINAL AND MULTIVISCERAL TRANSPLANT
For patients who develop permanent intestinal failure and require lifelong parenteral nutrition, and for patients who experience significant complications of parenteral nutrition, such as infections and liver disease,43 intestinal transplant has emerged as a way to restore clinical nutritional autonomy.44 In one study, the 1-year survival rate after intestinal transplant was approximately 90%.44
There are currently three transplant procedures: isolated intestine transplant, combined liver-intestine transplant, and multivisceral transplant with or without a liver, depending on the presence of parenteral nutrition-associated liver disease.42,45 Close postoperative care is required to help the patient transition from parenteral to enteral nutrition.42 An intestinal rehabilitation team is equipped to provide this level of postoperative care.42
Intestinal and multivisceral transplant gained momentum in the early 1960s in preclinical and clinical studies.46,47 Since that time, the field has experienced remarkable advances due to standardization of surgical techniques, novel immunosuppressive therapies and induction protocols, and improved postoperative care.48 With the advent of tacrolimus in 1989, the rates of allograft rejection improved significantly, and the field of transplant emerged as a potentially lifesaving therapy for patients with permanent intestinal failure.48
Since 1990, more than 2,300 intestinal transplant procedures have been performed for various etiologies of intestinal failure, with short-bowel syndrome being the most common.49
The indications for intestinal transplant approved by the US Centers for Medicare and Medicaid services are detailed in Table 6.50 Despite ongoing challenges of graft rejection and maintenance immunosuppression, posttransplant quality-of-life questionnaires have indicated a significant improvement in functional status and a decrease in depressive symptoms.51 As such, it is evident that intestinal and multivisceral transplant offers substantial promise in restoring a patient’s quality of life and nutritional status.
Intestinal failure, the inability of the gut to maintain nutritional homeostasis,1 is a complication of vascular thrombosis, inflammatory bowel disease, radiation enteritis, obstruction, and other conditions, and of removing segments of the small and large intestines in response to these diseases.1,2 Imbalances of fluids and electrolytes, dehydration, malabsorption, vitamin and mineral deficiencies, chronic diarrhea, and increased ostomy output contribute to a decline in the quality of life and in the survival rate in these patients.2,3
Referral to an intestinal rehabilitation program that combines gastroenterology, nutrition, pharmacy, nursing, and social work can improve nutritional status and quality of life.4 Whenever possible, the goal of rehabilitation is nutritional autonomy, helping the patient make the transition to an independent oral diet.4 In selected patients in whom rehabilitation is not effective, intestinal transplant may be an option.
In this article, we review the intestinal adaptations that follow surgical resection and provide an update on intestinal rehabilitation techniques such as dietary modification, drug therapy, and parenteral nutrition. We also review experience with intestinal transplant in patients with intestinal failure.
INTESTINAL FAILURE
Intestinal failure results from reduction in enterocyte cell mass, obstruction, dysmotility, surgical resection, congenital defects, or disease-associated loss of absorption with suboptimal nutritional autonomy.5 Patients often suffer from extensive nutrient, electrolyte, and fluid abnormalities proportional to the remnant length and part of the intestine removed.5
Epidemiologic studies have demonstrated that short-bowel syndrome is the most common cause of intestinal failure in adults and children.6,7 Short-bowel syndrome is defined as a small-bowel length less than 200 cm, most commonly from extensive resections for inflammatory bowel disease.6 In children, the syndrome is also defined by a residual small-bowel length of less than 25% expected for gestational age.7
Table 1 lists the frequencies of the underlying disorders leading to intestinal failure or short-bowel syndrome in one series.8
INTESTINAL ADAPTATION
The gastrointestinal tract is the only organ for nutrient, fluid, and electrolyte absorption.9 Every day, 8 to 9 L of fluids and secretions reach the small intestine, comprising about 2 to 3 L of oral fluids, 1 L of saliva, 2 L of gastric juices, 1 L of bile, and 2 L of pancreatic juices.9 Approximately 7 to 8 L are reabsorbed by the small intestine and 1 to 2 L by the colon.9
Although carbohydrates, lipids, and proteins are absorbed through the entire small intestine and colon, site-specific digestion and absorption of different nutrients occur in different parts of the gastrointestinal tract.10 Also, certain nutrients may need site-specific receptors or transporters for their absorption,10 for example:
- Iron in the duodenum and proximal jejunum1
- Lactose in the brush border membrane of the jejunum and proximal ileum, where most of the enzyme lactase is present
- Vitamin B12 and bile salts in the distal ileum.
Hence, resection of a specific part of the intestine may predict deficiencies the patient may encounter after surgery.
The diarrhea that occurs in short-bowel syndrome may be due partly to loss of neurohumoral mediators that govern gastrointestinal transit time, most importantly cholecystokinin, peptide YY, and glucagon-like peptide 1.11 After contact with lipid- or protein-rich nutrients, cholecystokinin is released from the proximal small intestine, which decreases the gastric emptying to maximize nutrient digestion.12 Additionally, release of peptide YY and glucagon-like peptide 1 from the ileal L cells decreases gastric and intestinal motility. These mediators prolong gastrointestinal transit, increase nutrient processing time, and enhance absorption.12
After massive intestinal resection, the remnant bowel undergoes physiologic and functional adaptation to maintain nutritional homeostasis.13 Enterocytes express membrane-bound transporters and undergo accelerated cell division to enhance the absorptive surface area.13 Intestinal hypertrophy, which includes an increase in villous diameter and crypt height, continues for 2 years or more after intestinal resection, leading to greater absorptive surface area.14 It is estimated that villous height may increase by as much as 80%, illustrating a dynamic process in response to intestinal stress.15
Luminal nutrients are essential to the stimulation of enterocyte cells through paracrine mechanisms as well as through the up-regulation of colonic peptide transporter PepT1.15 Furthermore, gut motility is initially decreased in order to increase the concentration of local luminal growth factors.16
Other factors that may affect intestinal adaptation are the length of the residual colon and small intestine, enteral growth, and enterotropic factors.16 And especially in patients with short-bowel syndrome, complications such as malabsorption secondary to pancreatic insufficiency or rapid transit, excessive gastric acid secretion, bile acid wasting due to terminal ileum resection, and bacterial overgrowth in the small intestine result in worsened nutritional status and poor quality of life.16
Key factors that affect the degree of nutritional deficiencies
The degree of nutritional deficiencies and fluid and electrolyte imbalances depends on the length and location of resection and whether the colon is still continuous with the small intestine.17 Normal small-bowel length in adults is highly variable and can be up to 600 cm. Malnutrition after surgical resection usually occurs when more than three-fourths of intestinal tissue is removed.17 However, because of intestinal adaptation, patients with 50% of remnant small bowel may be able to achieve nutritional autonomy.18 Furthermore, because absorption of nutrients occurs primarily in the first 150 cm of the small intestine, resections of this anatomic region have the highest probability of resulting in malnutrition.18
After extensive intestinal resection, absorption of water and electrolytes is better and intestinal transit time is longer if the colon is still continuous with the rest of the gastrointestinal system.19 Approximately 100 cm of remnant intestinal tissue without colonic continuity or 60 cm with colonic continuity is needed to ensure the possibility of nutritional autonomy and independence from parenteral nutrition.19 Severe malnutrition and fluid and electrolyte imbalances can be prevented by appropriate and timely multidisciplinary care and early referral for intestinal rehabilitation.
INTESTINAL REHABILITATION AND NUTRITIONAL AUTONOMY
The aim of intestinal rehabilitation is to improve quality of life by reversing malnutrition and promoting nutritional autonomy, ie, independence from parenteral nutrition (Table 2).20 The complex nature of intestinal failure necessitates collaboration of multiple specialists—gastroenterologists, surgeons, dietitians, nurses, psychiatrists or psychologists, pharmacists, and social workers.20
Although most patients with intestinal failure initially require parenteral nutrition to maintain nutritional homeostasis, progressive adaptation of the remnant intestine enables a transition to enteral nutrition.21 Stimulation of the remnant intestine by enteral feeding reduces the complications of parenteral nutrition and encourages intestinal adaptation.21
Outpatient participation in an intestinal rehabilitation program can facilitate weaning from parenteral nutrition. Patients are monitored and supported during dietary modification, pharmacologic interventions, and reconstructive surgeries.21 A study of 61 patients with short-bowel syndrome undergoing a 3-week program of intestinal rehabilitation (recombinant human growth hormone, glutamine, enteral nutrition, and parenteral nutrition) reported an overall survival rate of 95% with an 85% success rate in weaning from parenteral nutrition during a mean follow-up of 50 (± 24) months.22 Permanent dependence on parenteral nutrition despite rehabilitation was predicted by length of the small bowel less than 100 cm and by the absence of terminal ileum and colon.22
Permanent intestinal failure, defined by the inability to wean from parenteral nutrition and restore nutrition autonomy, may require early referral for evaluation for intestinal and multivisceral transplant. Early referral improves survival rates, possibly because of fewer complications from parenteral nutrition.4
DIETARY MODIFICATION
Dietary modification is the single most effective means of weaning patients safely from parenteral nutrition (Table 3).23,24 Small, frequent feedings help reduce symptoms associated with rapid intestinal transit and increase the activity of luminal growth factors.23 Likewise, limits on intake of simple sugars, stimulants such as caffeine or insoluble fiber, and hypo- or hypertonic fluids decrease intestinal losses and the risk of dehydration.23 Low sugar loads also aim to reduce the occurrence of d-lactic acidosis and bacterial overgrowth in the small intestine.23 Patients who cannot maintain positive fluid balance may require standardized oral rehydration (Table 4) to improve absorption by way of the sodium-glucose coupled transporters at the brush border membrane, or they may require intravenous fluid supplementation.25
Colonic continuity
Other dietary recommendations depend on colonic continuity. In 1994, Nordgaard et al26 compared the effects of high-carbohydrate and high-fat diets in eight patients with colonic continuity and six patients with jejunostomies. The authors noted that a high-carbohydrate diet (60% carbohydrate, 20% fat) reduced fecal loss of energy and increased energy absorption in patients with colonic continuity. However, patients with an end-jejunostomy experienced equal fecal losses of carbohydrates and fat proportional to the amount consumed. The authors concluded that the presence of colonic bacteria promoted carbohydrate salvage, ie, the fermentation of malabsorbed carbohydrates to easily absorbed short-chain fatty acids.26
The colon can salvage as much as 1,000 kcal/day in patients with less than 200 cm of small bowel, and the presence of at least 50% of colon in continuity has been shown to reduce parenteral nutrition requirements by half in patients with less than 100 cm of small bowel.27 As a result, a diet high in complex carbohydrates and soluble fiber supplements is recommended in cases of preserved colon to promote adaptation and nutritional autonomy.27
Another aim of a high-carbohydrate, low- fat diet is to prevent calcium oxalate-related nephrolithiasis and choleretic diarrhea.26
In summary, patients with short-bowel syndrome with or without colonic continuity need different dietary regimens to attain nutritional autonomy.
DRUG THERAPY
In addition to diet therapy, most patients with intestinal failure require pharmacologic therapy.28 High stool or stoma effluent is most commonly treated with an antidiarrheal to increase transit time; diphenoxylate-atropine, loperamide, codeine sulfate, paregoric, and opium tincture are commonly prescribed (Table 5).27 In severe high-output states, a somatostatin analogue (eg, octreotide) may be added.29
Postoperative increases in gastric secretion may be countered by histamine 2 receptor antagonists and proton pump inhibitors, but long-term use of these drugs may lead to nutritional deficiencies and bacterial overgrowth in the small intestine.29 Bile acid sequestrants (in cases of distal ileal resection) and pancreatic enzymes target fat malabsorption, resultant cases of choleretic diarrhea, deficiency of essential fatty acids, kidney stones, and deficiency of fat-soluble vitamins.29 Probiotics and antibiotics can also be given for prevention and treatment of small-intestinal bacterial overgrowth.29
When traditional dietary modification and medical therapy fail to achieve nutritional homeostasis, another option to consider is a glucagon-like peptide-2 analogue to enhance intestinal adaptation.30 Produced in the native distal ileum and colon, glucagon-like peptide 2 moderates the rate of gastric emptying and small-bowel transit and enhances epithelial cell proliferation, thereby promoting intestinal adaptation.30 Further, a randomized controlled trial of 83 patients reported efficacy of these agents in reducing parenteral nutrition requirements in patients with intestinal failure.31
Hence, in patients with intestinal failure who have increased stoma effluent, drug therapy may play an important role in maintaining fluid and nutritional homeostasis.
THE ROLE OF PARENTERAL NUTRITION IN INTESTINAL FAILURE
Despite the best efforts of an intestinal rehabilitation program, not all patients gain nutritional autonomy.32 Physiologic, psychological, social, and economic factors may contribute to dependence on parenteral nutrition.32 Currently, more than 40,000 US patients depend on it for survival.33
The need for short-term or long-term parenteral nutrition is determined by the patient’s medical needs.33 Patients requiring short-term parenteral nutrition (2–6 weeks) include those whose bowel function has not returned to normal postoperatively, and those who were severely malnourished preoperatively.34 Patients needing it long-term (from months to years to lifelong) are those with gastrointestinal dysmotility and short-bowel syndrome due to extensive bowel resections.33
Complications of parenteral nutrition
Catheter-related bloodstream infection is the most common complication and cause of hospitalization. Infection can be localized to the exit site or tunnel or can be systemic (eg, line sepsis).35Staphylococcus aureus and coagulase-negative staphylococci are most often implicated in catheter infection.35 When possible, catheter salvage is desirable, but the central venous catheter must be removed in cases of tunnel infection, port abscess, septic shock, paired blood cultures positive for fungi or highly virulent bacteria, endocarditis, septic thrombosis, and other conditions.35,36
Liver disease is a serious complication of long-term parenteral nutrition and may occur in up to 55% of patients on therapy for more than 2 years; it carries a mortality rate of 15%.37
Risk factors include younger age and use of excessive carbohydrate and fat compositions, mainly soybean-oil–based lipid emulsions.37 However, fish-oil–based lipid emulsions have recently shown promise in preventing and reversing parenteral nutrition-associated liver failure and cholestasis, especially in a pediatric population.38
Catheter thrombosis may occur in up to 30% of patients on long-term parenteral nutrition.39 However, this risk is reduced with appropriate positioning of the catheter tip in the mid or lower superior vena cava.37 Treatment of thrombosis of the central access includes either anticoagulation or thrombolysis.37
Hence, appropriate and timely follow-up of patients on parenteral nutrition is essential in reducing associated complications. Monitoring weight, fluid status, serum glucose, and patency of central access are critical to ensure that the patient maintains nutritional status effectively.40 To prevent complications, a specialized nutritional support team should monitor the patient’s parenteral nutrition both in the hospital and at home.
RECONSTRUCTIVE SURGERY
Patients with intestinal failure due to short- bowel syndrome should be considered for reconstructive surgery during different phases of the adaptation process. Options include reversed-segment procedures, stricturoplasty, bowel-lengthening procedures (eg, the Bianchi procedure), and serial transverse enteroplasty.41,42 If reconstructive surgery is ineffective, referral to an intestinal transplant program should be considered.
INTESTINAL AND MULTIVISCERAL TRANSPLANT
For patients who develop permanent intestinal failure and require lifelong parenteral nutrition, and for patients who experience significant complications of parenteral nutrition, such as infections and liver disease,43 intestinal transplant has emerged as a way to restore clinical nutritional autonomy.44 In one study, the 1-year survival rate after intestinal transplant was approximately 90%.44
There are currently three transplant procedures: isolated intestine transplant, combined liver-intestine transplant, and multivisceral transplant with or without a liver, depending on the presence of parenteral nutrition-associated liver disease.42,45 Close postoperative care is required to help the patient transition from parenteral to enteral nutrition.42 An intestinal rehabilitation team is equipped to provide this level of postoperative care.42
Intestinal and multivisceral transplant gained momentum in the early 1960s in preclinical and clinical studies.46,47 Since that time, the field has experienced remarkable advances due to standardization of surgical techniques, novel immunosuppressive therapies and induction protocols, and improved postoperative care.48 With the advent of tacrolimus in 1989, the rates of allograft rejection improved significantly, and the field of transplant emerged as a potentially lifesaving therapy for patients with permanent intestinal failure.48
Since 1990, more than 2,300 intestinal transplant procedures have been performed for various etiologies of intestinal failure, with short-bowel syndrome being the most common.49
The indications for intestinal transplant approved by the US Centers for Medicare and Medicaid services are detailed in Table 6.50 Despite ongoing challenges of graft rejection and maintenance immunosuppression, posttransplant quality-of-life questionnaires have indicated a significant improvement in functional status and a decrease in depressive symptoms.51 As such, it is evident that intestinal and multivisceral transplant offers substantial promise in restoring a patient’s quality of life and nutritional status.
- Parekh NR, Steiger E. Short bowel syndrome. Curr Treat Options Gastroenterol 2007; 10:10–23.
- Williamson RC. Intestinal adaptation (first of two parts). Structural, functional and cytokinetic changes. N Engl J Med 1978; 298:1393–1402.
- Vantini I, Benini L, Bonfante F, et al. Survival rate and prognostic factors in patients with intestinal failure. Dig Liver Dis 2004; 36:46–55.
- Abu-Elmagd KM, Bond GJ, Matarese L, et al. Gut rehabilitation and intestinal transplantation. Therapy 2005; 2:853–864.
- Nightingale JMD, Lennard-Jones JE. The short bowel syndrome: what’s new and old? Dig Dis 1993; 11:12–31.
- Parekh N, Seidner D, Steiger E. Managing short bowel syndrome: making the most of what the patient still has. Cleve Clin J Med 2005; 72:833–838.
- Wales PW. Surgical therapy for short bowel syndrome. Pediatr Surg Int 2004; 20:647–657.
- Parekh NR, Steiger E, Seidner DL. Determination of residual bowel length via surgical, radiological or historical data in patients with short bowel syndrome and intestinal failure (abstract). Gastroenterology 2006; 130:A605.
- Shatnawei A, Parekh NR, Rhoda KM, et al. Intestinal failure management at the Cleveland Clinic. Arch Surg 2010; 145:521–527.
- Kelly DG, Tappenden KA, Winkler MF. Short bowel syndrome: highlights of patient management, quality of life, and survival. JPEN J Parenter Enteral Nutr 2014; 38:427–437.
- Efsen E, Jeppesen PB. Modern treatment of adult short bowel syndrome patients. Minerva Gastroenterol Dietol 2011; 57:405–417.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Dowling RH, Booth DB. Functional compensation after small bowel resection in man. Lancet 1996; 2:146–147.
- Tappenden KA. Intestinal adaptation following resection. JPEN J Parenter Enteral Nutr 2014; 38(suppl 1):23S–31S.
- Friedman HI, Chandler JG, Peck CC, Nemeth TJ, Odum SK. Alterations in intestinal structure, fat absorption and body weight after intestinal bypass for morbid obesity. Surg Gynecol Obstet 1978; 146:757–767.
- O’Keefe SJ, Buchman AL, Fishbein TM, Jeejeebhoy KN, Jeppesen PB, Shaffer J. Short bowel syndrome and intestinal failure: consensus definitions and overview. Clin Gastroenterol Hepatol 2006; 4:6–10.
- Lennard-Jones JE. Review article: practical management of the short bowel. Aliment Pharmacol Ther 1994; 8:563–577.
- Goulet O, Colomb-Jung V, Joly F. Role of the colon in short bowel syndrome and intestinal transplantation. J Pediatr Gastroenterol Nutr 2009; 48(suppl 2):S66–S71.
- Jeppesen PB, Mortensen PB. Colonic digestion and absorption of energy from carbohydrates and medium-chain fat in small bowel failure. JPEN J Parenter Enteral Nutr 1999; 23(suppl 5):S101–S105.
- Buchman AL. Etiology and initial management of short bowel syndrome. Gastroenterology 2006; 130(suppl 1):S5–S15.
- Donohoe CL, Reynolds JV. Short bowel syndrome. Surgeon 2010; 8:270–279.
- Gong JF, Zhu WM, Yu WK, Li N, Li JS. Role of enteral nutrition in adult short bowel syndrome undergoing intestinal rehabilitation: the long-term outcome. Asia Pac J Clin Nutr 2009; 18:155–163.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol 2002; 34:207–220.
- Byrne TA, Wilmore DW, Iyer K, et al. Growth hormone, glutamine, and an optimal diet reduces parenteral nutrition in patients with short bowel syndrome: a prospective, randomized, placebo-controlled, double-blind clinical trial. Ann Surg 2005; 242:655–661.
- Matarese LE, Steiger E. Dietary and medical management of short bowel syndrome in adult patients. J Clin Gastroenterol 2006; 40(suppl 2):S85–S93.
- Nordgaard I, Hansen BS, Mortensen PB. Colon as a digestive organ in patients with short bowel. Lancet 1994; 343:373–376.
- Ukleja A, Scolapio JS, Buchman AL. Nutritional management of short bowel syndrome. Semin Gastrointest Dis 2002; 13:161–168.
- Jeejeebhoy KN. Short bowel syndrome: a nutritional and medical approach. CMAJ 2002; 166:1297–1302.
- Seetharam P, Rodrigues G. Short bowel syndrome: a review of management options. Saudi J Gastroenterol 2011; 17:229–235.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 2011; 60:902–914.
- Pironi L, Joly F, Forbes A, et al; Home Artificial Nutrition & Chronic Intestinal Failure Working Group of the European Society for Clinical Nutrition and Metabolism (ESPEN). Long-term follow-up of patients on home parenteral nutrition in Europe: implications for intestinal transplantation. Gut 2011; 60:17–25.
- Ekema G, Milianti S, Boroni G. Total parenteral nutrition in patients with short bowel syndrome. Minerva Pediatr 2009; 61:283–291.
- Messing B, Crenn P, Beau P, Boutron-Ruault MC, Rambaud JC, Matuchansky C. Long-term survival and parenteral nutrition dependence in adult patients with the short bowel syndrome. Gastroenterology 1999; 117:1043–1050.
- Opilla M. Epidemiology of bloodstream infection associated with parenteral nutrition. Am J Infect Control 2008; 36:S173.e5–e8.
- Ukleja A, Romano MM. Complications of parenteral nutrition. Gastroenterol Clin North Am 2007; 36:23–46.
- Buchman AI, Iyer K, Fryer J. Parenteral nutrition-associated liver disease and the role for isolated intestine and intestine/liver transplantation. Hepatology 2006; 43:9–19.
- Fürst P, Kuhn KS. Fish oil emulsions: what benefits can they bring? Clin Nutr 2000; 19:7–14.
- Verso M, Agnelli G. Venous thromboembolism associated with long-term use of central venous catheters in cancer patients. J Clin Oncol 2003; 21:3665–3675.
- McMahon MM, Nystrom E, Braunschweig C, Miles J, Compher C; American Society for Parenteral and Enteral Nutrition (ASPEN) Board of Directors; American Society for Parenteral and Enteral Nutrition. American Society of Parenteral and Enteral Nutrition (ASPEN) Board of Directors. A.S.P.E.N. clinical guidelines: nutrition support of adult patients with hyperglycemia. JPEN J Parenter Enteral Nutr 2013; 37:23–36.
- Kim HB, Fauza D, Garza J, Oh JT, Nurko S, Jaksic T. Serial transverse enteroplasty (STEP): a novel bowel lengthening procedure. J Pediatr Surg 2003; 38:425–429.
- King B, Carlson G, Khalil BA, Morabito A. Intestinal bowel lengthening in children with short bowel syndrome: systematic review of the Bianchi and STEP procedures. World J Surg 2013; 37:694–704.
- Matarese LE, O’Keefe SJ, Kandil HM, Costa G, Abu-Elmagd KM. Short bowel syndrome: clinical guidelines for nutrition management. Nutr Clin Pract 2005; 20:493–502.
- Abu-Elmagd KM, Costa G, Bond GJ, et al. Five hundred intestinal and multivisceral transplantations at a single center: major advances with new challenges. Ann Surg 2009; 250:567–581.
- Abu-Elmagd K. The concept of gut rehabilitation and the future of visceral transplantation. Nat Rev Gastroenterol Hepatol 2015; 12:108–120.
- Lillehei RC, Goott B, Miller FA. The physiological response of the small bowel of the dog to ischemia including prolonged in vitro preservation of the bowel with successful replacement and survival. Ann Surg 1959; 150:543–559.
- Starzl TE, Kaupp HA. Mass homotransplantation of abdominal organs in dogs. Surg Forum 1960; 11:28–30.
- O’Keefe SJ, Matarese L. Small bowel transplantation. Curr Gastroenterol Rep 2006; 8:360–366.
- Horslen SP. Optimal management of the post-intestinal transplant patient. Gastroenterology 2006; 130(suppl 1):S163–S169.
- Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003; 124:1111–1134.
- DiMartini A, Rovera GM, Graham TO, et al. Quality of life after small intestinal transplantation and among home parenteral nutrition patients. JPEN J Parenter Enteral Nutr 1998; 22:357–362.
- Parekh NR, Steiger E. Short bowel syndrome. Curr Treat Options Gastroenterol 2007; 10:10–23.
- Williamson RC. Intestinal adaptation (first of two parts). Structural, functional and cytokinetic changes. N Engl J Med 1978; 298:1393–1402.
- Vantini I, Benini L, Bonfante F, et al. Survival rate and prognostic factors in patients with intestinal failure. Dig Liver Dis 2004; 36:46–55.
- Abu-Elmagd KM, Bond GJ, Matarese L, et al. Gut rehabilitation and intestinal transplantation. Therapy 2005; 2:853–864.
- Nightingale JMD, Lennard-Jones JE. The short bowel syndrome: what’s new and old? Dig Dis 1993; 11:12–31.
- Parekh N, Seidner D, Steiger E. Managing short bowel syndrome: making the most of what the patient still has. Cleve Clin J Med 2005; 72:833–838.
- Wales PW. Surgical therapy for short bowel syndrome. Pediatr Surg Int 2004; 20:647–657.
- Parekh NR, Steiger E, Seidner DL. Determination of residual bowel length via surgical, radiological or historical data in patients with short bowel syndrome and intestinal failure (abstract). Gastroenterology 2006; 130:A605.
- Shatnawei A, Parekh NR, Rhoda KM, et al. Intestinal failure management at the Cleveland Clinic. Arch Surg 2010; 145:521–527.
- Kelly DG, Tappenden KA, Winkler MF. Short bowel syndrome: highlights of patient management, quality of life, and survival. JPEN J Parenter Enteral Nutr 2014; 38:427–437.
- Efsen E, Jeppesen PB. Modern treatment of adult short bowel syndrome patients. Minerva Gastroenterol Dietol 2011; 57:405–417.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Dowling RH, Booth DB. Functional compensation after small bowel resection in man. Lancet 1996; 2:146–147.
- Tappenden KA. Intestinal adaptation following resection. JPEN J Parenter Enteral Nutr 2014; 38(suppl 1):23S–31S.
- Friedman HI, Chandler JG, Peck CC, Nemeth TJ, Odum SK. Alterations in intestinal structure, fat absorption and body weight after intestinal bypass for morbid obesity. Surg Gynecol Obstet 1978; 146:757–767.
- O’Keefe SJ, Buchman AL, Fishbein TM, Jeejeebhoy KN, Jeppesen PB, Shaffer J. Short bowel syndrome and intestinal failure: consensus definitions and overview. Clin Gastroenterol Hepatol 2006; 4:6–10.
- Lennard-Jones JE. Review article: practical management of the short bowel. Aliment Pharmacol Ther 1994; 8:563–577.
- Goulet O, Colomb-Jung V, Joly F. Role of the colon in short bowel syndrome and intestinal transplantation. J Pediatr Gastroenterol Nutr 2009; 48(suppl 2):S66–S71.
- Jeppesen PB, Mortensen PB. Colonic digestion and absorption of energy from carbohydrates and medium-chain fat in small bowel failure. JPEN J Parenter Enteral Nutr 1999; 23(suppl 5):S101–S105.
- Buchman AL. Etiology and initial management of short bowel syndrome. Gastroenterology 2006; 130(suppl 1):S5–S15.
- Donohoe CL, Reynolds JV. Short bowel syndrome. Surgeon 2010; 8:270–279.
- Gong JF, Zhu WM, Yu WK, Li N, Li JS. Role of enteral nutrition in adult short bowel syndrome undergoing intestinal rehabilitation: the long-term outcome. Asia Pac J Clin Nutr 2009; 18:155–163.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol 2002; 34:207–220.
- Byrne TA, Wilmore DW, Iyer K, et al. Growth hormone, glutamine, and an optimal diet reduces parenteral nutrition in patients with short bowel syndrome: a prospective, randomized, placebo-controlled, double-blind clinical trial. Ann Surg 2005; 242:655–661.
- Matarese LE, Steiger E. Dietary and medical management of short bowel syndrome in adult patients. J Clin Gastroenterol 2006; 40(suppl 2):S85–S93.
- Nordgaard I, Hansen BS, Mortensen PB. Colon as a digestive organ in patients with short bowel. Lancet 1994; 343:373–376.
- Ukleja A, Scolapio JS, Buchman AL. Nutritional management of short bowel syndrome. Semin Gastrointest Dis 2002; 13:161–168.
- Jeejeebhoy KN. Short bowel syndrome: a nutritional and medical approach. CMAJ 2002; 166:1297–1302.
- Seetharam P, Rodrigues G. Short bowel syndrome: a review of management options. Saudi J Gastroenterol 2011; 17:229–235.
- Wallis K, Walters JR, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care 2009; 12:526–532.
- Jeppesen PB, Gilroy R, Pertkiewicz M, Allard JP, Messing B, O’Keefe SJ. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut 2011; 60:902–914.
- Pironi L, Joly F, Forbes A, et al; Home Artificial Nutrition & Chronic Intestinal Failure Working Group of the European Society for Clinical Nutrition and Metabolism (ESPEN). Long-term follow-up of patients on home parenteral nutrition in Europe: implications for intestinal transplantation. Gut 2011; 60:17–25.
- Ekema G, Milianti S, Boroni G. Total parenteral nutrition in patients with short bowel syndrome. Minerva Pediatr 2009; 61:283–291.
- Messing B, Crenn P, Beau P, Boutron-Ruault MC, Rambaud JC, Matuchansky C. Long-term survival and parenteral nutrition dependence in adult patients with the short bowel syndrome. Gastroenterology 1999; 117:1043–1050.
- Opilla M. Epidemiology of bloodstream infection associated with parenteral nutrition. Am J Infect Control 2008; 36:S173.e5–e8.
- Ukleja A, Romano MM. Complications of parenteral nutrition. Gastroenterol Clin North Am 2007; 36:23–46.
- Buchman AI, Iyer K, Fryer J. Parenteral nutrition-associated liver disease and the role for isolated intestine and intestine/liver transplantation. Hepatology 2006; 43:9–19.
- Fürst P, Kuhn KS. Fish oil emulsions: what benefits can they bring? Clin Nutr 2000; 19:7–14.
- Verso M, Agnelli G. Venous thromboembolism associated with long-term use of central venous catheters in cancer patients. J Clin Oncol 2003; 21:3665–3675.
- McMahon MM, Nystrom E, Braunschweig C, Miles J, Compher C; American Society for Parenteral and Enteral Nutrition (ASPEN) Board of Directors; American Society for Parenteral and Enteral Nutrition. American Society of Parenteral and Enteral Nutrition (ASPEN) Board of Directors. A.S.P.E.N. clinical guidelines: nutrition support of adult patients with hyperglycemia. JPEN J Parenter Enteral Nutr 2013; 37:23–36.
- Kim HB, Fauza D, Garza J, Oh JT, Nurko S, Jaksic T. Serial transverse enteroplasty (STEP): a novel bowel lengthening procedure. J Pediatr Surg 2003; 38:425–429.
- King B, Carlson G, Khalil BA, Morabito A. Intestinal bowel lengthening in children with short bowel syndrome: systematic review of the Bianchi and STEP procedures. World J Surg 2013; 37:694–704.
- Matarese LE, O’Keefe SJ, Kandil HM, Costa G, Abu-Elmagd KM. Short bowel syndrome: clinical guidelines for nutrition management. Nutr Clin Pract 2005; 20:493–502.
- Abu-Elmagd KM, Costa G, Bond GJ, et al. Five hundred intestinal and multivisceral transplantations at a single center: major advances with new challenges. Ann Surg 2009; 250:567–581.
- Abu-Elmagd K. The concept of gut rehabilitation and the future of visceral transplantation. Nat Rev Gastroenterol Hepatol 2015; 12:108–120.
- Lillehei RC, Goott B, Miller FA. The physiological response of the small bowel of the dog to ischemia including prolonged in vitro preservation of the bowel with successful replacement and survival. Ann Surg 1959; 150:543–559.
- Starzl TE, Kaupp HA. Mass homotransplantation of abdominal organs in dogs. Surg Forum 1960; 11:28–30.
- O’Keefe SJ, Matarese L. Small bowel transplantation. Curr Gastroenterol Rep 2006; 8:360–366.
- Horslen SP. Optimal management of the post-intestinal transplant patient. Gastroenterology 2006; 130(suppl 1):S163–S169.
- Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology 2003; 124:1111–1134.
- DiMartini A, Rovera GM, Graham TO, et al. Quality of life after small intestinal transplantation and among home parenteral nutrition patients. JPEN J Parenter Enteral Nutr 1998; 22:357–362.
KEY POINTS
- Some patients with intestinal failure require lifelong parenteral nutrition, which increases the risk of complications such as infection and liver disease. For these patients, intestinal transplant has emerged as a therapeutic option toward the goal of restoring nutritional autonomy.
- The complexities of intestinal failure require collaboration of multiple specialists—gastroenterologists, surgeons, dietitians, nurses, psychiatrists or psychologists, pharmacists, and social workers. This multidisciplinary team is essential to intestinal rehabilitation.
- Dietary modification is the single most effective means of weaning patients safely from parenteral nutrition.