User login
Help your patient with hoarding disorder move the clutter to the curb
Hoarding disorder (HD), categorized in DSM-5 under obsessive-compulsive and related disorders, is defined as the “persistent difficulty discarding or parting with possessions, regardless of their actual value.”1 Hoarders feel that they need to save items, and experience distress when discarding them. Prevalence of HD among the general population is 2% to 5%.
Compulsive hoarders usually keep old items in their home that they do not intend to use. In severe cases, the clutter is so great that areas of the home cannot be used or entered. Hoarders tend to isolate themselves and usually do not invite people home, perhaps because they are embarrassed about the clutter or anxious that someone might try to clean the house. Hoarders may travel long distances to collect items others have discarded.
Hoarding can lead to psychiatric disorders and social problems. Hoarders tend to not develop attachment with people because they are more attached to their possessions. They may avoid social interactions; in turn, others avoid them. This isolation can lead to depression, anxiety, and substance abuse. Hoarders may be evicted from their home if the clutter makes the house dangerous or unfit to live in it. Compulsive hoarding is detrimental to the hoarder and the health and well-being of family members. Hoarding can coexist or can be result of other psychiatric disorders (Table). 
Neural mechanism in hoarding
Hoarders may start to accumulate and store large quantities of items because of a cognitive deficit, such as trouble making decisions or poor recognition or acknowledgement of the situation, or maladaptive thoughts. Tolin et al1 found the anterior cingulate cortex and insula was stimulus-dependent in patients with HD. Functional MRI showed when patients with HD were shown an item that was their possession, they exhibited an abnormal brain activity, compared with low activity when the items shown were not theirs.
Interventions
Choice of treatment depends on the age of the patient and severity of illness: behavioral, medical, or a combination of both. For an uncomplicated case, management can begin with behavioral modification.
Behavioral modifications. HD can stem from any of several variables, including greater response latency for decision-making about possessions and maladaptive beliefs about, and emotional attachment to, possessions, which can lead to intense emotional experiences about the prospect of losing those possessions.2 Cognitive-behavioral therapy has shown promising results for treating HD by addressing the aforementioned factors. A step-by-step approach usually is feasible and convenient for the therapist and patient. It involves gradual mental detachment from items to accommodate the patient’s pace.2
Pharmacotherapy. There is no clear evidence for treating HD with any particular drug. Hoarders are less likely to use psychotropics, possibly because of poor insight (eg, they do not realize the potentially dangerous living conditions hoarding creates).3 Because HD is related to obsessive-compulsive disorder, it is intuitive to consider a selective serotonin reuptake inhibitor.
There is still a need for more research on management of HD.
Disclosure
Dr. Silman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tolin DF, Stevens MC, Villavicencio AL, et al. Neural mechanism of decision making in hoarding disorder. Arch Gen Psychiatry. 2012;69(8):832-841.
2. Tolin DF, Frost RO, Steketee G. An open trial of cognitivebehavioral therapy for compulsive hoarding. Behav Res Ther. 2007;45(7):1461-1470.
3. Brakoulias V, Starcevic V, Berle D, et al. The use of psychotropic agents for the symptoms of obsessivecompulsive disorder. Australas Psychiatry. 2013;21(2): 117-121.
Hoarding disorder (HD), categorized in DSM-5 under obsessive-compulsive and related disorders, is defined as the “persistent difficulty discarding or parting with possessions, regardless of their actual value.”1 Hoarders feel that they need to save items, and experience distress when discarding them. Prevalence of HD among the general population is 2% to 5%.
Compulsive hoarders usually keep old items in their home that they do not intend to use. In severe cases, the clutter is so great that areas of the home cannot be used or entered. Hoarders tend to isolate themselves and usually do not invite people home, perhaps because they are embarrassed about the clutter or anxious that someone might try to clean the house. Hoarders may travel long distances to collect items others have discarded.
Hoarding can lead to psychiatric disorders and social problems. Hoarders tend to not develop attachment with people because they are more attached to their possessions. They may avoid social interactions; in turn, others avoid them. This isolation can lead to depression, anxiety, and substance abuse. Hoarders may be evicted from their home if the clutter makes the house dangerous or unfit to live in it. Compulsive hoarding is detrimental to the hoarder and the health and well-being of family members. Hoarding can coexist or can be result of other psychiatric disorders (Table).
Neural mechanism in hoarding
Hoarders may start to accumulate and store large quantities of items because of a cognitive deficit, such as trouble making decisions or poor recognition or acknowledgement of the situation, or maladaptive thoughts. Tolin et al1 found the anterior cingulate cortex and insula was stimulus-dependent in patients with HD. Functional MRI showed when patients with HD were shown an item that was their possession, they exhibited an abnormal brain activity, compared with low activity when the items shown were not theirs.
Interventions
Choice of treatment depends on the age of the patient and severity of illness: behavioral, medical, or a combination of both. For an uncomplicated case, management can begin with behavioral modification.
Behavioral modifications. HD can stem from any of several variables, including greater response latency for decision-making about possessions and maladaptive beliefs about, and emotional attachment to, possessions, which can lead to intense emotional experiences about the prospect of losing those possessions.2 Cognitive-behavioral therapy has shown promising results for treating HD by addressing the aforementioned factors. A step-by-step approach usually is feasible and convenient for the therapist and patient. It involves gradual mental detachment from items to accommodate the patient’s pace.2
Pharmacotherapy. There is no clear evidence for treating HD with any particular drug. Hoarders are less likely to use psychotropics, possibly because of poor insight (eg, they do not realize the potentially dangerous living conditions hoarding creates).3 Because HD is related to obsessive-compulsive disorder, it is intuitive to consider a selective serotonin reuptake inhibitor.
There is still a need for more research on management of HD.
Disclosure
Dr. Silman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Hoarding disorder (HD), categorized in DSM-5 under obsessive-compulsive and related disorders, is defined as the “persistent difficulty discarding or parting with possessions, regardless of their actual value.”1 Hoarders feel that they need to save items, and experience distress when discarding them. Prevalence of HD among the general population is 2% to 5%.
Compulsive hoarders usually keep old items in their home that they do not intend to use. In severe cases, the clutter is so great that areas of the home cannot be used or entered. Hoarders tend to isolate themselves and usually do not invite people home, perhaps because they are embarrassed about the clutter or anxious that someone might try to clean the house. Hoarders may travel long distances to collect items others have discarded.
Hoarding can lead to psychiatric disorders and social problems. Hoarders tend to not develop attachment with people because they are more attached to their possessions. They may avoid social interactions; in turn, others avoid them. This isolation can lead to depression, anxiety, and substance abuse. Hoarders may be evicted from their home if the clutter makes the house dangerous or unfit to live in it. Compulsive hoarding is detrimental to the hoarder and the health and well-being of family members. Hoarding can coexist or can be result of other psychiatric disorders (Table).
Neural mechanism in hoarding
Hoarders may start to accumulate and store large quantities of items because of a cognitive deficit, such as trouble making decisions or poor recognition or acknowledgement of the situation, or maladaptive thoughts. Tolin et al1 found the anterior cingulate cortex and insula was stimulus-dependent in patients with HD. Functional MRI showed when patients with HD were shown an item that was their possession, they exhibited an abnormal brain activity, compared with low activity when the items shown were not theirs.
Interventions
Choice of treatment depends on the age of the patient and severity of illness: behavioral, medical, or a combination of both. For an uncomplicated case, management can begin with behavioral modification.
Behavioral modifications. HD can stem from any of several variables, including greater response latency for decision-making about possessions and maladaptive beliefs about, and emotional attachment to, possessions, which can lead to intense emotional experiences about the prospect of losing those possessions.2 Cognitive-behavioral therapy has shown promising results for treating HD by addressing the aforementioned factors. A step-by-step approach usually is feasible and convenient for the therapist and patient. It involves gradual mental detachment from items to accommodate the patient’s pace.2
Pharmacotherapy. There is no clear evidence for treating HD with any particular drug. Hoarders are less likely to use psychotropics, possibly because of poor insight (eg, they do not realize the potentially dangerous living conditions hoarding creates).3 Because HD is related to obsessive-compulsive disorder, it is intuitive to consider a selective serotonin reuptake inhibitor.
There is still a need for more research on management of HD.
Disclosure
Dr. Silman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Tolin DF, Stevens MC, Villavicencio AL, et al. Neural mechanism of decision making in hoarding disorder. Arch Gen Psychiatry. 2012;69(8):832-841.
2. Tolin DF, Frost RO, Steketee G. An open trial of cognitivebehavioral therapy for compulsive hoarding. Behav Res Ther. 2007;45(7):1461-1470.
3. Brakoulias V, Starcevic V, Berle D, et al. The use of psychotropic agents for the symptoms of obsessivecompulsive disorder. Australas Psychiatry. 2013;21(2): 117-121.
1. Tolin DF, Stevens MC, Villavicencio AL, et al. Neural mechanism of decision making in hoarding disorder. Arch Gen Psychiatry. 2012;69(8):832-841.
2. Tolin DF, Frost RO, Steketee G. An open trial of cognitivebehavioral therapy for compulsive hoarding. Behav Res Ther. 2007;45(7):1461-1470.
3. Brakoulias V, Starcevic V, Berle D, et al. The use of psychotropic agents for the symptoms of obsessivecompulsive disorder. Australas Psychiatry. 2013;21(2): 117-121.
8 tests rolled into a mnemonic to detect weakness in suspected conversion disorder
DSM-5 criteria for conversion disorder (or functional neurological symptom disorder) requires findings that are incompatible with recognized neurologic or medical conditions.1 Knowledge of signs specific to conversion disorder may help you diagnose the illness with confidence.
We review signs suggestive of conversion disorder. These can be remembered using the mnemonic How About Finding Some Conversion Weakness [in an otherwise] Strong Guy/Gal? (Table2).
Inconsistencies in motor function can be observed on examination. Signs may be consciously or unconsciously produced. Although most of the tests mentioned have high positive and negative predictive values (noted in the Table2) they have limited sensitivity and specificity,3 and the presence of a positive sign does not exclude the possibility of comorbid disease.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stone J, LaFrance WC Jr, Levenson JL, et al. Issues for DSM- 5: conversion disorder. Am J Psychiatry. 2010;167(6):626-627.
2. Daum C, Hubschmid M, Aybek S. The value of ‘positive’ clinical signs for weakness, sensory and gait disorders in conversion disorder: a systematic and narrative review. J Neurol Neurosurg Psychiatry. 2014;85(2):180-190.
3. Stone J, Carson A, Sharpe M. Functional symptoms and signs in neurology: assessment and diagnosis. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i2-i12.
DSM-5 criteria for conversion disorder (or functional neurological symptom disorder) requires findings that are incompatible with recognized neurologic or medical conditions.1 Knowledge of signs specific to conversion disorder may help you diagnose the illness with confidence.
We review signs suggestive of conversion disorder. These can be remembered using the mnemonic How About Finding Some Conversion Weakness [in an otherwise] Strong Guy/Gal? (Table2).
Inconsistencies in motor function can be observed on examination. Signs may be consciously or unconsciously produced. Although most of the tests mentioned have high positive and negative predictive values (noted in the Table2) they have limited sensitivity and specificity,3 and the presence of a positive sign does not exclude the possibility of comorbid disease.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
DSM-5 criteria for conversion disorder (or functional neurological symptom disorder) requires findings that are incompatible with recognized neurologic or medical conditions.1 Knowledge of signs specific to conversion disorder may help you diagnose the illness with confidence.
We review signs suggestive of conversion disorder. These can be remembered using the mnemonic How About Finding Some Conversion Weakness [in an otherwise] Strong Guy/Gal? (Table2).
Inconsistencies in motor function can be observed on examination. Signs may be consciously or unconsciously produced. Although most of the tests mentioned have high positive and negative predictive values (noted in the Table2) they have limited sensitivity and specificity,3 and the presence of a positive sign does not exclude the possibility of comorbid disease.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stone J, LaFrance WC Jr, Levenson JL, et al. Issues for DSM- 5: conversion disorder. Am J Psychiatry. 2010;167(6):626-627.
2. Daum C, Hubschmid M, Aybek S. The value of ‘positive’ clinical signs for weakness, sensory and gait disorders in conversion disorder: a systematic and narrative review. J Neurol Neurosurg Psychiatry. 2014;85(2):180-190.
3. Stone J, Carson A, Sharpe M. Functional symptoms and signs in neurology: assessment and diagnosis. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i2-i12.
1. Stone J, LaFrance WC Jr, Levenson JL, et al. Issues for DSM- 5: conversion disorder. Am J Psychiatry. 2010;167(6):626-627.
2. Daum C, Hubschmid M, Aybek S. The value of ‘positive’ clinical signs for weakness, sensory and gait disorders in conversion disorder: a systematic and narrative review. J Neurol Neurosurg Psychiatry. 2014;85(2):180-190.
3. Stone J, Carson A, Sharpe M. Functional symptoms and signs in neurology: assessment and diagnosis. J Neurol Neurosurg Psychiatry. 2005;76(suppl 1):i2-i12.
Treating methamphetamine abuse disorder: Experience from research and practice
Methamphetamine and other amphetamine-type stimulants are the world’s second most widely used group of illicit substances (after Cannabis), with prevalence of abuse varying by region and by locales within nations. As prescription use of stimulants has grown dramatically in recent years, so has abuse of these substances.
Given the widespread and growing misuse of amphetamine-type stimulants (Box,1-3), clinicians are faced with the need to learn how to recognize and manage methamphetamine abuse. Both prescribed and non-prescribed uses of stimulants present complex challenges; in this article, we examine effects, manifestations, and current evidence-based behavioral and medical treatments of methamphetamine misuse and abuse, and look ahead to investigational therapies that hold promise for improving the limited existing approaches to management.

Effects and manifestations of methamphetamine use
Different routes of administration produce different consequences, in terms of medical comorbidity and propensity to induce addiction. Smoked or injected, methamphetamine enters the brain in seconds; snorted or taken by mouth, the drug produces its effects in several minutes and a half hour, respectively.
Rapid uptake and effects of methamphetamine result from its ability to cross the blood−brain barrier. Its primary effects are caused by inhibition of dopamine storage and release of intracellular dopamine.
Methamphetamine stimulates the CNS and the cardiovascular system through release of dopamine and norepinephrine, which increases blood pressure, body temperature, and heart rate, and, occasionally, induces arrhythmia that can contribute to heart attack and stroke. Users experience euphoria, hypervigilance, suppressed appetite, and increased libido.
Binge use is common to sustain euphoria and other reinforcing effects, which subside with rapidly developing tolerance. After days of repeated dosing, elevated methamphetamine blood levels can lead to mood disturbances, repetitive motor activities, and psychotic symptoms such as hallucinations, delusions, and paranoia. Acute psychosis can bring on violence and other injurious behaviors that involve law enforcement and emergency medical services.
When methamphetamine is used over months or years, health consequences include anorexia, tremor, so-called meth mouth (broken teeth, infections, cavities, burns), insomnia, panic attacks, confusion, depression, irritability, and impaired memory and other cognitive processes.
Treating methamphetamine intoxication and withdrawal
At initial clinical contact with a person who abuses methamphetamine, practitioners may face several acute consequences requiring attention. Prominent among presenting conditions, especially during acute intoxication, are agitation, anxiety, and psychotic symptoms, which may improve by providing the patient with calming reassurance in a quiet space. In more severe cases, a benzodiazepine, antipsychotic, or both might be indicated4,5 (Table 1). 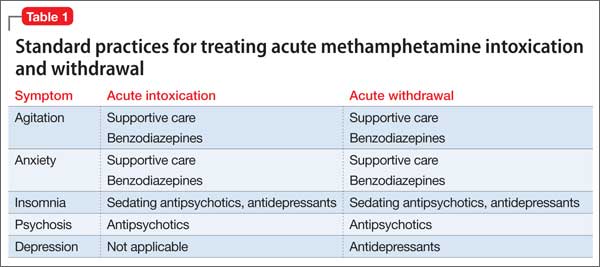
Methamphetamine withdrawal is characterized by anxiety, depression, and insomnia. These symptoms generally resolve in a matter of days after the start of withdrawal without pharmacotherapy. In some cases, depression or psychosis becomes chronic, as a result of methamphetamine use itself6 or as an emergent concomitant psychiatric condition.
A sedative-hypnotic medication or an anxiolytic can be used as necessary to ameliorate insomnia or anxiety, respectively. Prolonged depression can be treated with an antidepressant. An antipsychotic might be indicated for long-term management of patients who have persistent psychosis.
Therapy for methamphetamine abuse
Treatment of methamphetamine abuse— with the goal of stopping drug use—is a complicated matter on 2 counts:
• No medications are FDA-approved for treating methamphetamine addiction.
• There are no accepted substitution medications (ie, stimulants that can be used in place of methamphetamine, as is available for opioid addiction).
Pharmacotherapeutic possibilities. The rationale for considering replacement pharmacotherapy is that psychostimulants can counter the cravings, dysphoria, and fatigue produced by methamphetamine withdrawal and can alleviate methamphetamine-related cognitive impairment. Although dextroamphetamine and other psychostimulants have been evaluated in small trials as replacement medication, most countries are reluctant to consider their use, because of the potential for abuse and accompanying liability.
After decades of medication research, several drugs have shown promise for reducing methamphetamine abuse, although results have not been robust (Table 2):
• Bupropion has shown benefit in reducing methamphetamine use among users with less severe addiction.7,8
• Methylphenidate, a psychostimulant FDA-approved for attention-deficit/hyperactivity disorder, was found to reduce methamphetamine use compared with placebo in a European sample of amphetamine injectors who had attained abstinence in a residential program.9 Those results were not replicated in a recent study by Miles et al, however.10 A study with a more clinically realistic approach (ie, not requiring daily clinic attendance, as in the Miles trial) vs placebo for methamphetamine abuse was recently published, with promising results that require confirmation in further study.11
• Mirtazapine, an antidepressant, has demonstrated efficacy in reducing methamphetamine use compared with placebo.12
• Modafinil, another medication with stimulant properties, reduced methamphetamine use in a subgroup analysis of heavy users, compared with placebo.13
• Dextroamphetamine, 60 mg/d, showed no difference in reducing methamphetamine compared with placebo, but did diminish cravings and withdrawal symptoms.14
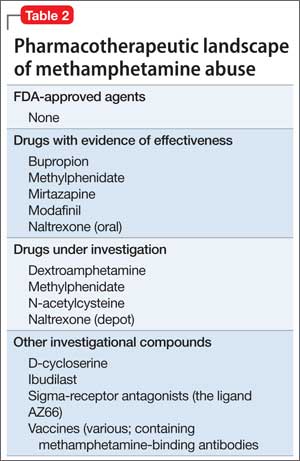
A trial of the phosphodiesterase inhibitor ibudilast (not available in the United States) for methamphetamine abuse is underway. Ibudilast has anti-inflammatory activity in the peripheral immune system and the central nervous system, including modulating the activity of glial cells.15
Many medications have yielded negligible results in studies: selegiline, baclofen, sertraline, topiramate, gabapentin, rivastigmine, risperidone, and ondansetron.16 Recent evaluation of disulfiram, vigabatrin, and lobeline also has yielded inconsistent findings.17
No drug has proved effective for preventing relapse; research continues, focusing on several types of compounds that target various mechanisms: the dopamine system, the opioid system (by way of the γ-aminobutyric acid inhibitory system), and cortico-limbic reward circuitry.
Once-monthly injectable naltrexone has potential for ameliorating craving and relapse by modulating the opioid receptor system. However, the drug has not been adequately explored in generalizable settings of methamphetamine users.
Trials of oral naltrexone in Sweden have shown encouraging results, including reduced subjective effects and amphetamine use in open-label trials18,19; results were replicated in a subsequent placebo-controlled trial.20 In an unpublished study, however, no differences in amphetamine use were found among users randomized to depot naltrexone or placebo.21
Depot naltrexone with assured dosing might have a role in treating methamphetamine abuse, however; a combination of depot naltrexone and oral bupropion is being examined in a National Institute on Drug Abuse Clinical Trials Networks study that commenced in 2013. Pairing medications that have different mechanistic targets might work toward promoting cessation of methamphetamine abuse and reducing relapse once patients are abstinent.
In an early phase of research, but showing promise based on their ability to target different systems, are:
• N-acetylcysteine, modulator of the glutamate system
• D3 antagonists and partial agonists22
• varenicline.23
Potential “vaccines” against methamphetamine are in preclinical development, including use of a protein carrier or other immune-stimulating molecule to create antibodies that bind methamphetamine in the bloodstream and block its psychoactive effects.24,25
Sigma receptor effects are being studied in rodents as potential targets to mitigate effects of methamphetamine. The ligand AZ66, a sigma receptor antagonist, has demonstrated efficacy in reducing methamphetamine-induced cognitive impairment—suggesting that the sigma receptor has a potential role in ameliorating methamphetamine-related neurotoxicity.26
Psychosocial and behavioral interventions. Among the non-drug treatments that have demonstrated efficacy for treating methamphetamine abuse, cognitive-behavioral therapy (CBT) and contingency management (CM) have been most widely studied and applied in treatment settings.
CBT involves individual or group counseling that focuses on relapse prevention skills, including identification of relapse triggers, strategies to diminish cravings, and engagement in alternative non-drug activities27,28 (Table 3). 
CM, which is based on positive reinforcement, offers tangible reinforcers, or rewards, for behaviors (eg, clinic attendance, providing a drug-free urine sample) according to guidelines set by the practitioner. CM-based interventions are the most reliably documented approaches for treating methamphetamine abuse,29,30 but their utility might prove to be most efficient in combination with medication— once suitable pharmacotherapeutic options emerge.
Although CBT and CM remain accepted standard treatments for methamphetamine abuse, outcomes are suboptimal.27 Both interventions have a high rate of dropout during the first month of treatment and a >50% relapse rate 6 to 19 months after treatment ends.31-33
As with treatment of other substance use disorders, patients who abuse methamphetamine can benefit from residential treatment in a drug-free setting for ≥30 days.34 In the residential approach, removing access to drugs, drug cues, and drug-using acquaintances combined with group and individual counseling reaches an inevitable end: discharge into the community. Then the patient’s battle to avoid relapse begins.
Because cognitive impairment is common among patients who abuse methamphetamine, even after they stop using,35 researchers have examined the potential for increasing participation in psychosocial interventions such as CBT by using medications that might have potential to increase cognitive function, such as modafinil.36 Increased attention and concentration afforded by medication could enhance efficacy of CBT. Results of trials and new drug development have been mixed37; no clear candidate for preventing relapse through any of the putative mechanisms of action has emerged.
Relapse is a problematic target for treatment
Ending methamphetamine abuse and sustaining abstinence from stimulants require a change in the cognitive associations that have been laid down in a drug user’s memory. Relapse occurs because of recalled memories that can be cued, or triggered, by internal or external stimuli. Eliminating drug memories, perhaps assisted by medications such as d-cycloserine (an antagonist of the N-methyl-d-aspartate receptor), could be useful for suppressing the inclination to relapse.
Last, alternative, non-drug forms of cognitive amendment have shown efficacy in preventing relapse: for example, incorporating mindfulness meditation, which has shown promise in managing craving for methamphetamine and decreasing reactivity to environmental cues for drug use.38
Bottom Line
Practitioners who work in emergency, inpatient, and outpatient settings will be called on more and more to treat acute stimulant intoxication and withdrawal, stimulant-induced psychosis, and methamphetamine abuse. Few evidence-based treatments and no FDA-approved medications are available to treat this addiction; many drugs and a few psychotherapeutic techniques have shown promise. Ongoing research promises to deliver medical and behavioral interventions to help patients quit using methamphetamine.
Related Resources
• Karch SB, Drummer O. Karch’s pathology of drug abuse, fifth ed. Boca Raton, FL: CRC Press/Taylor & David; 2013.
• Roll J, Rawson RA, Ling W, eds. Methamphetamine addiction: from basic science to treatment. New York, NY: Guilford Press; 2009.
• Sheff D. Beautiful boy: a father’s journey through his son’s addiction. New York, NY: Houghton Mifflin Harcourt Publishing Company; 2008.
• Sheff N. Tweak: growing up on methamphetamines. New York, NY: Antheneum Books for Young Readers; 2007.
• National Institute on Drug Abuse. Drugs of abuse. www. drugabuse.gov/drugs-abuse/methamphetamine.
Drug Brand Names
Baclofen • Lioresal Naltrexone (depot) • Vivitrol
Bupropion • Wellbutrin Naltrexone (oral) • ReVia
D-cycloserine • Seromycin Ondansetron • Zofran
Dexreoamphetaime • Adderall Risperidone • Risperadal
Disulfiram • Antabuse Rivastigimine • Exelon
Gabapentin • Neurontin Selegiline• EMSAM
Methylphenidate • Ritalin Sertraline • Zoloft
Mirtazapine • Remeron Topiramate • Topamax
Modafinil • Provigil Varenicline • Chantix
N-acetylcysteine • Mucomyst Vigabatrin • Sabril
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. UNODC. World Drug Report 2012 (United Nations publication, Sales No. E.12.XI.1). http://www.unodc. org/documents/data-and-analysis/WDR2012/ WDR_2012_web_small.pdf. Published 2012. Accessed August 4, 2014.
2. UNODC. World Drug Report 2010 (United Nations publication, Sales No. E.10.XI.13). http://www.unodc. org/documents/wdr/WDR_2010/World_Drug_ Report_2010_lo-res.pdf. Published 2010. Accessed August 4, 2014.
3. Rawson RA, Gonzales R, Brecht M, et al. Evaluation of the California Outcomes Measurement System (CalOMS): Final Report 2008. http://www.uclaisap.org/assets/documents/ California-ADP-DHCS-Evals/2007-2008_CalOMS%20 Report.pdf. Published 2008. Accessed August 4, 2014.
4. Shoptaw SJ, Kao U, Ling W. Treatment for amphetamine psychosis. Cochrane Database Syst Rev. 2009;(1):CD003026.
5. Leelahanaj T, Kongsakon R, Netrakom P. A 4-week, double-blind comparison of olanzapine with haloperidol in the treatment of amphetamine psychosis. J Med Assoc Thai. 2005;88(suppl 3):S43-S52.
6. McKetin R, McLaren J, Lubman D, et al. The prevalence of psychotic symptoms among methamphetamine users. Addiction. 2006;101(10):1473-1478.
7. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
8. McCann DJ, Li SH. A novel, nonbinary evaluation of success and failure reveals bupropion efficacy versus methamphetamine dependence: reanalysis of a multisite trial. CNS Neurosci Ther. 2012;18(5):414-418.
9. Tiihonen J, Kuoppasalmi K, Föhr J, et al. A comparison of aripiprazole, methylphenidate, and placebo for amphetamine dependence. Am J Psychiatry. 2007;164(1): 160-162.
10. Miles SW, Sheridan J, Russell B, et al. Extended-release methylphenidate for treatment of amphetamine/ methamphetamine dependence: a randomized, double-blind, placebo-controlled trial. Addiction. 2013;108(7): 1279-1286.
11. Ling W, Chang L, Hillhouse M, et al. Sustained-release methylphenidate in a randomized trial of treatment of methamphetamine use disorder. Addiction. 2014;109(9): 1489-1500.
12. Colfax GN, Santos GM, Das M, et al. Mirtazapine to reduce methamphetamine use: a randomized controlled trial. Arch Gen Psychiatry. 2011;68(11):1168-1175.
13. Heinzerling KG, Swanson AN, Kim S, et al. Randomized, double-blind, placebo-controlled trial of modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2010;109(1-3):20-29.
14. Galloway GP, Buscemi R, Coyle JR, et al. A randomized, placebo-controlled trial of sustained-release dextro-amphetamine for treatment of methamphetamine addiction. Clin Pharmacol Ther. 2011;89(2):276-282.
15. Snider SE, Hendrick ES, Beardsley PM. Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol. 2013;701(1-3):124-130.
16. Ling W, Rawson R, Shoptaw S. Management of methamphetamine abuse and dependence. Curr Psychiatry Rep. 2006;8(5):345-354.
17. Brackins T, Brahm NC, Kissack JC. Treatments for methamphetamine abuse: a literature review for the clinician. J Pharm Pract. 2011;24(6):541-550.
18. Jayaram-Lindström N, Wennberg P, Beck O, et al. An open clinical trial of naltrexone for amphetamine dependence: compliance and tolerability. Nord J Psychiatry. 2005;59(3):167-171.
19. Jayaram-Lindström N, Konstenius M, Eksborg S, et al. Naltrexone attenuates the subjective effects of amphetamine in patients with amphetamine dependence. Neuropsychopharmacology. 2007;33(8):1856-1863.
20. Jayaram-Lindström N, Hammarberg A, Beck O, et al. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry. 2008;165(11):1442-1448.
21. Woody GE, Tyrfingsoon P. Symposium XI: Emerging data on efficacy and clinical applications of extended-release naltrexone formulations. 75th Annual Meeting, College on Problems of Drug Dependence. June 19, 2013; San Diego, CA.
22. Newman AH, Blaylock BL, Nader MA, et al. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84(7):882-890.
23. Verrico CD, Mahoney JJ 3rd, Thompson-Lake DG, et al. Safety and efficacy of varenicline to reduce positive subjective effects produced by methamphetamine in methamphetamine-dependent volunteers. Int J Neuropsychopharmacol. 2014;17(2):223-233.
24. Miller ML, Moreno AY, Aarde S, et al. A methamphetamine vaccine attenuates methamphetamine-induced disruptions in thermoregulation and activity in rats. Biol Psychiatry. 2013;73(8):721-728.
25. Shen XY, Kosten TA, Lopez AY, et al. A vaccine against methamphetamine attenuates its behavioral effects in mice. Drug Alcohol Depend. 2013;129(1-2):41-48.
26. Seminerio MJ, Robson MJ, Abdelazeem AH, et al. Synthesis and pharmacological characterization of a novel sigma receptor ligand with improved metabolic stability and antagonistic effects against methamphetamine. AAPS J. 2012;14(1):43-51.
27. Rawson RA, Marinelli-Casey P, Anglin M, et al. A multi-site comparison of psychosocial approaches for the treatment of methamphetamine dependence. Addiction. 2004;99(6):708-717.
28. Vocci FJ, Montoya ID. Psychological treatments for stimulant misuse, comparing and contrasting those for amphetamine dependence and those for cocaine dependence. Curr Opin Psychiatry. 2009;22(3):263-268.
29. Rawson RA, McCann MJ, Flammino F, et al. A comparison of contingency management and cognitive-behavioral approaches for stimulant-dependent individuals. Addiction. 2006;101(2):267-274.
30. Roll JM, Petry NM, Stitzer ML, et al. Contingency management for the treatment of methamphetamine use disorders. Am J Psychiatry. 2006;163(11):1993-1999.
31. Brecht ML, von Mayrhauser C, Anglin MD. Predictors of relapse after treatment for methamphetamine use. J Psychoactive Drugs. 2000;32(2):211-220.
32. Smout MF, Longo M, Harrison S, et al. Psychosocial treatment for methamphetamine use disorders: a preliminary randomized controlled trial of cognitive behavior therapy and Acceptance and Commitment Therapy. Subst Abus. 2010;31(2):98-107.
33. Wang G, Shi J, Chen N, et al. Effects of length of abstinence on decision-making and craving in methamphetamine abusers. PLoS One. 2013;24;8(7):e68791. doi: 10.1371/ journal.pone.0068791.
34. McKetin R, Lubman DI, Baker AL, et al. Dose-related psychotic symptoms in chronic methamphetamine users: evidence from a prospective longitudinal study. JAMA Psychiatry. 2013;70(3):319-324.
35. Henry BL, Minassian A, Perry W. Effect of methamphetamine dependence on everyday functional ability. Addict Behav. 2010;35(6):593-598.
36. Dean AC, Sevak RJ, Monterosso JR, et al. Acute modafinil effects on attention and inhibitory control in methamphetamine-dependent humans. J Stud Alcohol Drugs. 2011;72(6):943-953.
37. Zullino DF, Benguettat D, Khazaal Y. Improvement of cognitive performance by topiramate: blockage of automatic processes may be the underlying mechanism [Comment on: Effects of topiramate on methamphetamine-induced changes in attentional and perceptual-motor skills of cognition in recently abstinent methamphetamine-dependent individuals. Prog Neuropsychopharmacol Biol Psychiatry. 2007.] Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):787.
38. Witkiewitz K, Lustyk M, Bowen S. Retraining the addicted brain: a review of hypothesized neurobiological mechanisms of mindfulness-based relapse prevention. Psychol Addict Behav. 2013;27(2):351-365.
Methamphetamine and other amphetamine-type stimulants are the world’s second most widely used group of illicit substances (after Cannabis), with prevalence of abuse varying by region and by locales within nations. As prescription use of stimulants has grown dramatically in recent years, so has abuse of these substances.
Given the widespread and growing misuse of amphetamine-type stimulants (Box,1-3), clinicians are faced with the need to learn how to recognize and manage methamphetamine abuse. Both prescribed and non-prescribed uses of stimulants present complex challenges; in this article, we examine effects, manifestations, and current evidence-based behavioral and medical treatments of methamphetamine misuse and abuse, and look ahead to investigational therapies that hold promise for improving the limited existing approaches to management.
Effects and manifestations of methamphetamine use
Different routes of administration produce different consequences, in terms of medical comorbidity and propensity to induce addiction. Smoked or injected, methamphetamine enters the brain in seconds; snorted or taken by mouth, the drug produces its effects in several minutes and a half hour, respectively.
Rapid uptake and effects of methamphetamine result from its ability to cross the blood−brain barrier. Its primary effects are caused by inhibition of dopamine storage and release of intracellular dopamine.
Methamphetamine stimulates the CNS and the cardiovascular system through release of dopamine and norepinephrine, which increases blood pressure, body temperature, and heart rate, and, occasionally, induces arrhythmia that can contribute to heart attack and stroke. Users experience euphoria, hypervigilance, suppressed appetite, and increased libido.
Binge use is common to sustain euphoria and other reinforcing effects, which subside with rapidly developing tolerance. After days of repeated dosing, elevated methamphetamine blood levels can lead to mood disturbances, repetitive motor activities, and psychotic symptoms such as hallucinations, delusions, and paranoia. Acute psychosis can bring on violence and other injurious behaviors that involve law enforcement and emergency medical services.
When methamphetamine is used over months or years, health consequences include anorexia, tremor, so-called meth mouth (broken teeth, infections, cavities, burns), insomnia, panic attacks, confusion, depression, irritability, and impaired memory and other cognitive processes.
Treating methamphetamine intoxication and withdrawal
At initial clinical contact with a person who abuses methamphetamine, practitioners may face several acute consequences requiring attention. Prominent among presenting conditions, especially during acute intoxication, are agitation, anxiety, and psychotic symptoms, which may improve by providing the patient with calming reassurance in a quiet space. In more severe cases, a benzodiazepine, antipsychotic, or both might be indicated4,5 (Table 1).
Methamphetamine withdrawal is characterized by anxiety, depression, and insomnia. These symptoms generally resolve in a matter of days after the start of withdrawal without pharmacotherapy. In some cases, depression or psychosis becomes chronic, as a result of methamphetamine use itself6 or as an emergent concomitant psychiatric condition.
A sedative-hypnotic medication or an anxiolytic can be used as necessary to ameliorate insomnia or anxiety, respectively. Prolonged depression can be treated with an antidepressant. An antipsychotic might be indicated for long-term management of patients who have persistent psychosis.
Therapy for methamphetamine abuse
Treatment of methamphetamine abuse— with the goal of stopping drug use—is a complicated matter on 2 counts:
• No medications are FDA-approved for treating methamphetamine addiction.
• There are no accepted substitution medications (ie, stimulants that can be used in place of methamphetamine, as is available for opioid addiction).
Pharmacotherapeutic possibilities. The rationale for considering replacement pharmacotherapy is that psychostimulants can counter the cravings, dysphoria, and fatigue produced by methamphetamine withdrawal and can alleviate methamphetamine-related cognitive impairment. Although dextroamphetamine and other psychostimulants have been evaluated in small trials as replacement medication, most countries are reluctant to consider their use, because of the potential for abuse and accompanying liability.
After decades of medication research, several drugs have shown promise for reducing methamphetamine abuse, although results have not been robust (Table 2):
• Bupropion has shown benefit in reducing methamphetamine use among users with less severe addiction.7,8
• Methylphenidate, a psychostimulant FDA-approved for attention-deficit/hyperactivity disorder, was found to reduce methamphetamine use compared with placebo in a European sample of amphetamine injectors who had attained abstinence in a residential program.9 Those results were not replicated in a recent study by Miles et al, however.10 A study with a more clinically realistic approach (ie, not requiring daily clinic attendance, as in the Miles trial) vs placebo for methamphetamine abuse was recently published, with promising results that require confirmation in further study.11
• Mirtazapine, an antidepressant, has demonstrated efficacy in reducing methamphetamine use compared with placebo.12
• Modafinil, another medication with stimulant properties, reduced methamphetamine use in a subgroup analysis of heavy users, compared with placebo.13
• Dextroamphetamine, 60 mg/d, showed no difference in reducing methamphetamine compared with placebo, but did diminish cravings and withdrawal symptoms.14
A trial of the phosphodiesterase inhibitor ibudilast (not available in the United States) for methamphetamine abuse is underway. Ibudilast has anti-inflammatory activity in the peripheral immune system and the central nervous system, including modulating the activity of glial cells.15
Many medications have yielded negligible results in studies: selegiline, baclofen, sertraline, topiramate, gabapentin, rivastigmine, risperidone, and ondansetron.16 Recent evaluation of disulfiram, vigabatrin, and lobeline also has yielded inconsistent findings.17
No drug has proved effective for preventing relapse; research continues, focusing on several types of compounds that target various mechanisms: the dopamine system, the opioid system (by way of the γ-aminobutyric acid inhibitory system), and cortico-limbic reward circuitry.
Once-monthly injectable naltrexone has potential for ameliorating craving and relapse by modulating the opioid receptor system. However, the drug has not been adequately explored in generalizable settings of methamphetamine users.
Trials of oral naltrexone in Sweden have shown encouraging results, including reduced subjective effects and amphetamine use in open-label trials18,19; results were replicated in a subsequent placebo-controlled trial.20 In an unpublished study, however, no differences in amphetamine use were found among users randomized to depot naltrexone or placebo.21
Depot naltrexone with assured dosing might have a role in treating methamphetamine abuse, however; a combination of depot naltrexone and oral bupropion is being examined in a National Institute on Drug Abuse Clinical Trials Networks study that commenced in 2013. Pairing medications that have different mechanistic targets might work toward promoting cessation of methamphetamine abuse and reducing relapse once patients are abstinent.
In an early phase of research, but showing promise based on their ability to target different systems, are:
• N-acetylcysteine, modulator of the glutamate system
• D3 antagonists and partial agonists22
• varenicline.23
Potential “vaccines” against methamphetamine are in preclinical development, including use of a protein carrier or other immune-stimulating molecule to create antibodies that bind methamphetamine in the bloodstream and block its psychoactive effects.24,25
Sigma receptor effects are being studied in rodents as potential targets to mitigate effects of methamphetamine. The ligand AZ66, a sigma receptor antagonist, has demonstrated efficacy in reducing methamphetamine-induced cognitive impairment—suggesting that the sigma receptor has a potential role in ameliorating methamphetamine-related neurotoxicity.26
Psychosocial and behavioral interventions. Among the non-drug treatments that have demonstrated efficacy for treating methamphetamine abuse, cognitive-behavioral therapy (CBT) and contingency management (CM) have been most widely studied and applied in treatment settings.
CBT involves individual or group counseling that focuses on relapse prevention skills, including identification of relapse triggers, strategies to diminish cravings, and engagement in alternative non-drug activities27,28 (Table 3).
CM, which is based on positive reinforcement, offers tangible reinforcers, or rewards, for behaviors (eg, clinic attendance, providing a drug-free urine sample) according to guidelines set by the practitioner. CM-based interventions are the most reliably documented approaches for treating methamphetamine abuse,29,30 but their utility might prove to be most efficient in combination with medication— once suitable pharmacotherapeutic options emerge.
Although CBT and CM remain accepted standard treatments for methamphetamine abuse, outcomes are suboptimal.27 Both interventions have a high rate of dropout during the first month of treatment and a >50% relapse rate 6 to 19 months after treatment ends.31-33
As with treatment of other substance use disorders, patients who abuse methamphetamine can benefit from residential treatment in a drug-free setting for ≥30 days.34 In the residential approach, removing access to drugs, drug cues, and drug-using acquaintances combined with group and individual counseling reaches an inevitable end: discharge into the community. Then the patient’s battle to avoid relapse begins.
Because cognitive impairment is common among patients who abuse methamphetamine, even after they stop using,35 researchers have examined the potential for increasing participation in psychosocial interventions such as CBT by using medications that might have potential to increase cognitive function, such as modafinil.36 Increased attention and concentration afforded by medication could enhance efficacy of CBT. Results of trials and new drug development have been mixed37; no clear candidate for preventing relapse through any of the putative mechanisms of action has emerged.
Relapse is a problematic target for treatment
Ending methamphetamine abuse and sustaining abstinence from stimulants require a change in the cognitive associations that have been laid down in a drug user’s memory. Relapse occurs because of recalled memories that can be cued, or triggered, by internal or external stimuli. Eliminating drug memories, perhaps assisted by medications such as d-cycloserine (an antagonist of the N-methyl-d-aspartate receptor), could be useful for suppressing the inclination to relapse.
Last, alternative, non-drug forms of cognitive amendment have shown efficacy in preventing relapse: for example, incorporating mindfulness meditation, which has shown promise in managing craving for methamphetamine and decreasing reactivity to environmental cues for drug use.38
Bottom Line
Practitioners who work in emergency, inpatient, and outpatient settings will be called on more and more to treat acute stimulant intoxication and withdrawal, stimulant-induced psychosis, and methamphetamine abuse. Few evidence-based treatments and no FDA-approved medications are available to treat this addiction; many drugs and a few psychotherapeutic techniques have shown promise. Ongoing research promises to deliver medical and behavioral interventions to help patients quit using methamphetamine.
Related Resources
• Karch SB, Drummer O. Karch’s pathology of drug abuse, fifth ed. Boca Raton, FL: CRC Press/Taylor & David; 2013.
• Roll J, Rawson RA, Ling W, eds. Methamphetamine addiction: from basic science to treatment. New York, NY: Guilford Press; 2009.
• Sheff D. Beautiful boy: a father’s journey through his son’s addiction. New York, NY: Houghton Mifflin Harcourt Publishing Company; 2008.
• Sheff N. Tweak: growing up on methamphetamines. New York, NY: Antheneum Books for Young Readers; 2007.
• National Institute on Drug Abuse. Drugs of abuse. www. drugabuse.gov/drugs-abuse/methamphetamine.
Drug Brand Names
Baclofen • Lioresal Naltrexone (depot) • Vivitrol
Bupropion • Wellbutrin Naltrexone (oral) • ReVia
D-cycloserine • Seromycin Ondansetron • Zofran
Dexreoamphetaime • Adderall Risperidone • Risperadal
Disulfiram • Antabuse Rivastigimine • Exelon
Gabapentin • Neurontin Selegiline• EMSAM
Methylphenidate • Ritalin Sertraline • Zoloft
Mirtazapine • Remeron Topiramate • Topamax
Modafinil • Provigil Varenicline • Chantix
N-acetylcysteine • Mucomyst Vigabatrin • Sabril
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Methamphetamine and other amphetamine-type stimulants are the world’s second most widely used group of illicit substances (after Cannabis), with prevalence of abuse varying by region and by locales within nations. As prescription use of stimulants has grown dramatically in recent years, so has abuse of these substances.
Given the widespread and growing misuse of amphetamine-type stimulants (Box,1-3), clinicians are faced with the need to learn how to recognize and manage methamphetamine abuse. Both prescribed and non-prescribed uses of stimulants present complex challenges; in this article, we examine effects, manifestations, and current evidence-based behavioral and medical treatments of methamphetamine misuse and abuse, and look ahead to investigational therapies that hold promise for improving the limited existing approaches to management.
Effects and manifestations of methamphetamine use
Different routes of administration produce different consequences, in terms of medical comorbidity and propensity to induce addiction. Smoked or injected, methamphetamine enters the brain in seconds; snorted or taken by mouth, the drug produces its effects in several minutes and a half hour, respectively.
Rapid uptake and effects of methamphetamine result from its ability to cross the blood−brain barrier. Its primary effects are caused by inhibition of dopamine storage and release of intracellular dopamine.
Methamphetamine stimulates the CNS and the cardiovascular system through release of dopamine and norepinephrine, which increases blood pressure, body temperature, and heart rate, and, occasionally, induces arrhythmia that can contribute to heart attack and stroke. Users experience euphoria, hypervigilance, suppressed appetite, and increased libido.
Binge use is common to sustain euphoria and other reinforcing effects, which subside with rapidly developing tolerance. After days of repeated dosing, elevated methamphetamine blood levels can lead to mood disturbances, repetitive motor activities, and psychotic symptoms such as hallucinations, delusions, and paranoia. Acute psychosis can bring on violence and other injurious behaviors that involve law enforcement and emergency medical services.
When methamphetamine is used over months or years, health consequences include anorexia, tremor, so-called meth mouth (broken teeth, infections, cavities, burns), insomnia, panic attacks, confusion, depression, irritability, and impaired memory and other cognitive processes.
Treating methamphetamine intoxication and withdrawal
At initial clinical contact with a person who abuses methamphetamine, practitioners may face several acute consequences requiring attention. Prominent among presenting conditions, especially during acute intoxication, are agitation, anxiety, and psychotic symptoms, which may improve by providing the patient with calming reassurance in a quiet space. In more severe cases, a benzodiazepine, antipsychotic, or both might be indicated4,5 (Table 1).
Methamphetamine withdrawal is characterized by anxiety, depression, and insomnia. These symptoms generally resolve in a matter of days after the start of withdrawal without pharmacotherapy. In some cases, depression or psychosis becomes chronic, as a result of methamphetamine use itself6 or as an emergent concomitant psychiatric condition.
A sedative-hypnotic medication or an anxiolytic can be used as necessary to ameliorate insomnia or anxiety, respectively. Prolonged depression can be treated with an antidepressant. An antipsychotic might be indicated for long-term management of patients who have persistent psychosis.
Therapy for methamphetamine abuse
Treatment of methamphetamine abuse— with the goal of stopping drug use—is a complicated matter on 2 counts:
• No medications are FDA-approved for treating methamphetamine addiction.
• There are no accepted substitution medications (ie, stimulants that can be used in place of methamphetamine, as is available for opioid addiction).
Pharmacotherapeutic possibilities. The rationale for considering replacement pharmacotherapy is that psychostimulants can counter the cravings, dysphoria, and fatigue produced by methamphetamine withdrawal and can alleviate methamphetamine-related cognitive impairment. Although dextroamphetamine and other psychostimulants have been evaluated in small trials as replacement medication, most countries are reluctant to consider their use, because of the potential for abuse and accompanying liability.
After decades of medication research, several drugs have shown promise for reducing methamphetamine abuse, although results have not been robust (Table 2):
• Bupropion has shown benefit in reducing methamphetamine use among users with less severe addiction.7,8
• Methylphenidate, a psychostimulant FDA-approved for attention-deficit/hyperactivity disorder, was found to reduce methamphetamine use compared with placebo in a European sample of amphetamine injectors who had attained abstinence in a residential program.9 Those results were not replicated in a recent study by Miles et al, however.10 A study with a more clinically realistic approach (ie, not requiring daily clinic attendance, as in the Miles trial) vs placebo for methamphetamine abuse was recently published, with promising results that require confirmation in further study.11
• Mirtazapine, an antidepressant, has demonstrated efficacy in reducing methamphetamine use compared with placebo.12
• Modafinil, another medication with stimulant properties, reduced methamphetamine use in a subgroup analysis of heavy users, compared with placebo.13
• Dextroamphetamine, 60 mg/d, showed no difference in reducing methamphetamine compared with placebo, but did diminish cravings and withdrawal symptoms.14
A trial of the phosphodiesterase inhibitor ibudilast (not available in the United States) for methamphetamine abuse is underway. Ibudilast has anti-inflammatory activity in the peripheral immune system and the central nervous system, including modulating the activity of glial cells.15
Many medications have yielded negligible results in studies: selegiline, baclofen, sertraline, topiramate, gabapentin, rivastigmine, risperidone, and ondansetron.16 Recent evaluation of disulfiram, vigabatrin, and lobeline also has yielded inconsistent findings.17
No drug has proved effective for preventing relapse; research continues, focusing on several types of compounds that target various mechanisms: the dopamine system, the opioid system (by way of the γ-aminobutyric acid inhibitory system), and cortico-limbic reward circuitry.
Once-monthly injectable naltrexone has potential for ameliorating craving and relapse by modulating the opioid receptor system. However, the drug has not been adequately explored in generalizable settings of methamphetamine users.
Trials of oral naltrexone in Sweden have shown encouraging results, including reduced subjective effects and amphetamine use in open-label trials18,19; results were replicated in a subsequent placebo-controlled trial.20 In an unpublished study, however, no differences in amphetamine use were found among users randomized to depot naltrexone or placebo.21
Depot naltrexone with assured dosing might have a role in treating methamphetamine abuse, however; a combination of depot naltrexone and oral bupropion is being examined in a National Institute on Drug Abuse Clinical Trials Networks study that commenced in 2013. Pairing medications that have different mechanistic targets might work toward promoting cessation of methamphetamine abuse and reducing relapse once patients are abstinent.
In an early phase of research, but showing promise based on their ability to target different systems, are:
• N-acetylcysteine, modulator of the glutamate system
• D3 antagonists and partial agonists22
• varenicline.23
Potential “vaccines” against methamphetamine are in preclinical development, including use of a protein carrier or other immune-stimulating molecule to create antibodies that bind methamphetamine in the bloodstream and block its psychoactive effects.24,25
Sigma receptor effects are being studied in rodents as potential targets to mitigate effects of methamphetamine. The ligand AZ66, a sigma receptor antagonist, has demonstrated efficacy in reducing methamphetamine-induced cognitive impairment—suggesting that the sigma receptor has a potential role in ameliorating methamphetamine-related neurotoxicity.26
Psychosocial and behavioral interventions. Among the non-drug treatments that have demonstrated efficacy for treating methamphetamine abuse, cognitive-behavioral therapy (CBT) and contingency management (CM) have been most widely studied and applied in treatment settings.
CBT involves individual or group counseling that focuses on relapse prevention skills, including identification of relapse triggers, strategies to diminish cravings, and engagement in alternative non-drug activities27,28 (Table 3).
CM, which is based on positive reinforcement, offers tangible reinforcers, or rewards, for behaviors (eg, clinic attendance, providing a drug-free urine sample) according to guidelines set by the practitioner. CM-based interventions are the most reliably documented approaches for treating methamphetamine abuse,29,30 but their utility might prove to be most efficient in combination with medication— once suitable pharmacotherapeutic options emerge.
Although CBT and CM remain accepted standard treatments for methamphetamine abuse, outcomes are suboptimal.27 Both interventions have a high rate of dropout during the first month of treatment and a >50% relapse rate 6 to 19 months after treatment ends.31-33
As with treatment of other substance use disorders, patients who abuse methamphetamine can benefit from residential treatment in a drug-free setting for ≥30 days.34 In the residential approach, removing access to drugs, drug cues, and drug-using acquaintances combined with group and individual counseling reaches an inevitable end: discharge into the community. Then the patient’s battle to avoid relapse begins.
Because cognitive impairment is common among patients who abuse methamphetamine, even after they stop using,35 researchers have examined the potential for increasing participation in psychosocial interventions such as CBT by using medications that might have potential to increase cognitive function, such as modafinil.36 Increased attention and concentration afforded by medication could enhance efficacy of CBT. Results of trials and new drug development have been mixed37; no clear candidate for preventing relapse through any of the putative mechanisms of action has emerged.
Relapse is a problematic target for treatment
Ending methamphetamine abuse and sustaining abstinence from stimulants require a change in the cognitive associations that have been laid down in a drug user’s memory. Relapse occurs because of recalled memories that can be cued, or triggered, by internal or external stimuli. Eliminating drug memories, perhaps assisted by medications such as d-cycloserine (an antagonist of the N-methyl-d-aspartate receptor), could be useful for suppressing the inclination to relapse.
Last, alternative, non-drug forms of cognitive amendment have shown efficacy in preventing relapse: for example, incorporating mindfulness meditation, which has shown promise in managing craving for methamphetamine and decreasing reactivity to environmental cues for drug use.38
Bottom Line
Practitioners who work in emergency, inpatient, and outpatient settings will be called on more and more to treat acute stimulant intoxication and withdrawal, stimulant-induced psychosis, and methamphetamine abuse. Few evidence-based treatments and no FDA-approved medications are available to treat this addiction; many drugs and a few psychotherapeutic techniques have shown promise. Ongoing research promises to deliver medical and behavioral interventions to help patients quit using methamphetamine.
Related Resources
• Karch SB, Drummer O. Karch’s pathology of drug abuse, fifth ed. Boca Raton, FL: CRC Press/Taylor & David; 2013.
• Roll J, Rawson RA, Ling W, eds. Methamphetamine addiction: from basic science to treatment. New York, NY: Guilford Press; 2009.
• Sheff D. Beautiful boy: a father’s journey through his son’s addiction. New York, NY: Houghton Mifflin Harcourt Publishing Company; 2008.
• Sheff N. Tweak: growing up on methamphetamines. New York, NY: Antheneum Books for Young Readers; 2007.
• National Institute on Drug Abuse. Drugs of abuse. www. drugabuse.gov/drugs-abuse/methamphetamine.
Drug Brand Names
Baclofen • Lioresal Naltrexone (depot) • Vivitrol
Bupropion • Wellbutrin Naltrexone (oral) • ReVia
D-cycloserine • Seromycin Ondansetron • Zofran
Dexreoamphetaime • Adderall Risperidone • Risperadal
Disulfiram • Antabuse Rivastigimine • Exelon
Gabapentin • Neurontin Selegiline• EMSAM
Methylphenidate • Ritalin Sertraline • Zoloft
Mirtazapine • Remeron Topiramate • Topamax
Modafinil • Provigil Varenicline • Chantix
N-acetylcysteine • Mucomyst Vigabatrin • Sabril
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. UNODC. World Drug Report 2012 (United Nations publication, Sales No. E.12.XI.1). http://www.unodc. org/documents/data-and-analysis/WDR2012/ WDR_2012_web_small.pdf. Published 2012. Accessed August 4, 2014.
2. UNODC. World Drug Report 2010 (United Nations publication, Sales No. E.10.XI.13). http://www.unodc. org/documents/wdr/WDR_2010/World_Drug_ Report_2010_lo-res.pdf. Published 2010. Accessed August 4, 2014.
3. Rawson RA, Gonzales R, Brecht M, et al. Evaluation of the California Outcomes Measurement System (CalOMS): Final Report 2008. http://www.uclaisap.org/assets/documents/ California-ADP-DHCS-Evals/2007-2008_CalOMS%20 Report.pdf. Published 2008. Accessed August 4, 2014.
4. Shoptaw SJ, Kao U, Ling W. Treatment for amphetamine psychosis. Cochrane Database Syst Rev. 2009;(1):CD003026.
5. Leelahanaj T, Kongsakon R, Netrakom P. A 4-week, double-blind comparison of olanzapine with haloperidol in the treatment of amphetamine psychosis. J Med Assoc Thai. 2005;88(suppl 3):S43-S52.
6. McKetin R, McLaren J, Lubman D, et al. The prevalence of psychotic symptoms among methamphetamine users. Addiction. 2006;101(10):1473-1478.
7. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
8. McCann DJ, Li SH. A novel, nonbinary evaluation of success and failure reveals bupropion efficacy versus methamphetamine dependence: reanalysis of a multisite trial. CNS Neurosci Ther. 2012;18(5):414-418.
9. Tiihonen J, Kuoppasalmi K, Föhr J, et al. A comparison of aripiprazole, methylphenidate, and placebo for amphetamine dependence. Am J Psychiatry. 2007;164(1): 160-162.
10. Miles SW, Sheridan J, Russell B, et al. Extended-release methylphenidate for treatment of amphetamine/ methamphetamine dependence: a randomized, double-blind, placebo-controlled trial. Addiction. 2013;108(7): 1279-1286.
11. Ling W, Chang L, Hillhouse M, et al. Sustained-release methylphenidate in a randomized trial of treatment of methamphetamine use disorder. Addiction. 2014;109(9): 1489-1500.
12. Colfax GN, Santos GM, Das M, et al. Mirtazapine to reduce methamphetamine use: a randomized controlled trial. Arch Gen Psychiatry. 2011;68(11):1168-1175.
13. Heinzerling KG, Swanson AN, Kim S, et al. Randomized, double-blind, placebo-controlled trial of modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2010;109(1-3):20-29.
14. Galloway GP, Buscemi R, Coyle JR, et al. A randomized, placebo-controlled trial of sustained-release dextro-amphetamine for treatment of methamphetamine addiction. Clin Pharmacol Ther. 2011;89(2):276-282.
15. Snider SE, Hendrick ES, Beardsley PM. Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol. 2013;701(1-3):124-130.
16. Ling W, Rawson R, Shoptaw S. Management of methamphetamine abuse and dependence. Curr Psychiatry Rep. 2006;8(5):345-354.
17. Brackins T, Brahm NC, Kissack JC. Treatments for methamphetamine abuse: a literature review for the clinician. J Pharm Pract. 2011;24(6):541-550.
18. Jayaram-Lindström N, Wennberg P, Beck O, et al. An open clinical trial of naltrexone for amphetamine dependence: compliance and tolerability. Nord J Psychiatry. 2005;59(3):167-171.
19. Jayaram-Lindström N, Konstenius M, Eksborg S, et al. Naltrexone attenuates the subjective effects of amphetamine in patients with amphetamine dependence. Neuropsychopharmacology. 2007;33(8):1856-1863.
20. Jayaram-Lindström N, Hammarberg A, Beck O, et al. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry. 2008;165(11):1442-1448.
21. Woody GE, Tyrfingsoon P. Symposium XI: Emerging data on efficacy and clinical applications of extended-release naltrexone formulations. 75th Annual Meeting, College on Problems of Drug Dependence. June 19, 2013; San Diego, CA.
22. Newman AH, Blaylock BL, Nader MA, et al. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84(7):882-890.
23. Verrico CD, Mahoney JJ 3rd, Thompson-Lake DG, et al. Safety and efficacy of varenicline to reduce positive subjective effects produced by methamphetamine in methamphetamine-dependent volunteers. Int J Neuropsychopharmacol. 2014;17(2):223-233.
24. Miller ML, Moreno AY, Aarde S, et al. A methamphetamine vaccine attenuates methamphetamine-induced disruptions in thermoregulation and activity in rats. Biol Psychiatry. 2013;73(8):721-728.
25. Shen XY, Kosten TA, Lopez AY, et al. A vaccine against methamphetamine attenuates its behavioral effects in mice. Drug Alcohol Depend. 2013;129(1-2):41-48.
26. Seminerio MJ, Robson MJ, Abdelazeem AH, et al. Synthesis and pharmacological characterization of a novel sigma receptor ligand with improved metabolic stability and antagonistic effects against methamphetamine. AAPS J. 2012;14(1):43-51.
27. Rawson RA, Marinelli-Casey P, Anglin M, et al. A multi-site comparison of psychosocial approaches for the treatment of methamphetamine dependence. Addiction. 2004;99(6):708-717.
28. Vocci FJ, Montoya ID. Psychological treatments for stimulant misuse, comparing and contrasting those for amphetamine dependence and those for cocaine dependence. Curr Opin Psychiatry. 2009;22(3):263-268.
29. Rawson RA, McCann MJ, Flammino F, et al. A comparison of contingency management and cognitive-behavioral approaches for stimulant-dependent individuals. Addiction. 2006;101(2):267-274.
30. Roll JM, Petry NM, Stitzer ML, et al. Contingency management for the treatment of methamphetamine use disorders. Am J Psychiatry. 2006;163(11):1993-1999.
31. Brecht ML, von Mayrhauser C, Anglin MD. Predictors of relapse after treatment for methamphetamine use. J Psychoactive Drugs. 2000;32(2):211-220.
32. Smout MF, Longo M, Harrison S, et al. Psychosocial treatment for methamphetamine use disorders: a preliminary randomized controlled trial of cognitive behavior therapy and Acceptance and Commitment Therapy. Subst Abus. 2010;31(2):98-107.
33. Wang G, Shi J, Chen N, et al. Effects of length of abstinence on decision-making and craving in methamphetamine abusers. PLoS One. 2013;24;8(7):e68791. doi: 10.1371/ journal.pone.0068791.
34. McKetin R, Lubman DI, Baker AL, et al. Dose-related psychotic symptoms in chronic methamphetamine users: evidence from a prospective longitudinal study. JAMA Psychiatry. 2013;70(3):319-324.
35. Henry BL, Minassian A, Perry W. Effect of methamphetamine dependence on everyday functional ability. Addict Behav. 2010;35(6):593-598.
36. Dean AC, Sevak RJ, Monterosso JR, et al. Acute modafinil effects on attention and inhibitory control in methamphetamine-dependent humans. J Stud Alcohol Drugs. 2011;72(6):943-953.
37. Zullino DF, Benguettat D, Khazaal Y. Improvement of cognitive performance by topiramate: blockage of automatic processes may be the underlying mechanism [Comment on: Effects of topiramate on methamphetamine-induced changes in attentional and perceptual-motor skills of cognition in recently abstinent methamphetamine-dependent individuals. Prog Neuropsychopharmacol Biol Psychiatry. 2007.] Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):787.
38. Witkiewitz K, Lustyk M, Bowen S. Retraining the addicted brain: a review of hypothesized neurobiological mechanisms of mindfulness-based relapse prevention. Psychol Addict Behav. 2013;27(2):351-365.
1. UNODC. World Drug Report 2012 (United Nations publication, Sales No. E.12.XI.1). http://www.unodc. org/documents/data-and-analysis/WDR2012/ WDR_2012_web_small.pdf. Published 2012. Accessed August 4, 2014.
2. UNODC. World Drug Report 2010 (United Nations publication, Sales No. E.10.XI.13). http://www.unodc. org/documents/wdr/WDR_2010/World_Drug_ Report_2010_lo-res.pdf. Published 2010. Accessed August 4, 2014.
3. Rawson RA, Gonzales R, Brecht M, et al. Evaluation of the California Outcomes Measurement System (CalOMS): Final Report 2008. http://www.uclaisap.org/assets/documents/ California-ADP-DHCS-Evals/2007-2008_CalOMS%20 Report.pdf. Published 2008. Accessed August 4, 2014.
4. Shoptaw SJ, Kao U, Ling W. Treatment for amphetamine psychosis. Cochrane Database Syst Rev. 2009;(1):CD003026.
5. Leelahanaj T, Kongsakon R, Netrakom P. A 4-week, double-blind comparison of olanzapine with haloperidol in the treatment of amphetamine psychosis. J Med Assoc Thai. 2005;88(suppl 3):S43-S52.
6. McKetin R, McLaren J, Lubman D, et al. The prevalence of psychotic symptoms among methamphetamine users. Addiction. 2006;101(10):1473-1478.
7. Elkashef AM, Rawson RA, Anderson AL, et al. Bupropion for the treatment of methamphetamine dependence. Neuropsychopharmacology. 2008;33(5):1162-1170.
8. McCann DJ, Li SH. A novel, nonbinary evaluation of success and failure reveals bupropion efficacy versus methamphetamine dependence: reanalysis of a multisite trial. CNS Neurosci Ther. 2012;18(5):414-418.
9. Tiihonen J, Kuoppasalmi K, Föhr J, et al. A comparison of aripiprazole, methylphenidate, and placebo for amphetamine dependence. Am J Psychiatry. 2007;164(1): 160-162.
10. Miles SW, Sheridan J, Russell B, et al. Extended-release methylphenidate for treatment of amphetamine/ methamphetamine dependence: a randomized, double-blind, placebo-controlled trial. Addiction. 2013;108(7): 1279-1286.
11. Ling W, Chang L, Hillhouse M, et al. Sustained-release methylphenidate in a randomized trial of treatment of methamphetamine use disorder. Addiction. 2014;109(9): 1489-1500.
12. Colfax GN, Santos GM, Das M, et al. Mirtazapine to reduce methamphetamine use: a randomized controlled trial. Arch Gen Psychiatry. 2011;68(11):1168-1175.
13. Heinzerling KG, Swanson AN, Kim S, et al. Randomized, double-blind, placebo-controlled trial of modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2010;109(1-3):20-29.
14. Galloway GP, Buscemi R, Coyle JR, et al. A randomized, placebo-controlled trial of sustained-release dextro-amphetamine for treatment of methamphetamine addiction. Clin Pharmacol Ther. 2011;89(2):276-282.
15. Snider SE, Hendrick ES, Beardsley PM. Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol. 2013;701(1-3):124-130.
16. Ling W, Rawson R, Shoptaw S. Management of methamphetamine abuse and dependence. Curr Psychiatry Rep. 2006;8(5):345-354.
17. Brackins T, Brahm NC, Kissack JC. Treatments for methamphetamine abuse: a literature review for the clinician. J Pharm Pract. 2011;24(6):541-550.
18. Jayaram-Lindström N, Wennberg P, Beck O, et al. An open clinical trial of naltrexone for amphetamine dependence: compliance and tolerability. Nord J Psychiatry. 2005;59(3):167-171.
19. Jayaram-Lindström N, Konstenius M, Eksborg S, et al. Naltrexone attenuates the subjective effects of amphetamine in patients with amphetamine dependence. Neuropsychopharmacology. 2007;33(8):1856-1863.
20. Jayaram-Lindström N, Hammarberg A, Beck O, et al. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry. 2008;165(11):1442-1448.
21. Woody GE, Tyrfingsoon P. Symposium XI: Emerging data on efficacy and clinical applications of extended-release naltrexone formulations. 75th Annual Meeting, College on Problems of Drug Dependence. June 19, 2013; San Diego, CA.
22. Newman AH, Blaylock BL, Nader MA, et al. Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol. 2012;84(7):882-890.
23. Verrico CD, Mahoney JJ 3rd, Thompson-Lake DG, et al. Safety and efficacy of varenicline to reduce positive subjective effects produced by methamphetamine in methamphetamine-dependent volunteers. Int J Neuropsychopharmacol. 2014;17(2):223-233.
24. Miller ML, Moreno AY, Aarde S, et al. A methamphetamine vaccine attenuates methamphetamine-induced disruptions in thermoregulation and activity in rats. Biol Psychiatry. 2013;73(8):721-728.
25. Shen XY, Kosten TA, Lopez AY, et al. A vaccine against methamphetamine attenuates its behavioral effects in mice. Drug Alcohol Depend. 2013;129(1-2):41-48.
26. Seminerio MJ, Robson MJ, Abdelazeem AH, et al. Synthesis and pharmacological characterization of a novel sigma receptor ligand with improved metabolic stability and antagonistic effects against methamphetamine. AAPS J. 2012;14(1):43-51.
27. Rawson RA, Marinelli-Casey P, Anglin M, et al. A multi-site comparison of psychosocial approaches for the treatment of methamphetamine dependence. Addiction. 2004;99(6):708-717.
28. Vocci FJ, Montoya ID. Psychological treatments for stimulant misuse, comparing and contrasting those for amphetamine dependence and those for cocaine dependence. Curr Opin Psychiatry. 2009;22(3):263-268.
29. Rawson RA, McCann MJ, Flammino F, et al. A comparison of contingency management and cognitive-behavioral approaches for stimulant-dependent individuals. Addiction. 2006;101(2):267-274.
30. Roll JM, Petry NM, Stitzer ML, et al. Contingency management for the treatment of methamphetamine use disorders. Am J Psychiatry. 2006;163(11):1993-1999.
31. Brecht ML, von Mayrhauser C, Anglin MD. Predictors of relapse after treatment for methamphetamine use. J Psychoactive Drugs. 2000;32(2):211-220.
32. Smout MF, Longo M, Harrison S, et al. Psychosocial treatment for methamphetamine use disorders: a preliminary randomized controlled trial of cognitive behavior therapy and Acceptance and Commitment Therapy. Subst Abus. 2010;31(2):98-107.
33. Wang G, Shi J, Chen N, et al. Effects of length of abstinence on decision-making and craving in methamphetamine abusers. PLoS One. 2013;24;8(7):e68791. doi: 10.1371/ journal.pone.0068791.
34. McKetin R, Lubman DI, Baker AL, et al. Dose-related psychotic symptoms in chronic methamphetamine users: evidence from a prospective longitudinal study. JAMA Psychiatry. 2013;70(3):319-324.
35. Henry BL, Minassian A, Perry W. Effect of methamphetamine dependence on everyday functional ability. Addict Behav. 2010;35(6):593-598.
36. Dean AC, Sevak RJ, Monterosso JR, et al. Acute modafinil effects on attention and inhibitory control in methamphetamine-dependent humans. J Stud Alcohol Drugs. 2011;72(6):943-953.
37. Zullino DF, Benguettat D, Khazaal Y. Improvement of cognitive performance by topiramate: blockage of automatic processes may be the underlying mechanism [Comment on: Effects of topiramate on methamphetamine-induced changes in attentional and perceptual-motor skills of cognition in recently abstinent methamphetamine-dependent individuals. Prog Neuropsychopharmacol Biol Psychiatry. 2007.] Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):787.
38. Witkiewitz K, Lustyk M, Bowen S. Retraining the addicted brain: a review of hypothesized neurobiological mechanisms of mindfulness-based relapse prevention. Psychol Addict Behav. 2013;27(2):351-365.

Can what we learned about reducing no-shows in our clinic work for you?
The no-show rate is high in ambulatory psychiatric clinics, especially those associated with academic medical institutions, which usually accept all public insurance providers and do not maintain a strict rule by which patients are charged a penalty when they fail to keep a scheduled appointment—a policy that, to the contrary, is customary in private practice. The University of Texas (UT) Health Sciences Center at Houston is primarily an academic medical center with resident-managed, faculty-supervised clinics that provide care to a large volume of patients.
At the UT clinics, we have struggled with a high no-show rate, and were challenged to reduce that rate. Our study of the problem, formulation and application of strategies to reduce that rate, and a discussion of our results are provided here for the benefit of psychiatric clinicians who struggle with this problem, to the detriment of their patients’ health and the financial well-being of the practice.
For patients who have a severe psychiatric illness, such as schizophrenia or bipolar disorder, 60% to 70% of the direct cost of their care is attributable to inpatient services.1,2 Poor medication adherence is a critical factor: It results in exacerbation of symptoms, relapse, and hospitalization. The matter is compounded by patients’ failure to show up for scheduled follow-up appointments.
Studies show that failure to attend routinely scheduled outpatient appointments increases the risk of hospitalization. Recent research has shown that, among all causes of hospitalization, length of stay and relapse hospitalization are increased in patients with low adherence to their treatment regimen.3 Patients who miss an appointment also are more unwell and more functionally impaired—also contributing to a higher risk and rate of rehospitalization.4,5
To begin to address the problem at UT, we acknowledged that an elevated no-show rate is linked to medication nonadherence, increased risk of re-hospitalization, and increased costs associated with poor care.
Impact of nonadherence
Significant evidence supports the efficacy of antipsychotic medications for treating schizophrenia, of course,6 but that success story is undermined by the mean rate of medication nonadherence among schizophrenia patients, which can be as high as 49% in studies.7 (The actual rate might be higher because those studies do not account for persons who refuse treatment or drop out.)
Nonadherence increases the risk of relapse 3.7-fold, compared with what is seen in patients who adhere to treatment.8 Nonadherence to a medication regimen also can increase patients’ risk of engaging in assault and other dangerous behaviors, especially during periods of psychosis.8 Variables consistently associated with nonadherence include poor insight, negative attitude or subjective response toward medication, previous nonadherence, substance abuse, shorter duration of illness, inadequate discharge planning or after-care environment, and poorer therapeutic alliance.7,8
Investigation of medication adherence in bipolar disorder suggests that 1 in 3 patients fail to take at least 30% of their medication.9 In such patients, medication nonadherence can lead to mania, depression, hospital readmission, suicide, increased substance abuse, and nonresponse to treatment.10,11
Depression also is associated with an increased rate of health care utilization and severe limitation in daily functioning.12 Compared with non-depressed patients, depressed patients are 3 times more likely to be nonadherent with medical treatment recommendations.13 Estimates of medication nonadherence for unipolar and bipolar disorders range from 10% to 60% (median, 40%). This prevalence has not changed significantly with the introduction of new medications.14
Our literature review of research devoted to reducing no-shows found that few studies have explored this critical treatment concern. The no-show rate was higher among younger patients and slightly higher among women, but varied by diagnosis.15 The most common reason psychiatric patients gave for missing an appointment was “forgetting”—a response heard twice as often among no-show patients in psychiatry than in other specialties.4
Little has been tried to solve the problem. Often, community mental health centers and private practices double-book appointments. Double-booking is intended to reduce the financial burden on the practice when a patient misses an appointment. This approach fails to address nonadherence or the poor care that usually results when a patient misses regular outpatient appointments.
Several methods have been employed to improve adherence, such as electronic pill dispensing.16 Increasing medication adherence appears to be a key factor in improving quality-of-life measures in patients with schizophrenia.6
The UT project
Methods. This project was completed at the ambulatory psychiatry clinic at the UT Medical School at Houston. The clinic staff comprises residents and faculty members who provide outpatient care. During the study period, the clinic was scheduling as many as 800 office visits a month, including a mix of new and follow-up appointments. Two weeks’ retrospective data revealed a no-show rate of 31%.
For the project, we defined no-show rate as the total number of patients who missed an appointment or canceled fewer than 24 hours before the scheduled time, divided by the total number of patients scheduled that day.
Table 1 demonstrates the no-show rate calculations for 1 of the weeks preceding the start of the project. Given approximately 800 patient appointments a month, a 31% no-show rate meant that, first, 248 patients failed to receive recommended care and, second, 248 appointment slots were wasted.
Besides undermining such components of quality care as patient safety and medication compliance, the high no-show rate also harms employee morale and productivity; impairs medical education; and, possibly, increases the use of emergency and after-hour services.
We agreed that our current no-show rate of 31% was too high.
We then formed a team of residents, faculty members, therapists, front office staff, an office manager, and an office nurse. We explored and hypothesized what could be contributing to the high no-show rate (Table 2). 
Several interventions were then devised and implemented:
• Patients. We increased patient education about 1) the need for regular follow-up and 2) risks associated with medication nonadherence.
• Environment. We explored environmental limitations to access and agreed that certain static factors could not be modified—eg, location of the clinic and lack of access to public transportation. We were able to make some changes to the environment (explained later) to reduce wait time.
• Staff. Some patients had complained of long wait times, which could hinder active participation in treatment. We agreed that the clinic nurse would make rounds through the waiting room every hour and talk to patients. The nurse would identify patients who had been waiting for longer than 30 minutes after their scheduled appointment time and notify the doctor accordingly. We also agreed to revise patient appointment reminder practices: instead of using an automated answering service, one of the staff members called patients personally to remind them about their appointments. (This also allowed us to update telephone numbers for many patients; numbers on record often were outdated.) We initially recruited summer interns and provided a written script to follow during calls to patients, which allowed patients to confirm, cancel, or reschedule their appointment. Once we demonstrated positive results from the change to personal calls, the department agreed to absorb the cost, and front desk personnel began making reminder calls.
• Policies and procedures. Although some practices are able to charge a small fine for missed appointments, this was not allowed at our institution. Instead, we had several departmental policies on the books, such as discharging patients from our clinics if they missed 3 consecutive appointments and limiting prescription refills to a maximum of 6 months. These policies were neither communicated to patients and staff, nor were they implemented. We decided to educate patients and staff and implement the policies.
• Transparency. We posted the no-show rate in common areas so that the team could review and follow the progression of that rate as we implemented the changes. This allowed team members to take ownership of the project and facilitated active participation.
By implementing these changes, we aimed to reduce the no-show rate to 20%.
Results. We were able to reduce the no-show rate from a documented average of 31% to an average of 12% during the study period after implementing all the proposed changes in the outpatient clinics.
We calculated the no-show rate (as shown in Table 1 for May 2013), then collected the daily no-show rate from June to September 2013 (Figure). With these calculations, we demonstrated a reduction in the no-show rate to 12%. Because of the time and effort required, we reduced data collection from daily to weekly, beginning in September.
Applying the changes required consistent effort and substantial input from various stakeholders—front desk staff, residents, the nurse, therapists, and faculty. Gradually, we were able to implement all the changes.
Keeping the no-show rate low required consistent effort and monitoring of the newly implemented procedures because even a slight change, such as failure to make reminder calls, resulted in a sudden increase in the no-show rate (that was the case in October of the study period, when we were short-staffed and could not call every patient). Patients told us that it was difficult to ignore a personal call; if they were not planning to keep the appointment, the call allowed them to reschedule on the spot.
We also made sure that current no-show rates were posted in common areas, visible to team members every day.
Discussion
We attempted a literature review of research exploring approaches to reducing the no-show rate but found few studies that explored this critical concern in patient treatment.15 Some data suggested that, in the setting studied, the no-show rate:
• was higher among younger patients (age 20 to 39) than older ones (age 60 to 79)
• was slightly higher in women than in men
• varied by diagnosis.
We found a paucity of data regarding interventions that can reduce the no-show rate.
Among the changes we made, the one that had the greatest impact was personalized appointment reminder calls, as evidenced by our patients’ reports and the increase in the no-show rate when personal calls were not made.
We also realized that, although we had several departmental policies in place regarding appointments, they were not being followed. Raising awareness among team members and their patients also was an effective deterrent to a no-show for an appointment. For example, patients were informed that 3 consecutive no-shows could lead to termination of care. Often, they reacted with surprise to this caution but also voiced a desire to improve their attendance to avoid such an outcome.
We found that establishing common operational definitions is important. It also was important to have a cohesive team, with every member agreeing on goals and changes to operational policies that needed to be implemented. Support from the department chair and the administration, we learned, is vital to the success of such an intervention.
A note about limitations. The goal of the project was limited to reducing the no-show rate. We demonstrated that this is possible among patients who have a severe mental illness, and that reducing the associated waste of time and resources can improve finances in an academic department of psychiatry. We would need additional measures, however, to quantify medication adherence and hospitalization; a larger, more inclusive project is needed to demonstrate that reducing the no-show rate reduces the symptomatic burden of psychiatric illness.
Comments in conclusion
This project was designed and conducted as a required part of a Clinical Safety and Effectiveness Program at Memorial Hermann Texas Medical Center and the UT Medical School at Houston.17 Although there was initial hesitancy about attempting to reduce the no-show rate in a chronically mentally ill population, the success of this project—indeed, it surpassed its proposed goals—demonstrates that operational changes in any clinic can reduce the no-show rate. It also is important to maintain operational changes, however; without consistent effort, desired results cannot be sustained.
Last, it is possible to replicate the methodology of this project and thereby attempt to reduce the no-show rate in other divisions of medicine that offer care to chronically ill patients, such as pediatrics and family medicine.
Bottom Line
Failure to attend routinely scheduled outpatient appointments increases a patient’s functional impairment and risk of hospitalization. Patient education, appointment reminder phone calls, revised policies and procedures, and transparency regarding the no-show rate can reduce the number of missed appointments and improve patient outcomes.
Related Resources
• Mitchell AJ, Selmes T. Why don’t patients attend their appointments? Maintaining engagement with psychiatric services. Advances in Psychiatric Treatment. 2007;13:423-434.
• Molfenter T. Reducing appointment no-shows: going from theory to practice. Subst Use Misuse. 2013;48(9):743-749.
• Williston MA, Block-Lerner J, Wolanin A, et al. Brief acceptance-based intervention for increasing intake attendance at a community mental health center. Psychol Serv. 2014;11(3):324-332.
Disclosure
Dr. Gajwani receives grant or research support from the National Institute on Mental Health, the National Institute of Drug Abuse, The Stanley Foundation, and Forest Laboratories, Inc. He is a member of the speakers’ bureau of AstraZeneca, Merck, Otsuka America Pharmaceutical, and Sunovion Pharmaceuticals.
1. Wyatt RJ, Henter I. An economic evaluation of manic-depressive illness—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5):213-219.
2. Wyatt RJ, Henter I, Leary MC, et al. An economic evaluation of schizophrenia—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5);196-205.
3. Offord S, Lin J, Wong B, et al. Impact of oral antipsychotic medication adherence on healthcare resource utilization among schizophrenic patients with medicare coverage. Community Ment Health J. 2013;49(6):625-629.
4. Killaspy H, Banerjee S, King M, et al. Prospective controlled study of psychiatric out-patient non-attendance: characteristics and outcome. Br J Psychiatry. 2000;176:160- 165.
5. Nelson EA, Maruish ME, Axler JL. Effects of discharge planning and compliance with outpatient appointments on readmission rates. Psychiatr Serv. 2000;51(7):885-889.
6. Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ. 1998;317(7167):1181-1184.
7. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
8. Fenton WS, Blyler C, Heinssen RK. Determinants of medication compliance in schizophrenia: empirical and clinical findings. Schizophr Bull. 1997;23(4):637-651.
9. Scott J, Pope M. Self-reported adherence to treatment with mood stabilizers, plasma levels, and psychiatric hospitalization. Am J Psychiatry. 2002;159(11):1927-1929.
10. Adams J, Scott J. Predicting medication adherence in severe mental disorders. Acta Psychiatr Scand. 2000;101(2):119-124.
11. Müller-Oerlinghausen B, Müser-Causemann B, Volk J. Suicides and parasuicides in a high-risk patient group on and off lithium long-term treatment. J Affect Disord. 1992;25(4):261-269.
12. Manning WG Jr, Wells KB. The effects of psychological distress and psychological well-being on use of medical services. Med Care. 1992;30(6):541-553.
13. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
14. Lingam R, Scott J. Treatment non‐adherence in affective disorders. Acta Psychiatr Scand. 2002;105(3):164-172.
15. Allan AT. No-shows at a community mental health clinic: a pilot study. Int J Soc Psychiatry. 1988;34(1):40-46.
16. Cramer JA, Rosenheck R. Compliance with medication regimens for mental and physical disorders. Psychiatr Serv. 1998;49(2):196-201.
17. Gajwani P. Improving quality of care: reducing no-show rate in ambulatory psychiatry clinic. Poster presented at: American Psychiatric Association 166th Annual Meeting; May 18-22, 2013; San Francisco, CA.
The no-show rate is high in ambulatory psychiatric clinics, especially those associated with academic medical institutions, which usually accept all public insurance providers and do not maintain a strict rule by which patients are charged a penalty when they fail to keep a scheduled appointment—a policy that, to the contrary, is customary in private practice. The University of Texas (UT) Health Sciences Center at Houston is primarily an academic medical center with resident-managed, faculty-supervised clinics that provide care to a large volume of patients.
At the UT clinics, we have struggled with a high no-show rate, and were challenged to reduce that rate. Our study of the problem, formulation and application of strategies to reduce that rate, and a discussion of our results are provided here for the benefit of psychiatric clinicians who struggle with this problem, to the detriment of their patients’ health and the financial well-being of the practice.
For patients who have a severe psychiatric illness, such as schizophrenia or bipolar disorder, 60% to 70% of the direct cost of their care is attributable to inpatient services.1,2 Poor medication adherence is a critical factor: It results in exacerbation of symptoms, relapse, and hospitalization. The matter is compounded by patients’ failure to show up for scheduled follow-up appointments.
Studies show that failure to attend routinely scheduled outpatient appointments increases the risk of hospitalization. Recent research has shown that, among all causes of hospitalization, length of stay and relapse hospitalization are increased in patients with low adherence to their treatment regimen.3 Patients who miss an appointment also are more unwell and more functionally impaired—also contributing to a higher risk and rate of rehospitalization.4,5
To begin to address the problem at UT, we acknowledged that an elevated no-show rate is linked to medication nonadherence, increased risk of re-hospitalization, and increased costs associated with poor care.
Impact of nonadherence
Significant evidence supports the efficacy of antipsychotic medications for treating schizophrenia, of course,6 but that success story is undermined by the mean rate of medication nonadherence among schizophrenia patients, which can be as high as 49% in studies.7 (The actual rate might be higher because those studies do not account for persons who refuse treatment or drop out.)
Nonadherence increases the risk of relapse 3.7-fold, compared with what is seen in patients who adhere to treatment.8 Nonadherence to a medication regimen also can increase patients’ risk of engaging in assault and other dangerous behaviors, especially during periods of psychosis.8 Variables consistently associated with nonadherence include poor insight, negative attitude or subjective response toward medication, previous nonadherence, substance abuse, shorter duration of illness, inadequate discharge planning or after-care environment, and poorer therapeutic alliance.7,8
Investigation of medication adherence in bipolar disorder suggests that 1 in 3 patients fail to take at least 30% of their medication.9 In such patients, medication nonadherence can lead to mania, depression, hospital readmission, suicide, increased substance abuse, and nonresponse to treatment.10,11
Depression also is associated with an increased rate of health care utilization and severe limitation in daily functioning.12 Compared with non-depressed patients, depressed patients are 3 times more likely to be nonadherent with medical treatment recommendations.13 Estimates of medication nonadherence for unipolar and bipolar disorders range from 10% to 60% (median, 40%). This prevalence has not changed significantly with the introduction of new medications.14
Our literature review of research devoted to reducing no-shows found that few studies have explored this critical treatment concern. The no-show rate was higher among younger patients and slightly higher among women, but varied by diagnosis.15 The most common reason psychiatric patients gave for missing an appointment was “forgetting”—a response heard twice as often among no-show patients in psychiatry than in other specialties.4
Little has been tried to solve the problem. Often, community mental health centers and private practices double-book appointments. Double-booking is intended to reduce the financial burden on the practice when a patient misses an appointment. This approach fails to address nonadherence or the poor care that usually results when a patient misses regular outpatient appointments.
Several methods have been employed to improve adherence, such as electronic pill dispensing.16 Increasing medication adherence appears to be a key factor in improving quality-of-life measures in patients with schizophrenia.6
The UT project
Methods. This project was completed at the ambulatory psychiatry clinic at the UT Medical School at Houston. The clinic staff comprises residents and faculty members who provide outpatient care. During the study period, the clinic was scheduling as many as 800 office visits a month, including a mix of new and follow-up appointments. Two weeks’ retrospective data revealed a no-show rate of 31%.
For the project, we defined no-show rate as the total number of patients who missed an appointment or canceled fewer than 24 hours before the scheduled time, divided by the total number of patients scheduled that day.
Table 1 demonstrates the no-show rate calculations for 1 of the weeks preceding the start of the project. Given approximately 800 patient appointments a month, a 31% no-show rate meant that, first, 248 patients failed to receive recommended care and, second, 248 appointment slots were wasted.
Besides undermining such components of quality care as patient safety and medication compliance, the high no-show rate also harms employee morale and productivity; impairs medical education; and, possibly, increases the use of emergency and after-hour services.
We agreed that our current no-show rate of 31% was too high.
We then formed a team of residents, faculty members, therapists, front office staff, an office manager, and an office nurse. We explored and hypothesized what could be contributing to the high no-show rate (Table 2).
Several interventions were then devised and implemented:
• Patients. We increased patient education about 1) the need for regular follow-up and 2) risks associated with medication nonadherence.
• Environment. We explored environmental limitations to access and agreed that certain static factors could not be modified—eg, location of the clinic and lack of access to public transportation. We were able to make some changes to the environment (explained later) to reduce wait time.
• Staff. Some patients had complained of long wait times, which could hinder active participation in treatment. We agreed that the clinic nurse would make rounds through the waiting room every hour and talk to patients. The nurse would identify patients who had been waiting for longer than 30 minutes after their scheduled appointment time and notify the doctor accordingly. We also agreed to revise patient appointment reminder practices: instead of using an automated answering service, one of the staff members called patients personally to remind them about their appointments. (This also allowed us to update telephone numbers for many patients; numbers on record often were outdated.) We initially recruited summer interns and provided a written script to follow during calls to patients, which allowed patients to confirm, cancel, or reschedule their appointment. Once we demonstrated positive results from the change to personal calls, the department agreed to absorb the cost, and front desk personnel began making reminder calls.
• Policies and procedures. Although some practices are able to charge a small fine for missed appointments, this was not allowed at our institution. Instead, we had several departmental policies on the books, such as discharging patients from our clinics if they missed 3 consecutive appointments and limiting prescription refills to a maximum of 6 months. These policies were neither communicated to patients and staff, nor were they implemented. We decided to educate patients and staff and implement the policies.
• Transparency. We posted the no-show rate in common areas so that the team could review and follow the progression of that rate as we implemented the changes. This allowed team members to take ownership of the project and facilitated active participation.
By implementing these changes, we aimed to reduce the no-show rate to 20%.
Results. We were able to reduce the no-show rate from a documented average of 31% to an average of 12% during the study period after implementing all the proposed changes in the outpatient clinics.
We calculated the no-show rate (as shown in Table 1 for May 2013), then collected the daily no-show rate from June to September 2013 (Figure). With these calculations, we demonstrated a reduction in the no-show rate to 12%. Because of the time and effort required, we reduced data collection from daily to weekly, beginning in September.
Applying the changes required consistent effort and substantial input from various stakeholders—front desk staff, residents, the nurse, therapists, and faculty. Gradually, we were able to implement all the changes.
Keeping the no-show rate low required consistent effort and monitoring of the newly implemented procedures because even a slight change, such as failure to make reminder calls, resulted in a sudden increase in the no-show rate (that was the case in October of the study period, when we were short-staffed and could not call every patient). Patients told us that it was difficult to ignore a personal call; if they were not planning to keep the appointment, the call allowed them to reschedule on the spot.
We also made sure that current no-show rates were posted in common areas, visible to team members every day.
Discussion
We attempted a literature review of research exploring approaches to reducing the no-show rate but found few studies that explored this critical concern in patient treatment.15 Some data suggested that, in the setting studied, the no-show rate:
• was higher among younger patients (age 20 to 39) than older ones (age 60 to 79)
• was slightly higher in women than in men
• varied by diagnosis.
We found a paucity of data regarding interventions that can reduce the no-show rate.
Among the changes we made, the one that had the greatest impact was personalized appointment reminder calls, as evidenced by our patients’ reports and the increase in the no-show rate when personal calls were not made.
We also realized that, although we had several departmental policies in place regarding appointments, they were not being followed. Raising awareness among team members and their patients also was an effective deterrent to a no-show for an appointment. For example, patients were informed that 3 consecutive no-shows could lead to termination of care. Often, they reacted with surprise to this caution but also voiced a desire to improve their attendance to avoid such an outcome.
We found that establishing common operational definitions is important. It also was important to have a cohesive team, with every member agreeing on goals and changes to operational policies that needed to be implemented. Support from the department chair and the administration, we learned, is vital to the success of such an intervention.
A note about limitations. The goal of the project was limited to reducing the no-show rate. We demonstrated that this is possible among patients who have a severe mental illness, and that reducing the associated waste of time and resources can improve finances in an academic department of psychiatry. We would need additional measures, however, to quantify medication adherence and hospitalization; a larger, more inclusive project is needed to demonstrate that reducing the no-show rate reduces the symptomatic burden of psychiatric illness.
Comments in conclusion
This project was designed and conducted as a required part of a Clinical Safety and Effectiveness Program at Memorial Hermann Texas Medical Center and the UT Medical School at Houston.17 Although there was initial hesitancy about attempting to reduce the no-show rate in a chronically mentally ill population, the success of this project—indeed, it surpassed its proposed goals—demonstrates that operational changes in any clinic can reduce the no-show rate. It also is important to maintain operational changes, however; without consistent effort, desired results cannot be sustained.
Last, it is possible to replicate the methodology of this project and thereby attempt to reduce the no-show rate in other divisions of medicine that offer care to chronically ill patients, such as pediatrics and family medicine.
Bottom Line
Failure to attend routinely scheduled outpatient appointments increases a patient’s functional impairment and risk of hospitalization. Patient education, appointment reminder phone calls, revised policies and procedures, and transparency regarding the no-show rate can reduce the number of missed appointments and improve patient outcomes.
Related Resources
• Mitchell AJ, Selmes T. Why don’t patients attend their appointments? Maintaining engagement with psychiatric services. Advances in Psychiatric Treatment. 2007;13:423-434.
• Molfenter T. Reducing appointment no-shows: going from theory to practice. Subst Use Misuse. 2013;48(9):743-749.
• Williston MA, Block-Lerner J, Wolanin A, et al. Brief acceptance-based intervention for increasing intake attendance at a community mental health center. Psychol Serv. 2014;11(3):324-332.
Disclosure
Dr. Gajwani receives grant or research support from the National Institute on Mental Health, the National Institute of Drug Abuse, The Stanley Foundation, and Forest Laboratories, Inc. He is a member of the speakers’ bureau of AstraZeneca, Merck, Otsuka America Pharmaceutical, and Sunovion Pharmaceuticals.
The no-show rate is high in ambulatory psychiatric clinics, especially those associated with academic medical institutions, which usually accept all public insurance providers and do not maintain a strict rule by which patients are charged a penalty when they fail to keep a scheduled appointment—a policy that, to the contrary, is customary in private practice. The University of Texas (UT) Health Sciences Center at Houston is primarily an academic medical center with resident-managed, faculty-supervised clinics that provide care to a large volume of patients.
At the UT clinics, we have struggled with a high no-show rate, and were challenged to reduce that rate. Our study of the problem, formulation and application of strategies to reduce that rate, and a discussion of our results are provided here for the benefit of psychiatric clinicians who struggle with this problem, to the detriment of their patients’ health and the financial well-being of the practice.
For patients who have a severe psychiatric illness, such as schizophrenia or bipolar disorder, 60% to 70% of the direct cost of their care is attributable to inpatient services.1,2 Poor medication adherence is a critical factor: It results in exacerbation of symptoms, relapse, and hospitalization. The matter is compounded by patients’ failure to show up for scheduled follow-up appointments.
Studies show that failure to attend routinely scheduled outpatient appointments increases the risk of hospitalization. Recent research has shown that, among all causes of hospitalization, length of stay and relapse hospitalization are increased in patients with low adherence to their treatment regimen.3 Patients who miss an appointment also are more unwell and more functionally impaired—also contributing to a higher risk and rate of rehospitalization.4,5
To begin to address the problem at UT, we acknowledged that an elevated no-show rate is linked to medication nonadherence, increased risk of re-hospitalization, and increased costs associated with poor care.
Impact of nonadherence
Significant evidence supports the efficacy of antipsychotic medications for treating schizophrenia, of course,6 but that success story is undermined by the mean rate of medication nonadherence among schizophrenia patients, which can be as high as 49% in studies.7 (The actual rate might be higher because those studies do not account for persons who refuse treatment or drop out.)
Nonadherence increases the risk of relapse 3.7-fold, compared with what is seen in patients who adhere to treatment.8 Nonadherence to a medication regimen also can increase patients’ risk of engaging in assault and other dangerous behaviors, especially during periods of psychosis.8 Variables consistently associated with nonadherence include poor insight, negative attitude or subjective response toward medication, previous nonadherence, substance abuse, shorter duration of illness, inadequate discharge planning or after-care environment, and poorer therapeutic alliance.7,8
Investigation of medication adherence in bipolar disorder suggests that 1 in 3 patients fail to take at least 30% of their medication.9 In such patients, medication nonadherence can lead to mania, depression, hospital readmission, suicide, increased substance abuse, and nonresponse to treatment.10,11
Depression also is associated with an increased rate of health care utilization and severe limitation in daily functioning.12 Compared with non-depressed patients, depressed patients are 3 times more likely to be nonadherent with medical treatment recommendations.13 Estimates of medication nonadherence for unipolar and bipolar disorders range from 10% to 60% (median, 40%). This prevalence has not changed significantly with the introduction of new medications.14
Our literature review of research devoted to reducing no-shows found that few studies have explored this critical treatment concern. The no-show rate was higher among younger patients and slightly higher among women, but varied by diagnosis.15 The most common reason psychiatric patients gave for missing an appointment was “forgetting”—a response heard twice as often among no-show patients in psychiatry than in other specialties.4
Little has been tried to solve the problem. Often, community mental health centers and private practices double-book appointments. Double-booking is intended to reduce the financial burden on the practice when a patient misses an appointment. This approach fails to address nonadherence or the poor care that usually results when a patient misses regular outpatient appointments.
Several methods have been employed to improve adherence, such as electronic pill dispensing.16 Increasing medication adherence appears to be a key factor in improving quality-of-life measures in patients with schizophrenia.6
The UT project
Methods. This project was completed at the ambulatory psychiatry clinic at the UT Medical School at Houston. The clinic staff comprises residents and faculty members who provide outpatient care. During the study period, the clinic was scheduling as many as 800 office visits a month, including a mix of new and follow-up appointments. Two weeks’ retrospective data revealed a no-show rate of 31%.
For the project, we defined no-show rate as the total number of patients who missed an appointment or canceled fewer than 24 hours before the scheduled time, divided by the total number of patients scheduled that day.
Table 1 demonstrates the no-show rate calculations for 1 of the weeks preceding the start of the project. Given approximately 800 patient appointments a month, a 31% no-show rate meant that, first, 248 patients failed to receive recommended care and, second, 248 appointment slots were wasted.
Besides undermining such components of quality care as patient safety and medication compliance, the high no-show rate also harms employee morale and productivity; impairs medical education; and, possibly, increases the use of emergency and after-hour services.
We agreed that our current no-show rate of 31% was too high.
We then formed a team of residents, faculty members, therapists, front office staff, an office manager, and an office nurse. We explored and hypothesized what could be contributing to the high no-show rate (Table 2).
Several interventions were then devised and implemented:
• Patients. We increased patient education about 1) the need for regular follow-up and 2) risks associated with medication nonadherence.
• Environment. We explored environmental limitations to access and agreed that certain static factors could not be modified—eg, location of the clinic and lack of access to public transportation. We were able to make some changes to the environment (explained later) to reduce wait time.
• Staff. Some patients had complained of long wait times, which could hinder active participation in treatment. We agreed that the clinic nurse would make rounds through the waiting room every hour and talk to patients. The nurse would identify patients who had been waiting for longer than 30 minutes after their scheduled appointment time and notify the doctor accordingly. We also agreed to revise patient appointment reminder practices: instead of using an automated answering service, one of the staff members called patients personally to remind them about their appointments. (This also allowed us to update telephone numbers for many patients; numbers on record often were outdated.) We initially recruited summer interns and provided a written script to follow during calls to patients, which allowed patients to confirm, cancel, or reschedule their appointment. Once we demonstrated positive results from the change to personal calls, the department agreed to absorb the cost, and front desk personnel began making reminder calls.
• Policies and procedures. Although some practices are able to charge a small fine for missed appointments, this was not allowed at our institution. Instead, we had several departmental policies on the books, such as discharging patients from our clinics if they missed 3 consecutive appointments and limiting prescription refills to a maximum of 6 months. These policies were neither communicated to patients and staff, nor were they implemented. We decided to educate patients and staff and implement the policies.
• Transparency. We posted the no-show rate in common areas so that the team could review and follow the progression of that rate as we implemented the changes. This allowed team members to take ownership of the project and facilitated active participation.
By implementing these changes, we aimed to reduce the no-show rate to 20%.
Results. We were able to reduce the no-show rate from a documented average of 31% to an average of 12% during the study period after implementing all the proposed changes in the outpatient clinics.
We calculated the no-show rate (as shown in Table 1 for May 2013), then collected the daily no-show rate from June to September 2013 (Figure). With these calculations, we demonstrated a reduction in the no-show rate to 12%. Because of the time and effort required, we reduced data collection from daily to weekly, beginning in September.
Applying the changes required consistent effort and substantial input from various stakeholders—front desk staff, residents, the nurse, therapists, and faculty. Gradually, we were able to implement all the changes.
Keeping the no-show rate low required consistent effort and monitoring of the newly implemented procedures because even a slight change, such as failure to make reminder calls, resulted in a sudden increase in the no-show rate (that was the case in October of the study period, when we were short-staffed and could not call every patient). Patients told us that it was difficult to ignore a personal call; if they were not planning to keep the appointment, the call allowed them to reschedule on the spot.
We also made sure that current no-show rates were posted in common areas, visible to team members every day.
Discussion
We attempted a literature review of research exploring approaches to reducing the no-show rate but found few studies that explored this critical concern in patient treatment.15 Some data suggested that, in the setting studied, the no-show rate:
• was higher among younger patients (age 20 to 39) than older ones (age 60 to 79)
• was slightly higher in women than in men
• varied by diagnosis.
We found a paucity of data regarding interventions that can reduce the no-show rate.
Among the changes we made, the one that had the greatest impact was personalized appointment reminder calls, as evidenced by our patients’ reports and the increase in the no-show rate when personal calls were not made.
We also realized that, although we had several departmental policies in place regarding appointments, they were not being followed. Raising awareness among team members and their patients also was an effective deterrent to a no-show for an appointment. For example, patients were informed that 3 consecutive no-shows could lead to termination of care. Often, they reacted with surprise to this caution but also voiced a desire to improve their attendance to avoid such an outcome.
We found that establishing common operational definitions is important. It also was important to have a cohesive team, with every member agreeing on goals and changes to operational policies that needed to be implemented. Support from the department chair and the administration, we learned, is vital to the success of such an intervention.
A note about limitations. The goal of the project was limited to reducing the no-show rate. We demonstrated that this is possible among patients who have a severe mental illness, and that reducing the associated waste of time and resources can improve finances in an academic department of psychiatry. We would need additional measures, however, to quantify medication adherence and hospitalization; a larger, more inclusive project is needed to demonstrate that reducing the no-show rate reduces the symptomatic burden of psychiatric illness.
Comments in conclusion
This project was designed and conducted as a required part of a Clinical Safety and Effectiveness Program at Memorial Hermann Texas Medical Center and the UT Medical School at Houston.17 Although there was initial hesitancy about attempting to reduce the no-show rate in a chronically mentally ill population, the success of this project—indeed, it surpassed its proposed goals—demonstrates that operational changes in any clinic can reduce the no-show rate. It also is important to maintain operational changes, however; without consistent effort, desired results cannot be sustained.
Last, it is possible to replicate the methodology of this project and thereby attempt to reduce the no-show rate in other divisions of medicine that offer care to chronically ill patients, such as pediatrics and family medicine.
Bottom Line
Failure to attend routinely scheduled outpatient appointments increases a patient’s functional impairment and risk of hospitalization. Patient education, appointment reminder phone calls, revised policies and procedures, and transparency regarding the no-show rate can reduce the number of missed appointments and improve patient outcomes.
Related Resources
• Mitchell AJ, Selmes T. Why don’t patients attend their appointments? Maintaining engagement with psychiatric services. Advances in Psychiatric Treatment. 2007;13:423-434.
• Molfenter T. Reducing appointment no-shows: going from theory to practice. Subst Use Misuse. 2013;48(9):743-749.
• Williston MA, Block-Lerner J, Wolanin A, et al. Brief acceptance-based intervention for increasing intake attendance at a community mental health center. Psychol Serv. 2014;11(3):324-332.
Disclosure
Dr. Gajwani receives grant or research support from the National Institute on Mental Health, the National Institute of Drug Abuse, The Stanley Foundation, and Forest Laboratories, Inc. He is a member of the speakers’ bureau of AstraZeneca, Merck, Otsuka America Pharmaceutical, and Sunovion Pharmaceuticals.
1. Wyatt RJ, Henter I. An economic evaluation of manic-depressive illness—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5):213-219.
2. Wyatt RJ, Henter I, Leary MC, et al. An economic evaluation of schizophrenia—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5);196-205.
3. Offord S, Lin J, Wong B, et al. Impact of oral antipsychotic medication adherence on healthcare resource utilization among schizophrenic patients with medicare coverage. Community Ment Health J. 2013;49(6):625-629.
4. Killaspy H, Banerjee S, King M, et al. Prospective controlled study of psychiatric out-patient non-attendance: characteristics and outcome. Br J Psychiatry. 2000;176:160- 165.
5. Nelson EA, Maruish ME, Axler JL. Effects of discharge planning and compliance with outpatient appointments on readmission rates. Psychiatr Serv. 2000;51(7):885-889.
6. Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ. 1998;317(7167):1181-1184.
7. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
8. Fenton WS, Blyler C, Heinssen RK. Determinants of medication compliance in schizophrenia: empirical and clinical findings. Schizophr Bull. 1997;23(4):637-651.
9. Scott J, Pope M. Self-reported adherence to treatment with mood stabilizers, plasma levels, and psychiatric hospitalization. Am J Psychiatry. 2002;159(11):1927-1929.
10. Adams J, Scott J. Predicting medication adherence in severe mental disorders. Acta Psychiatr Scand. 2000;101(2):119-124.
11. Müller-Oerlinghausen B, Müser-Causemann B, Volk J. Suicides and parasuicides in a high-risk patient group on and off lithium long-term treatment. J Affect Disord. 1992;25(4):261-269.
12. Manning WG Jr, Wells KB. The effects of psychological distress and psychological well-being on use of medical services. Med Care. 1992;30(6):541-553.
13. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
14. Lingam R, Scott J. Treatment non‐adherence in affective disorders. Acta Psychiatr Scand. 2002;105(3):164-172.
15. Allan AT. No-shows at a community mental health clinic: a pilot study. Int J Soc Psychiatry. 1988;34(1):40-46.
16. Cramer JA, Rosenheck R. Compliance with medication regimens for mental and physical disorders. Psychiatr Serv. 1998;49(2):196-201.
17. Gajwani P. Improving quality of care: reducing no-show rate in ambulatory psychiatry clinic. Poster presented at: American Psychiatric Association 166th Annual Meeting; May 18-22, 2013; San Francisco, CA.
1. Wyatt RJ, Henter I. An economic evaluation of manic-depressive illness—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5):213-219.
2. Wyatt RJ, Henter I, Leary MC, et al. An economic evaluation of schizophrenia—1991. Soc Psychiatry Psychiatr Epidemiol. 1995;30(5);196-205.
3. Offord S, Lin J, Wong B, et al. Impact of oral antipsychotic medication adherence on healthcare resource utilization among schizophrenic patients with medicare coverage. Community Ment Health J. 2013;49(6):625-629.
4. Killaspy H, Banerjee S, King M, et al. Prospective controlled study of psychiatric out-patient non-attendance: characteristics and outcome. Br J Psychiatry. 2000;176:160- 165.
5. Nelson EA, Maruish ME, Axler JL. Effects of discharge planning and compliance with outpatient appointments on readmission rates. Psychiatr Serv. 2000;51(7):885-889.
6. Thornley B, Adams C. Content and quality of 2000 controlled trials in schizophrenia over 50 years. BMJ. 1998;317(7167):1181-1184.
7. Lacro JP, Dunn LB, Dolder CR, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63(10):892-909.
8. Fenton WS, Blyler C, Heinssen RK. Determinants of medication compliance in schizophrenia: empirical and clinical findings. Schizophr Bull. 1997;23(4):637-651.
9. Scott J, Pope M. Self-reported adherence to treatment with mood stabilizers, plasma levels, and psychiatric hospitalization. Am J Psychiatry. 2002;159(11):1927-1929.
10. Adams J, Scott J. Predicting medication adherence in severe mental disorders. Acta Psychiatr Scand. 2000;101(2):119-124.
11. Müller-Oerlinghausen B, Müser-Causemann B, Volk J. Suicides and parasuicides in a high-risk patient group on and off lithium long-term treatment. J Affect Disord. 1992;25(4):261-269.
12. Manning WG Jr, Wells KB. The effects of psychological distress and psychological well-being on use of medical services. Med Care. 1992;30(6):541-553.
13. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160(14):2101-2107.
14. Lingam R, Scott J. Treatment non‐adherence in affective disorders. Acta Psychiatr Scand. 2002;105(3):164-172.
15. Allan AT. No-shows at a community mental health clinic: a pilot study. Int J Soc Psychiatry. 1988;34(1):40-46.
16. Cramer JA, Rosenheck R. Compliance with medication regimens for mental and physical disorders. Psychiatr Serv. 1998;49(2):196-201.
17. Gajwani P. Improving quality of care: reducing no-show rate in ambulatory psychiatry clinic. Poster presented at: American Psychiatric Association 166th Annual Meeting; May 18-22, 2013; San Francisco, CA.

Myelodysplastic Syndromes
Myelodysplastic syndromes (MDS) are a spectrum of clonal myeloid disorders characterized by ineffective hematopoiesis, cytopenias, qualitative disorders of blood cells, clonal chromosomal abnormalities, and the potential for clonal evolution to acute myeloid leukemia (AML). In this review, we discuss the various pathogenic conditions included in the spectrum of MDS and the associated risk stratification for these conditions. We further discuss the treatment recommendations based on the risk status and the expected prognosis.
To read the full article in PDF:
Myelodysplastic syndromes (MDS) are a spectrum of clonal myeloid disorders characterized by ineffective hematopoiesis, cytopenias, qualitative disorders of blood cells, clonal chromosomal abnormalities, and the potential for clonal evolution to acute myeloid leukemia (AML). In this review, we discuss the various pathogenic conditions included in the spectrum of MDS and the associated risk stratification for these conditions. We further discuss the treatment recommendations based on the risk status and the expected prognosis.
To read the full article in PDF:
Myelodysplastic syndromes (MDS) are a spectrum of clonal myeloid disorders characterized by ineffective hematopoiesis, cytopenias, qualitative disorders of blood cells, clonal chromosomal abnormalities, and the potential for clonal evolution to acute myeloid leukemia (AML). In this review, we discuss the various pathogenic conditions included in the spectrum of MDS and the associated risk stratification for these conditions. We further discuss the treatment recommendations based on the risk status and the expected prognosis.
To read the full article in PDF:
Primary Brain Tumors
Series Editor: Arthur T. Skarin, MD, FACP, FCCP
Primary central nervous system tumors are relatively rare, but they can cause significant morbidity. They are also among the most lethal of all neoplasms. Brain tumors are the second most common cause of death due to intracranial disease, second only to stroke. The estimated annual incidence of primary brain tumors is approximately 21 per 100,000 individuals in the United States. The incidence of brain tumors varies by gender, age, race, ethnicity, and geography and has increased over time. Gliomas and germ cell tumors are more common in men, whereas meningiomas are twice as common in women. The only validated environmental risk factor for primary brain tumors is exposure to ionizing radiation.
To read the full article in PDF:
Series Editor: Arthur T. Skarin, MD, FACP, FCCP
Primary central nervous system tumors are relatively rare, but they can cause significant morbidity. They are also among the most lethal of all neoplasms. Brain tumors are the second most common cause of death due to intracranial disease, second only to stroke. The estimated annual incidence of primary brain tumors is approximately 21 per 100,000 individuals in the United States. The incidence of brain tumors varies by gender, age, race, ethnicity, and geography and has increased over time. Gliomas and germ cell tumors are more common in men, whereas meningiomas are twice as common in women. The only validated environmental risk factor for primary brain tumors is exposure to ionizing radiation.
To read the full article in PDF:
Series Editor: Arthur T. Skarin, MD, FACP, FCCP
Primary central nervous system tumors are relatively rare, but they can cause significant morbidity. They are also among the most lethal of all neoplasms. Brain tumors are the second most common cause of death due to intracranial disease, second only to stroke. The estimated annual incidence of primary brain tumors is approximately 21 per 100,000 individuals in the United States. The incidence of brain tumors varies by gender, age, race, ethnicity, and geography and has increased over time. Gliomas and germ cell tumors are more common in men, whereas meningiomas are twice as common in women. The only validated environmental risk factor for primary brain tumors is exposure to ionizing radiation.
To read the full article in PDF:
New-onset epilepsy in the elderly: Challenges for the internist
Contrary to the popular belief that epilepsy is mainly a disease of youth, nearly 25% of new-onset seizures occur after age 65.1,2 The incidence of epilepsy in this age group is almost twice the rate in children, and in people over age 80, it is triple the rate in children.3 As our population ages, the burden of “elderly-onset” epilepsy will rise.
A seizure diagnosis carries significant implications in older people, who are already vulnerable to cognitive decline, loss of functional independence, driving restrictions, and risk of falls. Newly diagnosed epilepsy further worsens quality of life.4
The causes and clinical manifestations of seizures and epilepsy in the elderly differ from those in younger people.5 Hence, it is often difficult to make a diagnosis with certainty from a wide range of differential diagnoses. Older people are also more likely to have comorbidities, further complicating the situation.
Managing seizures in the elderly is also challenging, as age-associated physiologic changes can affect the pharmacokinetics and pharmacodynamics of antiepileptic drugs. Diagnosing and managing elderly-onset epilepsy can be challenging for a family physician, an internist, a geriatrician, or even a neurologist.
In this review, we emphasize the common causes of new-onset epilepsy in the elderly and the assessment of the clinical clues that are essential for making an accurate diagnosis. We also review the pharmacology of antiepileptic drugs used in old age and highlight the need for psychological support for patients and caregivers.
RISING PREVALENCE IN THE ELDERLY
In US Medicare beneficiaries age 65 and older, the average annual incidence rate of epilepsy in 2001 to 2005 was 10.8 per 1,000.6 A large study in Finland revealed falling incidence rates of epilepsy in childhood and middle age and rising trends in the elderly.7
In the United States, the rates are higher in African Americans (18.7 per 1,000) and lower in Asian Americans and Native Americans (5.5 and 7.7 per 1,000) than in whites (10.2 per 1,000).6 Incidence rates are slightly higher for women than for men and increase with age in both sexes and all racial groups.
Acute symptomatic seizure is also common in older patients. The incidence of acute seizures in patients over age 60 was estimated at 50 to 100 per 100,000 per year in one study.7 The rate was considerably higher in men than in women. The study also found a 3.6% risk of experiencing an acute symptomatic seizure in an 80-year lifespan, which approaches that of developing epilepsy.8 The major causes of acute symptomatic seizure were traumatic brain injury, cerebrovascular disease, drug withdrawal, and central nervous system infection.
CAUSES OF NEW-ONSET EPILEPSY IN THE ELDERLY
The most common causes of new-onset epilepsy in the elderly include cerebrovascular disease, metabolic disturbances, dementia, traumatic brain injury, tumors, and drugs.3,9–11
Cerebrovascular disease
In older adults, acute stroke is the most common cause, accounting for up to half of cases.5,12
Seizures occur in 4.4% to 8.9% of acute cerebrovascular events.13,14 The risk varies by stroke subtype, although all stroke subtypes, including transient ischemic attack, can be associated with seizure.15 For example, although 1% to 2% of patients experienced a seizure within 15 days of a transient ischemic attack or a lacunar infarct, this risk was 16.6% after an embolic stroke.15
Beyond this increased risk of “acute seizure” in the immediate poststroke period (usually defined as 1 week), the risk of epilepsy was also 20 times higher in the first year after a stroke.14 However, seizures tend to occur within the first 48 hours after the onset of ischemic stroke. In subarachnoid hemorrhage, seizures generally occur within hours.16
In a population-based study in Rochester, NY,17 epilepsy developed in two-thirds of patients with seizure related to acute stroke. Two factors that independently predicted the development of epilepsy were early seizure occurrence and recurrence of stroke.
Interestingly, the risk of stroke was three times higher in older patients who had new-onset seizure.18 Therefore, any elderly person with new-onset seizure should be assessed for cerebrovascular risk factors and treated accordingly for stroke prevention.
Metabolic disturbances
Acute metabolic disorders are common in elderly patients because of multiple comorbidities and polypharmacy. Hypoglycemia and hyponatremia need to be particularly considered in this population.19
Other well-documented metabolic causes of acute seizure, including nonketotic hyperglycemia, hypocalcemia, and uremic or hepatic encephalopathy, can all be considerations, albeit less specific to this age group.
Dementia
Primary neurodegenerative disorders associated with cognitive impairment, such as Alzheimer disease, are major risk factors for new-onset epilepsy in older patients.3,5 Seizures occur in about 10% of Alzheimer patients.20 Those who have brief periods of increased confusion may actually be experiencing unrecognized complex partial seizures.21
A case-control study discovered incidence rates of epilepsy almost 10 times higher in patients who had Alzheimer disease or vascular dementia than in nondemented patients.22 A prospective cohort study in patients with mild to moderate Alzheimer disease established that younger age, a greater degree of cognitive impairment, and a history of antipsychotic use were independent risk factors for new-onset seizures in the elderly.23 Preexisting dementia also increases the risk of poststroke epilepsy.24
Traumatic brain injury
The most common cause of brain trauma in the elderly is falls. Subdural hematoma, which can occur in the elderly with trivial trauma or sometimes even without it, needs to be considered. The risk of posttraumatic hemorrhage is especially relevant in patients taking anticoagulants.
Traumatic brain injury has a poorer prognosis in older people than in the young,25 and it accounts for up to 20% of cases of epilepsy in the elderly.26 Although no study has specifically addressed the longitudinal risk of epilepsy after traumatic brain injury in the elderly, a study in children and young adults revealed the risk was highest in the first year, with the increased risk persisting for more than 10 years.27
Brain tumors
Between 10% and 30% of new-onset seizures in the elderly are associated with tumor, typically glioma, meningioma, and brain metastasis.28,29 Seizures are usually associated more with primary than with secondary tumors, and more with low-grade tumors than high-grade ones.30
Drug-induced
Drugs and drug withdrawal can contribute to up to 10% of acute symptomatic seizures in the geriatric population.5,8,29 The elderly are susceptible to drug-induced seizure because of a higher prevalence of polypharmacy, impaired drug clearance, and heightened sensitivity to the proconvulsant side effects of medications.1 A number of commonly used drugs have been implicated,31 including:
Antibiotics such as carbapenems and high-dose penicillin
Antihistamines such as desloratadine (Clarinex)
Pain medications such as tramadol (Ul-tram) and high-dose opiates
Neuromodulators
Antidepressants such as clomipramine (Anafranil), maprotiline (Ludiomil), amoxapine (Asendin), and bupropion (Wellbutrin).32
Seizures also follow alcohol, benzodiazepine, and barbiturate withdrawal.33
Other causes
Paraneoplastic limbic encephalitis is a rare cause of seizures in the elderly.34 It can present with refractory seizures, confusion, and behavioral changes with or without a known concurrent neoplastic disease.
Posterior reversible leukoencephalopathy syndrome, another rare consideration, can particularly affect immunosuppressed elderly patients. This syndrome is characterized clinically by headache, confusion, seizures, vomiting, and visual disturbances with radiographic vasogenic edema.35
CLINICAL PRESENTATION
The signs and symptoms of a seizure may be atypical in the elderly. Seizures more often have a picture of “epileptic amnesia,” with confusion, sleepiness, or clumsiness, rather than motor manifestations such as tonic stiffening or automatism.36,37 Postictal states are also prolonged, particularly if there is underlying brain dysfunction.38 All these features render the clinical seizure manifestations more subtle and, as such, more difficult for the uninitiated caregiver to identify.
Convulsive and nonconvulsive status epilepticus
Status epilepticus is defined as a single generalized seizure lasting more than 5 minutes or a series of seizures lasting longer than 30 minutes without the patient’s regaining consciousness.39 The greatest increase in the incidence of status epilepticus occurs after age 60.40 It is the first seizure in about 30% of new-onset seizures in the elderly.41
Mortality rates increase with age, anoxia, and duration of status epilepticus and are over 50% in patients age 80 and older.40,42
Convulsive status epilepticus is most commonly caused by stroke.40
Absence status epilepticus can occur in elderly patients as a late complication of idiopathic generalized epilepsy related to benzodiazepine withdrawal, alcohol intoxication, or initiation of psychotropic drugs.42
Nonconvulsive status epilepticus manifests as altered mental status, psychosis, lethargy, or coma.42–44 Occasionally, it presents as a more focal cognitive disturbance with aphasia or a neglect syndrome.42,45 Electroencephalographic correlates of nonconvulsive status epilepticus include focal rhythmic discharges, often arising from frontal or temporal lobes, or generalized spike or sharp and slow-wave activity.46 Its management is challenging because of delayed diagnosis or misdiagnosis. The risk of death is higher in patients with severely impaired mental status or acute complications.47

Table 1 lists the typical seizure manifestations peculiar to the elderly.37,48
Differential diagnosis of new-onset epilepsy in the elderly
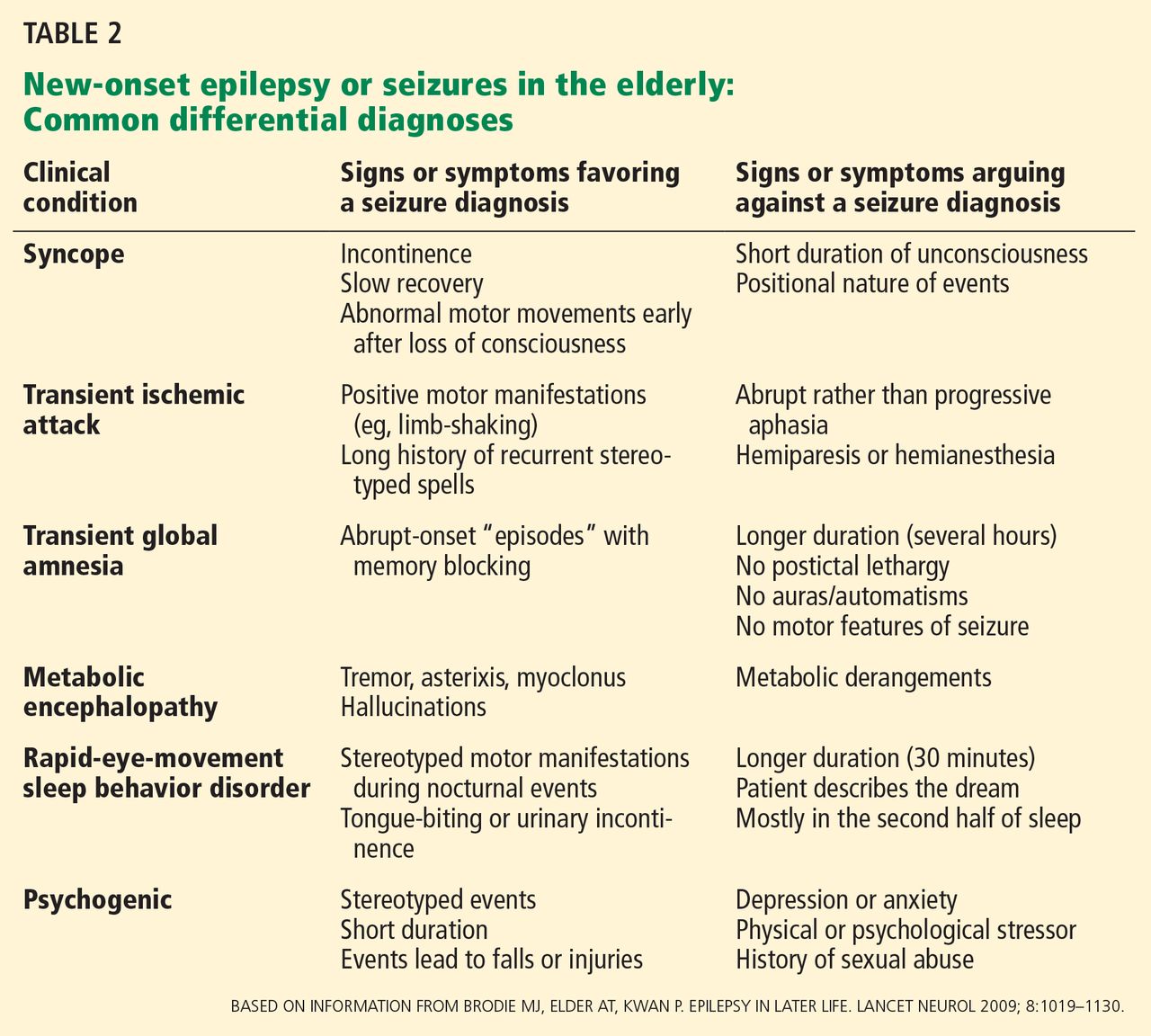
New-onset epilepsy in elderly patients can be confused with syncope, transient ischemic attack, cardiac arrhythmia, metabolic disturbances, transient global amnesia, neurodegenerative disease, rapid-eye-movement sleep behavior disorder, psychogenic disorders, and other conditions (Table 2). If there is a high clinical suspicion of seizure, the patient should undergo electroencephalography (EEG) and be referred to a neurologist or epileptologist.
KEYS TO THE DIAGNOSIS
Clinical history
A reliable history and description of the event from an eyewitness or a video recording of the event are invaluable to the diagnosis of epileptic seizure. Signs and symptoms that suggest the diagnosis include aura, ictal pallor, urinary incontinence, tongue-biting, and motor symptoms, as well as postictal confusion, drowsiness, and speech disturbance.
Electroencephalography
EEG is the most useful diagnostic tool in epilepsy. However, an interictal EEG reading (ie, between epileptic attacks) in an elderly patient has limited utility, showing epileptiform activity in only about one-fourth of patients.49 Nonspecific EEG abnormalities such as intermittent focal slowing are seen in many older people even without seizure.50 Also, normal findings on outpatient EEG do not rule out epilepsy, as EEG is normal in about one-third of patients with epilepsy, irrespective of age.1,49 Activation procedures such as hyperventilation and photic stimulation add little to the diagnosis in the elderly.49
On the other hand, video-EEG monitoring is an excellent tool for evaluating possible epilepsy, as it allows accurate assessment of brain electrical activity during the events in question. Moreover, studies of video-EEG recording of seizures in elderly patients demonstrated epileptiform discharges on EEG in 76% of clinical ictal events.50
Therefore, routine EEG is a useful screening tool, and inpatient video-EEG monitoring is the gold standard to characterize events of concern and distinguish between epileptic and nonepileptic or psychogenic seizures.
Other diagnostic studies
Brain imaging, preferably magnetic resonance imaging with contrast, should be done in every patient with possible epilepsy due to stroke, traumatic brain injury, or other structural brain disease.51
Electrocardiography helps exclude cardiac causes such as arrhythmia.
Blood testing. Metabolically provoked seizure can be distinguished by blood analysis for electrolytes, blood urea nitrogen, creatinine, glucose, calcium, magnesium, liver enzymes, and drug levels (eg, ethanol). A complete blood cell count with differential and platelets should also be done in anticipation of starting antiepileptic drug therapy.
Lumbar puncture for cell count, protein, glucose, stains, and cultures should be performed whenever meningitis or encephalitis is suspected.

A sleep study with concurrent video-EEG monitoring may be required to distinguish epileptic seizures from sleep disorders.
Neuropsychological testing may help account for the degree of cognitive impairment present.
Risk factors for stroke should be assessed in every elderly person who has new-onset seizures, because the risk of stroke is high.17
Figure 1 shows the workup for an elderly patient with suspected new-onset epilepsy.
TREATING EPILEPSY IN THE ELDERLY
Therapeutic challenges
Age-associated changes in drug absorption, protein binding, and distribution in body compartments require adjustments in drug selection and dosage. The causes and manifestations of these changes are typically multifactorial, mainly related to altered metabolism, declining plasma albumin concentrations, and increasing competition for protein binding by concomitantly used drugs.
The differences in the pharmacokinetics and pharmacodynamics of antiepileptic drugs depend on the patient’s physical status, relevant comorbidities, and concomitant medications.52 Renal and hepatic function may decline in an elderly patient; accordingly, precaution is needed in the prescribing and dosing of antiepileptic drugs.
Adverse effects from seizure medications are twice as common in elderly patients compared with younger patients. Ataxia, tremor, visual disturbance, and sedation are the most common.1 Antiepileptic drugs are also harmful to bone; induced abnormalities in bone metabolism include hypocalcemia, hypophosphatemia, decreased levels of active vitamin D metabolites, and hyperparathyroidism.53
Elderly patients tend to take multiple drugs, and some drugs can lower the seizure threshold, particularly antidepressants, anti-psychotics, and antibiotics.32 The herbal remedy ginkgo biloba can also precipitate seizure in this population.54
Antiepileptic drugs such as phenobarbital, primidone (Mysoline), phenytoin (Dilantin), and carbamazepine (Tegretol) can be broad-spectrum enzyme-inducers, increasing the metabolism of many drugs, including warfarin (Coumadin), cytotoxic agents, statins, cardiac antiarrhythmics, antihypertensives, corticosteroids, and other immunosuppressants.55 For example, carbamazepine can alter the metabolism of several hepatically metabolized drugs and cause significant hyponatremia. This is problematic in patients already taking sodium-depleting antihypertensives. Age-related cognitive decline can worsen the situation, often leading to misdiagnosis or patient noncompliance.

Table 3 profiles the interactions of commonly used antiepileptic drugs.
The ideal pharmacotherapy
No single drug is ideal for elderly patients with new-onset epilepsy. The choice mostly depends on the type of seizure and the patient’s comorbidities. The ideal antiepileptic drug would have minimal enzyme interaction, little protein binding, linear kinetics, a long half-life, a good safety profile, and a high therapeutic index. The goal of management should be to maintain the patient’s normal lifestyle with complete control of seizures and with minimal side effects.
The only randomized controlled trial in new-onset geriatric epilepsy concluded that gabapentin (Neurontin) and lamotrigine (Lamictal) should be the initial therapy in such patients.56 Trials indicate extended-release carbamazepine or levetiracetam (Keppra) can also be tried.57
The prescribing strategy includes lower initial dose, slower titration, and a lower target dose than for younger patients. Intense monitoring of dosing and drug levels is necessary to avoid toxicity. If the first drug is not tolerated well, another should be substituted. If seizures persist despite increasing dosage, a drug with a different mechanism of action should be tried.58 A patient with drug-resistant epilepsy (failure to respond to two adequate and appropriate antiepileptic drug trials59) should be referred to an epilepsy surgical center for reevaluation and consideration of epilepsy surgery.
Patient and caregiver support is an essential component of management. New-onset epilepsy in the elderly has a significant effect on quality of life, more so if the patient is already cognitively impaired. It erodes self-confidence, survival becomes difficult, and the condition is worse for patients who live alone. Driving restrictions further limit independence and increase isolation. Hence, psychological support programs can significantly boost the self-esteem and morale of such patients and their caregivers.
SPECIAL CONSIDERATION: EPILEPSY IN THE NURSING HOME
Certain points apply to the growing proportion of elderly who reside in nursing homes:
- Several studies in the United States and in Europe60–62 suggest that this subgroup is at higher risk of polypharmacy and more likely to be treated with older antiepileptic drugs.
- Only a minority of these patients (as low as 42% in one study60) received adequate monitoring of antiepileptic drug levels.
- The clinical characteristics and epileptic etiologies of these patients are less well defined.
Together, these observations highlight a particularly vulnerable population, at risk for medication toxicity as well as for undertreatment.
OUR KNOWLEDGE IS STILL GROWING
New-onset epilepsy, although common in the elderly, is difficult to diagnose because of its atypical presentation, concomitant cognitive impairment, and nonspecific abnormalities in routine investigations. But knowledge of its common causes and differential diagnoses makes the task easier. A high suspicion warrants referral to a neurologist or epileptologist.
Challenges to the management of seizures in the elderly include deranged physiologic processes, multiple comorbidities, and polypharmacy. No single drug is ideal for antiepileptic therapy in the elderly; the choice of drug is usually dictated by seizure type, comorbidities, and tolerance level. The treatment regimen in the elderly is more conservative, and the target dosage is lower than for younger adults. Emotional support of patient and caregivers should be an important aspect of management.
Our knowledge about new-onset epilepsy in the elderly is still growing, and future research should explore its diagnosis, treatment strategies, and care-delivery models.
- Ramsay RE, Rowan AJ, Pryor FM. Special considerations in treating the elderly patient with epilepsy. Neurology 2004; 62(suppl 2):S24–S29.
- Sander JW, Hart YM, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet 1990; 336:1267–1271.
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993; 34:453–468.
- Laccheo I, Ablah E, Heinrichs R, Sadler T, Baade L, Liow K. Assessment of quality of life among the elderly with epilepsy. Epilepsy Behav 2008; 12:257–261.
- Stephen LJ, Brodie MJ. Epilepsy in elderly people. Lancet 2000; 355:1441–1446.
- Faught E, Richman J, Martin R, et al. Incidence and prevalence of epilepsy among older US Medicare beneficiaries. Neurology 2012; 78:448–453.
- Sillanpää M, Lastunen S, Helenius H, Schmidt D. Regional differences and secular trends in the incidence of epilepsy in Finland: a nationwide 23-year registry study. Epilepsia 2011; 52:1857–1867.
- Annegers JF, Hauser WA, Lee JR, Rocca WA. Incidence of acute symptomatic seizures in Rochester, Minnesota, 1935–1984. Epilepsia 1995; 36:327–333.
- Lühdorf K, Jensen LK, Plesner AM. Etiology of seizures in the elderly. Epilepsia 1986; 27:458–463.
- Granger N, Convers P, Beauchet O, et al. First epileptic seizure in the elderly: electroclinical and etiological data in 341 patients [in French]. Rev Neurol (Paris) 2002; 158:1088–1095.
- Pugh MJ, Knoefel JE, Mortensen EM, Amuan ME, Berlowitz DR, Van Cott AC. New-onset epilepsy risk factors in older veterans. J Am Geriatr Soc 2009; 57:237–242.
- Brodie MJ, Elder AT, Kwan P. Epilepsy in later life. Lancet Neurol 2009; 8:1019–1030.
- Bladin CF, Alexandrov AV, Bellavance A, et al. Seizures after stroke: a prospective multicenter study. Arch Neurol 2000; 57:1617–1622.
- Kilpatrick CJ, Davis SM, Tress BM, Rossiter SC, Hopper JL, Vandendriesen ML. Epileptic seizures in acute stroke. Arch Neurol 1990; 47:157–160.
- Giroud M, Gras P, Fayolle H, André N, Soichot P, Dumas R. Early seizures after acute stroke: a study of 1,640 cases. Epilepsia 1994; 35:959–964.
- Asconapé JJ, Penry JK. Poststroke seizures in the elderly. Clin Geriatr Med 1991; 7:483–492.
- So EL, Annegers JF, Hauser WA, O’Brien PC, Whisnant JP. Population-based study of seizure disorders after cerebral infarction. Neurology 1996; 46:350–355.
- Cleary P, Shorvon S, Tallis R. Late-onset seizures as a predictor of subsequent stroke. Lancet 2004; 363:1184–1186.
- Loiseau P. Pathologic processes in the elderly and their association with seizures. In:Rowan AJ, Ramsay RE, editors. Seizures and epilepsy in the elderly. Boston, MA: Butterworth-Heinemann; 1997:63–86.
- Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986; 36:1226–1230.
- Leppik IE, Birnbaum AK. Epilepsy in the elderly. Ann N Y Acad Sci 2010; 1184:208–224.
- Imfeld P, Bodmer M, Schuerch M, Jick SS, Meier CR. Seizures in patients with Alzheimer’s disease or vascular dementia: a population-based nested case-control analysis. Epilepsia 2013; 54:700–707.
- Irizarry MC, Jin S, He F, et al. Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol 2012; 69:368–372.
- Cordonnier C, Hénon H, Derambure P, Pasquier F, Leys D. Influence of pre-existing dementia on the risk of post-stroke epileptic seizures. J Neurol Neurosurg Psychiatry 2005; 76:1649–1653.
- Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia 2003; 44(suppl 10):2–10.
- Hiyoshi T, Yagi K. Epilepsy in the elderly. Epilepsia 2000; 41(suppl 9):31–35.
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 2009; 373:1105–1110.
- Roberts MA, Godfrey JW, Caird FI. Epileptic seizures in the elderly: I. Aetiology and type of seizure. Age Ageing 1982; 11:24–28.
- Loiseau J, Loiseau P, Duché B, Guyot M, Dartigues JF, Aublet B. A survey of epileptic disorders in southwest France: seizures in elderly patients. Ann Neurol 1990; 27:232–237.
- Lote K, Stenwig AE, Skullerud K, Hirschberg H. Prevalence and prognostic significance of epilepsy in patients with gliomas. Eur J Cancer 1998; 34:98–102.
- Franson KL, Hay DP, Neppe V, et al. Drug-induced seizures in the elderly. Causative agents and optimal management. Drugs Aging 1995; 7:38–48.
- Starr P, Klein-Schwartz W, Spiller H, Kern P, Ekleberry SE, Kunkel S. Incidence and onset of delayed seizures after overdoses of extended-release bupropion. Am J Emerg Med 2009; 27:911–915.
- Hauser WA, Ng SK, Brust JC. Alcohol, seizures, and epilepsy. Epilepsia 1988; 29(suppl 2):S66–S78.
- Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014; 13:276–286.
- Ait S, Gilbert T, Cotton F, Bonnefoy M. Cortical blindness and posterior reversible encephalopathy syndrome in an older patient. BMJ Case Rep 2012;pii:bcr0920114782.
- Tinuper P, Provini F, Marini C, et al. Partial epilepsy of long duration: changing semiology with age. Epilepsia 1996; 37:162–164.
- Silveira DC, Jehi L, Chapin J, et al. Seizure semiology and aging. Epilepsy Behav 2011; 20:375–377.
- Theodore WH. The postictal state: effects of age and underlying brain dysfunction. Epilepsy Behav 2010; 19:118–120.
- Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998; 338:970–976.
- Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology 1998; 50:735–741.
- Sung CY, Chu NS. Status epilepticus in the elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989; 80:51–56.
- Pro S, Vicenzini E, Randi F, Pulitano P, Mecarelli O. Idiopathic late-onset absence status epilepticus: a case report with an electroclinical 14 years follow-up. Seizure 2011; 20:655–658.
- Martin Y, Artaz MA, Bornand-Rousselot A. Nonconvulsive status epilepticus in the elderly. J Am Geriatr Soc 2004; 52:476–477.
- Fernández-Torre JL, Díaz-Castroverde AG. Non-convulsive status epilepticus in elderly individuals: report of four representative cases. Age Ageing 2004; 33:78–81.
- Chung PW, Seo DW, Kwon JC, Kim H, Na DL. Nonconvulsive status epilepticus presenting as a subacute progressive aphasia. Seizure 2002; 11:449–454.
- Sheth RD, Drazkowski JF, Sirven JI, Gidal BE, Hermann BP. Protracted ictal confusion in elderly patients. Arch Neurol 2006; 63:529–532.
- Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003; 61:1066–1073.
- Kellinghaus C, Loddenkemper T, Dinner DS, Lachhwani D, Lüders HO. Seizure semiology in the elderly: a video analysis. Epilepsia 2004; 45:263–267.
- Drury I, Beydoun A. Interictal epileptiform activity in elderly patients with epilepsy. Electroencephalogr Clin Neurophysiol 1998; 106:369–373.
- McBride AE, Shih TT, Hirsch LJ. Video-EEG monitoring in the elderly: a review of 94 patients. Epilepsia 2002; 43:165–169.
- Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet 2006; 367:1087–1100.
- McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004; 56:163–184.
- Pack AM, Morrell MJ. Epilepsy and bone health in adults. Epilepsy Behav 2004; 5(suppl 2):S24–S29.
- Granger AS. Ginkgo biloba precipitating epileptic seizures. Age Ageing 2001; 30:523–525.
- Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol 2006; 61:246–255.
- Rowan AJ, Ramsay RE, Collins JF, et al; VA Cooperative Study 428 Group. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005; 64:1868–1673.
- Garnett WR. Optimizing antiepileptic drug therapy in the elderly. Ann Pharmacother 2005; 39:1852–1860.
- Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology 2002; 58(suppl 5):S2–S8.
- Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010; 51:1069–1077.
- Huying F, Klimpe S, Werhahn KJ. Antiepileptic drug use in nursing home residents: a cross-sectional, regional study. Seizure 2006; 15:194–197.
- Lackner TE, Cloyd JC, Thomas LW, Leppik IE. Antiepileptic drug use in nursing home residents: effect of age, gender, and comedication on patterns of use. Epilepsia 1998; 39:1083–1087.
- Galimberti CA, Magri F, Magnani B, et al. Antiepileptic drug use and epileptic seizures in elderly nursing home residents: a survey in the province of Pavia, Northern Italy. Epilepsy Res 2006; 68:1–8.
Contrary to the popular belief that epilepsy is mainly a disease of youth, nearly 25% of new-onset seizures occur after age 65.1,2 The incidence of epilepsy in this age group is almost twice the rate in children, and in people over age 80, it is triple the rate in children.3 As our population ages, the burden of “elderly-onset” epilepsy will rise.
A seizure diagnosis carries significant implications in older people, who are already vulnerable to cognitive decline, loss of functional independence, driving restrictions, and risk of falls. Newly diagnosed epilepsy further worsens quality of life.4
The causes and clinical manifestations of seizures and epilepsy in the elderly differ from those in younger people.5 Hence, it is often difficult to make a diagnosis with certainty from a wide range of differential diagnoses. Older people are also more likely to have comorbidities, further complicating the situation.
Managing seizures in the elderly is also challenging, as age-associated physiologic changes can affect the pharmacokinetics and pharmacodynamics of antiepileptic drugs. Diagnosing and managing elderly-onset epilepsy can be challenging for a family physician, an internist, a geriatrician, or even a neurologist.
In this review, we emphasize the common causes of new-onset epilepsy in the elderly and the assessment of the clinical clues that are essential for making an accurate diagnosis. We also review the pharmacology of antiepileptic drugs used in old age and highlight the need for psychological support for patients and caregivers.
RISING PREVALENCE IN THE ELDERLY
In US Medicare beneficiaries age 65 and older, the average annual incidence rate of epilepsy in 2001 to 2005 was 10.8 per 1,000.6 A large study in Finland revealed falling incidence rates of epilepsy in childhood and middle age and rising trends in the elderly.7
In the United States, the rates are higher in African Americans (18.7 per 1,000) and lower in Asian Americans and Native Americans (5.5 and 7.7 per 1,000) than in whites (10.2 per 1,000).6 Incidence rates are slightly higher for women than for men and increase with age in both sexes and all racial groups.
Acute symptomatic seizure is also common in older patients. The incidence of acute seizures in patients over age 60 was estimated at 50 to 100 per 100,000 per year in one study.7 The rate was considerably higher in men than in women. The study also found a 3.6% risk of experiencing an acute symptomatic seizure in an 80-year lifespan, which approaches that of developing epilepsy.8 The major causes of acute symptomatic seizure were traumatic brain injury, cerebrovascular disease, drug withdrawal, and central nervous system infection.
CAUSES OF NEW-ONSET EPILEPSY IN THE ELDERLY
The most common causes of new-onset epilepsy in the elderly include cerebrovascular disease, metabolic disturbances, dementia, traumatic brain injury, tumors, and drugs.3,9–11
Cerebrovascular disease
In older adults, acute stroke is the most common cause, accounting for up to half of cases.5,12
Seizures occur in 4.4% to 8.9% of acute cerebrovascular events.13,14 The risk varies by stroke subtype, although all stroke subtypes, including transient ischemic attack, can be associated with seizure.15 For example, although 1% to 2% of patients experienced a seizure within 15 days of a transient ischemic attack or a lacunar infarct, this risk was 16.6% after an embolic stroke.15
Beyond this increased risk of “acute seizure” in the immediate poststroke period (usually defined as 1 week), the risk of epilepsy was also 20 times higher in the first year after a stroke.14 However, seizures tend to occur within the first 48 hours after the onset of ischemic stroke. In subarachnoid hemorrhage, seizures generally occur within hours.16
In a population-based study in Rochester, NY,17 epilepsy developed in two-thirds of patients with seizure related to acute stroke. Two factors that independently predicted the development of epilepsy were early seizure occurrence and recurrence of stroke.
Interestingly, the risk of stroke was three times higher in older patients who had new-onset seizure.18 Therefore, any elderly person with new-onset seizure should be assessed for cerebrovascular risk factors and treated accordingly for stroke prevention.
Metabolic disturbances
Acute metabolic disorders are common in elderly patients because of multiple comorbidities and polypharmacy. Hypoglycemia and hyponatremia need to be particularly considered in this population.19
Other well-documented metabolic causes of acute seizure, including nonketotic hyperglycemia, hypocalcemia, and uremic or hepatic encephalopathy, can all be considerations, albeit less specific to this age group.
Dementia
Primary neurodegenerative disorders associated with cognitive impairment, such as Alzheimer disease, are major risk factors for new-onset epilepsy in older patients.3,5 Seizures occur in about 10% of Alzheimer patients.20 Those who have brief periods of increased confusion may actually be experiencing unrecognized complex partial seizures.21
A case-control study discovered incidence rates of epilepsy almost 10 times higher in patients who had Alzheimer disease or vascular dementia than in nondemented patients.22 A prospective cohort study in patients with mild to moderate Alzheimer disease established that younger age, a greater degree of cognitive impairment, and a history of antipsychotic use were independent risk factors for new-onset seizures in the elderly.23 Preexisting dementia also increases the risk of poststroke epilepsy.24
Traumatic brain injury
The most common cause of brain trauma in the elderly is falls. Subdural hematoma, which can occur in the elderly with trivial trauma or sometimes even without it, needs to be considered. The risk of posttraumatic hemorrhage is especially relevant in patients taking anticoagulants.
Traumatic brain injury has a poorer prognosis in older people than in the young,25 and it accounts for up to 20% of cases of epilepsy in the elderly.26 Although no study has specifically addressed the longitudinal risk of epilepsy after traumatic brain injury in the elderly, a study in children and young adults revealed the risk was highest in the first year, with the increased risk persisting for more than 10 years.27
Brain tumors
Between 10% and 30% of new-onset seizures in the elderly are associated with tumor, typically glioma, meningioma, and brain metastasis.28,29 Seizures are usually associated more with primary than with secondary tumors, and more with low-grade tumors than high-grade ones.30
Drug-induced
Drugs and drug withdrawal can contribute to up to 10% of acute symptomatic seizures in the geriatric population.5,8,29 The elderly are susceptible to drug-induced seizure because of a higher prevalence of polypharmacy, impaired drug clearance, and heightened sensitivity to the proconvulsant side effects of medications.1 A number of commonly used drugs have been implicated,31 including:
Antibiotics such as carbapenems and high-dose penicillin
Antihistamines such as desloratadine (Clarinex)
Pain medications such as tramadol (Ul-tram) and high-dose opiates
Neuromodulators
Antidepressants such as clomipramine (Anafranil), maprotiline (Ludiomil), amoxapine (Asendin), and bupropion (Wellbutrin).32
Seizures also follow alcohol, benzodiazepine, and barbiturate withdrawal.33
Other causes
Paraneoplastic limbic encephalitis is a rare cause of seizures in the elderly.34 It can present with refractory seizures, confusion, and behavioral changes with or without a known concurrent neoplastic disease.
Posterior reversible leukoencephalopathy syndrome, another rare consideration, can particularly affect immunosuppressed elderly patients. This syndrome is characterized clinically by headache, confusion, seizures, vomiting, and visual disturbances with radiographic vasogenic edema.35
CLINICAL PRESENTATION
The signs and symptoms of a seizure may be atypical in the elderly. Seizures more often have a picture of “epileptic amnesia,” with confusion, sleepiness, or clumsiness, rather than motor manifestations such as tonic stiffening or automatism.36,37 Postictal states are also prolonged, particularly if there is underlying brain dysfunction.38 All these features render the clinical seizure manifestations more subtle and, as such, more difficult for the uninitiated caregiver to identify.
Convulsive and nonconvulsive status epilepticus
Status epilepticus is defined as a single generalized seizure lasting more than 5 minutes or a series of seizures lasting longer than 30 minutes without the patient’s regaining consciousness.39 The greatest increase in the incidence of status epilepticus occurs after age 60.40 It is the first seizure in about 30% of new-onset seizures in the elderly.41
Mortality rates increase with age, anoxia, and duration of status epilepticus and are over 50% in patients age 80 and older.40,42
Convulsive status epilepticus is most commonly caused by stroke.40
Absence status epilepticus can occur in elderly patients as a late complication of idiopathic generalized epilepsy related to benzodiazepine withdrawal, alcohol intoxication, or initiation of psychotropic drugs.42
Nonconvulsive status epilepticus manifests as altered mental status, psychosis, lethargy, or coma.42–44 Occasionally, it presents as a more focal cognitive disturbance with aphasia or a neglect syndrome.42,45 Electroencephalographic correlates of nonconvulsive status epilepticus include focal rhythmic discharges, often arising from frontal or temporal lobes, or generalized spike or sharp and slow-wave activity.46 Its management is challenging because of delayed diagnosis or misdiagnosis. The risk of death is higher in patients with severely impaired mental status or acute complications.47
Table 1 lists the typical seizure manifestations peculiar to the elderly.37,48
Differential diagnosis of new-onset epilepsy in the elderly
New-onset epilepsy in elderly patients can be confused with syncope, transient ischemic attack, cardiac arrhythmia, metabolic disturbances, transient global amnesia, neurodegenerative disease, rapid-eye-movement sleep behavior disorder, psychogenic disorders, and other conditions (Table 2). If there is a high clinical suspicion of seizure, the patient should undergo electroencephalography (EEG) and be referred to a neurologist or epileptologist.
KEYS TO THE DIAGNOSIS
Clinical history
A reliable history and description of the event from an eyewitness or a video recording of the event are invaluable to the diagnosis of epileptic seizure. Signs and symptoms that suggest the diagnosis include aura, ictal pallor, urinary incontinence, tongue-biting, and motor symptoms, as well as postictal confusion, drowsiness, and speech disturbance.
Electroencephalography
EEG is the most useful diagnostic tool in epilepsy. However, an interictal EEG reading (ie, between epileptic attacks) in an elderly patient has limited utility, showing epileptiform activity in only about one-fourth of patients.49 Nonspecific EEG abnormalities such as intermittent focal slowing are seen in many older people even without seizure.50 Also, normal findings on outpatient EEG do not rule out epilepsy, as EEG is normal in about one-third of patients with epilepsy, irrespective of age.1,49 Activation procedures such as hyperventilation and photic stimulation add little to the diagnosis in the elderly.49
On the other hand, video-EEG monitoring is an excellent tool for evaluating possible epilepsy, as it allows accurate assessment of brain electrical activity during the events in question. Moreover, studies of video-EEG recording of seizures in elderly patients demonstrated epileptiform discharges on EEG in 76% of clinical ictal events.50
Therefore, routine EEG is a useful screening tool, and inpatient video-EEG monitoring is the gold standard to characterize events of concern and distinguish between epileptic and nonepileptic or psychogenic seizures.
Other diagnostic studies
Brain imaging, preferably magnetic resonance imaging with contrast, should be done in every patient with possible epilepsy due to stroke, traumatic brain injury, or other structural brain disease.51
Electrocardiography helps exclude cardiac causes such as arrhythmia.
Blood testing. Metabolically provoked seizure can be distinguished by blood analysis for electrolytes, blood urea nitrogen, creatinine, glucose, calcium, magnesium, liver enzymes, and drug levels (eg, ethanol). A complete blood cell count with differential and platelets should also be done in anticipation of starting antiepileptic drug therapy.
Lumbar puncture for cell count, protein, glucose, stains, and cultures should be performed whenever meningitis or encephalitis is suspected.
A sleep study with concurrent video-EEG monitoring may be required to distinguish epileptic seizures from sleep disorders.
Neuropsychological testing may help account for the degree of cognitive impairment present.
Risk factors for stroke should be assessed in every elderly person who has new-onset seizures, because the risk of stroke is high.17
Figure 1 shows the workup for an elderly patient with suspected new-onset epilepsy.
TREATING EPILEPSY IN THE ELDERLY
Therapeutic challenges
Age-associated changes in drug absorption, protein binding, and distribution in body compartments require adjustments in drug selection and dosage. The causes and manifestations of these changes are typically multifactorial, mainly related to altered metabolism, declining plasma albumin concentrations, and increasing competition for protein binding by concomitantly used drugs.
The differences in the pharmacokinetics and pharmacodynamics of antiepileptic drugs depend on the patient’s physical status, relevant comorbidities, and concomitant medications.52 Renal and hepatic function may decline in an elderly patient; accordingly, precaution is needed in the prescribing and dosing of antiepileptic drugs.
Adverse effects from seizure medications are twice as common in elderly patients compared with younger patients. Ataxia, tremor, visual disturbance, and sedation are the most common.1 Antiepileptic drugs are also harmful to bone; induced abnormalities in bone metabolism include hypocalcemia, hypophosphatemia, decreased levels of active vitamin D metabolites, and hyperparathyroidism.53
Elderly patients tend to take multiple drugs, and some drugs can lower the seizure threshold, particularly antidepressants, anti-psychotics, and antibiotics.32 The herbal remedy ginkgo biloba can also precipitate seizure in this population.54
Antiepileptic drugs such as phenobarbital, primidone (Mysoline), phenytoin (Dilantin), and carbamazepine (Tegretol) can be broad-spectrum enzyme-inducers, increasing the metabolism of many drugs, including warfarin (Coumadin), cytotoxic agents, statins, cardiac antiarrhythmics, antihypertensives, corticosteroids, and other immunosuppressants.55 For example, carbamazepine can alter the metabolism of several hepatically metabolized drugs and cause significant hyponatremia. This is problematic in patients already taking sodium-depleting antihypertensives. Age-related cognitive decline can worsen the situation, often leading to misdiagnosis or patient noncompliance.
Table 3 profiles the interactions of commonly used antiepileptic drugs.
The ideal pharmacotherapy
No single drug is ideal for elderly patients with new-onset epilepsy. The choice mostly depends on the type of seizure and the patient’s comorbidities. The ideal antiepileptic drug would have minimal enzyme interaction, little protein binding, linear kinetics, a long half-life, a good safety profile, and a high therapeutic index. The goal of management should be to maintain the patient’s normal lifestyle with complete control of seizures and with minimal side effects.
The only randomized controlled trial in new-onset geriatric epilepsy concluded that gabapentin (Neurontin) and lamotrigine (Lamictal) should be the initial therapy in such patients.56 Trials indicate extended-release carbamazepine or levetiracetam (Keppra) can also be tried.57
The prescribing strategy includes lower initial dose, slower titration, and a lower target dose than for younger patients. Intense monitoring of dosing and drug levels is necessary to avoid toxicity. If the first drug is not tolerated well, another should be substituted. If seizures persist despite increasing dosage, a drug with a different mechanism of action should be tried.58 A patient with drug-resistant epilepsy (failure to respond to two adequate and appropriate antiepileptic drug trials59) should be referred to an epilepsy surgical center for reevaluation and consideration of epilepsy surgery.
Patient and caregiver support is an essential component of management. New-onset epilepsy in the elderly has a significant effect on quality of life, more so if the patient is already cognitively impaired. It erodes self-confidence, survival becomes difficult, and the condition is worse for patients who live alone. Driving restrictions further limit independence and increase isolation. Hence, psychological support programs can significantly boost the self-esteem and morale of such patients and their caregivers.
SPECIAL CONSIDERATION: EPILEPSY IN THE NURSING HOME
Certain points apply to the growing proportion of elderly who reside in nursing homes:
- Several studies in the United States and in Europe60–62 suggest that this subgroup is at higher risk of polypharmacy and more likely to be treated with older antiepileptic drugs.
- Only a minority of these patients (as low as 42% in one study60) received adequate monitoring of antiepileptic drug levels.
- The clinical characteristics and epileptic etiologies of these patients are less well defined.
Together, these observations highlight a particularly vulnerable population, at risk for medication toxicity as well as for undertreatment.
OUR KNOWLEDGE IS STILL GROWING
New-onset epilepsy, although common in the elderly, is difficult to diagnose because of its atypical presentation, concomitant cognitive impairment, and nonspecific abnormalities in routine investigations. But knowledge of its common causes and differential diagnoses makes the task easier. A high suspicion warrants referral to a neurologist or epileptologist.
Challenges to the management of seizures in the elderly include deranged physiologic processes, multiple comorbidities, and polypharmacy. No single drug is ideal for antiepileptic therapy in the elderly; the choice of drug is usually dictated by seizure type, comorbidities, and tolerance level. The treatment regimen in the elderly is more conservative, and the target dosage is lower than for younger adults. Emotional support of patient and caregivers should be an important aspect of management.
Our knowledge about new-onset epilepsy in the elderly is still growing, and future research should explore its diagnosis, treatment strategies, and care-delivery models.
Contrary to the popular belief that epilepsy is mainly a disease of youth, nearly 25% of new-onset seizures occur after age 65.1,2 The incidence of epilepsy in this age group is almost twice the rate in children, and in people over age 80, it is triple the rate in children.3 As our population ages, the burden of “elderly-onset” epilepsy will rise.
A seizure diagnosis carries significant implications in older people, who are already vulnerable to cognitive decline, loss of functional independence, driving restrictions, and risk of falls. Newly diagnosed epilepsy further worsens quality of life.4
The causes and clinical manifestations of seizures and epilepsy in the elderly differ from those in younger people.5 Hence, it is often difficult to make a diagnosis with certainty from a wide range of differential diagnoses. Older people are also more likely to have comorbidities, further complicating the situation.
Managing seizures in the elderly is also challenging, as age-associated physiologic changes can affect the pharmacokinetics and pharmacodynamics of antiepileptic drugs. Diagnosing and managing elderly-onset epilepsy can be challenging for a family physician, an internist, a geriatrician, or even a neurologist.
In this review, we emphasize the common causes of new-onset epilepsy in the elderly and the assessment of the clinical clues that are essential for making an accurate diagnosis. We also review the pharmacology of antiepileptic drugs used in old age and highlight the need for psychological support for patients and caregivers.
RISING PREVALENCE IN THE ELDERLY
In US Medicare beneficiaries age 65 and older, the average annual incidence rate of epilepsy in 2001 to 2005 was 10.8 per 1,000.6 A large study in Finland revealed falling incidence rates of epilepsy in childhood and middle age and rising trends in the elderly.7
In the United States, the rates are higher in African Americans (18.7 per 1,000) and lower in Asian Americans and Native Americans (5.5 and 7.7 per 1,000) than in whites (10.2 per 1,000).6 Incidence rates are slightly higher for women than for men and increase with age in both sexes and all racial groups.
Acute symptomatic seizure is also common in older patients. The incidence of acute seizures in patients over age 60 was estimated at 50 to 100 per 100,000 per year in one study.7 The rate was considerably higher in men than in women. The study also found a 3.6% risk of experiencing an acute symptomatic seizure in an 80-year lifespan, which approaches that of developing epilepsy.8 The major causes of acute symptomatic seizure were traumatic brain injury, cerebrovascular disease, drug withdrawal, and central nervous system infection.
CAUSES OF NEW-ONSET EPILEPSY IN THE ELDERLY
The most common causes of new-onset epilepsy in the elderly include cerebrovascular disease, metabolic disturbances, dementia, traumatic brain injury, tumors, and drugs.3,9–11
Cerebrovascular disease
In older adults, acute stroke is the most common cause, accounting for up to half of cases.5,12
Seizures occur in 4.4% to 8.9% of acute cerebrovascular events.13,14 The risk varies by stroke subtype, although all stroke subtypes, including transient ischemic attack, can be associated with seizure.15 For example, although 1% to 2% of patients experienced a seizure within 15 days of a transient ischemic attack or a lacunar infarct, this risk was 16.6% after an embolic stroke.15
Beyond this increased risk of “acute seizure” in the immediate poststroke period (usually defined as 1 week), the risk of epilepsy was also 20 times higher in the first year after a stroke.14 However, seizures tend to occur within the first 48 hours after the onset of ischemic stroke. In subarachnoid hemorrhage, seizures generally occur within hours.16
In a population-based study in Rochester, NY,17 epilepsy developed in two-thirds of patients with seizure related to acute stroke. Two factors that independently predicted the development of epilepsy were early seizure occurrence and recurrence of stroke.
Interestingly, the risk of stroke was three times higher in older patients who had new-onset seizure.18 Therefore, any elderly person with new-onset seizure should be assessed for cerebrovascular risk factors and treated accordingly for stroke prevention.
Metabolic disturbances
Acute metabolic disorders are common in elderly patients because of multiple comorbidities and polypharmacy. Hypoglycemia and hyponatremia need to be particularly considered in this population.19
Other well-documented metabolic causes of acute seizure, including nonketotic hyperglycemia, hypocalcemia, and uremic or hepatic encephalopathy, can all be considerations, albeit less specific to this age group.
Dementia
Primary neurodegenerative disorders associated with cognitive impairment, such as Alzheimer disease, are major risk factors for new-onset epilepsy in older patients.3,5 Seizures occur in about 10% of Alzheimer patients.20 Those who have brief periods of increased confusion may actually be experiencing unrecognized complex partial seizures.21
A case-control study discovered incidence rates of epilepsy almost 10 times higher in patients who had Alzheimer disease or vascular dementia than in nondemented patients.22 A prospective cohort study in patients with mild to moderate Alzheimer disease established that younger age, a greater degree of cognitive impairment, and a history of antipsychotic use were independent risk factors for new-onset seizures in the elderly.23 Preexisting dementia also increases the risk of poststroke epilepsy.24
Traumatic brain injury
The most common cause of brain trauma in the elderly is falls. Subdural hematoma, which can occur in the elderly with trivial trauma or sometimes even without it, needs to be considered. The risk of posttraumatic hemorrhage is especially relevant in patients taking anticoagulants.
Traumatic brain injury has a poorer prognosis in older people than in the young,25 and it accounts for up to 20% of cases of epilepsy in the elderly.26 Although no study has specifically addressed the longitudinal risk of epilepsy after traumatic brain injury in the elderly, a study in children and young adults revealed the risk was highest in the first year, with the increased risk persisting for more than 10 years.27
Brain tumors
Between 10% and 30% of new-onset seizures in the elderly are associated with tumor, typically glioma, meningioma, and brain metastasis.28,29 Seizures are usually associated more with primary than with secondary tumors, and more with low-grade tumors than high-grade ones.30
Drug-induced
Drugs and drug withdrawal can contribute to up to 10% of acute symptomatic seizures in the geriatric population.5,8,29 The elderly are susceptible to drug-induced seizure because of a higher prevalence of polypharmacy, impaired drug clearance, and heightened sensitivity to the proconvulsant side effects of medications.1 A number of commonly used drugs have been implicated,31 including:
Antibiotics such as carbapenems and high-dose penicillin
Antihistamines such as desloratadine (Clarinex)
Pain medications such as tramadol (Ul-tram) and high-dose opiates
Neuromodulators
Antidepressants such as clomipramine (Anafranil), maprotiline (Ludiomil), amoxapine (Asendin), and bupropion (Wellbutrin).32
Seizures also follow alcohol, benzodiazepine, and barbiturate withdrawal.33
Other causes
Paraneoplastic limbic encephalitis is a rare cause of seizures in the elderly.34 It can present with refractory seizures, confusion, and behavioral changes with or without a known concurrent neoplastic disease.
Posterior reversible leukoencephalopathy syndrome, another rare consideration, can particularly affect immunosuppressed elderly patients. This syndrome is characterized clinically by headache, confusion, seizures, vomiting, and visual disturbances with radiographic vasogenic edema.35
CLINICAL PRESENTATION
The signs and symptoms of a seizure may be atypical in the elderly. Seizures more often have a picture of “epileptic amnesia,” with confusion, sleepiness, or clumsiness, rather than motor manifestations such as tonic stiffening or automatism.36,37 Postictal states are also prolonged, particularly if there is underlying brain dysfunction.38 All these features render the clinical seizure manifestations more subtle and, as such, more difficult for the uninitiated caregiver to identify.
Convulsive and nonconvulsive status epilepticus
Status epilepticus is defined as a single generalized seizure lasting more than 5 minutes or a series of seizures lasting longer than 30 minutes without the patient’s regaining consciousness.39 The greatest increase in the incidence of status epilepticus occurs after age 60.40 It is the first seizure in about 30% of new-onset seizures in the elderly.41
Mortality rates increase with age, anoxia, and duration of status epilepticus and are over 50% in patients age 80 and older.40,42
Convulsive status epilepticus is most commonly caused by stroke.40
Absence status epilepticus can occur in elderly patients as a late complication of idiopathic generalized epilepsy related to benzodiazepine withdrawal, alcohol intoxication, or initiation of psychotropic drugs.42
Nonconvulsive status epilepticus manifests as altered mental status, psychosis, lethargy, or coma.42–44 Occasionally, it presents as a more focal cognitive disturbance with aphasia or a neglect syndrome.42,45 Electroencephalographic correlates of nonconvulsive status epilepticus include focal rhythmic discharges, often arising from frontal or temporal lobes, or generalized spike or sharp and slow-wave activity.46 Its management is challenging because of delayed diagnosis or misdiagnosis. The risk of death is higher in patients with severely impaired mental status or acute complications.47
Table 1 lists the typical seizure manifestations peculiar to the elderly.37,48
Differential diagnosis of new-onset epilepsy in the elderly
New-onset epilepsy in elderly patients can be confused with syncope, transient ischemic attack, cardiac arrhythmia, metabolic disturbances, transient global amnesia, neurodegenerative disease, rapid-eye-movement sleep behavior disorder, psychogenic disorders, and other conditions (Table 2). If there is a high clinical suspicion of seizure, the patient should undergo electroencephalography (EEG) and be referred to a neurologist or epileptologist.
KEYS TO THE DIAGNOSIS
Clinical history
A reliable history and description of the event from an eyewitness or a video recording of the event are invaluable to the diagnosis of epileptic seizure. Signs and symptoms that suggest the diagnosis include aura, ictal pallor, urinary incontinence, tongue-biting, and motor symptoms, as well as postictal confusion, drowsiness, and speech disturbance.
Electroencephalography
EEG is the most useful diagnostic tool in epilepsy. However, an interictal EEG reading (ie, between epileptic attacks) in an elderly patient has limited utility, showing epileptiform activity in only about one-fourth of patients.49 Nonspecific EEG abnormalities such as intermittent focal slowing are seen in many older people even without seizure.50 Also, normal findings on outpatient EEG do not rule out epilepsy, as EEG is normal in about one-third of patients with epilepsy, irrespective of age.1,49 Activation procedures such as hyperventilation and photic stimulation add little to the diagnosis in the elderly.49
On the other hand, video-EEG monitoring is an excellent tool for evaluating possible epilepsy, as it allows accurate assessment of brain electrical activity during the events in question. Moreover, studies of video-EEG recording of seizures in elderly patients demonstrated epileptiform discharges on EEG in 76% of clinical ictal events.50
Therefore, routine EEG is a useful screening tool, and inpatient video-EEG monitoring is the gold standard to characterize events of concern and distinguish between epileptic and nonepileptic or psychogenic seizures.
Other diagnostic studies
Brain imaging, preferably magnetic resonance imaging with contrast, should be done in every patient with possible epilepsy due to stroke, traumatic brain injury, or other structural brain disease.51
Electrocardiography helps exclude cardiac causes such as arrhythmia.
Blood testing. Metabolically provoked seizure can be distinguished by blood analysis for electrolytes, blood urea nitrogen, creatinine, glucose, calcium, magnesium, liver enzymes, and drug levels (eg, ethanol). A complete blood cell count with differential and platelets should also be done in anticipation of starting antiepileptic drug therapy.
Lumbar puncture for cell count, protein, glucose, stains, and cultures should be performed whenever meningitis or encephalitis is suspected.
A sleep study with concurrent video-EEG monitoring may be required to distinguish epileptic seizures from sleep disorders.
Neuropsychological testing may help account for the degree of cognitive impairment present.
Risk factors for stroke should be assessed in every elderly person who has new-onset seizures, because the risk of stroke is high.17
Figure 1 shows the workup for an elderly patient with suspected new-onset epilepsy.
TREATING EPILEPSY IN THE ELDERLY
Therapeutic challenges
Age-associated changes in drug absorption, protein binding, and distribution in body compartments require adjustments in drug selection and dosage. The causes and manifestations of these changes are typically multifactorial, mainly related to altered metabolism, declining plasma albumin concentrations, and increasing competition for protein binding by concomitantly used drugs.
The differences in the pharmacokinetics and pharmacodynamics of antiepileptic drugs depend on the patient’s physical status, relevant comorbidities, and concomitant medications.52 Renal and hepatic function may decline in an elderly patient; accordingly, precaution is needed in the prescribing and dosing of antiepileptic drugs.
Adverse effects from seizure medications are twice as common in elderly patients compared with younger patients. Ataxia, tremor, visual disturbance, and sedation are the most common.1 Antiepileptic drugs are also harmful to bone; induced abnormalities in bone metabolism include hypocalcemia, hypophosphatemia, decreased levels of active vitamin D metabolites, and hyperparathyroidism.53
Elderly patients tend to take multiple drugs, and some drugs can lower the seizure threshold, particularly antidepressants, anti-psychotics, and antibiotics.32 The herbal remedy ginkgo biloba can also precipitate seizure in this population.54
Antiepileptic drugs such as phenobarbital, primidone (Mysoline), phenytoin (Dilantin), and carbamazepine (Tegretol) can be broad-spectrum enzyme-inducers, increasing the metabolism of many drugs, including warfarin (Coumadin), cytotoxic agents, statins, cardiac antiarrhythmics, antihypertensives, corticosteroids, and other immunosuppressants.55 For example, carbamazepine can alter the metabolism of several hepatically metabolized drugs and cause significant hyponatremia. This is problematic in patients already taking sodium-depleting antihypertensives. Age-related cognitive decline can worsen the situation, often leading to misdiagnosis or patient noncompliance.
Table 3 profiles the interactions of commonly used antiepileptic drugs.
The ideal pharmacotherapy
No single drug is ideal for elderly patients with new-onset epilepsy. The choice mostly depends on the type of seizure and the patient’s comorbidities. The ideal antiepileptic drug would have minimal enzyme interaction, little protein binding, linear kinetics, a long half-life, a good safety profile, and a high therapeutic index. The goal of management should be to maintain the patient’s normal lifestyle with complete control of seizures and with minimal side effects.
The only randomized controlled trial in new-onset geriatric epilepsy concluded that gabapentin (Neurontin) and lamotrigine (Lamictal) should be the initial therapy in such patients.56 Trials indicate extended-release carbamazepine or levetiracetam (Keppra) can also be tried.57
The prescribing strategy includes lower initial dose, slower titration, and a lower target dose than for younger patients. Intense monitoring of dosing and drug levels is necessary to avoid toxicity. If the first drug is not tolerated well, another should be substituted. If seizures persist despite increasing dosage, a drug with a different mechanism of action should be tried.58 A patient with drug-resistant epilepsy (failure to respond to two adequate and appropriate antiepileptic drug trials59) should be referred to an epilepsy surgical center for reevaluation and consideration of epilepsy surgery.
Patient and caregiver support is an essential component of management. New-onset epilepsy in the elderly has a significant effect on quality of life, more so if the patient is already cognitively impaired. It erodes self-confidence, survival becomes difficult, and the condition is worse for patients who live alone. Driving restrictions further limit independence and increase isolation. Hence, psychological support programs can significantly boost the self-esteem and morale of such patients and their caregivers.
SPECIAL CONSIDERATION: EPILEPSY IN THE NURSING HOME
Certain points apply to the growing proportion of elderly who reside in nursing homes:
- Several studies in the United States and in Europe60–62 suggest that this subgroup is at higher risk of polypharmacy and more likely to be treated with older antiepileptic drugs.
- Only a minority of these patients (as low as 42% in one study60) received adequate monitoring of antiepileptic drug levels.
- The clinical characteristics and epileptic etiologies of these patients are less well defined.
Together, these observations highlight a particularly vulnerable population, at risk for medication toxicity as well as for undertreatment.
OUR KNOWLEDGE IS STILL GROWING
New-onset epilepsy, although common in the elderly, is difficult to diagnose because of its atypical presentation, concomitant cognitive impairment, and nonspecific abnormalities in routine investigations. But knowledge of its common causes and differential diagnoses makes the task easier. A high suspicion warrants referral to a neurologist or epileptologist.
Challenges to the management of seizures in the elderly include deranged physiologic processes, multiple comorbidities, and polypharmacy. No single drug is ideal for antiepileptic therapy in the elderly; the choice of drug is usually dictated by seizure type, comorbidities, and tolerance level. The treatment regimen in the elderly is more conservative, and the target dosage is lower than for younger adults. Emotional support of patient and caregivers should be an important aspect of management.
Our knowledge about new-onset epilepsy in the elderly is still growing, and future research should explore its diagnosis, treatment strategies, and care-delivery models.
- Ramsay RE, Rowan AJ, Pryor FM. Special considerations in treating the elderly patient with epilepsy. Neurology 2004; 62(suppl 2):S24–S29.
- Sander JW, Hart YM, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet 1990; 336:1267–1271.
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993; 34:453–468.
- Laccheo I, Ablah E, Heinrichs R, Sadler T, Baade L, Liow K. Assessment of quality of life among the elderly with epilepsy. Epilepsy Behav 2008; 12:257–261.
- Stephen LJ, Brodie MJ. Epilepsy in elderly people. Lancet 2000; 355:1441–1446.
- Faught E, Richman J, Martin R, et al. Incidence and prevalence of epilepsy among older US Medicare beneficiaries. Neurology 2012; 78:448–453.
- Sillanpää M, Lastunen S, Helenius H, Schmidt D. Regional differences and secular trends in the incidence of epilepsy in Finland: a nationwide 23-year registry study. Epilepsia 2011; 52:1857–1867.
- Annegers JF, Hauser WA, Lee JR, Rocca WA. Incidence of acute symptomatic seizures in Rochester, Minnesota, 1935–1984. Epilepsia 1995; 36:327–333.
- Lühdorf K, Jensen LK, Plesner AM. Etiology of seizures in the elderly. Epilepsia 1986; 27:458–463.
- Granger N, Convers P, Beauchet O, et al. First epileptic seizure in the elderly: electroclinical and etiological data in 341 patients [in French]. Rev Neurol (Paris) 2002; 158:1088–1095.
- Pugh MJ, Knoefel JE, Mortensen EM, Amuan ME, Berlowitz DR, Van Cott AC. New-onset epilepsy risk factors in older veterans. J Am Geriatr Soc 2009; 57:237–242.
- Brodie MJ, Elder AT, Kwan P. Epilepsy in later life. Lancet Neurol 2009; 8:1019–1030.
- Bladin CF, Alexandrov AV, Bellavance A, et al. Seizures after stroke: a prospective multicenter study. Arch Neurol 2000; 57:1617–1622.
- Kilpatrick CJ, Davis SM, Tress BM, Rossiter SC, Hopper JL, Vandendriesen ML. Epileptic seizures in acute stroke. Arch Neurol 1990; 47:157–160.
- Giroud M, Gras P, Fayolle H, André N, Soichot P, Dumas R. Early seizures after acute stroke: a study of 1,640 cases. Epilepsia 1994; 35:959–964.
- Asconapé JJ, Penry JK. Poststroke seizures in the elderly. Clin Geriatr Med 1991; 7:483–492.
- So EL, Annegers JF, Hauser WA, O’Brien PC, Whisnant JP. Population-based study of seizure disorders after cerebral infarction. Neurology 1996; 46:350–355.
- Cleary P, Shorvon S, Tallis R. Late-onset seizures as a predictor of subsequent stroke. Lancet 2004; 363:1184–1186.
- Loiseau P. Pathologic processes in the elderly and their association with seizures. In:Rowan AJ, Ramsay RE, editors. Seizures and epilepsy in the elderly. Boston, MA: Butterworth-Heinemann; 1997:63–86.
- Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986; 36:1226–1230.
- Leppik IE, Birnbaum AK. Epilepsy in the elderly. Ann N Y Acad Sci 2010; 1184:208–224.
- Imfeld P, Bodmer M, Schuerch M, Jick SS, Meier CR. Seizures in patients with Alzheimer’s disease or vascular dementia: a population-based nested case-control analysis. Epilepsia 2013; 54:700–707.
- Irizarry MC, Jin S, He F, et al. Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol 2012; 69:368–372.
- Cordonnier C, Hénon H, Derambure P, Pasquier F, Leys D. Influence of pre-existing dementia on the risk of post-stroke epileptic seizures. J Neurol Neurosurg Psychiatry 2005; 76:1649–1653.
- Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia 2003; 44(suppl 10):2–10.
- Hiyoshi T, Yagi K. Epilepsy in the elderly. Epilepsia 2000; 41(suppl 9):31–35.
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 2009; 373:1105–1110.
- Roberts MA, Godfrey JW, Caird FI. Epileptic seizures in the elderly: I. Aetiology and type of seizure. Age Ageing 1982; 11:24–28.
- Loiseau J, Loiseau P, Duché B, Guyot M, Dartigues JF, Aublet B. A survey of epileptic disorders in southwest France: seizures in elderly patients. Ann Neurol 1990; 27:232–237.
- Lote K, Stenwig AE, Skullerud K, Hirschberg H. Prevalence and prognostic significance of epilepsy in patients with gliomas. Eur J Cancer 1998; 34:98–102.
- Franson KL, Hay DP, Neppe V, et al. Drug-induced seizures in the elderly. Causative agents and optimal management. Drugs Aging 1995; 7:38–48.
- Starr P, Klein-Schwartz W, Spiller H, Kern P, Ekleberry SE, Kunkel S. Incidence and onset of delayed seizures after overdoses of extended-release bupropion. Am J Emerg Med 2009; 27:911–915.
- Hauser WA, Ng SK, Brust JC. Alcohol, seizures, and epilepsy. Epilepsia 1988; 29(suppl 2):S66–S78.
- Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014; 13:276–286.
- Ait S, Gilbert T, Cotton F, Bonnefoy M. Cortical blindness and posterior reversible encephalopathy syndrome in an older patient. BMJ Case Rep 2012;pii:bcr0920114782.
- Tinuper P, Provini F, Marini C, et al. Partial epilepsy of long duration: changing semiology with age. Epilepsia 1996; 37:162–164.
- Silveira DC, Jehi L, Chapin J, et al. Seizure semiology and aging. Epilepsy Behav 2011; 20:375–377.
- Theodore WH. The postictal state: effects of age and underlying brain dysfunction. Epilepsy Behav 2010; 19:118–120.
- Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998; 338:970–976.
- Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology 1998; 50:735–741.
- Sung CY, Chu NS. Status epilepticus in the elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989; 80:51–56.
- Pro S, Vicenzini E, Randi F, Pulitano P, Mecarelli O. Idiopathic late-onset absence status epilepticus: a case report with an electroclinical 14 years follow-up. Seizure 2011; 20:655–658.
- Martin Y, Artaz MA, Bornand-Rousselot A. Nonconvulsive status epilepticus in the elderly. J Am Geriatr Soc 2004; 52:476–477.
- Fernández-Torre JL, Díaz-Castroverde AG. Non-convulsive status epilepticus in elderly individuals: report of four representative cases. Age Ageing 2004; 33:78–81.
- Chung PW, Seo DW, Kwon JC, Kim H, Na DL. Nonconvulsive status epilepticus presenting as a subacute progressive aphasia. Seizure 2002; 11:449–454.
- Sheth RD, Drazkowski JF, Sirven JI, Gidal BE, Hermann BP. Protracted ictal confusion in elderly patients. Arch Neurol 2006; 63:529–532.
- Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003; 61:1066–1073.
- Kellinghaus C, Loddenkemper T, Dinner DS, Lachhwani D, Lüders HO. Seizure semiology in the elderly: a video analysis. Epilepsia 2004; 45:263–267.
- Drury I, Beydoun A. Interictal epileptiform activity in elderly patients with epilepsy. Electroencephalogr Clin Neurophysiol 1998; 106:369–373.
- McBride AE, Shih TT, Hirsch LJ. Video-EEG monitoring in the elderly: a review of 94 patients. Epilepsia 2002; 43:165–169.
- Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet 2006; 367:1087–1100.
- McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004; 56:163–184.
- Pack AM, Morrell MJ. Epilepsy and bone health in adults. Epilepsy Behav 2004; 5(suppl 2):S24–S29.
- Granger AS. Ginkgo biloba precipitating epileptic seizures. Age Ageing 2001; 30:523–525.
- Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol 2006; 61:246–255.
- Rowan AJ, Ramsay RE, Collins JF, et al; VA Cooperative Study 428 Group. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005; 64:1868–1673.
- Garnett WR. Optimizing antiepileptic drug therapy in the elderly. Ann Pharmacother 2005; 39:1852–1860.
- Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology 2002; 58(suppl 5):S2–S8.
- Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010; 51:1069–1077.
- Huying F, Klimpe S, Werhahn KJ. Antiepileptic drug use in nursing home residents: a cross-sectional, regional study. Seizure 2006; 15:194–197.
- Lackner TE, Cloyd JC, Thomas LW, Leppik IE. Antiepileptic drug use in nursing home residents: effect of age, gender, and comedication on patterns of use. Epilepsia 1998; 39:1083–1087.
- Galimberti CA, Magri F, Magnani B, et al. Antiepileptic drug use and epileptic seizures in elderly nursing home residents: a survey in the province of Pavia, Northern Italy. Epilepsy Res 2006; 68:1–8.
- Ramsay RE, Rowan AJ, Pryor FM. Special considerations in treating the elderly patient with epilepsy. Neurology 2004; 62(suppl 2):S24–S29.
- Sander JW, Hart YM, Johnson AL, Shorvon SD. National General Practice Study of Epilepsy: newly diagnosed epileptic seizures in a general population. Lancet 1990; 336:1267–1271.
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia 1993; 34:453–468.
- Laccheo I, Ablah E, Heinrichs R, Sadler T, Baade L, Liow K. Assessment of quality of life among the elderly with epilepsy. Epilepsy Behav 2008; 12:257–261.
- Stephen LJ, Brodie MJ. Epilepsy in elderly people. Lancet 2000; 355:1441–1446.
- Faught E, Richman J, Martin R, et al. Incidence and prevalence of epilepsy among older US Medicare beneficiaries. Neurology 2012; 78:448–453.
- Sillanpää M, Lastunen S, Helenius H, Schmidt D. Regional differences and secular trends in the incidence of epilepsy in Finland: a nationwide 23-year registry study. Epilepsia 2011; 52:1857–1867.
- Annegers JF, Hauser WA, Lee JR, Rocca WA. Incidence of acute symptomatic seizures in Rochester, Minnesota, 1935–1984. Epilepsia 1995; 36:327–333.
- Lühdorf K, Jensen LK, Plesner AM. Etiology of seizures in the elderly. Epilepsia 1986; 27:458–463.
- Granger N, Convers P, Beauchet O, et al. First epileptic seizure in the elderly: electroclinical and etiological data in 341 patients [in French]. Rev Neurol (Paris) 2002; 158:1088–1095.
- Pugh MJ, Knoefel JE, Mortensen EM, Amuan ME, Berlowitz DR, Van Cott AC. New-onset epilepsy risk factors in older veterans. J Am Geriatr Soc 2009; 57:237–242.
- Brodie MJ, Elder AT, Kwan P. Epilepsy in later life. Lancet Neurol 2009; 8:1019–1030.
- Bladin CF, Alexandrov AV, Bellavance A, et al. Seizures after stroke: a prospective multicenter study. Arch Neurol 2000; 57:1617–1622.
- Kilpatrick CJ, Davis SM, Tress BM, Rossiter SC, Hopper JL, Vandendriesen ML. Epileptic seizures in acute stroke. Arch Neurol 1990; 47:157–160.
- Giroud M, Gras P, Fayolle H, André N, Soichot P, Dumas R. Early seizures after acute stroke: a study of 1,640 cases. Epilepsia 1994; 35:959–964.
- Asconapé JJ, Penry JK. Poststroke seizures in the elderly. Clin Geriatr Med 1991; 7:483–492.
- So EL, Annegers JF, Hauser WA, O’Brien PC, Whisnant JP. Population-based study of seizure disorders after cerebral infarction. Neurology 1996; 46:350–355.
- Cleary P, Shorvon S, Tallis R. Late-onset seizures as a predictor of subsequent stroke. Lancet 2004; 363:1184–1186.
- Loiseau P. Pathologic processes in the elderly and their association with seizures. In:Rowan AJ, Ramsay RE, editors. Seizures and epilepsy in the elderly. Boston, MA: Butterworth-Heinemann; 1997:63–86.
- Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986; 36:1226–1230.
- Leppik IE, Birnbaum AK. Epilepsy in the elderly. Ann N Y Acad Sci 2010; 1184:208–224.
- Imfeld P, Bodmer M, Schuerch M, Jick SS, Meier CR. Seizures in patients with Alzheimer’s disease or vascular dementia: a population-based nested case-control analysis. Epilepsia 2013; 54:700–707.
- Irizarry MC, Jin S, He F, et al. Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol 2012; 69:368–372.
- Cordonnier C, Hénon H, Derambure P, Pasquier F, Leys D. Influence of pre-existing dementia on the risk of post-stroke epileptic seizures. J Neurol Neurosurg Psychiatry 2005; 76:1649–1653.
- Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia 2003; 44(suppl 10):2–10.
- Hiyoshi T, Yagi K. Epilepsy in the elderly. Epilepsia 2000; 41(suppl 9):31–35.
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 2009; 373:1105–1110.
- Roberts MA, Godfrey JW, Caird FI. Epileptic seizures in the elderly: I. Aetiology and type of seizure. Age Ageing 1982; 11:24–28.
- Loiseau J, Loiseau P, Duché B, Guyot M, Dartigues JF, Aublet B. A survey of epileptic disorders in southwest France: seizures in elderly patients. Ann Neurol 1990; 27:232–237.
- Lote K, Stenwig AE, Skullerud K, Hirschberg H. Prevalence and prognostic significance of epilepsy in patients with gliomas. Eur J Cancer 1998; 34:98–102.
- Franson KL, Hay DP, Neppe V, et al. Drug-induced seizures in the elderly. Causative agents and optimal management. Drugs Aging 1995; 7:38–48.
- Starr P, Klein-Schwartz W, Spiller H, Kern P, Ekleberry SE, Kunkel S. Incidence and onset of delayed seizures after overdoses of extended-release bupropion. Am J Emerg Med 2009; 27:911–915.
- Hauser WA, Ng SK, Brust JC. Alcohol, seizures, and epilepsy. Epilepsia 1988; 29(suppl 2):S66–S78.
- Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol 2014; 13:276–286.
- Ait S, Gilbert T, Cotton F, Bonnefoy M. Cortical blindness and posterior reversible encephalopathy syndrome in an older patient. BMJ Case Rep 2012;pii:bcr0920114782.
- Tinuper P, Provini F, Marini C, et al. Partial epilepsy of long duration: changing semiology with age. Epilepsia 1996; 37:162–164.
- Silveira DC, Jehi L, Chapin J, et al. Seizure semiology and aging. Epilepsy Behav 2011; 20:375–377.
- Theodore WH. The postictal state: effects of age and underlying brain dysfunction. Epilepsy Behav 2010; 19:118–120.
- Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 1998; 338:970–976.
- Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Incidence of status epilepticus in Rochester, Minnesota, 1965–1984. Neurology 1998; 50:735–741.
- Sung CY, Chu NS. Status epilepticus in the elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989; 80:51–56.
- Pro S, Vicenzini E, Randi F, Pulitano P, Mecarelli O. Idiopathic late-onset absence status epilepticus: a case report with an electroclinical 14 years follow-up. Seizure 2011; 20:655–658.
- Martin Y, Artaz MA, Bornand-Rousselot A. Nonconvulsive status epilepticus in the elderly. J Am Geriatr Soc 2004; 52:476–477.
- Fernández-Torre JL, Díaz-Castroverde AG. Non-convulsive status epilepticus in elderly individuals: report of four representative cases. Age Ageing 2004; 33:78–81.
- Chung PW, Seo DW, Kwon JC, Kim H, Na DL. Nonconvulsive status epilepticus presenting as a subacute progressive aphasia. Seizure 2002; 11:449–454.
- Sheth RD, Drazkowski JF, Sirven JI, Gidal BE, Hermann BP. Protracted ictal confusion in elderly patients. Arch Neurol 2006; 63:529–532.
- Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003; 61:1066–1073.
- Kellinghaus C, Loddenkemper T, Dinner DS, Lachhwani D, Lüders HO. Seizure semiology in the elderly: a video analysis. Epilepsia 2004; 45:263–267.
- Drury I, Beydoun A. Interictal epileptiform activity in elderly patients with epilepsy. Electroencephalogr Clin Neurophysiol 1998; 106:369–373.
- McBride AE, Shih TT, Hirsch LJ. Video-EEG monitoring in the elderly: a review of 94 patients. Epilepsia 2002; 43:165–169.
- Duncan JS, Sander JW, Sisodiya SM, Walker MC. Adult epilepsy. Lancet 2006; 367:1087–1100.
- McLean AJ, Le Couteur DG. Aging biology and geriatric clinical pharmacology. Pharmacol Rev 2004; 56:163–184.
- Pack AM, Morrell MJ. Epilepsy and bone health in adults. Epilepsy Behav 2004; 5(suppl 2):S24–S29.
- Granger AS. Ginkgo biloba precipitating epileptic seizures. Age Ageing 2001; 30:523–525.
- Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol 2006; 61:246–255.
- Rowan AJ, Ramsay RE, Collins JF, et al; VA Cooperative Study 428 Group. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology 2005; 64:1868–1673.
- Garnett WR. Optimizing antiepileptic drug therapy in the elderly. Ann Pharmacother 2005; 39:1852–1860.
- Brodie MJ, Kwan P. Staged approach to epilepsy management. Neurology 2002; 58(suppl 5):S2–S8.
- Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010; 51:1069–1077.
- Huying F, Klimpe S, Werhahn KJ. Antiepileptic drug use in nursing home residents: a cross-sectional, regional study. Seizure 2006; 15:194–197.
- Lackner TE, Cloyd JC, Thomas LW, Leppik IE. Antiepileptic drug use in nursing home residents: effect of age, gender, and comedication on patterns of use. Epilepsia 1998; 39:1083–1087.
- Galimberti CA, Magri F, Magnani B, et al. Antiepileptic drug use and epileptic seizures in elderly nursing home residents: a survey in the province of Pavia, Northern Italy. Epilepsy Res 2006; 68:1–8.
KEY POINTS
- About 25% of new-onset seizures occur after the age of 65.
- Most new-onset cases of epilepsy in the elderly are secondary to cerebrovascular disease, metabolic disturbances, dementia, traumatic brain injury, tumor, or drug therapy.
- The diagnosis is challenging and can be confused with syncope, transient ischemic attack, cardiac arrhythmia, metabolic disturbances, transient global amnesia, neurodegenerative disease, rapid-eye-movement sleep behavior disorder, and psychogenic disorders.
- The clinical presentation of seizures in the elderly differs from that in younger patients.
- A detailed clinical history, blood tests, electrocardiography, magnetic resonance imaging, and EEG can be helpful in diagnosing.
- No single drug is ideal for new-onset epilepsy in the elderly; the choice depends mainly on the type of seizure and the comorbidities present.
Perioperative beta-blockers in noncardiac surgery: The evidence continues to evolve
Prophylactic use of beta-blockers in the perioperative period is highly controversial. Initial studies in the 1990s were favorable, but evidence has been conflicting since then.
The pendulum swung away from routinely recommending beta-blockers after the publication of negative results from several studies, including the Perioperative Ischemic Evaluation (POISE) trial in 2008.1 Highlighting this change in practice, a Canadian study2 found that the use of perioperative beta-blockade increased between 1999 and 2005 but subsequently declined from 2005 to 2010. However, there was no appreciable change in this pattern after the POISE trial or after changes in the American College of Cardiology guidelines in 2002 and 2006.3
In 2008, Harte and Jaffer reviewed the perioperative use of beta-blockers in noncardiac surgery in this journal.4 Since then, a number of meta-analyses and retrospective observational studies have reported variable findings related to specific beta-blockers and specific complications.
In this paper, we review the rationale and recent evidence for and against the perioperative use of beta-blockers as guidance for internists and hospitalists.
POTENTIAL CARDIOPROTECTIVE EFFECTS OF BETA-BLOCKERS
Myocardial infarction and unstable angina are the leading cardiovascular causes of death after surgery.5 These events are multifactorial. Some are caused by the stress of surgery, which precipitates physiologic changes related to inflammatory mediators, sympathetic tone, and oxygen supply and demand; others are caused by acute plaque rupture, thrombosis, and occlusion.6 Most perioperative infarcts are non-Q-wave events7 and occur within the first 2 days after the procedure, when the effects of anesthetics, pain, fluid shifts, and physiologic changes are greatest. Because multiple causes may contribute to perioperative myocardial infarction, a single preventive strategy may not be sufficient.8,9
Beta-blockers do several things that may be beneficial in the perioperative setting. They reduce myocardial oxygen demand by decreasing the force of contraction and by slowing the heart rate, and slowing the heart rate increases diastolic perfusion time.10 They suppress arrhythmias; they limit leukocyte recruitment, the production of free radicals, metalloproteinase activity, monocyte activation, release of growth factors, and inflammatory cytokine response; and they stabilize plaque.11 Their long-term use may also alter intracellular signaling processes, thus improving cell survival by decreasing the expression of receptors for substances that induce apoptosis.12
INITIAL POSITIVE TRIALS
Mangano et al13 began the beta-blocker trend in 1996 with a study in 200 patients known to have coronary artery disease or risk factors for it who were undergoing noncardiac surgery. Patients were randomized to receive either atenolol orally and intravenously, titrated to control the heart rate, or placebo in the immediate perioperative period.
The atenolol group had less perioperative ischemia but no difference in short-term rates of myocardial infarction and death. However, the death rate was lower in the atenolol group at 6 months after discharge and at 2 years, although patients who died in the immediate postoperative period were excluded from the analysis.
Although this finding did not appear to make sense physiologically, we now know that patients may experience myocardial injury without infarction after noncardiac surgery, a phenomenon associated with an increased risk of death in the short term and the long term.14 Preventing these episodes may be the explanation for the improved outcome.
The DECREASE trial15 (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) provided additional support for beta-blocker use. The patients were at high risk, had abnormal dobutamine stress echocardiograms, and were undergoing vascular surgery; 112 patients were randomized to receive either oral bisoprolol (started 1 month before surgery, titrated to control the heart rate, and continued for 1 month after surgery) or placebo.
The study was stopped early because the bisoprolol group reportedly had a 90% lower rate of myocardial infarction and cardiac death 1 month after surgery. However, the study was criticized because the total number of patients enrolled was small and the benefit was much greater than usual for any pharmacologic intervention, thus calling the results into question.
In a follow-up study,16 survivors continued to be followed while receiving bisoprolol or usual care. The incidence of myocardial infarction or cardiac death at 2 years was significantly lower in the group receiving bisoprolol (12% vs 32%, odds ratio [OR] 0.30, P = .025).
Boersma et al,17 in an observational study, analyzed data from all 1,351 patients scheduled for major vascular surgery being considered for enrollment in the DECREASE trial. The DECREASE protocol required patients to undergo dobutamine stress echocardiography if they had one or more risk factors (age 70 or older, angina, prior myocardial infarction, congestive heart failure, treatment for ventricular arrhythmia, treatment for diabetes mellitus, or limited exercise capacity) or if their physician requested it. Twenty-seven percent received beta-blockers.
In multivariate analysis, clinical predictors of adverse outcome were age 70 or older; current or prior history of angina; and prior myocardial infarction, heart failure, or cerebrovascular accident.
In patients who had fewer than three clinical risk factors, beta-blocker use was associated with a lower rate of complications (0.8% vs 2.3%). Dobutamine stress echocardiography had minimal predictive value in this lower-risk group, suggesting that stress testing may not be necessary in this group if beta-blockers are used appropriately. However, in patients who had three or more risk factors, this test did provide additional prognostic information; those without stress-induced ischemia had lower event rates than those with ischemia, and beta-blocker use further reduced those rates, except in patients with extensive ischemia (more than five left ventricular segments involved).
The Revised Cardiac Risk Index. Lee et al18 devised an index to assist in preoperative cardiac risk stratification that was subsequently incorporated into the 2007 American College of Cardiology/American Heart Association preoperative risk guidelines. (It does not, however, address the beta-blocker issue.) It consists of six independent risk-predictors of major cardiac complications derived from 4,315 patients over age 50 undergoing non-cardiac surgery. The risk factors, each of which is given 1 point, are:
- Congestive heart failure based on history or examination
- Renal insufficiency (serum creatinine level > 2 mg/dL)
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- History of transient ischemic attack or stroke
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with 3 or more points are considered to be at high risk, and those with 1 or 2 points are considered to be at intermediate risk. The American College of Cardiology/American Heart Association preoperative cardiac risk algorithm subsequently included five of these six risk factors (the type of surgery was considered separately) and made recommendations concerning noninvasive stress testing and heart rate control.
On the basis of these studies, specialty societies, guideline committees, and hospitals enthusiastically recommended the prophylactic use of beta-blockers to decrease postoperative cardiac complications.
THREE NEGATIVE TRIALS OF METOPROLOL
In 2005 and 2006, two studies in vascular surgery patients and another in patients with diabetes cast doubt on the role of beta-blockers when the results failed to show a benefit. The trials used metoprolol, started shortly before surgery, and with no titration to control the heart rate.
The MaVS study19 (Metoprolol After Vascular Surgery) randomized 496 patients to receive metoprolol or placebo 2 hours before surgery and until hospital discharge or a maximum of 5 days after surgery. The metoprolol dose varied by weight: patients weighing 75 kg or more got 100 mg, those weighing between 40 and 75 kg got 50 mg, and those weighing less than 40 kg got 25 mg. Overall effects at 6 months were not significantly different, but intraoperative bradycardia and hypotension requiring intervention were more frequent in the metoprolol group.
The POBBLE study20 (Perioperative Beta Blockade) randomized 103 patients who had no history of myocardial infarction to receive either metoprolol 50 mg twice daily or placebo from admission to 7 days after surgery. Myocardial ischemia was present in one-third of the patients after surgery. Metoprolol did not reduce the 30-day cardiac mortality rate, but it was associated with a shorter length of stay.
The DIPOM trial21 (Diabetic Postoperative Mortality and Morbidity) randomized 921 diabetic patients to receive long-acting metoprolol succinate controlled-release/extended release (CR/XL) or placebo. Patients in the metoprolol group received a test dose of 50 mg the evening before surgery, another dose 2 hours before surgery (100 mg if the heart rate was more than 65 bpm, or 50 mg if between 55 and 65 bpm), and daily thereafter until discharge or a maximum of 8 days. The dose was not titrated to heart-rate control.
Metoprolol had no statistically significant effect on the composite primary outcome measures of time to death from any cause, acute myocardial infarction, unstable angina, or congestive heart failure or on the secondary outcome measures of time to death from any cause, death from a cardiac cause, and nonfatal cardiac morbidity.
ADDITIONAL POSITIVE STUDIES
Lindenauer et al22 retrospectively evaluated the use of beta-blockers in the first 2 days after surgery in 782,969 patients undergoing non-cardiac surgery. Using propensity score matching and Revised Cardiac Risk Index scores, they found a lower rate of postoperative mortality in patients with three or more risk factors who received a beta-blocker. There was no significant difference in the group with two risk factors, but in the lowest-risk group (with a score of 0 to 1), beta-blockers were not beneficial and may have been associated with harm as evidenced by a higher odds ratio for death, although this was probably artifactual and reflecting database limitations.
Feringa et al,23 in an observational cohort study of 272 patients undergoing vascular surgery, reported that higher doses of beta-blockers and tight heart-rate control were associated with less perioperative myocardial ischemia, lower troponin T levels, and better long-term outcome.
THE POISE TRIAL: MIXED RESULTS
The randomized POISE trial,1 published in 2008, compared the effects of extended-release metoprolol succinate vs placebo on the 30-day risk of major cardiovascular events in 8,351 patients with or at risk of atherosclerotic disease who were undergoing noncardiac surgery. The metoprolol regimen was 100 mg 2 to 4 hours before surgery, another 100 mg by 6 hours after surgery, and then 200 mg 12 hours later and once daily for 30 days.
The incidence of the composite primary end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest at 30 days was lower in the metoprolol group than in the placebo group (5.8% vs 6.9%; P = .04), primarily because of fewer nonfatal myocardial infarctions. However, more patients in the metoprolol group died of any cause (3.1% vs 2.3% P = .03) or had a stroke (1.0% vs 0.5% P = .005) than in the placebo group.
The metoprolol group had a higher incidence of clinically significant hypotension, bradycardia, and stroke, which could account for much of the increase in the mortality rate. Sepsis was the major cause of death in this group; hypotension may have increased the risk of infection, and beta-blockers may have potentiated hypotension in patients who were already septic. Also, the bradycardic and negative inotropic effects of the beta-blocker could have masked the physiologic response to systemic infection, thereby delaying recognition and treatment or impeding the normal immune response.
One of the major criticisms of the POISE trial was its aggressive dosing regimen (200 to 400 mg within a 36-hour period) in patients who had not been on beta-blockers before then. Also, the drug was started only a few hours before surgery. In addition, these patients were at higher risk of death and stroke than those in other trials based on a high baseline rate of cerebrovascular disease, and inclusion of urgent and emergency surgical procedures.
STUDIES SINCE POISE
The POISE trial results1 prompted further questioning of the prophylactic perioperative use of beta-blockers. However, proponents of beta-blockers voiced serious criticisms of the trial, particularly the dosing regimen, and continued to believe that these drugs were beneficial if used appropriately.
The DECREASE IV trial. Dunkelgrun et al,24 in a study using bisoprolol started approximately 1 month before surgery and titrated to control the heart rate, reported beneficial results in intermediate-risk patients. In their randomized open-label study with a 2 × 2 factorial design, 1,066 patients at intermediate cardiac risk were assigned to receive bisoprolol, fluvastatin, combination treatment, or control therapy at least 34 days before surgery. Bisoprolol was started at 2.5 mg orally daily and slowly titrated up to a maximum dose of 10 mg to keep the heart rate between 50 and 70 beats per minute. The group of 533 patients randomized to receive bisoprolol had a lower incidence rate of cardiac death and nonfatal myocardial infarction than the control group (2.1% vs 6.0%, HR 0.34, P = .002). A potential limitation of this study was its open-label design, which might have led to treatment bias.
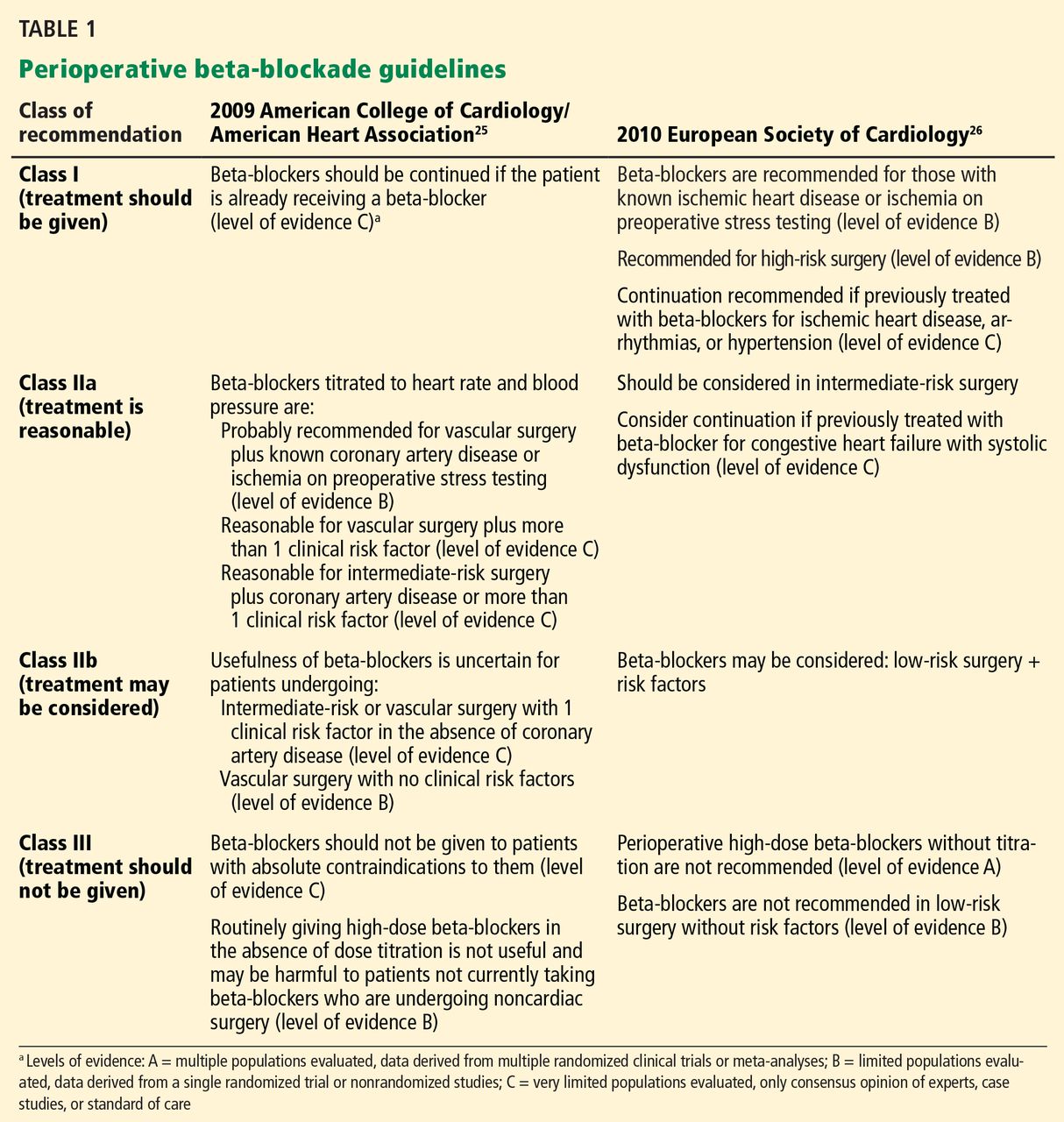
Updated guidelines. Based on the results from POISE and DECREASE IV, the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines25 published a focused update on beta-blockers in 2009 as an amendment to their 2007 guidelines on perioperative evaluation and care for noncardiac surgery. The European Society of Cardiology26 released similar but somewhat more liberal guidelines (Table 1).
London et al,27 in an observational study published in 2013, found a lower 30-day overall mortality rate with beta-blockers (relative risk [RR] 0.73, 95% confidence interval [CI] 0.65–0.83, P < .001, number needed to treat [NNT] 241), as well as a lower rate of cardiac morbidity (nonfatal myocardial infarction and cardiac death), but only in nonvascular surgery patients who were on beta-blockers within 7 days of scheduled surgery. Moreover, similar to the findings of Lindenauer et al,22 only patients with a Revised Cardiac Risk Index score of 2 or more benefited from beta-blocker use in terms of a lower risk of death, whereas the lower-risk patients did not:
- Risk score of 0 or 1—no association
- Score of 2—RR 0.63, 95% CI 0.50–0.80, P < .001, NNT 105
- Score of 3—RR 0.54, 95% CI 0.39–0.73, P < .001, NNT 41
- Score of 4 or more—RR 0.40, 95% CI 0.24–0.73, P < .001, NNT 18).
Beta-blocker exposure was associated with a significantly lower rate of cardiac complications (RR 0.67, 95% CI 0.57–0.79, P < .001, NNT 339), also limited to nonvascular surgery patients with a risk score of 2 or 3.
The Danish Nationwide Cohort Study28 examined the effect of beta-blockers on major adverse cardiac events (MACE, ie, myocardial infarction, cerebrovascular accident, and death) in 28,263 patients with ischemic heart disease undergoing noncardiac surgery; 7,990 with heart failure and 20,273 without. Beta-blockers were used in 53% of patients with heart failure and 36% of those without heart failure. Outcomes for all of the beta-blocker recipients:
- MACE—HR 0.90, 95% CI 0.79–1.02
- All-cause mortality—HR 0.95, 95% CI 0.85–1.06.
Outcomes for patients with heart failure if they received beta-blockers:
- MACE—HR 0.75, 95% CI 0.70–0.87
- All-cause mortality—HR 0.80, 95% CI 0.70–0.92.
There was no significant benefit from beta-blockers in patients without heart failure. Outcomes for those patients if they received beta-blockers:
- MACE—HR 1.11, 95% CI 0.92–1.33
- All-cause mortality—HR 1.15, 95% CI 0.98–1.35.
However, in patients without heart failure but with a history of myocardial infarction within the past 2 years, beta-blockers were associated with a lower risk of MACE and all-cause mortality. In patients with neither heart failure nor a recent myocardial infarction, beta-blockers were associated with an increased risk of MACE and all-cause mortality.
This difference in efficacy depending on the presence and timing of a prior myocardial infarction is consistent with the 2012 American College of Cardiology/American Heart Association guidelines for secondary prevention, in which beta-blockers are given a class I recommendation only for patients with a myocardial infarction within the past 3 years.
Meta-analyses and outcomes
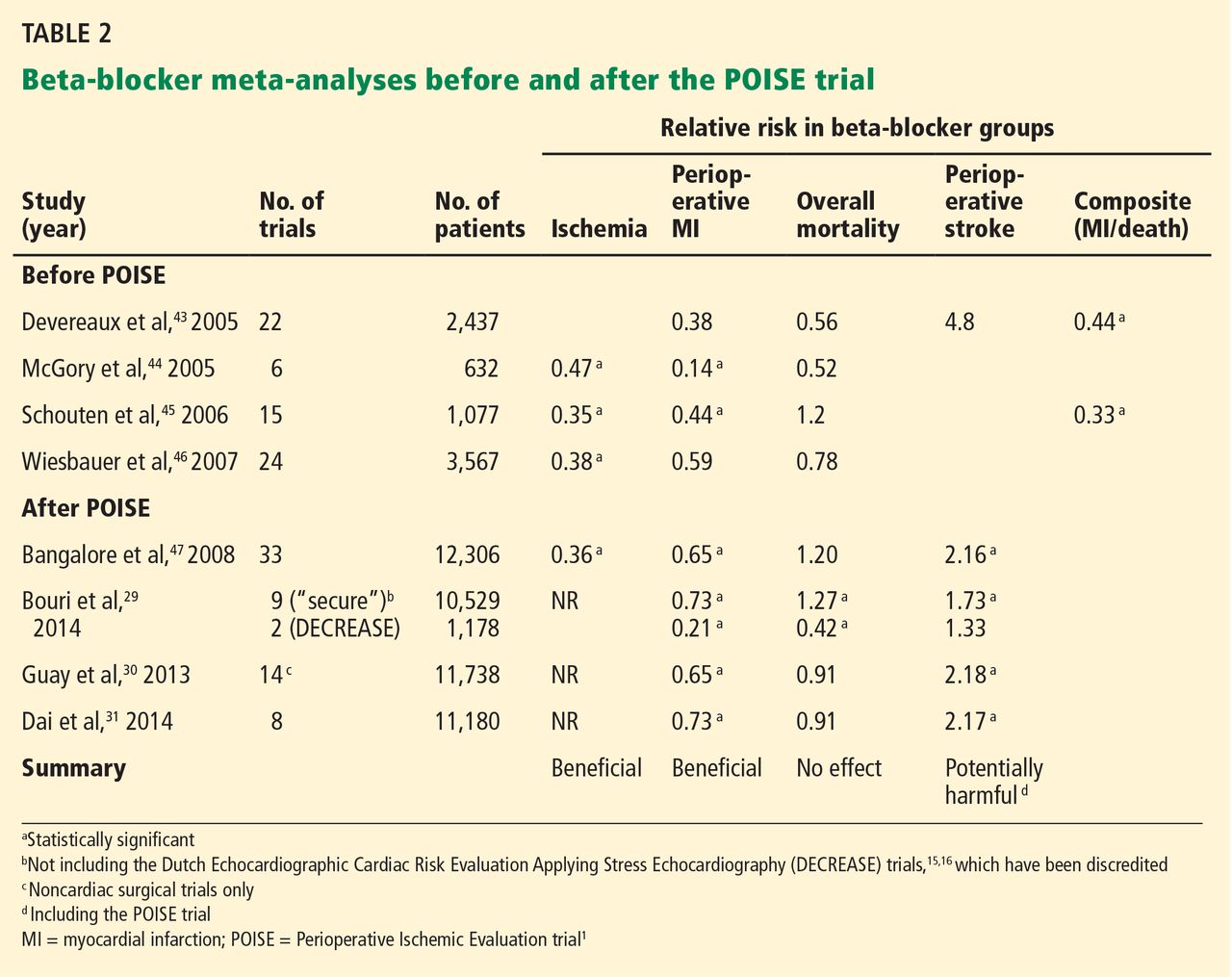
A number of meta-analyses have been published over the past 10 years, with conflicting results (Table 2). The divergent findings are primarily due to the different studies included in the analyses as well as the strong influence of the POISE trial.1 The studies varied in terms of the specific beta-blocker used, dose titration and heart rate control, time of initiation of beta-blocker use before surgery, type of surgery, patient characteristics, comorbidities, biomarkers and diagnosis of myocardial infarction, and clinical end points.
In general, these meta-analyses have found that prophylactic perioperative use of beta-blockers decreases ischemia and tends to reduce the risk of nonfatal myocardial infarction. They vary on whether the overall mortality risk is decreased. The meta-analyses that included POISE1 found an increased incidence of stroke, whereas those that excluded POISE found no significant difference, although there appeared to be slightly more strokes in the beta-blocker groups.
The beta-blocker controversy increased even further when Dr. Don Poldermans was fired by Erasmus Medical Center in November 2011 for violations of academic integrity involving his research, including the DECREASE trials. The most recent meta-analysis, by Bouri et al,29 included nine “secure trials” and excluded the DECREASE trials in view of the controversy about their authenticity. The analysis showed an increase in overall mortality as well as stroke, primarily because it was heavily influenced by POISE.1 In contrast, the DECREASE trials had reported a decreased risk of myocardial infarction and death, with no significant increase in stroke. The authors concluded that guideline bodies should “retract their recommendations based on the fictitious data without further delay.”29
Although the design of the DECREASE trials (in which beta-blockers were started well in advance of surgery and doses were titrated to achieve heart rate control) is physiologically more compelling than those of the negative trials, the results have been questioned in light of the integrity issue. However, to date, none of the published DECREASE trials have been retracted.
Two other meta-analyses,30,31 published in 2013, also found a decreased risk of myocardial infarction and increased risk of stroke but no significant difference in short-term all-cause mortality.
ARE ALL BETA-BLOCKERS EQUIVALENT?
In various studies evaluating specific beta-blockers, the more cardioselective agents bisoprolol and atenolol were associated with better outcomes than metoprolol. The affinity ratios for beta-1/beta-2 receptors range from 13.5 for bisoprolol to 4.7 for atenolol and 2.3 for metoprolol.32 Blocking beta-1 receptors blunts tachycardia, whereas blocking beta-2 receptors may block systemic or cerebral vasodilation.
In patients with anemia, beta-blockade in general may be harmful, but beta-2 blockade may be even worse. Beta-blockers were associated with an increased risk of MACE (6.5% vs 3.0%)33 in patients with acute surgical anemia if the hemoglobin concentration decreased to less than 35% of baseline, and increased risks of hospital death (OR 6.65) and multiorgan dysfunction syndrome (OR 4.18) with severe bleeding during aortic surgery.34
In addition, the pathway by which the beta-blocker is metabolized may also affect outcome, with less benefit from beta-blockers metabolized by the CYP2D6 isoenzyme of the cytochrome P450 system. Individual variations in CYP2D6 activity related to genetics or drug interactions may result in insufficient or excessive beta-blockade. Because metoprolol is the most dependent on this system, patients using it may be more susceptible to bradycardia.35
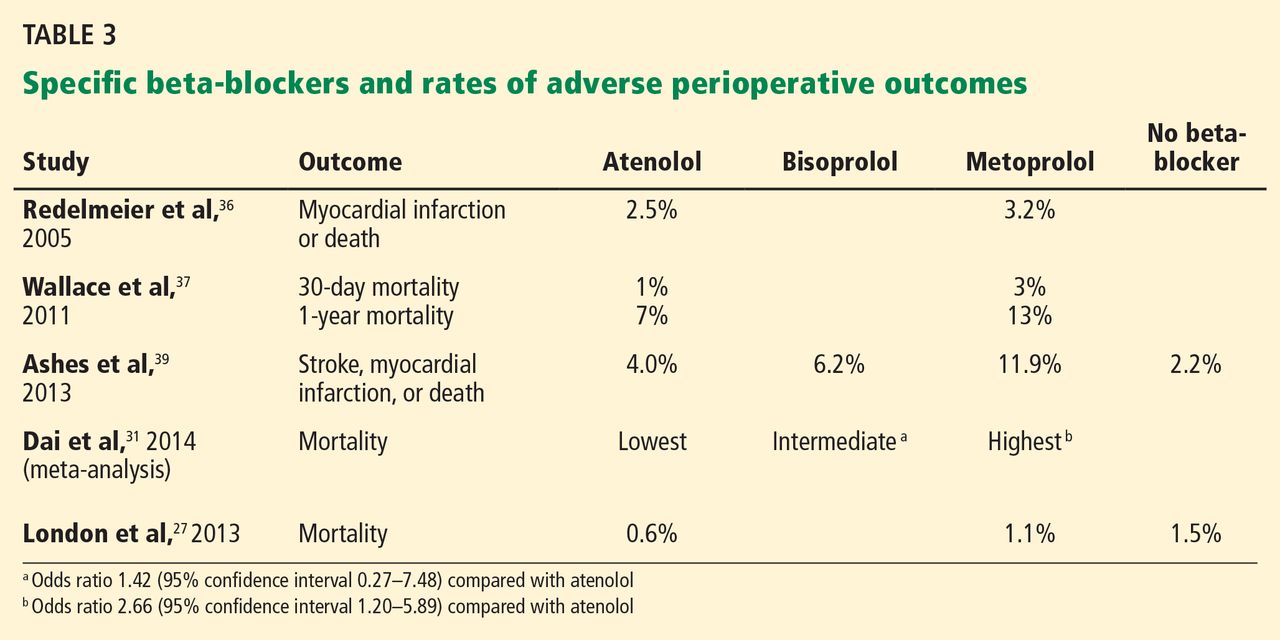
Studies comparing atenolol and metoprolol found that the atenolol groups had fewer myocardial infarctions and deaths36 and lower 30-day and 1-year mortality rates37 than the groups on metoprolol. Studies comparing the three beta-blockers found better outcomes with atenolol and bisoprolol than with metoprolol—fewer strokes,38,39 a lower mortality rate,31 and a better composite outcome39 (Table 3 and Table 4).
START THE BETA-BLOCKER EARLY, TITRATE TO CONTROL THE HEART RATE
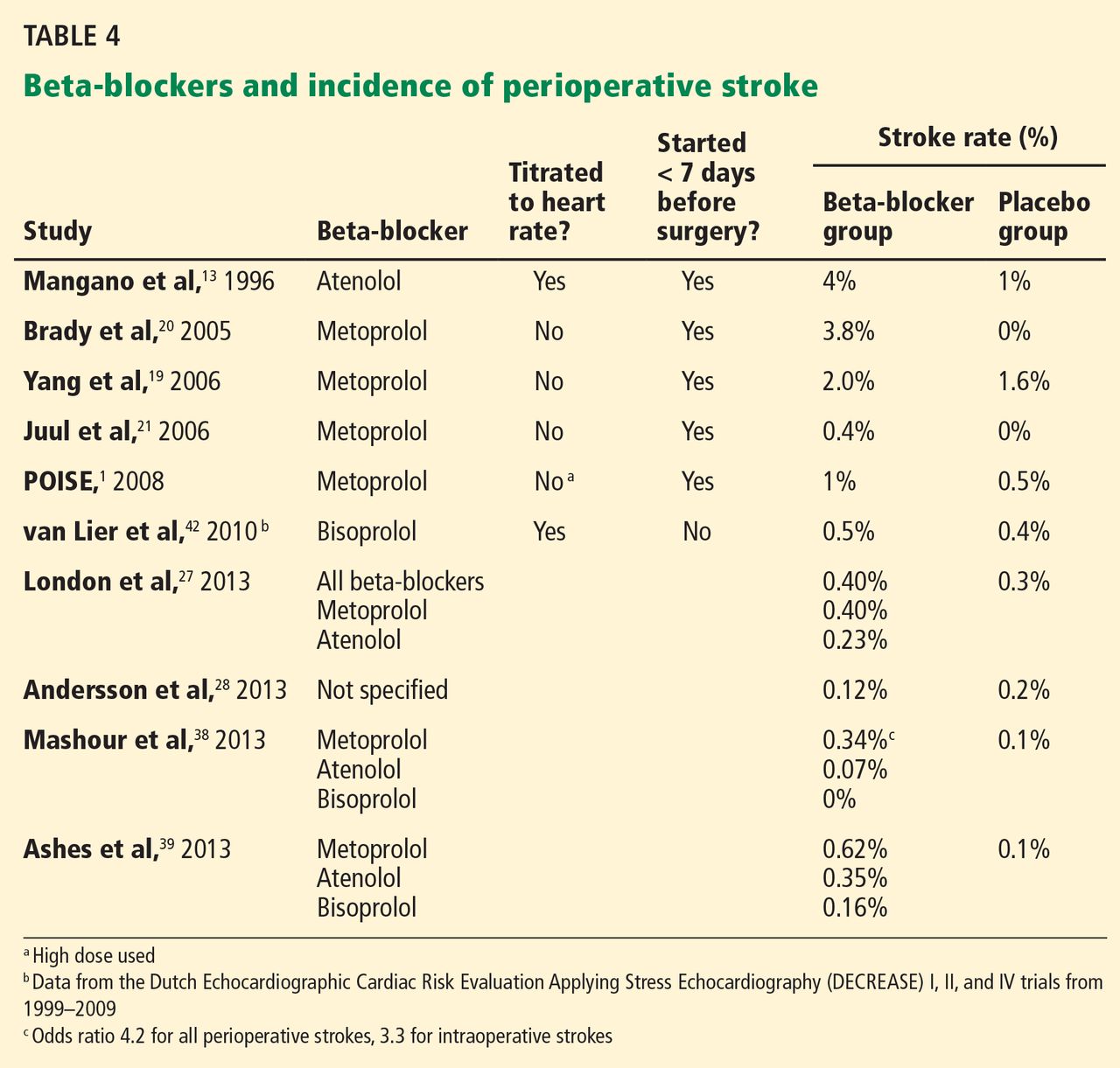
A number of studies suggest that how long the beta-blocker is given before surgery may influence the outcome (Table 5). The best results were achieved when beta-blockers were started approximately 1 month before surgery and titrated to control the heart rate.
Because this long lead-in time is not always practical, it is important to determine the shortest time before surgery in which starting beta-blockers may be beneficial and yet safe. Some evidence suggests that results are better when the beta-blocker is started more than 1 week preoperatively compared with less than 1 week, but it is unknown what the minimum or optimal time period should be.
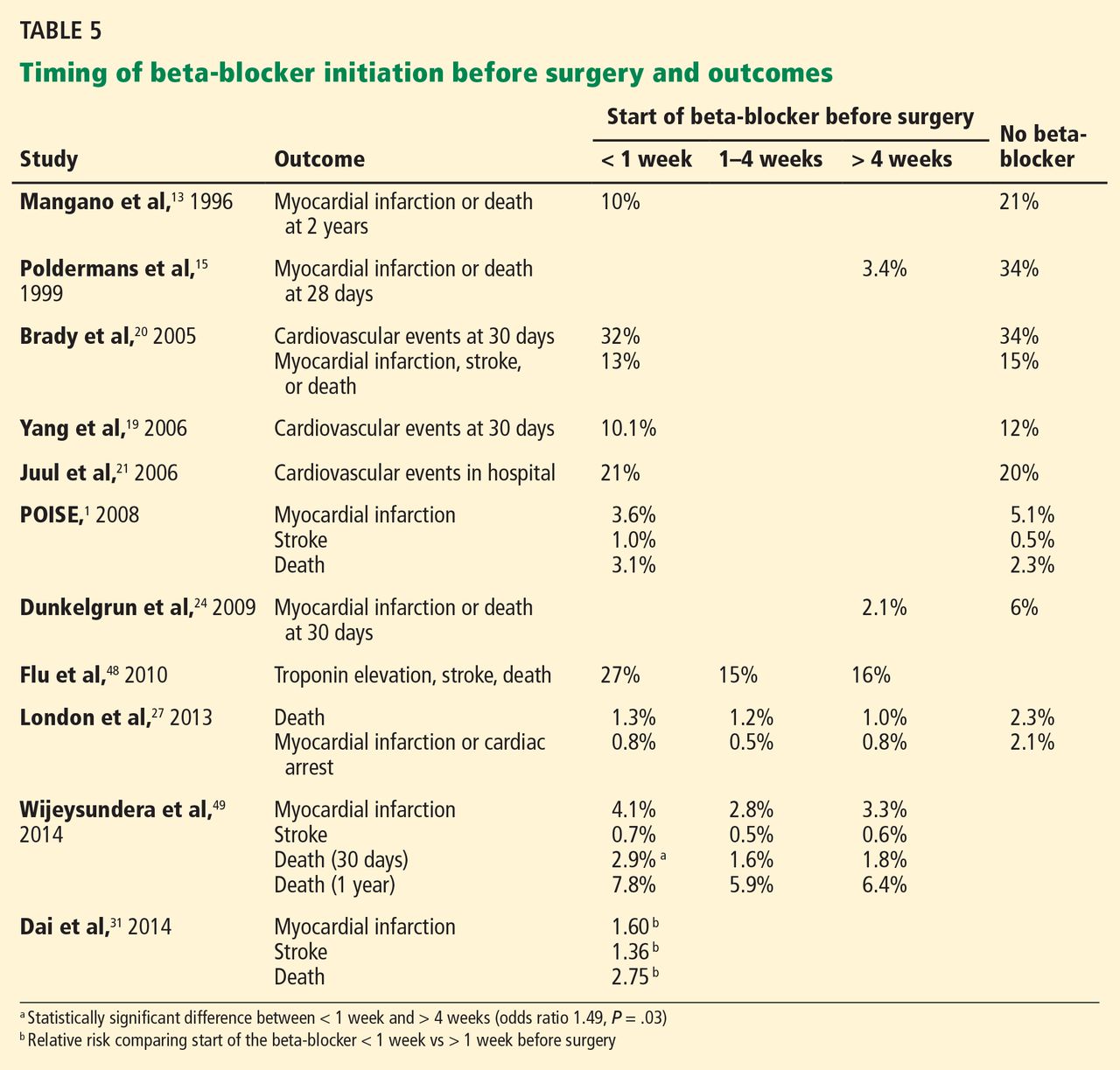
If a beta-blocker is started well in advance of the scheduled surgery, there is adequate time for dose titration and tighter heart rate control. Most of the studies demonstrating beneficial effects of perioperative beta-blockers used dose titration and achieved lower heart rates in the treatment group than in the control group. A criticism of the MaVs,19 POBBLE,20 and DIPOM21 trials was that the patients did not receive adequate beta-blockade. The POISE trial1 used a much higher dose of metoprolol in an attempt to assure beta-blockade without dose titration, and although the regimen decreased nonfatal myocardial infarctions, it increased strokes and the overall mortality rate, probably related to excess bradycardia and hypotension. The target heart rate should probably be between 55 and 70 beats per minute.
RISK OF STROKE
POISE1 was the first trial to note a clinically and statistically significant increase in strokes with perioperative beta-blocker use. Although no other study has shown a similar increased risk, almost all reported a higher number of strokes in the beta-blocker groups, although the absolute numbers and differences were small and not statistically significant. This risk may also vary from one beta-blocker to another (Table 4).
The usual incidence rate of postoperative stroke after noncardiac, noncarotid surgery is well under 1% in patients with no prior history of stroke but increases to approximately 3% in patients with a previous stroke.40 An observational study from the Dutch group reported a very low incidence of stroke overall (0.02%) in 186,779 patients undergoing noncardiac surgery with no significant difference in those on chronic beta-blocker therapy.41 The DECREASE trials, with a total of 3,884 patients, also found no statistically significant increase in stroke with beta-blocker use (0.46% overall vs 0.5% with a beta-blocker),42 which in this case was bisoprolol started well in advance of surgery and titrated to control the heart rate. Although the DECREASE data are under suspicion, they seem reasonable and consistent with those of observational studies.
Proposed mechanisms by which beta-blockers may increase stroke risk include the side effects of hypotension and bradycardia, particularly in the setting of anemia. They may also cause cerebral ischemia by blocking cerebral vasodilation. This effect on cerebral blood flow may be more pronounced with the less cardioselective beta-blockers, which may explain the apparent increased stroke risk associated with metoprolol.
WHAT SHOULD WE DO NOW?
The evidence for the safety and efficacy of beta-blockers in the perioperative setting continues to evolve, and new clinical trials are needed to clarify the ongoing controversy, particularly regarding the risk of stroke.
If patients have other indications for beta-blocker therapy, such as history of heart failure, myocardial infarction in the past 3 years, or atrial fibrillation for rate control, they should be receiving them if time permits.
If prophylactic beta-blockers are to be effective in minimizing perioperative complications, it appears that they may need to be more cardioselective, started at least 1 week before surgery, titrated to control heart rate, and used in high-risk patients (Revised Cardiac Risk Index score > 2 or 3) undergoing high-risk surgery.
Ideally, a large randomized controlled trial using a cardioselective beta-blocker started in advance of surgery in patients with a Revised Cardiac Risk Index score greater than 2, undergoing intermediate or high-risk procedures, is needed to fully answer the questions raised by the current data.
- POISE Study Group; Devereaux PJ, Yang H, Yusuf S, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Wijeysundera DN, Mamdani M, Laupacis A, et al. Clinical evidence, practice guidelines, and ß-blocker utilization before major noncardiac surgery. Circ Cardiovasc Qual Outcomes 2012; 5:558–565.
- American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery); American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2006 guideline update on perioperative cardiovascular evaluation for noncardiac surgery: focused update on perioperative beta-blocker therapy: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society for Vascular Medicine and Biology. J Am Coll Cardiol 2006; 47:2343–2355.
- Harte B, Jaffer AK. Perioperative beta-blockers in noncardiac surgery: evolution of the evidence. Cleve Clin J Med 2008; 75:513–519.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foëx P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Poldermans D, Boersma E, Bax JJ, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol reduces cardiac death and myocardial infarction in high-risk patients as long as 2 years after successful major vascular surgery. Eur Heart J 2001; 22:1353–1358.
- Boersma E, Poldermans D, Bax JJ, et al; DECREASE Study Group (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiogrpahy). Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR; POBBLE trial investigators. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al; DIPOM Trial Group. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major non-cardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114(suppl 1):1344–1349.
- Dunkelgrun M, Boersma E, Schouten O, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol and fluvastatin for the reduction of perioperative cardiac mortality and myocardial infarction in intermediate-risk patients undergoing noncardiovascular surgery: a randomized controlled trial (DECREASE-IV). Ann Surg 2009; 249:921–926.
- American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2009; 54:e13–e118.
- Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non-cardiac Surgery; European Society of Cardiology (ESC); Poldermans D, Bax JJ, Boersma E, et al. Guidelines for preoperative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery. Eur Heart J 2009; 30:2769–2812.
- London MJ, Hur K, Schwartz GG, Henderson WG. Association of perioperative beta-blockade with mortality and cardiovascular morbidity following major noncardiac surgery. JAMA 2013; 309:1704–1713.
- Andersson C, Mérie C, Jørgensen M, et al. Association of beta-blocker therapy with risks of adverse cardiovascular events and deaths in patients with ischemic heart disease undergoing noncardiac surgery: a Danish nationwide cohort study. JAMA Intern Med 2014; 174:336–344.
- Bouri S, Shun-Shin MJ, Cole GD, Mayet J, Francis DP. Meta-analysis of secure randomised controlled trials of beta-blockade to prevent perioperative death in non-cardiac surgery. Heart 2014; 100:456–464.
- Guay J, Ochroch EA. Beta-blocking agents for surgery: influence on mortality and major outcomes. A meta-analysis. J Cardiothorac Vasc Anesth 2013; 27:834–844.
- Dai N, Xu D, Zhang J, et al. Different beta-blockers and initiation time in patients undergoing noncardiac surgery: a meta-analysis. Am J Med Sci 2014; 347:235–244.
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol 2005; 144:317–322.
- Beattie WS, Wijeysundera DN, Karkouti K, et al. Acute surgical anemia influences the cardioprotective effects of beta-blockade: a single-center, propensity-matched cohort study. Anesthesiology 2010; 112:25–33.
- Le Manach Y, Collins GS, Ibanez C, et al. Impact of perioperative bleeding on the protective effect of beta-blockers during infrarenal aortic reconstruction. Anesthesiology 2012; 117:1203–1211.
- Badgett RG, Lawrence VA, Cohn SL. Variations in pharmacology of beta-blockers may contribute to heterogeneous results in trials of perioperative beta-blockade. Anesthesiology 2010; 113:585–592.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
- Wallace AW, Au S, Cason BA. Perioperative beta-blockade: atenolol is associated with reduced mortality when compared to metoprolol. Anesthesiology 2011; 114:824–836.
- Mashour GA, Sharifpour M, Freundlich RE, et al. Perioperative metoprolol and risk of stroke after noncardiac surgery. Anesthesiology 2013; 119:1340–1346.
- Ashes C, Judelman S, Wijeysundera DN, et al. Selective beta1-antagonism with bisoprolol is associated with fewer postoperative strokes than atenolol or metoprolol: a single-center cohort study of 44,092 consecutive patients. Anesthesiology 2013; 119:777–787.
- Selim M. Perioperative stroke. N Engl J Med 2007; 356:706–713.
- van Lier F, Schouten O, van Domburg RT, et al. Effect of chronic beta-blocker use on stroke after noncardiac surgery. Am J Cardiol 2009; 104:429–433.
- van Lier F, Schouten O, Hoeks SE, et al. Impact of prophylactic beta-blocker therapy to prevent stroke after noncardiac surgery. Am J Cardiol 2010; 105:43–47.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta-analysis of randomised controlled trials. BMJ 2005; 331:313–321.
- McGory ML, Maggard MA, Ko CY. A meta-analysis of perioperative beta blockade: what is the actual risk reduction? Surgery 2005; 138:171–179.
- Schouten O, Shaw LJ, Boersma E, et al. A meta-analysis of safety and effectiveness of perioperative beta-blocker use for the prevention of cardiac events in different types of noncardiac surgery. Coron Artery Dis 2006; 17:173–179.
- Wiesbauer F, Schlager O, Domanovits H, et al. Perioperative beta-blockers for preventing surgery-related mortality and morbidity: a systematic review and meta-analysis. Anesth Analg 2007; 104:27–41.
- Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet 2008; 372:1962–1976.
- Flu WJ, van Kuijk JP, Chonchol M, et al. Timing of preoperative beta-blocker treatment in vascular surgery patients: influence on postoperative outcome. J Am Coll Cardiol 2010; 56:1922–1929.
- Wijeysundera DN, Beattie WS, Wijeysundera HC, Yun L, Austin PC, Ko DT. Duration of preoperative beta-blockade and outcomes after major elective noncardiac surgery. Can J Cardiol 2014; 30:217–223.
Prophylactic use of beta-blockers in the perioperative period is highly controversial. Initial studies in the 1990s were favorable, but evidence has been conflicting since then.
The pendulum swung away from routinely recommending beta-blockers after the publication of negative results from several studies, including the Perioperative Ischemic Evaluation (POISE) trial in 2008.1 Highlighting this change in practice, a Canadian study2 found that the use of perioperative beta-blockade increased between 1999 and 2005 but subsequently declined from 2005 to 2010. However, there was no appreciable change in this pattern after the POISE trial or after changes in the American College of Cardiology guidelines in 2002 and 2006.3
In 2008, Harte and Jaffer reviewed the perioperative use of beta-blockers in noncardiac surgery in this journal.4 Since then, a number of meta-analyses and retrospective observational studies have reported variable findings related to specific beta-blockers and specific complications.
In this paper, we review the rationale and recent evidence for and against the perioperative use of beta-blockers as guidance for internists and hospitalists.
POTENTIAL CARDIOPROTECTIVE EFFECTS OF BETA-BLOCKERS
Myocardial infarction and unstable angina are the leading cardiovascular causes of death after surgery.5 These events are multifactorial. Some are caused by the stress of surgery, which precipitates physiologic changes related to inflammatory mediators, sympathetic tone, and oxygen supply and demand; others are caused by acute plaque rupture, thrombosis, and occlusion.6 Most perioperative infarcts are non-Q-wave events7 and occur within the first 2 days after the procedure, when the effects of anesthetics, pain, fluid shifts, and physiologic changes are greatest. Because multiple causes may contribute to perioperative myocardial infarction, a single preventive strategy may not be sufficient.8,9
Beta-blockers do several things that may be beneficial in the perioperative setting. They reduce myocardial oxygen demand by decreasing the force of contraction and by slowing the heart rate, and slowing the heart rate increases diastolic perfusion time.10 They suppress arrhythmias; they limit leukocyte recruitment, the production of free radicals, metalloproteinase activity, monocyte activation, release of growth factors, and inflammatory cytokine response; and they stabilize plaque.11 Their long-term use may also alter intracellular signaling processes, thus improving cell survival by decreasing the expression of receptors for substances that induce apoptosis.12
INITIAL POSITIVE TRIALS
Mangano et al13 began the beta-blocker trend in 1996 with a study in 200 patients known to have coronary artery disease or risk factors for it who were undergoing noncardiac surgery. Patients were randomized to receive either atenolol orally and intravenously, titrated to control the heart rate, or placebo in the immediate perioperative period.
The atenolol group had less perioperative ischemia but no difference in short-term rates of myocardial infarction and death. However, the death rate was lower in the atenolol group at 6 months after discharge and at 2 years, although patients who died in the immediate postoperative period were excluded from the analysis.
Although this finding did not appear to make sense physiologically, we now know that patients may experience myocardial injury without infarction after noncardiac surgery, a phenomenon associated with an increased risk of death in the short term and the long term.14 Preventing these episodes may be the explanation for the improved outcome.
The DECREASE trial15 (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) provided additional support for beta-blocker use. The patients were at high risk, had abnormal dobutamine stress echocardiograms, and were undergoing vascular surgery; 112 patients were randomized to receive either oral bisoprolol (started 1 month before surgery, titrated to control the heart rate, and continued for 1 month after surgery) or placebo.
The study was stopped early because the bisoprolol group reportedly had a 90% lower rate of myocardial infarction and cardiac death 1 month after surgery. However, the study was criticized because the total number of patients enrolled was small and the benefit was much greater than usual for any pharmacologic intervention, thus calling the results into question.
In a follow-up study,16 survivors continued to be followed while receiving bisoprolol or usual care. The incidence of myocardial infarction or cardiac death at 2 years was significantly lower in the group receiving bisoprolol (12% vs 32%, odds ratio [OR] 0.30, P = .025).
Boersma et al,17 in an observational study, analyzed data from all 1,351 patients scheduled for major vascular surgery being considered for enrollment in the DECREASE trial. The DECREASE protocol required patients to undergo dobutamine stress echocardiography if they had one or more risk factors (age 70 or older, angina, prior myocardial infarction, congestive heart failure, treatment for ventricular arrhythmia, treatment for diabetes mellitus, or limited exercise capacity) or if their physician requested it. Twenty-seven percent received beta-blockers.
In multivariate analysis, clinical predictors of adverse outcome were age 70 or older; current or prior history of angina; and prior myocardial infarction, heart failure, or cerebrovascular accident.
In patients who had fewer than three clinical risk factors, beta-blocker use was associated with a lower rate of complications (0.8% vs 2.3%). Dobutamine stress echocardiography had minimal predictive value in this lower-risk group, suggesting that stress testing may not be necessary in this group if beta-blockers are used appropriately. However, in patients who had three or more risk factors, this test did provide additional prognostic information; those without stress-induced ischemia had lower event rates than those with ischemia, and beta-blocker use further reduced those rates, except in patients with extensive ischemia (more than five left ventricular segments involved).
The Revised Cardiac Risk Index. Lee et al18 devised an index to assist in preoperative cardiac risk stratification that was subsequently incorporated into the 2007 American College of Cardiology/American Heart Association preoperative risk guidelines. (It does not, however, address the beta-blocker issue.) It consists of six independent risk-predictors of major cardiac complications derived from 4,315 patients over age 50 undergoing non-cardiac surgery. The risk factors, each of which is given 1 point, are:
- Congestive heart failure based on history or examination
- Renal insufficiency (serum creatinine level > 2 mg/dL)
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- History of transient ischemic attack or stroke
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with 3 or more points are considered to be at high risk, and those with 1 or 2 points are considered to be at intermediate risk. The American College of Cardiology/American Heart Association preoperative cardiac risk algorithm subsequently included five of these six risk factors (the type of surgery was considered separately) and made recommendations concerning noninvasive stress testing and heart rate control.
On the basis of these studies, specialty societies, guideline committees, and hospitals enthusiastically recommended the prophylactic use of beta-blockers to decrease postoperative cardiac complications.
THREE NEGATIVE TRIALS OF METOPROLOL
In 2005 and 2006, two studies in vascular surgery patients and another in patients with diabetes cast doubt on the role of beta-blockers when the results failed to show a benefit. The trials used metoprolol, started shortly before surgery, and with no titration to control the heart rate.
The MaVS study19 (Metoprolol After Vascular Surgery) randomized 496 patients to receive metoprolol or placebo 2 hours before surgery and until hospital discharge or a maximum of 5 days after surgery. The metoprolol dose varied by weight: patients weighing 75 kg or more got 100 mg, those weighing between 40 and 75 kg got 50 mg, and those weighing less than 40 kg got 25 mg. Overall effects at 6 months were not significantly different, but intraoperative bradycardia and hypotension requiring intervention were more frequent in the metoprolol group.
The POBBLE study20 (Perioperative Beta Blockade) randomized 103 patients who had no history of myocardial infarction to receive either metoprolol 50 mg twice daily or placebo from admission to 7 days after surgery. Myocardial ischemia was present in one-third of the patients after surgery. Metoprolol did not reduce the 30-day cardiac mortality rate, but it was associated with a shorter length of stay.
The DIPOM trial21 (Diabetic Postoperative Mortality and Morbidity) randomized 921 diabetic patients to receive long-acting metoprolol succinate controlled-release/extended release (CR/XL) or placebo. Patients in the metoprolol group received a test dose of 50 mg the evening before surgery, another dose 2 hours before surgery (100 mg if the heart rate was more than 65 bpm, or 50 mg if between 55 and 65 bpm), and daily thereafter until discharge or a maximum of 8 days. The dose was not titrated to heart-rate control.
Metoprolol had no statistically significant effect on the composite primary outcome measures of time to death from any cause, acute myocardial infarction, unstable angina, or congestive heart failure or on the secondary outcome measures of time to death from any cause, death from a cardiac cause, and nonfatal cardiac morbidity.
ADDITIONAL POSITIVE STUDIES
Lindenauer et al22 retrospectively evaluated the use of beta-blockers in the first 2 days after surgery in 782,969 patients undergoing non-cardiac surgery. Using propensity score matching and Revised Cardiac Risk Index scores, they found a lower rate of postoperative mortality in patients with three or more risk factors who received a beta-blocker. There was no significant difference in the group with two risk factors, but in the lowest-risk group (with a score of 0 to 1), beta-blockers were not beneficial and may have been associated with harm as evidenced by a higher odds ratio for death, although this was probably artifactual and reflecting database limitations.
Feringa et al,23 in an observational cohort study of 272 patients undergoing vascular surgery, reported that higher doses of beta-blockers and tight heart-rate control were associated with less perioperative myocardial ischemia, lower troponin T levels, and better long-term outcome.
THE POISE TRIAL: MIXED RESULTS
The randomized POISE trial,1 published in 2008, compared the effects of extended-release metoprolol succinate vs placebo on the 30-day risk of major cardiovascular events in 8,351 patients with or at risk of atherosclerotic disease who were undergoing noncardiac surgery. The metoprolol regimen was 100 mg 2 to 4 hours before surgery, another 100 mg by 6 hours after surgery, and then 200 mg 12 hours later and once daily for 30 days.
The incidence of the composite primary end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest at 30 days was lower in the metoprolol group than in the placebo group (5.8% vs 6.9%; P = .04), primarily because of fewer nonfatal myocardial infarctions. However, more patients in the metoprolol group died of any cause (3.1% vs 2.3% P = .03) or had a stroke (1.0% vs 0.5% P = .005) than in the placebo group.
The metoprolol group had a higher incidence of clinically significant hypotension, bradycardia, and stroke, which could account for much of the increase in the mortality rate. Sepsis was the major cause of death in this group; hypotension may have increased the risk of infection, and beta-blockers may have potentiated hypotension in patients who were already septic. Also, the bradycardic and negative inotropic effects of the beta-blocker could have masked the physiologic response to systemic infection, thereby delaying recognition and treatment or impeding the normal immune response.
One of the major criticisms of the POISE trial was its aggressive dosing regimen (200 to 400 mg within a 36-hour period) in patients who had not been on beta-blockers before then. Also, the drug was started only a few hours before surgery. In addition, these patients were at higher risk of death and stroke than those in other trials based on a high baseline rate of cerebrovascular disease, and inclusion of urgent and emergency surgical procedures.
STUDIES SINCE POISE
The POISE trial results1 prompted further questioning of the prophylactic perioperative use of beta-blockers. However, proponents of beta-blockers voiced serious criticisms of the trial, particularly the dosing regimen, and continued to believe that these drugs were beneficial if used appropriately.
The DECREASE IV trial. Dunkelgrun et al,24 in a study using bisoprolol started approximately 1 month before surgery and titrated to control the heart rate, reported beneficial results in intermediate-risk patients. In their randomized open-label study with a 2 × 2 factorial design, 1,066 patients at intermediate cardiac risk were assigned to receive bisoprolol, fluvastatin, combination treatment, or control therapy at least 34 days before surgery. Bisoprolol was started at 2.5 mg orally daily and slowly titrated up to a maximum dose of 10 mg to keep the heart rate between 50 and 70 beats per minute. The group of 533 patients randomized to receive bisoprolol had a lower incidence rate of cardiac death and nonfatal myocardial infarction than the control group (2.1% vs 6.0%, HR 0.34, P = .002). A potential limitation of this study was its open-label design, which might have led to treatment bias.
Updated guidelines. Based on the results from POISE and DECREASE IV, the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines25 published a focused update on beta-blockers in 2009 as an amendment to their 2007 guidelines on perioperative evaluation and care for noncardiac surgery. The European Society of Cardiology26 released similar but somewhat more liberal guidelines (Table 1).
London et al,27 in an observational study published in 2013, found a lower 30-day overall mortality rate with beta-blockers (relative risk [RR] 0.73, 95% confidence interval [CI] 0.65–0.83, P < .001, number needed to treat [NNT] 241), as well as a lower rate of cardiac morbidity (nonfatal myocardial infarction and cardiac death), but only in nonvascular surgery patients who were on beta-blockers within 7 days of scheduled surgery. Moreover, similar to the findings of Lindenauer et al,22 only patients with a Revised Cardiac Risk Index score of 2 or more benefited from beta-blocker use in terms of a lower risk of death, whereas the lower-risk patients did not:
- Risk score of 0 or 1—no association
- Score of 2—RR 0.63, 95% CI 0.50–0.80, P < .001, NNT 105
- Score of 3—RR 0.54, 95% CI 0.39–0.73, P < .001, NNT 41
- Score of 4 or more—RR 0.40, 95% CI 0.24–0.73, P < .001, NNT 18).
Beta-blocker exposure was associated with a significantly lower rate of cardiac complications (RR 0.67, 95% CI 0.57–0.79, P < .001, NNT 339), also limited to nonvascular surgery patients with a risk score of 2 or 3.
The Danish Nationwide Cohort Study28 examined the effect of beta-blockers on major adverse cardiac events (MACE, ie, myocardial infarction, cerebrovascular accident, and death) in 28,263 patients with ischemic heart disease undergoing noncardiac surgery; 7,990 with heart failure and 20,273 without. Beta-blockers were used in 53% of patients with heart failure and 36% of those without heart failure. Outcomes for all of the beta-blocker recipients:
- MACE—HR 0.90, 95% CI 0.79–1.02
- All-cause mortality—HR 0.95, 95% CI 0.85–1.06.
Outcomes for patients with heart failure if they received beta-blockers:
- MACE—HR 0.75, 95% CI 0.70–0.87
- All-cause mortality—HR 0.80, 95% CI 0.70–0.92.
There was no significant benefit from beta-blockers in patients without heart failure. Outcomes for those patients if they received beta-blockers:
- MACE—HR 1.11, 95% CI 0.92–1.33
- All-cause mortality—HR 1.15, 95% CI 0.98–1.35.
However, in patients without heart failure but with a history of myocardial infarction within the past 2 years, beta-blockers were associated with a lower risk of MACE and all-cause mortality. In patients with neither heart failure nor a recent myocardial infarction, beta-blockers were associated with an increased risk of MACE and all-cause mortality.
This difference in efficacy depending on the presence and timing of a prior myocardial infarction is consistent with the 2012 American College of Cardiology/American Heart Association guidelines for secondary prevention, in which beta-blockers are given a class I recommendation only for patients with a myocardial infarction within the past 3 years.
Meta-analyses and outcomes
A number of meta-analyses have been published over the past 10 years, with conflicting results (Table 2). The divergent findings are primarily due to the different studies included in the analyses as well as the strong influence of the POISE trial.1 The studies varied in terms of the specific beta-blocker used, dose titration and heart rate control, time of initiation of beta-blocker use before surgery, type of surgery, patient characteristics, comorbidities, biomarkers and diagnosis of myocardial infarction, and clinical end points.
In general, these meta-analyses have found that prophylactic perioperative use of beta-blockers decreases ischemia and tends to reduce the risk of nonfatal myocardial infarction. They vary on whether the overall mortality risk is decreased. The meta-analyses that included POISE1 found an increased incidence of stroke, whereas those that excluded POISE found no significant difference, although there appeared to be slightly more strokes in the beta-blocker groups.
The beta-blocker controversy increased even further when Dr. Don Poldermans was fired by Erasmus Medical Center in November 2011 for violations of academic integrity involving his research, including the DECREASE trials. The most recent meta-analysis, by Bouri et al,29 included nine “secure trials” and excluded the DECREASE trials in view of the controversy about their authenticity. The analysis showed an increase in overall mortality as well as stroke, primarily because it was heavily influenced by POISE.1 In contrast, the DECREASE trials had reported a decreased risk of myocardial infarction and death, with no significant increase in stroke. The authors concluded that guideline bodies should “retract their recommendations based on the fictitious data without further delay.”29
Although the design of the DECREASE trials (in which beta-blockers were started well in advance of surgery and doses were titrated to achieve heart rate control) is physiologically more compelling than those of the negative trials, the results have been questioned in light of the integrity issue. However, to date, none of the published DECREASE trials have been retracted.
Two other meta-analyses,30,31 published in 2013, also found a decreased risk of myocardial infarction and increased risk of stroke but no significant difference in short-term all-cause mortality.
ARE ALL BETA-BLOCKERS EQUIVALENT?
In various studies evaluating specific beta-blockers, the more cardioselective agents bisoprolol and atenolol were associated with better outcomes than metoprolol. The affinity ratios for beta-1/beta-2 receptors range from 13.5 for bisoprolol to 4.7 for atenolol and 2.3 for metoprolol.32 Blocking beta-1 receptors blunts tachycardia, whereas blocking beta-2 receptors may block systemic or cerebral vasodilation.
In patients with anemia, beta-blockade in general may be harmful, but beta-2 blockade may be even worse. Beta-blockers were associated with an increased risk of MACE (6.5% vs 3.0%)33 in patients with acute surgical anemia if the hemoglobin concentration decreased to less than 35% of baseline, and increased risks of hospital death (OR 6.65) and multiorgan dysfunction syndrome (OR 4.18) with severe bleeding during aortic surgery.34
In addition, the pathway by which the beta-blocker is metabolized may also affect outcome, with less benefit from beta-blockers metabolized by the CYP2D6 isoenzyme of the cytochrome P450 system. Individual variations in CYP2D6 activity related to genetics or drug interactions may result in insufficient or excessive beta-blockade. Because metoprolol is the most dependent on this system, patients using it may be more susceptible to bradycardia.35
Studies comparing atenolol and metoprolol found that the atenolol groups had fewer myocardial infarctions and deaths36 and lower 30-day and 1-year mortality rates37 than the groups on metoprolol. Studies comparing the three beta-blockers found better outcomes with atenolol and bisoprolol than with metoprolol—fewer strokes,38,39 a lower mortality rate,31 and a better composite outcome39 (Table 3 and Table 4).
START THE BETA-BLOCKER EARLY, TITRATE TO CONTROL THE HEART RATE
A number of studies suggest that how long the beta-blocker is given before surgery may influence the outcome (Table 5). The best results were achieved when beta-blockers were started approximately 1 month before surgery and titrated to control the heart rate.
Because this long lead-in time is not always practical, it is important to determine the shortest time before surgery in which starting beta-blockers may be beneficial and yet safe. Some evidence suggests that results are better when the beta-blocker is started more than 1 week preoperatively compared with less than 1 week, but it is unknown what the minimum or optimal time period should be.
If a beta-blocker is started well in advance of the scheduled surgery, there is adequate time for dose titration and tighter heart rate control. Most of the studies demonstrating beneficial effects of perioperative beta-blockers used dose titration and achieved lower heart rates in the treatment group than in the control group. A criticism of the MaVs,19 POBBLE,20 and DIPOM21 trials was that the patients did not receive adequate beta-blockade. The POISE trial1 used a much higher dose of metoprolol in an attempt to assure beta-blockade without dose titration, and although the regimen decreased nonfatal myocardial infarctions, it increased strokes and the overall mortality rate, probably related to excess bradycardia and hypotension. The target heart rate should probably be between 55 and 70 beats per minute.
RISK OF STROKE
POISE1 was the first trial to note a clinically and statistically significant increase in strokes with perioperative beta-blocker use. Although no other study has shown a similar increased risk, almost all reported a higher number of strokes in the beta-blocker groups, although the absolute numbers and differences were small and not statistically significant. This risk may also vary from one beta-blocker to another (Table 4).
The usual incidence rate of postoperative stroke after noncardiac, noncarotid surgery is well under 1% in patients with no prior history of stroke but increases to approximately 3% in patients with a previous stroke.40 An observational study from the Dutch group reported a very low incidence of stroke overall (0.02%) in 186,779 patients undergoing noncardiac surgery with no significant difference in those on chronic beta-blocker therapy.41 The DECREASE trials, with a total of 3,884 patients, also found no statistically significant increase in stroke with beta-blocker use (0.46% overall vs 0.5% with a beta-blocker),42 which in this case was bisoprolol started well in advance of surgery and titrated to control the heart rate. Although the DECREASE data are under suspicion, they seem reasonable and consistent with those of observational studies.
Proposed mechanisms by which beta-blockers may increase stroke risk include the side effects of hypotension and bradycardia, particularly in the setting of anemia. They may also cause cerebral ischemia by blocking cerebral vasodilation. This effect on cerebral blood flow may be more pronounced with the less cardioselective beta-blockers, which may explain the apparent increased stroke risk associated with metoprolol.
WHAT SHOULD WE DO NOW?
The evidence for the safety and efficacy of beta-blockers in the perioperative setting continues to evolve, and new clinical trials are needed to clarify the ongoing controversy, particularly regarding the risk of stroke.
If patients have other indications for beta-blocker therapy, such as history of heart failure, myocardial infarction in the past 3 years, or atrial fibrillation for rate control, they should be receiving them if time permits.
If prophylactic beta-blockers are to be effective in minimizing perioperative complications, it appears that they may need to be more cardioselective, started at least 1 week before surgery, titrated to control heart rate, and used in high-risk patients (Revised Cardiac Risk Index score > 2 or 3) undergoing high-risk surgery.
Ideally, a large randomized controlled trial using a cardioselective beta-blocker started in advance of surgery in patients with a Revised Cardiac Risk Index score greater than 2, undergoing intermediate or high-risk procedures, is needed to fully answer the questions raised by the current data.
Prophylactic use of beta-blockers in the perioperative period is highly controversial. Initial studies in the 1990s were favorable, but evidence has been conflicting since then.
The pendulum swung away from routinely recommending beta-blockers after the publication of negative results from several studies, including the Perioperative Ischemic Evaluation (POISE) trial in 2008.1 Highlighting this change in practice, a Canadian study2 found that the use of perioperative beta-blockade increased between 1999 and 2005 but subsequently declined from 2005 to 2010. However, there was no appreciable change in this pattern after the POISE trial or after changes in the American College of Cardiology guidelines in 2002 and 2006.3
In 2008, Harte and Jaffer reviewed the perioperative use of beta-blockers in noncardiac surgery in this journal.4 Since then, a number of meta-analyses and retrospective observational studies have reported variable findings related to specific beta-blockers and specific complications.
In this paper, we review the rationale and recent evidence for and against the perioperative use of beta-blockers as guidance for internists and hospitalists.
POTENTIAL CARDIOPROTECTIVE EFFECTS OF BETA-BLOCKERS
Myocardial infarction and unstable angina are the leading cardiovascular causes of death after surgery.5 These events are multifactorial. Some are caused by the stress of surgery, which precipitates physiologic changes related to inflammatory mediators, sympathetic tone, and oxygen supply and demand; others are caused by acute plaque rupture, thrombosis, and occlusion.6 Most perioperative infarcts are non-Q-wave events7 and occur within the first 2 days after the procedure, when the effects of anesthetics, pain, fluid shifts, and physiologic changes are greatest. Because multiple causes may contribute to perioperative myocardial infarction, a single preventive strategy may not be sufficient.8,9
Beta-blockers do several things that may be beneficial in the perioperative setting. They reduce myocardial oxygen demand by decreasing the force of contraction and by slowing the heart rate, and slowing the heart rate increases diastolic perfusion time.10 They suppress arrhythmias; they limit leukocyte recruitment, the production of free radicals, metalloproteinase activity, monocyte activation, release of growth factors, and inflammatory cytokine response; and they stabilize plaque.11 Their long-term use may also alter intracellular signaling processes, thus improving cell survival by decreasing the expression of receptors for substances that induce apoptosis.12
INITIAL POSITIVE TRIALS
Mangano et al13 began the beta-blocker trend in 1996 with a study in 200 patients known to have coronary artery disease or risk factors for it who were undergoing noncardiac surgery. Patients were randomized to receive either atenolol orally and intravenously, titrated to control the heart rate, or placebo in the immediate perioperative period.
The atenolol group had less perioperative ischemia but no difference in short-term rates of myocardial infarction and death. However, the death rate was lower in the atenolol group at 6 months after discharge and at 2 years, although patients who died in the immediate postoperative period were excluded from the analysis.
Although this finding did not appear to make sense physiologically, we now know that patients may experience myocardial injury without infarction after noncardiac surgery, a phenomenon associated with an increased risk of death in the short term and the long term.14 Preventing these episodes may be the explanation for the improved outcome.
The DECREASE trial15 (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography) provided additional support for beta-blocker use. The patients were at high risk, had abnormal dobutamine stress echocardiograms, and were undergoing vascular surgery; 112 patients were randomized to receive either oral bisoprolol (started 1 month before surgery, titrated to control the heart rate, and continued for 1 month after surgery) or placebo.
The study was stopped early because the bisoprolol group reportedly had a 90% lower rate of myocardial infarction and cardiac death 1 month after surgery. However, the study was criticized because the total number of patients enrolled was small and the benefit was much greater than usual for any pharmacologic intervention, thus calling the results into question.
In a follow-up study,16 survivors continued to be followed while receiving bisoprolol or usual care. The incidence of myocardial infarction or cardiac death at 2 years was significantly lower in the group receiving bisoprolol (12% vs 32%, odds ratio [OR] 0.30, P = .025).
Boersma et al,17 in an observational study, analyzed data from all 1,351 patients scheduled for major vascular surgery being considered for enrollment in the DECREASE trial. The DECREASE protocol required patients to undergo dobutamine stress echocardiography if they had one or more risk factors (age 70 or older, angina, prior myocardial infarction, congestive heart failure, treatment for ventricular arrhythmia, treatment for diabetes mellitus, or limited exercise capacity) or if their physician requested it. Twenty-seven percent received beta-blockers.
In multivariate analysis, clinical predictors of adverse outcome were age 70 or older; current or prior history of angina; and prior myocardial infarction, heart failure, or cerebrovascular accident.
In patients who had fewer than three clinical risk factors, beta-blocker use was associated with a lower rate of complications (0.8% vs 2.3%). Dobutamine stress echocardiography had minimal predictive value in this lower-risk group, suggesting that stress testing may not be necessary in this group if beta-blockers are used appropriately. However, in patients who had three or more risk factors, this test did provide additional prognostic information; those without stress-induced ischemia had lower event rates than those with ischemia, and beta-blocker use further reduced those rates, except in patients with extensive ischemia (more than five left ventricular segments involved).
The Revised Cardiac Risk Index. Lee et al18 devised an index to assist in preoperative cardiac risk stratification that was subsequently incorporated into the 2007 American College of Cardiology/American Heart Association preoperative risk guidelines. (It does not, however, address the beta-blocker issue.) It consists of six independent risk-predictors of major cardiac complications derived from 4,315 patients over age 50 undergoing non-cardiac surgery. The risk factors, each of which is given 1 point, are:
- Congestive heart failure based on history or examination
- Renal insufficiency (serum creatinine level > 2 mg/dL)
- Myocardial infarction, symptomatic ischemic heart disease, or a positive stress test
- History of transient ischemic attack or stroke
- Diabetes requiring insulin
- High-risk surgery (defined as intrathoracic, intra-abdominal, or suprainguinal vascular surgery).
Patients with 3 or more points are considered to be at high risk, and those with 1 or 2 points are considered to be at intermediate risk. The American College of Cardiology/American Heart Association preoperative cardiac risk algorithm subsequently included five of these six risk factors (the type of surgery was considered separately) and made recommendations concerning noninvasive stress testing and heart rate control.
On the basis of these studies, specialty societies, guideline committees, and hospitals enthusiastically recommended the prophylactic use of beta-blockers to decrease postoperative cardiac complications.
THREE NEGATIVE TRIALS OF METOPROLOL
In 2005 and 2006, two studies in vascular surgery patients and another in patients with diabetes cast doubt on the role of beta-blockers when the results failed to show a benefit. The trials used metoprolol, started shortly before surgery, and with no titration to control the heart rate.
The MaVS study19 (Metoprolol After Vascular Surgery) randomized 496 patients to receive metoprolol or placebo 2 hours before surgery and until hospital discharge or a maximum of 5 days after surgery. The metoprolol dose varied by weight: patients weighing 75 kg or more got 100 mg, those weighing between 40 and 75 kg got 50 mg, and those weighing less than 40 kg got 25 mg. Overall effects at 6 months were not significantly different, but intraoperative bradycardia and hypotension requiring intervention were more frequent in the metoprolol group.
The POBBLE study20 (Perioperative Beta Blockade) randomized 103 patients who had no history of myocardial infarction to receive either metoprolol 50 mg twice daily or placebo from admission to 7 days after surgery. Myocardial ischemia was present in one-third of the patients after surgery. Metoprolol did not reduce the 30-day cardiac mortality rate, but it was associated with a shorter length of stay.
The DIPOM trial21 (Diabetic Postoperative Mortality and Morbidity) randomized 921 diabetic patients to receive long-acting metoprolol succinate controlled-release/extended release (CR/XL) or placebo. Patients in the metoprolol group received a test dose of 50 mg the evening before surgery, another dose 2 hours before surgery (100 mg if the heart rate was more than 65 bpm, or 50 mg if between 55 and 65 bpm), and daily thereafter until discharge or a maximum of 8 days. The dose was not titrated to heart-rate control.
Metoprolol had no statistically significant effect on the composite primary outcome measures of time to death from any cause, acute myocardial infarction, unstable angina, or congestive heart failure or on the secondary outcome measures of time to death from any cause, death from a cardiac cause, and nonfatal cardiac morbidity.
ADDITIONAL POSITIVE STUDIES
Lindenauer et al22 retrospectively evaluated the use of beta-blockers in the first 2 days after surgery in 782,969 patients undergoing non-cardiac surgery. Using propensity score matching and Revised Cardiac Risk Index scores, they found a lower rate of postoperative mortality in patients with three or more risk factors who received a beta-blocker. There was no significant difference in the group with two risk factors, but in the lowest-risk group (with a score of 0 to 1), beta-blockers were not beneficial and may have been associated with harm as evidenced by a higher odds ratio for death, although this was probably artifactual and reflecting database limitations.
Feringa et al,23 in an observational cohort study of 272 patients undergoing vascular surgery, reported that higher doses of beta-blockers and tight heart-rate control were associated with less perioperative myocardial ischemia, lower troponin T levels, and better long-term outcome.
THE POISE TRIAL: MIXED RESULTS
The randomized POISE trial,1 published in 2008, compared the effects of extended-release metoprolol succinate vs placebo on the 30-day risk of major cardiovascular events in 8,351 patients with or at risk of atherosclerotic disease who were undergoing noncardiac surgery. The metoprolol regimen was 100 mg 2 to 4 hours before surgery, another 100 mg by 6 hours after surgery, and then 200 mg 12 hours later and once daily for 30 days.
The incidence of the composite primary end point of cardiovascular death, nonfatal myocardial infarction, and nonfatal cardiac arrest at 30 days was lower in the metoprolol group than in the placebo group (5.8% vs 6.9%; P = .04), primarily because of fewer nonfatal myocardial infarctions. However, more patients in the metoprolol group died of any cause (3.1% vs 2.3% P = .03) or had a stroke (1.0% vs 0.5% P = .005) than in the placebo group.
The metoprolol group had a higher incidence of clinically significant hypotension, bradycardia, and stroke, which could account for much of the increase in the mortality rate. Sepsis was the major cause of death in this group; hypotension may have increased the risk of infection, and beta-blockers may have potentiated hypotension in patients who were already septic. Also, the bradycardic and negative inotropic effects of the beta-blocker could have masked the physiologic response to systemic infection, thereby delaying recognition and treatment or impeding the normal immune response.
One of the major criticisms of the POISE trial was its aggressive dosing regimen (200 to 400 mg within a 36-hour period) in patients who had not been on beta-blockers before then. Also, the drug was started only a few hours before surgery. In addition, these patients were at higher risk of death and stroke than those in other trials based on a high baseline rate of cerebrovascular disease, and inclusion of urgent and emergency surgical procedures.
STUDIES SINCE POISE
The POISE trial results1 prompted further questioning of the prophylactic perioperative use of beta-blockers. However, proponents of beta-blockers voiced serious criticisms of the trial, particularly the dosing regimen, and continued to believe that these drugs were beneficial if used appropriately.
The DECREASE IV trial. Dunkelgrun et al,24 in a study using bisoprolol started approximately 1 month before surgery and titrated to control the heart rate, reported beneficial results in intermediate-risk patients. In their randomized open-label study with a 2 × 2 factorial design, 1,066 patients at intermediate cardiac risk were assigned to receive bisoprolol, fluvastatin, combination treatment, or control therapy at least 34 days before surgery. Bisoprolol was started at 2.5 mg orally daily and slowly titrated up to a maximum dose of 10 mg to keep the heart rate between 50 and 70 beats per minute. The group of 533 patients randomized to receive bisoprolol had a lower incidence rate of cardiac death and nonfatal myocardial infarction than the control group (2.1% vs 6.0%, HR 0.34, P = .002). A potential limitation of this study was its open-label design, which might have led to treatment bias.
Updated guidelines. Based on the results from POISE and DECREASE IV, the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines25 published a focused update on beta-blockers in 2009 as an amendment to their 2007 guidelines on perioperative evaluation and care for noncardiac surgery. The European Society of Cardiology26 released similar but somewhat more liberal guidelines (Table 1).
London et al,27 in an observational study published in 2013, found a lower 30-day overall mortality rate with beta-blockers (relative risk [RR] 0.73, 95% confidence interval [CI] 0.65–0.83, P < .001, number needed to treat [NNT] 241), as well as a lower rate of cardiac morbidity (nonfatal myocardial infarction and cardiac death), but only in nonvascular surgery patients who were on beta-blockers within 7 days of scheduled surgery. Moreover, similar to the findings of Lindenauer et al,22 only patients with a Revised Cardiac Risk Index score of 2 or more benefited from beta-blocker use in terms of a lower risk of death, whereas the lower-risk patients did not:
- Risk score of 0 or 1—no association
- Score of 2—RR 0.63, 95% CI 0.50–0.80, P < .001, NNT 105
- Score of 3—RR 0.54, 95% CI 0.39–0.73, P < .001, NNT 41
- Score of 4 or more—RR 0.40, 95% CI 0.24–0.73, P < .001, NNT 18).
Beta-blocker exposure was associated with a significantly lower rate of cardiac complications (RR 0.67, 95% CI 0.57–0.79, P < .001, NNT 339), also limited to nonvascular surgery patients with a risk score of 2 or 3.
The Danish Nationwide Cohort Study28 examined the effect of beta-blockers on major adverse cardiac events (MACE, ie, myocardial infarction, cerebrovascular accident, and death) in 28,263 patients with ischemic heart disease undergoing noncardiac surgery; 7,990 with heart failure and 20,273 without. Beta-blockers were used in 53% of patients with heart failure and 36% of those without heart failure. Outcomes for all of the beta-blocker recipients:
- MACE—HR 0.90, 95% CI 0.79–1.02
- All-cause mortality—HR 0.95, 95% CI 0.85–1.06.
Outcomes for patients with heart failure if they received beta-blockers:
- MACE—HR 0.75, 95% CI 0.70–0.87
- All-cause mortality—HR 0.80, 95% CI 0.70–0.92.
There was no significant benefit from beta-blockers in patients without heart failure. Outcomes for those patients if they received beta-blockers:
- MACE—HR 1.11, 95% CI 0.92–1.33
- All-cause mortality—HR 1.15, 95% CI 0.98–1.35.
However, in patients without heart failure but with a history of myocardial infarction within the past 2 years, beta-blockers were associated with a lower risk of MACE and all-cause mortality. In patients with neither heart failure nor a recent myocardial infarction, beta-blockers were associated with an increased risk of MACE and all-cause mortality.
This difference in efficacy depending on the presence and timing of a prior myocardial infarction is consistent with the 2012 American College of Cardiology/American Heart Association guidelines for secondary prevention, in which beta-blockers are given a class I recommendation only for patients with a myocardial infarction within the past 3 years.
Meta-analyses and outcomes
A number of meta-analyses have been published over the past 10 years, with conflicting results (Table 2). The divergent findings are primarily due to the different studies included in the analyses as well as the strong influence of the POISE trial.1 The studies varied in terms of the specific beta-blocker used, dose titration and heart rate control, time of initiation of beta-blocker use before surgery, type of surgery, patient characteristics, comorbidities, biomarkers and diagnosis of myocardial infarction, and clinical end points.
In general, these meta-analyses have found that prophylactic perioperative use of beta-blockers decreases ischemia and tends to reduce the risk of nonfatal myocardial infarction. They vary on whether the overall mortality risk is decreased. The meta-analyses that included POISE1 found an increased incidence of stroke, whereas those that excluded POISE found no significant difference, although there appeared to be slightly more strokes in the beta-blocker groups.
The beta-blocker controversy increased even further when Dr. Don Poldermans was fired by Erasmus Medical Center in November 2011 for violations of academic integrity involving his research, including the DECREASE trials. The most recent meta-analysis, by Bouri et al,29 included nine “secure trials” and excluded the DECREASE trials in view of the controversy about their authenticity. The analysis showed an increase in overall mortality as well as stroke, primarily because it was heavily influenced by POISE.1 In contrast, the DECREASE trials had reported a decreased risk of myocardial infarction and death, with no significant increase in stroke. The authors concluded that guideline bodies should “retract their recommendations based on the fictitious data without further delay.”29
Although the design of the DECREASE trials (in which beta-blockers were started well in advance of surgery and doses were titrated to achieve heart rate control) is physiologically more compelling than those of the negative trials, the results have been questioned in light of the integrity issue. However, to date, none of the published DECREASE trials have been retracted.
Two other meta-analyses,30,31 published in 2013, also found a decreased risk of myocardial infarction and increased risk of stroke but no significant difference in short-term all-cause mortality.
ARE ALL BETA-BLOCKERS EQUIVALENT?
In various studies evaluating specific beta-blockers, the more cardioselective agents bisoprolol and atenolol were associated with better outcomes than metoprolol. The affinity ratios for beta-1/beta-2 receptors range from 13.5 for bisoprolol to 4.7 for atenolol and 2.3 for metoprolol.32 Blocking beta-1 receptors blunts tachycardia, whereas blocking beta-2 receptors may block systemic or cerebral vasodilation.
In patients with anemia, beta-blockade in general may be harmful, but beta-2 blockade may be even worse. Beta-blockers were associated with an increased risk of MACE (6.5% vs 3.0%)33 in patients with acute surgical anemia if the hemoglobin concentration decreased to less than 35% of baseline, and increased risks of hospital death (OR 6.65) and multiorgan dysfunction syndrome (OR 4.18) with severe bleeding during aortic surgery.34
In addition, the pathway by which the beta-blocker is metabolized may also affect outcome, with less benefit from beta-blockers metabolized by the CYP2D6 isoenzyme of the cytochrome P450 system. Individual variations in CYP2D6 activity related to genetics or drug interactions may result in insufficient or excessive beta-blockade. Because metoprolol is the most dependent on this system, patients using it may be more susceptible to bradycardia.35
Studies comparing atenolol and metoprolol found that the atenolol groups had fewer myocardial infarctions and deaths36 and lower 30-day and 1-year mortality rates37 than the groups on metoprolol. Studies comparing the three beta-blockers found better outcomes with atenolol and bisoprolol than with metoprolol—fewer strokes,38,39 a lower mortality rate,31 and a better composite outcome39 (Table 3 and Table 4).
START THE BETA-BLOCKER EARLY, TITRATE TO CONTROL THE HEART RATE
A number of studies suggest that how long the beta-blocker is given before surgery may influence the outcome (Table 5). The best results were achieved when beta-blockers were started approximately 1 month before surgery and titrated to control the heart rate.
Because this long lead-in time is not always practical, it is important to determine the shortest time before surgery in which starting beta-blockers may be beneficial and yet safe. Some evidence suggests that results are better when the beta-blocker is started more than 1 week preoperatively compared with less than 1 week, but it is unknown what the minimum or optimal time period should be.
If a beta-blocker is started well in advance of the scheduled surgery, there is adequate time for dose titration and tighter heart rate control. Most of the studies demonstrating beneficial effects of perioperative beta-blockers used dose titration and achieved lower heart rates in the treatment group than in the control group. A criticism of the MaVs,19 POBBLE,20 and DIPOM21 trials was that the patients did not receive adequate beta-blockade. The POISE trial1 used a much higher dose of metoprolol in an attempt to assure beta-blockade without dose titration, and although the regimen decreased nonfatal myocardial infarctions, it increased strokes and the overall mortality rate, probably related to excess bradycardia and hypotension. The target heart rate should probably be between 55 and 70 beats per minute.
RISK OF STROKE
POISE1 was the first trial to note a clinically and statistically significant increase in strokes with perioperative beta-blocker use. Although no other study has shown a similar increased risk, almost all reported a higher number of strokes in the beta-blocker groups, although the absolute numbers and differences were small and not statistically significant. This risk may also vary from one beta-blocker to another (Table 4).
The usual incidence rate of postoperative stroke after noncardiac, noncarotid surgery is well under 1% in patients with no prior history of stroke but increases to approximately 3% in patients with a previous stroke.40 An observational study from the Dutch group reported a very low incidence of stroke overall (0.02%) in 186,779 patients undergoing noncardiac surgery with no significant difference in those on chronic beta-blocker therapy.41 The DECREASE trials, with a total of 3,884 patients, also found no statistically significant increase in stroke with beta-blocker use (0.46% overall vs 0.5% with a beta-blocker),42 which in this case was bisoprolol started well in advance of surgery and titrated to control the heart rate. Although the DECREASE data are under suspicion, they seem reasonable and consistent with those of observational studies.
Proposed mechanisms by which beta-blockers may increase stroke risk include the side effects of hypotension and bradycardia, particularly in the setting of anemia. They may also cause cerebral ischemia by blocking cerebral vasodilation. This effect on cerebral blood flow may be more pronounced with the less cardioselective beta-blockers, which may explain the apparent increased stroke risk associated with metoprolol.
WHAT SHOULD WE DO NOW?
The evidence for the safety and efficacy of beta-blockers in the perioperative setting continues to evolve, and new clinical trials are needed to clarify the ongoing controversy, particularly regarding the risk of stroke.
If patients have other indications for beta-blocker therapy, such as history of heart failure, myocardial infarction in the past 3 years, or atrial fibrillation for rate control, they should be receiving them if time permits.
If prophylactic beta-blockers are to be effective in minimizing perioperative complications, it appears that they may need to be more cardioselective, started at least 1 week before surgery, titrated to control heart rate, and used in high-risk patients (Revised Cardiac Risk Index score > 2 or 3) undergoing high-risk surgery.
Ideally, a large randomized controlled trial using a cardioselective beta-blocker started in advance of surgery in patients with a Revised Cardiac Risk Index score greater than 2, undergoing intermediate or high-risk procedures, is needed to fully answer the questions raised by the current data.
- POISE Study Group; Devereaux PJ, Yang H, Yusuf S, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Wijeysundera DN, Mamdani M, Laupacis A, et al. Clinical evidence, practice guidelines, and ß-blocker utilization before major noncardiac surgery. Circ Cardiovasc Qual Outcomes 2012; 5:558–565.
- American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery); American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2006 guideline update on perioperative cardiovascular evaluation for noncardiac surgery: focused update on perioperative beta-blocker therapy: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society for Vascular Medicine and Biology. J Am Coll Cardiol 2006; 47:2343–2355.
- Harte B, Jaffer AK. Perioperative beta-blockers in noncardiac surgery: evolution of the evidence. Cleve Clin J Med 2008; 75:513–519.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foëx P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Poldermans D, Boersma E, Bax JJ, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol reduces cardiac death and myocardial infarction in high-risk patients as long as 2 years after successful major vascular surgery. Eur Heart J 2001; 22:1353–1358.
- Boersma E, Poldermans D, Bax JJ, et al; DECREASE Study Group (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiogrpahy). Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR; POBBLE trial investigators. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al; DIPOM Trial Group. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major non-cardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114(suppl 1):1344–1349.
- Dunkelgrun M, Boersma E, Schouten O, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol and fluvastatin for the reduction of perioperative cardiac mortality and myocardial infarction in intermediate-risk patients undergoing noncardiovascular surgery: a randomized controlled trial (DECREASE-IV). Ann Surg 2009; 249:921–926.
- American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2009; 54:e13–e118.
- Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non-cardiac Surgery; European Society of Cardiology (ESC); Poldermans D, Bax JJ, Boersma E, et al. Guidelines for preoperative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery. Eur Heart J 2009; 30:2769–2812.
- London MJ, Hur K, Schwartz GG, Henderson WG. Association of perioperative beta-blockade with mortality and cardiovascular morbidity following major noncardiac surgery. JAMA 2013; 309:1704–1713.
- Andersson C, Mérie C, Jørgensen M, et al. Association of beta-blocker therapy with risks of adverse cardiovascular events and deaths in patients with ischemic heart disease undergoing noncardiac surgery: a Danish nationwide cohort study. JAMA Intern Med 2014; 174:336–344.
- Bouri S, Shun-Shin MJ, Cole GD, Mayet J, Francis DP. Meta-analysis of secure randomised controlled trials of beta-blockade to prevent perioperative death in non-cardiac surgery. Heart 2014; 100:456–464.
- Guay J, Ochroch EA. Beta-blocking agents for surgery: influence on mortality and major outcomes. A meta-analysis. J Cardiothorac Vasc Anesth 2013; 27:834–844.
- Dai N, Xu D, Zhang J, et al. Different beta-blockers and initiation time in patients undergoing noncardiac surgery: a meta-analysis. Am J Med Sci 2014; 347:235–244.
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol 2005; 144:317–322.
- Beattie WS, Wijeysundera DN, Karkouti K, et al. Acute surgical anemia influences the cardioprotective effects of beta-blockade: a single-center, propensity-matched cohort study. Anesthesiology 2010; 112:25–33.
- Le Manach Y, Collins GS, Ibanez C, et al. Impact of perioperative bleeding on the protective effect of beta-blockers during infrarenal aortic reconstruction. Anesthesiology 2012; 117:1203–1211.
- Badgett RG, Lawrence VA, Cohn SL. Variations in pharmacology of beta-blockers may contribute to heterogeneous results in trials of perioperative beta-blockade. Anesthesiology 2010; 113:585–592.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
- Wallace AW, Au S, Cason BA. Perioperative beta-blockade: atenolol is associated with reduced mortality when compared to metoprolol. Anesthesiology 2011; 114:824–836.
- Mashour GA, Sharifpour M, Freundlich RE, et al. Perioperative metoprolol and risk of stroke after noncardiac surgery. Anesthesiology 2013; 119:1340–1346.
- Ashes C, Judelman S, Wijeysundera DN, et al. Selective beta1-antagonism with bisoprolol is associated with fewer postoperative strokes than atenolol or metoprolol: a single-center cohort study of 44,092 consecutive patients. Anesthesiology 2013; 119:777–787.
- Selim M. Perioperative stroke. N Engl J Med 2007; 356:706–713.
- van Lier F, Schouten O, van Domburg RT, et al. Effect of chronic beta-blocker use on stroke after noncardiac surgery. Am J Cardiol 2009; 104:429–433.
- van Lier F, Schouten O, Hoeks SE, et al. Impact of prophylactic beta-blocker therapy to prevent stroke after noncardiac surgery. Am J Cardiol 2010; 105:43–47.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta-analysis of randomised controlled trials. BMJ 2005; 331:313–321.
- McGory ML, Maggard MA, Ko CY. A meta-analysis of perioperative beta blockade: what is the actual risk reduction? Surgery 2005; 138:171–179.
- Schouten O, Shaw LJ, Boersma E, et al. A meta-analysis of safety and effectiveness of perioperative beta-blocker use for the prevention of cardiac events in different types of noncardiac surgery. Coron Artery Dis 2006; 17:173–179.
- Wiesbauer F, Schlager O, Domanovits H, et al. Perioperative beta-blockers for preventing surgery-related mortality and morbidity: a systematic review and meta-analysis. Anesth Analg 2007; 104:27–41.
- Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet 2008; 372:1962–1976.
- Flu WJ, van Kuijk JP, Chonchol M, et al. Timing of preoperative beta-blocker treatment in vascular surgery patients: influence on postoperative outcome. J Am Coll Cardiol 2010; 56:1922–1929.
- Wijeysundera DN, Beattie WS, Wijeysundera HC, Yun L, Austin PC, Ko DT. Duration of preoperative beta-blockade and outcomes after major elective noncardiac surgery. Can J Cardiol 2014; 30:217–223.
- POISE Study Group; Devereaux PJ, Yang H, Yusuf S, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Wijeysundera DN, Mamdani M, Laupacis A, et al. Clinical evidence, practice guidelines, and ß-blocker utilization before major noncardiac surgery. Circ Cardiovasc Qual Outcomes 2012; 5:558–565.
- American College of Cardiology; American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery); American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2006 guideline update on perioperative cardiovascular evaluation for noncardiac surgery: focused update on perioperative beta-blocker therapy: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery) developed in collaboration with the American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Rhythm Society, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society for Vascular Medicine and Biology. J Am Coll Cardiol 2006; 47:2343–2355.
- Harte B, Jaffer AK. Perioperative beta-blockers in noncardiac surgery: evolution of the evidence. Cleve Clin J Med 2008; 75:513–519.
- Mangano DT. Perioperative cardiac morbidity. Anesthesiology 1990; 72:153–184.
- London MJ, Zaugg M, Schaub MC, Spahn DR. Perioperative beta-adrenergic receptor blockade: physiologic foundations and clinical controversies. Anesthesiology 2004; 100:170–175.
- Badner NH, Knill RL, Brown JE, Novick TV, Gelb AW. Myocardial infarction after noncardiac surgery. Anesthesiology 1998; 88:572–578.
- Priebe HJ. Triggers of perioperative myocardial ischaemia and infarction. Br J Anaesth 2004; 93:9–20.
- Zaugg M, Schaub MC, Foëx P. Myocardial injury and its prevention in the perioperative setting. Br J Anaesth 2004; 93:21–33.
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of beta-adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br J Anaesth 2002; 88:101–123.
- Landesberg G. The pathophysiology of perioperative myocardial infarction: facts and perspectives. J Cardiothorac Vasc Anesth 2003; 17:90–100.
- Yeager MP, Fillinger MP, Hettleman BD, Hartman GS. Perioperative beta-blockade and late cardiac outcomes: a complementary hypothesis. J Cardiothorac Vasc Anesth 2005; 19:237–241.
- Mangano DT, Layug EL, Wallace A, Tateo I. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. Multicenter Study of Perioperative Ischemia Research Group. N Engl J Med 1996; 335:1713–1720.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Poldermans D, Boersma E, Bax JJ, et al. The effect of bisoprolol on perioperative mortality and myocardial infarction in high-risk patients undergoing vascular surgery. Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. N Engl J Med 1999; 341:1789–1794.
- Poldermans D, Boersma E, Bax JJ, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol reduces cardiac death and myocardial infarction in high-risk patients as long as 2 years after successful major vascular surgery. Eur Heart J 2001; 22:1353–1358.
- Boersma E, Poldermans D, Bax JJ, et al; DECREASE Study Group (Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiogrpahy). Predictors of cardiac events after major vascular surgery: role of clinical characteristics, dobutamine echocardiography, and beta-blocker therapy. JAMA 2001; 285:1865–1873.
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Yang H, Raymer K, Butler R, Parlow J, Roberts R. The effects of perioperative beta-blockade: results of the Metoprolol after Vascular Surgery (MaVS) study, a randomized controlled trial. Am Heart J 2006; 152:983–990.
- Brady AR, Gibbs JS, Greenhalgh RM, Powell JT, Sydes MR; POBBLE trial investigators. Perioperative beta-blockade (POBBLE) for patients undergoing infrarenal vascular surgery: results of a randomized double-blind controlled trial. J Vasc Surg 2005; 41:602–609.
- Juul AB, Wetterslev J, Gluud C, et al; DIPOM Trial Group. Effect of perioperative beta blockade in patients with diabetes undergoing major non-cardiac surgery: randomised placebo controlled, blinded multicentre trial. BMJ 2006; 332:1482.
- Lindenauer PK, Pekow P, Wang K, Mamidi DK, Gutierrez B, Benjamin EM. Perioperative beta-blocker therapy and mortality after major non-cardiac surgery. N Engl J Med 2005; 353:349–361.
- Feringa HH, Bax JJ, Boersma E, et al. High-dose beta-blockers and tight heart rate control reduce myocardial ischemia and troponin T release in vascular surgery patients. Circulation 2006; 114(suppl 1):1344–1349.
- Dunkelgrun M, Boersma E, Schouten O, et al; Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography Study Group. Bisoprolol and fluvastatin for the reduction of perioperative cardiac mortality and myocardial infarction in intermediate-risk patients undergoing noncardiovascular surgery: a randomized controlled trial (DECREASE-IV). Ann Surg 2009; 249:921–926.
- American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Rhythm Society; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine; Society for Vascular Surgery; Fleisher LA, Beckman JA, Brown KA, et al. 2009 ACCF/AHA focused update on perioperative beta blockade incorporated into the ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2009; 54:e13–e118.
- Task Force for Preoperative Cardiac Risk Assessment and Perioperative Cardiac Management in Non-cardiac Surgery; European Society of Cardiology (ESC); Poldermans D, Bax JJ, Boersma E, et al. Guidelines for preoperative cardiac risk assessment and perioperative cardiac management in non-cardiac surgery. Eur Heart J 2009; 30:2769–2812.
- London MJ, Hur K, Schwartz GG, Henderson WG. Association of perioperative beta-blockade with mortality and cardiovascular morbidity following major noncardiac surgery. JAMA 2013; 309:1704–1713.
- Andersson C, Mérie C, Jørgensen M, et al. Association of beta-blocker therapy with risks of adverse cardiovascular events and deaths in patients with ischemic heart disease undergoing noncardiac surgery: a Danish nationwide cohort study. JAMA Intern Med 2014; 174:336–344.
- Bouri S, Shun-Shin MJ, Cole GD, Mayet J, Francis DP. Meta-analysis of secure randomised controlled trials of beta-blockade to prevent perioperative death in non-cardiac surgery. Heart 2014; 100:456–464.
- Guay J, Ochroch EA. Beta-blocking agents for surgery: influence on mortality and major outcomes. A meta-analysis. J Cardiothorac Vasc Anesth 2013; 27:834–844.
- Dai N, Xu D, Zhang J, et al. Different beta-blockers and initiation time in patients undergoing noncardiac surgery: a meta-analysis. Am J Med Sci 2014; 347:235–244.
- Baker JG. The selectivity of beta-adrenoceptor antagonists at the human beta1, beta2 and beta3 adrenoceptors. Br J Pharmacol 2005; 144:317–322.
- Beattie WS, Wijeysundera DN, Karkouti K, et al. Acute surgical anemia influences the cardioprotective effects of beta-blockade: a single-center, propensity-matched cohort study. Anesthesiology 2010; 112:25–33.
- Le Manach Y, Collins GS, Ibanez C, et al. Impact of perioperative bleeding on the protective effect of beta-blockers during infrarenal aortic reconstruction. Anesthesiology 2012; 117:1203–1211.
- Badgett RG, Lawrence VA, Cohn SL. Variations in pharmacology of beta-blockers may contribute to heterogeneous results in trials of perioperative beta-blockade. Anesthesiology 2010; 113:585–592.
- Redelmeier D, Scales D, Kopp A. Beta blockers for elective surgery in elderly patients: population based, retrospective cohort study. BMJ 2005; 331:932.
- Wallace AW, Au S, Cason BA. Perioperative beta-blockade: atenolol is associated with reduced mortality when compared to metoprolol. Anesthesiology 2011; 114:824–836.
- Mashour GA, Sharifpour M, Freundlich RE, et al. Perioperative metoprolol and risk of stroke after noncardiac surgery. Anesthesiology 2013; 119:1340–1346.
- Ashes C, Judelman S, Wijeysundera DN, et al. Selective beta1-antagonism with bisoprolol is associated with fewer postoperative strokes than atenolol or metoprolol: a single-center cohort study of 44,092 consecutive patients. Anesthesiology 2013; 119:777–787.
- Selim M. Perioperative stroke. N Engl J Med 2007; 356:706–713.
- van Lier F, Schouten O, van Domburg RT, et al. Effect of chronic beta-blocker use on stroke after noncardiac surgery. Am J Cardiol 2009; 104:429–433.
- van Lier F, Schouten O, Hoeks SE, et al. Impact of prophylactic beta-blocker therapy to prevent stroke after noncardiac surgery. Am J Cardiol 2010; 105:43–47.
- Devereaux PJ, Beattie WS, Choi PT, et al. How strong is the evidence for the use of perioperative beta blockers in non-cardiac surgery? Systematic review and meta-analysis of randomised controlled trials. BMJ 2005; 331:313–321.
- McGory ML, Maggard MA, Ko CY. A meta-analysis of perioperative beta blockade: what is the actual risk reduction? Surgery 2005; 138:171–179.
- Schouten O, Shaw LJ, Boersma E, et al. A meta-analysis of safety and effectiveness of perioperative beta-blocker use for the prevention of cardiac events in different types of noncardiac surgery. Coron Artery Dis 2006; 17:173–179.
- Wiesbauer F, Schlager O, Domanovits H, et al. Perioperative beta-blockers for preventing surgery-related mortality and morbidity: a systematic review and meta-analysis. Anesth Analg 2007; 104:27–41.
- Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet 2008; 372:1962–1976.
- Flu WJ, van Kuijk JP, Chonchol M, et al. Timing of preoperative beta-blocker treatment in vascular surgery patients: influence on postoperative outcome. J Am Coll Cardiol 2010; 56:1922–1929.
- Wijeysundera DN, Beattie WS, Wijeysundera HC, Yun L, Austin PC, Ko DT. Duration of preoperative beta-blockade and outcomes after major elective noncardiac surgery. Can J Cardiol 2014; 30:217–223.
KEY POINTS
- If patients have other indications for beta-blocker therapy, such as a history of heart failure, myocardial infarction in the past 3 years, or atrial fibrillation, they should be started on a beta-blocker before surgery if time permits.
- Of the various beta-blockers, the cardioselective ones appear to be preferable in the perioperative setting.
- Beta-blockers may need to be started at least 1 week before surgery, titrated to control the heart rate, and used only in patients at high risk (Revised Cardiac Risk Index score > 2 or 3) undergoing high-risk surgery.
- Further clinical trials are necessary to clarify the ongoing controversy, particularly regarding the risk of stroke, which was increased in the large Perioperative Ischemic Evaluation (POISE) trial.
Could thorough documentation have changed the outcome of this trial?
Case: Did the gynecologist have the right to not remove the ovaries?
A 36-year-old woman (G3 P3003) presented to her gynecologist with dysmenorrhea and abnormal uterine bleeding. She reported a family history of ovarian cancer for two generations. She was evaluated and underwent physical examination and preoperative ultrasound examination of pelvic organs. All findings were unremarkable. The gynecologist prescribed oral contraceptives (OCs). After an initial excellent response, the patient reported a reoccurrence of pelvic pain and abnormal bleeding 6 years later. The gynecologist suggested options including operative hysteroscopy, dilatation and curettage (D&C), endometrial ablation, off-label use of an intrauterine contraceptive system, or hysterectomy performed via a minimally invasive, vaginal, or abdominal approach. The patient opted for hysteroscopy, D&C, and endometrial ablation and operative laparoscopy. The patient received a diagnosis of stage I endometriosis, which was treated with fulguration.
Two years later, she reported menorrhagia and pelvic pain. The gynecologist suggested trying an OC again, and the patient was given a prescription for a low-dose estrogen/desogestrel combination pill. The patient then changed her mind within 72 hours, never took the OC, and contacted her gynecologist to schedule surgery with him. Upon a return visit to the office, the patient and gynecologist decided to proceed with laparoscopic-assisted vaginal hysterectomy (LAVH) with bilateral salpingo-oophorectomy (BSO). The written consent included laparoscopic hysterectomy with removal of ovary or ovaries and bilateral fallopian tubes, with a possibility of abdominal hysterectomy.
The gynecologist met with the patient preoperatively to update the history, which was unchanged from her prior office visit. In the operating room, “time out” occurred and was documented appropriately—concerns were to be provided to the gynecologist; none were noted.
Intraoperatively, the ovaries were normal in appearance and no endometriosis was noted. The gynecologist proceeded with LAVH and, because the ovaries were normal, did not remove them or the fallopian tubes.
The patient sued the gynecologist on the grounds that, because the originally planned BSO was not performed, she was fearful of developing ovarian cancer in the future.
Preoperative documentation was “sketchy”at best and did not reflect the preoperative discussion and options presented to the patient. There was no documentation of anyone accompanying the patient at the preoperative office visit.
The case went to trial.
What’s the verdict?
CASE: Final verdict was for the patient
The jury awarded damages to the patient based on the absence of adequate consent and failure to perform what was preoperatively agreed to in the consent form.
Legal takeaways from this case
This is an unusual case. Absent something else, it is unusual for there to be liability for not doing a procedure, where the procedure seemed medically unnecessary based on observations during surgery and where language of the signed written consent form created ambiguity about the plan for the removal of the ovaries. Here the patient alleged that her consent was not “informed.” Although informed consent claims are fairly common in malpractice litigation, they are generally appended to an underlying count (or counts) of negligent care; it is uncommon for there to be recovery of damages based solely on the absence of informed consent.
A signed consent form may not be sufficient. In general, a patient’s signature on a consent form alone is not sufficient evidence of informed consent. Whether the patient was truly informed will be judged by the circumstances during which the patient’s consent was obtained.
State laws vary on the specifics of informed consent. Many states have specific statutes or regulations dealing with informed consent. The “informed” part of informed consent generally requires that the patient be informed of:
- the nature of the procedure proposed
- the risks and benefits of the procedure
- the alternative forms of treatment
- the consequences of not undertaking the proposed procedure or an alternative.
In this case, the lawsuit alleges damages based on the fear of future ovarian cancer. It is likely that the patient offered credible testimony that she decided to proceed with surgery specifically because of her fear of ovarian cancer. The patient may have offered testimony about her specific request for her ovaries to be removed because of this fear, or she may have offered testimony about her belief or understanding that the ovaries were going to be removed based on her preoperative discussion with the gynecologist.
Related article: Sound strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Written consent must reflect the actual preoperative discussion
The written consent stated: “hysterectomy with removal of ovary or ovaries,” creating some ambiguity regarding whether the gynecologist had latitude in deciding whether or not to remove the ovaries. However, certain “facts” in this case scenario support the claim that the written consent form was meant to have reflected a decision and agreement between the doctor and patient that the ovaries were to be removed, including:
- the patient had a significant family history of ovarian cancer, making the fear of future ovarian cancer reasonable
- the patient opted out of a conservative treatment plan within 3 days and asked instead to schedule major surgery.
The gynecologist may have testified that the preoperative discussion included only the possibility of removing the ovaries, to be determined based upon what was observed in the course of the surgery. However, in the case description, we are told that the “preoperative documentation was ‘sketchy’ at best.” The jury may have concluded that the gynecologist did not know the wishes of the patient in the event that the ovaries appeared normal during the surgery.
We also know that when the patient returned to the gynecologist’s office after requesting surgery, a “discussion occurred to ‘proceed with LAVH with BSO.’” If this precise language was noted in the patient’s chart, the jury may have concluded that the gynecologist ignored the patient’s wishes!
A claim that the patient was adequately informed prior to treatment can be difficult to address if the informed consent process has not been adequately documented. Often in litigation the decisive question is not whether the right thing was done but whether that can be demonstrated. This case emphasizes the need for good documentation reflecting the specific discussions with the patient.
Litigation prevention rule #1: Thorough documentationVital elements to document | |
| Preoperative office-visit records
Written consent form
| Operative report
Postoperative office-visit records
|
*In the event of an adverse outcome, your actions will be judged on whether you were acting reasonably and using your best judgment. Your documentation needs to explain in detail what you did and why you did it that way. If what you did was a “variance,” explain why. | |
Clinical takeaways
The importance of a good rapport with patients as well as clear discussion of clinical findings, test results, and options for treatment remains paramount. This includes documentation of discussions, recording of who is present during the discussion (including witnesses), as well as the patient’s response to various treatment offerings.
The informed consent process from the clinician’s perspective should reflect discussion of risks, benefits, alternatives, sequelae of complications, and an appropriate risk of refusal. It is most important to pay attention to detail, and record that detail which will reflect your effort to be thorough. Make sure that the surgical consent form includes the operating physician’s name, the name(s) of the assisting physician(s), and no blank spaces.
Related article: More strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Open communication plus complete documentation are key
A well-designed history form—without blanks but with documentation of the physical examination and reflection of the impression and plan—can serve to avert litigation. Ideally, the operative report will reflect not only what was done but also the intraoperative decision-making process on the part of the gynecologist. Documentation of the physician’s thoroughness with intraoperative assessment may well avoid acceptance of a case by a patient’s attorney. Most importantly, transparent postoperative discussions with the patient and family detailing what occurred and the intraoperative decision-making process may convince the patient and family that the clinician has nothing to hide and has the patient’s best interest in mind.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
Please include the city and state in which you practice.
| Joseph S. Sanfilippo, MD, MBA Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors. |
| Steven R. Smith, JD |
| Shirley M. Pruitt, BSN, JD |



Disclosures
Dr. Sanfilippo reports being on the advisory board for Bayer Healthcare Pharmaceuticals and Smith and Nephew. Mr. Smith and Ms. Pruitt report no financial relationships relevant to this article.
| Joseph S. Sanfilippo, MD, MBA Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors. |
| Steven R. Smith, JD |
| Shirley M. Pruitt, BSN, JD |
Disclosures
Dr. Sanfilippo reports being on the advisory board for Bayer Healthcare Pharmaceuticals and Smith and Nephew. Mr. Smith and Ms. Pruitt report no financial relationships relevant to this article.
| Joseph S. Sanfilippo, MD, MBA Professor, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of Pittsburgh, and Director, Reproductive Endocrinology & Infertility at Magee-Womens Hospital, Pittsburgh, Pennsylvania. He also serves on the OBG Management Board of Editors. |
| Steven R. Smith, JD |
| Shirley M. Pruitt, BSN, JD |
Disclosures
Dr. Sanfilippo reports being on the advisory board for Bayer Healthcare Pharmaceuticals and Smith and Nephew. Mr. Smith and Ms. Pruitt report no financial relationships relevant to this article.
Case: Did the gynecologist have the right to not remove the ovaries?
A 36-year-old woman (G3 P3003) presented to her gynecologist with dysmenorrhea and abnormal uterine bleeding. She reported a family history of ovarian cancer for two generations. She was evaluated and underwent physical examination and preoperative ultrasound examination of pelvic organs. All findings were unremarkable. The gynecologist prescribed oral contraceptives (OCs). After an initial excellent response, the patient reported a reoccurrence of pelvic pain and abnormal bleeding 6 years later. The gynecologist suggested options including operative hysteroscopy, dilatation and curettage (D&C), endometrial ablation, off-label use of an intrauterine contraceptive system, or hysterectomy performed via a minimally invasive, vaginal, or abdominal approach. The patient opted for hysteroscopy, D&C, and endometrial ablation and operative laparoscopy. The patient received a diagnosis of stage I endometriosis, which was treated with fulguration.
Two years later, she reported menorrhagia and pelvic pain. The gynecologist suggested trying an OC again, and the patient was given a prescription for a low-dose estrogen/desogestrel combination pill. The patient then changed her mind within 72 hours, never took the OC, and contacted her gynecologist to schedule surgery with him. Upon a return visit to the office, the patient and gynecologist decided to proceed with laparoscopic-assisted vaginal hysterectomy (LAVH) with bilateral salpingo-oophorectomy (BSO). The written consent included laparoscopic hysterectomy with removal of ovary or ovaries and bilateral fallopian tubes, with a possibility of abdominal hysterectomy.
The gynecologist met with the patient preoperatively to update the history, which was unchanged from her prior office visit. In the operating room, “time out” occurred and was documented appropriately—concerns were to be provided to the gynecologist; none were noted.
Intraoperatively, the ovaries were normal in appearance and no endometriosis was noted. The gynecologist proceeded with LAVH and, because the ovaries were normal, did not remove them or the fallopian tubes.
The patient sued the gynecologist on the grounds that, because the originally planned BSO was not performed, she was fearful of developing ovarian cancer in the future.
Preoperative documentation was “sketchy”at best and did not reflect the preoperative discussion and options presented to the patient. There was no documentation of anyone accompanying the patient at the preoperative office visit.
The case went to trial.
What’s the verdict?
CASE: Final verdict was for the patient
The jury awarded damages to the patient based on the absence of adequate consent and failure to perform what was preoperatively agreed to in the consent form.
Legal takeaways from this case
This is an unusual case. Absent something else, it is unusual for there to be liability for not doing a procedure, where the procedure seemed medically unnecessary based on observations during surgery and where language of the signed written consent form created ambiguity about the plan for the removal of the ovaries. Here the patient alleged that her consent was not “informed.” Although informed consent claims are fairly common in malpractice litigation, they are generally appended to an underlying count (or counts) of negligent care; it is uncommon for there to be recovery of damages based solely on the absence of informed consent.
A signed consent form may not be sufficient. In general, a patient’s signature on a consent form alone is not sufficient evidence of informed consent. Whether the patient was truly informed will be judged by the circumstances during which the patient’s consent was obtained.
State laws vary on the specifics of informed consent. Many states have specific statutes or regulations dealing with informed consent. The “informed” part of informed consent generally requires that the patient be informed of:
- the nature of the procedure proposed
- the risks and benefits of the procedure
- the alternative forms of treatment
- the consequences of not undertaking the proposed procedure or an alternative.
In this case, the lawsuit alleges damages based on the fear of future ovarian cancer. It is likely that the patient offered credible testimony that she decided to proceed with surgery specifically because of her fear of ovarian cancer. The patient may have offered testimony about her specific request for her ovaries to be removed because of this fear, or she may have offered testimony about her belief or understanding that the ovaries were going to be removed based on her preoperative discussion with the gynecologist.
Related article: Sound strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Written consent must reflect the actual preoperative discussion
The written consent stated: “hysterectomy with removal of ovary or ovaries,” creating some ambiguity regarding whether the gynecologist had latitude in deciding whether or not to remove the ovaries. However, certain “facts” in this case scenario support the claim that the written consent form was meant to have reflected a decision and agreement between the doctor and patient that the ovaries were to be removed, including:
- the patient had a significant family history of ovarian cancer, making the fear of future ovarian cancer reasonable
- the patient opted out of a conservative treatment plan within 3 days and asked instead to schedule major surgery.
The gynecologist may have testified that the preoperative discussion included only the possibility of removing the ovaries, to be determined based upon what was observed in the course of the surgery. However, in the case description, we are told that the “preoperative documentation was ‘sketchy’ at best.” The jury may have concluded that the gynecologist did not know the wishes of the patient in the event that the ovaries appeared normal during the surgery.
We also know that when the patient returned to the gynecologist’s office after requesting surgery, a “discussion occurred to ‘proceed with LAVH with BSO.’” If this precise language was noted in the patient’s chart, the jury may have concluded that the gynecologist ignored the patient’s wishes!
A claim that the patient was adequately informed prior to treatment can be difficult to address if the informed consent process has not been adequately documented. Often in litigation the decisive question is not whether the right thing was done but whether that can be demonstrated. This case emphasizes the need for good documentation reflecting the specific discussions with the patient.
Litigation prevention rule #1: Thorough documentationVital elements to document | |
| Preoperative office-visit records
Written consent form
| Operative report
Postoperative office-visit records
|
*In the event of an adverse outcome, your actions will be judged on whether you were acting reasonably and using your best judgment. Your documentation needs to explain in detail what you did and why you did it that way. If what you did was a “variance,” explain why. | |
Clinical takeaways
The importance of a good rapport with patients as well as clear discussion of clinical findings, test results, and options for treatment remains paramount. This includes documentation of discussions, recording of who is present during the discussion (including witnesses), as well as the patient’s response to various treatment offerings.
The informed consent process from the clinician’s perspective should reflect discussion of risks, benefits, alternatives, sequelae of complications, and an appropriate risk of refusal. It is most important to pay attention to detail, and record that detail which will reflect your effort to be thorough. Make sure that the surgical consent form includes the operating physician’s name, the name(s) of the assisting physician(s), and no blank spaces.
Related article: More strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Open communication plus complete documentation are key
A well-designed history form—without blanks but with documentation of the physical examination and reflection of the impression and plan—can serve to avert litigation. Ideally, the operative report will reflect not only what was done but also the intraoperative decision-making process on the part of the gynecologist. Documentation of the physician’s thoroughness with intraoperative assessment may well avoid acceptance of a case by a patient’s attorney. Most importantly, transparent postoperative discussions with the patient and family detailing what occurred and the intraoperative decision-making process may convince the patient and family that the clinician has nothing to hide and has the patient’s best interest in mind.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
Please include the city and state in which you practice.
Case: Did the gynecologist have the right to not remove the ovaries?
A 36-year-old woman (G3 P3003) presented to her gynecologist with dysmenorrhea and abnormal uterine bleeding. She reported a family history of ovarian cancer for two generations. She was evaluated and underwent physical examination and preoperative ultrasound examination of pelvic organs. All findings were unremarkable. The gynecologist prescribed oral contraceptives (OCs). After an initial excellent response, the patient reported a reoccurrence of pelvic pain and abnormal bleeding 6 years later. The gynecologist suggested options including operative hysteroscopy, dilatation and curettage (D&C), endometrial ablation, off-label use of an intrauterine contraceptive system, or hysterectomy performed via a minimally invasive, vaginal, or abdominal approach. The patient opted for hysteroscopy, D&C, and endometrial ablation and operative laparoscopy. The patient received a diagnosis of stage I endometriosis, which was treated with fulguration.
Two years later, she reported menorrhagia and pelvic pain. The gynecologist suggested trying an OC again, and the patient was given a prescription for a low-dose estrogen/desogestrel combination pill. The patient then changed her mind within 72 hours, never took the OC, and contacted her gynecologist to schedule surgery with him. Upon a return visit to the office, the patient and gynecologist decided to proceed with laparoscopic-assisted vaginal hysterectomy (LAVH) with bilateral salpingo-oophorectomy (BSO). The written consent included laparoscopic hysterectomy with removal of ovary or ovaries and bilateral fallopian tubes, with a possibility of abdominal hysterectomy.
The gynecologist met with the patient preoperatively to update the history, which was unchanged from her prior office visit. In the operating room, “time out” occurred and was documented appropriately—concerns were to be provided to the gynecologist; none were noted.
Intraoperatively, the ovaries were normal in appearance and no endometriosis was noted. The gynecologist proceeded with LAVH and, because the ovaries were normal, did not remove them or the fallopian tubes.
The patient sued the gynecologist on the grounds that, because the originally planned BSO was not performed, she was fearful of developing ovarian cancer in the future.
Preoperative documentation was “sketchy”at best and did not reflect the preoperative discussion and options presented to the patient. There was no documentation of anyone accompanying the patient at the preoperative office visit.
The case went to trial.
What’s the verdict?
CASE: Final verdict was for the patient
The jury awarded damages to the patient based on the absence of adequate consent and failure to perform what was preoperatively agreed to in the consent form.
Legal takeaways from this case
This is an unusual case. Absent something else, it is unusual for there to be liability for not doing a procedure, where the procedure seemed medically unnecessary based on observations during surgery and where language of the signed written consent form created ambiguity about the plan for the removal of the ovaries. Here the patient alleged that her consent was not “informed.” Although informed consent claims are fairly common in malpractice litigation, they are generally appended to an underlying count (or counts) of negligent care; it is uncommon for there to be recovery of damages based solely on the absence of informed consent.
A signed consent form may not be sufficient. In general, a patient’s signature on a consent form alone is not sufficient evidence of informed consent. Whether the patient was truly informed will be judged by the circumstances during which the patient’s consent was obtained.
State laws vary on the specifics of informed consent. Many states have specific statutes or regulations dealing with informed consent. The “informed” part of informed consent generally requires that the patient be informed of:
- the nature of the procedure proposed
- the risks and benefits of the procedure
- the alternative forms of treatment
- the consequences of not undertaking the proposed procedure or an alternative.
In this case, the lawsuit alleges damages based on the fear of future ovarian cancer. It is likely that the patient offered credible testimony that she decided to proceed with surgery specifically because of her fear of ovarian cancer. The patient may have offered testimony about her specific request for her ovaries to be removed because of this fear, or she may have offered testimony about her belief or understanding that the ovaries were going to be removed based on her preoperative discussion with the gynecologist.
Related article: Sound strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Written consent must reflect the actual preoperative discussion
The written consent stated: “hysterectomy with removal of ovary or ovaries,” creating some ambiguity regarding whether the gynecologist had latitude in deciding whether or not to remove the ovaries. However, certain “facts” in this case scenario support the claim that the written consent form was meant to have reflected a decision and agreement between the doctor and patient that the ovaries were to be removed, including:
- the patient had a significant family history of ovarian cancer, making the fear of future ovarian cancer reasonable
- the patient opted out of a conservative treatment plan within 3 days and asked instead to schedule major surgery.
The gynecologist may have testified that the preoperative discussion included only the possibility of removing the ovaries, to be determined based upon what was observed in the course of the surgery. However, in the case description, we are told that the “preoperative documentation was ‘sketchy’ at best.” The jury may have concluded that the gynecologist did not know the wishes of the patient in the event that the ovaries appeared normal during the surgery.
We also know that when the patient returned to the gynecologist’s office after requesting surgery, a “discussion occurred to ‘proceed with LAVH with BSO.’” If this precise language was noted in the patient’s chart, the jury may have concluded that the gynecologist ignored the patient’s wishes!
A claim that the patient was adequately informed prior to treatment can be difficult to address if the informed consent process has not been adequately documented. Often in litigation the decisive question is not whether the right thing was done but whether that can be demonstrated. This case emphasizes the need for good documentation reflecting the specific discussions with the patient.
Litigation prevention rule #1: Thorough documentationVital elements to document | |
| Preoperative office-visit records
Written consent form
| Operative report
Postoperative office-visit records
|
*In the event of an adverse outcome, your actions will be judged on whether you were acting reasonably and using your best judgment. Your documentation needs to explain in detail what you did and why you did it that way. If what you did was a “variance,” explain why. | |
Clinical takeaways
The importance of a good rapport with patients as well as clear discussion of clinical findings, test results, and options for treatment remains paramount. This includes documentation of discussions, recording of who is present during the discussion (including witnesses), as well as the patient’s response to various treatment offerings.
The informed consent process from the clinician’s perspective should reflect discussion of risks, benefits, alternatives, sequelae of complications, and an appropriate risk of refusal. It is most important to pay attention to detail, and record that detail which will reflect your effort to be thorough. Make sure that the surgical consent form includes the operating physician’s name, the name(s) of the assisting physician(s), and no blank spaces.
Related article: More strategies to avoid malpractice hazards on labor and delivery. Martin L. Gimovsky, MD, and Alexis C. Gimosky, MD (Professional Liability; January 2011)
Open communication plus complete documentation are key
A well-designed history form—without blanks but with documentation of the physical examination and reflection of the impression and plan—can serve to avert litigation. Ideally, the operative report will reflect not only what was done but also the intraoperative decision-making process on the part of the gynecologist. Documentation of the physician’s thoroughness with intraoperative assessment may well avoid acceptance of a case by a patient’s attorney. Most importantly, transparent postoperative discussions with the patient and family detailing what occurred and the intraoperative decision-making process may convince the patient and family that the clinician has nothing to hide and has the patient’s best interest in mind.
WE WANT TO HEAR FROM YOU! Share your thoughts on this article. Send your Letter to the Editor to: [email protected]
Please include the city and state in which you practice.

Management of CCB Overdoses
The 2011 National Poison Data System (NPDS) of the American Association of Poison Control Centers reported that among the top 25 categories associated with mortality, cardiovascular medications were second to sedatives/hypnotics/antipsychotics in terms of the number of deaths resulting from overdose. Moreover, of cardiovascular medications, Calcium channel blockers (CCBs) were the most common agents associated with mortality.[1, 2] The 2012 NPDS report showed a similar trend, with cardiovascular drugs ranking among the top causes of overdoses, with an additional approximately 4614 cases in comparison to 2011.[3] In light of emerging strategies for the management of CCB overdoses, we sought to review the pathophysiology of CCB overdose and its management.
PATHOPHYSIOLOGY OF CCB OVERDOSE
CCBs are widely used in the management of various conditions such as hypertension, angina pectoris, atrial fibrillation, and other cardiac arrhythmias. CCBs block L‐type receptors on the cell surface.[4] Based on their predominant physiological effect, CCBs have been classified as dihydropyridines and nondihydropyridines (Table 1). Dihydropyridine overdose generally results in vasodilation with resultant hypotension and reflex tachycardia.[5] In comparison, nondihydropyridine overdose generally results in bradycardia and decreased cardiac contractility.[6] With high serum concentrations of either CCB class, however, selectivity is lost, and patients may presents with bradycardia, hypotension, and decreased cardiac contractility.[7, 8]
|
| Dihydropyridine |
| Short‐acting agents: nifedipine |
| Longer‐acting formulations: felodipine, isradipine, nicardipine, nifedipine, nisoldipine, amlodipinea |
| Nondihydropyridine |
| Verampamil and diltiazem |
CCBs show good oral bioavailability and undergo first‐pass metabolism. During an overdose, the enzymes involved in hepatic oxidation can become oversaturated, which reduces the effects of first‐pass metabolism, resulting in increased quantities of the active drug reaching the systemic circulation and a prolonged half‐life.[7] In addition, CCBs are highly protein bound and have large volumes of distribution.[9]
Calcium enters cells through specific channels and regulates various cell processes. In myocardial cells, calcium affects excitation‐contraction coupling and potential action generation in the sinoatrial node. Similarly, in the pancreas, calcium facilitates the release of insulin. CCB overdose can result in inhibition of insulin secretion from the pancreas and a state of hypoinsulinemia and insulin resistance.[10] Mtabolic acidosis is a common presentation noted in several published case reports.[11] Metabolic acidosis represents a combination of insulin dysregulation with ketoacidosis and hypoperfusion with lactic acidosis. In addition, because CCBs block the entry of calcium into the mitochondria,[12, 13] and because calcium is required for the normal enzymatic activity of the Krebs cycle, CCB overdose leads to lactic acid build‐up from its direct effects on aerobic metabolism.[14]
The clinical picture of CCB overdose is further complicated by the switch in the mechanism of adenosine triphosphate (ATP) generation in the myocardium from free fatty acid oxidation to carbohydrate metabolism.[15] In response to this stress, the liver increases glucose production via glycogenolysis. With concomitant hypoinsulinemia[10] and relative insulin resistance, intracellular glucose transport is disturbed, with a resultant decrease in ATP production that quickly leads to myocardial dysfunction and cardiogenic shock. The resultant clinical state of acidosis, hyperglycemia, and insulin deficiency is similar to diabetic ketoacidosis.[11, 14] A presentation of symptomatic bradycardia, hyperglycemia, and persistent hypotension, with signs of hypoperfusion usually manifested as altered mental status, clinically defines a severe overdose.
MANAGEMENT APPROACH
Maintenance of the airway and circulation is of primary importance in CCB overdose cases (Table 2). Hypotension and bradyarrhythmias are noted in cases of severe overdose, and some patients might require endotracheal intubation and mechanical ventilation very early in their management. The initial treatment strategy typically consists of the use of intravenous crystalloids and gastrointestinal (GI) decontamination; atropine is reserved for symptomatic bradycardia. Some patients may also require transcutaneous and transvenous pacing early and emergently due to complete cardiovascular collapse. Therefore, having a medical toxicologist or a regional poison control expert involved from the time of initial management is advised, especially for cases of severe overdose or consumption of extended‐release preparations.
|
| Initial resuscitation measures |
| Intravenous hydration with crystalloids, colloids. |
| Gastrointestinal decontamination |
| Activated charcoal 1 g/kg body weight in hemodynamically stable patients who can protect their airways.[1] Best administered within 2 hours. However, in poisoning from extended release preparations, it can be used beyond the 2‐hour window. Anecdotally, WBI has been utilized in calcium channel blocker overdose. However, it is not the recommended approach, especially in patients who are hemodynamically unstable. |
| Atropine |
| Reserved for bradycardia; 0.5 mg every 35 minutes, not to exceed a total of 3 mg or 0.04 mg/kg (per ACLS protocol). |
| Sodium bicarbonate |
| 12 mEq/kg boluses of hypertonic sodium bicarbonate when QRS widening is noted on the ECG.[46] For severe acidosis or persistent ECG changes, a sodium bicarbonate drip can be initiated with 150 mEq sodium bicarbonate in 1 L D5W to run at about 100125 mL per hour.[46] |
| Following intravenous hydration and GI decontamination (hyperinsulinemia‐euglycemia therapy) or vasopressors are usually initiated as resuscitation measures. |
| Agents used to reverse the calcium channel blocker poisoning |
| Hyperinsulinemia‐euglycemia therapy (refer to Table 33). |
| Glucagon |
| Initiated at 0.050.15 mg/kg as bolus dosing, with a repeat dosing in 35 minutes. An intravenous infusion can be initiated following this.[1] |
| Calcium salts |
| A bolus of 0.3 mEq/kg of calcium can be administered as intravenously over 510 minutes (0.6 mL/kg of 10% calcium gluconate solution or 0.2 mL/kg of 10% calcium chloride solution). |
| If beneficial response noted, an infusion of 0.3 mEq/kg per hour. |
| Titrate the infusion to obtain an adequate hemodynamic response. Serum ionized calcium levels should be monitored, and target ionized calcium levels should be less than twice the upper limit of normal.[2] |
| Adrenergic agents |
| Norepinephrine, dopamine, vasopressin. |
| Intravenous lipid emulsion therapy |
| 20% fat emulsion is what is usually used with 1 mL/kg given as a bolus followed by a continuous infusion of 0.250.5 mL/kg per hour. |
| Phosphodiesterase inhibitors |
| Amrinone, milrinone. |
| Invasive therapy |
| Transvenous and transcutaneous pacing for high‐grade atrioventricular dissociation. |
| Intra‐aortic balloon pump. |
| Extra corporeal membrane oxygenation. |
GI Decontamination
In cases of severe overdose, patients may present with lethargy from hypotension and poor cerebral flow, and the risk for aspiration and pneumonitis should be strongly considered in these patients if GI decontamination is considered. GI decontamination is best in cases where the patient is hemodynamically stable and presents early to the emergency department (ED), preferably within 2 hours[7, 9]; early use might decrease drug absorption and enterohepatic circulation, thus lowering the drug levels.[16] However, in cases in which the drug consumed was an extended‐release preparation, GI decontamination is beneficial even when the patient presents late to the ED.[17] GI decontamination is typically achieved using activated charcoal (1 g/kg body weight) or by performing whole bowel irrigation (WBI) with polyethylene glycol.[9] However, there is very little evidence that either approach changes the overall outcome, and WBI can be potentially harmful for patients with hemodynamic instability.[18] Therefore, airway and circulation maintenance is preferable to this approach.
Catecholamines
Catecholamines, such as dopamine, dobutamine, and norepinephrine, appear to be obvious choices in the management of cases of CCB overdose, because most patients present with hypotension and bradyarrhythmias.[19] However, there is no evidence to show the superiority of 1 agent over another in the management of CCB drug toxicity. Catecholamines increase the heart rate and blood pressure and increase systemic vascular resistance, which can potentially decrease the cardiac output by increasing the afterload.
Calcium Salts
In cases of severe overdose, the initial measures are typically not sufficient for stabilizing the patient. Intravenous (IV) calcium salts have been evaluated in animal models[20, 21] and, anecdotally, in human case reports.[22, 23, 24] However, the response to treatment has been mixed, with improvement in hemodynamic parameters in some cases and treatment failures in other cases. Moreover, the effects of these treatments are typically short lived, and repeated dosing might be required. Calcium salts are typically administered with the theoretical scheme of reversing antagonism with a higher calcium load and increasing cardiac inotropy. Calcium gluconate and calcium chloride are 2 frequently used agents, although no clear guidelines exist regarding this approach and the required dosage.[22] There are also published case reports in which refractory hypotension was treated with continuous calcium infusion in an attempt to reach predefined serum calcium levels.[24] However, the fear of iatrogenic hypercalcemia and its consequences is constant.[25] Calcium chloride contains 3 times the calcium for the identical volume compared to calcium gluconate and is more corrosive to the blood vessels; therefore, it is best administered through a central intravenous access. Although the evidence is limited to a few case reports, continuous calcium infusion appears effective and safe as an adjunctive therapy for patients with severe hypotension resulting from CCB overdose.[21, 22, 23, 24, 26]
Glucagon
Although insulin and glucagon are physiologically counter‐regulatory, they have a similar effect on heart stimulation. In animal models, the positive inotropic and chronotropic effects of glucagon have been clearly demonstrated.[27] Glucagon increases intracellular cyclic adenosine monophosphate (AMP) by stimulating adenylyl cyclase, a mechanism by which glucagon possibly exerts its inotropic effect.[7] Most studies conducted on the use of glucagon in the treatment of CCB overdose originated in an era in which bovine or porcine glucagon was used, and these animal glucagon products contained insulin.[9] Glucagon is typically initiated at 50 to 150 g/kg as bolus dosing, with a repeat dosing after 3 to 5 minutes.[9] A continuous IV infusion can then be administered following the initial treatment, because glucagon has a very short half‐life and works rapidly.[7, 9] However, there is no established maximum infusion dose of glucagon, and it should be titrated to the desired clinical outcome. IV glucagon therapy also carries a risk for nausea and vomiting,[7, 28] which in combination with lethargy may increase the risk for aspiration pneumonitis. The evidence for the use of glucagon in cases of CCB overdose is predominantly based on animal models[27]; evidence in human subjects is limited to case reports.[11, 28, 29] Some cases have demonstrated an improvement in hemodynamics with glucagon, whereas in a few cases, glucagon failed to result in such improvement.[30] In cases in which the ingestion history is unclear or there is polysubstance ingestion, as with ‐blockers and CCBs, glucagon is an ideal treatment agent[9]; in contrast, in single CCB overdose, glucagon might not be as helpful as more recent treatment modalities.
Hyperinsulinemia‐Euglycemia Therapy
In recent years, increasing evidence from multiple case reports and case series has shown the superiority of high‐dose insulin therapy over other treatment modalities (Table 3). Insulin acts as a potent inotrope[31, 32] and vasodilator. In their prospective observational series of 7 patients, Greene et al. report the successful use of hyperinsulinemia‐euglycemia therapy (HIET) with no significant adverse events when combined with conventional measures in a critical‐care setting.[33] Similarly, more than 50 cases have been reported in which HIET was used successfully in the management of CCB overdoses.[34]
| Bolus dosing |
| Check finger stick blood glucose, and 25 g dextrose can be given as a bolus, provided the patient is not markedly hyperglycemic[1] (eg, blood glucose >400 mg/dL). |
| 0.5 IU/kg of insulin given as bolus. An acceptable alternative would be to give 1 IU/kg as a bolus to saturate the receptors.[1, 3, 4] |
| Maintenance dose infusion |
| Short‐acting insulin initiated at 0.5 IU/kg per hour, and this dose can be titrated up to 2 IU/kg per hour. Doses as high as 10 IU/kg per hour have been tried and have been successful.[1, 4] |
| Continuous dextrose infusion might be required to maintain euglycemia (25 g per hour intravenous infusion would be a reasonable choice).[1] |
| Monitoring |
| Monitor blood glucose every 30 minutes for the first 4 hours and then hourly. Titrate dextrose infusion to maintain euglycemia.[1] |
| Dextrose containing fluid can be initiated at 0.51 g/kg per hour and titrated to maintain euglycemia.[10, 15] |
| Monitor potassium levels every 60 minutes and replace as needed to maintain at lower limits of normal (2.83.2 mEq/L). |
| Titration of the insulin infusion is usually to the resolution of hemodynamic parameters. |
| Discontinuation |
| No clear evidence to say if a weaning protocol is necessary. In several case reports, the protocol was discontinued after objective parameters of clinical resolution were achieved; however, continued dextrose infusion may be required despite the discontinuation of the insulin.[5] |
Although there is wide variation in the insulin dosing regimens in published case reports, hyperinsulinemia therapy is typically initiated with a 0.5 IU/kg to 1 IU/kg bolus, followed by a continuous drip of 0.5 UI/kg per hour to 1 IU/kg per hour. This dose is titrated every 15 to 20 minutes until satisfactory hemodynamic and clinical stability is noted. Titrations are usually avoided for a shorter time interval because insulin must enter cells and initiate intracellular signaling and metabolic activation. However, the response to HIET might be delayed, and other therapeutic modalities could be required simultaneously until the clinical effects of insulin are observed.
Euglycemia should be maintained by checking the blood glucose levels every 30 minutes and using a dextrose solution to maintain the blood glucose within the upper limits of normal.[35] Hyperglycemia noted in CCB overdose cases indicates the degree of insulin resistance and serves as a marker of the severity of the overdose.[14, 15] In particular, patients who are hyperglycemic at presentation may not require supplemental dextrose infusion despite the high‐dose insulin therapy. The blood glucose level should be checked every 30 minutes for the first 4 hours and then hourly to avoid overlooking hypoglycemia during the treatment regimen, especially in intubated and sedated patients. Fluids containing dextrose may be initiated at 0.5 to 1 g/kg per hour and titrated to maintain euglycemia.[9, 11]
However, there is no consensus as to how long the infusion should be continued once initiated. Although insulin has not been shown to induce tachyphylaxis in experimental animal models, many clinicians prefer to discontinue the infusion once hemodynamic stability has been achieved. There is also no evidence indicating whether a weaning protocol would make any difference over abrupt discontinuation.[36] The physiological effects of insulin persist for hours after the discontinuation of the infusion and will gradually taper down with time. Therefore, theoretically, an abrupt cessation should seldom cause any deleterious effects.[11] Dextrose supplementation may be required to maintain euglycemia for up to 24 hours following discontinuation of the insulin drip due to the elevated insulin levels.[11, 36]
Insulin is a potent vasodilator in the coronary and pulmonary vasculature but does not increase the requirement for myocardial oxygen. Instead, insulin facilitates endothelial nitric oxide activity through the phosphoinositide 3‐kinase (PI3K) pathway, which translates into vasodilatation of the capillary microvasculature and better perfusion at the capillary junction. As a result, insulin corrects the capillary dysfunction that is the major pathology in cardiogenic shock and the ultimate presentation in severe CCB overdose.
Gradinac et al. reported that patients with cardiogenic shock, in the postoperative period of coronary artery bypass grafting, showed a better cardiac index with the use of IV insulin therapy.[37] In an experiment on explanted human myocardium, von Lewinski et al. demonstrated the positive inotropic effect of insulin through calcium‐dependent pathways as well as PI3K pathways.[38] Moreover, Hsu et al. demonstrated with human myocardial cells that this inotropic property of insulin was dose dependent, with better responses observed after the use of higher doses of insulin; in addition, this effect was rapid (ie, as fast as 5 minutes after the infusion) and was sustained throughout the duration of insulin treatment.[39] The best clinical translation of this finding was demonstrated by Yuan et al.[11] in their case series of 5 patients with severe cardiogenic shock secondary to CCB overdoses.
There have also been cases of CCB overdoses in which insulin therapy has failed, which may be because the insulin protocol was initiated late as salvage therapy or because of the severity of the events.[35] Insulin therapy should be initiated early in the course of management rather than as salvage therapy.[7, 35] Agarwal et al. reported their experience in treating an patient on 3 separate occasions of CCB overdose. These authors reported rapid improvement on the third occasion, in which insulin therapy was initiated early during the course of management.[40] In recent years, HIET has been shown to be a promising approach in the management of CCB overdose. Patients with third‐degree heart blockage resulting from CCB overdose reverted to a normal sinus rhythm while on an insulin drip protocol without the intervention of a temporary pacemaker.[11]
High‐dose insulin therapy can also result in hypokalemia, which theoretically may represent a beneficial response in the management of CCB overdose, because it provides a membrane stabilizing effect by prolonging repolarization and allowing more calcium to enter the cytoplasm during cardiac systole.[11] Yuan et al. suggested a serum potassium range of 2.8 to 3.2 mEq/L during insulin‐glucose therapy.[11] Hypomagnesemia and hypophosphatemia are other electrolyte derangements reported during treatment that are similar to conditions observed in patients with diabetic ketoacidosis.[41, 42]
Intravenous Lipid Emulsion Therapy
CCBs are naturally lipophilic, and intravenous lipid emulsion (ILE) therapy has been attempted with success in cases of severe CCB overdose.[43, 44] A systematic review by Jamaty et al.[45] showed that, although the overall quality of the evidence for this modality was poor, ILE could be beneficial in the management of severe cases of CCB poisoning. ILE therapy was first described by Weinberg et al. for bupivacaine toxicity in the year 2003.[46] ILE is commonly utilized as part of total parenteral nutrition, and several case reports have shown the success of its use in the treatment of local anesthetic toxicity.[47] Although the mechanism remains to be clearly elucidated,[48] it is hypothesized that this emulsion in the circulation creates a lipid channel, which causes sequestration of lipophilic drugs, and stimulates the redistribution of lipophilic drugs from the tissues to this channel.[47] Recent data have further revealed the inotropic properties of lipid emulsion; when used for acute overdose, lipid emulsion improves ventricular contractility and diastolic relaxation, going beyond its role as a simple fuel for cardiac tissue or a lipid sink.[49] Lipid emulsion in the circulation also stimulates insulin secretion, which is beneficial in reversing the antagonism caused by CCB on the cells of the pancreas.[50] However, fat embolism, infection, and the development of acute respiratory distress syndrome have been reported as complications associated with this therapy.[51] Thus, it is prudent to involve a medical toxicologist or the regional poison center to decide whether a patient would be a candidate for this treatment approach. In most cases, this is reserved as a last resort in the management of CCB overdose. Typically, a 20% fat emulsion is used, with 1 mL/kg given as a bolus followed by a continuous infusion of 0.25 to 0.5 mL/kg per hour.[7]
Sodium Bicarbonate
Metabolic acidosis resulting from CCB overdose facilitates the binding of CCB to L‐type calcium channels; thus, correcting this acidemia might improve the hemodynamic profile. Sodium bicarbonate has been suggested as a useful adjunct because it decreases the affinity of the CCB for the calcium channel. In cases of severe toxicity, electrocardiogram (ECG) findings may show widening of the QRS complex; these ECG changes are mediated through the inhibitory action of CCB on fast sodium channels, similar to that observed in cases of overdose from tricyclic antidepressants.[9, 52]
Although the evidence is limited to a few case reports, treatment with 1 to 2 mEq/kg boluses of hypertonic sodium bicarbonate is recommended in cases in which QRS widening is noted on an ECG.[52] In cases of severe toxicity with severe acidosis, dysrhythmia, or persistent QRS widening, a sodium bicarbonate drip could be initiated, with 150 mEq of sodium bicarbonate in 1 L D5W to run at approximately 100 to 125 mL per hour.[52]
OTHER TREATMENT MODALITIES
Levosimendan has inotropic properties and is a calcium sensitizer to the myocardium. Although this drug has been used for CCB overdose,[53] it is not available in the United States. Temporary pacemakers and intra‐aortic balloon pump counter pulsation therapy are reserved for severe heart blocks and cases of refractory cardiogenic shock. The use of these 2 modalities is recommended only on a case‐by‐case basis. Wolf et al. demonstrated treatment success in a case of severe verapamil toxicity following the use of glucagon and amrinone.[54] However, there is the potential for hypotension, and this therapy is not routinely recommended. Considering that all CCBs are highly protein bound, with large volumes of distribution, extracorporeal measures such as hemodialysis and charcoal hemoperfusion have very limited roles in the management of an overdose.
CONCLUSION
There is no standardized approach for the management of patients with CCB overdose, and most of the existing evidence consists of case reports and case series. Calcium salts, glucagon, and vasopressors are common first‐line agents. In severe cases, HIET appears to be a promising treatment strategy, with several case reports reiterating its efficacy. However, euglycemia and a stable electrolyte panel should be maintained throughout the clinical course of management. Most of the benefits observed with HIET were noted in cases in which insulin therapy was initiated early in the course of management. ILE therapy, temporary pacemakers, and intra‐aortic balloon pump counter pulsation therapy are used on a case‐by‐case basis and best applied in consultation with a medical toxicologist or the regional poison control center.
Disclosure
Nothing to report.
- , , , , , . 2010 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 28th annual report. Clin Toxicol (Phila). 2011;49(10):910–941.
- , , , , . 2011 annual report of the American Association Of Poison Control Centers' National Poison Data System (NPDS): 29th annual report. Clin Toxicol (Phila). 2012;50(10):911–1164.
- , , , , . 2012 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 30th annual report. Clin Toxicol. 2013;51(10):949–1229.
- , , . Verapamil toxicity dysregulates the phosphatidylinositol 3‐kinase pathway. Acad Emerg Med. 2008;15(4):368–374.
- , , , . Adult toxicology in critical care: part II: specific poisonings. Chest. 2003;123(3):897–922.
- , , , . Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharmacol Exp Ther. 1993;267(2):744–750.
- , . Calcium channel blocker toxicity. Pediatr Emerg Care. 2009;25(8):532–538; quiz 539–540.
- , , . Severe intoxication after an intentional overdose of amlodipine. Acta Anaesthesiol Scand. 2003;47(8):1038–1040.
- . Management of beta‐adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am. 2007;25(2):309–331; abstract viii.
- , , , , . Effect of ca++ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J Pharmacol Exp Ther. 1993;264(1):35–40.
- , , , , . Insulin‐glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37(4):463–474.
- , . Binding of diltiazem and verapamil to isolated rat heart mitochondria. Basic Res Cardiol. 1987;82(3):246–251.
- , , , , . Effect of calcium channel antagonists on calcium uptake and release by isolated rat cardiac mitochondria. Eur J Pharmacol. 1988;152(3):247–253.
- , , , . The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol. 1997;145(2):357–362.
- , , , et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med. 2007;35(9):2071–2075.
- , , . Activated charcoal alone and followed by whole‐bowel irrigation in preventing the absorption of sustained‐release drugs. Clin Pharmacol Ther. 2001;70(3):255–260.
- , , , . Slow‐release verapamil poisoning. use of polyethylene glycol whole‐bowel lavage and high‐dose calcium. Med J Aust. 1993;158(3):202–204.
- , , , . Whole bowel irrigation and the hemodynamically unstable calcium channel blocker overdose: primum non nocere. J Emerg Med. 2010;38(2):171–174.
- , , , . Critical care management of verapamil and diltiazem overdose with a focus on vasopressors: a 25‐year experience at a single center. Ann Emerg Med. 2013;62(3):252–258.
- , , . Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med. 1995;23(7):1251–1263.
- , , , , , . Reversal of the cardiovascular effects of verapamil by calcium and sodium: differences between electrophysiologic and hemodynamic responses. Circulation. 1979;59(4):797–804.
- , , , . A novel dosing regimen for calcium infusion in a patient of massive overdose of sustained‐release nifedipine. Am J Med Sci. 2013;345(3):248–251.
- , , , , . Calcium gluconate in severe verapamil intoxication. N Engl J Med. 1994;330(10):718–720.
- , , . Continuous calcium chloride infusion for massive nifedipine overdose. Chest. 2001;119(4):1280–1282.
- , . A fatal case of iatrogenic hypercalcemia after calcium channel blocker overdose. J Med Toxicol. 2008;4(1):25–29.
- , . Acute amlodipine overdose treated by high dose intravenous calcium in a patient with severe renal insufficiency. Clin Toxicol (Phila). 2007;45(3):301–303.
- . Glucagon in beta‐blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41(5):595–602.
- , . Utilization of a glucagon infusion in the management of a massive nifedipine overdose. J Emerg Med. 2000;18(4):453–455.
- , , , , . A potential role for glucagon in the treatment of drug‐induced symptomatic bradycardia. Chest. 1998;114(1):323–326.
- , , , . Diltiazem overdose: case report and review. J Emerg Med. 1991;9(5):357–366.
- , , , , , . Haemodynamic effects of high doses of insulin during acute left ventricular failure in dogs. Eur Heart J. 1985;6(5):451–457.
- , . The actions of insulin on cardiac contractility. Life Sci. 1981;29(10):975–1000.
- , , , , . Relative safety of hyperinsulinaemia/euglycaemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med. 2007;33(11):2019–2024.
- , , . Hyperinsulin therapy for calcium channel antagonist poisoning: a seven‐year retrospective study. Am J Ther. 2013;20(1):29–31.
- , , , , . Bench‐to‐bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium‐channel blockers. Crit Care. 2006;10(3):212.
- , , , . High‐dose insulin therapy in beta‐blocker and calcium channel‐blocker poisoning. Clin Toxicol (Phila). 2011;49(4):277–283.
- , , , , . Improved cardiac function with glucose‐insulin‐potassium after aortocoronary bypass grafting. Ann Thorac Surg. 1989;48(4):484–489.
- , , , et al. Functional effects of glucose transporters in human ventricular myocardium. Eur J Heart Fail. 2010;12(2):106–113.
- , , , , , . Cellular mechanisms responsible for the inotropic action of insulin on failing human myocardium. J Heart Lung Transplant. 2006;25(9):1126–1134.
- , , , . Hyperinsulinemia euglycemia therapy for calcium channel blocker overdose: a case report. Tex Heart Inst J. 2012;39(4):575–578.
- , , , . Plasma phosphorus and magnesium values during treatment of severe diabetic ketoacidosis. Med Interne. 1981;19(1):63–68.
- , , . Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am J Med. 1985;79(5):571–576.
- , , . Diltiazem poisoning treated with hyperinsulinemic euglycemia therapy and intravenous lipid emulsion. Eur J Emerg Med. 2011;18(2):121–123.
- , , , , . Hemodynamic effects of intravenous fat emulsion in an animal model of severe verapamil toxicity resuscitated with atropine, calcium, and saline. Acad Emerg Med. 2007;14(2):105–111.
- , , , , , . Lipid emulsions in the treatment of acute poisoning: a systematic review of human and animal studies. Clin Toxicol (Phila). 2010;48(1):1–27.
- , , , . Lipid emulsion infusion rescues dogs from bupivacaine‐induced cardiac toxicity. Reg Anesth Pain Med. 2003;28(3):198–202.
- , . Use of lipid emulsion to reverse local anesthetic‐induced toxicity. Ann Pharmacother. 2007;41(11):1873–1877.
- . Lipid resuscitation: more than a sink. Crit Care Med. 2012;40(8):2521–2523.
- , , , et al. Rapid cardiotonic effects of lipid emulsion infusion. Crit Care Med. 2013;41(8):e156–e162.
- , , , . Intralipid prolongs survival in a rat model of verapamil toxicity. Acad Emerg Med. 2006;13(2):134–139.
- . Lipid emulsion for the treatment of local anesthetic toxicity: patient safety implications. Anesth Analg. 2008;106(5):1337–1339.
- , . Poisoning by sodium channel blocking agents. Crit Care Clin. 1997;13(4):829–848.
- , , , . Levosimendan as treatment option in severe verapamil intoxication: a case report and review of the literature. Case Rep Med. 2010;2010. pii: 546904.
- , , . Use of amrinone and glucagon in a case of calcium channel blocker overdose. Ann Emerg Med. 1993;22(7):1225–1228.
The 2011 National Poison Data System (NPDS) of the American Association of Poison Control Centers reported that among the top 25 categories associated with mortality, cardiovascular medications were second to sedatives/hypnotics/antipsychotics in terms of the number of deaths resulting from overdose. Moreover, of cardiovascular medications, Calcium channel blockers (CCBs) were the most common agents associated with mortality.[1, 2] The 2012 NPDS report showed a similar trend, with cardiovascular drugs ranking among the top causes of overdoses, with an additional approximately 4614 cases in comparison to 2011.[3] In light of emerging strategies for the management of CCB overdoses, we sought to review the pathophysiology of CCB overdose and its management.
PATHOPHYSIOLOGY OF CCB OVERDOSE
CCBs are widely used in the management of various conditions such as hypertension, angina pectoris, atrial fibrillation, and other cardiac arrhythmias. CCBs block L‐type receptors on the cell surface.[4] Based on their predominant physiological effect, CCBs have been classified as dihydropyridines and nondihydropyridines (Table 1). Dihydropyridine overdose generally results in vasodilation with resultant hypotension and reflex tachycardia.[5] In comparison, nondihydropyridine overdose generally results in bradycardia and decreased cardiac contractility.[6] With high serum concentrations of either CCB class, however, selectivity is lost, and patients may presents with bradycardia, hypotension, and decreased cardiac contractility.[7, 8]
|
| Dihydropyridine |
| Short‐acting agents: nifedipine |
| Longer‐acting formulations: felodipine, isradipine, nicardipine, nifedipine, nisoldipine, amlodipinea |
| Nondihydropyridine |
| Verampamil and diltiazem |
CCBs show good oral bioavailability and undergo first‐pass metabolism. During an overdose, the enzymes involved in hepatic oxidation can become oversaturated, which reduces the effects of first‐pass metabolism, resulting in increased quantities of the active drug reaching the systemic circulation and a prolonged half‐life.[7] In addition, CCBs are highly protein bound and have large volumes of distribution.[9]
Calcium enters cells through specific channels and regulates various cell processes. In myocardial cells, calcium affects excitation‐contraction coupling and potential action generation in the sinoatrial node. Similarly, in the pancreas, calcium facilitates the release of insulin. CCB overdose can result in inhibition of insulin secretion from the pancreas and a state of hypoinsulinemia and insulin resistance.[10] Mtabolic acidosis is a common presentation noted in several published case reports.[11] Metabolic acidosis represents a combination of insulin dysregulation with ketoacidosis and hypoperfusion with lactic acidosis. In addition, because CCBs block the entry of calcium into the mitochondria,[12, 13] and because calcium is required for the normal enzymatic activity of the Krebs cycle, CCB overdose leads to lactic acid build‐up from its direct effects on aerobic metabolism.[14]
The clinical picture of CCB overdose is further complicated by the switch in the mechanism of adenosine triphosphate (ATP) generation in the myocardium from free fatty acid oxidation to carbohydrate metabolism.[15] In response to this stress, the liver increases glucose production via glycogenolysis. With concomitant hypoinsulinemia[10] and relative insulin resistance, intracellular glucose transport is disturbed, with a resultant decrease in ATP production that quickly leads to myocardial dysfunction and cardiogenic shock. The resultant clinical state of acidosis, hyperglycemia, and insulin deficiency is similar to diabetic ketoacidosis.[11, 14] A presentation of symptomatic bradycardia, hyperglycemia, and persistent hypotension, with signs of hypoperfusion usually manifested as altered mental status, clinically defines a severe overdose.
MANAGEMENT APPROACH
Maintenance of the airway and circulation is of primary importance in CCB overdose cases (Table 2). Hypotension and bradyarrhythmias are noted in cases of severe overdose, and some patients might require endotracheal intubation and mechanical ventilation very early in their management. The initial treatment strategy typically consists of the use of intravenous crystalloids and gastrointestinal (GI) decontamination; atropine is reserved for symptomatic bradycardia. Some patients may also require transcutaneous and transvenous pacing early and emergently due to complete cardiovascular collapse. Therefore, having a medical toxicologist or a regional poison control expert involved from the time of initial management is advised, especially for cases of severe overdose or consumption of extended‐release preparations.
|
| Initial resuscitation measures |
| Intravenous hydration with crystalloids, colloids. |
| Gastrointestinal decontamination |
| Activated charcoal 1 g/kg body weight in hemodynamically stable patients who can protect their airways.[1] Best administered within 2 hours. However, in poisoning from extended release preparations, it can be used beyond the 2‐hour window. Anecdotally, WBI has been utilized in calcium channel blocker overdose. However, it is not the recommended approach, especially in patients who are hemodynamically unstable. |
| Atropine |
| Reserved for bradycardia; 0.5 mg every 35 minutes, not to exceed a total of 3 mg or 0.04 mg/kg (per ACLS protocol). |
| Sodium bicarbonate |
| 12 mEq/kg boluses of hypertonic sodium bicarbonate when QRS widening is noted on the ECG.[46] For severe acidosis or persistent ECG changes, a sodium bicarbonate drip can be initiated with 150 mEq sodium bicarbonate in 1 L D5W to run at about 100125 mL per hour.[46] |
| Following intravenous hydration and GI decontamination (hyperinsulinemia‐euglycemia therapy) or vasopressors are usually initiated as resuscitation measures. |
| Agents used to reverse the calcium channel blocker poisoning |
| Hyperinsulinemia‐euglycemia therapy (refer to Table 33). |
| Glucagon |
| Initiated at 0.050.15 mg/kg as bolus dosing, with a repeat dosing in 35 minutes. An intravenous infusion can be initiated following this.[1] |
| Calcium salts |
| A bolus of 0.3 mEq/kg of calcium can be administered as intravenously over 510 minutes (0.6 mL/kg of 10% calcium gluconate solution or 0.2 mL/kg of 10% calcium chloride solution). |
| If beneficial response noted, an infusion of 0.3 mEq/kg per hour. |
| Titrate the infusion to obtain an adequate hemodynamic response. Serum ionized calcium levels should be monitored, and target ionized calcium levels should be less than twice the upper limit of normal.[2] |
| Adrenergic agents |
| Norepinephrine, dopamine, vasopressin. |
| Intravenous lipid emulsion therapy |
| 20% fat emulsion is what is usually used with 1 mL/kg given as a bolus followed by a continuous infusion of 0.250.5 mL/kg per hour. |
| Phosphodiesterase inhibitors |
| Amrinone, milrinone. |
| Invasive therapy |
| Transvenous and transcutaneous pacing for high‐grade atrioventricular dissociation. |
| Intra‐aortic balloon pump. |
| Extra corporeal membrane oxygenation. |
GI Decontamination
In cases of severe overdose, patients may present with lethargy from hypotension and poor cerebral flow, and the risk for aspiration and pneumonitis should be strongly considered in these patients if GI decontamination is considered. GI decontamination is best in cases where the patient is hemodynamically stable and presents early to the emergency department (ED), preferably within 2 hours[7, 9]; early use might decrease drug absorption and enterohepatic circulation, thus lowering the drug levels.[16] However, in cases in which the drug consumed was an extended‐release preparation, GI decontamination is beneficial even when the patient presents late to the ED.[17] GI decontamination is typically achieved using activated charcoal (1 g/kg body weight) or by performing whole bowel irrigation (WBI) with polyethylene glycol.[9] However, there is very little evidence that either approach changes the overall outcome, and WBI can be potentially harmful for patients with hemodynamic instability.[18] Therefore, airway and circulation maintenance is preferable to this approach.
Catecholamines
Catecholamines, such as dopamine, dobutamine, and norepinephrine, appear to be obvious choices in the management of cases of CCB overdose, because most patients present with hypotension and bradyarrhythmias.[19] However, there is no evidence to show the superiority of 1 agent over another in the management of CCB drug toxicity. Catecholamines increase the heart rate and blood pressure and increase systemic vascular resistance, which can potentially decrease the cardiac output by increasing the afterload.
Calcium Salts
In cases of severe overdose, the initial measures are typically not sufficient for stabilizing the patient. Intravenous (IV) calcium salts have been evaluated in animal models[20, 21] and, anecdotally, in human case reports.[22, 23, 24] However, the response to treatment has been mixed, with improvement in hemodynamic parameters in some cases and treatment failures in other cases. Moreover, the effects of these treatments are typically short lived, and repeated dosing might be required. Calcium salts are typically administered with the theoretical scheme of reversing antagonism with a higher calcium load and increasing cardiac inotropy. Calcium gluconate and calcium chloride are 2 frequently used agents, although no clear guidelines exist regarding this approach and the required dosage.[22] There are also published case reports in which refractory hypotension was treated with continuous calcium infusion in an attempt to reach predefined serum calcium levels.[24] However, the fear of iatrogenic hypercalcemia and its consequences is constant.[25] Calcium chloride contains 3 times the calcium for the identical volume compared to calcium gluconate and is more corrosive to the blood vessels; therefore, it is best administered through a central intravenous access. Although the evidence is limited to a few case reports, continuous calcium infusion appears effective and safe as an adjunctive therapy for patients with severe hypotension resulting from CCB overdose.[21, 22, 23, 24, 26]
Glucagon
Although insulin and glucagon are physiologically counter‐regulatory, they have a similar effect on heart stimulation. In animal models, the positive inotropic and chronotropic effects of glucagon have been clearly demonstrated.[27] Glucagon increases intracellular cyclic adenosine monophosphate (AMP) by stimulating adenylyl cyclase, a mechanism by which glucagon possibly exerts its inotropic effect.[7] Most studies conducted on the use of glucagon in the treatment of CCB overdose originated in an era in which bovine or porcine glucagon was used, and these animal glucagon products contained insulin.[9] Glucagon is typically initiated at 50 to 150 g/kg as bolus dosing, with a repeat dosing after 3 to 5 minutes.[9] A continuous IV infusion can then be administered following the initial treatment, because glucagon has a very short half‐life and works rapidly.[7, 9] However, there is no established maximum infusion dose of glucagon, and it should be titrated to the desired clinical outcome. IV glucagon therapy also carries a risk for nausea and vomiting,[7, 28] which in combination with lethargy may increase the risk for aspiration pneumonitis. The evidence for the use of glucagon in cases of CCB overdose is predominantly based on animal models[27]; evidence in human subjects is limited to case reports.[11, 28, 29] Some cases have demonstrated an improvement in hemodynamics with glucagon, whereas in a few cases, glucagon failed to result in such improvement.[30] In cases in which the ingestion history is unclear or there is polysubstance ingestion, as with ‐blockers and CCBs, glucagon is an ideal treatment agent[9]; in contrast, in single CCB overdose, glucagon might not be as helpful as more recent treatment modalities.
Hyperinsulinemia‐Euglycemia Therapy
In recent years, increasing evidence from multiple case reports and case series has shown the superiority of high‐dose insulin therapy over other treatment modalities (Table 3). Insulin acts as a potent inotrope[31, 32] and vasodilator. In their prospective observational series of 7 patients, Greene et al. report the successful use of hyperinsulinemia‐euglycemia therapy (HIET) with no significant adverse events when combined with conventional measures in a critical‐care setting.[33] Similarly, more than 50 cases have been reported in which HIET was used successfully in the management of CCB overdoses.[34]
| Bolus dosing |
| Check finger stick blood glucose, and 25 g dextrose can be given as a bolus, provided the patient is not markedly hyperglycemic[1] (eg, blood glucose >400 mg/dL). |
| 0.5 IU/kg of insulin given as bolus. An acceptable alternative would be to give 1 IU/kg as a bolus to saturate the receptors.[1, 3, 4] |
| Maintenance dose infusion |
| Short‐acting insulin initiated at 0.5 IU/kg per hour, and this dose can be titrated up to 2 IU/kg per hour. Doses as high as 10 IU/kg per hour have been tried and have been successful.[1, 4] |
| Continuous dextrose infusion might be required to maintain euglycemia (25 g per hour intravenous infusion would be a reasonable choice).[1] |
| Monitoring |
| Monitor blood glucose every 30 minutes for the first 4 hours and then hourly. Titrate dextrose infusion to maintain euglycemia.[1] |
| Dextrose containing fluid can be initiated at 0.51 g/kg per hour and titrated to maintain euglycemia.[10, 15] |
| Monitor potassium levels every 60 minutes and replace as needed to maintain at lower limits of normal (2.83.2 mEq/L). |
| Titration of the insulin infusion is usually to the resolution of hemodynamic parameters. |
| Discontinuation |
| No clear evidence to say if a weaning protocol is necessary. In several case reports, the protocol was discontinued after objective parameters of clinical resolution were achieved; however, continued dextrose infusion may be required despite the discontinuation of the insulin.[5] |
Although there is wide variation in the insulin dosing regimens in published case reports, hyperinsulinemia therapy is typically initiated with a 0.5 IU/kg to 1 IU/kg bolus, followed by a continuous drip of 0.5 UI/kg per hour to 1 IU/kg per hour. This dose is titrated every 15 to 20 minutes until satisfactory hemodynamic and clinical stability is noted. Titrations are usually avoided for a shorter time interval because insulin must enter cells and initiate intracellular signaling and metabolic activation. However, the response to HIET might be delayed, and other therapeutic modalities could be required simultaneously until the clinical effects of insulin are observed.
Euglycemia should be maintained by checking the blood glucose levels every 30 minutes and using a dextrose solution to maintain the blood glucose within the upper limits of normal.[35] Hyperglycemia noted in CCB overdose cases indicates the degree of insulin resistance and serves as a marker of the severity of the overdose.[14, 15] In particular, patients who are hyperglycemic at presentation may not require supplemental dextrose infusion despite the high‐dose insulin therapy. The blood glucose level should be checked every 30 minutes for the first 4 hours and then hourly to avoid overlooking hypoglycemia during the treatment regimen, especially in intubated and sedated patients. Fluids containing dextrose may be initiated at 0.5 to 1 g/kg per hour and titrated to maintain euglycemia.[9, 11]
However, there is no consensus as to how long the infusion should be continued once initiated. Although insulin has not been shown to induce tachyphylaxis in experimental animal models, many clinicians prefer to discontinue the infusion once hemodynamic stability has been achieved. There is also no evidence indicating whether a weaning protocol would make any difference over abrupt discontinuation.[36] The physiological effects of insulin persist for hours after the discontinuation of the infusion and will gradually taper down with time. Therefore, theoretically, an abrupt cessation should seldom cause any deleterious effects.[11] Dextrose supplementation may be required to maintain euglycemia for up to 24 hours following discontinuation of the insulin drip due to the elevated insulin levels.[11, 36]
Insulin is a potent vasodilator in the coronary and pulmonary vasculature but does not increase the requirement for myocardial oxygen. Instead, insulin facilitates endothelial nitric oxide activity through the phosphoinositide 3‐kinase (PI3K) pathway, which translates into vasodilatation of the capillary microvasculature and better perfusion at the capillary junction. As a result, insulin corrects the capillary dysfunction that is the major pathology in cardiogenic shock and the ultimate presentation in severe CCB overdose.
Gradinac et al. reported that patients with cardiogenic shock, in the postoperative period of coronary artery bypass grafting, showed a better cardiac index with the use of IV insulin therapy.[37] In an experiment on explanted human myocardium, von Lewinski et al. demonstrated the positive inotropic effect of insulin through calcium‐dependent pathways as well as PI3K pathways.[38] Moreover, Hsu et al. demonstrated with human myocardial cells that this inotropic property of insulin was dose dependent, with better responses observed after the use of higher doses of insulin; in addition, this effect was rapid (ie, as fast as 5 minutes after the infusion) and was sustained throughout the duration of insulin treatment.[39] The best clinical translation of this finding was demonstrated by Yuan et al.[11] in their case series of 5 patients with severe cardiogenic shock secondary to CCB overdoses.
There have also been cases of CCB overdoses in which insulin therapy has failed, which may be because the insulin protocol was initiated late as salvage therapy or because of the severity of the events.[35] Insulin therapy should be initiated early in the course of management rather than as salvage therapy.[7, 35] Agarwal et al. reported their experience in treating an patient on 3 separate occasions of CCB overdose. These authors reported rapid improvement on the third occasion, in which insulin therapy was initiated early during the course of management.[40] In recent years, HIET has been shown to be a promising approach in the management of CCB overdose. Patients with third‐degree heart blockage resulting from CCB overdose reverted to a normal sinus rhythm while on an insulin drip protocol without the intervention of a temporary pacemaker.[11]
High‐dose insulin therapy can also result in hypokalemia, which theoretically may represent a beneficial response in the management of CCB overdose, because it provides a membrane stabilizing effect by prolonging repolarization and allowing more calcium to enter the cytoplasm during cardiac systole.[11] Yuan et al. suggested a serum potassium range of 2.8 to 3.2 mEq/L during insulin‐glucose therapy.[11] Hypomagnesemia and hypophosphatemia are other electrolyte derangements reported during treatment that are similar to conditions observed in patients with diabetic ketoacidosis.[41, 42]
Intravenous Lipid Emulsion Therapy
CCBs are naturally lipophilic, and intravenous lipid emulsion (ILE) therapy has been attempted with success in cases of severe CCB overdose.[43, 44] A systematic review by Jamaty et al.[45] showed that, although the overall quality of the evidence for this modality was poor, ILE could be beneficial in the management of severe cases of CCB poisoning. ILE therapy was first described by Weinberg et al. for bupivacaine toxicity in the year 2003.[46] ILE is commonly utilized as part of total parenteral nutrition, and several case reports have shown the success of its use in the treatment of local anesthetic toxicity.[47] Although the mechanism remains to be clearly elucidated,[48] it is hypothesized that this emulsion in the circulation creates a lipid channel, which causes sequestration of lipophilic drugs, and stimulates the redistribution of lipophilic drugs from the tissues to this channel.[47] Recent data have further revealed the inotropic properties of lipid emulsion; when used for acute overdose, lipid emulsion improves ventricular contractility and diastolic relaxation, going beyond its role as a simple fuel for cardiac tissue or a lipid sink.[49] Lipid emulsion in the circulation also stimulates insulin secretion, which is beneficial in reversing the antagonism caused by CCB on the cells of the pancreas.[50] However, fat embolism, infection, and the development of acute respiratory distress syndrome have been reported as complications associated with this therapy.[51] Thus, it is prudent to involve a medical toxicologist or the regional poison center to decide whether a patient would be a candidate for this treatment approach. In most cases, this is reserved as a last resort in the management of CCB overdose. Typically, a 20% fat emulsion is used, with 1 mL/kg given as a bolus followed by a continuous infusion of 0.25 to 0.5 mL/kg per hour.[7]
Sodium Bicarbonate
Metabolic acidosis resulting from CCB overdose facilitates the binding of CCB to L‐type calcium channels; thus, correcting this acidemia might improve the hemodynamic profile. Sodium bicarbonate has been suggested as a useful adjunct because it decreases the affinity of the CCB for the calcium channel. In cases of severe toxicity, electrocardiogram (ECG) findings may show widening of the QRS complex; these ECG changes are mediated through the inhibitory action of CCB on fast sodium channels, similar to that observed in cases of overdose from tricyclic antidepressants.[9, 52]
Although the evidence is limited to a few case reports, treatment with 1 to 2 mEq/kg boluses of hypertonic sodium bicarbonate is recommended in cases in which QRS widening is noted on an ECG.[52] In cases of severe toxicity with severe acidosis, dysrhythmia, or persistent QRS widening, a sodium bicarbonate drip could be initiated, with 150 mEq of sodium bicarbonate in 1 L D5W to run at approximately 100 to 125 mL per hour.[52]
OTHER TREATMENT MODALITIES
Levosimendan has inotropic properties and is a calcium sensitizer to the myocardium. Although this drug has been used for CCB overdose,[53] it is not available in the United States. Temporary pacemakers and intra‐aortic balloon pump counter pulsation therapy are reserved for severe heart blocks and cases of refractory cardiogenic shock. The use of these 2 modalities is recommended only on a case‐by‐case basis. Wolf et al. demonstrated treatment success in a case of severe verapamil toxicity following the use of glucagon and amrinone.[54] However, there is the potential for hypotension, and this therapy is not routinely recommended. Considering that all CCBs are highly protein bound, with large volumes of distribution, extracorporeal measures such as hemodialysis and charcoal hemoperfusion have very limited roles in the management of an overdose.
CONCLUSION
There is no standardized approach for the management of patients with CCB overdose, and most of the existing evidence consists of case reports and case series. Calcium salts, glucagon, and vasopressors are common first‐line agents. In severe cases, HIET appears to be a promising treatment strategy, with several case reports reiterating its efficacy. However, euglycemia and a stable electrolyte panel should be maintained throughout the clinical course of management. Most of the benefits observed with HIET were noted in cases in which insulin therapy was initiated early in the course of management. ILE therapy, temporary pacemakers, and intra‐aortic balloon pump counter pulsation therapy are used on a case‐by‐case basis and best applied in consultation with a medical toxicologist or the regional poison control center.
Disclosure
Nothing to report.
The 2011 National Poison Data System (NPDS) of the American Association of Poison Control Centers reported that among the top 25 categories associated with mortality, cardiovascular medications were second to sedatives/hypnotics/antipsychotics in terms of the number of deaths resulting from overdose. Moreover, of cardiovascular medications, Calcium channel blockers (CCBs) were the most common agents associated with mortality.[1, 2] The 2012 NPDS report showed a similar trend, with cardiovascular drugs ranking among the top causes of overdoses, with an additional approximately 4614 cases in comparison to 2011.[3] In light of emerging strategies for the management of CCB overdoses, we sought to review the pathophysiology of CCB overdose and its management.
PATHOPHYSIOLOGY OF CCB OVERDOSE
CCBs are widely used in the management of various conditions such as hypertension, angina pectoris, atrial fibrillation, and other cardiac arrhythmias. CCBs block L‐type receptors on the cell surface.[4] Based on their predominant physiological effect, CCBs have been classified as dihydropyridines and nondihydropyridines (Table 1). Dihydropyridine overdose generally results in vasodilation with resultant hypotension and reflex tachycardia.[5] In comparison, nondihydropyridine overdose generally results in bradycardia and decreased cardiac contractility.[6] With high serum concentrations of either CCB class, however, selectivity is lost, and patients may presents with bradycardia, hypotension, and decreased cardiac contractility.[7, 8]
|
| Dihydropyridine |
| Short‐acting agents: nifedipine |
| Longer‐acting formulations: felodipine, isradipine, nicardipine, nifedipine, nisoldipine, amlodipinea |
| Nondihydropyridine |
| Verampamil and diltiazem |
CCBs show good oral bioavailability and undergo first‐pass metabolism. During an overdose, the enzymes involved in hepatic oxidation can become oversaturated, which reduces the effects of first‐pass metabolism, resulting in increased quantities of the active drug reaching the systemic circulation and a prolonged half‐life.[7] In addition, CCBs are highly protein bound and have large volumes of distribution.[9]
Calcium enters cells through specific channels and regulates various cell processes. In myocardial cells, calcium affects excitation‐contraction coupling and potential action generation in the sinoatrial node. Similarly, in the pancreas, calcium facilitates the release of insulin. CCB overdose can result in inhibition of insulin secretion from the pancreas and a state of hypoinsulinemia and insulin resistance.[10] Mtabolic acidosis is a common presentation noted in several published case reports.[11] Metabolic acidosis represents a combination of insulin dysregulation with ketoacidosis and hypoperfusion with lactic acidosis. In addition, because CCBs block the entry of calcium into the mitochondria,[12, 13] and because calcium is required for the normal enzymatic activity of the Krebs cycle, CCB overdose leads to lactic acid build‐up from its direct effects on aerobic metabolism.[14]
The clinical picture of CCB overdose is further complicated by the switch in the mechanism of adenosine triphosphate (ATP) generation in the myocardium from free fatty acid oxidation to carbohydrate metabolism.[15] In response to this stress, the liver increases glucose production via glycogenolysis. With concomitant hypoinsulinemia[10] and relative insulin resistance, intracellular glucose transport is disturbed, with a resultant decrease in ATP production that quickly leads to myocardial dysfunction and cardiogenic shock. The resultant clinical state of acidosis, hyperglycemia, and insulin deficiency is similar to diabetic ketoacidosis.[11, 14] A presentation of symptomatic bradycardia, hyperglycemia, and persistent hypotension, with signs of hypoperfusion usually manifested as altered mental status, clinically defines a severe overdose.
MANAGEMENT APPROACH
Maintenance of the airway and circulation is of primary importance in CCB overdose cases (Table 2). Hypotension and bradyarrhythmias are noted in cases of severe overdose, and some patients might require endotracheal intubation and mechanical ventilation very early in their management. The initial treatment strategy typically consists of the use of intravenous crystalloids and gastrointestinal (GI) decontamination; atropine is reserved for symptomatic bradycardia. Some patients may also require transcutaneous and transvenous pacing early and emergently due to complete cardiovascular collapse. Therefore, having a medical toxicologist or a regional poison control expert involved from the time of initial management is advised, especially for cases of severe overdose or consumption of extended‐release preparations.
|
| Initial resuscitation measures |
| Intravenous hydration with crystalloids, colloids. |
| Gastrointestinal decontamination |
| Activated charcoal 1 g/kg body weight in hemodynamically stable patients who can protect their airways.[1] Best administered within 2 hours. However, in poisoning from extended release preparations, it can be used beyond the 2‐hour window. Anecdotally, WBI has been utilized in calcium channel blocker overdose. However, it is not the recommended approach, especially in patients who are hemodynamically unstable. |
| Atropine |
| Reserved for bradycardia; 0.5 mg every 35 minutes, not to exceed a total of 3 mg or 0.04 mg/kg (per ACLS protocol). |
| Sodium bicarbonate |
| 12 mEq/kg boluses of hypertonic sodium bicarbonate when QRS widening is noted on the ECG.[46] For severe acidosis or persistent ECG changes, a sodium bicarbonate drip can be initiated with 150 mEq sodium bicarbonate in 1 L D5W to run at about 100125 mL per hour.[46] |
| Following intravenous hydration and GI decontamination (hyperinsulinemia‐euglycemia therapy) or vasopressors are usually initiated as resuscitation measures. |
| Agents used to reverse the calcium channel blocker poisoning |
| Hyperinsulinemia‐euglycemia therapy (refer to Table 33). |
| Glucagon |
| Initiated at 0.050.15 mg/kg as bolus dosing, with a repeat dosing in 35 minutes. An intravenous infusion can be initiated following this.[1] |
| Calcium salts |
| A bolus of 0.3 mEq/kg of calcium can be administered as intravenously over 510 minutes (0.6 mL/kg of 10% calcium gluconate solution or 0.2 mL/kg of 10% calcium chloride solution). |
| If beneficial response noted, an infusion of 0.3 mEq/kg per hour. |
| Titrate the infusion to obtain an adequate hemodynamic response. Serum ionized calcium levels should be monitored, and target ionized calcium levels should be less than twice the upper limit of normal.[2] |
| Adrenergic agents |
| Norepinephrine, dopamine, vasopressin. |
| Intravenous lipid emulsion therapy |
| 20% fat emulsion is what is usually used with 1 mL/kg given as a bolus followed by a continuous infusion of 0.250.5 mL/kg per hour. |
| Phosphodiesterase inhibitors |
| Amrinone, milrinone. |
| Invasive therapy |
| Transvenous and transcutaneous pacing for high‐grade atrioventricular dissociation. |
| Intra‐aortic balloon pump. |
| Extra corporeal membrane oxygenation. |
GI Decontamination
In cases of severe overdose, patients may present with lethargy from hypotension and poor cerebral flow, and the risk for aspiration and pneumonitis should be strongly considered in these patients if GI decontamination is considered. GI decontamination is best in cases where the patient is hemodynamically stable and presents early to the emergency department (ED), preferably within 2 hours[7, 9]; early use might decrease drug absorption and enterohepatic circulation, thus lowering the drug levels.[16] However, in cases in which the drug consumed was an extended‐release preparation, GI decontamination is beneficial even when the patient presents late to the ED.[17] GI decontamination is typically achieved using activated charcoal (1 g/kg body weight) or by performing whole bowel irrigation (WBI) with polyethylene glycol.[9] However, there is very little evidence that either approach changes the overall outcome, and WBI can be potentially harmful for patients with hemodynamic instability.[18] Therefore, airway and circulation maintenance is preferable to this approach.
Catecholamines
Catecholamines, such as dopamine, dobutamine, and norepinephrine, appear to be obvious choices in the management of cases of CCB overdose, because most patients present with hypotension and bradyarrhythmias.[19] However, there is no evidence to show the superiority of 1 agent over another in the management of CCB drug toxicity. Catecholamines increase the heart rate and blood pressure and increase systemic vascular resistance, which can potentially decrease the cardiac output by increasing the afterload.
Calcium Salts
In cases of severe overdose, the initial measures are typically not sufficient for stabilizing the patient. Intravenous (IV) calcium salts have been evaluated in animal models[20, 21] and, anecdotally, in human case reports.[22, 23, 24] However, the response to treatment has been mixed, with improvement in hemodynamic parameters in some cases and treatment failures in other cases. Moreover, the effects of these treatments are typically short lived, and repeated dosing might be required. Calcium salts are typically administered with the theoretical scheme of reversing antagonism with a higher calcium load and increasing cardiac inotropy. Calcium gluconate and calcium chloride are 2 frequently used agents, although no clear guidelines exist regarding this approach and the required dosage.[22] There are also published case reports in which refractory hypotension was treated with continuous calcium infusion in an attempt to reach predefined serum calcium levels.[24] However, the fear of iatrogenic hypercalcemia and its consequences is constant.[25] Calcium chloride contains 3 times the calcium for the identical volume compared to calcium gluconate and is more corrosive to the blood vessels; therefore, it is best administered through a central intravenous access. Although the evidence is limited to a few case reports, continuous calcium infusion appears effective and safe as an adjunctive therapy for patients with severe hypotension resulting from CCB overdose.[21, 22, 23, 24, 26]
Glucagon
Although insulin and glucagon are physiologically counter‐regulatory, they have a similar effect on heart stimulation. In animal models, the positive inotropic and chronotropic effects of glucagon have been clearly demonstrated.[27] Glucagon increases intracellular cyclic adenosine monophosphate (AMP) by stimulating adenylyl cyclase, a mechanism by which glucagon possibly exerts its inotropic effect.[7] Most studies conducted on the use of glucagon in the treatment of CCB overdose originated in an era in which bovine or porcine glucagon was used, and these animal glucagon products contained insulin.[9] Glucagon is typically initiated at 50 to 150 g/kg as bolus dosing, with a repeat dosing after 3 to 5 minutes.[9] A continuous IV infusion can then be administered following the initial treatment, because glucagon has a very short half‐life and works rapidly.[7, 9] However, there is no established maximum infusion dose of glucagon, and it should be titrated to the desired clinical outcome. IV glucagon therapy also carries a risk for nausea and vomiting,[7, 28] which in combination with lethargy may increase the risk for aspiration pneumonitis. The evidence for the use of glucagon in cases of CCB overdose is predominantly based on animal models[27]; evidence in human subjects is limited to case reports.[11, 28, 29] Some cases have demonstrated an improvement in hemodynamics with glucagon, whereas in a few cases, glucagon failed to result in such improvement.[30] In cases in which the ingestion history is unclear or there is polysubstance ingestion, as with ‐blockers and CCBs, glucagon is an ideal treatment agent[9]; in contrast, in single CCB overdose, glucagon might not be as helpful as more recent treatment modalities.
Hyperinsulinemia‐Euglycemia Therapy
In recent years, increasing evidence from multiple case reports and case series has shown the superiority of high‐dose insulin therapy over other treatment modalities (Table 3). Insulin acts as a potent inotrope[31, 32] and vasodilator. In their prospective observational series of 7 patients, Greene et al. report the successful use of hyperinsulinemia‐euglycemia therapy (HIET) with no significant adverse events when combined with conventional measures in a critical‐care setting.[33] Similarly, more than 50 cases have been reported in which HIET was used successfully in the management of CCB overdoses.[34]
| Bolus dosing |
| Check finger stick blood glucose, and 25 g dextrose can be given as a bolus, provided the patient is not markedly hyperglycemic[1] (eg, blood glucose >400 mg/dL). |
| 0.5 IU/kg of insulin given as bolus. An acceptable alternative would be to give 1 IU/kg as a bolus to saturate the receptors.[1, 3, 4] |
| Maintenance dose infusion |
| Short‐acting insulin initiated at 0.5 IU/kg per hour, and this dose can be titrated up to 2 IU/kg per hour. Doses as high as 10 IU/kg per hour have been tried and have been successful.[1, 4] |
| Continuous dextrose infusion might be required to maintain euglycemia (25 g per hour intravenous infusion would be a reasonable choice).[1] |
| Monitoring |
| Monitor blood glucose every 30 minutes for the first 4 hours and then hourly. Titrate dextrose infusion to maintain euglycemia.[1] |
| Dextrose containing fluid can be initiated at 0.51 g/kg per hour and titrated to maintain euglycemia.[10, 15] |
| Monitor potassium levels every 60 minutes and replace as needed to maintain at lower limits of normal (2.83.2 mEq/L). |
| Titration of the insulin infusion is usually to the resolution of hemodynamic parameters. |
| Discontinuation |
| No clear evidence to say if a weaning protocol is necessary. In several case reports, the protocol was discontinued after objective parameters of clinical resolution were achieved; however, continued dextrose infusion may be required despite the discontinuation of the insulin.[5] |
Although there is wide variation in the insulin dosing regimens in published case reports, hyperinsulinemia therapy is typically initiated with a 0.5 IU/kg to 1 IU/kg bolus, followed by a continuous drip of 0.5 UI/kg per hour to 1 IU/kg per hour. This dose is titrated every 15 to 20 minutes until satisfactory hemodynamic and clinical stability is noted. Titrations are usually avoided for a shorter time interval because insulin must enter cells and initiate intracellular signaling and metabolic activation. However, the response to HIET might be delayed, and other therapeutic modalities could be required simultaneously until the clinical effects of insulin are observed.
Euglycemia should be maintained by checking the blood glucose levels every 30 minutes and using a dextrose solution to maintain the blood glucose within the upper limits of normal.[35] Hyperglycemia noted in CCB overdose cases indicates the degree of insulin resistance and serves as a marker of the severity of the overdose.[14, 15] In particular, patients who are hyperglycemic at presentation may not require supplemental dextrose infusion despite the high‐dose insulin therapy. The blood glucose level should be checked every 30 minutes for the first 4 hours and then hourly to avoid overlooking hypoglycemia during the treatment regimen, especially in intubated and sedated patients. Fluids containing dextrose may be initiated at 0.5 to 1 g/kg per hour and titrated to maintain euglycemia.[9, 11]
However, there is no consensus as to how long the infusion should be continued once initiated. Although insulin has not been shown to induce tachyphylaxis in experimental animal models, many clinicians prefer to discontinue the infusion once hemodynamic stability has been achieved. There is also no evidence indicating whether a weaning protocol would make any difference over abrupt discontinuation.[36] The physiological effects of insulin persist for hours after the discontinuation of the infusion and will gradually taper down with time. Therefore, theoretically, an abrupt cessation should seldom cause any deleterious effects.[11] Dextrose supplementation may be required to maintain euglycemia for up to 24 hours following discontinuation of the insulin drip due to the elevated insulin levels.[11, 36]
Insulin is a potent vasodilator in the coronary and pulmonary vasculature but does not increase the requirement for myocardial oxygen. Instead, insulin facilitates endothelial nitric oxide activity through the phosphoinositide 3‐kinase (PI3K) pathway, which translates into vasodilatation of the capillary microvasculature and better perfusion at the capillary junction. As a result, insulin corrects the capillary dysfunction that is the major pathology in cardiogenic shock and the ultimate presentation in severe CCB overdose.
Gradinac et al. reported that patients with cardiogenic shock, in the postoperative period of coronary artery bypass grafting, showed a better cardiac index with the use of IV insulin therapy.[37] In an experiment on explanted human myocardium, von Lewinski et al. demonstrated the positive inotropic effect of insulin through calcium‐dependent pathways as well as PI3K pathways.[38] Moreover, Hsu et al. demonstrated with human myocardial cells that this inotropic property of insulin was dose dependent, with better responses observed after the use of higher doses of insulin; in addition, this effect was rapid (ie, as fast as 5 minutes after the infusion) and was sustained throughout the duration of insulin treatment.[39] The best clinical translation of this finding was demonstrated by Yuan et al.[11] in their case series of 5 patients with severe cardiogenic shock secondary to CCB overdoses.
There have also been cases of CCB overdoses in which insulin therapy has failed, which may be because the insulin protocol was initiated late as salvage therapy or because of the severity of the events.[35] Insulin therapy should be initiated early in the course of management rather than as salvage therapy.[7, 35] Agarwal et al. reported their experience in treating an patient on 3 separate occasions of CCB overdose. These authors reported rapid improvement on the third occasion, in which insulin therapy was initiated early during the course of management.[40] In recent years, HIET has been shown to be a promising approach in the management of CCB overdose. Patients with third‐degree heart blockage resulting from CCB overdose reverted to a normal sinus rhythm while on an insulin drip protocol without the intervention of a temporary pacemaker.[11]
High‐dose insulin therapy can also result in hypokalemia, which theoretically may represent a beneficial response in the management of CCB overdose, because it provides a membrane stabilizing effect by prolonging repolarization and allowing more calcium to enter the cytoplasm during cardiac systole.[11] Yuan et al. suggested a serum potassium range of 2.8 to 3.2 mEq/L during insulin‐glucose therapy.[11] Hypomagnesemia and hypophosphatemia are other electrolyte derangements reported during treatment that are similar to conditions observed in patients with diabetic ketoacidosis.[41, 42]
Intravenous Lipid Emulsion Therapy
CCBs are naturally lipophilic, and intravenous lipid emulsion (ILE) therapy has been attempted with success in cases of severe CCB overdose.[43, 44] A systematic review by Jamaty et al.[45] showed that, although the overall quality of the evidence for this modality was poor, ILE could be beneficial in the management of severe cases of CCB poisoning. ILE therapy was first described by Weinberg et al. for bupivacaine toxicity in the year 2003.[46] ILE is commonly utilized as part of total parenteral nutrition, and several case reports have shown the success of its use in the treatment of local anesthetic toxicity.[47] Although the mechanism remains to be clearly elucidated,[48] it is hypothesized that this emulsion in the circulation creates a lipid channel, which causes sequestration of lipophilic drugs, and stimulates the redistribution of lipophilic drugs from the tissues to this channel.[47] Recent data have further revealed the inotropic properties of lipid emulsion; when used for acute overdose, lipid emulsion improves ventricular contractility and diastolic relaxation, going beyond its role as a simple fuel for cardiac tissue or a lipid sink.[49] Lipid emulsion in the circulation also stimulates insulin secretion, which is beneficial in reversing the antagonism caused by CCB on the cells of the pancreas.[50] However, fat embolism, infection, and the development of acute respiratory distress syndrome have been reported as complications associated with this therapy.[51] Thus, it is prudent to involve a medical toxicologist or the regional poison center to decide whether a patient would be a candidate for this treatment approach. In most cases, this is reserved as a last resort in the management of CCB overdose. Typically, a 20% fat emulsion is used, with 1 mL/kg given as a bolus followed by a continuous infusion of 0.25 to 0.5 mL/kg per hour.[7]
Sodium Bicarbonate
Metabolic acidosis resulting from CCB overdose facilitates the binding of CCB to L‐type calcium channels; thus, correcting this acidemia might improve the hemodynamic profile. Sodium bicarbonate has been suggested as a useful adjunct because it decreases the affinity of the CCB for the calcium channel. In cases of severe toxicity, electrocardiogram (ECG) findings may show widening of the QRS complex; these ECG changes are mediated through the inhibitory action of CCB on fast sodium channels, similar to that observed in cases of overdose from tricyclic antidepressants.[9, 52]
Although the evidence is limited to a few case reports, treatment with 1 to 2 mEq/kg boluses of hypertonic sodium bicarbonate is recommended in cases in which QRS widening is noted on an ECG.[52] In cases of severe toxicity with severe acidosis, dysrhythmia, or persistent QRS widening, a sodium bicarbonate drip could be initiated, with 150 mEq of sodium bicarbonate in 1 L D5W to run at approximately 100 to 125 mL per hour.[52]
OTHER TREATMENT MODALITIES
Levosimendan has inotropic properties and is a calcium sensitizer to the myocardium. Although this drug has been used for CCB overdose,[53] it is not available in the United States. Temporary pacemakers and intra‐aortic balloon pump counter pulsation therapy are reserved for severe heart blocks and cases of refractory cardiogenic shock. The use of these 2 modalities is recommended only on a case‐by‐case basis. Wolf et al. demonstrated treatment success in a case of severe verapamil toxicity following the use of glucagon and amrinone.[54] However, there is the potential for hypotension, and this therapy is not routinely recommended. Considering that all CCBs are highly protein bound, with large volumes of distribution, extracorporeal measures such as hemodialysis and charcoal hemoperfusion have very limited roles in the management of an overdose.
CONCLUSION
There is no standardized approach for the management of patients with CCB overdose, and most of the existing evidence consists of case reports and case series. Calcium salts, glucagon, and vasopressors are common first‐line agents. In severe cases, HIET appears to be a promising treatment strategy, with several case reports reiterating its efficacy. However, euglycemia and a stable electrolyte panel should be maintained throughout the clinical course of management. Most of the benefits observed with HIET were noted in cases in which insulin therapy was initiated early in the course of management. ILE therapy, temporary pacemakers, and intra‐aortic balloon pump counter pulsation therapy are used on a case‐by‐case basis and best applied in consultation with a medical toxicologist or the regional poison control center.
Disclosure
Nothing to report.
- , , , , , . 2010 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 28th annual report. Clin Toxicol (Phila). 2011;49(10):910–941.
- , , , , . 2011 annual report of the American Association Of Poison Control Centers' National Poison Data System (NPDS): 29th annual report. Clin Toxicol (Phila). 2012;50(10):911–1164.
- , , , , . 2012 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 30th annual report. Clin Toxicol. 2013;51(10):949–1229.
- , , . Verapamil toxicity dysregulates the phosphatidylinositol 3‐kinase pathway. Acad Emerg Med. 2008;15(4):368–374.
- , , , . Adult toxicology in critical care: part II: specific poisonings. Chest. 2003;123(3):897–922.
- , , , . Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharmacol Exp Ther. 1993;267(2):744–750.
- , . Calcium channel blocker toxicity. Pediatr Emerg Care. 2009;25(8):532–538; quiz 539–540.
- , , . Severe intoxication after an intentional overdose of amlodipine. Acta Anaesthesiol Scand. 2003;47(8):1038–1040.
- . Management of beta‐adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am. 2007;25(2):309–331; abstract viii.
- , , , , . Effect of ca++ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J Pharmacol Exp Ther. 1993;264(1):35–40.
- , , , , . Insulin‐glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37(4):463–474.
- , . Binding of diltiazem and verapamil to isolated rat heart mitochondria. Basic Res Cardiol. 1987;82(3):246–251.
- , , , , . Effect of calcium channel antagonists on calcium uptake and release by isolated rat cardiac mitochondria. Eur J Pharmacol. 1988;152(3):247–253.
- , , , . The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol. 1997;145(2):357–362.
- , , , et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med. 2007;35(9):2071–2075.
- , , . Activated charcoal alone and followed by whole‐bowel irrigation in preventing the absorption of sustained‐release drugs. Clin Pharmacol Ther. 2001;70(3):255–260.
- , , , . Slow‐release verapamil poisoning. use of polyethylene glycol whole‐bowel lavage and high‐dose calcium. Med J Aust. 1993;158(3):202–204.
- , , , . Whole bowel irrigation and the hemodynamically unstable calcium channel blocker overdose: primum non nocere. J Emerg Med. 2010;38(2):171–174.
- , , , . Critical care management of verapamil and diltiazem overdose with a focus on vasopressors: a 25‐year experience at a single center. Ann Emerg Med. 2013;62(3):252–258.
- , , . Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med. 1995;23(7):1251–1263.
- , , , , , . Reversal of the cardiovascular effects of verapamil by calcium and sodium: differences between electrophysiologic and hemodynamic responses. Circulation. 1979;59(4):797–804.
- , , , . A novel dosing regimen for calcium infusion in a patient of massive overdose of sustained‐release nifedipine. Am J Med Sci. 2013;345(3):248–251.
- , , , , . Calcium gluconate in severe verapamil intoxication. N Engl J Med. 1994;330(10):718–720.
- , , . Continuous calcium chloride infusion for massive nifedipine overdose. Chest. 2001;119(4):1280–1282.
- , . A fatal case of iatrogenic hypercalcemia after calcium channel blocker overdose. J Med Toxicol. 2008;4(1):25–29.
- , . Acute amlodipine overdose treated by high dose intravenous calcium in a patient with severe renal insufficiency. Clin Toxicol (Phila). 2007;45(3):301–303.
- . Glucagon in beta‐blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41(5):595–602.
- , . Utilization of a glucagon infusion in the management of a massive nifedipine overdose. J Emerg Med. 2000;18(4):453–455.
- , , , , . A potential role for glucagon in the treatment of drug‐induced symptomatic bradycardia. Chest. 1998;114(1):323–326.
- , , , . Diltiazem overdose: case report and review. J Emerg Med. 1991;9(5):357–366.
- , , , , , . Haemodynamic effects of high doses of insulin during acute left ventricular failure in dogs. Eur Heart J. 1985;6(5):451–457.
- , . The actions of insulin on cardiac contractility. Life Sci. 1981;29(10):975–1000.
- , , , , . Relative safety of hyperinsulinaemia/euglycaemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med. 2007;33(11):2019–2024.
- , , . Hyperinsulin therapy for calcium channel antagonist poisoning: a seven‐year retrospective study. Am J Ther. 2013;20(1):29–31.
- , , , , . Bench‐to‐bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium‐channel blockers. Crit Care. 2006;10(3):212.
- , , , . High‐dose insulin therapy in beta‐blocker and calcium channel‐blocker poisoning. Clin Toxicol (Phila). 2011;49(4):277–283.
- , , , , . Improved cardiac function with glucose‐insulin‐potassium after aortocoronary bypass grafting. Ann Thorac Surg. 1989;48(4):484–489.
- , , , et al. Functional effects of glucose transporters in human ventricular myocardium. Eur J Heart Fail. 2010;12(2):106–113.
- , , , , , . Cellular mechanisms responsible for the inotropic action of insulin on failing human myocardium. J Heart Lung Transplant. 2006;25(9):1126–1134.
- , , , . Hyperinsulinemia euglycemia therapy for calcium channel blocker overdose: a case report. Tex Heart Inst J. 2012;39(4):575–578.
- , , , . Plasma phosphorus and magnesium values during treatment of severe diabetic ketoacidosis. Med Interne. 1981;19(1):63–68.
- , , . Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am J Med. 1985;79(5):571–576.
- , , . Diltiazem poisoning treated with hyperinsulinemic euglycemia therapy and intravenous lipid emulsion. Eur J Emerg Med. 2011;18(2):121–123.
- , , , , . Hemodynamic effects of intravenous fat emulsion in an animal model of severe verapamil toxicity resuscitated with atropine, calcium, and saline. Acad Emerg Med. 2007;14(2):105–111.
- , , , , , . Lipid emulsions in the treatment of acute poisoning: a systematic review of human and animal studies. Clin Toxicol (Phila). 2010;48(1):1–27.
- , , , . Lipid emulsion infusion rescues dogs from bupivacaine‐induced cardiac toxicity. Reg Anesth Pain Med. 2003;28(3):198–202.
- , . Use of lipid emulsion to reverse local anesthetic‐induced toxicity. Ann Pharmacother. 2007;41(11):1873–1877.
- . Lipid resuscitation: more than a sink. Crit Care Med. 2012;40(8):2521–2523.
- , , , et al. Rapid cardiotonic effects of lipid emulsion infusion. Crit Care Med. 2013;41(8):e156–e162.
- , , , . Intralipid prolongs survival in a rat model of verapamil toxicity. Acad Emerg Med. 2006;13(2):134–139.
- . Lipid emulsion for the treatment of local anesthetic toxicity: patient safety implications. Anesth Analg. 2008;106(5):1337–1339.
- , . Poisoning by sodium channel blocking agents. Crit Care Clin. 1997;13(4):829–848.
- , , , . Levosimendan as treatment option in severe verapamil intoxication: a case report and review of the literature. Case Rep Med. 2010;2010. pii: 546904.
- , , . Use of amrinone and glucagon in a case of calcium channel blocker overdose. Ann Emerg Med. 1993;22(7):1225–1228.
- , , , , , . 2010 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 28th annual report. Clin Toxicol (Phila). 2011;49(10):910–941.
- , , , , . 2011 annual report of the American Association Of Poison Control Centers' National Poison Data System (NPDS): 29th annual report. Clin Toxicol (Phila). 2012;50(10):911–1164.
- , , , , . 2012 annual report of the American Association of Poison Control Centers' National Poison Data System (NPDS): 30th annual report. Clin Toxicol. 2013;51(10):949–1229.
- , , . Verapamil toxicity dysregulates the phosphatidylinositol 3‐kinase pathway. Acad Emerg Med. 2008;15(4):368–374.
- , , , . Adult toxicology in critical care: part II: specific poisonings. Chest. 2003;123(3):897–922.
- , , , . Insulin is a superior antidote for cardiovascular toxicity induced by verapamil in the anesthetized canine. J Pharmacol Exp Ther. 1993;267(2):744–750.
- , . Calcium channel blocker toxicity. Pediatr Emerg Care. 2009;25(8):532–538; quiz 539–540.
- , , . Severe intoxication after an intentional overdose of amlodipine. Acta Anaesthesiol Scand. 2003;47(8):1038–1040.
- . Management of beta‐adrenergic blocker and calcium channel antagonist toxicity. Emerg Med Clin North Am. 2007;25(2):309–331; abstract viii.
- , , , , . Effect of ca++ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J Pharmacol Exp Ther. 1993;264(1):35–40.
- , , , , . Insulin‐glucose as adjunctive therapy for severe calcium channel antagonist poisoning. J Toxicol Clin Toxicol. 1999;37(4):463–474.
- , . Binding of diltiazem and verapamil to isolated rat heart mitochondria. Basic Res Cardiol. 1987;82(3):246–251.
- , , , , . Effect of calcium channel antagonists on calcium uptake and release by isolated rat cardiac mitochondria. Eur J Pharmacol. 1988;152(3):247–253.
- , , , . The diabetogenic effects of acute verapamil poisoning. Toxicol Appl Pharmacol. 1997;145(2):357–362.
- , , , et al. Assessment of hyperglycemia after calcium channel blocker overdoses involving diltiazem or verapamil. Crit Care Med. 2007;35(9):2071–2075.
- , , . Activated charcoal alone and followed by whole‐bowel irrigation in preventing the absorption of sustained‐release drugs. Clin Pharmacol Ther. 2001;70(3):255–260.
- , , , . Slow‐release verapamil poisoning. use of polyethylene glycol whole‐bowel lavage and high‐dose calcium. Med J Aust. 1993;158(3):202–204.
- , , , . Whole bowel irrigation and the hemodynamically unstable calcium channel blocker overdose: primum non nocere. J Emerg Med. 2010;38(2):171–174.
- , , , . Critical care management of verapamil and diltiazem overdose with a focus on vasopressors: a 25‐year experience at a single center. Ann Emerg Med. 2013;62(3):252–258.
- , , . Beneficial myocardial metabolic effects of insulin during verapamil toxicity in the anesthetized canine. Crit Care Med. 1995;23(7):1251–1263.
- , , , , , . Reversal of the cardiovascular effects of verapamil by calcium and sodium: differences between electrophysiologic and hemodynamic responses. Circulation. 1979;59(4):797–804.
- , , , . A novel dosing regimen for calcium infusion in a patient of massive overdose of sustained‐release nifedipine. Am J Med Sci. 2013;345(3):248–251.
- , , , , . Calcium gluconate in severe verapamil intoxication. N Engl J Med. 1994;330(10):718–720.
- , , . Continuous calcium chloride infusion for massive nifedipine overdose. Chest. 2001;119(4):1280–1282.
- , . A fatal case of iatrogenic hypercalcemia after calcium channel blocker overdose. J Med Toxicol. 2008;4(1):25–29.
- , . Acute amlodipine overdose treated by high dose intravenous calcium in a patient with severe renal insufficiency. Clin Toxicol (Phila). 2007;45(3):301–303.
- . Glucagon in beta‐blocker and calcium channel blocker overdoses: a systematic review. J Toxicol Clin Toxicol. 2003;41(5):595–602.
- , . Utilization of a glucagon infusion in the management of a massive nifedipine overdose. J Emerg Med. 2000;18(4):453–455.
- , , , , . A potential role for glucagon in the treatment of drug‐induced symptomatic bradycardia. Chest. 1998;114(1):323–326.
- , , , . Diltiazem overdose: case report and review. J Emerg Med. 1991;9(5):357–366.
- , , , , , . Haemodynamic effects of high doses of insulin during acute left ventricular failure in dogs. Eur Heart J. 1985;6(5):451–457.
- , . The actions of insulin on cardiac contractility. Life Sci. 1981;29(10):975–1000.
- , , , , . Relative safety of hyperinsulinaemia/euglycaemia therapy in the management of calcium channel blocker overdose: a prospective observational study. Intensive Care Med. 2007;33(11):2019–2024.
- , , . Hyperinsulin therapy for calcium channel antagonist poisoning: a seven‐year retrospective study. Am J Ther. 2013;20(1):29–31.
- , , , , . Bench‐to‐bedside review: hyperinsulinaemia/euglycaemia therapy in the management of overdose of calcium‐channel blockers. Crit Care. 2006;10(3):212.
- , , , . High‐dose insulin therapy in beta‐blocker and calcium channel‐blocker poisoning. Clin Toxicol (Phila). 2011;49(4):277–283.
- , , , , . Improved cardiac function with glucose‐insulin‐potassium after aortocoronary bypass grafting. Ann Thorac Surg. 1989;48(4):484–489.
- , , , et al. Functional effects of glucose transporters in human ventricular myocardium. Eur J Heart Fail. 2010;12(2):106–113.
- , , , , , . Cellular mechanisms responsible for the inotropic action of insulin on failing human myocardium. J Heart Lung Transplant. 2006;25(9):1126–1134.
- , , , . Hyperinsulinemia euglycemia therapy for calcium channel blocker overdose: a case report. Tex Heart Inst J. 2012;39(4):575–578.
- , , , . Plasma phosphorus and magnesium values during treatment of severe diabetic ketoacidosis. Med Interne. 1981;19(1):63–68.
- , , . Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am J Med. 1985;79(5):571–576.
- , , . Diltiazem poisoning treated with hyperinsulinemic euglycemia therapy and intravenous lipid emulsion. Eur J Emerg Med. 2011;18(2):121–123.
- , , , , . Hemodynamic effects of intravenous fat emulsion in an animal model of severe verapamil toxicity resuscitated with atropine, calcium, and saline. Acad Emerg Med. 2007;14(2):105–111.
- , , , , , . Lipid emulsions in the treatment of acute poisoning: a systematic review of human and animal studies. Clin Toxicol (Phila). 2010;48(1):1–27.
- , , , . Lipid emulsion infusion rescues dogs from bupivacaine‐induced cardiac toxicity. Reg Anesth Pain Med. 2003;28(3):198–202.
- , . Use of lipid emulsion to reverse local anesthetic‐induced toxicity. Ann Pharmacother. 2007;41(11):1873–1877.
- . Lipid resuscitation: more than a sink. Crit Care Med. 2012;40(8):2521–2523.
- , , , et al. Rapid cardiotonic effects of lipid emulsion infusion. Crit Care Med. 2013;41(8):e156–e162.
- , , , . Intralipid prolongs survival in a rat model of verapamil toxicity. Acad Emerg Med. 2006;13(2):134–139.
- . Lipid emulsion for the treatment of local anesthetic toxicity: patient safety implications. Anesth Analg. 2008;106(5):1337–1339.
- , . Poisoning by sodium channel blocking agents. Crit Care Clin. 1997;13(4):829–848.
- , , , . Levosimendan as treatment option in severe verapamil intoxication: a case report and review of the literature. Case Rep Med. 2010;2010. pii: 546904.
- , , . Use of amrinone and glucagon in a case of calcium channel blocker overdose. Ann Emerg Med. 1993;22(7):1225–1228.