User login
Akathisia: Is restlessness a primary condition or an adverse drug effect?
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112). 
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
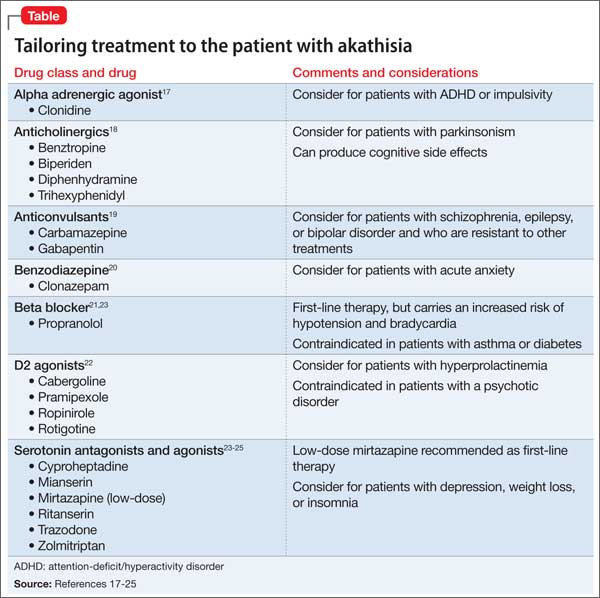
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Akathisia—from the Greek for “inability to sit”—is a neuropsychiatric syndrome characterized by subjective and objective psychomotor restlessness. Patients typically experience feelings of unease, inner restlessness mainly involving the legs, and a compulsion to move. Most engage in repetitive movement. They might swing or cross and uncross their legs, shift from one foot to the other, continuously pace, or persistently fidget.
In clinical settings, akathisia usually is a side effect of medication. Antipsychotics, serotonin reuptake inhibitors, and buspirone are common triggers, but akathisia also has been associated with some antiemetics, preoperative sedatives, calcium channel blockers, and antivertigo agents. It also can be caused by withdrawal from an antipsychotic or related to a substance use disorder, especially cocaine. Akathisia can be acute or chronic, occurring in a tardive form with symptoms that last >6 months.1-3
Much isn’t known about drug-induced akathisia
Our understanding of the pathophysiology of akathisia is incomplete. Some have suggested that it results from an imbalance between the dopaminergic/cholinergic and dopaminergic/serotonergic systems4; others, that the cause is a mismatch between the core and the shell of the nucleus accumbens, due in part to overstimulation of the locus ceruleus.5
More recently, researchers established a positive association between higher scores on the Liverpool University Neuroleptic Side Effects Rating Scale and D2/D3 receptor occupancy in the ventral striatum (nucleus accumbens and olfactory tubercle).6 The D2/D3 receptor occupancy model might explain withdrawal symptoms associated with cocaine,7 as well as relative worsening of symptoms after tapering or discontinuing stimulants in attention-deficit/hyperactivity disorder (ADHD).
Elements of a clinical evaluation
When akathisia is suspected, evaluation by a clinician familiar with its phenomenology is crucial. A validated tool, such as the Barnes Akathisia Rating Scale (at out cometracker.org/library/BAS.pdf) can aid in the detection and assessment of severity.8
In evaluating patients, keep in mind that the inner restlessness that characterizes akathisia can affect the trunk, hands, and arms, as well as the legs, and can cause dysphoria and anxiety. Akathisia has been linked to an increased likelihood of developing suicidal ideation and behavior.9
Less common subjective symptoms include rage, fear, nausea, and worsening of psychotic symptoms. Because of its association with aggression and agitation, drug-induced akathisia has been cited—with little success—as the basis for an insanity defense by people who have committed a violent act.10
Or is akathisia another psychiatric disorder?
Akathisia might go undetected for several reasons. One key factor: Its symptoms resemble and often overlap with those of other psychiatric disorders, such as mania, psychosis, agitated depression, and ADHD. In addition, akathisia often occurs concurrently with, and is masked by, akinesia, a common extrapyramidal side effect of many antipsychotics. Such patients might have the inner feeling of restlessness and urge to move but do not exhibit characteristic limb movements. In some cases, cognitive or intellectual limitations prevent patients from communicating the inner turmoil they feel.11
Medication nonadherence further complicates the picture, sometimes prompting a clinician to increase the dosage of the drug that is causing akathisia (Box 112).
Managing drug-induced akathisia
Akathisia usually resolves when the drug causing it is discontinued; decreasing the dosage might alleviate the symptoms. Whenever akathisia is detected, careful revision of the current drug regimen— substituting an antipsychotic with a lower prevalence of akathisia, for example— should be considered (Box 213-16). Treatment of drug-induced akathisia, which should be tailored to the patient’s psychopathology and comorbidities, is needed as well (Table17-25).
Beta blockers, particularly propranolol, are considered first-line therapy for drug-induced akathisia, with a dosage of 20 to 40 mg twice daily used to relieve symptoms26 The effect can be explained by adrenergic terminals in the locus ceruleus and ending in the nucleus accumbens and prefrontal cortex stimulate β adrenoreceptors.5,27 Although multiple small studies and case reports26,28-32 support the use of beta blockers to treat drug-induced akathisia, the quality of evidence of their efficacy is controversial.12,21,27 Consider the risk of hypotension and bradycardia and be aware of contraindications for patients with asthma or diabetes.
Low-dose mirtazapine (15 mg/d) was found to be as effective as propranolol, 80 mg/d, in a placebo-controlled study, and to be more effective than a beta blocker in treating akathisia induced by a first-generation antipsychotic. The authors concluded that both propranolol and mirtazapine should be first-line therapy.23 Others have suggested that these results be interpreted with caution because mirtazapine (at a higher dosage) has been linked to akathisia.33 Mirtazapine blocks α-adrenergic receptors, resulting in antagonism of 5-HT2 and 5-HT3 receptors and consequent enhancement of 5-HT1A serotonergic transmission.34 In one study, it was shown to reduce binding of the D2/D3 receptor agonist quinpirole.35
Serotonin antagonists and agonists. Blockade of 5-HT2 receptors can attenuate D2 blockade and mitigate akathisia symptoms. Mianserin, 15 mg/d, can be helpful, and ritanserin, 5 to 20 mg/d, produced about a 50% reduction in akathisia symptoms in 10 patients taking neuroleptics.36 Neither is available in the United States, however.
Cyproheptadine, a potent 5-HT2A and 5-HT2C antagonist with anticholinergic and antihistaminic action, improved akathisia symptoms in an open trial of 17 patients with antipsychotic-induced akathisia.37 The recommended dose is 8 to 16 mg/d.
A study using the selective inverse agonist pimavanserin (not FDA-approved) decreased akathisia in healthy volunteers taking haloperidol.14,24,33
Zolmitriptan, a 5-HT1D agonist, also can be used38; one study found that 7.5 mg/d of zolmitriptan is as effective as propranolol.39
A 2010 study showed a statistically significant improvement in 8 patients taking trazodone, compared with 5 patients on placebo, all of whom met criteria for at least mild akathisia. Trazodone’s antiakathitic effect is attributed to its 5-HT2A antagonism.25
Anticholinergics. Traditionally, benztropine, biperiden, diphenhydramine, and trihexyphenidyl have been used for prevention and treatment of extrapyramidal side effects. A Cochrane review concluded, however, that data are insufficient to support use of anticholinergics for akathisia.40 Although multiple case reports have shown anticholinergics to be effective in treating drug-induced akathisia,12,17,33 their association with cognitive side effects suggests a need for caution.18
Benzodiazepines. Through their sedative and anxiolytic properties, benzodiazepines are thought to partially alleviate akathisia symptoms. Two small trials found clonazepam helpful for akathisia symptoms2,20; and 1 case report revealed that a patient with akathisia improved after coadministration of clonazepam and baclofen.41
Anticonvulsants. Valproic acid has not been found to be useful in antipsychotic-induced tardive akathisia.42 However, a case report described a patient with schizophrenia whose akathisia symptoms improved after the dosage of gabapentin was increased.43 Last, carbamazepine was found to be effective in reducing akathisia symptoms in 3 patients with schizophrenia who were resistant to beta blockers, anticholinergics, antihistaminergics, and benzodiazepines.19
α-adrenergic agonists. In an open trial, akathisia symptoms in 6 patients improved with clonidine, 0.2 to 0.8 mg/d.17 Speculation is that strong α1 antagonism might help prevent akathisia, which could be why this condition is not associated with iloperidone.44
D2 agonists. Akathisia and restless legs syndrome have similar pathophysiology,1,2 and patients with akathisia could benefit from D2 agonists such as cabergoline, pramipexole, rotigotine, and ropinirole. One case study revealed that a patient with aripiprazole-induced akathisia improved with ropinirole.45 D2 agonists can precipitate or worsen psychosis, however, and would be a relative contraindication in patients with psychotic disorders.22
Bottom Line
Failure to detect drug-induced akathisia can increase morbidity and delay recovery in patients undergoing psychiatric care. Knowing what to look for and how to tailor treatment to the needs of a given patient is an essential component of good care.
Related Resources
• Ferrando SJ, Eisendrath SJ. Adverse neuropsychiatric effects of dopamine antagonist medications. Misdiagnosis in the medical setting. Psychosomatics. 1991;32(4):426-432.
• Vinson DR. Diphenhydramine in the treatment of akathisia induced by prochlorperazine. J Emerg Med. 2004;26(3):265-270.
Drug Brand Names
Aripiprazole • Abilify Haloperidol • Haldol
Baclofen • Lioresal Iloperidone • Fanapt
Benztropine • Cogentin Lurasidone • Latuda
Biperiden • Akineton Mirtazapine • Remeron
Buspirone • BuSpar Pramipexole • Mirapex
Cabergoline • Dostinex Propranolol • Inderal
Carbamazepine • Tegretol Quetiapine • Seroquel
Clonazepam • Klonopin Ropinirole • Requip
Clonidine • Catapres Rotigotine • Neupro
Clozapine • Clozaril Trazodone • Desyrel, Oleptro
Cyproheptadine • Periactin Trihexyphenidyl • Artane
Diphenhydramine • Benadryl Valproic acid • Depakene
Gabapentin • Neurontin Zolmitriptan • Zomig
Acknowledgement
Mandy Evans, MD, assisted with editing the manuscript of this article.
Disclosure
Dr. Forcen reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.
1. Sachdev P. Akathisia and restless legs. Cambridge, United Kingdom: Cambridge University Press; 1995.
2. Sachdev P, Longragan C. The present status of akathisia. J Nerv Ment Dis. 1991;179(7):381-391.
3. Poyurovsky M, Hermesh H, Weizman A. Severe withdrawal akathisia following neuroleptic discontinuation successfully controlled by clozapine. Int Clin Psychopharmacol. 1996;11(4):283-286.
4. Poyurovsky M, Weizman A. Serotonin-based pharma-cotherapy for acute neuroleptic-induced akathisia: a new approach to an old problem. Br J Psychiatry. 2001;179:4-8.
5. Loonen AJ, Stahl SM. The mechanism of drug-induced akathisia. CNS Spectr. 2011;16(1):7-10.
6. Kim JH, Son YD, Kim HK, et al. Antipsychotic-associated mental side effects and their relationship to dopamine D2 receptor occupancy in striatal subdivisions: a high-resolution PET study with [11C]raclopride. J Clin Psychopharmacol. 2011;31(4):507-511.
7. Dailey JW, Fryer TD, Brichard L, et al. Nucleus accumbens D2/3 receptor predict trait impulsivity and cocaine reinforcement. Science. 2007;315(5816):1267-1270.
8. Barnes TR, Braude WM. Akathisia variants and tardive dyskinesia. Arch Gen Psychiatry. 1985;42(9):874-878.
9. Seemüller F, Schennach R, Mayr A, et al. Akathisia and suicidal ideation in first-episode schizophrenia. J Clin Psychopharmacol. 2012;32(5):694-698.
10. Leong GB, Silva JA. Neuroleptic-induced akathisia and violence: a review. J Forensic Sci. 2003;48(1):187-189.
11. Hirose S. The causes of underdiagnosing akathisia. Schizophr Bull. 2003;29(3):547-558.
12. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):S1-S46; quiz 47-48.
13. Citrome L. A review of the pharmacology, efficacy and tolerability of recently approved and upcoming oral antipsychotics: an evidence-based medicine approach. CNS Drugs. 2013;27(11):879-911.
14. Poyurovsky M. Acute antipsychotic-induced akathisia revisited. Br J Psychiatry. 2010;196(2):89-91.
15. Saltz BL, Robinson DG, Woerner MG. Recognizing and managing antipsychotic drug treatment side effects in the elderly. Prim Care Companion J Clin Psychiatry. 2004;6(suppl 2):14-19.
16. Lieberman JA, Stroup TS. The NIMH-CATIE Schizophrenia Study: what did we learn? Am J Psychiatry. 2011;168(8):770-775.
17. Zubenko GS, Cohen BM, Lipinski JF Jr, et al. Use of clonidine in treating neuroleptic-induced akathisia. Psychiatry Res. 1984;13(3):253-259.
18. Vinogradov S, Fisher M, Warm H, et al. The cognitive cost of anticholinergic burden: decreased response to cognitive training in schizophrenia. Am J Psychiatry. 2009;166(9):1055-1062.
19. Masui T, Kusumi I, Takahashi Y, et al. Efficacy of carbamazepine against neuroleptic-induced akathisia in treatment with perospirone: case series. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):343-346.
20. Lima AR, Soares-Weiser K, Bacaltchuk J, et al. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2002;(1):CD001950.
21. Lima AR, Bacalcthuk J, Barnes TR, et al. Central action beta-blockers versus placebo for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2004;(4):CD001946.
22. Bilal L, Ching C. Cabergoline-induced psychosis in a patient with undiagnosed depression. J Neuropsychiatry Clin Neurosci. 2012;24(4):E54.
23. Poyurovsky M, Pashinian A, Weizman A, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry.
2006;59(11):1071-1077.
24. Maidment I. Use of serotonin antagonists in the treatment of neuroleptic-induced akathisia. Psychiatric Bulletin. 2000;24(9):348-351.
25. Stryjer R, Rosenzcwaig S, Bar F, et al. Trazodone for the treatment of neuroleptic-induced akathisia: a placebo-controlled, double-blind, crossover study. Clin Neuropharmacol. 2010;33(5):219-222.
26. Dumon JP, Catteau J, Lanvin F, et al. Randomized, double-blind, crossover, placebo-controlled comparison of propranolol and betaxolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1992;149(5):647-650.
27. van Waarde A, Vaalburg W, Doze P, et al. PET imaging of beta-adrenoceptors in the human brain: a realistic goal or a mirage? Curr Pharm Des. 2004;10(13):1519-1536.
28. Kurzthaler I, Hummer M, Kohl C, et al. Propranolol treatment of olanzapine-induced akathisia. Am J Psychiatry. 1997;154(9):1316.
29. Adler LA, Peselow E, Rosenthal MA, et al. A controlled comparison of the effects of propranolol, benztropine, and placebo on akathisia: an interim analysis. Psychopharmacol Bull. 1993;29(2):283-286.
30. Dorevitch A, Durst R, Ginath Y. Propranolol in the treatment of akathisia caused by antipsychotic drugs. South Med J. 1991;84(12):1505-1506.
31. Lipinski JF Jr, Zubenko GS, Cohen BM, et al. Propranolol in the treatment of neuroleptic-induced akathisia. Am J Psychiatry. 1984;141(3):412-415.
32. Adler L, Angrist B, Peselow E, et al. A controlled assessment of propranolol in the treatment of neuroleptic-induced akathisia. Br J Psychiatry. 1986;149:42-45.
33. Kumar R, Sachdev PS. Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry. 2009;22(3):293-299.
34. Anttila SA, Leinonen EV. A review of the pharmacological and clinical profile of mirtazapine. CNS Drug Rev. 2001;7(3):249-264.
35. Rogóz Z, Wróbel A, Dlaboga D, et al. Effect of repeated treatment with mirtazapine on the central dopaminergic D2/D3 receptors. Pol J Pharmacol. 2002;54(4):381-389.
36. Miller CH, Fleischhacker WW, Ehrmann H, et al. Treatment of neuroleptic induced akathisia with the 5-HT2 antagonist ritanserin. Psychopharmacol Bull. 1990;26(3):373-376.
37. Weiss D, Aizenberg D, Hermesh H, et al. Cyproheptadine treatment in neuroleptic-induced akathisia. Br J Psychiatry. 1995;167(4):483-486.
38. Gross-Isseroff R, Magen A, Shiloh R, et al. The 5-HT1D receptor agonist zolmitriptan for neuroleptic-induced akathisia: an open label preliminary study. Int Clin Psychopharmacol. 2005;20(1):23-25.
39. Avital A, Gross-Isseroff R, Stryjer R, et al. Zolmitriptan compared to propranolol in the treatment of acute neuroleptic-induced akathisia: a comparative double-blind study. Eur Neuropsychopharmacol. 2009;19(7):476-482.
40. Rathbone J, Soares-Weiser K. Anticholinergics for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev. 2006;(4):CD003727.
41. Sandyk R. Successful treatment of neuroleptic-induced akathisia with baclofen and clonazepam. A case report. Eur Neurol. 1985;24(4):286-288.
42. Miller CH, Fleischhacker W. Managing antipsychotic-induced acute and chronic akathisia. Drug Saf. 2000;22(1):73-81.
43. Pfeffer G, Chouinard G, Margolese HC. Gabapentin in the treatment of antipsychotic-induced akathisia in schizophrenia. Int Clin Psychopharmacol. 2005;20(3):179-181.
44. Stahl SM. Role of α1 adrenergic antagonism in the mechanism of action of iloperidone: reducing extrapyramidal symptoms. CNS Spectr. 2013;18(6):285-258.
45. Hettema JM, Ross DE. A case of aripiprazole-related tardive akathisia and its treatment with ropinirole. J Clin Psychiatry. 2007;68(11):1814-1815.

Using the Internet in your practice. Part 4: Reputation management—how to gather kudos and combat negative online reviews
“It takes 20 years to build a reputation and 5 minutes to ruin it. If you think about that, you’ll do things differently.”
—Warren Buffet
CASE: Decline in new patients
A well-respected physician—one of the best in his field—notices that the number of new patients in his practice has fallen off drastically over the past year. Baffled, he hires a consultant, who discovers that the doctor’s online reputation has plummeted, thanks to four negative reviews and no positive ones.
What can the physician do to remedy the situation and restore his reputation?
The problem can be fixed, but it takes time—like major surgery. Rather than wait until negative reviews are posted, we recommend that you become proactive and take steps as soon as possible to secure your online reputation. That way, you won’t get caught by surprise when one or two unhappy patients try to smear your good name. In this article, we step you through a number of remedies and proactive strategies for boosting positive online reviews and combating negative ones.

The Internet: A one-stop source of information
The Internet has become everyone’s go-to source for pretty much any kind of data, including details on products, services, and people. Anyone can access all kinds of information simply by asking.
Today, people research medical conditions on the Web, often using Google. If you have done your search engine optimization, your Web site will come up in the first page of search results, making it possible for prospective patients to click through to your homepage. (For the scoop on search engine optimization, see Part 3 of this series, “Maximizing your online reach through SEO and pay-per-click,” which appeared in the September 2014 issue of OBG Management.)
If visitors like what they see at your site, they may make an appointment. But they are more likely to visit three or four other sites before making a decision. And in all likelihood, they will research each physician to find out what patients have to say about her or him. It’s no different than looking at the reviews of hotels or products you are considering.
You are an open book on the Internet. Only a few short years ago, your peers and patients knew your reputation primarily through word of mouth, which traveled at the speed of molasses. For the most part, that information was favorable. Today your exposure is much greater, and negative comments about you can be viewed by thousands of potential patients. The speed of information has increased, as well. What is posted on the Internet can become readily available to hundreds, thousands, and even millions of Web users in a nanosecond.
The Internet provides a forum for people to say whatever they want about their experiences, both positive and negative. Regrettably, the positive experiences do not find their way online nearly as often as the negative ones!
The bottom line? In today’s Internet-savvy world, you need to pay regular attention to your online reputation. You need to take steps to ensure that your name and practice look their best and to negate any complaints that may appear.
What patients share about their experience with you
Many online review sites provide an opportunity for your patients to describe their experience with you and your practice. To name a few: RateMDs.com, Vitals.com, ZocDoc.com, healthgrades.com, UcompareHealth.com, Citysearch.com, yelp.com, and, of course, Google Plus reviews.
And when patients post comments on the Internet, you likely will be rated on:
- the patient’s wait time
- how your staff treated the patient
- the diagnosis
- your attitude
- the level of trust in your decisions
- treatment and outcome.
The online surfer searching for a reputable physician is likely to believe whatever he or she finds on the leading review sites.
The good news: Most physicians have a very favorable rating, averaging 9.3 out of 10 on a scale of 1 to 10. In fact, 70% of doctors have perfect scores!1
The bad news: Someone who is unhappy with her treatment or outcome will go out of her way to find every online review site possible and proclaim your faults to the cyber-world, using the Internet as a forum, whether her facts are straight or not. Patients who are pleased and satisfied rarely bother to place a positive review.
How you can control your online reputation
It is incumbent upon you to keep an eye on your online reputation at all times. Here are some tips for taking charge:
- If someone posts a negative review, respond to them directly in the review site. Doing so does not violate privacy laws as long as you do not mention the patient’s name or give other identifying details. Explain your side of the story without confirming or denying that the reviewer is or was a patient. Do not mention the specifics of any patient’s condition.
- If you feel that a negative review is completely unjustified, file a dispute with the review site. Many review sites will remove the unfavorable content if you can convince them that the patient is merely ranting.
- To protect your reputation over the long term, use your name or practice name to set up an alert with Google Alerts by visiting the site Google.com/alerts.
- Do a Google search of your name and the name of your practice at least once a month and check out all the review sites that come up. Read the comments!
Develop a proactive system
You have a lot of control when it comes to protecting your online reputation, provided you are willing to take the time to set up a system to regularly request feedback or testimonials from your patients.
Regrettably, this is where most medical practices fall short, by failing to establish a system to solicit positive reviews.
The process need not be complicated. Such a system can be set in motion by scheduling a quick meeting with your staff to announce your plans to solicit testimonials from patients. Often there will be a flurry of activity for a couple of weeks before the task is forgotten. To keep your system from falling through the cracks, make a checklist and decide who on your staff is responsible for each step in the process. Go over the results in your staff meetings on a regular basis—ie, at least monthly.
You want to solicit positive reviews for use in two places:
- your Web site
- the review sites we mentioned earlier.
Posting testimonials on your Web site
Your site is the place prospective patients visit when they are looking for information about you and your services. Here are a few tips on gathering and posting testimonials:
- The best time to solicit feedback from the patient is after the follow-up appointment, when her needs have been met and she has had at least two experiences with your practice. If she is happy with her outcome, she is likely to be receptive to the idea of providing a testimonial while the details are fresh in her mind.
- Post testimonials on your homepage and every other page at your site. They should be visible when each page loads without the need to scroll down. A testimonial is worthless if it can’t be easily seen.
- Post testimonials in italics, with quotation marks around the comments to distinguish them from other elements on the page.
- Give each testimonial a headline in bold italics. Use key words likely to resonate with the reader. For example, if the patient reports: “I had a surgical procedure and it was a game changer. You turned my life around! Thank you!” the headline might be: “You turned my life around.”
- Create a Web page just for testimonials and order the comments and headlines so that they will appeal to a diversity of prospective patients. The visitor may not read every testimonial, but she will at least read and scroll through the headlines.
Gathering feedback: Your options
- One option for automating the gathering of feedback is to include a patient feedback survey on your Web site. It’s a convenient way to ask for comments. When the patient is in the office, you or your staff can simply ask her to visit the survey page on your site and answer the questions. The problem with this approach is that many patients will agree to complete the survey but few will actually follow through.
- A far more effective way to get patients to complete a survey while they are still in your office is to have the receptionist hand the patient an iPad after her appointment and ask her to take a couple of minutes to complete the survey. You can then transcribe her comments and post them on your site.
- Asking patients to post positive comments on review sites such as healthgrades.com is another option—but, again, patients are unlikely to follow through unless you make it as easy and fast as possible. The best way to do this is to provide your patient with a blueprint for how to proceed. We offer a “patient feedback” form that contains four or five questions (FIGURE). The answers to these questions will provide a great testimonial for the doctor and the practice. Providing your patients with the right questions to elicit an emotional response will help them describe their experiences more fully. If you let the patient create a testimonial on her own, you’ll probably just receive comments such as, “I’m very happy with my results” or “She is a great doctor.”
- Also provide patients with a step-by-step process for entering their feedback on the desired review sites. This can be a daunting task for your patient, so your instructions should be clear and simple. Better yet, have someone on your staff sit with the patient at a computer or iPad to help her through the process.
- Another way to control your online reputation is to capture positive comments at the point of service. In our practice, we have a testimonial poster in every exam room as well as the reception area. It contains a quick response (QR) code that can be scanned to allow the patient to submit a testimonial about her experience with the practice. With this system, we are able to collect three to five positive reviews every day.
FIGURE: Patient follow-up satisfaction survey
| It is our intention to provide our patients with the absolute best medical care available to produce optimal results. Your feedback about your procedure and patient care is an important measure of our performance. Please take the time to let us know how you feel about your results:
Your name: _______________________________ Date: ________ Thank you for telling us about the results of your procedure. How you feel about your experience helps us better understand the physical and emotional needs of our patients. We would like to share your experience with others who might be struggling with the same issues. By signing this form, you agree to let us share this information on our Web site and informational material to help other patients understand the benefits of having these types of procedures performed. |
CASE: Resolved
The physician institutes a process in his practice to gather testimonials and positive feedback, and his staff takes time to help willing patients post their reviews online. He also disputes the negative comments that have already been posted online, offering an objective response to the complaints and asking the Web sites to take down the reviews that are merely ranting. In addition, he posts selected testimonials on the homepage of his Web site and adds a page that is just for testimonials.
Within a few weeks, the number of new patients scheduling appointments with him begins to increase until he once again enjoys a bustling practice.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Reference
- Schwartz SK. Online patient feedback: what to do. Physicianspractice.com. http://www.physicianspractice.com/health-it/online-patient-feedback-what-do. Published December 27, 2012. Accessed November 15, 2014.
Ron Romano and Neil H. Baum, MD

Ron Romano is President of www.YourInternetDoctor.com and CEO of Instant Marketing Systems. He co-authored The Internet Survival Guide for Doctors (2014, Instant Marketing Systems) and No B.S. Direct Marketing (2006, Entrepreneur Press) and contributed to the Walking with the Wise series (2004, Mentors Publishing). He is an Internet marketing consultant, speaker, and creator of “The Implementation Blueprint System.”

Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of several books, including Social Media for the Healthcare Professional (2012, Greenbranch), and Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett). Dr. Baum serves on the OBG Management Board of Contributing Editors.
Mr. Romano reports that he is CEO of Instant Marketing Systems, which provides consulting advice, marketing plans, and Internet marketing services for businesses and medical practices. Dr. Baum reports no financial relationships relevant to this article.
Ron Romano and Neil H. Baum, MD
Ron Romano is President of www.YourInternetDoctor.com and CEO of Instant Marketing Systems. He co-authored The Internet Survival Guide for Doctors (2014, Instant Marketing Systems) and No B.S. Direct Marketing (2006, Entrepreneur Press) and contributed to the Walking with the Wise series (2004, Mentors Publishing). He is an Internet marketing consultant, speaker, and creator of “The Implementation Blueprint System.”
Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of several books, including Social Media for the Healthcare Professional (2012, Greenbranch), and Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett). Dr. Baum serves on the OBG Management Board of Contributing Editors.
Mr. Romano reports that he is CEO of Instant Marketing Systems, which provides consulting advice, marketing plans, and Internet marketing services for businesses and medical practices. Dr. Baum reports no financial relationships relevant to this article.
Ron Romano and Neil H. Baum, MD
Ron Romano is President of www.YourInternetDoctor.com and CEO of Instant Marketing Systems. He co-authored The Internet Survival Guide for Doctors (2014, Instant Marketing Systems) and No B.S. Direct Marketing (2006, Entrepreneur Press) and contributed to the Walking with the Wise series (2004, Mentors Publishing). He is an Internet marketing consultant, speaker, and creator of “The Implementation Blueprint System.”
Neil H. Baum, MD, practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University School of Medicine, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of several books, including Social Media for the Healthcare Professional (2012, Greenbranch), and Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett). Dr. Baum serves on the OBG Management Board of Contributing Editors.
Mr. Romano reports that he is CEO of Instant Marketing Systems, which provides consulting advice, marketing plans, and Internet marketing services for businesses and medical practices. Dr. Baum reports no financial relationships relevant to this article.
“It takes 20 years to build a reputation and 5 minutes to ruin it. If you think about that, you’ll do things differently.”
—Warren Buffet
CASE: Decline in new patients
A well-respected physician—one of the best in his field—notices that the number of new patients in his practice has fallen off drastically over the past year. Baffled, he hires a consultant, who discovers that the doctor’s online reputation has plummeted, thanks to four negative reviews and no positive ones.
What can the physician do to remedy the situation and restore his reputation?
The problem can be fixed, but it takes time—like major surgery. Rather than wait until negative reviews are posted, we recommend that you become proactive and take steps as soon as possible to secure your online reputation. That way, you won’t get caught by surprise when one or two unhappy patients try to smear your good name. In this article, we step you through a number of remedies and proactive strategies for boosting positive online reviews and combating negative ones.
The Internet: A one-stop source of information
The Internet has become everyone’s go-to source for pretty much any kind of data, including details on products, services, and people. Anyone can access all kinds of information simply by asking.
Today, people research medical conditions on the Web, often using Google. If you have done your search engine optimization, your Web site will come up in the first page of search results, making it possible for prospective patients to click through to your homepage. (For the scoop on search engine optimization, see Part 3 of this series, “Maximizing your online reach through SEO and pay-per-click,” which appeared in the September 2014 issue of OBG Management.)
If visitors like what they see at your site, they may make an appointment. But they are more likely to visit three or four other sites before making a decision. And in all likelihood, they will research each physician to find out what patients have to say about her or him. It’s no different than looking at the reviews of hotels or products you are considering.
You are an open book on the Internet. Only a few short years ago, your peers and patients knew your reputation primarily through word of mouth, which traveled at the speed of molasses. For the most part, that information was favorable. Today your exposure is much greater, and negative comments about you can be viewed by thousands of potential patients. The speed of information has increased, as well. What is posted on the Internet can become readily available to hundreds, thousands, and even millions of Web users in a nanosecond.
The Internet provides a forum for people to say whatever they want about their experiences, both positive and negative. Regrettably, the positive experiences do not find their way online nearly as often as the negative ones!
The bottom line? In today’s Internet-savvy world, you need to pay regular attention to your online reputation. You need to take steps to ensure that your name and practice look their best and to negate any complaints that may appear.
What patients share about their experience with you
Many online review sites provide an opportunity for your patients to describe their experience with you and your practice. To name a few: RateMDs.com, Vitals.com, ZocDoc.com, healthgrades.com, UcompareHealth.com, Citysearch.com, yelp.com, and, of course, Google Plus reviews.
And when patients post comments on the Internet, you likely will be rated on:
- the patient’s wait time
- how your staff treated the patient
- the diagnosis
- your attitude
- the level of trust in your decisions
- treatment and outcome.
The online surfer searching for a reputable physician is likely to believe whatever he or she finds on the leading review sites.
The good news: Most physicians have a very favorable rating, averaging 9.3 out of 10 on a scale of 1 to 10. In fact, 70% of doctors have perfect scores!1
The bad news: Someone who is unhappy with her treatment or outcome will go out of her way to find every online review site possible and proclaim your faults to the cyber-world, using the Internet as a forum, whether her facts are straight or not. Patients who are pleased and satisfied rarely bother to place a positive review.
How you can control your online reputation
It is incumbent upon you to keep an eye on your online reputation at all times. Here are some tips for taking charge:
- If someone posts a negative review, respond to them directly in the review site. Doing so does not violate privacy laws as long as you do not mention the patient’s name or give other identifying details. Explain your side of the story without confirming or denying that the reviewer is or was a patient. Do not mention the specifics of any patient’s condition.
- If you feel that a negative review is completely unjustified, file a dispute with the review site. Many review sites will remove the unfavorable content if you can convince them that the patient is merely ranting.
- To protect your reputation over the long term, use your name or practice name to set up an alert with Google Alerts by visiting the site Google.com/alerts.
- Do a Google search of your name and the name of your practice at least once a month and check out all the review sites that come up. Read the comments!
Develop a proactive system
You have a lot of control when it comes to protecting your online reputation, provided you are willing to take the time to set up a system to regularly request feedback or testimonials from your patients.
Regrettably, this is where most medical practices fall short, by failing to establish a system to solicit positive reviews.
The process need not be complicated. Such a system can be set in motion by scheduling a quick meeting with your staff to announce your plans to solicit testimonials from patients. Often there will be a flurry of activity for a couple of weeks before the task is forgotten. To keep your system from falling through the cracks, make a checklist and decide who on your staff is responsible for each step in the process. Go over the results in your staff meetings on a regular basis—ie, at least monthly.
You want to solicit positive reviews for use in two places:
- your Web site
- the review sites we mentioned earlier.
Posting testimonials on your Web site
Your site is the place prospective patients visit when they are looking for information about you and your services. Here are a few tips on gathering and posting testimonials:
- The best time to solicit feedback from the patient is after the follow-up appointment, when her needs have been met and she has had at least two experiences with your practice. If she is happy with her outcome, she is likely to be receptive to the idea of providing a testimonial while the details are fresh in her mind.
- Post testimonials on your homepage and every other page at your site. They should be visible when each page loads without the need to scroll down. A testimonial is worthless if it can’t be easily seen.
- Post testimonials in italics, with quotation marks around the comments to distinguish them from other elements on the page.
- Give each testimonial a headline in bold italics. Use key words likely to resonate with the reader. For example, if the patient reports: “I had a surgical procedure and it was a game changer. You turned my life around! Thank you!” the headline might be: “You turned my life around.”
- Create a Web page just for testimonials and order the comments and headlines so that they will appeal to a diversity of prospective patients. The visitor may not read every testimonial, but she will at least read and scroll through the headlines.
Gathering feedback: Your options
- One option for automating the gathering of feedback is to include a patient feedback survey on your Web site. It’s a convenient way to ask for comments. When the patient is in the office, you or your staff can simply ask her to visit the survey page on your site and answer the questions. The problem with this approach is that many patients will agree to complete the survey but few will actually follow through.
- A far more effective way to get patients to complete a survey while they are still in your office is to have the receptionist hand the patient an iPad after her appointment and ask her to take a couple of minutes to complete the survey. You can then transcribe her comments and post them on your site.
- Asking patients to post positive comments on review sites such as healthgrades.com is another option—but, again, patients are unlikely to follow through unless you make it as easy and fast as possible. The best way to do this is to provide your patient with a blueprint for how to proceed. We offer a “patient feedback” form that contains four or five questions (FIGURE). The answers to these questions will provide a great testimonial for the doctor and the practice. Providing your patients with the right questions to elicit an emotional response will help them describe their experiences more fully. If you let the patient create a testimonial on her own, you’ll probably just receive comments such as, “I’m very happy with my results” or “She is a great doctor.”
- Also provide patients with a step-by-step process for entering their feedback on the desired review sites. This can be a daunting task for your patient, so your instructions should be clear and simple. Better yet, have someone on your staff sit with the patient at a computer or iPad to help her through the process.
- Another way to control your online reputation is to capture positive comments at the point of service. In our practice, we have a testimonial poster in every exam room as well as the reception area. It contains a quick response (QR) code that can be scanned to allow the patient to submit a testimonial about her experience with the practice. With this system, we are able to collect three to five positive reviews every day.
FIGURE: Patient follow-up satisfaction survey
| It is our intention to provide our patients with the absolute best medical care available to produce optimal results. Your feedback about your procedure and patient care is an important measure of our performance. Please take the time to let us know how you feel about your results:
Your name: _______________________________ Date: ________ Thank you for telling us about the results of your procedure. How you feel about your experience helps us better understand the physical and emotional needs of our patients. We would like to share your experience with others who might be struggling with the same issues. By signing this form, you agree to let us share this information on our Web site and informational material to help other patients understand the benefits of having these types of procedures performed. |
CASE: Resolved
The physician institutes a process in his practice to gather testimonials and positive feedback, and his staff takes time to help willing patients post their reviews online. He also disputes the negative comments that have already been posted online, offering an objective response to the complaints and asking the Web sites to take down the reviews that are merely ranting. In addition, he posts selected testimonials on the homepage of his Web site and adds a page that is just for testimonials.
Within a few weeks, the number of new patients scheduling appointments with him begins to increase until he once again enjoys a bustling practice.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
“It takes 20 years to build a reputation and 5 minutes to ruin it. If you think about that, you’ll do things differently.”
—Warren Buffet
CASE: Decline in new patients
A well-respected physician—one of the best in his field—notices that the number of new patients in his practice has fallen off drastically over the past year. Baffled, he hires a consultant, who discovers that the doctor’s online reputation has plummeted, thanks to four negative reviews and no positive ones.
What can the physician do to remedy the situation and restore his reputation?
The problem can be fixed, but it takes time—like major surgery. Rather than wait until negative reviews are posted, we recommend that you become proactive and take steps as soon as possible to secure your online reputation. That way, you won’t get caught by surprise when one or two unhappy patients try to smear your good name. In this article, we step you through a number of remedies and proactive strategies for boosting positive online reviews and combating negative ones.
The Internet: A one-stop source of information
The Internet has become everyone’s go-to source for pretty much any kind of data, including details on products, services, and people. Anyone can access all kinds of information simply by asking.
Today, people research medical conditions on the Web, often using Google. If you have done your search engine optimization, your Web site will come up in the first page of search results, making it possible for prospective patients to click through to your homepage. (For the scoop on search engine optimization, see Part 3 of this series, “Maximizing your online reach through SEO and pay-per-click,” which appeared in the September 2014 issue of OBG Management.)
If visitors like what they see at your site, they may make an appointment. But they are more likely to visit three or four other sites before making a decision. And in all likelihood, they will research each physician to find out what patients have to say about her or him. It’s no different than looking at the reviews of hotels or products you are considering.
You are an open book on the Internet. Only a few short years ago, your peers and patients knew your reputation primarily through word of mouth, which traveled at the speed of molasses. For the most part, that information was favorable. Today your exposure is much greater, and negative comments about you can be viewed by thousands of potential patients. The speed of information has increased, as well. What is posted on the Internet can become readily available to hundreds, thousands, and even millions of Web users in a nanosecond.
The Internet provides a forum for people to say whatever they want about their experiences, both positive and negative. Regrettably, the positive experiences do not find their way online nearly as often as the negative ones!
The bottom line? In today’s Internet-savvy world, you need to pay regular attention to your online reputation. You need to take steps to ensure that your name and practice look their best and to negate any complaints that may appear.
What patients share about their experience with you
Many online review sites provide an opportunity for your patients to describe their experience with you and your practice. To name a few: RateMDs.com, Vitals.com, ZocDoc.com, healthgrades.com, UcompareHealth.com, Citysearch.com, yelp.com, and, of course, Google Plus reviews.
And when patients post comments on the Internet, you likely will be rated on:
- the patient’s wait time
- how your staff treated the patient
- the diagnosis
- your attitude
- the level of trust in your decisions
- treatment and outcome.
The online surfer searching for a reputable physician is likely to believe whatever he or she finds on the leading review sites.
The good news: Most physicians have a very favorable rating, averaging 9.3 out of 10 on a scale of 1 to 10. In fact, 70% of doctors have perfect scores!1
The bad news: Someone who is unhappy with her treatment or outcome will go out of her way to find every online review site possible and proclaim your faults to the cyber-world, using the Internet as a forum, whether her facts are straight or not. Patients who are pleased and satisfied rarely bother to place a positive review.
How you can control your online reputation
It is incumbent upon you to keep an eye on your online reputation at all times. Here are some tips for taking charge:
- If someone posts a negative review, respond to them directly in the review site. Doing so does not violate privacy laws as long as you do not mention the patient’s name or give other identifying details. Explain your side of the story without confirming or denying that the reviewer is or was a patient. Do not mention the specifics of any patient’s condition.
- If you feel that a negative review is completely unjustified, file a dispute with the review site. Many review sites will remove the unfavorable content if you can convince them that the patient is merely ranting.
- To protect your reputation over the long term, use your name or practice name to set up an alert with Google Alerts by visiting the site Google.com/alerts.
- Do a Google search of your name and the name of your practice at least once a month and check out all the review sites that come up. Read the comments!
Develop a proactive system
You have a lot of control when it comes to protecting your online reputation, provided you are willing to take the time to set up a system to regularly request feedback or testimonials from your patients.
Regrettably, this is where most medical practices fall short, by failing to establish a system to solicit positive reviews.
The process need not be complicated. Such a system can be set in motion by scheduling a quick meeting with your staff to announce your plans to solicit testimonials from patients. Often there will be a flurry of activity for a couple of weeks before the task is forgotten. To keep your system from falling through the cracks, make a checklist and decide who on your staff is responsible for each step in the process. Go over the results in your staff meetings on a regular basis—ie, at least monthly.
You want to solicit positive reviews for use in two places:
- your Web site
- the review sites we mentioned earlier.
Posting testimonials on your Web site
Your site is the place prospective patients visit when they are looking for information about you and your services. Here are a few tips on gathering and posting testimonials:
- The best time to solicit feedback from the patient is after the follow-up appointment, when her needs have been met and she has had at least two experiences with your practice. If she is happy with her outcome, she is likely to be receptive to the idea of providing a testimonial while the details are fresh in her mind.
- Post testimonials on your homepage and every other page at your site. They should be visible when each page loads without the need to scroll down. A testimonial is worthless if it can’t be easily seen.
- Post testimonials in italics, with quotation marks around the comments to distinguish them from other elements on the page.
- Give each testimonial a headline in bold italics. Use key words likely to resonate with the reader. For example, if the patient reports: “I had a surgical procedure and it was a game changer. You turned my life around! Thank you!” the headline might be: “You turned my life around.”
- Create a Web page just for testimonials and order the comments and headlines so that they will appeal to a diversity of prospective patients. The visitor may not read every testimonial, but she will at least read and scroll through the headlines.
Gathering feedback: Your options
- One option for automating the gathering of feedback is to include a patient feedback survey on your Web site. It’s a convenient way to ask for comments. When the patient is in the office, you or your staff can simply ask her to visit the survey page on your site and answer the questions. The problem with this approach is that many patients will agree to complete the survey but few will actually follow through.
- A far more effective way to get patients to complete a survey while they are still in your office is to have the receptionist hand the patient an iPad after her appointment and ask her to take a couple of minutes to complete the survey. You can then transcribe her comments and post them on your site.
- Asking patients to post positive comments on review sites such as healthgrades.com is another option—but, again, patients are unlikely to follow through unless you make it as easy and fast as possible. The best way to do this is to provide your patient with a blueprint for how to proceed. We offer a “patient feedback” form that contains four or five questions (FIGURE). The answers to these questions will provide a great testimonial for the doctor and the practice. Providing your patients with the right questions to elicit an emotional response will help them describe their experiences more fully. If you let the patient create a testimonial on her own, you’ll probably just receive comments such as, “I’m very happy with my results” or “She is a great doctor.”
- Also provide patients with a step-by-step process for entering their feedback on the desired review sites. This can be a daunting task for your patient, so your instructions should be clear and simple. Better yet, have someone on your staff sit with the patient at a computer or iPad to help her through the process.
- Another way to control your online reputation is to capture positive comments at the point of service. In our practice, we have a testimonial poster in every exam room as well as the reception area. It contains a quick response (QR) code that can be scanned to allow the patient to submit a testimonial about her experience with the practice. With this system, we are able to collect three to five positive reviews every day.
FIGURE: Patient follow-up satisfaction survey
| It is our intention to provide our patients with the absolute best medical care available to produce optimal results. Your feedback about your procedure and patient care is an important measure of our performance. Please take the time to let us know how you feel about your results:
Your name: _______________________________ Date: ________ Thank you for telling us about the results of your procedure. How you feel about your experience helps us better understand the physical and emotional needs of our patients. We would like to share your experience with others who might be struggling with the same issues. By signing this form, you agree to let us share this information on our Web site and informational material to help other patients understand the benefits of having these types of procedures performed. |
CASE: Resolved
The physician institutes a process in his practice to gather testimonials and positive feedback, and his staff takes time to help willing patients post their reviews online. He also disputes the negative comments that have already been posted online, offering an objective response to the complaints and asking the Web sites to take down the reviews that are merely ranting. In addition, he posts selected testimonials on the homepage of his Web site and adds a page that is just for testimonials.
Within a few weeks, the number of new patients scheduling appointments with him begins to increase until he once again enjoys a bustling practice.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Reference
- Schwartz SK. Online patient feedback: what to do. Physicianspractice.com. http://www.physicianspractice.com/health-it/online-patient-feedback-what-do. Published December 27, 2012. Accessed November 15, 2014.
Reference
- Schwartz SK. Online patient feedback: what to do. Physicianspractice.com. http://www.physicianspractice.com/health-it/online-patient-feedback-what-do. Published December 27, 2012. Accessed November 15, 2014.

Management of Bleeding Complications in Patients with Cancer
Patients with cancer can have many hematologic complications. One of the most serious is bleeding, which can range in severity from laboratory abnormalities to life-threatening hemorrhage. The bleeding can be due to complications of the cancer, its therapy, or treatment for complications of cancer such as thrombosis. This manual discusses an approach to the cancer patient with bleeding, with a specific focus on issues such as coagulation defects, thrombocytopenia, and platelet dysfunction. Bleeding complications of specific cancers and their treatment will be discussed as well.
To read the full article in PDF:
Patients with cancer can have many hematologic complications. One of the most serious is bleeding, which can range in severity from laboratory abnormalities to life-threatening hemorrhage. The bleeding can be due to complications of the cancer, its therapy, or treatment for complications of cancer such as thrombosis. This manual discusses an approach to the cancer patient with bleeding, with a specific focus on issues such as coagulation defects, thrombocytopenia, and platelet dysfunction. Bleeding complications of specific cancers and their treatment will be discussed as well.
To read the full article in PDF:
Patients with cancer can have many hematologic complications. One of the most serious is bleeding, which can range in severity from laboratory abnormalities to life-threatening hemorrhage. The bleeding can be due to complications of the cancer, its therapy, or treatment for complications of cancer such as thrombosis. This manual discusses an approach to the cancer patient with bleeding, with a specific focus on issues such as coagulation defects, thrombocytopenia, and platelet dysfunction. Bleeding complications of specific cancers and their treatment will be discussed as well.
To read the full article in PDF:
Metastatic Prostate Cancer: A Case Study
Prostate cancer remains the second leading cause of death in men in the United States as of 2012. It is estimated that prostate cancer affected more than 241,000 new men in 2012, with 15% of these patients presenting with advanced disease. As one would expect, compared to localized prostate cancer, metastatic disease remains the more challenging type to treat. In 1941 Huggins and Hodges demonstrated the dependence of prostatic tissues on androgens and from this work hormonal therapy was developed as the primary treatment for metastatic prostate cancer. Since then, significant progress has been made in the treatment of metastatic prostate cancer, including advances in androgen deprivation therapy and in the treatment of castrationresistant prostate cancer (CRPC), with many advances yet to come. CPRC has been an exciting topic for recent research and advancement, as our understanding of how prostate cancer utilizes very low levels of androgen has evolved considerably.
To read the full article in PDF:
Prostate cancer remains the second leading cause of death in men in the United States as of 2012. It is estimated that prostate cancer affected more than 241,000 new men in 2012, with 15% of these patients presenting with advanced disease. As one would expect, compared to localized prostate cancer, metastatic disease remains the more challenging type to treat. In 1941 Huggins and Hodges demonstrated the dependence of prostatic tissues on androgens and from this work hormonal therapy was developed as the primary treatment for metastatic prostate cancer. Since then, significant progress has been made in the treatment of metastatic prostate cancer, including advances in androgen deprivation therapy and in the treatment of castrationresistant prostate cancer (CRPC), with many advances yet to come. CPRC has been an exciting topic for recent research and advancement, as our understanding of how prostate cancer utilizes very low levels of androgen has evolved considerably.
To read the full article in PDF:
Prostate cancer remains the second leading cause of death in men in the United States as of 2012. It is estimated that prostate cancer affected more than 241,000 new men in 2012, with 15% of these patients presenting with advanced disease. As one would expect, compared to localized prostate cancer, metastatic disease remains the more challenging type to treat. In 1941 Huggins and Hodges demonstrated the dependence of prostatic tissues on androgens and from this work hormonal therapy was developed as the primary treatment for metastatic prostate cancer. Since then, significant progress has been made in the treatment of metastatic prostate cancer, including advances in androgen deprivation therapy and in the treatment of castrationresistant prostate cancer (CRPC), with many advances yet to come. CPRC has been an exciting topic for recent research and advancement, as our understanding of how prostate cancer utilizes very low levels of androgen has evolved considerably.
To read the full article in PDF:
Syncope: Etiology and diagnostic approach
Syncope is a transient loss of consciousness and postural tone with spontaneous, complete recovery. There are three major types: neurally mediated, orthostatic, and cardiac (Table 1).
NEURALLY MEDIATED SYNCOPE
Neurally mediated (reflex) syncope is the most common type, accounting for two-thirds of cases.1–3 It results from autonomic reflexes that respond inappropriately, leading to vasodilation and bradycardia.
See related patient-education handout

Neurally mediated syncope is usually preceded by premonitory symptoms such as lightheadedness, diaphoresis, nausea, malaise, abdominal discomfort, and tunnel vision. However, this may not be the case in one-third of patients, especially in elderly patients, who may not recognize or remember the warning symptoms. Palpitations are frequently reported with neurally mediated syncope and do not necessarily imply that the syncope is due to an arrhythmia.4,5 Neurally mediated syncope does not usually occur in the supine position4,5 but can occur in the seated position.6
Subtypes of neurally mediated syncope are as follows:
Vasovagal syncope
Vasovagal syncope is usually triggered by sudden emotional stress, prolonged sitting or standing, dehydration, or a warm environment, but it can also occur without a trigger. It is the most common type of syncope in young patients (more so in females than in males), but contrary to a common misconception, it can also occur in the elderly.7 Usually, it is not only preceded by but also followed by nausea, malaise, fatigue, and diaphoresis4,5,8; full recovery may be slow. If the syncope lasts longer than 30 to 60 seconds, clonic movements and loss of bladder control are common.9
Mechanism. Vasovagal syncope is initiated by anything that leads to strong myocardial contractions in an "empty" heart. Emotional stress, reduced venous return (from dehydration or prolonged standing), or vasodilation (caused by a hot environment) stimulates the sympathetic nervous system and reduces the left ventricular cavity size, which leads to strong hyperdynamic contractions in a relatively empty heart. This hyperdynamic cavity obliteration activates myocardial mechanoreceptors, initiating a paradoxical vagal reflex with vasodilation and relative bradycardia.10 Vasodilation is usually the predominant mechanism (vasodepressor response), particularly in older patients, but severe bradycardia is also possible (cardioinhibitory response), particularly in younger patients.7 Diuretic and vasodilator therapies increase the predisposition to vasovagal syncope, particularly in the elderly.
On tilt-table testing, vasovagal syncope is characterized by hypotension and relative bradycardia, sometimes severe (see Note on Tilt-Table Testing).10–12
Situational syncope
Situational syncope is caused by a reflex triggered in specific circumstances such as micturition, defecation, coughing, weight-lifting, laughing, or deglutition. The reflex may be initiated by a receptor on the visceral wall (eg, the bladder wall) or by straining that reduces venous return.
Carotid sinus hypersensitivity

Carotid sinus hypersensitivity is an abnormal response to carotid massage, predominantly occurring in patients over the age of 50. In spontaneous carotid sinus syndrome, syncope clearly occurs in a situation that stimulates the carotid sinus, such as head rotation, head extension, shaving, or wearing a tight collar. It is a rare cause of syncope, responsible for about 1% of cases. Conversely, induced carotid sinus syndrome is much more common and represents carotid sinus hypersensitivity in a patient with unexplained syncope and without obvious triggers; the abnormal response is mainly induced during carotid massage rather than spontaneously. In the latter case, carotid sinus hypersensitivity is a marker of a diseased sinus node or atrioventricular node that cannot withstand any inhibition. This diseased node is the true cause of syncope rather than carotid sinus hypersensitivity per se, and carotid massage is a "stress test" that unveils conduction disease.
Thus, carotid massage is indicated in cases of unexplained syncope regardless of circumstantial triggers. This test consists of applying firm pressure over each carotid bifurcation (just below the angle of the jaw) consecutively for 10 seconds. It is performed at the bedside, and may be performed with the patient in both supine and erect positions during tilt-table testing; erect positioning of the patient increases the sensitivity of this test.
An abnormal response to carotid sinus massage is defined as any of the following13–15:
- Vasodepressor response: the systolic blood pressure decreases by at least 50 mm Hg
- Cardioinhibitory response: sinus or atrioventricular block causes the heartbeat to pause for 3 or more seconds
- Mixed vasodepressor and cardioinhibitory response.
Overall, a cardioinhibitory component is present in about two-thirds of cases of carotid sinus hypersensitivity.
Carotid sinus hypersensitivity is found in 25% to 50% of patients over age 50 who have had unexplained syncope or a fall, and it is seen almost equally in men and women.13
One study correlated carotid sinus hypersensitivity with the later occurrence of asystolic syncope during prolonged internal loop monitoring; subsequent pacemaker therapy reduced the burden of syncope.14 Another study, in patients over 50 years old with unexplained falls, found that 16% had cardioinhibitory carotid sinus hypersensitivity. Pacemaker placement reduced falls and syncope by 70% compared with no pacemaker therapy in these patients.15
On the other hand, carotid sinus hypersensitivity can be found in 39% of elderly patients who do not have a history of fainting or falling, so it is important to rule out other causes of syncope before attributing it to carotid sinus hypersensitivity.
Postexertional syncope
While syncope on exertion raises the worrisome possibility of a cardiac cause, postexertional syncope is usually a form of vasovagal syncope. When exercise ceases, venous blood stops getting pumped back to the heart by peripheral muscular contraction. Yet the heart is still exposed to the catecholamine surge induced by exercising, and it hypercontracts on an empty cavity. This triggers a vagal reflex.
Postexertional syncope may also be seen in hypertrophic obstructive cardiomyopathy or aortic stenosis, in which the small left ventricular cavity is less likely to tolerate the reduced preload after exercise and is more likely to obliterate.
ORTHOSTATIC HYPOTENSION
Orthostatic hypotension accounts for about 10% of cases of syncope.1–3
Normally, after the first few minutes of standing, about 25% to 30% of the blood pools in the veins of the pelvis and the lower extremities, strikingly reducing venous return and stroke volume. Upon more prolonged standing, more blood leaves the vascular space and collects in the extravascular space, further reducing venous return. This normally leads to a reflex increase in sympathetic tone, peripheral and splanchnic vasoconstriction, and an increase in heart rate of 10 to 15 beats per minute. Overall, cardiac output is reduced and vascular resistance is increased while blood pressure is maintained, blood pressure being equal to cardiac output times vascular resistance.
Orthostatic hypotension is characterized by autonomic failure, with a lack of compensatory increase in vascular resistance or heart rate upon orthostasis, or by significant hypovolemia that cannot be overcome by sympathetic mechanisms. It is defined as a drop in systolic blood pressure of 20 mm Hg or more or a drop in diastolic pressure of 10 mm Hg or more after 30 seconds to 5 minutes of upright posture. Blood pressure is checked immediately upon standing and at 3 and 5 minutes. This may be done at the bedside or during tilt-table testing.2,4
Some patients have an immediate drop in blood pressure of more than 40 mm Hg upon standing, with a quick return to normal within 30 seconds. This "initial orthostatic hypotension" may be common in elderly patients taking antihypertensive drugs and may elude detection during standard blood pressure measurement.2 Other patients with milder orthostatic hypotension may develop a more delayed hypotension 10 to 15 minutes later, as more blood pools in the periphery.16
Along with the drop in blood pressure, a failure of the heart rate to increase identifies autonomic dysfunction. On the other hand, an increase in the heart rate of more than 20 to 30 beats per minute may signify a hypovolemic state even if blood pressure is maintained, the lack of blood pressure drop being related to the excessive heart rate increase.
Orthostatic hypotension is the most common cause of syncope in the elderly and may be due to autonomic dysfunction (related to age, diabetes, uremia, or Parkinson disease), volume depletion, or drugs that block autonomic effects or cause hypovolemia, such as vasodilators, beta-blockers, diuretics, neuropsychiatric medications, and alcohol.
Since digestion leads to peripheral vasodilation and splanchnic blood pooling, syncope that occurs within 1 hour after eating has a mechanism similar to that of orthostatic syncope.
Supine hypertension with orthostatic hypotension. Some patients with severe autonomic dysfunction and the inability to regulate vascular tone have severe hypertension when supine and significant hypotension when upright.
Postural orthostatic tachycardia syndrome, another form of orthostatic failure, occurs most frequently in young women (under the age of 50). In this syndrome, autonomic dysfunction affects peripheral vascular resistance, which fails to increase in response to orthostatic stress. This autonomic dysfunction does not affect the heart, which manifests a striking compensatory increase in rate of more than 30 beats per minute within the first 10 minutes of orthostasis, or an absolute heart rate greater than 120 beats per minute. Unlike in orthostatic hypotension, blood pressure and cardiac output are maintained through this increase in heart rate, although the patient still develops symptoms of severe fatigue or near-syncope, possibly because of flow maldistribution and reduced cerebral flow.2
While postural orthostatic tachycardia syndrome per se does not induce syncope,2 it may be associated with a vasovagal form of syncope that occurs beyond the first 10 minutes of orthostasis in up to 38% of these patients.17
In a less common, hyperadrenergic form of postural orthostatic tachycardia syndrome, there is no autonomic failure but the sympathetic system is overly activated, with orthostasis leading to excessive tachycardia.10,18
CARDIAC SYNCOPE
Accounting for 10% to 20% of cases of syncope, a cardiac cause is the main concern in patients presenting with syncope, as cardiac syncope predicts an increased risk of death and may herald sudden cardiac death.1,2,8,19,20 It often occurs suddenly without any warning signs, in which case it is called malignant syncope. Unlike what occurs in neurally mediated syncope, the postrecovery period is not usually marked by lingering malaise.
There are three forms of cardiac syncope:
Syncope due to structural heart disease with cardiac obstruction
In cases of aortic stenosis, hypertrophic obstructive cardiomyopathy, or severe pulmonary arterial hypertension, peripheral vasodilation occurs during exercise, but cardiac output cannot increase because of the fixed or dynamic obstruction to the ventricular outflow. Since blood pressure is equal to cardiac output times peripheral vascular resistance, pressure drops with the reduction in peripheral vascular resistance. Exertional ventricular arrhythmias may also occur in these patients. Conversely, postexertional syncope is usually benign.
Syncope due to ventricular tachycardia
Ventricular tachycardia can be secondary to underlying structural heart disease, with or without reduced ejection fraction, such as coronary arterial disease, hypertrophic cardiomyopathy, hypertensive cardiomyopathy, or valvular disease. It can also be secondary to primary electrical disease (eg, long QT syndrome, Wolff-Parkinson-White syndrome, Brugada syndrome, arrhythmogenic right ventricular dysplasia, sarcoidosis).
Occasionally, fast supraventricular tachycardia causes syncope at its onset, before vascular compensation develops. This occurs in patients with underlying heart disease.2,8,19
Syncope from bradyarrhythmias
Bradyarrhythmias can occur with or without underlying structural heart disease. They are most often related to degeneration of the conduction system or to medications rather than to cardiomyopathy.
Caveats
When a patient with a history of heart failure presents with syncope, the top considerations are ventricular tachycardia and bradyarrhythmia. Nevertheless, about half of cases of syncope in patients with cardiac disease have a noncardiac cause,19 including the hypotensive or bradycardiac side effect of drugs.
As noted above, most cases of syncope are neurally mediated. However, long asystolic pauses due to sinus or atrioventricular nodal block are the most frequent mechanism of unexplained syncope and are seen in more than 50% of syncope cases on prolonged rhythm monitoring.1,21 These pauses may be related to intrinsic sinus or atrioventricular nodal disease or, more commonly, to extrinsic effects such as the vasovagal mechanism. Some experts favor classifying and treating syncope on the basis of the final mechanism rather than the initiating process, but this is not universally accepted.1,22
OTHER CAUSES OF SYNCOPE
Acute medical or cardiovascular illnesses can cause syncope and are looked for in the appropriate clinical context: severe hypovolemia or gastrointestinal bleeding, large pulmonary embolus with hemodynamic compromise, tamponade, aortic dissection, or hypoglycemia.
Bilateral critical carotid disease or severe vertebrobasilar disease very rarely cause syncope, and, when they do, they are associated with focal neurologic deficits.2 Vertebrobasilar disease may cause "drop attacks," ie, a loss of muscular tone with falling but without loss of consciousness.23
Severe proximal subclavian disease leads to reversal of the flow in the ipsilateral vertebral artery as blood is shunted toward the upper extremity. It manifests as dizziness and syncope during the ipsilateral upper extremity activity, usually with focal neurologic signs (subclavian steal syndrome).2
Psychogenic pseudosyncope is characterized by frequent attacks that typically last longer than true syncope and occur multiple times per day or week, sometimes with a loss of motor tone.2 It occurs in patients with anxiety or somatization disorders.
SEIZURE: A SYNCOPE MIMIC
Certain features differentiate seizure from syncope:
- In seizure, unconsciousness often lasts longer than 5 minutes
- After a seizure, the patient may experience postictal confusion or paralysis
- Seizure may include prolonged tonic-clonic movements; although these movements may be seen with any form of syncope lasting more than 30 seconds, the movements during syncope are more limited and brief, lasting less than 15 seconds
- Tongue biting strongly suggests seizure.
Urinary incontinence does not help distinguish the two, as it frequently occurs with syncope as well as seizure.
DIAGNOSTIC EVALUATION OF SYNCOPE

Table 2 lists clinical clues to the type of syncope.2–5,8
Underlying structural heart disease is the most important predictor of ventricular arrhythmia and death.20,24–26 Thus, the primary goal of the evaluation is to rule out structural heart disease by history, examination, electrocardiography, and echocardiography (Figure 1).
Initial strategy for finding the cause
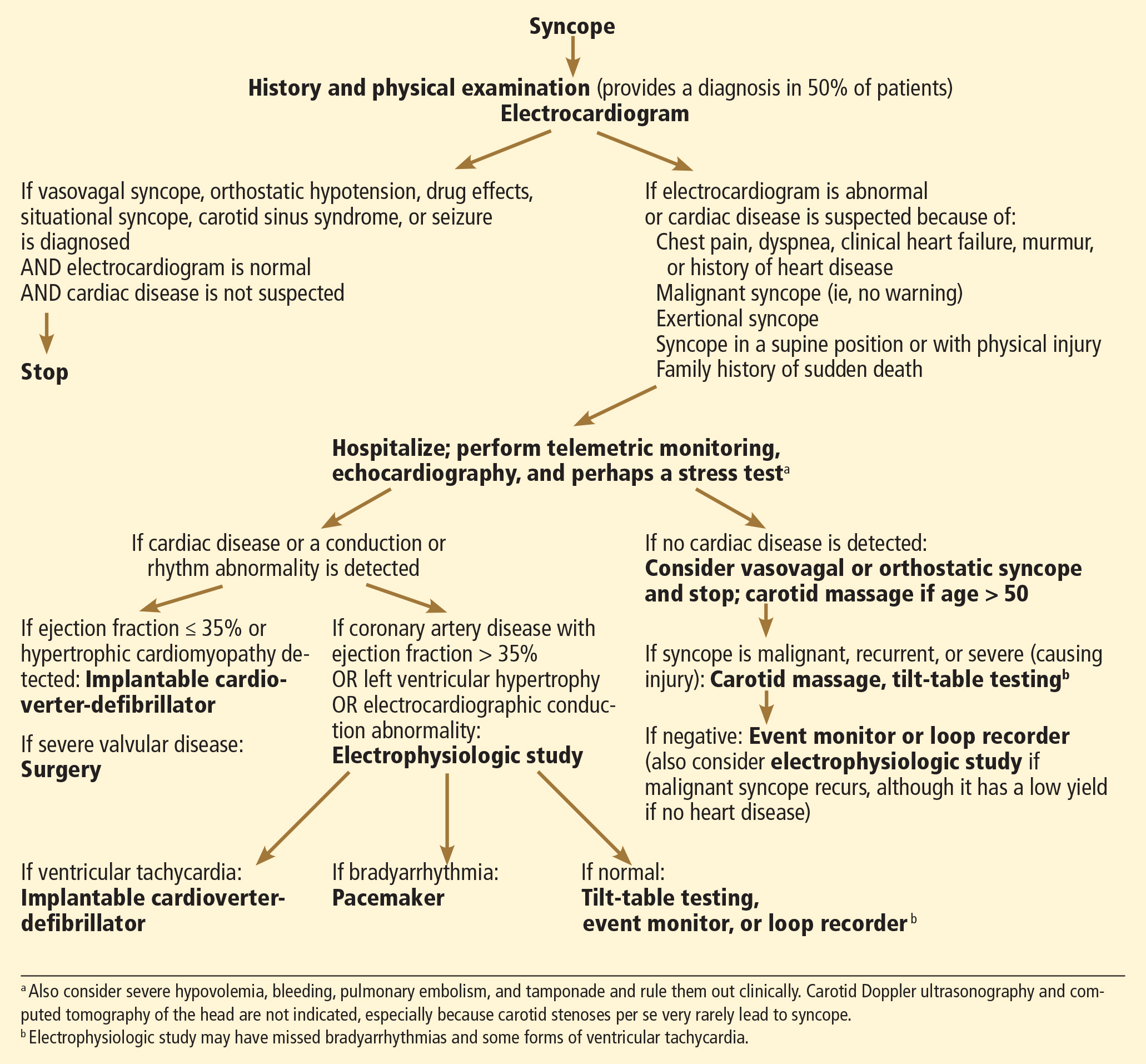
The cause of syncope is diagnosed by history and physical examination alone in up to 50% of cases, mainly neurally mediated syncope, orthostatic syncope, or seizure.2,3,19
Always check blood pressure with the patient both standing and sitting and in both arms, and obtain an electrocardiogram.
Perform carotid massage in all patients over age 50 if syncope is not clearly vasovagal or orthostatic and if cardiac syncope is not likely. Carotid massage is contraindicated if the patient has a carotid bruit or a history of stroke.

Electrocardiography establishes or suggests a diagnosis in 10% of patients (Table 3, Figure 2).1,2,8,19 A normal electrocardiogram or a mild nonspecific ST-T abnormality suggests a low likelihood of cardiac syncope and is associated with an excellent prognosis. Abnormal electrocardiographic findings are seen in 90% of cases of cardiac syncope and in only 6% of cases of neurally mediated syncope.27 In one study of syncope patients with normal electrocardiograms and negative cardiac histories, none had an abnormal echocardiogram.28
If the heart is normal
If the history suggests neurally mediated syncope or orthostatic hypotension and the history, examination, and electrocardiogram do not suggest coronary artery disease or any other cardiac disease, the workup is stopped.
If the patient has signs or symptoms of heart disease
If the patient has signs or symptoms of heart disease (angina, exertional syncope, dyspnea, clinical signs of heart failure, murmur), a history of heart disease, or exertional, supine, or malignant features, heart disease should be looked for and the following performed:
- Echocardiography to assess left ventricular function, severe valvular disease, and left ventricular hypertrophy
- A stress test (possibly) in cases of exertional syncope or associated angina; however, the overall yield of stress testing in syncope is low (< 5%).29
If electrocardiography and echocardiography do not suggest heart disease
Often, in this situation, the workup can be stopped and syncope can be considered neurally mediated. The likelihood of cardiac syncope is very low in patients with normal findings on electrocardiography and echocardiography, and several studies have shown that patients with syncope who have no structural heart disease have normal long-term survival rates.20,26,30
The following workup may, however, be ordered if the presentation is atypical and syncope is malignant, recurrent, or associated with physical injury, or occurs in the supine position19:
Carotid sinus massage in patients over age 50, if not already performed. Up to 50% of these patients with unexplained syncope have carotid sinus hypersensitivity.13
24-hour Holter monitoring rarely detects significant arrhythmias, but if syncope or dizziness occurs without any arrhythmia, Holter monitoring rules out arrhythmia as the cause of the symptoms.31 The diagnostic yield of Holter monitoring is low (1% to 2%) in patients with infrequent symptoms1,2 and is not improved with 72-hour monitoring.30 The yield is higher in patients with very frequent daily symptoms, many of whom have psychogenic pseudosyncope.2
Tilt-table testing to diagnose vasovagal syncope. This test is positive for a vasovagal response in up to 66% of patients with unexplained syncope.1,19 Patients with heart disease taking vasodilators or beta-blockers may have abnormal baroreflexes. Therefore, a positive tilt test is less specific in these patients and does not necessarily indicate vasovagal syncope.
Event monitoring. If the etiology remains unclear or there are some concerns about arrhythmia, an event monitor (4 weeks of external rhythm monitoring) or an implantable loop recorder (implanted subcutaneously in the prepectoral area for 1 to 2 years) is placed. These monitors record the rhythm when the rate is lower or higher than predefined cutoffs or when the rhythm is irregular, regardless of symptoms. The patient or an observer can also activate the event monitor during or after an event, which freezes the recording of the 2 to 5 minutes preceding the activation and the 1 minute after it.
In a patient who has had syncope, a pacemaker is indicated for episodes of high-grade atrioventricular block, pauses longer than 3 seconds while awake, or bradycardia (< 40 beats per minute) while awake, and an implantable cardioverter-defibrillator is indicated for sustained ventricular tachycardia, even if syncope does not occur concomitantly with these findings. The finding of nonsustained ventricular tachycardia on monitoring increases the suspicion of ventricular tachycardia as the cause of syncope but does not prove it, nor does it necessarily dictate implantation of a cardioverter-defibrillator device.
An electrophysiologic study has a low yield in patients with normal electrocardiographic and echocardiographic studies. Bradycardia is detected in 10%.31
If heart disease or a rhythm abnormality is found
If heart disease is diagnosed by echocardiography or if significant electrocardiographic abnormalities are found, perform the following:
Pacemaker placement for the following electrocardiographic abnormalities1,2,19:
- Second-degree Mobitz II or third-degree atrioventricular block
- Sinus pause (> 3 seconds) or bradycardia (< 40 beats per minute) while awake
- Alternating left bundle branch block and right bundle branch block on the same electrocardiogram or separate ones.
Telemetric monitoring (inpatient).
An electrophysiologic study is valuable mainly for patients with structural heart disease, including an ejection fraction 36% to 49%, coronary artery disease, or left ventricular hypertrophy with a normal ejection fraction.32 Overall, in patients with structural heart disease and unexplained syncope, the yield is 55% (inducible ventricular tachycardia in 21%, abnormal indices of bradycardia in 34%).31
However, the yield of electrophysiologic testing is low in bradyarrhythmia and in patients with an ejection fraction of 35% or less.33 In the latter case, the syncope is often arrhythmia-related and the patient often has an indication for an implantable cardioverter-defibrillator regardless of electrophysiologic study results, especially if the low ejection fraction has persisted despite medical therapy.32
If the electrophysiologic study is negative
If the electrophysiologic study is negative, the differential diagnosis still includes arrhythmia, as the yield of electrophysiologic study is low for bradyarrhythmias and some ventricular tachycardias, and the differential diagnosis also includes, at this point, neurally mediated syncope.
The next step may be either prolonged rhythm monitoring or tilt-table testing. An event monitor or an implantable loop recorder can be placed for prolonged monitoring. The yield of the 30-day event monitor is highest in patients with frequently recurring syncope, in whom it reaches a yield of up to 40% (10% to 20% will have a positive diagnosis of arrhythmia, while 15% to 20% will have symptoms with a normal rhythm).31,34 The implantable recorder has a high overall diagnostic yield and is used in patients with infrequent syncopal episodes (yield up to 50%).1,35,36
In brief, there are two diagnostic approaches to unexplained syncope: the monitoring approach (loop recorder) and the testing approach (tilt-table testing). A combination of both strategies is frequently required in patients with unexplained syncope, and, according to some investigators, a loop recorder may be implanted early on.21
Heart disease with left ventricular dysfunction and low ejection fraction
In patients with heart disease with left ventricular dysfunction and an ejection fraction of 35% or less, an implantable cardioverter-defibrillator can be placed without the need for an electrophysiologic study. These patients need these devices anyway to prevent sudden death, even if the cause of syncope is not an arrhythmia. Patients with a low ejection fraction and a history of syncope are at a high risk of sudden cardiac death.32 Yet in some patients with newly diagnosed cardiomyopathy, left ventricular function may improve with medical therapy. Because the arrhythmic risk is essentially high during the period of ventricular dysfunction, a wearable external defibrillator may be placed while the decision about an implantable defibrillator is finalized within the ensuing months.
In patients with hypertrophic cardiomyopathy, place an implantable cardioverter-defibrillator after any unexplained syncopal episode.
Valvular heart disease needs surgical correction.
If ischemic heart disease is suspected, coronary angiography is indicated, with revascularization if appropriate. An implantable cardioverter-defibrillator should be placed if the ejection fraction is lower than 35%. Except in a large acute myocardial infarction, the substrate for ventricular tachycardia is not ameliorated with revascularization.32,37 Consider an electrophysiologic study when syncope occurs with coronary artery disease and a higher ejection fraction.
A note on left or right bundle branch block
Patients with left or right bundle branch block and unexplained syncope (not clearly vasovagal or orthostatic) likely have syncope related to intermittent high-grade atrioventricular block.38
One study monitored these patients with an implanted loop recorder and showed that about 40% had a recurrence of syncope within 48 days, often concomitantly with complete atrioventricular block. About 55% of these patients had a major event (syncope or high-grade atrioventricular block).39 Many of the patients had had a positive tilt test; thus, tilt testing is not specific for vasovagal syncope in these patients and should not be used to exclude a bradyarrhythmic syncope. Also, patients selected for this study had undergone carotid sinus massage and an electrophysiology study with a negative result.
In another analysis, an electrophysiologic study detected a proportion of the bradyarrhythmias but, more importantly, it induced ventricular tachycardia in 14% of patients with right or left bundle branch block. Although it is not sensitive enough for bradyarrhythmia, electrophysiologic study was highly specific and fairly sensitive for the occurrence of ventricular tachycardia on follow-up.38 Thus, unexplained syncope in a patient with right or left bundle branch block may warrant carotid sinus massage, then an electrophysiologic study to rule out ventricular tachycardia, followed by placement of a dual-chamber pacemaker if the study is negative for ventricular tachycardia, or at least placement of a loop recorder.
INDICATIONS FOR HOSPITALIZATION
Patients should be hospitalized if they have severe hypovolemia or bleeding, or if there is any suspicion of heart disease by history, examination, or electrocardiography, including:
- History of heart failure, low ejection fraction, or coronary artery disease
- An electrocardiogram suggestive of arrhythmia (Table 3)
- Family history of sudden death
- Lack of prodromes; occurrence of physical injury, exertional syncope, syncope in a supine position, or syncope associated with dyspnea or chest pain.2,40
In these situations, there is concern about arrhythmia, structural heart disease, or acute myocardial ischemia. The patient is admitted for immediate telemetric monitoring. Echocardiography and sometimes stress testing are performed. The patient is discharged if this initial workup does not suggest underlying heart disease. Alternatively, an electrophysiologic study is performed or a device is placed in patients found to have structural heart disease. Prolonged rhythm monitoring or tilt-table testing may be performed when syncope with underlying heart disease or worrisome features remains unexplained.
Several Web-based interactive algorithms have been used to determine the indication for hospitalization. They incorporate the above clinical, electrocardiographic, and sometimes echocardiographic features.2,24,25,40–42 A cardiology consultation is usually necessary in patients with the above features, as they frequently require specialized cardiac testing.
Among high-risk patients, the risk of sudden death, a major cardiovascular event, or significant arrhythmia is high in the first few days after the index syncopal episode, justifying the hospitalization and inpatient rhythm monitoring and workup in the presence of the above criteria.24,40,42
SYNCOPE AND DRIVING
A study has shown that the most common cause of syncope while driving is vasovagal syncope.6 In all patients, the risk of another episode of syncope was relatively higher during the first 6 months after the event, with a 12% recurrence rate during this period. However, recurrences were often also seen more than 6 months later (12% recurrence between 6 months and the following few years).6 Fortunately, those episodes rarely occurred while the patient was driving. In a study in survivors of ventricular arrhythmia, the risk of recurrence of arrhythmic events was highest during the first 6 to 12 months after the event.43
Thus, in general, patients with syncope should be prohibited from driving for at least the period of time (eg, 6 months) during which the risk of a recurrent episode of syncope is highest and during which serious cardiac disease or arrhythmia, if present, would emerge. Recurrence of syncope is more likely and more dangerous for commercial drivers who spend a significant proportion of their time driving; individualized decisions are made in these cases.
- Brignole M, Hamdan MH. New concepts in the assessment of syncope. J Am Coll Cardiol 2012; 59:1583–1591.
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS); Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009 Eur Heart J 2009; 30:2631–2671.
- Kapoor WN. Syncope. N Engl J Med 2000; 343:1856–1862.
- Graham LA, Kenny RA. Clinical characteristics of patients with vasovagal reactions presenting as unexplained syncope. Europace 2001; 3:141–146.
- Calkins H, Shyr Y, Frumin H, Schork A, Morady F. The value of the clinical history in the differentiation of syncope due to ventricular tachycardia, atrioventricular block, and neurocardiogenic syncope. Am J Med 1995; 98:365–373.
- Sorajja D, Nesbitt GC, Hodge DO, et al. Syncope while driving: clinical characteristics, causes, and prognosis. Circulation 2009; 120:928–934.
- Kochiadakis GE, Papadimitriou EA, Marketou ME, Chrysostomakis SI, Simantirakis EN, Vardas PE. Autonomic nervous system changes in vasovagal syncope: is there any difference between young and older patients? Pacing Clin Electrophysiol 2004; 27:1371–1377.
- Alboni P, Brignole M, Menozzi C, et al. Diagnostic value of history in patients with syncope with or without heart disease. J Am Coll Cardiol 2001; 37:1921–1928.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Task force on syncope, European Society of Cardiology. Part 1. The initial evaluation of patients with syncope. Europace 2001; 3:253–260.
- Grubb BP. Neurocardiogenic syncope and related disorders of orthostatic intolerance. Circulation 2005; 111:2997–3006.
- Brignole M, Menozzi C, Del Rosso A, et al. New classification of haemodynamics of vasovagal syncope: beyond the VASIS classification. Analysis of the pre-syncopal phase of the tilt test without and with nitroglycerin challenge. Vasovagal Syncope International Study. Europace 2000; 2:66–76.
- Grubb BP, Kosinski D. Tilt table testing: concepts and limitations. Pacing Clin Electrophysiol 1997; 20:781–787.
- Brignole M, Menozzi C, Gianfranchi L, Oddone D, Lolli G, Bertulla A. Carotid sinus massage, eyeball compression, and head-up tilt test in patients with syncope of uncertain origin and in healthy control subjects. Am Heart J 1991; 122:1644–1651.
- Maggi R, Menozzi C, Brignole M, et al. Cardioinhibitory carotid sinus hypersensitivity predicts an asystolic mechanism of spontaneous neurally mediated syncope. Europace 2007; 9:563–567.
- Kenny RA, Richardson DA, Steen N, Bexton RS, Shaw FE, Bond J. Carotid sinus syndrome: a modifiable risk factor for nonaccidental falls in older adults (SAFE PACE). J Am Coll Cardiol 2001; 38:1491–1496.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Ojha A, McNeeley K, Heller E, Alshekhlee A, Chelimsky G, Chelimsky TC. Orthostatic syndromes differ in syncope frequency. Am J Med 2010; 123:245–249.
- Kanjwal Y, Kosinski D, Grubb BP. The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management. Pacing Clin Electrophysiol 2003; 26:1747–1757.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Guidelines on management (diagnosis and treatment) of syncope. Eur Heart J 2001; 22:1256–1306.
- Soteriades ES, Evans JC, Larson MG, et al. Incidence and prognosis of syncope. N Engl J Med 2002; 347:878–885.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology 3 (ISSUE-3) Investigators. Pacemaker therapy in patients with neurally mediated syncope and documented asystole: Third International Study on Syncope of Uncertain Etiology (ISSUE-3): a randomized trial. Circulation 2012; 125:2566–2571.
- Kubak MJ, Millikan CH. Diagnosis, pathogenesis, and treatment of "drop attacks." Arch Neurol 1964; 11:107–113.
- Quinn J, McDermott D, Stiell I, Kohn M, Wells G. Prospective validation of the San Francisco Syncope Rule to predict patients with serious outcomes. Ann Emerg Med 2006; 47:448–454.
- Colivicchi F, Ammirati F, Melina D, Guido V, Imperoli G, Santini M; OESIL (Osservatorio Epidemiologico sulla Sincope nel Lazio) Study Investigators. Development and prospective validation of a risk stratification system for patients with syncope in the emergency department: the OESIL risk score. Eur Heart J 2003; 24:811–819.
- Kapoor WN, Hanusa BH. Is syncope a risk factor for poor outcomes? Comparison of patients with and without syncope. Am J Med 1996; 100:646–655.
- Sarasin FP, Louis-Simonet M, Carballo D, et al. Prospective evaluation of patients with syncope: a population-based study. Am J Med 2001; 111:177–184.
- Sarasin FP, Junod AF, Carballo D, Slama S, Unger PF, Louis-Simonet M. Role of echocardiography in the evaluation of syncope: a prospective study. Heart 2002; 88:363–367.
- AlJaroudi WA, Alraies MC, Wazni O, Cerqueira MD, Jaber WA. Yield and diagnostic value of stress myocardial perfusion imaging in patients without known coronary artery disease presenting with syncope. Circ Cardiovasc Imaging 2013; 6:384–391.
- Ungar A, Del Rosso A, Giada F, et al; Evaluation of Guidelines in Syncope Study 2 Group. Early and late outcome of treated patients referred for syncope to emergency department: the EGSYS 2 follow-up study. Eur Heart J 2010; 31:2021–2026.
- Linzer M, Yang EH, Estes NA 3rd, Wang P, Vorperian VR, Kapoor WN. Diagnosing syncope. Part 2: Unexplained syncope. Clinical Efficacy Assessment Project of the American College of Physicians. Ann Intern Med 1997; 127:76–86.
- Strickberger SA, Benson DW, Biaggioni I, et al; American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke; Quality of Care and Outcomes Research Interdisciplinary Working Group; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF scientific statement on the evaluation of syncope: from the American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke, and the Quality of Care and Outcomes Research Interdisciplinary Working Group; and the American College of Cardiology Foundation In Collaboration With the Heart Rhythm Society. J Am Coll Cardiol 2006; 47:473–484.
- Fujimura O, Yee R, Klein GJ, Sharma AD, Boahene KA. The diagnostic sensitivity of electrophysiologic testing in patients with syncope caused by transient bradycardia. N Engl J Med 1989; 321:1703–1707.
- Linzer M, Pritchett EL, Pontinen M, McCarthy E, Divine GW. Incremental diagnostic yield of loop electrocardiographic recorders in unexplained syncope. Am J Cardiol 1990; 66:214–219.
- Edvardsson N, Frykman V, van Mechelen R, et al; PICTURE Study Investigators. Use of an implantable loop recorder to increase the diagnostic yield in unexplained syncope: results from the PICTURE registry. Europace 2011; 13:262–269.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brugada J, Aguinaga L, Mont L, Betriu A, Mulet J, Sanz G. Coronary artery revascularization in patients with sustained ventricular arrhythmias in the chronic phase of a myocardial infarction: effects on the electrophysiologic substrate and outcome. J Am Coll Cardiol 2001; 37:529–533.
- Moya A, García-Civera R, Croci F, et al; Bradycardia detection in Bundle Branch Block (B4) study. Diagnosis, management, and outcomes of patients with syncope and bundle branch block. Eur Heart J 2011; 32:1535–1541.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology (ISSUE) Investigators. Mechanism of syncope in patients with bundle branch block and negative electrophysiological test. Circulation 2001; 104:2045–2050.
- Brignole M, Shen WK. Syncope management from emergency department to hospital. J Am Coll Cardiol 2008; 51:284–287.
- Daccarett M, Jetter TL, Wasmund SL, Brignole M, Hamdan MH. Syncope in the emergency department: comparison of standardized admission criteria with clinical practice. Europace 2011; 13:1632–1638.
- Costantino G, Perego F, Dipaola F, et al; STePS Investigators. Short- and long-term prognosis of syncope, risk factors, and role of hospital admission: results from the STePS (Short-Term Prognosis of Syncope) study. J Am Coll Cardiol 2008; 51:276–283.
- Larsen GC, Stupey MR, Walance CG, et al. Recurrent cardiac events in survivors of ventricular fibrillation or tachycardia. Implications for driving restrictions. JAMA 1994; 271:1335–1339.
Syncope is a transient loss of consciousness and postural tone with spontaneous, complete recovery. There are three major types: neurally mediated, orthostatic, and cardiac (Table 1).
NEURALLY MEDIATED SYNCOPE
Neurally mediated (reflex) syncope is the most common type, accounting for two-thirds of cases.1–3 It results from autonomic reflexes that respond inappropriately, leading to vasodilation and bradycardia.
See related patient-education handout
Neurally mediated syncope is usually preceded by premonitory symptoms such as lightheadedness, diaphoresis, nausea, malaise, abdominal discomfort, and tunnel vision. However, this may not be the case in one-third of patients, especially in elderly patients, who may not recognize or remember the warning symptoms. Palpitations are frequently reported with neurally mediated syncope and do not necessarily imply that the syncope is due to an arrhythmia.4,5 Neurally mediated syncope does not usually occur in the supine position4,5 but can occur in the seated position.6
Subtypes of neurally mediated syncope are as follows:
Vasovagal syncope
Vasovagal syncope is usually triggered by sudden emotional stress, prolonged sitting or standing, dehydration, or a warm environment, but it can also occur without a trigger. It is the most common type of syncope in young patients (more so in females than in males), but contrary to a common misconception, it can also occur in the elderly.7 Usually, it is not only preceded by but also followed by nausea, malaise, fatigue, and diaphoresis4,5,8; full recovery may be slow. If the syncope lasts longer than 30 to 60 seconds, clonic movements and loss of bladder control are common.9
Mechanism. Vasovagal syncope is initiated by anything that leads to strong myocardial contractions in an "empty" heart. Emotional stress, reduced venous return (from dehydration or prolonged standing), or vasodilation (caused by a hot environment) stimulates the sympathetic nervous system and reduces the left ventricular cavity size, which leads to strong hyperdynamic contractions in a relatively empty heart. This hyperdynamic cavity obliteration activates myocardial mechanoreceptors, initiating a paradoxical vagal reflex with vasodilation and relative bradycardia.10 Vasodilation is usually the predominant mechanism (vasodepressor response), particularly in older patients, but severe bradycardia is also possible (cardioinhibitory response), particularly in younger patients.7 Diuretic and vasodilator therapies increase the predisposition to vasovagal syncope, particularly in the elderly.
On tilt-table testing, vasovagal syncope is characterized by hypotension and relative bradycardia, sometimes severe (see Note on Tilt-Table Testing).10–12
Situational syncope
Situational syncope is caused by a reflex triggered in specific circumstances such as micturition, defecation, coughing, weight-lifting, laughing, or deglutition. The reflex may be initiated by a receptor on the visceral wall (eg, the bladder wall) or by straining that reduces venous return.
Carotid sinus hypersensitivity
Carotid sinus hypersensitivity is an abnormal response to carotid massage, predominantly occurring in patients over the age of 50. In spontaneous carotid sinus syndrome, syncope clearly occurs in a situation that stimulates the carotid sinus, such as head rotation, head extension, shaving, or wearing a tight collar. It is a rare cause of syncope, responsible for about 1% of cases. Conversely, induced carotid sinus syndrome is much more common and represents carotid sinus hypersensitivity in a patient with unexplained syncope and without obvious triggers; the abnormal response is mainly induced during carotid massage rather than spontaneously. In the latter case, carotid sinus hypersensitivity is a marker of a diseased sinus node or atrioventricular node that cannot withstand any inhibition. This diseased node is the true cause of syncope rather than carotid sinus hypersensitivity per se, and carotid massage is a "stress test" that unveils conduction disease.
Thus, carotid massage is indicated in cases of unexplained syncope regardless of circumstantial triggers. This test consists of applying firm pressure over each carotid bifurcation (just below the angle of the jaw) consecutively for 10 seconds. It is performed at the bedside, and may be performed with the patient in both supine and erect positions during tilt-table testing; erect positioning of the patient increases the sensitivity of this test.
An abnormal response to carotid sinus massage is defined as any of the following13–15:
- Vasodepressor response: the systolic blood pressure decreases by at least 50 mm Hg
- Cardioinhibitory response: sinus or atrioventricular block causes the heartbeat to pause for 3 or more seconds
- Mixed vasodepressor and cardioinhibitory response.
Overall, a cardioinhibitory component is present in about two-thirds of cases of carotid sinus hypersensitivity.
Carotid sinus hypersensitivity is found in 25% to 50% of patients over age 50 who have had unexplained syncope or a fall, and it is seen almost equally in men and women.13
One study correlated carotid sinus hypersensitivity with the later occurrence of asystolic syncope during prolonged internal loop monitoring; subsequent pacemaker therapy reduced the burden of syncope.14 Another study, in patients over 50 years old with unexplained falls, found that 16% had cardioinhibitory carotid sinus hypersensitivity. Pacemaker placement reduced falls and syncope by 70% compared with no pacemaker therapy in these patients.15
On the other hand, carotid sinus hypersensitivity can be found in 39% of elderly patients who do not have a history of fainting or falling, so it is important to rule out other causes of syncope before attributing it to carotid sinus hypersensitivity.
Postexertional syncope
While syncope on exertion raises the worrisome possibility of a cardiac cause, postexertional syncope is usually a form of vasovagal syncope. When exercise ceases, venous blood stops getting pumped back to the heart by peripheral muscular contraction. Yet the heart is still exposed to the catecholamine surge induced by exercising, and it hypercontracts on an empty cavity. This triggers a vagal reflex.
Postexertional syncope may also be seen in hypertrophic obstructive cardiomyopathy or aortic stenosis, in which the small left ventricular cavity is less likely to tolerate the reduced preload after exercise and is more likely to obliterate.
ORTHOSTATIC HYPOTENSION
Orthostatic hypotension accounts for about 10% of cases of syncope.1–3
Normally, after the first few minutes of standing, about 25% to 30% of the blood pools in the veins of the pelvis and the lower extremities, strikingly reducing venous return and stroke volume. Upon more prolonged standing, more blood leaves the vascular space and collects in the extravascular space, further reducing venous return. This normally leads to a reflex increase in sympathetic tone, peripheral and splanchnic vasoconstriction, and an increase in heart rate of 10 to 15 beats per minute. Overall, cardiac output is reduced and vascular resistance is increased while blood pressure is maintained, blood pressure being equal to cardiac output times vascular resistance.
Orthostatic hypotension is characterized by autonomic failure, with a lack of compensatory increase in vascular resistance or heart rate upon orthostasis, or by significant hypovolemia that cannot be overcome by sympathetic mechanisms. It is defined as a drop in systolic blood pressure of 20 mm Hg or more or a drop in diastolic pressure of 10 mm Hg or more after 30 seconds to 5 minutes of upright posture. Blood pressure is checked immediately upon standing and at 3 and 5 minutes. This may be done at the bedside or during tilt-table testing.2,4
Some patients have an immediate drop in blood pressure of more than 40 mm Hg upon standing, with a quick return to normal within 30 seconds. This "initial orthostatic hypotension" may be common in elderly patients taking antihypertensive drugs and may elude detection during standard blood pressure measurement.2 Other patients with milder orthostatic hypotension may develop a more delayed hypotension 10 to 15 minutes later, as more blood pools in the periphery.16
Along with the drop in blood pressure, a failure of the heart rate to increase identifies autonomic dysfunction. On the other hand, an increase in the heart rate of more than 20 to 30 beats per minute may signify a hypovolemic state even if blood pressure is maintained, the lack of blood pressure drop being related to the excessive heart rate increase.
Orthostatic hypotension is the most common cause of syncope in the elderly and may be due to autonomic dysfunction (related to age, diabetes, uremia, or Parkinson disease), volume depletion, or drugs that block autonomic effects or cause hypovolemia, such as vasodilators, beta-blockers, diuretics, neuropsychiatric medications, and alcohol.
Since digestion leads to peripheral vasodilation and splanchnic blood pooling, syncope that occurs within 1 hour after eating has a mechanism similar to that of orthostatic syncope.
Supine hypertension with orthostatic hypotension. Some patients with severe autonomic dysfunction and the inability to regulate vascular tone have severe hypertension when supine and significant hypotension when upright.
Postural orthostatic tachycardia syndrome, another form of orthostatic failure, occurs most frequently in young women (under the age of 50). In this syndrome, autonomic dysfunction affects peripheral vascular resistance, which fails to increase in response to orthostatic stress. This autonomic dysfunction does not affect the heart, which manifests a striking compensatory increase in rate of more than 30 beats per minute within the first 10 minutes of orthostasis, or an absolute heart rate greater than 120 beats per minute. Unlike in orthostatic hypotension, blood pressure and cardiac output are maintained through this increase in heart rate, although the patient still develops symptoms of severe fatigue or near-syncope, possibly because of flow maldistribution and reduced cerebral flow.2
While postural orthostatic tachycardia syndrome per se does not induce syncope,2 it may be associated with a vasovagal form of syncope that occurs beyond the first 10 minutes of orthostasis in up to 38% of these patients.17
In a less common, hyperadrenergic form of postural orthostatic tachycardia syndrome, there is no autonomic failure but the sympathetic system is overly activated, with orthostasis leading to excessive tachycardia.10,18
CARDIAC SYNCOPE
Accounting for 10% to 20% of cases of syncope, a cardiac cause is the main concern in patients presenting with syncope, as cardiac syncope predicts an increased risk of death and may herald sudden cardiac death.1,2,8,19,20 It often occurs suddenly without any warning signs, in which case it is called malignant syncope. Unlike what occurs in neurally mediated syncope, the postrecovery period is not usually marked by lingering malaise.
There are three forms of cardiac syncope:
Syncope due to structural heart disease with cardiac obstruction
In cases of aortic stenosis, hypertrophic obstructive cardiomyopathy, or severe pulmonary arterial hypertension, peripheral vasodilation occurs during exercise, but cardiac output cannot increase because of the fixed or dynamic obstruction to the ventricular outflow. Since blood pressure is equal to cardiac output times peripheral vascular resistance, pressure drops with the reduction in peripheral vascular resistance. Exertional ventricular arrhythmias may also occur in these patients. Conversely, postexertional syncope is usually benign.
Syncope due to ventricular tachycardia
Ventricular tachycardia can be secondary to underlying structural heart disease, with or without reduced ejection fraction, such as coronary arterial disease, hypertrophic cardiomyopathy, hypertensive cardiomyopathy, or valvular disease. It can also be secondary to primary electrical disease (eg, long QT syndrome, Wolff-Parkinson-White syndrome, Brugada syndrome, arrhythmogenic right ventricular dysplasia, sarcoidosis).
Occasionally, fast supraventricular tachycardia causes syncope at its onset, before vascular compensation develops. This occurs in patients with underlying heart disease.2,8,19
Syncope from bradyarrhythmias
Bradyarrhythmias can occur with or without underlying structural heart disease. They are most often related to degeneration of the conduction system or to medications rather than to cardiomyopathy.
Caveats
When a patient with a history of heart failure presents with syncope, the top considerations are ventricular tachycardia and bradyarrhythmia. Nevertheless, about half of cases of syncope in patients with cardiac disease have a noncardiac cause,19 including the hypotensive or bradycardiac side effect of drugs.
As noted above, most cases of syncope are neurally mediated. However, long asystolic pauses due to sinus or atrioventricular nodal block are the most frequent mechanism of unexplained syncope and are seen in more than 50% of syncope cases on prolonged rhythm monitoring.1,21 These pauses may be related to intrinsic sinus or atrioventricular nodal disease or, more commonly, to extrinsic effects such as the vasovagal mechanism. Some experts favor classifying and treating syncope on the basis of the final mechanism rather than the initiating process, but this is not universally accepted.1,22
OTHER CAUSES OF SYNCOPE
Acute medical or cardiovascular illnesses can cause syncope and are looked for in the appropriate clinical context: severe hypovolemia or gastrointestinal bleeding, large pulmonary embolus with hemodynamic compromise, tamponade, aortic dissection, or hypoglycemia.
Bilateral critical carotid disease or severe vertebrobasilar disease very rarely cause syncope, and, when they do, they are associated with focal neurologic deficits.2 Vertebrobasilar disease may cause "drop attacks," ie, a loss of muscular tone with falling but without loss of consciousness.23
Severe proximal subclavian disease leads to reversal of the flow in the ipsilateral vertebral artery as blood is shunted toward the upper extremity. It manifests as dizziness and syncope during the ipsilateral upper extremity activity, usually with focal neurologic signs (subclavian steal syndrome).2
Psychogenic pseudosyncope is characterized by frequent attacks that typically last longer than true syncope and occur multiple times per day or week, sometimes with a loss of motor tone.2 It occurs in patients with anxiety or somatization disorders.
SEIZURE: A SYNCOPE MIMIC
Certain features differentiate seizure from syncope:
- In seizure, unconsciousness often lasts longer than 5 minutes
- After a seizure, the patient may experience postictal confusion or paralysis
- Seizure may include prolonged tonic-clonic movements; although these movements may be seen with any form of syncope lasting more than 30 seconds, the movements during syncope are more limited and brief, lasting less than 15 seconds
- Tongue biting strongly suggests seizure.
Urinary incontinence does not help distinguish the two, as it frequently occurs with syncope as well as seizure.
DIAGNOSTIC EVALUATION OF SYNCOPE
Table 2 lists clinical clues to the type of syncope.2–5,8
Underlying structural heart disease is the most important predictor of ventricular arrhythmia and death.20,24–26 Thus, the primary goal of the evaluation is to rule out structural heart disease by history, examination, electrocardiography, and echocardiography (Figure 1).
Initial strategy for finding the cause
The cause of syncope is diagnosed by history and physical examination alone in up to 50% of cases, mainly neurally mediated syncope, orthostatic syncope, or seizure.2,3,19
Always check blood pressure with the patient both standing and sitting and in both arms, and obtain an electrocardiogram.
Perform carotid massage in all patients over age 50 if syncope is not clearly vasovagal or orthostatic and if cardiac syncope is not likely. Carotid massage is contraindicated if the patient has a carotid bruit or a history of stroke.
Electrocardiography establishes or suggests a diagnosis in 10% of patients (Table 3, Figure 2).1,2,8,19 A normal electrocardiogram or a mild nonspecific ST-T abnormality suggests a low likelihood of cardiac syncope and is associated with an excellent prognosis. Abnormal electrocardiographic findings are seen in 90% of cases of cardiac syncope and in only 6% of cases of neurally mediated syncope.27 In one study of syncope patients with normal electrocardiograms and negative cardiac histories, none had an abnormal echocardiogram.28
If the heart is normal
If the history suggests neurally mediated syncope or orthostatic hypotension and the history, examination, and electrocardiogram do not suggest coronary artery disease or any other cardiac disease, the workup is stopped.
If the patient has signs or symptoms of heart disease
If the patient has signs or symptoms of heart disease (angina, exertional syncope, dyspnea, clinical signs of heart failure, murmur), a history of heart disease, or exertional, supine, or malignant features, heart disease should be looked for and the following performed:
- Echocardiography to assess left ventricular function, severe valvular disease, and left ventricular hypertrophy
- A stress test (possibly) in cases of exertional syncope or associated angina; however, the overall yield of stress testing in syncope is low (< 5%).29
If electrocardiography and echocardiography do not suggest heart disease
Often, in this situation, the workup can be stopped and syncope can be considered neurally mediated. The likelihood of cardiac syncope is very low in patients with normal findings on electrocardiography and echocardiography, and several studies have shown that patients with syncope who have no structural heart disease have normal long-term survival rates.20,26,30
The following workup may, however, be ordered if the presentation is atypical and syncope is malignant, recurrent, or associated with physical injury, or occurs in the supine position19:
Carotid sinus massage in patients over age 50, if not already performed. Up to 50% of these patients with unexplained syncope have carotid sinus hypersensitivity.13
24-hour Holter monitoring rarely detects significant arrhythmias, but if syncope or dizziness occurs without any arrhythmia, Holter monitoring rules out arrhythmia as the cause of the symptoms.31 The diagnostic yield of Holter monitoring is low (1% to 2%) in patients with infrequent symptoms1,2 and is not improved with 72-hour monitoring.30 The yield is higher in patients with very frequent daily symptoms, many of whom have psychogenic pseudosyncope.2
Tilt-table testing to diagnose vasovagal syncope. This test is positive for a vasovagal response in up to 66% of patients with unexplained syncope.1,19 Patients with heart disease taking vasodilators or beta-blockers may have abnormal baroreflexes. Therefore, a positive tilt test is less specific in these patients and does not necessarily indicate vasovagal syncope.
Event monitoring. If the etiology remains unclear or there are some concerns about arrhythmia, an event monitor (4 weeks of external rhythm monitoring) or an implantable loop recorder (implanted subcutaneously in the prepectoral area for 1 to 2 years) is placed. These monitors record the rhythm when the rate is lower or higher than predefined cutoffs or when the rhythm is irregular, regardless of symptoms. The patient or an observer can also activate the event monitor during or after an event, which freezes the recording of the 2 to 5 minutes preceding the activation and the 1 minute after it.
In a patient who has had syncope, a pacemaker is indicated for episodes of high-grade atrioventricular block, pauses longer than 3 seconds while awake, or bradycardia (< 40 beats per minute) while awake, and an implantable cardioverter-defibrillator is indicated for sustained ventricular tachycardia, even if syncope does not occur concomitantly with these findings. The finding of nonsustained ventricular tachycardia on monitoring increases the suspicion of ventricular tachycardia as the cause of syncope but does not prove it, nor does it necessarily dictate implantation of a cardioverter-defibrillator device.
An electrophysiologic study has a low yield in patients with normal electrocardiographic and echocardiographic studies. Bradycardia is detected in 10%.31
If heart disease or a rhythm abnormality is found
If heart disease is diagnosed by echocardiography or if significant electrocardiographic abnormalities are found, perform the following:
Pacemaker placement for the following electrocardiographic abnormalities1,2,19:
- Second-degree Mobitz II or third-degree atrioventricular block
- Sinus pause (> 3 seconds) or bradycardia (< 40 beats per minute) while awake
- Alternating left bundle branch block and right bundle branch block on the same electrocardiogram or separate ones.
Telemetric monitoring (inpatient).
An electrophysiologic study is valuable mainly for patients with structural heart disease, including an ejection fraction 36% to 49%, coronary artery disease, or left ventricular hypertrophy with a normal ejection fraction.32 Overall, in patients with structural heart disease and unexplained syncope, the yield is 55% (inducible ventricular tachycardia in 21%, abnormal indices of bradycardia in 34%).31
However, the yield of electrophysiologic testing is low in bradyarrhythmia and in patients with an ejection fraction of 35% or less.33 In the latter case, the syncope is often arrhythmia-related and the patient often has an indication for an implantable cardioverter-defibrillator regardless of electrophysiologic study results, especially if the low ejection fraction has persisted despite medical therapy.32
If the electrophysiologic study is negative
If the electrophysiologic study is negative, the differential diagnosis still includes arrhythmia, as the yield of electrophysiologic study is low for bradyarrhythmias and some ventricular tachycardias, and the differential diagnosis also includes, at this point, neurally mediated syncope.
The next step may be either prolonged rhythm monitoring or tilt-table testing. An event monitor or an implantable loop recorder can be placed for prolonged monitoring. The yield of the 30-day event monitor is highest in patients with frequently recurring syncope, in whom it reaches a yield of up to 40% (10% to 20% will have a positive diagnosis of arrhythmia, while 15% to 20% will have symptoms with a normal rhythm).31,34 The implantable recorder has a high overall diagnostic yield and is used in patients with infrequent syncopal episodes (yield up to 50%).1,35,36
In brief, there are two diagnostic approaches to unexplained syncope: the monitoring approach (loop recorder) and the testing approach (tilt-table testing). A combination of both strategies is frequently required in patients with unexplained syncope, and, according to some investigators, a loop recorder may be implanted early on.21
Heart disease with left ventricular dysfunction and low ejection fraction
In patients with heart disease with left ventricular dysfunction and an ejection fraction of 35% or less, an implantable cardioverter-defibrillator can be placed without the need for an electrophysiologic study. These patients need these devices anyway to prevent sudden death, even if the cause of syncope is not an arrhythmia. Patients with a low ejection fraction and a history of syncope are at a high risk of sudden cardiac death.32 Yet in some patients with newly diagnosed cardiomyopathy, left ventricular function may improve with medical therapy. Because the arrhythmic risk is essentially high during the period of ventricular dysfunction, a wearable external defibrillator may be placed while the decision about an implantable defibrillator is finalized within the ensuing months.
In patients with hypertrophic cardiomyopathy, place an implantable cardioverter-defibrillator after any unexplained syncopal episode.
Valvular heart disease needs surgical correction.
If ischemic heart disease is suspected, coronary angiography is indicated, with revascularization if appropriate. An implantable cardioverter-defibrillator should be placed if the ejection fraction is lower than 35%. Except in a large acute myocardial infarction, the substrate for ventricular tachycardia is not ameliorated with revascularization.32,37 Consider an electrophysiologic study when syncope occurs with coronary artery disease and a higher ejection fraction.
A note on left or right bundle branch block
Patients with left or right bundle branch block and unexplained syncope (not clearly vasovagal or orthostatic) likely have syncope related to intermittent high-grade atrioventricular block.38
One study monitored these patients with an implanted loop recorder and showed that about 40% had a recurrence of syncope within 48 days, often concomitantly with complete atrioventricular block. About 55% of these patients had a major event (syncope or high-grade atrioventricular block).39 Many of the patients had had a positive tilt test; thus, tilt testing is not specific for vasovagal syncope in these patients and should not be used to exclude a bradyarrhythmic syncope. Also, patients selected for this study had undergone carotid sinus massage and an electrophysiology study with a negative result.
In another analysis, an electrophysiologic study detected a proportion of the bradyarrhythmias but, more importantly, it induced ventricular tachycardia in 14% of patients with right or left bundle branch block. Although it is not sensitive enough for bradyarrhythmia, electrophysiologic study was highly specific and fairly sensitive for the occurrence of ventricular tachycardia on follow-up.38 Thus, unexplained syncope in a patient with right or left bundle branch block may warrant carotid sinus massage, then an electrophysiologic study to rule out ventricular tachycardia, followed by placement of a dual-chamber pacemaker if the study is negative for ventricular tachycardia, or at least placement of a loop recorder.
INDICATIONS FOR HOSPITALIZATION
Patients should be hospitalized if they have severe hypovolemia or bleeding, or if there is any suspicion of heart disease by history, examination, or electrocardiography, including:
- History of heart failure, low ejection fraction, or coronary artery disease
- An electrocardiogram suggestive of arrhythmia (Table 3)
- Family history of sudden death
- Lack of prodromes; occurrence of physical injury, exertional syncope, syncope in a supine position, or syncope associated with dyspnea or chest pain.2,40
In these situations, there is concern about arrhythmia, structural heart disease, or acute myocardial ischemia. The patient is admitted for immediate telemetric monitoring. Echocardiography and sometimes stress testing are performed. The patient is discharged if this initial workup does not suggest underlying heart disease. Alternatively, an electrophysiologic study is performed or a device is placed in patients found to have structural heart disease. Prolonged rhythm monitoring or tilt-table testing may be performed when syncope with underlying heart disease or worrisome features remains unexplained.
Several Web-based interactive algorithms have been used to determine the indication for hospitalization. They incorporate the above clinical, electrocardiographic, and sometimes echocardiographic features.2,24,25,40–42 A cardiology consultation is usually necessary in patients with the above features, as they frequently require specialized cardiac testing.
Among high-risk patients, the risk of sudden death, a major cardiovascular event, or significant arrhythmia is high in the first few days after the index syncopal episode, justifying the hospitalization and inpatient rhythm monitoring and workup in the presence of the above criteria.24,40,42
SYNCOPE AND DRIVING
A study has shown that the most common cause of syncope while driving is vasovagal syncope.6 In all patients, the risk of another episode of syncope was relatively higher during the first 6 months after the event, with a 12% recurrence rate during this period. However, recurrences were often also seen more than 6 months later (12% recurrence between 6 months and the following few years).6 Fortunately, those episodes rarely occurred while the patient was driving. In a study in survivors of ventricular arrhythmia, the risk of recurrence of arrhythmic events was highest during the first 6 to 12 months after the event.43
Thus, in general, patients with syncope should be prohibited from driving for at least the period of time (eg, 6 months) during which the risk of a recurrent episode of syncope is highest and during which serious cardiac disease or arrhythmia, if present, would emerge. Recurrence of syncope is more likely and more dangerous for commercial drivers who spend a significant proportion of their time driving; individualized decisions are made in these cases.
Syncope is a transient loss of consciousness and postural tone with spontaneous, complete recovery. There are three major types: neurally mediated, orthostatic, and cardiac (Table 1).
NEURALLY MEDIATED SYNCOPE
Neurally mediated (reflex) syncope is the most common type, accounting for two-thirds of cases.1–3 It results from autonomic reflexes that respond inappropriately, leading to vasodilation and bradycardia.
See related patient-education handout
Neurally mediated syncope is usually preceded by premonitory symptoms such as lightheadedness, diaphoresis, nausea, malaise, abdominal discomfort, and tunnel vision. However, this may not be the case in one-third of patients, especially in elderly patients, who may not recognize or remember the warning symptoms. Palpitations are frequently reported with neurally mediated syncope and do not necessarily imply that the syncope is due to an arrhythmia.4,5 Neurally mediated syncope does not usually occur in the supine position4,5 but can occur in the seated position.6
Subtypes of neurally mediated syncope are as follows:
Vasovagal syncope
Vasovagal syncope is usually triggered by sudden emotional stress, prolonged sitting or standing, dehydration, or a warm environment, but it can also occur without a trigger. It is the most common type of syncope in young patients (more so in females than in males), but contrary to a common misconception, it can also occur in the elderly.7 Usually, it is not only preceded by but also followed by nausea, malaise, fatigue, and diaphoresis4,5,8; full recovery may be slow. If the syncope lasts longer than 30 to 60 seconds, clonic movements and loss of bladder control are common.9
Mechanism. Vasovagal syncope is initiated by anything that leads to strong myocardial contractions in an "empty" heart. Emotional stress, reduced venous return (from dehydration or prolonged standing), or vasodilation (caused by a hot environment) stimulates the sympathetic nervous system and reduces the left ventricular cavity size, which leads to strong hyperdynamic contractions in a relatively empty heart. This hyperdynamic cavity obliteration activates myocardial mechanoreceptors, initiating a paradoxical vagal reflex with vasodilation and relative bradycardia.10 Vasodilation is usually the predominant mechanism (vasodepressor response), particularly in older patients, but severe bradycardia is also possible (cardioinhibitory response), particularly in younger patients.7 Diuretic and vasodilator therapies increase the predisposition to vasovagal syncope, particularly in the elderly.
On tilt-table testing, vasovagal syncope is characterized by hypotension and relative bradycardia, sometimes severe (see Note on Tilt-Table Testing).10–12
Situational syncope
Situational syncope is caused by a reflex triggered in specific circumstances such as micturition, defecation, coughing, weight-lifting, laughing, or deglutition. The reflex may be initiated by a receptor on the visceral wall (eg, the bladder wall) or by straining that reduces venous return.
Carotid sinus hypersensitivity
Carotid sinus hypersensitivity is an abnormal response to carotid massage, predominantly occurring in patients over the age of 50. In spontaneous carotid sinus syndrome, syncope clearly occurs in a situation that stimulates the carotid sinus, such as head rotation, head extension, shaving, or wearing a tight collar. It is a rare cause of syncope, responsible for about 1% of cases. Conversely, induced carotid sinus syndrome is much more common and represents carotid sinus hypersensitivity in a patient with unexplained syncope and without obvious triggers; the abnormal response is mainly induced during carotid massage rather than spontaneously. In the latter case, carotid sinus hypersensitivity is a marker of a diseased sinus node or atrioventricular node that cannot withstand any inhibition. This diseased node is the true cause of syncope rather than carotid sinus hypersensitivity per se, and carotid massage is a "stress test" that unveils conduction disease.
Thus, carotid massage is indicated in cases of unexplained syncope regardless of circumstantial triggers. This test consists of applying firm pressure over each carotid bifurcation (just below the angle of the jaw) consecutively for 10 seconds. It is performed at the bedside, and may be performed with the patient in both supine and erect positions during tilt-table testing; erect positioning of the patient increases the sensitivity of this test.
An abnormal response to carotid sinus massage is defined as any of the following13–15:
- Vasodepressor response: the systolic blood pressure decreases by at least 50 mm Hg
- Cardioinhibitory response: sinus or atrioventricular block causes the heartbeat to pause for 3 or more seconds
- Mixed vasodepressor and cardioinhibitory response.
Overall, a cardioinhibitory component is present in about two-thirds of cases of carotid sinus hypersensitivity.
Carotid sinus hypersensitivity is found in 25% to 50% of patients over age 50 who have had unexplained syncope or a fall, and it is seen almost equally in men and women.13
One study correlated carotid sinus hypersensitivity with the later occurrence of asystolic syncope during prolonged internal loop monitoring; subsequent pacemaker therapy reduced the burden of syncope.14 Another study, in patients over 50 years old with unexplained falls, found that 16% had cardioinhibitory carotid sinus hypersensitivity. Pacemaker placement reduced falls and syncope by 70% compared with no pacemaker therapy in these patients.15
On the other hand, carotid sinus hypersensitivity can be found in 39% of elderly patients who do not have a history of fainting or falling, so it is important to rule out other causes of syncope before attributing it to carotid sinus hypersensitivity.
Postexertional syncope
While syncope on exertion raises the worrisome possibility of a cardiac cause, postexertional syncope is usually a form of vasovagal syncope. When exercise ceases, venous blood stops getting pumped back to the heart by peripheral muscular contraction. Yet the heart is still exposed to the catecholamine surge induced by exercising, and it hypercontracts on an empty cavity. This triggers a vagal reflex.
Postexertional syncope may also be seen in hypertrophic obstructive cardiomyopathy or aortic stenosis, in which the small left ventricular cavity is less likely to tolerate the reduced preload after exercise and is more likely to obliterate.
ORTHOSTATIC HYPOTENSION
Orthostatic hypotension accounts for about 10% of cases of syncope.1–3
Normally, after the first few minutes of standing, about 25% to 30% of the blood pools in the veins of the pelvis and the lower extremities, strikingly reducing venous return and stroke volume. Upon more prolonged standing, more blood leaves the vascular space and collects in the extravascular space, further reducing venous return. This normally leads to a reflex increase in sympathetic tone, peripheral and splanchnic vasoconstriction, and an increase in heart rate of 10 to 15 beats per minute. Overall, cardiac output is reduced and vascular resistance is increased while blood pressure is maintained, blood pressure being equal to cardiac output times vascular resistance.
Orthostatic hypotension is characterized by autonomic failure, with a lack of compensatory increase in vascular resistance or heart rate upon orthostasis, or by significant hypovolemia that cannot be overcome by sympathetic mechanisms. It is defined as a drop in systolic blood pressure of 20 mm Hg or more or a drop in diastolic pressure of 10 mm Hg or more after 30 seconds to 5 minutes of upright posture. Blood pressure is checked immediately upon standing and at 3 and 5 minutes. This may be done at the bedside or during tilt-table testing.2,4
Some patients have an immediate drop in blood pressure of more than 40 mm Hg upon standing, with a quick return to normal within 30 seconds. This "initial orthostatic hypotension" may be common in elderly patients taking antihypertensive drugs and may elude detection during standard blood pressure measurement.2 Other patients with milder orthostatic hypotension may develop a more delayed hypotension 10 to 15 minutes later, as more blood pools in the periphery.16
Along with the drop in blood pressure, a failure of the heart rate to increase identifies autonomic dysfunction. On the other hand, an increase in the heart rate of more than 20 to 30 beats per minute may signify a hypovolemic state even if blood pressure is maintained, the lack of blood pressure drop being related to the excessive heart rate increase.
Orthostatic hypotension is the most common cause of syncope in the elderly and may be due to autonomic dysfunction (related to age, diabetes, uremia, or Parkinson disease), volume depletion, or drugs that block autonomic effects or cause hypovolemia, such as vasodilators, beta-blockers, diuretics, neuropsychiatric medications, and alcohol.
Since digestion leads to peripheral vasodilation and splanchnic blood pooling, syncope that occurs within 1 hour after eating has a mechanism similar to that of orthostatic syncope.
Supine hypertension with orthostatic hypotension. Some patients with severe autonomic dysfunction and the inability to regulate vascular tone have severe hypertension when supine and significant hypotension when upright.
Postural orthostatic tachycardia syndrome, another form of orthostatic failure, occurs most frequently in young women (under the age of 50). In this syndrome, autonomic dysfunction affects peripheral vascular resistance, which fails to increase in response to orthostatic stress. This autonomic dysfunction does not affect the heart, which manifests a striking compensatory increase in rate of more than 30 beats per minute within the first 10 minutes of orthostasis, or an absolute heart rate greater than 120 beats per minute. Unlike in orthostatic hypotension, blood pressure and cardiac output are maintained through this increase in heart rate, although the patient still develops symptoms of severe fatigue or near-syncope, possibly because of flow maldistribution and reduced cerebral flow.2
While postural orthostatic tachycardia syndrome per se does not induce syncope,2 it may be associated with a vasovagal form of syncope that occurs beyond the first 10 minutes of orthostasis in up to 38% of these patients.17
In a less common, hyperadrenergic form of postural orthostatic tachycardia syndrome, there is no autonomic failure but the sympathetic system is overly activated, with orthostasis leading to excessive tachycardia.10,18
CARDIAC SYNCOPE
Accounting for 10% to 20% of cases of syncope, a cardiac cause is the main concern in patients presenting with syncope, as cardiac syncope predicts an increased risk of death and may herald sudden cardiac death.1,2,8,19,20 It often occurs suddenly without any warning signs, in which case it is called malignant syncope. Unlike what occurs in neurally mediated syncope, the postrecovery period is not usually marked by lingering malaise.
There are three forms of cardiac syncope:
Syncope due to structural heart disease with cardiac obstruction
In cases of aortic stenosis, hypertrophic obstructive cardiomyopathy, or severe pulmonary arterial hypertension, peripheral vasodilation occurs during exercise, but cardiac output cannot increase because of the fixed or dynamic obstruction to the ventricular outflow. Since blood pressure is equal to cardiac output times peripheral vascular resistance, pressure drops with the reduction in peripheral vascular resistance. Exertional ventricular arrhythmias may also occur in these patients. Conversely, postexertional syncope is usually benign.
Syncope due to ventricular tachycardia
Ventricular tachycardia can be secondary to underlying structural heart disease, with or without reduced ejection fraction, such as coronary arterial disease, hypertrophic cardiomyopathy, hypertensive cardiomyopathy, or valvular disease. It can also be secondary to primary electrical disease (eg, long QT syndrome, Wolff-Parkinson-White syndrome, Brugada syndrome, arrhythmogenic right ventricular dysplasia, sarcoidosis).
Occasionally, fast supraventricular tachycardia causes syncope at its onset, before vascular compensation develops. This occurs in patients with underlying heart disease.2,8,19
Syncope from bradyarrhythmias
Bradyarrhythmias can occur with or without underlying structural heart disease. They are most often related to degeneration of the conduction system or to medications rather than to cardiomyopathy.
Caveats
When a patient with a history of heart failure presents with syncope, the top considerations are ventricular tachycardia and bradyarrhythmia. Nevertheless, about half of cases of syncope in patients with cardiac disease have a noncardiac cause,19 including the hypotensive or bradycardiac side effect of drugs.
As noted above, most cases of syncope are neurally mediated. However, long asystolic pauses due to sinus or atrioventricular nodal block are the most frequent mechanism of unexplained syncope and are seen in more than 50% of syncope cases on prolonged rhythm monitoring.1,21 These pauses may be related to intrinsic sinus or atrioventricular nodal disease or, more commonly, to extrinsic effects such as the vasovagal mechanism. Some experts favor classifying and treating syncope on the basis of the final mechanism rather than the initiating process, but this is not universally accepted.1,22
OTHER CAUSES OF SYNCOPE
Acute medical or cardiovascular illnesses can cause syncope and are looked for in the appropriate clinical context: severe hypovolemia or gastrointestinal bleeding, large pulmonary embolus with hemodynamic compromise, tamponade, aortic dissection, or hypoglycemia.
Bilateral critical carotid disease or severe vertebrobasilar disease very rarely cause syncope, and, when they do, they are associated with focal neurologic deficits.2 Vertebrobasilar disease may cause "drop attacks," ie, a loss of muscular tone with falling but without loss of consciousness.23
Severe proximal subclavian disease leads to reversal of the flow in the ipsilateral vertebral artery as blood is shunted toward the upper extremity. It manifests as dizziness and syncope during the ipsilateral upper extremity activity, usually with focal neurologic signs (subclavian steal syndrome).2
Psychogenic pseudosyncope is characterized by frequent attacks that typically last longer than true syncope and occur multiple times per day or week, sometimes with a loss of motor tone.2 It occurs in patients with anxiety or somatization disorders.
SEIZURE: A SYNCOPE MIMIC
Certain features differentiate seizure from syncope:
- In seizure, unconsciousness often lasts longer than 5 minutes
- After a seizure, the patient may experience postictal confusion or paralysis
- Seizure may include prolonged tonic-clonic movements; although these movements may be seen with any form of syncope lasting more than 30 seconds, the movements during syncope are more limited and brief, lasting less than 15 seconds
- Tongue biting strongly suggests seizure.
Urinary incontinence does not help distinguish the two, as it frequently occurs with syncope as well as seizure.
DIAGNOSTIC EVALUATION OF SYNCOPE
Table 2 lists clinical clues to the type of syncope.2–5,8
Underlying structural heart disease is the most important predictor of ventricular arrhythmia and death.20,24–26 Thus, the primary goal of the evaluation is to rule out structural heart disease by history, examination, electrocardiography, and echocardiography (Figure 1).
Initial strategy for finding the cause
The cause of syncope is diagnosed by history and physical examination alone in up to 50% of cases, mainly neurally mediated syncope, orthostatic syncope, or seizure.2,3,19
Always check blood pressure with the patient both standing and sitting and in both arms, and obtain an electrocardiogram.
Perform carotid massage in all patients over age 50 if syncope is not clearly vasovagal or orthostatic and if cardiac syncope is not likely. Carotid massage is contraindicated if the patient has a carotid bruit or a history of stroke.
Electrocardiography establishes or suggests a diagnosis in 10% of patients (Table 3, Figure 2).1,2,8,19 A normal electrocardiogram or a mild nonspecific ST-T abnormality suggests a low likelihood of cardiac syncope and is associated with an excellent prognosis. Abnormal electrocardiographic findings are seen in 90% of cases of cardiac syncope and in only 6% of cases of neurally mediated syncope.27 In one study of syncope patients with normal electrocardiograms and negative cardiac histories, none had an abnormal echocardiogram.28
If the heart is normal
If the history suggests neurally mediated syncope or orthostatic hypotension and the history, examination, and electrocardiogram do not suggest coronary artery disease or any other cardiac disease, the workup is stopped.
If the patient has signs or symptoms of heart disease
If the patient has signs or symptoms of heart disease (angina, exertional syncope, dyspnea, clinical signs of heart failure, murmur), a history of heart disease, or exertional, supine, or malignant features, heart disease should be looked for and the following performed:
- Echocardiography to assess left ventricular function, severe valvular disease, and left ventricular hypertrophy
- A stress test (possibly) in cases of exertional syncope or associated angina; however, the overall yield of stress testing in syncope is low (< 5%).29
If electrocardiography and echocardiography do not suggest heart disease
Often, in this situation, the workup can be stopped and syncope can be considered neurally mediated. The likelihood of cardiac syncope is very low in patients with normal findings on electrocardiography and echocardiography, and several studies have shown that patients with syncope who have no structural heart disease have normal long-term survival rates.20,26,30
The following workup may, however, be ordered if the presentation is atypical and syncope is malignant, recurrent, or associated with physical injury, or occurs in the supine position19:
Carotid sinus massage in patients over age 50, if not already performed. Up to 50% of these patients with unexplained syncope have carotid sinus hypersensitivity.13
24-hour Holter monitoring rarely detects significant arrhythmias, but if syncope or dizziness occurs without any arrhythmia, Holter monitoring rules out arrhythmia as the cause of the symptoms.31 The diagnostic yield of Holter monitoring is low (1% to 2%) in patients with infrequent symptoms1,2 and is not improved with 72-hour monitoring.30 The yield is higher in patients with very frequent daily symptoms, many of whom have psychogenic pseudosyncope.2
Tilt-table testing to diagnose vasovagal syncope. This test is positive for a vasovagal response in up to 66% of patients with unexplained syncope.1,19 Patients with heart disease taking vasodilators or beta-blockers may have abnormal baroreflexes. Therefore, a positive tilt test is less specific in these patients and does not necessarily indicate vasovagal syncope.
Event monitoring. If the etiology remains unclear or there are some concerns about arrhythmia, an event monitor (4 weeks of external rhythm monitoring) or an implantable loop recorder (implanted subcutaneously in the prepectoral area for 1 to 2 years) is placed. These monitors record the rhythm when the rate is lower or higher than predefined cutoffs or when the rhythm is irregular, regardless of symptoms. The patient or an observer can also activate the event monitor during or after an event, which freezes the recording of the 2 to 5 minutes preceding the activation and the 1 minute after it.
In a patient who has had syncope, a pacemaker is indicated for episodes of high-grade atrioventricular block, pauses longer than 3 seconds while awake, or bradycardia (< 40 beats per minute) while awake, and an implantable cardioverter-defibrillator is indicated for sustained ventricular tachycardia, even if syncope does not occur concomitantly with these findings. The finding of nonsustained ventricular tachycardia on monitoring increases the suspicion of ventricular tachycardia as the cause of syncope but does not prove it, nor does it necessarily dictate implantation of a cardioverter-defibrillator device.
An electrophysiologic study has a low yield in patients with normal electrocardiographic and echocardiographic studies. Bradycardia is detected in 10%.31
If heart disease or a rhythm abnormality is found
If heart disease is diagnosed by echocardiography or if significant electrocardiographic abnormalities are found, perform the following:
Pacemaker placement for the following electrocardiographic abnormalities1,2,19:
- Second-degree Mobitz II or third-degree atrioventricular block
- Sinus pause (> 3 seconds) or bradycardia (< 40 beats per minute) while awake
- Alternating left bundle branch block and right bundle branch block on the same electrocardiogram or separate ones.
Telemetric monitoring (inpatient).
An electrophysiologic study is valuable mainly for patients with structural heart disease, including an ejection fraction 36% to 49%, coronary artery disease, or left ventricular hypertrophy with a normal ejection fraction.32 Overall, in patients with structural heart disease and unexplained syncope, the yield is 55% (inducible ventricular tachycardia in 21%, abnormal indices of bradycardia in 34%).31
However, the yield of electrophysiologic testing is low in bradyarrhythmia and in patients with an ejection fraction of 35% or less.33 In the latter case, the syncope is often arrhythmia-related and the patient often has an indication for an implantable cardioverter-defibrillator regardless of electrophysiologic study results, especially if the low ejection fraction has persisted despite medical therapy.32
If the electrophysiologic study is negative
If the electrophysiologic study is negative, the differential diagnosis still includes arrhythmia, as the yield of electrophysiologic study is low for bradyarrhythmias and some ventricular tachycardias, and the differential diagnosis also includes, at this point, neurally mediated syncope.
The next step may be either prolonged rhythm monitoring or tilt-table testing. An event monitor or an implantable loop recorder can be placed for prolonged monitoring. The yield of the 30-day event monitor is highest in patients with frequently recurring syncope, in whom it reaches a yield of up to 40% (10% to 20% will have a positive diagnosis of arrhythmia, while 15% to 20% will have symptoms with a normal rhythm).31,34 The implantable recorder has a high overall diagnostic yield and is used in patients with infrequent syncopal episodes (yield up to 50%).1,35,36
In brief, there are two diagnostic approaches to unexplained syncope: the monitoring approach (loop recorder) and the testing approach (tilt-table testing). A combination of both strategies is frequently required in patients with unexplained syncope, and, according to some investigators, a loop recorder may be implanted early on.21
Heart disease with left ventricular dysfunction and low ejection fraction
In patients with heart disease with left ventricular dysfunction and an ejection fraction of 35% or less, an implantable cardioverter-defibrillator can be placed without the need for an electrophysiologic study. These patients need these devices anyway to prevent sudden death, even if the cause of syncope is not an arrhythmia. Patients with a low ejection fraction and a history of syncope are at a high risk of sudden cardiac death.32 Yet in some patients with newly diagnosed cardiomyopathy, left ventricular function may improve with medical therapy. Because the arrhythmic risk is essentially high during the period of ventricular dysfunction, a wearable external defibrillator may be placed while the decision about an implantable defibrillator is finalized within the ensuing months.
In patients with hypertrophic cardiomyopathy, place an implantable cardioverter-defibrillator after any unexplained syncopal episode.
Valvular heart disease needs surgical correction.
If ischemic heart disease is suspected, coronary angiography is indicated, with revascularization if appropriate. An implantable cardioverter-defibrillator should be placed if the ejection fraction is lower than 35%. Except in a large acute myocardial infarction, the substrate for ventricular tachycardia is not ameliorated with revascularization.32,37 Consider an electrophysiologic study when syncope occurs with coronary artery disease and a higher ejection fraction.
A note on left or right bundle branch block
Patients with left or right bundle branch block and unexplained syncope (not clearly vasovagal or orthostatic) likely have syncope related to intermittent high-grade atrioventricular block.38
One study monitored these patients with an implanted loop recorder and showed that about 40% had a recurrence of syncope within 48 days, often concomitantly with complete atrioventricular block. About 55% of these patients had a major event (syncope or high-grade atrioventricular block).39 Many of the patients had had a positive tilt test; thus, tilt testing is not specific for vasovagal syncope in these patients and should not be used to exclude a bradyarrhythmic syncope. Also, patients selected for this study had undergone carotid sinus massage and an electrophysiology study with a negative result.
In another analysis, an electrophysiologic study detected a proportion of the bradyarrhythmias but, more importantly, it induced ventricular tachycardia in 14% of patients with right or left bundle branch block. Although it is not sensitive enough for bradyarrhythmia, electrophysiologic study was highly specific and fairly sensitive for the occurrence of ventricular tachycardia on follow-up.38 Thus, unexplained syncope in a patient with right or left bundle branch block may warrant carotid sinus massage, then an electrophysiologic study to rule out ventricular tachycardia, followed by placement of a dual-chamber pacemaker if the study is negative for ventricular tachycardia, or at least placement of a loop recorder.
INDICATIONS FOR HOSPITALIZATION
Patients should be hospitalized if they have severe hypovolemia or bleeding, or if there is any suspicion of heart disease by history, examination, or electrocardiography, including:
- History of heart failure, low ejection fraction, or coronary artery disease
- An electrocardiogram suggestive of arrhythmia (Table 3)
- Family history of sudden death
- Lack of prodromes; occurrence of physical injury, exertional syncope, syncope in a supine position, or syncope associated with dyspnea or chest pain.2,40
In these situations, there is concern about arrhythmia, structural heart disease, or acute myocardial ischemia. The patient is admitted for immediate telemetric monitoring. Echocardiography and sometimes stress testing are performed. The patient is discharged if this initial workup does not suggest underlying heart disease. Alternatively, an electrophysiologic study is performed or a device is placed in patients found to have structural heart disease. Prolonged rhythm monitoring or tilt-table testing may be performed when syncope with underlying heart disease or worrisome features remains unexplained.
Several Web-based interactive algorithms have been used to determine the indication for hospitalization. They incorporate the above clinical, electrocardiographic, and sometimes echocardiographic features.2,24,25,40–42 A cardiology consultation is usually necessary in patients with the above features, as they frequently require specialized cardiac testing.
Among high-risk patients, the risk of sudden death, a major cardiovascular event, or significant arrhythmia is high in the first few days after the index syncopal episode, justifying the hospitalization and inpatient rhythm monitoring and workup in the presence of the above criteria.24,40,42
SYNCOPE AND DRIVING
A study has shown that the most common cause of syncope while driving is vasovagal syncope.6 In all patients, the risk of another episode of syncope was relatively higher during the first 6 months after the event, with a 12% recurrence rate during this period. However, recurrences were often also seen more than 6 months later (12% recurrence between 6 months and the following few years).6 Fortunately, those episodes rarely occurred while the patient was driving. In a study in survivors of ventricular arrhythmia, the risk of recurrence of arrhythmic events was highest during the first 6 to 12 months after the event.43
Thus, in general, patients with syncope should be prohibited from driving for at least the period of time (eg, 6 months) during which the risk of a recurrent episode of syncope is highest and during which serious cardiac disease or arrhythmia, if present, would emerge. Recurrence of syncope is more likely and more dangerous for commercial drivers who spend a significant proportion of their time driving; individualized decisions are made in these cases.
- Brignole M, Hamdan MH. New concepts in the assessment of syncope. J Am Coll Cardiol 2012; 59:1583–1591.
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS); Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009 Eur Heart J 2009; 30:2631–2671.
- Kapoor WN. Syncope. N Engl J Med 2000; 343:1856–1862.
- Graham LA, Kenny RA. Clinical characteristics of patients with vasovagal reactions presenting as unexplained syncope. Europace 2001; 3:141–146.
- Calkins H, Shyr Y, Frumin H, Schork A, Morady F. The value of the clinical history in the differentiation of syncope due to ventricular tachycardia, atrioventricular block, and neurocardiogenic syncope. Am J Med 1995; 98:365–373.
- Sorajja D, Nesbitt GC, Hodge DO, et al. Syncope while driving: clinical characteristics, causes, and prognosis. Circulation 2009; 120:928–934.
- Kochiadakis GE, Papadimitriou EA, Marketou ME, Chrysostomakis SI, Simantirakis EN, Vardas PE. Autonomic nervous system changes in vasovagal syncope: is there any difference between young and older patients? Pacing Clin Electrophysiol 2004; 27:1371–1377.
- Alboni P, Brignole M, Menozzi C, et al. Diagnostic value of history in patients with syncope with or without heart disease. J Am Coll Cardiol 2001; 37:1921–1928.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Task force on syncope, European Society of Cardiology. Part 1. The initial evaluation of patients with syncope. Europace 2001; 3:253–260.
- Grubb BP. Neurocardiogenic syncope and related disorders of orthostatic intolerance. Circulation 2005; 111:2997–3006.
- Brignole M, Menozzi C, Del Rosso A, et al. New classification of haemodynamics of vasovagal syncope: beyond the VASIS classification. Analysis of the pre-syncopal phase of the tilt test without and with nitroglycerin challenge. Vasovagal Syncope International Study. Europace 2000; 2:66–76.
- Grubb BP, Kosinski D. Tilt table testing: concepts and limitations. Pacing Clin Electrophysiol 1997; 20:781–787.
- Brignole M, Menozzi C, Gianfranchi L, Oddone D, Lolli G, Bertulla A. Carotid sinus massage, eyeball compression, and head-up tilt test in patients with syncope of uncertain origin and in healthy control subjects. Am Heart J 1991; 122:1644–1651.
- Maggi R, Menozzi C, Brignole M, et al. Cardioinhibitory carotid sinus hypersensitivity predicts an asystolic mechanism of spontaneous neurally mediated syncope. Europace 2007; 9:563–567.
- Kenny RA, Richardson DA, Steen N, Bexton RS, Shaw FE, Bond J. Carotid sinus syndrome: a modifiable risk factor for nonaccidental falls in older adults (SAFE PACE). J Am Coll Cardiol 2001; 38:1491–1496.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Ojha A, McNeeley K, Heller E, Alshekhlee A, Chelimsky G, Chelimsky TC. Orthostatic syndromes differ in syncope frequency. Am J Med 2010; 123:245–249.
- Kanjwal Y, Kosinski D, Grubb BP. The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management. Pacing Clin Electrophysiol 2003; 26:1747–1757.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Guidelines on management (diagnosis and treatment) of syncope. Eur Heart J 2001; 22:1256–1306.
- Soteriades ES, Evans JC, Larson MG, et al. Incidence and prognosis of syncope. N Engl J Med 2002; 347:878–885.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology 3 (ISSUE-3) Investigators. Pacemaker therapy in patients with neurally mediated syncope and documented asystole: Third International Study on Syncope of Uncertain Etiology (ISSUE-3): a randomized trial. Circulation 2012; 125:2566–2571.
- Kubak MJ, Millikan CH. Diagnosis, pathogenesis, and treatment of "drop attacks." Arch Neurol 1964; 11:107–113.
- Quinn J, McDermott D, Stiell I, Kohn M, Wells G. Prospective validation of the San Francisco Syncope Rule to predict patients with serious outcomes. Ann Emerg Med 2006; 47:448–454.
- Colivicchi F, Ammirati F, Melina D, Guido V, Imperoli G, Santini M; OESIL (Osservatorio Epidemiologico sulla Sincope nel Lazio) Study Investigators. Development and prospective validation of a risk stratification system for patients with syncope in the emergency department: the OESIL risk score. Eur Heart J 2003; 24:811–819.
- Kapoor WN, Hanusa BH. Is syncope a risk factor for poor outcomes? Comparison of patients with and without syncope. Am J Med 1996; 100:646–655.
- Sarasin FP, Louis-Simonet M, Carballo D, et al. Prospective evaluation of patients with syncope: a population-based study. Am J Med 2001; 111:177–184.
- Sarasin FP, Junod AF, Carballo D, Slama S, Unger PF, Louis-Simonet M. Role of echocardiography in the evaluation of syncope: a prospective study. Heart 2002; 88:363–367.
- AlJaroudi WA, Alraies MC, Wazni O, Cerqueira MD, Jaber WA. Yield and diagnostic value of stress myocardial perfusion imaging in patients without known coronary artery disease presenting with syncope. Circ Cardiovasc Imaging 2013; 6:384–391.
- Ungar A, Del Rosso A, Giada F, et al; Evaluation of Guidelines in Syncope Study 2 Group. Early and late outcome of treated patients referred for syncope to emergency department: the EGSYS 2 follow-up study. Eur Heart J 2010; 31:2021–2026.
- Linzer M, Yang EH, Estes NA 3rd, Wang P, Vorperian VR, Kapoor WN. Diagnosing syncope. Part 2: Unexplained syncope. Clinical Efficacy Assessment Project of the American College of Physicians. Ann Intern Med 1997; 127:76–86.
- Strickberger SA, Benson DW, Biaggioni I, et al; American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke; Quality of Care and Outcomes Research Interdisciplinary Working Group; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF scientific statement on the evaluation of syncope: from the American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke, and the Quality of Care and Outcomes Research Interdisciplinary Working Group; and the American College of Cardiology Foundation In Collaboration With the Heart Rhythm Society. J Am Coll Cardiol 2006; 47:473–484.
- Fujimura O, Yee R, Klein GJ, Sharma AD, Boahene KA. The diagnostic sensitivity of electrophysiologic testing in patients with syncope caused by transient bradycardia. N Engl J Med 1989; 321:1703–1707.
- Linzer M, Pritchett EL, Pontinen M, McCarthy E, Divine GW. Incremental diagnostic yield of loop electrocardiographic recorders in unexplained syncope. Am J Cardiol 1990; 66:214–219.
- Edvardsson N, Frykman V, van Mechelen R, et al; PICTURE Study Investigators. Use of an implantable loop recorder to increase the diagnostic yield in unexplained syncope: results from the PICTURE registry. Europace 2011; 13:262–269.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brugada J, Aguinaga L, Mont L, Betriu A, Mulet J, Sanz G. Coronary artery revascularization in patients with sustained ventricular arrhythmias in the chronic phase of a myocardial infarction: effects on the electrophysiologic substrate and outcome. J Am Coll Cardiol 2001; 37:529–533.
- Moya A, García-Civera R, Croci F, et al; Bradycardia detection in Bundle Branch Block (B4) study. Diagnosis, management, and outcomes of patients with syncope and bundle branch block. Eur Heart J 2011; 32:1535–1541.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology (ISSUE) Investigators. Mechanism of syncope in patients with bundle branch block and negative electrophysiological test. Circulation 2001; 104:2045–2050.
- Brignole M, Shen WK. Syncope management from emergency department to hospital. J Am Coll Cardiol 2008; 51:284–287.
- Daccarett M, Jetter TL, Wasmund SL, Brignole M, Hamdan MH. Syncope in the emergency department: comparison of standardized admission criteria with clinical practice. Europace 2011; 13:1632–1638.
- Costantino G, Perego F, Dipaola F, et al; STePS Investigators. Short- and long-term prognosis of syncope, risk factors, and role of hospital admission: results from the STePS (Short-Term Prognosis of Syncope) study. J Am Coll Cardiol 2008; 51:276–283.
- Larsen GC, Stupey MR, Walance CG, et al. Recurrent cardiac events in survivors of ventricular fibrillation or tachycardia. Implications for driving restrictions. JAMA 1994; 271:1335–1339.
- Brignole M, Hamdan MH. New concepts in the assessment of syncope. J Am Coll Cardiol 2012; 59:1583–1591.
- Task Force for the Diagnosis and Management of Syncope; European Society of Cardiology (ESC); European Heart Rhythm Association (EHRA); Heart Failure Association (HFA); Heart Rhythm Society (HRS); Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009 Eur Heart J 2009; 30:2631–2671.
- Kapoor WN. Syncope. N Engl J Med 2000; 343:1856–1862.
- Graham LA, Kenny RA. Clinical characteristics of patients with vasovagal reactions presenting as unexplained syncope. Europace 2001; 3:141–146.
- Calkins H, Shyr Y, Frumin H, Schork A, Morady F. The value of the clinical history in the differentiation of syncope due to ventricular tachycardia, atrioventricular block, and neurocardiogenic syncope. Am J Med 1995; 98:365–373.
- Sorajja D, Nesbitt GC, Hodge DO, et al. Syncope while driving: clinical characteristics, causes, and prognosis. Circulation 2009; 120:928–934.
- Kochiadakis GE, Papadimitriou EA, Marketou ME, Chrysostomakis SI, Simantirakis EN, Vardas PE. Autonomic nervous system changes in vasovagal syncope: is there any difference between young and older patients? Pacing Clin Electrophysiol 2004; 27:1371–1377.
- Alboni P, Brignole M, Menozzi C, et al. Diagnostic value of history in patients with syncope with or without heart disease. J Am Coll Cardiol 2001; 37:1921–1928.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Task force on syncope, European Society of Cardiology. Part 1. The initial evaluation of patients with syncope. Europace 2001; 3:253–260.
- Grubb BP. Neurocardiogenic syncope and related disorders of orthostatic intolerance. Circulation 2005; 111:2997–3006.
- Brignole M, Menozzi C, Del Rosso A, et al. New classification of haemodynamics of vasovagal syncope: beyond the VASIS classification. Analysis of the pre-syncopal phase of the tilt test without and with nitroglycerin challenge. Vasovagal Syncope International Study. Europace 2000; 2:66–76.
- Grubb BP, Kosinski D. Tilt table testing: concepts and limitations. Pacing Clin Electrophysiol 1997; 20:781–787.
- Brignole M, Menozzi C, Gianfranchi L, Oddone D, Lolli G, Bertulla A. Carotid sinus massage, eyeball compression, and head-up tilt test in patients with syncope of uncertain origin and in healthy control subjects. Am Heart J 1991; 122:1644–1651.
- Maggi R, Menozzi C, Brignole M, et al. Cardioinhibitory carotid sinus hypersensitivity predicts an asystolic mechanism of spontaneous neurally mediated syncope. Europace 2007; 9:563–567.
- Kenny RA, Richardson DA, Steen N, Bexton RS, Shaw FE, Bond J. Carotid sinus syndrome: a modifiable risk factor for nonaccidental falls in older adults (SAFE PACE). J Am Coll Cardiol 2001; 38:1491–1496.
- Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology 2006; 67:28–32.
- Ojha A, McNeeley K, Heller E, Alshekhlee A, Chelimsky G, Chelimsky TC. Orthostatic syndromes differ in syncope frequency. Am J Med 2010; 123:245–249.
- Kanjwal Y, Kosinski D, Grubb BP. The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management. Pacing Clin Electrophysiol 2003; 26:1747–1757.
- Brignole M, Alboni P, Benditt D, et al; Task Force on Syncope; European Society of Cardiology. Guidelines on management (diagnosis and treatment) of syncope. Eur Heart J 2001; 22:1256–1306.
- Soteriades ES, Evans JC, Larson MG, et al. Incidence and prognosis of syncope. N Engl J Med 2002; 347:878–885.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology 3 (ISSUE-3) Investigators. Pacemaker therapy in patients with neurally mediated syncope and documented asystole: Third International Study on Syncope of Uncertain Etiology (ISSUE-3): a randomized trial. Circulation 2012; 125:2566–2571.
- Kubak MJ, Millikan CH. Diagnosis, pathogenesis, and treatment of "drop attacks." Arch Neurol 1964; 11:107–113.
- Quinn J, McDermott D, Stiell I, Kohn M, Wells G. Prospective validation of the San Francisco Syncope Rule to predict patients with serious outcomes. Ann Emerg Med 2006; 47:448–454.
- Colivicchi F, Ammirati F, Melina D, Guido V, Imperoli G, Santini M; OESIL (Osservatorio Epidemiologico sulla Sincope nel Lazio) Study Investigators. Development and prospective validation of a risk stratification system for patients with syncope in the emergency department: the OESIL risk score. Eur Heart J 2003; 24:811–819.
- Kapoor WN, Hanusa BH. Is syncope a risk factor for poor outcomes? Comparison of patients with and without syncope. Am J Med 1996; 100:646–655.
- Sarasin FP, Louis-Simonet M, Carballo D, et al. Prospective evaluation of patients with syncope: a population-based study. Am J Med 2001; 111:177–184.
- Sarasin FP, Junod AF, Carballo D, Slama S, Unger PF, Louis-Simonet M. Role of echocardiography in the evaluation of syncope: a prospective study. Heart 2002; 88:363–367.
- AlJaroudi WA, Alraies MC, Wazni O, Cerqueira MD, Jaber WA. Yield and diagnostic value of stress myocardial perfusion imaging in patients without known coronary artery disease presenting with syncope. Circ Cardiovasc Imaging 2013; 6:384–391.
- Ungar A, Del Rosso A, Giada F, et al; Evaluation of Guidelines in Syncope Study 2 Group. Early and late outcome of treated patients referred for syncope to emergency department: the EGSYS 2 follow-up study. Eur Heart J 2010; 31:2021–2026.
- Linzer M, Yang EH, Estes NA 3rd, Wang P, Vorperian VR, Kapoor WN. Diagnosing syncope. Part 2: Unexplained syncope. Clinical Efficacy Assessment Project of the American College of Physicians. Ann Intern Med 1997; 127:76–86.
- Strickberger SA, Benson DW, Biaggioni I, et al; American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke; Quality of Care and Outcomes Research Interdisciplinary Working Group; American College of Cardiology Foundation; Heart Rhythm Society. AHA/ACCF scientific statement on the evaluation of syncope: from the American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke, and the Quality of Care and Outcomes Research Interdisciplinary Working Group; and the American College of Cardiology Foundation In Collaboration With the Heart Rhythm Society. J Am Coll Cardiol 2006; 47:473–484.
- Fujimura O, Yee R, Klein GJ, Sharma AD, Boahene KA. The diagnostic sensitivity of electrophysiologic testing in patients with syncope caused by transient bradycardia. N Engl J Med 1989; 321:1703–1707.
- Linzer M, Pritchett EL, Pontinen M, McCarthy E, Divine GW. Incremental diagnostic yield of loop electrocardiographic recorders in unexplained syncope. Am J Cardiol 1990; 66:214–219.
- Edvardsson N, Frykman V, van Mechelen R, et al; PICTURE Study Investigators. Use of an implantable loop recorder to increase the diagnostic yield in unexplained syncope: results from the PICTURE registry. Europace 2011; 13:262–269.
- Brignole M, Sutton R, Menozzi C, et al; International Study on Syncope of Uncertain Etiology 2 (ISSUE 2) Group. Early application of an implantable loop recorder allows effective specific therapy in patients with recurrent suspected neurally mediated syncope. Eur Heart J 2006; 27:1085–1092.
- Brugada J, Aguinaga L, Mont L, Betriu A, Mulet J, Sanz G. Coronary artery revascularization in patients with sustained ventricular arrhythmias in the chronic phase of a myocardial infarction: effects on the electrophysiologic substrate and outcome. J Am Coll Cardiol 2001; 37:529–533.
- Moya A, García-Civera R, Croci F, et al; Bradycardia detection in Bundle Branch Block (B4) study. Diagnosis, management, and outcomes of patients with syncope and bundle branch block. Eur Heart J 2011; 32:1535–1541.
- Brignole M, Menozzi C, Moya A, et al; International Study on Syncope of Uncertain Etiology (ISSUE) Investigators. Mechanism of syncope in patients with bundle branch block and negative electrophysiological test. Circulation 2001; 104:2045–2050.
- Brignole M, Shen WK. Syncope management from emergency department to hospital. J Am Coll Cardiol 2008; 51:284–287.
- Daccarett M, Jetter TL, Wasmund SL, Brignole M, Hamdan MH. Syncope in the emergency department: comparison of standardized admission criteria with clinical practice. Europace 2011; 13:1632–1638.
- Costantino G, Perego F, Dipaola F, et al; STePS Investigators. Short- and long-term prognosis of syncope, risk factors, and role of hospital admission: results from the STePS (Short-Term Prognosis of Syncope) study. J Am Coll Cardiol 2008; 51:276–283.
- Larsen GC, Stupey MR, Walance CG, et al. Recurrent cardiac events in survivors of ventricular fibrillation or tachycardia. Implications for driving restrictions. JAMA 1994; 271:1335–1339.
KEY POINTS
- Neurally mediated forms of syncope, such as vasovagal, result from autonomic reflexes that respond inappropriately, leading to vasodilation and relative bradycardia.
- Orthostatic hypotension is the most common cause of syncope in the elderly and may be due to autonomic dysfunction, volume depletion, or drugs that block autonomic effects or cause hypovolemia, such as vasodilators, beta-blockers, diuretics, neuropsychiatric medications, and alcohol.
- The likelihood of cardiac syncope is low in patients with normal electrocardiographic and echocardiographic findings.
- Hospitalization is indicated in patients with syncope who have or are suspected of having structural heart disease.
Updated guidelines on cardiovascular evaluation before noncardiac surgery: A view from the trenches
Guidelines jointly issued by the American College of Cardiology and American Heart Association (ACC/AHA)1 provide a framework for evaluating and managing perioperative cardiac risk in noncardiac surgery. An overriding theme in successive documents from these organizations through the years has been that preoperative intervention, coronary artery bypass grafting, or percutaneous coronary intervention is rarely necessary just to get the patient through surgery, unless it is otherwise indicated independent of the need for surgery.
This article highlights some of the key recommendations in the 2014 updates to these guidelines,1–3 how they differ from previous guidelines,4 and the ongoing challenges and unresolved issues facing physicians involved in perioperative care.
Of note, while these guidelines were being updated, Erasmus University5 expressed concern about the scientific integrity of some of the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE) trials. As a result, the evidence review committee included these trials in its analysis but not in a systematic review of beta-blockers.2 These trials were not included in the clinical practice guideline supplements and tables but were cited in the text if relevant.
The European Society of Cardiology and European Society of Anesthesiology6 revised their guidelines concurrently with but independently of the ACC/AHA, and although they discussed and aligned some recommendations, many differences remain between the two sets of guidelines. Readers should consult the full guidelines for more detailed information.1
THE ROLE OF THE PREOPERATIVE CARDIAC EVALUATION
The purpose of preoperative medical evaluation is not to "get medical clearance" but rather to evaluate the patient’s medical status and risk of complications. The process includes:
- Identifying risk factors and assessing their severity and stability
- Establishing a clinical risk profile for informed and shared decision-making
- Recommending needed changes in management, further testing, or specialty consultation.
The updated guidelines emphasize the importance of communication among the perioperative team and with the patient. They reiterate the focus on appropriateness of care and cost containment—one should order a test only if the result may change the patient’s management.
HOW URGENT IS SURGERY? HOW RISKY?
The new guidelines classify the urgency of surgery as follows:
- Emergency (necessary within 6 hours)
- Urgent (necessary within 6–24 hours)
- Time-sensitive (can delay 1–6 weeks)
- Elective (can delay up to 1 year).
Surgical risk is now classified as either low (< 1% risk of major adverse cardiac events) or elevated (≥ 1%) on the basis of surgical and patient characteristics. Previous schemas included an intermediate-risk category. Low-risk procedures include endoscopic procedures, superficial procedures, cataract surgery, breast surgery, and ambulatory surgery. Elevated-risk procedures include vascular surgery, intraperitoneal and intrathoracic surgery, head and neck surgery, orthopedic surgery, and prostate surgery.
Risk calculators and biomarkers
To estimate the perioperative risk of major adverse cardiac events, the guidelines suggest incorporating the Revised Cardiac Risk Index (RCRI)7 with an estimation of surgical risk or using a newer surgical risk calculator derived from a database of the American College of Surgeons’ National Surgical Quality Improvement Project (ACS NSQIP).
The RCRI is based on six risk factors, each worth 1 point:
- High-risk surgery
- Ischemic heart disease
- Heart failure
- Stroke or transient ischemic attack
- Diabetes requiring insulin
- Renal insufficiency (serum creatinine > 2.0 mg/dL).7
MICA. The Myocardial Infarction or Cardiac Arrest (MICA) calculator8 has a narrower focus and was validated in only one center.
ACS NSQIP. The recommended newer ACS NSQIP surgical risk calculator9 provides an estimate of procedure-specific risk based on Current Procedural Terminology code and includes 21 patient-specific variables to predict death, major adverse cardiac events, and eight other outcomes. While more comprehensive, this risk calculator has yet to be validated outside of the ACS NSQIP database.
Reconstructed RCRI. The RCRI has been externally validated, but it underestimates risk in major vascular surgery and was outperformed by the MICA calculator. Although not discussed in the new guidelines, a recently published "reconstructed RCRI,"10 in which a serum creatinine level greater than 2 mg/dL in the original RCRI is replaced by a glomerular filtration rate less than 30 mL/min and diabetes is eliminated, may outperform the standard RCRI. A patient with either an RCRI score or a reconstructed RCRI score of 0 or 1 would be considered to be at low risk, whereas patients with two or more risk factors would have an elevated risk.
Cardiac biomarkers, primarily B-type natriuretic peptide (BNP) and N-terminal (NT) proBNP, are independent predictors of cardiac risk, and their addition to preoperative risk indices may provide incremental predictive value. However, how to use these biomarkers and whether any treatment aimed at them will reduce risk is unclear, and the new guidelines did not recommend their routine use.
CLINICAL RISK FACTORS
Coronary artery disease
Ischemic symptoms, a history of myocardial infarction, and elevated cardiac biomarkers are individually associated with perioperative risk of morbidity and death. The risk is modified by how long ago the infarction occurred, whether the patient underwent coronary revascularization, and if so, what type (bypass grafting or percutaneous coronary intervention). A patient with acute coronary syndrome (currently or in the recent past) is at higher risk, and should have elective surgery delayed and be referred for cardiac evaluation and management according to guidelines.
Heart failure
In terms of posing a risk for major adverse cardiac events, heart failure is at least equal to coronary artery disease, and is possibly worse. Its impact depends on its stability, its symptoms, and the patient’s left ventricular function. Symptomatic decompensated heart failure and depressed left ventricular function (ejection fraction < 30% or 40%) confer higher risk than asymptomatic heart failure and preserved left ventricular function. However, evidence is limited with respect to asymptomatic left ventricular dysfunction and diastolic dysfunction. Patients with stable heart failure treated according to guidelines may have better perioperative outcomes.
Valvular heart disease
Significant valvular heart disease is associated with increased risk of postoperative cardiac complications. This risk depends on the type and severity of the valvular lesion and type of noncardiac surgery, but can be minimized by clinical and echocardiographic assessment, choosing appropriate anesthesia, and closer perioperative monitoring. Aortic and mitral stenosis are associated with greater risk of perioperative adverse cardiac events than regurgitant valvular disease.
Echocardiography is recommended in patients suspected of having moderate to severe stenotic or regurgitant lesions if it has not been done within the past year or if the patient’s clinical condition has worsened.
If indicated, valvular intervention can reduce perioperative risk in these patients. Even if the planned noncardiac surgery is high-risk, it may be reasonable to proceed with it (using appropriate perioperative hemodynamic monitoring, which is not specified but typically would be with an arterial line, central line, and possibly a pulmonary arterial catheter) in patients who have asymptomatic severe aortic or mitral regurgitation or aortic stenosis. Surgery may also be reasonable in patients with asymptomatic severe mitral stenosis who are not candidates for repair.
Arrhythmias
Cardiac arrhythmias and conduction defects are often seen in the perioperative period, but there is only limited evidence as to how they affect surgical risk. In addition to their hemodynamic effects, certain arrhythmias (atrial fibrillation, ventricular tachycardia) often indicate underlying structural heart disease, which requires further evaluation before surgery.
The new guidelines refer the reader to previously published clinical practice guidelines for atrial fibrillation,11 supraventricular arrhythmias,12 and device-based therapy.13
ALGORITHM FOR PREOPERATIVE CARDIAC ASSESSMENT
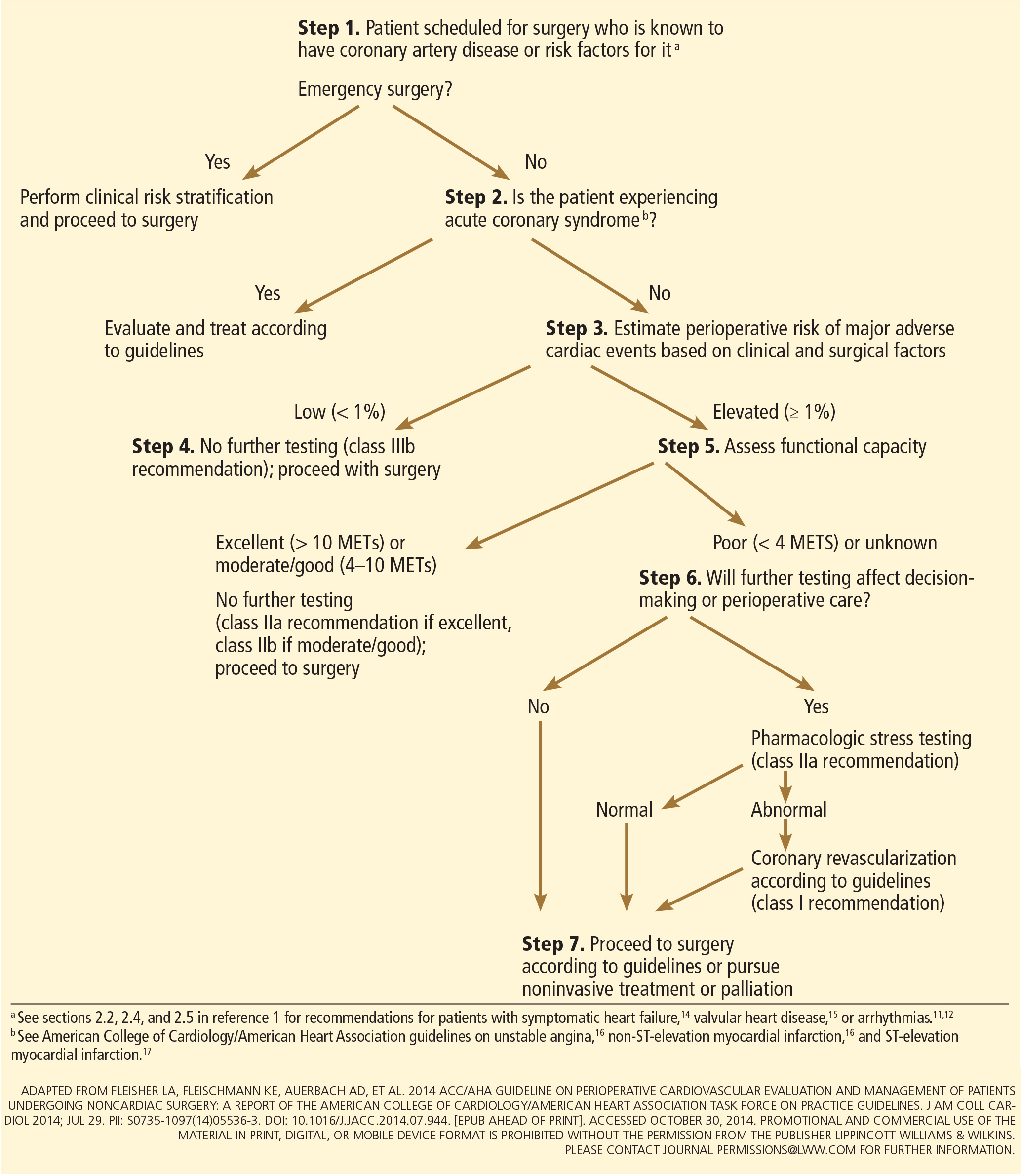
The new algorithm for evaluating a patient who is known to have coronary artery disease or risk factors for it has seven steps (Figure 1).1,11,12,14–17 It differs from the previous algorithm in several details:
- Instead of listing the four active cardiac conditions for which elective surgery should be delayed while the patient is being evaluated and treated (unstable coronary syndrome, decompensated heart failure, significant arrhythmias, severe valvular heart disease), the new version specifically asks about acute coronary syndrome and recommends cardiac evaluation and treatment according to guidelines. A footnote directs readers to other clinical practice guidelines for symptomatic heart failure,14 valvular heart disease,15 and arrhythmias.11,12
- Instead of asking if the procedure is low-risk, the guidelines recommend estimating risk of major adverse cardiac events on the basis of combined clinical and surgical risk and define only two categories: low or elevated. Patients at low risk proceed to surgery with no further testing, as in the earlier algorithm.
- "Excellent" exercise capacity (> 10 metabolic equivalents of task [METs]) is separated from "moderate/good" (4–10 METs), presumably to indicate a stronger recommendation, but patients in both categories proceed to surgery as before.
- If the patient cannot exercise to at least 4 METs, the new algorithm asks whether further testing will affect decision-making or perioperative care (an addition to the previous algorithm). This entails discussing with the patient and perioperative team whether the original surgery will be performed and whether the patient is willing to undergo revascularization if indicated. If so, pharmacologic stress testing is recommended. Previously, this decision also included the number of RCRI factors as well as the type of surgery (vascular or nonvascular).
- If testing will not affect the decision or if the stress test is normal, in addition to recommending proceeding to surgery according to guidelines the new algorithm also lists an option for alternative strategies, including palliation.
- If the stress test is abnormal, especially with left main disease, it recommends coronary revascularization according to the 2011 clinical practice guidelines.18,19
TESTING FOR LEFT VENTRICULAR DYSFUNCTION OR ISCHEMIA
In patients with dyspnea of unexplained cause or worsening dyspnea, assessment of left ventricular function is reasonable, but this is not part of a routine preoperative evaluation.
Pharmacologic stress testing is reasonable for patients at elevated risk with poor functional capacity if the results will change their management, but it is not useful for patients undergoing low-risk surgery. Although dobutamine stress echocardiography may be slightly superior to pharmacologic myocardial perfusion imaging, there are no head-to-head randomized controlled trials, and the guidelines suggest considering local expertise in deciding which test to use.
The presence of moderate to large areas of ischemia (reversible perfusion defects or new wall-motion abnormalities) is associated with risk of perioperative myocardial infarction or death, whereas evidence of an old infarction is associated with long-term but not short-term risk. The negative predictive value of these tests in predicting postoperative cardiac events is high (> 90%), but the positive predictive value is low.
CORONARY REVASCULARIZATION
Coronary artery bypass grafting and percutaneous coronary intervention
The guidelines recommend coronary revascularization before noncardiac surgery only when it is indicated anyway, on the basis of existing clinical practice guidelines.
Whether performing percutaneous coronary intervention before surgery will reduce perioperative cardiac complications is uncertain, and coronary revascularization should not be routinely performed solely to reduce perioperative cardiac events. The only two randomized controlled trials, Coronary Artery Revascularization Prophylaxis (CARP)20 and DECREASE V21 evaluating prophylactic coronary revascularization before noncardiac surgery found no difference in either short-term or long-term outcomes, although subgroup analysis found a survival benefit in patients with left main disease who underwent bypass grafting. Preoperative percutaneous coronary intervention should be limited to patients with left main disease in whom comorbidities preclude bypass surgery and those with unstable coronary disease who may benefit from early invasive management.
The urgency and timing of the noncardiac surgery needs to be taken into account if percutaneous coronary intervention is being considered because of the need for antiplatelet therapy after the procedure, and the potential risks of bleeding and stent thrombosis. If the planned surgery is deemed time-sensitive, then balloon angioplasty or bare-metal stenting is preferred over placement of a drug-eluting stent.
The new guidelines continue to recommend that elective noncardiac surgery be delayed at least 14 days after balloon angioplasty, 30 days after bare-metal stent implantation, and ideally 365 days after drug-eluting stent placement, and reiterate that it is potentially harmful to perform elective surgery within these time frames without any antiplatelet therapy. However, a new class IIb recommendation (benefit ≥ risk) states that "elective noncardiac surgery after [drug-eluting stent] implantation may be considered after 180 days if the risk of further delay is greater than the expected risks of ischemia and stent thrombosis."
This is an important addition to the guidelines because we are often faced with patients needing to undergo surgery in the 6 to 12 months after placement of a drug-eluting stent. Based on previous guidelines, whether it was safe to proceed in this setting created controversy among the perioperative team caring for the patient, and surgery was often delayed unnecessarily. Recent studies22,23 suggest that the newer drug-eluting stents may require a shorter duration of dual antiplatelet therapy, at least in the nonsurgical setting.
MEDICAL THERAPY
Antiplatelet therapy: Stop or continue?
The risk of perioperative bleeding if antiplatelet drugs are continued must be weighed against the risk of stent thrombosis and ischemia if they are stopped before the recommended duration of therapy. Ideally, some antiplatelet therapy should be continued perioperatively in these situations, but the guidelines recommend that a consensus decision among the treating physicians should be made regarding the relative risks of surgery and discontinuation or continuation of antiplatelet therapy. Whenever possible, aspirin should be continued in these patients.
Although the Perioperative Ischemic Evaluation (POISE)-2 trial24 found that perioperative aspirin use was not associated with lower rates of postoperative myocardial infarction or death, it increased bleeding. Patients with stents who had not completed the recommended duration of antiplatelet therapy were excluded from the trial. Additionally, only 5% of the study patients had undergone percutaneous coronary intervention.
According to the guidelines and package inserts, if antiplatelet agents need to be discontinued before surgery, aspirin can be stopped 3 to 7 days before, clopidogrel and ticagrelor 5 days before, and prasugrel 7 days before. In patients without stents, it may be reasonable to continue aspirin perioperatively if the risk of cardiac events outweighs the risk of bleeding, but starting aspirin is not beneficial for patients undergoing elective noncardiac noncarotid surgery unless the risk of ischemic events outweighs the risk of bleeding.
Beta-blockers
In view of the issue of scientific integrity of the DECREASE trials, a separately commissioned systematic review2 of perioperative beta-blocker therapy was performed. This review suggested that giving beta-blockers before surgery was associated with fewer postoperative cardiac events, primarily ischemia and nonfatal myocardial infarction, but few data supported their use to reduce postoperative mortality. Beta-blocker use was associated with adverse outcomes that included bradycardia and stroke. These findings were similar with the inclusion or exclusion of the DECREASE trials in question or of the POISE trial.25
In addition to recommending continuing beta-blockers in patients already on them (class I—the highest recommendation), the guidelines say that it may be reasonable to start them in patients with intermediate- or high-risk ischemia on stress tests as well as in patients with three or more RCRI risk factors (class IIb). In the absence of these indications, initiating beta-blockers preoperatively to reduce risk even in patients with long-term indications is of uncertain benefit. They also recommended starting beta-blockers more than 1 day preoperatively, preferably at least 2 to 7 days before, and note that it was harmful to start them on the day of surgery, particularly at high doses, and with long-acting formulations.
Additionally, there is evidence of differences in outcome within the class of beta-blockers, with the more cardioselective drugs bisoprolol and atenolol being associated with more favorable outcomes than metoprolol in observational studies.
Statins
Multiple observational trials have reported that statins are associated with decreased perioperative morbidity and mortality. Limited evidence from three randomized controlled trials (including two from the discredited DECREASE group) suggests that there is a benefit in patients undergoing vascular surgery, but it is unclear for nonvascular surgery.26–30
The ACC/AHA guidelines again give a class I recommendation to continue statin therapy perioperatively in patients already taking statins and undergoing noncardiac surgery, as there is some evidence that statin withdrawal is associated with increased risk. The guidelines comment that starting statin therapy perioperatively is reasonable for patients undergoing vascular surgery (class IIa) and may be considered in patients with other clinical guideline indications who are undergoing elevated-risk surgery (class IIb).
The mechanism of this benefit is unclear and may relate to the pleotropic as well as the lipid-lowering effects of the statins. Statins may also have beneficial effects in reducing the incidence of acute kidney injury and postoperative atrial fibrillation.
Whether a particular statin, dose, or time of initiation before surgery affects risk is also unknown at this time. The European guidelines6 recommend starting a longer-acting statin ideally at least 2 weeks before surgery for maximal plaque-stabilizing effects.
The risk of statin-induced myopathy, rhabdomyolysis, and hepatic injury appears to be minimal.
Other medications
Of note, the new guidelines do not recommend starting alpha-2 agonists for preventing cardiac events in patients undergoing noncardiac surgery. Despite previous evidence from smaller studies suggesting a benefit, the POISE-2 trial31 demonstrated that perioperative use of clonidine did not reduce cardiac events and was associated with a significant increase in hypotension and nonfatal cardiac arrest. However, clonidine should be continued in patients already taking it.
A somewhat surprising recommendation is that it is reasonable to continue angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs), and if they are held before surgery, to restart them as soon as possible postoperatively (class IIa). The guidelines note reports of increased hypotension associated with induction of anesthesia in patients taking these drugs but also note that there was no change in important postoperative cardiac and other outcomes. Although evidence of harm if these drugs are temporarily discontinued before surgery is sparse, the guidelines advocate continuing them in patients with heart failure or hypertension.
ANESTHESIA AND INTRAOPERATIVE MANAGEMENT
The classes of anesthesia include local, regional (nerve block or neuraxial), monitored anesthesia care (ie, intravenous sedation), and general (volatile agent, total intravenous, or a combination). The guideline committee found no evidence to support the use of neuraxial over general anesthesia, volatile over total intravenous anesthesia, or monitored anesthesia care over general anesthesia. Neuraxial anesthesia for postoperative pain relief in patients undergoing abdominal aortic surgery did reduce the incidence of myocardial infarction.
The guidelines do not recommend routinely using intraoperative transesophageal echocardiography during noncardiac surgery to screen for cardiac abnormalities or to monitor for myocardial ischemia in patients without risk factors or procedural risks for significant hemodynamic, pulmonary, or neurologic compromise. Only in emergency settings do they deem perioperative transesophageal echocardiography reasonable to determine the cause of hemodynamic instability when it persists despite attempted corrective therapy.
Maintenance of normothermia is reasonable, as studies evaluating hypothermia or use of warmed air did not find a lower rate of cardiac events.32,33
POSTOPERATIVE SURVEILLANCE
In observational studies, elevated troponin levels, and even detectable levels within the normal range, have been associated with adverse outcomes and predict mortality after noncardiac surgery—the higher the level, the higher the mortality rate.34 Elevated troponins have many potential causes, both cardiac and noncardiac.
An entity termed myocardial injury after noncardiac surgery (MINS)35 was described as prognostically relevant myocardial injury with a troponin T level higher than 0.03 ng/mL in the absence of a nonischemic etiology but not requiring the presence of ischemic features. Patients who had MINS had a higher 30-day mortality rate (9.8% vs 1.1%) and were also at higher risk of nonfatal cardiac arrest, heart failure, and stroke compared with patients who did not.
The guidelines recommend obtaining an electrocardiogram and troponin levels if there are signs or symptoms suggesting myocardial ischemia or infarction. However, despite the association between troponin and mortality, the guidelines state that "the usefulness of postoperative screening with troponin levels (and electrocardiograms) in patients at high risk for perioperative myocardial infarction, but without signs or symptoms suggestive of myocardial ischemia or infarction, is uncertain in the absence of established risks and benefits of a defined management strategy." They also recommend against routinely measuring postoperative troponins in unselected patients without signs or symptoms suggestive of myocardial ischemia or infarction, stating it is not useful for guiding perioperative management.
Although there was a suggestion that patients in the POISE trial36 who suffered postoperative myocardial infarction had better outcomes if they had received aspirin and statins, and another study37 showed that intensification of cardiac therapy in patients with elevated postoperative troponin levels after vascular surgery led to better 1-year outcomes, there are no randomized controlled trials at this time to support any specific plan or intervention.
IMPACT ON CLINICAL PRACTICE: A PERIOPERATIVE HOSPITALIST'S VIEW
Regarding testing
Although the updated guidelines provide some novel concepts in risk stratification, the new algorithm still leaves many patients in a gray zone with respect to noninvasive testing. Patients with heart failure, valvular heart disease, and arrhythmias appear to be somewhat disconnected from the algorithm in this version, and management according to clinical practice guidelines is recommended.
Patients with acute coronary syndrome remain embedded in the algorithm, with recommendations for cardiology evaluation and management according to standard guidelines before proceeding to elective surgery.
The concept of a combined risk based on clinical factors along with the surgical procedure is important, and an alternative to the RCRI factors is offered. However, while this new NSQIP surgical risk calculator is more comprehensive, it may be too time-consuming for routine clinical use and still needs to be externally validated.
The concept of shared decision-making and team communication is stressed, but the physician may still have difficulty deciding when further testing may influence management. The guidelines remain somewhat vague, and many physicians may be uncomfortable and will continue to look for further guidance in this area.
Without more specific recommendations, this uncertainty may result in more stress tests being ordered—often inappropriately, as they rarely change management. Future prospective studies using biomarkers in conjunction with risk calculators may shed some light on this decision.
The new perioperative guidelines incorporate other ACC/AHA guidelines for valvular heart disease15 and heart failure.14 Some of their recommendations, in my opinion, may lead to excessive testing (eg, repeat echocardiograms) that will not change perioperative management.
Regarding revascularization
The ACC/AHA guidelines continue to emphasize the important concept that coronary revascularization is rarely indicated just to get the patient through surgery.
The new guidelines give physicians some leeway in allowing patients with drug-eluting stents to undergo surgery after 6 rather than 12 months of dual antiplatelet therapy if they believe that delaying surgery would place the patient at more risk than that of stent thrombosis. There is evidence in the nonsurgical setting that the newer stents currently being used may require no more than 6 months of therapy. In my opinion it was never clear that there was a statistically significant benefit in delaying surgery more than 6 months after placement of a drug-eluting stent, so this is a welcome addition.
Regarding beta-blockers
The systematic review of beta-blockers reinforces the importance of continuing them preoperatively while downgrading recommendations for their prophylactic use in patients who are not at increased risk.
Although the debate continues, there is no doubt that beta-blockers are associated with a decrease in myocardial ischemia and infarction but an increase in bradycardia and hypotension. They probably are associated with some increased risk of stroke, although this may be related to the specific beta-blocker used as well as the time of initiation before surgery. Evidence of a possible effect on mortality depends on whether the DECREASE and POISE trials are included or excluded in the analysis.
In the absence of new large-scale randomized controlled trials, we are forced to rely on observational trials and expert opinion in the meantime. I think that if a beta-blocker is to be started preoperatively, it should be done at least 1 week before surgery, and a more cardioselective beta-blocker should be used.
Regarding other drugs and tests
I agree with the recommendation to continue ACE inhibitors and ARBs preoperatively in patients with heart failure and poorly controlled hypertension. Although somewhat contrary to current practice, continuance of these drugs has not been associated with an increase in myocardial infarction or death despite concern about intraoperative hypotension.
Data from randomized controlled trials of perioperative statins are limited, but the information from observational studies is favorable, and I see little downside to initiating statins preoperatively in patients who otherwise have indications for their use, particularly if undergoing vascular or other high-risk noncardiac surgery. It is not known whether the specific drug, dose, or timing of initiation of statins influences outcome.
Although multiple studies of biomarkers suggest that there is an association with outcome, there are no randomized controlled trials or specific interventions shown to improve outcome.
Some of the recommended interventions have included various cardiac medications, stress testing, possible coronary angiography, and revascularization, which are not without risk. In the absence of data and following the directive to "first do no harm," the ACC/AHA has been appropriately cautious in not recommending them for routine use at this time.
The updated guidelines have summarized the new evidence in perioperative cardiac evaluation and management. Many of their recommendations were reinforced by this information and remain essentially unchanged. Several new recommendations will lead to changes in management going forward. Unfortunately, we lack the evidence to answer many questions that arise in routine practice and are therefore forced to rely on expert opinion and our clinical judgment in these cases. The ACC/AHA guidelines do provide a framework for our evaluation and management and help keep clinicians up-to-date with the latest evidence.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05536-3. doi: 10.1016/j.jacc.2014.07.944. [Epub ahead of print].
- Wijeysundera DN, Duncan D, Nkonde-Price C, et al. Perioperative beta blockade in noncardiac surgery: a systematic review for the 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05528-4. doi: 10.1016/j.jacc.2014.07.939. [Epub ahead of print].
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05537-5. doi: 10.1016/j.jacc.2014.07.945. [Epub ahead of print].
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2007; 50:e159–e242.
- Erasmus MC Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. September 30, 2012. http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Accessed October 30, 2014.
- Anderson JL, Antman EM, Harold JG, et al. Clinical practice guidelines on perioperative cardiovascular evaluation: collaborative efforts among the ACC, AHA, and ESC. J Am Coll Cardiol 2014 Jul 29. pii: S0735-1097(14)05527-2. doi: 10.1016/j.jacc.2014.07.938. [Epub ahead of print].
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Gupta PK, Gupta H, Sundaram A, et al. Development and validation of a risk calculator for prediction of cardiac risk after surgery. Circulation 2011; 124:381–387.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surg 2013; 217:833–842. e1-3.
- Davis C, Tait G, Carroll J, Wijeysundera DN, Beattie WS. The Revised Cardiac Risk Index in the new millennium: a single-centre prospective cohort re-evaluation of the original variables in 9,519 consecutive elective surgical patients. Can J Anaesth 2013; 60:855–863.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; e-pub before print. doi:10.1016/j.jacc.2014.03.022.
- Aliot EM, Alpert JS, Calkins H, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias. http://www.escardio.org/guidelines-surveys/esc-guidelines/GuidelinesDocuments/guidelines-SVA-FT.pdf. Accessed October 30,2014.
- Crossley GH, Poole JE, Rozner MA, et al. The Heart Rhythm Society (HRS)/American Society of Anesthesiologists (ASA) Expert Consensus Statement on the perioperative management of patients with implantable defibrillators, pacemakers and arrhythmia monitors: facilities and patient management. Developed as a joint project with the American Society of Anesthesiologists (ASA), and in collaboration with the American Heart Association (AHA), and the Society of Thoracic Surgeons (STS). Heart Rhythm 2011; 8:1114–1154.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:e57–e185.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645-681.
- O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Hillis LD, Smith PK, Anderson JL, et al. 2011 ACCF/AHA guideline for coronary artery bypass graft surgery: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, Society of Cardiovascular Anesthesiologists, and Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 58:e123–e210.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- McFalls EO, Ward HB, Krupski WC, et al. Prophylactic coronary artery revascularization for elective vascular surgery: study design. Veterans Affairs Cooperative Study Group on Coronary Artery Revascularization Prophylaxis for Elective Vascular Surgery. Control Clin Trials 1999; 20:297–308.
- Schouten O, van Kuijk JP, Flu WJ, et al. Long-term outcome of prophylactic coronary revascularization in cardiac high-risk patients undergoing major vascular surgery (from the randomized DECREASE-V Pilot Study). Am J Cardiol 2009; 103:897–901.
- Wijeysundera DN, Wijeysundera HC, Yun L, et al. risk of elective major noncardiac surgery after coronary stent insertion: a population-based study. Circulation 2012; 126:1355-1362.
- Hawn MT, Graham LA, Richman JS, et al. Risk of major adverse cardiac events following noncardiac surgery in patients with coronary stents. JAMA 2013; 310:1462–1472.
- Devereaux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
- Group PS, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Lindenauer PK, Pekow P, Wang K, et al. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- Kennedy J, Quan H, Buchan AM, et al. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- Raju MG, Pachika A, Punnam SR, et al. Statin therapy in the reduction of cardiovascular events in patients undergoing intermediate-risk noncardiac, nonvascular surgery. Clin Cardiol 2013; 36:456–461.
- Desai H, Aronow WS, Ahn C, et al. Incidence of perioperative myocardial infarction and of 2-year mortality in 577 elderly patients undergoing noncardiac vascular surgery treated with and without statins. Arch Gerontol Geriatr 2010; 51:149–151.
- Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1504–1513.
- Nguyen HP, Zaroff JG, Bayman EO, et al. Perioperative hypothermia (33 degrees C) does not increase the occurrence of cardiovascular events in patients undergoing cerebral aneurysm surgery: findings from the Intraoperative Hypothermia for Aneurysm Surgery Trial. Anesthesiology 2010; 113:327–342.
- Frank SM, Fleisher LA, Breslow MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277:1127–1134.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Foucrier A, Rodseth R, Aissaoui M, Ibanes C, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
Guidelines jointly issued by the American College of Cardiology and American Heart Association (ACC/AHA)1 provide a framework for evaluating and managing perioperative cardiac risk in noncardiac surgery. An overriding theme in successive documents from these organizations through the years has been that preoperative intervention, coronary artery bypass grafting, or percutaneous coronary intervention is rarely necessary just to get the patient through surgery, unless it is otherwise indicated independent of the need for surgery.
This article highlights some of the key recommendations in the 2014 updates to these guidelines,1–3 how they differ from previous guidelines,4 and the ongoing challenges and unresolved issues facing physicians involved in perioperative care.
Of note, while these guidelines were being updated, Erasmus University5 expressed concern about the scientific integrity of some of the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE) trials. As a result, the evidence review committee included these trials in its analysis but not in a systematic review of beta-blockers.2 These trials were not included in the clinical practice guideline supplements and tables but were cited in the text if relevant.
The European Society of Cardiology and European Society of Anesthesiology6 revised their guidelines concurrently with but independently of the ACC/AHA, and although they discussed and aligned some recommendations, many differences remain between the two sets of guidelines. Readers should consult the full guidelines for more detailed information.1
THE ROLE OF THE PREOPERATIVE CARDIAC EVALUATION
The purpose of preoperative medical evaluation is not to "get medical clearance" but rather to evaluate the patient’s medical status and risk of complications. The process includes:
- Identifying risk factors and assessing their severity and stability
- Establishing a clinical risk profile for informed and shared decision-making
- Recommending needed changes in management, further testing, or specialty consultation.
The updated guidelines emphasize the importance of communication among the perioperative team and with the patient. They reiterate the focus on appropriateness of care and cost containment—one should order a test only if the result may change the patient’s management.
HOW URGENT IS SURGERY? HOW RISKY?
The new guidelines classify the urgency of surgery as follows:
- Emergency (necessary within 6 hours)
- Urgent (necessary within 6–24 hours)
- Time-sensitive (can delay 1–6 weeks)
- Elective (can delay up to 1 year).
Surgical risk is now classified as either low (< 1% risk of major adverse cardiac events) or elevated (≥ 1%) on the basis of surgical and patient characteristics. Previous schemas included an intermediate-risk category. Low-risk procedures include endoscopic procedures, superficial procedures, cataract surgery, breast surgery, and ambulatory surgery. Elevated-risk procedures include vascular surgery, intraperitoneal and intrathoracic surgery, head and neck surgery, orthopedic surgery, and prostate surgery.
Risk calculators and biomarkers
To estimate the perioperative risk of major adverse cardiac events, the guidelines suggest incorporating the Revised Cardiac Risk Index (RCRI)7 with an estimation of surgical risk or using a newer surgical risk calculator derived from a database of the American College of Surgeons’ National Surgical Quality Improvement Project (ACS NSQIP).
The RCRI is based on six risk factors, each worth 1 point:
- High-risk surgery
- Ischemic heart disease
- Heart failure
- Stroke or transient ischemic attack
- Diabetes requiring insulin
- Renal insufficiency (serum creatinine > 2.0 mg/dL).7
MICA. The Myocardial Infarction or Cardiac Arrest (MICA) calculator8 has a narrower focus and was validated in only one center.
ACS NSQIP. The recommended newer ACS NSQIP surgical risk calculator9 provides an estimate of procedure-specific risk based on Current Procedural Terminology code and includes 21 patient-specific variables to predict death, major adverse cardiac events, and eight other outcomes. While more comprehensive, this risk calculator has yet to be validated outside of the ACS NSQIP database.
Reconstructed RCRI. The RCRI has been externally validated, but it underestimates risk in major vascular surgery and was outperformed by the MICA calculator. Although not discussed in the new guidelines, a recently published "reconstructed RCRI,"10 in which a serum creatinine level greater than 2 mg/dL in the original RCRI is replaced by a glomerular filtration rate less than 30 mL/min and diabetes is eliminated, may outperform the standard RCRI. A patient with either an RCRI score or a reconstructed RCRI score of 0 or 1 would be considered to be at low risk, whereas patients with two or more risk factors would have an elevated risk.
Cardiac biomarkers, primarily B-type natriuretic peptide (BNP) and N-terminal (NT) proBNP, are independent predictors of cardiac risk, and their addition to preoperative risk indices may provide incremental predictive value. However, how to use these biomarkers and whether any treatment aimed at them will reduce risk is unclear, and the new guidelines did not recommend their routine use.
CLINICAL RISK FACTORS
Coronary artery disease
Ischemic symptoms, a history of myocardial infarction, and elevated cardiac biomarkers are individually associated with perioperative risk of morbidity and death. The risk is modified by how long ago the infarction occurred, whether the patient underwent coronary revascularization, and if so, what type (bypass grafting or percutaneous coronary intervention). A patient with acute coronary syndrome (currently or in the recent past) is at higher risk, and should have elective surgery delayed and be referred for cardiac evaluation and management according to guidelines.
Heart failure
In terms of posing a risk for major adverse cardiac events, heart failure is at least equal to coronary artery disease, and is possibly worse. Its impact depends on its stability, its symptoms, and the patient’s left ventricular function. Symptomatic decompensated heart failure and depressed left ventricular function (ejection fraction < 30% or 40%) confer higher risk than asymptomatic heart failure and preserved left ventricular function. However, evidence is limited with respect to asymptomatic left ventricular dysfunction and diastolic dysfunction. Patients with stable heart failure treated according to guidelines may have better perioperative outcomes.
Valvular heart disease
Significant valvular heart disease is associated with increased risk of postoperative cardiac complications. This risk depends on the type and severity of the valvular lesion and type of noncardiac surgery, but can be minimized by clinical and echocardiographic assessment, choosing appropriate anesthesia, and closer perioperative monitoring. Aortic and mitral stenosis are associated with greater risk of perioperative adverse cardiac events than regurgitant valvular disease.
Echocardiography is recommended in patients suspected of having moderate to severe stenotic or regurgitant lesions if it has not been done within the past year or if the patient’s clinical condition has worsened.
If indicated, valvular intervention can reduce perioperative risk in these patients. Even if the planned noncardiac surgery is high-risk, it may be reasonable to proceed with it (using appropriate perioperative hemodynamic monitoring, which is not specified but typically would be with an arterial line, central line, and possibly a pulmonary arterial catheter) in patients who have asymptomatic severe aortic or mitral regurgitation or aortic stenosis. Surgery may also be reasonable in patients with asymptomatic severe mitral stenosis who are not candidates for repair.
Arrhythmias
Cardiac arrhythmias and conduction defects are often seen in the perioperative period, but there is only limited evidence as to how they affect surgical risk. In addition to their hemodynamic effects, certain arrhythmias (atrial fibrillation, ventricular tachycardia) often indicate underlying structural heart disease, which requires further evaluation before surgery.
The new guidelines refer the reader to previously published clinical practice guidelines for atrial fibrillation,11 supraventricular arrhythmias,12 and device-based therapy.13
ALGORITHM FOR PREOPERATIVE CARDIAC ASSESSMENT
The new algorithm for evaluating a patient who is known to have coronary artery disease or risk factors for it has seven steps (Figure 1).1,11,12,14–17 It differs from the previous algorithm in several details:
- Instead of listing the four active cardiac conditions for which elective surgery should be delayed while the patient is being evaluated and treated (unstable coronary syndrome, decompensated heart failure, significant arrhythmias, severe valvular heart disease), the new version specifically asks about acute coronary syndrome and recommends cardiac evaluation and treatment according to guidelines. A footnote directs readers to other clinical practice guidelines for symptomatic heart failure,14 valvular heart disease,15 and arrhythmias.11,12
- Instead of asking if the procedure is low-risk, the guidelines recommend estimating risk of major adverse cardiac events on the basis of combined clinical and surgical risk and define only two categories: low or elevated. Patients at low risk proceed to surgery with no further testing, as in the earlier algorithm.
- "Excellent" exercise capacity (> 10 metabolic equivalents of task [METs]) is separated from "moderate/good" (4–10 METs), presumably to indicate a stronger recommendation, but patients in both categories proceed to surgery as before.
- If the patient cannot exercise to at least 4 METs, the new algorithm asks whether further testing will affect decision-making or perioperative care (an addition to the previous algorithm). This entails discussing with the patient and perioperative team whether the original surgery will be performed and whether the patient is willing to undergo revascularization if indicated. If so, pharmacologic stress testing is recommended. Previously, this decision also included the number of RCRI factors as well as the type of surgery (vascular or nonvascular).
- If testing will not affect the decision or if the stress test is normal, in addition to recommending proceeding to surgery according to guidelines the new algorithm also lists an option for alternative strategies, including palliation.
- If the stress test is abnormal, especially with left main disease, it recommends coronary revascularization according to the 2011 clinical practice guidelines.18,19
TESTING FOR LEFT VENTRICULAR DYSFUNCTION OR ISCHEMIA
In patients with dyspnea of unexplained cause or worsening dyspnea, assessment of left ventricular function is reasonable, but this is not part of a routine preoperative evaluation.
Pharmacologic stress testing is reasonable for patients at elevated risk with poor functional capacity if the results will change their management, but it is not useful for patients undergoing low-risk surgery. Although dobutamine stress echocardiography may be slightly superior to pharmacologic myocardial perfusion imaging, there are no head-to-head randomized controlled trials, and the guidelines suggest considering local expertise in deciding which test to use.
The presence of moderate to large areas of ischemia (reversible perfusion defects or new wall-motion abnormalities) is associated with risk of perioperative myocardial infarction or death, whereas evidence of an old infarction is associated with long-term but not short-term risk. The negative predictive value of these tests in predicting postoperative cardiac events is high (> 90%), but the positive predictive value is low.
CORONARY REVASCULARIZATION
Coronary artery bypass grafting and percutaneous coronary intervention
The guidelines recommend coronary revascularization before noncardiac surgery only when it is indicated anyway, on the basis of existing clinical practice guidelines.
Whether performing percutaneous coronary intervention before surgery will reduce perioperative cardiac complications is uncertain, and coronary revascularization should not be routinely performed solely to reduce perioperative cardiac events. The only two randomized controlled trials, Coronary Artery Revascularization Prophylaxis (CARP)20 and DECREASE V21 evaluating prophylactic coronary revascularization before noncardiac surgery found no difference in either short-term or long-term outcomes, although subgroup analysis found a survival benefit in patients with left main disease who underwent bypass grafting. Preoperative percutaneous coronary intervention should be limited to patients with left main disease in whom comorbidities preclude bypass surgery and those with unstable coronary disease who may benefit from early invasive management.
The urgency and timing of the noncardiac surgery needs to be taken into account if percutaneous coronary intervention is being considered because of the need for antiplatelet therapy after the procedure, and the potential risks of bleeding and stent thrombosis. If the planned surgery is deemed time-sensitive, then balloon angioplasty or bare-metal stenting is preferred over placement of a drug-eluting stent.
The new guidelines continue to recommend that elective noncardiac surgery be delayed at least 14 days after balloon angioplasty, 30 days after bare-metal stent implantation, and ideally 365 days after drug-eluting stent placement, and reiterate that it is potentially harmful to perform elective surgery within these time frames without any antiplatelet therapy. However, a new class IIb recommendation (benefit ≥ risk) states that "elective noncardiac surgery after [drug-eluting stent] implantation may be considered after 180 days if the risk of further delay is greater than the expected risks of ischemia and stent thrombosis."
This is an important addition to the guidelines because we are often faced with patients needing to undergo surgery in the 6 to 12 months after placement of a drug-eluting stent. Based on previous guidelines, whether it was safe to proceed in this setting created controversy among the perioperative team caring for the patient, and surgery was often delayed unnecessarily. Recent studies22,23 suggest that the newer drug-eluting stents may require a shorter duration of dual antiplatelet therapy, at least in the nonsurgical setting.
MEDICAL THERAPY
Antiplatelet therapy: Stop or continue?
The risk of perioperative bleeding if antiplatelet drugs are continued must be weighed against the risk of stent thrombosis and ischemia if they are stopped before the recommended duration of therapy. Ideally, some antiplatelet therapy should be continued perioperatively in these situations, but the guidelines recommend that a consensus decision among the treating physicians should be made regarding the relative risks of surgery and discontinuation or continuation of antiplatelet therapy. Whenever possible, aspirin should be continued in these patients.
Although the Perioperative Ischemic Evaluation (POISE)-2 trial24 found that perioperative aspirin use was not associated with lower rates of postoperative myocardial infarction or death, it increased bleeding. Patients with stents who had not completed the recommended duration of antiplatelet therapy were excluded from the trial. Additionally, only 5% of the study patients had undergone percutaneous coronary intervention.
According to the guidelines and package inserts, if antiplatelet agents need to be discontinued before surgery, aspirin can be stopped 3 to 7 days before, clopidogrel and ticagrelor 5 days before, and prasugrel 7 days before. In patients without stents, it may be reasonable to continue aspirin perioperatively if the risk of cardiac events outweighs the risk of bleeding, but starting aspirin is not beneficial for patients undergoing elective noncardiac noncarotid surgery unless the risk of ischemic events outweighs the risk of bleeding.
Beta-blockers
In view of the issue of scientific integrity of the DECREASE trials, a separately commissioned systematic review2 of perioperative beta-blocker therapy was performed. This review suggested that giving beta-blockers before surgery was associated with fewer postoperative cardiac events, primarily ischemia and nonfatal myocardial infarction, but few data supported their use to reduce postoperative mortality. Beta-blocker use was associated with adverse outcomes that included bradycardia and stroke. These findings were similar with the inclusion or exclusion of the DECREASE trials in question or of the POISE trial.25
In addition to recommending continuing beta-blockers in patients already on them (class I—the highest recommendation), the guidelines say that it may be reasonable to start them in patients with intermediate- or high-risk ischemia on stress tests as well as in patients with three or more RCRI risk factors (class IIb). In the absence of these indications, initiating beta-blockers preoperatively to reduce risk even in patients with long-term indications is of uncertain benefit. They also recommended starting beta-blockers more than 1 day preoperatively, preferably at least 2 to 7 days before, and note that it was harmful to start them on the day of surgery, particularly at high doses, and with long-acting formulations.
Additionally, there is evidence of differences in outcome within the class of beta-blockers, with the more cardioselective drugs bisoprolol and atenolol being associated with more favorable outcomes than metoprolol in observational studies.
Statins
Multiple observational trials have reported that statins are associated with decreased perioperative morbidity and mortality. Limited evidence from three randomized controlled trials (including two from the discredited DECREASE group) suggests that there is a benefit in patients undergoing vascular surgery, but it is unclear for nonvascular surgery.26–30
The ACC/AHA guidelines again give a class I recommendation to continue statin therapy perioperatively in patients already taking statins and undergoing noncardiac surgery, as there is some evidence that statin withdrawal is associated with increased risk. The guidelines comment that starting statin therapy perioperatively is reasonable for patients undergoing vascular surgery (class IIa) and may be considered in patients with other clinical guideline indications who are undergoing elevated-risk surgery (class IIb).
The mechanism of this benefit is unclear and may relate to the pleotropic as well as the lipid-lowering effects of the statins. Statins may also have beneficial effects in reducing the incidence of acute kidney injury and postoperative atrial fibrillation.
Whether a particular statin, dose, or time of initiation before surgery affects risk is also unknown at this time. The European guidelines6 recommend starting a longer-acting statin ideally at least 2 weeks before surgery for maximal plaque-stabilizing effects.
The risk of statin-induced myopathy, rhabdomyolysis, and hepatic injury appears to be minimal.
Other medications
Of note, the new guidelines do not recommend starting alpha-2 agonists for preventing cardiac events in patients undergoing noncardiac surgery. Despite previous evidence from smaller studies suggesting a benefit, the POISE-2 trial31 demonstrated that perioperative use of clonidine did not reduce cardiac events and was associated with a significant increase in hypotension and nonfatal cardiac arrest. However, clonidine should be continued in patients already taking it.
A somewhat surprising recommendation is that it is reasonable to continue angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs), and if they are held before surgery, to restart them as soon as possible postoperatively (class IIa). The guidelines note reports of increased hypotension associated with induction of anesthesia in patients taking these drugs but also note that there was no change in important postoperative cardiac and other outcomes. Although evidence of harm if these drugs are temporarily discontinued before surgery is sparse, the guidelines advocate continuing them in patients with heart failure or hypertension.
ANESTHESIA AND INTRAOPERATIVE MANAGEMENT
The classes of anesthesia include local, regional (nerve block or neuraxial), monitored anesthesia care (ie, intravenous sedation), and general (volatile agent, total intravenous, or a combination). The guideline committee found no evidence to support the use of neuraxial over general anesthesia, volatile over total intravenous anesthesia, or monitored anesthesia care over general anesthesia. Neuraxial anesthesia for postoperative pain relief in patients undergoing abdominal aortic surgery did reduce the incidence of myocardial infarction.
The guidelines do not recommend routinely using intraoperative transesophageal echocardiography during noncardiac surgery to screen for cardiac abnormalities or to monitor for myocardial ischemia in patients without risk factors or procedural risks for significant hemodynamic, pulmonary, or neurologic compromise. Only in emergency settings do they deem perioperative transesophageal echocardiography reasonable to determine the cause of hemodynamic instability when it persists despite attempted corrective therapy.
Maintenance of normothermia is reasonable, as studies evaluating hypothermia or use of warmed air did not find a lower rate of cardiac events.32,33
POSTOPERATIVE SURVEILLANCE
In observational studies, elevated troponin levels, and even detectable levels within the normal range, have been associated with adverse outcomes and predict mortality after noncardiac surgery—the higher the level, the higher the mortality rate.34 Elevated troponins have many potential causes, both cardiac and noncardiac.
An entity termed myocardial injury after noncardiac surgery (MINS)35 was described as prognostically relevant myocardial injury with a troponin T level higher than 0.03 ng/mL in the absence of a nonischemic etiology but not requiring the presence of ischemic features. Patients who had MINS had a higher 30-day mortality rate (9.8% vs 1.1%) and were also at higher risk of nonfatal cardiac arrest, heart failure, and stroke compared with patients who did not.
The guidelines recommend obtaining an electrocardiogram and troponin levels if there are signs or symptoms suggesting myocardial ischemia or infarction. However, despite the association between troponin and mortality, the guidelines state that "the usefulness of postoperative screening with troponin levels (and electrocardiograms) in patients at high risk for perioperative myocardial infarction, but without signs or symptoms suggestive of myocardial ischemia or infarction, is uncertain in the absence of established risks and benefits of a defined management strategy." They also recommend against routinely measuring postoperative troponins in unselected patients without signs or symptoms suggestive of myocardial ischemia or infarction, stating it is not useful for guiding perioperative management.
Although there was a suggestion that patients in the POISE trial36 who suffered postoperative myocardial infarction had better outcomes if they had received aspirin and statins, and another study37 showed that intensification of cardiac therapy in patients with elevated postoperative troponin levels after vascular surgery led to better 1-year outcomes, there are no randomized controlled trials at this time to support any specific plan or intervention.
IMPACT ON CLINICAL PRACTICE: A PERIOPERATIVE HOSPITALIST'S VIEW
Regarding testing
Although the updated guidelines provide some novel concepts in risk stratification, the new algorithm still leaves many patients in a gray zone with respect to noninvasive testing. Patients with heart failure, valvular heart disease, and arrhythmias appear to be somewhat disconnected from the algorithm in this version, and management according to clinical practice guidelines is recommended.
Patients with acute coronary syndrome remain embedded in the algorithm, with recommendations for cardiology evaluation and management according to standard guidelines before proceeding to elective surgery.
The concept of a combined risk based on clinical factors along with the surgical procedure is important, and an alternative to the RCRI factors is offered. However, while this new NSQIP surgical risk calculator is more comprehensive, it may be too time-consuming for routine clinical use and still needs to be externally validated.
The concept of shared decision-making and team communication is stressed, but the physician may still have difficulty deciding when further testing may influence management. The guidelines remain somewhat vague, and many physicians may be uncomfortable and will continue to look for further guidance in this area.
Without more specific recommendations, this uncertainty may result in more stress tests being ordered—often inappropriately, as they rarely change management. Future prospective studies using biomarkers in conjunction with risk calculators may shed some light on this decision.
The new perioperative guidelines incorporate other ACC/AHA guidelines for valvular heart disease15 and heart failure.14 Some of their recommendations, in my opinion, may lead to excessive testing (eg, repeat echocardiograms) that will not change perioperative management.
Regarding revascularization
The ACC/AHA guidelines continue to emphasize the important concept that coronary revascularization is rarely indicated just to get the patient through surgery.
The new guidelines give physicians some leeway in allowing patients with drug-eluting stents to undergo surgery after 6 rather than 12 months of dual antiplatelet therapy if they believe that delaying surgery would place the patient at more risk than that of stent thrombosis. There is evidence in the nonsurgical setting that the newer stents currently being used may require no more than 6 months of therapy. In my opinion it was never clear that there was a statistically significant benefit in delaying surgery more than 6 months after placement of a drug-eluting stent, so this is a welcome addition.
Regarding beta-blockers
The systematic review of beta-blockers reinforces the importance of continuing them preoperatively while downgrading recommendations for their prophylactic use in patients who are not at increased risk.
Although the debate continues, there is no doubt that beta-blockers are associated with a decrease in myocardial ischemia and infarction but an increase in bradycardia and hypotension. They probably are associated with some increased risk of stroke, although this may be related to the specific beta-blocker used as well as the time of initiation before surgery. Evidence of a possible effect on mortality depends on whether the DECREASE and POISE trials are included or excluded in the analysis.
In the absence of new large-scale randomized controlled trials, we are forced to rely on observational trials and expert opinion in the meantime. I think that if a beta-blocker is to be started preoperatively, it should be done at least 1 week before surgery, and a more cardioselective beta-blocker should be used.
Regarding other drugs and tests
I agree with the recommendation to continue ACE inhibitors and ARBs preoperatively in patients with heart failure and poorly controlled hypertension. Although somewhat contrary to current practice, continuance of these drugs has not been associated with an increase in myocardial infarction or death despite concern about intraoperative hypotension.
Data from randomized controlled trials of perioperative statins are limited, but the information from observational studies is favorable, and I see little downside to initiating statins preoperatively in patients who otherwise have indications for their use, particularly if undergoing vascular or other high-risk noncardiac surgery. It is not known whether the specific drug, dose, or timing of initiation of statins influences outcome.
Although multiple studies of biomarkers suggest that there is an association with outcome, there are no randomized controlled trials or specific interventions shown to improve outcome.
Some of the recommended interventions have included various cardiac medications, stress testing, possible coronary angiography, and revascularization, which are not without risk. In the absence of data and following the directive to "first do no harm," the ACC/AHA has been appropriately cautious in not recommending them for routine use at this time.
The updated guidelines have summarized the new evidence in perioperative cardiac evaluation and management. Many of their recommendations were reinforced by this information and remain essentially unchanged. Several new recommendations will lead to changes in management going forward. Unfortunately, we lack the evidence to answer many questions that arise in routine practice and are therefore forced to rely on expert opinion and our clinical judgment in these cases. The ACC/AHA guidelines do provide a framework for our evaluation and management and help keep clinicians up-to-date with the latest evidence.
Guidelines jointly issued by the American College of Cardiology and American Heart Association (ACC/AHA)1 provide a framework for evaluating and managing perioperative cardiac risk in noncardiac surgery. An overriding theme in successive documents from these organizations through the years has been that preoperative intervention, coronary artery bypass grafting, or percutaneous coronary intervention is rarely necessary just to get the patient through surgery, unless it is otherwise indicated independent of the need for surgery.
This article highlights some of the key recommendations in the 2014 updates to these guidelines,1–3 how they differ from previous guidelines,4 and the ongoing challenges and unresolved issues facing physicians involved in perioperative care.
Of note, while these guidelines were being updated, Erasmus University5 expressed concern about the scientific integrity of some of the Dutch Echocardiographic Cardiac Risk Evaluation Applying Stress Echocardiography (DECREASE) trials. As a result, the evidence review committee included these trials in its analysis but not in a systematic review of beta-blockers.2 These trials were not included in the clinical practice guideline supplements and tables but were cited in the text if relevant.
The European Society of Cardiology and European Society of Anesthesiology6 revised their guidelines concurrently with but independently of the ACC/AHA, and although they discussed and aligned some recommendations, many differences remain between the two sets of guidelines. Readers should consult the full guidelines for more detailed information.1
THE ROLE OF THE PREOPERATIVE CARDIAC EVALUATION
The purpose of preoperative medical evaluation is not to "get medical clearance" but rather to evaluate the patient’s medical status and risk of complications. The process includes:
- Identifying risk factors and assessing their severity and stability
- Establishing a clinical risk profile for informed and shared decision-making
- Recommending needed changes in management, further testing, or specialty consultation.
The updated guidelines emphasize the importance of communication among the perioperative team and with the patient. They reiterate the focus on appropriateness of care and cost containment—one should order a test only if the result may change the patient’s management.
HOW URGENT IS SURGERY? HOW RISKY?
The new guidelines classify the urgency of surgery as follows:
- Emergency (necessary within 6 hours)
- Urgent (necessary within 6–24 hours)
- Time-sensitive (can delay 1–6 weeks)
- Elective (can delay up to 1 year).
Surgical risk is now classified as either low (< 1% risk of major adverse cardiac events) or elevated (≥ 1%) on the basis of surgical and patient characteristics. Previous schemas included an intermediate-risk category. Low-risk procedures include endoscopic procedures, superficial procedures, cataract surgery, breast surgery, and ambulatory surgery. Elevated-risk procedures include vascular surgery, intraperitoneal and intrathoracic surgery, head and neck surgery, orthopedic surgery, and prostate surgery.
Risk calculators and biomarkers
To estimate the perioperative risk of major adverse cardiac events, the guidelines suggest incorporating the Revised Cardiac Risk Index (RCRI)7 with an estimation of surgical risk or using a newer surgical risk calculator derived from a database of the American College of Surgeons’ National Surgical Quality Improvement Project (ACS NSQIP).
The RCRI is based on six risk factors, each worth 1 point:
- High-risk surgery
- Ischemic heart disease
- Heart failure
- Stroke or transient ischemic attack
- Diabetes requiring insulin
- Renal insufficiency (serum creatinine > 2.0 mg/dL).7
MICA. The Myocardial Infarction or Cardiac Arrest (MICA) calculator8 has a narrower focus and was validated in only one center.
ACS NSQIP. The recommended newer ACS NSQIP surgical risk calculator9 provides an estimate of procedure-specific risk based on Current Procedural Terminology code and includes 21 patient-specific variables to predict death, major adverse cardiac events, and eight other outcomes. While more comprehensive, this risk calculator has yet to be validated outside of the ACS NSQIP database.
Reconstructed RCRI. The RCRI has been externally validated, but it underestimates risk in major vascular surgery and was outperformed by the MICA calculator. Although not discussed in the new guidelines, a recently published "reconstructed RCRI,"10 in which a serum creatinine level greater than 2 mg/dL in the original RCRI is replaced by a glomerular filtration rate less than 30 mL/min and diabetes is eliminated, may outperform the standard RCRI. A patient with either an RCRI score or a reconstructed RCRI score of 0 or 1 would be considered to be at low risk, whereas patients with two or more risk factors would have an elevated risk.
Cardiac biomarkers, primarily B-type natriuretic peptide (BNP) and N-terminal (NT) proBNP, are independent predictors of cardiac risk, and their addition to preoperative risk indices may provide incremental predictive value. However, how to use these biomarkers and whether any treatment aimed at them will reduce risk is unclear, and the new guidelines did not recommend their routine use.
CLINICAL RISK FACTORS
Coronary artery disease
Ischemic symptoms, a history of myocardial infarction, and elevated cardiac biomarkers are individually associated with perioperative risk of morbidity and death. The risk is modified by how long ago the infarction occurred, whether the patient underwent coronary revascularization, and if so, what type (bypass grafting or percutaneous coronary intervention). A patient with acute coronary syndrome (currently or in the recent past) is at higher risk, and should have elective surgery delayed and be referred for cardiac evaluation and management according to guidelines.
Heart failure
In terms of posing a risk for major adverse cardiac events, heart failure is at least equal to coronary artery disease, and is possibly worse. Its impact depends on its stability, its symptoms, and the patient’s left ventricular function. Symptomatic decompensated heart failure and depressed left ventricular function (ejection fraction < 30% or 40%) confer higher risk than asymptomatic heart failure and preserved left ventricular function. However, evidence is limited with respect to asymptomatic left ventricular dysfunction and diastolic dysfunction. Patients with stable heart failure treated according to guidelines may have better perioperative outcomes.
Valvular heart disease
Significant valvular heart disease is associated with increased risk of postoperative cardiac complications. This risk depends on the type and severity of the valvular lesion and type of noncardiac surgery, but can be minimized by clinical and echocardiographic assessment, choosing appropriate anesthesia, and closer perioperative monitoring. Aortic and mitral stenosis are associated with greater risk of perioperative adverse cardiac events than regurgitant valvular disease.
Echocardiography is recommended in patients suspected of having moderate to severe stenotic or regurgitant lesions if it has not been done within the past year or if the patient’s clinical condition has worsened.
If indicated, valvular intervention can reduce perioperative risk in these patients. Even if the planned noncardiac surgery is high-risk, it may be reasonable to proceed with it (using appropriate perioperative hemodynamic monitoring, which is not specified but typically would be with an arterial line, central line, and possibly a pulmonary arterial catheter) in patients who have asymptomatic severe aortic or mitral regurgitation or aortic stenosis. Surgery may also be reasonable in patients with asymptomatic severe mitral stenosis who are not candidates for repair.
Arrhythmias
Cardiac arrhythmias and conduction defects are often seen in the perioperative period, but there is only limited evidence as to how they affect surgical risk. In addition to their hemodynamic effects, certain arrhythmias (atrial fibrillation, ventricular tachycardia) often indicate underlying structural heart disease, which requires further evaluation before surgery.
The new guidelines refer the reader to previously published clinical practice guidelines for atrial fibrillation,11 supraventricular arrhythmias,12 and device-based therapy.13
ALGORITHM FOR PREOPERATIVE CARDIAC ASSESSMENT
The new algorithm for evaluating a patient who is known to have coronary artery disease or risk factors for it has seven steps (Figure 1).1,11,12,14–17 It differs from the previous algorithm in several details:
- Instead of listing the four active cardiac conditions for which elective surgery should be delayed while the patient is being evaluated and treated (unstable coronary syndrome, decompensated heart failure, significant arrhythmias, severe valvular heart disease), the new version specifically asks about acute coronary syndrome and recommends cardiac evaluation and treatment according to guidelines. A footnote directs readers to other clinical practice guidelines for symptomatic heart failure,14 valvular heart disease,15 and arrhythmias.11,12
- Instead of asking if the procedure is low-risk, the guidelines recommend estimating risk of major adverse cardiac events on the basis of combined clinical and surgical risk and define only two categories: low or elevated. Patients at low risk proceed to surgery with no further testing, as in the earlier algorithm.
- "Excellent" exercise capacity (> 10 metabolic equivalents of task [METs]) is separated from "moderate/good" (4–10 METs), presumably to indicate a stronger recommendation, but patients in both categories proceed to surgery as before.
- If the patient cannot exercise to at least 4 METs, the new algorithm asks whether further testing will affect decision-making or perioperative care (an addition to the previous algorithm). This entails discussing with the patient and perioperative team whether the original surgery will be performed and whether the patient is willing to undergo revascularization if indicated. If so, pharmacologic stress testing is recommended. Previously, this decision also included the number of RCRI factors as well as the type of surgery (vascular or nonvascular).
- If testing will not affect the decision or if the stress test is normal, in addition to recommending proceeding to surgery according to guidelines the new algorithm also lists an option for alternative strategies, including palliation.
- If the stress test is abnormal, especially with left main disease, it recommends coronary revascularization according to the 2011 clinical practice guidelines.18,19
TESTING FOR LEFT VENTRICULAR DYSFUNCTION OR ISCHEMIA
In patients with dyspnea of unexplained cause or worsening dyspnea, assessment of left ventricular function is reasonable, but this is not part of a routine preoperative evaluation.
Pharmacologic stress testing is reasonable for patients at elevated risk with poor functional capacity if the results will change their management, but it is not useful for patients undergoing low-risk surgery. Although dobutamine stress echocardiography may be slightly superior to pharmacologic myocardial perfusion imaging, there are no head-to-head randomized controlled trials, and the guidelines suggest considering local expertise in deciding which test to use.
The presence of moderate to large areas of ischemia (reversible perfusion defects or new wall-motion abnormalities) is associated with risk of perioperative myocardial infarction or death, whereas evidence of an old infarction is associated with long-term but not short-term risk. The negative predictive value of these tests in predicting postoperative cardiac events is high (> 90%), but the positive predictive value is low.
CORONARY REVASCULARIZATION
Coronary artery bypass grafting and percutaneous coronary intervention
The guidelines recommend coronary revascularization before noncardiac surgery only when it is indicated anyway, on the basis of existing clinical practice guidelines.
Whether performing percutaneous coronary intervention before surgery will reduce perioperative cardiac complications is uncertain, and coronary revascularization should not be routinely performed solely to reduce perioperative cardiac events. The only two randomized controlled trials, Coronary Artery Revascularization Prophylaxis (CARP)20 and DECREASE V21 evaluating prophylactic coronary revascularization before noncardiac surgery found no difference in either short-term or long-term outcomes, although subgroup analysis found a survival benefit in patients with left main disease who underwent bypass grafting. Preoperative percutaneous coronary intervention should be limited to patients with left main disease in whom comorbidities preclude bypass surgery and those with unstable coronary disease who may benefit from early invasive management.
The urgency and timing of the noncardiac surgery needs to be taken into account if percutaneous coronary intervention is being considered because of the need for antiplatelet therapy after the procedure, and the potential risks of bleeding and stent thrombosis. If the planned surgery is deemed time-sensitive, then balloon angioplasty or bare-metal stenting is preferred over placement of a drug-eluting stent.
The new guidelines continue to recommend that elective noncardiac surgery be delayed at least 14 days after balloon angioplasty, 30 days after bare-metal stent implantation, and ideally 365 days after drug-eluting stent placement, and reiterate that it is potentially harmful to perform elective surgery within these time frames without any antiplatelet therapy. However, a new class IIb recommendation (benefit ≥ risk) states that "elective noncardiac surgery after [drug-eluting stent] implantation may be considered after 180 days if the risk of further delay is greater than the expected risks of ischemia and stent thrombosis."
This is an important addition to the guidelines because we are often faced with patients needing to undergo surgery in the 6 to 12 months after placement of a drug-eluting stent. Based on previous guidelines, whether it was safe to proceed in this setting created controversy among the perioperative team caring for the patient, and surgery was often delayed unnecessarily. Recent studies22,23 suggest that the newer drug-eluting stents may require a shorter duration of dual antiplatelet therapy, at least in the nonsurgical setting.
MEDICAL THERAPY
Antiplatelet therapy: Stop or continue?
The risk of perioperative bleeding if antiplatelet drugs are continued must be weighed against the risk of stent thrombosis and ischemia if they are stopped before the recommended duration of therapy. Ideally, some antiplatelet therapy should be continued perioperatively in these situations, but the guidelines recommend that a consensus decision among the treating physicians should be made regarding the relative risks of surgery and discontinuation or continuation of antiplatelet therapy. Whenever possible, aspirin should be continued in these patients.
Although the Perioperative Ischemic Evaluation (POISE)-2 trial24 found that perioperative aspirin use was not associated with lower rates of postoperative myocardial infarction or death, it increased bleeding. Patients with stents who had not completed the recommended duration of antiplatelet therapy were excluded from the trial. Additionally, only 5% of the study patients had undergone percutaneous coronary intervention.
According to the guidelines and package inserts, if antiplatelet agents need to be discontinued before surgery, aspirin can be stopped 3 to 7 days before, clopidogrel and ticagrelor 5 days before, and prasugrel 7 days before. In patients without stents, it may be reasonable to continue aspirin perioperatively if the risk of cardiac events outweighs the risk of bleeding, but starting aspirin is not beneficial for patients undergoing elective noncardiac noncarotid surgery unless the risk of ischemic events outweighs the risk of bleeding.
Beta-blockers
In view of the issue of scientific integrity of the DECREASE trials, a separately commissioned systematic review2 of perioperative beta-blocker therapy was performed. This review suggested that giving beta-blockers before surgery was associated with fewer postoperative cardiac events, primarily ischemia and nonfatal myocardial infarction, but few data supported their use to reduce postoperative mortality. Beta-blocker use was associated with adverse outcomes that included bradycardia and stroke. These findings were similar with the inclusion or exclusion of the DECREASE trials in question or of the POISE trial.25
In addition to recommending continuing beta-blockers in patients already on them (class I—the highest recommendation), the guidelines say that it may be reasonable to start them in patients with intermediate- or high-risk ischemia on stress tests as well as in patients with three or more RCRI risk factors (class IIb). In the absence of these indications, initiating beta-blockers preoperatively to reduce risk even in patients with long-term indications is of uncertain benefit. They also recommended starting beta-blockers more than 1 day preoperatively, preferably at least 2 to 7 days before, and note that it was harmful to start them on the day of surgery, particularly at high doses, and with long-acting formulations.
Additionally, there is evidence of differences in outcome within the class of beta-blockers, with the more cardioselective drugs bisoprolol and atenolol being associated with more favorable outcomes than metoprolol in observational studies.
Statins
Multiple observational trials have reported that statins are associated with decreased perioperative morbidity and mortality. Limited evidence from three randomized controlled trials (including two from the discredited DECREASE group) suggests that there is a benefit in patients undergoing vascular surgery, but it is unclear for nonvascular surgery.26–30
The ACC/AHA guidelines again give a class I recommendation to continue statin therapy perioperatively in patients already taking statins and undergoing noncardiac surgery, as there is some evidence that statin withdrawal is associated with increased risk. The guidelines comment that starting statin therapy perioperatively is reasonable for patients undergoing vascular surgery (class IIa) and may be considered in patients with other clinical guideline indications who are undergoing elevated-risk surgery (class IIb).
The mechanism of this benefit is unclear and may relate to the pleotropic as well as the lipid-lowering effects of the statins. Statins may also have beneficial effects in reducing the incidence of acute kidney injury and postoperative atrial fibrillation.
Whether a particular statin, dose, or time of initiation before surgery affects risk is also unknown at this time. The European guidelines6 recommend starting a longer-acting statin ideally at least 2 weeks before surgery for maximal plaque-stabilizing effects.
The risk of statin-induced myopathy, rhabdomyolysis, and hepatic injury appears to be minimal.
Other medications
Of note, the new guidelines do not recommend starting alpha-2 agonists for preventing cardiac events in patients undergoing noncardiac surgery. Despite previous evidence from smaller studies suggesting a benefit, the POISE-2 trial31 demonstrated that perioperative use of clonidine did not reduce cardiac events and was associated with a significant increase in hypotension and nonfatal cardiac arrest. However, clonidine should be continued in patients already taking it.
A somewhat surprising recommendation is that it is reasonable to continue angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs), and if they are held before surgery, to restart them as soon as possible postoperatively (class IIa). The guidelines note reports of increased hypotension associated with induction of anesthesia in patients taking these drugs but also note that there was no change in important postoperative cardiac and other outcomes. Although evidence of harm if these drugs are temporarily discontinued before surgery is sparse, the guidelines advocate continuing them in patients with heart failure or hypertension.
ANESTHESIA AND INTRAOPERATIVE MANAGEMENT
The classes of anesthesia include local, regional (nerve block or neuraxial), monitored anesthesia care (ie, intravenous sedation), and general (volatile agent, total intravenous, or a combination). The guideline committee found no evidence to support the use of neuraxial over general anesthesia, volatile over total intravenous anesthesia, or monitored anesthesia care over general anesthesia. Neuraxial anesthesia for postoperative pain relief in patients undergoing abdominal aortic surgery did reduce the incidence of myocardial infarction.
The guidelines do not recommend routinely using intraoperative transesophageal echocardiography during noncardiac surgery to screen for cardiac abnormalities or to monitor for myocardial ischemia in patients without risk factors or procedural risks for significant hemodynamic, pulmonary, or neurologic compromise. Only in emergency settings do they deem perioperative transesophageal echocardiography reasonable to determine the cause of hemodynamic instability when it persists despite attempted corrective therapy.
Maintenance of normothermia is reasonable, as studies evaluating hypothermia or use of warmed air did not find a lower rate of cardiac events.32,33
POSTOPERATIVE SURVEILLANCE
In observational studies, elevated troponin levels, and even detectable levels within the normal range, have been associated with adverse outcomes and predict mortality after noncardiac surgery—the higher the level, the higher the mortality rate.34 Elevated troponins have many potential causes, both cardiac and noncardiac.
An entity termed myocardial injury after noncardiac surgery (MINS)35 was described as prognostically relevant myocardial injury with a troponin T level higher than 0.03 ng/mL in the absence of a nonischemic etiology but not requiring the presence of ischemic features. Patients who had MINS had a higher 30-day mortality rate (9.8% vs 1.1%) and were also at higher risk of nonfatal cardiac arrest, heart failure, and stroke compared with patients who did not.
The guidelines recommend obtaining an electrocardiogram and troponin levels if there are signs or symptoms suggesting myocardial ischemia or infarction. However, despite the association between troponin and mortality, the guidelines state that "the usefulness of postoperative screening with troponin levels (and electrocardiograms) in patients at high risk for perioperative myocardial infarction, but without signs or symptoms suggestive of myocardial ischemia or infarction, is uncertain in the absence of established risks and benefits of a defined management strategy." They also recommend against routinely measuring postoperative troponins in unselected patients without signs or symptoms suggestive of myocardial ischemia or infarction, stating it is not useful for guiding perioperative management.
Although there was a suggestion that patients in the POISE trial36 who suffered postoperative myocardial infarction had better outcomes if they had received aspirin and statins, and another study37 showed that intensification of cardiac therapy in patients with elevated postoperative troponin levels after vascular surgery led to better 1-year outcomes, there are no randomized controlled trials at this time to support any specific plan or intervention.
IMPACT ON CLINICAL PRACTICE: A PERIOPERATIVE HOSPITALIST'S VIEW
Regarding testing
Although the updated guidelines provide some novel concepts in risk stratification, the new algorithm still leaves many patients in a gray zone with respect to noninvasive testing. Patients with heart failure, valvular heart disease, and arrhythmias appear to be somewhat disconnected from the algorithm in this version, and management according to clinical practice guidelines is recommended.
Patients with acute coronary syndrome remain embedded in the algorithm, with recommendations for cardiology evaluation and management according to standard guidelines before proceeding to elective surgery.
The concept of a combined risk based on clinical factors along with the surgical procedure is important, and an alternative to the RCRI factors is offered. However, while this new NSQIP surgical risk calculator is more comprehensive, it may be too time-consuming for routine clinical use and still needs to be externally validated.
The concept of shared decision-making and team communication is stressed, but the physician may still have difficulty deciding when further testing may influence management. The guidelines remain somewhat vague, and many physicians may be uncomfortable and will continue to look for further guidance in this area.
Without more specific recommendations, this uncertainty may result in more stress tests being ordered—often inappropriately, as they rarely change management. Future prospective studies using biomarkers in conjunction with risk calculators may shed some light on this decision.
The new perioperative guidelines incorporate other ACC/AHA guidelines for valvular heart disease15 and heart failure.14 Some of their recommendations, in my opinion, may lead to excessive testing (eg, repeat echocardiograms) that will not change perioperative management.
Regarding revascularization
The ACC/AHA guidelines continue to emphasize the important concept that coronary revascularization is rarely indicated just to get the patient through surgery.
The new guidelines give physicians some leeway in allowing patients with drug-eluting stents to undergo surgery after 6 rather than 12 months of dual antiplatelet therapy if they believe that delaying surgery would place the patient at more risk than that of stent thrombosis. There is evidence in the nonsurgical setting that the newer stents currently being used may require no more than 6 months of therapy. In my opinion it was never clear that there was a statistically significant benefit in delaying surgery more than 6 months after placement of a drug-eluting stent, so this is a welcome addition.
Regarding beta-blockers
The systematic review of beta-blockers reinforces the importance of continuing them preoperatively while downgrading recommendations for their prophylactic use in patients who are not at increased risk.
Although the debate continues, there is no doubt that beta-blockers are associated with a decrease in myocardial ischemia and infarction but an increase in bradycardia and hypotension. They probably are associated with some increased risk of stroke, although this may be related to the specific beta-blocker used as well as the time of initiation before surgery. Evidence of a possible effect on mortality depends on whether the DECREASE and POISE trials are included or excluded in the analysis.
In the absence of new large-scale randomized controlled trials, we are forced to rely on observational trials and expert opinion in the meantime. I think that if a beta-blocker is to be started preoperatively, it should be done at least 1 week before surgery, and a more cardioselective beta-blocker should be used.
Regarding other drugs and tests
I agree with the recommendation to continue ACE inhibitors and ARBs preoperatively in patients with heart failure and poorly controlled hypertension. Although somewhat contrary to current practice, continuance of these drugs has not been associated with an increase in myocardial infarction or death despite concern about intraoperative hypotension.
Data from randomized controlled trials of perioperative statins are limited, but the information from observational studies is favorable, and I see little downside to initiating statins preoperatively in patients who otherwise have indications for their use, particularly if undergoing vascular or other high-risk noncardiac surgery. It is not known whether the specific drug, dose, or timing of initiation of statins influences outcome.
Although multiple studies of biomarkers suggest that there is an association with outcome, there are no randomized controlled trials or specific interventions shown to improve outcome.
Some of the recommended interventions have included various cardiac medications, stress testing, possible coronary angiography, and revascularization, which are not without risk. In the absence of data and following the directive to "first do no harm," the ACC/AHA has been appropriately cautious in not recommending them for routine use at this time.
The updated guidelines have summarized the new evidence in perioperative cardiac evaluation and management. Many of their recommendations were reinforced by this information and remain essentially unchanged. Several new recommendations will lead to changes in management going forward. Unfortunately, we lack the evidence to answer many questions that arise in routine practice and are therefore forced to rely on expert opinion and our clinical judgment in these cases. The ACC/AHA guidelines do provide a framework for our evaluation and management and help keep clinicians up-to-date with the latest evidence.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05536-3. doi: 10.1016/j.jacc.2014.07.944. [Epub ahead of print].
- Wijeysundera DN, Duncan D, Nkonde-Price C, et al. Perioperative beta blockade in noncardiac surgery: a systematic review for the 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05528-4. doi: 10.1016/j.jacc.2014.07.939. [Epub ahead of print].
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05537-5. doi: 10.1016/j.jacc.2014.07.945. [Epub ahead of print].
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2007; 50:e159–e242.
- Erasmus MC Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. September 30, 2012. http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Accessed October 30, 2014.
- Anderson JL, Antman EM, Harold JG, et al. Clinical practice guidelines on perioperative cardiovascular evaluation: collaborative efforts among the ACC, AHA, and ESC. J Am Coll Cardiol 2014 Jul 29. pii: S0735-1097(14)05527-2. doi: 10.1016/j.jacc.2014.07.938. [Epub ahead of print].
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Gupta PK, Gupta H, Sundaram A, et al. Development and validation of a risk calculator for prediction of cardiac risk after surgery. Circulation 2011; 124:381–387.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surg 2013; 217:833–842. e1-3.
- Davis C, Tait G, Carroll J, Wijeysundera DN, Beattie WS. The Revised Cardiac Risk Index in the new millennium: a single-centre prospective cohort re-evaluation of the original variables in 9,519 consecutive elective surgical patients. Can J Anaesth 2013; 60:855–863.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; e-pub before print. doi:10.1016/j.jacc.2014.03.022.
- Aliot EM, Alpert JS, Calkins H, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias. http://www.escardio.org/guidelines-surveys/esc-guidelines/GuidelinesDocuments/guidelines-SVA-FT.pdf. Accessed October 30,2014.
- Crossley GH, Poole JE, Rozner MA, et al. The Heart Rhythm Society (HRS)/American Society of Anesthesiologists (ASA) Expert Consensus Statement on the perioperative management of patients with implantable defibrillators, pacemakers and arrhythmia monitors: facilities and patient management. Developed as a joint project with the American Society of Anesthesiologists (ASA), and in collaboration with the American Heart Association (AHA), and the Society of Thoracic Surgeons (STS). Heart Rhythm 2011; 8:1114–1154.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:e57–e185.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645-681.
- O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Hillis LD, Smith PK, Anderson JL, et al. 2011 ACCF/AHA guideline for coronary artery bypass graft surgery: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, Society of Cardiovascular Anesthesiologists, and Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 58:e123–e210.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- McFalls EO, Ward HB, Krupski WC, et al. Prophylactic coronary artery revascularization for elective vascular surgery: study design. Veterans Affairs Cooperative Study Group on Coronary Artery Revascularization Prophylaxis for Elective Vascular Surgery. Control Clin Trials 1999; 20:297–308.
- Schouten O, van Kuijk JP, Flu WJ, et al. Long-term outcome of prophylactic coronary revascularization in cardiac high-risk patients undergoing major vascular surgery (from the randomized DECREASE-V Pilot Study). Am J Cardiol 2009; 103:897–901.
- Wijeysundera DN, Wijeysundera HC, Yun L, et al. risk of elective major noncardiac surgery after coronary stent insertion: a population-based study. Circulation 2012; 126:1355-1362.
- Hawn MT, Graham LA, Richman JS, et al. Risk of major adverse cardiac events following noncardiac surgery in patients with coronary stents. JAMA 2013; 310:1462–1472.
- Devereaux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
- Group PS, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Lindenauer PK, Pekow P, Wang K, et al. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- Kennedy J, Quan H, Buchan AM, et al. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- Raju MG, Pachika A, Punnam SR, et al. Statin therapy in the reduction of cardiovascular events in patients undergoing intermediate-risk noncardiac, nonvascular surgery. Clin Cardiol 2013; 36:456–461.
- Desai H, Aronow WS, Ahn C, et al. Incidence of perioperative myocardial infarction and of 2-year mortality in 577 elderly patients undergoing noncardiac vascular surgery treated with and without statins. Arch Gerontol Geriatr 2010; 51:149–151.
- Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1504–1513.
- Nguyen HP, Zaroff JG, Bayman EO, et al. Perioperative hypothermia (33 degrees C) does not increase the occurrence of cardiovascular events in patients undergoing cerebral aneurysm surgery: findings from the Intraoperative Hypothermia for Aneurysm Surgery Trial. Anesthesiology 2010; 113:327–342.
- Frank SM, Fleisher LA, Breslow MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277:1127–1134.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Foucrier A, Rodseth R, Aissaoui M, Ibanes C, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05536-3. doi: 10.1016/j.jacc.2014.07.944. [Epub ahead of print].
- Wijeysundera DN, Duncan D, Nkonde-Price C, et al. Perioperative beta blockade in noncardiac surgery: a systematic review for the 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05528-4. doi: 10.1016/j.jacc.2014.07.939. [Epub ahead of print].
- Fleisher LA, Fleischmann KE, Auerbach AD, et al. 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; Jul 29. pii: S0735-1097(14)05537-5. doi: 10.1016/j.jacc.2014.07.945. [Epub ahead of print].
- Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery. J Am Coll Cardiol 2007; 50:e159–e242.
- Erasmus MC Follow-up Investigation Committee. Report on the 2012 follow-up investigation of possible breaches of academic integrity. September 30, 2012. http://cardiobrief.files.wordpress.com/2012/10/integrity-report-2012-10-english-translation.pdf. Accessed October 30, 2014.
- Anderson JL, Antman EM, Harold JG, et al. Clinical practice guidelines on perioperative cardiovascular evaluation: collaborative efforts among the ACC, AHA, and ESC. J Am Coll Cardiol 2014 Jul 29. pii: S0735-1097(14)05527-2. doi: 10.1016/j.jacc.2014.07.938. [Epub ahead of print].
- Lee TH, Marcantonio ER, Mangione CM, et al. Derivation and prospective validation of a simple index for prediction of cardiac risk of major noncardiac surgery. Circulation 1999; 100:1043–1049.
- Gupta PK, Gupta H, Sundaram A, et al. Development and validation of a risk calculator for prediction of cardiac risk after surgery. Circulation 2011; 124:381–387.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surg 2013; 217:833–842. e1-3.
- Davis C, Tait G, Carroll J, Wijeysundera DN, Beattie WS. The Revised Cardiac Risk Index in the new millennium: a single-centre prospective cohort re-evaluation of the original variables in 9,519 consecutive elective surgical patients. Can J Anaesth 2013; 60:855–863.
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2014; e-pub before print. doi:10.1016/j.jacc.2014.03.022.
- Aliot EM, Alpert JS, Calkins H, et al. ACC/AHA/ESC guidelines for the management of patients with supraventricular arrhythmias. http://www.escardio.org/guidelines-surveys/esc-guidelines/GuidelinesDocuments/guidelines-SVA-FT.pdf. Accessed October 30,2014.
- Crossley GH, Poole JE, Rozner MA, et al. The Heart Rhythm Society (HRS)/American Society of Anesthesiologists (ASA) Expert Consensus Statement on the perioperative management of patients with implantable defibrillators, pacemakers and arrhythmia monitors: facilities and patient management. Developed as a joint project with the American Society of Anesthesiologists (ASA), and in collaboration with the American Heart Association (AHA), and the Society of Thoracic Surgeons (STS). Heart Rhythm 2011; 8:1114–1154.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62:e147–e239.
- Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63:e57–e185.
- Jneid H, Anderson JL, Wright RS, et al. 2012 ACCF/AHA focused update of the guideline for the management of patients with unstable angina/non-ST-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2012; 60:645-681.
- O’Gara PT, Kushner FG, Ascheim DD, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61:e78–e140.
- Hillis LD, Smith PK, Anderson JL, et al. 2011 ACCF/AHA guideline for coronary artery bypass graft surgery: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, Society of Cardiovascular Anesthesiologists, and Society of Thoracic Surgeons. J Am Coll Cardiol 2011; 58:e123–e210.
- Levine GN, Bates ER, Blankenship JC, et al. 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. J Am Coll Cardiol 2011; 58:e44–e122.
- McFalls EO, Ward HB, Krupski WC, et al. Prophylactic coronary artery revascularization for elective vascular surgery: study design. Veterans Affairs Cooperative Study Group on Coronary Artery Revascularization Prophylaxis for Elective Vascular Surgery. Control Clin Trials 1999; 20:297–308.
- Schouten O, van Kuijk JP, Flu WJ, et al. Long-term outcome of prophylactic coronary revascularization in cardiac high-risk patients undergoing major vascular surgery (from the randomized DECREASE-V Pilot Study). Am J Cardiol 2009; 103:897–901.
- Wijeysundera DN, Wijeysundera HC, Yun L, et al. risk of elective major noncardiac surgery after coronary stent insertion: a population-based study. Circulation 2012; 126:1355-1362.
- Hawn MT, Graham LA, Richman JS, et al. Risk of major adverse cardiac events following noncardiac surgery in patients with coronary stents. JAMA 2013; 310:1462–1472.
- Devereaux PJ, Mrkobrada M, Sessler DI, et al. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
- Group PS, Devereaux PJ, Yang H, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet 2008; 371:1839–1847.
- Lindenauer PK, Pekow P, Wang K, et al. Lipid-lowering therapy and in-hospital mortality following major noncardiac surgery. JAMA 2004; 291:2092–2099.
- Kennedy J, Quan H, Buchan AM, et al. Statins are associated with better outcomes after carotid endarterectomy in symptomatic patients. Stroke 2005; 36:2072–2076.
- Raju MG, Pachika A, Punnam SR, et al. Statin therapy in the reduction of cardiovascular events in patients undergoing intermediate-risk noncardiac, nonvascular surgery. Clin Cardiol 2013; 36:456–461.
- Desai H, Aronow WS, Ahn C, et al. Incidence of perioperative myocardial infarction and of 2-year mortality in 577 elderly patients undergoing noncardiac vascular surgery treated with and without statins. Arch Gerontol Geriatr 2010; 51:149–151.
- Durazzo AES, Machado FS, Ikeoka DT, et al. Reduction in cardiovascular events after vascular surgery with atorvastatin: a randomized trial. J Vasc Surg 2004; 39:967–975.
- Devereaux PJ, Sessler DI, Leslie K, et al. Clonidine in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1504–1513.
- Nguyen HP, Zaroff JG, Bayman EO, et al. Perioperative hypothermia (33 degrees C) does not increase the occurrence of cardiovascular events in patients undergoing cerebral aneurysm surgery: findings from the Intraoperative Hypothermia for Aneurysm Surgery Trial. Anesthesiology 2010; 113:327–342.
- Frank SM, Fleisher LA, Breslow MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277:1127–1134.
- Vascular Events In Noncardiac Surgery Patients Cohort Evaluation Study I, Devereaux PJ, Chan MT, Alonso-Coello P, et al. Association between postoperative troponin levels and 30-day mortality among patients undergoing noncardiac surgery. JAMA 2012; 307:2295–2304.
- Botto F, Alonso-Coello P, Chan MT, et al. Myocardial injury after noncardiac surgery: a large, international, prospective cohort study establishing diagnostic criteria, characteristics, predictors, and 30-day outcomes. Anesthesiology 2014; 120:564–578.
- Devereaux PJ, Xavier D, Pogue J, et al. Characteristics and short-term prognosis of perioperative myocardial infarction in patients undergoing noncardiac surgery: a cohort study. Ann Intern Med 2011; 154:523–528.
- Foucrier A, Rodseth R, Aissaoui M, Ibanes C, et al. The long-term impact of early cardiovascular therapy intensification for postoperative troponin elevation after major vascular surgery. Anesth Analg 2014; 119:1053–1063.
KEY POINTS
- Like earlier guidelines, the update recommends preoperative cardiac testing only when the results may influence the patient’s management.
- Preoperative intervention is rarely necessary just to get the patient through surgery, unless it is otherwise indicated independent of the need for surgery.
- The update proposes a modified algorithm for preoperative risk assessment and management and suggests using a new calculator of surgical risk.
- The report also updates information on the timing of surgery after percutaneous coronary intervention, as well as on antiplatelet therapy, other medical therapy, and biomarkers.
Identifying statin-associated autoimmune necrotizing myopathy
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS

Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS
Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
Statins are among the most widely prescribed drugs, as they reduce cardiovascular risk very effectively. They work by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), a key enzyme in cholesterol biosynthesis. Although most patients tolerate statins well, muscle-related toxicity can limit the use of these drugs.
Recently, progressive necrotizing myopathy leading to profound weakness has been directly linked to statin therapy. Proper recognition of this ominous complication is important to prevent further damage from statin use.
STATIN-ASSOCIATED MUSCLE EFFECTS: A SPECTRUM
Muscle symptoms are among the best known and most important side effects of statins, ranging from asymptomatic, mild elevation of creatine kinase and benign myalgias to life-threatening rhabdomyolysis. As the various terms are often used inconsistently in the literature, we will briefly review each entity to put immune-necrotizing myopathy in its proper context on the spectrum of statin-associated muscle symptoms.
Myalgia
Myalgia, ie, muscle pain without elevation of muscle enzymes, is the most common side effect of statins. The incidence is about 10% in observational studies1,2; however, the incidence in the real world of clinical practice seems much higher. Myalgias can present as widespread pain or as localized pain, usually in the lower extremities. Muscle cramps and tendonitis-related pain are commonly reported.
Several clinical predictors of increased risk of statin-associated muscle pain were noted in the Prediction of Muscular Risk in Observational Conditions (PRIMO) study,2 assessing mild to moderate muscular symptoms in patients on high-dose statins. These included a history of muscle pain with another lipid-lowering agent, a history of cramps, a history of elevated creatine kinase levels, a personal or family history of muscle symptoms, untreated hypothyroidism, and a background of fibromyalgia-like symptoms. The incidence of muscle symptoms increased with the level of physical activity, and the median time of symptom onset was 1 month after starting statin therapy or titrating to a high dose.
In patients with myalgia alone, symptoms often improve when the statin is stopped.
Myopathy, myositis
The general terms myopathy and myositis have been used to refer to elevated muscle enzymes together with the muscle symptoms of pain, cramps, soreness, or weakness. An analysis of 21 clinical trials of statin therapy found that myopathy (creatine kinase level more than 10 times the upper limit of normal) occurred in 5 patients per 100,000 person-years.3 The incidence of myopathy increases when statins are used in high doses.
In another cohort of patients seen for statin intolerance,1 conventional risk factors for overt myositis included renal disease, diabetes, and thyroid disease. Electrolyte abnormalities did not differ between statin-tolerant and statin-intolerant patients.1
The search for genetic indicators of risk
In 2008, the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group conducted a genome-wide association study to identify genetic variants associated with myopathy with high-dose statin therapy.4 Eighty-five patients who had developed definite myopathy (muscle symptoms with enzyme levels more than 10 times the upper limit of normal) or incipient myopathy (asymptomatic or symptomatic, but enzyme levels at least three times the upper limit of normal) while taking simvastatin 80 mg were compared with 90 controls taking the same daily dose. The study reported variants in the SLCO1B1 gene as “strongly associated with an increased risk of statin-associated myopathy.”4
SLCO1B1 encodes the peptide responsible for hepatic uptake of statins, thus affecting the blood level of these drugs. Patients in the study who had the C variant, which predisposes to higher blood levels of statins, were at higher risk of myopathy than those with the T variant. The odds ratio for myopathy increased from 4.4 in heterozygotes for the C allele to 17.4 for homozygotes. This effect was similar even with lower doses of statins.4
We believe that these findings provide a strong basis for genetic testing of patients who may be at risk of statin-associated myopathy.
Rechallenging with a different statin
Often, patients with myopathy can be rechallenged with a different statin agent. In a study done in a lipid clinic, patients identified as having simvastatin-associated myopathy were given another statin.5 Between 15% and 42% tolerated the second statin, with no statistically significant difference between the tolerability rates with the different agents used (atorvastatin, rosuvastatin, pravastatin, and fluvastatin).
Rhabdomyolysis
Rhabdomyolysis, the most devastating complication of statin use, is marked by a creatine kinase level more than 10 times the upper limit of normal or greater than 10,000 IU/L, resulting from acute and massive destruction of muscle fibers and release of their contents into the bloodstream.
But rhabdomyolysis is a clinical syndrome, not solely an alarming increase in muscle enzyme levels. It can include renal failure and death. The rate of occurrence is low (1/100,000), but it can occur at any point in treatment.6 An analysis of Canadian and US case reports of statin-associated rhabdomyolysis showed an average of 824 cases each year.7 A dose-response relationship was observed with higher statin doses.
But statin-associated myopathy may not stop when the drug is stopped
The types of muscle toxicity discussed above stem from direct myotoxic effects of statins and are thought to be related to the blood concentration of the statin.6,8 They may be limited by genetic susceptibility. Mechanisms may include a change in muscle membrane excitability caused by modulation in membrane cholesterol levels, impaired mitochondrial function and calcium signaling, induction of apoptosis, and increased lipid peroxidation. Stopping the offending drug can often halt these downstream effects—hence the dictum that statin myopathy is self-limiting and should resolve with cessation of the drug.
But as we discuss in the following sections, some patients on statin therapy develop an immune-mediated myopathy that does not resolve with discontinuation of the statin and that may only resolve with immunosuppressive therapy.
STATINS AND AUTOIMMUNE NECROTIZING MYOPATHY
At the Johns Hopkins Myositis Center, a group of patients was identified who had necrotizing myopathy on biopsy but no known underlying condition or associated autoantibodies. In an attempt to establish an autoimmune basis for their disease, the sera from 26 patients were screened for novel antibodies.9 Sera from 16 of the patients immunoprecipitated a pair of proteins (sizes 100 kd and 200 kd), indicating these patients had an antibody to these proteins. This finding was highly specific for necrotizing myopathy when compared with controls, ie, other patients with myositis. Patients who had this finding displayed proximal muscle weakness, elevated muscle enzyme levels, and myopathic findings on electromyography; 63% had been exposed to statins before the onset of weakness, and when only patients over age 50 were included, the number rose to 83%.
This association of statins with necrotizing myopathy had been previously noted by two other groups. Needham et al10 described eight patients who, while on statins, developed myopathy that continued to worsen despite cessation of the drugs. An analysis of their muscle pathology revealed myofiber necrosis with little inflammatory infiltrate, as well as widespread up-regulation of expression of major histocompatibility complex class 1. These patients required immunosuppressive treatment (prednisone and methotrexate) to control their disease.
Grable-Esposito et al11 corroborated this finding by identifying 25 additional patients who developed a similar necrotizing myopathy while on statins.11 They also noted a significantly higher frequency of statin use in patients with necrotizing myopathy than in age-matched controls with polymyositis or inclusion-body myositis.
The researchers at the Johns Hopkins Myositis Center noted the similarity between these two patient groups and their own group of patients with necrotizing myopathy. Thus, a follow-up study was done to identify the 100-kg and 200-kd autoantigens observed in their earlier study. Exposure to a statin was found to up-regulate the expression of the two molecules.12 HMGCR was hypothesized as being the 100-kd antigen, because of its 97-kd molecular weight, and also because statin treatment had already been shown to up-regulate the expression of HMGCR.13 The researchers concluded that HMGCR was indeed the 100-kd antigen, with no distinctive antibodies recognizing the 200-kd protein. Although the 200-kd protein was once postulated to be a dimer of the 100-kd protein, its identity remains unknown.
The anti-HMGCR antibody was then screened for in a cohort of 750 myositis patients. The 16 patients previously found to have anti-200/100 were all positive for anti-HMGCR antibody. An additional 45 patients from the cohort (6%) were anti-HMGCR-positive by enzyme-linked immunosorbent assay, and all had necrotizing myopathy. Patients with other types of myopathy, including inflammatory myopathy, do not possess this antibody.12
The HMGCR antibody was quite specific for immune-mediated necrotizing myopathy, and this suggested that statins were capable of triggering an immune-mediated myopathy that is then perpetuated even if the drug is discontinued. As it was also demonstrated that statins increase the expression of HMGCR in muscle as well as in regenerating cells, the process may be sustained through persistently increased HMGCR expression associated with muscle repair.12
The C allele of the SLCO1B1 gene, which has been associated with statin-associated myopathy, was not increased in this population of patients positive for anti-HMGCR. Follow-up studies of the prevalence of anti-HMGCR in statin users in the Atherosclerosis Risk in Communities (ARIC) cohort, including those with self-limited statin myotoxicity, have also shown the absence of this antibody.14 This shows that anti-HMGCR is not found in the majority of statin-exposed patients and is highly specific for autoimmune myopathy. This also suggests that statin-associated autoimmune myopathy represents a pathologic process that is distinct from self-limited statin intolerance.
HOW THE CONDITION PRESENTS
Immune-mediated statin myopathy presents similarly to other idiopathic inflammatory myopathies such as polymyositis (Table 1). Symptoms often develop in a subacute to chronic course and can occur at any time with statin treatment. In one study,10 the average duration of statin use before the onset of weakness was 3 years (range 2 months to 10 years). In some patients whose statin had been stopped because of abnormal creatine kinase levels, weakness developed later, at a range of 0.5 to 20 months. Even low doses of statins (such as 10 mg of simvastatin) have been found to trigger this condition.10
Patients uniformly develop symmetric proximal arm and leg weakness, and distal weakness can also occur.11 Other features have included dysphagia, arthralgias, myalgias, and Raynaud phenomenon.9 Men and women are represented in roughly equal numbers.
The muscle enzymes are strikingly elevated in this disease, with a mean creatine kinase value of 10,333 IU/L at initial presentation.9 Although the creatine kinase level may be very elevated, patients often do not present with weakness until a certain threshold value is reached, in contrast with patients with anti-signal recognition particle necrotizing myopathy, who can present with profound weakness at a lower level. Hence, by the time patients are clinically symptomatic, the process may have been going on for some time. Despite the seemingly massive leak in muscle enzymes, the patients do not develop rhabdomyolysis. Inflammatory markers need not be elevated, and an association with other antibodies such as antinuclear antibody is not often seen.
Magnetic resonance imaging of the thigh has shown muscle edema in all patients.9 In decreasing order of frequency, other findings are atrophy, fatty replacement, and fascial edema.
Electromyography of involved muscle has shown irritable myopathy in most patients (88%) and nonirritable myopathy in a few.
Muscle biopsy studies have shown prominent necrotic and regenerating fibers without significant inflammatory infiltrate.11 There is also myophagocytosis of necrotic fibers and diffuse or focal up-regulation of major histocompatability complex class I expression.10
PATIENTS WITH ANTI-HMGCR WHO HAVE AND WHO HAVE NEVER TAKEN STATINS
When the anti-HMGCR antibody was tested for in the Johns Hopkins cohort,12 33% of patients with a necrotizing myopathy associated with this antibody had never taken a statin. The two groups were clinically indistinguishable, save for a few aspects. Compared with patients who had taken a statin, those who had never taken a statin were younger (mean age 37 vs 59), had higher levels of creatine kinase (13,392 vs 7,881 IU/L), and had a different race distribution (46.7% vs 86.7% white). Initial HMGCR antibody levels were also noted to correlate with creatine kinase levels and strength in statin-exposed patients but not in those who had never taken a statin.15
We hypothesize that in patients who have never taken a statin, other genetic or environmental factors may be the cause of the increased HMGCR expression, which then triggers the autoimmune response. Until further data are gathered, we should probably treat these patients as we treat those who develop this disease after taking a statin, and avoid giving them statins altogether.
MANAGEMENT OF STATIN-ASSOCIATED AUTOIMMUNE NECROTIZING MYOPATHY
Treatment of statin-associated autoimmune necrotizing myopathy can be challenging and requires immunosuppressive drugs.
Statin therapy should be stopped once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy. Electromyography detects myopathy and shows chronicity, distribution, and degree of severity. Although not necessary for diagnosis, magnetic resonance imaging helps to evaluate the extent of muscle involvement and damage and provides guidance when choosing a site for muscle biopsy. Muscle biopsy is necessary to determine the actual pathology and to exclude mimics such as dystrophy or metabolic myopathies.
When an immune-mediated myopathy is confirmed, prompt referral to a rheumatologist or a neuromuscular specialist is recommended.
Steroids are usually the first-line treatment for this disease. Other immunosuppressives, such as methotrexate, azathioprine, mycophenolate mofetil, and rituximab have been used with varying levels of success. In our experience, intravenous immunoglobulin has been particularly beneficial for refractory cases. With treatment, muscle enzyme levels and weakness improve, but relapses can occur. The ideal choice of immunosuppressive therapy and the duration of therapy are currently under investigation.
Rechallenge with another statin
At this time, the issue of rechallenging the patient with another statin has not been clarified. Given the autoimmune nature of the disease, we would avoid exposing the patient to a known trigger. However, this may be a difficult decision in patients with cardiovascular risk factors who require statins for primary or secondary prevention. We suggest using alternative cholesterol-lowering agents first and using them in combination if needed.
We have had some success in maintaining a handful of patients on a statin while treating them concurrently with immunosuppression. This is not ideal because they are constantly being exposed to the likely trigger for their disease, but it may be unavoidable if statins are deemed absolutely necessary. We have also had a patient with known statin-associated immune-mediated necrotizing myopathy who later became profoundly weak after another physician started her on a newer-generation agent, pitavastatin. This suggests to us that rechallenging patients, even with a different statin, can have deleterious effects.
IMPLICATIONS FOR CLINICAL PRACTICE
The true prevalence of statin-associated autoimmune myopathy in practice is unknown. In the Johns Hopkins Myositis Center cohort of patients with suspected myopathy, anti-HMGCR was found in 6% of the patients and was the second most frequent antibody found after anti-Jo1.12
Given the frequency of muscle-related complaints in patients on statins, we recommend obtaining baseline muscle enzyme measurements before starting statin therapy. As recommended by the National Lipid Association Statin Safety Assessment Task Force, the creatine kinase level should be measured when a patient develops muscular complaints, to help gauge the severity of the disease and to help decide whether to continue therapy.8 Random testing of the creatine kinase level in asymptomatic patients is not recommended.
At present, the diagnosis of statin-associated autoimmune necrotizing myopathy is based on a combination of findings—elevated muscle enzyme levels, muscle weakness, irritable findings on electromyography, and necrotizing myopathy on biopsy in a patient on a statin. The finding of the HMGCR antibody confirms the diagnosis. A test for this antibody is now commercially available in the United States. We suggest testing for the antibody in the following scenarios:
- A persistently elevated or rising creatine kinase, aspartate aminotransferase, alanine aminotransferase, or aldolase level after the statin is stopped; although no fixed creatine kinase level has been determined, a level above 1,000 U/L would be a reasonable cutoff at which to test
- Muscle symptoms (proximal or distal weakness) that persist 12 weeks after statin cessation regardless of the creatine kinase level, especially if the patient has dysphagia
- The finding of muscle irritability on electromyography or diffuse muscle edema on magnetic resonance imaging when testing for other myositis-specific antibodies is negative
- Muscle biopsy showing necrotizing myopathy with little or no inflammation.
In addition, since necrotizing myopathy is known to be associated with malignancy and since necrotizing myopathy is more common in older people, who are also more likely to be taking a statin, an age-appropriate malignancy evaluation is warranted as well.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
- Harris LJ, Thapa R, Brown M, et al. Clinical and laboratory phenotype of patients experiencing statin intolerance attributable to myalgia. J Clin Lipidol 2011; 5:299–307.
- Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther 2005; 19:403–414.
- Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol 2006; 97:52C–60C.
- SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med 2008; 359:789–799.
- Fung EC, Crook MA. Statin myopathy: a lipid clinic experience on the tolerability of statin rechallenge. Cardiovasc Ther 2012; 30:e212–e218.
- Sirvent P, Mercier J, Lacampagne A. New insights into mechanisms of statin-associated myotoxicity. Curr Opin Pharmacol 2008; 8:333–338.
- Holbrook A, Wright M, Sung M, Ribic C, Baker S. Statin-associated rhabdomyolysis: is there a dose-response relationship? Can J Cardiol 2011; 27:146–151.
- McKenney JM, Davidson MH, Jacobson TA, Guyton JR; National Lipid Association Statin Safety Assessment Task Force. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol 2006; 97:89C–94C.
- Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62:2757–2766.
- Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord 2007; 17:194–200.
- Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve 2010; 41:185–190.
- Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63:713–721.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature 1990; 343:425–430.
- Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012; 64:269–272.
- Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Rheum 2012; 64:4087–4093.
KEY POINTS
- Most cases of muscle symptoms associated with statin use are a direct effect of the statin on the muscle and resolve after the statin is discontinued.
- In contrast to simple myalgia or myositis, statin-associated autoimmune necrotizing myopathy can persist or even arise de novo after the statin is stopped.
- This condition presents with symmetric proximal arm and leg weakness and striking elevations of muscle enzymes such as creatine kinase.
- Treatment can be challenging and requires immunosuppressive drugs; referral to a specialist is recommended.
- Statin therapy should be discontinued once this condition is suspected. Patients who continue to have elevated muscle enzymes or weakness should undergo further testing with electromyography, magnetic resonance imaging, and muscle biopsy.
After substance withdrawal, underlying psychiatric symptoms emerge
When treating patients who abuse substances, it is important to watch for underlying clinical conditions that have been suppressed, relieved, or muted by alcohol or drugs. Many of these conditions can be mistaken for signs of withdrawal, drug-seeking, or new conditions arising from loss of euphoria from the drug. Prompt recognition of these disorders and use of appropriate non-addictive treatments can prevent “against medical advice” discharges, relapses, and unneeded suffering in many cases.
Because the brain is the target organ, these conditions are either neurologic or psychiatric in nosology. Although psychiatric clinicians might not be familiar with neurologic conditions, quick recognition and treatment is necessary.
Restless legs syndrome and periodic limb movements of sleep
Restless legs syndrome (RLS) has 2 key components: paresthesia and akathisia. Although primarily involving the lower extremities, involvement also can include the upper extremities, torso, and head.
Paresthesia differs from typical neuropathies in that it usually is not painful; rather, patients describe an odd sensation using terms such as ticklish, “creepy-crawly,” and other uncomfortable sensations.
Akathisia is a motor restlessness and need to move. The patient might feel momentary relief by moving or rubbing the extremities, only to have the paresthesia return quickly followed by the akathisia. Generally, reclining is the most prominent position that produces symptoms, but they can occur while sitting.
The cause of RLS is an abnormality of central dopamine or iron, or both, in the substantia nigra; iron is a cofactor in dopamine synthesis. All RLS patients should have a serum ferritin level drawn and if <50 μg/dL, be treated with iron supplementation. Dopamine agonists, such as ropinirole, pramipexole, and carbidopa/levodopa, are effective (Table 1); other useful agents include benzodiazepines such as clonazepam and opioids such as hydrocodone.

When a patient withdraws from benzodiazepines or narcotics, RLS can emerge and cause suffering until it is diagnosed and treated. Typical myalgia in opioid withdrawal can confound the diagnosis. The immediate-release (IR) and extended-release (ER) formulations of gabapentin often are a good choice when treating benzodiazepine or narcotic withdrawal. The side effect profile of gabapentin is relatively benign, with somnolence often reported by non-substance abusers, but it is unlikely that addicts, who have grown tolerant to more potent agents such as benzodiazepines and opioids, will complain of sleepiness. Studies have shown that gabapentin is useful in managing withdrawal as well as anxiety and insomnia.1,2 A randomized trial showed that gabapentin increases abstinence rates and decreases heavy drinking.2 The agent has a short half-life (5 to 7 hours); the IR form needs to be dosed at least 3 times a day to be effective. An ER formulation of gabapentin was released in 2013 with the sole indication for RLS.
Gabapentin is not significantly metabolized by the liver, has a 3% rate of protein binding, and is excreted by the kidneys—making it safe for patients who abuse alcohol or opioids and have impaired hepatic function. Typical starting dosages of IR gabapentin are 100 to 300 mg, 3 times daily, if symptoms are present in the daytime. Asymmetric dosing can be helpful, with larger or single dosages given at bedtime (eg, 100 mg in morning, 100 mg in afternoon, 300 mg at bedtime). Dosing varies from patient to patient, from 300 mg to 3,600 mg/d. Increasing dosages produce lower bioavailability because of saturation in absorption or at the blood-brain barrier. At 100 mg every 8 hours, bioavailability is 80% but at 1,600 mg every 8 hours it drops to 27%.3
Periodic limb movements of sleep (PLMS) essentially is akathisia during sleep, and occurs in most patients with RLS. The patient feels tired in the morning because of lack of deep stage-N3 sleep. Because of the inverse relationship between serotonin and dopamine, most selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors can exacerbate RLS and PLMS.4,5 Other culprits include antipsychotics, antiemetics, and antihistamines. The differential diagnosis includes withdrawal from opioids and attention-deficit/hyperactivity disorder (ADHD), which may be comorbid with RLS. There are many causes of secondary RLS including renal failure, pregnancy, varicose veins, and neuropathy.
Tremor
Benign familial, or essential, tremor is a fine intention tremor that can be suppressed by alcohol or benzodiazepines. After detoxification from either of these substances, persistent tremor can re-emerge; often, it is benign, although cerebellar and parkinsonian tremors must be ruled out. Essential tremor can be treated with gabapentin or beta blockers such as propranolol or metoprolol (Table 2).
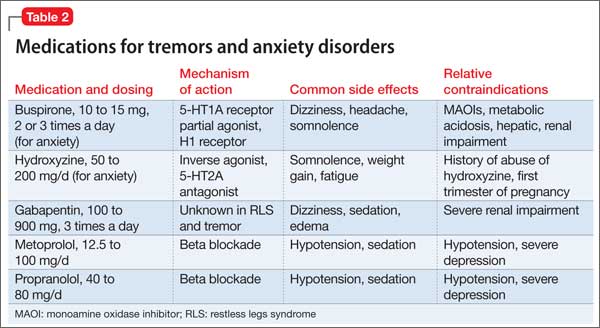
Anxiety and panic disorder
Social anxiety often presents in addiction treatment centers in the context of group therapy, speaking in 12-step meetings, and having the patient describe his (her) autobiography and history of addiction. Because social anxiety disorder is the third most common psychiatric disorder after simple phobia and major depressive disorder,6 it is not surprising that it emerges after withdrawal.
Patients with social anxiety disorder might self-medicate with alcohol or drugs, especially benzodiazepines (Box). Residential treatment presents an excellent environment for desensitization to fears of public speaking; early recognition is key. Apprehension about group therapy, presenting a substance abuse history, or speaking at a 12-step meeting can lead to premature or “against medical advice” discharge.

Panic disorder commonly is comorbid with substance abuse. Many patients will arrive at treatment with a prescription for benzodiazepines. Because the risk of cross-addiction is high among recovering addicts, benzodiazepines should be avoided. Treating underlying anxiety is crucial for fostering sobriety. Generalized anxiety disorder is common among patients with an addiction, and can lead to relapse if not addressed. Use of non-addictive medications and cognitive therapy is useful in addressing this condition.
A quandary might arise in states where medical marijuana is legal, because Cannabis can be prescribed for anxiety disorders and posttraumatic stress disorder (PTSD). Promoting abstinence from all substances can present a challenge in patients with anxiety disorders who live in these states.
Medications for anxiety and panic disorder include gabapentin, buspirone, hydroxyzine, beta blockers, and atypical antipsychotics (Table 2). Only buspirone and hydroxyzine are FDA-approved for anxiety; buspirone monotherapy generally is ineffective for panic disorder.
Explaining to patients how anxiety arises, such as how classical conditioning leads to specific phobias, can be therapeutic. Describing Klein’s false suffocation alarm theory of panic attacks can illustrate the importance of practicing slow, deep breathing to prevent hyperventilation.7 Also, relabeling a panic attack with self-talk statements such as “I know what this is. It’s just a panic attack” can be helpful. Smartphone apps are available to help patients cope with anxiety and acute panic.8
Mood disorders
Many patients with bipolar disorder experience substance abuse at some point; estimates are that up to 57% of patients have a comorbid addiction.6,9 Persons with a mood disorder are at high risk of substance abuse because of genetic factors; patients also might self-medicate their mood symptoms.
After alcohol or drugs are withdrawn, mood disorders can emerge or resurge. Often, patients enter treatment taking antidepressants and mood stabilizers and usually haven’t been truthful with their treatment provider about their substance abuse. Care must be taken to ascertain whether mood symptoms are secondary to substance abuse. Asking “What’s the longest period of abstinence you’ve had in 2 years and how did you feel emotionally?” often will help you identify a secondary mood disorder. For example, a response of “6 months and I felt really depressed the entire time” would indicate a primary depressive disorder.
Because CNS depressants, such as alcohol and benzodiazepines, can exacerbate a mood disorder, consider continuing or resuming a mood stabilizer or antidepressant during substance abuse treatment. When meeting a new patient, perform an independent evaluation, because substance use can mimic bipolar and depressive disorders. Careful assessment of suicidal ideation is necessary for all patients.
Sleep disorders
Insomnia—as a primary or secondary disorder—is common among patients with a substance use disorder. Insomnia always needs to be addressed. Not sleeping well interferes with cognition and energy and makes depression and bipolar disorder worse. Some experts recommend “waiting out” the insomnia, hoping that sobriety will resolve it—but it might not.
Initial insomnia can be treated with melatonin, 3 to 6 mg at bedtime or earlier in the evening.10-12 Melatonin acts by regulating circadian rhythms, but can cause increased dreaming and nightmares; therefore, it should be avoided in patients who struggle with nightmares. Trazodone, 50 to 150 mg at bedtime, is an inexpensive sleep aid for initial insomnia and doesn’t cause weight gain, which many drugs with antihistaminic properties can. Prazosin, 1 to 2 mg initially, for nightmares in PTSD is effective.13
Antipsychotics might be necessary if nothing else works; quetiapine is effective for sleep and the ER form is FDA-approved as an add-on agent in major depression. Low-dose doxepin (≤10 mg) is effective for middle insomnia.14 At these low dosages, troublesome side effects of tricyclic antidepressants can be avoided.
As many as 40% of adults with ADHD have a delayed sleep-phase disorder. Ask your patient if she is a “night owl,” how chronic the condition is, and when her best sleep occurs.15-17 Morning light and evening melatonin can help, but often are insufficient. Many patients present with undiagnosed or untreated sleep apnea, which can cause excessive daytime sleepiness. Referral to a sleep center is prudent; use of the Epworth Sleepiness Scale is a quick way to assess excessive daytime sleepiness.18
ADHD
ADHD commonly is comorbid with a substance use disorder. Patients might present with an earlier diagnosis, including treatment. Several drugs of abuse can alleviate ADHD symptoms, including amphetamines, opioids, cocaine, and Cannabis; self-medicating is common. Because opioids increase dopamine release, a report of improved work and school performance while taking opioids early in addiction can be a clue to an ADHD diagnosis.
Explaining ADHD as a syndrome of “interest-based attention” helps. If a residential treatment program uses reading and writing assignments, a patient with ADHD might struggle and will need extra help and time and a quiet place to do assignments.19,20 A non-addictive medication, such as atomoxetine, can help, but has an antidepressant-like delay of 3 to 5 weeks until onset of symptom relief. Using a long-acting stimulant can be effective and quick, with an effect size 3 to 4 times higher than atomoxetine; such agents should be avoided in patients who abuse amphetamines.
Studies show that treating ADHD, even with stimulants, neither helps nor hurts outcomes in substance use. Lisdexamfetamine is difficult to abuse and is an inactive prodrug (a bond of lysine and dextroamphetamine) that requires enzymatic cleavage and activation by red blood cells; these characteristics creates a long-acting medication that has a lower abuse liability than other drugs for ADHD. However, abuse can occur and the drug must be used cautiously. Earley’s medication guide referenced below recommends that lisdexamfetamine and other stimulants should be avoided if possible in patients in recovery. However, it adds that specialists in treating ADHD in substance-abusing patients should weigh the potential benefits of stimulant use against the risk of relapse.17 Many patients enter treatment with a diagnosis of bipolar disorder that might, in fact, be comorbid with ADHD.
Chronic pain
Many substance abuse patients began taking opioids for acute, then chronic, pain before their use escalated to addiction. These are challenging patients; often, they are referred for treatment without true addiction.
Keep in mind that dependence is not addiction. Pseudo-addiction is a condition in which pain is undertreated and the patient takes more medication to obtain relief, calls for early refills, and displays drug-seeking behavior but is not using drugs to achieve euphoria. A thorough history and physical and referrals to specialists such as orthopedic surgeons and pain specialists are necessary. Explaining opioid-induced hyperalgesia is important to help the patient understand that (1) pain can be made worse by increasing the dosage of an opioid because of supersensitivity and (2) many patients who are weaned off these drugs will experience a decrease or complete relief of pain.21
Gabapentin, duloxetine, or amitriptyline can be beneficial for chronic pain, as well as mindfulness techniques, physical therapy, and complementary and alternative medicine. Pregabalin can produce euphoria and often should be avoided.
A medication guide for recovery
Paul Earley, MD, former medical director at Talbott Recovery in Atlanta, Georgia, publishes an online guide that classifies medications into categories:
• A: safe
• B: use only under the supervision of an addiction medicine specialist or doctor
• C: completely avoid if the patient is in recovery.17
The Talbott guide lists all stimulants in category C, (except for atomoxetine, which is category A). Hydroxyzine is listed under category B. Many programs for impaired professionals and state medical boards use the Guide, and will question the prescribing of any medication from categories B and C.17
Related Resources
• Spiegel DR, Kumari N, Petri JD. Safer use of benzodiazepines for alcohol detoxification. Current Psychiatry. 2012;11(10):10-15.
• Kelly TM, Daley DC, Douaihy AB. Treatment of substance abusing patients with comorbid psychiatric disorders. Addict Behav. 2012;37(1):11-24.
Drug Brand Names
Amitriptyline • Elavil Hydrocodone • Vicodin
Atenolol • Tenormin Hydroxyzine • Vistaril, Atarax
Atomoxetine • Strattera Lisdexamfetamine • Vyvanse
Buprenorphine/ naloxone • Suboxone Metoprolol • Lopressor, Toprol
Buspirone • BuSpar Paroxetine • Paxil
Carbidopa-levodopa • Sinemet Pramipexole • Mirapex
Clonazepam • Klonopin Prazosin • Minipress
Diazepam • Valium Pregabalin • Lyrica
Doxepin • Silenor, Adapin, Sinequan Propranolol • Inderal
Duloxetine • Cymbalta Quetiapine • Seroquel
Escitalopram • Lexapro Ropinirole • Requip
Gabapentin • Neurontin, Horizant Sertraline • Zoloft
Trazodone • Desyrel
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stock CJ, Carpenter L, Ying J, et al. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother. 2013;47(7-8):961-969.
2. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
3. Bockbrader HN, Wesche D, Miller R, et al. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin Pharmacokinet. 2010; 49(10):661-669.
4. Yang C, White DP, Winkelman JW. Antidepressants and periodic leg movements of sleep. Biol Psychiatry. 2005;58(6):510-514.
5. Hoque R, Chesson AL Jr. Pharmacologically induced/ exacerbated restless legs syndrome, periodic limb movements of sleep, and REM behavior disorder/ REM sleep without atonia: literature review, qualitative scoring, and comparative analysis. J Clin Sleep Med. 2010; 6(1):79-83.
6. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
7. Klein DF. False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry. 1993;50(4):306-317.
8. Holland K. The 17 best anxiety iPhone & Android apps of 2014. http://www.healthline.com/health-slideshow/top-anxiety-iphone-android-apps. Accessed October 28, 2014.
9. Chengappa KN, Levine J, Gershon S, et al. Lifetime prevalence of substance or alcohol abuse and dependence among subjects with bipolar I and II disorders in a voluntary registry. Bipolar Disord. 2000;2(3 Pt 1):191-195.
10. Ferracioli-Oda E, Qawasmi A, Bloch MH. Meta-analysis: melatonin for the treatment of primary sleep disorders [published online May 17, 2013]. PLoS One. 2013;8(5):e63773. doi: 10.1371/journal.pone.0063773.
11. Wade AG, Ford I, Crawford G, et al. Efficacy of prolonged release melatonin in insomnia patients aged 55-80 years: quality of sleep and next-day alertness outcomes. Curr Med Res Opin. 2007;23(10):2597-2605.
12. Srinivasan V, Brzezinski A, Pandi-Perumal SR, et al. Melatonin agonists in primary insomnia and depression-associated insomnia: are they superior to sedative-hypnotics? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):913-923.
13. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
14. Scharf M, Rogowski R, Hull S, et al. Efficacy and safety of doxepin 1 mg, 3 mg, and 6 mg in elderly patients with primary insomnia: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry. 2008;69(10):1557-1564.
15. Baird AL, Coogan AN, Siddiqui A, et al. Adult attention-deficit hyperactivity disorder is associated with alterations in circadian rhythms at the behavioural, endocrine and molecular levels. Mol Psychiatry. 2012;17(10):988-995.
16. Yoon SY, Jain U, Shapiro C. Sleep in attention-deficit/ hyperactivity disorder in children and adults: past, present, and future. Sleep Med Rev. 2012;16(4):371-388.
17. Earley PH, Merkin B, Skipper G. The medication guide for safe recovery. Revision 1.7. http://paulearley.net/index. php?option=com_docman&Itemid=239. Published March 2014. Accessed October 28, 2014.
18. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540-545.
19. Dodson W. Secrets of the ADHD brain. ADDitude. http:// www.additudemag.com/adhd/article/10117.html. Accessed October 28, 2014.
20. Wilens TE, Dodson W. A clinical perspective of attention-deficit/hyperactivity disorder into adulthood. J Clin Psychiatry. 2004;65(10):1301-1313.
21. Lee M, Silverman SM, Hansen H, et al. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145-161.
When treating patients who abuse substances, it is important to watch for underlying clinical conditions that have been suppressed, relieved, or muted by alcohol or drugs. Many of these conditions can be mistaken for signs of withdrawal, drug-seeking, or new conditions arising from loss of euphoria from the drug. Prompt recognition of these disorders and use of appropriate non-addictive treatments can prevent “against medical advice” discharges, relapses, and unneeded suffering in many cases.
Because the brain is the target organ, these conditions are either neurologic or psychiatric in nosology. Although psychiatric clinicians might not be familiar with neurologic conditions, quick recognition and treatment is necessary.
Restless legs syndrome and periodic limb movements of sleep
Restless legs syndrome (RLS) has 2 key components: paresthesia and akathisia. Although primarily involving the lower extremities, involvement also can include the upper extremities, torso, and head.
Paresthesia differs from typical neuropathies in that it usually is not painful; rather, patients describe an odd sensation using terms such as ticklish, “creepy-crawly,” and other uncomfortable sensations.
Akathisia is a motor restlessness and need to move. The patient might feel momentary relief by moving or rubbing the extremities, only to have the paresthesia return quickly followed by the akathisia. Generally, reclining is the most prominent position that produces symptoms, but they can occur while sitting.
The cause of RLS is an abnormality of central dopamine or iron, or both, in the substantia nigra; iron is a cofactor in dopamine synthesis. All RLS patients should have a serum ferritin level drawn and if <50 μg/dL, be treated with iron supplementation. Dopamine agonists, such as ropinirole, pramipexole, and carbidopa/levodopa, are effective (Table 1); other useful agents include benzodiazepines such as clonazepam and opioids such as hydrocodone.
When a patient withdraws from benzodiazepines or narcotics, RLS can emerge and cause suffering until it is diagnosed and treated. Typical myalgia in opioid withdrawal can confound the diagnosis. The immediate-release (IR) and extended-release (ER) formulations of gabapentin often are a good choice when treating benzodiazepine or narcotic withdrawal. The side effect profile of gabapentin is relatively benign, with somnolence often reported by non-substance abusers, but it is unlikely that addicts, who have grown tolerant to more potent agents such as benzodiazepines and opioids, will complain of sleepiness. Studies have shown that gabapentin is useful in managing withdrawal as well as anxiety and insomnia.1,2 A randomized trial showed that gabapentin increases abstinence rates and decreases heavy drinking.2 The agent has a short half-life (5 to 7 hours); the IR form needs to be dosed at least 3 times a day to be effective. An ER formulation of gabapentin was released in 2013 with the sole indication for RLS.
Gabapentin is not significantly metabolized by the liver, has a 3% rate of protein binding, and is excreted by the kidneys—making it safe for patients who abuse alcohol or opioids and have impaired hepatic function. Typical starting dosages of IR gabapentin are 100 to 300 mg, 3 times daily, if symptoms are present in the daytime. Asymmetric dosing can be helpful, with larger or single dosages given at bedtime (eg, 100 mg in morning, 100 mg in afternoon, 300 mg at bedtime). Dosing varies from patient to patient, from 300 mg to 3,600 mg/d. Increasing dosages produce lower bioavailability because of saturation in absorption or at the blood-brain barrier. At 100 mg every 8 hours, bioavailability is 80% but at 1,600 mg every 8 hours it drops to 27%.3
Periodic limb movements of sleep (PLMS) essentially is akathisia during sleep, and occurs in most patients with RLS. The patient feels tired in the morning because of lack of deep stage-N3 sleep. Because of the inverse relationship between serotonin and dopamine, most selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors can exacerbate RLS and PLMS.4,5 Other culprits include antipsychotics, antiemetics, and antihistamines. The differential diagnosis includes withdrawal from opioids and attention-deficit/hyperactivity disorder (ADHD), which may be comorbid with RLS. There are many causes of secondary RLS including renal failure, pregnancy, varicose veins, and neuropathy.
Tremor
Benign familial, or essential, tremor is a fine intention tremor that can be suppressed by alcohol or benzodiazepines. After detoxification from either of these substances, persistent tremor can re-emerge; often, it is benign, although cerebellar and parkinsonian tremors must be ruled out. Essential tremor can be treated with gabapentin or beta blockers such as propranolol or metoprolol (Table 2).
Anxiety and panic disorder
Social anxiety often presents in addiction treatment centers in the context of group therapy, speaking in 12-step meetings, and having the patient describe his (her) autobiography and history of addiction. Because social anxiety disorder is the third most common psychiatric disorder after simple phobia and major depressive disorder,6 it is not surprising that it emerges after withdrawal.
Patients with social anxiety disorder might self-medicate with alcohol or drugs, especially benzodiazepines (Box). Residential treatment presents an excellent environment for desensitization to fears of public speaking; early recognition is key. Apprehension about group therapy, presenting a substance abuse history, or speaking at a 12-step meeting can lead to premature or “against medical advice” discharge.
Panic disorder commonly is comorbid with substance abuse. Many patients will arrive at treatment with a prescription for benzodiazepines. Because the risk of cross-addiction is high among recovering addicts, benzodiazepines should be avoided. Treating underlying anxiety is crucial for fostering sobriety. Generalized anxiety disorder is common among patients with an addiction, and can lead to relapse if not addressed. Use of non-addictive medications and cognitive therapy is useful in addressing this condition.
A quandary might arise in states where medical marijuana is legal, because Cannabis can be prescribed for anxiety disorders and posttraumatic stress disorder (PTSD). Promoting abstinence from all substances can present a challenge in patients with anxiety disorders who live in these states.
Medications for anxiety and panic disorder include gabapentin, buspirone, hydroxyzine, beta blockers, and atypical antipsychotics (Table 2). Only buspirone and hydroxyzine are FDA-approved for anxiety; buspirone monotherapy generally is ineffective for panic disorder.
Explaining to patients how anxiety arises, such as how classical conditioning leads to specific phobias, can be therapeutic. Describing Klein’s false suffocation alarm theory of panic attacks can illustrate the importance of practicing slow, deep breathing to prevent hyperventilation.7 Also, relabeling a panic attack with self-talk statements such as “I know what this is. It’s just a panic attack” can be helpful. Smartphone apps are available to help patients cope with anxiety and acute panic.8
Mood disorders
Many patients with bipolar disorder experience substance abuse at some point; estimates are that up to 57% of patients have a comorbid addiction.6,9 Persons with a mood disorder are at high risk of substance abuse because of genetic factors; patients also might self-medicate their mood symptoms.
After alcohol or drugs are withdrawn, mood disorders can emerge or resurge. Often, patients enter treatment taking antidepressants and mood stabilizers and usually haven’t been truthful with their treatment provider about their substance abuse. Care must be taken to ascertain whether mood symptoms are secondary to substance abuse. Asking “What’s the longest period of abstinence you’ve had in 2 years and how did you feel emotionally?” often will help you identify a secondary mood disorder. For example, a response of “6 months and I felt really depressed the entire time” would indicate a primary depressive disorder.
Because CNS depressants, such as alcohol and benzodiazepines, can exacerbate a mood disorder, consider continuing or resuming a mood stabilizer or antidepressant during substance abuse treatment. When meeting a new patient, perform an independent evaluation, because substance use can mimic bipolar and depressive disorders. Careful assessment of suicidal ideation is necessary for all patients.
Sleep disorders
Insomnia—as a primary or secondary disorder—is common among patients with a substance use disorder. Insomnia always needs to be addressed. Not sleeping well interferes with cognition and energy and makes depression and bipolar disorder worse. Some experts recommend “waiting out” the insomnia, hoping that sobriety will resolve it—but it might not.
Initial insomnia can be treated with melatonin, 3 to 6 mg at bedtime or earlier in the evening.10-12 Melatonin acts by regulating circadian rhythms, but can cause increased dreaming and nightmares; therefore, it should be avoided in patients who struggle with nightmares. Trazodone, 50 to 150 mg at bedtime, is an inexpensive sleep aid for initial insomnia and doesn’t cause weight gain, which many drugs with antihistaminic properties can. Prazosin, 1 to 2 mg initially, for nightmares in PTSD is effective.13
Antipsychotics might be necessary if nothing else works; quetiapine is effective for sleep and the ER form is FDA-approved as an add-on agent in major depression. Low-dose doxepin (≤10 mg) is effective for middle insomnia.14 At these low dosages, troublesome side effects of tricyclic antidepressants can be avoided.
As many as 40% of adults with ADHD have a delayed sleep-phase disorder. Ask your patient if she is a “night owl,” how chronic the condition is, and when her best sleep occurs.15-17 Morning light and evening melatonin can help, but often are insufficient. Many patients present with undiagnosed or untreated sleep apnea, which can cause excessive daytime sleepiness. Referral to a sleep center is prudent; use of the Epworth Sleepiness Scale is a quick way to assess excessive daytime sleepiness.18
ADHD
ADHD commonly is comorbid with a substance use disorder. Patients might present with an earlier diagnosis, including treatment. Several drugs of abuse can alleviate ADHD symptoms, including amphetamines, opioids, cocaine, and Cannabis; self-medicating is common. Because opioids increase dopamine release, a report of improved work and school performance while taking opioids early in addiction can be a clue to an ADHD diagnosis.
Explaining ADHD as a syndrome of “interest-based attention” helps. If a residential treatment program uses reading and writing assignments, a patient with ADHD might struggle and will need extra help and time and a quiet place to do assignments.19,20 A non-addictive medication, such as atomoxetine, can help, but has an antidepressant-like delay of 3 to 5 weeks until onset of symptom relief. Using a long-acting stimulant can be effective and quick, with an effect size 3 to 4 times higher than atomoxetine; such agents should be avoided in patients who abuse amphetamines.
Studies show that treating ADHD, even with stimulants, neither helps nor hurts outcomes in substance use. Lisdexamfetamine is difficult to abuse and is an inactive prodrug (a bond of lysine and dextroamphetamine) that requires enzymatic cleavage and activation by red blood cells; these characteristics creates a long-acting medication that has a lower abuse liability than other drugs for ADHD. However, abuse can occur and the drug must be used cautiously. Earley’s medication guide referenced below recommends that lisdexamfetamine and other stimulants should be avoided if possible in patients in recovery. However, it adds that specialists in treating ADHD in substance-abusing patients should weigh the potential benefits of stimulant use against the risk of relapse.17 Many patients enter treatment with a diagnosis of bipolar disorder that might, in fact, be comorbid with ADHD.
Chronic pain
Many substance abuse patients began taking opioids for acute, then chronic, pain before their use escalated to addiction. These are challenging patients; often, they are referred for treatment without true addiction.
Keep in mind that dependence is not addiction. Pseudo-addiction is a condition in which pain is undertreated and the patient takes more medication to obtain relief, calls for early refills, and displays drug-seeking behavior but is not using drugs to achieve euphoria. A thorough history and physical and referrals to specialists such as orthopedic surgeons and pain specialists are necessary. Explaining opioid-induced hyperalgesia is important to help the patient understand that (1) pain can be made worse by increasing the dosage of an opioid because of supersensitivity and (2) many patients who are weaned off these drugs will experience a decrease or complete relief of pain.21
Gabapentin, duloxetine, or amitriptyline can be beneficial for chronic pain, as well as mindfulness techniques, physical therapy, and complementary and alternative medicine. Pregabalin can produce euphoria and often should be avoided.
A medication guide for recovery
Paul Earley, MD, former medical director at Talbott Recovery in Atlanta, Georgia, publishes an online guide that classifies medications into categories:
• A: safe
• B: use only under the supervision of an addiction medicine specialist or doctor
• C: completely avoid if the patient is in recovery.17
The Talbott guide lists all stimulants in category C, (except for atomoxetine, which is category A). Hydroxyzine is listed under category B. Many programs for impaired professionals and state medical boards use the Guide, and will question the prescribing of any medication from categories B and C.17
Related Resources
• Spiegel DR, Kumari N, Petri JD. Safer use of benzodiazepines for alcohol detoxification. Current Psychiatry. 2012;11(10):10-15.
• Kelly TM, Daley DC, Douaihy AB. Treatment of substance abusing patients with comorbid psychiatric disorders. Addict Behav. 2012;37(1):11-24.
Drug Brand Names
Amitriptyline • Elavil Hydrocodone • Vicodin
Atenolol • Tenormin Hydroxyzine • Vistaril, Atarax
Atomoxetine • Strattera Lisdexamfetamine • Vyvanse
Buprenorphine/ naloxone • Suboxone Metoprolol • Lopressor, Toprol
Buspirone • BuSpar Paroxetine • Paxil
Carbidopa-levodopa • Sinemet Pramipexole • Mirapex
Clonazepam • Klonopin Prazosin • Minipress
Diazepam • Valium Pregabalin • Lyrica
Doxepin • Silenor, Adapin, Sinequan Propranolol • Inderal
Duloxetine • Cymbalta Quetiapine • Seroquel
Escitalopram • Lexapro Ropinirole • Requip
Gabapentin • Neurontin, Horizant Sertraline • Zoloft
Trazodone • Desyrel
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
When treating patients who abuse substances, it is important to watch for underlying clinical conditions that have been suppressed, relieved, or muted by alcohol or drugs. Many of these conditions can be mistaken for signs of withdrawal, drug-seeking, or new conditions arising from loss of euphoria from the drug. Prompt recognition of these disorders and use of appropriate non-addictive treatments can prevent “against medical advice” discharges, relapses, and unneeded suffering in many cases.
Because the brain is the target organ, these conditions are either neurologic or psychiatric in nosology. Although psychiatric clinicians might not be familiar with neurologic conditions, quick recognition and treatment is necessary.
Restless legs syndrome and periodic limb movements of sleep
Restless legs syndrome (RLS) has 2 key components: paresthesia and akathisia. Although primarily involving the lower extremities, involvement also can include the upper extremities, torso, and head.
Paresthesia differs from typical neuropathies in that it usually is not painful; rather, patients describe an odd sensation using terms such as ticklish, “creepy-crawly,” and other uncomfortable sensations.
Akathisia is a motor restlessness and need to move. The patient might feel momentary relief by moving or rubbing the extremities, only to have the paresthesia return quickly followed by the akathisia. Generally, reclining is the most prominent position that produces symptoms, but they can occur while sitting.
The cause of RLS is an abnormality of central dopamine or iron, or both, in the substantia nigra; iron is a cofactor in dopamine synthesis. All RLS patients should have a serum ferritin level drawn and if <50 μg/dL, be treated with iron supplementation. Dopamine agonists, such as ropinirole, pramipexole, and carbidopa/levodopa, are effective (Table 1); other useful agents include benzodiazepines such as clonazepam and opioids such as hydrocodone.
When a patient withdraws from benzodiazepines or narcotics, RLS can emerge and cause suffering until it is diagnosed and treated. Typical myalgia in opioid withdrawal can confound the diagnosis. The immediate-release (IR) and extended-release (ER) formulations of gabapentin often are a good choice when treating benzodiazepine or narcotic withdrawal. The side effect profile of gabapentin is relatively benign, with somnolence often reported by non-substance abusers, but it is unlikely that addicts, who have grown tolerant to more potent agents such as benzodiazepines and opioids, will complain of sleepiness. Studies have shown that gabapentin is useful in managing withdrawal as well as anxiety and insomnia.1,2 A randomized trial showed that gabapentin increases abstinence rates and decreases heavy drinking.2 The agent has a short half-life (5 to 7 hours); the IR form needs to be dosed at least 3 times a day to be effective. An ER formulation of gabapentin was released in 2013 with the sole indication for RLS.
Gabapentin is not significantly metabolized by the liver, has a 3% rate of protein binding, and is excreted by the kidneys—making it safe for patients who abuse alcohol or opioids and have impaired hepatic function. Typical starting dosages of IR gabapentin are 100 to 300 mg, 3 times daily, if symptoms are present in the daytime. Asymmetric dosing can be helpful, with larger or single dosages given at bedtime (eg, 100 mg in morning, 100 mg in afternoon, 300 mg at bedtime). Dosing varies from patient to patient, from 300 mg to 3,600 mg/d. Increasing dosages produce lower bioavailability because of saturation in absorption or at the blood-brain barrier. At 100 mg every 8 hours, bioavailability is 80% but at 1,600 mg every 8 hours it drops to 27%.3
Periodic limb movements of sleep (PLMS) essentially is akathisia during sleep, and occurs in most patients with RLS. The patient feels tired in the morning because of lack of deep stage-N3 sleep. Because of the inverse relationship between serotonin and dopamine, most selective serotonin reuptake inhibitors and serotonin-norepinephrine reuptake inhibitors can exacerbate RLS and PLMS.4,5 Other culprits include antipsychotics, antiemetics, and antihistamines. The differential diagnosis includes withdrawal from opioids and attention-deficit/hyperactivity disorder (ADHD), which may be comorbid with RLS. There are many causes of secondary RLS including renal failure, pregnancy, varicose veins, and neuropathy.
Tremor
Benign familial, or essential, tremor is a fine intention tremor that can be suppressed by alcohol or benzodiazepines. After detoxification from either of these substances, persistent tremor can re-emerge; often, it is benign, although cerebellar and parkinsonian tremors must be ruled out. Essential tremor can be treated with gabapentin or beta blockers such as propranolol or metoprolol (Table 2).
Anxiety and panic disorder
Social anxiety often presents in addiction treatment centers in the context of group therapy, speaking in 12-step meetings, and having the patient describe his (her) autobiography and history of addiction. Because social anxiety disorder is the third most common psychiatric disorder after simple phobia and major depressive disorder,6 it is not surprising that it emerges after withdrawal.
Patients with social anxiety disorder might self-medicate with alcohol or drugs, especially benzodiazepines (Box). Residential treatment presents an excellent environment for desensitization to fears of public speaking; early recognition is key. Apprehension about group therapy, presenting a substance abuse history, or speaking at a 12-step meeting can lead to premature or “against medical advice” discharge.
Panic disorder commonly is comorbid with substance abuse. Many patients will arrive at treatment with a prescription for benzodiazepines. Because the risk of cross-addiction is high among recovering addicts, benzodiazepines should be avoided. Treating underlying anxiety is crucial for fostering sobriety. Generalized anxiety disorder is common among patients with an addiction, and can lead to relapse if not addressed. Use of non-addictive medications and cognitive therapy is useful in addressing this condition.
A quandary might arise in states where medical marijuana is legal, because Cannabis can be prescribed for anxiety disorders and posttraumatic stress disorder (PTSD). Promoting abstinence from all substances can present a challenge in patients with anxiety disorders who live in these states.
Medications for anxiety and panic disorder include gabapentin, buspirone, hydroxyzine, beta blockers, and atypical antipsychotics (Table 2). Only buspirone and hydroxyzine are FDA-approved for anxiety; buspirone monotherapy generally is ineffective for panic disorder.
Explaining to patients how anxiety arises, such as how classical conditioning leads to specific phobias, can be therapeutic. Describing Klein’s false suffocation alarm theory of panic attacks can illustrate the importance of practicing slow, deep breathing to prevent hyperventilation.7 Also, relabeling a panic attack with self-talk statements such as “I know what this is. It’s just a panic attack” can be helpful. Smartphone apps are available to help patients cope with anxiety and acute panic.8
Mood disorders
Many patients with bipolar disorder experience substance abuse at some point; estimates are that up to 57% of patients have a comorbid addiction.6,9 Persons with a mood disorder are at high risk of substance abuse because of genetic factors; patients also might self-medicate their mood symptoms.
After alcohol or drugs are withdrawn, mood disorders can emerge or resurge. Often, patients enter treatment taking antidepressants and mood stabilizers and usually haven’t been truthful with their treatment provider about their substance abuse. Care must be taken to ascertain whether mood symptoms are secondary to substance abuse. Asking “What’s the longest period of abstinence you’ve had in 2 years and how did you feel emotionally?” often will help you identify a secondary mood disorder. For example, a response of “6 months and I felt really depressed the entire time” would indicate a primary depressive disorder.
Because CNS depressants, such as alcohol and benzodiazepines, can exacerbate a mood disorder, consider continuing or resuming a mood stabilizer or antidepressant during substance abuse treatment. When meeting a new patient, perform an independent evaluation, because substance use can mimic bipolar and depressive disorders. Careful assessment of suicidal ideation is necessary for all patients.
Sleep disorders
Insomnia—as a primary or secondary disorder—is common among patients with a substance use disorder. Insomnia always needs to be addressed. Not sleeping well interferes with cognition and energy and makes depression and bipolar disorder worse. Some experts recommend “waiting out” the insomnia, hoping that sobriety will resolve it—but it might not.
Initial insomnia can be treated with melatonin, 3 to 6 mg at bedtime or earlier in the evening.10-12 Melatonin acts by regulating circadian rhythms, but can cause increased dreaming and nightmares; therefore, it should be avoided in patients who struggle with nightmares. Trazodone, 50 to 150 mg at bedtime, is an inexpensive sleep aid for initial insomnia and doesn’t cause weight gain, which many drugs with antihistaminic properties can. Prazosin, 1 to 2 mg initially, for nightmares in PTSD is effective.13
Antipsychotics might be necessary if nothing else works; quetiapine is effective for sleep and the ER form is FDA-approved as an add-on agent in major depression. Low-dose doxepin (≤10 mg) is effective for middle insomnia.14 At these low dosages, troublesome side effects of tricyclic antidepressants can be avoided.
As many as 40% of adults with ADHD have a delayed sleep-phase disorder. Ask your patient if she is a “night owl,” how chronic the condition is, and when her best sleep occurs.15-17 Morning light and evening melatonin can help, but often are insufficient. Many patients present with undiagnosed or untreated sleep apnea, which can cause excessive daytime sleepiness. Referral to a sleep center is prudent; use of the Epworth Sleepiness Scale is a quick way to assess excessive daytime sleepiness.18
ADHD
ADHD commonly is comorbid with a substance use disorder. Patients might present with an earlier diagnosis, including treatment. Several drugs of abuse can alleviate ADHD symptoms, including amphetamines, opioids, cocaine, and Cannabis; self-medicating is common. Because opioids increase dopamine release, a report of improved work and school performance while taking opioids early in addiction can be a clue to an ADHD diagnosis.
Explaining ADHD as a syndrome of “interest-based attention” helps. If a residential treatment program uses reading and writing assignments, a patient with ADHD might struggle and will need extra help and time and a quiet place to do assignments.19,20 A non-addictive medication, such as atomoxetine, can help, but has an antidepressant-like delay of 3 to 5 weeks until onset of symptom relief. Using a long-acting stimulant can be effective and quick, with an effect size 3 to 4 times higher than atomoxetine; such agents should be avoided in patients who abuse amphetamines.
Studies show that treating ADHD, even with stimulants, neither helps nor hurts outcomes in substance use. Lisdexamfetamine is difficult to abuse and is an inactive prodrug (a bond of lysine and dextroamphetamine) that requires enzymatic cleavage and activation by red blood cells; these characteristics creates a long-acting medication that has a lower abuse liability than other drugs for ADHD. However, abuse can occur and the drug must be used cautiously. Earley’s medication guide referenced below recommends that lisdexamfetamine and other stimulants should be avoided if possible in patients in recovery. However, it adds that specialists in treating ADHD in substance-abusing patients should weigh the potential benefits of stimulant use against the risk of relapse.17 Many patients enter treatment with a diagnosis of bipolar disorder that might, in fact, be comorbid with ADHD.
Chronic pain
Many substance abuse patients began taking opioids for acute, then chronic, pain before their use escalated to addiction. These are challenging patients; often, they are referred for treatment without true addiction.
Keep in mind that dependence is not addiction. Pseudo-addiction is a condition in which pain is undertreated and the patient takes more medication to obtain relief, calls for early refills, and displays drug-seeking behavior but is not using drugs to achieve euphoria. A thorough history and physical and referrals to specialists such as orthopedic surgeons and pain specialists are necessary. Explaining opioid-induced hyperalgesia is important to help the patient understand that (1) pain can be made worse by increasing the dosage of an opioid because of supersensitivity and (2) many patients who are weaned off these drugs will experience a decrease or complete relief of pain.21
Gabapentin, duloxetine, or amitriptyline can be beneficial for chronic pain, as well as mindfulness techniques, physical therapy, and complementary and alternative medicine. Pregabalin can produce euphoria and often should be avoided.
A medication guide for recovery
Paul Earley, MD, former medical director at Talbott Recovery in Atlanta, Georgia, publishes an online guide that classifies medications into categories:
• A: safe
• B: use only under the supervision of an addiction medicine specialist or doctor
• C: completely avoid if the patient is in recovery.17
The Talbott guide lists all stimulants in category C, (except for atomoxetine, which is category A). Hydroxyzine is listed under category B. Many programs for impaired professionals and state medical boards use the Guide, and will question the prescribing of any medication from categories B and C.17
Related Resources
• Spiegel DR, Kumari N, Petri JD. Safer use of benzodiazepines for alcohol detoxification. Current Psychiatry. 2012;11(10):10-15.
• Kelly TM, Daley DC, Douaihy AB. Treatment of substance abusing patients with comorbid psychiatric disorders. Addict Behav. 2012;37(1):11-24.
Drug Brand Names
Amitriptyline • Elavil Hydrocodone • Vicodin
Atenolol • Tenormin Hydroxyzine • Vistaril, Atarax
Atomoxetine • Strattera Lisdexamfetamine • Vyvanse
Buprenorphine/ naloxone • Suboxone Metoprolol • Lopressor, Toprol
Buspirone • BuSpar Paroxetine • Paxil
Carbidopa-levodopa • Sinemet Pramipexole • Mirapex
Clonazepam • Klonopin Prazosin • Minipress
Diazepam • Valium Pregabalin • Lyrica
Doxepin • Silenor, Adapin, Sinequan Propranolol • Inderal
Duloxetine • Cymbalta Quetiapine • Seroquel
Escitalopram • Lexapro Ropinirole • Requip
Gabapentin • Neurontin, Horizant Sertraline • Zoloft
Trazodone • Desyrel
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Stock CJ, Carpenter L, Ying J, et al. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother. 2013;47(7-8):961-969.
2. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
3. Bockbrader HN, Wesche D, Miller R, et al. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin Pharmacokinet. 2010; 49(10):661-669.
4. Yang C, White DP, Winkelman JW. Antidepressants and periodic leg movements of sleep. Biol Psychiatry. 2005;58(6):510-514.
5. Hoque R, Chesson AL Jr. Pharmacologically induced/ exacerbated restless legs syndrome, periodic limb movements of sleep, and REM behavior disorder/ REM sleep without atonia: literature review, qualitative scoring, and comparative analysis. J Clin Sleep Med. 2010; 6(1):79-83.
6. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
7. Klein DF. False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry. 1993;50(4):306-317.
8. Holland K. The 17 best anxiety iPhone & Android apps of 2014. http://www.healthline.com/health-slideshow/top-anxiety-iphone-android-apps. Accessed October 28, 2014.
9. Chengappa KN, Levine J, Gershon S, et al. Lifetime prevalence of substance or alcohol abuse and dependence among subjects with bipolar I and II disorders in a voluntary registry. Bipolar Disord. 2000;2(3 Pt 1):191-195.
10. Ferracioli-Oda E, Qawasmi A, Bloch MH. Meta-analysis: melatonin for the treatment of primary sleep disorders [published online May 17, 2013]. PLoS One. 2013;8(5):e63773. doi: 10.1371/journal.pone.0063773.
11. Wade AG, Ford I, Crawford G, et al. Efficacy of prolonged release melatonin in insomnia patients aged 55-80 years: quality of sleep and next-day alertness outcomes. Curr Med Res Opin. 2007;23(10):2597-2605.
12. Srinivasan V, Brzezinski A, Pandi-Perumal SR, et al. Melatonin agonists in primary insomnia and depression-associated insomnia: are they superior to sedative-hypnotics? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):913-923.
13. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
14. Scharf M, Rogowski R, Hull S, et al. Efficacy and safety of doxepin 1 mg, 3 mg, and 6 mg in elderly patients with primary insomnia: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry. 2008;69(10):1557-1564.
15. Baird AL, Coogan AN, Siddiqui A, et al. Adult attention-deficit hyperactivity disorder is associated with alterations in circadian rhythms at the behavioural, endocrine and molecular levels. Mol Psychiatry. 2012;17(10):988-995.
16. Yoon SY, Jain U, Shapiro C. Sleep in attention-deficit/ hyperactivity disorder in children and adults: past, present, and future. Sleep Med Rev. 2012;16(4):371-388.
17. Earley PH, Merkin B, Skipper G. The medication guide for safe recovery. Revision 1.7. http://paulearley.net/index. php?option=com_docman&Itemid=239. Published March 2014. Accessed October 28, 2014.
18. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540-545.
19. Dodson W. Secrets of the ADHD brain. ADDitude. http:// www.additudemag.com/adhd/article/10117.html. Accessed October 28, 2014.
20. Wilens TE, Dodson W. A clinical perspective of attention-deficit/hyperactivity disorder into adulthood. J Clin Psychiatry. 2004;65(10):1301-1313.
21. Lee M, Silverman SM, Hansen H, et al. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145-161.
1. Stock CJ, Carpenter L, Ying J, et al. Gabapentin versus chlordiazepoxide for outpatient alcohol detoxification treatment. Ann Pharmacother. 2013;47(7-8):961-969.
2. Mason BJ, Quello S, Goodell V, et al. Gabapentin treatment for alcohol dependence: a randomized clinical trial. JAMA Intern Med. 2014;174(1):70-77.
3. Bockbrader HN, Wesche D, Miller R, et al. A comparison of the pharmacokinetics and pharmacodynamics of pregabalin and gabapentin. Clin Pharmacokinet. 2010; 49(10):661-669.
4. Yang C, White DP, Winkelman JW. Antidepressants and periodic leg movements of sleep. Biol Psychiatry. 2005;58(6):510-514.
5. Hoque R, Chesson AL Jr. Pharmacologically induced/ exacerbated restless legs syndrome, periodic limb movements of sleep, and REM behavior disorder/ REM sleep without atonia: literature review, qualitative scoring, and comparative analysis. J Clin Sleep Med. 2010; 6(1):79-83.
6. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21(3):169-184.
7. Klein DF. False suffocation alarms, spontaneous panics, and related conditions. An integrative hypothesis. Arch Gen Psychiatry. 1993;50(4):306-317.
8. Holland K. The 17 best anxiety iPhone & Android apps of 2014. http://www.healthline.com/health-slideshow/top-anxiety-iphone-android-apps. Accessed October 28, 2014.
9. Chengappa KN, Levine J, Gershon S, et al. Lifetime prevalence of substance or alcohol abuse and dependence among subjects with bipolar I and II disorders in a voluntary registry. Bipolar Disord. 2000;2(3 Pt 1):191-195.
10. Ferracioli-Oda E, Qawasmi A, Bloch MH. Meta-analysis: melatonin for the treatment of primary sleep disorders [published online May 17, 2013]. PLoS One. 2013;8(5):e63773. doi: 10.1371/journal.pone.0063773.
11. Wade AG, Ford I, Crawford G, et al. Efficacy of prolonged release melatonin in insomnia patients aged 55-80 years: quality of sleep and next-day alertness outcomes. Curr Med Res Opin. 2007;23(10):2597-2605.
12. Srinivasan V, Brzezinski A, Pandi-Perumal SR, et al. Melatonin agonists in primary insomnia and depression-associated insomnia: are they superior to sedative-hypnotics? Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):913-923.
13. Raskind MA, Peterson K, Williams T, et al. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry. 2013;170(9):1003-1010.
14. Scharf M, Rogowski R, Hull S, et al. Efficacy and safety of doxepin 1 mg, 3 mg, and 6 mg in elderly patients with primary insomnia: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry. 2008;69(10):1557-1564.
15. Baird AL, Coogan AN, Siddiqui A, et al. Adult attention-deficit hyperactivity disorder is associated with alterations in circadian rhythms at the behavioural, endocrine and molecular levels. Mol Psychiatry. 2012;17(10):988-995.
16. Yoon SY, Jain U, Shapiro C. Sleep in attention-deficit/ hyperactivity disorder in children and adults: past, present, and future. Sleep Med Rev. 2012;16(4):371-388.
17. Earley PH, Merkin B, Skipper G. The medication guide for safe recovery. Revision 1.7. http://paulearley.net/index. php?option=com_docman&Itemid=239. Published March 2014. Accessed October 28, 2014.
18. Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540-545.
19. Dodson W. Secrets of the ADHD brain. ADDitude. http:// www.additudemag.com/adhd/article/10117.html. Accessed October 28, 2014.
20. Wilens TE, Dodson W. A clinical perspective of attention-deficit/hyperactivity disorder into adulthood. J Clin Psychiatry. 2004;65(10):1301-1313.
21. Lee M, Silverman SM, Hansen H, et al. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14(2):145-161.

A guide to the mysteries of maintenance of certification
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.

Is your patient using cocaine to self-medicate undiagnosed ADHD?
Attention-deficit/hyperactivity disorder (ADHD) often persists beyond childhood into adulthood. One of the therapeutic challenges of treating ADHD is identifying comorbidities, including underlying mood and anxiety disorders, and ongoing substance abuse. Effective treatment modalities tend to prioritize management of substance abuse, but the patient’s age may dictate the overall assessment plan.
So-called 'reward' center
Treating childhood ADHD with stimulants might reduce the risk for future drug abuse.1 It is estimated that approximately 10 million people with ADHD are undiagnosed in the United States2; characteristic ADHD symptoms—inattention, hyperactivity, impulsivity—can persist in adulthood, and affected persons might not meet societal expectations. Previously unidentified attention difficulties may emerge during early adulthood because of increasingly complex tasks at school and work.
Persons with undiagnosed ADHD might turn to potentially self-destructive means of placating inner tension. Cocaine has pharmacological properties in common with stimulants such as methylphenidate, which often is prescribed for ADHD. Cocaine and methylphenidate both work on altering brain chemistry with a similar mechanism of action, allowing for increased dopamine in the nucleus accumbens, also known as the “reward center” of the brain.
Adults with ADHD have a 300% higher risk of developing a substance use disorder than adults without ADHD.3 An estimated 15% to 25% of adults with substance abuse have comorbid ADHD. Although these patients abuse of a variety of substances including Cannabis and alcohol, cocaine is one of the most commonly abused substances among this population. These observations could point to a self-medication hypothesis.
Why self-medicate?
The self-medication hypothesis, formulated by Khantzian in 1985, was based on several clinical observations. Khantzian stated that an abuser’s drug of choice is not selected at random but, rather, by an inherent desire to suppress the attributes of the condition that seems to otherwise wreak havoc on his (her) life. Almost a century earlier, Freud mentioned that cocaine is an antidepressant. Among persons with ADHD who have not been given that diagnosis, or treated for the disorder, cocaine is a popular drug. Because of the antidepressant features of cocaine and its ability to produce a rapid increase of dopamine levels that exert a pro-euphoric effect, coupled with a seemingly paradoxical calming influence that leads to increased productivity, it is not surprising to find that cocaine is abused. Reportedly, persons who have not been treated because their ADHD is undiagnosed turn to cocaine because it improves attention, raises self-esteem, and allows users to harness a level of focus that they could not otherwise achieve.4
Mechanism of action
Methylphenidate reduces ADHD symptoms by increasing extracellular dopamine in the brain, acting by means of a mechanism that is similar to that of cocaine.5 By blocking reuptake of dopamine and allowing an extracellular surplus, users continue to experience the pleasurable effect the neuro-transmitter produces. Methylphenidate has been shown to be an even more potent inhibitor of the same autoreceptors. Injecting methylphenidate has been shown to produce a rapid release of dopamine similar to that of cocaine.5
However, methylphenidate causes a much slower increase in dopamine; its effect on the brain has been shown to be similar to that of cocaine without the increased abuse potential. Cocaine use remodels the brain by reconfiguring connections that are essential for craving and self-control.5 Therefore, substituting methylphenidate for cocaine could help ADHD patients by:
• improving overall executive functioning
• decreasing feelings of low self-worth
• increasing daily functioning
• minimizing craving and the risk of subsequent cocaine abuse.
Treatment recommendations
Carefully consider pharmacodynamics and pharmacokinetics when prescribing ADHD medication. In general, children and adolescents with ADHD respond more favorably to stimulants than adults do. In children, the mainstay of treatment is slow-dose stimulants such as methylphenidate; second-line treatments are immediate-release stimulants and atomoxetine, a selective norepinephrine reuptake inhibitor.6 Adults with ADHD might benefit from a nonstimulant, in part because of the presence of complex comorbidities.6 Modafinil often is prescribed for adults with ADHD.
Atomoxetine readily increases norepinephrine and dopamine in the prefrontal cortex as it bypasses the nucleus accumbens. Although atomoxetine is not a stimulant, the efficacy of the drug is based on its ability to increase norepinephrine through selective inhibition of the norepinephrine transporter. Norepinephrine modulates higher cortical functions—attention, executive function, arousal—that lead to a reduction in hyperactivity, inattention, and impulsivity.
Because dopamine is released in the prefrontal cortex—not in the nucleus accumbens—the addiction potential of atomoxetine is low.7 The drug might be an effective intervention for patients who are using cocaine to self-medicate. Stimulants such as methylphenidate have proven effective in safely mimicking the mechanism of action of cocaine. Nonstimulants, such as atomoxetine and modafinil, lack abuse potential and are excellent options for treating adults with ADHD.
Clinicians generally are advised to treat a patient’s underlying ADHD symptoms before addressing ongoing substance abuse. If a patient abruptly discontinues cocaine use before ADHD symptoms are properly controlled, her (his) condition might deteriorate further and the treatment plan might fail to progress. Some patients have experienced a reduction in craving for cocaine after they began stimulant therapy; these people no longer felt a need to self-medicate because their symptoms were being addressed.4
1. Jain S, Jain R, Islam J. Do stimulants for ADHD increase the risk of substance use disorders? Current Psychiatry. 2011;10(8):20-24.
2. Baskin S. Adult ADHD—A common disorder, often missed. http://www.stevebaskinmd.com/articles-about-adultadhd.html. Published 2009. Accessed November 5, 2014.
3. Tuzee M. Many adults who have ADHD go undiagnosed.
http://abclocal.go.com/kabc/story?section=news/health/your_health&id=7657326. Published September 8, 2010. Accessed October 9, 2014.
4. Plume D. The self medication hypothesis: ADHD & chronic cocaine abuse. A literature review. http://www.addcentre.co.uk/selfmedcocaine.htm. Published April 1995. Accessed October 9, 2014.
5. Searight HR, Burke JM. Adult attention deficit hyperactivity disorder. UpToDate. Updated Feb 2011. Accessed November 5, 2014.
6. Stahl SM. Attention deficit disorder and its treatment. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:884-897.
7. Michelson D, Adler L, Spencer T, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53(2):112-120.
Attention-deficit/hyperactivity disorder (ADHD) often persists beyond childhood into adulthood. One of the therapeutic challenges of treating ADHD is identifying comorbidities, including underlying mood and anxiety disorders, and ongoing substance abuse. Effective treatment modalities tend to prioritize management of substance abuse, but the patient’s age may dictate the overall assessment plan.
So-called 'reward' center
Treating childhood ADHD with stimulants might reduce the risk for future drug abuse.1 It is estimated that approximately 10 million people with ADHD are undiagnosed in the United States2; characteristic ADHD symptoms—inattention, hyperactivity, impulsivity—can persist in adulthood, and affected persons might not meet societal expectations. Previously unidentified attention difficulties may emerge during early adulthood because of increasingly complex tasks at school and work.
Persons with undiagnosed ADHD might turn to potentially self-destructive means of placating inner tension. Cocaine has pharmacological properties in common with stimulants such as methylphenidate, which often is prescribed for ADHD. Cocaine and methylphenidate both work on altering brain chemistry with a similar mechanism of action, allowing for increased dopamine in the nucleus accumbens, also known as the “reward center” of the brain.
Adults with ADHD have a 300% higher risk of developing a substance use disorder than adults without ADHD.3 An estimated 15% to 25% of adults with substance abuse have comorbid ADHD. Although these patients abuse of a variety of substances including Cannabis and alcohol, cocaine is one of the most commonly abused substances among this population. These observations could point to a self-medication hypothesis.
Why self-medicate?
The self-medication hypothesis, formulated by Khantzian in 1985, was based on several clinical observations. Khantzian stated that an abuser’s drug of choice is not selected at random but, rather, by an inherent desire to suppress the attributes of the condition that seems to otherwise wreak havoc on his (her) life. Almost a century earlier, Freud mentioned that cocaine is an antidepressant. Among persons with ADHD who have not been given that diagnosis, or treated for the disorder, cocaine is a popular drug. Because of the antidepressant features of cocaine and its ability to produce a rapid increase of dopamine levels that exert a pro-euphoric effect, coupled with a seemingly paradoxical calming influence that leads to increased productivity, it is not surprising to find that cocaine is abused. Reportedly, persons who have not been treated because their ADHD is undiagnosed turn to cocaine because it improves attention, raises self-esteem, and allows users to harness a level of focus that they could not otherwise achieve.4
Mechanism of action
Methylphenidate reduces ADHD symptoms by increasing extracellular dopamine in the brain, acting by means of a mechanism that is similar to that of cocaine.5 By blocking reuptake of dopamine and allowing an extracellular surplus, users continue to experience the pleasurable effect the neuro-transmitter produces. Methylphenidate has been shown to be an even more potent inhibitor of the same autoreceptors. Injecting methylphenidate has been shown to produce a rapid release of dopamine similar to that of cocaine.5
However, methylphenidate causes a much slower increase in dopamine; its effect on the brain has been shown to be similar to that of cocaine without the increased abuse potential. Cocaine use remodels the brain by reconfiguring connections that are essential for craving and self-control.5 Therefore, substituting methylphenidate for cocaine could help ADHD patients by:
• improving overall executive functioning
• decreasing feelings of low self-worth
• increasing daily functioning
• minimizing craving and the risk of subsequent cocaine abuse.
Treatment recommendations
Carefully consider pharmacodynamics and pharmacokinetics when prescribing ADHD medication. In general, children and adolescents with ADHD respond more favorably to stimulants than adults do. In children, the mainstay of treatment is slow-dose stimulants such as methylphenidate; second-line treatments are immediate-release stimulants and atomoxetine, a selective norepinephrine reuptake inhibitor.6 Adults with ADHD might benefit from a nonstimulant, in part because of the presence of complex comorbidities.6 Modafinil often is prescribed for adults with ADHD.
Atomoxetine readily increases norepinephrine and dopamine in the prefrontal cortex as it bypasses the nucleus accumbens. Although atomoxetine is not a stimulant, the efficacy of the drug is based on its ability to increase norepinephrine through selective inhibition of the norepinephrine transporter. Norepinephrine modulates higher cortical functions—attention, executive function, arousal—that lead to a reduction in hyperactivity, inattention, and impulsivity.
Because dopamine is released in the prefrontal cortex—not in the nucleus accumbens—the addiction potential of atomoxetine is low.7 The drug might be an effective intervention for patients who are using cocaine to self-medicate. Stimulants such as methylphenidate have proven effective in safely mimicking the mechanism of action of cocaine. Nonstimulants, such as atomoxetine and modafinil, lack abuse potential and are excellent options for treating adults with ADHD.
Clinicians generally are advised to treat a patient’s underlying ADHD symptoms before addressing ongoing substance abuse. If a patient abruptly discontinues cocaine use before ADHD symptoms are properly controlled, her (his) condition might deteriorate further and the treatment plan might fail to progress. Some patients have experienced a reduction in craving for cocaine after they began stimulant therapy; these people no longer felt a need to self-medicate because their symptoms were being addressed.4
Attention-deficit/hyperactivity disorder (ADHD) often persists beyond childhood into adulthood. One of the therapeutic challenges of treating ADHD is identifying comorbidities, including underlying mood and anxiety disorders, and ongoing substance abuse. Effective treatment modalities tend to prioritize management of substance abuse, but the patient’s age may dictate the overall assessment plan.
So-called 'reward' center
Treating childhood ADHD with stimulants might reduce the risk for future drug abuse.1 It is estimated that approximately 10 million people with ADHD are undiagnosed in the United States2; characteristic ADHD symptoms—inattention, hyperactivity, impulsivity—can persist in adulthood, and affected persons might not meet societal expectations. Previously unidentified attention difficulties may emerge during early adulthood because of increasingly complex tasks at school and work.
Persons with undiagnosed ADHD might turn to potentially self-destructive means of placating inner tension. Cocaine has pharmacological properties in common with stimulants such as methylphenidate, which often is prescribed for ADHD. Cocaine and methylphenidate both work on altering brain chemistry with a similar mechanism of action, allowing for increased dopamine in the nucleus accumbens, also known as the “reward center” of the brain.
Adults with ADHD have a 300% higher risk of developing a substance use disorder than adults without ADHD.3 An estimated 15% to 25% of adults with substance abuse have comorbid ADHD. Although these patients abuse of a variety of substances including Cannabis and alcohol, cocaine is one of the most commonly abused substances among this population. These observations could point to a self-medication hypothesis.
Why self-medicate?
The self-medication hypothesis, formulated by Khantzian in 1985, was based on several clinical observations. Khantzian stated that an abuser’s drug of choice is not selected at random but, rather, by an inherent desire to suppress the attributes of the condition that seems to otherwise wreak havoc on his (her) life. Almost a century earlier, Freud mentioned that cocaine is an antidepressant. Among persons with ADHD who have not been given that diagnosis, or treated for the disorder, cocaine is a popular drug. Because of the antidepressant features of cocaine and its ability to produce a rapid increase of dopamine levels that exert a pro-euphoric effect, coupled with a seemingly paradoxical calming influence that leads to increased productivity, it is not surprising to find that cocaine is abused. Reportedly, persons who have not been treated because their ADHD is undiagnosed turn to cocaine because it improves attention, raises self-esteem, and allows users to harness a level of focus that they could not otherwise achieve.4
Mechanism of action
Methylphenidate reduces ADHD symptoms by increasing extracellular dopamine in the brain, acting by means of a mechanism that is similar to that of cocaine.5 By blocking reuptake of dopamine and allowing an extracellular surplus, users continue to experience the pleasurable effect the neuro-transmitter produces. Methylphenidate has been shown to be an even more potent inhibitor of the same autoreceptors. Injecting methylphenidate has been shown to produce a rapid release of dopamine similar to that of cocaine.5
However, methylphenidate causes a much slower increase in dopamine; its effect on the brain has been shown to be similar to that of cocaine without the increased abuse potential. Cocaine use remodels the brain by reconfiguring connections that are essential for craving and self-control.5 Therefore, substituting methylphenidate for cocaine could help ADHD patients by:
• improving overall executive functioning
• decreasing feelings of low self-worth
• increasing daily functioning
• minimizing craving and the risk of subsequent cocaine abuse.
Treatment recommendations
Carefully consider pharmacodynamics and pharmacokinetics when prescribing ADHD medication. In general, children and adolescents with ADHD respond more favorably to stimulants than adults do. In children, the mainstay of treatment is slow-dose stimulants such as methylphenidate; second-line treatments are immediate-release stimulants and atomoxetine, a selective norepinephrine reuptake inhibitor.6 Adults with ADHD might benefit from a nonstimulant, in part because of the presence of complex comorbidities.6 Modafinil often is prescribed for adults with ADHD.
Atomoxetine readily increases norepinephrine and dopamine in the prefrontal cortex as it bypasses the nucleus accumbens. Although atomoxetine is not a stimulant, the efficacy of the drug is based on its ability to increase norepinephrine through selective inhibition of the norepinephrine transporter. Norepinephrine modulates higher cortical functions—attention, executive function, arousal—that lead to a reduction in hyperactivity, inattention, and impulsivity.
Because dopamine is released in the prefrontal cortex—not in the nucleus accumbens—the addiction potential of atomoxetine is low.7 The drug might be an effective intervention for patients who are using cocaine to self-medicate. Stimulants such as methylphenidate have proven effective in safely mimicking the mechanism of action of cocaine. Nonstimulants, such as atomoxetine and modafinil, lack abuse potential and are excellent options for treating adults with ADHD.
Clinicians generally are advised to treat a patient’s underlying ADHD symptoms before addressing ongoing substance abuse. If a patient abruptly discontinues cocaine use before ADHD symptoms are properly controlled, her (his) condition might deteriorate further and the treatment plan might fail to progress. Some patients have experienced a reduction in craving for cocaine after they began stimulant therapy; these people no longer felt a need to self-medicate because their symptoms were being addressed.4
1. Jain S, Jain R, Islam J. Do stimulants for ADHD increase the risk of substance use disorders? Current Psychiatry. 2011;10(8):20-24.
2. Baskin S. Adult ADHD—A common disorder, often missed. http://www.stevebaskinmd.com/articles-about-adultadhd.html. Published 2009. Accessed November 5, 2014.
3. Tuzee M. Many adults who have ADHD go undiagnosed.
http://abclocal.go.com/kabc/story?section=news/health/your_health&id=7657326. Published September 8, 2010. Accessed October 9, 2014.
4. Plume D. The self medication hypothesis: ADHD & chronic cocaine abuse. A literature review. http://www.addcentre.co.uk/selfmedcocaine.htm. Published April 1995. Accessed October 9, 2014.
5. Searight HR, Burke JM. Adult attention deficit hyperactivity disorder. UpToDate. Updated Feb 2011. Accessed November 5, 2014.
6. Stahl SM. Attention deficit disorder and its treatment. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:884-897.
7. Michelson D, Adler L, Spencer T, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53(2):112-120.
1. Jain S, Jain R, Islam J. Do stimulants for ADHD increase the risk of substance use disorders? Current Psychiatry. 2011;10(8):20-24.
2. Baskin S. Adult ADHD—A common disorder, often missed. http://www.stevebaskinmd.com/articles-about-adultadhd.html. Published 2009. Accessed November 5, 2014.
3. Tuzee M. Many adults who have ADHD go undiagnosed.
http://abclocal.go.com/kabc/story?section=news/health/your_health&id=7657326. Published September 8, 2010. Accessed October 9, 2014.
4. Plume D. The self medication hypothesis: ADHD & chronic cocaine abuse. A literature review. http://www.addcentre.co.uk/selfmedcocaine.htm. Published April 1995. Accessed October 9, 2014.
5. Searight HR, Burke JM. Adult attention deficit hyperactivity disorder. UpToDate. Updated Feb 2011. Accessed November 5, 2014.
6. Stahl SM. Attention deficit disorder and its treatment. In: Stahl’s essential psychopharmacology. 3rd ed. New York, NY: Cambridge University Press; 2008:884-897.
7. Michelson D, Adler L, Spencer T, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry. 2003;53(2):112-120.