User login
Novel therapy shows promise for treating skin-predominant dermatomyositis
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.

Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.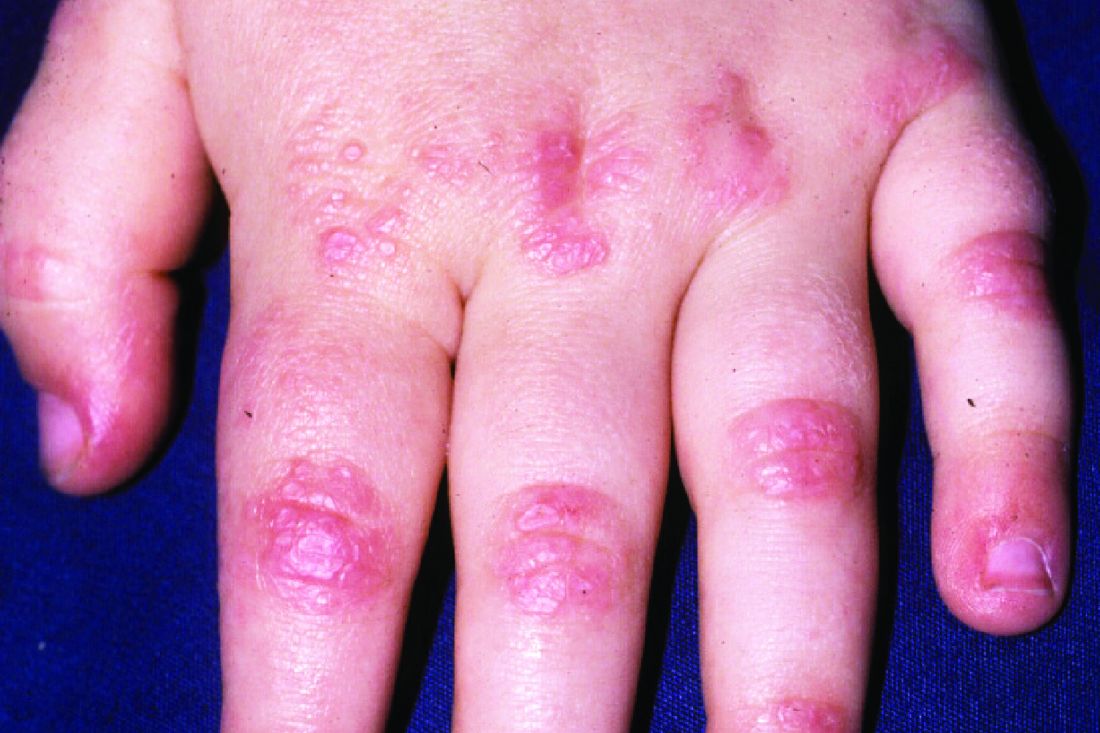
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.
Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – in a double-blind, placebo-controlled phase 2 trial, according to results presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
“These findings support the inhibition of IFN-beta as a promising therapeutic strategy in skin-predominant disease,” said principal investigator Aaron Mangold, MD, associate professor of dermatology, Mayo Clinic, Scottsdale, Ariz.
Dermatomyositis, a rare autoimmune inflammatory condition that typically involves both skeletal muscles and skin, is a challenging disease with a diverse set of potential complications.
Immunosuppressive and immunomodulatory agents are used with mixed success for myositis, but skin manifestations, which include papular eruptions, heliotrope rash, photoerythema, burning, and pruritus, are often the most troublesome and the most difficult to control. Treatment options other than immunomodulators that target cutaneous involvement – which include steroids, emollients, and photoprotection – are generally modestly effective, according to Dr. Mangold.
Targeting an elevated cytokine
Interest in IFN-beta, which is elevated in the blood of individuals with dermatomyositis, was triggered by evidence that this cytokine plays an important role in driving the skin inflammation, Dr. Mangold explained.
“The blood concentrations of IFN-beta are positively correlated with cutaneous disease activity and severity,” he said.
The study drug, currently known as PF-06823859 (Dazukibart), “is a potent, selective humanized IgG1-neutralizing antibody directed at IFN-beta,” Dr. Mangold said. A dose-ranging phase 1 study published 2 years ago provided evidence of acceptable pharmacokinetics and safety in healthy individuals to support treatment studies for disorders associated with elevated IFN-beta levels. In addition to dermatomyositis, this includes systemic lupus erythematosus.
In this phase 2 trial, patients whose condition was not improved by at least one standard-care therapy for skin manifestations of dermatomyositis were eligible if they had moderate to severe disease as measured with the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), according to Dr. Mangold. During the study, patients were allowed to remain on a disease modifying antirheumatic drug and/or prednisone if they had been on stable doses and did not change the dose.
After a screening run-in, the trial had two blinded stages. In stage 1, 30 patients were randomly assigned either to 600 mg of PF-06823859 or to placebo, both administered intravenously every 4 weeks. A second cohort of 25 patients was randomly assigned in stage 2 to placebo, 150 mg of PF-06823859, or 600 mg of PF-06823859. The primary endpoint assessed at 12 weeks was a greater than 5-point reduction in CDASI score or greater than 40% CDASI improvement from baseline.
Both endpoints are associated with a clinically meaningful response in regard to an improved quality of life, Dr. Mangold noted.
Both doses better than placebo
In results from the stage 1 portion, the mean reduction in CDASI at 12 weeks after three doses of the assigned therapy was 18.8 points in the active-treatment group versus 3.9 points in the placebo group. In pooled data from stage 1 and 2, the reductions were 16.6 points, 19.2 points, and 2.9 points for the 150-mg, 600-mg, and placebo arms, respectively. Both doses achieved a highly significant advantage over placebo.
For both stages and doses, the response curves of the active-treatment groups and the placebo group diverged almost immediately. By 4 weeks, both measures of CDASI reductions on active therapy were significantly improved relative to placebo, and the response curves had a consistent downward slope through the end of the 12-week study, Dr. Mangold reported.
The majority of patients responded by either of the primary endpoint criteria. For a CDASI reduction of greater than 5 points, the response rates were 100% and 96% for the 150-mg and 600-mg doses of PF-06823859, respectively. The placebo response was 35.7%. For the CDASI reduction of greater than 40%, the rates were 80%, 82.1%, and 7.1% for the 150-mg, 600-mg, and placebo arms, respectively.
“There were no major safety concerns. Most of the treatment-emergent adverse events were mild, and adverse events did not have a relationship to dose,” Dr. Mangold said. Notably, there were no cases of herpes zoster, and infections of any kind were low in all study groups.
A phase 3 study is being planned with the 600-mg dose, according to Dr. Mangold, but he acknowledged that regulatory authorities have generally required endpoints for both cutaneous and muscle manifestations in previous trials of therapies for dermatomyositis.
It is not yet certain that “there will be a carve-out for skin,” he said in answer to a question about investigations moving forward. So far, studies have been focused on skin response. However, a meaningful degree of benefit against muscle involvement, which has not yet been well studied, has not been ruled out.
Even though this is a phase 2 trial with small numbers, it was controlled and blinded, and the potential of an inhibitor of IFN-beta to control the skin manifestations of dermatomyositis “is kind of a big deal,” said Paul Nghiem, MD, PhD, professor of dermatology, University of Washington, Seattle.
“There is definitely an unmet need for better therapies to control the skin involvement,” Dr. Nghiem said.
Hensin Tsao, MD, PhD, clinical director of the Melanoma and Pigmented Lesion Center at Massachusetts General Hospital, Boston, agreed. Like Dr. Nghiem, Dr. Tsao was a panelist during the late-breaker session where the study was presented, and he was impressed by the data.
“This is something that is definitely newsworthy,” Dr. Tsao said.
Dr. Mangold reports financial relationships with Actelion, Amgen, Corbus, Eli Lilly, Incyte, miRagen, Novartis, Regeneron, Solagenix, Sun Pharmaceuticals, Teva, and Pfizer, which provided funding for this trial. Both Dr. Nghiem and Dr. Tsao reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT AAD 2023
Another FDA class I recall of Cardiosave Hybrid/Rescue IABPs
Datascope/Getinge is recalling certain Cardiosave Hybrid and Cardiosave Rescue Intra-Aortic Balloon Pumps (IABPs) because the coiled cable connecting the display and base on some units may fail, causing an unexpected shutdown without warnings or alarms to alert the user.
The U.S. Food and Drug Administration has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The FDA warns that an unexpected pump shutdown and any interruption to therapy that occurs can lead to hemodynamic instability, organ damage, and/or death, especially in patients who are critically ill and most likely to receive therapy using these devices.

The devices are indicated for acute coronary syndrome, cardiac and noncardiac surgery, and complications of heart failure in adults.
From June 2019 to August 2022, Datascope/Getinge reported 44 complaints about damaged coiled cords resulting in unexpected shutdowns. There have been no reports of injuries or deaths related to this issue, according to the recall notice posted on the FDA’s website.
The recall includes a total of 2,300 CardioSave Hybrid or Rescue IABP units distributed prior to July 24, 2017, and/or coiled cord part number 0012-00-1801. Product model numbers for the recalled Cardiosave Hybrid and Cardiosave Rescue are available online.
The Cardiosave IABPs have previously been flagged by the FDA for subpar battery performance and fluid leaks.
To address the cable issue, Datascope/Getinge sent an urgent medical device correction letter to customers recommending that the coiled cable cord of the Cardiosave IABP be inspected for visible damage prior to use.
If an unexpected shutdown occurs, an attempt should be made to restart the Cardiosave IABP until an alternative pump is available. If the restart attempt is unsuccessful, an alternative IABP should be used. Any device that remains inoperable after a shutdown should be removed from patient care.
Customers should inspect their inventory to identify any Cardiosave Hybrid and/or Rescue IABPs that have the recalled coiled cord.
The company also asks customers to complete and sign the Medical Device Correction-Response form included with the letter and return it to Datascope/Getinge by emailing a scanned copy to [email protected] or by faxing the form to 1-877-660-5841.
Customers with questions about this recall should contact their Datascope/Getinge representative or call Datascope/Getinge technical support at 1-888-943-8872, Monday through Friday, between 8:00 AM and 6:00 PM ET.
The company has developed a hardware correction to address this issue and says a service representative will contact customers to schedule installation of the correction when the correction kit is available.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
Datascope/Getinge is recalling certain Cardiosave Hybrid and Cardiosave Rescue Intra-Aortic Balloon Pumps (IABPs) because the coiled cable connecting the display and base on some units may fail, causing an unexpected shutdown without warnings or alarms to alert the user.
The U.S. Food and Drug Administration has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The FDA warns that an unexpected pump shutdown and any interruption to therapy that occurs can lead to hemodynamic instability, organ damage, and/or death, especially in patients who are critically ill and most likely to receive therapy using these devices.
The devices are indicated for acute coronary syndrome, cardiac and noncardiac surgery, and complications of heart failure in adults.
From June 2019 to August 2022, Datascope/Getinge reported 44 complaints about damaged coiled cords resulting in unexpected shutdowns. There have been no reports of injuries or deaths related to this issue, according to the recall notice posted on the FDA’s website.
The recall includes a total of 2,300 CardioSave Hybrid or Rescue IABP units distributed prior to July 24, 2017, and/or coiled cord part number 0012-00-1801. Product model numbers for the recalled Cardiosave Hybrid and Cardiosave Rescue are available online.
The Cardiosave IABPs have previously been flagged by the FDA for subpar battery performance and fluid leaks.
To address the cable issue, Datascope/Getinge sent an urgent medical device correction letter to customers recommending that the coiled cable cord of the Cardiosave IABP be inspected for visible damage prior to use.
If an unexpected shutdown occurs, an attempt should be made to restart the Cardiosave IABP until an alternative pump is available. If the restart attempt is unsuccessful, an alternative IABP should be used. Any device that remains inoperable after a shutdown should be removed from patient care.
Customers should inspect their inventory to identify any Cardiosave Hybrid and/or Rescue IABPs that have the recalled coiled cord.
The company also asks customers to complete and sign the Medical Device Correction-Response form included with the letter and return it to Datascope/Getinge by emailing a scanned copy to [email protected] or by faxing the form to 1-877-660-5841.
Customers with questions about this recall should contact their Datascope/Getinge representative or call Datascope/Getinge technical support at 1-888-943-8872, Monday through Friday, between 8:00 AM and 6:00 PM ET.
The company has developed a hardware correction to address this issue and says a service representative will contact customers to schedule installation of the correction when the correction kit is available.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
Datascope/Getinge is recalling certain Cardiosave Hybrid and Cardiosave Rescue Intra-Aortic Balloon Pumps (IABPs) because the coiled cable connecting the display and base on some units may fail, causing an unexpected shutdown without warnings or alarms to alert the user.
The U.S. Food and Drug Administration has identified this as a class I recall, the most serious type of recall, because of the risk for serious injury or death.
The FDA warns that an unexpected pump shutdown and any interruption to therapy that occurs can lead to hemodynamic instability, organ damage, and/or death, especially in patients who are critically ill and most likely to receive therapy using these devices.
The devices are indicated for acute coronary syndrome, cardiac and noncardiac surgery, and complications of heart failure in adults.
From June 2019 to August 2022, Datascope/Getinge reported 44 complaints about damaged coiled cords resulting in unexpected shutdowns. There have been no reports of injuries or deaths related to this issue, according to the recall notice posted on the FDA’s website.
The recall includes a total of 2,300 CardioSave Hybrid or Rescue IABP units distributed prior to July 24, 2017, and/or coiled cord part number 0012-00-1801. Product model numbers for the recalled Cardiosave Hybrid and Cardiosave Rescue are available online.
The Cardiosave IABPs have previously been flagged by the FDA for subpar battery performance and fluid leaks.
To address the cable issue, Datascope/Getinge sent an urgent medical device correction letter to customers recommending that the coiled cable cord of the Cardiosave IABP be inspected for visible damage prior to use.
If an unexpected shutdown occurs, an attempt should be made to restart the Cardiosave IABP until an alternative pump is available. If the restart attempt is unsuccessful, an alternative IABP should be used. Any device that remains inoperable after a shutdown should be removed from patient care.
Customers should inspect their inventory to identify any Cardiosave Hybrid and/or Rescue IABPs that have the recalled coiled cord.
The company also asks customers to complete and sign the Medical Device Correction-Response form included with the letter and return it to Datascope/Getinge by emailing a scanned copy to [email protected] or by faxing the form to 1-877-660-5841.
Customers with questions about this recall should contact their Datascope/Getinge representative or call Datascope/Getinge technical support at 1-888-943-8872, Monday through Friday, between 8:00 AM and 6:00 PM ET.
The company has developed a hardware correction to address this issue and says a service representative will contact customers to schedule installation of the correction when the correction kit is available.
Any adverse events or suspected adverse events related to the recalled CardioSave Hybrid/Rescue IABPs should be reported to the FDA through MedWatch, its adverse event reporting program.
A version of this article first appeared on Medscape.com.
Novel single-use patch shows promise for primary axillary hyperhidrosis
NEW ORLEANS – , results from a pivotal randomized trial showed.
“This is a new kind of device that is going to be a nice tool to have for treating patients who have hyperhidrosis of the axilla,” the study’s lead investigator, David M. Pariser, MD, who practices dermatology in Norfolk, Va., said during a late-breaking abstract session at the annual meeting of the American Academy of Dermatology.

In a study known as SAHARA, investigators at 11 sites evaluated the efficacy of the targeted alkali thermolysis (TAT) patch, a single-use disposable device. The patch consists of a thin sodium layer on an adhesive overlay. It’s applied to the dry axilla, and as the patient sweats during treatment, the sweat reacts with the sodium. According to Dr. Pariser, this interaction generates precisely targeted thermal energy that targets sweat glands, leading to a reduction in excessive sweat production for up to three months.
The researchers enrolled 110 individuals with Hyperhidrosis Disease Severity Scale (HDSS) scores of 3 or 4 and randomized them to either an active TAT or a sham patch for up to 3 minutes. Their mean age was about 33 years, and slightly more than half were women. “If significant discomfort or pain was noted, [the patch] treatment was halted; otherwise, it was left on for 3 minutes,” said Dr. Pariser, professor of dermatology at Eastern Virginia Medical School, Norfolk. “The treated area was thoroughly cleaned after treatment, and the TAT patch was deactivated. This process was repeated on the other axilla.”
The HDSS, Gravimetric Sweat Production (GSP), and quality of life assessments for bother and impact were measured through 12 weeks. The quality of life assessments were an exploratory endpoint and scored from 0 to 4, with 4 being extremely bothered or impacted and 0 not being bothered or impacted at all. The primary efficacy endpoint was the proportion of treated patients achieving a 1 or 2 on the HDSS at week 4, compared with sham treatment.
Secondary endpoints included the proportion of patients with an improvement of at least 2 grades from baseline to 4 weeks in HDSS by treatment group; mean improvement in the quality of life scale bother by treatment group; mean improvement in the quality of life scale impact by treatment group; and the proportion of subjects with at least 50% improvement in GSP from baseline to 4 weeks in the active patch group only.

Adverse events (AEs) were divided into 3 categories: AEs at the treatment site (or skin reactions within the treated part of the axilla); procedure-related AEs (those that are the result of treatment, but not in the treated part of the axilla), and non-axillary AEs.
Dr. Pariser reported that at 4 weeks, 63.6% of patients in the active patch group versus 44.2% of those in the sham group improved to an HDSS score of 1 or 2 (P = .0332) and that 43.2% of those in the active patch group versus 16.3% of those in the sham group (P = .0107) achieved a 2-point or greater HDSS improvement. In addition, 9.1% of those in the active patch group achieved a 3-point improvement on the HDSS, compared with none in the sham group. “That’s an amazing improvement; you’re basically going from moderate or severe to none,” he commented.
In other findings, 60.5% of patients in the active patch group showed at least a 50% reduction in GSP, compared with 32.6% of those in the sham group (P = .0102), with mean reductions of 57.3 mg/5min and 18.2 mg/5min, respectively (P = .0036). As for quality-of-life outcome scores, bother associated with hyperhidrosis was reduced by 1.52 points in active versus 0.61 in sham subjects (P = .0005), while impact was reduced by 1.44 in active versus 0.57 in sham subjects (P = .0004).
Adverse events
A total of 13 patients in the active patch group experienced AEs at the treatment site, including six with erythema; four with erosion; two with burning, itching or stinging; and one with underarm odor. “The two procedure-related AEs in the TAT-treated group were compensatory sweating and irritant contact dermatitis due to the adhesive,” said Dr. Pariser said.
Most adverse events resolved in fewer than 2 weeks, and all were mild to moderate. No serious adverse events occurred. Only five adverse events occurred in the sham group.
The TAT patch is currently undergoing review by the Food and Drug Administration, and according to Dr. Pariser, no other body sites have been treated with the device.
Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, characterized hyperhidrosis as “an exceedingly common medical condition that is commonly overlooked even though it has a tremendous burden on quality of life. I should know, as both someone who manages a large cohort of these patients but also as someone who suffers from it.”
Treatment options “have historically been limited, many of which are off-label and some which are difficult to access due to cost and/or duration/frequency of treatment,” added Dr. Friedman, who was not involved with the study. “The TAT patch offers a new, targeted, in-office, practical procedure-based approach to treat primary axillary hyperhidrosis. Innovation is certainly welcomed and needed, and I am curious to see how this technology is employed in practice once approved.”
The device is being developed by Candesant Biomedical. Dr. Pariser disclosed that he is a consultant or investigator for Bickel Biotechnology, Biofrontera AG, Bristol Myers Squibb, Celgene Corporation, Novartis Pharmaceuticals, Pfizer, Regeneron, and Sanofi.
Dr. Friedman reported having no relevant disclosures.
NEW ORLEANS – , results from a pivotal randomized trial showed.
“This is a new kind of device that is going to be a nice tool to have for treating patients who have hyperhidrosis of the axilla,” the study’s lead investigator, David M. Pariser, MD, who practices dermatology in Norfolk, Va., said during a late-breaking abstract session at the annual meeting of the American Academy of Dermatology.
In a study known as SAHARA, investigators at 11 sites evaluated the efficacy of the targeted alkali thermolysis (TAT) patch, a single-use disposable device. The patch consists of a thin sodium layer on an adhesive overlay. It’s applied to the dry axilla, and as the patient sweats during treatment, the sweat reacts with the sodium. According to Dr. Pariser, this interaction generates precisely targeted thermal energy that targets sweat glands, leading to a reduction in excessive sweat production for up to three months.
The researchers enrolled 110 individuals with Hyperhidrosis Disease Severity Scale (HDSS) scores of 3 or 4 and randomized them to either an active TAT or a sham patch for up to 3 minutes. Their mean age was about 33 years, and slightly more than half were women. “If significant discomfort or pain was noted, [the patch] treatment was halted; otherwise, it was left on for 3 minutes,” said Dr. Pariser, professor of dermatology at Eastern Virginia Medical School, Norfolk. “The treated area was thoroughly cleaned after treatment, and the TAT patch was deactivated. This process was repeated on the other axilla.”
The HDSS, Gravimetric Sweat Production (GSP), and quality of life assessments for bother and impact were measured through 12 weeks. The quality of life assessments were an exploratory endpoint and scored from 0 to 4, with 4 being extremely bothered or impacted and 0 not being bothered or impacted at all. The primary efficacy endpoint was the proportion of treated patients achieving a 1 or 2 on the HDSS at week 4, compared with sham treatment.
Secondary endpoints included the proportion of patients with an improvement of at least 2 grades from baseline to 4 weeks in HDSS by treatment group; mean improvement in the quality of life scale bother by treatment group; mean improvement in the quality of life scale impact by treatment group; and the proportion of subjects with at least 50% improvement in GSP from baseline to 4 weeks in the active patch group only.
Adverse events (AEs) were divided into 3 categories: AEs at the treatment site (or skin reactions within the treated part of the axilla); procedure-related AEs (those that are the result of treatment, but not in the treated part of the axilla), and non-axillary AEs.
Dr. Pariser reported that at 4 weeks, 63.6% of patients in the active patch group versus 44.2% of those in the sham group improved to an HDSS score of 1 or 2 (P = .0332) and that 43.2% of those in the active patch group versus 16.3% of those in the sham group (P = .0107) achieved a 2-point or greater HDSS improvement. In addition, 9.1% of those in the active patch group achieved a 3-point improvement on the HDSS, compared with none in the sham group. “That’s an amazing improvement; you’re basically going from moderate or severe to none,” he commented.
In other findings, 60.5% of patients in the active patch group showed at least a 50% reduction in GSP, compared with 32.6% of those in the sham group (P = .0102), with mean reductions of 57.3 mg/5min and 18.2 mg/5min, respectively (P = .0036). As for quality-of-life outcome scores, bother associated with hyperhidrosis was reduced by 1.52 points in active versus 0.61 in sham subjects (P = .0005), while impact was reduced by 1.44 in active versus 0.57 in sham subjects (P = .0004).
Adverse events
A total of 13 patients in the active patch group experienced AEs at the treatment site, including six with erythema; four with erosion; two with burning, itching or stinging; and one with underarm odor. “The two procedure-related AEs in the TAT-treated group were compensatory sweating and irritant contact dermatitis due to the adhesive,” said Dr. Pariser said.
Most adverse events resolved in fewer than 2 weeks, and all were mild to moderate. No serious adverse events occurred. Only five adverse events occurred in the sham group.
The TAT patch is currently undergoing review by the Food and Drug Administration, and according to Dr. Pariser, no other body sites have been treated with the device.
Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, characterized hyperhidrosis as “an exceedingly common medical condition that is commonly overlooked even though it has a tremendous burden on quality of life. I should know, as both someone who manages a large cohort of these patients but also as someone who suffers from it.”
Treatment options “have historically been limited, many of which are off-label and some which are difficult to access due to cost and/or duration/frequency of treatment,” added Dr. Friedman, who was not involved with the study. “The TAT patch offers a new, targeted, in-office, practical procedure-based approach to treat primary axillary hyperhidrosis. Innovation is certainly welcomed and needed, and I am curious to see how this technology is employed in practice once approved.”
The device is being developed by Candesant Biomedical. Dr. Pariser disclosed that he is a consultant or investigator for Bickel Biotechnology, Biofrontera AG, Bristol Myers Squibb, Celgene Corporation, Novartis Pharmaceuticals, Pfizer, Regeneron, and Sanofi.
Dr. Friedman reported having no relevant disclosures.
NEW ORLEANS – , results from a pivotal randomized trial showed.
“This is a new kind of device that is going to be a nice tool to have for treating patients who have hyperhidrosis of the axilla,” the study’s lead investigator, David M. Pariser, MD, who practices dermatology in Norfolk, Va., said during a late-breaking abstract session at the annual meeting of the American Academy of Dermatology.
In a study known as SAHARA, investigators at 11 sites evaluated the efficacy of the targeted alkali thermolysis (TAT) patch, a single-use disposable device. The patch consists of a thin sodium layer on an adhesive overlay. It’s applied to the dry axilla, and as the patient sweats during treatment, the sweat reacts with the sodium. According to Dr. Pariser, this interaction generates precisely targeted thermal energy that targets sweat glands, leading to a reduction in excessive sweat production for up to three months.
The researchers enrolled 110 individuals with Hyperhidrosis Disease Severity Scale (HDSS) scores of 3 or 4 and randomized them to either an active TAT or a sham patch for up to 3 minutes. Their mean age was about 33 years, and slightly more than half were women. “If significant discomfort or pain was noted, [the patch] treatment was halted; otherwise, it was left on for 3 minutes,” said Dr. Pariser, professor of dermatology at Eastern Virginia Medical School, Norfolk. “The treated area was thoroughly cleaned after treatment, and the TAT patch was deactivated. This process was repeated on the other axilla.”
The HDSS, Gravimetric Sweat Production (GSP), and quality of life assessments for bother and impact were measured through 12 weeks. The quality of life assessments were an exploratory endpoint and scored from 0 to 4, with 4 being extremely bothered or impacted and 0 not being bothered or impacted at all. The primary efficacy endpoint was the proportion of treated patients achieving a 1 or 2 on the HDSS at week 4, compared with sham treatment.
Secondary endpoints included the proportion of patients with an improvement of at least 2 grades from baseline to 4 weeks in HDSS by treatment group; mean improvement in the quality of life scale bother by treatment group; mean improvement in the quality of life scale impact by treatment group; and the proportion of subjects with at least 50% improvement in GSP from baseline to 4 weeks in the active patch group only.
Adverse events (AEs) were divided into 3 categories: AEs at the treatment site (or skin reactions within the treated part of the axilla); procedure-related AEs (those that are the result of treatment, but not in the treated part of the axilla), and non-axillary AEs.
Dr. Pariser reported that at 4 weeks, 63.6% of patients in the active patch group versus 44.2% of those in the sham group improved to an HDSS score of 1 or 2 (P = .0332) and that 43.2% of those in the active patch group versus 16.3% of those in the sham group (P = .0107) achieved a 2-point or greater HDSS improvement. In addition, 9.1% of those in the active patch group achieved a 3-point improvement on the HDSS, compared with none in the sham group. “That’s an amazing improvement; you’re basically going from moderate or severe to none,” he commented.
In other findings, 60.5% of patients in the active patch group showed at least a 50% reduction in GSP, compared with 32.6% of those in the sham group (P = .0102), with mean reductions of 57.3 mg/5min and 18.2 mg/5min, respectively (P = .0036). As for quality-of-life outcome scores, bother associated with hyperhidrosis was reduced by 1.52 points in active versus 0.61 in sham subjects (P = .0005), while impact was reduced by 1.44 in active versus 0.57 in sham subjects (P = .0004).
Adverse events
A total of 13 patients in the active patch group experienced AEs at the treatment site, including six with erythema; four with erosion; two with burning, itching or stinging; and one with underarm odor. “The two procedure-related AEs in the TAT-treated group were compensatory sweating and irritant contact dermatitis due to the adhesive,” said Dr. Pariser said.
Most adverse events resolved in fewer than 2 weeks, and all were mild to moderate. No serious adverse events occurred. Only five adverse events occurred in the sham group.
The TAT patch is currently undergoing review by the Food and Drug Administration, and according to Dr. Pariser, no other body sites have been treated with the device.
Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, who was asked to comment on the study, characterized hyperhidrosis as “an exceedingly common medical condition that is commonly overlooked even though it has a tremendous burden on quality of life. I should know, as both someone who manages a large cohort of these patients but also as someone who suffers from it.”
Treatment options “have historically been limited, many of which are off-label and some which are difficult to access due to cost and/or duration/frequency of treatment,” added Dr. Friedman, who was not involved with the study. “The TAT patch offers a new, targeted, in-office, practical procedure-based approach to treat primary axillary hyperhidrosis. Innovation is certainly welcomed and needed, and I am curious to see how this technology is employed in practice once approved.”
The device is being developed by Candesant Biomedical. Dr. Pariser disclosed that he is a consultant or investigator for Bickel Biotechnology, Biofrontera AG, Bristol Myers Squibb, Celgene Corporation, Novartis Pharmaceuticals, Pfizer, Regeneron, and Sanofi.
Dr. Friedman reported having no relevant disclosures.
AT AAD 2023
JAK inhibitor safety warnings drawn from rheumatologic data may be misleading in dermatology
NEW ORLEANS – , even though the basis for all the risks is a rheumatoid arthritis study, according to a critical review at the annual meeting of the American Academy of Dermatology.
Given the fact that the postmarketing RA study was specifically enriched with high-risk patients by requiring an age at enrollment of at least 50 years and the presence of at least one cardiovascular risk factor, the extrapolation of these risks to dermatologic indications is “not necessarily data-driven,” said Brett A. King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
The recently approved deucravacitinib is the only JAK inhibitor that has so far been exempt from these warnings. Instead, based on the ORAL Surveillance study, published in the New England Journal of Medicine, the Food and Drug Administration requires a boxed warning in nearly identical language for all the other JAK inhibitors. Relative to tofacitinib, the JAK inhibitor tested in ORAL Surveillance, many of these drugs differ by JAK selectivity and other characteristics that are likely relevant to risk of adverse events, Dr. King said. The same language has even been applied to topical ruxolitinib cream.
Basis of boxed warnings
In ORAL Surveillance, about 4,300 high-risk patients with RA were randomized to one of two doses of tofacitinib (5 mg or 10 mg) twice daily or a tumor necrosis factor (TNF) inhibitor. All patients in the trial were taking methotrexate, and almost 60% were taking concomitant corticosteroids. The average body mass index of the study population was about 30 kg/m2.
After a median 4 years of follow-up (about 5,000 patient-years), the incidence of many of the adverse events tracked in the study were higher in the tofacitinib groups, including serious infections, MACE, thromboembolic events, and cancer. Dr. King did not challenge the importance of these data, but he questioned whether they are reasonably extrapolated to dermatologic indications, particularly as many of those treated are younger than those common to an RA population.
In fact, despite a study enriched for a higher risk of many events tracked, most adverse events were only slightly elevated, Dr. King pointed out. For example, the incidence of MACE over the 4 years of follow-up was 3.4% among those taking any dose of tofacitinib versus 2.5% of those randomized to TNF inhibitor. Rates of cancer were 4.2% versus 2.9%, respectively. There were also absolute increases in the number of serious infections and thromboembolic events for tofacitinib relative to TNF inhibitor.
Dr. King acknowledged that the numbers in ORAL Surveillance associated tofacitinib with a higher risk of serious events than TNF inhibitor in patients with RA, but he believes that “JAK inhibitor safety is almost certainly not the same in dermatology as it is in rheumatology patients.”
Evidence of difference in dermatology
There is some evidence to back this up. Dr. King cited a recently published study in RMD Open that evaluated the safety profile of the JAK inhibitor upadacitinib in nearly 7,000 patients over 15,000 patient-years of follow-up. Drug safety data were evaluated with up to 5.5 years of follow-up from 12 clinical trials of the four diseases for which upadacitinib is now indicated. Three were rheumatologic (RA, psoriatic arthritis, and ankylosing spondylitis), and the fourth was atopic dermatitis (AD). Fourteen outcomes, including numerous types of infection, MACE, hepatic complications, and malignancy, were compared with methotrexate and the TNF inhibitor adalimumab.
For the RA diseases, upadacitinib was associated with a greater risk than comparators for several outcomes, including serious infections. But in AD, there was a smaller increased risk of adverse outcomes for the JAK inhibitor relative to comparators.
When evaluated by risk of adverse events across indications, for MACE, the exposure-adjusted event rates for upadacitinib were less than 0.1 in patients treated for AD over the observation period versus 0.3 and 0.4 for RA and psoriatic arthritis, respectively. Similarly, for venous thromboembolism, the rates for upadacitinib were again less than 0.1 in patients with AD versus 0.4 and 0.2 in RA and psoriatic arthritis, respectively.
Referring back to the postmarketing study, Dr. King emphasized that it is essential to consider how the boxed warning for JAK inhibitors was generated before applying them to dermatologic indications.
“Is a 30-year-old patient with a dermatologic disorder possibly at the same risk as the patients in the study from which we got the boxed warning? The answer is simply no,” he said.
Like the tofacitinib data in the ORAL Surveillance study, the upadacitinib clinical trial data are not necessarily relevant to other JAK inhibitors. In fact, Dr. King pointed out that the safety profiles of the available JAK inhibitors are not identical, an observation that is consistent with differences in JAK inhibitor selectivity that has implications for off-target events.
Dr. King does not dismiss the potential risks outlined in the current regulatory cautions about the use of JAK inhibitors, but he believes that dermatologists should be cognizant of “where the black box warning comes from.”
“We need to think carefully about the risk-to-benefit ratio in older patients or patients with risk factors, such as obesity and diabetes,” he said. But the safety profile of JAK inhibitors “is almost certainly better” than the profile suggested in boxed warnings applied to JAK inhibitors for dermatologic indications, he advised.
Risk-benefit considerations in dermatology
This position was supported by numerous other experts when asked for their perspectives. “I fully agree,” said Emma Guttman-Yassky, MD, PhD, system chair of dermatology and immunology, Icahn School of Medicine, Mount Sinai, New York.
Like Dr. King, Dr. Guttman-Yassky did not dismiss the potential risks of JAK inhibitors when treating dermatologic diseases.
“While JAK inhibitors need monitoring as advised, adopting a boxed warning from an RA study for patients who are older [is problematic],” she commented. A study with the nonselective tofacitinib in this population “cannot be compared to more selective inhibitors in a much younger population, such as those treated [for] alopecia areata or atopic dermatitis.”
George Z. Han, MD, PhD, an associate professor of dermatology, Zucker School of Medicine, Hofstra, Northwell Medical Center, New Hyde Park, New York, also agreed but added some caveats.
“The comments about the ORAL Surveillance study are salient,” he said in an interview. “This kind of data should not directly be extrapolated to other patient types or to other medications.” However, one of Dr. Han’s most important caveats involves long-term use.
“JAK inhibitors are still relatively narrow-therapeutic-window drugs that in a dose-dependent fashion could lead to negative effects, including thromboembolic events, abnormalities in red blood cells, white blood cells, platelets, and lipids,” he said. While doses used in dermatology “are generally below the level of any major concern,” Dr. Han cautioned that “we lack definitive data” on long-term use, and this is important for understanding “any potential small risk of rare events, such as malignancy or thromboembolism.”
Saakshi Khattri, MD, a colleague of Dr. Guttman-Yassky at Mount Sinai, said the risks of JAK inhibitors should not be underestimated, but she also agreed that risk “needs to be delivered in the right context.” Dr. Khattri, who is board certified in both dermatology and rheumatology, noted the safety profiles of available JAK inhibitors differ and that extrapolating safety from an RA study to dermatologic indications does not make sense. “Different diseases, different age groups,” she said.
Dr. King has reported financial relationships with more than 15 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Han reports financial relationships with Amgen, Athenex, Boehringer Ingelheim, Bond Avillion, Bristol-Myers Squibb, Celgene, Janssen, Lilly, Novartis, PellePharm, Pfizer, and UCB. Dr. Khattri has reported financial relationships with AbbVie, Arcutis, Bristol-Myers Squibb, Janssen, Leo, Lilly, Novartis, Pfizer, and UCB.
A version of this article originally appeared on Medscape.com.
NEW ORLEANS – , even though the basis for all the risks is a rheumatoid arthritis study, according to a critical review at the annual meeting of the American Academy of Dermatology.
Given the fact that the postmarketing RA study was specifically enriched with high-risk patients by requiring an age at enrollment of at least 50 years and the presence of at least one cardiovascular risk factor, the extrapolation of these risks to dermatologic indications is “not necessarily data-driven,” said Brett A. King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
The recently approved deucravacitinib is the only JAK inhibitor that has so far been exempt from these warnings. Instead, based on the ORAL Surveillance study, published in the New England Journal of Medicine, the Food and Drug Administration requires a boxed warning in nearly identical language for all the other JAK inhibitors. Relative to tofacitinib, the JAK inhibitor tested in ORAL Surveillance, many of these drugs differ by JAK selectivity and other characteristics that are likely relevant to risk of adverse events, Dr. King said. The same language has even been applied to topical ruxolitinib cream.
Basis of boxed warnings
In ORAL Surveillance, about 4,300 high-risk patients with RA were randomized to one of two doses of tofacitinib (5 mg or 10 mg) twice daily or a tumor necrosis factor (TNF) inhibitor. All patients in the trial were taking methotrexate, and almost 60% were taking concomitant corticosteroids. The average body mass index of the study population was about 30 kg/m2.
After a median 4 years of follow-up (about 5,000 patient-years), the incidence of many of the adverse events tracked in the study were higher in the tofacitinib groups, including serious infections, MACE, thromboembolic events, and cancer. Dr. King did not challenge the importance of these data, but he questioned whether they are reasonably extrapolated to dermatologic indications, particularly as many of those treated are younger than those common to an RA population.
In fact, despite a study enriched for a higher risk of many events tracked, most adverse events were only slightly elevated, Dr. King pointed out. For example, the incidence of MACE over the 4 years of follow-up was 3.4% among those taking any dose of tofacitinib versus 2.5% of those randomized to TNF inhibitor. Rates of cancer were 4.2% versus 2.9%, respectively. There were also absolute increases in the number of serious infections and thromboembolic events for tofacitinib relative to TNF inhibitor.
Dr. King acknowledged that the numbers in ORAL Surveillance associated tofacitinib with a higher risk of serious events than TNF inhibitor in patients with RA, but he believes that “JAK inhibitor safety is almost certainly not the same in dermatology as it is in rheumatology patients.”
Evidence of difference in dermatology
There is some evidence to back this up. Dr. King cited a recently published study in RMD Open that evaluated the safety profile of the JAK inhibitor upadacitinib in nearly 7,000 patients over 15,000 patient-years of follow-up. Drug safety data were evaluated with up to 5.5 years of follow-up from 12 clinical trials of the four diseases for which upadacitinib is now indicated. Three were rheumatologic (RA, psoriatic arthritis, and ankylosing spondylitis), and the fourth was atopic dermatitis (AD). Fourteen outcomes, including numerous types of infection, MACE, hepatic complications, and malignancy, were compared with methotrexate and the TNF inhibitor adalimumab.
For the RA diseases, upadacitinib was associated with a greater risk than comparators for several outcomes, including serious infections. But in AD, there was a smaller increased risk of adverse outcomes for the JAK inhibitor relative to comparators.
When evaluated by risk of adverse events across indications, for MACE, the exposure-adjusted event rates for upadacitinib were less than 0.1 in patients treated for AD over the observation period versus 0.3 and 0.4 for RA and psoriatic arthritis, respectively. Similarly, for venous thromboembolism, the rates for upadacitinib were again less than 0.1 in patients with AD versus 0.4 and 0.2 in RA and psoriatic arthritis, respectively.
Referring back to the postmarketing study, Dr. King emphasized that it is essential to consider how the boxed warning for JAK inhibitors was generated before applying them to dermatologic indications.
“Is a 30-year-old patient with a dermatologic disorder possibly at the same risk as the patients in the study from which we got the boxed warning? The answer is simply no,” he said.
Like the tofacitinib data in the ORAL Surveillance study, the upadacitinib clinical trial data are not necessarily relevant to other JAK inhibitors. In fact, Dr. King pointed out that the safety profiles of the available JAK inhibitors are not identical, an observation that is consistent with differences in JAK inhibitor selectivity that has implications for off-target events.
Dr. King does not dismiss the potential risks outlined in the current regulatory cautions about the use of JAK inhibitors, but he believes that dermatologists should be cognizant of “where the black box warning comes from.”
“We need to think carefully about the risk-to-benefit ratio in older patients or patients with risk factors, such as obesity and diabetes,” he said. But the safety profile of JAK inhibitors “is almost certainly better” than the profile suggested in boxed warnings applied to JAK inhibitors for dermatologic indications, he advised.
Risk-benefit considerations in dermatology
This position was supported by numerous other experts when asked for their perspectives. “I fully agree,” said Emma Guttman-Yassky, MD, PhD, system chair of dermatology and immunology, Icahn School of Medicine, Mount Sinai, New York.
Like Dr. King, Dr. Guttman-Yassky did not dismiss the potential risks of JAK inhibitors when treating dermatologic diseases.
“While JAK inhibitors need monitoring as advised, adopting a boxed warning from an RA study for patients who are older [is problematic],” she commented. A study with the nonselective tofacitinib in this population “cannot be compared to more selective inhibitors in a much younger population, such as those treated [for] alopecia areata or atopic dermatitis.”
George Z. Han, MD, PhD, an associate professor of dermatology, Zucker School of Medicine, Hofstra, Northwell Medical Center, New Hyde Park, New York, also agreed but added some caveats.
“The comments about the ORAL Surveillance study are salient,” he said in an interview. “This kind of data should not directly be extrapolated to other patient types or to other medications.” However, one of Dr. Han’s most important caveats involves long-term use.
“JAK inhibitors are still relatively narrow-therapeutic-window drugs that in a dose-dependent fashion could lead to negative effects, including thromboembolic events, abnormalities in red blood cells, white blood cells, platelets, and lipids,” he said. While doses used in dermatology “are generally below the level of any major concern,” Dr. Han cautioned that “we lack definitive data” on long-term use, and this is important for understanding “any potential small risk of rare events, such as malignancy or thromboembolism.”
Saakshi Khattri, MD, a colleague of Dr. Guttman-Yassky at Mount Sinai, said the risks of JAK inhibitors should not be underestimated, but she also agreed that risk “needs to be delivered in the right context.” Dr. Khattri, who is board certified in both dermatology and rheumatology, noted the safety profiles of available JAK inhibitors differ and that extrapolating safety from an RA study to dermatologic indications does not make sense. “Different diseases, different age groups,” she said.
Dr. King has reported financial relationships with more than 15 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Han reports financial relationships with Amgen, Athenex, Boehringer Ingelheim, Bond Avillion, Bristol-Myers Squibb, Celgene, Janssen, Lilly, Novartis, PellePharm, Pfizer, and UCB. Dr. Khattri has reported financial relationships with AbbVie, Arcutis, Bristol-Myers Squibb, Janssen, Leo, Lilly, Novartis, Pfizer, and UCB.
A version of this article originally appeared on Medscape.com.
NEW ORLEANS – , even though the basis for all the risks is a rheumatoid arthritis study, according to a critical review at the annual meeting of the American Academy of Dermatology.
Given the fact that the postmarketing RA study was specifically enriched with high-risk patients by requiring an age at enrollment of at least 50 years and the presence of at least one cardiovascular risk factor, the extrapolation of these risks to dermatologic indications is “not necessarily data-driven,” said Brett A. King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
The recently approved deucravacitinib is the only JAK inhibitor that has so far been exempt from these warnings. Instead, based on the ORAL Surveillance study, published in the New England Journal of Medicine, the Food and Drug Administration requires a boxed warning in nearly identical language for all the other JAK inhibitors. Relative to tofacitinib, the JAK inhibitor tested in ORAL Surveillance, many of these drugs differ by JAK selectivity and other characteristics that are likely relevant to risk of adverse events, Dr. King said. The same language has even been applied to topical ruxolitinib cream.
Basis of boxed warnings
In ORAL Surveillance, about 4,300 high-risk patients with RA were randomized to one of two doses of tofacitinib (5 mg or 10 mg) twice daily or a tumor necrosis factor (TNF) inhibitor. All patients in the trial were taking methotrexate, and almost 60% were taking concomitant corticosteroids. The average body mass index of the study population was about 30 kg/m2.
After a median 4 years of follow-up (about 5,000 patient-years), the incidence of many of the adverse events tracked in the study were higher in the tofacitinib groups, including serious infections, MACE, thromboembolic events, and cancer. Dr. King did not challenge the importance of these data, but he questioned whether they are reasonably extrapolated to dermatologic indications, particularly as many of those treated are younger than those common to an RA population.
In fact, despite a study enriched for a higher risk of many events tracked, most adverse events were only slightly elevated, Dr. King pointed out. For example, the incidence of MACE over the 4 years of follow-up was 3.4% among those taking any dose of tofacitinib versus 2.5% of those randomized to TNF inhibitor. Rates of cancer were 4.2% versus 2.9%, respectively. There were also absolute increases in the number of serious infections and thromboembolic events for tofacitinib relative to TNF inhibitor.
Dr. King acknowledged that the numbers in ORAL Surveillance associated tofacitinib with a higher risk of serious events than TNF inhibitor in patients with RA, but he believes that “JAK inhibitor safety is almost certainly not the same in dermatology as it is in rheumatology patients.”
Evidence of difference in dermatology
There is some evidence to back this up. Dr. King cited a recently published study in RMD Open that evaluated the safety profile of the JAK inhibitor upadacitinib in nearly 7,000 patients over 15,000 patient-years of follow-up. Drug safety data were evaluated with up to 5.5 years of follow-up from 12 clinical trials of the four diseases for which upadacitinib is now indicated. Three were rheumatologic (RA, psoriatic arthritis, and ankylosing spondylitis), and the fourth was atopic dermatitis (AD). Fourteen outcomes, including numerous types of infection, MACE, hepatic complications, and malignancy, were compared with methotrexate and the TNF inhibitor adalimumab.
For the RA diseases, upadacitinib was associated with a greater risk than comparators for several outcomes, including serious infections. But in AD, there was a smaller increased risk of adverse outcomes for the JAK inhibitor relative to comparators.
When evaluated by risk of adverse events across indications, for MACE, the exposure-adjusted event rates for upadacitinib were less than 0.1 in patients treated for AD over the observation period versus 0.3 and 0.4 for RA and psoriatic arthritis, respectively. Similarly, for venous thromboembolism, the rates for upadacitinib were again less than 0.1 in patients with AD versus 0.4 and 0.2 in RA and psoriatic arthritis, respectively.
Referring back to the postmarketing study, Dr. King emphasized that it is essential to consider how the boxed warning for JAK inhibitors was generated before applying them to dermatologic indications.
“Is a 30-year-old patient with a dermatologic disorder possibly at the same risk as the patients in the study from which we got the boxed warning? The answer is simply no,” he said.
Like the tofacitinib data in the ORAL Surveillance study, the upadacitinib clinical trial data are not necessarily relevant to other JAK inhibitors. In fact, Dr. King pointed out that the safety profiles of the available JAK inhibitors are not identical, an observation that is consistent with differences in JAK inhibitor selectivity that has implications for off-target events.
Dr. King does not dismiss the potential risks outlined in the current regulatory cautions about the use of JAK inhibitors, but he believes that dermatologists should be cognizant of “where the black box warning comes from.”
“We need to think carefully about the risk-to-benefit ratio in older patients or patients with risk factors, such as obesity and diabetes,” he said. But the safety profile of JAK inhibitors “is almost certainly better” than the profile suggested in boxed warnings applied to JAK inhibitors for dermatologic indications, he advised.
Risk-benefit considerations in dermatology
This position was supported by numerous other experts when asked for their perspectives. “I fully agree,” said Emma Guttman-Yassky, MD, PhD, system chair of dermatology and immunology, Icahn School of Medicine, Mount Sinai, New York.
Like Dr. King, Dr. Guttman-Yassky did not dismiss the potential risks of JAK inhibitors when treating dermatologic diseases.
“While JAK inhibitors need monitoring as advised, adopting a boxed warning from an RA study for patients who are older [is problematic],” she commented. A study with the nonselective tofacitinib in this population “cannot be compared to more selective inhibitors in a much younger population, such as those treated [for] alopecia areata or atopic dermatitis.”
George Z. Han, MD, PhD, an associate professor of dermatology, Zucker School of Medicine, Hofstra, Northwell Medical Center, New Hyde Park, New York, also agreed but added some caveats.
“The comments about the ORAL Surveillance study are salient,” he said in an interview. “This kind of data should not directly be extrapolated to other patient types or to other medications.” However, one of Dr. Han’s most important caveats involves long-term use.
“JAK inhibitors are still relatively narrow-therapeutic-window drugs that in a dose-dependent fashion could lead to negative effects, including thromboembolic events, abnormalities in red blood cells, white blood cells, platelets, and lipids,” he said. While doses used in dermatology “are generally below the level of any major concern,” Dr. Han cautioned that “we lack definitive data” on long-term use, and this is important for understanding “any potential small risk of rare events, such as malignancy or thromboembolism.”
Saakshi Khattri, MD, a colleague of Dr. Guttman-Yassky at Mount Sinai, said the risks of JAK inhibitors should not be underestimated, but she also agreed that risk “needs to be delivered in the right context.” Dr. Khattri, who is board certified in both dermatology and rheumatology, noted the safety profiles of available JAK inhibitors differ and that extrapolating safety from an RA study to dermatologic indications does not make sense. “Different diseases, different age groups,” she said.
Dr. King has reported financial relationships with more than 15 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Han reports financial relationships with Amgen, Athenex, Boehringer Ingelheim, Bond Avillion, Bristol-Myers Squibb, Celgene, Janssen, Lilly, Novartis, PellePharm, Pfizer, and UCB. Dr. Khattri has reported financial relationships with AbbVie, Arcutis, Bristol-Myers Squibb, Janssen, Leo, Lilly, Novartis, Pfizer, and UCB.
A version of this article originally appeared on Medscape.com.
AT AAD 2023
Lanolin gets nod for Allergen of the Year
Lanolin is a complex and varying mixture of high molecular weight esters, aliphatic alcohols, sterols, fatty acids, and hydrocarbons, but the allergic components are mainly the free lanolin alcohols, especially alkanediols, said Donald V. Belsito, MD, professor of dermatology, Columbia University, New York, who announced the Allergen of the Year at the society’s annual meeting.
Criteria for selection can include a known allergen with a new twist or increasing frequency or a newly reported allergen with mini-epidemics that may have been missed for years, Dr. Belsito said.
“The prevalence and severity of allergy to ‘lanolin’ have been hotly debated” since a potential case was first reported in the 1920s, wrote Dr. Belsito and Blair A. Jenkins, MD, PhD, a dermatology resident at New York–Presbyterian Hospital, Columbia Campus, in a review published in Dermatitis.
“ ‘Lanolin’ is indeed a paradox allergen,” wrote Dr. Jenkins and Dr. Belsito. “The most appropriate patch test preparation(s) for detecting allergy remain disputed. Detection of lanolin-induced contact dermatitis in diseased skin by patch testing on normal skin may lead to false negative results.”
And those who test positive for a lanolin allergy on diseased skin may be able to use lanolin products on normal skin, they wrote.
“From my perspective, this was a timely year to think about lanolin, as there is significant ongoing controversy about whether it is allergenic,” Dr. Jenkins said in an interview. “Numerous companies market lanolin-containing topicals as safe and effective emollients,” she said.
Medical grade and highly purified anhydrous lanolin, which contain less than 2.5% and less than 1.5% of free alcohols, respectively, can still elicit or induce a contact allergy, Dr. Belsito said in his presentation. Hydrogenated lanolin has shown more allergenicity than lanolin alcohol, while lanolin wax, lanolin acid, and lanolin esters possess lower allergenicity than lanolin alcohol, he said.
Notably, modern wool textiles do not contain lanolin, and lanolin-allergic patients need not avoid wool, Dr. Belsito added.
Amerchol L-101, a common trade name on products containing lanolin, contains 10% wool wax alcohols obtained from the hydrolysis of wool fat dissolved in mineral oil at a 1:1 ratio, said Dr. Belsito. He recommended testing lanolin alcohols (in 30% petrolatum) and Amerchol L-101 (in 50% petrolatum) simultaneously with or without other lanolin derivatives and/or the patient’s products in cases of possible allergy, he said.
Consider high-risk groups
Current evidence suggests that the prevalence of contact allergy in the western European population is 0.4%, wrote Dr. Jenkins and Dr. Belsito.
Although the frequency of lanolin allergy is relatively low, certain conditions convey greater risk, such as stasis dermatitis, leg ulcers, perianal/genital dermatitis, and atopic dermatitis, they wrote. Older adults and children are at increased risk because they are more likely to have these conditions. Demographic data also suggest that lanolin allergy is more common in non-Hispanic Whites than in non-Hispanic Blacks, they wrote.
Looking ahead, “I think further exploration of allergy across different skin types and ethnicities is warranted,” Dr. Jenkins said. “Further investigation of ideal [lanolin] allergens for patch testing is also needed.”
Dr. Jenkins and Dr. Belsito said they had no relevant financial conflicts to disclose.
Lanolin is a complex and varying mixture of high molecular weight esters, aliphatic alcohols, sterols, fatty acids, and hydrocarbons, but the allergic components are mainly the free lanolin alcohols, especially alkanediols, said Donald V. Belsito, MD, professor of dermatology, Columbia University, New York, who announced the Allergen of the Year at the society’s annual meeting.
Criteria for selection can include a known allergen with a new twist or increasing frequency or a newly reported allergen with mini-epidemics that may have been missed for years, Dr. Belsito said.
“The prevalence and severity of allergy to ‘lanolin’ have been hotly debated” since a potential case was first reported in the 1920s, wrote Dr. Belsito and Blair A. Jenkins, MD, PhD, a dermatology resident at New York–Presbyterian Hospital, Columbia Campus, in a review published in Dermatitis.
“ ‘Lanolin’ is indeed a paradox allergen,” wrote Dr. Jenkins and Dr. Belsito. “The most appropriate patch test preparation(s) for detecting allergy remain disputed. Detection of lanolin-induced contact dermatitis in diseased skin by patch testing on normal skin may lead to false negative results.”
And those who test positive for a lanolin allergy on diseased skin may be able to use lanolin products on normal skin, they wrote.
“From my perspective, this was a timely year to think about lanolin, as there is significant ongoing controversy about whether it is allergenic,” Dr. Jenkins said in an interview. “Numerous companies market lanolin-containing topicals as safe and effective emollients,” she said.
Medical grade and highly purified anhydrous lanolin, which contain less than 2.5% and less than 1.5% of free alcohols, respectively, can still elicit or induce a contact allergy, Dr. Belsito said in his presentation. Hydrogenated lanolin has shown more allergenicity than lanolin alcohol, while lanolin wax, lanolin acid, and lanolin esters possess lower allergenicity than lanolin alcohol, he said.
Notably, modern wool textiles do not contain lanolin, and lanolin-allergic patients need not avoid wool, Dr. Belsito added.
Amerchol L-101, a common trade name on products containing lanolin, contains 10% wool wax alcohols obtained from the hydrolysis of wool fat dissolved in mineral oil at a 1:1 ratio, said Dr. Belsito. He recommended testing lanolin alcohols (in 30% petrolatum) and Amerchol L-101 (in 50% petrolatum) simultaneously with or without other lanolin derivatives and/or the patient’s products in cases of possible allergy, he said.
Consider high-risk groups
Current evidence suggests that the prevalence of contact allergy in the western European population is 0.4%, wrote Dr. Jenkins and Dr. Belsito.
Although the frequency of lanolin allergy is relatively low, certain conditions convey greater risk, such as stasis dermatitis, leg ulcers, perianal/genital dermatitis, and atopic dermatitis, they wrote. Older adults and children are at increased risk because they are more likely to have these conditions. Demographic data also suggest that lanolin allergy is more common in non-Hispanic Whites than in non-Hispanic Blacks, they wrote.
Looking ahead, “I think further exploration of allergy across different skin types and ethnicities is warranted,” Dr. Jenkins said. “Further investigation of ideal [lanolin] allergens for patch testing is also needed.”
Dr. Jenkins and Dr. Belsito said they had no relevant financial conflicts to disclose.
Lanolin is a complex and varying mixture of high molecular weight esters, aliphatic alcohols, sterols, fatty acids, and hydrocarbons, but the allergic components are mainly the free lanolin alcohols, especially alkanediols, said Donald V. Belsito, MD, professor of dermatology, Columbia University, New York, who announced the Allergen of the Year at the society’s annual meeting.
Criteria for selection can include a known allergen with a new twist or increasing frequency or a newly reported allergen with mini-epidemics that may have been missed for years, Dr. Belsito said.
“The prevalence and severity of allergy to ‘lanolin’ have been hotly debated” since a potential case was first reported in the 1920s, wrote Dr. Belsito and Blair A. Jenkins, MD, PhD, a dermatology resident at New York–Presbyterian Hospital, Columbia Campus, in a review published in Dermatitis.
“ ‘Lanolin’ is indeed a paradox allergen,” wrote Dr. Jenkins and Dr. Belsito. “The most appropriate patch test preparation(s) for detecting allergy remain disputed. Detection of lanolin-induced contact dermatitis in diseased skin by patch testing on normal skin may lead to false negative results.”
And those who test positive for a lanolin allergy on diseased skin may be able to use lanolin products on normal skin, they wrote.
“From my perspective, this was a timely year to think about lanolin, as there is significant ongoing controversy about whether it is allergenic,” Dr. Jenkins said in an interview. “Numerous companies market lanolin-containing topicals as safe and effective emollients,” she said.
Medical grade and highly purified anhydrous lanolin, which contain less than 2.5% and less than 1.5% of free alcohols, respectively, can still elicit or induce a contact allergy, Dr. Belsito said in his presentation. Hydrogenated lanolin has shown more allergenicity than lanolin alcohol, while lanolin wax, lanolin acid, and lanolin esters possess lower allergenicity than lanolin alcohol, he said.
Notably, modern wool textiles do not contain lanolin, and lanolin-allergic patients need not avoid wool, Dr. Belsito added.
Amerchol L-101, a common trade name on products containing lanolin, contains 10% wool wax alcohols obtained from the hydrolysis of wool fat dissolved in mineral oil at a 1:1 ratio, said Dr. Belsito. He recommended testing lanolin alcohols (in 30% petrolatum) and Amerchol L-101 (in 50% petrolatum) simultaneously with or without other lanolin derivatives and/or the patient’s products in cases of possible allergy, he said.
Consider high-risk groups
Current evidence suggests that the prevalence of contact allergy in the western European population is 0.4%, wrote Dr. Jenkins and Dr. Belsito.
Although the frequency of lanolin allergy is relatively low, certain conditions convey greater risk, such as stasis dermatitis, leg ulcers, perianal/genital dermatitis, and atopic dermatitis, they wrote. Older adults and children are at increased risk because they are more likely to have these conditions. Demographic data also suggest that lanolin allergy is more common in non-Hispanic Whites than in non-Hispanic Blacks, they wrote.
Looking ahead, “I think further exploration of allergy across different skin types and ethnicities is warranted,” Dr. Jenkins said. “Further investigation of ideal [lanolin] allergens for patch testing is also needed.”
Dr. Jenkins and Dr. Belsito said they had no relevant financial conflicts to disclose.
FROM ACDS 2023
Study finds quality of topical steroid withdrawal videos on YouTube subpar
NEW ORLEANS –

“Video-sharing platforms such as YouTube are a great place for patients to connect and find community with others dealing with the same conditions,” senior author Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. “There is no doubt tremendous value in viewing the shared experience; however, it is important that medical advice be evidence based and validated. Seeking said advice from a medical professional such as a board-certified dermatologist will no doubt increase the likelihood that said guidance is supported by the literature and most importantly, will do no harm.”
Noting a trend of increased user-created content on social media and Internet sites about topical steroid withdrawal in recent years, Dr. Friedman, first author Erika McCormick, a fourth-year medical student at George Washington University, and colleagues used the keywords “topical steroid withdrawal” on YouTube to search for and analyze the top 10 most viewed videos on the subject.
Two independent reviewers used the modified DISCERN (mDISCERN) tool and the Global Quality Scale (GQS) to assess reliability and quality/scientific accuracy of videos, respectively. Average scores were generated for each video and the researchers used one way ANOVA, unpaired t-tests, and linear regression to analyze the ratings. For mDISCERN criteria, a point is given per each of five criteria for a possible score between 0 and 5. Examples of criteria included “Are the aims clear and achieved?” and “Is the information presented both balanced and unbiased”? For GQS, a score from 1 to 5 is designated based on criteria ranging from “poor quality, poor flow, most information missing” to “excellent quality and flow, very useful for patients.”
The researchers found that the mean combined mDISCERN score of the 10 videos was a 2, which indicates poor reliability and shortcomings. Similarly, the combined mean GQS score was 2.5, which suggests poor to moderate quality of videos, missing discussion of important topics, and limited use to patients. The researchers found no correlation between mDISCERN or GQS scores and length of video, duration on YouTube, or number of views, subscribers, or likes.

“We were disheartened that patient testimonial videos had the poorest quality and reliability of the information sources,” Ms. McCormick said in an interview. “Videos that included medical research and information from dermatologists had significantly higher quality and reliability scores than the remainder of videos.” Accurate information online is essential to help patients recognize topical steroid withdrawal and seek medical care, she continued.
Conversely, wide viewership of unreliable information “may contribute to fear of topical corticosteroids and dissuade use in patients with primary skin diseases that may benefit from this common treatment,” Dr. Friedman said. “Dermatologists must be aware of the content patients are consuming online, should guide patients in appraising quality and reliability of online resources, and must provide valid sources of additional information for their patients.” One such resource he recommended is the National Eczema Association, which has created online content for patients about topical steroid withdrawal.

Doris Day, MD, a New York–based dermatologist who was asked to comment on the study, said that many patients rely on YouTube as a go-to resource, with videos that can be watched at times of their choosing. “Oftentimes, the person on the video is relatable and has some general knowledge but is lacking the information that would be relevant and important for the individual patient,” said Dr. Day, who was not involved with the study. “The downside of this is that the person who takes that advice may not use the prescription properly or for the correct amount of time, which can lead to either undertreating or, even worse, overtreatment, which can have permanent consequences.”
One possible solution is for more doctors to create videos for YouTube, she added, “but that doesn’t guarantee that those would be the ones patients would choose to watch.” Another solution “is to have YouTube add qualifiers indicating that the information being discussed is not medical,” she suggested. “Ideally, patients will get all the information they need while they are in the office and also have clear written instructions and even a video they can review at a later time, made by the office, to help them feel they are getting personalized care and the attention they need.”
Ms. McCormick’s research is funded by a grant from Galderma. Dr. Friedman and Dr. Day had no relevant disclosures to report.
NEW ORLEANS –
“Video-sharing platforms such as YouTube are a great place for patients to connect and find community with others dealing with the same conditions,” senior author Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. “There is no doubt tremendous value in viewing the shared experience; however, it is important that medical advice be evidence based and validated. Seeking said advice from a medical professional such as a board-certified dermatologist will no doubt increase the likelihood that said guidance is supported by the literature and most importantly, will do no harm.”
Noting a trend of increased user-created content on social media and Internet sites about topical steroid withdrawal in recent years, Dr. Friedman, first author Erika McCormick, a fourth-year medical student at George Washington University, and colleagues used the keywords “topical steroid withdrawal” on YouTube to search for and analyze the top 10 most viewed videos on the subject.
Two independent reviewers used the modified DISCERN (mDISCERN) tool and the Global Quality Scale (GQS) to assess reliability and quality/scientific accuracy of videos, respectively. Average scores were generated for each video and the researchers used one way ANOVA, unpaired t-tests, and linear regression to analyze the ratings. For mDISCERN criteria, a point is given per each of five criteria for a possible score between 0 and 5. Examples of criteria included “Are the aims clear and achieved?” and “Is the information presented both balanced and unbiased”? For GQS, a score from 1 to 5 is designated based on criteria ranging from “poor quality, poor flow, most information missing” to “excellent quality and flow, very useful for patients.”
The researchers found that the mean combined mDISCERN score of the 10 videos was a 2, which indicates poor reliability and shortcomings. Similarly, the combined mean GQS score was 2.5, which suggests poor to moderate quality of videos, missing discussion of important topics, and limited use to patients. The researchers found no correlation between mDISCERN or GQS scores and length of video, duration on YouTube, or number of views, subscribers, or likes.
“We were disheartened that patient testimonial videos had the poorest quality and reliability of the information sources,” Ms. McCormick said in an interview. “Videos that included medical research and information from dermatologists had significantly higher quality and reliability scores than the remainder of videos.” Accurate information online is essential to help patients recognize topical steroid withdrawal and seek medical care, she continued.
Conversely, wide viewership of unreliable information “may contribute to fear of topical corticosteroids and dissuade use in patients with primary skin diseases that may benefit from this common treatment,” Dr. Friedman said. “Dermatologists must be aware of the content patients are consuming online, should guide patients in appraising quality and reliability of online resources, and must provide valid sources of additional information for their patients.” One such resource he recommended is the National Eczema Association, which has created online content for patients about topical steroid withdrawal.
Doris Day, MD, a New York–based dermatologist who was asked to comment on the study, said that many patients rely on YouTube as a go-to resource, with videos that can be watched at times of their choosing. “Oftentimes, the person on the video is relatable and has some general knowledge but is lacking the information that would be relevant and important for the individual patient,” said Dr. Day, who was not involved with the study. “The downside of this is that the person who takes that advice may not use the prescription properly or for the correct amount of time, which can lead to either undertreating or, even worse, overtreatment, which can have permanent consequences.”
One possible solution is for more doctors to create videos for YouTube, she added, “but that doesn’t guarantee that those would be the ones patients would choose to watch.” Another solution “is to have YouTube add qualifiers indicating that the information being discussed is not medical,” she suggested. “Ideally, patients will get all the information they need while they are in the office and also have clear written instructions and even a video they can review at a later time, made by the office, to help them feel they are getting personalized care and the attention they need.”
Ms. McCormick’s research is funded by a grant from Galderma. Dr. Friedman and Dr. Day had no relevant disclosures to report.
NEW ORLEANS –
“Video-sharing platforms such as YouTube are a great place for patients to connect and find community with others dealing with the same conditions,” senior author Adam Friedman, MD, professor and chair of dermatology at George Washington University, Washington, said in an interview in advance of the annual meeting of the American Academy of Dermatology, where the study was presented during an e-poster session. “There is no doubt tremendous value in viewing the shared experience; however, it is important that medical advice be evidence based and validated. Seeking said advice from a medical professional such as a board-certified dermatologist will no doubt increase the likelihood that said guidance is supported by the literature and most importantly, will do no harm.”
Noting a trend of increased user-created content on social media and Internet sites about topical steroid withdrawal in recent years, Dr. Friedman, first author Erika McCormick, a fourth-year medical student at George Washington University, and colleagues used the keywords “topical steroid withdrawal” on YouTube to search for and analyze the top 10 most viewed videos on the subject.
Two independent reviewers used the modified DISCERN (mDISCERN) tool and the Global Quality Scale (GQS) to assess reliability and quality/scientific accuracy of videos, respectively. Average scores were generated for each video and the researchers used one way ANOVA, unpaired t-tests, and linear regression to analyze the ratings. For mDISCERN criteria, a point is given per each of five criteria for a possible score between 0 and 5. Examples of criteria included “Are the aims clear and achieved?” and “Is the information presented both balanced and unbiased”? For GQS, a score from 1 to 5 is designated based on criteria ranging from “poor quality, poor flow, most information missing” to “excellent quality and flow, very useful for patients.”
The researchers found that the mean combined mDISCERN score of the 10 videos was a 2, which indicates poor reliability and shortcomings. Similarly, the combined mean GQS score was 2.5, which suggests poor to moderate quality of videos, missing discussion of important topics, and limited use to patients. The researchers found no correlation between mDISCERN or GQS scores and length of video, duration on YouTube, or number of views, subscribers, or likes.
“We were disheartened that patient testimonial videos had the poorest quality and reliability of the information sources,” Ms. McCormick said in an interview. “Videos that included medical research and information from dermatologists had significantly higher quality and reliability scores than the remainder of videos.” Accurate information online is essential to help patients recognize topical steroid withdrawal and seek medical care, she continued.
Conversely, wide viewership of unreliable information “may contribute to fear of topical corticosteroids and dissuade use in patients with primary skin diseases that may benefit from this common treatment,” Dr. Friedman said. “Dermatologists must be aware of the content patients are consuming online, should guide patients in appraising quality and reliability of online resources, and must provide valid sources of additional information for their patients.” One such resource he recommended is the National Eczema Association, which has created online content for patients about topical steroid withdrawal.
Doris Day, MD, a New York–based dermatologist who was asked to comment on the study, said that many patients rely on YouTube as a go-to resource, with videos that can be watched at times of their choosing. “Oftentimes, the person on the video is relatable and has some general knowledge but is lacking the information that would be relevant and important for the individual patient,” said Dr. Day, who was not involved with the study. “The downside of this is that the person who takes that advice may not use the prescription properly or for the correct amount of time, which can lead to either undertreating or, even worse, overtreatment, which can have permanent consequences.”
One possible solution is for more doctors to create videos for YouTube, she added, “but that doesn’t guarantee that those would be the ones patients would choose to watch.” Another solution “is to have YouTube add qualifiers indicating that the information being discussed is not medical,” she suggested. “Ideally, patients will get all the information they need while they are in the office and also have clear written instructions and even a video they can review at a later time, made by the office, to help them feel they are getting personalized care and the attention they need.”
Ms. McCormick’s research is funded by a grant from Galderma. Dr. Friedman and Dr. Day had no relevant disclosures to report.
AT AAD 2023
Upadacitinib shows positive endoscopic outcomes in Crohn’s disease at 1 year
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The findings of this subanalysis come from two phase 3 induction trials (U-EXCEL and U-EXCEED) and one maintenance study (U-ENDURE) of upadacitinib in this patient population.
“Upadacitinib shows large differences relative to placebo in endoscopic response and remission ... in a difficult-to-treat population of patients, the majority of whom had failed an advanced therapy,” lead investigator Brian Feagan, MD, senior scientific director of the GI contract research firm Alimentiv in London, Ontario, said in an interview.
“The absolute magnitude of the finding was unanticipated – a greater treatment effect than might be anticipated for these outcomes compared with other advanced treatments for Crohn’s disease in these higher-risk patients,” he said.
Dr. Feagan presented the research at the annual congress of the European Crohn’s and Colitis Organisation.
Research methodology
At baseline, participants had an average daily stool frequency of 4 or more and/or an abdominal pain score of 2 or greater. They also had a Simple Endoscopic Score for Crohn’s disease of 6 or more, excluding a narrowing component, or a score of 4 or more for isolated ileal Crohn’s disease.
In the treatment induction phase, patients were randomly assigned 2:1, with 674 people receiving 45 mg upadacitinib and 347 taking a placebo once daily for 12 weeks.
Participants who experienced at least a 30% decrease in stool frequency and/or daily abdominal pain scores were enrolled in the maintenance phase of the study. For this phase, patients were randomly assigned again, with 168 receiving 30 mg upadacitinib, 169 receiving 15 mg upadacitinib, and 165 taking a placebo once daily for 52 weeks.
In each induction and maintenance cohort, more than 70% of patients had failed one prior biologic therapy, with failure defined as inadequate response or intolerance. Among those who failed a previous biologic in induction, 96% had also failed prior treatment with an anti–tumor necrosis factor (anti-TNF) inhibitor.
Participants’ mean age was 38-40 years, and 52%-55% were men. Patients who had not failed previous therapy had Crohn’s disease for a median of 6-7 years. In contrast, the prior-failure group had Crohn’s disease for a median of 9-10 years.
Key outcomes
At 12 weeks, endoscopic response among patients who had not failed a prior biologic was 52% in the treatment group versus 16% of the placebo group. In the prior-failure group, endoscopic response was observed in 36% and 5%, respectively.
Endoscopic remission at 12 weeks among patients who had not failed a prior biologic was 36% in the treatment group versus 10% in the placebo group. In the prior-failure group, endoscopic remission was 20% in the treatment group versus 3% in those who took placebo.
Participants in the treatment groups of the 52-week maintenance phase of the study experienced higher endoscopic response and endoscopic remission rates compared with those who received placebo.
Endoscopic response in the group without prior biologic failure was 44% in the 30-mg upadacitinib group, 40% in the 15-mg group, and 18% in the placebo group. Among those with prior biologic failure, endoscopic response was seen in 39% of the 30-mg upadacitinib group, 23% of the 15-mg group, and 4% of the placebo group.
There is a “very striking difference in endoscopic response rates between the high dose and placebo,” Dr. Feagan said. “That difference here is in the response rate. You see dose separation.”
Endoscopic remission among those without prior biologic failure was observed in 34% of the 30-mg upadacitinib group, 27% of the 15-mg group, and 16% of the placebo group. Among those with prior biologic failure, endoscopic remission was seen in 27% of the 30-mg upadacitinib group, 16% of the 15-mg group, and 2% of the placebo group.
The results show “a clear advantage for the 30-mg dose versus the 15-mg in the maintenance component, especially in patients who had failed an advanced therapy,” Dr. Feagan said.
Safety signals
Upadacitinib was well tolerated in the induction and maintenance phases, and no new safety risks were observed compared with the known safety profile of the drug, the researchers noted.
For example, during the induction studies, the rate of any adverse event among patients without prior biologic failure was 60% in the 45-mg upadacitinib group and 53% in the placebo group. Among those who failed a prior biologic, the rates were 67% in the 45-mg upadacitinib group and 66% in the placebo group.
The adverse events were “issues that have already been identified with JAK inhibitors, the biochemical abnormalities with CPK [creatine phosphokinase] elevations and transaminase elevations,” Dr. Feagan said.
There were no cases of herpes zoster among patients who received placebo compared with five cases in the 45-mg upadacitinib group without prior biologic failure and 10 cases in the prior biologic failure group.
“The zoster signal is there even at induction with the 45-mg dose versus placebo,” Dr. Feagan said.
‘Encouraging’ results
The study indicates that upadacitinib is effective in improving endoscopic outcomes for patients with Crohn’s disease, regardless of their prior biologic treatments, Robin L. Dalal, MD, assistant professor of medicine at Vanderbilt University in Nashville, Tenn., said when asked to comment on the study.
“This is important because, as the treatment landscape for Crohn’s disease has expanded, sequencing of therapies has become more complex,” added Dr. Dalal, who was not involved in the research. “For upadacitinib in Crohn’s disease, prior biologic use may not be a factor in endoscopic response rates.”
The findings are “very encouraging for physicians and practitioners who treat IBD [inflammatory bowel disease] patients,” Maithili Chitnavis, MD, of the inflammatory bowel disease section at Atrium Health Gastroenterology in Charlotte, N.C., said when asked for comment.
“We clearly care about how patients feel overall, but endoscopic and histologic outcomes are important to investigate because we want to ensure there is internal healing to prevent a lot of the longstanding complications of Crohn’s disease, such as malignancy, strictures, fistulizing/penetrating disease, and need for surgery,” said Dr. Chitnavis, who was not involved with the study.
Upadacitinib is an oral agent, which distinguishes it from the injectable or infusion-based biologic therapies for Crohn’s disease, Dr. Chitnavis noted.
The finding that the medication works in patients with or without prior biologic failure is important, she said.
“With its anticipated ... approval for Crohn’s disease [by the Food and Drug Administration], it is expected that patients will have had to have demonstrated a lack of or loss of response to another biologic, specifically in the anti-TNF category (for example, infliximab, adalimumab, certolizumab) prior to starting upadacitinib due to concerns of potential side effects associated with the class of medications to which it belongs,” Dr. Chitnavis said. “Therefore, it makes it even more relevant to know how patients who have failed a prior biologic respond to this therapy.”
Dr. Feagan has reported serving as a consultant and speaker for AbbVie. Dr. Dalal has reported being a consultant for AbbVie in 2021. Dr. Chitnavis has reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM ECCO 2023
Oral PCSK9 inhibitor shows encouraging LDL lowering
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.

“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.

“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.
“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
A new oral formulation of a PCSK9-inhibiting, cholesterol-lowering drug in development by Merck has shown encouraging results in a phase 2 study.
The study was presented by Christie Ballantyne, MD, Baylor College of Medicine, Houston, at the joint scientific sessions of the American College of Cardiology and the World Heart Federation.
“In this diverse population of hypercholesterolemic patients, all doses of MK-0616 showed superior reduction of LDL vs. placebo up to a 60.9% placebo-adjusted reduction from baseline to week 8, which was consistent across subgroups,” Dr. Ballantyne reported.
“Reduction in ApoB and non-HDL cholesterol were consistent with that of LDL cholesterol, with up to a 51.8% reduction in ApoB and a 55.8% reduction in non-HDL,” he noted.
He added that the drug was well tolerated with no difference in adverse events across the treatment groups, compared with placebo.
“These data support the further development of MK-0616, an oral PCSK9 inhibitor that may improve access to effective LDL-cholesterol lowering therapies and improve attainment of guideline-recommended LDL goals aimed at reducing cardiovascular risk,” Dr. Ballantyne concluded. “The results are encouraging for a phase 3 program that is now being designed.”
He explained that elevated LDL is a primary causative factor for atherosclerotic cardiovascular disease (ASCVD), and despite effective treatments (statins), a large proportion of patients fail to achieve guideline-recommended LDL levels. Injectable treatments targeting PCSK9 have demonstrated large reductions in LDL and decreased risk of ASCVD events, but access barriers and need for repeat injections have led to poor adoption. An oral PCSK9 inhibitor may widen access and improve attainment of guideline-recommended treatment goals.
Dr. Ballantyne described the new drug, MK-0616, as a “macrocyclic peptide that can bind PCSK9 with monoclonal antibody-like affinity at 1/100th of the molecular weight.”
The current phase 2 study included 381 adult patients (49% female; median age 62 years) with a wide range of ASCVD risk. Average LDL-C level was 119.5 mg/dL at baseline. Around 40% of patients were not taking statins, 35% were on low- to moderate-intensity statin therapy, and 26% were on high-intensity statin therapy.
They were randomly assigned to four different doses of MK-0616 (6, 12, 18, or 30 mg once daily) or matching placebo.
Results showed that all doses of MK-0616 demonstrated statistically significant differences in percentage change in LDL-C from baseline to week 8 vs. placebo: –41.2% (6 mg), –55.7% (12 mg), –59.1% (18 mg), and –60.9% (30 mg).
The mean percentage changes in ApoB from baseline vs. placebo were –32.8%, –45.8%, –48.7%, and 51.8% for the four escalating doses of the drug. And non-HDL cholesterol changes were –35.9%, –50.5%, –53.2%, and –55.8% respectively.
The proportion of participants at protocol-defined goals for LDL reduction was 80.5%, 85.5%, 90.8%, and 90.8% with MK-0616 at the 6-mg, 12-mg, 18-mg, and 30-mg doses, compared with 9.3% with placebo.
Dr. Ballantyne reported that the efficacy looked similar in all subgroups, and regardless of baseline therapy.
“This was a dose-finding study, which will help select a dose to be taken forward in larger studies, and it looks from these results as though you get most of the efficacy by 12 mg,” he added.
Adverse events occurred in a proportion of participants in the MK-0616 groups (39.5% to 43.4%) similar to that of placebo (44.0%), and discontinuations as a result of adverse events occurred in two or fewer participants in any treatment group.
‘Super exciting’
Putting the results of his study into perspective at an ACC press conference, Rhonda Cooper-DeHoff, PharmD, associate professor in the department of pharmacotherapy and translational research at the University of Florida in Gainesville, commented.
“For the last quarter of a century we have had statins available to treat elevated LDL and atherosclerosis and despite that we have many patients who refuse to take statins or are afraid to take statins,” she said. “This is not about cost as the statins are all available generically now. But many patients claim to be intolerant or unresponsive.”
She noted that in 2015/2016 the first injectable PCSK9 inhibitors became available “which really were very exciting molecules, but they have a high cost and access issues, and patients often do not like injections so there are still a lot of issues.”
Dr. Cooper-DeHoff pointed out that this oral PCSK9 inhibitor seems to be as effective at lowering LDL as the injectable products regardless of whether statins are on board or not, which she said was “super exciting.”
She added: “We are all going to be waiting excitedly for the outcome data with this oral PCSK9 inhibitor.”
She also noted that another study (CLEAR Outcomes) presented at the ACC meeting showed good lipid-lowering results and a reduction in cardiovascular outcomes in statin-intolerant patients with another oral lipid lowering drug, bempedoic acid (Nexletol).
She said the two oral drugs promised a “very bright for the future for LDL lowering and the treatment of atherosclerosis in our patients,” adding that “we are now really chipping away at the barriers to achieving the holy grail of guideline-directed LDL lowering to prevent hard outcomes.”
The results were published online in the Journal of the American College of Cardiology at the time of presentation.
This study was funded by Merck. Dr. Ballantyne has received grant/research support through his institution from Abbott Diagnostic, Akcea, Amgen, Arrowhead, Esperion, Ionis, Merck, New Amsterdam, Novartis, Novo Nordisk, Regeneron, and Roche Diagnostics and has been a consultant for 89Bio, Abbott Diagnostics, Alnylam Pharmaceuticals, Althera, Amarin, Amgen, Arrowhead, AstraZeneca, Denka Seiken, Esperion, Genentech, Gilead, Illumina, Ionis, Matinas BioPharma, Merck, New Amsterdam, Novartis, Novo Nordisk, Pfizer, Regeneron, and Roche Diagnostics.
A version of this article first appeared on Medscape.com.
FROM ACC 2023
Spironolactone: an ‘inexpensive, effective’ option for acne in women
HONOLULU – In the clinical experience of Julie C. Harper, MD, an increasing number of women with acne are turning to off-label, long-term treatment with spironolactone.
“Spironolactone is fairly accessible, inexpensive, and effective for our patients,” Dr. Harper, a dermatologist who practices in Birmingham, Ala., said at the Hawaii Dermatology Seminar provided by MedscapeLIVE!
An aldosterone receptor antagonist commonly used to treat high blood pressure and heart failure, spironolactone also has antiandrogenic properties with a proven track record for treating acne and hirsutism. It reduces androgen production, inhibits 5-alpha reductase, and increases sex hormone binding globulin. The dosing range for treating acne is 25 mg to 200 mg per day, but Dr. Harper prefers a maximum dose of 100 mg per day.
According to a systematic review of its use for acne in adult women, the most common side effect is menstrual irregularity, while other common side effects include breast tenderness/swelling, fatigue, and headaches.
“The higher the dose, the higher the rate of side effects,” she said. Concomitant use of an oral contraceptive lessens menstrual irregularities and prevents pregnancies, to avoid exposure during pregnancy and the hypothetical risk of feminization of the male fetus with exposure late in the first trimester. “Early in my career, I used to say if you’re going to be on spironolactone you’re also going to be on an oral contraceptive. But the longer I’ve practiced, I’ve learned that women who have a contraindication to birth control pills or who don’t want to take it can still benefit from an oral antiandrogen by being on spironolactone.”
A large retrospective analysis of 14-year data concluded that routine potassium monitoring is unnecessary for healthy women taking spironolactone for acne. “If you’re between the ages of 18 and 45, healthy, and not taking other medications where I’m worried about potassium levels, I’m not checking those levels at all,” Dr. Harper said.
Spironolactone labeling includes a boxed warning regarding the potential for tumorigenicity based on rat studies, but the dosages used in those studies were 25-250 times higher than the exposure dose in humans, Dr. Harper said.
Results from a systematic review and meta-analysis of seven studies in the medical literature found no evidence of an increased risk of breast cancer in women with exposure to spironolactone. “However, the certainty of the evidence was low and future studies are needed, including among diverse populations such as younger individuals and those with acne or hirsutism,” the study authors wrote.
In a separate study, researchers drew from patients in the Humana Insurance database from 2005 to 2017 to address whether spironolactone is associated with an increased risk of recurrence of breast cancer. Recurrent breast cancer was examined in 29,146 women with continuous health insurance for 2 years after a diagnosis of breast cancer. Of these, 746 were prescribed spironolactone, and the remainder were not. The researchers found that 123 women (16.5%) who were prescribed spironolactone had a breast cancer recurrence, compared with 3,649 women (12.8%) with a breast cancer recurrence who had not been prescribed spironolactone (P = .004). Adjusted Cox regression analysis following propensity matching showed no association between spironolactone and increased breast cancer recurrence (adjusted hazard ratio, 0.966; P = .953).
According to Dr. Harper, spironolactone may take about 3 months to kick in. “Likely this is a long-term treatment, and most of the time we’re going to be using it in combination with other acne treatments such as topical retinoids or topical benzoyl peroxide, oral antibiotics, or even isotretinoin.”
A study of long-term spironolactone use in 403 women found that the most common dose prescribed was 100 mg/day, and 68% of the women were concurrently prescribed a topical retinoid, 2.2% an oral antibiotic, and 40.7% an oral contraceptive.
The study population included 32 patients with a history of polycystic ovarian syndrome, 1 with a history of breast cancer, and 5 were hypercoagulable. Patients took the drug for a mean of 471 days. “As opposed to our antibiotics, where the course for patients is generally 3-4 months, when you start someone on spironolactone, they may end up staying on it,” Dr. Harper said.
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, Cassiopeia, Cutera, EPI, Galderma, L’Oreal, Ortho Dermatologics, Sol Gel, and Vyne. She also serves as a speaker or member of a speaker’s bureau for Almirall, Cassiopeia, Cutera, EPI, Galderma, Journey Almirall, L’Oreal, Ortho Dermatologics, Sun Pharmaceutical Industries, and Vyne.
Medscape and this news organization are owned by the same parent company.
HONOLULU – In the clinical experience of Julie C. Harper, MD, an increasing number of women with acne are turning to off-label, long-term treatment with spironolactone.
“Spironolactone is fairly accessible, inexpensive, and effective for our patients,” Dr. Harper, a dermatologist who practices in Birmingham, Ala., said at the Hawaii Dermatology Seminar provided by MedscapeLIVE!
An aldosterone receptor antagonist commonly used to treat high blood pressure and heart failure, spironolactone also has antiandrogenic properties with a proven track record for treating acne and hirsutism. It reduces androgen production, inhibits 5-alpha reductase, and increases sex hormone binding globulin. The dosing range for treating acne is 25 mg to 200 mg per day, but Dr. Harper prefers a maximum dose of 100 mg per day.
According to a systematic review of its use for acne in adult women, the most common side effect is menstrual irregularity, while other common side effects include breast tenderness/swelling, fatigue, and headaches.
“The higher the dose, the higher the rate of side effects,” she said. Concomitant use of an oral contraceptive lessens menstrual irregularities and prevents pregnancies, to avoid exposure during pregnancy and the hypothetical risk of feminization of the male fetus with exposure late in the first trimester. “Early in my career, I used to say if you’re going to be on spironolactone you’re also going to be on an oral contraceptive. But the longer I’ve practiced, I’ve learned that women who have a contraindication to birth control pills or who don’t want to take it can still benefit from an oral antiandrogen by being on spironolactone.”
A large retrospective analysis of 14-year data concluded that routine potassium monitoring is unnecessary for healthy women taking spironolactone for acne. “If you’re between the ages of 18 and 45, healthy, and not taking other medications where I’m worried about potassium levels, I’m not checking those levels at all,” Dr. Harper said.
Spironolactone labeling includes a boxed warning regarding the potential for tumorigenicity based on rat studies, but the dosages used in those studies were 25-250 times higher than the exposure dose in humans, Dr. Harper said.
Results from a systematic review and meta-analysis of seven studies in the medical literature found no evidence of an increased risk of breast cancer in women with exposure to spironolactone. “However, the certainty of the evidence was low and future studies are needed, including among diverse populations such as younger individuals and those with acne or hirsutism,” the study authors wrote.
In a separate study, researchers drew from patients in the Humana Insurance database from 2005 to 2017 to address whether spironolactone is associated with an increased risk of recurrence of breast cancer. Recurrent breast cancer was examined in 29,146 women with continuous health insurance for 2 years after a diagnosis of breast cancer. Of these, 746 were prescribed spironolactone, and the remainder were not. The researchers found that 123 women (16.5%) who were prescribed spironolactone had a breast cancer recurrence, compared with 3,649 women (12.8%) with a breast cancer recurrence who had not been prescribed spironolactone (P = .004). Adjusted Cox regression analysis following propensity matching showed no association between spironolactone and increased breast cancer recurrence (adjusted hazard ratio, 0.966; P = .953).
According to Dr. Harper, spironolactone may take about 3 months to kick in. “Likely this is a long-term treatment, and most of the time we’re going to be using it in combination with other acne treatments such as topical retinoids or topical benzoyl peroxide, oral antibiotics, or even isotretinoin.”
A study of long-term spironolactone use in 403 women found that the most common dose prescribed was 100 mg/day, and 68% of the women were concurrently prescribed a topical retinoid, 2.2% an oral antibiotic, and 40.7% an oral contraceptive.
The study population included 32 patients with a history of polycystic ovarian syndrome, 1 with a history of breast cancer, and 5 were hypercoagulable. Patients took the drug for a mean of 471 days. “As opposed to our antibiotics, where the course for patients is generally 3-4 months, when you start someone on spironolactone, they may end up staying on it,” Dr. Harper said.
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, Cassiopeia, Cutera, EPI, Galderma, L’Oreal, Ortho Dermatologics, Sol Gel, and Vyne. She also serves as a speaker or member of a speaker’s bureau for Almirall, Cassiopeia, Cutera, EPI, Galderma, Journey Almirall, L’Oreal, Ortho Dermatologics, Sun Pharmaceutical Industries, and Vyne.
Medscape and this news organization are owned by the same parent company.
HONOLULU – In the clinical experience of Julie C. Harper, MD, an increasing number of women with acne are turning to off-label, long-term treatment with spironolactone.
“Spironolactone is fairly accessible, inexpensive, and effective for our patients,” Dr. Harper, a dermatologist who practices in Birmingham, Ala., said at the Hawaii Dermatology Seminar provided by MedscapeLIVE!
An aldosterone receptor antagonist commonly used to treat high blood pressure and heart failure, spironolactone also has antiandrogenic properties with a proven track record for treating acne and hirsutism. It reduces androgen production, inhibits 5-alpha reductase, and increases sex hormone binding globulin. The dosing range for treating acne is 25 mg to 200 mg per day, but Dr. Harper prefers a maximum dose of 100 mg per day.
According to a systematic review of its use for acne in adult women, the most common side effect is menstrual irregularity, while other common side effects include breast tenderness/swelling, fatigue, and headaches.
“The higher the dose, the higher the rate of side effects,” she said. Concomitant use of an oral contraceptive lessens menstrual irregularities and prevents pregnancies, to avoid exposure during pregnancy and the hypothetical risk of feminization of the male fetus with exposure late in the first trimester. “Early in my career, I used to say if you’re going to be on spironolactone you’re also going to be on an oral contraceptive. But the longer I’ve practiced, I’ve learned that women who have a contraindication to birth control pills or who don’t want to take it can still benefit from an oral antiandrogen by being on spironolactone.”
A large retrospective analysis of 14-year data concluded that routine potassium monitoring is unnecessary for healthy women taking spironolactone for acne. “If you’re between the ages of 18 and 45, healthy, and not taking other medications where I’m worried about potassium levels, I’m not checking those levels at all,” Dr. Harper said.
Spironolactone labeling includes a boxed warning regarding the potential for tumorigenicity based on rat studies, but the dosages used in those studies were 25-250 times higher than the exposure dose in humans, Dr. Harper said.
Results from a systematic review and meta-analysis of seven studies in the medical literature found no evidence of an increased risk of breast cancer in women with exposure to spironolactone. “However, the certainty of the evidence was low and future studies are needed, including among diverse populations such as younger individuals and those with acne or hirsutism,” the study authors wrote.
In a separate study, researchers drew from patients in the Humana Insurance database from 2005 to 2017 to address whether spironolactone is associated with an increased risk of recurrence of breast cancer. Recurrent breast cancer was examined in 29,146 women with continuous health insurance for 2 years after a diagnosis of breast cancer. Of these, 746 were prescribed spironolactone, and the remainder were not. The researchers found that 123 women (16.5%) who were prescribed spironolactone had a breast cancer recurrence, compared with 3,649 women (12.8%) with a breast cancer recurrence who had not been prescribed spironolactone (P = .004). Adjusted Cox regression analysis following propensity matching showed no association between spironolactone and increased breast cancer recurrence (adjusted hazard ratio, 0.966; P = .953).
According to Dr. Harper, spironolactone may take about 3 months to kick in. “Likely this is a long-term treatment, and most of the time we’re going to be using it in combination with other acne treatments such as topical retinoids or topical benzoyl peroxide, oral antibiotics, or even isotretinoin.”
A study of long-term spironolactone use in 403 women found that the most common dose prescribed was 100 mg/day, and 68% of the women were concurrently prescribed a topical retinoid, 2.2% an oral antibiotic, and 40.7% an oral contraceptive.
The study population included 32 patients with a history of polycystic ovarian syndrome, 1 with a history of breast cancer, and 5 were hypercoagulable. Patients took the drug for a mean of 471 days. “As opposed to our antibiotics, where the course for patients is generally 3-4 months, when you start someone on spironolactone, they may end up staying on it,” Dr. Harper said.
Dr. Harper disclosed that she serves as an advisor or consultant for Almirall, Cassiopeia, Cutera, EPI, Galderma, L’Oreal, Ortho Dermatologics, Sol Gel, and Vyne. She also serves as a speaker or member of a speaker’s bureau for Almirall, Cassiopeia, Cutera, EPI, Galderma, Journey Almirall, L’Oreal, Ortho Dermatologics, Sun Pharmaceutical Industries, and Vyne.
Medscape and this news organization are owned by the same parent company.
AT THE MEDSCAPELIVE! HAWAII DERMATOLOGY SEMINAR
Lebrikizumab monotherapy for AD found safe, effective during induction
, researchers reported in the New England Journal of Medicine.
The identically designed, 52-week, randomized, double-blind, placebo-controlled trials enrolled 851 adolescents and adults with moderate to severe AD and included a 16-week induction period followed by a 36-week maintenance period. At week 16, the results “show a rapid onset of action in multiple domains of the disease, such as skin clearance and itch,” wrote lead author Jonathan Silverberg, MD, PhD, director of clinical research and contact dermatitis, at George Washington University, Washington, and colleagues. “Although 16 weeks of treatment with lebrikizumab is not sufficient to assess its long-term safety, the results from the induction period of these two trials suggest a safety profile that is consistent with findings in previous trials,” they added.
Results presented at the European Academy of Dermatology and Venereology 2022 annual meeting, but not yet published, showed similar efficacy maintained through the end of the trial.
Eligible patients were randomly assigned to receive either lebrikizumab 250 mg (with a 500-mg loading dose given at baseline and at week 2) or placebo, administered subcutaneously every 2 weeks, with concomitant topical or systemic treatments prohibited through week 16 except when deemed appropriate as rescue therapy. In such cases, moderate-potency topical glucocorticoids were preferred as first-line rescue therapy, while the study drug was discontinued if systemic therapy was needed.
In both trials, the primary efficacy outcome – a score of 0 or 1 on the Investigator’s Global Assessment (IGA) – and a reduction of at least 2 points from baseline at week 16, was met by more patients treated with lebrikizumab than with placebo: 43.1% vs. 12.7% respectively in trial 1 (P < .001); and 33.2% vs. 10.8% in trial 2 (P < .001).
Similarly, in both trials, a higher percentage of the lebrikizumab than placebo patients had an EASI-75 response (75% improvement in the Eczema Area and Severity Index score): 58.8% vs. 16.2% (P < .001) in trial 1 and 52.1% vs. 18.1% (P < .001) in trial 2.
Improvement in itch was also significantly better in patients treated with lebrikizumab, compared with placebo. This was measured by a reduction of at least 4 points in the Pruritus NRS from baseline to week 16 and a reduction in the Sleep-Loss Scale score of at least 2 points from baseline to week 16 (P < .001 for both measures in both trials).
A higher percentage of placebo vs. lebrikizumab patients discontinued the trials during the induction phases (14.9% vs. 7.1% in trial 1 and 11.0% vs. 7.8% in trial 2), and the use of rescue medication was approximately three times and two times higher in both placebo groups respectively.
Conjunctivitis was the most common adverse event, occurring consistently more frequently in patients treated with lebrikizumab, compared with placebo (7.4% vs. 2.8% in trial 1 and 7.5% vs. 2.1% in trial 2).
“Although several theories have been proposed for the pathogenesis of conjunctivitis in patients with atopic dermatitis treated with this class of biologic agents, the mechanism remains unclear and warrants further study,” the investigators wrote.
Asked to comment on the new results, Zelma Chiesa Fuxench, MD, who was not involved in the research, said they “continue to demonstrate the superior efficacy and favorable safety profile” of lebrikizumab in adolescents and adults and support the results of earlier phase 2 studies. “The results of these studies thus far continue to offer more hope and the possibility of a better future for our patients with atopic dermatitis who are still struggling to achieve control of their disease.”
Dr. Chiesa Fuxench from the department of dermatology at the University of Pennsylvania, Philadelphia, said she looks forward to reviewing the full study results in which patients who achieved the primary outcomes of interest were then rerandomized to either placebo, or lebrikizumab every 2 weeks or every 4 weeks for the 36-week maintenance period “because we know that there is data for other biologics in atopic dermatitis (such as tralokinumab) that demonstrate that a decrease in the frequency of injections may be possible for patients who achieve disease control after an initial 16 weeks of therapy every 2 weeks.”
The research was supported by Dermira, a wholly owned subsidiary of Eli Lilly. Dr. Silverberg disclosed he is a consultant for Dermira and Eli Lilly, as are other coauthors on the paper who additionally disclosed grants from Dermira and other relationships with Eli Lilly such as advisory board membership and having received lecture fees. Three authors are Eli Lilly employees. Dr. Chiesa Fuxench disclosed that she is a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, Pfizer, Abbvie, and Incyte for which she has received honoraria for work related to AD. Dr. Chiesa Fuxench has also been a recipient of research grants from Regeneron, Sanofi, Tioga, Vanda, Menlo Therapeutics, Leo Pharma, and Eli Lilly for work related to AD as well as honoraria for continuing medical education work related to AD sponsored through educational grants from Regeneron/Sanofi and Pfizer.
, researchers reported in the New England Journal of Medicine.
The identically designed, 52-week, randomized, double-blind, placebo-controlled trials enrolled 851 adolescents and adults with moderate to severe AD and included a 16-week induction period followed by a 36-week maintenance period. At week 16, the results “show a rapid onset of action in multiple domains of the disease, such as skin clearance and itch,” wrote lead author Jonathan Silverberg, MD, PhD, director of clinical research and contact dermatitis, at George Washington University, Washington, and colleagues. “Although 16 weeks of treatment with lebrikizumab is not sufficient to assess its long-term safety, the results from the induction period of these two trials suggest a safety profile that is consistent with findings in previous trials,” they added.
Results presented at the European Academy of Dermatology and Venereology 2022 annual meeting, but not yet published, showed similar efficacy maintained through the end of the trial.
Eligible patients were randomly assigned to receive either lebrikizumab 250 mg (with a 500-mg loading dose given at baseline and at week 2) or placebo, administered subcutaneously every 2 weeks, with concomitant topical or systemic treatments prohibited through week 16 except when deemed appropriate as rescue therapy. In such cases, moderate-potency topical glucocorticoids were preferred as first-line rescue therapy, while the study drug was discontinued if systemic therapy was needed.
In both trials, the primary efficacy outcome – a score of 0 or 1 on the Investigator’s Global Assessment (IGA) – and a reduction of at least 2 points from baseline at week 16, was met by more patients treated with lebrikizumab than with placebo: 43.1% vs. 12.7% respectively in trial 1 (P < .001); and 33.2% vs. 10.8% in trial 2 (P < .001).
Similarly, in both trials, a higher percentage of the lebrikizumab than placebo patients had an EASI-75 response (75% improvement in the Eczema Area and Severity Index score): 58.8% vs. 16.2% (P < .001) in trial 1 and 52.1% vs. 18.1% (P < .001) in trial 2.
Improvement in itch was also significantly better in patients treated with lebrikizumab, compared with placebo. This was measured by a reduction of at least 4 points in the Pruritus NRS from baseline to week 16 and a reduction in the Sleep-Loss Scale score of at least 2 points from baseline to week 16 (P < .001 for both measures in both trials).
A higher percentage of placebo vs. lebrikizumab patients discontinued the trials during the induction phases (14.9% vs. 7.1% in trial 1 and 11.0% vs. 7.8% in trial 2), and the use of rescue medication was approximately three times and two times higher in both placebo groups respectively.
Conjunctivitis was the most common adverse event, occurring consistently more frequently in patients treated with lebrikizumab, compared with placebo (7.4% vs. 2.8% in trial 1 and 7.5% vs. 2.1% in trial 2).
“Although several theories have been proposed for the pathogenesis of conjunctivitis in patients with atopic dermatitis treated with this class of biologic agents, the mechanism remains unclear and warrants further study,” the investigators wrote.
Asked to comment on the new results, Zelma Chiesa Fuxench, MD, who was not involved in the research, said they “continue to demonstrate the superior efficacy and favorable safety profile” of lebrikizumab in adolescents and adults and support the results of earlier phase 2 studies. “The results of these studies thus far continue to offer more hope and the possibility of a better future for our patients with atopic dermatitis who are still struggling to achieve control of their disease.”
Dr. Chiesa Fuxench from the department of dermatology at the University of Pennsylvania, Philadelphia, said she looks forward to reviewing the full study results in which patients who achieved the primary outcomes of interest were then rerandomized to either placebo, or lebrikizumab every 2 weeks or every 4 weeks for the 36-week maintenance period “because we know that there is data for other biologics in atopic dermatitis (such as tralokinumab) that demonstrate that a decrease in the frequency of injections may be possible for patients who achieve disease control after an initial 16 weeks of therapy every 2 weeks.”
The research was supported by Dermira, a wholly owned subsidiary of Eli Lilly. Dr. Silverberg disclosed he is a consultant for Dermira and Eli Lilly, as are other coauthors on the paper who additionally disclosed grants from Dermira and other relationships with Eli Lilly such as advisory board membership and having received lecture fees. Three authors are Eli Lilly employees. Dr. Chiesa Fuxench disclosed that she is a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, Pfizer, Abbvie, and Incyte for which she has received honoraria for work related to AD. Dr. Chiesa Fuxench has also been a recipient of research grants from Regeneron, Sanofi, Tioga, Vanda, Menlo Therapeutics, Leo Pharma, and Eli Lilly for work related to AD as well as honoraria for continuing medical education work related to AD sponsored through educational grants from Regeneron/Sanofi and Pfizer.
, researchers reported in the New England Journal of Medicine.
The identically designed, 52-week, randomized, double-blind, placebo-controlled trials enrolled 851 adolescents and adults with moderate to severe AD and included a 16-week induction period followed by a 36-week maintenance period. At week 16, the results “show a rapid onset of action in multiple domains of the disease, such as skin clearance and itch,” wrote lead author Jonathan Silverberg, MD, PhD, director of clinical research and contact dermatitis, at George Washington University, Washington, and colleagues. “Although 16 weeks of treatment with lebrikizumab is not sufficient to assess its long-term safety, the results from the induction period of these two trials suggest a safety profile that is consistent with findings in previous trials,” they added.
Results presented at the European Academy of Dermatology and Venereology 2022 annual meeting, but not yet published, showed similar efficacy maintained through the end of the trial.
Eligible patients were randomly assigned to receive either lebrikizumab 250 mg (with a 500-mg loading dose given at baseline and at week 2) or placebo, administered subcutaneously every 2 weeks, with concomitant topical or systemic treatments prohibited through week 16 except when deemed appropriate as rescue therapy. In such cases, moderate-potency topical glucocorticoids were preferred as first-line rescue therapy, while the study drug was discontinued if systemic therapy was needed.
In both trials, the primary efficacy outcome – a score of 0 or 1 on the Investigator’s Global Assessment (IGA) – and a reduction of at least 2 points from baseline at week 16, was met by more patients treated with lebrikizumab than with placebo: 43.1% vs. 12.7% respectively in trial 1 (P < .001); and 33.2% vs. 10.8% in trial 2 (P < .001).
Similarly, in both trials, a higher percentage of the lebrikizumab than placebo patients had an EASI-75 response (75% improvement in the Eczema Area and Severity Index score): 58.8% vs. 16.2% (P < .001) in trial 1 and 52.1% vs. 18.1% (P < .001) in trial 2.
Improvement in itch was also significantly better in patients treated with lebrikizumab, compared with placebo. This was measured by a reduction of at least 4 points in the Pruritus NRS from baseline to week 16 and a reduction in the Sleep-Loss Scale score of at least 2 points from baseline to week 16 (P < .001 for both measures in both trials).
A higher percentage of placebo vs. lebrikizumab patients discontinued the trials during the induction phases (14.9% vs. 7.1% in trial 1 and 11.0% vs. 7.8% in trial 2), and the use of rescue medication was approximately three times and two times higher in both placebo groups respectively.
Conjunctivitis was the most common adverse event, occurring consistently more frequently in patients treated with lebrikizumab, compared with placebo (7.4% vs. 2.8% in trial 1 and 7.5% vs. 2.1% in trial 2).
“Although several theories have been proposed for the pathogenesis of conjunctivitis in patients with atopic dermatitis treated with this class of biologic agents, the mechanism remains unclear and warrants further study,” the investigators wrote.
Asked to comment on the new results, Zelma Chiesa Fuxench, MD, who was not involved in the research, said they “continue to demonstrate the superior efficacy and favorable safety profile” of lebrikizumab in adolescents and adults and support the results of earlier phase 2 studies. “The results of these studies thus far continue to offer more hope and the possibility of a better future for our patients with atopic dermatitis who are still struggling to achieve control of their disease.”
Dr. Chiesa Fuxench from the department of dermatology at the University of Pennsylvania, Philadelphia, said she looks forward to reviewing the full study results in which patients who achieved the primary outcomes of interest were then rerandomized to either placebo, or lebrikizumab every 2 weeks or every 4 weeks for the 36-week maintenance period “because we know that there is data for other biologics in atopic dermatitis (such as tralokinumab) that demonstrate that a decrease in the frequency of injections may be possible for patients who achieve disease control after an initial 16 weeks of therapy every 2 weeks.”
The research was supported by Dermira, a wholly owned subsidiary of Eli Lilly. Dr. Silverberg disclosed he is a consultant for Dermira and Eli Lilly, as are other coauthors on the paper who additionally disclosed grants from Dermira and other relationships with Eli Lilly such as advisory board membership and having received lecture fees. Three authors are Eli Lilly employees. Dr. Chiesa Fuxench disclosed that she is a consultant for the Asthma and Allergy Foundation of America, National Eczema Association, Pfizer, Abbvie, and Incyte for which she has received honoraria for work related to AD. Dr. Chiesa Fuxench has also been a recipient of research grants from Regeneron, Sanofi, Tioga, Vanda, Menlo Therapeutics, Leo Pharma, and Eli Lilly for work related to AD as well as honoraria for continuing medical education work related to AD sponsored through educational grants from Regeneron/Sanofi and Pfizer.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE