User login
3-Month paliperidone palmitate for preventing relapse in schizophrenia
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.

How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
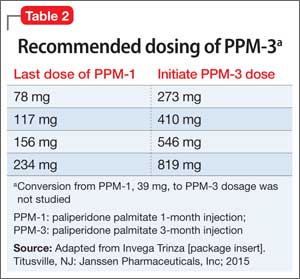
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
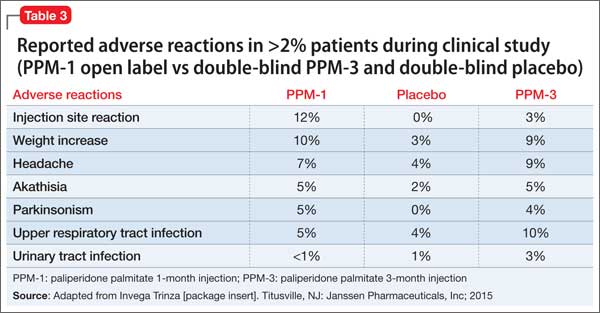
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.
How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
A 3-month paliperidone palmitate (PPM-3) extended-release injectable suspension was approved by the FDA in May 2015 for preventing relapse among patients with schizophrenia, under the brand name Invega Trinza (Table 1). Administered 4 times a year, PPM-3 provides the longest interval of any approved long-acting injectable antipsychotic (LAIA). PPM-3 can be administered to patients with schizophrenia who have been taking 1-month paliperidone palmitate (PPM-1) extended-release injectable suspension (brand name, Invega Sustenna), once a month, for at least 4 months.
How it works
PPM-3 is a LAIA injection. Because of its low solubility in water, paliperidone palmitate dissolves slowly once injected before being hydrolyzed as paliperidone and absorbed into the bloodstream. From time of release on Day 1, PPM-3 remains active for as long 18 months.
PPM-3 reaches a maximum plasma concentration between Day 30 and Day 33. In clinical trials, PPM-3 had a median half-life of 84 to 95 days when injected into the deltoid muscle and a median half-life of 118 to 139 days when injected into the gluteal muscle.
Paliperidone is not extensively metabolized in the liver. Although results of a study suggest that cytochrome P450 (CYP) 2D6 and CYP3A4 might play a role in metabolizing paliperidone, there is no evidence that it has a significant role.
Dosing and administration
PPM-3 is administered intramuscularly by a licensed health care professional, once every 3 months. The recommended dosage is based on the patient’s previous dosage of PPM-1 (Table 2).
See the prescribing information for administration instructions.
Efficacy
The efficacy of PPM-3 was assessed in a long-term double-blind, placebo-controlled, randomized-withdrawal trial in adult patients with acute symptoms (previously treated with an oral antipsychotic) or adequately treated with a LAIA, either PPM-1 or another agent; patients receiving PPM-1, 39 mg, injections were ineligible. All patients entering the study received PPM-1 in place of the next scheduled injection.
The study comprised 3 treatment periods:
• 17-Week flexible-dose open-label period with PPM-1 (ie, first part of a 29-week open-label stabilization phase): Patients (N = 506) received PPM-1 with a flexible dose based on symptom response, tolerability, and medication history. Patients had to achieve a Positive and Negative Syndrome Scale (PANSS) total score of <70 at Week 17 to enter the second phase.
• 12-Week open-label with PPM-3 (ie, second part of the 29-week open-label stabilization phase): Patients (N = 379) received a single injection of PPM-3 that was 3.5 times the last dose of PPM-1. Patients had to achieve a PANSS total score of <70 and ≤4 for 7 specific PANSS items.
• A variable length double-blind treatment period: Patients (N = 305) were randomized 1:1 to continue treatment with PPM-3 (273 mg, 410 mg, 546 mg, or 819 mg) or placebo (administered once every 12 weeks) until relapse, early withdrawal, or end of the study. The primary efficacy measure was time to first relapse, defined as psychiatric hospitalization, ≥25% increase or a 10-point increase in total PANSS score on 2 consecutive assessments, deliberate self-injury, violent behavior, suicidal or homicidal ideation, or a score of ≥5 (if the maximum baseline score was ≤3) or ≥6 (if the maximum baseline score was 4) on 2 consecutive assessments of the specific PANSS items.
Among the patients in the third treatment period, 23% of those who received placebo and 7.4% of those who received PPM-3 experienced a relapse event. The time to relapse was significantly longer for patients who received PPM-3 than for those who received placebo.
See Table 3 for adverse reactions reported in patients who received PPM-3 and those taking placebo in the study.
Contraindications
Allergic reactions. Patients who have a hypersensitivity to paliperidone, risperidone, or their components should not receive PPM-3. Anaphylactic reactions have been reported in patients who previously tolerated risperidone or oral paliperidone, which could be significant because the drug is slowly released over 3 months. Other adverse reactions, including angioedema, ileus, swollen tongue, thrombotic thrombocytopenic purpura, urinary incontinence, and urinary retention, were reported post-approval of paliperidone; however, these adverse effects were reported voluntarily from an unknown population size and, therefore, it is unknown whether there is a causal relationship to the drug or its frequency.
Drug-drug interactions. Although paliperidone is not expected to cause drug– drug interactions with medications that are metabolized by CYP isoenzymes, it is recommended to avoid using a strong inducer of CYP3A4 and/or P-glycoprotein.
Overdose. When assessing treatment options and recovery, consider the half-life of PPM-3 and its long-lasting effects.
Because PPM-3 is administered by a licensed health care provider, the potential for overdose is low. However, if overdose occurs, general treatment and management measures should be employed as with overdose of any drug and the possibility of multiple drug overdose should be considered. There is no specific antidote to paliperidone. Contact a certified poison control center for guidance on managing paliperidone and PPM-3 overdose. Generally, management consists of supportive care.
Black-box warning in dementia. As with all atypical antipsychotics, the black-box warning for PPM-3 states that it is not approved for, and should not be used in, patients with dementia-related psychosis. An analysis of placebo-controlled studies revealed that patients taking an antipsychotic had (1) 1.6 to 1.7 times the risk of death than those who received placebo and (2) a higher incidence of cerebrovascular adverse reactions.
Adverse reactions
The safety profile of PPM-3 is similar to that of PPM-1. The most common adverse reactions are:
• reaction at the injection site
• weight gain
• headache
• upper respiratory tract infection
• akathisia
• parkinsonism.
See the full prescribing information for a complete list of adverse effects.
Related Resources
• Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-49.
• Berwaerts J, Liu Y, Gopal S, et al. Efficacy and safety of the 3-month formulation of paliperidone palmitate vs placebo for relapse prevention of schizophrenia: a randomized clinical trial [published online March 29, 2015]. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2015.0241.
Drug Brand Names
Paliperidone palmitate • Invega Sustenna, Invega Trinza
Risperidone • Risperdal
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
Source: Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2015.
Asenapine for pediatric bipolar disorder: New indication
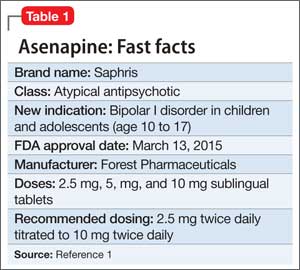
Asenapine an atypical antipsychotic sold under the brand name Saphris, was granted a second, pediatric indication by the FDA in March 2015 as monotherapy for acute treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17 (Table 1).1 (Asenapine was first approved in August 2009 as monotherapy or adjunctive therapy to lithium or valproate in adults for schizophrenia and bipolar I disorder.1,2)
Dosage and administration
Asenapine is available as 2.5-, 5-, and 10-mg sublingual tablets, the only atypical antipsychotic with this formulation.1 The recommended dosage for the new indication is 2.5 mg twice daily for 3 days, titrated to 5 mg twice daily, titrated again to 10 mg twice daily after 3 days.3 In a phase I study, pediatric patients appeared to be more sensitive to dystonia when the recommended dosage escalation schedule was not followed.3
In clinical trials, drinking water 2 to 5 minutes after taking asenapine decreased exposure to the drug. Instruct patients not to swallow the tablet and to avoid eating and drinking for 10 minutes after administration.3
For full prescribing information for pediatric and adult patients, see Reference 3.
Safety and efficacy
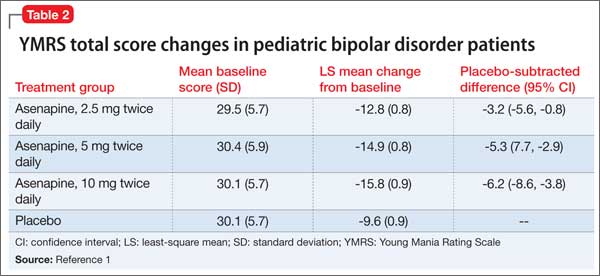
In a 3-week, placebo-controlled, double-blind trial of 403 patients, 302 children and adolescents age 10 to 17 received asenapine at fixed dosages of 2.5 to 10 mg twice daily; the remainder were given placebo. The Young Mania Rating Scale (YMRS) total score and Clinical Global Impressions Severity of Illness scores of patients who received asenapine improved significantly compared with those who received placebo, as measured by change from baseline to week 3 (Table 2).1
The safety and efficacy of asenapine has not been evaluated in pediatric bipolar disorder patients age ≤10 or pediatric schizophrenia patients age ≤12, or as an adjunctive therapy in pediatric bipolar disorder patients.
Asenapine was not shown to be effective in pediatric patients with schizophrenia in an 8-week, placebo-controlled, double-blind trial.
The pharmacokinetics of asenapine in pediatric patients are similar to those seen in adults.
Adverse effects
In pediatric patients, the most common reported adverse effects of asenapine are:
• dizziness
• dysgeusia
• fatigue
• increased appetite
• increased weight
• nausea
• oral paresthesia
• somnolence.
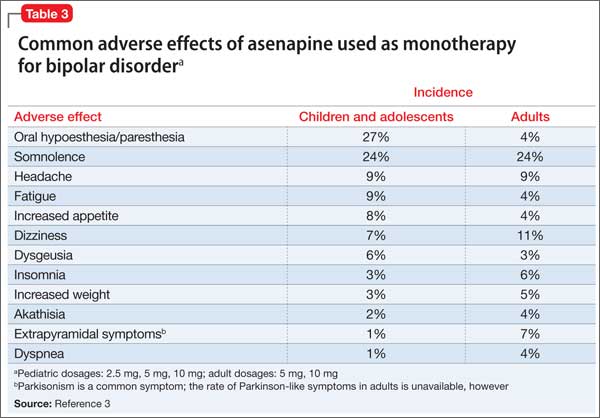
Similar adverse effects were reported in the pediatric bipolar disorder and adult bipolar disorder clinical trials (Table 3).3 A complete list of reported adverse effects is given in the package insert.3
When treating pediatric patients, monitor the child’s weight gain against expected normal weight gain.
Asenapine is contraindicated in patients with hepatic impairment and those who have a hypersensitivity to asenapine or any components in its formulation.3
1. Actavis receives FDA approval of Saphris for pediatric patients with bipolar I disorder. Drugs.com. http://www.drugs.com/newdrugs/actavis-receivesfda-
approval-saphris-pediatric-patients-bipolardisorder-4188.html. Published March 2015. Accessed June 19, 2015.
2. Lincoln J, Preskon S. Asenapine for schizophrenia and bipolar I disorder. Current Psychiatry. 2009;12(8):75-76,83-85.
3. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2015.
Asenapine an atypical antipsychotic sold under the brand name Saphris, was granted a second, pediatric indication by the FDA in March 2015 as monotherapy for acute treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17 (Table 1).1 (Asenapine was first approved in August 2009 as monotherapy or adjunctive therapy to lithium or valproate in adults for schizophrenia and bipolar I disorder.1,2)
Dosage and administration
Asenapine is available as 2.5-, 5-, and 10-mg sublingual tablets, the only atypical antipsychotic with this formulation.1 The recommended dosage for the new indication is 2.5 mg twice daily for 3 days, titrated to 5 mg twice daily, titrated again to 10 mg twice daily after 3 days.3 In a phase I study, pediatric patients appeared to be more sensitive to dystonia when the recommended dosage escalation schedule was not followed.3
In clinical trials, drinking water 2 to 5 minutes after taking asenapine decreased exposure to the drug. Instruct patients not to swallow the tablet and to avoid eating and drinking for 10 minutes after administration.3
For full prescribing information for pediatric and adult patients, see Reference 3.
Safety and efficacy
In a 3-week, placebo-controlled, double-blind trial of 403 patients, 302 children and adolescents age 10 to 17 received asenapine at fixed dosages of 2.5 to 10 mg twice daily; the remainder were given placebo. The Young Mania Rating Scale (YMRS) total score and Clinical Global Impressions Severity of Illness scores of patients who received asenapine improved significantly compared with those who received placebo, as measured by change from baseline to week 3 (Table 2).1
The safety and efficacy of asenapine has not been evaluated in pediatric bipolar disorder patients age ≤10 or pediatric schizophrenia patients age ≤12, or as an adjunctive therapy in pediatric bipolar disorder patients.
Asenapine was not shown to be effective in pediatric patients with schizophrenia in an 8-week, placebo-controlled, double-blind trial.
The pharmacokinetics of asenapine in pediatric patients are similar to those seen in adults.
Adverse effects
In pediatric patients, the most common reported adverse effects of asenapine are:
• dizziness
• dysgeusia
• fatigue
• increased appetite
• increased weight
• nausea
• oral paresthesia
• somnolence.
Similar adverse effects were reported in the pediatric bipolar disorder and adult bipolar disorder clinical trials (Table 3).3 A complete list of reported adverse effects is given in the package insert.3
When treating pediatric patients, monitor the child’s weight gain against expected normal weight gain.
Asenapine is contraindicated in patients with hepatic impairment and those who have a hypersensitivity to asenapine or any components in its formulation.3
Asenapine an atypical antipsychotic sold under the brand name Saphris, was granted a second, pediatric indication by the FDA in March 2015 as monotherapy for acute treatment of manic or mixed episodes of bipolar I disorder in children and adolescents age 10 to 17 (Table 1).1 (Asenapine was first approved in August 2009 as monotherapy or adjunctive therapy to lithium or valproate in adults for schizophrenia and bipolar I disorder.1,2)
Dosage and administration
Asenapine is available as 2.5-, 5-, and 10-mg sublingual tablets, the only atypical antipsychotic with this formulation.1 The recommended dosage for the new indication is 2.5 mg twice daily for 3 days, titrated to 5 mg twice daily, titrated again to 10 mg twice daily after 3 days.3 In a phase I study, pediatric patients appeared to be more sensitive to dystonia when the recommended dosage escalation schedule was not followed.3
In clinical trials, drinking water 2 to 5 minutes after taking asenapine decreased exposure to the drug. Instruct patients not to swallow the tablet and to avoid eating and drinking for 10 minutes after administration.3
For full prescribing information for pediatric and adult patients, see Reference 3.
Safety and efficacy
In a 3-week, placebo-controlled, double-blind trial of 403 patients, 302 children and adolescents age 10 to 17 received asenapine at fixed dosages of 2.5 to 10 mg twice daily; the remainder were given placebo. The Young Mania Rating Scale (YMRS) total score and Clinical Global Impressions Severity of Illness scores of patients who received asenapine improved significantly compared with those who received placebo, as measured by change from baseline to week 3 (Table 2).1
The safety and efficacy of asenapine has not been evaluated in pediatric bipolar disorder patients age ≤10 or pediatric schizophrenia patients age ≤12, or as an adjunctive therapy in pediatric bipolar disorder patients.
Asenapine was not shown to be effective in pediatric patients with schizophrenia in an 8-week, placebo-controlled, double-blind trial.
The pharmacokinetics of asenapine in pediatric patients are similar to those seen in adults.
Adverse effects
In pediatric patients, the most common reported adverse effects of asenapine are:
• dizziness
• dysgeusia
• fatigue
• increased appetite
• increased weight
• nausea
• oral paresthesia
• somnolence.
Similar adverse effects were reported in the pediatric bipolar disorder and adult bipolar disorder clinical trials (Table 3).3 A complete list of reported adverse effects is given in the package insert.3
When treating pediatric patients, monitor the child’s weight gain against expected normal weight gain.
Asenapine is contraindicated in patients with hepatic impairment and those who have a hypersensitivity to asenapine or any components in its formulation.3
1. Actavis receives FDA approval of Saphris for pediatric patients with bipolar I disorder. Drugs.com. http://www.drugs.com/newdrugs/actavis-receivesfda-
approval-saphris-pediatric-patients-bipolardisorder-4188.html. Published March 2015. Accessed June 19, 2015.
2. Lincoln J, Preskon S. Asenapine for schizophrenia and bipolar I disorder. Current Psychiatry. 2009;12(8):75-76,83-85.
3. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2015.
1. Actavis receives FDA approval of Saphris for pediatric patients with bipolar I disorder. Drugs.com. http://www.drugs.com/newdrugs/actavis-receivesfda-
approval-saphris-pediatric-patients-bipolardisorder-4188.html. Published March 2015. Accessed June 19, 2015.
2. Lincoln J, Preskon S. Asenapine for schizophrenia and bipolar I disorder. Current Psychiatry. 2009;12(8):75-76,83-85.
3. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2015.
Liraglutide for obesity: New indication
Liraglutide (rDNA origin) injection, approved by the FDA in 2010 for managing type 2 diabetes mellitus (T2DM), has a new formulation and indication for treating obesity in adults as an adjunct to a reduced-calorie diet and increased physical activity (Table 1).1
Liraglutide, recommended dosage 3 mg/d (under the brand name Saxenda), is approved for adults with a body mass index (BMI) ≥30, or those with a BMI of ≥27 and a weight-related condition such as hypertension, T2DM, or high cholesterol.1 (A 1.8-mg formulation, under the brand name Victoza, is FDA-approved for managing T2DM, but is not indicated for weight management.)
How it works
Liraglutide is a glucagon-like peptide-1 (GLP-1) receptor agonist. GLP-1, which regulates appetite and calorie intake, is found in several regions of the brain that are involved in regulating appetite. Patients taking liraglutide lose weight because of decreased calorie intake, not increased energy expenditure.
Liraglutide is endogenously metabolized without a specific organ as a major route of elimination.1
Dosage and administration
Liraglutide is administered using a prefilled, multi-dose pen that can be injected in the abdomen, thigh, or upper arm. Recommended dosage is 3 mg/d, administered any time of day. Initiate dosage at 0.6 mg/d the first week, then titrate by 0.6 mg a week—to reduce the likelihood of adverse gastrointestinal symptoms—until 3 mg/d is reached.
Discontinue liraglutide if a patient has not lost at least 4% of body weight after 16 weeks of treatment, because it is unlikely the patient will achieve and sustain weight loss.
Efficacy
Liraglutide was studied in 3 clinical trials of obese and overweight participants who had a weight-related condition. Patients who had a history of major depressive disorder or suicide attempt were excluded from the studies. All participants in Studies 1 and 2 received instruction about following a reduced-calorie diet and increasing physical activity. In Study 3, patients were randomized to treatment after losing >5% of their body weight through reduced calorie intake and exercise; those who did not meet the required weight loss were excluded from the study. In these 56-week clinical studies:
• of 3,731 participants without diabetes or a weight-related comorbidity, such as high blood pressure or high cholesterol, 62% of patients (n = 2,313) who took liraglutide lost ≥5% of their body weight from baseline, compared with 34% of participants who received placebo
• of 635 participants with T2DM, 49% of patients (n = 311) treated with liraglutide lost ≥5% of their body weight compared with 16% placebo patients
• of 422 participants with a weight-related comorbidity, 42% of patients (n = 177) lost ≥5% of their body weight compared with 21.7% of placebo patients.
Improvements in some cardiovascular disease risk factors were observed. Long-term follow up was not studied.
Contraindictations
Liraglutide is contraindicated in patients who have a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2. In a 104-week study, malignant thyroid C-cell carcinomas were detected in rats and mice given liraglutide, 1 and 3 mg/kg/d; however, it was not detected in groups given 0.03 and 0.2 mg/kg/d. It isn’t known whether liraglutide can cause thyroid C-cell tumors in humans.
Patients should not take liraglutide if they have hypersensitivity to liraglutide or any product components, are using insulin, are taking any other GLP-1 receptor agonist, or are pregnant.
Adverse effects
The most common reported adverse effects are nausea (39.3%), hypoglycemia in patients with T2DM (23%), diarrhea (20.9%), constipation (19.4%), and vomiting (15.7%) (Table 2). In clinical trials, 9.8% of patients discontinued treatment because of adverse effects, compared with 4.3% of those receiving placebo.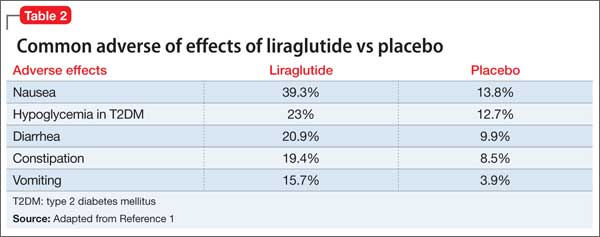
Liraglutide has low potential for pharmacokinetic drug-drug interactions related to cytochrome P450 and plasma protein binding. For a full list of drug-drug interactions, see the full prescribing information.1
Reference
1. Saxenda [package insert]. Plainsboro, NJ: Novo Nordisk A/S; 2015.
Liraglutide (rDNA origin) injection, approved by the FDA in 2010 for managing type 2 diabetes mellitus (T2DM), has a new formulation and indication for treating obesity in adults as an adjunct to a reduced-calorie diet and increased physical activity (Table 1).1
Liraglutide, recommended dosage 3 mg/d (under the brand name Saxenda), is approved for adults with a body mass index (BMI) ≥30, or those with a BMI of ≥27 and a weight-related condition such as hypertension, T2DM, or high cholesterol.1 (A 1.8-mg formulation, under the brand name Victoza, is FDA-approved for managing T2DM, but is not indicated for weight management.)
How it works
Liraglutide is a glucagon-like peptide-1 (GLP-1) receptor agonist. GLP-1, which regulates appetite and calorie intake, is found in several regions of the brain that are involved in regulating appetite. Patients taking liraglutide lose weight because of decreased calorie intake, not increased energy expenditure.
Liraglutide is endogenously metabolized without a specific organ as a major route of elimination.1
Dosage and administration
Liraglutide is administered using a prefilled, multi-dose pen that can be injected in the abdomen, thigh, or upper arm. Recommended dosage is 3 mg/d, administered any time of day. Initiate dosage at 0.6 mg/d the first week, then titrate by 0.6 mg a week—to reduce the likelihood of adverse gastrointestinal symptoms—until 3 mg/d is reached.
Discontinue liraglutide if a patient has not lost at least 4% of body weight after 16 weeks of treatment, because it is unlikely the patient will achieve and sustain weight loss.
Efficacy
Liraglutide was studied in 3 clinical trials of obese and overweight participants who had a weight-related condition. Patients who had a history of major depressive disorder or suicide attempt were excluded from the studies. All participants in Studies 1 and 2 received instruction about following a reduced-calorie diet and increasing physical activity. In Study 3, patients were randomized to treatment after losing >5% of their body weight through reduced calorie intake and exercise; those who did not meet the required weight loss were excluded from the study. In these 56-week clinical studies:
• of 3,731 participants without diabetes or a weight-related comorbidity, such as high blood pressure or high cholesterol, 62% of patients (n = 2,313) who took liraglutide lost ≥5% of their body weight from baseline, compared with 34% of participants who received placebo
• of 635 participants with T2DM, 49% of patients (n = 311) treated with liraglutide lost ≥5% of their body weight compared with 16% placebo patients
• of 422 participants with a weight-related comorbidity, 42% of patients (n = 177) lost ≥5% of their body weight compared with 21.7% of placebo patients.
Improvements in some cardiovascular disease risk factors were observed. Long-term follow up was not studied.
Contraindictations
Liraglutide is contraindicated in patients who have a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2. In a 104-week study, malignant thyroid C-cell carcinomas were detected in rats and mice given liraglutide, 1 and 3 mg/kg/d; however, it was not detected in groups given 0.03 and 0.2 mg/kg/d. It isn’t known whether liraglutide can cause thyroid C-cell tumors in humans.
Patients should not take liraglutide if they have hypersensitivity to liraglutide or any product components, are using insulin, are taking any other GLP-1 receptor agonist, or are pregnant.
Adverse effects
The most common reported adverse effects are nausea (39.3%), hypoglycemia in patients with T2DM (23%), diarrhea (20.9%), constipation (19.4%), and vomiting (15.7%) (Table 2). In clinical trials, 9.8% of patients discontinued treatment because of adverse effects, compared with 4.3% of those receiving placebo.
Liraglutide has low potential for pharmacokinetic drug-drug interactions related to cytochrome P450 and plasma protein binding. For a full list of drug-drug interactions, see the full prescribing information.1
Liraglutide (rDNA origin) injection, approved by the FDA in 2010 for managing type 2 diabetes mellitus (T2DM), has a new formulation and indication for treating obesity in adults as an adjunct to a reduced-calorie diet and increased physical activity (Table 1).1
Liraglutide, recommended dosage 3 mg/d (under the brand name Saxenda), is approved for adults with a body mass index (BMI) ≥30, or those with a BMI of ≥27 and a weight-related condition such as hypertension, T2DM, or high cholesterol.1 (A 1.8-mg formulation, under the brand name Victoza, is FDA-approved for managing T2DM, but is not indicated for weight management.)
How it works
Liraglutide is a glucagon-like peptide-1 (GLP-1) receptor agonist. GLP-1, which regulates appetite and calorie intake, is found in several regions of the brain that are involved in regulating appetite. Patients taking liraglutide lose weight because of decreased calorie intake, not increased energy expenditure.
Liraglutide is endogenously metabolized without a specific organ as a major route of elimination.1
Dosage and administration
Liraglutide is administered using a prefilled, multi-dose pen that can be injected in the abdomen, thigh, or upper arm. Recommended dosage is 3 mg/d, administered any time of day. Initiate dosage at 0.6 mg/d the first week, then titrate by 0.6 mg a week—to reduce the likelihood of adverse gastrointestinal symptoms—until 3 mg/d is reached.
Discontinue liraglutide if a patient has not lost at least 4% of body weight after 16 weeks of treatment, because it is unlikely the patient will achieve and sustain weight loss.
Efficacy
Liraglutide was studied in 3 clinical trials of obese and overweight participants who had a weight-related condition. Patients who had a history of major depressive disorder or suicide attempt were excluded from the studies. All participants in Studies 1 and 2 received instruction about following a reduced-calorie diet and increasing physical activity. In Study 3, patients were randomized to treatment after losing >5% of their body weight through reduced calorie intake and exercise; those who did not meet the required weight loss were excluded from the study. In these 56-week clinical studies:
• of 3,731 participants without diabetes or a weight-related comorbidity, such as high blood pressure or high cholesterol, 62% of patients (n = 2,313) who took liraglutide lost ≥5% of their body weight from baseline, compared with 34% of participants who received placebo
• of 635 participants with T2DM, 49% of patients (n = 311) treated with liraglutide lost ≥5% of their body weight compared with 16% placebo patients
• of 422 participants with a weight-related comorbidity, 42% of patients (n = 177) lost ≥5% of their body weight compared with 21.7% of placebo patients.
Improvements in some cardiovascular disease risk factors were observed. Long-term follow up was not studied.
Contraindictations
Liraglutide is contraindicated in patients who have a personal or family history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2. In a 104-week study, malignant thyroid C-cell carcinomas were detected in rats and mice given liraglutide, 1 and 3 mg/kg/d; however, it was not detected in groups given 0.03 and 0.2 mg/kg/d. It isn’t known whether liraglutide can cause thyroid C-cell tumors in humans.
Patients should not take liraglutide if they have hypersensitivity to liraglutide or any product components, are using insulin, are taking any other GLP-1 receptor agonist, or are pregnant.
Adverse effects
The most common reported adverse effects are nausea (39.3%), hypoglycemia in patients with T2DM (23%), diarrhea (20.9%), constipation (19.4%), and vomiting (15.7%) (Table 2). In clinical trials, 9.8% of patients discontinued treatment because of adverse effects, compared with 4.3% of those receiving placebo.
Liraglutide has low potential for pharmacokinetic drug-drug interactions related to cytochrome P450 and plasma protein binding. For a full list of drug-drug interactions, see the full prescribing information.1
Reference
1. Saxenda [package insert]. Plainsboro, NJ: Novo Nordisk A/S; 2015.
Reference
1. Saxenda [package insert]. Plainsboro, NJ: Novo Nordisk A/S; 2015.
Lisdexamfetamine for binge eating disorder: New indication
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.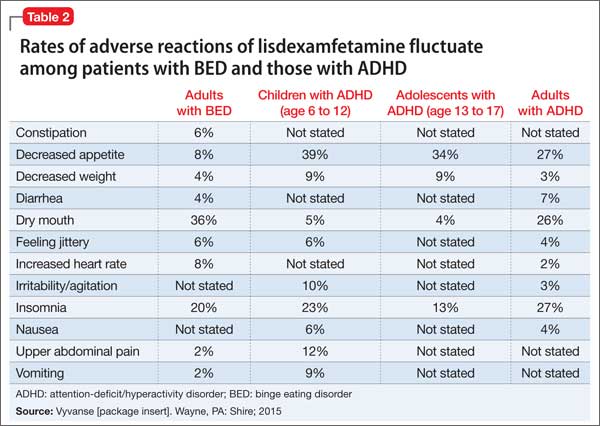
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Lisdexamfetamine, approved by the FDA in 2007 for attention-deficit/hyperactivity disorder (ADHD), has a new indication: binge eating disorder (BED) (Table 1). BED is characterized by recurrent episodes of consuming a large amount of food in a short time. A prodrug of amphetamine, lisdexamfetamine is a Schedule-II controlled substance, with a high potential for abuse and the risk of severe psychological or physical dependence.
Lisdexamfetamine is not indicated for weight loss or obesity.
Dosage
For BED, the initial dosage of lisdexamfetamine is 30 mg/d in the morning, titrated by 20 mg/d per week to the target dosage of 50 to 70 mg/d. Maximum dosage is 70 mg/d. Morning dosing is recommended to avoid sleep disturbance.
Efficacy
The clinical efficacy of lisdexamfetamine was assessed in two 12-week parallel group, flexible-dose, placebo-controlled trials in adults with BED (age 18 to 55). Primary efficacy measure was the number of binge days per week. Both studies had a 4-week dose-optimization period and an 8-week dose-maintenance period and followed the same dosage protocol. Patients began treatment at 30 mg/d and after 1 week were titrated to 50 mg/d; increases to 70 mg/d were made if clinically necessary and well tolerated. Patients were maintained on the optimized dosage during the 8-week dose-maintenance period. A dosage of 30 mg/d did not produce a statistically significant effect, but 50 mg/d and 70 mg/d dosages were statistically superior to placebo. Patients taking lisdexamfetamine also had greater improvement on the Clinical Global Impression—Improvement scores, 4-week binge cessation, and greater reduction in the Yale-Brown Obsessive Compulsive Scale Modified for Binge Eating score.
The prescribing information does not state if lisdexamfetamine should be continued long-term for treating BED.
Adverse reactions
In controlled trials, 5.1% of patients receiving lisdexamfetamine for BED discontinued the drug because of an adverse event, compared with 2.4% of patients receiving placebo. The most common adverse reactions in BED studies were dry mouth (36%), insomnia (20%), decreased appetite (8%), increased heart rate (8%), constipation (6%), and feeling jittery (6%). In trials of children, adolescents, and adults with ADHD, decreased appetite was more common (39%, 34%, and 27%, respectively) than in BED trials (Table 2). Anaphylactic reactions, Stevens-Johnson syndrome, angioedema, and urticaria have been described in postmarketing reports.
The safety of lisdexamfetamine for BED has not been studied in patients age <18, but has been studied in patients with ADHD.
Contraindications
Do not give lisdexamfetamine to patients who have a known hypersensitivity to amphetamine products or other ingredients in lisdexamfetamine capsules.
Lisdexamfetamine is contraindicated in patients who are taking a monoamine oxidase inhibitor, because of a risk of hypertensive crisis.
Related Resources
• Wilens TE. Lisdexamfetamine for ADHD. Current Psychiatry. 2007;6(6):96-98,105.
• Peat CM, Brownley KA, Berkman ND, et al. Binge eating disorder: evidence-based treatments. Current Psychiatry. 2012; 11(5):32-39.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Source: Vyvanse [package insert]. Wayne, PA: Shire; 2015.
Suvorexant for sleep-onset insomnia or sleep-maintenance insomnia, or both
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2

Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
Suvorexant, FDA-approved to treat insomnia, has demonstrated efficacy in helping patients with insomnia improve their ability to fall asleep and remain asleep (Table 1).1 This first-in-class compound represents a novel mechanism of action to promoting sleep that may avoid some problems associated with other hypnotics.2
Clinical implications
Insomnia is among the most common clinical complaints in psychiatry and medicine. The FDA-approved insomnia medications include several benzodiazepine-receptor agonists (zolpidem, eszopiclone, zaleplon), a melatonin-receptor agonist (ramelteon), and a histamine-receptor antagonist (low-dose doxepin). Suvorexant joins these drugs and is an entirely novel compound that is the first orexin- (also called hypocretin) receptor antagonist approved by the FDA for any indication.
Through a highly targeted mechanism of action, suvorexant could enhance sleep for patients with insomnia, while maintaining an acceptable safety profile.3 The drug should help patients with chronic insomnia, particularly those who have difficulty maintaining sleep—the sleep disturbance pattern that is most challenging to treat pharmacotherapeutically.
Because orexin antagonists have not been used outside of clinical trials, it is too soon to tell whether suvorexant will have the ideal real-world efficacy and safety profile to make it a first-line treatment for insomnia patients, or if it will be reserved for those who have failed a trial of several other treatments.4
In theory, the orexin antagonist approach to treating insomnia could represent a major advance that modulates the fundamental pathology of the disorder.5 The syndrome of chronic insomnia encompasses not just the nighttime sleep disturbance but also an assortment of daytime symptoms that can include fatigue, poor concentration, irritability, and decreased school or work performance but usually not sleepiness. This constellation of nighttime and daytime symptoms could be conceptualized as a manifestation of persistent CNS hyperarousal. Because the orexin system promotes and reinforces arousal, perhaps an orexin antagonist that dampens the level of orexin activity will ameliorate the full spectrum of insomnia symptoms—not simply sedate patients.6
How suvorexant works
Suvorexant is a potent and reversible dual orexin-receptor antagonist. The orexin system, first described in 1998, has a key role in promoting and stabilizing wakefulness.7 Evidence suggests that people with chronic insomnia exhibit a central hyperarousal that perpetuates their sleep difficulty. Accordingly, a targeted pharmaceutical approach that reduces orexin activity should facilitate sleep onset and sleep maintenance for these patients. It is well known that the regulation of sleep and wakefulness depends on the interaction of multiple nuclei within the hypothalamus. Orexinergic neurons in the perifornical-lateral hypothalamic region project widely in the CNS and have especially dense connections with wake-promoting cholinergic, serotonergic, noradrenergic, and histaminergic neurons.6
A precursor prepro-orexin peptide is split into 2 orexin neurotransmitters (orexin A and orexin B). These 2 orexins bind with 2 G-protein-coupled receptors (OX1R and OX2R) that have both overlapping and distinct distributions.7 Suvorexant is highly selective and has similar affinity for OX1R and OX2R, functioning as an antagonist for both.8 Fundamentally, suvorexant enhances sleep by dampening the arousing wake drive.
Pharmacokinetics
Suvorexant is available as an immediate-release tablet with pharmacokinetic properties that offer benefits for sleep onset and maintenance.9 Ingestion under fasting conditions results in a median time to maximum concentration (Tmax) of approximately 2 hours, although the Tmax values vary widely from patient to patient (range 30 minutes to 6 hours). Although suvorexant can be taken with food, there is a modest absorption delay after a high-fat meal, resulting in a further Tmax delay of approximately 1.5 hours.
Suvorexant is primarily metabolized through the cytochrome P450 (CYP) 3A pathway, with limited contribution by CYP2C19. There are no active metabolites. The suvorexant blood level and risk of side effects will be higher with concomitant use of CYP3A inhibitors. The drug should not be administered with strong CYP3A inhibitors; the initial dosage should be reduced with moderate CYP3A inhibitors. Concomitant use of strong CYP3A inducers can result in a low suvorexant level and reduced efficacy.
Suvorexant has little effect on other medications, although a person taking digoxin might experience intestinal P-glycoprotein inhibition with a slight rise in the digoxin level. In a patient taking both medications, monitoring of the digoxin level is recommended.
The elimination half-life of suvorexant is approximately 12 hours, with a steady state in approximately 3 days. Because the half-life of suvorexant is moderately long for a sleep-promoting medication, use of the drug might be associated with residual sleepiness the morning after bedtime dosing. The risk for next-morning sleepiness or impairment should be minimized, however, when using the recommended dosages. Elimination is approximately two-thirds through feces and one-third in the urine.
Suvorexant metabolism can be affected by sex and body mass index. Females and obese people have a modestly elevated exposure to suvorexant, as reflected by the area under the curve and maximum concentration (Cmax). These patients might not require dosage adjustments unless they are obese and female, in which case they should take a lower dosage.
Age and race have not been shown to influence suvorexant metabolism to a significant degree. Patients with renal impairment and those with mild or moderate hepatic impairment do not need dosage adjustment. Suvorexant has not been evaluated in patients with severe hepatic impairment.
Efficacy
Suvorexant showed significant evidence of improved sleep onset and sleep maintenance in patients with insomnia in clinical trials. The key efficacy clinical trials with insomnia patients included a phase-IIb dose-finding study,10 2 similar 3-month phase-III studies,11 and one 12-month phase-III safety study that incorporated efficacy outcomes.12 All these trials included subjective sleep measures and all except for the long-term safety study also incorporated polysomnographic assessment. The specific sleep laboratory outcomes were latency to persistent sleep (LPS), wake after the onset of persistent sleep (WASO), total sleep time (TST), and sleep efficiency (SE). Subjective sleep outcomes were time to sleep onset (sTSO), wake after sleep onset (sWASO), and total sleep time (sTST). Other exploratory endpoints also were assessed. These efficacy and safety studies mostly were performed at dosages considerably higher than those approved by the FDA.
The dose-finding (phase-IIb) trial was conducted with non-geriatric (age 18 to 64) patients with insomnia in a randomized, double-blind, crossover design of two 4-week periods with subjects given a nightly placebo or suvorexant (10 mg, 20 mg, 40 mg, or 80 mg).10 Each of the 4 groups included approximately 60 subjects. The 2 co-primary endpoints were SE at Night 1 and the end of Week 4; secondary endpoints were LPS and WASO. Suvorexant was associated with dosage-related improvements in SE and WASO compared with placebo at both time points. Carryover effects from the period-1 active drug group complicated the analysis of LPS.
The phase-III efficacy and safety trials were performed with 40 mg high dosage (HD) and 20 mg low dosage (LD) groups for adults and with 30 mg HD and 15 mg LD groups for geriatric (age ≥65) patients.11 Two similarly designed 3-month randomized, double-blind, placebo-controlled pivotal efficacy studies assessed objective and subjective sleep measures in 4 groups with non-geriatric (HD and LD) and geriatric (HD and LD) insomnia patients.
After baseline assessment, patients took nightly bedtime doses of placebo; suvorexant, 40 mg or 20 mg (non-geriatric individuals); or suvorexant, 30 mg or 15 mg (geriatric individuals). All subjects kept a daily electronic diary and had polysomnographic recordings performed on Night 1, at the end of Month 1, and at the end of Month 3. Both the individual studies and combined analyses (2,030 subjects) showed that, in non-geriatric and geriatric patients, HD suvorexant resulted in significantly greater improvement in key subjective and objective measures throughout the study (Table 2,9 and Table 3,9), with the exception of a single LPS outcome in 1 study, compared with placebo. The LD dosages also demonstrated efficacy, but to a reduced extent.
Subjective sleep outcomes were assessed in a 1-year randomized, placebo-controlled trial with nightly placebo, suvorexant, 40 mg, for non-geriatric, or suvorexant, 30 mg, for geriatric insomnia patients.12 The 1-year phase was completed with 484 subjects. Key efficacy outcomes were sTST and sTSO changes from baseline during the first month of treatment. Compared with placebo, suvorexant dosages demonstrated significantly greater efficacy, improvements that were sustained throughout the year.
Clinical trials found suvorexant to be generally safe and well tolerated.13 However, specific safety concerns led the FDA to approve the medication at dosages lower than those assessed in the phase-III studies.1
Somnolence was the most common adverse event in clinical trials. In the phase- IIb dose-finding study, somnolence was reported in <1% in the placebo group, but was associated with suvorexant in 2% of the 10 mg group, 5% with 20 mg, 12% with 40 mg, and 11% with 80 mg.9 In the phase-III combined analysis of the 3-month studies, somnolence was reported by 3% in the placebo group and 7% of non-geriatric patients taking 20 mg or geriatric patients taking 15 mg. Somnolence was reported in 8% of women and 3% of men taking the 15 mg or 20 mg dosage in these studies. The 1-year study was performed only with higher suvorexant dosages (30 mg and 40 mg), in comparison with placebo. In this long-term trial, somnolence was reported by 13% of subjects taking suvorexant and 3% taking placebo.
Additional safety issues in trials included excessive daytime sleepiness, impaired driving, suicidal ideation, sleep paralysis, hypnagogic/hypnopompic hallucinations, and cataplexy-like symptoms.9 Occurrences of these events are rare but have been reported more often among patients taking suvorexant than among those taking placebo.
Unique clinical issues
The U.S. Drug Enforcement Agency has categorized suvorexant as a Schedule IV controlled substance. Although there is no evidence of physiological dependence or withdrawal symptoms with suvorexant, studies with recreational substance abusers have shown that the likeability rating is similar to that of zolpidem.13
Contraindication
Suvorexant is contraindicated in patients with narcolepsy.9 The underlying pathology of narcolepsy involves a marked reduction in orexin functioning with corresponding excessive sleepiness and related symptoms, such as cataplexy, hypnagogic hallucinations, and sleep paralysis. Although suvorexant has not been evaluated in patients with narcolepsy, the drug might, hypothetically, put patients at higher risk of the full spectrum of narcolepsy symptoms.
There are no other contraindications for suvorexant.
Dosing
Suvorexant should be taken no more than once a night within 30 minutes of bedtime and with at least 7 hours before the planned wake time.9 The recommended starting dosage is 10 mg. If this dosage is well tolerated but insufficiently effective, the dosage can be increased to a maximum of 20 mg. The 5-mg dosage is recommended for individuals taking a moderate CYP3A inhibitor. Generally, patients should take the lowest effective dosage.
There are no specified limitations on the duration of suvorexant use. There is no evidence of withdrawal effects when discontinuing the medication. Patients taking suvorexant should be educated about possible next-day effects that might impair driving or other activities that require full mental alertness, especially if they are taking the 20-mg dosage.
Bottom Line
Suvorexant is FDA-approved for treating sleep onset and sleep maintenance insomnia. The drug is a dual orexin-receptor antagonist, which targets persistent CNS hyperarousal. In clinical trials, suvorexant improved the ability to fall asleep and remain asleep in patients with insomnia. It is generally safe and well tolerated. However, these studies evaluated dosages higher than those approved by the FDA.
Related Resources
• Jacobson LH, Callander GE, Hoyer D. Suvorexant for the treatment of insomnia. Expert Rev Clin Pharmacol. 2014; 7(6):711-730.
• Neubauer DN. New and emerging pharmacotherapeutic approaches for insomnia. Int Rev Psychiatry. 2014;26(2): 214-224.
Drug Brand Names
Doxepin • Silenor Suvorexant • Belsomra
Digoxin • Lanoxin Zaleplon • Sonata
Eszopiclone • Lunesta Zolpidem • Ambien,
Ramelteon • Rozerem Edluar, Intermezzo
Disclosure
Dr. Neubauer is a consultant to Ferring Pharmaceuticals and Vanda Pharmaceuticals.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
1. U.S. Food and Drug Administration. Survorexant (orexin receptor antagonist). For insomnia characterized by difficulties with sleep onset and/or maintenance. http:// www.fda.gov/downloads/AdvisoryCommittees/ CommitteesMeetingMaterials/Drugs/Peripheraland CentralNervousSystemDrugsAdvisoryCommittee/ UCM352969.pdf. Published May 22, 2013. Accessed November 24, 2014.
2. Mignot E. Sleep, sleep disorders and hypocretin (orexin). Sleep Med. 2004;5(suppl 1):S2-S8.
3. Nishino S. The hypocretin/orexin receptor: therapeutic prospective in sleep disorders. Expert Opin Investig Drugs. 2007;16(11):1785-1797.
4. Citrome L. Suvorexant for insomnia: a systematic review of the efficacy and safety profile for this newly approved hypnotic - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2014;68(12):1429-1441.
5. Winrow CJ, Gotter AL, Cox CD, et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J Neurogenet. 2011;25(1-2):52-61.
6. Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24(12):726-731.
7. Sakurai T, Amemiya A, Ishii M, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92(4):573-585.
8. Winrow CJ, Renger JJ. Discovery and development of orexin receptor antagonists as therapeutics for insomnia. Br J Pharmacol. 2014;171(2):283-293.
9. Belsomra [package insert]. Whitehouse Station, NJ: Merck; 2014.
10. Herring WJ, Snyder E, Budd K, et al. Orexin receptor antagonism for treatment of insomnia: a randomized clinical trial of suvorexant. Neurology. 2012;79(23):2265-2274.
11. Ivgy-May N, Snavely D, Minigh J, et al. Efficacy of suvorexant, an orexin receptor antagonist, in patients with primary insomnia: integrated results from 2 similarly designed phase 3 trials. Sleep. 2013;36(abstract supplement): A192.
12. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1-year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(5):461-471.
13. Merck Sharp and Dohme Corporation. Suvorexant advisory committee meeting briefing document. http:// www.fda.govdownloadsadvisorycommittees/committee smeetingmaterials/drugsperipheralandcentralnervous systemdrugsadvisorycommittee/ucm352970.pdf. Published May 22, 2013. Accessed November 24, 2014.
Vortioxetine for major depressive disorder
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
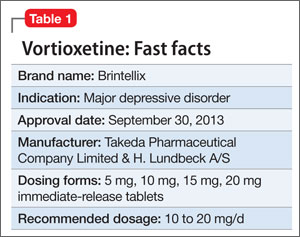
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.


Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Vortioxetine is FDA-approved to treat major depressive disorder (MDD) (Table 1), having shown efficacy in relieving depressive symptoms in clinical trials.1 Vortioxetine’s mechanism of action enhances CNS serotonergic activity through inhibiting serotonin (5-HT) reuptake, agonizing the 5-HT1A receptor, partially agonizing the 5-HT1B receptor, and antagonizing the 5-HT3, 5-HT1D, and 5-HT7 receptors.
Clinical implications
It is hypothesized that depression is a heterogeneous disease caused by dysregulation of serotonin, norepinephrine, and dopamine, subsequently producing mood and neurovegetative symptoms of depression. Preclinical, in vivo studies indicate that vortioxetine enhances levels of serotonin, norepinephrine, dopamine, acetylcholine, and histamine in specific areas of the brain with the ability to improve depressive symptoms. Vortioxetine’s multimodal activity can be a useful alternative to other serotonergic antidepressants for some patients who are partial responders or non-responders to other treatment options. In addition, vortioxetine appears to have minimal effect on weight2 and sexual function—the latter being dose-dependent.3
How does it work?
Vortioxetine differs from other antidepressants in its multimodal activity (ie, affecting G-protein mode receptors, ion channel mode receptors, and neurotransmitter transporters). It inhibits the serotonin transporter (Ki = 1.6 nM), causing subsequent inhibition of serotonin reuptake into presynaptic neurons as well as selectively acting on the other subtypes of serotonergic receptors; however, activity on the norepinephrine transporter (Ki = 113 nM) and dopamine transporter (Ki > 1000 nM) is minimal. It is believed that mood-regulating effects of vortioxetine are caused by inhibition of serotonin reuptake, prolonged availability of serotonin to the postsynaptic neurons, its agonist activity on the 5-HT1A receptor (Ki = 15 nM), and partial agonist activity on the 5-HT1B receptor (Ki = 33 nM). Vortioxetine has strong affinity for the 5-HT3 receptor (Ki = 3.7 nM), which plays a role in modulation of centrally mediated nausea and vomiting. Positron emission tomography studies in humans determined that the occupancy of 5-HT transporter was 50% at 5 mg/d, 65% at 10 mg/d, and 80% at 20 mg/d.1,4 Human studies did not show that vortioxetine causes QTc prolongation.
Pharmacokinetics
Therapeutic activity of vortioxetine is thought to be due to the parent drug. It has a half-life of approximately 66 hours, and achieves steady state in 13.5 to 19 days. Bioavailability of vortioxetine is 75%; absorption does not depend on food; and 98% of drug is bound on plasma proteins.
Vortioxetine has linear pharmacokinetics, with maximum plasma concentration 7 to 11 hours after ingestion. The medication is metabolized primarily by oxidation through cytochrome P (CYP) 450: CYP2D6 (primary), CYP 3A4/5, CYP 2C19, CYP 2C9, CYP2A6, CYP2C8, and CYP2B6 with subsequent glucuronidation. This predisposes vortioxetine to potential pharmacokinetic drug-drug interaction warranting dose adjustment consideration when vortioxetine is coadministered with compounds inhibiting CYP2D6 or inducing CYP3A4 for ≥14 days, or for patients identified as poor 2D6 metabolizers.
In addition, coadministration of vortioxetine with serotonergic medications such as triptans, other antidepressants, and tramadol can cause potentially life-threatening serotonin syndrome, characterized by mental status changes, autonomic instability, neuromuscular aberrations, and GI symptoms. Concomitant use of vortioxetine and a nonsteroidal anti-inflammatory drug, aspirin, or warfarin can result in abnormal bleeding. Coadministration of vortioxetine with another highly protein-bound drug may increase or decrease the free concentration of either drug depending on the binding affinity of the drug for the protein.
Efficacy
Vortioxetine reduced depressive symptoms in 6 positive, 6- to 8-week, double-blind, placebo controlled and randomized studies and 1 maintenance study.1 Subjects were adults (Studies 1 to 5) and geriatric patients from age 64 to 88 who had ≥1 depressive episode before age 60 (Study 6). All met DSM-IV-TR criteria for MDD. Subjects with cognitive impairment scoring <24 on the Mini-Mental Status Examination and children were excluded. Depending on the study, response to the treatment was primarily measured on the Montgomery-Åsberg Depression Rating Scale (MADRS) or Hamilton Depression Rating Scale (HAM-D).
See Table 2 for a description of the positive studies, including dosages. In all studies, vortioxetine was superior to placebo at least one dosage for treating depression. In the 6- to 8-week placebo-controlled studies, an effect of vortioxetine based on the primary efficacy measure was generally observed starting at Week 2; that effect increased in subsequent weeks with the full antidepressant effect of vortioxetine generally not seen until study Week 4 or later.1
The maintenance treatment study included 639 patients who met DSM-IV-TR criteria for MDD. This study lasted for as long as 64 weeks. The first 12-week period was open-label, during which patients were treated with vortioxetine, 5 mg/d or 10 mg/d, with a possibility to adjust the dosage in the first 8 weeks. By the end of Week 12, 396 subjects achieved remission (MADRS <10), 75% of whom were taking vortioxetine, 10 mg/d. These patients were then randomly assigned to placebo or the dosage of vortioxetine to which they had responded, and continued the study for as long as 64 weeks. Time to relapse (MADRS total score ≥22) or an insufficient therapeutic response (as judged by the investigator) was the primary efficacy outcome, and demonstrated that vortioxetine was superior to placebo.
Tolerability
The tolerability of vortioxetine is comparable with other serotonergic antidepressants. In pooled analysis of pre-marketing studies, 5% to 8% of patients receiving vortioxetine (5 to 20 mg/d) discontinued treatment because of adverse effects (AEs), compared with 4% in the placebo group. Nausea was the most commonly reported AE leading to discontinuation and appeared to be dose dependent.
AEs, such as nausea, constipation, and vomiting, most commonly occurred in the first week of treatment, with a median duration of 2 weeks.5 In the 6- to 8-week trials, the most common AEs were nausea, constipation, and vomiting. In longer trials (24 to 64 weeks), the most common AE was nausea.
In 6- to 8-week placebo-controlled studies, vortioxetine was not associated with any clinically significant effect on vital signs or laboratory values in hematology, urinalysis, or serum chemistry (except sodium). Hyponatremia, the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH), has occurred. The risk of developing SIADH and resultant hyponatremia is greater in geriatric patients and patients taking a diuretic.
Abruptly discontinuing vortioxetine can cause transient withdrawal symptoms, including headache and muscle tension, especially at a higher dosage (15 to 20 mg/d). Gradual tapering can reduce withdrawal symptoms.
Specific clinical issues
All antidepressants have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation and behaviors during the first months of treatment and with dosage changes.
Vortioxetine is categorized as pregnancy category C. Newborns exposed to a selective serotonin reuptake inhibitor (SSRI) in pregnancy may have an increased risk of persistent pulmonary hypertension during the neonatal period. When taken during the third trimester of pregnancy, SSRIs and serotonin-norepinephrine reuptake inhibitors can cause serious neonatal complications, including respiratory distress, cyanosis, apnea, and seizures, which may require longer hospitalization, respiratory support, or tube feeding for the infant. Consider risks and benefits of third-trimester use of an antidepressant. It is not known if vortioxetine is present in human breast milk.
Clinical studies on vortioxetine in pediatric patients have not been conducted.
No dosage adjustment is recommended on the basis of age for geriatric patients. No dose adjustment of vortioxetine is necessary on the basis of race, sex, ethnicity, renal function, or mild to moderate hepatic impairment. See Table 3 for practice points when prescribing vortioxetine. See Table 4 for contraindications to vortioxetine.
Dosing
The recommended starting dosage is 10 mg, administered orally once daily without regard to meals. Dosage should then be increased to 20 mg/d, as clinically warranted and tolerated. Consider a dosage decrease to 5 mg/d in patients who do not tolerate higher dosages or require drug adjustment because of drug-drug interaction or poor 2D6 metabolizer status.
Bottom Line
FDA-approved for major depressive disorder in adults, vortioxetine reduced depressive symptoms in 6 positive, double-blind, placebo-controlled, and randomized studies. The multimodal activity of vortioxeine can be a useful alternative to serotonergic antidepressants for some patients who are partial responders or nonresponders. Tolerability is comparable with other serotonergic antidepressants.
Related Resources
- Alam MY, Jacobsen PL, Chen Y, et al. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: results of an open-label, flexible-dose, 52-week extension study. Int Clin Psychopharmacol. 2014; 29(1):36-44.
- Mahableshwarkar AR, Jacobsen PL, Chen Y. A randomized, double-blind trial of 2.5 mg and 5 mg vortioxetine (Lu AA21004) versus placebo for 8 weeks in adults with major depressive disorder. Curr Med Res Opin. 2013;29(3):217-226.
Drug Brand Names
Linezolid • Zyvox Vortioxetine • Brintellix
Methylene blue • Urolene Blue Warfarin • Coumadin
Tramadol • Ultram
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
1. Vortioxetine [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2013.
2. Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. J Clin Psychiatry. 2010;71(10):1259-1272.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a comprehensive review and meta-analysis. J Clin Psychopharmacol. 2009; 29(3):259-266.
4. Chen G, Lee R, Højer A, et al. Pharmacokinetic drug interactions involving vortioxetine (LU AA 21004), a multimodal antidepressant. Clin Drug Invetig. 2013; 33(10):727-736.
5. Citrome L. Vortioxetine for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Prac. 2014;68(1):60-82.
Levomilnacipran for the treatment of major depressive disorder
In July 2013, the FDA approved levomilnacipran for the treatment of major depressive disorder (MDD) in adults.1 It is available in a once-daily, extended-release formulation (Table 1).1 The drug is the fifth serotonin-norepinephrine reuptake inhibitor (SNRI) to be sold in the United States and the fourth to receive FDA approval for treating MDD.

Levomilnacipran is believed to be the more active enantiomer of milnacipran, which has been available in Europe for years and was approved by the FDA in 2009 for treating fibromyalgia. Efficacy of levomilnacipran for treating patients with MDD was established in three 8-week randomized controlled trials (RCTs).1
Clinical implications
Levomilnacipran is indicated for treating MDD in adults and is unique compared with other SNRIs because it is relatively more selective for norepinephrine reuptake inhibition (NRI) compared with serotonin reuptake inhibition (SRI).1 In vitro studies demonstrate that the drug has >10-fold greater selectivity for norepinephrine reuptake inhibition than it does for serotonin reuptake inhibition, compared with duloxetine or venlafaxine.2
This difference in selectivity could lend itself to treating symptoms of MDD that might be related to norepinephrine deficiency; these include decreased concentration, lassitude, mental and physical slowing, and decreased self-care.3,4 Some authors claim that individual patients could experience improvement in their social and occupational functioning in addition to improvement in the core symptoms of depression.5
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved for treating fibromyalgia in the United States and approved for treating depression in many other countries.6 In general, enantiomeric formulations are believed to have advantages over racemic formulations because they are less complex and have a more selective pharmacodynamic profile, better therapeutic index, lower liability for drug-drug interactions (DDIs), and a less complicated relationship between plasma concentration and pharmacodynamic effect.6 In addition, regulatory guidelines in the United States recommend development of enantiomers over racemates where appropriate.7
How it works
Levomilnacipran’s exact mechanism of action is unknown. Similar to other SNRIs, it binds with high affinity to the serotonin (5-HT) and norepinephrine (NE) transporters and potently inhibits 5-HT and NE reuptake. Levomilnacipran lacks significant affinity for any other receptors, ion channels, or transporters tested in vitro.2 It differs from other SNRIs such as venlafaxine and duloxetine in having higher selectivity for norepinephrine vs serotonin reuptake inhibition. In vitro studies demonstrated a 2-fold preference for NE over 5-HT reuptake inhibition.2
Pharmacokinetics
Levomilnacipran reaches maximum plasma concentration within 6 to 8 hours of oral administration and has a half-life of approximately 12 hours, which makes it suitable for once-daily dosing. The concentration of levomilnacipran at steady state is proportional to the dosage of the drug when administered within the range of 25 to 300 mg once daily.1
The drug’s mean apparent total clearance is 21 to 29 liters/hour and its bioavailability is not significantly affected when taken with food. The drug is widely distributed in the body and is converted primarily to 2 metabolites: desethy levomilnacipran and p-hydroxy-levomilnacipran. Both metabolites are inactive and undergo further conjugation with glucuronide. The drug is eliminated primarily via renal excretion.1
The major enzyme that catalyzes metabolism of levomilnacipram is cytochrome P 450 (CYP) 3A4, which makes it susceptible to DDIs with drugs that inhibit or induce this enzyme. For example, a person taking levomilnacipran with a potent CYP3A4 inhibitor, such as ketoconazole, may require a dosage adjustment. No dosage adjustment is needed when given with a CYP3A4 inducer or substrate. Drinking alcohol with levomilnacipran may cause more rapid release of the drug into the blood stream.1
Efficacy
Levomilnacipran decreased core symptoms of MDD and showed a statistically significant separation from placebo in 2 phase III RCTs (Table 2).3,5 The first study was a 10-week flexible dose (75 or 100 mg) trial in 563 outpatients age 18 to 70 who met DSM-IV-TR criteria of MDD for >1 month and had a 17-item Hamilton Depression Rating Scale (HDRS-17) score >22 and a Sheehan Disability Scale (SDS) score >10.3 The primary efficacy measure was change in Montgomery-Åsberg Depression Rating Scale (MADRS) score from baseline to week 10. Secondary efficacy measures included the HDRS-17, SDS, and Clinical Global Impressions-improvement scale. Efficacy analyses included 276 subjects treated with levomilnacipran and 277 treated with placebo.3
Levomilnacipran was significantly superior to placebo on the MADRS and HDRS-17 from baseline to week 10. Response and remission rates were
significantly greater for the levomilnacipran group compared with placebo. Response exceeded the 10% average advantage for drug vs placebo and 46% of levomilnacipran-treated patients achieved remission.3
The number needed to treat (NNT), based on the MADRS scores for the levomilnacipran group compared with the placebo group, was 6 for response and 5 for remission.3 By comparison, most studies of venlafaxine demonstrate a NNT of 8.3
Levomilnacipran generally was reported to be safe and well tolerated. The most common adverse events leading to discontinuation in the levomilnacipran group were nausea, vomiting, change in systolic and diastolic blood pressure, and increase in heart rate. The favorable tolerability profile of levomilnacipran may relate to the 2-fold greater potency for NE reuptake inhibition compared with 5-HT reuptake inhibition.3
The second study was an 11-week, fixed-dose trial of levomilnacipran using 40, 80, or 120 mg. A total of 724 outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD and who had an ongoing episode of depression lasting >8 weeks were randomly assigned to receive placebo (n = 179) or levomilnacipran at 40 mg (n = 181), 80 mg (n = 181), or 120 mg (n = 183) once daily for 8 weeks of double-blind treatment followed by a 2-week, double-blind taper of the drug.5 The primary efficacy parameter was change from baseline on the MADRS and the secondary efficacy parameter was change from baseline in SDS total score. HDRS-17, CGI-I, and CGI-S were included as secondary outcome measures.5
Significant difference in MADRS total score were seen in the levomilnacipran group compared with the placebo group (least mean squared difference: 40 mg/d, −3.23; 80 mg/d, −3.99; and 120 mg/d, −4.86). Higher dosages produced a numerically greater change and significant separation from placebo occurred sooner in the 80-mg and 120-mg groups compared with the 40-mg group.5
Significant differences vs placebo were consistently observed across secondary outcome measures for the higher dosages of levomilnacipran, and improvement in SDS total score was noted in all levomilnacipran groups compared with the placebo group. When dosed at 120 mg/d, levomilnacipran produced significant improvement vs placebo on all SDS subscales and was as well tolerated as the 80 mg dosage.5
No new safety concerns were observed in the study. A dose-response relationship in tolerability was not demonstrated and the number of patients reporting adverse events and who discontinued participation because of adverse events was higher in the 80-mg group than in the 40-mg and 120-mg groups.5
Tolerability
Overall, levomilnacipran was well tolerated in clinical trials, during which 2,673 subjects were exposed to the drug—translating to 942 patient-years of exposure. These patients ranged in age from 18 to 78; 825 of these subjects were enrolled in long-term studies for 1 year. Dosing of levomilnacipran during these studies ranged from 40 to 120 mg once daily, without regard to food.1
Nine percent of patients who received levomilnacipran in short-term studies discontinued because of adverse events, compared with 3% of patients who received placebo. The most common adverse event reported was nausea; other common adverse events reported included constipation, hyperhidrosis, elevated heart rate, erectile dysfunction, tachycardia, palpitations, and vomiting. Of these events, only erectile dysfunction and urinary hesitation were dose-related.1 Among levomilnacipran-treated female patients, <2% reported adverse events related to sexual dysfunction.
All SNRIs have well established associations with elevation in blood pressure and heart rate. Levomilnacipran resulted in a mean increase of 3 mm HG in systolic and 3.2 mm Hg in diastolic blood pressure in short-term, placebo-controlled trials.1
Orthostatic hypotension was observed in 11.6% of patients in the levomilnacipran groups, compared with 9.7% in placebo groups in all short-term studies. Orthostatic reductions of blood pressure occurred in 5.8%, 6.1%, and 9.8% of levomilnacipran-treated patients with dosages of 40, 80, and 120 mg/d respectively, indicating a dose-dependent adverse event. A mean increase in heart rate of 7.2 beats per minute (bpm) also was seen in short-term studies in the levomilnacipran-treated group compared with 0.3 bpm in the placebo-treated group.1 Clinicians should monitor blood pressure and heart rate routinely because of potential increases seen in some subjects in these studies, which excluded those who had significant cardiovascular disease.
Unique clinical issues
Both in-vitro and in-vivo studies found that levomilnacipran exhibited more potency for NE reuptake inhibition than for 5-HT reuptake inhibition at the lowest effective dosage (10 mg/kg). However as the dosage was increased (20 mg/kg and 40 mg/kg), it was equally potent at NE and 5-HT reuptake inhibition. This is in contrast to venlafaxine, which demonstrates a similar, but opposite, effect in terms of potentiation at the 5-HT and NE reuptake pumps.2
The greater noradrenergic effect of levomilnacipran could lend itself to treating certain subgroups of patients whose symptoms are believed to be related to deficiencies in NE (eg, lassitude).4 This concept is theoretical, and was not explicitly studied in clinical trials and the drug is not labeled in this way.
Contraindications
Contraindications to levomilnacipran are similar to those seen with SSRIs and SNRIs, including concomitant use of a monoamine oxidase inhibitor (MAOI) and the use of the levomilnacipran within 14 days of stopping an MAOI. Contraindications unique to levomilnacipran include hypersensitivity to levomilnacipran, milnacipran, or any component specific to the formulation; and uncontrolled narrow-angle glaucoma.1
Dosing
The recommended dosage range of levomilnacipran is 40 to 120 mg once
daily with or without food. The capsules should be swallowed whole and should not be opened or crushed. As with most psychotropics, levomilnacipran should be taken at approximately the same time each day.1
The manufacturer recommends an initial dose of levomilnacipran of 20 mg once daily for 2 days, increased to 40 mg once daily. Based on efficacy and tolerably, levomilnacipran can be increased in increments of 40 mg every 2 days.
Dosage adjustment is recommended for patients with moderate or severe renal impairment; and the maintenance dosage should not exceed 80 mg and 40 mg respectively in these populations. As with many antidepressants, gradual dosage reduction is recommended to avoid discontinuation symptoms.
Bottom Line
Levomilnacipran is FDA-approved for treating major depressive disorder in adults. In 2 randomized controlled trials, the drug showed a significant separation from placebo. Levomilnacipran generally was reported to be safe and well tolerated; common adverse events were nausea, vomiting, changes in blood pressure, and an increase in heart rate.
Related Resources
- Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? [published online September 8, 2013]. Int J Clin Pract. doi: 10.1111/ijcp.12298.
- Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
Drug Brand Names
Duloxetine • Cymbalta Milnacipran • Savella
Ketoconazole • Nizoral Venlafaxine • Effexor Levomilnacipran • Fetzima
Disclosures
Dr. Macaluso has been the principal investigator for clinical trials for AbbVie, Eisai, Envivo, Janssen, Naurex, and Pfizer. All clinical trial and study contracts and payments were made through the Kansas University Medical Center Research Institute. Drs. Kazanchi and Malhotra report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Fetzima [package insert]. St. Louis, MO: Forest Laboratories; 2013.
2. Auclair AL, Martel JC, Assié MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338-347.
3. Montgomery SA, Mansuy L, Ruth A, et al. Efficacy and Safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74(4):363-369.
4. Kasper S, Meshkat D, Kutzelnigg A. Improvement of the noradrenergic symptom cluster following treatment with milnacipran. Neuropsychiatric Dis Treat. 2011; 7(suppl 1):21-27.
5. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
6. Hutt AJ, Vanetová J. The chiral switch: the development of single enantiomer drugs from racemates. Acta Facultatis Pharmaceuticae Universitatis Comenianae. 2003; 50(7):23.
7. U.S. Food and Drug Administration. Development of new stereoisomeric drugs. Published May 1, 1992. http://www.fda.gov/drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122883.htm#.UKHEWm4ZyYE.email. Accessed October 8, 2013.
In July 2013, the FDA approved levomilnacipran for the treatment of major depressive disorder (MDD) in adults.1 It is available in a once-daily, extended-release formulation (Table 1).1 The drug is the fifth serotonin-norepinephrine reuptake inhibitor (SNRI) to be sold in the United States and the fourth to receive FDA approval for treating MDD.
Levomilnacipran is believed to be the more active enantiomer of milnacipran, which has been available in Europe for years and was approved by the FDA in 2009 for treating fibromyalgia. Efficacy of levomilnacipran for treating patients with MDD was established in three 8-week randomized controlled trials (RCTs).1
Clinical implications
Levomilnacipran is indicated for treating MDD in adults and is unique compared with other SNRIs because it is relatively more selective for norepinephrine reuptake inhibition (NRI) compared with serotonin reuptake inhibition (SRI).1 In vitro studies demonstrate that the drug has >10-fold greater selectivity for norepinephrine reuptake inhibition than it does for serotonin reuptake inhibition, compared with duloxetine or venlafaxine.2
This difference in selectivity could lend itself to treating symptoms of MDD that might be related to norepinephrine deficiency; these include decreased concentration, lassitude, mental and physical slowing, and decreased self-care.3,4 Some authors claim that individual patients could experience improvement in their social and occupational functioning in addition to improvement in the core symptoms of depression.5
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved for treating fibromyalgia in the United States and approved for treating depression in many other countries.6 In general, enantiomeric formulations are believed to have advantages over racemic formulations because they are less complex and have a more selective pharmacodynamic profile, better therapeutic index, lower liability for drug-drug interactions (DDIs), and a less complicated relationship between plasma concentration and pharmacodynamic effect.6 In addition, regulatory guidelines in the United States recommend development of enantiomers over racemates where appropriate.7
How it works
Levomilnacipran’s exact mechanism of action is unknown. Similar to other SNRIs, it binds with high affinity to the serotonin (5-HT) and norepinephrine (NE) transporters and potently inhibits 5-HT and NE reuptake. Levomilnacipran lacks significant affinity for any other receptors, ion channels, or transporters tested in vitro.2 It differs from other SNRIs such as venlafaxine and duloxetine in having higher selectivity for norepinephrine vs serotonin reuptake inhibition. In vitro studies demonstrated a 2-fold preference for NE over 5-HT reuptake inhibition.2
Pharmacokinetics
Levomilnacipran reaches maximum plasma concentration within 6 to 8 hours of oral administration and has a half-life of approximately 12 hours, which makes it suitable for once-daily dosing. The concentration of levomilnacipran at steady state is proportional to the dosage of the drug when administered within the range of 25 to 300 mg once daily.1
The drug’s mean apparent total clearance is 21 to 29 liters/hour and its bioavailability is not significantly affected when taken with food. The drug is widely distributed in the body and is converted primarily to 2 metabolites: desethy levomilnacipran and p-hydroxy-levomilnacipran. Both metabolites are inactive and undergo further conjugation with glucuronide. The drug is eliminated primarily via renal excretion.1
The major enzyme that catalyzes metabolism of levomilnacipram is cytochrome P 450 (CYP) 3A4, which makes it susceptible to DDIs with drugs that inhibit or induce this enzyme. For example, a person taking levomilnacipran with a potent CYP3A4 inhibitor, such as ketoconazole, may require a dosage adjustment. No dosage adjustment is needed when given with a CYP3A4 inducer or substrate. Drinking alcohol with levomilnacipran may cause more rapid release of the drug into the blood stream.1
Efficacy
Levomilnacipran decreased core symptoms of MDD and showed a statistically significant separation from placebo in 2 phase III RCTs (Table 2).3,5 The first study was a 10-week flexible dose (75 or 100 mg) trial in 563 outpatients age 18 to 70 who met DSM-IV-TR criteria of MDD for >1 month and had a 17-item Hamilton Depression Rating Scale (HDRS-17) score >22 and a Sheehan Disability Scale (SDS) score >10.3 The primary efficacy measure was change in Montgomery-Åsberg Depression Rating Scale (MADRS) score from baseline to week 10. Secondary efficacy measures included the HDRS-17, SDS, and Clinical Global Impressions-improvement scale. Efficacy analyses included 276 subjects treated with levomilnacipran and 277 treated with placebo.3
Levomilnacipran was significantly superior to placebo on the MADRS and HDRS-17 from baseline to week 10. Response and remission rates were
significantly greater for the levomilnacipran group compared with placebo. Response exceeded the 10% average advantage for drug vs placebo and 46% of levomilnacipran-treated patients achieved remission.3
The number needed to treat (NNT), based on the MADRS scores for the levomilnacipran group compared with the placebo group, was 6 for response and 5 for remission.3 By comparison, most studies of venlafaxine demonstrate a NNT of 8.3
Levomilnacipran generally was reported to be safe and well tolerated. The most common adverse events leading to discontinuation in the levomilnacipran group were nausea, vomiting, change in systolic and diastolic blood pressure, and increase in heart rate. The favorable tolerability profile of levomilnacipran may relate to the 2-fold greater potency for NE reuptake inhibition compared with 5-HT reuptake inhibition.3
The second study was an 11-week, fixed-dose trial of levomilnacipran using 40, 80, or 120 mg. A total of 724 outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD and who had an ongoing episode of depression lasting >8 weeks were randomly assigned to receive placebo (n = 179) or levomilnacipran at 40 mg (n = 181), 80 mg (n = 181), or 120 mg (n = 183) once daily for 8 weeks of double-blind treatment followed by a 2-week, double-blind taper of the drug.5 The primary efficacy parameter was change from baseline on the MADRS and the secondary efficacy parameter was change from baseline in SDS total score. HDRS-17, CGI-I, and CGI-S were included as secondary outcome measures.5
Significant difference in MADRS total score were seen in the levomilnacipran group compared with the placebo group (least mean squared difference: 40 mg/d, −3.23; 80 mg/d, −3.99; and 120 mg/d, −4.86). Higher dosages produced a numerically greater change and significant separation from placebo occurred sooner in the 80-mg and 120-mg groups compared with the 40-mg group.5
Significant differences vs placebo were consistently observed across secondary outcome measures for the higher dosages of levomilnacipran, and improvement in SDS total score was noted in all levomilnacipran groups compared with the placebo group. When dosed at 120 mg/d, levomilnacipran produced significant improvement vs placebo on all SDS subscales and was as well tolerated as the 80 mg dosage.5
No new safety concerns were observed in the study. A dose-response relationship in tolerability was not demonstrated and the number of patients reporting adverse events and who discontinued participation because of adverse events was higher in the 80-mg group than in the 40-mg and 120-mg groups.5
Tolerability
Overall, levomilnacipran was well tolerated in clinical trials, during which 2,673 subjects were exposed to the drug—translating to 942 patient-years of exposure. These patients ranged in age from 18 to 78; 825 of these subjects were enrolled in long-term studies for 1 year. Dosing of levomilnacipran during these studies ranged from 40 to 120 mg once daily, without regard to food.1
Nine percent of patients who received levomilnacipran in short-term studies discontinued because of adverse events, compared with 3% of patients who received placebo. The most common adverse event reported was nausea; other common adverse events reported included constipation, hyperhidrosis, elevated heart rate, erectile dysfunction, tachycardia, palpitations, and vomiting. Of these events, only erectile dysfunction and urinary hesitation were dose-related.1 Among levomilnacipran-treated female patients, <2% reported adverse events related to sexual dysfunction.
All SNRIs have well established associations with elevation in blood pressure and heart rate. Levomilnacipran resulted in a mean increase of 3 mm HG in systolic and 3.2 mm Hg in diastolic blood pressure in short-term, placebo-controlled trials.1
Orthostatic hypotension was observed in 11.6% of patients in the levomilnacipran groups, compared with 9.7% in placebo groups in all short-term studies. Orthostatic reductions of blood pressure occurred in 5.8%, 6.1%, and 9.8% of levomilnacipran-treated patients with dosages of 40, 80, and 120 mg/d respectively, indicating a dose-dependent adverse event. A mean increase in heart rate of 7.2 beats per minute (bpm) also was seen in short-term studies in the levomilnacipran-treated group compared with 0.3 bpm in the placebo-treated group.1 Clinicians should monitor blood pressure and heart rate routinely because of potential increases seen in some subjects in these studies, which excluded those who had significant cardiovascular disease.
Unique clinical issues
Both in-vitro and in-vivo studies found that levomilnacipran exhibited more potency for NE reuptake inhibition than for 5-HT reuptake inhibition at the lowest effective dosage (10 mg/kg). However as the dosage was increased (20 mg/kg and 40 mg/kg), it was equally potent at NE and 5-HT reuptake inhibition. This is in contrast to venlafaxine, which demonstrates a similar, but opposite, effect in terms of potentiation at the 5-HT and NE reuptake pumps.2
The greater noradrenergic effect of levomilnacipran could lend itself to treating certain subgroups of patients whose symptoms are believed to be related to deficiencies in NE (eg, lassitude).4 This concept is theoretical, and was not explicitly studied in clinical trials and the drug is not labeled in this way.
Contraindications
Contraindications to levomilnacipran are similar to those seen with SSRIs and SNRIs, including concomitant use of a monoamine oxidase inhibitor (MAOI) and the use of the levomilnacipran within 14 days of stopping an MAOI. Contraindications unique to levomilnacipran include hypersensitivity to levomilnacipran, milnacipran, or any component specific to the formulation; and uncontrolled narrow-angle glaucoma.1
Dosing
The recommended dosage range of levomilnacipran is 40 to 120 mg once
daily with or without food. The capsules should be swallowed whole and should not be opened or crushed. As with most psychotropics, levomilnacipran should be taken at approximately the same time each day.1
The manufacturer recommends an initial dose of levomilnacipran of 20 mg once daily for 2 days, increased to 40 mg once daily. Based on efficacy and tolerably, levomilnacipran can be increased in increments of 40 mg every 2 days.
Dosage adjustment is recommended for patients with moderate or severe renal impairment; and the maintenance dosage should not exceed 80 mg and 40 mg respectively in these populations. As with many antidepressants, gradual dosage reduction is recommended to avoid discontinuation symptoms.
Bottom Line
Levomilnacipran is FDA-approved for treating major depressive disorder in adults. In 2 randomized controlled trials, the drug showed a significant separation from placebo. Levomilnacipran generally was reported to be safe and well tolerated; common adverse events were nausea, vomiting, changes in blood pressure, and an increase in heart rate.
Related Resources
- Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? [published online September 8, 2013]. Int J Clin Pract. doi: 10.1111/ijcp.12298.
- Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
Drug Brand Names
Duloxetine • Cymbalta Milnacipran • Savella
Ketoconazole • Nizoral Venlafaxine • Effexor Levomilnacipran • Fetzima
Disclosures
Dr. Macaluso has been the principal investigator for clinical trials for AbbVie, Eisai, Envivo, Janssen, Naurex, and Pfizer. All clinical trial and study contracts and payments were made through the Kansas University Medical Center Research Institute. Drs. Kazanchi and Malhotra report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
In July 2013, the FDA approved levomilnacipran for the treatment of major depressive disorder (MDD) in adults.1 It is available in a once-daily, extended-release formulation (Table 1).1 The drug is the fifth serotonin-norepinephrine reuptake inhibitor (SNRI) to be sold in the United States and the fourth to receive FDA approval for treating MDD.
Levomilnacipran is believed to be the more active enantiomer of milnacipran, which has been available in Europe for years and was approved by the FDA in 2009 for treating fibromyalgia. Efficacy of levomilnacipran for treating patients with MDD was established in three 8-week randomized controlled trials (RCTs).1
Clinical implications
Levomilnacipran is indicated for treating MDD in adults and is unique compared with other SNRIs because it is relatively more selective for norepinephrine reuptake inhibition (NRI) compared with serotonin reuptake inhibition (SRI).1 In vitro studies demonstrate that the drug has >10-fold greater selectivity for norepinephrine reuptake inhibition than it does for serotonin reuptake inhibition, compared with duloxetine or venlafaxine.2
This difference in selectivity could lend itself to treating symptoms of MDD that might be related to norepinephrine deficiency; these include decreased concentration, lassitude, mental and physical slowing, and decreased self-care.3,4 Some authors claim that individual patients could experience improvement in their social and occupational functioning in addition to improvement in the core symptoms of depression.5
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved for treating fibromyalgia in the United States and approved for treating depression in many other countries.6 In general, enantiomeric formulations are believed to have advantages over racemic formulations because they are less complex and have a more selective pharmacodynamic profile, better therapeutic index, lower liability for drug-drug interactions (DDIs), and a less complicated relationship between plasma concentration and pharmacodynamic effect.6 In addition, regulatory guidelines in the United States recommend development of enantiomers over racemates where appropriate.7
How it works
Levomilnacipran’s exact mechanism of action is unknown. Similar to other SNRIs, it binds with high affinity to the serotonin (5-HT) and norepinephrine (NE) transporters and potently inhibits 5-HT and NE reuptake. Levomilnacipran lacks significant affinity for any other receptors, ion channels, or transporters tested in vitro.2 It differs from other SNRIs such as venlafaxine and duloxetine in having higher selectivity for norepinephrine vs serotonin reuptake inhibition. In vitro studies demonstrated a 2-fold preference for NE over 5-HT reuptake inhibition.2
Pharmacokinetics
Levomilnacipran reaches maximum plasma concentration within 6 to 8 hours of oral administration and has a half-life of approximately 12 hours, which makes it suitable for once-daily dosing. The concentration of levomilnacipran at steady state is proportional to the dosage of the drug when administered within the range of 25 to 300 mg once daily.1
The drug’s mean apparent total clearance is 21 to 29 liters/hour and its bioavailability is not significantly affected when taken with food. The drug is widely distributed in the body and is converted primarily to 2 metabolites: desethy levomilnacipran and p-hydroxy-levomilnacipran. Both metabolites are inactive and undergo further conjugation with glucuronide. The drug is eliminated primarily via renal excretion.1
The major enzyme that catalyzes metabolism of levomilnacipram is cytochrome P 450 (CYP) 3A4, which makes it susceptible to DDIs with drugs that inhibit or induce this enzyme. For example, a person taking levomilnacipran with a potent CYP3A4 inhibitor, such as ketoconazole, may require a dosage adjustment. No dosage adjustment is needed when given with a CYP3A4 inducer or substrate. Drinking alcohol with levomilnacipran may cause more rapid release of the drug into the blood stream.1
Efficacy
Levomilnacipran decreased core symptoms of MDD and showed a statistically significant separation from placebo in 2 phase III RCTs (Table 2).3,5 The first study was a 10-week flexible dose (75 or 100 mg) trial in 563 outpatients age 18 to 70 who met DSM-IV-TR criteria of MDD for >1 month and had a 17-item Hamilton Depression Rating Scale (HDRS-17) score >22 and a Sheehan Disability Scale (SDS) score >10.3 The primary efficacy measure was change in Montgomery-Åsberg Depression Rating Scale (MADRS) score from baseline to week 10. Secondary efficacy measures included the HDRS-17, SDS, and Clinical Global Impressions-improvement scale. Efficacy analyses included 276 subjects treated with levomilnacipran and 277 treated with placebo.3
Levomilnacipran was significantly superior to placebo on the MADRS and HDRS-17 from baseline to week 10. Response and remission rates were
significantly greater for the levomilnacipran group compared with placebo. Response exceeded the 10% average advantage for drug vs placebo and 46% of levomilnacipran-treated patients achieved remission.3
The number needed to treat (NNT), based on the MADRS scores for the levomilnacipran group compared with the placebo group, was 6 for response and 5 for remission.3 By comparison, most studies of venlafaxine demonstrate a NNT of 8.3
Levomilnacipran generally was reported to be safe and well tolerated. The most common adverse events leading to discontinuation in the levomilnacipran group were nausea, vomiting, change in systolic and diastolic blood pressure, and increase in heart rate. The favorable tolerability profile of levomilnacipran may relate to the 2-fold greater potency for NE reuptake inhibition compared with 5-HT reuptake inhibition.3
The second study was an 11-week, fixed-dose trial of levomilnacipran using 40, 80, or 120 mg. A total of 724 outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD and who had an ongoing episode of depression lasting >8 weeks were randomly assigned to receive placebo (n = 179) or levomilnacipran at 40 mg (n = 181), 80 mg (n = 181), or 120 mg (n = 183) once daily for 8 weeks of double-blind treatment followed by a 2-week, double-blind taper of the drug.5 The primary efficacy parameter was change from baseline on the MADRS and the secondary efficacy parameter was change from baseline in SDS total score. HDRS-17, CGI-I, and CGI-S were included as secondary outcome measures.5
Significant difference in MADRS total score were seen in the levomilnacipran group compared with the placebo group (least mean squared difference: 40 mg/d, −3.23; 80 mg/d, −3.99; and 120 mg/d, −4.86). Higher dosages produced a numerically greater change and significant separation from placebo occurred sooner in the 80-mg and 120-mg groups compared with the 40-mg group.5
Significant differences vs placebo were consistently observed across secondary outcome measures for the higher dosages of levomilnacipran, and improvement in SDS total score was noted in all levomilnacipran groups compared with the placebo group. When dosed at 120 mg/d, levomilnacipran produced significant improvement vs placebo on all SDS subscales and was as well tolerated as the 80 mg dosage.5
No new safety concerns were observed in the study. A dose-response relationship in tolerability was not demonstrated and the number of patients reporting adverse events and who discontinued participation because of adverse events was higher in the 80-mg group than in the 40-mg and 120-mg groups.5
Tolerability
Overall, levomilnacipran was well tolerated in clinical trials, during which 2,673 subjects were exposed to the drug—translating to 942 patient-years of exposure. These patients ranged in age from 18 to 78; 825 of these subjects were enrolled in long-term studies for 1 year. Dosing of levomilnacipran during these studies ranged from 40 to 120 mg once daily, without regard to food.1
Nine percent of patients who received levomilnacipran in short-term studies discontinued because of adverse events, compared with 3% of patients who received placebo. The most common adverse event reported was nausea; other common adverse events reported included constipation, hyperhidrosis, elevated heart rate, erectile dysfunction, tachycardia, palpitations, and vomiting. Of these events, only erectile dysfunction and urinary hesitation were dose-related.1 Among levomilnacipran-treated female patients, <2% reported adverse events related to sexual dysfunction.
All SNRIs have well established associations with elevation in blood pressure and heart rate. Levomilnacipran resulted in a mean increase of 3 mm HG in systolic and 3.2 mm Hg in diastolic blood pressure in short-term, placebo-controlled trials.1
Orthostatic hypotension was observed in 11.6% of patients in the levomilnacipran groups, compared with 9.7% in placebo groups in all short-term studies. Orthostatic reductions of blood pressure occurred in 5.8%, 6.1%, and 9.8% of levomilnacipran-treated patients with dosages of 40, 80, and 120 mg/d respectively, indicating a dose-dependent adverse event. A mean increase in heart rate of 7.2 beats per minute (bpm) also was seen in short-term studies in the levomilnacipran-treated group compared with 0.3 bpm in the placebo-treated group.1 Clinicians should monitor blood pressure and heart rate routinely because of potential increases seen in some subjects in these studies, which excluded those who had significant cardiovascular disease.
Unique clinical issues
Both in-vitro and in-vivo studies found that levomilnacipran exhibited more potency for NE reuptake inhibition than for 5-HT reuptake inhibition at the lowest effective dosage (10 mg/kg). However as the dosage was increased (20 mg/kg and 40 mg/kg), it was equally potent at NE and 5-HT reuptake inhibition. This is in contrast to venlafaxine, which demonstrates a similar, but opposite, effect in terms of potentiation at the 5-HT and NE reuptake pumps.2
The greater noradrenergic effect of levomilnacipran could lend itself to treating certain subgroups of patients whose symptoms are believed to be related to deficiencies in NE (eg, lassitude).4 This concept is theoretical, and was not explicitly studied in clinical trials and the drug is not labeled in this way.
Contraindications
Contraindications to levomilnacipran are similar to those seen with SSRIs and SNRIs, including concomitant use of a monoamine oxidase inhibitor (MAOI) and the use of the levomilnacipran within 14 days of stopping an MAOI. Contraindications unique to levomilnacipran include hypersensitivity to levomilnacipran, milnacipran, or any component specific to the formulation; and uncontrolled narrow-angle glaucoma.1
Dosing
The recommended dosage range of levomilnacipran is 40 to 120 mg once
daily with or without food. The capsules should be swallowed whole and should not be opened or crushed. As with most psychotropics, levomilnacipran should be taken at approximately the same time each day.1
The manufacturer recommends an initial dose of levomilnacipran of 20 mg once daily for 2 days, increased to 40 mg once daily. Based on efficacy and tolerably, levomilnacipran can be increased in increments of 40 mg every 2 days.
Dosage adjustment is recommended for patients with moderate or severe renal impairment; and the maintenance dosage should not exceed 80 mg and 40 mg respectively in these populations. As with many antidepressants, gradual dosage reduction is recommended to avoid discontinuation symptoms.
Bottom Line
Levomilnacipran is FDA-approved for treating major depressive disorder in adults. In 2 randomized controlled trials, the drug showed a significant separation from placebo. Levomilnacipran generally was reported to be safe and well tolerated; common adverse events were nausea, vomiting, changes in blood pressure, and an increase in heart rate.
Related Resources
- Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant - what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? [published online September 8, 2013]. Int J Clin Pract. doi: 10.1111/ijcp.12298.
- Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
Drug Brand Names
Duloxetine • Cymbalta Milnacipran • Savella
Ketoconazole • Nizoral Venlafaxine • Effexor Levomilnacipran • Fetzima
Disclosures
Dr. Macaluso has been the principal investigator for clinical trials for AbbVie, Eisai, Envivo, Janssen, Naurex, and Pfizer. All clinical trial and study contracts and payments were made through the Kansas University Medical Center Research Institute. Drs. Kazanchi and Malhotra report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Fetzima [package insert]. St. Louis, MO: Forest Laboratories; 2013.
2. Auclair AL, Martel JC, Assié MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338-347.
3. Montgomery SA, Mansuy L, Ruth A, et al. Efficacy and Safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74(4):363-369.
4. Kasper S, Meshkat D, Kutzelnigg A. Improvement of the noradrenergic symptom cluster following treatment with milnacipran. Neuropsychiatric Dis Treat. 2011; 7(suppl 1):21-27.
5. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
6. Hutt AJ, Vanetová J. The chiral switch: the development of single enantiomer drugs from racemates. Acta Facultatis Pharmaceuticae Universitatis Comenianae. 2003; 50(7):23.
7. U.S. Food and Drug Administration. Development of new stereoisomeric drugs. Published May 1, 1992. http://www.fda.gov/drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122883.htm#.UKHEWm4ZyYE.email. Accessed October 8, 2013.
1. Fetzima [package insert]. St. Louis, MO: Forest Laboratories; 2013.
2. Auclair AL, Martel JC, Assié MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013;70:338-347.
3. Montgomery SA, Mansuy L, Ruth A, et al. Efficacy and Safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013;74(4):363-369.
4. Kasper S, Meshkat D, Kutzelnigg A. Improvement of the noradrenergic symptom cluster following treatment with milnacipran. Neuropsychiatric Dis Treat. 2011; 7(suppl 1):21-27.
5. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
6. Hutt AJ, Vanetová J. The chiral switch: the development of single enantiomer drugs from racemates. Acta Facultatis Pharmaceuticae Universitatis Comenianae. 2003; 50(7):23.
7. U.S. Food and Drug Administration. Development of new stereoisomeric drugs. Published May 1, 1992. http://www.fda.gov/drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122883.htm#.UKHEWm4ZyYE.email. Accessed October 8, 2013.
Long-acting injectable aripiprazole for adult schizophrenia
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
In February 2013, the FDA approved a long-acting IM aripiprazole formulation for treating adult schizophrenia (Table 1).1 It is the fourth second-generation antipsychotic (SGA) depot formulation approved for treating schizophrenia, and the sixth depot antipsychotic if haloperidol and fluphenazine decanoate are considered.
Table 1
Depot aripiprazole: Fast facts
| Brand name: Abilify Maintena |
| Class: Atypical antipsychotic |
| Indication: Adult schizophrenia |
| Approval date: February 28, 2013 |
| Availability date: March 18, 2013 |
| Manufacturer: Otsuka Pharmaceutical and Lundbeck |
| Dosing form: IM long-acting injection |
| Recommended dosage: 400 mg IM once a month; 200 to 300 mg IM if drug-drug interactions, poor cytochrome P450 2D6 metabolism, or adverse effects |
| Source: Reference 1 |
Clinical implications
Depot medications can improve treatment adherence2; however, long-term antipsychotic use can lead to irreversible adverse effects (dyskinesias), which in some cases were reduced by using newer antipsychotics.3
How it works
Similar to other SGAs, aripiprazole’s mechanism of action is unknown. Aripiprazole was developed based on the dopamine theory, in which dopamine hyperactivity in mesolimbic pathways of the brain leads to hallucinations, delusions, disorganization, and catatonia, and dopamine hypoactivity in mesocortical pathways and the prefrontal cortex causes alogia, anhedonia, autism, avolition, and problems with attention and abstract thinking.
Aripiprazole’s proposed mechanism of action on dopamine receptors is that of partial agonism,1 rather than antagonism, as is the case for other SGAs. In theory, aripiprazole antagonizes postsynaptic D2 receptors and activates presynaptic D2 autoreceptors, with subsequently decreased dopamine production and further stabilization of the dopamine system.4 Its antagonism of 5-HT2A is similar to other SGAs.5
Pharmacokinetics
After depot aripiprazole is injected into the gluteal muscle, the active moiety slowly is released into circulation. The effectiveness of depot aripiprazole is attributable to its active parent drug, aripiprazole monohydrate, and its active metabolite, dehydro-aripiprazole, which is the same as oral aripiprazole. Depot aripiprazole reaches maximum concentration in 5 to 7 days. The elimination half-life of depot aripiprazole is 29.9 days for a 300-mg dose and 46.5 days for a 400-mg dose if administered monthly.1
Aripiprazole does not undergo direct glucuronidation. It is metabolized predominantly through cytochrome P450 (CYP) 2D6 and 3A4 enzymes, which predisposes it to significant drug-drug interactions and may require dose adjustment (Table 2).1
Table 2
Dose adjustments of depot aripiprazole
| Drug-drug interaction | Adjusted dose |
|---|---|
| CYP2D6 poor metabolizers | 300 mg |
| CYP2D6 poor metabolizers taking CYP3A4 inhibitors (ketoconazole, itraconazole, erythromycin, grapefruit juice) | 200 mg |
| Lithium, valproate, desvenlafaxine, venlafaxine, escitalopram, dextromethorphan, omeprazole, warfarin | No significant interaction No dose adjustment |
| Sex, race, liver impairment, renal impairment, tobacco smokers | No dose adjustment |
| Patients taking 400 mg of depot aripiprazole with: | |
| 300 mg |
| 200 mg |
| Avoid use |
| Patients taking 300 mg of depot aripiprazole with: | |
| 200 mg |
| 160 mg |
| Avoid use |
| CYP: cytochrome P450 Source: Adapted from reference 1 | |
Efficacy
The ability of depot aripiprazole to sustain long-term symptom control in adult patients with schizophrenia was demonstrated in a randomized-withdrawal, double-blind, placebo-controlled trial.1 Adults included had a DSM-IV-TR diagnosis of schizophrenia, had ≥3-year history of the illness, had undergone treatment with ≥1 antipsychotic, and had a history of relapse or symptom exacerbation when not receiving antipsychotics. Psychopathology was measured by the Positive and Negative Syndrome Scale (PANSS), the Clinical Global Impression-Severity scale, the Clinical Global Impression-Improvement (CGI-I) scale, and the Clinical Global Impression-Severity of Suicide (CGI-SS) scale.1
The trial lasted 52 weeks, was divided into 4 phases, and concluded early because of demonstrated efficacy.
Phase I: Conversion phase switched patients from a different antipsychotic to oral aripiprazole. This phase lasted 4 to 6 weeks and included 633 patients. An additional 210 patients already receiving aripiprazole were entered directly into Phase II.
Phase II: Open-label, oral stabilization phase included 710 patients (60% males) age 18 to 60 who had a mean PANSS score 66. Patients received 10 to 20 mg/d of oral aripiprazole until they achieved stabilization, defined as PANSS score
Phase III: IM depot stabilization (uncontrolled single blind) included 576 patients. Patients were started on depot aripiprazole, 400 mg monthly, and continued to take 10 to 20 mg/d of oral aripiprazole for 14 consecutive days. Depot aripiprazole was decreased to 300 mg monthly if a patient developed adverse effects. Patients continued to the double-blind phase when stabilization was achieved, as evidenced by PANSS score
Phase IV: Maintenance (double-blind, randomized, placebo-controlled) included 403 patients. Two-thirds of patients continued to take the same dose of depot aripiprazole they took in Phase III. One-third of patients were switched to placebo. The primary efficacy endpoint was time to impending relapse, defined as the first occurrence of ≥1 criteria: hospitalization due to psychosis; violence toward self, others, or property; CGI-SS score ≥4 on part I or ≥7 on part II; or CGI-I score ≥5 and any individual PANSS score >4 for disorganization, hallucinations, suspiciousness, or abnormal thought content.1
Patients randomized to continue depot aripiprazole took longer to relapse or worsening of symptoms compared with the placebo group. Of 403 patients, 10% taking an active drug and 39.6% taking placebo relapsed within 360 days of randomization. This difference was statistically significant (P 1
Tolerability
One possible problem with any long-acting medication is increased duration of adverse effects (AEs), if they develop. Therefore, assessment of safety and tolerability is more important in depot formulations than in oral drugs. During the clinical trial, depot aripiprazole was well tolerated.6
During clinical trials, the most common AEs—insomnia (>5%), anxiety, and tremors—were mild to moderate and occurred within the first 4 weeks. Discontinuation of the medication because of AEs was low, and pain at the injection site was minimal.6 There were 2 deaths during the trial, which were unrelated to depot aripiprazole.6
Aripiprazole’s activity on the D2 receptor can cause extrapyramidal AEs. In head-to-head trials, patients taking aripiprazole had fewer extrapyramidal AEs than those taking risperidone or ziprasidone, but more than patients receiving olanzapine.7 Its moderate antagonism on α-adrenergic and histamine 1 (H1) receptors translates to low orthostatic hypotension, H1-mediated weight gain, and sedation. In clinical trials, weight gain and metabolic changes were comparable with placebo. In head-to-head trials, aripiprazole caused less weight gain and a higher incidence of increased cholesterol than olanzapine and risperidone, and less increase in blood glucose than olanzapine, but more than risperidone.8 Muscarinic 1-mediated cognitive impairment, dry mouth, constipation, urinary retention, and increased intraocular pressure were low.8 See Table 3 for aripiprazole's receptor binding profile.
Table 3
Aripiprazole’s receptor binding profile
| Affinity | Ki (nM)a | Effects associated with activity on the receptor | |
|---|---|---|---|
| D2 | High | 0.34 | Partial agonist |
| D3 | High | 0.8 | Partial agonist |
| 5-HT1A | High | 1.7 | Partial agonist |
| 5-HT2A | High | 3.4 | Antagonist |
| 5-HT2C | Moderate | 15 | Partial agonist |
| 5-HT7 | Moderate | 39 | Antagonist |
| D4 | Moderate | 44 | Partial agonist |
| α1-adrenergic | Moderate | 57 | Antagonist |
| H1 | Moderate | 61 | Antagonist |
| M1 | No appreciable activity | >1,000 | No appreciable activity |
| aKi dissociation constant: lower numbers indicate higher affinity of the compound for the receptor H1: histamine 1; M1: muscarinic 1 Source: References 1,6 | |||
Unique clinical issues
Clinical features for depot aripiprazole can be partially deduced based on data on oral aripiprazole. Advantages over other depot SGAs might include aripiprazole’s more favorable weight and metabolic profile.
Contraindications
Depot aripiprazole is contraindicated in patients with known sensitivity to aripiprazole or other components of the formulation. Because of pharmacokinetic drug-drug interactions, using depot aripiprazole should be avoided in patients taking strong CYP3A4 inducers (eg, rifampin and carbamazepine). Dose adjustment is recommended in patients who are taking moderate CYP2D6 and 3A4 inhibitors, such as paroxetine, fluoxetine, ketoconazole, or erythromycin.1 A “black-box” warning of increased mortality in older patients with dementia-related psychosis applies for depot aripiprazole as well as for other atypical antipsychotics.1
Depot aripiprazole is pregnancy category C and should be used in pregnant or breastfeeding mothers only when benefits outweigh the risks. Use of depot aripiprazole in geriatric and pediatric populations has not been studied; however, patients age ≥65 who received oral aripiprazole, 15 mg/d, showed decreased clearance by 20%.1
Dosing
Depot aripiprazole is available as a lyophilized powder that needs to be reconstituted in sterile water. The drug can be stored at room temperature. The kit includes two 21-gauge needles, a 1.5-inch needle for non-obese patients and a 2-inch needle for obese patients. Depot aripiprazole should be given to patients who demonstrate tolerability to oral aripiprazole. The starting and maintenance dose of depot aripiprazole is 400 mg injected into the gluteal muscle, once a month. If a patient develops an AE, decrease the monthly dose to 300 mg. Rotate the injection site between gluteal muscles to reduce AEs from injection.
Because of the potential for significant pharmacokinetic drug-drug interactions, dose adjustment is recommended for patients who are CYP2D6 poor metabolizers and those taking certain other medications (Table 4).1 See Table 4 for the recommended dosage adjustment in the case of missed doses.
Table 4
Adjusting depot aripiprazole after missed doses
| Doses missed since last injection | ||||
|---|---|---|---|---|
| Second or third dose | Fourth or subsequent dose | |||
| >4 weeks and | >5 weeks | >4 weeks and | >6 weeks | |
| Oral aripiprazole | Administer for 14 days | Administer for 14 days | ||
| Depot aripiprazole | Administer as soon as possible | Administer next injection | Administer as soon as possible | Administer next injection |
| Source: Reference 1 | ||||
After depot aripiprazole is injected into the gluteal muscle, the patient receives 10 to 20 mg/d of oral aripiprazole for 14 consecutive days to avoid a drop in plasma concentrations into subtherapeutic levels.
Related Resource
- Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
Drug Brand Names
- Aripiprazole • Abilify
- Aripiprazole depot • Maintena
- Carbamazepine • Tegretol
- Desvenlafaxine • Pristiq
- Dextromethorphan • Delsym
- Erythromycin • E-Mycin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Ketoconazole • Nizoral
- Lithium • Eskalith, Lithobid
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Paliperidone • Invega
- Paroxetine • Paxil
- Quinidine • Quinidex
- Rifampin • Rifadin
- Risperidone • Risperdal
- Valproate • Depakote
- Venlafaxine • Effexor
- Warfarin • Coumadin
- Ziprasidone • Geodon
Disclosure
Dr. Lincoln receives grant or research support from the Wichita Center for Graduate Medical Education.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
1. Abilify Maintena [package insert]. Tokyo, Japan: Otsuka Pharmaceutical Company; 2013.
2. Leucht C, Heres S, Kane JM, et al. Oral versus depot antipsychotic drugs for schizophrenia—a critical systematic review and meta-analysis of randomized long-term trials. Schizophr Res. 2011;127(1-3):83-92.
3. de Araújo AN, de Sena EP, de Oliveira IR, et al. Antipsychotic agents: efficacy and safety in schizophrenia. Drug Healthc Patient Saf. 2012;4:173-180.
4. Mailman RB, Murty V. Third generation antipsychotic drugs: partial agonism or receptor functional selectivity? Curr Pharm Des. 2010;16(5):488-501.
5. Roth BL, Meltzer HY. The role of serotonin in schizophrenia. http://www.acnp.org/g4/GN401000117/CH115.html. Published 2000. Accessed March 27 2013.
6. Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617-624.
7. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167-177.
8. Rummel-Kluge C, Komossa K, Schwarz S, et al. Head-to-head comparisons of metabolic side effects of second generation antipsychotics in the treatment of schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2010;123(2-3):225-233.
Inhaled loxapine for agitation
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approved by the FDA on December 21, 2012, loxapine inhalation powder is the newest agent commercialized for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults (Table 1).1,2 Loxapine is a first-generation antipsychotic that garnered newfound interest because of its potential atypical properties.3 Loxapine’s reformulation allows for direct administration to the lungs, resulting in rapid absorption into systemic circulation. This formulation offers a different method to manage agitation, for which IM formulations of other antipsychotics have been approved.4
Inhaled loxapine is delivered using a handheld device that produces a thermally-generated condensation aerosol.5,6 A single inhalation is sufficient to activate the controlled rapid heating (300 to 500°C in approximately 100 ms) of a thin layer of excipient-free loxapine on a metal substrate. Once vaporized, the medication cools down rapidly and aggregates into particles. The 1- to 3.5-micron aerosol particles of loxapine enter the respiratory track in 7
Table 1
Inhaled loxapine: Fast facts
| Brand name: Adasuve |
| Class: Dibenzoxazepine antipsychotic |
| Indication: Acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults |
| FDA approval date: December 21, 2012 |
| Availability date: Third quarter of 2013 |
| Manufacturer: Alexza Pharmaceuticals |
| Dosing forms: Single-dose inhaler, 10 mg |
| Recommended dose: 10 mg; only a single dose within a 24-hour period is recommended |
| Source: References 1,2 |
How it works
As with all antipsychotics, loxapine is an antagonist at the dopamine D2 receptor. However, loxapine also has clinically relevant serotonin-2A antagonism.3 Pharmacologic effects for loxapine and its metabolites include biogenic amine transporter inhibitor activity, alpha adrenergic blocking effects, and histaminergic and muscarinic receptor affinity.3,8
Clinical pharmacokinetics
In a phase I study of healthy volunteers, inhaled loxapine produced IV administration-type kinetics, with maximum plasma concentration achieved in approximately 2 minutes.6 Plasma exposure to loxapine was dose-proportional. Half-life for the 5- and 10-mg doses was approximately 6 hours. In these patients, exposure to loxapine’s metabolites as a percentage of exposure to the parent compound were 8.79% for 7-OH loxapine, 52.6% for 8-OH loxapine, and 3.96% for amoxapine (all produced as a result of metabolism via liver cytochrome P450 [CYP] enzymes CYP1A2, CYP2D6, and/or CYP3A46). 7-OH loxapine has a 5-fold higher affinity for the dopamine D2 receptor compared with loxapine, and may contribute to the drug’s clinical effect.6
Based on loxapine levels observed in the pharmacokinetic study,6 loxapine is not extensively metabolized in the lungs. Peak plasma concentrations immediately after inhalation are higher than for oral loxapine, but concentration of loxapine and its metabolites after the initial distribution phase is similar to that of oral loxapine.6 Loxapine and its metabolites are excreted through the kidneys.
Efficacy
Three efficacy studies were completed (Table 2)9-11; all were double-blind randomized controlled trials that compared inhaled loxapine, 5 or 10 mg, with placebo. Patients were required to be clinically agitated at baseline, with a score of ≥14 on the Positive and Negative Syndrome Scale Excited Component (PANSS-EC)—which consists of the PANSS items of tension, excitement, hostility, uncooperativeness, and poor impulse control; each item is rated from 1 (absent) to 7 (extreme)—and a score of ≥4 (moderate) on ≥1 item. Patients who were intoxicated or had a positive drug screen for psychostimulants were excluded. Lorazepam was allowed ≥2 hours after the study drug was administered. Change in the PANSS-EC was measured 10 minutes to 24 hours post-dose. The primary endpoint used to statistically test loxapine vs placebo was 2 hours post-dose.
In the initial phase II trial, loxapine 10 mg, but not 5 mg, was superior to placebo on the PANSS-EC at 2 hours.9 The authors described the 5-mg dose effect size as intermediate between placebo and the 10-mg dose, suggesting a possible dose response relationship. The 10-mg dose did separate from placebo as early as 20 minutes post-dose. The small number of patients enrolled is a limitation of this trial, but this was addressed in studies in the phase III program, which were considerably larger. For each of the 2 phase III trials—1 for patients with schizophrenia10 and the other for those with bipolar disorder (BD)11—both doses of loxapine were superior to placebo starting at 10 minutes post-dose. The number needed to treat (NNT) for response—as defined by a Clinical Global Impressions-Improvement score of much improved or very much improved—for loxapine vs placebo is included in Table 2.9-11 NNT for other outcomes, such as reduction on the PANSS-EC by at least 40% from baseline, demonstrated similar results.
12 The lower the NNT, the stronger the effect size.13 See the Box for an explanation of NNT. NNTs in the range of 3 to 5 are comparable to other agents used to treat agitation.4
When examining each individual item on the PANSS-EC in each of the phase III trials, every item improved with treatment, starting 10 to 20 minutes after dosing.14 Each item improved an average of 1 to 2 units from baseline over the first 2 hours post-dose. Moreover, inhaled loxapine appears to reduce agitation equally well in patients with higher or lower levels of agitation at baseline.
Another clinically relevant outcome is whether or not a patient required an additional dose or rescue medication within 24 hours. In the phase III schizophrenia trial,10 60.9% of patients randomized to loxapine, 10 mg, did not require an additional dose or rescue medication, compared with 54.4% and 46.1% for loxapine, 5 mg, and placebo, respectively. This yielded an NNT of 7 when comparing loxapine, 10 mg, with placebo.12 In the BD study,10 61.5%, 41.3%, and 26.7% did not require an additional dose or rescue medication within 24 hours for loxapine, 10 mg, 5 mg, and placebo, respectively. In this study, the NNT for loxapine, 10 mg, vs placebo was 3.12
In general, there appears to be a dose response for efficacy with inhaled loxapine, and therefore the FDA approved the 10-mg dose.2
Table 2
Summary of double-blind RCTs for inhaled loxapine vs inhaled placebo
| Study | Diagnosis | Loxapine | Placebo | Outcomes | Loxapine vs placebo NNT for response at 2 hoursa | ||
|---|---|---|---|---|---|---|---|
| 5 mg | 10 mg | 5 mg | 10 mg | ||||
| Allen et al, 20119 (Phase II) | Agitation associated with schizophrenia | n=45 | n=41 | n=43 | On the PANSS-EC score at 2 hours, loxapine, 10 mg, but not 5 mg, was superior to placebo. Loxapine, 10 mg, separated from placebo at 20 minutes, and control was sustained. On the CGI-I at 2 hours, both doses of loxapine were superior to placebo. Using the BARS, loxapine, 10 mg, was superior to placebo starting at 30 minutes and this effect was sustained. Dysgeusia was observed in 4% and 17% for loxapine, 5 mg and 10 mg, respectively, and 9% for placebo | 4 | 3 |
| Lesem et al, 201110 (Phase III) | Agitation associated with schizophrenia | n=116 | n=113 | n=115 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 9% and 11% for loxapine 5 mg and 10 mg, respectively, and 3% for placebo | 5 | 4 |
| Kwentus et al, 201211 (Phase III) | Agitation associated with bipolar I disorder (manic or mixed episode) | n=104 | n=105 | n=105 | On the PANSS-EC score and CGI-I at 2 hours, both doses of loxapine were superior to placebo. Loxapine separated from placebo at 10 minutes. Sustained control was observed over 24 hours. Dysgeusia was observed in 17% for either loxapine 5 mg or 10 mg, respectively, and 6% for placebo | 3 | 3 |
| aas measured by a CGI-I score of 1 or 2 BARS: Behavioral Activity Rating Scale; CGI-I: Clinical Global Impression Improvement Scale; NNT: number needed to treat; PANSS-EC: Positive and Negative Syndrome Scale Excited Component; RCTs: randomized controlled trials | |||||||
Clinical trials produce a mountain of data that can be difficult to interpret and apply to clinical practice. When reading about studies you may wonder:
- How large is the effect being measured?
- Is it clinically important?
- Are we dealing with a result that may be statistically significant but irrelevant for day-to-day patient care?
Number needed to treat (NNT) and number needed to harm (NNH)—2 tools of evidence-based medicine—can help answer these questions. NNT helps us gauge effect size—or clinical significance. It is different from knowing if a clinical trial result is statistically significant. NNT allows us to place a number on how often we can expect to encounter a difference between 2 interventions. If we see a therapeutic difference once every 100 patients (an NNT of 100), the difference between 2 treatments is not of great concern under most circumstances. But if a difference in outcome is seen once in every 5 patients being treated with 1 intervention vs another (an NNT of 5), the result likely will influence day-to-day practice.
How to calculate NNT (or NNH)
What is the NNT for an outcome for drug A vs drug B?
fA= frequency of outcome for drug A
fB= frequency of outcome for drug B
NNT = 1/[ fA - fB]
By convention, we round up the NNT to the next higher whole number.
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to encounter a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75 - 0.55 = 0.20
- NNT = 1 / 0.20 = 5.
Source: Adapted from Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3): 66-71 and Citrome L. Can you interpret confidence intervals? It’s not that difficult. Current Psychiatry. 2007;6(8):77-82
Tolerability and safety
Combined safety results from phase III trials10,11 as well as information about a phase I ECG QT interval study were presented in a poster.15 Among 524 patients receiving loxapine vs 263 receiving placebo, there were no significant differences in the likelihood of experiencing any adverse event, a nervous system adverse event, sedation, sedation or somnolence, or sedation, somnolence or dizziness, when stratified by lorazepam rescue.16 Adverse events that were more frequently encountered with both doses of loxapine (ie, 5 and 10 mg) than placebo are listed in Table 3,15 along with the number needed to harm (NNH). The most commonly encountered adverse event was dysgeusia. The NNH of 10 for dysgeusia for loxapine, 10 mg, vs placebo means that for every 10 patients receiving inhaled loxapine, 10 mg, instead of inhaled placebo, you would encounter 1 additional case of dysgeusia. This contrasts with the NNT for response of 4 and 3 for agitation associated with schizophrenia and BD, respectively. Therefore, one would encounter response more often than dysgeusia when comparing loxapine with placebo.
No important changes in the ECG QT interval after inhaled loxapine, 10 mg, were observed in a phase I study with healthy volunteers.15 Difference from placebo in change from baseline for QTc was
Additional details regarding overall safety and tolerability can be found in a previously published review.17
Table 3
Inhaled loxapine: Incidence of adverse events
| Adverse event | Placebo (n=220) | Loxapine | |||
|---|---|---|---|---|---|
| 5 mg (n=220) | 10 mg (n=218) | ||||
| Rate | Rate | NNH vs placebo | Rate | NNH vs placebo | |
| Dysgeusia | 4% | 13% | 12 | 14% | 10 |
| Sedation or somnolence | 8% | 11% | 34 | 10% | 50 |
| Oral hypoesthesia | 0% | 200 | 2% | 50 | |
| NNH: number needed to harm Source: Reference 15 | |||||
Pulmonary safety
Because this product is inhaled, additional information on pulmonary safety was gathered.18,19 Among 1,095 patients without active airways disease, 1 (0.09%) required treatment for post-treatment airway-related symptoms (bronchospasm). In the agitated patient population, the rate of airway adverse events was 0.4% of loxapine exposures among 524 patients, in which 6.7% had a history of asthma or chronic obstructive pulmonary disease (COPD). Others were likely to have some respiratory impairment because of a history of cigarette smoking, but they did not have active respiratory symptoms that required treatment because such patients were excluded from the trials.12 Phase I spirometry-based studies also were completed in healthy nonsmoking volunteers, in patients with asthma, and in patients with COPD. No clinically relevant effects were observed in healthy volunteers, but in patients with asthma or COPD a reduction in forced expiratory volume was observed. In patients with asthma, rates of bronchospasm as an adverse event were 26.9% for loxapine vs 3.8% for placebo, for a NNH of 5.12 Bronchospasm was not reported for patients with COPD receiving loxapine but was observed in 1 patient who received placebo. All airway adverse events in patients with asthma or COPD were mild or moderate. All respiratory signs or symptoms requiring treatment in the phase I asthma and COPD studies were managed with an inhaled bronchodilator.
Product labeling notes in a warning that inhaled loxapine can cause bronchospasm that has the potential to lead to respiratory distress and respiratory arrest.2 Therefore, inhaled loxapine is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the “ADASUVE REMS.” Enrolled health care facilities are required to have immediate, on-site access to equipment and personnel trained to manage acute bronchospasm, including advanced airway management (intubation and mechanical ventilation). Inhaled loxapine is contraindicated in patients with a current diagnosis or history of asthma, COPD, or other lung diseases associated with bronchospasm; acute respiratory signs or symptoms such as wheezing; current use of medications to treat airway diseases such as asthma or COPD; history of bronchospasm following inhaled loxapine treatment; or known hypersensitivity to loxapine and amoxapine.
Only a single dose within a 24-hour period is recommended. Before administration, patients should be screened for a history of pulmonary disease and examined (including chest auscultation) for respiratory abnormalities (eg, wheezing). After administration, patients require monitoring for signs and symptoms of bronchospasm at least every 15 minutes for ≥1 hour.
Related Resource
- Dinh K, Myers DJ, Glazer M, et al. In vitro aerosol characterization of Staccato(®) Loxapine. Int J Pharm. 2011; 403(1-2):101-108.
Drug Brand Names
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Loxapine inhalation powder • Adasuve
Disclosure
In the past 36 months, Dr. Citrome has engaged in collaborative research with or received consulting or speaking fees from Alexza Pharmaceuticals, Alkermes, AstraZeneca, Avanir Pharmaceuticals, Bristol-Myers Squibb, Eli Lilly and Company, EnVivo Pharmaceuticals, Forest Pharmaceuticals, Genentech, Janssen, L.P., Lundbeck, Merck, Mylan, Novartis, Noven, Otsuka, Pfizer Inc., Shire, Sunovion, and Valeant.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
1. Alexza Pharmaceuticals U.S. FDA Approves Alexza’s ADASUVE (loxapine) inhalation powder for the acute treatment of agitation associated with schizophrenia or bipolar I disorder in adults. http://nocache-phx.corporate-ir.net/phoenix.zhtml?c=196151
&p=RssLanding&cat=news&id=1769476. Published December 21, 2012. Accessed January 2, 2013.
2. ADASUVE [package insert]. Mountain View, CA: Alexza Pharmaceuticals; 2012.
3. Ereshefsky L. Pharmacologic and pharmacokinetic considerations in choosing an antipsychotic. J Clin Psychiatry. 1999;60(suppl 10):20-30.
4. Citrome L. Comparison of intramuscular ziprasidone olanzapine, or aripiprazole for agitation: a quantitative review of efficacy and safety. J Clin Psychiatry. 2007;68(12):1876-1885.
5. Noymer P, Myers D, Glazer M, et al. The staccato system: inhaler design characteristics for rapid treatment of CNS disorders. Respiratory Drug Delivery. 2010;1(1):11-20.
6. Spyker DA, Munzar P, Cassella JV. Pharmacokinetics of loxapine following inhalation of a thermally generated aerosol in healthy volunteers. J Clin Pharmacol. 2010;50(2):169-179.
7. Dinh KV, Myers DJ, Noymer PD, et al. In vitro aerosol deposition in the oropharyngeal region for Staccato Loxapine. J Aerosol Med Pulm Drug Deliv. 2010;23(4):253-260.
8. Brunton LL, Lazo JS, Parker KL. eds. Goodman & Gilman’s: the pharmacological basis of therapeutics. 11th ed. New York, NY: McGraw-Hill; 2005:472.
9. Allen MH, Feifel DA, Lesem MD, et al. Efficacy and safety of loxapine for inhalation in the treatment of agitation in patients with schizophrenia: a randomized, double-blind, placebo controlled trial. J Clin Psychiatry. 2011;72(10):1313-1321.
10. Lesem MD, Tran-Johnson TK, Riesenberg RA, et al. Rapid acute treatment of agitation in individuals with schizophrenia: multicentre, randomised, placebo-controlled study of inhaled loxapine. Br J Psychiatry. 2011;198(1):51-58.
11. Kwentus J, Riesenberg RA, Marandi M, et al. Rapid acute treatment of agitation in patients with bipolar I disorder: a multicenter, randomized, placebo-controlled clinical trial with inhaled loxapine. Bipolar Disord. 2012;14(1):31-40.
12. Citrome L. Inhaled loxapine for agitation revisited: focus on effect sizes from 2 Phase III randomised controlled trials in persons with schizophrenia or bipolar disorder. Int J Clin Pract. 2012;66(3):318-325.
13. Citrome L. Compelling or irrelevant? Using number needed to treat can help decide. Acta Psychiatr Scand. 2008;117(6):412-419.
14. Cassella J, Spyker D, Kwentus J, et al. Rapid improvement in the five-item Positive and Negative Syndrome-Excited Component (PANSS-EC) scale for agitation with inhaled loxapine. Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
15. Fishman R, Gottwald M, Cassella J. Inhaled loxapine (AZ-004) rapidly and effectively reduces agitation in patients with schizophrenia and bipolar disorder. Poster presented at: 13th annual meeting of the College of Psychiatric and Neurologic Pharmacists; April 18-21 2010; San Antonio, TX.
16. Fishman R, Spyker D, Cassella J. The safety of concomitant use of lorazepam rescue in treating agitation with inhaled loxapine (AZ-004). Poster presented at: 50th meeting of New Research Approaches for Mental Health Interventions; June 14-17, 2010; Boca Raton, FL.
17. Citrome L. Aerosolised antipsychotic assuages agitation: inhaled loxapine for agitation associated with schizophrenia or bipolar disorder. Int J Clin Pract. 2011;65(3):330-340.
18. Alexza Pharmaceuticals. Adasuve (loxapine) inhalation powder NDA 022549. Psychopharmacologic drug advisory committee briefing document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282900.pdf. Published December 12, 2011. Accessed January 2, 2013.
19. Food and Drug Administration Briefing document for NDA 022549. Psychopharmacologic Drug Advisory Committee Briefing Document. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharma
cologicDrugsAdvisoryCommittee/UCM282897.pdf. Accessed January 2, 2013.
Vilazodone for major depressive disorder
In January 2011, the FDA approved vilazodone for the treatment of major depressive disorder (MDD) (Table 1).
Vilazodone was discovered by Merck KGaA in Germany.1 In February 2001, Merck KGaA licensed vilazodone to GlaxoSmithKline. In April 2003, GlaxoSmithKline returned all rights to Merck KGaA because phase IIb clinical data did not support progression to phase III clinical trials. In September 2004, Genaissance Pharmaceuticals Inc. acquired an exclusive worldwide license from Merck KGaA to develop and commercialize vilazodone for depression treatment.2 Subsequently, Clinical Data Inc. acquired Genaissance Pharmaceuticals Inc., including vilazodone, and proceeded with 2 phase III trials and a large safety trial resulting in FDA approval. In February 2011, Forest Laboratories Inc. acquired Clinical Data Inc. and will launch vilazodone in second quarter of 2011.
Table 1
Vilazodone: Fast facts
| Brand name: Viibryd |
| Class: Serotonin reuptake inhibitor and 5-HT1A receptor partial agonist |
| Indication: Major depressive disorder |
| Approval date: January 24, 2011 |
| Availability date: Second quarter of 2011 |
| Manufacturer: Forest Laboratories Inc. |
| Dosage forms: 10 mg, 20 mg, and 40 mg tablets |
| Starting dose: 10 mg/d |
| Target dose: 40 mg/d |
How it works
Similar to all antidepressants, vilazodone’s mechanism of action is not fully understood, but is thought to be related to its inhibition of serotonin (ie, 5-HT) reuptake and partial agonism of 5-HT1A receptors.3 Vilazodone technically is not a selective serotonin reuptake inhibitor (SSRI) because it has greater affinity for the 5-HT1A receptor (0.2nM) than it does for the 5-HT reuptake pump (0.5nM).4
Vilazodone was developed based on the theory that inhibition of 5-HT1A autoreceptor inhibition was responsible for SSRIs’ delayed (approximately 2 weeks) onset of antidepressant efficacy. Briefly, this theory is as follows: In humans, 5-HT1A receptors are primarily presynaptic in the raphe nuclei and postsynaptic 5-HT1A receptors predominate in the neocortex and limbic regions of the brain.5 Presynaptically, 5-HT1A are autoreceptors, ie, serotonin stimulation of these receptors results in inhibition of firing of 5-HT neurons, while postsynaptically they may be involved in downstream serotonergic effects such as sexual function.5 SSRIs are thought to work as antidepressants by increasing 5-HT concentration in the synapse but their initial effect is to turn off 5-HT neuronal firing as a result of increased concentration of 5-HT at the presynaptic 5-HT1A autoreceptor. Subsequently, these 5-HT1A autoreceptors subsensitize such that 5-HT neuronal firing rate returns to normal. The time course for this subsensitization parallels the onset of SSRI antidepressant efficacy. For several years, efforts have been made to antagonize the 5-HT1A presynaptic autoreceptors as a means of potentially shortening SSRIs’ onset of efficacy.6-8
Pharmacokinetics
Vilazodone is absorbed in the gastrointestinal tract and reaches peak concentration at a median of 4 to 5 hours. Its bioavailability increases when taken with food such that Cmax (maximum concentration) is increased by 147% to 160%, and area under the curve is increased by 64% to 85%. Its absolute bioavailability in the presence of food is 72%.4 In systemic circulation, the drug is 96% to 99% protein-bound.3 Vilazodone is eliminated primarily through cytochrome P450 (CYP) 3A4 metabolism in the liver.3
Terminal half-life of vilazodone is 25 hours. In general, steady state is achieved in 4 to 5 times the half-life at a stable dose. However, dosing guidelines for vilazodone recommend titration over 2 weeks to achieve a target of 40 mg/d. Thus, steady state will not be achieved until the patient has been on the stable target dose for approximately 2.5 weeks.3
Efficacy
Vilazodone’ efficacy for MDD treatment was established in 2 pivotal 8-week, randomized, double-blind, placebo-controlled, but not active-controlled, trials (Table 2).9-11 Study participants were outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD. Patients were required to have a 17-item Hamilton Rating Scale for Depression (HAM-D-17) score >22 and a HAM-D-17 item 1 (depressed mood) score >2.
In the first clinical trial, 410 patients were randomly assigned to vilazodone or placebo. In the vilazodone group, patients were started on 10 mg/d for 1 week, titrated to 20 mg/d for a second week, and then 40 mg/d for the remainder of the study. At week 8, the mean change from baseline on the Montgomery-Åsberg Depression Rating Scale (MADRS), HAM-D-17, Clinical Global Impression-Improvement scale (CGI-I), Clinical Global Impression-Severity scale (CGI-S), and Hamilton Anxiety scale (HAM-A) was statistically greater with vilazodone than placebo (P <.05).9 Compared with placebo, vilazodone-treated patients showed a statistically significant (P <. 05) improvement in MADRS and HAM-D-17 scores at week 1. Approximately 12% more vilazodone-treated patients achieved response (defined as ≥50% decrease in total score at end of treatment) on the primary efficacy measure, which was MADRS (40.4% vs 28.1%, P=.007), and the 2 secondary efficacy measures, which were HAM-D-17 (44.4% vs 32.7%, P =.011) and CGI-I (48.0 vs 32.7, P =.001). Remission rates (MADRS <10) were not reported in this study, but the authors stated that there was no statistical difference in remission rates between the vilazodone and placebo groups.9
In a second trial, which featured design and titration schedule identical to that of the first study, 481 patients were randomized to vilazodone or placebo.10 At week 8, the vilazodone-treated patients had significantly greater improvement in MADRS, HAM-D-17, HAM-A, CGI-S, and CGI-I score compared with the placebo group (P <.05). Approximately 14% more patients in the vilazodone group were MADRS responders compared with placebo (44% vs 30%, P =.002). Remission rates were not statistically different between patients taking vilazodone vs placebo (27% vs 20% respectively).10 Demonstrating a statistically significant difference between a 27% vs 20% remission rate would require a much larger number of patients than were included in this study.
Table 2
Efficacy of vilazodone in phase III clinical trials
| Trial | Drug response rate | Placebo response rate | Drug-specific response rate* | NNT† | Average reduction in MADRS change (drug minus placebo) (mean) | Average reduction in HAM-D change (drug minus placebo) (mean) |
|---|---|---|---|---|---|---|
| Rickels et al9 | 40% | 28% | 12% | 100/12=8 | 12.9 to 9.6 (3.3) | 10.4 to 8.6 (1.8) |
| Khan et al10 | 44% | 30% | 14% | 100/14=7 | 13.3 to 10.8 (2.5) | 10.7 to 9.1 (1.6) |
| HAM-D: Hamilton Rating Scale for Depression; MADRS: Montgomery-Åsburg Depression Rating Scale; NNT: number needed to treat *Difference in response rate between the drug and placebo groups. This rate is what the drug added to the treatment effects seen as a result of time and clinical management provided in the trial †The number of patients who need to be treated to benefit (ie, achieve response) one additional patient compared with placebo Source: Reference 11. Table reproduced with permission from Sheldon H. Preskorn, MD | ||||||
Tolerability
Vilazodone’s safety was evaluated in 2, 177 patients (age 18 to 70) diagnosed with MDD who participated in clinical studies, including the two 8-week, randomized, doubleblind, placebo-controlled studies (N=891) and a 52-week, open-label study of 599 patients.12 Overall, 7.1% of patients who received vilazodone discontinued treatment because of an adverse reaction, compared with 3.2% of placebo-treated patients in the double-blind studies.3 Diarrhea, nausea, and headache were the most commonly reported adverse events; the incidence of headache was similar to that in the placebo group (13.2% vs 14.2%).10 These adverse events are consistent with serotonin agonism, mild to moderate intensity, and occurred mainly during the first week of treatment.3
Doses up to 80 mg/d have not been associated with clinically significant changes in ECG parameters or laboratory parameters in serum chemistry hematology and urine analysis.9,10 The drug had no effect on weight as measured by mean change from baseline.9,10
In one 8-week trial, there were no substantial differences between vilazodone and placebo in Arizona Sexual Experience Scale (ASEX) scores at treatment end for either sex.9 ASEX is a 5-item scale used to assess sexual dysfunction; a score >18 indicates clinically significant sexual dysfunction. At baseline, mean ASEX scores among men were 15.8 in the placebo group and 16.5 in the vilazodone group. Among women, the mean ASEX score was 21.2 in both groups.9 Overall sexual function for men and women was similar for vilazodone and placebo, as measured by the Changes in Sexual Function Questionnaire.10 The most commonly reported sexual adverse effect was decreased libido.10
Contraindications
Vilazodone is contraindicated for concomitant use with monoamine oxidase inhibitors (MAOIs) or within 14 days of stopping or starting an MAOI. Vilazodone is contraindicated in patients taking strong CYP3A4 inhibitors (eg, ketoconazole) because of increased vilazodone concentrations and resulting concentration-dependent adverse effects.3 Concomitant administration of strong CYP3A4 inducers (eg, rifampin) might result in a reduction in vilazodone levels leading to lack or loss of efficacy.13
As with other antidepressants, vilazodone carries a black-box warning about increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants for MDD and other psychiatric disorders.3 Vilazodone showed evidence of developmental toxicity in rats, but was not teratogenic in rats or rabbits. There are no adequate, well-controlled studies of vilazodone in pregnant women and no human data regarding vilazodone concentrations in breast milk.3 Women taking vilazodone are advised to breastfeed only if the potential benefits outweigh the risks. Vilazodone is not recommended for use in pediatric patients.3
Similar to other antidepressants, vilazodone labeling carries warnings about serotonin syndrome, seizures, abnormal bleeding, activation of mania/hypomania, and hyponatremia.4
Dosing
Vilazodone is available as 10 mg, 20 mg, and 40 mg tablets. The recommended target dose for vilazodone is 40 mg/d, with a starting dose of 10 mg/d for 7 days, followed by 20 mg/d for 7 days, then 40 mg/d.3 The drug should be taken with food, but unlike other psychotropics, the manufacturer does not recommended a specific calorie amount.3 Dose tapering is recommended when the drug is discontinued.3
Dose should be reduced to 20 mg/d when vilazodone is coadministered with strong CYP3A4 inhibitors, such as azole antifungals, macrolides, protease inhibitors, and verapamil.13 No dosage adjustment is recommended based on age, mild or moderate liver impairment, or renal impairment of any severity.3 Vilazodone has not been studied in patients with severe hepatic impairment.3
How does vilazodone compare?
Ideally, it would be good to know how vilazodone compares with other marketed antidepressants. Unfortunately, there are no published head-to-head comparison data to address this matter. The number needed to treat with vilazodone is between 7 and 8 based on the data from the 2 phase III trials (Table 2).9-11 This is comparable to other antidepressants. One phase III study showed a statistically greater reduction in depressive symptomatology in vilazodone-treated patients after 1 week, 9 but that was not replicated in the second trial.10
Related Resources
- Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
Drug Brand Names
- Ketoconazole • Nizoral
- Rifampin • Rifadin
- Verapamil • Isoptin
- Vilazodone • Viibryd
Disclosures
Drs. Kalia and Mittal report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Since January 2010, Dr. Preskorn has received grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Ipsen, Link Medicine, Pfizer Inc, Sunovion, Takeda, and Targacept. He has been a consultant to Abbott Laboratories, Allergan, Biovail, Boehringer Ingelheim, Eisai, Eli Lilly and Company, Evotec Johnson and Johnson, Labopharm, Merck, NovaDel Pharma, Orexigen, Prexa, Psylin Neurosciences Inc., and Sunovion. He is on the speakers bureau of Eisai, Pfizer Inc., and Sunovion.
1. Bartoszyk GD, Hegenbart R, Ziegler H. EMD 68843 a serotonin reuptake inhibitor with selective presynaptic 5-HT1A receptor agonistic properties. Eur J Pharmacol. 1997;322(2-3):147-153.
2. de Paulis T. Drug evaluation: vilazodone—a combined SSRI and 5-HT1A partial agonist for the treatment of depression. IDrugs 2007;10(3):193-201.
3. Viibryd [package insert]. New Haven CT: Trovis Pharmaceuticals, LLC; 2011.
4. Heinrich T, Böttcher H, Schiemann K, et al. Dual 5-HT1A agonists and 5-HT re-uptake inhibitors by combination of indole-butyl-amine and chromenonyl-piperazine structural elements in a single molecular entity. Bioorg Med Chem. 2004;12(18):4843-4852.
5. Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry. 2003;53:193-203.
6. Bielski RJ, Cunningham L, Horrigan JP, et al. Gepirone extended-release in the treatment of adult outpatients with major depressive disorder: a double-blind, randomized, placebo-controlled, parallel-group study. J Clin Psychiatry. 2008;69:571-577.
7. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25:193-201.
8. Robinson DS, Rickels K, Feighner J, et al. Clinical effects of the 5-HT1A partial agonists in depression: a composite analysis of buspirone in the treatment of depression. J Clin Psychopharmacol. 1990;10(3 suppl):67S-76S.
9. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326-333.
10. Khan A, Cutler AJ, Kajdasz DK, et al. Efficacy and tolerability of vilazodone, a dual-acting serotonergic antidepressant, in the treatment of patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting; New Orleans, LA; May 25, 2010.
11. Mittal MS, Kalia R, Preskorn SH. Vilazodone a 5-HT1A receptor partial agonist and serotonin reuptake pump inhibitor: what can be gleaned from its history and its clinical data? J Psychiatr Pract. In press.
12. Robinson D, Kajdasz D, Gallipoli S, et al. A one-year open-label study assessing the safety and tolerability of vilazodone in patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting: New Orleans, LA; May 25, 2010.
13. Preskorn SH, Flockhart D. 2010 guide to psychiatric drug interactions. Primary Psychiatry. 2009;16(12):45-74.
In January 2011, the FDA approved vilazodone for the treatment of major depressive disorder (MDD) (Table 1).
Vilazodone was discovered by Merck KGaA in Germany.1 In February 2001, Merck KGaA licensed vilazodone to GlaxoSmithKline. In April 2003, GlaxoSmithKline returned all rights to Merck KGaA because phase IIb clinical data did not support progression to phase III clinical trials. In September 2004, Genaissance Pharmaceuticals Inc. acquired an exclusive worldwide license from Merck KGaA to develop and commercialize vilazodone for depression treatment.2 Subsequently, Clinical Data Inc. acquired Genaissance Pharmaceuticals Inc., including vilazodone, and proceeded with 2 phase III trials and a large safety trial resulting in FDA approval. In February 2011, Forest Laboratories Inc. acquired Clinical Data Inc. and will launch vilazodone in second quarter of 2011.
Table 1
Vilazodone: Fast facts
| Brand name: Viibryd |
| Class: Serotonin reuptake inhibitor and 5-HT1A receptor partial agonist |
| Indication: Major depressive disorder |
| Approval date: January 24, 2011 |
| Availability date: Second quarter of 2011 |
| Manufacturer: Forest Laboratories Inc. |
| Dosage forms: 10 mg, 20 mg, and 40 mg tablets |
| Starting dose: 10 mg/d |
| Target dose: 40 mg/d |
How it works
Similar to all antidepressants, vilazodone’s mechanism of action is not fully understood, but is thought to be related to its inhibition of serotonin (ie, 5-HT) reuptake and partial agonism of 5-HT1A receptors.3 Vilazodone technically is not a selective serotonin reuptake inhibitor (SSRI) because it has greater affinity for the 5-HT1A receptor (0.2nM) than it does for the 5-HT reuptake pump (0.5nM).4
Vilazodone was developed based on the theory that inhibition of 5-HT1A autoreceptor inhibition was responsible for SSRIs’ delayed (approximately 2 weeks) onset of antidepressant efficacy. Briefly, this theory is as follows: In humans, 5-HT1A receptors are primarily presynaptic in the raphe nuclei and postsynaptic 5-HT1A receptors predominate in the neocortex and limbic regions of the brain.5 Presynaptically, 5-HT1A are autoreceptors, ie, serotonin stimulation of these receptors results in inhibition of firing of 5-HT neurons, while postsynaptically they may be involved in downstream serotonergic effects such as sexual function.5 SSRIs are thought to work as antidepressants by increasing 5-HT concentration in the synapse but their initial effect is to turn off 5-HT neuronal firing as a result of increased concentration of 5-HT at the presynaptic 5-HT1A autoreceptor. Subsequently, these 5-HT1A autoreceptors subsensitize such that 5-HT neuronal firing rate returns to normal. The time course for this subsensitization parallels the onset of SSRI antidepressant efficacy. For several years, efforts have been made to antagonize the 5-HT1A presynaptic autoreceptors as a means of potentially shortening SSRIs’ onset of efficacy.6-8
Pharmacokinetics
Vilazodone is absorbed in the gastrointestinal tract and reaches peak concentration at a median of 4 to 5 hours. Its bioavailability increases when taken with food such that Cmax (maximum concentration) is increased by 147% to 160%, and area under the curve is increased by 64% to 85%. Its absolute bioavailability in the presence of food is 72%.4 In systemic circulation, the drug is 96% to 99% protein-bound.3 Vilazodone is eliminated primarily through cytochrome P450 (CYP) 3A4 metabolism in the liver.3
Terminal half-life of vilazodone is 25 hours. In general, steady state is achieved in 4 to 5 times the half-life at a stable dose. However, dosing guidelines for vilazodone recommend titration over 2 weeks to achieve a target of 40 mg/d. Thus, steady state will not be achieved until the patient has been on the stable target dose for approximately 2.5 weeks.3
Efficacy
Vilazodone’ efficacy for MDD treatment was established in 2 pivotal 8-week, randomized, double-blind, placebo-controlled, but not active-controlled, trials (Table 2).9-11 Study participants were outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD. Patients were required to have a 17-item Hamilton Rating Scale for Depression (HAM-D-17) score >22 and a HAM-D-17 item 1 (depressed mood) score >2.
In the first clinical trial, 410 patients were randomly assigned to vilazodone or placebo. In the vilazodone group, patients were started on 10 mg/d for 1 week, titrated to 20 mg/d for a second week, and then 40 mg/d for the remainder of the study. At week 8, the mean change from baseline on the Montgomery-Åsberg Depression Rating Scale (MADRS), HAM-D-17, Clinical Global Impression-Improvement scale (CGI-I), Clinical Global Impression-Severity scale (CGI-S), and Hamilton Anxiety scale (HAM-A) was statistically greater with vilazodone than placebo (P <.05).9 Compared with placebo, vilazodone-treated patients showed a statistically significant (P <. 05) improvement in MADRS and HAM-D-17 scores at week 1. Approximately 12% more vilazodone-treated patients achieved response (defined as ≥50% decrease in total score at end of treatment) on the primary efficacy measure, which was MADRS (40.4% vs 28.1%, P=.007), and the 2 secondary efficacy measures, which were HAM-D-17 (44.4% vs 32.7%, P =.011) and CGI-I (48.0 vs 32.7, P =.001). Remission rates (MADRS <10) were not reported in this study, but the authors stated that there was no statistical difference in remission rates between the vilazodone and placebo groups.9
In a second trial, which featured design and titration schedule identical to that of the first study, 481 patients were randomized to vilazodone or placebo.10 At week 8, the vilazodone-treated patients had significantly greater improvement in MADRS, HAM-D-17, HAM-A, CGI-S, and CGI-I score compared with the placebo group (P <.05). Approximately 14% more patients in the vilazodone group were MADRS responders compared with placebo (44% vs 30%, P =.002). Remission rates were not statistically different between patients taking vilazodone vs placebo (27% vs 20% respectively).10 Demonstrating a statistically significant difference between a 27% vs 20% remission rate would require a much larger number of patients than were included in this study.
Table 2
Efficacy of vilazodone in phase III clinical trials
| Trial | Drug response rate | Placebo response rate | Drug-specific response rate* | NNT† | Average reduction in MADRS change (drug minus placebo) (mean) | Average reduction in HAM-D change (drug minus placebo) (mean) |
|---|---|---|---|---|---|---|
| Rickels et al9 | 40% | 28% | 12% | 100/12=8 | 12.9 to 9.6 (3.3) | 10.4 to 8.6 (1.8) |
| Khan et al10 | 44% | 30% | 14% | 100/14=7 | 13.3 to 10.8 (2.5) | 10.7 to 9.1 (1.6) |
| HAM-D: Hamilton Rating Scale for Depression; MADRS: Montgomery-Åsburg Depression Rating Scale; NNT: number needed to treat *Difference in response rate between the drug and placebo groups. This rate is what the drug added to the treatment effects seen as a result of time and clinical management provided in the trial †The number of patients who need to be treated to benefit (ie, achieve response) one additional patient compared with placebo Source: Reference 11. Table reproduced with permission from Sheldon H. Preskorn, MD | ||||||
Tolerability
Vilazodone’s safety was evaluated in 2, 177 patients (age 18 to 70) diagnosed with MDD who participated in clinical studies, including the two 8-week, randomized, doubleblind, placebo-controlled studies (N=891) and a 52-week, open-label study of 599 patients.12 Overall, 7.1% of patients who received vilazodone discontinued treatment because of an adverse reaction, compared with 3.2% of placebo-treated patients in the double-blind studies.3 Diarrhea, nausea, and headache were the most commonly reported adverse events; the incidence of headache was similar to that in the placebo group (13.2% vs 14.2%).10 These adverse events are consistent with serotonin agonism, mild to moderate intensity, and occurred mainly during the first week of treatment.3
Doses up to 80 mg/d have not been associated with clinically significant changes in ECG parameters or laboratory parameters in serum chemistry hematology and urine analysis.9,10 The drug had no effect on weight as measured by mean change from baseline.9,10
In one 8-week trial, there were no substantial differences between vilazodone and placebo in Arizona Sexual Experience Scale (ASEX) scores at treatment end for either sex.9 ASEX is a 5-item scale used to assess sexual dysfunction; a score >18 indicates clinically significant sexual dysfunction. At baseline, mean ASEX scores among men were 15.8 in the placebo group and 16.5 in the vilazodone group. Among women, the mean ASEX score was 21.2 in both groups.9 Overall sexual function for men and women was similar for vilazodone and placebo, as measured by the Changes in Sexual Function Questionnaire.10 The most commonly reported sexual adverse effect was decreased libido.10
Contraindications
Vilazodone is contraindicated for concomitant use with monoamine oxidase inhibitors (MAOIs) or within 14 days of stopping or starting an MAOI. Vilazodone is contraindicated in patients taking strong CYP3A4 inhibitors (eg, ketoconazole) because of increased vilazodone concentrations and resulting concentration-dependent adverse effects.3 Concomitant administration of strong CYP3A4 inducers (eg, rifampin) might result in a reduction in vilazodone levels leading to lack or loss of efficacy.13
As with other antidepressants, vilazodone carries a black-box warning about increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants for MDD and other psychiatric disorders.3 Vilazodone showed evidence of developmental toxicity in rats, but was not teratogenic in rats or rabbits. There are no adequate, well-controlled studies of vilazodone in pregnant women and no human data regarding vilazodone concentrations in breast milk.3 Women taking vilazodone are advised to breastfeed only if the potential benefits outweigh the risks. Vilazodone is not recommended for use in pediatric patients.3
Similar to other antidepressants, vilazodone labeling carries warnings about serotonin syndrome, seizures, abnormal bleeding, activation of mania/hypomania, and hyponatremia.4
Dosing
Vilazodone is available as 10 mg, 20 mg, and 40 mg tablets. The recommended target dose for vilazodone is 40 mg/d, with a starting dose of 10 mg/d for 7 days, followed by 20 mg/d for 7 days, then 40 mg/d.3 The drug should be taken with food, but unlike other psychotropics, the manufacturer does not recommended a specific calorie amount.3 Dose tapering is recommended when the drug is discontinued.3
Dose should be reduced to 20 mg/d when vilazodone is coadministered with strong CYP3A4 inhibitors, such as azole antifungals, macrolides, protease inhibitors, and verapamil.13 No dosage adjustment is recommended based on age, mild or moderate liver impairment, or renal impairment of any severity.3 Vilazodone has not been studied in patients with severe hepatic impairment.3
How does vilazodone compare?
Ideally, it would be good to know how vilazodone compares with other marketed antidepressants. Unfortunately, there are no published head-to-head comparison data to address this matter. The number needed to treat with vilazodone is between 7 and 8 based on the data from the 2 phase III trials (Table 2).9-11 This is comparable to other antidepressants. One phase III study showed a statistically greater reduction in depressive symptomatology in vilazodone-treated patients after 1 week, 9 but that was not replicated in the second trial.10
Related Resources
- Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
Drug Brand Names
- Ketoconazole • Nizoral
- Rifampin • Rifadin
- Verapamil • Isoptin
- Vilazodone • Viibryd
Disclosures
Drs. Kalia and Mittal report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Since January 2010, Dr. Preskorn has received grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Ipsen, Link Medicine, Pfizer Inc, Sunovion, Takeda, and Targacept. He has been a consultant to Abbott Laboratories, Allergan, Biovail, Boehringer Ingelheim, Eisai, Eli Lilly and Company, Evotec Johnson and Johnson, Labopharm, Merck, NovaDel Pharma, Orexigen, Prexa, Psylin Neurosciences Inc., and Sunovion. He is on the speakers bureau of Eisai, Pfizer Inc., and Sunovion.
In January 2011, the FDA approved vilazodone for the treatment of major depressive disorder (MDD) (Table 1).
Vilazodone was discovered by Merck KGaA in Germany.1 In February 2001, Merck KGaA licensed vilazodone to GlaxoSmithKline. In April 2003, GlaxoSmithKline returned all rights to Merck KGaA because phase IIb clinical data did not support progression to phase III clinical trials. In September 2004, Genaissance Pharmaceuticals Inc. acquired an exclusive worldwide license from Merck KGaA to develop and commercialize vilazodone for depression treatment.2 Subsequently, Clinical Data Inc. acquired Genaissance Pharmaceuticals Inc., including vilazodone, and proceeded with 2 phase III trials and a large safety trial resulting in FDA approval. In February 2011, Forest Laboratories Inc. acquired Clinical Data Inc. and will launch vilazodone in second quarter of 2011.
Table 1
Vilazodone: Fast facts
| Brand name: Viibryd |
| Class: Serotonin reuptake inhibitor and 5-HT1A receptor partial agonist |
| Indication: Major depressive disorder |
| Approval date: January 24, 2011 |
| Availability date: Second quarter of 2011 |
| Manufacturer: Forest Laboratories Inc. |
| Dosage forms: 10 mg, 20 mg, and 40 mg tablets |
| Starting dose: 10 mg/d |
| Target dose: 40 mg/d |
How it works
Similar to all antidepressants, vilazodone’s mechanism of action is not fully understood, but is thought to be related to its inhibition of serotonin (ie, 5-HT) reuptake and partial agonism of 5-HT1A receptors.3 Vilazodone technically is not a selective serotonin reuptake inhibitor (SSRI) because it has greater affinity for the 5-HT1A receptor (0.2nM) than it does for the 5-HT reuptake pump (0.5nM).4
Vilazodone was developed based on the theory that inhibition of 5-HT1A autoreceptor inhibition was responsible for SSRIs’ delayed (approximately 2 weeks) onset of antidepressant efficacy. Briefly, this theory is as follows: In humans, 5-HT1A receptors are primarily presynaptic in the raphe nuclei and postsynaptic 5-HT1A receptors predominate in the neocortex and limbic regions of the brain.5 Presynaptically, 5-HT1A are autoreceptors, ie, serotonin stimulation of these receptors results in inhibition of firing of 5-HT neurons, while postsynaptically they may be involved in downstream serotonergic effects such as sexual function.5 SSRIs are thought to work as antidepressants by increasing 5-HT concentration in the synapse but their initial effect is to turn off 5-HT neuronal firing as a result of increased concentration of 5-HT at the presynaptic 5-HT1A autoreceptor. Subsequently, these 5-HT1A autoreceptors subsensitize such that 5-HT neuronal firing rate returns to normal. The time course for this subsensitization parallels the onset of SSRI antidepressant efficacy. For several years, efforts have been made to antagonize the 5-HT1A presynaptic autoreceptors as a means of potentially shortening SSRIs’ onset of efficacy.6-8
Pharmacokinetics
Vilazodone is absorbed in the gastrointestinal tract and reaches peak concentration at a median of 4 to 5 hours. Its bioavailability increases when taken with food such that Cmax (maximum concentration) is increased by 147% to 160%, and area under the curve is increased by 64% to 85%. Its absolute bioavailability in the presence of food is 72%.4 In systemic circulation, the drug is 96% to 99% protein-bound.3 Vilazodone is eliminated primarily through cytochrome P450 (CYP) 3A4 metabolism in the liver.3
Terminal half-life of vilazodone is 25 hours. In general, steady state is achieved in 4 to 5 times the half-life at a stable dose. However, dosing guidelines for vilazodone recommend titration over 2 weeks to achieve a target of 40 mg/d. Thus, steady state will not be achieved until the patient has been on the stable target dose for approximately 2.5 weeks.3
Efficacy
Vilazodone’ efficacy for MDD treatment was established in 2 pivotal 8-week, randomized, double-blind, placebo-controlled, but not active-controlled, trials (Table 2).9-11 Study participants were outpatients age 18 to 65 who met DSM-IV-TR criteria for MDD. Patients were required to have a 17-item Hamilton Rating Scale for Depression (HAM-D-17) score >22 and a HAM-D-17 item 1 (depressed mood) score >2.
In the first clinical trial, 410 patients were randomly assigned to vilazodone or placebo. In the vilazodone group, patients were started on 10 mg/d for 1 week, titrated to 20 mg/d for a second week, and then 40 mg/d for the remainder of the study. At week 8, the mean change from baseline on the Montgomery-Åsberg Depression Rating Scale (MADRS), HAM-D-17, Clinical Global Impression-Improvement scale (CGI-I), Clinical Global Impression-Severity scale (CGI-S), and Hamilton Anxiety scale (HAM-A) was statistically greater with vilazodone than placebo (P <.05).9 Compared with placebo, vilazodone-treated patients showed a statistically significant (P <. 05) improvement in MADRS and HAM-D-17 scores at week 1. Approximately 12% more vilazodone-treated patients achieved response (defined as ≥50% decrease in total score at end of treatment) on the primary efficacy measure, which was MADRS (40.4% vs 28.1%, P=.007), and the 2 secondary efficacy measures, which were HAM-D-17 (44.4% vs 32.7%, P =.011) and CGI-I (48.0 vs 32.7, P =.001). Remission rates (MADRS <10) were not reported in this study, but the authors stated that there was no statistical difference in remission rates between the vilazodone and placebo groups.9
In a second trial, which featured design and titration schedule identical to that of the first study, 481 patients were randomized to vilazodone or placebo.10 At week 8, the vilazodone-treated patients had significantly greater improvement in MADRS, HAM-D-17, HAM-A, CGI-S, and CGI-I score compared with the placebo group (P <.05). Approximately 14% more patients in the vilazodone group were MADRS responders compared with placebo (44% vs 30%, P =.002). Remission rates were not statistically different between patients taking vilazodone vs placebo (27% vs 20% respectively).10 Demonstrating a statistically significant difference between a 27% vs 20% remission rate would require a much larger number of patients than were included in this study.
Table 2
Efficacy of vilazodone in phase III clinical trials
| Trial | Drug response rate | Placebo response rate | Drug-specific response rate* | NNT† | Average reduction in MADRS change (drug minus placebo) (mean) | Average reduction in HAM-D change (drug minus placebo) (mean) |
|---|---|---|---|---|---|---|
| Rickels et al9 | 40% | 28% | 12% | 100/12=8 | 12.9 to 9.6 (3.3) | 10.4 to 8.6 (1.8) |
| Khan et al10 | 44% | 30% | 14% | 100/14=7 | 13.3 to 10.8 (2.5) | 10.7 to 9.1 (1.6) |
| HAM-D: Hamilton Rating Scale for Depression; MADRS: Montgomery-Åsburg Depression Rating Scale; NNT: number needed to treat *Difference in response rate between the drug and placebo groups. This rate is what the drug added to the treatment effects seen as a result of time and clinical management provided in the trial †The number of patients who need to be treated to benefit (ie, achieve response) one additional patient compared with placebo Source: Reference 11. Table reproduced with permission from Sheldon H. Preskorn, MD | ||||||
Tolerability
Vilazodone’s safety was evaluated in 2, 177 patients (age 18 to 70) diagnosed with MDD who participated in clinical studies, including the two 8-week, randomized, doubleblind, placebo-controlled studies (N=891) and a 52-week, open-label study of 599 patients.12 Overall, 7.1% of patients who received vilazodone discontinued treatment because of an adverse reaction, compared with 3.2% of placebo-treated patients in the double-blind studies.3 Diarrhea, nausea, and headache were the most commonly reported adverse events; the incidence of headache was similar to that in the placebo group (13.2% vs 14.2%).10 These adverse events are consistent with serotonin agonism, mild to moderate intensity, and occurred mainly during the first week of treatment.3
Doses up to 80 mg/d have not been associated with clinically significant changes in ECG parameters or laboratory parameters in serum chemistry hematology and urine analysis.9,10 The drug had no effect on weight as measured by mean change from baseline.9,10
In one 8-week trial, there were no substantial differences between vilazodone and placebo in Arizona Sexual Experience Scale (ASEX) scores at treatment end for either sex.9 ASEX is a 5-item scale used to assess sexual dysfunction; a score >18 indicates clinically significant sexual dysfunction. At baseline, mean ASEX scores among men were 15.8 in the placebo group and 16.5 in the vilazodone group. Among women, the mean ASEX score was 21.2 in both groups.9 Overall sexual function for men and women was similar for vilazodone and placebo, as measured by the Changes in Sexual Function Questionnaire.10 The most commonly reported sexual adverse effect was decreased libido.10
Contraindications
Vilazodone is contraindicated for concomitant use with monoamine oxidase inhibitors (MAOIs) or within 14 days of stopping or starting an MAOI. Vilazodone is contraindicated in patients taking strong CYP3A4 inhibitors (eg, ketoconazole) because of increased vilazodone concentrations and resulting concentration-dependent adverse effects.3 Concomitant administration of strong CYP3A4 inducers (eg, rifampin) might result in a reduction in vilazodone levels leading to lack or loss of efficacy.13
As with other antidepressants, vilazodone carries a black-box warning about increased risk of suicidal thinking and behavior in children, adolescents, and young adults taking antidepressants for MDD and other psychiatric disorders.3 Vilazodone showed evidence of developmental toxicity in rats, but was not teratogenic in rats or rabbits. There are no adequate, well-controlled studies of vilazodone in pregnant women and no human data regarding vilazodone concentrations in breast milk.3 Women taking vilazodone are advised to breastfeed only if the potential benefits outweigh the risks. Vilazodone is not recommended for use in pediatric patients.3
Similar to other antidepressants, vilazodone labeling carries warnings about serotonin syndrome, seizures, abnormal bleeding, activation of mania/hypomania, and hyponatremia.4
Dosing
Vilazodone is available as 10 mg, 20 mg, and 40 mg tablets. The recommended target dose for vilazodone is 40 mg/d, with a starting dose of 10 mg/d for 7 days, followed by 20 mg/d for 7 days, then 40 mg/d.3 The drug should be taken with food, but unlike other psychotropics, the manufacturer does not recommended a specific calorie amount.3 Dose tapering is recommended when the drug is discontinued.3
Dose should be reduced to 20 mg/d when vilazodone is coadministered with strong CYP3A4 inhibitors, such as azole antifungals, macrolides, protease inhibitors, and verapamil.13 No dosage adjustment is recommended based on age, mild or moderate liver impairment, or renal impairment of any severity.3 Vilazodone has not been studied in patients with severe hepatic impairment.3
How does vilazodone compare?
Ideally, it would be good to know how vilazodone compares with other marketed antidepressants. Unfortunately, there are no published head-to-head comparison data to address this matter. The number needed to treat with vilazodone is between 7 and 8 based on the data from the 2 phase III trials (Table 2).9-11 This is comparable to other antidepressants. One phase III study showed a statistically greater reduction in depressive symptomatology in vilazodone-treated patients after 1 week, 9 but that was not replicated in the second trial.10
Related Resources
- Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
Drug Brand Names
- Ketoconazole • Nizoral
- Rifampin • Rifadin
- Verapamil • Isoptin
- Vilazodone • Viibryd
Disclosures
Drs. Kalia and Mittal report no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Since January 2010, Dr. Preskorn has received grant/research support from Abbott Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Ipsen, Link Medicine, Pfizer Inc, Sunovion, Takeda, and Targacept. He has been a consultant to Abbott Laboratories, Allergan, Biovail, Boehringer Ingelheim, Eisai, Eli Lilly and Company, Evotec Johnson and Johnson, Labopharm, Merck, NovaDel Pharma, Orexigen, Prexa, Psylin Neurosciences Inc., and Sunovion. He is on the speakers bureau of Eisai, Pfizer Inc., and Sunovion.
1. Bartoszyk GD, Hegenbart R, Ziegler H. EMD 68843 a serotonin reuptake inhibitor with selective presynaptic 5-HT1A receptor agonistic properties. Eur J Pharmacol. 1997;322(2-3):147-153.
2. de Paulis T. Drug evaluation: vilazodone—a combined SSRI and 5-HT1A partial agonist for the treatment of depression. IDrugs 2007;10(3):193-201.
3. Viibryd [package insert]. New Haven CT: Trovis Pharmaceuticals, LLC; 2011.
4. Heinrich T, Böttcher H, Schiemann K, et al. Dual 5-HT1A agonists and 5-HT re-uptake inhibitors by combination of indole-butyl-amine and chromenonyl-piperazine structural elements in a single molecular entity. Bioorg Med Chem. 2004;12(18):4843-4852.
5. Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry. 2003;53:193-203.
6. Bielski RJ, Cunningham L, Horrigan JP, et al. Gepirone extended-release in the treatment of adult outpatients with major depressive disorder: a double-blind, randomized, placebo-controlled, parallel-group study. J Clin Psychiatry. 2008;69:571-577.
7. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25:193-201.
8. Robinson DS, Rickels K, Feighner J, et al. Clinical effects of the 5-HT1A partial agonists in depression: a composite analysis of buspirone in the treatment of depression. J Clin Psychopharmacol. 1990;10(3 suppl):67S-76S.
9. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326-333.
10. Khan A, Cutler AJ, Kajdasz DK, et al. Efficacy and tolerability of vilazodone, a dual-acting serotonergic antidepressant, in the treatment of patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting; New Orleans, LA; May 25, 2010.
11. Mittal MS, Kalia R, Preskorn SH. Vilazodone a 5-HT1A receptor partial agonist and serotonin reuptake pump inhibitor: what can be gleaned from its history and its clinical data? J Psychiatr Pract. In press.
12. Robinson D, Kajdasz D, Gallipoli S, et al. A one-year open-label study assessing the safety and tolerability of vilazodone in patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting: New Orleans, LA; May 25, 2010.
13. Preskorn SH, Flockhart D. 2010 guide to psychiatric drug interactions. Primary Psychiatry. 2009;16(12):45-74.
1. Bartoszyk GD, Hegenbart R, Ziegler H. EMD 68843 a serotonin reuptake inhibitor with selective presynaptic 5-HT1A receptor agonistic properties. Eur J Pharmacol. 1997;322(2-3):147-153.
2. de Paulis T. Drug evaluation: vilazodone—a combined SSRI and 5-HT1A partial agonist for the treatment of depression. IDrugs 2007;10(3):193-201.
3. Viibryd [package insert]. New Haven CT: Trovis Pharmaceuticals, LLC; 2011.
4. Heinrich T, Böttcher H, Schiemann K, et al. Dual 5-HT1A agonists and 5-HT re-uptake inhibitors by combination of indole-butyl-amine and chromenonyl-piperazine structural elements in a single molecular entity. Bioorg Med Chem. 2004;12(18):4843-4852.
5. Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry. 2003;53:193-203.
6. Bielski RJ, Cunningham L, Horrigan JP, et al. Gepirone extended-release in the treatment of adult outpatients with major depressive disorder: a double-blind, randomized, placebo-controlled, parallel-group study. J Clin Psychiatry. 2008;69:571-577.
7. Gammans RE, Stringfellow JC, Hvizdos AJ, et al. Use of buspirone in patients with generalized anxiety disorder and coexisting depressive symptoms. A meta-analysis of eight randomized, controlled studies. Neuropsychobiology. 1992;25:193-201.
8. Robinson DS, Rickels K, Feighner J, et al. Clinical effects of the 5-HT1A partial agonists in depression: a composite analysis of buspirone in the treatment of depression. J Clin Psychopharmacol. 1990;10(3 suppl):67S-76S.
9. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326-333.
10. Khan A, Cutler AJ, Kajdasz DK, et al. Efficacy and tolerability of vilazodone, a dual-acting serotonergic antidepressant, in the treatment of patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting; New Orleans, LA; May 25, 2010.
11. Mittal MS, Kalia R, Preskorn SH. Vilazodone a 5-HT1A receptor partial agonist and serotonin reuptake pump inhibitor: what can be gleaned from its history and its clinical data? J Psychiatr Pract. In press.
12. Robinson D, Kajdasz D, Gallipoli S, et al. A one-year open-label study assessing the safety and tolerability of vilazodone in patients with MDD. Poster presented at: American Psychiatric Association Annual Meeting: New Orleans, LA; May 25, 2010.
13. Preskorn SH, Flockhart D. 2010 guide to psychiatric drug interactions. Primary Psychiatry. 2009;16(12):45-74.